
Gene Distribution in Pediatric-Onset Inherited Peripheral Neuropathy: A Single Tertiary Center in Thailand
Abstract
Background:
Inherited peripheral neuropathy presents a diagnostic and therapeutic challenge due to its association with mutations in over 100 genes. This condition leads to long-term disability and poses a substantial healthcare burden on society.
Objective:
This study aimed to investigate the distribution of genes and establish the genotype-phenotype correlations, focusing on pediatric-onset cases.
Methods:
Exome sequencing and other analytical techniques were employed to identify pathogenic variants, including duplication analysis of the PMP22 gene. Each patient underwent physical examination and electrophysiological studies. Genotypes were correlated with phenotypic features, such as age at disease onset and ulnar motor nerve conduction velocity.
Results:
We identified 35 patients with pediatric-onset inherited peripheral neuropathy. Pathogenic or likely pathogenic variants were confirmed in 24 out of 35 (68.6%) patients, with 4 of these variants being novel. A confirmed molecular diagnosis was achieved in 90.9% (10/11) of patients with demyelinating Charcot-Marie-Tooth disease (CMT) and 56.3% (9/16) of patients with axonal CMT. Among patients with infantile-onset CMT (≤2 years), the most common causative genes were MFN2 and NEFL, while GDAP1 and MFN2 were frequent causes among patients with childhood- or adolescent-onset CMT (3–9 years).
Conclusions:
The MFN2 gene was the most commonly implicated gene, and the axonal type was predominant in this cohort of Thai patients with pediatric-onset inherited peripheral neuropathy.
INTRODUCTION
Inherited peripheral neuropathies (IPNs) are complex diseases with diverse variations, particularly in pediatric cases, and result from mutations in over 100 genes [1–3]. These conditions lead to long-term disability and impose a substantial burden on healthcare systems. Among them, hereditary sensory and motor neuropathy (HSMN), also known as Charcot-Marie-Tooth disease (CMT), is a prevalent form with an estimated frequency of 1 in 2500 individuals [4, 5]. However, accurately determining this figure is challenging due to clinical and geographical heterogeneity [6]. CMT is characterized by progressive muscle weakness, sensory loss, foot deformities, and reduced tendon reflexes. Classification is based on upper limb motor nerve conduction velocities (MNCVs): MNCV < 35 m/s as demyelinating, MNCV > 35 m/s as axonal, and MNCVs between 35 m/s and 45 m/s as intermediate [7, 8].
Accurate diagnoses, prognoses, and targeted treatments rely on genetic testing results. Existing studies on gene distribution and genotype-phenotype correlations in pediatric CMT patients are scarce compared to those in adults. Most studies have focused on Caucasian [9–11], Japanese [12, 13], and Chinese [14, 15] populations. A study conducted in the Chinese Han population in Taiwan reported genotype-phenotype correlations for pediatric-onset CMT based on age of onset and ulnar MNCV [15]. Therefore, population-specific investigations are imperative to elucidate the genetic landscape, identify prevalent genes associated with pediatric-onset IPN, and facilitate prognostication, particularly in limited-resource settings.
In Thailand and other developing countries, access to genetic testing, including gene panel testing and exome sequencing, is limited and costly. Therefore, the diagnosis of CMT often relies on clinical presentation and neurophysiological studies. In case where physicians suspect CMT, PMP22 duplication analysis is performed. If the result is negative, further investigation may be hindered by limited access to gene panel testing and exome sequencing. Consequently, diagnosing this disease in developing countries poses significant challenges.
In this study, we investigated the gene distribution in pediatric-onset IPN within the Thai population. Through the utilization of next-generation sequencing and other analytical techniques, we sought to identify pathogenic variants, study the associated phenotype, and assess neurophysiological characteristics. These findings contribute to a better understanding of the genetic landscape of pediatric-onset IPN and have implications for improved diagnostic and therapeutic approaches in this population.
MATERIALS AND METHODS
Patient recruitment
Patients with clinically suspected IPN who had received medical follow-up at the pediatric neuromuscular clinic of Siriraj hospital were enrolled in the study from January 2017 to December 2021. Cross-sectional assessments were performed, including the patient’s clinical history, physical examination, and electrophysiological study, which were meticulously recorded. The clinical diagnosis needed to be established before the age of 18, and genetic testing was mandatory.
Clinical assessment
Demographic and clinical data included sex, age of clinical onset, current age, affected family members, pedigree, motor and sensory symptoms in the lower and upper limbs, ambulatory status, and use of mobility aids. Additionally, data on associated symptoms, such as scoliosis, hearing and visual impairment, and hand or foot surgery, were collected.
Muscle strength was evaluated using the Medical Research Council Muscle Testing scale, ranging from 0 to 5, for both proximal and distal muscles. Other collected data encompassed bone deformations, skin lesions, deep tendon reflexes, and the presence of pyramidal signs, ophthalmic, and hearing problems.
Pediatric neurologists collected the clinical data and performed the IPN diagnoses. The inclusion criteria required (1) clinical suspicion of IPN and (2) demonstration of evidence of peripheral polyneuropathy through electrophysiological testing or confirmation of neuropathy through nerve biopsy. Patients were excluded if they had a condition associated with acquired neuropathy, such as chemical toxicity, metabolic diseases, immune-mediated neuropathies, neuropathy related to leukodystrophy, and congenital muscular dystrophy. Investigations to exclude acquired neuropathy were conducted based on clinical suspicion.
Electrophysiological testing
The results of nerve conduction studies were collected for both the upper and lower limbs. The data were limited by each child’s tolerability, which restricted the number of nerves and muscles tested. Age-standardized reference tools were used to compare the obtained values. Based on compound muscle action potential (CMAP), sensory nerve action potential (SNAP), and conduction velocity (CV), patients were categorized into demyelinating, axonal, and intermediate types. A CV of < 35 m/s in the upper extremity nerve indicated the demyelinating type, while a CV of 35–45 m/s was classified as intermediate, and a CV of > 45 m/s was considered axonal. Absent or no responses of CMAP and SNAP were classified as undetermined. In patients who were non-reactive (NR) on nerve conduction studies, examiners proceeded to stimulate from the proximal sites. The data presented in table S1 represent values obtained from the distal stimulation.
Genetic testing
Genomic DNA was extracted from leukocytes using a Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). Initially, all samples were screened for PMP22 duplication using semiquantitative multiplex PCR, followed by DNA separation on a 5200 Fragment Analyzer System (Agilent). For patients without PMP22 duplication, further mutational analysis was conducted using exome sequencing to detect variants in other IPN-related genes. Exome sequencing, variant calling, filtering and pathogenicity analysis were conducted at the Sungkyunkwan University School of Medicine, 3Billion Co Ltd, University of South Florida in the United States, and the Faculty of Medicine Siriraj Hospital in Bangkok, Thailand. In this study, genetic testing refers to PMP22 duplication analysis and exome sequencing.
All candidate variants underwent assessment by clinical geneticists and neurologists, who classified them based on the interpretation guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP criteria) [16]. The analysis primarily utilized GENESIS software [17] and Varsome [18], which aggregated data from multiple databases and provided variant pathogenicity using an automatic variant classifier [19]. Additional evidence was obtained from ClinVar [20], the Inherited Neuropathy variant browser [20], and a literature review. Variants were labeled as “novel” if they had not been previously reported in PubMed (https://pubmed.ncbi.nlm.nih.gov/), Clinvar, Varsome, or dbSNP. Segregation analysis of the variants in parents was performed to evaluate the cis/trans phase in the case of autosomal recessive inheritance. It was also used to ascertain the de novo origin or inheritance from parents in autosomal dominant cases and to identify cases with multiple potential pathogenic variants or variants of uncertain significance or novel variants.
Cases with autosomal dominant inheritance were designated as “assumed de novo” when only one of the patient’s parents could undergo targeted sequencing and the variant found in their child was not present in the tested parent.
Genotype-phenotype correlation
To explore the correlations between genotypes and phenotypes, we analyzed the range of variants in patients with different ages at disease onset (infantile onset≤2 years and childhood-/adolescent-onset disease 3–19 years) and ulnar MNCVs (<15 m/sec, 15–25 m/sec, 25–38 m/sec, and > 38 m/sec). The age at onset was based on when parents noticed motor abnormalities in their children or when the patients themselves became aware of their motor or sensory dysfunctions.
Ethical considerations
The Siriraj Institutional Review Board authorized the study protocol (approval number 343/2560[EC4]), and the research was conducted in accordance with the principles of the Declaration of Helsinki. Informed consent for the collection of 3 to 5 mL of peripheral blood from the participants was obtained from the subjects and their parents. Written informed consent for publication was also obtained.
RESULTS
Patient characteristics and clinical description
Of the 36 enrolled patients, one was excluded due to Friedreich ataxia, leaving 35 patients with IPNs from 34 families. Among these, there were 16 males and 19 females (Table 1). The median age was 14 years, with disease onset occurring at a median age of 2 years (range: 3 months–12 years). A family history of IPN was present in 25.7% of patients. Consanguinity was reported in the family of 2 brothers diagnosed with hereditary sensory neuropathy (HSN; Patients No. 33 and 34; Table S1).
Table 1
Demographic data
| Characteristics | N = 35 |
| Male Sex, n (%) | 16 (45.7%) |
| Age (years), median (IQR) | |
| Age at enrolment | 14 (8.5, 20.5) |
| Age at disease onset | 2 (1.0, 3.5) |
| Family history of IPN, n (%) | 9 (25.7%) |
| Clinical diagnosis, n (%) | |
| HSMN/CMT | 32 (91.4%) |
| HSN | 3 (8.6%) |
| Electrophysiologic subtype, n (%) | |
| Demyelinating | 11 (31.4%) |
| Axonal | 19 (54.3%) |
| Intermediate | 2 (5.7%) |
| Undetermined | 3 (8.6%) |
Abbreviations: CMT: Charcot-Marie-Tooth disease; HSMN: hereditary sensorimotor neuropathy; HSN: hereditary sensory neuropathy. Data are presented as median (interquartile range; IQR) or n (%).
At the time of recruitment, most patients had already developed motor symptoms. Specifically, foot deformities were observed in 97.1% (34/35) of cases, difficulty walking in 91.4% (32/35) of cases, difficulty with balance in 68.6% (24/35) of cases, and delayed walking after 15 months of age in 42.9% (15/35) of cases. Unfortunately, among the 34 patients with foot deformities, 3 were initially misdiagnosed with isolated clubfeet.
Furthermore, 17 out of 35 patients experienced impairment in hand motor function, resulting in difficulties with buttons, zippers, fasteners, and bottles. Additionally, 31.4% (11/35) of patients reported difficulties with using eating utensils. Among the 20 patients who exhibited sensory symptoms (57.1%, with 3 missing values), 19 experienced a decreased ability to feel, 3 had a burning or tingling sensation, and 4 had ulcers on their feet or hands. The clinical phenotypes of the patients based on the common genes involved are detailed in Table 2.
Table 2
Clinical phenotype of patients based on the common genes involved
| No | Age at | Age of | Weakness | Sensory | DTRd | Foot | Scoliosis | Ambulation status | Mobility aids | Visual | Hearing | Pyramidal | ||
| study (year) | onset (year) | UEa | LEb | lossc | UE | LE | deformity | problem | problem | sign | ||||
| MFN2 | ||||||||||||||
| 16 | 16 | 3 | + | ++ | P, V | N | A | pes planus | N | Ambulatory | AFO, toeoff | N | N | N |
| 17 | 20 | 7 | + | ++ | P | N | A | pes cavus | N | Ambulatory | AFO | N | N | N |
| 18 | 20 | 2 | ++ | +++ | P, V | A | A | pes cavus | N | Non-ambulatory | wheelchair since 18 years | Y | N | N |
| 19 | 8 | 1 | ++ | ++ | P, V | A | A | pes cavus | N | Ambulatory | AFO | N | N | N |
| 20 | 4 | 0.9 | ++ | ++ | P, V | A | A | pes planus | N | Ambulatory | AFO, hand splint | N | N | N |
| 21 | 35 | 8 | ++ | +++ | P, V | A | A | pes cavus | N | Non-ambulatory | KO with axillary crutch, walker | N | Y | N |
| GDAP1 | ||||||||||||||
| 11 | 14 | 8 | + | + | P, V | N | A | pes cavus | N | Ambulatory | TCO, orthopedic shoes | N | N | N |
| 12 | 13 | 6 | + | ++ | nl | D | A | pes cavus | N | Ambulatory | AFO, orthopedic shoes | N | N | N |
| 22 | 9 | 8 | + | ++ | nl | A | A | pes cavus | mild | Ambulatory | AFO | N | N | N |
| NEFL | ||||||||||||||
| 5 | 13 | 2 | + | ++ | P, V | A | A | pes cavus | mild | Ambulatory | AFO, SMO | N | Y | N |
| 6 | 6 | 2 | + | ++ | P, V | N | A | pes cavus | N | Ambulatory | AFO | N | Y | N |
| 7 | 21 | 2 | + | ++ | P | A | A | pes cavus | N | Ambulatory | AFO | N | Y | N |
aWeakness in LE: +: ankle dorsiflexion 4/5 on MRC scale; ++: ankle dorsiflexion < 4/5 on MRC scale; +++: proximal weakness and wheelchair-dependent. bWeakness in UE:+: intrinsic hand weakness 4/5 on MRC scale; ++: intrinsic hand weakness < 4/5 on MRC scale; –: no symptoms. cSensory loss: P = pain sense; V = vibration sense; nl = normal sense. dDTR: N = normal; D = diminished; A = absent. AFO: ankle-foot orthosis; DTR: deep tendon reflex; LE: lower extremities; KO: knee orthosis; N: no; N/A: not available; SMO: supramalleolar orthosis; TCO: total-contact orthosis; UE: upper extremities; Y: yes.
Clinical examinations also revealed scoliosis in 7 patients within the cohort. Hearing loss was described in 8 patients, with 3 associated with NEFL and 1 each associated with MFN2, GDAP1, IGHMBP2, PMP22 small deletion, and an unknown gene. Optic atrophy was exclusively found in 1 patient with MFN2.
Most of the patients were able to walk with the assistance of orthosis aids, while a small minority required a wheelchair (17.1%). Approximately one-third of the patients (11/35) underwent foot surgery, including procedures such as Achilles tendon lengthening, tendon transfer, osteotomy, and tibio-talo-calcaneal fusion.
Three patients from 2 families were diagnosed with HSN. Patient No.32, the only affected member in her family, experienced a gradual onset of symptoms, including tongue mutilation, finger amputation, and corneal ulcer. The physical examination revealed chronic scarring wounds extending from the forehead to the middle of the nose, loss of portions of the lower lip due to self-biting, and loss of fingertips. Patients No.33 and 34, the only affected children in a consanguineous family, presented with corneal ulcer, later diagnosed as neuropathic keratitis, and wounds on their fingers and toes at 6 months and 4 months of age, respectively. They did not exhibit motor weakness and later reported a loss of sensation in their fingers and toes.
Electrophysiological results
Of the 35 IPN patients, 34 were diagnosed based on electrophysiological testing (Table S1), and 1 patient (Patient No. 14) was diagnosed based on nerve pathology. Patient No. 14 showed a marked reduction in myelinated axons and an increase in endoneurial connective tissue. Electron microscopy confirmed the reduction in axons with an increase in endoneurial collagen fibers, indicating axonal neuropathy. Therefore, we classified this patient as having the axonal form.
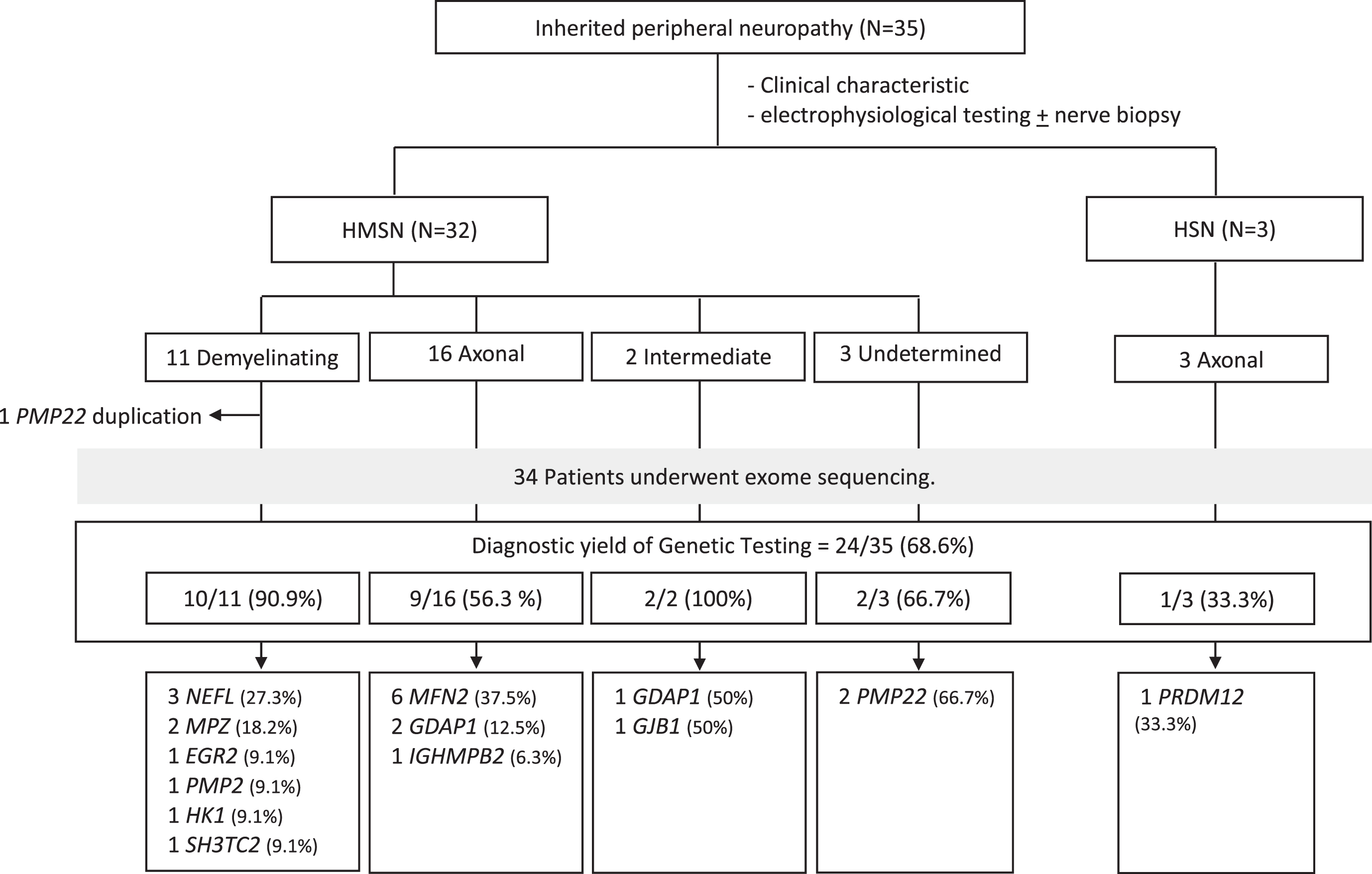
Among the 35 IPN patients, 32 were categorized as having CMT (11 demyelinating, 16 axonal, 2 intermediate, and 3 undetermined types), while 3 were diagnosed with HSN of the axonal type (Fig. 1).
Fig. 1
Distribution of disease-causing genes in pediatric-onset inherited peripheral neuropathy. Abbreviations: HSMN: hereditary sensorimotor neuropathy; HSN: hereditary sensory neuropathy.
Molecular findings
The molecular and electrophysiological findings of patients without PMP22 duplication are summarized in Table S1. Genetic testing identified disease-causing variants in 24 patients (68.6%; 24/35). Among them, 6 had MFN2 variants (17.1%), 3 had GDAP1 and NEFL variants each (8.6%), and 2 had MPZ and PMP22 variants each (5.7%). The remaining IPN-related genes (EGR2, GJB1, HK1, IGHMBP2, PMP22 duplication, PMP2, PRDM12 and SH3TC2) individually accounted for approximately 3% of the total IPN patients.
In patients with demyelinating CMT, 90.9% (10/11) had a confirmed disease-causing variant, as did 56.3% (9/16) of axonal CMT patients and both patients with the intermediate form (Fig. 1). Among demyelinating CMT patients, the most common genetic causes were variants in NEFL (27.2%; 3/11) and MPZ (18.2%; 2/11), which accounted for 50% (5/10) of the cases.
For axonal CMT patients, the most prevalent genetic causes were MFN2 (37.5%; 6/16) and GDAP1 (12.5%; 2/16). The variants in these genes encompassed 50% (8/16) of the axonal CMT patients.
In HSN patients, we identified PRDM12 as a pathogenic gene in 1 out of 3 patients. However, 2 patients from the same family (Patients No. 33 and 34) did not show a pathogenic variant.
Patients without PMP22 duplication had 25 pathogenic/likely pathogenic variants and 2 variants of uncertain significance across 12 genes. Among 34 patients without PMP22 duplication, we identified the following 25 pathogenic/likely pathogenic variants were discovered: ERG2 (c.925 C>T), GDAP1 (c.368A>G, c.563A>G), GJB1 (c.223 C>T), HK1 (c.1A>G), IGHMBP2 (c.547 + 1 G>A, c.983_987del, c.2362 C>T, c.2773del), MFN2 (c.272T>G, c.280 C>T, c.617 C>T, c.707 C>T, c.1090 C>T, c.1091 G>C), MPZ (c.245A>G, c.551del), NEFL (c.280 C>T, c.293A>G), PMP2 (c.155T>C), PMP22 (c.215 C>T, c.251_253del), PRDM12 (c.570 + 2T>G, c.796A>C), and SH3TC2 (c.929dup). Of these 27 variants, 20 were missense, 4 were frameshift deletions, 2 were splice-sites, and 1 was in-frame deletion.
Among the identified variants, 4 were novel, and all were classified as pathogenic or likely pathogenic (Patients No. 2, 9, and 32; Table S1). These variants were absent in both the Genome Aggregation Database (gnomAD) and the Thai reference exome (T-Rex) database [21]. Additionally, their pathogenicity was supported by in silico prediction, utilizing REVEL [22] for missense variants and splice AI [23] for splice site analysis. Segregation analysis was conducted for patients No. 9 and 32, as depicted in Supplementary Figure 1 as shown. Unfortunately, we were unable to collect samples from the parents of patient No. 2 due to relocation. The pedigrees of patients harboring novel variants and undergoing segregation analysis are illustrated in Supplementary Figure 1, indicating that each parent of patients No. 9 and 32 carries one variant found in his/her child.
Targeted sequencing was performed in 17 families, and specimens from both parents were obtained in most cases. However, in 4 cases, we could investigate only 1 parent due to divorce, relocation, or death. Parental mutation status was confirmed in 7 de novo variants (Table S1). In situations where 1 parent did not have the same variant as their child (Patients No. 8, 16, and 22), we assumed a de novo occurrence of the variant in the context of autosomal dominant inheritance, as the parents were asymptomatic. Notably, 2 parents without clinical neuropathy had the same variant as their children (Patients No. 12 and 29), and their variants were inherited in an autosomal dominant manner.
Genotype-phenotype correlations
In our cohort, 62.9% (22/35) of the IPN patients experienced infantile onset, while another 47.1% (13/35) had childhood- or adolescent-onset disease. Among patients with infantile-onset CMT, the most frequent disease causes were mutations in MFN2 (13.6%; 3/22) and NEFL (13.6%; 3/22). For patients with childhood- or adolescent-onset CMT, both MFN2 and GDAP1 were common causes of the disease, accounting for 23.1% (3/13) of cases each.
When CMT patients were grouped based on ulnar MNCV, 19.4% had MNCV < 15 m/sec, 9.7% had MNCV between 15 and 25 m/sec, 22.6% had MNCV between 25 and 38 m/sec, and 38.7% had MNCV > 38 m/sec. Among CMT patients with ulnar MNCV < 15 m/sec, the major causes were PMP22 and MPZ variants. For patients with ulnar MNCV between 15 and 25 m/sec, no specific etiology was identified. NEFL was the major etiology for patients with ulnar MNCV between 25 and 38 m/sec, while mutations in MFN2 were the major disease causes for patients with ulnar MNCV > 38 m/sec.
DISCUSSION
This study focused on 35 pediatric patients with IPNs, including CMT and HSN. The patients underwent initial testing for PMP22 gene duplication, followed by exome sequencing. The overall diagnostic yield was 68.6%, providing valuable insights into the pathogenic variants associated with pediatric early-onset IPN.
Clinically, IPN in children may present differently from that in adults, with manifestations such as orthopedic issues, including foot and spine deformities, occurring before the onset of clinical weakness and sensory impairment. Notably, 3 patients initially misdiagnosed with clubfeet underwent unnecessary orthopedic surgeries. They were later found to have distinct features of IPN, such as bilateral diminished or absent ankle reflex, progressive symptoms, and abnormal electrophysiological test results. Patient No. 32, diagnosed with CMT, also exhibited midface toddler excoriation syndrome. This condition is characterized by severe midfacial lesions with little evidence of generalized pain insensitivity, as described by Moss et al. [24].
Pathogenic or likely pathogenic variants were identified in 68.6% (24/35) of patients. This proportion is consistent with previous reports on exome sequencing in pediatric IPN patients, with diagnostic yields ranging from 66.7% to 80.6% [9–11, 15, 25]. We observed a dominant prevalence of the axonal type of IPN in the children included in our study, in contrast to previous studies by Fridman et al., [25] Abe et al., [26] Hsu et al., [15] and other studies in children [9, 11, 15, 27, 28]. The most common genes identified in our study were MFN2, and NEFL, which differ from previous reports on pediatric CMT, where most patients had PMP22 duplication (Table S2). Several explanations could account for the discrepancy, including an earlier age of symptom onset, a lower proportion of PMP22 duplication cases, and differences in referral institutions.
The lower proportion of PMP22 duplication cases in our study can be attributed to multiple factors. Firstly, patients with PMP22 duplication often present with milder symptoms and a slower clinical progression, potentially leading to delayed medical attention until adulthood when symptoms significantly disrupt daily life. Furthermore, in developing countries, access to genetic testing may be hindered by resource limitations in hospitals, causing some patients seeking medical attention in the first decade of life to face challenges in obtaining genetic testing. It is essential to note that our study was conducted in a pediatric center, and our center primarily receives referrals for severe CMT cases from across the country, potentially excluding individuals with milder CMT symptoms from our study cohort. Consequently, the proportion of PMP22 duplication cases among children may appear lower than previous studies. The relatively young median age of symptom onset in our study, at 2 years, can be explained by the predominance of severe cases, primarily associated with the axonal type where MFN2 is the most responsible gene.
Our findings underscore the importance of prioritizing genetic testing for Thai patients with CMT, taking into account the age of disease onset to classify patients into more specific subgroups. In our cohort, the most common genetic causes of infantile-onset CMT were mutations in MFN2 and NEFL, aligning with findings observed among the Han Chinese population in Taiwan (Table S2) [15]. The clinical symptoms of patients with common genes in our cohort were similar to previous descriptions. We identified 2 pathogenic NEFL variants associated with CMT and deafness. All 3 patients with the NEFL variants had sensorineural hearing loss. One patient has c.293A>G, p.Asn98Ser (Patient No.5) and 2 unrelated patients have c.280 C>T, p.Leu94Phe (Patient No. 6 and 7). Abe et al., [29] found that the former variant was associated with hearing disturbance, but the latter variant had not been previously reported.
Regarding the autosomal dominant inheritance in Patients No. 12 and 29, where both parents lacked clinical neuropathy but shared the same variant as their children, we considered the possibility of variable expression, incomplete penetrance, or modifier genes contributing to this scenario.
We acknowledge that additional functional experiments are necessary to confirm the causal roles of the 4 novel mutations observed in our cohort. Although their pathogenicity can be partially supported by in silico analysis and population data, uncertainties remain.
We also recognize potential limitations of our study. Firstly, this study was conducted at a single center, which may limit the generalizability of our findings to the broader Thai population. This limitation arises because our center primarily serves as a tertiary referral center, attracting the most severe cases from around the country. Consequently, patients with milder symptoms may not have been adequately represented in our cohort. Additionally, there is a possibility of recall bias regarding the onset of clinical symptoms, a factor we recognize as another limitation. We did not include data on CMT assessment tools due to a high proportion of missing information.
In conclusion, this study presents the mutational spectrum and genotype-phenotype correlations of pediatric-onset IPN. Differences in the frequencies of PMP22 duplication and NEFL mutations were observed between the Thai population and Caucasian populations. These findings expand the spectrum of mutations causing CMT and provide valuable insights for optimal strategies in mutational analysis and genetic counseling of CMT for patients of Thai origin. We plan to perform genome sequencing in undiagnosed cases of genetic origin. Additionally, we will conduct a national and regional research study to obtain a more accurate understanding of the genetic prevalence and to study the natural history of this disease in a larger population.
ACKNOWLEDGMENTS
We extend our gratitude to Cheewasan Apirukphanakhet for his valuable contribution, and we acknowledge the support of the Siriraj Institute of Clinical Research with the development of this manuscript. Most importantly, we sincerely thank all the children and families who participated in this study for their invaluable contribution.
FUNDING
This research received supported from Specific League Funds from Mahidol University (R016421009) and The Royal College of Pediatricians of Thailand (R2021.2.4).
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available within the article and its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230174.
REFERENCES
[1] | Timmerman V , Strickland AV , Züchner S . Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes (2014) ;5: (1). |
[2] | Gutmann L , Shy M Update on Charcot-Marie-Tooth disease. Curr Opin Neurol. 2015. |
[3] | Benarroch L , Bonne G , Rivier F , Hamroun D The 2021 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2020. |
[4] | Skre H . Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet (1974) ;6: (2):98–118. |
[5] | Reilly MM , Murphy SM , Laurá M Charcot-Marie-Tooth disease. J Peripher Nerv Syst (2011) ;16: (1):1–14. |
[6] | Barreto LCLS , Oliveira FS , Nunes PS , de França Costa IMP , Garcez CA , Goes GM et al. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology (2016) ;46: (3):157–65. |
[7] | Harding AE , Thomas PK . The clinical features of hereditary motor and sensory neuropathy types I and II. Brain (1980) ;103: (2):259–80. |
[8] | Saporta AS , Sottile SL , Miller LJ , Feely SM , Siskind CE , Shy ME . Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol (2011) ;69: (1):22–33. |
[9] | Hoebeke C , Bonello-Palot N , Audic F , Boulay C , Tufod D , Attarian S , et al. Retrospective study of 75 children with peripheral inherited neuropathy: Genotype–phenotype correlations. Archives de Pédiatrie (2018) ;25: (8):452–8. |
[10] | Argente-Escrig H , Frasquet M , Vázquez-Costa JF , Millet-Sancho E , Pitarch I , Tomás-Vila M , et al. Pediatric inherited peripheral neuropathy: a prospective study at a Spanish referral center. Ann Clin Transl Neurol (2021) ;8: (9):1809–16. |
[11] | Cornett KMD , Menezes MP , Bray P , Halaki M , Shy RR , Yum SW , et al. Phenotypic Variability of Childhood Charcot-Marie-Tooth Disease. JAMA Neurology (2016) ;73: (6):645–51. |
[12] | Abe A , Numakura C , Kijima K , Hayashi M , Hashimoto T , Hayasaka K . Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J Hum Genet (2011) ;56: (5):364–8. |
[13] | Yoshimura A , Yuan JH , Hashiguchi A , Ando M , Higuchi Y , Nakamura T , et al. Genetic profile and onset features of patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry (2019) ;90: (2):195–202. |
[14] | Sun B , Chen Z , Ling L , Yang F , Huang X . Clinical and genetic spectra of Charcot-Marie-Tooth disease in Chinese Han patients. J Peripher Nerv Syst (2017) ;22: (1):13–8. |
[15] | Hsu YH , Lin K-P , Guo YC , Tsai YS , Liao Y-C , Lee Y-C Mutation spectrum of Charcot-Marie-Tooth disease among the Han Chinese in Taiwan. Annals of Clinical and Translational Neurology. 2019;6. |
[16] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine (2015) ;17: (5):405–23. |
[17] | Gonzalez M FM , Gai X , Postrel R , Schüle R , Zuchner S . Innovative Genomic Collaboration Using the GENESIS (GEM.app) Platform. Boycott K, Hamosh A, Rehm H, eds. Hum Mutat (2015) ;36: (10):950–6. |
[18] | Kopanos C , Tsiolkas V , Kouris A , Chapple CE , Albarca Aguilera M , Meyer R , et al. VarSome: the human genomic variant search engine. Bioinformatics (2018) ;35: (11):1978–80. |
[19] | Kopanos C , Tsiolkas V , Kouris A , Chapple CE , Albarca Aguilera M , Meyer R , et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019. |
[20] | Saghira C , Bis D , Stanek D , Strickland A , Herrmann D , Reilly M , et al. Variant pathogenicity evaluation in the community-driven Inherited Neuropathy Variant Browser. Human Mutation. 2018;39. |
[21] | Shotelersuk V , Wichadakul D , Ngamphiw C , Srichomthong C , Phokaew C , Wilantho A , et al. The Thai reference exome (T-REx) variant database. Clin Genet (2021) ;100: (6):703–12. |
[22] | Ioannidis NM , Rothstein JH , Pejaver V , Middha S , McDonnell SK , Baheti S , et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet (2016) ;99: (4):877–85. |
[23] | de Sainte Agathe J-M , Filser M , Isidor B , Besnard T , Gueguen P , Perrin A , et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Human Genomics (2023) ;17: (1):7. |
[24] | Moss C , Srinivas SM , Sarveswaran N , Nahorski M , Gowda VK , Browne FM , et al. Midface toddler excoriation syndrome (MiTES) can be caused by autosomal recessive biallelic mutations in a gene for congenital insensitivity to pain, PRDM12. Br J Dermatol (2018) ;179: (5):1135–40. |
[25] | Fridman V , Bundy B , Reilly MM , Pareyson D , Bacon C , Burns J , et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. Journal of Neurology, Neurosurgery & Psychiatry (2015) ;86: (8):873. |
[26] | Abe A , Numakura C , Kijima K , Hayashi M , Hashimoto T , Hayasaka K Molecular diagnosis and clinical onset of Charcot–Marie–Tooth disease in Japan. Journal of Human Genetics (2011) ;56: (5):364–8. |
[27] | Fernández-Ramos JA , López-Laso E , Fau-Camino-León R , Camino-León R , Fau - Gascón-Jiménez FJ , Gascón-Jiménez FJ , Fau-Jiménez-González MD , Jimenez-González MD Experience in molecular diagnostic in hereditary neuropathies in a pediatric tertiary hospital. Rev Neurol. 2015. |
[28] | Thongsing A , Pho-iam T , Limwongse C , Likasitwattanakul S , Sanmaneechai O . Case series: Childhood Charcot-Marie-Tooth: Predominance of axonal subtype. eNeurologicalSci (2019) ;16: :100200. |
[29] | Abe A , Numakura C , Saito K , Koide H , Oka N , Honma A , et al. Neurofilament light chain polypeptide gene mutations in Charcot–Marie–Tooth disease: nonsense mutation probably causes a recessive phenotype. Journal of Human Genetics (2009) ;54: (2):94–7. |


