
Contribution of Complement, Microangiopathy and Inflammation in Idiopathic Inflammatory Myopathies
Abstract
Purpose of review:
Idiopathic inflammatory myopathies (IIMs) are a heterogeneous group characterized by muscle weakness and skin symptoms and are categorized into six subtypes: dermatomyositis (DM), polymyositis (PM), anti-synthetase syndrome (ASS), immune-mediated myopathy (IMNM), inclusion body myopathy (IBM), and overlap myositis. Myositis-specific autoantibodies were detected for the diagnosis and classification of IIM. This review highlights the pathogenic contributions of the complement system, microangiopathy, and inflammation in IIM.
Recent findings:
Deposition of complement around capillaries and/or the sarcolemma was observed in muscle biopsy specimens from patients with DM, ASS, and IMNM, suggesting the pathomechanism of complement-dependent muscle and endothelial cell injury. A recent study using human muscle microvascular endothelial cells showed that Jo-1 antibodies from ASS induce complement-dependent cellular cytotoxicity in vitro. Based on both clinical and pathological observations, antibody- and complement-mediated microangiopathy may contribute to the development of DM and anti-Jo-1 ASS. Juvenile DM is characterized by the loss of capillaries, perivascular inflammation, and small-vessel angiopathies, which may be related to microinfarction and perifascicular atrophy. Several serum biomarkers that reflect the IFN1 signature and microangiopathy are elevated in patients with DM. The pathological observation of myxovirus resistance protein A (MxA), which suggests a type 1 interferon (IFN1) signature in DM, supports the diagnosis and further understanding of the pathomechanism of IIM. A recent report showed that an increase in triggering receptor expressed on myeloid cells (TREM-1) around perimysial blood vessels and muscles in patients with IIM plays a role in triggering inflammation and promoting the migration of inflammatory cells by secreting proinflammatory cytokines, such as tumor necrosis factor α.
Summary:
The deposition of complement in muscles and capillaries is a characteristic feature of DM, ASS, and IMNM. Microangiopathy plays a pathogenic role in DM, possibly resulting in perifascicular atrophy. Further understanding of the detailed pathomechanism regarding complement, microangiopathy, and inflammation may lead to novel therapeutic approaches for IIM.
INTRODUCTION
Idiopathic inflammatory myopathies (IIMs) are classified into six subtypes: dermatomyositis (DM), polymyositis (PM), antisynthetase syndrome (ASS), immune-mediated necrotizing myopathy (IMNM), sporadic inclusion body myositis (sIBM), and overlap myositis [1–8]. The clinical findings, organ manifestations, treatment responses, and prognoses differ among these groups [9, 10]. The clinical features of ASS include myositis, interstitial pneumonia, arthritis, Raynaud phenomenon, and mechanical hands [11–13]. DM is often associated with interstitial pneumonia and malignancy [1, 4, 14]. The diagnosis of IIMs is based on the assessment of clinical symptoms, elevated serum creatine kinase levels, electromyography findings, muscle magnetic resonance imaging (MRI) changes, myositis-specific autoantibodies, and skeletal muscle biopsy findings [15–19]. Recent evidence suggests that complement, microangiopathy, and inflammation are involved in the pathomechanism of several types of IIM [4, 11, 20], and our recent data also demonstrated the importance of complement-induced microangiopathy in IIMs [21].
We herein review the pathogenic roles of complement and inflammation in IIMs, particularly DM, ASS, and IMNM.
MYOSITIS-SPECIFIC AUTOANTIBODY PROFILES
The discovery of myositis-specific antibodies and accumulated evidence of their association with relatively specific clinical features have changed the classification of IIM [22]. Myositis-specific autoantibodies include antibodies against anti-complex nucleosome remodeling histone deacetylase (anti-Mi-2), anti-melanoma differentiation-associated gene 5 (anti-MDA5), anti-transcription intermediary factor 1-γ (anti-TIF1-γ), anti-antinuclear matrix protein 2 (anti-NXP2), and anti-small ubiquitin-like modifier-activating enzyme (anti-SAE) in DM; anti-ARS antibody (anti-Jo-1, anti-OJ, anti-EJ, anti-PL7, anti-PL12, anti-Ha, and anti-Zo antibodies) in ASS; and antibodies against anti-signal recognition particle (anti-SRP) and anti-3-hydroxy-3-methylglutaryl coenzyme A reductase antibodies (anti-HMGCR) in IMNM (Table 1) [10, 11, 22]. For the detection of anti-OJ antibodies, immunoprecipitation is preferred over ELISA or line blot [11, 23]. Generally, one patient rarely has multiple myositis-specific autoantibodies simultaneously [11, 22]. Clinically, anti-TIF1-γ and anti-NXP2 DM are highly associated with malignancy, anti-Mi-2 DM with dysphagia, anti-NXP2 DM with skin edema, muscle ischemia, and subcutaneous calcification, anti-Mi-2 DM with apparent muscle weakness with less association with malignancy, and anti-MDA5 DM with clinically amyotrophic DM with rapidly progressive and fatal interstitial pneumonia and arthritis [10, 11, 14, 22]. Anti-SRP and anti-HMGCR antibodies are myositis-specific and clinically valuable in the diagnosis of IMNM (Table 1) [10, 11, 22].
Table 1
Pathological and clinical features in subgroups of IIMs
| Subgroup | Myositis antibody | Histological features | IHC features | Clinical features | ||
| HLA-ABC | HLA-DR | C5b-9 | ||||
| DM | Anti-TIF1-γ | PFA, Punched-out vacuoles | + | -/+ | Capillaries>sarcolemma | CAM Skin erythema |
| Anti-Mi-2 | PFA, PFN, Promiment inflammation | + | -/+ | Sarcolemma>Capillaries | ||
| Anti-NXP2 | Microinfarction Muscle ischemia | + | -/+ | Capillaries>sarcolemma | JDM CAM Calcification | |
| Anti-MDA5 | Non-PFA | + | -/+ | Capillaries>sarcolemma | ADM | |
| Anti-SAE | PFA | + | -/+ | Capillaries | ILD | |
| ASS | Anti-Jo-1 | PFA Myofiber necrosis and regederation | + | +(PF) | sarcolemma | Myositis>ILD With Raynaud, Mechanic hands, Joint involvement |
| Anti-PL-7 | + | +(PF) | sarcolemma | ILD>myositis | ||
| Anti-PL-12 | + | +(PF) | sarcolemma | ILD>myositis | ||
| Anti-OJ | + | +(PF) | sarcolemma | Severe muscle involvement | ||
| IMNM | Anti-SRP | Myofiber necrosis and regeneration Less inflammation (macrophage>lymphocyte) | + | – | sarcolemma | Cardiac involvement |
| Anti-HMGCR | + | – | sarcolemma | |||
IIMs, idiopathic inflammatory myopathies; DM, dermatomyositis; ASS, anti-synthetase syndrome; IMNM, immune-mediated myopathy; anti-Mi-1, anti-complex nucleosome remodeling histone deacetylase antibodies; anti-MDA5, anti-melanoma differentiation associated gene 5 antibodies; anti-TIF1γ, anti-transcription intermediary factor 1 γ antibodies; anti-NXP2, anti-antinuclear matrix protein 2 antibodies; anti-SAE, anti-small ubiquitin-like modifier-activating enzyme antibodies; anti-SRP, anti-signal recognition particle antibodies; anti-HMGCR, anti-3-hydroxy-3-methylglutaryl coenzyme A reductase antibodies. PFA, perifascicular atrophy; PFN, perifascicular necrosis; IHC, immunohistochemistry.
CLINICOPATHOLOGY AND ROLE OF COMPLEMENT IN DM, ASS, AND IMNM
Complement pathways
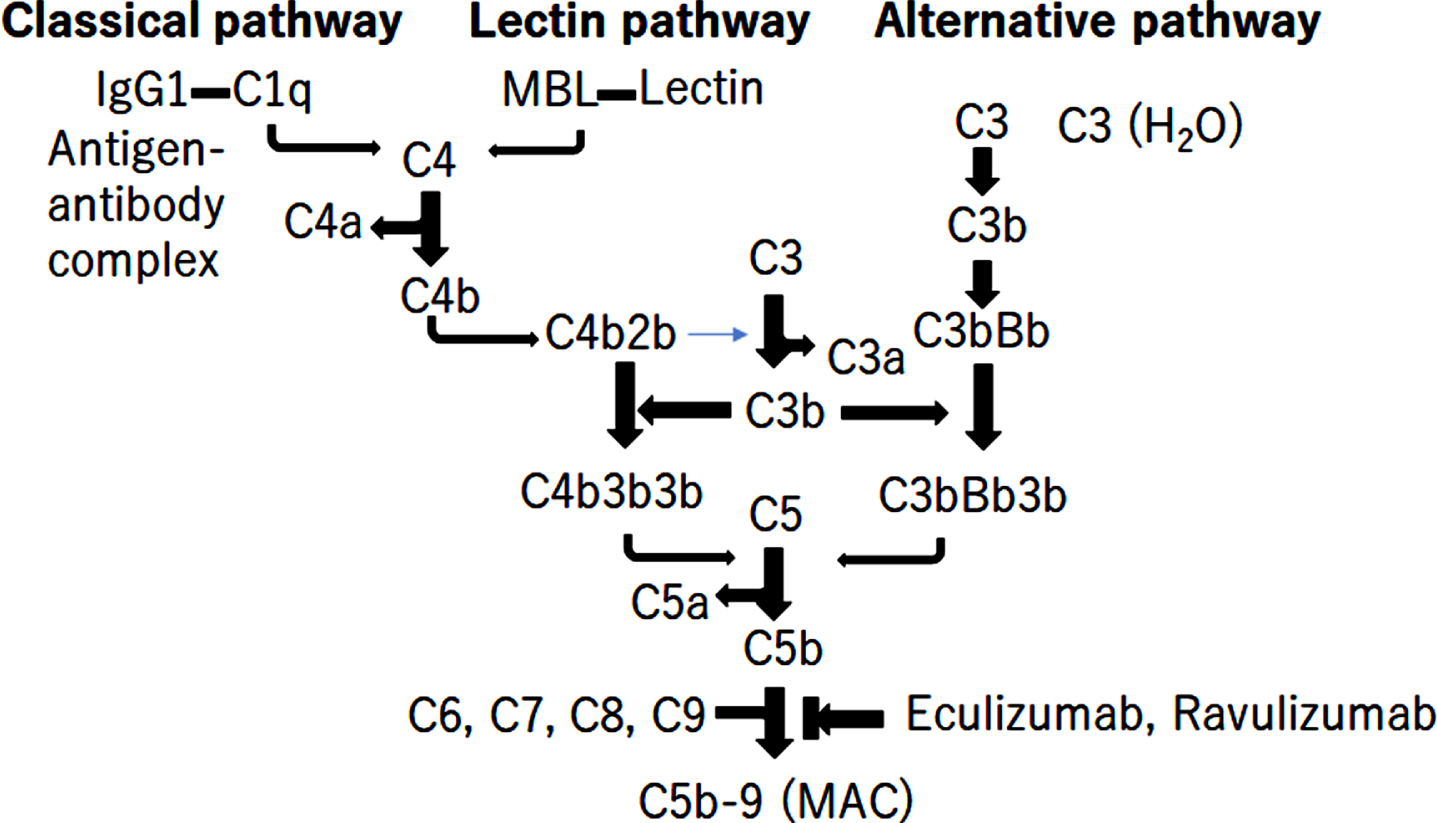
Complement system activation is associated with the pathomechanism of IIM. Three pathways of complement activation have been elucidated: the classical pathway (CP), alternative pathway (AP), and lectin pathway (LP) (Fig. 1) [24, 25]. Each of the three pathways is activated by antigen/antibody complex binding to the C1q of the C1 complex in the classical pathway, spontaneous hydrolysis of C3 in alternative pathways, or binding of foreign carbohydrate moieties to mannose binding lectin or ficolin in the lectin pathways, causing the cleavage of C3 into C3a and C3b25-27. C3b then binds to C5 convertase C4b1a3b for CP, LP, or C3bBb3b for AP, leading to cleavage of C5 into C5a and C5b. C5b initiates the lytic pathway and forms the C5b-9 complex, the membrane attack complex (MAC), causing the direct lysis of target cells (Fig. 1) [24, 26]. Eculizumab and Ravulizumab are complement-targeted monoclonal antibodies that inhibit the cleavage of C5 into C5a and C5b, approved for the treatment of myasthenia gravis and neuromyelitis optica (Fig. 1) [27].
Fig. 1
The activation of complement pathway. There are 3 different pathways for complement activation: the classical pathway, the alternative pathway, and the lectin pathway. The triggers of activation are: as follows: (1) the antigen/antibody complex binding to the C1q of the C1 complex in the classical pathway; (2) foreign carbohydrate moieties binding to mannose binding lectin or ficolin in the lectin pathway; and (3) spontaneous hydrolysis of C3 in the alternative pathway. These 3 pathways cause the cleavage of C3 into C3a and C3b. C3b binds to C5 convertase C4b1a3b or C3bBb3b, leading to the cleavage of C5 into C5a and C5b. C5b initiates the lytic pathway and form C5b-9 the membrane attack complex (MAC), leading to direct lysis. Eculizumab and ravulizumab are complement-targeted monoclonal antibodies for inhibiting the cleavage of C5 into C5a and C5b.
DM
Morphological characteristics of muscle biopsy specimens (Fig. 2)
Perifascicular atrophy (PFA) and myofiber expression of myxovirus-resistant protein A (MxA), which reflect the activation of IFN1 signaling, are now included as definite muscle biopsy criteria in the 2018 ENMC-DM consensus [28]. Human leukocyte antigen (HLA)-ABC is a marker of myositis, including DM, PM, ASS, IMNM, and IBM (Table 1) [11, 29].
DM is associated with five specific autoantibodies, each with characteristic clinical symptoms. It is becoming increasingly evident that there are also differences in the muscle histopathological findings. For example, cases that are positive for anti-TIF1-γ, anti-NXP2, and anti-SAE antibodies typically exhibit PFA, whereas cases that are positive for anti-Mi-2 antibodies exhibit perifascicular necrosis (PFN), similar to anti-synthetase syndrome-associated myopathy (ASS-OM) [30]. Vacuolated punched-out fibers are characteristic features of anti-TIF1-γ DM [14]. Severe muscle pathology, prominent inflammation, and perifascicular necrosis are associated with Mi-2 DM [11, 14, 29]. Microinfarction in anti-NXP2 DM and mild nonspecific myopathic changes in anti-MDA5 DM have been observed [11, 14, 29]. However, the pathological features of anti-SAE DM remain unclear.
Furthermore, complement deposition on small blood vessels is commonly observed in cases positive for anti-TIF1-γ and anti-NXP2 antibodies [31, 32]. In cases that are positive for anti-Mi-2 antibodies, complement deposition on the sarcolemma is frequently observed [11].
The role of complement in DM pathogenesis
In DM, the membranolytic attack complex (MAC) composed of C5b-9 primarily targets the endothelium of the capillaries. This complex is deposited on endothelial cells at an early stage of the disease, even before any apparent damage to muscle fibers occurs [4, 17, 33, 34]. Complement deposition leads to the necrosis of endothelial cells and a decrease in the number of capillaries in the endomysium. As a result, ischemia and microinfarcts occur, particularly at the periphery of the fascicles, which explains the observed PFA.
Key observations of complements in DM based on the effectiveness of intravenous immunoglobulin (IVIg)
One of the mechanisms of action of IVIg, which is commonly used in refractory cases, is its effective binding to C1q, which prevents initiation of the complement cascade by pathogenic antibodies. In addition, IVIg binds to activated C3 and C4 proteins, inhibiting their deposition in tissues [35, 36]. The most compelling evidence for the effectiveness of IVIg in inhibiting complement can be seen both in vivo and in vitro. A double-blind placebo-controlled study conducted in patients with treatment-resistant DM clearly demonstrated the clinical efficacy of IVIg by inhibiting complement at the C3 level [37]. In that study, IVIg exhibited rapid formation of complexes with C3b and effectively inhibited the consumption of C3 as early as two days after infusion, intercepting the formation of MAC in the muscles of patients by reducing the assembly of C5 convertase. These effects were demonstrated in the serum of patients with DM who were randomly assigned to receive IVIg and repeated muscle biopsies, in correlation with clinical improvement [36, 37].
ASS-overlapping myositis (ASS-OM)
Clinical features
ASS-OM is defined by the presence of antibodies against aminoacyl tRNA synthetases (ARSs). ARS enzymes are responsible for catalyzing protein translation. Anti-ARS antibodies have been identified, including eight types of ARS (Jo-1, PL-7, PL-12, OJ, EJ, KS, Zo, and Ha). Cases positive for these antibodies exhibit a set of common clinical symptoms, such as muscle involvement, interstitial lung disease, and mechanic hands. As a result, they are collectively referred to as antisynthetase syndrome.
Morphological characteristics of the muscle biopsy specimens (Fig. 2)
ASS-OM is a distinct entity with unique histological characteristics, exhibiting necrotizing features in the perifascicular areas that differ from perifascicular lesions commonly observed in DM. This finding is referred to as perifascicular necrosis. In addition, it is often accompanied by perimysial connective tissue fragmentation and alkaline phosphatase activity in the perifascicular sheath [38, 39]. However, these pathological features have also been observed in DM, especially anti-Mi-2 DM [40–43]. Therefore, the detection of anti-ARS or DM-specific autoantibodies supports the differential diagnosis between ASS and DM. MHC class I is strongly expressed in the periphery of muscle fibers, and there are cases associated with a high expression of MHC class II. A recent report suggested that myofiber HLA-DR expression is a diagnostic marker in ASS (36.8% in ASS), suggesting the activation of IFN type 2 signaling [43–45] (Table 1). Complement deposition is often observed in cases with sarcolemma [46].
Pathogenesis role of complement and anti-Jo-1 antibody in ASS
Antibodies targeting ARS are found in approximately 20% -30% of patients with IIM, with histidyl-transfer RNA synthetase (anti-Jo-1) being the most prevalent, accounting for 75% of cases. However, the pathogenic mechanisms underlying the activities of these antibodies, which target widely distributed cytoplasmic antigens, remain unclear.
Using the human muscle microvascular endothelial cell (HMMEC) line, our recent study suggested that IgG from Jo-1 antibody-positive patients caused complement-dependent cellular cytotoxicity in HMMECs [21]. The attachment of Jo-1 antibodies to Jo-1 embedded in the cell membrane of muscle capillaries can lead to complement deposition, resulting in the lysis of endomysial capillaries and muscle ischemia. This process ultimately causes perifascicular necrosis, which is a characteristic feature observed in patients with myositis who are positive for Jo-1 antibodies. Interestingly, complement-mediated cell death in HMMECs does not occur with IgG in DM patients. This suggests that the pathogeneses of DM and ASS-OM may differ.
IMNM
Concept of disease
IMNM is a disease concept based on muscle pathology, characterized by muscle fiber atrophy and size variation as well as prominent muscle fiber necrosis and regeneration, with minimal inflammatory cell infiltration. This was proposed in the 2004 ENMC workshop [5]. Various disorders can cause necrotizing myopathy, but the majority are immune-mediated, with anti-SRP and anti-HMGCR antibodies being the representative autoantibodies.
Role of complements in IMNM
In the muscle pathology of SRP antibody-positive and HMGCR antibody-positive myopathies, complement deposition on the muscle fibers and the expression of SRP and HMGCR were observed on the sarcolemma (Fig. 2). It has been speculated that complement activation plays a significant role in pathogenesis based on the predominance of the IgG1 isotype, which is involved in complement activation via the classical pathway, accounting for approximately 80% of anti-SRP antibodies, and is present in all cases of anti-HMGCR antibodies [47, 48]. Recently, further investigations and verifications of this hypothesis have been conducted.
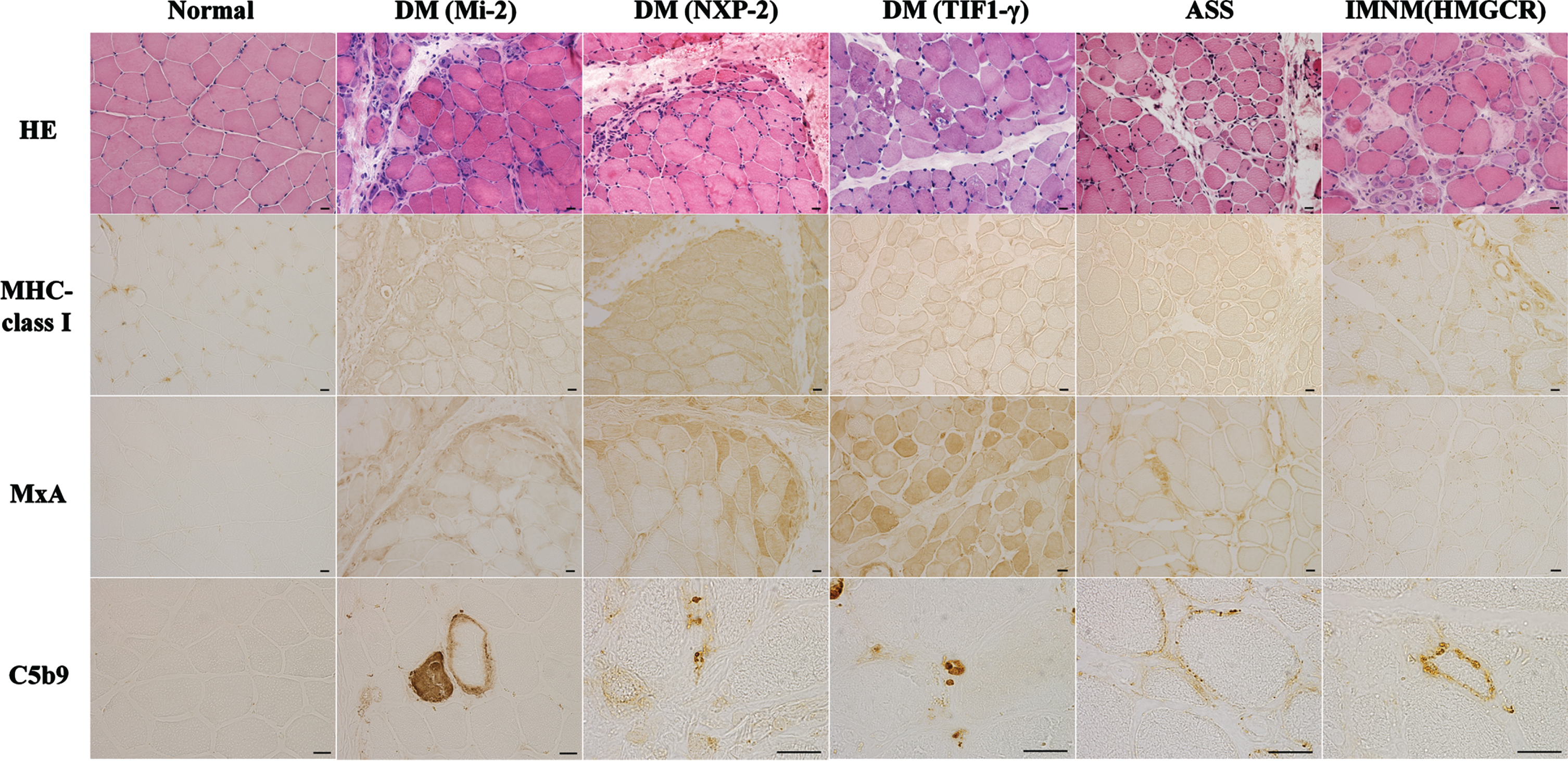
Fig. 2
Represent muscle biopsy findings in different idiopathic inflammatory myopathies subtypes based on antibody subtypes. Hemotoxylin and eosin (HE) staining shows perifascicular necrosis and atrophy in anti-Mi-2 dermatomyositis (DM) and anti-NXP2 DM, perifascicular atrophy and vacuolated punched-out fibers in anti-TIF1-γ DM, myofiber necrosis and regeneration in anti-synthetase syndrome (ASS) and immune-mediated necrotizing myopathy (IMNM). Positivity of HLA-ABC immunostaining is observed in anti-Mi-2 DM, anti-NXP2 DM, anti-TIF1-γ DM, ASS and IMNM. Sarcoplasmic Myxovirus Resistance Protein A (MxA) positivity in perifascicular is observed in anti-Mi-2 DM, anti-NXP2 DM and anti-TIF1-γ DM. Deposition of C5b9 is present in capillaries in anti-NXP2 DM and anti-TIF1-γ DM and in muscle fiber membrane in anti-Mi-2 DM, ASS and anti-HMGCR IMNM. Scale bar = 20μm.
When purified IgG from patient sera is administered to complement C3-deficient and wild-type mice, there is a significant reduction in muscle weakness in C3-deficient mice, and the co-administration of purified IgG with human C3 exacerbates muscle weakness [49]. These findings suggest that SRP and HMGCR antibodies recognize SRP and HMGCR expressed on the muscle membrane and directly impair muscle fibers through complement activation via the classical pathway. Based on this mechanism, therapies that target plasma exchange and complement inhibitors may be effective.
In experiments where zilucoplan, a targeted peptide inhibitor of complement component 5, was administered to humanized mouse models of IMNM, early administration prevented muscle weakness and reduced C5b-9 deposition on the muscle fibers [50]. Based on these findings, a phase 2 randomized placebo-controlled clinical trial of zilucoplan was conducted [ClinicalTrials.gov identifier: NCT04025632]. However, it did not demonstrate significant clinical efficacy in patients with IMNM, and the study was terminated early.
RECENT FINDINGS OF MICROANGIOPATHY IN JUVENILE DM (JDM)
JDM is the most common idiopathic inflammatory myopathy affecting the skin, muscles, and vital organs. Vasculopathic changes, including loss of capillaries, perivascular inflammation, and small vessel angiopathies, have been observed [51, 52]. The deposition of anti-endothelial cell antibodies, immune complexes, and complement is considered to play a prominent role in endothelial damage and subsequent vasculopathy [53–58]. The severity of vasculopathy in the muscle reflects the severity of systemic vasculopathy and the clinical severity in JDM patients [56, 59].
The co-expression of MxA and VCAM-1 in muscle endothelial cells suggests the activation of the IFN1 pathway in JDM patients with vasculopathy [60, 61]. After the overexpression of MHC-1 and MHC-2 on monocyte and endothelial cell activation, several cytokines and chemokines are released, resulting in inflammatory cell infiltration, including CD4 + T cells, CD8 + T cells, B cells, mature plasmacytoid dendritic cells (pDCs), monocytes, muscle compartments, and the formation of lymphoid-like structures [62–65]. pDCs are the major source of IFN1, which explains the IFN1 signature in biopsied muscles in JDM. Typical IFN-inducible chemokines, such as CXCL10 and CXCL11, are secreted by muscle endothelial cells, subsequently inducing inflammatory cell infiltration into the muscle, and are associated with the severity of vasculopathy [20, 66–69]. CXCR3, VCAM-1, and ICAM-1 expression in muscle endothelial cells reflects endothelial cell activation, facilitating the attraction and invasion of immune cells into the muscle [65].
In addition to JDM, vasculopathic changes have been observed in adults with DM. Ultrastructural studies have shown abnormalities in capillary endothelial cells in both the arterioles and veins [34, 70]. Capillary density loss in the area of perifascicular atrophy and C5-9 complement MAC deposits in capillary endothelial cells have been observed [34]. Focal vasculopathy and capillary loss cause ischemia in the perifascicular lesions, possibly resulting in perifascicular atrophy [34, 71].
Several serum biomarkers have been investigated to detect the disease activity and tissue involvement in JDM, as follows: (1) markers related to the IFN1 signature, including Eotaxin, MCP-1, CXCL10, CXCL11, IL-10, galectin-9 and neopterin [72–75]; (2) markers related to endothelial activation, including vWF, C3d, fibrinopepetide A, sVCAM-1 and sICAM-1 [76–80]; (3) other inflammatory mediators, such as soluble IL-2 R, IL-6, IL-8, TNF-α and ΔBAFF [81–84]; and (4) circulating immune cell subsets, including CD4 + T cells, CD8 + T cells, B cells, Th17 cells, and follicular helper T cells [85, 86], which reflect the disease activity and were increased in comparison to the controls (Fig. 3).
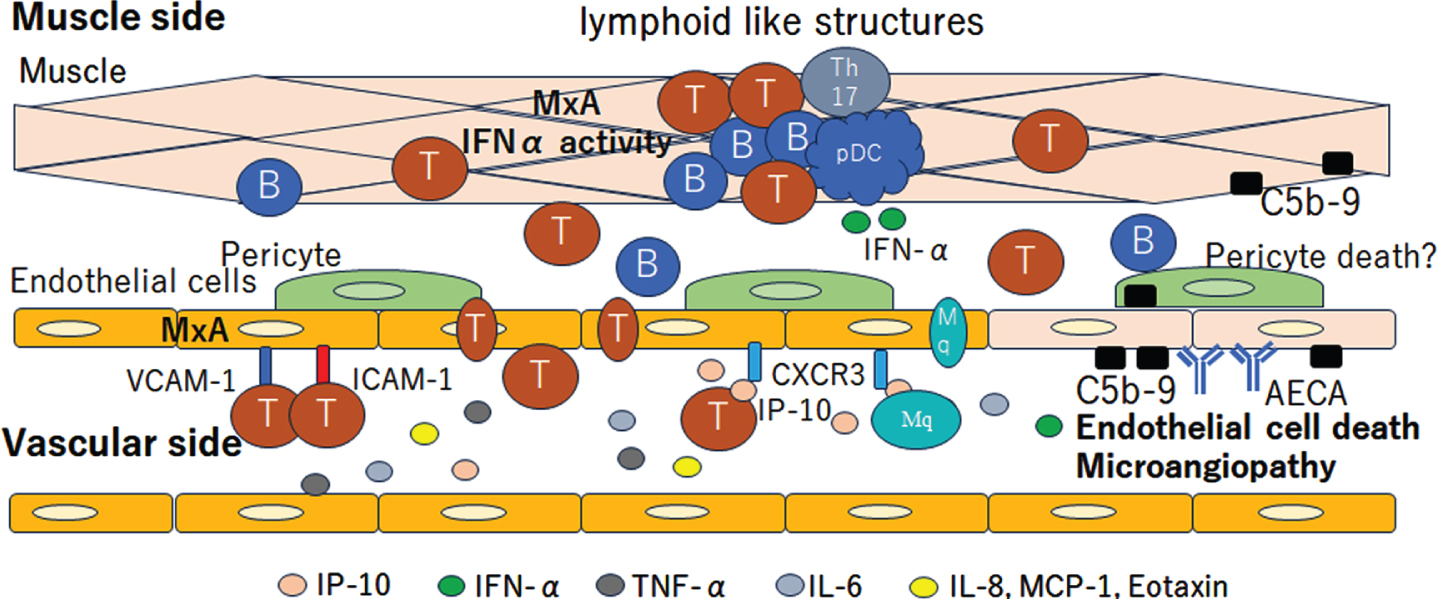
Fig. 3
Vasculopathic change in Juvenile dermatomyositis (JDM). The deposition of anti-endothelial cell antibodies (AECA) and C5b-9 complement complex cause endothelial damage and subsequent vasculopathy. The co-expression of MxA and VCAM-1 on muscle endothelial cells suggest the activation of the IFN1 pathway. The CXCR3, VCAM-1 and ICAM-1 expression in muscle endothelial cells reflects the endothelial cell activation, facilitating the attraction and invasion of immune cells into the muscle. CD4+ T cells, CD8+ T cells, B cells, Th17 cells, mature plasmacytoid dendritic cells (pDC) and monocytes infiltrate into the muscle compartments and form lymphoid like structures. The pDC is the major source of IFN1, which explains the IFN1 signature in biopsied muscle in JDM. Typical IFN-inducible chemokines, such as CXCL10 (IP-10) and CXCL11, are secreted by muscle endothelial cells. Cytokines related to the IFN1 signature, including Eotaxin, MCP-1, CXCL10, CXCL11, IL-10, galectin-9 and neopterin, are increased on the vascular side. T, T cells; B, B cells; Th17, T helper 17 cell.
INFLAMMATORY MARKERS OF IIM
MxA
Over a decade ago, interferon-stimulated genes associated with type I interferon (IFN-I) were found to be upregulated in the muscle tissues of patients with DM, accompanied by elevated levels of IFN-β and IFN-α in the serum [87, 88]. The severity of DM was also correlated with serum levels of IFN-inducible proteins. Immunostaining with antibodies against proteins, such as MxA, induced by IFN-I revealed positive staining in the muscle fibers and small blood vessels in the perifascicular and PFA regions of DM cases [89]. In contrast, PFN was observed in cases that were positive for anti-ARS antibodies. In PFN, muscle fibers and blood vessels in the perifascicular regions do not exhibit staining for MxA, and an enhanced expression of non-necrotic muscle fiber membrane-associated MHC class II, which is not observed in DM, has been observed [37, 39]. Exceptionally, anti-Mi-2 positive DM patients exhibit PFN, but myofibers are positive for MxA.
Based on a recent comprehensive gene expression analysis in peripheral blood, skeletal muscles, and skin, it was revealed that DM is a type I interferonopathy characterized by the upregulation of downstream genes of IFN-I, regardless of whether it occurs in adults or children [90]. Furthermore, tubuloreticular inclusions (TRIs) have been observed in endothelial cells by electron microscope [91]. TRIs are formed through the action of IFN-I. In contrast, in anti-ASS-OM patients, although an increased expression of IFN-I-related genes is observed in some cases and tissues, activation of IFN-II genes, centered around IFN-γ, has been observed [43, 92].
TREM-1
Recently, we first reported that IgGs from patients with IIM, including Jo-1-ASS, SRP-IMNM, and PM, were found to induce upregulation of triggering receptor expressed on myeloid cells (TREM)-1 and its expression in the perimysial blood vessels of DM patients as well as in the muscle of patients with DM, SRP-IMNM, and Jo-1-ASS [21]. This suggests that TREM-1 plays a role in triggering inflammation and promoting the migration of inflammatory cells by inducing the secretion of proinflammatory cytokines, such as TNF-α and chemokines, in individuals with IIM associated with Jo-1-ASS, DM, or SRP-IMNM.
TREM-1 is a receptor expressed in myeloid cells, such as macrophages and neutrophils, and enhances inflammatory responses by collaborating with Toll-like receptors [93, 94]. The expression of TREM-1 is dramatically increased in skin, body fluids, and tissues infected with fungi or bacteria [95]. Furthermore, its expression is elevated in non-infectious diseases, such as pancreatitis, inflammatory bowel disease, gout, and rheumatoid arthritis. In addition, elevated levels of TREM-1 have been linked to neurodegenerative disorders, such as Alzheimer’s disease, and cerebrovascular disease [96].
The activation of TREM-1 and its signaling pathways contributes to inflammation by inducing nuclear factor-κB, cyclic adenosine monophosphate-responsive element-binding protein, activator protein 1, ETS like-1 protein signaling, and the transcription of various proinflammatory cytokines, such as TNF-α, interferon type 1, and chemokines [97]. TREM-1 exists in two forms: a membrane-bound receptor and soluble protein. The membrane-bound form of TREM-1 consists of three distinct domains: an Ig-like structure (presumably responsible for ligand binding), a transmembrane region, and a cytoplasmic tail that interacts with the adapter molecule DNAX-activating protein of 12 kDa [98]. In contrast, soluble TREM-1 (sTREM-1) lacks transmembrane and intracellular domains, making it incapable of signal transduction. The primary function of sTREM-1 is to neutralize the inflammatory activity of TREM-1 [94]. Increased levels of sTREM-1 in the bloodstream have been observed in certain diseases, including myocardial infarction, inflammatory bowel disease, and rheumatoid arthritis [96, 97].
EXPERIMENTAL MODELS FOR ANALYZING THE IIM PATHOGENESIS
As in vivo models, several animal models of experimental myositis, including experimental autoimmune myositis (EAM) and C protein-induced myositis (CIM), have been established by immunization with muscle-specific antigens, myosin, and skeletal C protein [99]. Myositis is induced in the quadriceps femoris muscles with infiltration of CD4 + T cells in the EAM or CD8 + T cells in the CIM in muscle tissue [100–105]. A Jo-1 antigen-induced ASS murine model exhibiting muscle and lung inflammation [106–108] and a TIF-1γ antigen-induced DM murine model [109] were also established. Recently, the transfer of human IgGs from patients with IMNM (HMGCR antibody-positive myopathies) was found to induce complement-mediated myositis in recipient mice [51].
A novel in vitro model was established to investigate the invasion of CD8 + T cells into myotube [110]. We recently established a novel HMMEC line and pericyte cell line from human skeletal muscle harboring the temperature-sensitive SV40 T antigen and telomerase genes. HMMECs form the blood-muscle barrier (BMB) by having as high barrier function as the human brain microvascular endothelial cell lines and expressing tight junction proteins, including claudin-5, occludin, ZO-1, and transporter [111]. Our human skeletal muscle pericyte cell (HSMPCT) line secreted GNNF and bFGF and showed an increased barrier function in HMMEC cells [112]. Organoid models consisting of skeletal muscle cells, endothelial cells, and pericytes are promising tools for analyzing the pathogenesis of IIM.
CONCLUSION
In this review, we summarize the pathogenic contributions of the complement system, microangiopathy, and inflammation in IIM. Microangiopathy plays a pathogenic role in DM, possibly causing perifascicular atrophy. The deposition of complement in muscles and capillaries is a characteristic feature of IIM, and inhibition of complement activation is a therapeutic target. The investigation of additional biomarkers, reflecting complement activation, microangiopathy, and inflammation, may lead to the discovery of novel serum surrogate markers for the diagnosis of IIM, which may have therapeutic implications. Complement-targeted therapeutics are now being developed for NMO and MG and may be promising for DM and ASS. A greater understanding of the detailed pathomechanism may help lead to the development of novel therapeutic approaches for IIM.
AUTHOR CONTRIBUTION
Masaya Honda and Fumitaka Shimizu reviewed the reference and wrote the manuscript. Masayuki Nakamori edited the manuscript. Ryota Sato prepared the pathological pictures (Figure) and review the pathological findings of IIMs. Fumitaka Shimizu and Masayuki Nakamori were responsible for planning conception and design of this review.
COMPETING INTERESTS
None.
FUNDING
Funding had no influence on the design of this research. Grants-in-Aid for Scientific Research (Kakenhi from the Japan Society for the Promotion of Science, Tokyo, Japan, Nos. 21K07416 and 21H02839).
REFERENCES
[1] | Dalakas MC . Polymyositis, dermatomyositis and inclusion-bodymyositis. N Engl J Med. (1991) ;325: :1487–98. |
[2] | Griggs RC , Askanas V , DiMauro S , Engel A , Karpati G , Mendell JR , et al., Inclusion body myositis and myopathies. Ann Neurol. (1995) ;38: :705–13. |
[3] | Targoff IN , Miller FW , Medsger TA , Oddis CV . Classification criteriafor the idiopathic inflammatory myopathies. Curr Opin Rheumatol. (1997) ;9: :527–35. |
[4] | Dalakas MC , Hohlfeld R . Polymyositis and dermatomyositis. Lancet. (2003) ;362: :971–82. |
[5] | Hoogendijk JE , Amato AA , Lecky BR , Choy EH , Lundberg IE , Rose MR , et al., 119th ENMC international workshop:trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October. 2003, Naarden, The Netherlands. Neuromuscul Disord. (2004) ;14: :337–45. |
[6] | Lloyd TE , Mammen AL , Amato AA , Weiss MD , Needham M , Greenberg SA . Evaluation and construction of diagnostic criteria for inclusionbody myositis. Neurology. (2014) ;83: :426–33. |
[7] | Lundberg IE , Tj"arnlund A , Bottai M , Werth VP , Pilkington C , Visser M , et al., 2017 European League Against Rheumatism/AmericanCollege of Rheumatology Classification Criteria for Adult andJuvenile Idiopathic Inflammatory Myopathies and Their MajorSubgroups. Ann Rheum Dis. (2017) ;76: :1955–64. |
[8] | Pestronk A . Acquired immune and inflammatory myopathies: pathologicclassification. Curr Opin Rheumatol. (2011) ;23: :595–604. |
[9] | Mariampillai K , Granger B , Amelin D , Guiguet M , Hachulla E , MaurierF , et al., Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. (2018) ;75: (12):1528–37. |
[10] | Mandel DE , Malemud CJ , Askari AD . Idiopathic InflammatoryMyopathies: A Review of the Classification and Impact of Pathogenesis. Int J Mol Sci. (2017) ;18: :1084. |
[11] | Tanboon J , Uruha A , Stenzel W , Nishino I . Where are we moving in theclassification of idiopathic inflammatory myopathies? Curr Opin Neurol. (2020) ;33: (5):590–603. |
[12] | Connors GR , Christopher-Stine L , Oddis CV , Danoff SK . Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. (2010) ;138: (6):1464–74. |
[13] | Solomon J , Swigris JJ , Brown KK . Myositis-related interstitial lungdisease and antisynthetase syndrome. J Bras Pneumol. (2011) ;37: (1):100–9. |
[14] | Tanboon J , Inoue M , Saito Y , Tachimori H , Hayashi S , Noguchi S , et al., Dermatomyositis: Muscle Pathology According to AntibodySubtypes. Neurology. (2022) ;98: (7):e739–49. |
[15] | Lundberg I . New ways to subclassify patients with myositis. J Intern Med. (2016) ;280: (1):4–7. |
[16] | Findlay AR , Goyal NA , Mozaffar T . An overview of polymyositis anddermatomyositis. Muscle Nerve. (2015) ;51: (5):638–56. |
[17] | Dalakas M.C . Inflammatory muscle diseases. N Engl J Med. (2015) ;372: (18):1734–47. |
[18] | Day J , Patel S , Limaye V . The role of magnetic resonance imagingtechniques in evaluation and management of the idiopathicinflammatory myopathies. Semin Arthritis Rheum. (2017) ;46: (5):642–9. |
[19] | Zubair AS , Salam S , Dimachkie MM , Machado PM , Roy B . Imagingbiomarkers in the idiopathic inflammatory myopathies. Front Neurol. (2023) ;14: :1146015. |
[20] | Wienke J , Deakin CT , Wedderburn LR , van Wijk F , van Royen-Kerkhof A . Systemic and Tissue Inflammation in Juvenile Dermatomyositis: FromPathogenesis to the Quest for Monitoring Tools. Front Immunol. (2018) ;9: :2951. |
[21] | Honda M , Shimizu F , Sato R , Mizukami Y , Watanabe K , Takeshita Y , et al., Jo-1 antibodies from myositis induce complement-dependentcytotoxicity and TREM-1 upregulation in muscle endothelial cells. Neurol Neuroimmunol Neuroinflamm. (2023) ;10: (4):e200116. |
[22] | Halilu F , Christopher-Stine L . Myositis-specific Antibodies:Overview and Clinical Utilization. Rheumatol Immunol Res. (2022) ;3: (1):1–10. |
[23] | Vulsteke JB , Satoh M , Malyavantham K , Bossuyt X , De Langhe E , Mahler M . Anti-OJ autoantibodies: Rare or underdetected? Autoimmun Rev. (2019) ;18: (7):658–64. |
[24] | Walport MJ . Complement. First of two parts. N Engl J Med. (2001) ;344: (14):1058–66. |
[25] | Walport MJ . Complement. Second of two parts. N Engl J Med. (2001) ;344: (15):1140–4. |
[26] | Coss SL , Zhou D , Chua GT , Aziz RA , Hoffman RP , Wu YL , et al., Thecomplement system and human autoimmune diseases. J Autoimmun. (2023) ;137: :102979. |
[27] | Dalakas MC . Complement in autoimmune inflammatory myopathies, the role of myositis-associated antibodies, COVID-19 associations, and muscle amyloid deposits. Expert Rev Clin Immunol. (2022) ;18: (4):413–423. |
[28] | Mammen AL , Allenbach Y , Stenzel W , Benveniste O . 239th ENMC International Workshop: classification ofdermatomyositis, Amsterdam, the Netherlands, 14–16 December. 2018. Neuromuscul Disord. (2020) ;30: :70–92. |
[29] | Tanboon J , Nishino I . Update on dermatomyositis. Curr Opin Neurol. (2022) ;35: (5):611–21. |
[30] | Tanboon J , Inoue M , Hirakawa S , Tachimori H , Hayashi S , Noguchi S , et al., Pathologic Features of Anti-Mi-2 Dermatomyositis. Neurology. (2021) ;96: (3):e448–e459. |
[31] | Hida A , Yamashita T , Hosono Y , Inoue M , Kaida K , Kadoya M , et al., Anti-TIF1-γ antibody and cancer-associated myositis: aclinicohistopathologic study. Neurology. (2016) ;87: (3):299–308. |
[32] | Benveniste O , Goebel HH , Stenzel W . Biomarkers in inflammatorymyopathies-an expanded definition. Front Neurol. (2019) ;10: :554. |
[33] | Kissel JT , Mendell JR , Rammohan KW . Microvascular deposition ofcomplement membrane attack complex in dermatomyositis. N Engl J Med. (1986) ;314: (6):329–34. |
[34] | Lahoria R , Selcen D , Engel AG . Microvascular alterations and therole of complement in dermatomyositis. Brain. (2016) ;139: (Pt 7):1891–903. |
[35] | Basta M , Dalakas MC . Increased in vitro uptake of thecomplement C3b in the serum of patients with Guillain-Barré syndrome, myasthenia gravis anddermatomyositis. J Neuroimmunol. (1996) ;71: (1-2):227–229. |
[36] | Basta M , Dalakas MC . High-dose intravenous immunoglobulin exerts itsbeneficial effect in patients with dermatomyositis by blockingendomysial deposition of activated complement fragments. J Clin Invest. (1994) ;94: (5):1729–1735. |
[37] | Dalakas MC , Illa I , Dambrosia JM , Soueidan SA , Stein DP , Otero C , et al., A controlled trial of high-doseintravenous immune globulininfusions as treatment for dermatomyositis. N Engl J Med. (1993) ;329: (27):1993–2000. |
[38] | Mescam-Mancini L , Allenbach Y , Hervier B , Devilliers H , Mariampillay K , Dubourg O , et al., Anti-Jo-1 antibody-positive patients show acharacteristic necrotizing perifascicular myositis. Brain. (2015) ;138: (Pt 9):2485–92. |
[39] | Stenzel W , Preusse C , Allenbach Y , Pehl D , Junckerstorff R , Heppner FL ,et al., Nuclear actin aggregation is a hallmark ofanti-synthetase syndrome-induced dysimmune myopathy. Neurolgy. (2015) ;84: (13):1346–1354. |
[40] | Uruha A , Suzuki S , Suzuki N , Nishino I . Perifascicular necrosis inantisynthetase syndrome beyond anti-Jo-1. Brain. (2016) ;139: :e50. |
[41] | Tumasyan A , Adam W , Andrejkovic JW , Bergauer T , Chatterjee S , Damanakis K , et al., Distinct tissue injury patterns in juveniledermatomyositis auto-antibody subgroups. Acta Neuropathol Commun. (2020) ;8: (1):125. |
[42] | Pestronk A , Choksi R . Pathology features of immune and inflammatorymyopathies, including a polymyositis pattern, relate strongly toserum autoantibodies. J Neuropathol Exp Neurol. (2021) ;80: (9):812–20. |
[43] | Tanboon J , Inoue M , Hirakawa S , Tachimori H , Hayashi S , Noguchi S , et al., Muscle pathology of antisynthetase syndrome according toantibody subtypes. Brain Pathol. (2023) ;33: (4):e13155. |
[44] | Rigolet M , Hou C , Baba Amer Y , Aouizerate J , Periou B , Gherardi RK , et al., Distinct interferon signatures stratify inflammatory anddysimmune myopathies. RMD Open. (2019) ;5: :e000811. |
[45] | Pinal-Fernandez I , Casal-Dominguez M , Derfoul A , Pak K , Plotz P , Miller FW , et al., Identification of distinctive interferon genesignatures in different types of myositis. Neurology. (2019) ;93: :e1193–204. |
[46] | Aouizerate J , De Antonio M , Bassez G , Gherardi RK , Berenbaum F , Guillevin L , et al., Myofiber HLA-DR expression is a distinctivebiomarker for antisynthetase-associated myopathy. Acta NeuropatholCommun. (2014) ;2: :154. |
[47] | Benveniste O , Drouot L , Jouen F , Charuel JL , Bloch-Queyrat C , Behin A , et al., Correlation of anti-signal recognition particleautoantibody levels with creatine kinase activity in patients withnecrotizing myopathy. Arthritis Rheum. (2011) ;63: (7):1961–71. |
[48] | Drouot L , Allenbach Y , Jouen F , Charuel JL , Martinet J , Meyer A , et al., Exploring necrotizing autoimmune myopathies with a novelimmunoassay for anti-3-hydroxy-3-methyl-glutaryl-CoA reductaseautoantibodies. Arthritis Res Ther. (2014) ;16: (1):R39. |
[49] | Bergua C , Chiavelli H , Allenbach Y , Arouche-Delaperche L , Arnoult C , Bourdenet G , et al., In vivo pathogenicity of IgG from patients withanti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotisingmyopathy. Ann Rheum Dis. (2019) ;78: (1):131–139. |
[50] | Julien S , Vadysirisack D , Sayegh C , Ragunathan S , Tang Y , Briand E , et al., Prevention of anti-HMGCR immune-mediated necrotising myopathyby C5 complement inhibition in a humanised mouse model. Biomedicines. (2022) ;10: (8):2036. |
[51] | Wedderburn LR , Rider LG . Juvenile dermatomyositis: new developmentsin pathogenesis, assessment and treatment. Best Pract Res ClinRheumatol. (2009) ;23: (5):665–78. |
[52] | Lundberg IE , Tjarnlund A , Bottai M , Werth VP , Pilkington C , deVisser M , et al., 2017 European League Against Rheumatism/AmericanCollege of Rheumatology classification criteria for adult andjuvenile idiopathic inflammatory myopathies and their majorsubgroups. Arthritis Rheumatol. (2017) ;69: (12):2271–82. |
[53] | Goncalves FGP , Chimelli L , Sallum AME , Marie SKN , Kiss MHB , Ferriani VPL . Immunohistological analysis of CD59 and membrane attackcomplexof complement in muscle in juvenile dermatomyositis. J Rheumatol. (2002) ;29: (6):1301–7. |
[54] | Yu HH , Chang HM , Chiu CJ , Yang YH , Lee JH , Wang LC , et al., Detectionof anti-p155/140, anti-p140, and antiendothelial cellsautoantibodies in patients with juvenile dermatomyositis. J Microbiol Immunol Infect. (2016) ;49: (2):264–70. |
[55] | Karasawa R , Tamaki M , Sato T , Tanaka M , Nawa M , Yudoh K , et al., Multiple target autoantigens on endothelial cells identified injuvenile dermatomyositis using proteomics. Rheumatology (Oxford). (2018) ;57: (4):671–6. |
[56] | Whitaker JN , Engel WK . Vascular deposits of immunoglobulin andcomplement in idiopathic inflammatory myopathy. N Engl J Med. (1972) ;286: (7):333–8. |
[57] | Kissel JT , Halterman RK , Rammohan KW , Mendell JR . The relationshipof complement-mediated microvasculopathy to the histologic featuresand clinical duration of disease in dermatomyositis. Arch Neurol. (1991) ;48: (1):26–30. |
[58] | Emslie-Smith AM , Engel AG . Microvascular changes in early andadvanced dermatomyositis: a quantitative study. Ann Neurol. (1990) ;27: (4):343–56. |
[59] | Gitiaux C , De Antonio M , Aouizerate J , Gherardi RK , Guilbert T , Barnerias C , et al., Vasculopathy-related clinical and pathologicalfeatures are associated with severe juvenile dermatomyositis. Rheumatology (Oxford). (2016) ;55: (3):470–9. |
[60] | Soponkanaporn S , Deakin CT , Schutz PW , Marshall LR , Yasin SA , Johnson CM , et al., Expression of myxovirus-resistance protein A: apossible marker of muscle disease activity and autoantibodyspecificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol. (2019) ;45: (4):410–20. |
[61] | Kim E , Cook-Mills J , Morgan G , Sredni ST , Pachman LM . Increasedexpression of vascular cell adhesion molecule 1 in muscle biopsysamples from juvenile dermatomyositis patients with short durationof untreated disease is regulated by miR-126. Arthritis Rheum. (2012) ;64: (11):3809–17. |
[62] | Li CKC , Varsani H , Holton JL , Gao B , Woo P , Wedderburn LR . MHC ClassI overexpression on muscles in early juvenile dermatomyositis. J Rheumatol. (2004) ;31: :605–9. |
[63] | Englund P , Nennesmo I , Klareskog L , Lundberg IE . Interleukin-1alphaexpression in capillaries and major histocompatibility complex classI expression in type II muscle fibers from polymyositis anddermatomyositis patients: important pathogenic features independentof inflammatory cell clusters in muscle. J Rheumatol. (2004) ;31: (3):605–9. |
[64] | Fall N , Bove KE , Stringer K , Lovell DJ , Brunner HI , Weiss J , et al., Association between lack of angiogenic response in muscle tissue andhigh expression of angiostatic ELR-negative CXC chemokines inpatients with juvenile dermatomyositis: possible link tovasculopathy. Arthritis Rheum. (2005) ;52: (10):3175–80. |
[65] | O’Connor KA , Abbott KA , Sabin B , Kuroda M , Pachman LM . MxA geneexpression in juvenile dermatomyositis peripheral blood mononuclearcells: association with muscle involvement. Clin Immunol. (2006) ;120: (3):319–25. |
[66] | Piper CJM , Wilkinson MGL , Deakin CT , Otto GW , Dowle S , Duurland CL , et al., CD19(+)CD24(hi)CD38(hi) B cells are expanded in juveniledermatomyositis and exhibit a pro-inflammatory phenotype afteractivation through toll-like receptor 7 and interferon-alpha. Front Immunol. (2018) ;9: :1372. |
[67] | Gitiaux C , Latroche C , Weiss-Gayet M , Rodero MP , Duffy D , Bader-Meunier B , et al., Myogenic progenitor cells exhibit type 1interferon-driven proangiogenic properties and molecular signatureduring juvenile dermatomyositis. Arthritis Rheumatol. (2018) ;70: (1):134–45. |
[68] | Ladislau L , Suarez-Calvet X , Toquet S , Landon-Cardinal O , Amelin D , Depp M , et al., JAK inhibitor improves type I interferon induceddamage: proof of concept in dermatomyositis. Brain. (2018) ;141: (6):1609–1621. |
[69] | Crescioli C , Sottili M , Bonini P , Cosmi L , Chiarugi P , Romagnani P , et al., Inflammatory response in human skeletal muscle cells: CXCL10as a potential therapeutic target. Eur J Cell Biol. (2012) ;91: (2):139–49. |
[70] | Lemmer D , Schmidt J , Kummer K , Lemmer B , Wrede A , Seitz C , et al., Impairment of muscular endothelial cell regeneration indermatomyositis. Front Neurol. (2022) ;13: :952699. |
[71] | De Paepe B . Vascular changes and perifascicular muscle fiber damagein dermatomyositis: another question of the chicken or the egg thatis on our mind. Ann Transl Med. (2017) ;5: (1):22. |
[72] | Sanner H , Schwartz T , Flato B , Vistnes M , Christensen G , Sjaastad I . Increased levels of eotaxin and MCP-1 in juvenile dermatomyositis median 16.8 years after disease onset; associations with disease activity, duration and organ damage. PLoS ONE. (2014) ;9: :e92171. |
[73] | Bellutti Enders F , vanWijk F , Scholman R , Hofer M , Prakken BJ , vanRoyen-Kerkhof A ,et al., Correlation of CXCL10, tumor necrosis factorreceptor type II, and galectin 9 with disease activity in juveniledermatomyositis. Arthritis Rheumatol. (2014) ;66: (8):2281–9. |
[74] | Enders FB , Delemarre EM , Kuemmerle-Deschner J , van der Torre P , Wulffraat NM , Prakken BP , et al., Autologous stem celltransplantation leads to a change in proinflammatory plasma cytokineprofile of patients with juvenile dermatomyositis correlating with disease activity. Ann Rheum Dis. (2015) ;74: (1):315–7. |
[75] | Kobayashi I , Ono S , Kawamura N , Okano M , Kobayashi K . Elevated serumlevels of soluble interleukin-2 receptor in juveniledermatomyositis. Pediatr Int. (2001) ;43: (1):109–11. |
[76] | Guzman J , Petty RE , Malleson PN . Monitoring disease activity injuvenile dermatomyositis: the role of von Willebrand factor andmuscle enzymes. J Rheumatol. (1994) ;21: (4):739–43. |
[77] | Bloom BJ , Tucker LB , Miller LC , Schaller JG . von Willebrand factorin juvenile dermatomyositis. J Rheumatol. (1995) ;22: (2):320–5. |
[78] | Scott JP , Arroyave C . Activation of complement and coagulation injuvenile dermatomyositis. Arthritis Rheum. (1987) ;30: (5):572–6. |
[79] | Xu D , Huang C-C , Kachaochana A , Morgan GA , Bonaldo MF , Soares MB , et al., MicroRNA-10a regulation of proinflammatory mediators: animportant component of untreated juvenile dermatomyositis. J Rheumatol. (2016) ;43: (1):161–8. |
[80] | Xu D , Kacha-Ochana A , Morgan GA , Huang C-C , Pachman LM . Endothelialprogenitor cell number is not decreased in 34 children with JuvenileDermatomyositis: a pilot study. Pediatr Rheumatol Online J. (2017) ;15: (1):42. |
[81] | Reed AM , Peterson E , Bilgic H , Ytterberg SR , Amin S , Hein MS , et al., Changes in novel biomarkers of disease activity in juvenile andadult dermatomyositis are sensitive biomarkers of disease course. Arthritis Rheum. (2012) ;64: (12):4078–86. |
[82] | Gabay C , Gay-Croisier F , Roux-Lombard P , Meyer O , Maineti C , Guerne PA , et al., Elevated serum levels ofinterleukin-1 receptor antagonistin polymyositis/dermatomyositis. A biologic marker of diseaseactivity with apossible role in the lack of acute-phase proteinresponse. Arthritis Rheum. (1994) ;37: (12):1744–51. |
[83] | Krystufkova O , Vallerskog T , Helmers SB , Mann H , Putova I , Belacek J , et al., Increased serum levels of B cell activating factor (BAFF)in subsets of patients with idiopathic inflammatory myopathies. AnnRheum Dis. (2009) ;68: (6):836–43. |
[84] | Lopez De Padilla CM , McNallan KT , Crowson CS , Bilgic H , Bram RJ , Hein MS , et al., BAFF expression correlates with idiopathicinflammatory myopathy disease activity measures and autoantibodies. J Rheumatol. (2013) ;40: (3):294–302. |
[85] | Ishida T , Matsumoto Y , Ohashi M , Sasaki R . Analysis of lymphocytesubpopulations in peripheral blood in adult and juvenile cases ofdermatomyositis. J Dermatol. (1993) ;20: (1):30–4. |
[86] | Aleksza M , Szegedi A , Antal-Szalmas P , Irinyi B , Gergely L , Ponyi A , et al., Altered cytokine expression of peripheral blood lymphocytesin polymyositis and dermatomyositis. Ann Rheum Dis. (2005) ;64: (10):1485–9. |
[87] | Greenberg SA , Pinkus JL , Pinkus GS , Burleson T , Sanoudou D , Tawil R , et al., Interferon-alpha/beta-mediated innate immune mechanisms indermatomyositis. Ann Neurol. (2005) ;57: (5):664–678. |
[88] | Suarez-Calvet X , Gallardo E , Nogales-Gadea G , Querol L , Navas M , Diaz-Manera J , et al., Altered RIG-I/DDX58-mediated innate immunityin dermatomyositis. J Pathol. (2014) ;233: (3):258–268. |
[89] | Uruha A , Nishikawa A , Tsuburaya R , Hamanaka K , Kuwana M , Watanabe Y , et al., Sarcoplasmic MxA expression: A valuable marker ofdermatomyositis. Neurology. (2017) ;88: (5):493–500. |
[90] | Bolko L , Jiang W , Tawara N , Landon-Cardinal O , Anquetil C , Benvensite O , et al., The role of interferons type I, II and III inmyositis: A review. Brain Pathol. (2021) ;21: (3):e12955. |
[91] | Visser MD , Emslie-Smith AM , Engel AG , et al., Early ultrastructuralalterations in adult dermatomyositis. Capillary abnormalitiesprecede other structural changes in muscle. J Neurol Sci. (1989) ;94: :181–192. |
[92] | Gallay L , Mouchiroud G , Chazaud B . Interferon-signature inidiopathic inflammatory myopathies. Curr Opin Rheumatol. (2019) ;31: (6):634–642. |
[93] | Bouchon A , Dietrich J , Colonna M . Cutting edge: inflammatoryresponses can be triggered by TREM-1, a novel receptor expressed onneutrophils and monocytes. J Immunol. (2000) ;164: (10):4991–4995. |
[94] | Klesney-Tait J , Turnbull IR , Colonna M . The TREM receptor family andsignal integration. Nat Immunol. (2006) ;7: (12):1266–1273. |
[95] | Bouchon A , Facchetti F , Weigand MA , Colonna M . TREM-1 amplifiesinflammation and is a crucial mediator of septic shock. Nature. (2001) ;410: (6832):1103–1107. |
[96] | Natale G , Biagioni F , Busceti CL , Gambardella S , Limanaqi F , Fornai F . TREM receptors connecting bowel inflammation to neurodegenerativedisorders. Cells. (2019) ;8: (10):1124. |
[97] | Tammaro A , Derive M , Gibot S , Leemans JC , Florquin S , Dessing MC . TREM-1 and its potential ligands in non-infectious diseases: frombiology to clinical perspectives. Pharmacol Ther. (2017) ;177: :81–95. |
[98] | Colonna M . TREMs in the immune system and beyond. Nat Rev Immunol. (2003) ;3: (6):445–453. |
[99] | Konishi R , Ichimura Y , Okiyama N . Murine models of idiopathicinflammatory myopathy. Immunol Med. (2023) ;46: (1):9–14. |
[100] | Matsubara S , Okumura S . Experimental autoimmune myositis in SJL/J mice produced by immunization with syngeneic myosin B fraction. Transfer by both immunoglobulin G and T cells. J Neurol Sci. (1996) ;144: (1–2):171–5. |
[101] | Rosenberg NL , Ringel SP , Kotzin BL . Experimental autoimmune myositisin SJL/J mice. Clin Exp Immunol. (1987) ;68: (1):117–29. |
[102] | Rosenberg NL , Kotzin BL . Aberrant expression of class II MHCantigens by skeletal muscle endothelial cells in experimentalautoimmune myositis. J Immunol. (1989) ;142: (12):4289–94. |
[103] | Kohyama K , Matsumoto Y . C-protein in the skeletal muscle inducessevere autoimmune polymyositis in lewis rats. J Neuroimmunol. (1999) ;98: (2):130–5. |
[104] | Sugihara T , Okiyama N , Suzuki M , Kohyama K , Matsumoto Y , Miyasaka N , et al., Definitive engagement of cytotoxic CD8 T cells in Cprotein-induced myositis, a murine model of polymyositis. ArthritisRheum. (2010) ;62: (10):3088–92. |
[105] | Okiyama N , Hasegawa H , Oida T , et al., Experimental myositisinducible with transfer of dendritic cells presenting a skeletalmuscle C protein-derived CD8 epitope peptide. Int Immunol. (2015) ;27: (7):327–32. |
[106] | Katsumata Y , Ridgway WM , Oriss T , Gu X , Chin D , Wu Y , et al., Species-specific immune responses generated by histidyl-tRNAsynthetase immunization are associated with muscle and lunginflammation. J Autoimmun. (2007) ;29: (2-3):174–86. |
[107] | Soejima M , Kang EH , Gu X , Katsumata Y , Clemens PR , Ascherman DP . Role of innate immunity in a murine model ofhistidyl-transfer RNAsynthetase (Jo-1)-mediated myositis. Arthritis Rheum. (2011) ;63: (2):479–87. |
[108] | Galindo-Feria AS , Albrecht I , Fernandes-Cerqueira C , Notarnicola A , James EA , Herrath J , et al., ProinflammatoryHistidyl-transfer RNAsynthetase-specific CD4lb T cells in the blood and lungs of patients with idiopathicinflammatory myopathies. ArthritisRheumatol. (2020) ;72: (1):179–91. |
[109] | Okiyama N , Ichimura Y , Shobo M , Tanaka R , Kubota N , Saito A , et al., Immune response to dermatomyositis-specific autoantigen, transcriptional intermediary factor 1gamma can result inexperimental myositis. Ann Rheum Dis. (2021) ;80: (9):1201–8. |
[110] | Kamiya M , Mizoguchi F , Takamura A , Kimura N , Kawahata K , Kohsaka H . A new in vitro model of polymyositis reveals CD8+T cellinvasion into muscle cells and its cytotoxic role. Rheumatology (Oxford). (2020) ;59: (1):224–32. |
[111] | Sano H , Sano Y , Ishiguchi E , Shimizu F , Omoto M , Maeda T , et al., Establishment of a new conditionally immortalized human skeletalmuscle microvascular endothelial cell line. J Cell Physiol. (2017) ;232: (12):3286–95. |
[112] | Ishiguchi E , Sano Y , Maeda T , Shimizu F , Fujisawa M , Honda M , et al., Glial cell line-derived neurotrophic factor and basic fibroblastgrowth factor derived from skeletal musclepericytes increase the barrier function of endothelial cells in theendomysium. Clin Exp Neuroimmunol. (2021) ;12: :258–67. |


