
Soluble endogenous oligomeric α-synuclein species in neurodegenerative diseases: Expression, spreading, and cross-talk
Abstract
There is growing recognition in the field of neurodegenerative diseases that mixed proteinopathies are occurring at greater frequency than originally thought. This is particularly true for three amyloid proteins defining most of these neurological disorders, amyloid-beta (Aβ), tau, and alpha-synuclein (αSyn). The co-existence and often co-localization of aggregated forms of these proteins has led to the emergence of concepts positing molecular interactions and cross-seeding between Aβ, tau, and αSyn aggregates. Amongst this trio, αSyn has received particular attention in this context during recent years due to its ability to modulate Aβ and tau aggregation in vivo, to interact at a molecular level with Aβ and tau in vivo and to cross-seed tau in mice. Here we provide a comprehensive, critical, and accessible review about the expression, role and nature of endogenous soluble αSyn oligomers because of recent developments in the understanding of αSyn multimerization, misfolding, aggregation, cross-talk, spreading and cross-seeding in neurodegenerative disorders, including Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, Alzheimer’s disease, and Huntington’s disease. We will also discuss our current understanding about the relative toxicity of endogenous αSyn oligomers in vivo and in vitro, and introduce potential opportunities to counter their deleterious effects.
INTRODUCTION
Neurodegenerative disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), dementia with Lewy body (DLB), multiple system atrophy (MSA), and Huntington’s disease (HD) are characterized by progressive dysfunction and death of neurons. Within each disease type, this degeneration affects specific neural systems implying selective vulnerability and is associated with the abnormal accumulation of distinct proteins, e.g., amyloid-beta (Aβ), tau, and alpha-synuclein (αSyn), in the brain defining each disease neuropathologically [1, 2]. Classically, deposition of Aβ and tau respectively known as Aβ plaques and neurofibrillary tangles characterize AD, inclusions of αSyn as Lewy body (LB) or glial cytoplasmic inclusion (GCI) define PD, DLB, and MSA (collectively known as synucleinopathies), and inclusions of huntingtin (Htt) neuropathologically define HD.
Recent studies are however providing new evidence for prevalent mixed proteinopathies across these neurodegenerative diseases, with aging and apolipoprotein E (ApoE) ɛ4 genotype constituting risk factors [3]. While tau pathology proved ubiquitous across the neurodegenerative diseases analyzed, mixed pathologies were observed in 38–81% of synucleinopathy cases including neocortical Lewy body disease as a stark example with 80% increases in Aβ load and 22% increases in TAR DNA-binding protein 43 (TDP-43) burden [3]. Although HD cases were not included in the latter study, prior work indicated that αSyn pathology is also increased in HD and colocalizes with Htt inclusions [4–6]. These observations echo prior work showing that mixed neuropathologies are the most common cause of the clinical syndrome of dementia and are also common among persons with mild cognitive impairment or cognitive decline [7–12]. Furthermore, two recent longitudinal clinical-pathologic studies of aging reported that only 9% presented pure AD pathology and 78% included more than two neuropathologies [12]. AD pathology and neocortical intracellular αSyn inclusions had the strongest effect accounting for 58 and 46% of cognitive loss, respectively [12]. Combined, these findings extend earlier observations indicating that a more rapid rate of cognitive decline is observed in 30–40% of AD cases presenting with LB and Lewy neurites (LN) [13, 14] compared to subjects with AD free of αSyn pathology.
The co-existence of these proteinopathies provides additional support to accumulating observations documenting molecular interactions and cross-seeding between aggregates of Aβ, tau, Htt and αSyn [15–18]. In this particular context, αSyn, the amyloid protein genetically linked to synucleinopathies, stands out for several reasons. First, synucleinopathies appear to exhibit distinct prevalence of Aβ burden and Htt compared to other neurodegenerative diseases [3, 6]. Second, αSyn expression modulates Aβ aggregation and deposition in mice [19, 20]. Third, αSyn oligomers can either form hybrid soluble oligomeric aggregates or interact at a molecular level with Aβ [21] and tau [22, 23] in vivo. Fourth, short preformed fibrils of αSyn (αSyn PFFs) can cross-seed tau in vitro and in vivo [24]. Thus, all of these points infer a central role of pathological oligomeric αSyn aggregates as key determinants in influencing or controlling aggregation of other amyloid proteins in the brain.
Beyond a potential contribution of αSyn in shaping inter-relationships with other amyloid proteins in neurodegenerative disease, a now large body of evidence from biochemical, biophysical, genetic and functional studies supports the hypothesis that the processes of αSyn oligomerization and fibrilization occupy central roles in the pathogenesis of synucleinopathies [25–29]. Because endogenous aggregated species of amyloid proteins are formed in cells or brain tissues where they are exposed to cell- and environment-specific variables (e.g., post-translational modifications) and because these endogenous species have been consistently reported to be more potent than their recombinant forms [30–34], a specific focus will be brought in this review on soluble endogenous oligomeric assemblies defined by the following criteria: (i) soluble in aqueous buffers following ultracentrifugation, (ii) SDS-resistant following tissue or cell lysis, (iii) segregated from monomeric species in liquid-phase chromatography, and (iv) immunoreactive to at least two different antibodies for that amyloid molecule and oligomer-selective antibodies. The importance of the endogenous milieu for αSyn aggregation was well-illustrated by Strohäker and coworkers who rigorously demonstrated that brain-derived αSyn fibrillar aggregates were structurally different to all recombinant αSyn fibrillar polymorphs tested [35]. For these reasons, our review will focus on endogenous brain-derived αSyn oligomers (o-αSyn).
Despite this greater understanding, many fundamental questions remain unanswered: 1) which endogenous forms of αSyn are toxic to neurons and glial cells, 2) when are αSyn species prevalent in the pathophysiology of neurodegenerative diseases, 3) what molecular mechanisms induced by endogenous αSyn aggregates (oligomers, protofibrils, fibrils) contribute to neurodegeneration, 4) how are aggregated endogenous αSyn species impacting the physiological cellular localization and function of physiological forms of αSyn (monomeric and recently proposed native multimeric species), 5) what is the conformation of brain-derived o-αSyn? Given the exponential complication of the landscape related to amyloid protein aggregation due to the intrinsic identification of new assemblies and due to the emergence of prion-like propagation of pathological aggregates, we provide an overview of the current state of knowledge regarding the detection and role of physiological and pathological αSyn assemblies across neurodegenerative diseases, the spreading of oligomeric αSyn species and the molecular interactions between oligomeric αSyn and other amyloid proteins. We finally discuss the implications of these findings for potential therapeutic interventions in the contexts of mono- and multi-therapies.
αSYN FUNCTION AND STRUCTURE
In the past 5–7 years, growing evidence indicates that αSyn (gene name: SNCA), a synaptically-enriched protein traditionally linked to PD, may act as a regulator of synaptic vesicle release. Despite the original observation that αSyn knockout (αSyn-KO) mice display only subtle ‘functional deficits in the nigrostriatal dopamine system’ [36], many research groups have been seeking to identify the exact physiological function of αSyn with a special focus on the pre-synapse (reviewed, e.g., in [37, 38]). In a recent study, the gene deletion of SNCA (together with its homologs SNCB and SNCG) promotes the dilation of the exocytotic fusion in cultured rodent neurons [39], potentially explaining previous findings related to αSyn’s role in exocytosis [37, 38]. In addition, αSyn may also affect endocytosis by promoting clathrin-dependent endocytosis [40] and/or by regulating the kinetics of synaptic vesicle endocytosis [41]. Synucleins may have multiple effects on presynaptic architecture [42], and it was suggested that αSyn can ‘cluster’ vesicles, thereby preventing free dispersal between neighboring en-passant boutons [43].
In search of a mechanism for how exactly αSyn may affect vesicle biology, the Südhof group proposed that αSyn acts as a chaperone for Synaptobrevin-2 (VAMP-2) [44], thereby increasing the number of assembled soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes. However, it was also shown that αSyn can inhibit membrane fusion by directly affecting the lipid bilayer, rather than indirectly via SNARE proteins [45, 46].
Beyond neurons, larger amounts of αSyn are also found in red blood cells, and its function there is even more enigmatic [37]. Red blood cells, however, must be kept in mind as a relevant source for αSyn in body fluids, e.g., as a potential contaminant of cerebrospinal fluid when measuring αSyn in longitudinal and cross-sectional studies.
Structure-function relationships of αSyn
Lipid binding
Consistent with its proposed function(s), αSyn is found mostly at presynaptic terminals, both in brain [47, 48] and in mature cultured neurons [49]. The driver of its localization may be the attraction between αSyn and synaptic vesicles as biophysical studies have shown that the relatively small synaptic vesicles may possess the optimal membrane curvature to promote αSyn binding [50]. Early characterization of αSyn already suggested that its membrane binding goes hand in hand with the formation of amphipathic helices (so-called ‘11/3 helices’: 11 aa/3 turns) [51]. The helix formation is dictated by an 11-aa repeat with the core motif KTKEGV, which appears imperfectly 6–9 times in the first two-thirds of αSyn [48]. Nonpolar aa in the lower half of the helix ‘dip’ into the membrane bilayer (∼1–5 Å below lipid head groups) [52–55] while hydrophilic residues in the upper half interact with the cytoplasm. In addition, positively-charged lysine residues (K) in the KTKEGV motifs interact with negatively-charged membrane lipid head groups [56]. Yet, the helix formation is imperfect and only transient because, e.g., polar threonine (T) residues are located in the hydrophobic half of the helix [57–59]. A recent in vitro study suggests that αSyn coming off the membrane may retain its fold and assemble into helical multimers [60] (see next chapter). This notion is in contrast to the classical view of soluble αSyn as an unfolded monomer [61]. A model has been proposed in which αSyn constantly cycles through at least three different states: unfolded cytosolic monomer, folded monomer at membranes, and (soluble?) folded multimer [62].
Multimeric vs. oligomeric αSyn
Definitions. ‘Oligo-’ (ancient Greek) and ‘multi-’ (Latin) both mean ‘several’ or ‘a few’, and ‘meros’ is ancient Greek for ‘part’ or ‘unit’. Thus, multimer and oligomer could be used interchangeably to describe small assemblies. In the case of αSyn, however, the term oligomer is mostly used to describe abnormal assemblies (aggregates), while several studies have now established the term multimer for the new concept of native αSyn assembly [43, 62–67]. In the following two paragraphs we will briefly contrast αSyn oligomers vs. multimers and focus on the evidence for the (putatively) physiological αSyn multimers, while the rest of our review will be dedicated to the pathological oligomers.
Shared properties and differences. Endogenous αSyn multimers and oligomers have in common that two or more synuclein molecules are directly interacting. Multimeric, native αSyn is typically described as tetrameric [68–73], sometimes octameric [67]. Oligomeric αSyn aggregates in contrast seems to exist in a wide variety of different conformations (reviewed below). Multimeric αSyn is considered to be helical [63, 68, 69] and aggregation-resistant [68], while αSyn aggregates would traditionally be expected to be rich in β-sheet (new insight that indicates bigger complexity is emerging, see below). Multimeric αSyn has been proposed to have functional roles in the cells, but there is no consensus yet on the details: multimers may cluster vesicles [43], promote SNARE assembly [67] or be a passive storage form [74]. Multimeric αSyn species were shown to be sensitive to temperature and heat denaturation, to cell lysis and to the presence of the anionic detergent sodium dodecyl sulfate (SDS) [75] while oligomeric αSyn aggregates are typically, but maybe not in all cases, more stable (see below).
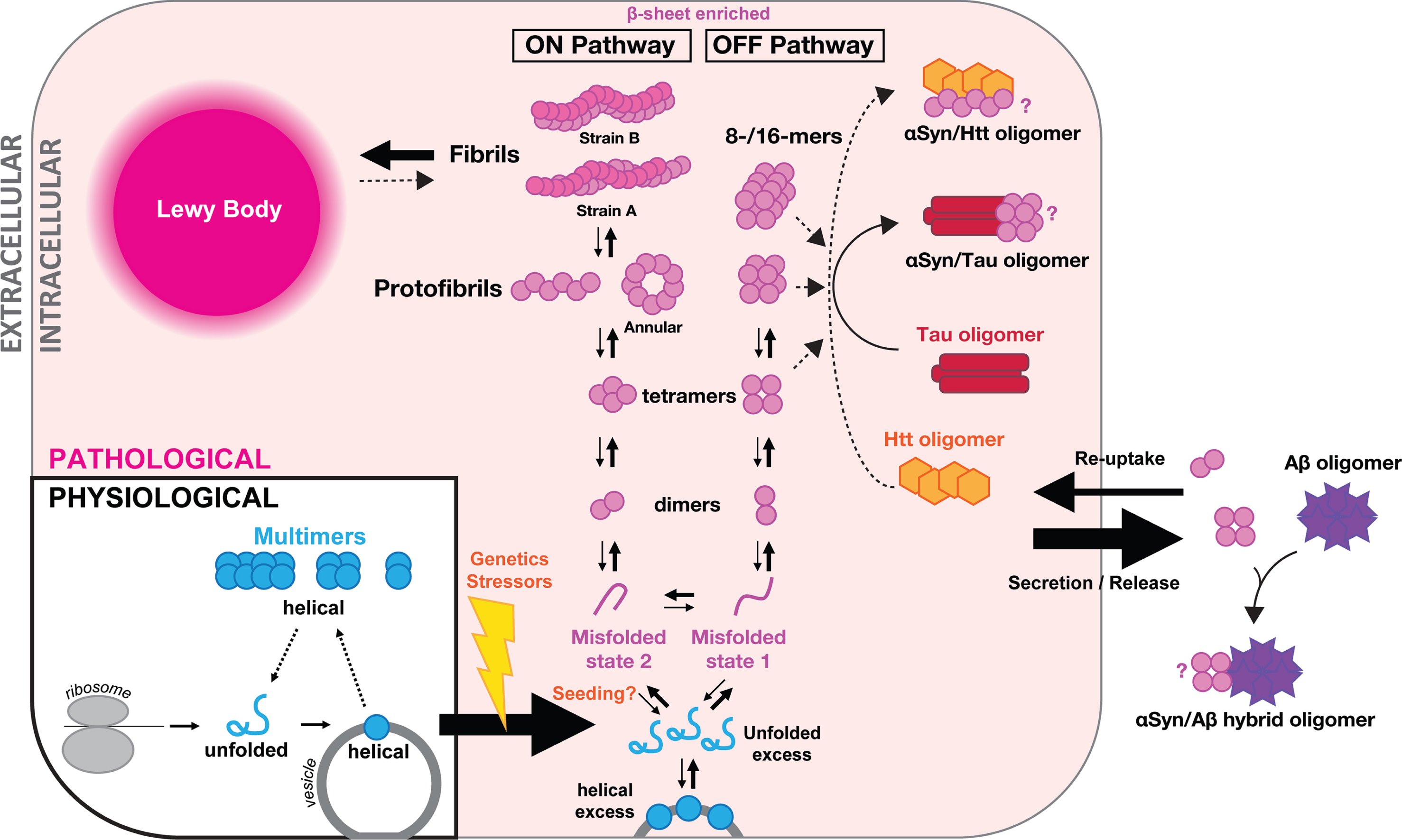
Evidence for native αSyn assembly (multimers). Bartels, Choi and Selkoe proposed in 2011 that αSyn assembles naturally inside cells and occurs as a helical tetramer that resists aggregation [68]. αSyn was purified from various human sources, including red blood cells, under non-denaturing conditions. Several methods, most prominently analytical ultracentrifugation, sized the purified multimers as principally tetramers while circular dichroism characterized them as helical. Around the same time, tetrameric αSyn was purified from bacteria under non-denaturing conditions [69]. αSyn N-acetylation may be critical for the successful purification of helical αSyn multimers from bacteria [76]. Subsequent attempts of purifying helical αSyn assemblies led to different results: unsuccessful in red blood cells [77], successful in red blood cells [74], largely unsuccessful in human brain (only evidence for small amounts of multimer) [78], partially successful in human brain (multimeric αSyn in fractions of lower purity, but not in the highly pure material) [66]. Another study probed the native state of αSyn from human brain by gel filtration coupled with native gradient gel separation, an array of antibodies with non-overlapping epitopes, and mass spectrometry. The authors concluded that metastable higher-molecular weight ‘conformers’ and stable monomers co-exist in the human brain [79] (Fig. 1). Such a model had heretofore been suggested by a nuclear magnetic resonance (NMR)/computational study [64].
Fig. 1
Proposed model of αSyn assemblies in health (‘multimers’) and disease (‘oligomers’). Schematic of one cell and extracellular space. Under physiological conditions (in blue, bottom left), αSyn comes off the ribosome as a soluble unfolded monomer (blue ‘αSyn’), which forms helical monomers (blue circle) upon getting in contact with vesicle membranes. Helical monomers at membranes may assemble into metastable physiological multimers (low-n assemblies in blue) that eventually fall apart again, initiating a new cycle. Under stress of pathological conditions (in pink shading, starting bottom middle), excess of unfolded or helical membrane-bound αSyn has been proposed to be the starting point for pathological aggregation into oligomers including dimers, tetramers, annular protofibrils and larger oligomers. We propose that two distinct misfolded states of αSyn lead to two distinct aggregation pathways, one forming amyloid fibrils (ON pathway; A11–/OC+), and one forming large non-fibrillar αSyn oligomers (OFF pathway, A11+/OC–). Endogenous oligomeric αSyn are likely to be enriched for β-sheet motifs. The endpoint of intracellular aggregation is the so-called Lewy body (top left). Mixed intracellular aggregates have also been reported in particular with tau (red baton), although the exact αSyn oligomer interacting with tau is unknown (indicated by ‘?’). Cross-talks between αSyn species and huntingtin oligomers (Htt, orange hexagons) have also been advanced but require confirmation. Oligomers of αSyn may be released into the extracellular space by an ill-defined mechanism (right). There, they may further aggregate, which may include the formation of mixed aggregates, with amyloid-β peptides (Aβ, purple stars) in particular with which αSyn can form hybrid αSyn/Aβ oligomers. Extracellular aggregates may be taken up by cells and released into the cytosol where they may in turn ‘seed’ αSyn aggregation. Release, uptake and seeding of intracellular pathology might be underlying the phenomenon of αSyn spread and release might correspond to a possible protective mechanism adopted by cells to maintain cellular proteostasis.
Given the complexity of purification, intact-cell methods became increasingly important. A live-cell crosslinking study provided a large variety of controls to suggest abundant physiological, not pathological αSyn assemblies [75]. A follow-up study [71] revealed not only that αSyn in fresh human brain was shown to be multimeric by crosslinking but also that familial PD-linked mutations such as A30P and E46K decrease multimer:monomer ratios. In another study [70], overexpression of strategic αSyn variants that render αSyn largely monomeric (plus some dimers) were cytotoxic and prone to forming inclusions in primary rat neurons. Subsequently, it was suggested that lysosomal glucocerebrosidase deficiency overall reduces αSyn multimers and increases monomers [65]. Intact-cell crosslinking typically traps 60, 80 and 100 kDa species of αSyn (αS60, -80 and -100), and their identity is not yet clear. They may be tetramer, hexamer and octamer or could all be tetramers that differ by the exact mode of crosslinking. The 60 kDa αSyn multimer is typically the most abundant defined multimeric species of intact-cell crosslinking, consistent with a tetramer (monomeric αS: 14.5 kDa; 4× 14.5 kDa = 58 kDa) [75]. Burré et al. sized crosslinked αSyn multimers in brain slices, brain homogenates and recombinant αSyn on charged liposomes as tetramers, octamers and other multimers, including prominent dimers [67]. This pattern differed from that typically reported for intact-cell crosslinking (see above). The same group postulated that the loss of αSyn multimerization may occur upstream of αSyn aggregation and neurotoxicity [80]. Importantly, defined point mutations that abolish αSyn multimer intact-cell crosslinking, also prevented αSyn-mediated Venus/Yellow fluorescent protein (YFP) complementation [71], an orthogonal intact-cell method. Wang et al. used αSyn-mediated Venus YFP complementation in live neurons to demonstrate that αSyn multimers cluster synaptic vesicles and attenuate recycling [43]. Similarly, Burré et al. used fluorescence resonance energy transfer revealing the detection of normal αSyn multimers at membranes [67].
A key concept to understanding αSyn cellular behavior may be context-dependent folding [72]. For example, αSyn may exists primarily as a monomer in intact enteric neurons while αSyn may be present in the brain in a different conformation [81]. This view is further supported by the finding that αSyn (and βSyn) multimers were only efficiently trapped by crosslinking of intact neurons, whereas crosslinking of lysates largely yielded monomers [75]. This observation was unique to the synuclein isoforms (control multimeric proteins were trapped both in cells and lysates) and in line with the concept of metastable or ‘dynamic’ multimers, a model also postulated by others [64, 69]. The requirement of molecular crowding or a limiting factor that holds intracellular multimers together would be a possible explanation for the lysis sensitivity and difficulties in purifying multimeric αSyn. Of note, several independent reports suggest that (re)folding and (re)assembly of helical αSyn in vitro might be possible: 1) Killinger et al. suggested that αSyn forms multimers during electrophoresis, possibly due to crowding effects [82], 2) Iljina et al. used arachidonic acid to reconstitute αSyn assemblies in vitro that sized as tetramers [63] and 3) Rovere et al. proposed a mechanism in which helical αSyn folding at membranes is preserved when αSyn comes off membranes and hydrophobic interactions may drive cytosolic assembly that is stable for an undetermined amount of time [60].
The list of relevant publications on the topic of native αSyn multimers would be incomplete without documenting reports that are skeptical about the relevance of native αSyn assemblies. Fauvet et al. reported their inability to purify folded multimers from a variety of available sources [77]. Bacterial [83] and mammalian [84] in-cell NMR analyses were consistent with largely disordered, monomeric states of αSyn. Despite the rapidly growing appeal of this novel adaptation of NMR, it is however worth mentioning here some of the limitations of the technique including an inability to detect αSyn assemblies due to their size, the necessity to perform measurements below body and even room temperature, and a reliance on proteofection of large amounts of labeled αSyn into mammalian cells, which might raise caution towards drawing definite conclusions. Neither can the existence of smaller, but relevant, amounts of folded and assembled αSyn be ruled out by the technique [85].
All in all, there is accumulating evidence from multiple independent laboratories to support the existence of native αSyn assembly(ies) through a variety of techniques, but also reports that deny their existence and/or relevance. The debate about multimer abundance, exact location (membrane vs. soluble), stability (stable vs. dynamic) and function (storage form vs. active role) will continue and promises to be intellectually stimulating for the field.
ENDOGENOUS OLIGOMERIC αSyn ASSEMBLIES IN NEURODEGENERATIVE DISEASES
Endogenous oligomeric αSyn assemblies in neurodegenerative diseases
Detection of oligomeric αSyn species by disease
Parkinson’s disease. An emerging view emphasizes o-αSyn as the key mediators in synucleinopathies. Soluble αSyn oligomeric species associated with polyunsaturated fatty acids have been detected in human PD and DLB brain tissues, as well as in human αSyn-expressing transgenic mouse brain tissues. It has been suggested that unlike saturated fatty acids, increased levels of polyunsaturated fatty acids promote αSyn oligomerization in PD brains [86]. Elevated levels of αSyn oligomeric assemblies have been detected in the postmortem brain tissues of PD [87–89], notably putative ∼60 kDa αSyn tetramers [88]. Oligomers of αSyn were also detected from the blood plasma and CSF of PD patients present at an increased level compared to controls [90–92]. Hence, αSyn oligomers, as well as a ratio of αSyn oligomers to total αSyn have been proposed as biomarkers for early diagnosis of PD and PD with dementia [93, 94]. Oligomeric αSyn has also been detected at an increased level from CSF of PD patients by using a highly sensitive electronic biosensor. This technique enabled the distinction of AD and PD pathologies from cognitively normal control subjects based on the detection of CSF αSyn oligomer levels [95]. Although the exact mechanism by which αSyn oligomers exert their toxic effects is still unknown, the presence of these oligomeric assemblies in the extracellular body fluids indicates their release from affected brain regions and opens up the intriguing possibility of subsequent propagation of these assemblies between cells. Exocytosis via exosomes has been implicated in the spreading of αSyn in cell culture [96]. The cellular release of αSyn oligomers via exosomes was suggested as an alternate pathway for survival upon failure of their clearance via autophagosomes in vitro [97].
All known SNCA missense mutations associated with familial forms of PD (A30P, E46K, A53T, A53E, H50Q and G51D) reside in the amino-terminal domain of αSyn responsible for membrane binding. Amongst those, TgA53T transgenic mice overexpressing the A53T mutant of human αSyn have been extensively studied as models of familial PD due to their relatively robust phenotype [98]. In TgA53T mice from the G2.3 line [98], oligomeric αSyn was shown to accumulate in the mitochondrial membrane and impair complex I function [99]. Treating these animals with an anti-ER stress molecule, salubrinal, reduced accumulation of such toxic oligomers in the ER and delayed disease onset [99], suggesting that αSyn oligomer-dependent ER stress may constitute a key event in the pathophysiology of PD.
Dementia with Lewy bodies. Elevated levels of oligomeric αSyn were detected in the postmortem brain tissues of DLB patients by western blotting and ELISA assays [89, 100]. However, CSF o-αSyn levels in DLB patients were comparable to that measured in age-matched control subjects contrary to subjects with PD dementia (PDD) or to individuals with AD [94].
In mice, a transgenic animal model expressing mutant E46K αSyn associated with the familial form of DLB [101] showed deposition of Lewy-like structures and motor impairment [102]. Western blot analysis of brain lysates from TgE46K mice (line 47) revealed bands in the stacking gel in the ultracentrifugated high salt fractions, suggestive of the presence of large soluble αSyn oligomers [102]. These presumably large (>250 kDa) putative αSyn oligomers were immunoreactive to antibody Syn211 detecting human αSyn and to antibodies detecting phosphorylated αSyn at Serine 129 (pS129-αSyn), a post-translational modification inducing a higher propensity for αSyn to aggregate [103].
In both systems, it thus remains clear that additional studies are needed to fully assess the nature of o-αSyn species in DLB samples and the contribution of o-αSyn to DLB.
Multiple system atrophy. Using homotypic ELISA assays for total o-αSyn (with antibody Syn211) or for S129-phosphorylated o-αSyn, Foulds and colleagues reported no differences in total CSF o-αSyn concentration in MSA patients (n = 8) compared to control subjects. Of note, none of the tested groups (PD, DLB, PSP, MSA and controls) differed for total CSF αSyn, total CSF o-αSyn and CSF pS129-αSyn. However, the concentration of CSF pS129-αSyn oligomers was sharply elevated in the postmortem CSF samples of MSA patients compared to all other clinical groups [104]. Moreover, recent studies described that o-αSyn was abundantly distributed in neurons and oligodendrocytes in MSA brain tissues using homotypic proximity ligation assay detection [105].
Alzheimer’s disease. Accumulating evidence demonstrates the occurrence of αSyn pathology, LB and Lewy neurites (LN), in 40–55% of AD brains [3, 13, 15, 106]. Despite this observation, very few groups have measured the relative abundance of soluble αSyn aggregates in postmortem AD brain tissue. Using an agnostic view about the nature of the apparent inter-relationship between αSyn and AD pathology, Larson and colleagues found an approximate two-fold increase over controls in soluble monomeric αSyn levels in intracellular-enriched (IC) lysates from AD brains in the absence of LB cytopathology [107]. Importantly, the abundance of soluble αSyn monomers in temporal cortices translated into a better biological correlate of AD-associated cognitive impairment than soluble Aβ and tau levels [107]. Because the abundance and concentration of amyloid proteins directly regulate the aggregation state of said molecules, the same group hypothesized that oligomeric αSyn might be elevated in parallel to the increase in IC monomeric αSyn. Using a biochemical approach relying on homotypic and heterotypic enzyme-linked immunosorbent assays (ELISA), denaturing and non-denaturing electrophoreses, and size-exclusion chromatography combined with extensive immunoreactivity profiling, the same group identified increases in αSyn oligomers in AD brain tissue by 25–75% with the largest accumulation in αSyn oligomers corresponding to ∼35 kDa species (68% increase vs. controls) and to ∼56 kDa species (44% increase vs. controls) [108]. The nature of these dimeric and tetrameric assemblies was further confirmed using antibodies differentially detecting aggregated forms of various amyloid proteins (i.e., A11, OC, Officer) or antibodies specific to αSyn oligomers (i.e., Syn33 and F8H7). Importantly, the elevation of αSyn oligomers in temporal cortex was also observed in additional cortical domains, namely angular, calcarine and entorhinal cortices although with distinct observed profiles for each αSyn oligomer [108].
Functionally, soluble αSyn IC oligomers were associated with changes in cognitive function and synaptic expression in AD but not in age-matched control subjects [108]. Multi-variate analyses revealed that this was particularly true for episodic memory performance whereby 28-, 35- and 56 kDa αSyn oligomers inversely correlated with z-scores for this AD-defining memory modality. In a similar fashion, soluble αSyn IC oligomers inversely correlated with the abundance of synapsin isoforms [108], presynaptic proteins previously identified to be selectively down-regulated in mice overexpressing human wild-type (WT) αSyn [109]. This relationship was specific to synapsins but not to synaptophysin, another presynaptic protein decreased in AD. Overall, these findings infer a potential contribution of αSyn oligomers to AD-associated cognitive decline and synaptic loss.
Contrasting with the paucity of studies which used postmortem brain tissue to detect and measure αSyn oligomers, several groups have used CSF to measure αSyn in AD. Contrary to PD and DLB in which CSF αSyn is reliably lowered compared to control subjects [110], several reports have documented increased concentrations of CSF αSyn in AD vs. age-matched controls [90, 94, 111]. It is worth noting that CSF αSyn concentrations between sporadic AD and controls were unaltered in a longitudinal cohort [112], while the same group found elevated CSF αSyn concentrations in familial AD subjects vs. noncarriers [112]. However, the diagnostic sensitivity/specificity of classical AD CSF biomarkers can be improved when incorporating CSF αSyn [113, 114]. Overall, this lack of consensus regarding elevated CSF αSyn in AD is likely due to several variables, including differences in the ELISA platform used, possible contamination of the samples with erythrocytes, heterogeneity of the subjects included in the respective studies, and specimen storage and preparation.
Despite these issues, a few studies have measured oligomeric αSyn species in CSF from subjects with AD [90, 94, 115]. Hansson and colleagues reported decreased CSF concentrations of o-αSyn in AD compared to controls using a homotypic ELISA based on the 211 antibody, whereas total CSF αSyn concentration was elevated in the AD cohort [94]. In contrast, in their 2017 study, the El-Agnaf group found no differences in CSF concentrations of o-αSyn or pS129-αSyn between the AD and other neurodegenerative disease (OND) groups while total CSF αSyn concentration was markedly elevated in the AD cohort compared to the OND cohort [90]. The authors also concluded that inclusion of CSF o-αSyn measured by the antibody Syn-O2, detecting αSyn fibrils [116], did not improve the global diagnostic performance of total CSF αSyn in detecting AD [90]. Finally in the latter work, neither CSF total αSyn nor CSF o-αSyn discriminated AD from cognitively normal controls [115]. In summary, one study reported a reduction in CSF o-αSyn concentration in AD subjects and two other studies found no differences. Like for total CSF αSyn, these differences prevent the formation of a clear consensus about the abundance of o-αSyn in human CSF and are likely due to the ELISA platform used (homotypic ELISA with 211 vs. heterotypic ELISA with Syn-O2) and heterogeneity of the subjects considering that all three studies were generated in the same laboratory.
Huntington’s disease. Huntington’s disease (HD) is the most common neurological disorder caused by trinucleotide repeat expansions. In HD, the expanded CAG repeats encode polyglutamine (polyQ) sequences in the huntingtin (Htt) protein thereby conferring toxic gain-of-function and loss-of-function properties to the mutant protein [117]. Based on a case report documenting the colocalization of αSyn with Htt inclusions in the striatum of a male subject with HD and in the brain of transgenic mouse models of HD (HD89, HD94 and R6/1), an aberrant interaction of mutant Htt with αSyn has been proposed [4, 5]. The exact nature of this inter-relationship is still under investigation with studies indicating that αSyn forms independent fibrillar aggregates in neurons harboring Htt deposits in vivo [5] while other work relying on bimolecular fluorescence complementation (BiFC) assays found that αSyn oligomerizes and co-aggregates with N-terminally truncated Htt (Httexon1) in human H4 neuroglioma cells [17]. More recent work expanded these in vitro studies by reporting the colocalization and co-aggregation of αSyn and mutant Htt in the fly brain [18]. Despite the mention of αSyn oligomerization, it is worth noting that none of these studies provided direct evidence of a contribution of o-αSyn to the interaction with Htt. Overall, additional studies are warranted to better understand the inter-relationship between αSyn and Htt in vivo.
Detection of oligomeric αSyn species by selective brain areas
Neurodegenerative diseases are also defined by the neurodegeneration of distinct brain regions or nuclei, implying a selective vulnerability of neuronal subtypes to the accumulating amyloid protein [1]. Across the studies aforementioned above which have measured o-αSyn in post-mortem brain tissue, rare are those that analyzed multiple brain regions [5, 88, 108].
Roberts and colleagues detected αSyn assemblies using proximity ligation assay (PLA), a technique relying on the close proximity of two molecules in fixed samples using two primary antibodies, in three brain regions, cingulate cortex, midbrain and medulla from control subjects and subjects with PD (Braak stages 3–6, n = 8 cases per group) [88]. Within each region, three to six areas were examined including the substantia nigra pars compacta and raphe nuclei. The αSyn species revealed by PLA (αSyn-PLA) indicated lightly compacted pathological aggregates in only three sub-regions of PD brains, namely the medullary intermediary reticular zone, reticular formation and pyramidal cortical neurons, which were different from age-matched control subjects [88]. The exact nature of the species reactive to αSyn-PLA and the relevance of their spatial distribution remains unclear.
In their analyses of AD brain tissues, Larson and coworkers also detected o-αSyn across five brain regions, including the inferior temporal gyrus (ITG), entorhinal cortex, the midfrontal gyrus (MF), the angular or inferior parietal gyrus (AG), and the calcarine cortex (CALC), encompassing all five cerebral lobes [108]. The accumulation of o-αSyn principally observed in the ITG, was not limited to this gyrus as other brain regions (parieto-occipital and entorhinal cortices) also displayed marked increases in o-αSyn despite a relatively-low number of specimen per clinical group per area (n = 5–6) [108]. It is interesting to note that all brain areas displaying elevations of o-αSyn are part of the default mode network linked to AD, where Aβ and tau pathologies are prevalent [118–120]. Clearly, larger studies will be needed in the future to extensively compare regional differences within each subject.
Lastly, the Lucas group observed αSyn-positive inclusions in the striatum and the cerebral cortex of subjects with HD (grade 4 following the criteria of Vonsattel) [5]. The group sizes were statistically minimum (n = 3) and the report does not indicate which domain of the cerebral cortex was used for these studies, thereby preventing any rigorous conclusion.
Overall, extensive work remains to be done to determine the existence, expression, accumulation and potential spreading of o-αSyn in disease-specific vulnerable brain areas.
Current understanding of oligomeric αSyn production and toxicity in vivo
Dimers
Studies by several groups showed that chemical or photochemical crosslinking of αSyn in mammalian cells or purified αSyn from E. coli bacteria results in the formation of a ladder of αSyn species ranging from monomers to hexamers, with 35 kDa dimers being the major species [25, 43, 69, 77, 121]. Of note, the 35 kDa αSyn dimers seem to be the most commonly detected form of αSyn assemblies in denaturing polyacrylamide gels and appear to be stable in the presence of conditions that would disrupt their oligomerization state including chemical denaturants, suggesting that they might be covalently crosslinked [77, 86, 122, 123].
Similarly, analysis of protein lysates from human brain tissue also yield equivalent electrophoretic profiles [21, 67, 107, 108, 124, 125]. Amongst those reports, Larson and colleagues readily detected the presence of apparent 35 kDa αSyn dimers in temporal cortices of subjects with no cognitive impairment (NCI), mild cognitive impairment (MCI) and AD by western blotting using LB509, an antibody specific to human αSyn [107]. In follow-up studies, the same group reported similar SDS-PAGE profiles for αSyn using the monoclonal antibody 4D6 to αSyn, which displayed enhanced sensitivity towards oligomeric αSyn [108]. Importantly the oligomeric nature of these 35 kDa αSyn dimers was confirmed by several approaches including homotypic LB509-LB509 ELISA detection, size-exclusion chromatography (SEC) combined with denaturing and non-denaturing analyses of SEC eluates using antibodies detecting various oligomers of amyloid proteins (A11, OC, Officer), as well as antibodies specific to o-αSyn (Syn33 and F8H7) [108]. In addition to the 35 kDa αSyn dimers, another putative αSyn dimer of ∼28 kDa was also detected in postmortem human brain tissue and in brain tissue from transgenic TgI2.2 mice overexpressing WT human αSyn [19, 98, 107, 108]. Whereas OC and Officer antibodies detected fibrillar amyloid species co-segregating with the 35 kDa and 72 kDa αSyn molecules in both AD and TgI2.2 samples suggesting that these αSyn forms are prefibrillar oligomeric αSyn assemblies, the 28 kDa αSyn dimers were instead detected with A11 and F8H7 in AD brain tissue and to a lesser extent in TgI2.2 mice, indicating that this αSyn species is a nonfibrillar oligomer [108]. As a reminder, OC antibodies detects in-register parallel β-sheets while A11 antibodies recognize out-of-register anti-parallel β-sheet motifs [126–128]. To our knowledge, these findings were the first to provide indirect evidence of the existence of two separate dimeric assemblies of αSyn with distinct conformations, which had been previously suggested [25]. It is unclear whether these dimeric forms are apparent multimers of 14- and 17 kDa αSyn monomers or conformation variants from the same molecular αSyn unit and further studies will be needed to investigate these hypotheses.
Functionally, multivariate regression analyses revealed inferior temporal gyrus levels of putative 28- and 35 kDa αSyn dimers were inversely correlated with episodic memory deficits [108]. Beyond these correlative observations, proteo-transduction of SEC-segregated 28 k-Da αSyn dimers isolated from TgI2.2 mice onto wild-type primary neurons resulted in a marked suppression of synapsin-I and synapsin-II levels whereas parallel experiments using SEC-segregated monomeric αSyn had no effect on synapsins. Note that because the 35 kDa αSyn dimers co-segregated with larger αSyn assemblies (>70 kDa), it was not possible for the authors to assess a contribution of that assembly unlike the SEC-segregated 28 kDa αSyn dimers. Remarkably, the changes induced by SEC-segregated 28 kDa αSyn dimers were also observed in SNCA-null primary neurons indicating that these species do not dependent on endogenous αSyn expression to mediate their effects on synapsins. Promoter activity assays and cellular signaling studies further revealed: 1) that SEC-segregated 28 kDa αSyn dimers suppress the activation of two αSyn-regulated signaling pathways controlled by the cAMP responsive element-binding protein (CREB) and the nuclear receptor related 1 protein (Nurr1) and 2) novel responsive elements to CREB and Nurr1 in the 5′UTR/promoter region of SYN1 and SYN2 genes, two transcription factors known to be suppressed by αSyn [129, 130]. Finally, independent studies from the Björklund and Perlmann groups also demonstrated that lentivirus-mediated overexpression of full-length human WT αSyn (αSynWT) in midbrain dopaminergic neurons resulted in the detection of 28 kDa αSyn dimers and reductions in CREB and Nurr1 signaling [130]. Taken together, these results provide molecular insights in neuronal changes induced by isolated αSyn dimers and warrant additional work comparing the functional roles of these two distinct dimeric αSyn species.
Trimers
Rare studies have documented the existence of putative αSyn trimers with a theorical molecular weight ranging between 42–51 kDa based on 14- or 17 kDa αSyn monomeric units in protein lysates from brain tissue [86]. One could argue that a potential reason for the apparent absence of evidence for trimeric αSyn assemblies could be related to an observation made by the Haass group years ago. Kahle and colleagues [125] noted that putative αSyn trimers migrating between the 44 and 71 kDa molecular standards were present in ultracentrifugation supernatants of in vitro formed aggregates of recombinant αSyn partially solubilized in Tris-buffered saline (TBS). However, addition of 5% SDS to fully solubilize αSyn resulted in a nearly complete disappearance of these assemblies in favor of monomeric and 35 kDa dimeric αSyn species. It is however possible that, given the low resolution of the immunoreactive band(s) combined with that of the gel used as evidenced by the small distance separating the 44 and 71 kDa standards, the putative trimer might in fact correspond to a tetrameric assembly or both. Another potential confounding factor preventing reliable identification of putative ∼42–51 kDa αSyn trimers in lysates from brain tissue relies on inconsistent electrophoretic migration across sample types, suboptimum gel resolution and overreliance on western-blot detection using a single antibody. Accordingly, it is clear that further work will be needed to validate the existence of trimeric αSyn in brain tissue.
Tetramers
Contrasting with the anecdotal reports related to possible αSyn trimers, several groups have detected putative tetrameric assemblies of αSyn in brain lysates [67, 71, 88, 108]. The experimental observation of putative tetramers is conceptually appealing considering the prominent detection of αSyn dimers and considering the potential diversity of dimeric assemblies (see dedicated chapter above). Based on the same model relying either on 14- or on 17 kDa αSyn monomeric units, homomeric αSyn tetramers can thus encompass multimeric species whose molecular weights range between 56 to 68 kDa. Like αSyn dimers, two tetrameric αSyn assemblies may exist, i.e., ∼58–60 kDa αSyn species (proposed tetramer of 14.5 kDa αSyn) sensitive to SDS [71] and ∼56 kDa αSyn species (possible tetramer of 14 kDa αSyn or compact tetramer of 17 kDa αSyn) resistant to SDS [88, 108], based on the conflicting sensitivity of putative physiological tetramers and oligomeric tetramers to this anionic detergent. Despite the apparent resistance to SDS, the 4D6-immunoreactive ∼56 kDa αSyn species identified by Larson and colleagues are not covalently linked as 10 to 20% hexafluoroisopropanol (HFIP) trigger its disassembly in favor of monomeric αSyn [108]. This finding thus opens the door to the hypothesis that this oligomeric assembly corresponds to a possible homomeric tetramer of 14 kDa αSyn, a compact homomeric tetramer of 17 kDa αSyn molecular units or a heteromeric tetramer composed of both molecular building blocks. To attempt getting at this question, it can be postulated that, if the 28 kDa or the 35 kDa αSyn dimers were the nuclei of the ∼55–56 kDa αSyn assembly, then their relative abundance in vivo might be related in brain tissue. Leveraging existing data from the Religious Orders Study cohort initially reported by Larson and coworkers [108], we performed pair-wise regression analyses which unexpectedly revealed strong positive correlations between the 35 kDa and 55 kDa αSyn species in intracellular-enriched protein fractions from individuals with no cognitive impairment (NCI) and from subjects with AD (Fig. 2A). Quantile density contours readily illustrated the parallel elevation of both species in AD tissue compared to the NCI group (Fig. 2A). By contrast, there was no apparent relationship between the 28 kDa and 55 kDa αSyn species in the same biological samples (Fig. 2B). Together, these new results thus favor the notion that the increased amounts of intracellular 55 kDa αSyn species in AD brain tissue might depend on the availability of 35 kDa αSyn dimers, as opposed to the smaller 28 kDa αSyn dimers. This hypothesis is further supported by SEC profiles of similar AD lysates in which the 35 kDa αSyn dimers reliably co-eluted with larger αSyn species including 55–56 kDa assemblies while the 28 kDa αSyn dimers displayed an elution profile consistent with a globular molecule allowing its isolation from other αSyn species [108].
Fig. 2
Relationships between 28-, 35- and 55 kDa αSyn oligomers in human brain tissue from cognitively intact subjects and subjects with AD within the ROS cohort. Regression analyses and quantile density contours for pair-wise comparisons between 35 kDa (A) or 28 kDa (B) dimeric αSyn and the ∼55–56 kDa putative αSyn tetramer detected in the intracellular-enriched fraction (IC). All species were detected with the monoclonal antibody 4D6. NCI, no cognitive impairment; AD, Alzheimer’s disease; ROS, Religious Orders Study.
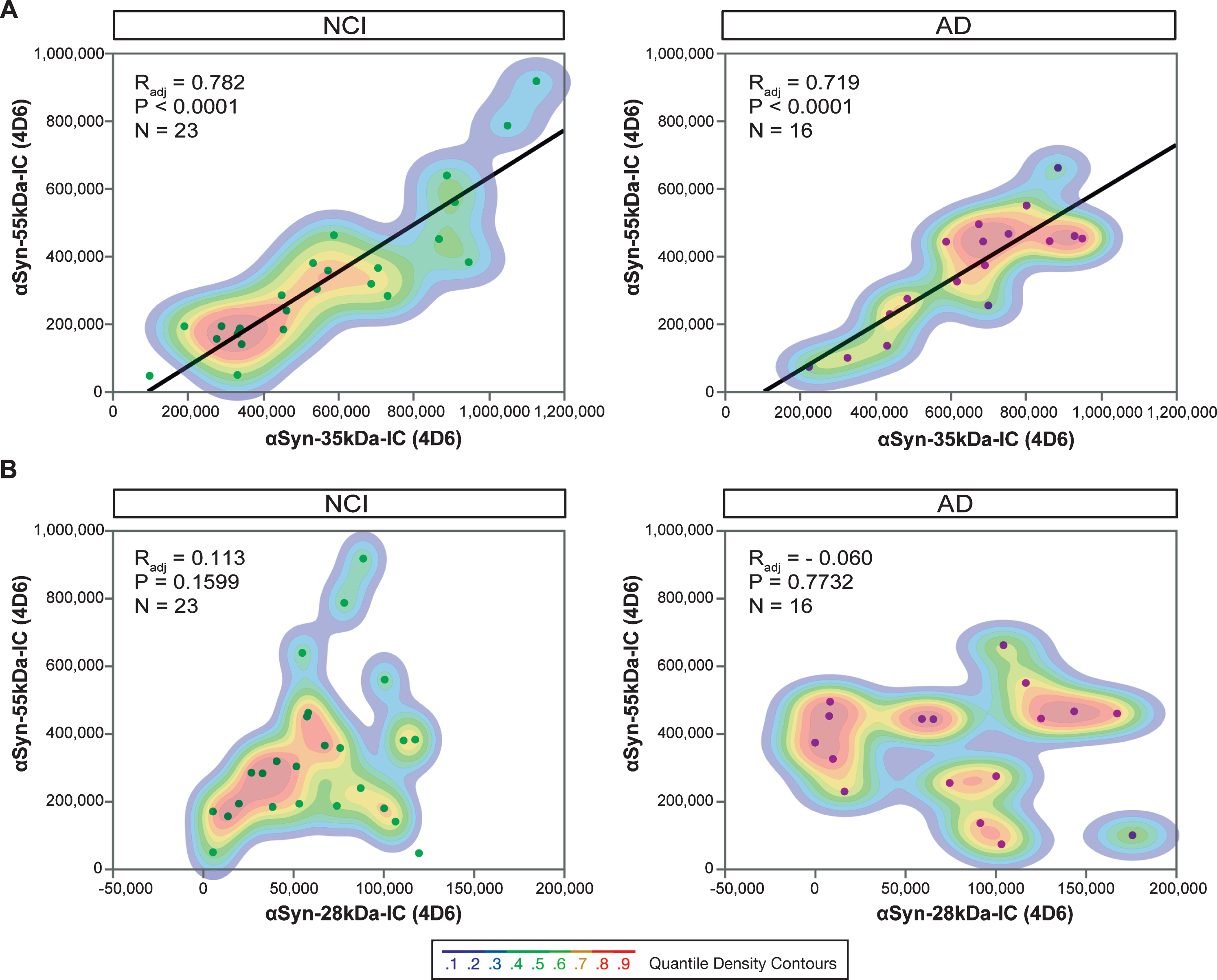
Beyond the differences in their respective intrinsic biochemical properties, the SDS-sensitive ∼58–60 kDa αSyn tetramers, also called αSyn ‘multimers’ [71], and the SDS-resistant ∼55–60 kDa αSyn species [108] also appear to possess markedly different functions. As a brief reminder, genetic destabilization of αSyn multimers in mice was recently shown to produce a phenotype reminiscent of PD [73], indicating a supportive or physiological function of αSyn multimers in healthy conditions [58]. Contrasting with this role, brain levels of SDS-resistant ∼55 kDa αSyn oligomers inversely correlated with episodic memory and semantic memory performance in subjects with AD [108]. Moreover, SEC-segregated αSyn species containing SDS-resistant ∼55 kDa αSyn oligomers caused a selective suppression of synapsin-1 and synapsin-2 proteins in cultured cortical neurons by a prion-like mechanism requiring αSyn expression [108]. These results therefore indicate divergent functions of distinct αSyn tetramers in health and disease.
Annular protofibrils
Earlier it was shown that A30P and A53T mutants can form pore-like annular protofibrils faster than αSynWT and were capable of permeabilizing in vitro prepared phosphatidylglycerol (PG) vesicles. It was proposed that an increase in αSyn protofibril structures might occur in an early stage of PD, thus suggesting a possible mechanism of toxicity in PD [131]. Moreover, the same study had also demonstrated that in addition to annular protofibrils, A53T mutant synuclein can form tubular protofibrils. An ordered assembly of αSynWT oligomers into protofibrils significantly permeabilized synthetic lipid vesicles, implying their ability to disrupt cellular membranes [132]. Using multiple techniques, including cryo-EM, another study has demonstrated the formation of two types of annular assemblies of αSyn, one with a typical ring shape and another with a cylindrical shape. Addition of these assemblies into rat midbrain neuronal cultures not only generated reactive oxygen species but also significantly disrupted synthetic lipid vesicles compared to monomeric or fibrillar αSyn [133]. However, in another study, αSyn pre-fibrillar oligomers (PFOs) showed comparatively stronger membrane-permeabilizing activity than the annular protofibrils of αSyn. Furthermore, αSyn PFOs were significantly more toxic when applied exogenously to SH-SY5Y cells compared to αSyn annular protofibrils [134]. As different types of o-αSyn were shown to cause cellular toxicity at different degrees, such mixed observations with annular protofibrils toxicity imply the complex nature of these amyloid assemblies.
Other assemblies
Large oligomers of αSyn preferentially bind to the SNARE protein synaptobrevin-2 and inhibit SNARE complex formation, thus impeding vesicle docking and neurotransmitter release [135]. Small diffused granular αSyn aggregates were enormously detected in the presynaptic terminals of neurons in DLB brain tissues along with significant loss of postsynaptic dendritic spines, suggesting the role of such granular αSyn aggregates in synaptic dysfunction [136].
Using newly generated transgenic mice expressing split luciferase αSyn probes (the constitutive S1/S2 mice and the inducible VS1/VS2 mice), recent work from the Danzer group demonstrated an age-dependent accumulation of endogenous 8–16-mer o-αSyn species, which reached its maximum levels in 24-month-old animals [137]. At a subcellular level, these o-αSyn species appear to accumulate preferentially within the presynaptic compartments and not within the neuronal soma, perhaps due to the enrichment of synucleins in this element. Importantly, 24-month-old S1/S2 mice displayed a reduced anxiety-like behavior, a phenotype also observed in transgenic mice overexpressing mutant A53T αSyn [138].
Future directions
As summarized above, many forms of putative oligomeric assemblies of αSyn have been detected across several neurological diseases. To improve the reproducibility of o-αSyn detection and to facilitate comparisons across independent groups, we here advocate for a reduced reliance on single antibody detection following western blotting and for increasing the necessity to include biochemical analyses comprising combinations of non-denaturing (e.g., homotypic ELISA, heterotypic ELISA with oligomer-specific antibodies, liquid-phase chromatography separation, clear native-PAGE, dot/spot blotting) and denaturing approaches (western-blotting, co-immunoprecipitations) with extensive panels of pan-, conformation- or oligomer-specific antibodies. In particular, we would like to draw attention to characterizing all brain-derived αSyn oligomers using A11 and OC due to their demonstrated preference in detecting distinct β-sheet conformers [126–128]. Given the heterogeneity among the oligomeric assemblies of αSyn and pending an amelioration of the isolation techniques for oligomeric αSyn from brain tissues or cells (typically generating low to very low yields), cryo-electron microscopy (Cryo-EM) and NMR studies will likely prove essential to dissect out the structural and conformational variants of various oligomers.
Post-translational modifications including truncation [139], nitration [140] and phosphorylation [141, 142] might play important roles in αSyn aggregation and hence its toxicity (see for review [143]). Paleologou and colleagues showed oligomeric pS129-αSyn in the membrane fractions of DLB and MSA brain tissues ranging from ∼28 to 98 kDa molecular weights. Similarly, monomeric (∼17 kDa) and oligomeric (∼42 kDa) pS129-αSyn were also detected in the membrane fractions of brain tissues from transgenic mouse models of synucleinopathies [141]. Finally, studies in flies demonstrated that soluble endogenous o-αSyn was increased by pS129 and decreased by phosphorylation at tyrosine 125 (pY125) [142]. Moreover, biochemical analysis of postmortem human brain tissues revealed a reduction of pY125-αSyn with aging and in subjects with DLB [142]. These findings suggest that αSyn-induced toxicity may result from a dysregulation between an oligomer-promoting effect of pS129 and an oligomer-inhibiting effect of pY125. Therefore, mass spectrometry-based analysis of post-translational modifications of in vivo occurring αSyn aggregates (including o-αSyn) would be important to gain more understanding of the pathogenic αSyn assemblies.
If achieved, these largely technical improvements should allow the field to compare the function of multiple αSyn species in parallel, reminiscent of the work done for Aβ oligomers in the AD field. It would be indeed surprising to find that each o-αSyn member of this pleiotropic ensemble acts similarly on neuronal and glial cells in the brain.
Lastly, two very recent studies from the Holtzman and Bu groups investigated whether APOE ɛ4 genotype, a major risk factor for neurodegenerative diseases, affected αSyn pathology in mice and subjects with PD [144, 145]. While both studies relied on APOE knock-in (E2/E3/E4) backgrounds to compare the impact of APOE ɛ2/ɛ3/ɛ4 alleles in mouse models of synucleinopathy, Davis and colleagues used G2.3 TgA53T mice [145]; Zhao and coworkers opted for an AAV-mediated overexpression of human αSynWT, which does not cause amyloid inclusions as defined by a lack of Thioflavin-S positivity [144, 146]. In both experimental settings, E4 exacerbated αSyn pathology, including pathological conformational changes detected by the antibody 5G4 and pS129-αSyn accumulation, and worsened motor deficits. However, none of these reports assessed the exact nature of the accumulating αSyn. A qualitative western blot analysis using Syn1 and MJFR-13 antibodies detecting total αSyn and pS129-αSyn respectively revealed ∼23 kDa and ∼30 kDa bands, in addition to monomeric ∼14 kDa αSyn, in the SDS-soluble fraction of symptomatic G2.3 TgA53T mice which the authors interpreted as o-αSyn oligomers [145]. Several points are problematic with this analysis: (i) there are no quantitative measurements provided; (ii) these additional bands are detected in symptomatic TgA53T/E4, TgA53T/E3 and TgA53T lacking murine APOE (TgA53T/EKO) with no apparent difference across APOE genotype; (iii) the molecular weights (∼23 kDa and ∼30 kDa) are inconsistent with those previously reported for o-αSyn by electrophoretic separation as discussed above in this section. Based on these findings, further work is needed to determine if αSyn oligomerization is altered by APOE ɛ4 and, if it were, what o-αSyn species are differentially accumulating across APOE genotypes.
SPREADING OF OLIGOMERIC αSYN SPECIES
Introduction to prion-like propagation of αSyn
A group of highly infectious neurodegenerative diseases, known as transmissible spongiform encephalopathy (TSE) or prion diseases is caused by the infectious prion protein or PrP [147]. Animals affected with prion diseases exhibit different phenotypes. One of the most astonishing phenomena in prion diseases is that animals infected with the same pathogenic infectious agent displayed different clinical phenotypes [148–150]. Moreover, when such infectious protein aggregates were isolated and inoculated in identical hosts, they induced distinct prion disease specific phenotypes [151]. Growing experimental evidence suggests that many neurodegenerative diseases share common characteristics with prion diseases. Indeed, the pathogenic protein conformers of amyloid proteins defining neurodegenerative diseases can seed and promote the aggregation of their natively-folded counterparts in a prion-like manner [152].
Based on the course of the Lewy pathology in the enteric nervous system (ENS), peripheral nervous system (PNS) and central nervous system (CNS) together with clinical symptoms of patients with sporadic PD, Del Tredici and Braak have proposed the following staging procedure [153]. At stage 1, the Lewy pathology initially develops somewhere in ENS, PNS or CNS, then is detected in olfactory structures (bulb and nucleus) and in the medulla oblongata, specifically in the dorsal motor nucleus of the vagal nerve (CN X) and/or intermediate reticular zone. At stage 2, LNs and LBs are found in peripheral parasympathetic and sympathetic nerves, medullary nuclei including the locus coeruleus and lower raphe nuclei. Stage 3 is characterized by αSyn pathology in the substantia nigra, pars compacta and the magnocellular nuclei of the basal forebrain. At stage 4, intralaminar thalamic nuclei and anteromedial temporal cortex (including entorhinal cortex and hippocampus) are affected. At stage 5, the high-order sensory association areas of the neocortex and prefrontal fields are affected while at stage 6, first-order sensory association, premotor, primary motor and sensory areas are also affected, ultimately coinciding with dementia. This mostly ascending caudo-rostral neuropathological pattern is suggesting that, once formed, αSyn aggregates seem capable of propagating between neurons synaptically interconnected.
In the late 2000s, several reports documented host-to-graft propagation of αSyn pathology in fetal mesencephalic transplants of subjects with PD [154, 155]. Shortly thereafter, Desplats and colleagues demonstrated that neurons overexpressing αSyn can transmit the protein to neighboring cortical neuronal stem cells expressing the green fluorescent protein (GFP) in vitro and in vivo [156]. In the accompanying commentary, Olanow and Prusiner openly questioned whether PD was a prion disorder [157] promoting vast interest in the field to entertain the hypothesis that some forms of αSyn, perhaps including oligomeric or small fibrillar assemblies, might act as prion-like conformers and promote the misfolding of additional wild-type proteins. By extension, these studies also suggested that αSyn aggregates may propagate in anatomically connected neural networks.
Spreading of endogenous oligomeric αSyn species in vitro
Although the exact mechanism by which o-αSyn exert their toxic effects is still unknown, the presence of these oligomeric assemblies in the extracellular body fluids suggests their release from affected brain regions and subsequent potential spreading between cells. The cell-to-cell propagation of o-αSyn could occur through the internalization of these species via receptor-mediated endocytosis as demonstrated in several cell lines and primary neurons [158–161]. In addition, exogenously added o-αSyn can act as seeds promoting the aggregation of endogenous αSyn in primary neuronal cells as well as in neuronal cell lines, suggesting that extracellular release of o- αSyn coupled with reuptake as a plausible mechanism for the spreading of αSyn pathology [162]. Specifically, secretion of αSyn within exosomes has been proposed to amplify and propagate PD-associated αSyn pathology in cell culture systems [96, 97]. It has been suggested that effective propagation of αSyn aggregates is dependent on both the templated seeding process and the in vivo cellular condition [163].
Spreading of endogenous oligomeric αSyn species in vivo
To our knowledge, experimental evidence demonstrating a spreading of endogenous o-αSyn is rare [137] in contrast to strategies relying on exogenous injection of recombinant aggregated αSyn preparations in rodent brains [164–169]. Leveraging newly created conditional S1/S2 and VS1/VS2 transgenic mice in which protein-fragment complementation of tagged αSyn occurs, Kiechle and coworkers provided novel evidence of o-αSyn formation at presynaptic terminals, age-dependent accumulation of 8–16-mer αSyn oligomeric assemblies and trans-synaptic spreading of endogenous o-αSyn species from transgene-expressing neurons to synaptically-connected nuclei which did not express the transgene [137]. These changes were accompanied by reduced locomotor deficits, behavioral abnormalities (e.g., reduced anxiety-like behavior), olfactory dysfunction, reduced striatal dopamine concentrations and ultimately loss of dopaminergic neurons in the substantia nigra [137]. While further elaboration on such elegant study is necessary to better understand the extent of oligomeric αSyn-induced synaptotoxicity, these findings add weight to prior work demonstrating that endogenous o-αSyn species alter synaptic structure and function in vivo [108].
INTERACTIONS BETWEEN OLIGOMERIC αSYN ASSEMBLIES AND OTHER AMYLOID PROTEINS
Although amyloidogenic proteins might follow unique cascades of pathological events, the majority of them shares certain common mechanisms for toxicity. Membrane permeabilization is one of such mechanisms that has been suggested for many amyloid proteins [170]. Co-occurrence of multiple protein pathologies represents a wide range of neurodegenerative disorders where comorbidity and overlap between the diseases are frequently observed [171]. In addition to LB, senile plaques of Aβ, neurofibrillary tangles (NFTs) and neuropil threads composed of tau were observed in the cerebral cortex of PD brain tissues [172]. Senile plaques and NFTs are often found together in DLB cases [173]. Thus, there is a considerable overlap between αSyn, tau and Aβ protein pathologies in synucleinopathies, suggesting their potential synergistic effects on pathogenesis.
For the context of this review, we will differentiate the terms “cross-talk” as defined by direct or indirect molecular interactions in absence of enhanced fibrillization from “cross-seeding” which occurs when oligomers composed by one misfolded amyloid protein induce the oligomerization of a distinct amyloid protein.
αSyn/Aβ hybrid species
Cross-talk
While many studies have suggested that αSyn and Aβ are capable of direct and indirect molecular cross-talks in the brain along with hybrid oligomer formation [21, 174–179], few studies have demonstrated that endogenous Aβ and αSyn directly interact in cultured cells or in vivo [21]. Specifically, Tsigelny and colleagues reported that αSyn and Aβ directly interact in the brains of small (n = 4) groups of patients with AD, DLB and in APP/α-syn tg mice (Thy1-APPmut tg [line 41] x PDGFβ-α-syn tg mice [line D]) using co-immunoprecipitation analyses of membrane fractions. Although immunoreactive bands at ∼28, ∼40 and ∼70 kDa labeled as αSyn dimers, trimers and pentamers by the authors were readily detected by western blotting using unspecified αSyn antibodies in the membrane fractions of AD, DLB and APP/α-syn tg brain tissue, only monomeric αSyn (or Aβ) and no oligomeric species were identified by coimmunoprecipitation. By contrast, two independent studies from the Lesné group failed to observe putative αSyn/Aβ hybrid species in AD brain lysates and in double transgenic APP/αSyn mice (PDGFβ-APPmut tg [line J20] ×PrP-αSyn tg mice [line TgI2.2]) using non-denaturing dot blotting following size exclusion chromatography with several monoclonal αSyn antibodies (LB509, 4B12 and 4D6) or using coimmunoprecipitation with the LB509 αSyn antibody [19, 108]. All three studies used the monoclonal antibody 6E10 to detect human Aβ. Overall, these findings are not consistent and additional work is needed to confirm the presence of endogenous αSyn/Aβ hybrid assemblies and to assess whether differences in the approaches and/or in the biological specimens might be responsible for these diverging observations.
To assess whether Aβ and αSyn aggregation co-occurs or whether a potential cross-talk between these two amyloid precursors, Bassil and coworkers (2020) injected mouse αSyn preformed fibrils (αSyn-mPFFs) into young adult 5XFAD mice harboring amyloid plaques. The presence of Aβ deposits enhanced αSyn pathology and spreading throughout the brain [180]. Mechanistically, the injection of αSyn-mPFFs altered APP processing favoring Aβ production resulting from a decrease in ADAM-10 protein abundance coupled to an increase in presenilin-2 and nicastrin, two members of the γ-secretase complex. These findings have led the authors to suggest a “feed-forward” mechanism whereby Aβ plaques potentiate αSyn seeding and spreading over time [180]. It is however worth noting that soluble APP-α (sAPPα) levels were remarkably unchanged despite a 50% reduction in ADAM-10, a proponent candidate of α-secretase activity, arguing against an overall decrease in α-secretase activity. In addition, these putative alterations of APP processing induced by αSyn were not observed in two other recent studies examining the interaction between Aβ and αSyn [19, 20]. Furthermore, these latter two studies actually reported effects of αSyn on Aβ deposition opposite to that described by Bassil and colleagues, whereby overexpression of either mutant human αSynA30P or αSynWT in two distinct APP transgenic mice led to a reduction in Aβ burden [19, 20]. Both independent groups validated these in vivo findings with in vitro approaches relying on thioflavin-T analyses with recombinant proteins [20] and relying on measurement of on-pathway amyloid oligomers using conformation-specific OC antibodies [19]. Of particular interest, the forebrain abundance of o-αSyn detected with oligomeric αSyn-specific antibody Syn33 was highest in bigenic APP/αSyn mice when Aβ deposits were the lowest [19]. Furthermore, ablation of the SNCA gene encoding for αSyn in the same APP transgenic model led Khan and colleagues to witness a bidirectional effect of αSyn on Aβ deposition, by which the genetic removal of αSyn elevated Aβ deposition [19] reminiscent of prior work by the Zheng group [181]. While differences in APP transgenic mouse models could in principle account for these differences, it is more likely that the nature of the αSyn molecules introduced in these various models is contributing to the diverging observations. Indeed, while αSyn-mPFFs are created by sonicating long, pre-formed fibrils are injected in young mice, transgene-derived expressions of human αSyn used by the Meyer-Luehmann and Lesné groups do not induce αSyn fibrillization. Therefore, it is likely that the crosstalk between Aβ and αSyn differs based on the conformational nature of each component, i.e., oligomers vs. fibrils, and with dramatically distinct functional consequences.
αSyn/tau species
While αSyn aggregates are considered as the main pathogenic entities in synucleinopathies, tau pathology is also abundantly detected in these diseases (in > 92% of subjects with synucleinopathies [3]). Abnormal accumulation of the microtubule binding protein tau has been implicated in a group of neurodegenerative diseases known as tauopathies [182]. However, an increasing amount of studies has demonstrated the possible interaction between αSyn and tau. NFTs consisting of hyperphosphorylated tau have long been studied in PD and DLB cases. Genome-wide association studies (GWAS) have indicated a strong association between SNCA and MAPT genes coding for αSyn and tau, respectively, in PD and DLB pathogenesis [183, 184]. Moreover, tau aggregates have been reported in several transgenic mouse models of PD and DLB [102, 185–189].
There is also accumulating evidence that spreading of pathogenic protein aggregates is a necessary event in the progression of neurodegenerative diseases [190–193]. To this end, studies have also been directed to dissecting out the mechanisms by which amyloidogenic protein aggregates spread from cell to cell [190, 191]. Walker et al. have suggested a templating mechanism of misfolded protein in which once a small amount of aggregate is formed, it serves as “seed” for further aggregation. Such seeds can recruit newly-formed protein by templating their conformation to the newly added proteins [194], including αSyn [195] and tau [196, 197].
Cross-talk
Using human brain cytosol from unspecified source and characterization, early studies reported that αSyn directly interacts with the microtubule binding region of tau thereby modulating the phosphorylation state of tau proteins at least at two serine residues (S262 and S356), suggesting a possibility of direct physical interaction between αSyn and tau [198]. In situ, LBs found in PD and DLB cases also contain hyperphosphorylated tau [199, 200]. A few years later, transgenic TgA53T mice overexpressing mutant human αSynA53T (M83 line) were shown to display abundant tau pathology as threads, grains, spheroids and pre-tangles in several brain regions [201, 202], whereas transgenic mice overexpressing human wildtype αSynWT (unspecified line from PDGF-αSyn mice [203]) develop spontaneous pathological tau changes including tau hyperphosphorylation and tau misfolding [189]. Profound neuronal tau inclusions resembling NFTs were also found in transgenic mice overexpressing another human αSyn mutation, E46K [102]. Despite the demonstration that mutant αSynE46K is less efficient than αSynWT at promoting tau inclusions in cultured QBI293 cells, it is worth noting that TgE46K mice displayed a greater number of tau inclusions compared to TgA53T mice [102], suggesting the marked effects of the environment, species and cell type differences on potential molecular interactions between αSyn and tau. To date, the reason for the accumulation of hyperphosphorylated tau inclusions in TgE46K mice remains unclear and may warrant additional work, specifically with respect to αSyn and tau oligomers. Beyond the detection of co-occurring aggregates of tau and αSyn in neurons from M83 TgA53T mice, this co-aggregation was also exacerbated in oligodendrocytes from a bigenic CNP-TauP301L/αSynWT mouse model overexpressing mutant P301L tau and human αSynWT driven by an oligodendrocyte-specific promoter (2′,3′-cyclic nucleotide 3′-phosphodiesterase or CNP) [201]. In these cells, inclusions were positive to Thioflavin S [201], denoting advanced fibrillization.
However, these seminal studies focused on fibril formation as opposed to oligomerization per se. Consequently, the functional role of oligomeric αSyn/tau cross-talks in vivo was investigated by several groups. In recent studies, the co-occurrence of αSyn and tau in their toxic oligomeric forms was observed in postmortem brain tissues of PD and DLB patients [89]. In this study, Sengupta and colleagues demonstrated a co-immunoprecipitation of o-αSyn with tau oligomers (o-Tau) coupled with a cytoplasmic colocalization of these species in brain tissue from individuals with PD [89]. Recognizing that mixed protein pathologies of αSyn and tau are observed in PD whereas tau pathology alone is found without any documented αSyn pathology in progressive supranuclear palsy (PSP), Castillo-Carranza and coworkers investigated the toxicity of hybrid o-αSyn/o-Tau assemblies by injecting either complexes of oligomeric αSyn and tau hybrid species derived from PD brain tissues or PSP-derived o-Tau alone in human tau transgenic Htau animals overexpressing human tau isoforms in a murine tau background [23]. Hybrid o-αSyn/o-Tau complexes accelerated endogenous tau aggregation along with memory deficits in Htau animals compared to o-Tau [23]. Importantly, the functional interaction of αSyn/tau cross-talks in vivo was further investigated by Gerson and coworkers using passive immunotherapy with the tau oligomer-specific monoclonal antibody (TOMA) in M83 TgA53T mice [204]. Compared to mice injected with control immunoglobulins, TgA53T mice treated with TOMA displayed reduced synaptic loss, premature deaths and cognitive and motor deficits [204]. Importantly, TOMA-mediated lowering of tau oligomers in TgA53T mice was accompanied by a marked reduction in LB-like pathology and by an increase in o-αSyn detected by F8H7 and Syn33 antibodies. Based on the properties of TOMA antibodies, which preferentially detect off-pathway o-Tau [205], and based on those for F8H7 and Syn33, which preferentially bind off-pathway o-αSyn [89], these findings highlight complex molecular interactions between o-αSyn and o-Tau, which are not fully clear at this time.
Furthermore, the role of tau in mediating impairment of hippocampal neurotransmission and memory deficits in a similar transgenic animal model, TgA53T mice overexpressing mutant A53T human αSyn (line G2.3), has been recently demonstrated suggesting a tau-dependent mechanism of αSyn pathology [206]. However, this work from Singh and colleagues suggests that the role of tau in young and middle-aged adult G2.3 TgA53T mice is independent of o-αSyn/o-Tau cross-talks because no differences in o-αSyn nor o-Tau were found in absence or presence of MAPT deletion [206]. We speculate that this molecular cross-talk would strengthen with aging and have an impact on the phenotype of this PD mouse model.
Combined, these findings thus suggest the existence of hybrid o-αSyn/o-Tau complexes and of an influence of these oligomeric assemblies on each other’s aggregation via an interface in synucleinopathies. The exact nature of this interface and the exact functional impact of these o-αSyn/o-Tau hybrid forms remain unknown to date and gaining a better understanding of this molecular interaction could lead to the identification of novel therapeutic compounds for subjects affected by synucleinopathies.
Beyond a potentially direct effect of αSyn on tau hyperphosphorylation and/or aggregation, several studies from the Sidhu group indicate that αSyn may regulate the activity of glycogen synthase kinase-3β (GSK3β), a major tau kinase, in cell models of PD, signifying a possible indirect functional cross-talk between these two proteins [187, 188]. In this potential heteromeric αSyn/Tau/GSK3β complex, GSK3b could trigger the co-aggregation of αSyn and Tau into early oligomeric assemblies upon phosphorylation of both amyloid proteins thereby creating an interface permitting both proteins to influence each other’s aggregation in a discrete subcellular compartment.
Overall, these studies strongly support the importance of functional crosstalk between αSyn and tau.
Cross-seeding
As overlapping protein pathologies are frequently observed in multiple neurodegenerative diseases, cross-seeding between disparate proteins has been suggested as a possible mechanism underlying the detection of mixed pathologies in the same brain tissue [207, 208]. While it has been proposed that both tau and αSyn exert synergistic effects on each other’s aggregation, resulting in fibrillar amyloid structure formation in vitro and in vivo, most of the studies supporting this claim relied on recombinant forms of αSyn (oligomeric or fibrillar) with unclear disease relevance [24, 201, 209]. Few studies have assessed the cross-seeding propensity of endogenous or transgene-derived αSyn and tau in vivo. Clinton and colleagues demonstrated that crossing M83 TgA53T mice with the Alzheimer’s disease transgenic mouse model 3×Tg-AD [210] led to a reciprocal enhancement of the aggregation of all three transgene-derived amyloid proteins, i.e., Aβ, tau and αSyn, accompanied by an acceleration of cognitive deficits [211]. These findings suggested a synergistic interaction between αSyn, tau and Aβ (i.e., exacerbating each other’s aggregation) even though the aggregation nature of these amyloid proteins in this interaction remains unclear.
In more recent studies however, the bilateral intracerebroventricular injection of o-αSyn/o-Tau hybrid complexes, immunocaptured from PD brain tissue using a combination of o-Tau specific antibodies T22 and o-αSyn specific antibodies F8H7, in young Htau mice led to a delay in NFT formation compared to animals injected with o-Tau alone [23]. Supported by in vitro studies using recombinant proteins, Castillo-Carranza and coworkers proposed that o-αSyn/o-Tau complexes extend the lifespan of toxic tau conformers, leading to enhanced tau oligomerization and hippocampal neuron loss in vivo [23]. These results therefore suggest that: (i) the cross-seeding of tau induced by off-pathway F8H7-positive o-αSyn occurs in PD and (ii) αSyn oligomers enhance the deleterious effects of tau in synucleinopathies by reducing tau deposition.
Together, these αSyn/tau cross-seeding studies appear to indicate that the state of αSyn aggregates might be an important contributor in determining the fate of its interaction with tau.
αSyn/HTT species
Cross-talk
As discussed earlier, an aberrant interaction of mutant Htt with αSyn has been proposed based on the colocalization of αSyn with Htt within striatal inclusions of a male subject with HD and in the brain of transgenic mouse models of HD [4, 5]. Disparate conclusions have questioned the exact nature of a putative cross-talk between endogenous αSyn and Htt: 1) studies noted that αSyn forms independent fibrillar aggregates in neurons harboring Htt deposits in vivo [5] and 2) other work using BiFC assays found that αSyn oligomerizes and co-aggregates with N-terminally truncated Htt (Httexon1) in human H4 neuroglioma cells [17]. More recently, expansion of these in vitro BiFC studies led to the identification of colocalized and co-aggregated αSyn and mutant Htt in the fly brain [18]. Again, it is worth noting that none of these studies provided direct evidence of a contribution of o-αSyn to the interaction with Htt.
Overall, further investigation is needed to better understand the molecular inter-relationship between αSyn and Htt aggregates (including oligomers) in vivo. This task might be challenging in light of experimental studies performed in HD mice in which genetic modulation of αSyn expression bidirectionally affected autophagy and HD phenotypes [212].
OLIGOMERIC αSYN SPECIES AS DRUG TARGET(S)
Monotherapy
With the discovery of cell-to-cell spreading of αSyn oligomers, targeting extracellular protein aggregates to promote their clearance has been an active area of research. Studies involving both active and passive immunotherapy are being performed in preclinical settings. Developing antibodies that will specifically target the oligomeric forms of αSyn and using them for passive immunotherapy are getting more attraction. However, obtaining highly specific antibodies is a challenging task raising important questions to consider: would antibodies detecting common structural elements of αSyn oligomers be more beneficial? Or should developing antibodies specific to distinct o-αSyn assemblies be favored? In addition, several key variables can affect the outcome of such approaches including the delivery route of immunotherapy, internalization or cellular uptake of the antibody and the in vivo localization of the targeted oligomeric species [213]. Detection of o-αSyn in the CSF and blood plasma raises the possibility of early detection of pathological changes occurring in the CNS. To this end, probes specifically targeting o-αSyn with high sensitivity and high affinity hold great promise [214]. Of note, it is important to indicate here that higher CSF αSyn concentrations can be linked to blood contamination due to the expression of αSyn in erythrocytes [215], thereby requiring strict and rigorous markers for blood contamination and/or a standardized grade system for blood levels in CSF samples. Studies are also being performed to find molecules blocking the endocytosis of αSyn oligomers, thus preventing healthy cells from taking up pathogenic αSyn species, which could serve as a potential strategy for reducing oligomeric αSyn-mediated toxicity [214].
Molecules that can modulate the aggregation process by decreasing the load of αSyn oligomers have also been considered as therapeutic potential. The diphenyl-pyrazole molecule anle138b [3-(1,3-benzodioxol-5-yl)-5-(3-bromophenyl)-1H-pyrazole], an oligomer modulator was shown to inhibit αSyn oligomer formation both in vitro and in vivo [216]. This molecule strongly inhibited the formation and accumulation of o-αSyn and neuronal degeneration thereby suppressing disease progression in the TgA30P PD transgenic mouse model [216, 217]. Based on spectral emission properties in presence of o-αSyn, the authors proposed that this molecule targets directly o-αSyn in vivo [216], a hypothesis which was further supported by all-atom molecular dynamic simulations [218]. Both studies suggest Anle138b as a promising disease-modifying agent not only for synucleinopathies, but also for other amyloid forming proteins as reported recently for PrPC, Aβ and tau [216, 218–221]. Several other molecules such as NPT100-18A and NPT200-11 have also been reported as promising therapeutic agents that modulate o-αSyn formation. Among these, the small molecule αSyn misfolding inhibitor, NPT200-11 (Neuropore Therapies Inc./UCB), produced multiple benefits in the TgWT mice overexpressing αSynWT (line 61) used as model of PD [203], including attenuation of motor deficits and rescue of striatal dopamine transporter expression [222]. While the phase 1 clinical trial assessing safety, tolerability and pharmacokinetics of NPT200-11 was completed on February 2016 (NCT02606682; see for review [223]), no results have been reported as of yet. Other promising inhibitors of αSyn aggregation currently tested in phase 1 clinical trials include PBT434 (Alterity Therapeutic; U1111–1211-0052) and ENT-01 (Enterin; NCT03938922); both started last year. Curcumin, a natural polyphenolic antioxidant derived from turmeric, has demonstrated anti-inflammatory and neuroprotective effects in the Tg2576 AD mouse model despite poor bioavailability and presumably-poor blood-brain barrier penetrance of the parent compound [224–226]. In vitro, curcumin and its analogs have been shown to bind to pre-formed αSyn oligomers and to reduce their toxicity by modulating their morphologies and biophysical properties [227, 228]. Molecular chaperones, including heat shock proteins (HSPs), have also been considered as potential therapeutic alternatives against oligomeric αSyn-mediated toxicity [229]. Proof of principle studies in which overexpressing the chaperone HSP104 proved to be a safe approach in protecting dopaminergic neurons by disassembling o-αSyn in a lentivirus-mediated A30P-αSyn rat model of PD supported this view [230]. Molecules regulating HSP70 or HSP90 activity were also shown to be effective by lowering o-αSyn levels and their associated toxicity in a rat model of parkinsonism [231]. Such molecules might serve as effective therapeutic strategy for synucleinopathies.
Multi-therapy with other amyloid proteins
Most neurodegenerative diseases, including synucleinopathies, encompass various protein pathologies. Passive TOMA immunotherapy targeting tau oligomers in the M83 Tg53T animal model of PD prevents cognitive and motor deficits as well as brain protein pathology [204]. The importance of synergistic effects of αSyn and tau as outlined in above sections, indicates that a combinatorial immunotherapy against both αSyn and tau oligomers may constitute a better option to protect neurons from their toxicity. Given the complexity of synucleinopathies, multi-therapy that targets multiple proteins at their oligomeric states as well as altered biological pathways might turn out to be superior to monotherapies. Lastly, immunotherapies may be combined with modifiers of intracellular αSyn dynamics [232, 233] or lipid pathways that affect αSyn biology [234–236].
CONCLUSIONS
Aggregation of αSyn is a hallmark feature in synucleinopathies. Since the discovery of αSyn protein as the main component of the neuropathological lesions defining synucleinopathies, LB and LN, there has been a continuous effort to better understand the underlying mechanisms of these debilitating diseases. Despite significant advances made in recent years, the exact reasons behind the selective vulnerability of different neuronal and non-neuronal cell populations as well as brain regions to the aggregation of αSyn remain unknown. The implication of soluble αSyn oligomers in impairing multiple cellular processes has led to the consideration of such oligomeric assemblies as the main pathogenic entity in synucleinopathies. Hence, better understanding the molecular events leading to αSyn oligomer formation, seeding and propagation in vivo are of utmost importance. Unexpected findings such as the identification of native αSyn assembly [68] or the presence of a strong lipidic component in LB [237] will have to be critically investigated. Intensive and collaborative studies involving multi-disciplinary approaches will be necessary to further our understanding of these debilitating diseases and identify disease-modifying interventions.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NIH) to SEL (RF1-AG044342, R21-AG065693, R01-NS092918, R01-AG062135 and R56-NS113549), to RK (R01-AG054025, R01-NS094557 and RF1-AG055771) and to UD (R01-NS099328). Additional support included start-up funds from the University of Minnesota Foundation and bridge funds from the Institute of Translational Neuroscience to SEL, grants from Gilson Longenbaugh Foundation and Mitchell Center for Neurodegenerative Diseases to RK. We thank the study participants and staff of the Rush Alzheimer’s Disease Center and its director Dr. David Bennett at Rush University.
REFERENCES
[1] | Ross CA , Poirier MA ((2004) ) Protein aggregation and neurodegenerative disease. Nat Med 10: , S10–S17. |
[2] | Arnold SE , Toledo JB , Appleby DH , Xie SX , Wang L-S , Baek Y , Wolk DA , Lee EB , Miller BL , Lee VM-Y , Trojanowski JQ ((2013) ) Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J Comp Neurol 521: , 4339–4355. |
[3] | Robinson JL , Lee EB , Xie SX , Rennert L , Suh E , Bredenberg C , Caswell C , Van Deerlin VM , Yan N , Yousef A , Hurtig HI , Siderowf A , Grossman M , McMillan CT , Miller B , Duda JE , Irwin DJ , Wolk D , Elman L , McCluskey L , Chen-Plotkin A , Weintraub D , Arnold SE , Brettschneider J , Lee VM-Y , Trojanowski JQ ((2018) ) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141: , 2181–2193. |
[4] | Charles V , Mezey E , Reddy PH , Dehejia A , Young TA , Polymeropoulos MH , Brownstein MJ , Tagle DA ((2000) ) Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett 289: , 29–32. |
[5] | Tomás-Zapico C , Díez-Zaera M , Ferrer I , Gómez-Ramos P , Morán MA , Miras-Portugal MT , Díaz-Hernández M , Lucas JJ ((2012) ) α-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington’s disease. Hum Mol Genet 21: , 495–510. |
[6] | St-Amour I , Turgeon A , Goupil C , Planel E , Hébert SS ((2018) ) Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol 135: , 249–265. |
[7] | Schneider JA , Arvanitakis Z , Leurgans SE , Bennett DA ((2009) ) The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 66: , 200–208. |
[8] | Schneider JA , Arvanitakis Z , Bang W , Bennett DA ((2007) ) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69: , 2197–2204. |
[9] | Kapasi A , DeCarli C , Schneider JA ((2017) ) Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134: , 171–186. |
[10] | Bennett DA , Wilson RS , Boyle PA , Buchman AS , Schneider JA ((2012) ) Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 72: , 599–609. |
[11] | Boyle PA , Yu L , Wilson RS , Schneider JA , Bennett DA ((2013) ) Relation of neuropathology with cognitive decline among older persons without dementia. Front Aging Neurosci 5: , 50. |
[12] | Boyle PA , Yu L , Wilson RS , Leurgans SE , Schneider JA , Bennett DA ((2018) ) Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83: , 74–83. |
[13] | Hamilton RL ((2000) ) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol 10: , 378–384. |
[14] | Trojanowski JQ ((2002) ) Emerging Alzheimer’s disease therapies: focusing on the future. Neurobiol Aging 23: , 985–990. |
[15] | Spires-Jones TL , Attems J , Thal DR ((2017) ) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134: , 187–205. |
[16] | Gerson JE , Mudher A , Kayed R ((2016) ) Potential mechanisms and implications for the formation of tau oligomeric strains. Crit Rev Biochem Mol Biol 51: , 482–496. |
[17] | Herrera F , Outeiro TF ((2012) ) α-Synuclein modifies huntingtin aggregation in living cells. FEBS Lett 586: , 7–12. |
[18] | Poças GM , Branco-Santos J , Herrera F , Outeiro TF , Domingos PM ((2015) ) α-Synuclein modifies mutant huntingtin aggregation and neurotoxicity in Drosophila. Hum Mol Genet 24: , 1898–1907. |
[19] | Khan SS , LaCroix M , Boyle G , Sherman MA , Brown JL , Amar F , Aldaco J , Lee MK , Bloom GS , Lesné SE ((2018) ) Bidirectional modulation of Alzheimer phenotype by alpha-synuclein in mice and primary neurons. Acta Neuropathol 136: , 589–605. |
[20] | Bachhuber T , Katzmarski N , McCarter JF , Loreth D , Tahirovic S , Kamp F , Abou-Ajram C , Nuscher B , Serrano-Pozo A , Müller A , Prinz M , Steiner H , Hyman BT , Haass C , Meyer-Luehmann M ((2015) ) Inhibition of amyloid-β plaque formation by α-synuclein. Nat Med 21: , 802–807. |
[21] | Tsigelny IF , Crews L , Desplats P , Shaked GM , Sharikov Y , Mizuno H , Spencer B , Rockenstein E , Trejo M , Platoshyn O , Yuan JX-J , Masliah E ((2008) ) Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS One 3: , e3135. |
[22] | Gerson JE , Sengupta U , Kayed R ((2017) ) Tau oligomers as pathogenic seeds: preparation and propagation in vitro and in vivo. Methods Mol Biol 1523: , 141–157. |
[23] | Castillo-Carranza DL , Guerrero-Muñoz MJ , Sengupta U , Gerson JE , Kayed R ((2018) ) α-Synuclein oligomers induce a unique toxic tau strain. Biol Psychiatry 84: , 499–508. |
[24] | Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , Riddle DM , Kwong LK , Xu Y , Trojanowski JQ , Lee VMY ((2013) ) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154: , 103–117. |
[25] | Lashuel HA , Overk CR , Oueslati A , Masliah E ((2013) ) α-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. Nat Rev Neurosci 14: , 38–48. |
[26] | Ingelsson M ((2016) ) Alpha-synuclein oligomers—neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front Neurosci 10: , 408. |
[27] | Alam P , Bousset L , Melki R , Otzen DE ((2019) ) α-synuclein oligomers and fibrils: a spectrum of species, a of toxicities. J Neurochem 150: , 522–534. |
[28] | Melki R ((2015) ) Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J Parkinsons Dis 5: , 217–227. |
[29] | Bengoa-Vergniory N , Roberts RF , Wade-Martins R , Alegre-Abarrategui J ((2017) ) Alpha-synuclein oligomers: a new hope. Acta Neuropathol 134: , 819–838. |
[30] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Guerrero-Munoz MJ , Kiritoshi T , Neugebauer V , Jackson GR , Kayed R ((2012) ) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2: , 700. |
[31] | Larson ME , Lesné SE ((2012) ) Soluble Aβ oligomer production and toxicity. J Neurochem 120 Suppl 1: , 125–139. |
[32] | Sherman MA , LaCroix M , Amar F , Larson ME , Forster C , Aguzzi A , Bennett DA , Ramsden M , Lesné SE ((2016) ) Soluble conformers of Aβ and tau alter selective proteins governing axonal transport. J Neurosci 36: , 9647–9658. |
[33] | Larson M , Sherman MA , Amar F , Nuvolone M , Schneider JA , Bennett DA , Aguzzi A , Lesné SE ((2012) ) The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci 32: , 16857–16871a. |
[34] | Amar F , Sherman MA , Rush T , Larson M , Boyle G , Chang L , Götz J , Buisson A , Lesné SE ((2017) ) The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal 10: , eaal2021. |
[35] | Strohäker T , Jung BC , Liou S-H , Fernandez CO , Riedel D , Becker S , Halliday GM , Bennati M , Kim WS , Lee S-J , Zweckstetter M ((2019) ) Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat Commun 10: , 5535. |
[36] | Abeliovich A , Schmitz Y , Fariñas I , Choi-Lundberg D , Ho WH , Castillo PE , Shinsky N , Verdugo JM , Armanini M , Ryan A , Hynes M , Phillips H , Sulzer D , Rosenthal A ((2000) ) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: , 239–252. |
[37] | Bendor JT , Logan TP , Edwards RH ((2013) ) The function of α-synuclein. Neuron 79: , 1044–1066. |
[38] | Sulzer D , Edwards RH ((2019) ) The physiological role of α-synuclein and its relationship to Parkinson’s disease. J Neurochem 150: , 475–486. |
[39] | Logan T , Bendor J , Toupin C , Thorn K , Edwards RH ((2017) ) α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 20: , 681–689. |
[40] | Ben Gedalya T , Loeb V , Israeli E , Altschuler Y , Selkoe DJ , Sharon R ((2009) ) Alpha-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 10: , 218–234. |
[41] | Vargas KJ , Makani S , Davis T , Westphal CH , Castillo PE , Chandra SS ((2014) ) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34: , 9364–9376. |
[42] | Vargas KJ , Schrod N , Davis T , Fernandez-Busnadiego R , Taguchi YV , Laugks U , Lucic V , Chandra SS ((2017) ) Synucleins have multiple effects on presynaptic architecture. Cell Rep 18: , 161–173. |
[43] | Wang L , Das U , Scott DA , Tang Y , McLean PJ , Roy S ((2014) ) α-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24: , 2319–2326. |
[44] | Burré J , Sharma M , Tsetsenis T , Buchman V , Etherton MR , Südhof TC ((2010) ) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329: , 1663–1667. |
[45] | DeWitt DC , Rhoades E ((2013) ) α-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 52: , 2385–2387. |
[46] | Nuscher B , Kamp F , Mehnert T , Odoy S , Haass C , Kahle PJ , Beyer K ((2004) ) Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J Biol Chem 279: , 21966–75. |
[47] | George JM , Jin H , Woods WS , Clayton DF ((1995) ) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15: , 361–372. |
[48] | Maroteaux L , Scheller RH ((1991) ) The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 11: , 335–343. |
[49] | Withers GS , George JM , Banker GA , Clayton DF ((1997) ) Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res 99: , 87–94. |
[50] | Jensen MB , Bhatia VK , Jao CC , Rasmussen JE , Pedersen SL , Jensen KJ , Langen R , Stamou D ((2011) ) Membrane curvature sensing by amphipathic helices: a single liposome study using alpha-synuclein and annexin B12. J Biol Chem 286: , 42603–42614. |
[51] | Alderson TR , Markley JL ((2013) ) Biophysical characterization of α-synuclein and its controversial structure. Intrinsically Disord Proteins 1: , e26255. |
[52] | Wietek J , Haralampiev I , Amoussouvi A , Herrmann A , Stöckl M ((2013) ) Membrane bound α-synuclein is fully embedded in the lipid bilayer while segments with higher flexibility remain. FEBS Lett 587: , 2572–2577. |
[53] | Tsigelny IF , Sharikov Y , Wrasidlo W , Gonzalez T , Desplats PA , Crews L , Spencer B , Masliah E ((2012) ) Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J 279: , 1000–1013. |
[54] | West A , Brummel BE , Braun AR , Rhoades E , Sachs JN ((2016) ) Membrane remodeling and mechanics: Experiments and simulations of α-Synuclein. Biochim Biophys Acta 1858: , 1594–1609. |
[55] | Cheng C-Y , Varkey J , Ambroso MR , Langen R , Han S ((2013) ) Hydration dynamics as an intrinsic ruler for refining protein structure at lipid membrane interfaces. Proc Natl Acad Sci U S A 110: , 16838–16843. |
[56] | Jao CC , Hegde BG , Chen J , Haworth IS , Langen R ((2008) ) Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A 105: , 19666–19671. |
[57] | Wang C , Zhao C , Li D , Tian Z , Lai Y , Diao J , Liu C ((2016) ) Versatile structures of α-synuclein. Front Mol Neurosci 9: , 48. |
[58] | Dettmer U ((2018) ) Rationally designed variants of α-synuclein illuminate its in vivo structural properties in health and disease. Front Neurosci 12: , 623. |
[59] | Yeboah F , Kim T-E , Bill A , Dettmer U ((2019) ) Dynamic behaviors of α-synuclein and tau in the cellular context: New mechanistic insights and therapeutic opportunities in neurodegeneration. Neurobiol Dis 132: , 104543. |
[60] | Rovere M , Sanderson JB , Fonseca-Ornelas L , Patel DS , Bartels T ((2018) ) Refolding of helical soluble α-synuclein through transient interaction with lipid interfaces. FEBS Lett 592: , 1464–1472. |
[61] | Weinreb PH , Zhen W , Poon AW , Conway KA , Lansbury PT Jr ((1996) ) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35: , 13709–13715. |
[62] | Dettmer U , Ramalingam N , von Saucken VE , Kim T-E , Newman AJ , Terry-Kantor E , Nuber S , Ericsson M , Fanning S , Bartels T , Lindquist S , Levy OA , Selkoe D ((2017) ) Loss of native α-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum Mol Genet 26: , 3466–3481. |
[63] | Iljina M , Tosatto L , Choi ML , Sang JC , Ye Y , Hughes CD , Bryant CE , Gandhi S , Klenerman D ((2016) ) Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein. Sci Rep 6: , 33928. |
[64] | Gurry T , Ullman O , Fisher CK , Perovic I , Pochapsky T , Stultz CM ((2013) ) The dynamic structure of α-synuclein multimers. J Am Chem Soc 135: , 3865–3872. |
[65] | Kim S , Yun SP , Lee S , Umanah GE , Bandaru VVR , Yin X , Rhee P , Karuppagounder SS , Kwon S-H , Lee H , Mao X , Kim D , Pandey A , Lee G , Dawson VL , Dawson TM , Ko HS ((2018) ) GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 115: , 798–803. |
[66] | Luth ES , Bartels T , Dettmer U , Kim NC , Selkoe DJ ((2015) ) Purification of α-synuclein from human brain reveals an instability of endogenous multimers as the protein approaches purity. Biochemistry 54: , 279–292. |
[67] | Burré J , Sharma M , Südhof TC ((2014) ) α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111: , E4274–4283. |
[68] | Bartels T , Choi JG , Selkoe DJ ((2011) ) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477: , 107–110. |
[69] | Wang W , Perovic I , Chittuluru J , Kaganovich A , Nguyen LTT , Liao J , Auclair JR , Johnson D , Landeru A , Simorellis AK , Ju S , Cookson MR , Asturias FJ , Agar JN , Webb BN , Kang C , Ringe D , Petsko GA , Pochapsky TC , Hoang QQ ((2011) ) A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108: , 17797–17802. |
[70] | Dettmer U , Newman AJ , von Saucken VE , Bartels T , Selkoe D ((2015) ) KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci U S A 112: , 9596–9601. |
[71] | Dettmer U , Newman AJ , Soldner F , Luth ES , Kim NC , von Saucken VE , Sanderson JB , Jaenisch R , Bartels T , Selkoe D ((2015) ) Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6: , 7314. |
[72] | Pochapsky TC ((2015) ) From intrinsically disordered protein to context-dependent folding: The α-synuclein tetramer is teased out of hiding. Proc Natl Acad Sci U S A 112: , 9502–9503. |
[73] | Nuber S , Rajsombath M , Minakaki G , Winkler J , Müller CP , Ericsson M , Caldarone B , Dettmer U , Selkoe DJ ((2018) ) Abrogating native α-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron 100: , 75–90. |
[74] | Westphal CH , Chandra SS ((2013) ) Monomeric synucleins generate membrane curvature. J Biol Chem 288: , 1829–1840. |
[75] | Dettmer U , Newman AJ , Luth ES , Bartels T , Selkoe D ((2013) ) In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J Biol Chem 288: , 6371–6385. |
[76] | Trexler AJ , Rhoades E ((2012) ) N-Terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci 21: , 601–605. |
[77] | Fauvet B , Mbefo MK , Fares M-B , Desobry C , Michael S , Ardah MT , Tsika E , Coune P , Prudent M , Lion N , Eliezer D , Moore DJ , Schneider B , Aebischer P , El-Agnaf OM , Masliah E , Lashuel HA ((2012) ) α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem 287: , 15345–15364. |
[78] | Burré J , Vivona S , Diao J , Sharma M , Brunger AT , Südhof TC ((2013) ) Properties of native brain α-synuclein. Nature 498: , E4–6; discussion E6-7. |
[79] | Gould N , Mor DE , Lightfoot R , Malkus K , Giasson B , Ischiropoulos H ((2014) ) Evidence of native alpha-synuclein conformers in the human brain. J Biol Chem 289: , 7929–7934. |
[80] | Burré J , Sharma M , Südhof TC ((2015) ) Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci 35: , 5221–5232. |
[81] | Corbillé A-G , Neunlist M , Derkinderen P ((2016) ) Cross-linking for the analysis of α-synuclein in the enteric nervous system. J Neurochem 139: , 839–847. |
[82] | Killinger BA , Moszczynska A ((2016) ) Characterization of α-synuclein multimer stoichiometry in complex biological samples by electrophoresis. Anal Chem 88: , 4071–4084. |
[83] | Binolfi A , Theillet F-X , Selenko P ((2012) ) Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature? Biochem Soc Trans 40: , 950–954. |
[84] | Theillet F-X , Binolfi A , Bekei B , Martorana A , Rose HM , Stuiver M , Verzini S , Lorenz D , van Rossum M , Goldfarb D , Selenko P ((2016) ) Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530: , 45–50. |
[85] | Alderson TR , Bax A ((2016) ) Parkinson’s disease: Disorder in the court. Nature 530: , 38–39. |
[86] | Sharon R , Bar-Joseph I , Frosch MP , Walsh DM , Hamilton JA , Selkoe DJ ((2003) ) The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37: , 583–595. |
[87] | Roberts RF , Bengoa-Vergniory N , Alegre-Abarrategui J ((2019) ) Alpha-synuclein proximity ligation assay (AS-PLA) in brain sections to probe for alpha-synuclein oligomers. Methods Mol Biol 1948: , 69–76. |
[88] | Roberts RF , Wade-Martins R , Alegre-Abarrategui J ((2015) ) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138: , 1642–1657. |
[89] | Sengupta U , Guerrero-Muñoz MJ , Castillo-Carranza DL , Lasagna-Reeves CA , Gerson JE , Paulucci-Holthauzen AA , Krishnamurthy S , Farhed M , Jackson GR , Kayed R ((2015) ) Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol Psychiatry 78: , 672–683. |
[90] | Majbour NK , Chiasserini D , Vaikath NN , Eusebi P , Tokuda T , Berg W van de , Parnetti L , Calabresi P , El-Agnaf OMA ((2017) ) Increased levels of CSF total but not oligomeric or phosphorylated forms of alpha-synuclein in patients diagnosed with probable Alzheimer’s disease. Sci Rep 7: , 40263. |
[91] | Park MJ , Cheon SM , Bae HR , Kim SH , Kim JW ((2011) ) Elevated levels of alpha-synuclein oligomer in the cerebrospinal fluid of drug-naive patients with Parkinson’s disease. J Clin Neurol 7: , 215–222. |
[92] | El-Agnaf OM , Salem SA , Paleologou KE , Curran MD , Gibson MJ , Court JA , Schlossmacher MG , Allsop D ((2006) ) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20: , 419–425. |
[93] | Parnetti L , Chiasserini D , Bellomo G , Giannandrea D , De Carlo C , Qureshi MM , Ardah MT , Varghese S , Bonanni L , Borroni B , Tambasco N , Eusebi P , Rossi A , Onofrj M , Padovani A , Calabresi P , El-Agnaf O ((2011) ) Cerebrospinal fluid Tau/alpha-synuclein ratio in Parkinson’s disease and degenerative dementias. Mov Disord 26: , 1428–1435. |
[94] | Hansson O , Hall S , Öhrfelt A , Zetterberg H , Blennow K , Minthon L , Nägga K , Londos E , Varghese S , Majbour NK , Al-Hayani A , El-Agnaf OM ((2014) ) Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res Ther 6: , 25. |
[95] | Sierks MR , Chatterjee G , McGraw C , Kasturirangan S , Schulz P , Prasad S ((2011) ) CSF levels of oligomeric alpha-synuclein and beta-amyloid as biomarkers for neurodegenerative disease. Integr Biol (Camb) 3: , 1188–1196. |
[96] | Emmanouilidou E , Melachroinou K , Roumeliotis T , Garbis SD , Ntzouni M , Margaritis LH , Stefanis L , Vekrellis K ((2010) ) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30: , 6838–6851. |
[97] | Danzer KM , Haasen D , Karow AR , Moussaud S , Habeck M , Giese A , Kretzschmar H , Hengerer B , Kostka M ((2007) ) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 27: , 9220–9232. |
[98] | Lee MK , Stirling W , Xu Y , Xu X , Qui D , Mandir AS , Dawson TM , Copeland NG , Jenkins NA , Price DL ((2002) ) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –>Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A 99: , 8968–8973. |
[99] | Colla E , Jensen PH , Pletnikova O , Troncoso JC , Glabe C , Lee MK ((2012) ) Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy {in vivo. J Neurosci 32: , 3301–3305. |
[100] | Paleologou KE , Kragh CL , Mann DM , Salem SA , Al-Shami R , Allsop D , Hassan AH , Jensen PH , El-Agnaf OM ((2009) ) Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 132: , 1093–1101. |
[101] | Zarranz JJ , Alegre J , Gómez-Esteban JC , Lezcano E , Ros R , Ampuero I , Vidal L , Hoenicka J , Rodriguez O , Atarés B , Llorens V , Gomez Tortosa E , del Ser T , Muñoz DG , de Yebenes JG ((2004) ) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: , 164–173. |
[102] | Emmer KL , Waxman EA , Covy JP , Giasson BI ((2011) ) E46K human α-synuclein transgenic mice develop lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem 286: , 35104–35118. |
[103] | Samuel F , Flavin WP , Iqbal S , Pacelli C , Sri Renganathan SD , Trudeau L-E , Campbell EM , Fraser PE , Tandon A ((2016) ) Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J Biol Chem 291: , 4374–4385. |
[104] | Foulds PG , Yokota O , Thurston A , Davidson Y , Ahmed Z , Holton J , Thompson JC , Akiyama H , Arai T , Hasegawa M , Gerhard A , Allsop D , Mann DM ((2012) ) Post mortem cerebrospinal fluid alpha-synuclein levels are raised in multiple system atrophy and distinguish this from the other alpha-synucleinopathies, Parkinson’s disease and dementia with Lewy bodies. Neurobiol Dis 45: , 188–195. |
[105] | Sekiya H , Kowa H , Koga H , Takata M , Satake W , Futamura N , Funakawa I , Jinnai K , Takahashi M , Kondo T , Ueno Y , Kanagawa M , Kobayashi K , Toda T ((2019) ) Wide distribution of alpha-synuclein oligomers in multiple system atrophy brain detected by proximity ligation. Acta Neuropathol 137: , 455–466. |
[106] | Lippa CF , Fujiwara H , Mann DM , Giasson B , Baba M , Schmidt ML , Nee LE , O’Connell B , Pollen DA , St George-Hyslop P , Ghetti B , Nochlin D , Bird TD , Cairns NJ , Lee VM , Iwatsubo T , Trojanowski JQ ((1998) ) Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153: , 1365–1370. |
[107] | Larson ME , Sherman MA , Greimel S , Kuskowski M , Schneider JA , Bennett DA , Lesné SE ((2012) ) Soluble α-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci 32: , 10253–10266. |
[108] | Larson ME , Greimel SJ , Amar F , LaCroix M , Boyle G , Sherman MA , Schley H , Miel C , Schneider JA , Kayed R , Benfenati F , Lee MK , Bennett DA , Lesné SE ((2017) ) Selective lowering of synapsins induced by oligomeric α-synuclein exacerbates memory deficits. Proc Natl Acad Sci U S A 114: , E4648–E4657. |
[109] | Nemani VM , Lu W , Berge V , Nakamura K , Onoa B , Lee MK , Chaudhry FA , Nicoll RA , Edwards RH ((2010) ) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65: , 66–79. |
[110] | Parnetti L , Gaetani L , Eusebi P , Paciotti S , Hansson O , El-Agnaf O , Mollenhauer B , Blennow K , Calabresi P ((2019) ) CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol 18: , 573–586. |
[111] | Mollenhauer B , Locascio JJ , Schulz-Schaeffer W , Sixel-Döring F , Trenkwalder C , Schlossmacher MG ((2011) ) α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10: , 230–240. |
[112] | Twohig D , Rodriguez-Vieitez E , Sando SB , Berge G , Lauridsen C , Møller I , Grøntvedt GR , Bråthen G , Patra K , Bu G , Benzinger TLS , Karch CM , Fagan A , Morris JC , Bateman RJ , Nordberg A , White LR , Nielsen HM , Dominantly Inherited Alzheimer Network (DIAN) ((2018) ) The relevance of cerebrospinal fluid α-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol Commun 6: , 130. |
[113] | Toledo JB , Korff A , Shaw LM , Trojanowski JQ , Zhang J ((2013) ) CSF α-synuclein improves diagnostic and prognostic performance of CSF tau and Aβ in Alzheimer’s disease. Acta Neuropathol 126: , 683–697. |
[114] | Slaets S , Vanmechelen E , Le Bastard N , Decraemer H , Vandijck M , Martin J-J , De Deyn PP , Engelborghs S ((2014) ) Increased CSF α-synuclein levels in Alzheimer’s disease: correlation with tau levels. Alzheimers Dement 10: , S290–298. |
[115] | van Steenoven I , Majbour NK , Vaikath NN , Berendse HW , van der Flier WM , van de Berg WDJ , Teunissen CE , Lemstra AW , El-Agnaf OMA ((2018) ) α-Synuclein species as potential cerebrospinal fluid biomarkers for dementia with lewy bodies. Mov Disord 33: , 1724–1733. |
[116] | Vaikath NN , Majbour NK , Paleologou KE , Ardah MT , van Dam E , van de Berg WDJ , Forrest SL , Parkkinen L , Gai W-P , Hattori N , Takanashi M , Lee S-J , Mann DMA , Imai Y , Halliday GM , Li J-Yi , El-Agnaf OMA ((2015) ) Generation and characterization of novel conformation-specific monoclonal antibodies for α-synuclein pathology. Neurobiol Dis 79: , 81–99. |
[117] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , Nance M , Ross CA , Scahill RI , Wetzel R , Wild EJ , Tabrizi SJ ((2015) ) Huntington disease. Nat Rev Dis Primers 1: , 15005. |
[118] | Chhatwal JP , Schultz AP , Johnson K , Benzinger TLS , Jack C , Ances BM , Sullivan CA , Salloway SP , Ringman JM , Koeppe RA , Marcus DS , Thompson P , Saykin AJ , Correia S , Schofield PR , Rowe CC , Fox NC , Brickman AM , Mayeux R , McDade E , Bateman R , Fagan AM , Goate AM , Xiong C , Buckles VD , Morris JC , Sperling RA ((2013) ) Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81: , 736–744. |
[119] | Schultz AP , Chhatwal JP , Hedden T , Mormino EC , Hanseeuw BJ , Sepulcre J , Huijbers W , LaPoint M , Buckley RF , Johnson KA , Sperling RA ((2017) ) Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J Neurosci 37: , 4323–4331. |
[120] | Ward AM , Mormino EC , Huijbers W , Schultz AP , Hedden T , Sperling RA ((2015) ) Relationships between default-mode network connectivity, medial temporal lobe structure, and age-related memory deficits. Neurobiol Aging 36: , 265–272. |
[121] | Medeiros AT , Soll LG , Tessari I , Bubacco L , Morgan JR ((2017) ) α-Synuclein dimers impair vesicle fission during clathrin-mediated synaptic vesicle recycling. Front Cell Neurosci 11: , 388. |
[122] | Souza JM , Giasson BI , Chen Q , Lee VM-Y , Ischiropoulos H ((2000) ) Dityrosine cross-linking promotes formation of stable α-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem 275: , 18344–18349. |
[123] | Masuda M , Suzuki N , Taniguchi S , Oikawa T , Nonaka T , Iwatsubo T , Hisanaga S , Goedert M , Hasegawa M ((2006) ) Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 45: , 6085–6094. |
[124] | Rockenstein E , Nuber S , Overk CR , Ubhi K , Mante M , Patrick C , Adame A , Trejo-Morales M , Gerez J , Picotti P , Jensen PH , Campioni S , Riek R , Winkler J , Gage FH , Winner B , Masliah E ((2014) ) Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137: , 1496–1513. |
[125] | Kahle PJ , Neumann M , Ozmen L , Müller V , Odoy S , Okamoto N , Jacobsen H , Iwatsubo T , Trojanowski JQ , Takahashi H , Wakabayashi K , Bogdanovic N , Riederer P , Kretzschmar HA , Haass C ((2001) ) Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol 159: , 2215–2225. |
[126] | Wu JW , Breydo L , Isas JM , Lee J , Kuznetsov YG , Langen R , Glabe C ((2010) ) Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J Biol Chem 285: , 6071–6079. |
[127] | Laganowsky A , Liu C , Sawaya MR , Whitelegge JP , Park J , Zhao M , Pensalfini A , Soriaga AB , Landau M , Teng PK , Cascio D , Glabe C , Eisenberg D ((2012) ) Atomic view of a toxic amyloid small oligomer. Science 335: , 1228–1231. |
[128] | Liu P , Reed MN , Kotilinek LA , Grant MKO , Forster CL , Qiang W , Shapiro SL , Reichl JH , Chiang ACA , Jankowsky JL , Wilmot CM , Cleary JP , Zahs KR , Ashe KH ((2015) ) Quaternary structure defines a large class of amyloid-β oligomers neutralized by sequestration. Cell Rep 11: , 1760–1771. |
[129] | Decressac M , Kadkhodaei B , Mattsson B , Laguna A , Perlmann T , Björklund A ((2012) ) α-Synuclein–induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 4: , 163ra156–163ra156. |
[130] | Volakakis N , Tiklova K , Decressac M , Papathanou M , Mattsson B , Gillberg L , Nobre A , Björklund A , Perlmann T ((2015) ) Nurr1 and retinoid X receptor ligands stimulate ret signaling in dopamine neurons and can alleviate α-synuclein disrupted gene expression. J. Neurosci 35: , 14370–14385. |
[131] | Lashuel HA , Petre BM , Wall J , Simon M , Nowak RJ , Walz T , Lansbury PT ((2002) ) Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322: , 1089–102. |
[132] | Volles MJ , Lee SJ , Rochet JC , Shtilerman MD , Ding TT , Kessler JC , Lansbury PT ((2001) ) Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40: , 7812–7819. |
[133] | Chen SW , Drakulic S , Deas E , Ouberai M , Aprile FA , Arranz R , Ness S , Roodveldt C , Guilliams T , De-Genst EJ , Klenerman D , Wood NW , Knowles TPJ , Alfonso C , Rivas G , Abramov AY , Valpuesta JM , Dobson CM , Cremades N ((2015) ) Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci U S A 112: , E1994–2003. |
[134] | Kayed R , Pensalfini A , Margol L , Sokolov Y , Sarsoza F , Head E , Hall J , Glabe C ((2009) ) Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J Biol Chem 284: , 4230–4237. |
[135] | Choi BK , Choi MG , Kim JY , Yang Y , Lai Y , Kweon DH , Lee NK , Shin YK ((2013) ) Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A 110: , 4087–4092. |
[136] | Kramer ML , Schulz-Schaeffer WJ ((2007) ) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27: , 1405–1410. |
[137] | Kiechle M , von Einem B , Höfs L , Voehringer P , Grozdanov V , Markx D , Parlato R , Wiesner D , Mayer B , Sakk O , Baumann B , Lukassen S , Liss B , Ekici AB , Ludolph AC , Walther P , Ferger B , McLean PJ , Falkenburger BH , Weishaupt JH , Danzer KM ((2019) ) In vivo protein complementation demonstrates presynaptic α-synuclein oligomerization and age-dependent accumulation of 8-16-mer oligomer species. Cell Rep 29: , 2862–2874.e9. |
[138] | George S , van den Buuse M , San Mok S , Masters CL , Li Q-X , Culvenor JG ((2008) ) Alpha-synuclein transgenic mice exhibit reduced anxiety-like behaviour. Exp Neurol 210: , 788–792. |
[139] | Sung JY , Park SM , Lee C-H , Um JW , Lee HJ , Kim J , Oh YJ , Lee S-T , Paik SR , Chung KC ((2005) ) Proteolytic cleavage of extracellular secreted alpha-synuclein via matrix metalloproteinases. J Biol Chem 280: , 25216–25224. |
[140] | Hodara R , Norris EH , Giasson BI , Mishizen-Eberz AJ , Lynch DR , Lee VM-Y , Ischiropoulos H ((2004) ) Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem 279: , 47746–47753. |
[141] | Paleologou KE , Oueslati A , Shakked G , Rospigliosi CC , Kim H-Y , Lamberto GR , Fernandez CO , Schmid A , Chegini F , Gai WP , Chiappe D , Moniatte M , Schneider BL , Aebischer P , Eliezer D , Zweckstetter M , Masliah E , Lashuel HA ((2010) ) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci 30: , 3184–3198. |
[142] | Chen L , Periquet M , Wang X , Negro A , McLean PJ , Hyman BT , Feany MB ((2009) ) Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest 119: , 3257–3265. |
[143] | Barrett PJ , Timothy Greenamyre J ((2015) ) Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res 1628: , 247–253. |
[144] | Zhao N , Attrebi ON , Ren Y , Qiao W , Sonustun B , Martens YA , Meneses AD , Li F , Shue F , Zheng J , Van Ingelgom AJ , Davis MD , Kurti A , Knight JA , Linares C , Chen Y , Delenclos M , Liu C-C , Fryer JD , Asmann YW , McLean PJ , Dickson DW , Ross OA , Bu G ((2020) ) APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid.eaay. Sci Transl Med 12: , 1809. |
[145] | Davis AA , Inman CE , Wargel ZM , Dube U , Freeberg BM , Galluppi A , Haines JN , Dhavale DD , Miller R , Choudhury FA , Sullivan PM , Cruchaga C , Perlmutter JS , Ulrich JD , Benitez BA , Kotzbauer PT , Holtzman DM ((2020) ) APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med 12: , eaay3069. |
[146] | Delenclos M , Faroqi AH , Yue M , Kurti A , Castanedes-Casey M , Rousseau L , Phillips V , Dickson DW , Fryer JD , McLean PJ ((2017) ) Neonatal AAV delivery of alpha-synuclein induces pathology in the adult mouse brain. Acta Neuropathol Commun 5: , 1–14. |
[147] | Collinge J , Sidle KC , Meads J , Ironside J , Hill AF ((1996) ) Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383: , 685–690. |
[148] | Bessen RA , Marsh RF ((1992) ) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 73: , 329–334. |
[149] | Bartz JC , Bessen RA , McKenzie D , Marsh RF , Aiken JM ((2000) ) Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol 74: , 5542–5547. |
[150] | Klimova N , Makarava N , Baskakov IV ((2015) ) The diversity and relationship of prion protein self-replicating states. Virus Res 207: , 113–119. |
[151] | Morales R ((2017) ) Prion strains in mammals: Different conformations leading to disease. PLoS Pathog 13: , e1006323. |
[152] | Polymenidou M , Cleveland DW ((2011) ) The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147: , 498–508. |
[153] | Tredici KD , Braak H ((2016) ) Review: Sporadic Parkinson’s disease: development and distribution of α-synuclein pathology. Neuropathol Appl Neurobiol 42: , 33–50. |
[154] | Kordower JH , Chu Y , Hauser RA , Freeman TB , Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14: , 504–506. |
[155] | Li J-Y , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Björklund A , Widner H , Revesz T , Lindvall O , Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14: , 501–503. |
[156] | Desplats P , Lee H-J , Bae E-J , Patrick C , Rockenstein E , Crews L , Spencer B , Masliah E , Lee S-J ((2009) ) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci U S A 106: , 13010–13015. |
[157] | Olanow CW , Prusiner SB ((2009) ) Is Parkinson’s disease a prion disorder? Proc Natl Acad Sci U S A 106: , 12571–12572. |
[158] | Lee HJ , Khoshaghideh F , Patel S , Lee SJ ((2004) ) Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24: , 1888–1896. |
[159] | Lee HJ , Suk JE , Bae EJ , Lee JH , Paik SR , Lee SJ ((2008) ) Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 40: , 1835–1849. |
[160] | Domert J , Sackmann C , Severinsson E , Agholme L , Bergstrom J , Ingelsson M , Hallbeck M ((2016) ) Aggregated alpha-synuclein transfer efficiently between cultured human neuron-like cells and localize to lysosomes. PloS One 11: , e0168700. |
[161] | Hoffmann AC , Minakaki G , Menges S , Salvi R , Savitskiy S , Kazman A , Vicente Miranda H , Mielenz D , Klucken J , Winkler J , Xiang W ((2019) ) Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep 9: , 544. |
[162] | Danzer KM , Krebs SK , Wolff M , Birk G , Hengerer B ((2009) ) Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 111: , 192–203. |
[163] | Iljina M , Garcia GA , Horrocks MH , Tosatto L , Choi ML , Ganzinger KA , Abramov AY , Gandhi S , Wood NW , Cremades N , Dobson CM , Knowles TPJ , Klenerman D ((2016) ) Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci U S A 113: , E1206–1215. |
[164] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[165] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[166] | Rey NL , Bousset L , George S , Madaj Z , Meyerdirk L , Schulz E , Steiner JA , Melki R , Brundin P ((2019) ) α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun 7: , 221. |
[167] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VM-Y ((2011) ) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[168] | Luk KC , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , Lee VM-Y ((2012) ) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[169] | Luk KC , Kehm VM , Zhang B , O’Brien P , Trojanowski JQ , Lee VMY ((2012) ) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209: , 975–986. |
[170] | Glabe CG , Kayed R ((2006) ) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66: , S74–78. |
[171] | Moussaud S , Jones DR , Moussaud-Lamodière EL , Delenclos M , Ross OA , McLean PJ ((2014) ) Alpha-synuclein and tau: Teammates in neurodegeneration? Mol Neurodegener 9: , 43. |
[172] | Braak H , Braak E ((1990) ) Cognitive impairment in Parkinson’s disease: amyloid plaques, neurofibrillary tangles, and neuropil threads in the cerebral cortex. J Neural Transm Park Dis Dement Sect 2: , 45–57. |
[173] | Jacobson SA , Morshed T , Dugger BN , Beach TG , Hentz JG , Adler CH , Shill HA , Sabbagh MN , Belden CM , Sue LI , Caviness JN , Hu C , Arizona Parkinson’s Disease Consortium ((2014) ) Plaques and tangles as well as Lewy-type alpha synucleinopathy are associated with formed visual hallucinations. Parkinsonism Relat Disord 20: , 1009–1014. |
[174] | Masliah E , Rockenstein E , Veinbergs I , Sagara Y , Mallory M , Hashimoto M , Mucke L ((2001) ) Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 12245–12250. |
[175] | Mandal PK , Pettegrew JW , Masliah E , Hamilton RL , Mandal R ((2006) ) Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res 31: , 1153–1162. |
[176] | Ono K , Takahashi R , Ikeda T , Yamada M ((2012) ) Cross-seeding effects of amyloid β-protein and α-synuclein. J Neurochem 122: , 883–890. |
[177] | Marsh SE , Blurton-Jones M ((2012) ) Examining the mechanisms that link β-amyloid and α-synuclein pathologies. Alzheimers Res Ther 4: , 11. |
[178] | Chia S , Flagmeier P , Habchi J , Lattanzi V , Linse S , Dobson CM , Knowles TPJ , Vendruscolo M ((2017) ) Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc Natl Acad Sci U S A 114: , 8005–8010. |
[179] | Köppen J , Schulze A , Machner L , Wermann M , Eichentopf R , Guthardt M , Hähnel A , Klehm J , Kriegeskorte M-C , Hartlage-Rübsamen M , Morawski M , von Hörsten S , Demuth H-U , Roßner S , Schilling S ((2020) ) Amyloid-beta peptides trigger aggregation of alpha-synuclein in vitro. Molecules 25: , E580. |
[180] | Bassil F , Brown HJ , Pattabhiraman S , Iwasyk JE , Maghames CM , Meymand ES , Cox TO , Riddle DM , Zhang B , Trojanowski JQ , Lee VM-Y ((2020) ) Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron 105: , 260–275.e6. |
[181] | Kallhoff V , Peethumnongsin E , Zheng H ((2007) ) Lack of alpha-synuclein increases amyloid plaque accumulation in a transgenic mouse model of Alzheimer’s disease. Mol Neurodegener 2: , 6. |
[182] | Orr ME , Sullivan AC , Frost B ((2017) ) A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol Sci 38: , 637–648. |
[183] | Simon-Sanchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Kruger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[184] | Colom-Cadena M , Gelpi E , Martí MJ , Charif S , Dols-Icardo O , Blesa R , Clarimón J , Lleó A ((2013) ) MAPT H1 haplotype is associated with enhanced α-synuclein deposition in dementia with Lewy bodies. Neurobiol Aging 34: , 936–942. |
[185] | Kaul T , Credle J , Haggerty T , Oaks AW , Masliah E , Sidhu A ((2011) ) Region-specific tauopathy and synucleinopathy in brain of the alpha-synuclein overexpressing mouse model of Parkinson’s disease. BMC Neurosci 12: , 79. |
[186] | Frasier M , Walzer M , McCarthy L , Magnuson D , Lee JM , Haas C , Kahle P , Wolozin B ((2005) ) Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol 192: , 274–287. |
[187] | Duka T , Duka V , Joyce JN , Sidhu A ((2009) ) Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J 23: , 2820–2830. |
[188] | Duka T , Rusnak M , Drolet RE , Duka V , Wersinger C , Goudreau JL , Sidhu A ((2006) ) Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J 20: , 2302–2312. |
[189] | Haggerty T , Credle J , Rodriguez O , Wills J , Oaks AW , Masliah E , Sidhu A ((2011) ) Hyperphosphorylated Tau in an alpha-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci 33: , 1598–1610. |
[190] | Peng C , Trojanowski JQ , Lee VM-Y ((2020) ) Protein transmission in neurodegenerative disease. Nat Rev Neurol 16: , 199–212. |
[191] | Jucker M , Walker LC ((2011) ) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70: , 532–540. |
[192] | Goedert M ((2015) ) Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349: , 1255555. |
[193] | Caughey B , Kraus A ((2019) ) Transmissibility versus pathogenicity of self-propagating protein aggregates. Viruses 11: , E1044. |
[194] | Walker LC , Diamond MI , Duff KE , Hyman BT ((2013) ) Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol 70: , 304–310. |
[195] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , Giles K ((2015) ) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112: , E5308–5317. |
[196] | Clavaguera F , Akatsu H , Fraser G , Crowther RA , Frank S , Hench J , Probst A , Winkler DT , Reichwald J , Staufenbiel M , Ghetti B , Goedert M , Tolnay M ((2013) ) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110: , 9535–9540. |
[197] | Clavaguera F , Hench J , Goedert M , Tolnay M ((2015) ) Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 41: , 47–58. |
[198] | Jensen PH , Hager H , Nielsen MS , Højrup P , Gliemann J , Jakes R ((1999) ) α-Synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem 274: , 25481–25489. |
[199] | Iseki E , Takayama N , Marui W , Ueda K , Kosaka K ((2002) ) Relationship in the formation process between neurofibrillary tangles and Lewy bodies in the hippocampus of dementia with Lewy bodies brains. J Neurol Sci 195: , 85–91. |
[200] | Arima K , Hirai S , Sunohara N , Aoto K , Izumiyama Y , Ueda K , Ikeda K , Kawai M ((1999) ) Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res 843: , 53–61. |
[201] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM-Y ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[202] | Wills J , Credle J , Haggerty T , Lee J-H , Oaks AW , Sidhu A ((2011) ) Tauopathic changes in the striatum of A53T α-synuclein mutant mouse model of Parkinson’s disease. PLoS One 6: , e17953. |
[203] | Rockenstein E , Mallory M , Hashimoto M , Song D , Shults CW , Lang I , Masliah E ((2002) ) Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 68: , 568–578. |
[204] | Gerson JE , Farmer KM , Henson N , Castillo-Carranza DL , Carretero Murillo M , Sengupta U , Barrett A , Kayed R ((2018) ) Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol Neurodegener 13: , 13. |
[205] | Castillo-Carranza DL , Sengupta U , Guerrero-Muñoz MJ , Lasagna-Reeves CA , Gerson JE , Singh G , Estes DM , Barrett ADT , Dineley KT , Jackson GR , Kayed R ((2014) ) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34: , 4260–4272. |
[206] | Singh B , Covelo A , Martell-Martínez H , Nanclares C , Sherman MA , Okematti E , Meints J , Teravskis PJ , Gallardo C , Savonenko AV , Benneyworth MA , Lesné SE , Liao D , Araque A , Lee MK ((2019) ) Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy. Acta Neuropathol 138: , 551–574. |
[207] | Chaudhuri P , Prajapati KP , Anand BG , Dubey K , Kar K ((2019) ) Amyloid cross-seeding raises new dimensions to understanding of amyloidogenesis mechanism. Ageing Res Rev 56: , 100937. |
[208] | Morales R , Moreno-Gonzalez I , Soto C ((2013) ) Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog 9: , e1003537. |
[209] | Lasagna-Reeves CA , Castillo-Carranza DL , Guerrero-Munoz MJ , Jackson GR , Kayed R ((2010) ) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49: , 10039–10041. |
[210] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[211] | Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM ((2010) ) Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 30: , 7281–7289. |
[212] | Corrochano S , Renna M , Carter S , Chrobot N , Kent R , Stewart M , Cooper J , Brown SDM , Rubinsztein DC , Acevedo-Arozena A ((2012) ) α-Synuclein levels modulate Huntington’s disease in mice. Hum Mol Genet 21: , 485–494. |
[213] | Bittar A , Sengupta U , Kayed R ((2018) ) Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 3: , 9. |
[214] | Brundin P , Dave KD , Kordower JH ((2017) ) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298: , 225–235. |
[215] | Barkovits K , Kruse N , Linden A , Tönges L , Pfeiffer K , Mollenhauer B , Marcus K ((2020) ) Blood contamination in CSF and its impact on quantitative analysis of alpha-synuclein. Cells 9: , E370. |
[216] | Wagner J , Ryazanov S , Leonov A , Levin J , Shi S , Schmidt F , Prix C , Pan-Montojo F , Bertsch U , Mitteregger-Kretzschmar G , Geissen M , Eiden M , Leidel F , Hirschberger T , Deeg AA , Krauth JJ , Zinth W , Tavan P , Pilger J , Zweckstetter M , Frank T , Bähr M , Weishaupt JH , Uhr M , Urlaub H , Teichmann U , Samwer M , Bötzel K , Groschup M , Kretzschmar H , Griesinger C , Giese A ((2013) ) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 125: , 795–813. |
[217] | Neumann M , Kahle PJ , Giasson BI , Ozmen L , Borroni E , Spooren W , Müller V , Odoy S , Fujiwara H , Hasegawa M , Iwatsubo T , Trojanowski JQ , Kretzschmar HA , Haass C ((2002) ) Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest 110: , 1429–1439. |
[218] | Matthes D , Gapsys V , Griesinger C , de Groot BL ((2017) ) Resolving the atomistic modes of Anle138b inhibitory action on peptide oligomer formation. ACS Chem Neurosci 8: , 2791–2808. |
[219] | Martinez Hernandez A , Urbanke H , Gillman AL , Lee J , Ryazanov S , Agbemenyah HY , Benito E , Jain G , Kaurani L , Grigorian G , Leonov A , Rezaei-Ghaleh N , Wilken P , Arce FT , Wagner J , Fuhrmann M , Caruana M , Camilleri A , Vassallo N , Zweckstetter M , Benz R , Giese A , Schneider A , Korte M , Lal R , Griesinger C , Eichele G , Fischer A ((2018) ) The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol Med 10: , 32–47. |
[220] | Brendel M , Deussing M , Blume T , Kaiser L , Probst F , Overhoff F , Peters F , von Ungern-Sternberg B , Ryazanov S , Leonov A , Griesinger C , Zwergal A , Levin J , Bartenstein P , Yakushev I , Cumming P , Boening G , Ziegler S , Herms J , Giese A , Rominger A ((2019) ) Late-stage Anle138b treatment ameliorates tau pathology and metabolic decline in a mouse model of human Alzheimer’s disease tau. Alzheimers Res Ther 11: , 67. |
[221] | Wagner J , Krauss S , Shi S , Ryazanov S , Steffen J , Miklitz C , Leonov A , Kleinknecht A , Göricke B , Weishaupt JH , Weckbecker D , Reiner AM , Zinth W , Levin J , Ehninger D , Remy S , Kretzschmar HA , Griesinger C , Giese A , Fuhrmann M ((2015) ) Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol 130: , 619–631. |
[222] | Price DL , Koike MA , Khan A , Wrasidlo W , Rockenstein E , Masliah E , Bonhaus D ((2018) ) The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci Rep 8: , 16165. |
[223] | Fields CR , Bengoa-Vergniory N , Wade-Martins R ((2019) ) Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci 12: , 299. |
[224] | Yang F , Lim GP , Begum AN , Ubeda OJ , Simmons MR , Ambegaokar SS , Chen PP , Kayed R , Glabe CG , Frautschy SA , Cole GM ((2005) ) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280: , 5892–5901. |
[225] | Begum AN , Jones MR , Lim GP , Morihara T , Kim P , Heath DD , Rock CL , Pruitt MA , Yang F , Hudspeth B , Hu S , Faull KF , Teter B , Cole GM , Frautschy SA ((2008) ) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326: , 196–208. |
[226] | Hu S , Maiti P , Ma Q , Zuo X , Jones MR , Cole GM , Frautschy SA ((2015) ) Clinical development of curcumin in neurodegenerative disease. Expert Rev Neurother 15: , 629–637. |
[227] | Singh PK , Kotia V , Ghosh D , Mohite GM , Kumar A , Maji SK ((2013) ) Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem Neurosci 4: , 393–407. |
[228] | Jha NN , Ghosh D , Das S , Anoop A , Jacob RS , Singh PK , Ayyagari N , Namboothiri IN , Maji SK ((2016) ) Effect of curcumin analogs on alpha-synuclein aggregation and cytotoxicity. Sci Rep 6: , 28511. |
[229] | Dimant H , Ebrahimi-Fakhari D , McLean PJ ((2012) ) Molecular chaperones and co-chaperones in Parkinson disease. Neuroscientist 18: , 589–601. |
[230] | Lo Bianco C , Shorter J , xE , gulier E , Lashuel H , Iwatsubo T , Lindquist S , Aebischer P ((2008) ) Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 118: , 3087–3097. |
[231] | Putcha P , Danzer KM , Kranich LR , Scott A , Silinski M , Mabbett S , Hicks CD , Veal JM , Steed PM , Hyman BT , McLean PJ ((2010) ) Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exp Ther 332: , 849–857. |
[232] | Fonseca-Ornelas L , Eisbach SE , Paulat M , Giller K , Fernández CO , Outeiro TF , Becker S , Zweckstetter M ((2014) ) Small molecule-mediated stabilization of vesicle-associated helical α-synuclein inhibits pathogenic misfolding and aggregation. Nat Commun 5: , 5857. |
[233] | Collier TJ , Srivastava KR , Justman C , Grammatopoulous T , Hutter-Paier B , Prokesch M , Havas D , Rochet J-C , Liu F , Jock K , de Oliveira P , Stirtz GL , Dettmer U , Sortwell CE , Feany MB , Lansbury P , Lapidus L , Paumier KL ((2017) ) Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol Dis 106: , 191–204. |
[234] | Fanning S , Haque A , Imberdis T , Baru V , Barrasa MI , Nuber S , Termine D , Ramalingam N , Ho GPH , Noble T , Sandoe J , Lou Y , Landgraf D , Freyzon Y , Newby G , Soldner F , Terry-Kantor E , Kim T-E , Hofbauer HF , Becuwe M , Jaenisch R , Pincus D , Clish CB , Walther TC , Farese RV , Srinivasan S , Welte MA , Kohlwein SD , Dettmer U , Lindquist S , Selkoe D ((2018) ) Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell 73: , 1001–1014. |
[235] | Imberdis T , Negri J , Ramalingam N , Terry-Kantor E , Ho GPH , Fanning S , Stirtz G , Kim T-E , Levy OA , Young-Pearse TL , Selkoe D , Dettmer U ((2019) ) Cell models of lipid-rich α-synuclein aggregation validate known modifiers of α-synuclein biology and identify stearoyl-CoA desaturase. Proc Natl Acad Sci U S A 116: , 20760–20769. |
[236] | Vincent BM , Tardiff DF , Piotrowski JS , Aron R , Lucas MC , Chung CY , Bacherman H , Chen Y , Pires M , Subramaniam R , Doshi DB , Sadlish H , Raja WK , Solís EJ , Khurana V , Le Bourdonnec B , Scannevin RH , Rhodes KJ ((2018) ) Inhibiting stearoyl-CoA desaturase ameliorates α-synuclein cytotoxicity. Cell Rep 25: , 2742–2754. |
[237] | Shahmoradian SH , Lewis AJ , Genoud C , Hench J , Moors TE , Navarro PP , Castaño-Díez D , Schweighauser G , Graff-Meyer A , Goldie KN , Sütterlin R , Huisman E , Ingrassia A , Gier Y de , Rozemuller AJM , Wang J , Paepe AD , Erny J , Staempfli A , Hoernschemeyer J , Großerüschkamp F , Niedieker D , El-Mashtoly SF , Quadri M , Van IJcken WFJ , Bonifati V , Gerwert K , Bohrmann B , Frank S , Britschgi M , Stahlberg H , Van de Berg WDJ , Lauer ME ((2019) ) Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 22: , 1099–1109. |


