
Modeling α-Synucleinopathy in Organotypic Brain Slice Culture with Preformed α-Synuclein Amyloid Fibrils
Abstract
Background:
Synucleinopathy is a group of neurodegenerative disorders characterized by neurodegeneration and accumulation of alpha-synuclein (α-syn) aggregates in various brain regions. The detailed mechanism of α-syn-caused neurotoxicity remains obscure, which is partly due to the lack of a suitable model that retains the in vivo three-dimensional cellular network and allows a convenient dissection of the neurotoxic pathways. Recent studies revealed that the pre-formed recombinant α-syn amyloid fibrils (PFFs) induce a robust accumulation of pathogenic α-syn species in cultured cells and animals.
Objective:
Our goal is to determine whether PFFs are able to induce the pathogenic α-syn accumulation and neurotoxicity in organotypic brain slice culture, an ex vivo system that retains the in vivo three-dimensional cell-cell connections.
Methods/Results:
Adding PFFs to cultured wild-type rat or mouse brain slices induced a time-dependent accumulation of pathogenic α-syn species, which was indicated by α-syn phosphorylated at serine 129 (pα-syn). The PFF-induced pα-syn was abolished in brain slices prepared from α-syn null mice, suggesting that the pα-syn is from the phosphorylation of endogenous α-syn. Human PFFs also induced pα-syn in brain slices prepared from mice expressing human α-syn on a mouse α-syn-null background. Furthermore, the synaptophysin immunoreactivity was inversely associated with pα-syn accumulation and an increase of neuronal loss was detected.
Conclusion:
PFF-treatment of brain slices is able to induce key pathological features of synucleinopathy: pα-syn accumulation and neurotoxicity. This model will be useful for investigating the neurotoxic mechanism and evaluating efficacy of therapeutic approaches.
INTRODUCTION
Alpha-synuclein (α-syn) is generally believed to be an unfolded protein that adopts various folds when it binds other partners [1, 2]. In vitro, the unfolded recombinant α-syn spontaneously forms oligomers and fibrils over time [1, 3] and in vivo, certain forms of stress and disruption of proteostasis are able to induce α-syn to aggregate [4]. In patients, the misfolded α-syn forms the distinct pathological structures called Lewy bodies or Lewy neurites, which are hallmarks for many synucleinopathies [5, 6].
Synucleinopathies include Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) [7, 8]. PD is the most common synucleinopathy characterized by nigral dopaminergic neuron degeneration and the presence of Lewy bodies and Lewy neurites [9]. The α-syn aggregates in PD are widely distributed and spread in a caudal-rostral pattern as the disease progresses [10, 11]. DLB is the second most common cause of dementia in elderly [12, 13] and α-syn aggregates in DLB are commonly detected in the cortex, hippocampus, limbic system and brainstem [12, 14]. In MSA, aggregated α-syn is characteristically detected in oligodendrocytes forming the glial cytoplasmic inclusions (GCIs), which are deposited in various brain regions including cerebellum and striatum [15]. Despite the differences in clinical manifestation and pathology, amyloid fibrillary α-syn aggregates are detected in all synucleinopathies [6] and pα-syn is commonly used as a marker for the pathogenic α-syn species [16].
Although the association of α-syn aggregation and synucleinopathies is well established, the detailed neurotoxic mechanism remains elusive, which is partly due to the lack of a suitable model that recapitulates the pathogenic changes and can be conveniently studied with modern molecular biological approaches. Recently developed PFF models are based on the prion-like property of misfolded α-syn [17] and represent a major advance in this field. In these models, PFFs are added to primary neurons [18, 19] or injected into animals [20], which is sufficient to induce a robust α-syn aggregation and neuronal loss. Because the neurotoxicity directly results from α-syn aggregation, these models are well suited for studying the neurotoxic mechanism. The limitation for the PFF/primary neuron model is the lack of the three-dimensional cellular environment and the incapability to evaluate the role of cell types other than neurons. The PFF/animal model overcomes these drawbacks, but the utilization of a whole animal makes it difficult to dissect the molecular mechanism.
The organotypic brain slice culture is a commonly used model that retains the three-dimensional brain architecture, allows relatively easy manipulations and has been used extensively in studying neurodegenerative diseases [21–39]. Using an astrocytes and hippocampal slice co-culture model, Loria et al. determined the contribution of astrocytes to the degradation and spread of α-syn [37]. Very recently, two groups reported that PFFs induce α-syn aggregation and inter-neuronal spreading in hippocampal slices [38, 39]. In these studies, the slices were analyzed relatively shortly after PFF-treatment (3 days to 2 weeks) and no neurotoxicity was reported. Here we showed that adding PFFs to organotypic cerebellar slice culture was able to induce a time-dependent pα-syn accumulation. Importantly, neurotoxicity was detected 2–4 weeks after PFF-treatment, which correlates with PFF-induced α-syn aggregation.
MATERIALS AND METHODS
Animals
Rats and mice were housed and sacrificed according to the IACUC-approved protocol at the Van Andel Institute (VAI). Wild-type C57BL/6 mice were generated by breeding in the VAI vivarium. Pregnant Sprague Dawley rats were purchased from Charles River. Three lines of genetically modified mice used in this study, α-syn knock-out mice (KO), transgenic mice overexpressing human α-syn carrying A53T mutation (A53T) [40], and Bac transgenic mice expressing human α-syn on mouse α-syn null background (OVX) [41], were all on C57BL/6 background.
Organotypic brain slice culture
Postnatal day 10–12 (P10–12) pups were euthanized. Mouse or rat brains were rapidly dissected and immersed for one minute in the ice-cold dissecting medium enriched with 95% O2–5% CO2 (4 mM KCl, 26 mM NaHCO3, 5 mM MgCl2, 1 mM CaCl, 246 mM sucrose, 10 mM D-glucose, phenol red (1/1000), filter sterilized). The level of oxygenation was indicated by the color change of phenol red. Desired brain regions were separated from the rest of the brain. The orientation of slice was chosen based on the α-syn expression pattern, the reproducibility and practicality of slice preparation. Frontal cortex was sliced coronally. Cerebellum was sagittally cut with a razor blade to obtain two halves. Dissected frontal cortex or half cerebellum was glued on the cutting surface of a vibratome (Leica VT1200) and immersed in the ice-cold dissecting medium that was continuously oxygenated with 95% O2–5% CO2. Sagittal cerebellar or coronal cortical slices (400 μm thick) were generated with the vibratome and kept in the ice-cold, oxygenated medium during the procedure.
Cerebellar slices were counted from medial (slice 1) to lateral (slice 9) (Supplementary Figure 1). Only the most medial slices (slices 1 and 2) were used for the study. After removing the glue, the half-cerebellar or cortical slices were transferred to 6-wells plates containing the Millicell CM insert membrane (Millipore) pre-balanced with the warm culture medium (50% Earle’s minimum essential medium (MEM), 25% Hank solution (HBSS), 25% inactivated horse serum, 25 mM HEPES, 6.5 mg/ml high-glucose, 1 mM L-glutamine, 1×antibiotic-antimycotic (Gibco)). Depending on the size of the slice, 2 to 4 slices were deposited in the middle of each well. The cerebellar or coronal cortical slices were incubated at 37°C (95% O2–5% CO2) for a few weeks (2 to 5 weeks). Every 2–3 days, half of the medium was replaced. Excess medium was removed to ensure that the slices were wet but directly exposed to air.
Lactate dehydrogenase activity
Medium was collected and kept at –20°C until analysis. Cell viability was assessed by the lactate dehydrogenase (LDH) calorimetric assay (Sigma-Aldrich), which was performed according to the protocol suggested by the manufacturer [42].
PFFs or α-syn monomer treatment
PFFs or α-syn monomer treatments were performed after the brain slices were cultured in vitro for 5 days. Recombinant mouse or human α-syn was purified following the protocol reported previously [43, 44]. To prepare amyloid fibrils, purified α-syn was dialyzed against phosphate-buffered saline (PBS), concentrated to 350 μM and shaken at 37°C, 1,000 rpm for 7 days. PFFs were diluted to 7 μM in sterilized PBS and sonicated at 50% power for 5 minutes (Misonix XL2020). The morphology of PFFs was imaged by a Tecnai G2 Spirit TWIN transmission electron microscopy (Supplementary Figure 2). Immediately after sonication, 7 μM (0.1 μg/ μl) PFFs were added onto the slices (15 μl/a half rat cerebellar slice; 5 μl/a half mouse cerebellar slice; 40 μl/a rat coronal cortical slice). For α-syn monomer treatment, the same amount of soluble α-syn monomer was added onto the slices. After 2 days, PFFs/PBS solution was aspirated and half of the culture medium was replaced with fresh medium. The slices were collected 2, 3, or 4 weeks after PFFs or monomer treatment as indicated.
Immunofluorescence staining
Slices were fixed in 4% paraformaldehyde (PFA)/PBS solution for 2 days at 4°C and then kept in PBS until the last time point. All slices were rotated at 37°C for 2 days in the ScaleSQ (0) solution (22.5% D(–)-sorbitol (w/v), 9.1 M urea, pH 8.4) [45] to clarify the tissue. After clarification, the slices were washed in PBS, permeabilized overnight in 0.5% Triton X-100 in PBS, blocked with 20% bovine serum albumin (BSA) in PBS for 4 h, and stained with indicated primary and secondary antibodies in the staining solution (2% normal goat serum and 4% BSA in PBS).
The primary antibodies used in this study were antibodies against synaptophysin (Sigma, 1:500), pα-syn (Abcam, EP1536Y 1:1000), Iba1 (Wako, 1:2000), NeuN (Millipore, 1:200), MAP2 (Sigma, 1:500), GFAP (Dako, 1:1000), and synapsin (Chemicon, 1:500). After overnight incubation with primary antibody at 4°C, slices were washed 3 times in PBS and incubated with Alexa 594-conjugated goat anti-mouse IgG and/or Alexa 488-conjugated goat anti-rabbit IgG (ThermoFisher, 1:500) for 2 h at 4°C. After the incubation, slices were washed 3 times in PBS. DAPI (1:10,000) was included in the washing buffer during second wash. Slices were mounted onto glass microscope slide with Prolong Diamond Antifade Mountant (ThermoFisher), coverslipped and dried at room temperature. The stained slices were kept at 4°C before analyses with epifluorescence (Olympus BX63 microscope) and confocal microscopes (Nikon A1plus-RSi scanning confocal microscope).
Immunohistochemistry (IHC) staining
Slices were fixed for 2 days in 4% PFA at 4°C, followed by one week in 30% sucrose at 4°C, embedded in O.C.T. (Optimal Cutting Temperature) and frozen at –20°C. The frozen thick slices were sectioned with a cryostat to obtain slices with a thickness of 40 μm. Then the slices were incubated with an anti-NeuN antibody and processed with the standard IHC staining procedure.
Biochemical analyses
Slices were collected at 2, 3, or 4 weeks after the indicated treatment and kept at –20°C. For protein extraction, frozen slices were weighed, homogenized with plastic pestles in 10% w/v TBS (Tris-buffered saline) with cOmplete™ EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich) and phosphatase inhibitors (30 mM NaF, 30 mM β-glycerophosphate, 0.2 mM Na3VO4, 5 mM Na-pyrophosphate, and 30 nM okadaic acid) [46], and incubated on ice for 15 min. Afterwards, samples were sonicated for 10 s (Branson Digital 450; Amplitude 10%) to obtain total homogenates. The protein concentrations were determined with the Bradford Assay (Bio-Rad) and samples were normalized according their protein concentrations. To determine the solubility of proteins, normalized samples were centrifuged at 110,000 g for 1 h at 4°C. Supernatants were collected as soluble fractions and stored at –20°C. Each pellet was resuspended in 50 μl of TBS containing 5% SDS and 8 M urea, centrifuged at 110,000 g for 1 h at 25°C, and the supernatant was collected as the insoluble fraction [46]. For western blots, samples were separated by 12% SDS-PAGE and transferred overnight to a nitrocellulose membrane. The membrane was incubated in TBST (10% Tween 20 in TBS) containing 4% PFA and 0.01% glutaraldehyde for 30 min at room temperature, and then washed 3 times with TBST for 10 min each. The membrane was blocked in 5% milk in TBST for 1 h at room temperature, and then incubated overnight at 4°C with indicated primary antibody in 2.5% milk in TBST. After 3 washes in 5% milk in TBST, the membrane was incubated with a secondary antibody in 5% milk in TBST for 1 hour at room temperature. After 3 washes in TBST for 10 min each, the membrane was incubated with enhanced chemiluminescence reagent (Pierce) and the signal was detected with luminescent image analyzer (LAS-3000, Fujifilm).
Antibodies used for western blots were antibodies against α-syn (BD, mouse 1:1000), pα-syn (Abcam, EP1536Y 1:1000), β-actin (Sigma, 1:50,000), and HRP-conjugated goat anti-mouse or rabbit IgG (H + L) (Bio-Rad).
Quantification of pα-Syn
The signal of pα-syn was normalized against the β-actin. Quantification of pα-syn/actin was quantified with the ImageJ software: (pα-syn - background)/(Actin - background), n = 4–7 independent brain slice cultures.
Quantification of synaptophysin staining
The images of the punctate synaptophysin staining were acquired by confocal microscopy with a 20× objective (Emission wavelength = 595 nm, Excitation wavelength = 561 nm; Image resolution = 1024×1024; Pinhole size = 24.27 μm). The synaptophysin signals were quantified with the IMARIS software through z-stacks of images (Z-step = 1.50 μm). We selected the same cerebellar areas containing cerebellar lobules IX-X. The punctate synaptophysin signals were analyzed through the “mean” with the diameter of the spots fixed at 4 and a selected area of 200×200 on the whole z-stacks. The quantification included 3 –4 slices for each condition and 3 selected areas per slice.
Quantification of NeuN staining
Immunofluorescence images were acquired by confocal microscopy with a 10× objective (Emission wavelength = 595 nm, Excitation wavelength = 561 nm; Image resolution = 1024×1024; Pinhole size = 29.37 μm). The density of NeuN staining on the whole slice was quantified with the ImageJ software (3–4 slices for each condition).
Statistical analyses
Statistical analyses were performed with GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
RESULTS
Establishing the organotypic cerebellar slice culture
Because cerebellar slice culture has a long in vitro viability [25, 29, 30] and α-syn aggregation is known to occur in the cerebella of PD, DLB, and MSA patients [47, 48], we chose cerebellar slice to study PFF-induced effects. P10 pups were sacrificed to prepare 400 μm-thick cerebellar slices. LDH activity in the medium was low (Supplementary Figure 3A, B) and stable from 2 h to 11 days in culture, indicating no increase of cell death during the culture. Immunofluorescence staining revealed the presence of various brain cell types in cultured slices, including neurons (NeuN staining), microglia (Iba1 staining), astrocytes (GFAP staining), and specific neuronal structures including dendrites (MAP2 staining) and synapses (synaptophysin and synapsin staining) (Supplementary Figure 3C).
The expression of α-syn is known to be uneven in the cerebellum [49]. We performed immunoblot analysis to determine the level of α-syn in each cerebellar slice and observed a gradient of α-syn expression, with highest levels in the medial slices (Supplementary Figure 1). To ensure that the results are comparable, in all our studies, we only used the 2 most medial slices (slice 1 and 2 in Supplementary Figure 1), which express the highest levels of α-syn.
PFFs induce pα-syn accumulation in rat brain slices
Rat cerebellar slices were cultured for 5 days and then mouse PFFs or equivalent amount of mouse α-syn monomer were added onto the slices. Untreated cerebellar slices were used as another set of control. After treatment, slices were cultured with the standard protocol and collected at the end of 4 weeks for analyses (Fig. 1A).
Fig. 1
Exogenous PFFs induce pα-syn accumulation in cerebellar slice culture. A) Timeline for the experiment. B) Rat cerebellar slices were treated with PFFs, α-syn monomer (M) or untreated (C). Four weeks after the treatment, slices were lysed and the presence of pα-syn, total α-syn and β-actin were detected by immunoblot analyses. C) Representative epifluorescence (top panel) and confocal images (bottom panel) of pα-syn in slices that were untreated, α-syn monomer or PFF-treated for 4 weeks. Scale bars represent 400 μm (top panel) and 100 μm (bottom panel). D) A representative high magnification image of pα-syn in PFF-treated slice. Scale bar represents 20 μm. E) The schematic of brain slices prepared from pre-frontal cortex (PFC) is on the left. The mouse PFC slices were treated with PFFs, α-syn monomer (M) or no treatment (C) for 4 weeks and the accumulation of pα-syn was detected by immunoblot analysis. Equal loading was verified by Coomassie Blue stain of the blot.
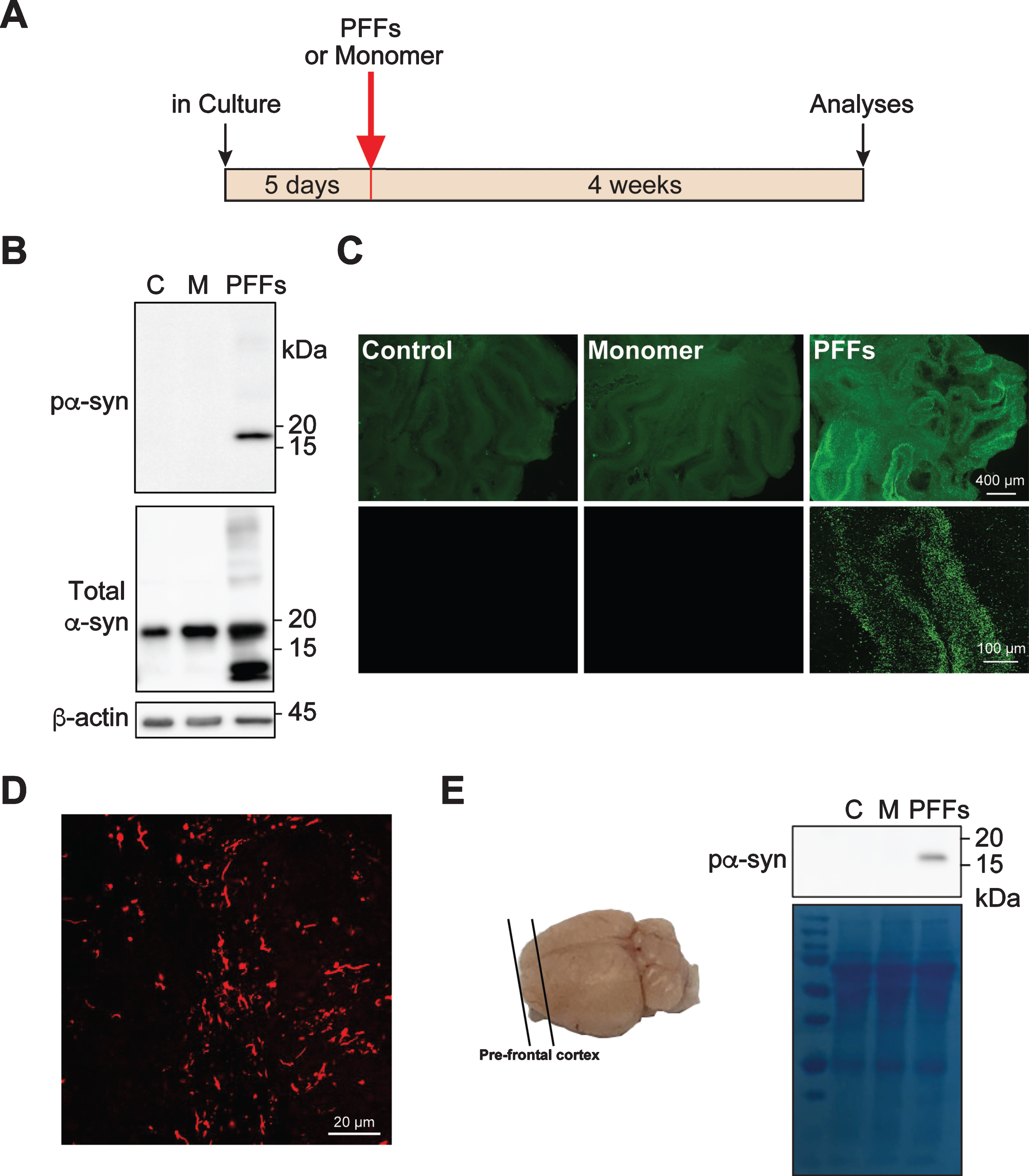
We detected a significant amount of pα-syn in slices treated with PFFs, but not in α-syn monomer treated or untreated control slices (Fig. 1B). Using antibody against total α-syn, we did not detect a significant increase of the full-length α-syn in slices treated with PFFs or α-syn monomer. Only in PFF-treated slices, truncated α-syn was detected (Fig. 1B) and was likely derived from PFFs. Immunofluorescence staining revealed that the pα-syn was unevenly distributed in the cerebellar slice and the majority was in the granular layer (Fig. 1C, D). Our results revealed that PFFs are able to induce pα-syn accumulation in rat cerebellar slice culture.
The PFF-treatment was also tested in slices prepared from pre-frontal cortical region of mouse brains (Fig. 1E). Four weeks after PFF-treatment, pα-syn was detected in PFF-treated slices, but not in untreated or monomer treated control slices. Thus, the PFF-induced pα-syn accumulation is not limited to slices prepared from cerebellum.
Time-dependent pα-syn accumulation in PFF-treated rat cerebellar slices
According to the prion-like seeding theory [17], the PFF-induced pα-syn accumulation is expected to be a time-dependent process. To test this possibility, we collected slices 2, 3, and 4 weeks after the treatments. As expected, no pα-syn was detected in either untreated or α-syn monomer treated control slices (Fig. 2A, B). The pα-syn was detected in slices collected 2 weeks after PFF-treatment and its signal increased over time. Immunofluorescence staining confirmed the gradual increase of pα-syn (Fig. 2C).
Fig. 2
Time-dependent accumulation of pα-syn in PFF-treated cerebellar slices. A) Rat cerebellar slices were treated with PFFs, α-syn monomer (M) or untreated (C) for 2, 3, or 4 weeks as indicated. The presence of pα-syn, total α-syn and β-actin were detected by immunoblot analyses. B) Quantification of the results from immunoblot analyses. Error bar represents standard deviation. Statistical analysis was performed by two-way ANOVA with Tukey post hoc test (n = 4–7 independent cultures). ****p < 0.0001, ***p < 0.001, **p < 0.01. C) Immunofluorescence staining of pα-syn in slices treated with PFFs, α-syn monomer or untreated for 2, 3, or 4 weeks as indicated. Scale bar represents 600 μm. D) Cerebellar slices were untreated or treated with α-syn monomer or PFFs for 2, 3, and 4 weeks as indicated. Lysates were centrifuged at 110,000 g for 1 h at 4°C to separate soluble and insoluble fractions. Total α-syn and pα-syn were determined by immunoblot analyses.
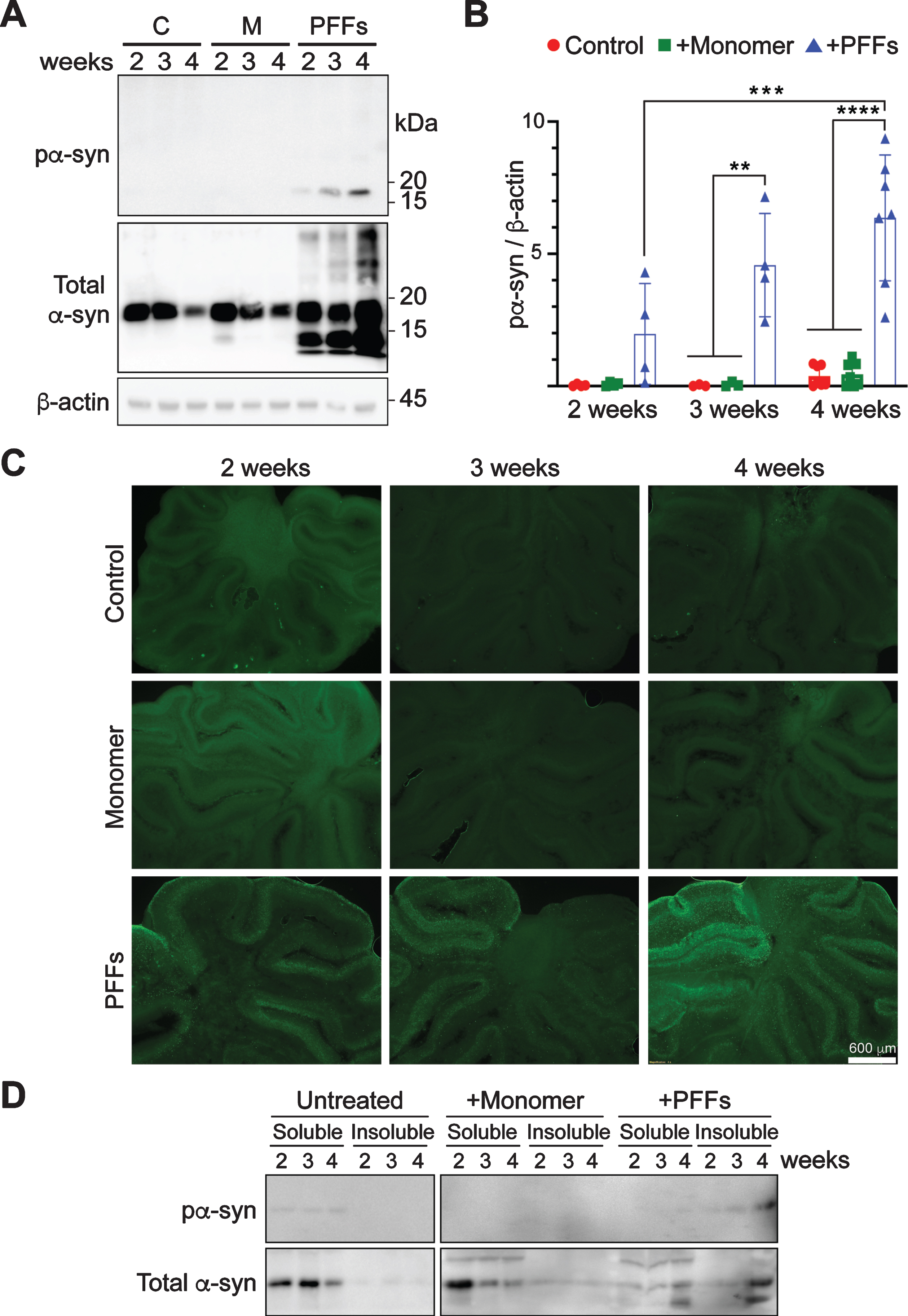
Solubility of α-syn in these slices was also determined (Fig. 2D). As expected, almost all endogenous α-syn in untreated slices was soluble. In α-syn monomer-treated slices, the majority of α-syn was soluble as well and no pα-syn was detected. In PFF-treated slices, a significant portion of α-syn appeared in the insoluble fraction and almost all pα-syn was in the insoluble fraction.
These results led us to conclude that mouse PFFs induce pα-syn to accumulate in a time-dependent manner, which is consistent with the interpretation that PFFs seeded the endogenous α-syn to misfold, resulting in a gradual increase of pα-syn.
PFF-induced pα-syn accumulation in cerebellar slices prepared from genetically engineered mice
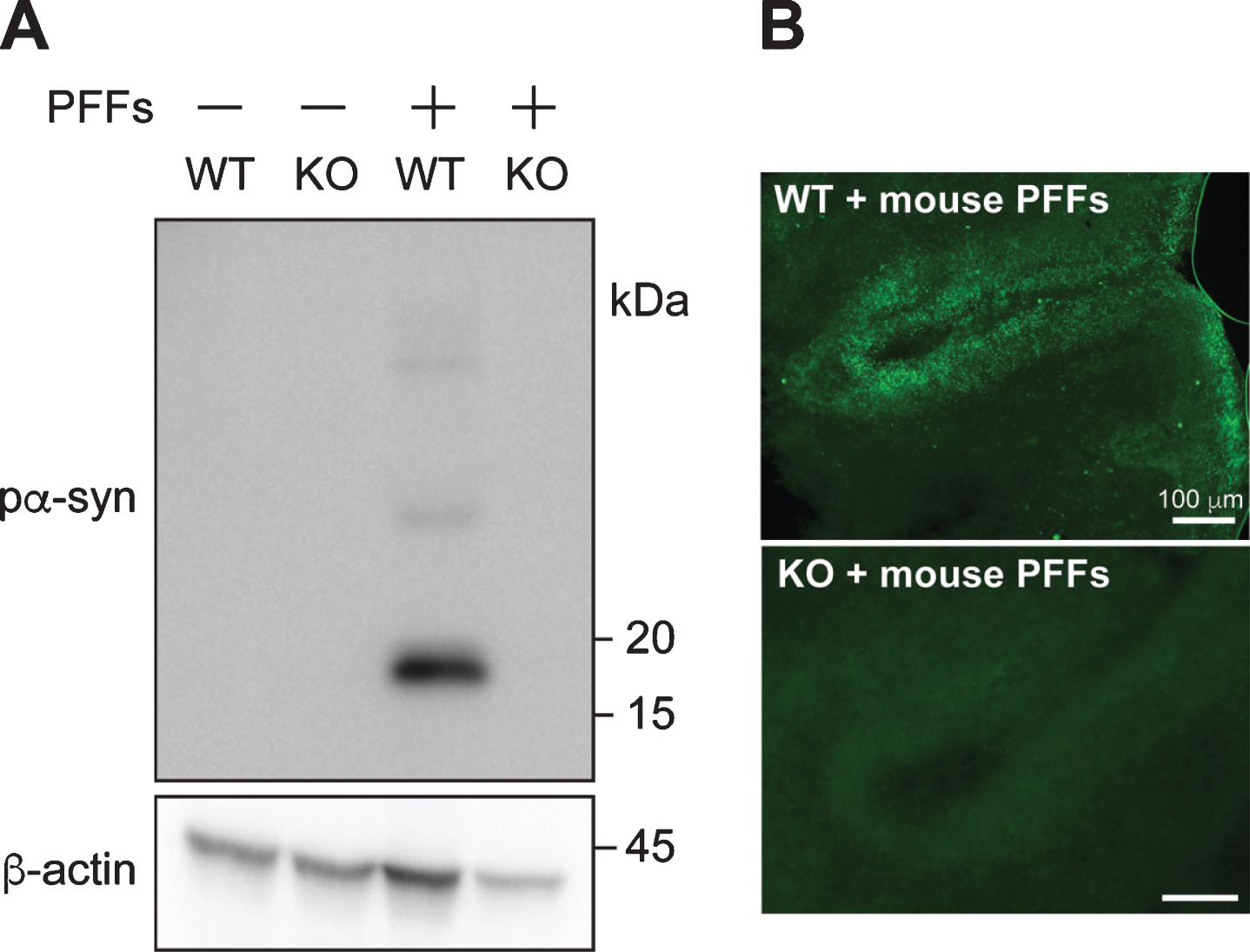
To determine whether PFF-induced pα-syn is indeed derived from PFF-seeded conversion of endogenous α-syn, we prepared cerebellar slices from α-syn knockout or wild-type mouse pups. Without PFF-treatment, no pα-syn was detected in slices prepared from either wild-type or α-syn-null pups (Fig. 3). After PFF-treatment, a significant amount of pα-syn was detected in wild-type mouse brain slices, but not in the slices prepared from α-syn-null mice. Together with the observation that similar amount of α-syn monomer did not induce pα-syn in α-syn-null slices (Supplementary Figure 4), these results indicated that the PFF-treatment induced endogenous α-syn to misfold and become phosphorylated at serine 129 residue.
Fig. 3
PFF-induced pα-syn is derived from endogenous α-syn. A) Cerebellar slices from wild-type (WT) or α-syn knock-out (KO) mice were treated with or without PFFs for 4 weeks. The presence of pα-syn and β-actin were detected by immunoblot analyses. B) Immunofluorescence staining of pα-syn in wild-type (WT) or α-syn knock-out (KO) cerebellar slices treated with mouse PFFs as indicated. Scale bar represents 100 μm.
Next, we tested whether human PFFs were able to induce pα-syn in cerebellar slices derived from genetically engineered mice that express human α-syn. We chose two mouse lines: the transgenic mice overexpressing human α-syn carrying A53T mutation (A53T mice) [40] and the BAC transgenic mice expressing human wild-type α-syn (OVX mice). In OVX mice, the human α-syn expression is under the control of endogenous human α-syn promoter, at 2 fold of the endogenous mouse α-syn level and on the mouse α-syn-null background [41].
In cerebellar slices prepared from A53T mice, a background level of pα-syn was detected in untreated slices (Fig. 4A), which was likely due to the overexpression of the aggregation-prone A53T mutant α-syn [40]. When A53T mouse cerebellar slices were treated with human PFFs, a significant increase of pα-syn was detected (Fig. 4A), consistent with the idea that PFFs seeded additional α-syn to misfold and become phosphorylated at serine 129.
Fig. 4
Human α-syn PFFs induce pα-syn accumulation in cerebellar slices prepared from transgenic mice expressing human α-syn transgene. Cerebellar slices were prepared from transgenic mice expressing human α-syn carrying A53T mutation (A) or wild-type α-syn (B). Slices were treated with human α-syn PFFs (PFFs), human α-syn monomer (M) or untreated (–). Four weeks after the treatment, the presence of pα-syn in these slices was determined by immunoblot analysis. C) Immunofluorescence staining of pα-syn in cerebellar slices prepared from transgenic mice overexpressing wild-type α-syn (OVX) or α-syn knock-out mice (KO) that received human α-syn PFFs or monomer treatment as indicated. Scale bar represents 100 μm.
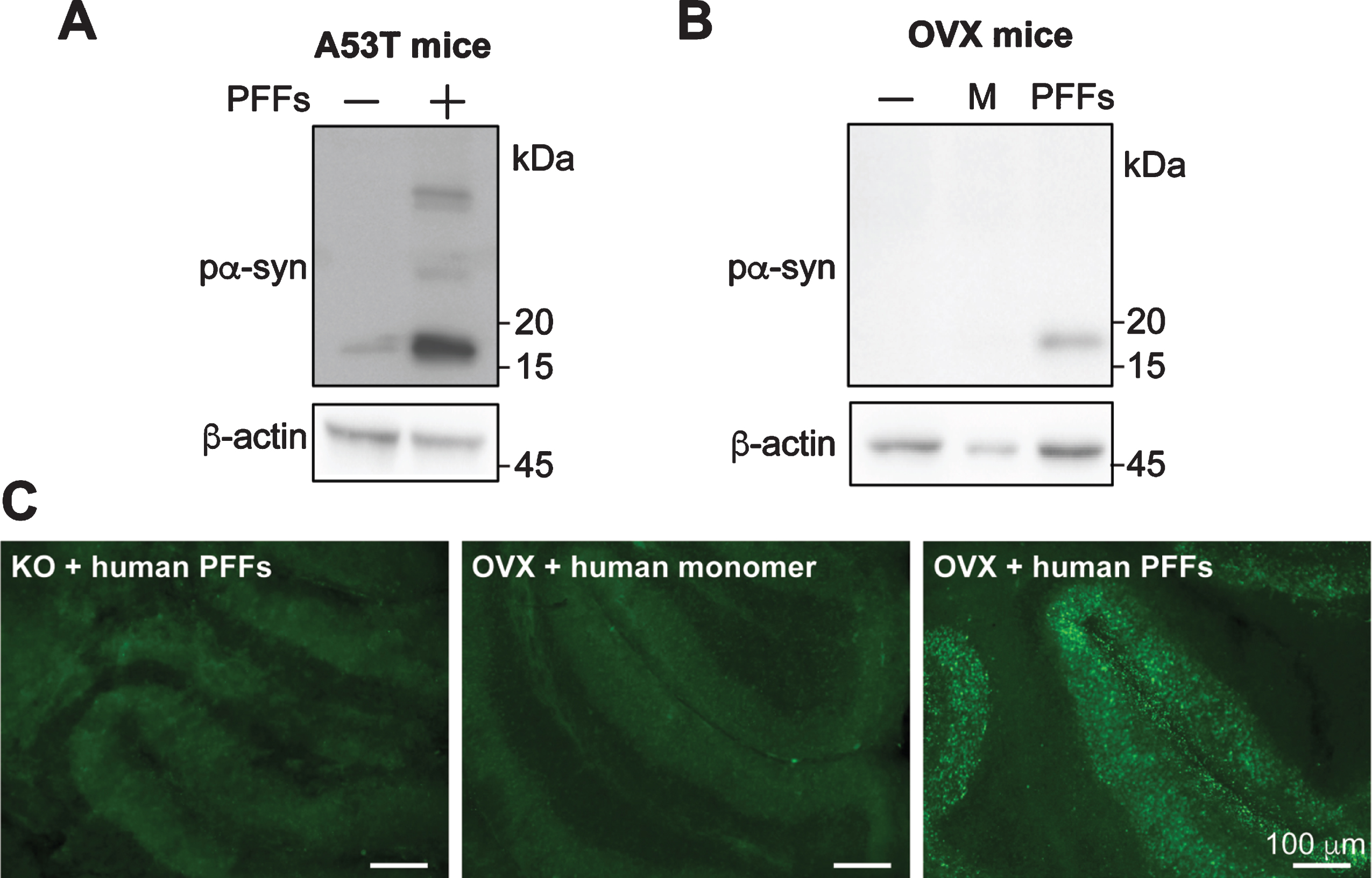
For slices prepared from OVX mice, there was much less background pα-syn signal (Fig. 4B). Human PFF-induced pα-syn accumulation was detected by both western blot (Fig. 4B) and immunofluorescence (Fig. 4C). Together, our results supported that human PFFs are able to induce pα-syn accumulation in cerebellar slices prepared from genetically engineered mice expressing human α-syn.
The PFF-induced pα-syn accumulation is accompanied with neurotoxicity
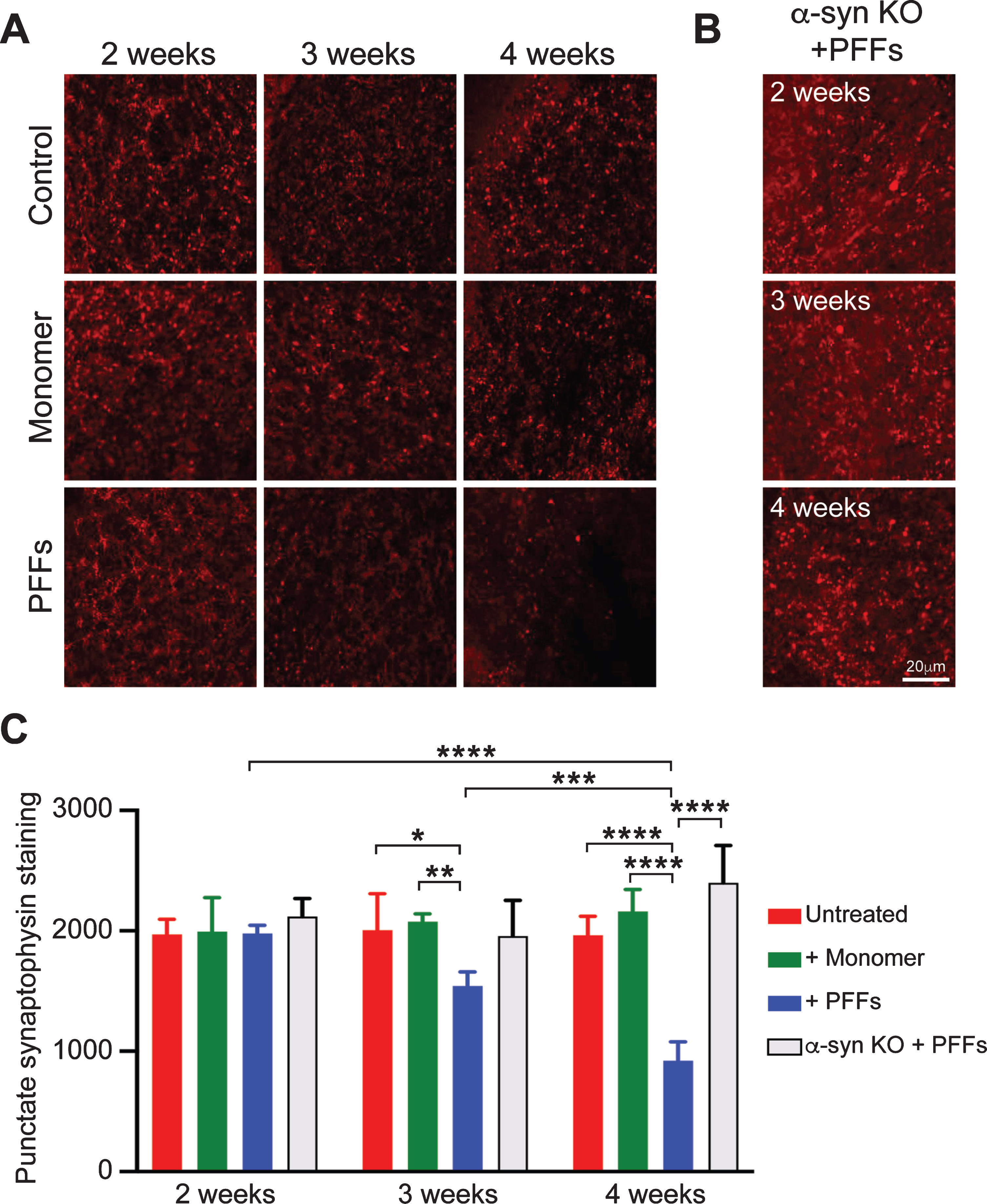
To determine whether PFF-induced pα-syn accumulation is accompanied with neurotoxicity, we performed immunofluorescence staining of synaptophysin, a marker of the synaptic network that has been used to demonstrate neurotoxicity in various neurodegeneration models [50–52]. At 3 and 4 weeks after the treatment with PFFs or α-syn monomer, the punctate synaptophysin staining showed a time-dependent reduction in PFF-treated slices, but not in untreated or α-syn monomer treated slices (Fig. 5A, 5C and Supplementary Figure 5). In slices prepared from α-syn-null mice, however, the PFF treatment did not cause an obvious loss of synaptophysin staining (Fig. 5B), suggesting that the reduction in synaptophysin signal was due to PFF-induced endogenous α-syn aggregation, but not due to the exposure to exogenously added PFFs. Quantification of the synaptophysin signal was performed with the Imaris Software. A similar amount of synaptophysin staining was observed among untreated and α-syn monomer-treated slices at 2, 3, or 4 weeks after the treatment (Fig. 5C), which indicated that the neuronal structure was stable during the experiment. For PFF-treated slices, the synaptophysin staining was at a similar level of untreated or monomer treated controls at 2 weeks, but significantly reduced at 3 and 4 weeks after PFF-treatment. Notably, in slices prepared from α-syn-null mice, the synaptophysin staining remained constant 4 weeks after PFF-treatment (Fig. 5B, C). These results suggested that the synaptoxicity was associated with PFF-induced misfolding of endogenous α-syn.
Fig. 5
Time-dependent reduction in synaptophysin immunoreactivity in PFF-treated slices. A) Cerebellar slices from wild-type mice received no treatment (Control), α-syn monomer (Monomer) or PFF-treatment for 2, 3, or 4 weeks as indicated. Synaptophysin was stained by immunofluorescence. B) Immunofluorescence staining of synaptophysin was performed on cerebellar slices prepared from α-syn-null mice received PFF-treatment for 2, 3, or 4 weeks as indicated. C) Quantification of synaptophysin staining. Error bar represents standard deviation. Images were captured by a Nikon A1plus-RSi scanning confocal microscope. Statistical analysis was performed by two-way ANOVA with Tukey post hoc test (n = 5 independent cultures). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. Scale bar represents 20 μm.
Immunofluorescence staining with an anti-NeuN antibody was performed to determine whether the PFF-induced α-syn aggregation resulted in neuronal loss. Indeed, although the PFF-treated slices at 2 or 3 weeks did not show a significant reduction in NeuN stain, at 4 weeks, the PFF-treated slice displayed a significant decrease in NeuN staining (Fig. 6). Similar result was obtained by IHC staining of brain slices with an anti-NeuN antibody (Supplementary Figure 6).
Fig. 6
Time-dependent reduction in NeuN immunoreactivity in PFF-treated slices. A) Wild-type mouse cerebellar slices received no treatment (Control) or PFF-treatment for 2, 3 or 4 weeks as indicated. The immunofluorescence staining of NeuN was imaged by a Nikon A1plus-RSi scanning confocal microscope. B) Quantification of NeuN staining. Error bar represents standard deviation. Statistical analysis was performed by two-way ANOVA with Tukey post hoc test (n = 3 independent cultures). ****p < 0.0001. Scale bar represents 200 μm.
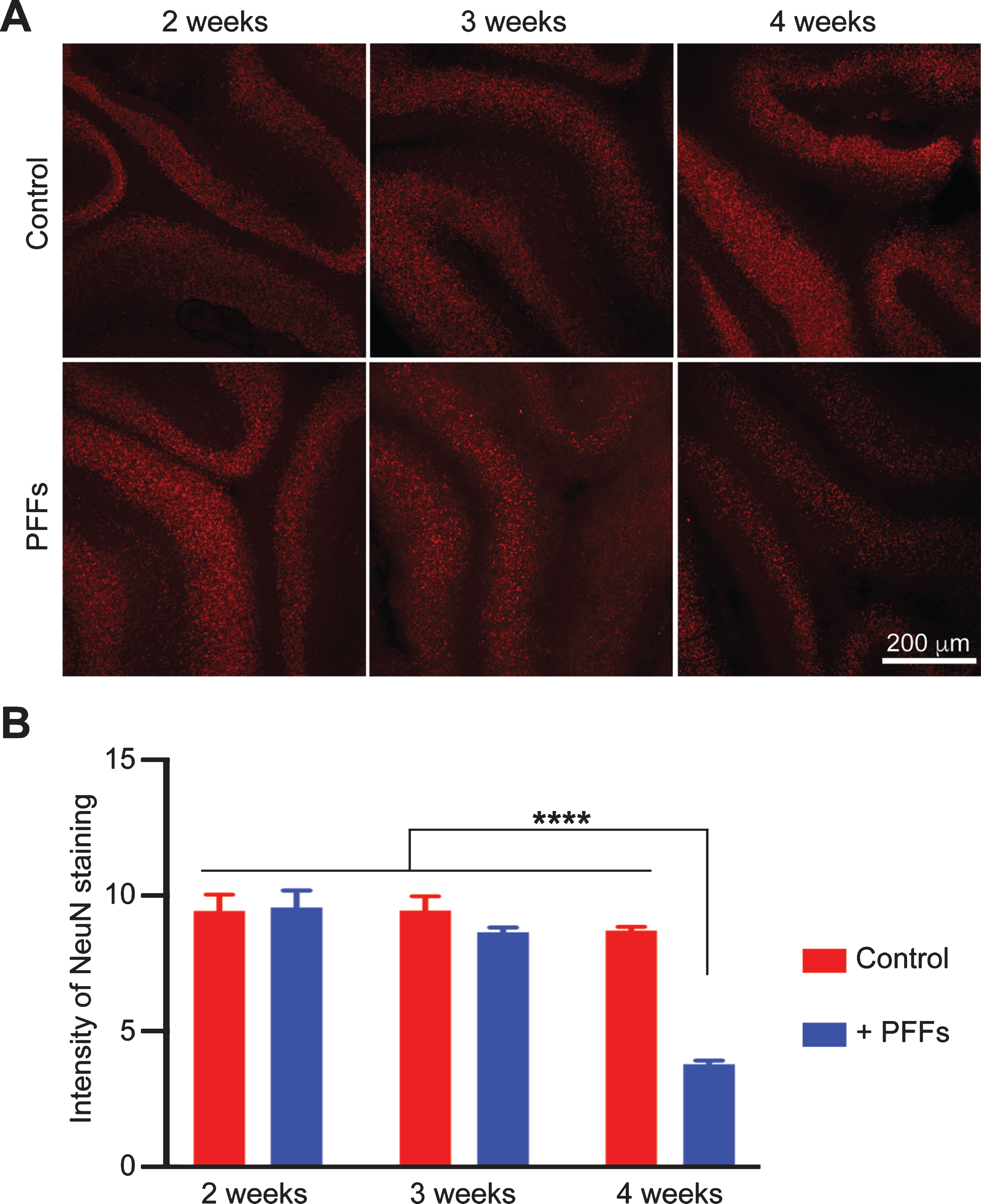
Together, our results suggested that PFF-treatment caused neurotoxicity in cerebellar slices. The toxicity appeared to be time-dependent and occurred at 3–4 weeks after PFF-treatment.
DISCUSSION
Our study established an organotypic brain slice culture model that displays the key features of synucleinopathies, α-syn aggregation and neurotoxicity. The synucleinopathy in our model is induced by addition of exogenously prepared PFFs, which is consistent with the prion-like seeding theory and indicates that the aggregated α-syn is able to seed the endogenous α-syn in a manner similar to prions [17]. The in vivo like three-dimensional environment of the brain slice culture, the time-dependent α-syn aggregation and neurotoxicity, and the relative ease for manipulation are the advantages of this model.
In this model, the accumulation of pα-syn was first detected at 2 weeks after PFF-treatment, while a significant decrease of synaptophysin stain was at 3 weeks after PFF-treatment and the loss of NeuN stain was at 4 weeks. Together with the observation that PFFs failed to induce a reduction in synaptophysin stain in slices prepared from α-syn-null mice, the timeline of these PFF-induced events suggests sequential changes in these neurons that starts with α-syn. That is, the PFF-induced endogenous α-syn misfolding occurs first, then the accumulation of pα-syn results in the disruption of synaptic structures, and the last event is the loss of cell body. The sequence of these changes fits the “dying-back degeneration” theory of PD [53], which is supported by observations from decades of post-mortem and neuroimaging studies [54]. Notably, using the PFF/primary neuron model, Volpicelli-Daley et al. also showed that PFF-induced α-syn aggregation was initiated at the synaptic terminal and gradually spread to cell body; the reduction of synaptic proteins proceeds neuronal death [18, 55]. Thus, PFF-induced changes are similar in primary neurons and brain slice models, which are similar to the pathogenic changes in synucleinopathy patients.
The pathogenesis of synucleinopathies is known to involve different cell types. In MSA, misfolded α-syn accumulates in oligodendrocytes [15]. In other synucleinopathies, although α-syn aggregates are mainly accumulated in neurons, the involvement of other cell types is well documented [56], including microglia [57, 58], astrocytes [59, 60], infiltrated monocytes and macrophages [61, 62], T cells [63–65], etc. Although the peripheral immune cells were not examined in this study, our results revealed that brain resident cell types are present in brain slices, which offers us an opportunity to dissect the contribution of various cell types to the pathogenic process.
The involvement of various types of brain cells can also be modeled in the co-culture system. Indeed, using the astrocytes/primary neuron co-culture system, Loria et al. showed that astrocytes plays a role in degradation rather than spreading of misfolded α-syn, and they confirmed their finding with an astrocytes/hippocampal slice co-culture system [37]. The advantage of our PFF/brain slice model, at least theoretically, is the presence of all brain cell types with native three-dimensional connections, which creates an “in vivo-like” environment and allows it to recapitulate the pathogenic process. This point is exemplified by the brain slice model of prion disease [30, 31]. Although many cultured cell lines can be infected with prion, none of them exhibit toxicity. In contrast, neurotoxicity associated with prion-infection is faithfully recapitulated in the organotypic brain slice culture [31], which allowed the identification of the neurotoxic pathways and testing for effective therapies [32, 33, 66].
Our results are consistent with the prion-like spreading theory of α-syn aggregates [17]. PFFs, the in vitro prepared recombinant α-syn aggregates, are able to enter the neurons in the brain slices and induce the aggregation of host-encoded endogenous α-syn, resulting in neurotoxicity. The use of human α-syn PFFs and mouse expressing human α-syn transgene makes the model more relevant to human disease. Future studies are required to determine whether α-syn aggregates in diseased brain homogenates are able to induce such a change in the slice culture. If successful, that will be an excellent model for testing new therapies.
Despite the advantages, this model also has some potential shortcomings. We choose cerebellar slice culture because of its long-term viability. Although α-syn aggregation is documented in the cerebellum in PD, DLB and MSA [47, 48], the relatively lower level and uneven α-syn expression in the cerebellum may limit the extent of α-syn aggregation. Because of the uneven α-syn expression, immunofluorescence is probably more sensitive than western blot to detect PFF-induced pα-syn. In this study, we used medial cerebellar slices with higher α-syn expression level to limit its influence and showed PFF-seeded endogenous α-syn misfolding by both immunofluorescence and western blot (Figs. 1 and 2). In the future, using transgenic mice with more robust and even α-syn expression in the cerebellum should overcome this potential pitfall. Another concern is the potential variability in the status of slice culture, which may affect the detection of neurotoxicity. As shown in our study, proper controls are important to mitigate this concern. Although using the cerebellar slices prepared from young pups is not ideal to model late-age-onset neurodegenerative diseases, it is the best choice to maintain cell viability in culture. Several groups reported their trials of culturing brain slices from adult mice [67–70], but the neuronal viability appears to be a major challenge [21]. Further technical development is needed to overcome this hurdle.
While we were preparing this manuscript, Elfarrash et al. reported PFF-induced α-syn aggregation in hippocampal slice model [38]. Using microinjection to apply PFFs to the slices, they elegantly demonstrated inter-neuronal spreading of α-syn aggregates. More recently, Shrivastava et al. reported differential seeding capabilities of various polymorphs of PFFs in their hippocampal slice model [39]. Different from their hippocampal models, our cerebellar slice model offers a longer time period to observe PFF-induced effects, which allowed us to detect robust pα-syn accumulation and the resulting synaptoxicity and neurotoxicity. All three studies support that the synthetically generated PFFs are able to induce α-syn to aggregate in organotypic brain slice cultures. Together with previous reported approach of viral vector mediated transgene expression in brain slices [35, 36], this method opens new avenues to study synucleinopathies.
CONFLICT OF INTEREST
All authors declare no competing interests.
ACKNOWLEDGMENTS
Confocal microscopic images were acquired at Van Andel Institute Optical Imaging Core – Grand Rapids, Michigan. We thank the core manager, Dr. Corinne Esquibel, for her assistance in image capture and analysis. This work was supported in part by NIH grants R01NS060729 and R01NS071035.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-202026.
REFERENCES
[1] | Conway KA , Harper JD , Lansbury PT ((1998) ) Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 4: , 1318–1320. |
[2] | Lashuel HA , Overk CR , Oueslati A , Masliah E ((2013) ) The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat Rev Neurosci 14: , 38–48. |
[3] | El-Agnaf OM , Jakes R , Curran MD , Middleton D , Ingenito R , Bianchi E , Pessi A , Neill D , Wallace A ((1998) ) Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett 440: , 71–75. |
[4] | Luna E , Luk KC ((2015) ) Bent out of shape: Alpha-Synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease. FEBS Lett 589: , 3749–3759. |
[5] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[6] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95: , 6469–6473. |
[7] | Goedert M , Jakes R , Spillantini MG ((2017) ) The synucleinopathies: Twenty years on. J Parkinsons Dis 7: , S53–S71. |
[8] | Peng C , Gathagan RJ , Lee VM ((2018) ) Distinct alpha-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol Dis 109: , 209–218. |
[9] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 17013. |
[10] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[11] | Del Tredici K , Rub U , De Vos RA , Bohl JR , Braak H ((2002) ) Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 61: , 413–426. |
[12] | McKeith I , Mintzer J , Aarsland D , Burn D , Chiu H , Cohen-Mansfield J , Dickson D , Dubois B , Duda JE , Feldman H , Gauthier S , Halliday G , Lawlor B , Lippa C , Lopez OL , Carlos Machado J , O’Brien J , Playfer J , Reid W , International Psychogeriatric Association Expert Meeting on DLB ((2004) ) Dementia with Lewy bodies. Lancet Neurol 3: , 19–28. |
[13] | Walker Z , Possin KL , Boeve BF , Aarsland D ((2015) ) Lewy body dementias. Lancet 386: , 1683–1697. |
[14] | Fujishiro H , Ferman TJ , Boeve BF , Smith GE , Graff-Radford NR , Uitti RJ , Wszolek ZK , Knopman DS , Petersen RC , Parisi JE , Dickson DW ((2008) ) Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 67: , 649–656. |
[15] | Fanciulli A , Wenning GK ((2015) ) Multiple-system atrophy. N Engl J Med 372: , 249–263. |
[16] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , Iwatsubo T ((2002) ) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4: , 160–164. |
[17] | Brundin P , Melki R , Kopito R ((2010) ) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11: , 301–307. |
[18] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VM ((2011) ) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[19] | Wang X , Becker K , Levine N , Zhang M , Lieberman AP , Moore DJ , Ma J ((2019) ) Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol Commun 7: , 41. |
[20] | Luk KC , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , Lee VM ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[21] | Croft CL , Futch HS , Moore BD , Golde TE ((2019) ) Organotypic brain slice cultures to model neurodegenerative proteinopathies. Mol Neurodegener 14: , 45. |
[22] | Linsley JW , Tripathi A , Epstein I , Schmunk G , Mount E , Campioni M , Oza V , Barch M , Javaherian A , Nowakowski TJ , Samsi S , Finkbeiner S ((2019) ) Automated four-dimensional long term imaging enables single cell tracking within organotypic brain slices to study neurodevelopment and degeneration. Commun Biol 2: , 155. |
[23] | Croft CL , Noble W ((2018) ) Preparation of organotypic brain slice cultures for the study of Alzheimer’s disease. F1000Res 7: , 592. |
[24] | McCaughey-Chapman A , Connor B ((2017) ) Rat brain sagittal organotypic slice cultures as an ex vivo dopamine cell loss system. J Neurosci Methods 277: , 83–87. |
[25] | Humpel C ((2015) ) Organotypic brain slice cultures: A review. Neuroscience 305: , 86–98. |
[26] | Marschalek N , Albert F , Meske V , Ohm TG ((2014) ) The natural history of cerebellar degeneration of Niemann-Pick C mice monitored in vitro. Neuropathol Appl Neurobiol 40: , 933–945. |
[27] | Ravikumar M , Jain S , Miller RH , Capadona JR , Selkirk SM ((2012) ) An organotypic spinal cord slice culture model to quantify neurodegeneration. J Neurosci Methods 211: , 280–288. |
[28] | Gerace E , Landucci E , Scartabelli T , Moroni F , Pellegrini-Giampietro DE ((2012) ) Rat hippocampal slice culture models for the evaluation of neuroprotective agents. Methods Mol Biol 846: , 343–354. |
[29] | Falsig J , Aguzzi A ((2008) ) The prion organotypic slice culture assay–POSCA. Nat Protoc 3: , 555–562. |
[30] | Falsig J , Julius C , Margalith I , Schwarz P , Heppner FL , Aguzzi A ((2008) ) A versatile prion replication assay in organotypic brain slices. Nat Neurosci 11: , 109–117. |
[31] | Falsig J , Sonati T , Herrmann US , Saban D , Li B , Arroyo K , Ballmer B , Liberski PP , Aguzzi A ((2012) ) Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog 8: , e1002985. |
[32] | Herrmann US , Sonati T , Falsig J , Reimann RR , Dametto P , O’Connor T , Li B , Lau A , Hornemann S , Sorce S , Wagner U , Sanoudou D , Aguzzi A ((2015) ) Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 11: , e1004662. |
[33] | Sorce S , Nuvolone M , Keller A , Falsig J , Varol A , Schwarz P , Bieri M , Budka H , Aguzzi A ((2014) ) The role of the NADPH oxidase NOX2 in prion pathogenesis. PLoS Pathog 10: , e1004531. |
[34] | Ullrich C , Daschil N , Humpel C ((2011) ) Organotypic vibrosections: Novel whole sagittal brain cultures. J Neurosci Methods 201: , 131–141. |
[35] | Croft CL , Cruz PE , Ryu DH , Ceballos-Diaz C , Strang KH , Woody BM , Lin WL , Deture M , Rodriguez-Lebron E , Dickson DW , Chakrabarty P , Levites Y , Giasson BI , Golde TE ((2019) ) rAAV-based brain slice culture models of Alzheimer’s and Parkinson’s disease inclusion pathologies. J Exp Med 216: , 539–555. |
[36] | Zach S , Bueler H , Hengerer B , Gillardon F ((2007) ) Predominant neuritic pathology induced by viral overexpression of alpha-synuclein in cell culture. Cell Mol Neurobiol 27: , 505–515. |
[37] | Loria F , Vargas JY , Bousset L , Syan S , Salles A , Melki R , Zurzolo C ((2017) ) alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134: , 789–808. |
[38] | Elfarrash S , Jensen NM , Ferreira N , Betzer C , Thevathasan JV , Diekmann R , Adel M , Omar NM , Boraie MZ , Gad S , Ries J , Kirik D , Nabavi S , Jensen PH ((2019) ) Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol Commun 7: , 213. |
[39] | Shrivastava AN , Bousset L , Renner M , Redeker V , Savistchenko J , Triller A , Melki R ((2020) ) Differential membrane binding and seeding of distinct alpha-synuclein fibrillar polymorphs. Biophys J 118: , 1301–1320. |
[40] | Lee MK , Stirling W , Xu Y , Xu X , Qui D , Mandir AS , Dawson TM , Copeland NG , Jenkins NA , Price DL ((2002) ) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –>Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A 99: , 8968–8973. |
[41] | Janezic S , Threlfell S , Dodson PD , Dowie MJ , Taylor TN , Potgieter D , Parkkinen L , Senior SL , Anwar S , Ryan B , Deltheil T , Kosillo P , Cioroch M , Wagner K , Ansorge O , Bannerman DM , Bolam JP , Magill PJ , Cragg SJ , Wade-Martins R ((2013) ) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A 110: , E4016–4025. |
[42] | Ding Y , Li L ((2008) ) Lipopolysaccharide preconditioning induces protection against lipopolysaccharide-induced neurotoxicity in organotypic midbrain slice culture. Neurosci Bull 24: , 209–218. |
[43] | Volpicelli-Daley LA , Luk KC , Lee VM ((2014) ) Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9: , 2135–2146. |
[44] | Becker K , Wang X , Vander Stel K , Chu Y , Kordower J , Ma J ((2018) ) Detecting alpha synuclein seeding activity in formaldehyde-fixed MSA patient tissue by PMCA. Mol Neurobiol 55: , 8728–8737. |
[45] | Hama H , Hioki H , Namiki K , Hoshida T , Kurokawa H , Ishidate F , Kaneko T , Akagi T , Saito T , Saido T , Miyawaki A ((2015) ) ScaleS: An optical clearing palette for biological imaging. Nat Neurosci 18: , 1518–1529. |
[46] | Fournier M , Roux A , Garrigue J , Muriel MP , Blanche P , Lashuel HA , Anderson JP , Barbour R , Huang J , du Montcel ST , Brice A , Corti O ((2013) ) Parkin depletion delays motor decline dose-dependently without overtly affecting neuropathology in alpha-synuclein transgenic mice. BMC Neurosci 14: , 135. |
[47] | Seidel K , Bouzrou M , Heidemann N , Kruger R , Schols L , den Dunnen WFA , Korf HW , Rub U ((2017) ) Involvement of the cerebellum in Parkinson disease and dementia with Lewy bodies. Ann Neurol 81: , 898–903. |
[48] | Burn DJ , Jaros E ((2001) ) Multiple system atrophy: Cellular and molecular pathology. Mol Pathol 54: , 419–426. |
[49] | Lee SK , Sillitoe RV , Silva C , Martina M , Sekerkova G ((2015) ) alpha-Synuclein expression in the mouse cerebellum is restricted to VGluT1 excitatory terminals and is enriched in unipolar brush cells. Cerebellum 14: , 516–527. |
[50] | Tampellini D , Capetillo-Zarate E , Dumont M , Huang Z , Yu F , Lin MT , Gouras GK ((2010) ) Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer’s disease transgenic mice. J Neurosci 30: , 14299–14304. |
[51] | Yuki D , Sugiura Y , Zaima N , Akatsu H , Takei S , Yao I , Maesako M , Kinoshita A , Yamamoto T , Kon R , Sugiyama K , Setou M ((2014) ) DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer’s disease. Sci Rep 4: , 7130. |
[52] | Harwell CS , Coleman MP ((2016) ) Synaptophysin depletion and intraneuronal Abeta in organotypic hippocampal slice cultures from huAPP transgenic mice. Mol Neurodegener 11: , 44. |
[53] | Hornykiewicz O ((1998) ) Biochemical aspects of Parkinson’s disease. Neurology 51: , S2–9. |
[54] | Burke RE , O’Malley K ((2013) ) Axon degeneration in Parkinson’s disease. Exp Neurol 246: , 72–83. |
[55] | Froula JM , Henderson BW , Gonzalez JC , Vaden JH , McLean JW , Wu Y , Banumurthy G , Overstreet-Wadiche L , Herskowitz JH , Volpicelli-Daley LA ((2018) ) alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun 6: , 35. |
[56] | Gelders G , Baekelandt V , Van der Perren A ((2018) ) Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res 2018: , 4784268. |
[57] | Gerhard A , Pavese N , Hotton G , Turkheimer F , Es M , Hammers A , Eggert K , Oertel W , Banati RB , Brooks DJ ((2006) ) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21: , 404–412. |
[58] | Hirsch EC , Hunot S ((2009) ) Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol 8: , 382–397. |
[59] | Booth HDE , Hirst WD , Wade-Martins R ((2017) ) The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci 40: , 358–370. |
[60] | Yun SP , Kam TI , Panicker N , Kim S , Oh Y , Park JS , Kwon SH , Park YJ , Karuppagounder SS , Park H , Kim S , Oh N , Kim NA , Lee S , Brahmachari S , Mao X , Lee JH , Kumar M , An D , Kang SU , Lee Y , Lee KC , Na DH , Kim D , Lee SH , Roschke VV , Liddelow SA , Mari Z , Barres BA , Dawson VL , Lee S , Dawson TM , Ko HS ((2018) ) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24: , 931–938. |
[61] | Harms AS , Delic V , Thome AD , Bryant N , Liu Z , Chandra S , Jurkuvenaite A , West AB ((2017) ) alpha-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol Commun 5: , 85. |
[62] | Harms AS , Thome AD , Yan Z , Schonhoff AM , Williams GP , Li X , Liu Y , Qin H , Benveniste EN , Standaert DG ((2018) ) Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp Neurol 300: , 179–187. |
[63] | Brochard V , Combadiere B , Prigent A , Laouar Y , Perrin A , Beray-Berthat V , Bonduelle O , Alvarez-Fischer D , Callebert J , Launay JM , Duyckaerts C , Flavell RA , Hirsch EC , Hunot S ((2009) ) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119: , 182–192. |
[64] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546: , 656–661. |
[65] | Sommer A , Maxreiter F , Krach F , Fadler T , Grosch J , Maroni M , Graef D , Eberhardt E , Riemenschneider MJ , Yeo GW , Kohl Z , Xiang W , Gage FH , Winkler J , Prots I , Winner B ((2018) ) Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23: , 123–131 e126. |
[66] | Goniotaki D , Lakkaraju AKK , Shrivastava AN , Bakirci P , Sorce S , Senatore A , Marpakwar R , Hornemann S , Gasparini F , Triller A , Aguzzi A ((2017) ) Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog 13: , e1006733. |
[67] | Su T , Paradiso B , Long YS , Liao WP , Simonato M ((2011) ) Evaluation of cell damage in organotypic hippocampal slice culture from adult mouse: A potential model system to study neuroprotection. Brain Res 1385: , 68–76. |
[68] | Wilhelmi E , Schoder UH , Benabdallah A , Sieg F , Breder J , Reymann KG ((2002) ) Organotypic brain-slice cultures from adult rats: Approaches for a prolonged culture time. Altern Lab Anim 30: , 275–283. |
[69] | Xiang Z , Hrabetova S , Moskowitz SI , Casaccia-Bonnefil P , Young SR , Nimmrich VC , Tiedge H , Einheber S , Karnup S , Bianchi R , Bergold PJ ((2000) ) Long-term maintenance of mature hippocampal slices in vitro. J Neurosci Methods 98: , 145–154. |
[70] | Humpel C ((2015) ) Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front Aging Neurosci 7: , 47. |


