
Risdiplam Real World Data – Looking Beyond Motor Neurons and Motor Function Measures
Abstract
Background:
Risdiplam is an orally administered treatment for spinal muscular atrophy which leads to an improvement in motor function as measured by functional motor scales compared with placebo. Although risdiplam has been registered since 2020, real-world data in adults is still scarce. There have been no new safety signals so far, with some results pointing that risdiplam may be effective
Objective:
The objective was to present real-world data of 31 adult patients with spinal muscular atrophy type 2 and type 3 treated with risdiplam in the Republic of Croatia
Methods:
Treatment effects were assessed with motor function tests and patient reported outcome measures, including Individualized Neuromuscular Quality of Life questionnaire, and Jaw Functional Limitation Scale. Side effects, as well as subjective improvements and symptoms, were noted.
Results:
Majority of patients did not report any side effects. During treatment, we have observed clinically meaningful improvements in some patients, with stabilization of motor functions in the remaining patients. The majority of patients with bulbar function impairment experienced bulbar function improvement, all patients reported an increased quality of life with treatment. An unexpected observed treatment effect was weight gain in a third of all patients with some patients reporting an increase in appetite and subjective improvement in digestion.
Conclusions:
Risdiplam treatment was well tolerated with subjective and objective positive outcomes registered as measured by functional motor scales and patient-reported outcomes. Since risdiplam is administered orally and acts as a systemic therapy for a multisystemic disorder, effects in systems other than neuromuscular can be expected and should be monitored. Due to systemic nature of the disease patients need multidisciplinary monitoring.
INTRODUCTION
Spinal muscular atrophy (SMA) is a progressive, autosomal recessive motor neuron disease caused by mutation of survival motor neuron 1 (SMN1) gene. The SMN protein is ubiquitously expressed but its dominant function is in spinal motor neurons. Deletions or loss of function mutations of SMN1gene lead to insufficient SMN protein expression resulting in severe and progressive muscular atrophy and weakness [1, 2]. SMA severity is mainly determined by the number of copies of the SMN2 gene, a paralogous gene that also encodes for a truncated SMN protein that is dysfunctional due to the exclusion of exon 7 from mature RNA. Because SMN2 can produce some functional protein, number of SMN2 copies determines SMA severity, with more copies leading to a less severe disease [3, 4].
Based on the age of symptom onset, achieved motor development milestones and life expectancy, SMA is classified into 5 subtypes (0–4, 0 being the most severe and 4 the mildest). SMA type 2 most often presents between 6 and 18 months of age after the patient has achieved motor milestone of independent sitting (which can be lost later with the disease progression), independent standing and walking is never achieved [5]. Tongue atrophy with fasciculations is also characteristic as is impaired swallowing and ventilatory insufficiency, particularly in patients at the severe end of the type 2 spectrum. The survival probability in the pre-treatment era at 20 years of age was 77% [6, 7].
SMA type 3 (SMA3) constitutes milder end of the spectrum with age of onset after 18 months (3a) or 3 years (3b) and patients achieving the motor milestone of walking for at least some time in life [8]. Natural history studies have shown slowly progressive decline of motor function in all SMA subtypes [9–12]. Until recently SMA treatment was symptomatic, however in the year 2016 first specific therapy for SMA, nusinersen, was approved (in 2017 in Europe). Following registration of risdiplam with the U.S. Food and Drug Administration in 2020 (and the European Medicines Agency in 2021), adult patients with SMA have two treatment options. Risdiplam is an orally administered small molecule SMN2 splicing modifier [13] with safety and efficacy demonstrated in individuals with type 2 and 3 SMA [14]. Third registered treatment onasemnogen abeparvovec is currently approved for individuals less than 24 months of age. Both treatments approved for adults with SMA target SMN2 gene
The natural history of type 2 and 3 SMA involves progression of the disease and continued loss of function [15].
Mercuri et al. have shown in a pivotal trial that risdiplam treatment led to a significant improvement in motor function (measured by functional motor scales) compared with placebo in patients aged 2–25 years with type 2 or non-ambulant type 3 spinal muscular atrophy. Younger patients had an improvement of motor function while older patients experienced stabilization [14].
Although a 3-point change in the functional motor scales has been highlighted as a clinically meaningful change, stabilization of motor function is an important goal identified by patients with type 2 and 3 SMA and is considered a clinically meaningful outcome in this population [5, 16–18].
Understanding the many impacts of disease is essential to providing optimal patient care. Patient Reported Outcome Measures (PROM) are valuable tools clinicians and researchers can use to capture patient changes and quantify patient experiences which may otherwise be missed [19].
The Individualized Neuromuscular Quality of Life questionnaire (INQoL) is a validated muscle disease specific measure of quality of life developed from the experiences of patients with muscle disease to specifically detect functional limitations relevant to neuromuscular patients [20]. INQoL has already been used in patients with SMA [21]. It consists of 45 self-administered questions within 10 sections related to the physical health domain, and the areas of life and psycho-social aspects. The physical health domain pertains to the impact of common neuromuscular symptoms (i.e., weakness, locking, pain and fatigue) on QoL, while activities, dependence, body image, relationships and emotions evaluate the impact of the disease on psychological and social functioning [20, 21].
SMA type 2 is frequently associated with bulbar impairment, which is not captured in the currently used outcome measures. Malocclusion disorders are frequent in patients with SMA type 2 which together with the different degree of TMJ contractures creates an additional burden on an already impaired bulbar function. The Jaw Functional Limitation Scale (JFLS) is a questionnaire focusing on mastication, mobility, and communication. This scale measures the global functional limitation of the jaw and has been used in groups of patients with a range of functional limitations of the jaw [22].
MATERIALS AND METHODS
All adult patients with SMA in Croatia are treated at Referral Centre for neuromuscular disorders and clinical electromyoneurography, Clinical hospital Centre Zagreb. Adult patients with a clinical diagnosis of 5q-associated spinal muscular atrophy type 1, 2, or 3 SMA and one to four copies of the SMN2 gene could opt for risdiplam treatment since risdiplam became available in Croatia in January 2022. Prior to risdiplam registration, treatment-naïve adult patients with SMA type 2 were able to participate in the compassionate use program since November 2020.
The compassionate use program was conducted in full conformance with the International Council for Harmonization Guideline for Good Clinical Practice; principles of the Declaration of Helsinki; and applicable state law. Written informed consent was provided by the patient or the patient’s legally authorized representative before participation in the program. The protocol and the informed consent form were submitted and approved by the Ethics Committee and the Ministry of Health of the Republic of Croatia.
The study conforms with World Medical Association Declaration of Helsinki published on the website of the Journal of American Medical Association. Ethical approval was waived by the local Ethics Committee of Clinical Hospital Centre Zagreb in view of the retrospective nature of the study and all the procedures being performed being part of the routine care. Informed consent was obtained from all individual participants included in the study.
Validated spinal muscular atrophy (SMA) outcome measures - Revised Hammersmith score (RHS), Revised Upper Limb Module (RULM), 6-minute walk test (6MWT) were prospectively collected during risdiplam treatment. We used RHS in place of more frequently used Hammersmith functional motor scale-expanded (HFMSE) because it has been shown to be more sensitive to changes in the functionally stronger SMA3 patients [23]. Since individual patients in our group had dissimilar baselines, outcome measures were tailored to the specific patient’s functional abilities.
All patients had motor function evaluation every 6 months during treatment. Functionally stronger patients were assessed with RHS, RULM and in some cases (4 out of 31 patients) with 6MWT. Functionally weaker patients were assessed with RULM. The majority of patients (30/31) reported on quality of life before and after starting treatment. For assessing quality of life change we used Individualized Neuromuscular Quality of Life questionnaire (INQoL). Half of our cohort were patients with SMA type 2 which is frequently associated with bulbar impairment; in these patients we used another PROM - Jaw Functional Limitation Scale (JFLS) to assess changes in bulbar function [22, 24].
Routine laboratory tests and adverse events were monitored; patient reported experiences noted as well as their demographics and clinical characteristics
RESULTS
Baseline demographics and characteristics
Our cohort consists of 31 treatment-naïve adult patients with a genetically confirmed diagnosis of 5q-autosomal recessive SMA and clinical symptoms attributable to type 2 or type 3 SMA. 16 patients have SMA type 3 (SMA3), 15 patients have SMA type 2 (SMA2), 16 patients are female, 15 are male. All patients have been treated with risdiplam 5 mg daily for at least a year. Over a third of patients (12/31, 38.7%) have been on treatment for 2 and a half years (29–31 months) through compassionate use program (since December 2020) (Fig. 1)
Fig. 1
Risdiplam treatment timeline.

Patient demographics and baseline characteristics are presented in Table 1. Our group consists of patients with dissimilar baselines with some patients on the weakest end of the SMA spectrum and some on the functionally strong end, which is why the choice of outcome measures was individualized considering the functional abilities of the patient. All patients were assessed with RULM, baseline values ranged from 0 to 37 (maximal and the best score is 37), average score 14, median 9. Five patients had an initial RULM scale score of 0. Out of five SMA type 3 patients that are considered ambulant (defined as able to perform 5 steps unaided) four were able to perform 6-minute walk test. Baseline 6MWT ranged from 92 to 394 m. Six patients with SMA 3 were assessed with RHS with baseline values ranging from 21 to 47 (maximal and the best score is 69) (Fig. 2).
Table 1
Patient demographics and baseline characteristics
| Characteristic | |
| Age at symptom onset, median (range) | 2 years (0.5–18 years) |
| Sex, female/male, n (%) | 16/15 (51.6% /48.4%) |
| SMA type, n (%) | |
| Type 2 | 15 (48.4%) |
| Type 3 | 16 (51.6%) |
| SMN2 copy number, n (%) | |
| 2 copies | 5 (16.12%) |
| 3 copies | 18 (58.06%) |
| 4 copies | 8 (25.8%) |
| Disease duration prior to treatment, median (range) | 29 years (11–62 years) |
| Scoliosis surgery, n (%) | 12 (38.7%) |
| Age at treatment start, median (range) | 30 years (18–65 years) |
| Functional status, n (%) | |
| non-sitter1 | 17 (54.8%) |
| sitter2 | 8 (25.8%) |
| walker3 | 6 (19.4%) |
| Baseline RULM score (best motor function), mean (range) | 9 (0–37) |
| Baseline weight, kilograms | |
| SMA type 2 | 20–30 kg 6pt |
| 30–40 kg 5pt | |
| 40–60 kg 1pt | |
| >60 kg 3pt | |
| SMA type 3 | <65 kg 10pt |
| >65 kg 6 pt |
1non sitter: unable to sit without support. 2sitter: able to sit unsupported. 3walker: able to walk 5 steps unaided.
Motor function outcomes
As it has been decided by consensus and used in previous clinical studies, a change of three points in HFMSE and RULM is considered to be clinically meaningful [2, 25] which is why we have used the change of three points in RHS and RULM as a cut-off for a “clinically significant change” [26]. In 6MWT we considered a change of 30 m to be clinically meaningful [11, 12, 27]. Changes in motor functions measures are presented in Table 2 and Fig. 2
Fig. 2
RHS and 6 minute walk test (baseline and after treatment).
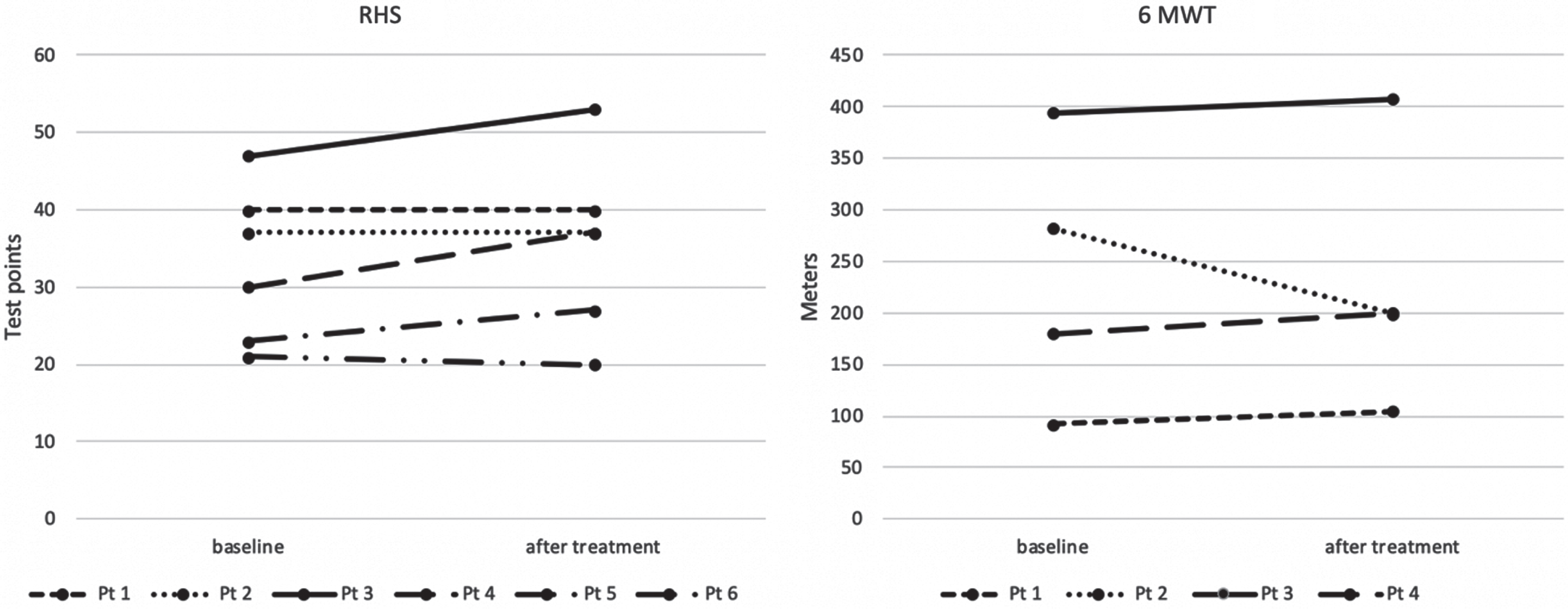
Table 2
Changes in motor function measures and 6 minute walk test
| SMA 2, treatment duration median 30 months, n = 12 | SMA 2, treatment duration median 16 months, n = 3 | SMA 3, treatment duration median 16 months, n = 16 | |
| RULM, difference from baseline, average (SD) | +0.33 (SD 1.3) | 0 (SD 1) | +1.625 (SD 3.61) |
| RHS, difference from baseline, average (SD) | +2.67 (SD 3.45) | ||
| 6MWT, difference from baseline, average (SD) | –9.25m (SD 46.3) |
Meaningful improvement on at least one motor scale was achieved by seven patients (7/31, 22.6%). In this group, five patients had an increase in RULM, additional two patients an increase in RHS. All of these patients have SMA 3, three of them are ambulant, four sitters.
Looking at functionally stronger SMA patients, out of six patients that were tested with RHS, three had a meaningful improvement. Four of the functionally weaker patients had a meaningful improvement in upper limb function.
Two patients had a meaningful worsening in one of the motor function outcome measures but remained stable in other two tested outcomes. Three additional patients have lost more than 1 point on RULM, of which one patient lost a point on RHS as well. It is likely that if these patients’ motor function doesn’t stabilize or improve in the future they will be considered non-responders. (5/31, 16.1% of patients in our cohort, one SMA2 and four SMA3).
Patient-reported outcomes: Bulbar function and quality of life
All SMA type 2 patients were assessed for bulbar function changes using JFLS. All patients reported mouth opening limitations indicative of temporomandibular joint (TMJ) contracture of a different degree, 13 patients reported swallowing difficulties. None of the patients reported bulbar function worsening over the course of treatment. Nine patients (60% SMA 2 patients) reported improvement in at least one tested item. Improvements were most frequently registered in swallowing, talking and chewing. Further improvements were registered in facial expressions, easier singing followed by better yawning and easier laughter (Fig. 3).
Fig. 3
Improvement in Jaw Functional Limitation Scale after risdiplam treatment.

Two patients with SMA type 3 reported swallowing difficulties during QOL evaluation, of which one reported improvement with treatment,
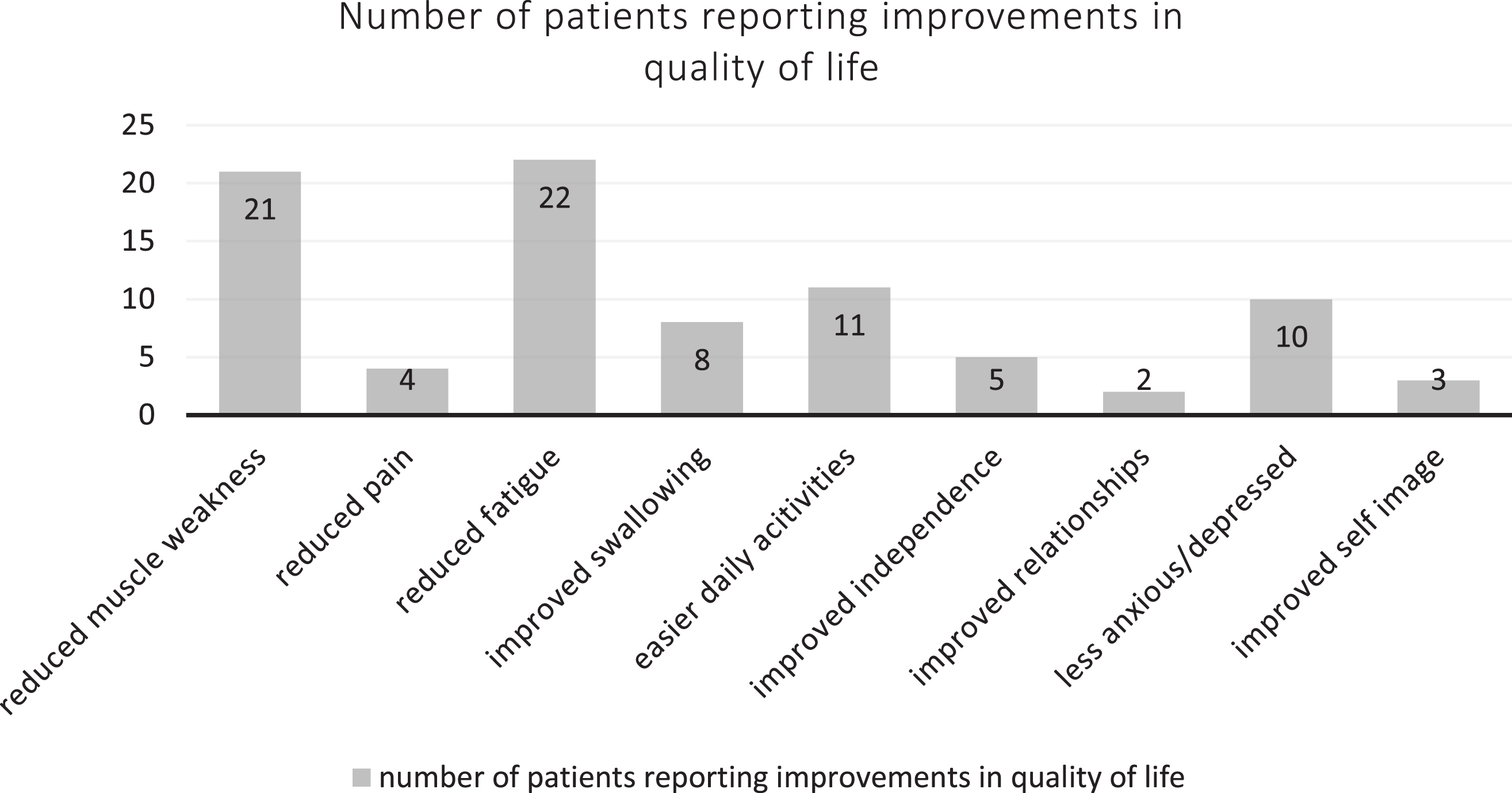
Thirty patients were assessed using the INQoL questionnaire (Fig. 4). None of the patients reported worsening in the quality of life following treatment All of the patients reported having some degree of muscle weakness on the initial testing with 21 patients noticing improvements after treatment. Of the 26 patients who initially reported fatigue as a symptom, 22 patients (85%) reported a reduction in the fatigue after treatment. Four out of seven patients reported reduction of pain as a result of treatment. Improvements with treatment were also observed in the ability to perform daily activities, increased independence, improved relationships, and self-image. When asked about the effects of the treatment, all the patients reported noticing beneficial treatment effects, only three patients thought that the treatment had some harmful side effects as well. All patients reported that the importance of the beneficial effect of the treatment outweighed the importance of the possible harmful side effects.
Fig. 4
Patient reported quality of life improvements after treatment.
Side effects/adverse events and systemic treatment effects
Most frequently reported side effects were headaches and mouth aphthous ulcers, both were transient and mild. One patient reported insomnia (Table 3).
Table 3
Treatment side effect/adverse events
| Aphthous ulcers | Nausea | Headache | Ramsey hunt | Hyperuri-caemia | Macrohe-maturia | Anaemia | Feeling bloated | Insomnia | Death | No side effects | |
| Number of patients | 2 | 1 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 23 |
One patient had temporary treatment discontinuation due to anaemia and another because of macrohematuria. Treatment was restarted in both cases after further tests showed menorrhagia and recurrent urinary tract infections associated with permanent urinary catheter as the cause.
During risdiplam treatment one patient had Ramsay Hunt syndrome, another patient had hyperuricemia, treatment was not paused or discontinued in these cases.
A 42-year-old male patient with SMA type 2 died in another hospital while being treated for a respiratory infection. Medical records were not available, and we were informed of the outcome by the family members. His previous medical history included anaemia for which he periodically took iron supplements. No autopsy was performed. This patient was on risdiplam for 6 months, his data is not included in the group analysis due to short treatment period.
Patients reported additional subjective improvements that were not captured in the used outcome measures (Fig. 5). Most frequent improvements were increased endurance and increased head and trunk stability and control. Two-thirds (10/15) of SMA type 2 patients reported subjective respiratory improvement, most frequently easier breathing (5 patients), better cough (3 patients), stronger voice (3 patients), and reduced frequency of respiratory infections.
Fig. 5
Other subjective improvements after treatment.
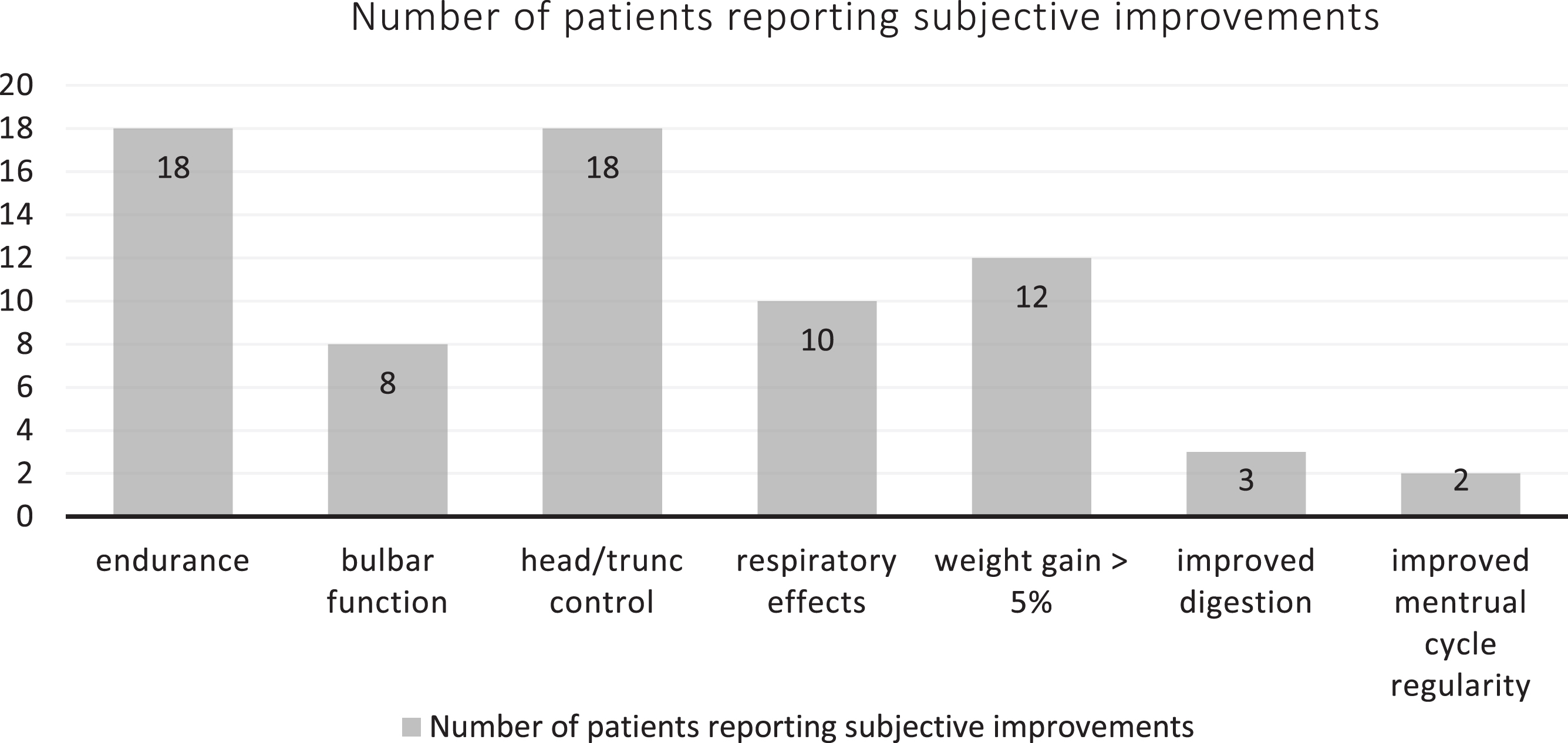
Almost a third of all patients (9/31) had a significant weight gain (more than 5% of body weight) with 6 patients reporting a subjective feeling of an increased appetite. We found no correlation between initial body weight and risdiplam efficacy (Pearson correlation coefficient, PCC –0.02), or SMA3 weight change and RULM change (PCC –0.062). There was moderate negative correlation between SMA2 weight change and RULM change (PCC –0.545). We did not register weight gain in walkers. 40% of SMA2 patients gained weight (6/15) and 18.8% of SMA3 patients (3/16, all sitters).
Another unexpected treatment effect was subjective digestion improvement in three patients and restoration of a regular menstrual cycle in two patients
DISCUSSION
In this study we present real world data for adult patients with SMA treated with risdiplam for 2.5 years. Real world data in adults is still scarce, so far there has been no new safety signals and some results point that risdiplam may be effective [28–31]. Nungo Garzon et al. reported on 6 adult non-sitter patients, two of which had an increase of >5% in BMI and another two with an increase in RULM after one year of treatment. McCluskey et al. reported on 6 adult patients treated with risdiplam over 9 months with subjective improvements and no change in RULM.
In our experience, risdiplam was well tolerated with majority of patients (74%) not reporting any side effects. Treatment effects were assessed using motor function outcome measures (RHS, RULM, 6MWT) and patient reported outcome measures (INQoL and JFLS) both of which registered improvements with treatment. During treatment we have observed clinically meaningful improvements in some patients (22.6%) with stabilization of motor functions in the remaining patients (61%).
Taking into account the natural history of SMA, we regard stabilization in the motor function outcome measures as a positive treatment effect, especially since in this age group, natural history studies predict only continued loss of function over the years [9, 10].
Adult study participants in the pivotal risdiplam trial were a minority (4/51 in SUNFISH part 1 aged 18–25, 10/180 non-ambulant patients in SUNFISH part 2) [14, 32]. In SUNFISH part 1, younger patients showed greater improvements in all exploratory efficacy endpoints, while patients aged 12–25 had a 1.7 point increase in RULM and 0.7 decrease in HMFSE after 24 months of treatment. Direct comparison with our adult cohort is not possible because SUNFISH didn’t report on adults but on a group of 12–25 year old patients. However, looking at the percentages of responders, even with older patients and more strict meaningful change criteria (≥3 points in RULM/RHS), our group still had 22.6% of responders. SUNFISH had 26.3% of patients with an increase of ≥2 points on HMFSE and 57.9 % with an increase of ≥2 points in RULM. Considering long disease duration in our adult patient group, it is possible that an even longer treatment period is needed to fully assess treatment effects, or lack thereof.
All of the patients in our group reported an increased quality of life, 59% of patients with bulbar symptoms reported improvements in bulbar function. An unexpected observed treatment effect was weight gain in a third of all patients with some patients reporting an increase in appetite and subjective improvement in digestion.
The etiology of weight gain in our group of patients might have more to it than simply being a result of improved bulbar function. It has been shown that the lack of SMN protein is associated with changes in fatty acids metabolism, impaired glucose tolerance and muscle mitochondria function abnormalities [33–35]. Increased leptin levels have been observed in underweight and functionally weaker SMA patients [36]. An increase in SMN protein in treated patients with SMA could lead to changes in metabolism, including a reduction in leptin, leading to an increased appetite and the resulting increase in body weight. This weight gain can in some cases be detrimental to motor function, as it can overburden functionally weaker muscles. Indeed, in our group there was a moderate negative correlation between weight gain in SMA2 patients and treatment efficacy. SMA treatment could have other systemic effects as well, with restoration of a regular menstrual cycle in two patients being another possible effect.
Limitation of this study is patient group heterogeneity with very different levels of baseline motor functions which makes intragroup comparisons impossible.
In conclusion, in our experience risdiplam treatment was well tolerated with registered subjective and objective improvements measured by functional motor scales and patient reported outcomes. Since risdiplam is administered orally and acts as a systemic therapy for a multisystemic disorder, effects in systems other than neuromuscular can be expected and should be monitored. More research is needed on metabolic markers that could potentially be used as SMA biomarkers. Due to systemic nature of the disease patients need multidisciplinary monitoring.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Authors Barbara Sitas, Mirea Hancevic, Hrvoje Bilic, and Ervina Bilic have received speaking fees from Biogen and Roche. The remaining authors have no conflicts of interest.
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
AUTHOR CONTRIBUTIONS
Barbara Sitas: conceptualization; investigation; methodology; resources; writing original draft; writing-review and editing.
Mirea Hancevic: conceptualization; investigation; methodology; resources; writing original draft; writing-review and editing.
Katarina Bilic: conceptualization; investigation; methodology; writing-review and editing.
Hrvoje Bilic: conceptualization; investigation; methodology; resources; writing-review and editing.
Ervina Bilic: conceptualization; supervision; writing-review and editing.
REFERENCES
[1] | Nance JR . Spinal muscular atrophy. CONTINUUM: Lifelong Learning in Neurology. (2020) ;26: :1348–68. https://doi.org/10.1212/CON.0000000000000918. |
[2] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus sham control in later-onset spinal muscular atrophy, N Engl J Med (2018) ;378: :625–35. https://doi.org/10.1056/NEJMoa1710504 |
[3] | Cartwright MS , Upadhya S Selecting disease-modifying medications in 5q spinal muscular atrophy, Muscle and Nerve (2021) ;64: :404–12. https://doi.org/10.1002/mus.27358 |
[4] | Feldkötter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy, The American Journal of Human Genetics (2002) ;70: :358–68. https://doi.org/10.1086/338627 |
[5] | Mercuri E , Sansone V . Nusinersen in adults with spinal muscular atrophy: New challenges, The Lancet Neurology (2020) ;19: :283–4. https://doi.org/10.1016/S1474-4422(20)30068-5 |
[6] | Arnold WD , Kassar D , Kissel JT . Spinal muscular atrophy: Diagnosis and management in a new therapeutic era: Spinal Muscular Atrophy, Muscle Nerve (2015) ;51: :157–67. https://doi.org/10.1002/mus.24497. |
[7] | Zerres K Natural history in proximal spinal muscular atrophy: Clinical analysis of 445 patients and suggestions for a modification of existing classifications, Arch Neurol. (1995) ;52: :518. https://doi.org/10.1001/archneur.1995.00540290108025 |
[8] | Zerres K , Rudnik-Schöneborn S , Forrest E , Lusakowska A , Borkowska J , Hausmanowa-Petrusewicz I A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients, Journal of the Neurological Sciences (1997) ;146: :67–72. https://doi.org/10.1016/S0022-510X(96)00284-5 |
[9] | Wadman RI , Wijngaarde CA , Stam M , Bartels B , Otto LAM , Lemmink HH , et al. Muscle strength and motor function throughout life in a cross-sectional cohort of 180 patients with spinal muscular atrophy types 1c–4, Euro J of Neurology (2018) ;25: :512–8. https://doi.org/10.1111/ene.13534 |
[10] | Montes J , McDermott MP , Mirek E , Mazzone ES , Main M , Glanzman AM , et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression, PLoS ONE (2018) ;13: :e0199657. https://doi.org/10.1371/journal.pone.0199657 |
[11] | Dunaway Young S , Montes J , Kramer SS , Marra J , Salazar R , Cruz R , et al. Six-minute walk test is reliable and valid in spinal muscular atrophy: 6MWT in SMA, Muscle Nerve (2016) ;54: :836–42. https://doi.org/10.1002/mus.25120. |
[12] | Mcdonald CM , Henricson EK , Abresch RT , Florence JM , Eagle M , Gappmaier E , et al. THE 6-minute walk test and other endpoints in Duchenne muscular dystrophy: Longitudinal natural history observations over 48 weeks from a multicenter study, Muscle Nerve (2013) ;48: :343–56. https://doi.org/10.1002/mus.23902 |
[13] | Ratni H , Ebeling M , Baird J , Bendels S , Bylund J , Chen KS , et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA), J Med Chem (2018) ;61: :6501–17. https://doi.org/10.1021/acs.jmedchem.8b00741 |
[14] | Mercuri E , Deconinck N , Mazzone ES , Nascimento A , Oskoui M , Saito K , et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial, The Lancet Neurology (2022) ;21: :42–52. https://doi.org/10.1016/S1474-4422(21)00367-7 |
[15] | Kaufmann P , McDermott MP , Darras BT , Finkel RS , Sproule DM , Kang PB , et al. Prospective cohort study of spinal muscular atrophy types 2 and 3, Neurology (2012) ;79: :1889–97. https://doi.org/10.1212/WNL.0b013e318271f7e4 |
[16] | Oskoui M , Day JW , Deconinck N , Mazzone ES , Nascimento A , Saito K , et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA), J Neurol (2023) ;270: :2531–46. https://doi.org/10.1007/s00415-023-11560-1 |
[17] | Gusset N , Stalens C , Stumpe E , Klouvi L , Mejat A , Ouillade M-C , et al. Understanding European patient expectations towards current therapeutic development in spinal muscular atrophy, Neuromuscular Disorders (2021) ;31: :419–30. https://doi.org/10.1016/j.nmd.2021.01.012 |
[18] | Osmanovic A , Ranxha G , Kumpe M , Müschen L , Binz C , Wiehler F , et al. Treatment expectations and patient-reported outcomes of nusinersen therapy in adult spinal muscular atrophy, J Neurol (2020) ;267: :2398–407. https://doi.org/10.1007/s00415-020-09847-8 |
[19] | Nelson EC , Eftimovska E , Lind C , Hager A , Wasson JH , Lindblad S Patient reported outcome measures in practice. BMJ. 2015:g7818. https://doi.org/10.1136/bmj.g7818. |
[20] | Vincent KA , Carr AJ , Walburn J , Scott DL , Rose MR . Construction and validation of a quality of life questionnaire for neuromuscular disease (INQoL), Neurology (2007) ;68: :1051–7. https://doi.org/10.1212/01.wnl.0000257819.47628.41 |
[21] | Bonanno S , Zanin R , Bello L , Tramacere I , Bozzoni V , Caumo L , et al. Quality of life assessment in adult spinal muscular atrophy patients treated with nusinersen, J Neurol (2022) ;269: :3264–75. https://doi.org/10.1007/s00415-021-10954-3 |
[22] | Ohrbach R , Larsson P , List T . The jaw functional limitation scale: Development, reliability, and validity of 8-item and 20-item versions, J Orofac Pain (2008) ;22: , 219–30. |
[23] | Ramsey D , Scoto M , Mayhew A , Main M , Mazzone ES , Montes J , et al. Revised Hammersmith Scale for spinal muscular atrophy: A SMA specific clinical outcome assessment tool, PLoS ONE (2017) ;12: :e0172346. https://doi.org/10.1371/journal.pone.0172346 |
[24] | Riera-Punet N , Martinez-Gomis J , Willaert E , Povedano M , Peraire M . Functional limitation of the masticatory system in patients with bulbar involvement in amyotrophic lateral sclerosis, J Oral Rehabil (2018) ;45: :204–10. https://doi.org/10.1111/joor.12597 |
[25] | Swoboda KJ , Scott CB , Crawford TO , Simard LR , Reyna SP , Krosschell KJ , et al. SMA CARNI-VAL trial part I: Double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy, PLoS ONE (2010) ;5: :e12140. https://doi.org/10.1371/journal.pone.0012140 |
[26] | Mazzone ES , Mayhew A , Montes J , Ramsey D , Fanelli L , Young SD , et al. Revised upper limb module for spinal muscular atrophy: Development of a new module, Muscle and Nerve (2017) ;55: :869–74. https://doi.org/10.1002/mus.25430 |
[27] | Montes J , Dunaway Young S , Mazzone ES , Pasternak A , Glanzman AM , Finkel RS , et al. Nusinersen improves walking distance and reduces fatigue in later-onset spinal muscular atrophy, Muscle Nerve (2019) ;60: :409–14. https://doi.org/10.1002/mus.26633 |
[28] | Kwon JM , Arya K , Kuntz N , Phan HC , Sieburg C , Swoboda KJ , et al. An expanded access program of risdiplam for patients with Type 1 or 2 spinal muscular atrophy, Ann Clin Transl Neurol (2022) ;9: :810–8. https://doi.org/10.1002/acn3.51560 |
[29] | Hahn A , Günther R , Ludolph A , Schwartz O , Trollmann R , Weydt P , et al. Short-term safety results from compassionate use of risdiplam in patients with spinal muscular atrophy in Germany, Orphanet J Rare Dis. (2022) ;17: :276. https://doi.org/10.1186/s13023-022-02420-8 |
[30] | Ñungo Garzón NC , Pitarch Castellano I , Sevilla T , Vázquez-Costa JF . Risdiplam in non-sitter patients aged 16 years and older with 5q spinal muscular atrophy, Muscle and Nerve (2023) ;67: :407–11. https://doi.org/10.1002/mus.27804 |
[31] | McCluskey G , Lamb S , Mason S , NicFhirleinn G , Douglas I , Tirupathi S , Morrison KE , McConville J . Risdiplam for the treatment of adults with spinal muscular atrophy: Experience of the Northern Ireland neuromuscular service, Muscle Nerve (2023) ;67: (2):157–61. https://doi.org/10.1002/mus.27755. Epub 2022 Dec 5. PMID: 36382958 |
[32] | Oskoui M , Day JW , Deconinck N , Mazzone ES , Nascimento A , Saito K , Vuillerot C , Baranello G , Goemans N , Kirschner J , Kostera-Pruszczyk A , Servais L , Papp G , Gorni K , Kletzl H , Martin C , McIver T , Scalco RS , Staunton H , Yeung WY , Fontoura P , Mercuri E . SUNFISH Working GrouTwo-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA), J Neurol (2023) ;270: (5):2531–46. https://doi.org/10.1007/s00415-023-11560-1. Epub 2023 Feb 3. Erratum in: J Neurol. 2023 Apr 18;: PMID: 36735057; PMCID: PMC9897618. |
[33] | Crawford TO , Sladky JT , Hurko O , Besner-Johnston A , Kelley RI . Abnormal fatty acid metabolism in childhood spinal muscular atrophy, Ann Neurol (1999) ;45: :337–43. https://doi.org/10.1002/1531-8249(199903)45:3<337::AID-ANA9>3.0.CO;2-U |
[34] | Bowerman M , Swoboda KJ , Michalski J-P , Wang G-S , Reeks C , Beauvais A , et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy, Ann Neurol (2012) ;72: :256–68. https://doi.org/10.1002/ana.23582 |
[35] | Ripolone M , Ronchi D , Violano R , Vallejo D , Fagiolari G , Barca E , et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy, JAMA Neurol. (2015) ;72: :666. https://doi.org/10.1001/jamaneurol.2015.0178 |
[36] | Kölbel H , Hauffa BP , Wudy SA , Bouikidis A , Della Marina A , Schara U . Hyperleptinemia in children with autosomal recessive spinal muscular atrophy type I-III, PLoS ONE (2017) ;12: :e0173144. https://doi.org/10.1371/journal.pone.0173144 |


