
Induced Muscle and Liver Absence of Gne in Postnatal Mice Does Not Result in Structural or Functional Muscle Impairment
Abstract
Background:
GNE Myopathy is a unique recessive neuromuscular disorder characterized by adult-onset, slowly progressive distal and proximal muscle weakness, caused by mutations in the GNE gene which is a key enzyme in the biosynthesis of sialic acid. To date, the precise pathophysiology of the disease is not well understood and no reliable animal model is available. Gne KO is embryonically lethal in mice.
Objective:
To gain insights into GNE function in muscle, we have generated an inducible muscle Gne KO mouse. To minimize the contribution of the liver to the availability of sialic acid to muscle via the serum, we have also induced combined Gne KO in liver and muscle.
Methods:
A mouse carrying loxp sequences flanking Gne exon3 was generated by Crispr/Cas9 and bred with a human skeletal actin (HSA) promoter driven CreERT mouse. Gne muscle knock out was induced by tamoxifen injection of the resulting homozygote GneloxpEx3loxp/HSA Cre mouse. Liver Gne KO was induced by systemic injection of AAV8 vectors carrying the Cre gene driven by the hepatic specific promoter of the thyroxine binding globulin gene.
Results:
Characterization of these mice for a 12 months period showed no significant changes in their general behaviour, motor performance, muscle mass and structure in spite of a dramatic reduction in sialic acid content in both muscle and liver.
Conclusions:
We conclude that post weaning lack of Gne and sialic acid in muscle and liver have no pathologic effect in adult mice. These findings could reflect a strong interspecies versatility, but also raise questions about the loss of function hypothesis in Gne Myopathy. If these findings apply to humans they have a major impact on therapeutic strategies.
INTRODUCTION
GNE Myopathy is a unique recessive neuromuscular disorder characterized by adult-onset, slowly progressive distal and proximal muscle weakness, and a typical muscle pathology [1]. Although the disease occurs worldwide, clear clusters have been recognized, in the Middle East, with M743T as the founder mutation and also in Japan, with two main mutations D207V and V603L [2]. Although GNE has been identified as the disease-causing gene in 2001 [3], there is still no therapy for this disorder, for two main reasons: the pathophysiological mechanism leading from GNE mutations to the muscle phenotype in GNE Myopathy is still unclear, and there is a lack of reliable animal models for this disorder. The product of the GNE gene is UDP-N-acetylglucosamine 2-epimerase/N-acetyl-mannosamine kinase (UDP-GlcNAc 2-epimerase/ManNAc kinase, GNE), a 753 aa very highly conserved protein in mammals. GNE consists of two functional domains, an N-terminal epimerase domain and a C-terminal kinase domain [4, 5]. More than 400 mutations reported to date in patients with GNE Myopathy are dispersed along the entire gene, both in the epimerase and in the kinase coding sequences, and very often one mutation in each domain are combined in the compound heterozygous patients (for the recent updates of GNE myopathy mutations see the online GNE variation database http://www.dmd.nl/nmdb2/home.php?select_db=GNE). GNE is the key enzyme in the metabolic pathway leading to the synthesis of N-acetylneuraminic acid (Neu5Ac) [4, 6], a cytosolic pathway of four consecutive reactions. Neu5Ac is the biosynthetic precursor of virtually all the naturally occurring sialic acids, the most abundant terminal monosaccharides on glycoconjugates in eukaryotic cells [7, 8]. Sialylation of glycoproteins and glycolipids on cell surfaces has an important role during development and regeneration, particularly in the brain, and in the pathogenesis of several diseases [9]. The biological importance of GNE is further reflected by a drastic reduction of cellular sialylation upon loss of enzyme activity [6] and by the fact that a knockout of the gene in mice is lethal to the embryo at day E8.5 [10]. Wedekind et al. [11] showed the cause of intra uterine death to be brain hemorrhages in these embryos, consistent with the fact that sialic acid is particularly needed for brain development during the embryonic stages. Sialic acid is also a crucial nutrient during periods of rapid neural growth and brain development in the newborn, as reflected by the fact that sialic acid is present in large amounts in human and mouse feeding milk [12, 13]. Recently Huang et al. [14] reported that specific GNE missense variants impair angiogenesis in the developing brain in mice. Also, in two knock-in mouse models (Gne M743T/M743T and Gne V603L/V603L mice), a severe renal phenotype leading to early death (without muscle symptoms) was observed [15–17], suggesting a role for sialylation in early kidney development too. In addition, it seems that GNE is important in humans for the development of thrombocytes, since recent reports of congenital thrombocytopenia have been related to mutations in the GNE gene [18].
Typically, the first symptoms of GNE Myopathy are related to muscle weakness and appear during the third decade of life or later (there are rare cases of teenage onset but no congenital myopathies). Only partial GNE deficient activity has been observed in GNE Myopathy patients [19]. The pathophysiological pathway leading from bi allelic GNE mutations to the adult onset muscle phenotype in GNE Myopathy is still unresolved. The currently leading hypothesis of impaired sialylation in patients’ muscle cells as the cause of the muscle pathology is still controversial. Some patients have reduced (but not complete) hyposialylation (general or of specific proteins) and others have not [20–25]. Furthermore, clinical trials delivering slow release sialic acid orally to patients were not successful [26], although the efficacy of supplementation treatments is still controversial as recent studies report some efficacy with aceneuramic acid administration [27] and with sialyllactose in a pilot trial [28].
Established mouse models for elucidating the human disease mechanism are also difficult to interpret. Hyposialylation was detected in the two knock-in mouse models [15–17] which had no apparent muscle symptoms. Although treatment with sialic acid or its metabolic precursors, (e.g.ManNAc) could rescue (at least in part) the renal failure in all mouse models, the lack of renal involvement in human GNE Myopathy and the lack of muscle phenotype in these mice, hampers conclusions on GNE Myopathy mechanism as simply lack of sialylation and complicates metabolic treatment approaches. Finally, a transgenic mouse overexpressing the human GNE epimerase D207V mutation was generated on a Gne endogenous knock out background [29]. These Gne(–/–) hGNED207V-Tg mice were reported to show hyposialylation in most organs, and also a muscle phenotype, that could be partly improved by administration of ManNAc, the product of the epimerase activity in the sialic acid pathway [30]. However, this phenotype could not be reproduced in other laboratories [31, 32], and these mice are no longer available.
Since to date, cellular and mouse models for human adult onset GNE Myopathy have not been informative enough to understand its pathophysiology, we decided to further explore the function(s) of GNE in adult muscle, by completely deleting this gene in the postnatal period.
We hypothesized that these studies will reveal such function(s) that could then be investigated in patients’ cells and tissues for their role in muscle pathology. In order to have more insights of the Gne function in muscle, we have generated a loxp GneFLAG loxp conditional mouse. We have used this mouse platform to generate post weaning Gne KO in muscle. In order to minimize the potential supply of sialic acid to muscles through the serum, and since liver is the main source of sialylated glycoconjugates in the serum, we have also generated a double tissue post weaning Gne KO in muscle and in liver.
MATERIALS AND METHODS
Mice maintenance
Animals were maintained at the facilities of the Authority for Biological and Biomedical Models at the Faculty of Medicine of the Hebrew University of Jerusalem. Mice were housed at a specific pathogen-free facility in single aged occupancy cages, under conditions of constant photoperiod (12 : 12 L:D) with free access to food and water ad libitum. All mice were on a standard diet (Cat # 2918, Teklad Global Rodent diet, 18% protein, Harlan, Wisconsin, USA-which contains approximately 6μg of Neu5Gc per gram of chow) [33]. All animal procedures were performed in accordance with institutional guidelines under protocols approved by the Institutional Committee for Animal Care of the Hebrew University-Hadassah Medical Center.
Mice generation
Our previously established GneFlag mice [34] were used as a platform for the insertion of the loxp sequences flanking exon3, by Crispr/Cas9. To generate the GneFlag loxpexon3loxp mouse two gRNAs targeting intron 2 and intron 3 of the Gne gene were designed, using CrisprGold and IDT gRNA designer softwares, which predict the most suitable targets and minimize off target outcomes. The well-established crRNA:tracrRNA Crispr/Cas complex strategy was followed (idtdna.com). Synthetic crRNA sequence of either intron2/intron3 were annealed with universal tracrRNA to generate the crRNA:tracrRNA complex. For HDR we designed a symmetrical, single-strand oligonucleotide donor containing the forward 5’ – 3’ consensus 34bp lox sequence with upstream and downstream homologous “tails” (ssODN; IDT Technologies) The ssODN was of the same strand of the gRNA to prevent hybridization of the two in the injected embryo. Approximately 200 GneFLAG zygotes were microinjected with 0.61μM guide RNA (cr+tracr complex), 0.3μM Cas9 IDT protein and 15 ng/μl ssDNA loxp donor. Viable 2-cell stage embryos were transferred to pseudopregnant CB6/F1 females. The final lineage was obtained by 2 consecutive steps, first loxp sequences were introduced into intron 2. After establishing a loxp2 homozygote lineage, zygotes from those mice were used for the insertion of loxp3 sequences in intron 3. Heterozygotes mice for both loxp sites were identified by genotyping and bred with the human skeletal actin (HSA) CreERT lineage, purchased from Jackson Laboratories. Eventually we established a mouse lineage homozygote for loxp sequences flanking exon 3 carrying a muscle specific CreERT hemizygote transgene. The actual zygotes injections were performed at the Weizmann Institute of Science, Department of Veterinary Resources.
Mice injections
The Gne loxp ex3 FLAG CreHSA colony was expanded. To generate muscle specific GneKO, 9 male and 9 female 6 week old mice were injected subcutaneously with tamoxifen (Sigma, #T2859) at a dose of 8–10 mg per mouse (according to weight) for 2 non consecutive days (total of 16–20 mg/mouse) [35]. Controls for muscle specific Gne KO were homozygotes for the loxp exon3 loxp insertion but did not carry the muscle specific Cre transgene.
To generate muscle and liver Gne KO, 9 male and 9 female 6 week old mice were injected with a liver specific promoter driven Cre gene viral vector -AAV8 TBG Cre- at a dose of 1.1011 vg per mouse. Viral vectors were diluted in PBS and injected in the tail vein in a 250 microliters solution volume. TBG is the thyroxine binding globulin promoter, a liver specific promoter. The plasmid was a gift from Prof Pikarsky laboratory. The viral vectors were produced at the ELSC Vector Core Facility, The Hebrew University of Jerusalem. Twenty four hours after viral vector injection, the tamoxifen injection protocol was applied as described above. Controls for the double tissue Gne KO were also homozygotes for the loxp exon3 loxp sequences, did not carry the muscle specific Cre transgene and were injected with PBS instead of AAV TBG Cre viral vector. All mice were injected with tamoxifen.
Mice follow up
The mice were followed for diverse parameters: weight (once a week); Rotarod performance measurements (time in seconds till fall) in a USB Rota-Rod treadmill, every month up to 3 months and then every 3 months. Open Field tests were carried out at Hadassah Brain labs, 6 and 12 months after GneKO: each animal is introduced to a plastic arena 50X50X30 (height) cm and monitored during the 6 minutes trial for the distance moved, velocity, time and frequency of the animal in the different arena locations using the tracking system -a digital GigE monochrome camera set connected to a computer with the EthoVision tracking software XT version 17. Measurements for each test described above were done at a similar time of day in identical environmental conditions.
In vivo MRI scanning
MRI images were acquired on a 7T 24 cm bore, cryogen-free MR scanner based on the proprietary dry magnet technology (MR Solutions, Guildford, UK) using 38-mm inner diameter, 70 mm length mouse whole body volume coil. The mice were positioned on a heated bed, which allowed for continuous anesthesia and breathing rate monitoring throughout the entire scan period. Coronal and axial T1-weighted (T1 W) spin-echo images were acquired (repetition time = 1100 milliseconds, echo time = 11 milliseconds, FOV = 60 mm, in-plane resolution = 230μm, slice thickness = 1 mm). Muscle volume of hindlimbs was analyzed using VivoQuant 4.0 software from Invicro.
Ultrasound (US) and photoacoustic (PA) imaging
The high-resolution ultrasound imaging was performed using a Vevo3100- LAZRX small animal US combined with photoacoustic (PA) imaging system (Visualsonics, Toronto, Canada), with a MX-550D linear-array transducer (40-MHz center frequency) used to acquire all images. The tunable laser supplied 10– 20 mJ per pulse over the 680–970-nm wavelength range, with a pulse repetition frequency of 20 Hz. Once initialized, the system was switched to the oxy/hemo mode to measure sO2 using the following parameters: depth, 10.00 mm; width, 14.08 mm; wavelength, 750 and 850 nm for the total hemoglobin concentration threshold (Hbt), and sO2, respectively. All images were acquired by placing the probe directly over the right hind limb. Before sO2 measurement, B-mode and Doppler US images were acquired to evaluate femoral artery blood flow and identify the region of interest in the hind limb muscle. Hind limb sO2 mapping were pseudo-colorized. For imaging, mice were anesthetized with isoflurane vaporized with O2. Isoflurane is used at 3.0% for induction and at 1.0–2.0% for maintenance. MRI and PA imaging experiments were performed at the Wohl Institute for Translational Medicine at Hadassah Hebrew University Medical Center.
Tissue specimens
At various time points mice were euthanized, blood was collected from retroorbital sinus, and different tissues (liver, spleen, kidney, brain, heart, quadriceps, tibialis anterior,gastrocnemius and upper limbs) were partly snap frozen in liquid nitrogen and stored at –80°C till further processing, and partly fixed in 4% paraformaldehyde.
Molecular analyses
– Genotyping
Four-week-old pups were ear punched and genomic DNA was extracted using a D-tail kit (Syntezza, USA, #DE-11). Genotyping for loxp sequences was performed by PCR with primers loxp2: Int2F- 5’ctgccctcaaagaaaattccc3’ & Ex3R – 5’ctaccgactctaccatggcc3’, loxp3 – int3-425F – 5’ cagacatgaacacttaaccataacttcg3’ and int3-425R – 5’cagaaggccatcgataacttcg3’. Cre sequences were assessed with primers HSAmCREF– 5’gcatggtggagatctttga3’ and HSAmCRER – 5’cgaccggcaaacggacagaagc3’. DNA from the different tissues collected at various timepoints was extracted by Wizard purification kit (Promega, USA, #A1120). Presence or absence of exon3 was evaluated by PCR with primers Int2F- 5’ctgccctcaaagaaaattccc3’ and int3-248R – 5’gatgcccatttctggtgtgt3’.
– Western blot
Frozen tissues were homogenized using the TissueLyser LT (Qiagen) for 3 minutes at 50 Hz in RIPA lysis buffer (50 mM Tris buffer pH 7.5, 5 mM EDTA, 150 mM NaCl, tritonX100 1%, 1 mM DTT, 1 mM PMSF, 17μg/ml Aprotinin, 10μg/ml leupeptin, 1 mM Vanadate), followed by 20 strokes through a syringe with a 26 gauge needle, incubation on ice for 10 min., and centrifugation (14,000 rpm for 30 min. at 4°C). Protein concentration was determined using Bradford Reagent (Sigma, #B6916). Protein lysates were separated on a precast 8% SDS-PAGE (NB10-008, Nusep, Australia, #10401206) and transferred to 0.2μm nitrocellulose membrane (PROTRAN, Germany, #BA85) according to manufacturer instructions (Trans-Blot Electrophoretic Transfer Cell; Bio-Rad Laboratories, Hercules, CA, USA). Nitrocellulose blots were blocked in 5% milk for 1 h. GneFlag was detected by incubation of the membrane in anti-FLAG M2 mouse monoclonal primary antibody (Sigma, # F3165) diluted in 2% BSA/PBS-Tween 20 (0.3%) for 3 hrs at RT, followed by incubation in HRP-conjugated goat anti mouse secondary antibody (Jackson ImmunoResearch, #115-035-008). Protein expression was visualized using the EZ-ECL (Biological Industries, Israel, #20-500-500).
Sialic acid measurements
– Sialic acid hydrolysis
Frozen dried tissues (Liver, Kidney, Quadriceps and Gastrocnemius muscles) were gently homogenized in 2 ml of 2M propionic acid in a MACS Dissociator (Miltenyi Biotec, Teterow, Germany) and incubated for 4 h at 80°C shaking. The samples were cooled down for 10 min on ice, followed by centrifugation for 20 min, 13200 rpm at 4°C. The supernatants were frozen dried overnight. Samples were resuspended in 0.5 ml aquadest. Protein concentrations were measured twice for every sample.
– DMB Labelling of sialic acids
Sample and reference solutions were labelled (2.5 h, 50°C, shaking) by DMB-labelling reagent (1,2-diamino-4,5-methylenedioxybenzene.2HCl (DMB), 1.6 mg/ml, DMB Sialic Acid Labelling Kit, #LT-DMB-01, QA Bio, Palm Desert, CA, USA). Samples were diluted in 2 : 1 ratio with water and measured as duplicates.
– HPLC
To measure the sialic acid concentration, 20μL of labelled sample and reference were injected into the device. A LiChroCART®250-4 LiChrospher®100 RP-18e (5μm) column (Merck KGaA, Darmstadt, Germany) was used. Reference panel (1.25nmol, AdvanceBio Sialic Acid reference panel, #GKRP-2503, Agilent, Santa Clara, CA, USA) was used as reference for every measurement. Solvent A (acetonitrile: methanol: water 9 : 7:84) and solvent B (acetonitrile) were used with a flow of 0.5 mL/min as flux. The concentrations were determined using diluted standard of the according sialic acid – 50 ng/mL to 10μg/mL of N-Glycolylneuraminic acid (Cay30283, Biomol GmbH, Hamburg, Germany) and of N-Acetylneuraminic acid (Cay16091, Biomol GmbH, Hamburg, Germany).
Histology
Paraffin-embedded tissue sections were stained with hematoxylin and eosin (H&E) for routine histologic examination.
Statistical analysis
Statistical analysis for the comparison between groups was performed by the Student’s two tailed unpaired t test.
RESULTS
Generation and validation of Gne KO mice lineages
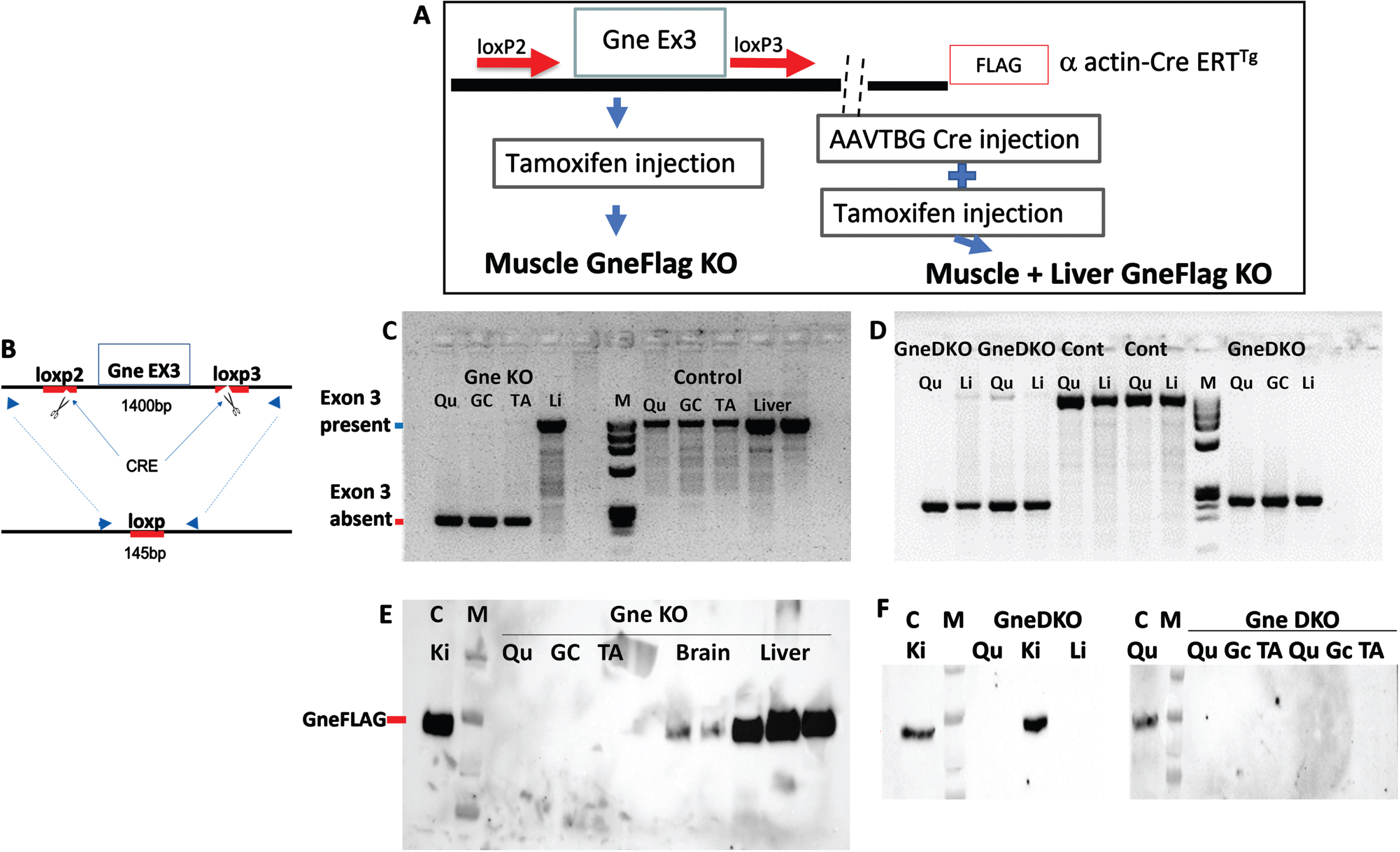
Using Crispr/Cas9 methodology, Loxp sequences were introduced consecutively at intron 2 and intron 3 to flank exon 3 of the Gne gene in the GneFLAG mice we previously developed [34]. This colony was then crossed with muscle specific (human skeletal actin) promoter driven HSA-CreERT mice and finally a lineage of Cre transgenic mice homozygotes for the loxp GneEx3loxp locus was established (Fig. 1A) and termed Gne loxp FLAG/HSACreERT. To induce Gne KO in skeletal muscle, tamoxifen was injected subcutaneously at the age of 6 weeks. To prevent the potential supply of sialic acid to muscles from the serum, we knocked out Gne from the liver by systemically injecting AAV8 TBG Cre (the Cre gene driven by a liver specific promoter TBG) viral vector 24 hours before tamoxifen injection. To validate the efficacy of these treatments in knocking out Gne, mice were sacrificed 30 days after treatment. Figure 1 depicts the absence of exon 3 and of GneFLAG only in muscles of tamoxifen treated mice (B, C, E). Similarly, mice injected with both the liver specific derived Cre viral vector AAVTBGCre and tamoxifen showed no FlagGne in both muscles and liver (D, F). To note, tests performed at 2 weeks post treatment showed traces of both exon 3 and GneFLAG protein in either muscles and liver (Suppl. Fig. 1).
Fig. 1
Generation of a double tissue GneKO mouse. A. Schematic representation of the design strategy to generate muscle GneKO and muscle+liver GneKO mice. B. Schematic representation of the PCR assay to assess GneKO status; blue arrows illustrate the primers location before and after excision of exon3 by Cre. C, D: PCR assessment of Gne exon3 KO in tissues of muscle specific GneKO (C)and muscle+liver GneKO (D), and controls (Cont) mice, 30 days after treatment. E, F: Western blot assessment with anti FLAG antibody of Gne protein KO in tissues of muscle specific GneKO (E), and muscle+liver GneKO (F) and control mice. Qu, quadriceps; Gc, gastrocnemius, TA, tibialis anterior; Li, liver; Ki, kidney; C, control. M, size marker.
Follow up of Gne KO lineages
Both Gne KO mice lineages were characterized during a period of 12 months after de facto Gne abrogation (that is 1 month after tamoxifen and AAVTBGCre injection) for general behavior, locomotor abilities and muscle function by various performance tests, and imaging. No changes were observed in general behavior, survival or weight gain during a 12 months period in either males or females (Fig. 2) both in muscle only or muscle and liver Gne KO mice, compared to their respective control littermates.
Fig. 2
Effect of GneKO on motor performance of mice. Weight (in grams), rotarod performance (in seconds to fall) and open field (distance moved in cms) of GneKO mice were monitored at different timepoints after Gne KO was effective for both muscle GneKO and muscle+liver GneKO mice. Nine mice per group were included in the assays up to 6 months, 7 up to 9 months and 5 up to 12 months. Blue, GneKO; orange, Controls.
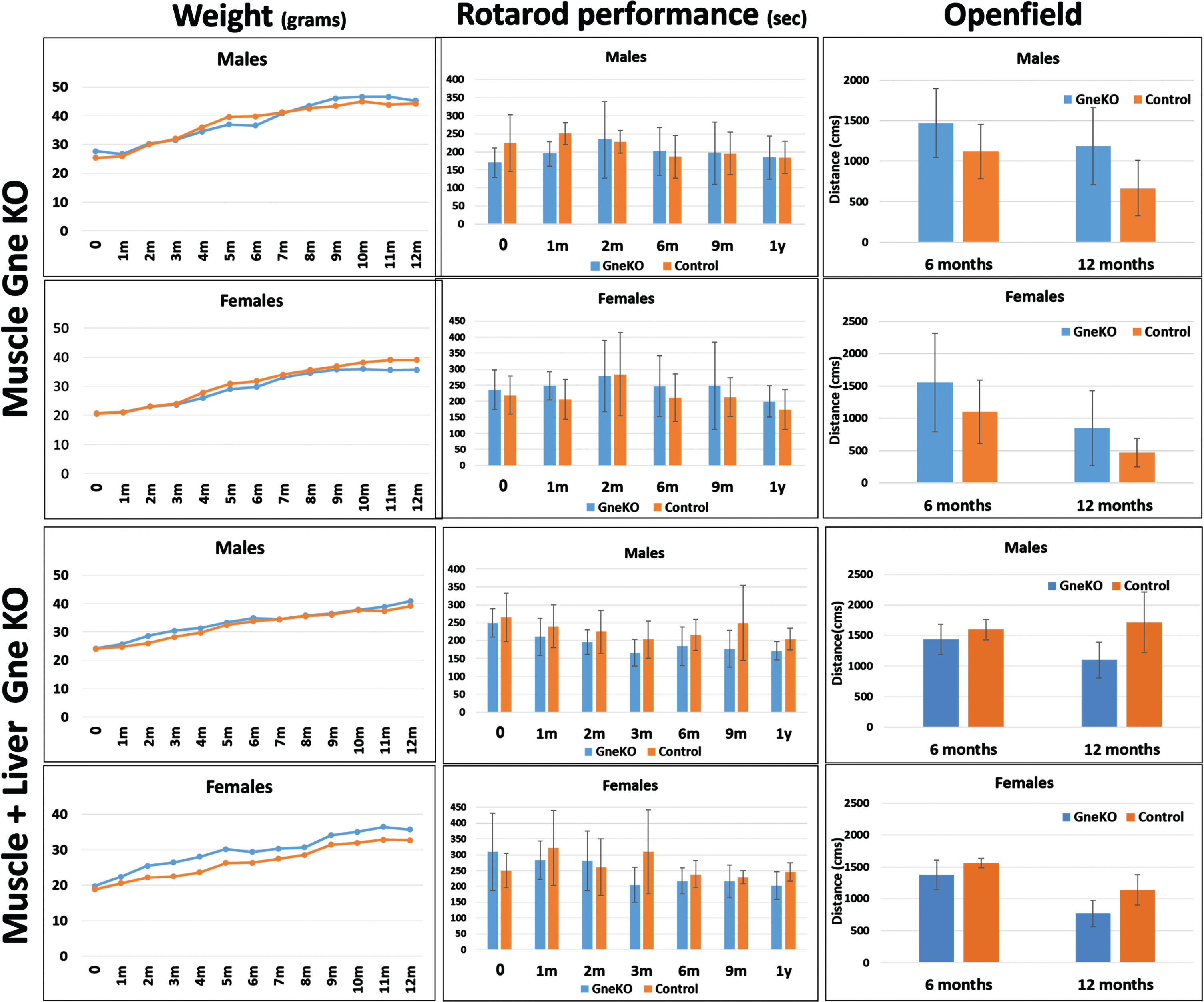
Locomotor (motility) assays showed no statistically significant changes in any of the 2 models (Fig. 2): rotarod performance and openfield tests did not point to consistent changes between the GneKO mice and the control animals during the 1year period examined.
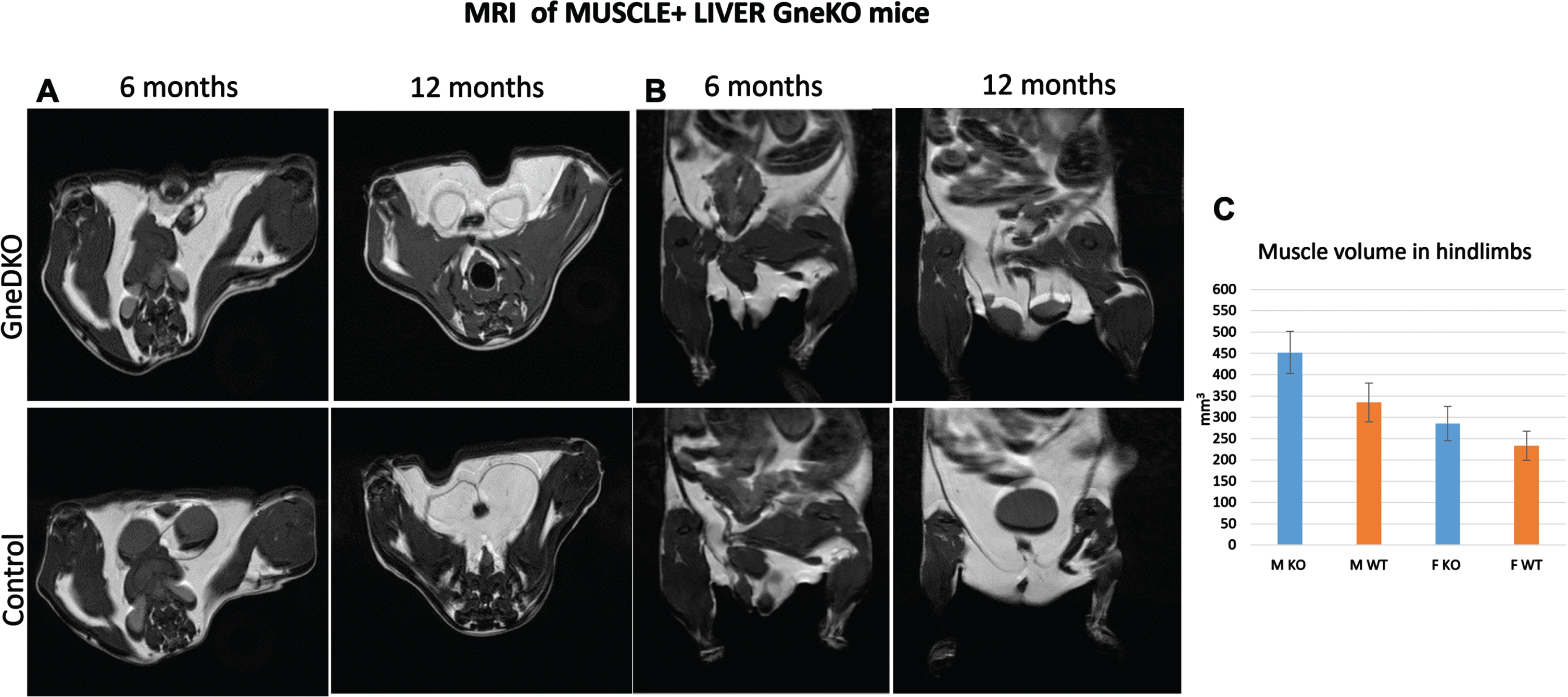
Photoacoustics were also applied to the double tissue Gne KO mice, for measuring saturated oxygen levels in muscle (Suppl Fig. 2). At 6 months post GneKO, SO2 levels in muscle were slightly reduced. However, at 1 year post Gne KO, these differences disappeared. Muscle MRI imaging of these mice (Fig. 3) showed no difference in muscle tissue volume, either after 6 or 12 months post Gne KO. No special features were observed in the liver of these mice either (data not shown).
Fig. 3
Muscle MRI. Representative figures of axial (A) and coronal planes (B) of Gne double tissue KO mice and controls. C, quantitation of the total hindlimb muscle volume (M, males; F, females).
Gne KO status assessment
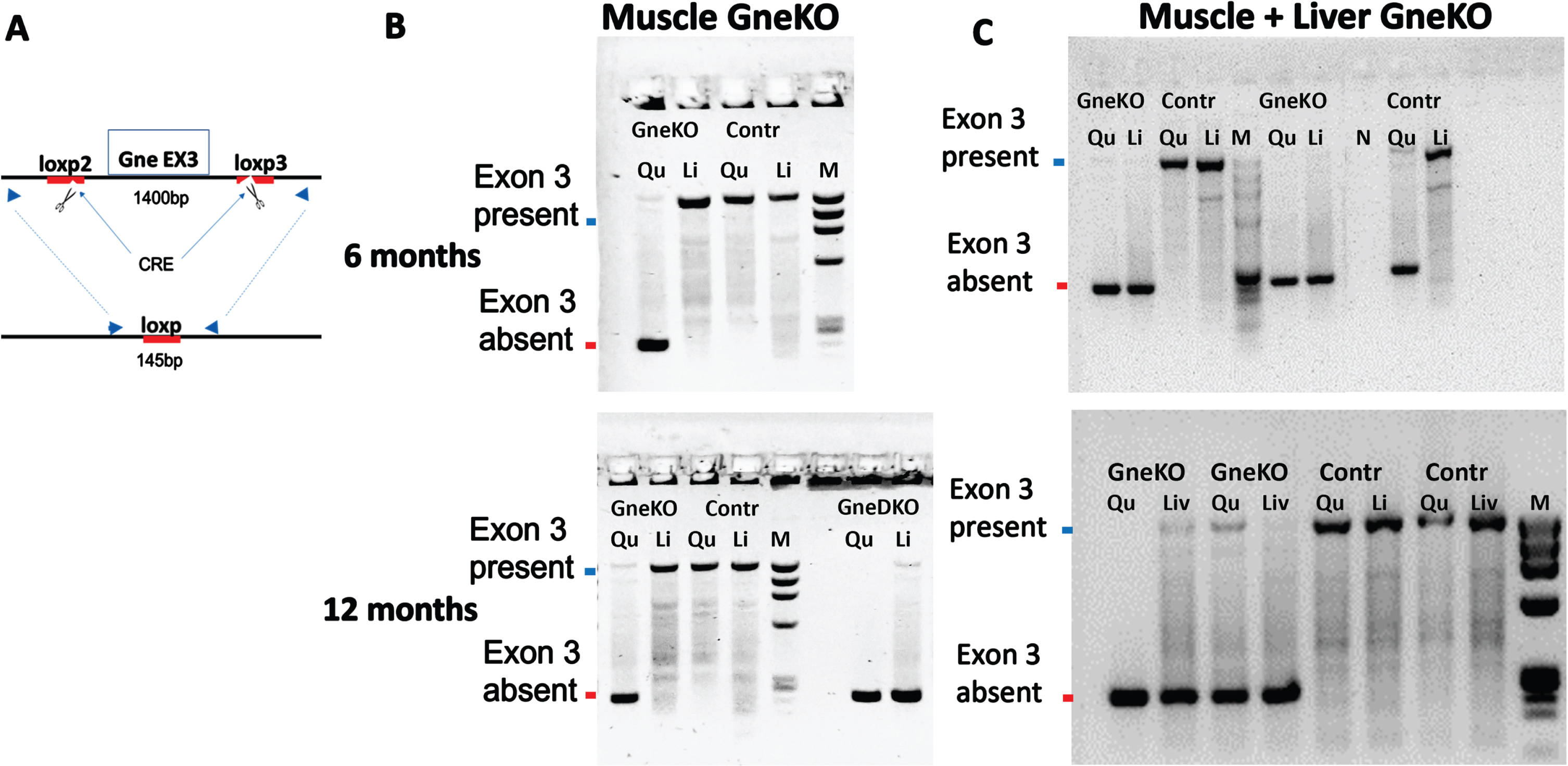
Mice were sacrificed at 6 and 12 months after Gne KO was originally achieved. DNA of various tissues was examined for the assessment of the Gne KO status by analyzing the presence or absence of exon3. Figure 4 shows the absence of exon3 in muscle (Fig. 4B) and in muscle and liver (Fig. 4C) as expected, 6 months and 12 months after Gne KO status was primarily achieved.
Fig. 4
Validation of GneKO status with time. A. Schematic representation of the PCR assay to assess GneKO status; blue arrows illustrate the primers location before and after excision of exon3 by Cre. B, C: PCR assessment of Gne exon3 KO in tissues of muscle specific GneKO (B) and muscle+liver Gne double KO (C), and controls (Cont) mice at 6 months and at 12 months after GneKO was effective. Qu, quadriceps; Li, liver;M, size marker. GneDKO, Gne double tissue KO.
General blood count and a full serum chemistry panel were performed. Serum biochemical analysis (Suppl Tables 1–4) was mostly normal, in particular CPK was in the normal range in all mice. Liver enzymes were slightly higher in the double tissue Gne KO mice. This could be due to the AAVTBG Cre viral infection, as previously described [36]. There was no difference in the platelets count.
Sialic acid measurements
Various organs including several skeletal muscle types were collected and processed for total bound sialic acid measurements by HPLC. This method enables also to discriminate between 2 forms of sialic acid, N-glycolylneuraminic acid (SiaGc), which is the major sialic acid form in mice, from N-acetylneuraminic acid (SiaAc) which is the major sialic acid in humans. As shown in Fig. 5, muscle specific GneKO mice show a strong reduction of SiaGc in muscle only, while double tissue muscle and liver Gne KO mice show a strong reduction of this sialic acid in both muscle and liver. Although the absolute level of sialic acid in control mice goes down with age, the reduction ratio between the controls and the GneKO mice stays stable at 6 and 12months post GneKO. Notably, the levels of SiaAc stayed essentially unchanged.
Fig. 5
Sialic acid content in various tissues of Gne KO mice. SiaGc and SiaAc content were quantitated in quadriceps, liver and kidney of muscle Gne KO (A) and muscle+liver Gne double KO (B) mice at 6 and 12 months after Gne KO was effective. n.s, not significant; *p < 0.05; **p < 0.01; ***p < 0.005.
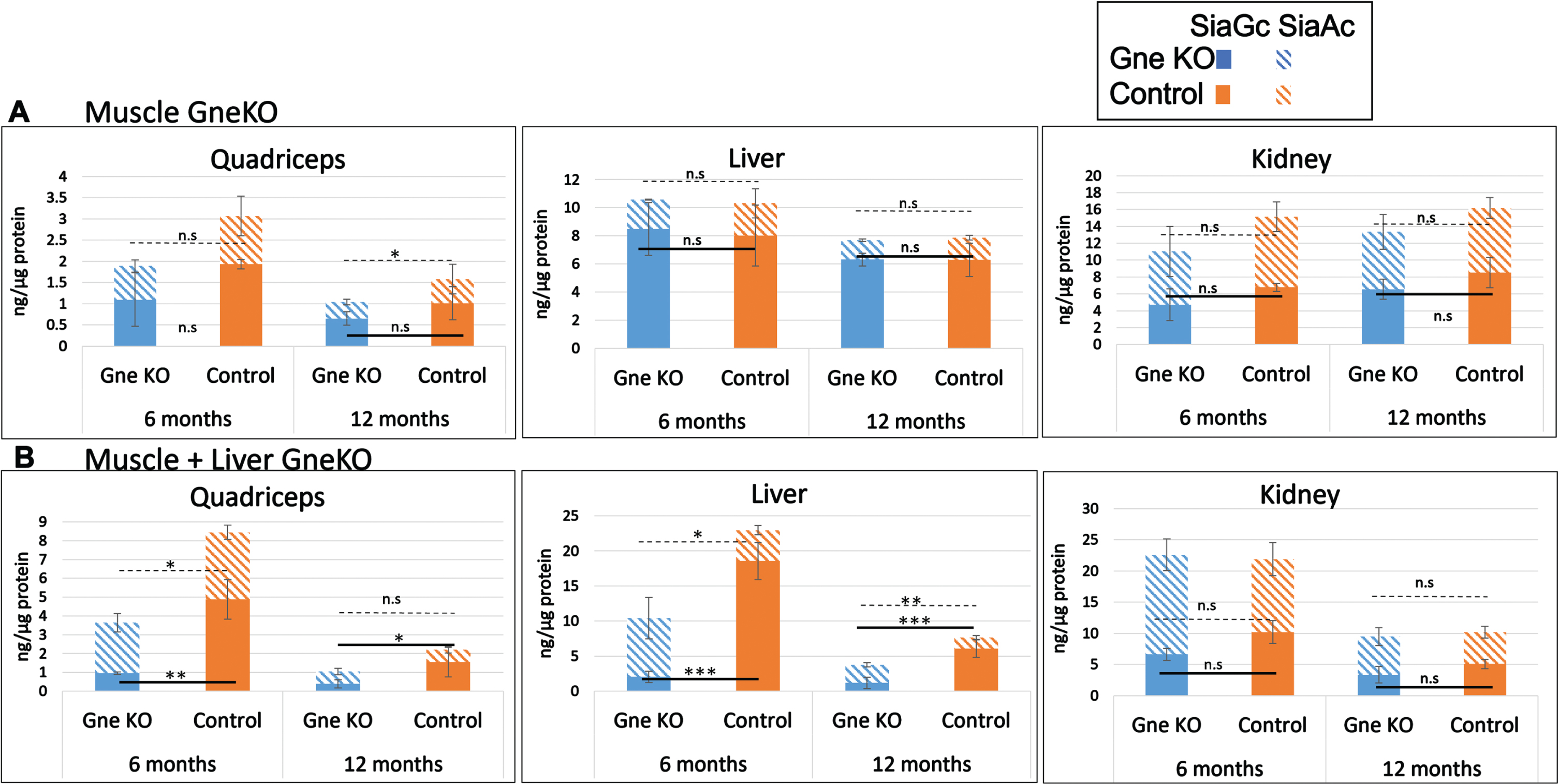
Interestingly, the level of SiaGc in muscle specific GneKO mice was the same as in the double Gne KO mice. This significant sialic acid reduction was maintained during the 1year period, thus confirming the Gne KO status in muscle and liver of the mice during the entire study.
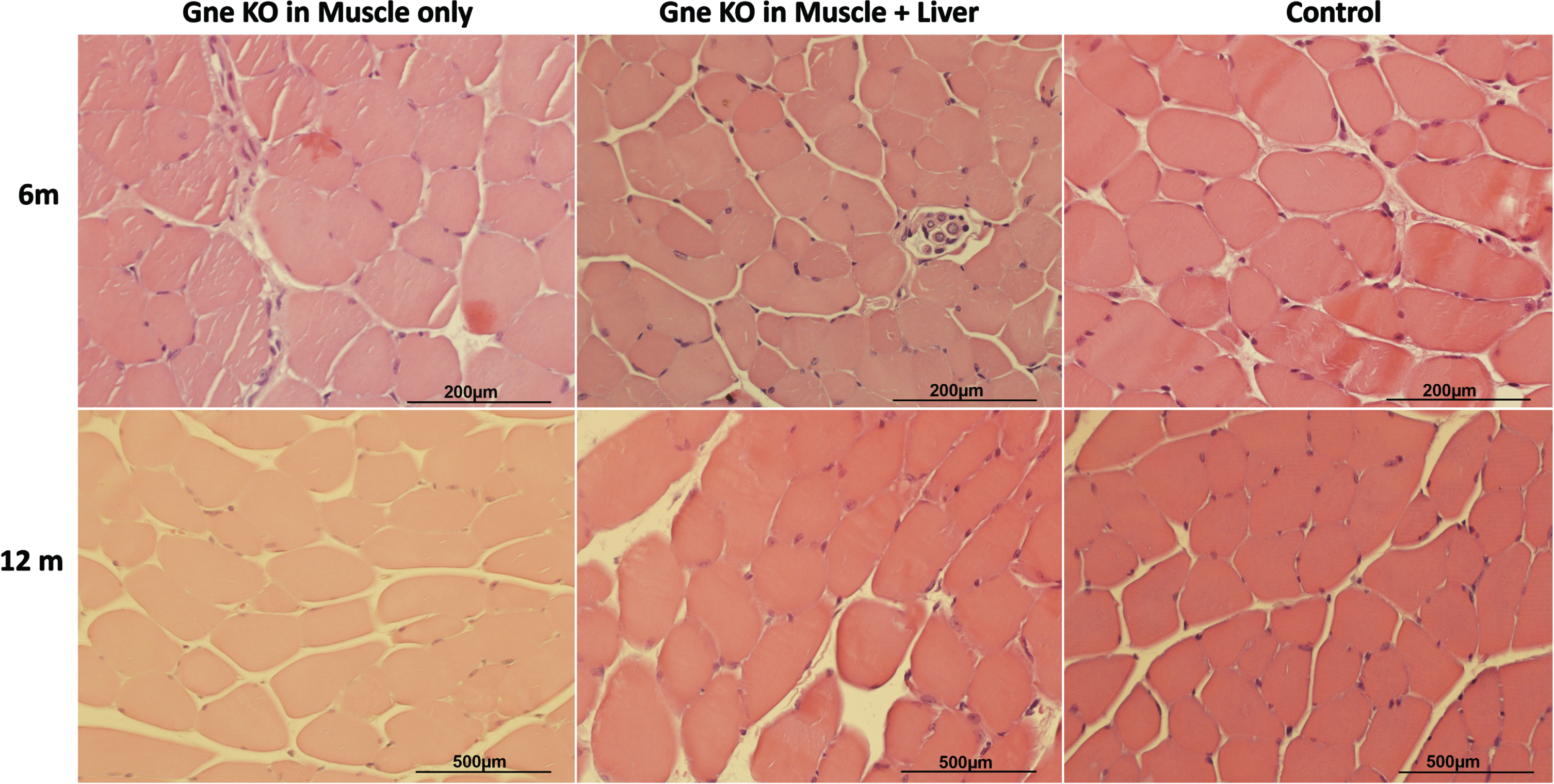
Histology
Histology sections of muscles did not point to any significant change in these tissues in the Gne KO mice compared to their control littermates at either 6 or 12months post GneKO (Fig. 6). Liver was not affected either (Suppl. Fig. 3).
Fig. 6
Muscle Histological Sections. Representative H&E tibialis anterior histological sections of muscle Gne KO, muscle+liver Gne double KO and control mice 6 and 12 months after GneKO was effective.
DISCUSSION
Gne and sialic acid are essential for embryonal development in mice, and most likely in all mammalians. Although the importance of sialic acid to the immune system during the entire lifespan of mammals and in humans in particular, is well described [8, 9], the role of sialic acid in different tissues, and in particular in muscle in postnatal life has not been evaluated. GNE Myopathy is a rare recessive, adult onset, progressive neuromuscular disorder. Although reduction of sialic acid synthesis has been thought to be the obvious hypothesis for the pathomechanism of GNE myopathy, the muscle pathophysiology of this disease is not fully understood.
The lack of suitable animal models for this myopathy severely impairs the comprehensive understanding of GNE functions in tissues, in particular muscle. Thus, viable models with a complete lack of the Gne protein could be more informative. To investigate the role of Gne and sialic acid in this adult human disease and since Gne KO is embryonically lethal, we have induced Gne knock out in mice postnatally, by establishing a loxp GneExon3 loxp mouse lineage. In addition to a postnatal muscle specific Gne KO that examines its effect on muscle we have also generated a double tissue, post weaning combined Gne KO in liver and muscle. The reason for the generation of the double GNE KO model is because the liver is the main source of sialylated glycoconjugates in the serum that could be incorporated in muscle tissue.
Surprisingly, these two mouse models did not present any sign of muscle impairment either structural or functional. Despite lack of Gne and severe reduction in sialic acid in muscle and liver these mice behaved normally and did not suffer from any detectable abnormality in these two organs and also not in basic renal tests and thrombocytes count. These findings indicate that in contrast to the period of fetal development, GNE and sialic acid are not needed postnatally for muscle and for liver function in mice. If this is true for adult humans, the late onset myopathy cannot be explained by a simple biochemical disturbance- the lack of sialylation. Still, it must be considered that the sialic acid reduction observed in mice comes from SiaGc and that SiaAc levels remained unchanged. Although these levels are rather low, it could be that they are able to function as and compensate for the lacking SiaGc. It could also be that SiaAc plays a different role than SiaGc. SiaAc could be responsible for muscle pathology in humans and not be relevant in mice since there is almost no sialic acid of this type in their organism. Also, we cannot rule out that the sialic acid form relevant to GNE Myopathy is one of the few rare forms of sialic acid, but since to date reliable techniques to quantify them are not available it is not possible to find out and most studies in mammals focus on SiaGc and SiaAc major sialic acid forms.
Interestingly though, according to the canonical biosynthetic pathway for SiaGc in mice, which results from the hydroxylation of SiaAc by the Cmah enzymatic activity, it is not clear why the SiaAc present in muscle is not catalyzed to SiaGc in these mice which have an intact Cmah activity. Furthermore, it is not clear how in the total absence of Gne in muscle and liver, the SiaAc levels are maintained. The fact that the same reduced levels of sialic acid were found in skeletal muscles of the muscle specific Gne KO and of the muscle+liver Gne KO mice could indicate that most sialic acid in muscle is not dependent on the circulating free sialic acid. If this is true for humans too then oral therapy leading to increased serum sialic acid may not suffice.
From all these considerations, we can conclude that lack of sialic acid in adult muscle and liver tissue of mice does not result in muscle impairment. This observation should be taken into account when potential treatment evaluations for GNE Myopathy are based on mouse models. A simple explanation for the results of our studies could be that Gne and sialic acid have a different importance in skeletal muscle physiology in mice than in humans and therefore their response to Gne KO is not useful to unravel the disease mechanism in humans. Indeed there are other examples of mice models for several neuromuscular and other disorders which do not recapitulate the disease symptoms, such as in DMD mice models [37]. In some cases, the use of different animal models such as rats have proved to be more relevant [38–42] and therefore could be considered also for GNE Myopathy.
If lack of Gne in adult muscle and liver tissues does not impair muscle function at least in mice, other mechanisms for GNE myopathy could be postulated. In particular, several studies converge on the role of Gne in myogenesis at least in vitro [34, 43, 44] and in muscle regeneration in mice [45], but the present findings suggest that Gne is not critical for these processes. As we have considered previously it could be that the potential additional function(s) of Gne in muscle alone is not enough and needs the support of other tissues for the maintenance of muscle structure and function. To address both this and the more general sialic acid supplementation hypotheses, we are currently generating a whole body conditional Gne KO lineage using our loxpGneExon3 loxp mouse platform and a ubiquitin promoter driven CreERT lineage acquired from Jackson Laboratories.
In the present studies we see that lack of Gne has no pathogenic effect in muscle and liver in adult mice. In contrast recessive missense mutations in the embryo have a strong effect that often result in early death [15–17]. This could suggest that missense mutations result in a form of toxic gain of function. Although the vast majority of gain-of function mutations are dominant, there are some rare examples of recessive gain of function [46–48]. The hypothesis of gain of function of GNE recessive mutations for GNE Myopathy could explain the present results as well as the pathology observed in the Gne(–/–) hGNED207V-Tg mice, which were generated based on over supplementation of the D207V mutated GNE. Therefore, this hypothesis should be explored as it would open new avenues for the understanding of GNE Myopathy pathophysiology and would have critical implications in the design of therapeuticstrategies.
ACKNOWLEDGMENTS
This work was supported by the Neuromuscular Disease Foundation (NDF), Los Angeles, California (202201 & 202303 to S.M-R), by the Israel Science Foundation (1442/18 to S.M-R), by the Deutsche Forschungsgemeinschaft (460683975 to RH, as part of the European Union’s Horizon 2020 research and innovation programme under the EJP RD COFUND-EJP No. 825575).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Stella Mitrani-Rosenbaum is an Editorial Board member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer review.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study. The data supporting the findings of this study are available within the article and/or its supplementary material, or on request from the corresponding author.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-240056.
REFERENCES
[1] | Argov Z , Mitrani-Rosenbaum S . GNE Myopathy. In: Narayanaswami, P., Liewluck, T. (eds) Principles and Practice of the Muscular Dystrophies. Current Clinical Neurology. Humana, Cham. (2023) . https://doi.org/10.1007/978-3-031-44009-0_9. |
[2] | Argov Z , Mitrani-Rosenbaum S . GNE myopathy: Two clusters with history and several founder mutations. J Neuromuscul Dis. (2015) ;2: :S73–6. |
[3] | Eisenberg I , Avidan N , Potikha T , Hochner H , Chen M , Olender T , et al. The UDP-N -Acetylglucosamine 2-epimerase/N-Acetylemannosamine kinase is mutated in recessive hereditary inclusion body myopathy. Nat Genet. (2001) ;29: :83–7. |
[4] | Hinderlich S , Stasche R , Zeitler R , Reutter W . A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver: purification and characterization of UDPN-acetylglucosamine 2 epimerase/N-acetylmannosamine kinase. J Biol Chem. (1997) ;272: :24313–8. |
[5] | Effertz K , Hinderlich S , Reutter W . Selective loss of either the epimerase or kinase activity of UDP-N-acetylglucosamine 2 epimerase/N-acetylmannosamine kinase due to site-directed mutagenesis based on sequence alignments. J Biol Chem. (1999) ;274: :28771–8. |
[6] | Keppler T , Hinderlich S , Langner J , Schwartz-Albiez R , Reutter W , Pawlita M . UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science. (1999) ;284: :1372–6. |
[7] | Chen X , Varki A . Advances in the biology and chemistry of sialic acids. ACS Chem Biol. (2010) ;19: :163–76. |
[8] | Varki A , Gagneux P . Multifarious roles of sialic acids in immunity. Ann NY Acad Sci. (2012) ;1253: :16–36. |
[9] | Varki A . Sialic acids in human health and disease. Trends Mol Med. (2008) ;14: :351–60. |
[10] | Schwarzkopf M , Knobeloch KP , Rohde E , Hinderlich S , Wiechens N , Lucka L , et al. Sialylation is essential for early development in mice. Proc Natl Acad Sci USA. (2002) ;99: :5267–70. |
[11] | Wedekind H , Kats E , Weiss AC , Thiesler H , Klaus C , Kispert A , et al. Uridine diphosphate-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase deletion in mice leads to lethal intracerebral hemorrhage during embryonic development. Glycobiology. (2021) ;31: :1478–89. |
[12] | Wang B . Molecular mechanism underlying sialic acid as an essential nutrient for brain development and cognition. Adv Nutr. (2012) ;3: :465S–72S. |
[13] | Sprenger N , Duncan PI . Sialic acid utilization. Adv Nutr. (2012) ;3: :392S–7S. |
[14] | Huang L , Kondo Y , Cao L , Han J , Li T , Zuo B , et al. Novel GNE missense variants impair de novo sialylation and cause defective angiogenesis in the developing brain in mice. Blood Adv. (2024) ;8: :991–1001. |
[15] | Galeano B , Klootwijk R , Manoli I , Sun M , Ciccone C , Darvish D , et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. (2007) ;117: :1585–94. |
[16] | Sela I , Yakovlev L , Becker Cohen M , Elbaz M , Yanay N , Ben Shlomo U , et al. Variable phenotypes of knockin mice carrying the M712T Gne mutation. Neuromolecular Med. (2013) ;15: :180–91. |
[17] | Ito M , Sugihara K , Asaka T , Toyama T , Yoshihara T , Furuichi K , et al. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS ONE. (2012) ;7: (1):e29873. |
[18] | Marín-Quílez A , Díaz-Ajenjo L , Di Buduo CA , Zamora-Cánovas A , Lozano ML , Benito R , González-Porras JR , Balduini A , Rivera J , Bastida JM . Inherited Thrombocytopenia Caused by Variants in Crucial Genes for Glycosylation. Int J Mol Sci. (2023) ;24: :5109. |
[19] | Krause S , Aleo A , Hinderlich S , Merlini L , Tournev I , Walter MC , et al. GNE protein expression and subcellular distribution are unaltered in HIBM. Neurology. (2007) ;69: :655–9. |
[20] | Hinderlich S , Salama I , Eisenberg I , Potikha T , Mantey L , Yarema K , et al. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered cellular sialylation in hereditary inclusion body myopathy. FEBS Letters. (2004) ;566: :105–9. |
[21] | Noguchi S , Keira Y , Murayama K , Ogawa M , Fujita M , Kawahara G , et al. Reduction of UDP-N acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem. (2004) ;279: :11402–7. |
[22] | Saito F , Tomimitsu H , Arai K , Nakai S , Kanda T , Shimizu T , et al. A Japanese patient with distal myopathy with rimmed vacuoles: missense mutations in the epimerase domain of the UDP-N-acetylglucosamine 2-epimerase N-acetylmannosaminekinase (GNE) gene accompanied by hyposialylation of skeletal muscle glycoproteins. Neuromuscul Disord. (2004) ;14: :158–61. |
[23] | Salama I , Hinderlich S , Shlomai Z , Eisenberg I , Krause S , Yarema K , et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. (2005) ;328: :221–6. |
[24] | Broccolini A , Gidaro T , Tasca G , Morosetti R , Rodolico C , Ricci E , Mirabella M . Analysis of NCAM helps identify unusual phenotypes of hereditary inclusion-body myopathy. Neurology. (2010) ;75: :265–72. |
[25] | Sela I , Goss V , Becker-Cohen M , Dell A , Haslam MS , Mitrani-Rosenbaum S . The glycomic sialylation profile of GNE Myopathy muscle cells does not point to consistent hyposialylation of individual glycoconjugates. Neuromuscul Disord. (2020) ;30: :621–30. |
[26] | Lochmüller H , Behin A , Caraco Y , Lau H , Mirabella M , Tournev I , et al. A phase 3 randomized study evaluating sialic acid extended-release for GNE myopathy. Neurology. (2019) ;92: :e2109–17. |
[27] | Suzuki N , Mori-Yoshimura M , Katsuno M , Takahashi MP , Yamashita S , Oya Y , et al. Phase II/III Study of Aceneuramic Acid Administration for GNE Myopathy in Japan. J Neuromuscul Dis. (2023) ;10: :555–66. |
[28] | Park YE , Park E , Choi J , Go H , Park DB , Kim MY , et al. Pharmacokinetics and clinical efficacy of 6’-sialyllactose in patients with GNE myopathy: Randomized pilot trial. Biomed Pharmacother. (2023) ;168: :115689. |
[29] | Malicdan MC , Noguchi S , Nonaka I , Hayashi YK , Nishino I . A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. (2007) ;16: :2669–82. |
[30] | Malicdan MC , Noguchi S , Hayashi YK , Nonaka I , Nishino I . Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. (2009) ;15: :690–5. |
[31] | Crowe KE , Zygmunt DA , Heller K , Rodino-Klapac L , Noguchi S , Nishino I , Martin PT . Visualizing Muscle Sialic Acid Expression in the GNED207VTgGne-/- Cmah-/- Model of GNE Myopathy: A Comparison of Dietary and Gene Therapy Approaches, J Neuromuscul Dis. (2022) ;9: :53–71. |
[32] | Mitrani-Rosenbaum S , Yakovlev L , Becker Cohen M , Argov Z , Fellig Y , Harazi A . Pre Clinical Assessment of AAVrh74.MCK.GNE Viral Vector Therapeutic Potential: Robust Activity Despite Lack of Consistent Animal Model for GNE Myopathy. J Neuromuscul Dis. (2022) ;9: :179–92. |
[33] | Chandrasekharan K , Yoon JH , Xu Y , deVries S , Camboni M , Janssen PM , et al. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci Transl Med. (2010) ;2: :42ra54. |
[34] | Ilouz N , Harazi A , Guttman M , Daya A , Ruppo S , Yakovlev L , Mitrani-Rosenbaum S . In vivo and in vitro genome editing to explore GNE functions. Front Genome Ed. (2022) ;4: :930110. 10.3389/fgeed.2022.930110. |
[35] | Schlesinger Y , Yosefov-Levi O , Kolodkin-Gal D , Granit RZ , Peters L , Kalifa R , et al. Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat Commun. (2020) ;11: :4516. |
[36] | Hinderer C , Katz N , Buza EL , Dyer C , Goode T , Bell P , et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Human Gene Therapy. (2018) ;29, 10.1089/hum.2018.015j 285-298. |
[37] | Hu X , Charles JP , Akay T , Hutchinson JR , Blemker SS . Are mice good models for human neuromuscular disease? Comparing muscle excursions in walking between mice and humans. Skeletal Muscle. (2017) ;7: :26. |
[38] | Lostal W , Roudaut C , Faivre M , Lafoux A , Huet CD , Richard I . development of a new CAPN3 deficient rodent animal model. 6th International Congress of Myology, (2019) .P03-030-#153. |
[39] | Iyer SR , Xu S , Shah SB , Lovering RM . Muscle phenotype of a rat model of Duchenne muscular dystrophy. Muscle Nerve. (2020) ;62: :757–61. |
[40] | Larcher T , Lafoux A , Tesson L , Remy S , Thepenier V , François V , et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One. (2014) ;9: (10):e110371. |
[41] | Nakamura K , Fujii W , Tsuboi M , Tanihata J , Teramoto N , Takeuchi S , Larcher T , Lafoux A , Tesson L , et al. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep. (2014) ;4: :5635. |
[42] | Muñoz S , Bertolin J , Jimenez V , Jaén ML , Garcia M , Pujol A , et al. Treatment of infantile-onset Pompe disease in a rat model with muscle-directed AAV gene therapy. Molecular Metabolism. (2024) ;81: :101899. |
[43] | Schmitt RE , Smith DY 4th , Cho DS , Kirkeby LA , Resch ZT , Liewluck T , et al. Myogenesis defects in a patient-derived iPSC model of hereditary GNE myopathy. NPJ Regen Med. (2022) ;7: :48. |
[44] | Neu CT , Weilepp L , Bork K , Gesper A , Horstkorte R . GNE deficiency impairs Myogenesis in C2C12 cells and cannot be rescued by ManNAc supplementation. Glycobiology. (2024) ;34: :cwae004. |
[45] | Nakamura K , Tsukamoto Y , Hijiya N , Higuchi Y , Yano S , Yokoyama S , et al. Induction of GNE in myofibers after muscle injury. Pathobiology. (2010) ;77: :191–99. |
[46] | Backwell L , Marsh JA . Diverse molecular mechanisms underlying pathogenic protein mutations: Beyond the Loss-of-Function Paradigm. Annu Rev Genomics Hum Genet. (2022) ;23: :475–98. |
[47] | Cavaco BM , Canaff L , Nolin-Lapalme A , Vieira M , Silva TN , Saramago A , et al. Homozygous calcium-sensing receptor polymorphism R544Q presents as hypocalcemic hypoparathyroidism. J Clin Endocrinol Metab. (2018) ;103: :2879–88. |
[48] | Drutman SB , Haerynck F , Zhong FL , Hum D , Hernandez NJ , Belkaya S , et al. Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci USA. (2019) ;116: :19055–63. |


