
Considering the Promise of Vamorolone for Treating Duchenne Muscular Dystrophy
Abstract
This commentary provides an independent consideration of data related to the drug vamorolone (VBP15) as an alternative steroid proposed for treatment of Duchenne muscular dystrophy (DMD). Glucocorticoids such as prednisone and deflazacort have powerful anti-inflammatory benefits and are the standard of care for DMD, but their long-term use can result in severe adverse side effects; thus, vamorolone was designed as a unique dissociative steroidal anti-inflammatory drug, to retain efficacy and minimise these adverse effects. Extensive clinical trials (ongoing) have investigated the use of vamorolone for DMD, with two trials also for limb-girdle muscular dystrophies including dysferlinopathy (current), plus a variety of pre-clinical trials published. Vamorolone looks very promising, with similar efficacy and some reduced adverse effects (e.g., related to height) compared with other glucocorticoids, specifically prednisone/prednisolone, although it has not yet been directly compared with deflazacort. Of particular interest to clarify is the optimal clinical dose and other aspects of vamorolone that are proposed to provide additional benefits for membranes of dystrophic muscle: to stabilise and protect the sarcolemma from damage and enhance repair. The use of vamorolone (and other glucocorticoids) needs to be evaluated in terms of overall long-term efficacy and cost, and also in comparison with many candidate non-steroidal drugs with anti-inflammatory and other benefits for DMD.
INTRODUCTION
Vamorolone is a new dissociative steroid drug that aims to retain or improve the therapeutic benefit of traditional glucocorticoids while reducing severe adverse side effects associated with long-term administration. This topic is of much interest for young people with Duchenne muscular dystrophy (DMD) who are typically treated with glucocorticoids over many years. DMD is an inherited X chromosome-linked muscle disorder, manifesting in young children and affecting mainly males, with rapid loss of mass and function of most skeletal muscles. Symptoms are evident around 2–3 years of age and many boys become wheelchair dependent around 10–12 years. Further complications include progressive denervation of muscles, scoliosis, contractures, and increasing respiratory and cardiac complications with age, but with modern interventions and care the lifespan of patients with DMD can be extended to about 20–40 years [1]. The affected DMD gene codes for a muscle isoform of the protein dystrophin (Dp427m). In skeletal muscles, dystrophin is located beneath the cell membrane (sarcolemma) and is the major intracellular component of the transmembrane dystrophin-glycoprotein complex that mechanically links the actin cytoskeleton of muscle fibres (myofibres) to the surrounding extracellular matrix (ECM), particularly laminin and various collagens [1]. This mechanical linkage enables the transmission of force generated by contractile proteins (organised into sarcomeres) within individual myofibres to the whole muscle and tendons, resulting in the movement of different body parts. In the absence of a functional dystrophin protein, the sarcolemma of dystrophic myofibres is vulnerable to mechanical and other damage in response to force generated by the contractile filaments; this damage escalates to myofibre breakdown and necrosis (myonecrosis), which is pronounced in fast contracting myofibres [2]. There is also a much milder form of DMD, with later onset called Becker muscular dystrophy (BMD), that results from different mutations of the DMD gene [1].
Myonecrosis and dystropathology
A key cause of the severe dystropathology of DMD is the high incidence of intrinsic myonecrosis, which is also a feature of many animal models of DMD, including dystrophic mdx mice, rats, dogs, and other species [3]. The onset of myonecrosis is linked with calcium dysregulation and rapid accumulation of neutrophils (that are transitory) followed by other inflammatory cells including macrophages, associated with high levels of the major pro-inflammatory cytokine tumour necrosis factor [4, 5]. Inflammatory cells produce high levels of reactive oxygen species that oxidise diverse proteins and other macromolecules and can be very damaging to cellular components; for example, neutrophils produce hypochlorous acid that is far more cytotoxic than oxidants like hydrogen peroxide [6]. Myonecrosis is characterised by complete breakdown of segments of myofibres (clustered in small groups, called focal necrosis) and the presence of many inflammatory cells within these necrotic myofibres [6, 7]. Increased reactive oxygen species results in oxidation of many proteins, observed to be localised specifically in foci of myonecrosis in dystrophic mdx muscles [8]. When myonecrosis occurs, inflammatory cells are essential for phagocytosis of the necrotic muscle tissue, remodelling of ECM, and activation of myogenesis, all required for successful regeneration and formation of new muscle cells to repair the damaged myofibre segment. Such myonecrosis/regeneration requires high energy and takes between 1–2 weeks to restore function of the affected myofibres [9]. Intrinsic myonecrosis occurs repeatedly in dystrophic muscles, with associated asynchronous bouts of inflammation progressively increasing fibrosis [10]; this myonecrosis appears to be exacerbated by growth (where energy demands are already high) [11]. Thus, a primary objective of drugs that aim to reduce the severity of dystropathology in DMD, is to protect dystrophic myofibres from myonecrosis, by stabilising the sarcolemma and calcium dysregulation, with a particular focus on preventing excessive inflammation and oxidative stress. There is strong support for inflammation as a key drug target for DMD from many studies in humans and dystrophic mice and dogs [5, 7, 12]. Studies using RNA analyses of human DMD and control muscles from foetopsies, infants (aged 8–10 months), and symptomatic patients (aged 5–12 years), emphasise that the inflammatory pathways with strong induction of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) are pronounced in the early pre-symptomatic stages of DMD [13].
Therapies for DMD
The ideal therapy for DMD and BMD is to restore functional dystrophin protein in the muscles with many strategies under investigation. One approach is to replace or correct the defective DMD gene (theoretically applicable to most DMD boys), others target gene transcription to normalise the production of dystrophin protein using specifically engineered molecules to target different mutations (e.g., for exon-skipping strategy) and thus is applicable to only sub-groups of DMD boys initially [1, 14, 15]. While many of these potential therapies produce promising results in pre-clinical studies in dystrophic animals, successful clinical translation remains challenging, and they are not yet available for the wide DMD population [14]. Hence, the quest for drugs to ameliorate the severity of this progressive disease continues to attract much attention. The classic anti-inflammatory drugs to treat DMD are glucocorticoid steroids, such as prednisone/prednisolone and deflazacort, that can extend muscle function, ambulation, and life expectancy, although such chronic long-term use for DMD has severe adverse side effects [16, 17]. Thus, the question arises: ‘is there a better drug for DMD?’ [18].
This commentary is focussed on a new modified steroid drug vamorolone, also known as VBP15, that was selected for its strong anti-inflammatory activity with reduced adverse side effects, compared with the classic glucocorticoids like prednisone/prednisolone and deflazacort [19]. Vamorolone is structurally related to glucocorticoids but contains an essential D-9,11 double bond modification to the steroid C-ring, with distinct differences in sub-activity profiles, as described previously [19, 20]. In brief, like other glucocorticoids, vamorolone binds to the glucocorticoid receptor and suppresses transcription of the NF-κB signalling pathway (transrepression). Importantly, vamorolone appears to avoid much of the broad transcriptional activity (transactivation) associated with the many adverse side effects of other glucocorticoids, and vamorolone also functions as an antagonist (instead of an agonist) of mineralocorticoid receptor activity [21]: discussed further below.
GLUCOCORTICOID STEROIDS
The glucocorticoid steroids (e.g., prednisone/prednisolone, deflazacort, and dexamethasone) diffuse through the cell membrane and bind to the cytoplasmic glucocorticoid receptor, this complex interacts with other proteins with non-genomic and genomic consequences. Genomic effects appear after the glucocorticoid receptor complex translocates into the nucleus where it binds glucocorticoid response elements (GREs) in DNA, either by itself or in concert with co-factors, to activate (transactivate) or repress (transrepress) expression of many (∼10–20%) genes [22]: for example, transrepression suppresses transcription of NF-κB signalling to exert the well-known potent anti-inflammatory effects. In addition, many glucocorticoids can interact with the structurally similar mineralocorticoid receptor and androgen receptor: the great complexity of this topic is covered by many excellent reviews [17, 22–24]. Unfortunately, long-term use of such synthetic steroids (e.g., throughout the lifespan for DMD) can have severe adverse side effects, including growth impairment, osteoporosis and poor bone health, hyperglycaemia and metabolic syndrome, hypertension, arrhythmias, skin thinning, cushingoid appearance, and development of tissue-specific glucocorticoid resistance [17, 22]. Both vamorolone and deflazacort were designed to avoid/reduce these major adverse effects.
To clarify the use of the terms prednisone and prednisolone, it is noted that prednisone is converted by the liver to the active form prednisolone (provided the liver is functional) with in vivo administration of prednisone and prednisolone having similar effects in mdx mice [25]. Prednisone is widely administered clinically, whereas prednisolone (or methylprednisolone) is more routinely used for animal studies.
Both prednisone and deflazacort have been used extensively to treat DMD, and a recent review [26] concluded that “patients receiving deflazacort experience similar or slower rates of functional decline compared with those receiving prednisone/prednisolone. Regarding side-effects, weight gain and behavior side effects appear to be greater with prednisone/prednisolone than with deflazacort, whereas bone health, growth parameters, and cataracts appear worse with deflazacort.” (abstract). An earlier review of deflazacort for DMD [27] concluded that “deflazacort presents an additional, FDA-approved corticosteroid option for patients that offers improved quality of life for DMD patients. However, there is weak evidence to support these benefits . . . ” (abstract). A third study that compared the effectiveness and cost of deflazacort and prednisone concluded that “Current evidence demonstrates comparable or perhaps better clinical benefit of deflazacort versus prednisone” (p. 365) [28].
Frequency of glucocorticoid administration: daily vs intermittently
There is much interest in different dosing regimens for glucocorticoids through either daily or intermittent (once every 7–10 days) administration [17, 26], with controversy related to balancing efficacy with adverse effects and recent clinical trials endorsing daily administration [29, 30]. In marked contrast with the benefits of prednisone and deflazacort for DMD, in dysferlinopathy, known as limb-girdle muscular dystrophy (LGMD) type R2 dysferlin-related (LGMDR2) and Miyoshi myopathy, daily glucocorticoid administration has unexpected adverse effects [31–33]. The clinical trial for dysferlinopathy used deflazacort treatment (1 mg/kg/d) versus placebo over 6 months (NCT00527228, completed), with daily administration in the first month and then every second day for the remaining 5 months [32]. Another clinical trial is testing daily deflazacort (0.6 mg/kg/d) versus placebo in LGMDR13 fukutin-related (NCT03783923, in progress). To specifically evaluate the proposed benefits of once-weekly glucocorticoid treatment (rather than daily), a clinical trial (NCT04054375, completed) administered oral prednisone (0.75 mg/kg) once a week (in the evening) to patients where glucocorticoids are not routinely administered therapeutically, including BMD, dysferlinopathy, and six other forms of LGMD. Benefits of such once-a-week delivery of glucocorticoids in various muscular dystrophies are supported by pre-clinical studies in the mdx mouse model of DMD/BMD and two models of LGMD, dysferlinopathy (LGMDR2) and sarcoglycanopathy (LGMDR5 γ-sarcoglycan-related) [34–36]: these studies provide valuable insight into the mechanistic basis for the benefits of once-a-week glucocorticoid treatment.
Timing of drug delivery in human and rodent studies: day or night?
Another issue to consider is the time of day when the drug is delivered, which can influence the pharmacokinetics of drug absorption/persistence and breakdown, with possible impact on both beneficial and adverse effects, due to strong circadian rhythms that regulate activity, feeding, metabolism, and other signalling (chronobiology). A study using an algorithm to evaluate the dosing time for prednisolone [37], emphasised that circadian patterns are also observed for cortisol and blood lymphocytes in plasma (two routine biomarkers of glucocorticoid activity), and that such dosage timing can influence clinical outcomes with fewer side effects of prednisolone. For most clinical trials, glucocorticoids are provided in the morning; however, for the clinical trial where prednisone was administered once a week (NCT04054375), this was done in the evening. Mice have the opposite circadian rhythms to humans (since they are nocturnal with active/feeding phase at night), so the consequences of drug delivery in the morning differ between mice and men; this lack of equivalence between many clinical and pre-clinical studies needs to be considered.
CRITICAL EVALUATION OF VAMOROLONE FOR DMD
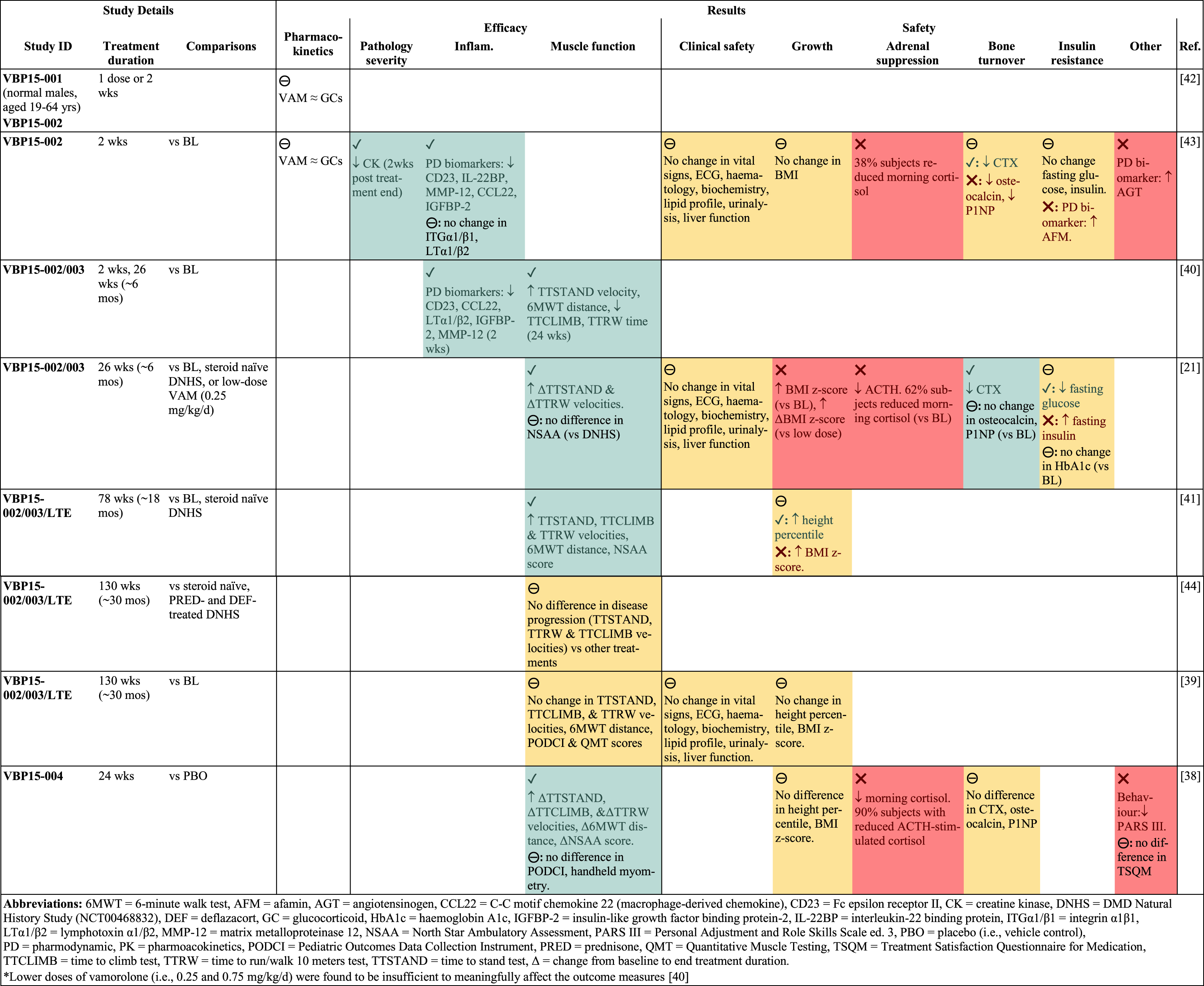
Vamorolone/VBP15 was developed in collaboration with scientists based in Washington USA and the company ReveraGen, with a series of recent clinical trials for DMD (four completed to date) and associated publications [21, 38–45]; summarised in Table 1. These trials have produced promising results; discussed below and summarised in Table 2. There are also strong data from pre-clinical studies with vamorolone (often compared with prednisolone) in the classic mdx mouse model for DMD [10, 20, 46–48], along with models of other diseases [49–56]; summarised in Table 3.
Table 1
Clinical trials design for vamorolone in healthy and Duchenne and Becker muscular dystrophy subjects. In all trials, vamorolone was administered orally as a cherry-flavoured suspension (4% by weight), and in the Phase 2 trials this vamorolone was administered along with 8 oz of whole milk (or equivalent high-fat food portion). * indicates completed. Published clinical studies arising from these trials are shown (see Ref.)
| Study ID (NCT number) | Title | Details | Ref. |
| VBP15-001* (NCT02415439) | A Phase 1 SAD and MAD Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of VBP15 in Healthy Adult Subjects | Phase 1. SAD: Male healthy adults aged 18–65 yrs (n = 54)Treatment: Vamorolone single dose 0.1, 0.3, 1, 3, 8, 20 mg/kg or placebo.Phase 1. MAD: Male healthy adults aged 18–65 yrs (n = 32)Treatment: daily vamorolone 1, 3, 9, 20 mg/kg/d or placeboDuration: 2 wks | [42, 57] |
| VBP15-002* (NCT02760264) | A Study to Assess Vamorolone in Boys With DMD | Phase 2A. Male DMD patients aged 4-<7 yrs (n = 48): steroid naïveTreatment: daily vamorolone 0.25, 0.75, 2, 6 mg/kg/dDuration: 2 wks | [39, 40, 42–45] |
| VBP15-003* (NCT02760277) | An Extension Study to Assess Vamorolone in Boys With DMD | Phase 2A. Male DMD patients aged 4–7 yrs (n = 48): steroid naïveIntervention: daily vamorolone 0.25, 0.75, 2, 6 mg/kg/dDuration: 24 wks (∼6 mos) | [21, 39–41, 44] |
| VBP15-LTE* (NCT03038399) | Long-term Extension Study to Assess Vamorolone in Boys With DMD | Phase 2A. Male DMD patients aged 4–7 yrs (n = 46): steroid naïveTreatment: daily vamorolone 2, 4, 6 mg/kg/dDuration: 24 mos (2 yrs) | [39, 41, 44] |
| VBP15-004* (NCT03439670) | A Study to Assess the Efficacy and Safety of Vamorolone in Boys With DMD | Phase 2B. Male DMD patients aged 4–7 yrs (n = 121): steroid naïveIntervention: daily vamorolone 2 or 6 mg/kg/d, prednisone 0.75 mg/kg/d, or placebo.Duration: 48 wks (∼1 yr): 48 wks vamorolone, or 24 wks prednisone or placebo, 4 wks transition, then 20 wks vamorolone | [38] |
| VBP15-006 (NCT05185622) | A Study to Assess Vamorolone in Boys Ages 2 to < 4 Years and 7 to < 18 Years With DMD | Phase 2. Male DMD patients (n = 44): young steroid naïve boys aged 2–4 yrs; older boys aged 7–18 yrs without and with current steroid treatment.Intervention: daily vamorolone 2 or 6 mg/kg/dDuration: 12 wks | |
| VBP15-EAP (NCT03863119) | Expanded Access Protocol for Boys with DMD | Expanded access. Applies to DMD patients who have completed the VBP15-LTE or VBP15-004 studies (above).Intervention: daily vamorolone 2, 4, 6 mg/kg/dDuration: ongoing | |
| VBP15-BMD-001 (NCT05166109) | A Study to Assess Vamorolone in Becker Muscular Dystrophy (BMD) | Phase 2A. Male BMD patients aged 18–64 yrs (n = 39): no recent/current steroidIntervention: daily vamorolone 500 mg/d (250 mg if < 50 kg body weight) or placeboDuration: 12 wks |
Abbreviations: BMD = Becker muscular dystrophy, DMD = Duchenne muscular dystrophy, MAD = multiple ascending dose, mos = months, Ref.=References, SAD = single ascending dose, wks = weeks, yrs = years.
Table 2
Clinical trial results. Visual summary to illustrate results of completed vamorolone trials in DMD boys (shown only for vamorolone dose > 2 mg/kg/d*). Results are presented as the effect of vamorolone on outcome measures, compared to the specified groups. Beneficial effects indicated by green shading and/or green text and a check mark (✓). Adverse effects indicated by red shading and/or red text and a cross (❌). Mixed effects or no effect indicated by yellow shading and/or black text and a circled dash (⊖). Completed clinical trials involved DMD steroid naïve males aged 4–7 yrs; with exception of VBP15-001 using normal adult males aged 19–64 yrs. For greater detail of results, see Supplementary Table S2. For design of these clinical trials see summary in Table 1 and details in Supplementary Table S1. For interpretation of the references to colour in this caption, please refer to the online version of this article.
 |
Table 3
Pre-clinical results. Visual summary to illustrate differences in effects between (i) vamorolone/VBP15 and (ii) other glucocorticoid treatment in pre-clinical studies. 3A. Studies in rodent models of Duchenne muscular dystrophy (DMD; mdx) and dysferlinopathy (BLAJ). 3B. Studies in rodent models of other disorders. Beneficial effects indicated by green shading and/or green text and a check mark (✓). Adverse effects indicated by red shading and/or red text and a cross (❌). Mixed effects or no effect indicated by yellow shading and/or black text and a circled dash (⊖). For full study details see Supplementary Table S3. For interpretation of the references to colour in this caption, please refer to the online version of this article.
 |
Clinical trials of vamorolone
There have been eight registered clinical trials of vamorolone since 2015, with five completed and three ongoing; these are outlined in Table 1, with further detail in Supplementary Table S1. The first trial (VBP15-001, NCT02415439; 2015–2016), was a Phase 1 dose escalation study in normal adult males with oral administration of vamorolone (0.1–20 mg/kg/d) for 2 weeks [57]. This was followed by three progressive Phase 2A trials in young steroid naïve DMD boys (aged 4–7 years) from 2016 to 2020, with (i) an initial 2-week study of daily vamorolone (0.25, 0.75, 2, or 6 mg/kg/d; VBP15-002, NCT02760264), (ii) a 24-week extension (VBP15-003, NCT02760277), and then (iii) a 24-month long term extension study (VBP15-LTE, NCT03038399). A Phase 2B trial was initiated in 2018 (completed in 2021) in young steroid naïve DMD boys (aged 4–7 years) that directly compared vamorolone (2 or 6 mg/kg/d) with prednisone (0.75 mg/kg/d) or placebo (vehicle) for 24 weeks, followed by a 4-week transition and then vamorolone for all subjects for a further 20 weeks (VBP15-004, NCT03439670). A meta-analysis of data from the 4 completed trials in DMD was recently published [58].
Another Phase 2 trial (initiated in 2022) is in progress for both young steroid naïve (2–4 years) and older DMD boys (7–18 years) without and with current glucocorticoid treatment (VBP15-006, NCT05185622). An expanded access protocol is now ongoing for participants who completed the VBP-LTE, VBP15-004, or VBP15-006 trials. Finally, another Phase 2 trial (initiated in 2022) is in progress for BMD (VBP15-BMD-001, NCT05166109). These clinical trials with vamorolone in DMD/BMD, involve a large Cooperative International Neuromuscular Research Group (CINRG) across many centres.
Clinical trial design
For the Phase 2 clinical trials, daily vamorolone was administered, in the morning, orally as a 4% suspension in syrup, with 8 oz of whole milk or equivalent high-fat food to increase the time for absorption. The detailed inclusion and exclusion criteria for the respective clinical trials are outlined in Supplementary Table S1.
The initial trials (VBP15-002/003/LTE), focused only on vamorolone treatment of young DMD boys; they did not administer prednisone nor deflazacort for comparison, yet this would have ensured the same selection criteria for all young DMD boys, with the same conditions for all analyses at a similar time. Instead, the effects of vamorolone were mainly compared with archived data from a large prospective cohort study of DMD boys from the DMD Natural History Study (DNHS; NCT00468832). This multicentre study by CINRG (2006–2016), collected data from > 400 boys and young men aged 2–28 years, including recruitment of an additional cohort of DMD participants aged 4–7 years. In addition, the effects of vamorolone were also compared with archived data from other DMD studies: a CINRG clinical trial of prednisone (0.75 mg/kg/d) in young steroid naïve DMD boys (4-<8 years; NCT00110669) [59] and the NorthStar United Kingdom Network database [60].
These DNHS data have been reported in many studies, here we comment briefly on two papers published in 2018. McDonald et al. [61] describe this large complex trial (and inclusion criteria) and evaluate the impact of different glucocorticoids (prednisone, prednisolone, and deflazacort) on 440 DMD boys across the age-range, over 10 years. This study compared two groups: (i) those with no or less than 1 month of cumulative GC treatment duration and (ii) those with more than 1 year of cumulative GC treatment duration, and assessed the progression of clinical mobility, upper limb, and respiratory function measures, and also participants’ wellbeing. One conclusion from these studies was that the data “suggest that the specific glucocorticoid regimens have less impact on the earliest milestone lost (specific supine to standing times)” and that “incremental benefits of deflazacort versus prednisolone or prednisone might require longer follow-up in older age groups”: such comparisons with deflazacort are discussed further by Bello et al. [62] and a recent review [26]. A second study focussed on parent/caregiver reported neurodevelopmental needs, from a cohort of 124 DMD boys aged 4–9 years, with 79 boys (39%) either on prednisone or prednisolone and 44 (22%) on deflazacort; no marked difference was evident between the different glucocorticoid treatments [63]. These papers indicate the breadth of this large natural history study and the populations of DMD boys and normal controls used as a source of archived data for comparison with the first clinical trials with vamorolone.
When considering the comparison of data between the vamorolone studies and the DNHS it is important to note that the DNHS study was initiated about 10 years before the onset of the vamorolone studies. During this decade it is not clear what divergence may have occurred for inclusion criteria for DMD participants, plus variations in routine measures for data collection across different international centres over time: presenting possible uncertainty about the equivalence of experimental conditions used to compare effects of vamorolone and other glucocorticoids on these cohorts of young DMD boys. However, as mentioned, two subsequent trials do directly compare effects of vamorolone and prednisone for new cohorts of DMD boys: VBP15-004 and VBP15-006, see Table 1 and Supplementary Table S1 for details. None of these clinical trials directly compared vamorolone with deflazacort.
Outcome measures
Major readouts of these vamorolone clinical trials included regular measurements of physical parameters (e.g., height, weight), vital signs, and standard blood tests. Pharmacodynamic safety biomarkers included measures of adrenal axis suppression (first-in-morning cortisol), bone turnover (serum osteocalcin, procollagen type 1 N-terminal propeptide, C-terminal telopeptide of type 1 collagen), and insulin resistance (fasting glucose and insulin). The impact of vamorolone was further assessed by exploratory safety and efficacy biomarkers in serum, for blood sampled at the end of the trial, plus muscle function via well-validated timed function tests, including time to stand (TTSTAND), time to run/walk 10 metres (TTRW), and time to climb 4 steps (TTCLIMB) [64]. For more detail of the study objective-specific outcome measures see Supplementary Table S1.
Results of clinical trials
Overview Results of the completed vamorolone clinical trials are simplistically presented in Table 2: they include eight published studies investigating aspects of pharmacokinetics, dosage, efficacy, and safety of vamorolone. A more detailed description of data from these published studies is provided in Supplementary Table S2.
Data analyses for the published clinical studies varied between clinical trials and for different outcome measures. As mentioned, the initial DMD vamorolone trial data (i.e., VBP15-002/003/LTE) were compared with archived DNHS and CINRG data. In addition, specific functional data using the NorthStar Ambulatory Assessment were compared with archived data from the NorthStar United Kingdom Network [60], since this measure was not routinely assessed in the DNHS. In general, the efficacy of vamorolone (e.g., muscle function) was assessed by comparing vamorolone (mid- and endpoint) treatment data with the individual’s baseline or archived untreated (steroid naïve) DMD boys. Yet, there were few comparisons with other glucocorticoids from the archived datasets. In contrast, the safety of vamorolone was mainly compared with glucocorticoid data.
Similarly, for the dedicated trial with vamorolone, prednisone, or placebo treatments for 6 months (i.e., VBP15-004), the efficacy of vamorolone was mainly compared with placebo in results, with information for prednisone provided only as supplementary data; whereas measures of safety were directly compared between vamorolone and prednisone treatment [38].
Pharmacokinetics The pharmacokinetic properties of vamorolone appear similar to prednisone and deflazacort as first shown by pre-clinical studies [19] and subsequently by the Phase 1 clinical trial (VBP15-001) [42, 57]. These steroids have low solubility and thus vamorolone (similar to most glucocorticoids) is delivered in a suspension (as 4% by weight). Moreover, vamorolone is administered along with full-fat milk or equivalent high-fat food (in DMD boys and normal adult males) to extend the bioavailability of the drug [42]. The clinical pharmacokinetic studies emphasise that delivery with food influences clearance [42]; this is important to consider in the context of the time of day for drug delivery in clinical trials, compared with pre-clinical studies in rodents (discussed above under Timing of drug delivery in human and rodent studies: day or night? section).
Dosage Results of the initial clinical trials of vamorolone in DMD (VBP15-002/003) showed that the lower doses (i.e., 0.25 and 0.75 mg/kg/d) had very little efficacy relative to the higher doses (i.e., 2 and 6 mg/kg/d) [21, 40]. As such, a dosage of 2 or 6 mg/kg/d was used for subsequent trials, including the extension (VBP15-LTE), where occasional participants were reduced from the highest dose to 4 mg/kg/d to address undesirable weight gain. Overall, the highest dose of vamorolone (6 mg/kg/d) tended to have slightly more efficacy, but also more pronounced adverse effects (e.g., adrenal suppression, see Safety below).
In this context, it is of interest to relate the doses of vamorolone administered in the clinical trials (e.g., 2 or 6 mg/kg/d) to the doses of vamorolone tested in mice, and also those used in tissue culture. For in vivo studies, allometric conversion of dose from mice (×0.081) to humans is often used, which is related to the different body surface area of species [65]: accordingly, a human dose of 2 or 6 mg/kg/d corresponds to 25 and 75 mg/kg/d in mice. The in vivo pre-clinical studies with mice used doses of vamorolone that ranged from 5–45 mg/kg/d [20, 46], none as high as the 75 mg/kg/day: so the high 6 mg/kg/d human dose might be expected to have more pronounced effects (beneficial and adverse). However, it is stated that the pharmacokinetic measure NOAEL (no observed adverse effect level) was used to select the dosages used in the early clinical studies based on dosage in mice [42].
It is much more complex to relate the range of doses of VBP15 tested in tissue culture conditions (e.g., 0.01, 1 and 10 mM to assess short-term impact on receptor binding in different cell types) to the in vivo situation at the tissue level for different dosages, due to the in vivo pharmacokinetics and dispersal of the drug in different tissues; however, it would be useful to have insight into this comparison. Such in vitro studies show that higher VBP15 concentrations increase the response of GRE-mediated transcription, for example, for ACTH expression related to adrenal suppression (e.g., see Fig. 3C of Heier et al. [20]).
Efficacy Results of the extended trial (VBP15-LTE) for 46 DMD boys showed continued efficacy of vamorolone after 30 months of treatment [39], with maintenance of muscle strength and function (compared with glucocorticoid treatment from historical control cohorts). The later clinical trial that directly compared vamorolone (2 or 6 mg/kg/d) with prednisone and placebo (vehicle control), for young DMD boys (n = 114) with analyses at 6 months (VBP15-004), showed improved muscle function with vamorolone compared with placebo, similar to prednisone treatment [38].
A recent meta-analysis [58] of data from the first 4 major clinical studies [21, 38, 39, 41] (including data from VBP15-002/003/LTE and VBP15-004) demonstrated a significant association between vamorolone treatment and improvement (i.e., increased velocity) of the timed function tests (TTSTAND, TTRW, TTCLIMB) compared with the placebo (steroid naïve, i.e., young untreated DMD boys). In addition, vamorolone increased TTRW velocity and height percentile, compared with glucocorticoid treatment [58].
Of interest with respect to relative clinical efficacy of different glucocorticoids for DMD boys, a recent study by Fang et al. [44] reported no significant differences in disease progression between data for three functional measures for DMD boys aged 4–10 years, treated with vamorolone (VBP15-LTE), compared with similar measurements (DNHS data) for DMD boys either steroid naïve, or treated with prednisone/prednisolone and/or deflazacort. This accords with conclusions of Mah et al. [39], for no difference between functional efficacy measures with vamorolone or glucocorticoid treatments.
In conclusion, the clinical trials with vamorolone in young DMD boys, with studies published for VBP15-LTE up to 30 months and a dedicated trial of vamorolone compared with prednisone for 6 months (VBP15-004), indicate very similar efficacy of vamorolone and other glucocorticoids. Data from longer-term studies and in older DMD boys will provide more vital information.
Safety The adverse effects of glucocorticoid treatment are a major problem for DMD. Some adverse effects seen with glucocorticoids particularly related to restricted height, increased bone turnover, insulin resistance, and weight gain, were less pronounced with Vamorolone. Vamorolone after 30 months of treatment (compared with glucocorticoid treatment from historical control cohorts) improved height velocity [39], in contrast with growth deceleration associated with glucocorticoid treatment, especially deflazacort [26, 66, 67]. The later clinical trial that directly compared vamorolone (2 or 6 mg/kg/d) with prednisone and placebo (vehicle control), for young DMD boys (n = 114) with analyses at 6 months (VBP15-004), showed no decline in height nor bone turnover markers, compared with placebo control, whereas adverse effects were observed for prednisone where these values decreased [38]. Of some concern for these results from VBP15-002/003/LTE and VBP15-004, is that all three glucocorticoid treatment groups increased adrenal insufficiency; this was particularly pronounced for the highest dose of vamorolone (6 mg/kg/d) that reduced first-in-morning cortisol to a greater extent than prednisone and 2 mg/kg/d vamorolone [38].
Pre-clinical studies using vamorolone in mdx mice and other rodent models
Pre-clinical studies comparing vamorolone and other glucocorticoids (each relative to vehicle control) are simplistically summarised in Table 3: they include five studies using mdx mouse models of DMD, one study using the BLAJ mouse model of the muscular dystrophy dysferlinopathy (Table 3A), plus seven other rodent models for different conditions (Table 3B). A more detailed comparison of data from all these pre-clinical studies is provided in Supplementary Table S3. On the path to selecting vamorolone as the candidate drug for treating DMD, an early study tested the pro-drug anecortave (VBP1, a precursor of VBP15) in mdx mice and the SJL/J model of dysferlinopathy [68]; this study is shown only in Supplementary Table S3.
For most of these pre-clinical studies, vamorolone was administered in cherry syrup by gavage at a dose ranging from 5–45 mg/kg, in the morning, with treatment duration ranging from 3 days (acute) to 6 months (chronic), where a 6-week treatment period was most common. Prednisolone and vehicle control (i.e., cherry syrup) were similarly administered. Most (12) of these pre-clinical studies directly compared vamorolone/VBP15 with prednisolone, and one compared vamorolone with dexamethasone [51]; however, there is no comparison with deflazacort (the major alternative steroid drug for DMD).
Overall, these studies endorse observations that vamorolone has beneficial anti-inflammatory effects and improves muscle function, compared with untreated/vehicle-treated mdx mice, with the extent of such benefits generally being similar to prednisone/prednisolone and mediated by the glucocorticoid receptor (See Table 3 and discussion for myonecrosis below). Of particular interest is that vamorolone reduced the extent of adverse glucocorticoid-associated side effects including stunted growth and bone loss that are linked with prednisone/prednisolone. However, with respect to adverse effects, vamorolone increased glucocorticoid-induced liver toxicity in the sickle cell disease model beyond the effects of prednisolone [56]. While this does not seem to be a concern for liver function in the young DMD boys treated with vamorolone for up to 30 months so far [39], this needs careful consideration for long-term use since liver damage and disease is also a feature of DMD (reviewed in Ohlendieck and Swandulla [7]).
For the 13 pre-clinical studies with vamorolone (there may be others beyond those outlined in Table 3), all of these (with the exception of Akkad et al. [52]) have one or more co-authors in common with the large group of collaborators associated with publications resulting from the clinical trials. Since it is strongly recommended that pre-clinical studies should also be conducted by independent groups, some additional studies by independent investigators would be welcomed (also perhaps using a larger animal model of DMD [3]), to help validate the main observations, describe additional mechanisms, and generally strengthen the data.
Consideration of ‘other effects’ of glucocorticoids that may benefit dystrophic muscles
Beyond the well-characterised capacity of vamorolone to decrease inflammation, it is proposed that vamorolone/VBP15 has additional benefits due to properties that directly stabilise and protect the sarcolemma from damage and enhance repair, in marked contrast with prednisolone. These interesting properties warrant further consideration. In this context, two assays are discussed: the first uses ProSense 680 for in vivo quantification of inflammation, extrapolated to the incidence of intrinsic myonecrosis in mdx muscles. The second assay involves experimental perforation/wounding of the sarcolemma of muscle cells, to evaluate the impact of different glucocorticoids on the fluidity of lipids in the sarcolemma and membrane resealing. These data are considered with respect to in vivo translation for the situations of DMD and dysferlinopathy, based mainly on the papers by Heier et al. [20] and Sreetama et al. [49], respectively (for details of these results see Supplementary Table S3). Here we emphasise the central importance of myonecrosis in DMD and mdx mice and also discuss the possible mechanistic basis for loss of myofibres (with adipocyte replacement) in dysferlinopathies.
Impact of VBP15 on myonecrosis in mdx mice
Since myonecrosis of dystrophic mdx muscles is closely associated with increased inflammation and oxidative stress (see Myonecrosis and dystropathology section), a non-invasive live imaging technique using ProSense 680 (a substrate for cathepsin protease present in inflammatory cells) was used to visualise and quantify inflammation, as a surrogate marker of active myonecrosis in limb muscles of mice [20, 68]. The studies of VBP15 in mdx mice [20] demonstrated various benefits of VBP15 and concluded that both “VBP15 and prednisolone decreased cathepsin activity towards WT levels” (p. 1574) [20]; a similar result was seen in an earlier study of anecortave (VBP1 pro-drug) and prednisolone [68]. It is proposed that this measure of ‘cathepsin activity/inflammation’ correlates with the incidence of myonecrosis: yet this can be hard to determine.
In the main experiment of Heier et al. [20] where drug treatment began at 2 weeks of age (to prevent/reduce the acute onset of myonecrosis at ∼3 weeks), imaging was at 6–7 weeks and muscles sampled at 8 weeks. For the three doses of VBP15 (5, 15, 30 mg/kg/d), only the 15 mg/kg dose showed a significant (-38%) reduction in inflammation in the hindlimb, compared with control (vehicle-treated) mdx mice (see Fig. 4E’ in Heier et al. [20]). Prednisolone (0.75 mg/kg/d) had the largest reduction in cathepsin activity (–52%) compared with any dose of VBP15, however, the statistical impact of prednisolone was not reported. Similarly, in the forelimb of mdx mice, the most marked reduction of inflammation was seen for prednisolone (similar to WT levels) with, unfortunately, statistical significance not shown for any groups (see Supporting Information Fig. 1C in Heier et al. [20]).
Histological images of muscle sections stained by haematoxylin and eosin (H&E) are ideal to test for (i) prior bouts of myonecrosis/regeneration identified by the presence of centrally-located myonuclei in regenerated myofibres (usually pronounced in mdx limb muscles by 8 weeks of age), and to demonstrate (ii) the incidence of inflammation and active myonecrosis. While H&E-stained diaphragm muscle show few inflammatory cells in the VBP15-treated mdx muscle (for all three doses), there was clear evidence of centrally-located myonuclei, indicating that some prior myonecrosis and regeneration had occurred (see Fig. 4F in Heier et al. [20]). This questions the extent to which VBP15 may have delayed or prevented the onset of myonecrosis. Images of some H&E-stained limb muscles (e.g., the gastrocnemius) could help to clarify this. Such histology is also essential to clearly illustrate and define the appearance of the ‘degenerating myofibres’ (that may represent myonecrosis) that were quantified for the gastrocnemius muscle (see Supporting Information Fig. 2C in Heier et al. [20]); this analysis showed no benefit of VBP15, but a significant increase in degenerating myofibres with prednisolone (∼2-fold greater than untreated mdx). Clearly, this result for prednisolone in the mdx gastrocnemius contrasts with the reduced inflammation observed by the cathepsin assay in the mdx hindlimb (See Fig. 4E in Heier et al. [20]). Such discrepancies raise concerns about the relationship between these different measures and confound conclusions concerning the impact of these steroids on the incidence of ‘acute bouts of myonecrosis’.
Of interest is a similar study using Compound A (a non-steroidal drug that also binds the glucocorticoid receptor) in mdx mice, which was conducted in parallel to the study by Heier et al. [20] (using these assays in the same laboratories and at the same time). Compound A showed similar anti-inflammatory benefits to prednisolone and fewer adverse effects (compared with prednisolone) related to reduced body and muscle mass and growth stunting [69].
A second study in Heier et al. [20] used mdx mice subjected to treadmill exercise twice a week (to increase myonecrosis), with daily treatment of vamorolone (5, 15, 45 mg/kg/d) or prednisolone (5 mg/kg/d), from 6 weeks of age for 4 months. Analyses at 23 weeks of age showed no significant impact for either drug for the cathepsin assay (see Fig. 4G’ in Heier et al. [20]). For the H&E-stained diaphragm, there was statistically reduced (-47%) inflammation only for the highest (45 mg/kg/d) dose of VBP15; prednisolone had a similar-sized effect (-41%) although no statistical significance was indicated (see Fig. 4I in Heier et al. [20]). Since reduction/prevention of myonecrosis is such a key aspect of therapies for DMD, it seems that more direct histological analyses of limb muscles are warranted to help clarify the relative benefit of VBP15 (at different doses) and prednisolone on myonecrosis.
Effect of VBP15 on sarcolemma lesions, resealing, and lipid dynamics
In tissue culture and other in vitro studies, a small disruption of the cell (plasma) membrane (by laser injury or use of glass beads) has been widely used to assess the process of vesicle trafficking to reseal the perforated membrane of various cells [70]. Using this single-cell wounding model on ‘normal’ C2C12 (immortalised mouse) myoblasts it was reported that VBP15 reduced laser-induced membrane injury and enhanced the speed of membrane repair in a dose-dependent manner, while prednisolone worsened these outcomes [20]. It is noted that myoblasts are not equivalent to mature myofibres, which have more complex sarcolemma composition. In the context of DMD, the effect of drugs on this experimentally-induced membrane repair assay [20] can be hard to extrapolate mechanistically to the in vivo reality of high intrinsic myonecrosis in mdx and DMD muscles, where the early cellular events have been well described (see Myonecrosis and dystropathology section).
The membrane wounding/repair model was also used in the context of dysferlinopathy to investigate the impact of vamorolone compared with prednisolone [49]. While this disease has a very different pathomechanism to DMD, with late-onset, typically post-growth, and pronounced adipocyte replacement of myofibres (but little myonecrosis) that initially manifests only in limb-girdle muscles (e.g., psoas and quadriceps) [71], it provides useful data related to the impact of these drugs in a wider context. Vamorolone was shown to improve membrane repair in dysferlin-deficient BLAJ mouse muscles (i.e., model of dysferlinopathy) [49], with similar benefits for vamorolone observed using immortalised myoblasts derived from patients with dysferlinopathy; whereas prednisolone was not beneficial. Similarly, vamorolone (compared with vehicle) improved repair following laser-induced perforation of myofibres isolated from muscles of dysferlin-deficient BLAJ mice [49]: this study used myofibres (from biceps) treated with vamorolone (50 mM) ex vivo for 30 min, or myofibres from mice with an acute in vivo vamorolone treatment (twice daily at 30 mg/kg for 2 days). Such benefits of vamorolone were also evident after a long-term study using BLAJ mice, treated daily for 3 months with vamorolone (30 mg/kg/d), prednisolone (30 mg/kg/d), or vehicle, with ex vivo muscle injury and analyses at 10 months of age (when histopathology is evident in specific muscles of the limb-girdle region). Myofibres isolated from biceps muscle from these vamorolone-treated mice had improved repair following laser wounding (whereas slower repair was seen with prednisolone) and vamorolone-treated intact EDL muscles (from the same study) had some protection from ex vivo eccentric contraction-induced force loss. Other experiments showed that the dysferlinopathy patient myoblasts had increased plasma membrane lipid mobility (compared with normal myoblasts), and this was decreased (normalised) by vamorolone but increased further by prednisolone [49]. The intrinsic increased membrane lipid mobility for dysferlin-deficient muscles and the benefits of vamorolone are interesting observations for this disease.
Such membrane resealing assays have been a powerful tool in earlier studies to describe molecules associated with dysferlin protein and vesicle trafficking after induced sarcolemma wounding and have provided much valuable information [70]. However, this ex vivo sarcolemmal repair process does not appear to relate closely to the in vivo mechanistic basis for muscle dystropathology and disease progression of dysferlinopathy [72]. This is not surprising, since most dysferlin-deficient mouse limb muscles are relatively unaffected initially, even by one year of age, in contrast with the pronounced dystropathology in limb-girdle muscles like quadriceps and psoas [71]. Thus, care is needed when extrapolating the significance of such ex vivo assays to the clinical reality for various muscles in different muscular dystrophies. However, the observation that vamorolone significantly reduced adipogenic replacement of myofibres (see Fig. 5K in Sreetama et al. [49]), compared with vehicle control and prednisolone in BLAJ gastrocnemius muscles at 10 months, seems of direct relevance to the pathology of dysferlinopathy [73].
Pre-clinical studies in animal models are essential to test and validate the mechanistic targets of drug activity, provide new insights to identify potential additional drug applications and form a firm foundation for future clinical translation in many muscular dystrophies and other disorders. Thus, more independent pre-clinical studies with vamorolone are welcomed.
OTHER CONSIDERATIONS
To help select the best drug for future clinical use, brief comments are made below concerning the use of blood biomarkers, costs of drugs, and alternative anti-inflammatory drugs with possible combination therapies.
Biomarkers to rapidly assess drug efficacy
To determine efficacy of drugs that aim to protect dystrophic muscles from myonecrosis in DMD boys (and animal studies), the key aspects to measure initially are (i) the extent to which the incidence of myonecrosis is reduced, along with associated reduced inflammation and oxidative stress, and (ii) demonstrated engagement of the drug with its specific molecular target: this information can allow the rapid evaluation of likely drug efficacy and go/no-go decisions (well before any functional indication of benefit). Such analyses can be readily conducted in pre-clinical trials with dystrophic animals where sampling of many muscles and other tissues is feasible, yet clearly such invasive tissue sampling is not an option for DMD boys. However, small samples of blood from humans (e.g., finger or ear prick, with ease of repeated sampling) are very suitable for some analyses.
Measuring levels of specific blood biomarkers is potentially a powerful tool to assess systemically the extent of active myonecrosis (closely associated with dysregulated inflammation and protein oxidation) occurring in the many muscles throughout the body [6]. Recent studies have used a range of tools to identify robust blood biomarkers (e.g., proteins, RNA, metabolites) that are sensitive and specific to myonecrosis and inflammation across species, including humans [74, 75], and dystrophic mice [76] and dogs [77, 78]. Some examples of promising blood protein biomarkers that are linked to the extent of myonecrosis/inflammation/oxidation in dystrophic muscles are osteopontin [79], the chemokine CCL2 [75, 78], and oxidised albumen [80]. The value of blood biomarkers for quick feedback was demonstrated by a study using SOMAscan aptamer panels (to assay 1,310 proteins) to compare the impact of various combinations of four drugs, including vamorolone and prednisolone, after 4 weeks of treatment in young mdx mice [47]. There is now a need to focus on confirmatory studies by different labs, testing a short list of the most promising blood biomarker candidates in various clinical and pre-clinical trials for DMD, especially biomarkers that can be measured by a simple standardised technique suitable for routine (inexpensive) analyses.
Use of such informative blood biomarkers complements the use of non-invasive measures to determine the benefits of a drug on DMD muscle function. Demonstration of improved or stabilised muscle function is highly desirable (to reflect reduced myonecrosis), although function is usually a longer-term consequence of drug benefit in clinical trials. Ideally, this is done in conjunction with some analysis of muscle composition to quantify the progression of the dystropathology over time: this can be achieved using non-invasive magnetic resonance imaging [81], although specialised facilities are required and this can be expensive and time consuming.
Considering the relative cost of drug therapies for DMD
Due to the recent very high price of some new drugs, the potential cost of long-term vamorolone treatment for DMD is of much interest and would be useful to clarify; however, if vamorolone proves to have wide use for many other conditions, this should reduce the general clinical price. Prednisone is relatively inexpensive (∼$135/month) and deflazacort was originally similarly affordable (∼$1,500/year). However, when deflazacort (now known as Emflaza) was approved by the Food and Drug Administration (FDA) for use in the USA in 2017, the company Marathon Pharmaceuticals initiated a 60-fold price increase, so that annual treatment became US$89,000 per year [82]; later this was reduced to about $35,000 after purchase of deflazacort/Emflaza by PTC Therapeutics. This expense presents problems (compared with relatively inexpensive prednisone), with an analysis of the relative affordability of drugs for DMD concluding that “the long-term value for money of deflazacort versus prednisone is ‘low”’ (p. 364) [28]. Two striking examples of high cost are provided for gene therapy drugs for DMD (both owned by Sarepta Therapeutics): Eteplirsen (Exondys 51) costs about $300,000 annually per patient [28], and Elevidys (SRP-9001), with functional efficacy yet to be demonstrated, received accelerated approval from the FDA in June 2023 and will cost $3.2 million for each patient (announced by Sarepta Therapeutics). While it is recognised that drug development is a very expensive process, such extraordinarily high prices for consumers present difficulties for patient access to such treatments, and for global health systems; this emerging reality requires balanced discussion [83].
Benefits of steroids compared with non-steroidal anti-inflammatory drugs
Because of the well-documented adverse effects of glucocorticoids, it is essential to also compare the benefits of steroids with other non-steroidal drugs, or nutraceuticals, that target inflammation and may be equally efficacious (with different and fewer adverse side effects), and may be capable of addressing different pathogenic features of DMD to elicit therapeutic advantages [18].
The complex signalling interactions associated with the use of steroids and many other drugs that target anti-inflammatory activity via inhibition of NF-κB pathways and molecules in pro-inflammatory pathways, are summarised and discussed in-depth in the context of DMD in many reviews [5, 17, 22, 84]. Of interest is a recent study showing that silencing the receptor activator of the NF-κB ligand (RANKL) alone (and combined with deflazacort) has major benefits for bone health and muscle function, along with reduced myonecrosis and fibrosis in mdx mice [85]. In addition, some nutraceuticals such as amino acids and their derivatives have striking benefits on dystrophic muscles, with decreased myonecrosis, along with reduction of inflammation, oxidative damage, and fibrosis, and are suitable for combination therapies [86]. Glucocorticoids act through multiple mechanisms, although the precise molecular pathways that provide efficacy in DMD (see also discussion in Consideration of ‘other effects’ of glucocorticoids that may benefit dystrophic muscles section), and those that are responsible for detrimental effects are not fully understood. When considering a polypharmaceutical approach for DMD, it is of the utmost importance to know which molecules should not be combined with glucocorticoids [22]. An excellent historical review provides insight into a wide range of steroidal and non-steroidal drugs that target the glucocorticoid receptor, with merit observed for non-steroid drugs such as Compound A, and a useful discussion of possible combination therapies [87]. This emphasises that a deeper understanding of the specific molecular and cellular mechanisms involved in the actions of these different drugs on the dystropathology in vivo is essential, in the context of potential future clinical applications to DMD.
CONCLUSIONS
The data available from the (ongoing) DMD clinical and pre-clinical trials indicate that vamorolone has similar protective effects, with reduced inflammation to maintain muscle function, and generally fewer adverse effects (especially related to growth) compared with prednisone/prednisolone; this is very promising. However, there are few direct comparisons of vamorolone with deflazacort, the closest competitor drug to prednisone, and it seems important to address this. The optimal dose of vamorolone for clinical and pre-clinical applications also needs to be clarified. Additionally, some estimate of the cost of vamorolone will be useful since the comparative expense of these drugs is a key factor for wide clinical accessibility and use.
The pre-clinical studies raise questions about the precise mechanistic basis for the benefits of vamorolone in vivo, which would be useful to clarify. One key aspect is the capacity of vamorolone to reduce myonecrosis: this could be addressed by more histological analyses of limb muscles of dystrophic animals, plus blood biomarkers that systemically reflect myonecrosis throughout the body (and are also relevant for clinical trials). The interesting ‘additional benefits of vamorolone’ related to sarcolemmal membrane stability and repair and lipid dynamics also warrant more intense consideration, since the relevance of these ex vivo observations to the in vivo situation is unclear and needs to be carefully evaluated for different muscular dystrophies, including DMD and dysferlinopathies. Pre-clinical studies by independent groups could help to strengthen data related to these in vivo mechanistic aspects of vamorolone.
AUTHOR CONTRIBUTIONS
MDG constructed the overall commentary, EML scrutinised publications and data analyses, and both contributed to writing.
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230161.
REFERENCES
[1] | Duan D , Goemans N , Takeda Si , Mercuri E , Aartsma-Rus A . Duchenne muscular dystrophy, Nature Reviews Disease Primers (2021) ;7: (1):13. doi: 10.1038/s41572-021-00248-3. |
[2] | Lloyd EM , Pinniger GJ , Murphy RM , Grounds MD Slow or fast: implications of myofibre-type and associated differences for manifestation of neuromuscular disorders, Acta Physiol. (2023) ;238: (4):e14012. doi: 10.1111/apha.14012. |
[3] | Zaynitdinova MI , Lavrov AV , Smirnikhina SA Animal models for researching approaches to therapy of Duchenne muscular dystrophy, Transgenic Res (2021) ;30: (6):709–25. doi: 10.1007/s11248-021-00278-3. |
[4] | Hodgetts S , Radley H , Davies M , Grounds MD Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNF alpha function with Etanercept in mdx mice, Neuromuscul Disord (2006) ;16: (9-10):591–602. doi: 10.1016/j.nmd.2006.06.011. |
[5] | Tidball JG , Welc SS , Wehling-Henricks M . Immunobiology of inherited muscular dystrophies, Compr Physiol (2018) ;8: (4):1313–56. doi: 10.1002/cphy.c170052. |
[6] | Grounds MD , Terrill JR , Al-Mshhdani BA , Duong MN , Radley-Crabb HG , Arthur PG . Biomarkers for Duchenne muscular dystrophy: myonecrosis, inflammation and oxidative stress, Dis Model Mech. (2020) ;13: (2):dmm043638. doi: 10.1242/dmm.043638. |
[7] | Ohlendieck K , Swandulla D . Complexity of skeletal muscle degeneration: multi-systems pathophysiology and organ crosstalk in dystrophinopathy, Pflug Arch Eur J Phy (2021) ;473: (12):1813–39. doi: 10.1007/s00424-021-02623-1. |
[8] | Iwasaki T , Terrill JR , Kawarai K , Miyata Y , Tagami T , Maeda N , Hasegawa Y , Watanabe T , Grounds MD , Arthur PG The location of protein oxidation in dystrophic skeletal muscle from the mdx mouse model of Duchenne muscular dystrophy, Acta Histochem (2022) ;124: (8):151959. doi: 10.1016/j.acthis.2022.151959. |
[9] | Grounds MD . The need to more precisely define aspects of skeletal muscle regeneration, Int J Biochem Cell Biol (2014) ;56: :56–65. doi: 10.1016/j.biocel.2014.09.010. |
[10] | Dadgar S , Wang ZY , Johnston H , Kesari A , Nagaraju K , Chen YW , Hill DA , Partridge TA , Giri M , Freishtat RJ , Nazarian J , Xuan JH , Wang Y , Hoffman EP . Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy, J Cell Biol (2014) ;207: (1):139–58. doi: 10.1083/jcb.201402079. |
[11] | Radley-Crabb HG , Marini JC , Sosa HA , Castillo LI , Grounds MD , Fiorotto ML . Dystropathology increases energy expenditure and protein turnover in the mdx mouse model of duchenne muscular dystrophy, PLoS One. (2014) ;9: (2):e89277. doi: 10.1371/journal.pone.0089277. |
[12] | Terrill JR , Duong MN , Turner R , Le Guiner C , Boyatzis A , Kettle AJ , Grounds MD , Arthur PG . Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy, Redox Biol (2016) ;9: :276–86. doi: 10.1016/j.redox.2016.08.016. |
[13] | Chen Y-W , Nagaraju K , Bakay M , McIntyre O , Rawat R , Shi R , Hoffman EP Early onset of inflammation and later involvement of TGFβ in Duchenne muscular dystrophy, Neurology (2005) ;65: (6):826–34. doi: 10.1212/01.wnl.0000173836.09176.c4. |
[14] | Markati T , Oskoui M , Farrar MA , Duong T , Goemans N , Servais L . Emerging therapies for Duchenne muscular dystrophy, Lancet Neurol (2022) ;21: (9):814–29. doi: 10.1016/S1474-4422(22)00125-9. |
[15] | van Deutekom J , Beekman C , Bijl S , Bosgra S , van den Eijnde R , Franken D , Groenendaal B , Harquouli B , Janson A , Koevoets P , Mulder M , Muilwijk D , Peterburgska G , Querido B , Testerink J , Verheul R , de Visser P , Weij R , Aartsma-Rus A , Puolivali J , Bragge T , O’Neill C , Datson NA Next generation exon 51 skipping antisense oligonucleotides for Duchenne muscular dystrophy, Nucleic Acid Ther (2023) ;33: (3):193–208. doi: 10.1089/nat.2022.0063. |
[16] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY . Corticosteroids for the treatment of Duchenne muscular dystrophy, Cochrane Db Syst Rev (2016) ;5:CD003725. doi 10.1002/14651858.CD003725.pub4. |
[17] | Quattrocelli M , Zelikovich AS , Salamone IM , Fischer JA , McNally EM . Mechanisms and clinical applications of glucocorticoid steroids in muscular dystrophy, J Neuromuscul Dis (2021) ;8: :39–52. doi: 10.3233/JND-200556. |
[18] | Kourakis S , Timpani CA , Campelj DG , Hafner P , Gueven N , Fischer D , Rybalka E . Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: Can we do better? Orphanet J Rare Dis (2021) ;16: (1):117. doi: 10.1186/s13023-021-01758-9. |
[19] | Reeves EKM , Hoffman EP , Nagaraju K , Damsker JM , McCall JM . VBP Preclinical characterization of a novel anti-inflammatory delta 9,11 steroid, Bioorg Med Chem (2013) ;21: (8):2241–9. doi: 10.1016/j.bmc.2013.02.009. |
[20] | Heier CR , Damsker JM , Yu Q , Dillingham BC , Huynh T , Van der Meulen JH , Sali A , Miller BK , Phadke A , Scheffer L , Quinn J , Tatem K , Jordan S , Dadgar S , Rodriguez OC , Albanese C , Calhoun M , Gordish-Dressman H , Jaiswal JK , Connor EM , McCall JM , Hoffman EP , Reeves EKM , Nagaraju K . VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects, EMBO Mol Med (2013) ;5: (10):1569–85. doi: 10.1002/emmm.201302621. |
[21] | Hoffman EP , Schwartz BD , Mengle-Gaw LJ , Smith EC , Castro D , Mah JK , McDonald CM , Kuntz NL , Finkel RS , Guglieri M , Bushby K , Tulinius M , Nevo Y , Ryan MM , Webster R , Smith AL , Morgenroth LP , Arrieta A , Shimony M , Siener C , Jaros M , Shale P , McCall JM , Nagaraju K , van den Anker J , Conklin LS , Cnaan A , Gordish-Dressman H , Damsker JM , Clemens PR , Res CIN . Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function, Neurology (2019) ;93: (13):E1312–E23. doi: 10.1212/Wnl.0000000000008168. |
[22] | Herbelet S , Rodenbach A , De Paepe B , De Bleecker JL . Anti-inflammatory and general glucocorticoid physiology in skeletal muscles affected by duchenne muscular dystrophy: exploration of steroid-sparing agents, Int J Mol Sci (2020) ;21: (13):4596. doi: 10.3390/ijms21134596. |
[23] | Timmermans S , Souffriau J , Libert C . A general introduction to glucocorticoid biology, Front Immunol (2019) ;10: ;1545. doi: 10.3389/fimmu.2019.01545. |
[24] | Hapgood JP , Avenant C , Moliki JM Glucocorticoid-independent modulation of GR activity: Implications for immunotherapy, Pharmacol Ther (2016) ;165: :93–113. doi: 10.1016/j.pharmthera.2016.06.002. |
[25] | Sali A , Guerron AD , Gordish-Dressman H , Spurney CF , Iantorno M , Hoffman EP , Nagaraju K . Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse, PLoS One (2012) ;7: (4):e34204. doi: 10.1371/journal.pone.0034204. |
[26] | Biggar WD , Skalsky A , McDonald CM . Comparing deflazacort and prednisone in Duchenne muscular dystrophy, J Neuromuscul Dis (2022) ;9: (4):463–76. doi: 10.3233/Jnd-210776. |
[27] | Bylo M , Farewell R , Coppenrath VA , Yogaratnam D . A review of deflazacort for patients With Duchenne muscular dystrophy, Ann Pharmacother (2020) ;54: (8):788–94. doi: 10.1177/1060028019900500. |
[28] | Agboola F , Lin GA , Fluetsch N , Walton SM , Rind DM , Pearson SD . The effectiveness and value of deflazacort and exon-skipping therapies for the management of Duchenne muscular dystrophy a summary from the Institute for Clinical and Economic Review’s New England Comparative Effectiveness Public Advisory Council, Journal of Managed Care & Specialty Pharmacy (2020) ;26: (4):361–6. |
[29] | Araujo APDC , Saute JAM , Fortes CPDD , Franca MC , Pereira JA , de Albuquerque MAV , Carvalho AAD , Cavalcanti EBU , Covaleski APPM , Fagondes SC , Gurgel-Giannetti J , Goncalves MVM , Martinez ARM , Neto ARC , Neves FR , Nucci A , Nucera APCD , Pessoa ALS , Rebel MF , dos Santos FN , Scola RH , Sobreira CFD . Update of the Brazilian consensus recommendations on Duchenne muscular dystrophy, Arq Neuropsiquiatr (2023) ;81: (1):81–94. doi: 10.1055/s-0043-1761466.ISSN0004-282X. |
[30] | Guglieri M , Bushby K , McDermott MP , Hart KA , Tawil R , Martens WB , Herr BE , McColl E , Speed C , Wilkinson J , Kirschner J , King WM , Eagle M , Brown MW , Willis T , Griggs RC , Muscle FDI . Effect of different corticosteroid dosing regimens on clinical outcomes in boys with Duchenne muscular dystrophy a randomized clinical trial, Jama-Journal of the American Medical Association (2022) ;327: (15):1456–68. doi: 10.1001/jama.2022.4315. |
[31] | Hoffman EP , Rao D , Pachman LM . Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostics and microarrays, Rheum Dis Clin N Am (2002) ;28: (4):743–57. doi: 10.1016/S0889-857x(02)00031-5. |
[32] | Walter MC , Reilich P , Thiele S , Schessl J , Schreiber H , Reiners K , Kress W , Muller-Reible C , Vorgerd M , Urban P , Schrank B , Deschauer M , Schlotter-Weigel B , Kohnen R , Lochmuller H . Treatment of dysferlinopathy with deflazacort: a double-blind, placebo-controlled clinical trial, Orphanet J Rare Dis (2013) ;8: :26. doi: 10.1186/1750-1172-8-26. |
[33] | Alharbi N , Matar R , Cupler E , Al-Hindi H , Murad H , Alhomud I , Monies D , Alshehri A , Alyahya M , Meyer B , Bohlega S . Clinical, neurophysiological, radiological, pathological, and genetic features of dysferlinopathy in Saudi Arabia, Front Neurosci (2022) ;16: :815556. doi: 10.3389/fnins.2022.815556. |
[34] | Wintzinger M , Miz K , York A , Demonbreun AR , Molkentin JD , McNally EM , Quattrocelli M Effects of glucocorticoids in murine models of Duchenne and limb-girdle muscular dystrophy. In: Maruyama R, Yokota T, editors. Muscular Dystrophy Therapeutics: Methods and Protocols. New York, NY: Springer US; 2023. p. 467-78. |
[35] | Quattrocelli M , Salamone IM , Page PG , Warner JL , Demonbreun AR , McNally EM . Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy, Am J Pathol (2017) ;187: (11):2520–35. doi: 10.1016/j.ajpath.2017.07.017. |
[36] | Quattrocelli M , Barefield DY , Warner JL , Vo AH , Hadhazy M , Earley JU , Demonbreun AR , McNally EM . Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy, J Clin Investig (2017) ;127: (6):2418–32. doi: 10.1172/Jci91445. |
[37] | Xu J , Winkler J , Sabarinath SN , Derendorf H . Assessment of the impact of dosing time on the pharmacokinetics/pharmacodynamics of prednisolone, AAPS Journal (2008) ;10: (2):331–41. doi: 10.1208/s12248-008-9038-3. |
[38] | Guglieri M , Clemens PR , Perlman SJ , Smith EC , Horrocks I , Finkel RS , Mah JK , Deconinck N , Goemans N , Haberlova J , Straub V , Mengle-Gaw LJ , Schwartz BD , Harper AD , Shieh PB , De Waele L , Castro D , Yang ML , Ryan MM , McDonald CM , Tulinius M , Webster R , McMillan HJ , Kuntz NL , Rao VK , Baranello G , Spinty S , Childs AM , Sbrocchi AM , Selby KA , Monduy M , Nevo Y , Vilchez-Padilla JJ , Nascimento-Osorio A , Niks EH , de Groot IJM , Katsalouli M , James MK , van den Anker J , Damsker JM , Ahmet A , Ward LM , Jaros M , Shale P , Dang UJ , Hoffman EP . Efficacy and safety of vamorolone vs placebo and prednisone among boys with Duchenne muscular dystrophy, Jama Neurology (2022) ;79: (10):1005–14. doi: 10.1001/jamaneurol.2022.2480. |
[39] | Mah JK , Clemens PR , Guglieri M , Smith EC , Finkel RS , Tulinius M , Nevo Y , Ryan MM , Webster R , Castro D , Kuntz NL , McDonald CM , Damsker JM , Schwartz BD , Mengle-Gaw LJ , Jackowski S , Stimpson G , Ridout DA , Ayyar-Gupta V , Baranello G , Manzur AY , Muntoni F , Gordish-Dressman H , Leinonen M , Ward LM , Hoffman EP , Dang UTJ , I NUNCD Efficacy and safety of vamorolone in Duchenne muscular dystrophy a 30-month nonrandomized controlled open-label extension trial, Jama Network Open (2022) ;5: (1):e2144178. doi: 10.1001/jamanetworkopen.2021.44178. |
[40] | Li XN , Conklin LS , van den Anker J , Hoffman EP , Clemens PR , Jusko WJ . Exposure-response analysis of Vamorolone (VBP15) in boys with Duchenne muscular dystrophy, J Clin Pharmacol (2020) ;60: (10):1385–96. doi: 10.1002/jcph.1632. |
[41] | Smith EC , Conklin LS , Hoffman EP , Clemens PR , Mah JK , Finkel RS , Guglieri M , Tulinius M , Nevo Y , Ryan MM , Webster R , Castro D , Kuntz NL , Kerchner L , Morgenroth LP , Arrieta A , Shimony M , Jaros M , Shale P , Gordish-Dressman H , Hagerty L , Dang UJ , Damsker JM , Schwartz BD , Mengle-Gaw LJ , McDonald CM , Investigators CVD Efficacy and safety of vamorolone in Duchenne muscular dystrophy: An 18-month interim analysis of a non-randomized open-label extension study, PLoS Med (2020) ;17: (9):e1003222. doi: 10.1371/journal.pmed.1003222. |
[42] | Mavroudis PD , van den Anker J , Conklin LS , Damsker JM , Hoffman EP , Nagaraju K , Clemens PR , Jusko WJ . Population pharmacokinetics of vamorolone (VBP15) in healthy men and boys with Duchenne muscular dystrophy, J Clin Pharmacol (2019) ;59: (7):979–88. doi: 10.1002/jcph.1388. |
[43] | Conklin LS , Damsker JM , Hoffman EP , Jusko WJ , Mavroudis PD , Schwartz BD , Mengle-Gaw LJ , Smith EC , Mah JK , Guglieri M , Nevo Y , Kuntz N , McDonald CM , Tulinius M , Ryan MM , Webster R , Castro D , Finkel RS , Smith AL , Morgenroth LP , Arrieta A , Shimony M , Jaros M , Shale P , McCall JM , Hathout Y , Nagaraju K , van den Anker J , Ward LM , Ahmet A , Cornish MR , Clemens PR Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug, Pharmacol Res (2018) ;136: :140–50. doi: 10.1016/j.phrs.2018.09.007. |
[44] | Fang Y , McDonald CM , Clemens PR , Gordish-Dressman H , Illei K , Hoffman EP , CINRG DNHS and Vamorolone 002/003/LTE Investigators Dang UJ Modeling early heterogeneous rates of progression in boys with Duchenne muscular dystrophy, J Neuromuscul Dis (2023) ;10: (3):349–64. doi: 10.3233/Jnd-221527. |
[45] | Dang UJ , Ziemba M , Clemens PR , Hathout Y , Conklin LS , CINRG Vamorolone 002/003 Investigators Hoffman EP . Serum biomarkers associated with baseline clinical severity in young steroid-naive Duchenne muscular dystrophy boys, Hum Mol Genet (2020) ;29: (15):2481–95. doi: 10.1093/hmg/ddaa132. |
[46] | Heier CR , Yu Q , Fiorillo AA , Tully CB , Tucker A , Mazala DA , Uaesoontrachoon K , Srinivassane S , Damsker JM , Hoffman EP , Nagaraju K , Spurney CF . Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy, Life Science Alliance. (2019) ;2: (1):e00186. doi: 10.26508/lsa.201800186. |
[47] | Ziemba M , Barkhouse M , Uaesoontrachoon K , Giri M , Hathout Y , Dang UJ , Gordish-Dressman H , Nagaraju K , Hoffman EP . Biomarker-focused multi-drug combination therapy and repurposing trial in mdx mice, PLoS One (2021) ;16: (2):e0246507. doi: 10.1371/journal.pone.0246507. |
[48] | Fiorillo AA , Tully CR , Damsker JM , Nagaraju K , Hoffman EP , Heier CR Muscle miRNAome shows suppression of chronic inflammatory miRNAs with both prednisone and vamorolone, Physiol Genomics (2018) ;50: (9):735–45. doi: 10.1152/physiolgenomics.00134.2017. |
[49] | Sreetama SC , Chandra G , Van der Meulen JH , Ahmad MM , Suzuki P , Bhuvanendran S , Nagaraju K , Hoffman EP , Jaiswal JK Membrane stabilization by modified steroid offers a potential therapy for muscular dystrophy due to dysferlin deficit, Mol Ther (2018) ;26: (9):2231–42. doi: 10.1016/j.ymthe.2018.07.021. |
[50] | Damsker JM , Dillingham BC , Rose MC , Balsley MA , Heier CR , Watson AM , Stemmy EJ , Jurjus RA , Huynh T , Tatem K , Uaesoontrachoon K , Berry DM , Benton AS , Freishtat RJ , Hoffman EP , McCall JM , Gordish-Dressman H , Constant SL , Reeves EKM , Nagaraju K . VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice, PLoS One (2013) ;8: (5):e63871γ. doi: 10.1371/journal.pone.0063871. |
[51] | Wells E , Kambhampati M , Damsker JM , Gordish-Dressman H , Yadavilli S , Becher OJ , Gittens J , Stampar M , Packer RJ , Nazarian J . Vamorolone, a dissociative steroidal compound, reduces pro-inflammatory cytokine expression in glioma cells and increases activity and survival in a murine model of cortical tumor, Oncotarget (2017) ;8: (6):9366–74. doi: 10.18632/oncotarget.14070. |
[52] | Akkad H , Cacciani N , Llano-Diez M , Kalamgi RC , Tchkonia T , Kirkland JL , Larsson L . Vamorolone treatment improves skeletal muscle outcome in a critical illness myopathy rat model, Acta Physiol. (2019) ;225: (2):e13172γ. doi: 10.1111/apha.13172. |
[53] | Damsker JM , Conklin LS , Sadri S , Dillingham BC , Panchapakesan K , Heier CR , McCall JM , Sandler AD . VBP15, a novel dissociative steroid compound, reduces NFκB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis, Inflammation Research (2016) ;65: (9):737–43. doi: 10.1007/s00011-016-0956-8. |
[54] | Dillingham BC , Knoblach SM , Many GM , Harmon BT , Mullen AM , Heier CR , Bello L , McCall JM , Hoffman EP , Connor EM , Nagaraju K , Reeves EKM , Damsker JM VBP15, a novel anti-inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis, Cell Mol Neurobiol (2015) ;35: (3):377–87. doi: 10.1007/s10571-014-0133-y. |
[55] | Damsker JM , Cornish MR , Kanneboyina P , Kanneboyina I , Yu Q , Lipson R , Phadke A , Knoblach SM , Panchapakesan K , Morales M , Fiorillo AA , Partridge T , Nagaraju K Vamorolone, a dissociative steroidal compound, reduces collagen antibody-induced joint damage and inflammation when administered after disease onset, Inflammation Research (2019) ;68: (11):969–80. doi: 10.1007/s00011-019-01279-z. |
[56] | Almeida LEF , Damsker JM , Albani S , Afsar N , Kamimura S , Pratt D , Kleiner DE , Quezado M , Gordish-Dressman H , Quezado ZMN The corticosteroid compounds prednisolone and vamorolone do not alter the nociception phenotype and exacerbate liver injury in sickle cell mice, Sci Rep (2018) ;8: (1):6081. doi: 10.1038/s41598-018-24274-6. |
[57] | Hoffman EP , Riddle V , Siegler MA , Dickerson D , Backonja M , Kramer WG , Nagaraju K , Gordish-Dressman H , Damsker JM , McCall JM Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes, Steroids (2018) ;134: :43–52. doi: 10.1016/j.steroids.2018.02.010. |
[58] | Elhalag RH , Motawea KR , Talat NE , Rouzan SS , Shah JF . Efficacy of vamorolone in treatment of Duchene muscle dystrophy, A meta-analysis. Front Neurol. (2023) ;14: :1107474γ. doi: 10.3389/fneur.2023.1107474. |
[59] | Escolar DM , Hache LP , Clemens PR , Cnaan A , McDonald CM , Viswanathan V , Kornberg AJ , Bertorini TE , Nevo Y , Lotze T , Pestronk A , Ryan MM , Monasterio E , Day JW , Zimmerman A , Arrieta A , Henricson E , Mayhew J , Florence J , Hu F , Connolly AM . Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy, Neurology (2011) ;77: (5):444–52. doi: 10.1212/WNL.0b013e318227b164. |
[60] | Ricotti V , Ridout DA , Pane M , Main M , Mayhew A , Mercuri E , Manzur AY , Muntoni F , Robb S , Quinlivan R , Sarkozy A , Butler J , Bushby K , Straub V , Guglieri M , Eagle M , Roper H , McMurchie H , Childs A , Pysden K , Pallant L , Spinty S , Peachey G , Shillington A , Wraige E , Jungbluth H , Sheehan J , Spahr R , Hughes I , Bateman E , Cammiss C , Willis T , Groves L , Emery N , Baxter P , Senior M , Scott E , Hartley L , Parsons B , Majumdar A , Jenkins L , Toms B , Naismith K , Keddie A , Horrocks I , Di Marco M , Chow G , Miah A , de Goede C , Thomas N , Geary M , Palmer J , White C , Greenfield K , Wilson I , Messina S , Berardinelli A , Comi G , D’Amico A , Bertini E , Bruno C , Politano L , Battini R , Pegoraro E , Pini A , Mongini T , Morandi L , Network UNC . The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials, J Neurol Neurosur Ps (2016) ;87: (2):149–55. doi: 10.1136/jnnp-2014-309405. |
[61] | McDonald CM , Henricson EK , Abresch RT , Duong TN , Joyce NC , Hu FM , Clemens PR , Hoffman EP , Cnaan A , Gordish-Dressman H , Investigators C . Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. (2018) ;391: (10119):451–61. doi: 10.1016/S0140-6736(17)32160-8. |
[62] | Bello L , Gordish-Dressman H , Morgenroth LP , Henricson EK , Duong T , Hoffman EP , Cnaan A , McDonald CM , Investigators C Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study, Neurology (2018) ;85: (12):1048–55. doi: 10.1212/Wnl.0000000000001950. |
[63] | Thangarajh M , Spurney CF , Gordish-Dressman H , Clemens PR , Hoffman EP , McDonald CM , Henricson EK , Investigators C Neurodevelopmental Needs in Young Boys with Duchenne Muscular Dystrophy (DMD): Observations from the Cooperative International Neuromuscular Research Group (CINRG) DMD Natural History Study (DNHS). PLoS Curr. 2018;10. doi: 10.1371/currents.md.4cdeb6970e54034db2bc3dfa54b4d987. |
[64] | McDonald CM Timed function tests have withstood the test of time as clinically meaningful and responsive endpoints in duchenne muscular dystrophy, Muscle Nerve (2018) ;58: (5):614–7. doi: 10.1002/mus.26334. |
[65] | Nair AB , Jacob S A simple practice guide for dose conversion between animals and human, Journal of Basic and Clinical Pharmacy (2016) ;7: (2):27–31. doi: 10.4103/0976-0105.177703. |
[66] | Wood CL , Straub V , Guglieri M , Bushby K , Cheetham T Short stature and pubertal delay in Duchenne muscular dystrophy, Arch Dis Child (2016) ;101: (1):101–6. doi: 10.1136/archdischild-2015-308654. |
[67] | Joseph S , Wang C , Bushby K , Guglieri M , Horrocks I , Straub V , Ahmed SF , Wong SC Network ftUNC Fractures and linear growth in a nationwide cohort of boys with Duchenne muscular dystrophy with and without glucocorticoid treatment: Results from the UK NorthStar database, JAMA Neurology (2019) ;76: (6):701–9. doi: 10.1001/jamaneurol.2019.0242. |
[68] | Baudy AR , Reeves EKM , Damsker JM , Heier C , Garvin LM , Dillingham BC , McCall J , Rayavarapu S , Wang Z , Vandermeulen JH , Sali A , Jahnke V , Duguez S , DuBois D , Rose MC , Nagaraju K , Hoffman EP Δ-9,11 modification of glucocorticoids dissociates nuclear factor-κB inhibitory efficacy from glucocorticoid response element-associated side effects, Journal of Pharmacology and Experimental Therapeutics (2012) ;343: (1):225–32. doi: 10.1124/jpet.112.194340. |
[69] | Huynh T , Uaesoontrachoon K , Quinn JL , Tatem KS , Heier CR , Van Der Meulen J H , Yu Q , Harris M , Nolan CJ , Haegeman G , Grounds MD , Nagaraju K Selective modulation through the glucocorticoid receptor ameliorates muscle pathology in mdx mice, The Journal of Pathology (2013) ;231: (2):223–35. doi: 10.1002/path.4231. |
[70] | Han R , Campbell KP Dysferlin and muscle membrane repair, Curr Opin Cell Biol (2007) ;19: (4):409–16. doi: 10.1016/j.ceb.2007.07.001. |
[71] | van Putten M , Lloyd EM , de Greef JC , Raz V , Willmann R , Grounds MD . Mouse models for muscular dystrophies: an overview, Dis Model Mech (2020) ;13: (2):dmm043562. doi: 10.1242/dmm.043562. |
[72] | Lostal W , Bartoli M , Roudaut C , Bourg N , Krahn M , Pryadkina M , Borel P , Suel L , Roche JA , Stockholm D , Bloch RJ , Levy N , Bashir R , Richard I Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy, PLoS One (2012) ;7: (5):e38036γ. doi: 10.1371/journal.pone.0038036. |
[73] | Grounds MD , Terrill JR , Radley-Crabb HG , Robertson T , Papadimitriou J , Spuler S , Shavlakadze T Lipid accumulation in dysferlin-deficient muscles, Am J Pathol (2014) ;184: (6):1668–76. doi: 10.1016/j.ajpath.2014.02.005. |
[74] | Zweyer M , Sabir H , Dowling P , Gargan S , Murphy S , Swandulla D , Ohlendieck K Histopathology of Duchenne muscular dystrophy in correlation with changes in proteomic biomarkers, Histol Histopathol (2022) ;37: (2):101–16. doi: 10.14670/Hh-18-403. |
[75] | Ogundele M , Zhang JS , Goswami MV , Barbieri ML , Dang UJ , Novak JS , Hoffman EP , Nagaraju K , Hathout Y , Investigators C-D . Validation of chemokine biomarkers in Duchenne muscular dystrophy, Life-Basel (2021) ;11: (8):827. doi: 10.3390/life11080827. |
[76] | Signorelli M , Tsonaka R , Aartsma-Rus A , Spitali P . Multiomic characterization of disease progression in mice lacking dystrophin, PLoS One (2023) ;18: (3):e0283869. doi: 10.1371/journal.pone.0283869. |
[77] | Riddell D , Hildyard J , Harron R , Wells D , Piercy R Longitudinal assessment of blood-borne musculoskeletal disease biomarkers in the DE50-MD dog model of Duchenne muscular dystrophy [version 2; peer review: 2 approved], Wellcome Open Research (2022) ;6: (354):354. doi: 10.12688/wellcomeopenres.17398.2. |
[78] | Riddell DO , Hildyard JCW , Harron RCM , Hornby NL , Wells DJ , Piercy RJ Serum inflammatory cytokines as disease biomarkers in the DE50-MD dog model of Duchenne muscular dystrophy, Dis Model Mech. (2022) ;15: (12):dmm049394γ. doi: 10.1242/dmm.049394. |
[79] | Kuraoka M , Aoki Y , Takeda S . Development of outcome measures according to dystrophic phenotypes in canine X-linked muscular dystrophy in Japan, Exp Anim (2021) ;70: (4):419–30. doi: 10.1538/expanim.21-0072. |
[80] | Al-Mshhdani BA , Grounds MD , Arthur PG , Terrill JR A blood biomarker for Duchenne muscular dystrophy shows that oxidation state of albumin correlates with protein oxidation and damage in mdx muscle, Antioxidants (2021) ;10: (8):1241. doi: 10.3390/antiox10081241. |
[81] | Alic L , Griffin JF , Eresen A , Kornegay JN , Ji JX Using MRI to quantify skeletal muscle pathology in Duchenne muscular dystrophy: A systematic mapping review, Muscle Nerve (2021) ;64: (1):8–22. doi: 10.1002/mus.27133. |
[82] | David F , Dixit R How to prevent the next Marathon Pharmaceuticals [version 1; peer review: 2 approved, 1 approved with reservations], FRes (2018) ;7: (74):74. doi: 10.12688/f1000research.13678.1. |
[83] | Robbins R , Nolen S A Dilemma for Governments: How to Pay for Million-Dollar Therapies. The New York Times. 2023 25 Jan;Sect. A. |
[84] | Miyatake S , Shimizu-Motohashi Y , Takeda S , Aoki Y . Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors, Drug Design Development and Therapy (2016) ;10: :2745–58. doi: 10.2147/Dddt.S110163. |
[85] | Jayash SN , Hamoudi D , Stephen LA , Argaw A , Huesa C , Joseph S , Wong SC , Frenette J , Farquharson C RANKL therapy prevents glucocorticoid-induced bone loss and promotes muscle function in a mouse model of Duchenne muscular dystrophy. Calcif Tissue Int. 2023;epub ahead of print. doi: 10.1007/s00223-023-01116-w. |
[86] | De Paepe B . What Nutraceuticals Can Do for Duchenne Muscular Dystrophy: Lessons Learned from Amino Acid Supplementation in Mouse Models, Biomedicines (2023) ;11: (7):2033. doi: 10.3390/biomedicines11072033. |
[87] | Lesovaya EA , Chudakova D , Baida G , Zhidkova EM , Kirsanov KI , Yakubovskaya MG , Budunova IV . The long winding road to the safer glucocorticoid receptor (GR) targeting therapies, Oncotarget (2022) ;13: (1):408–24. doi: 10.18632/oncotarget.28191. |


