
SERCA1 Overexpression in Skeletal Muscle Attenuates Muscle Atrophy and Improves Motor Function in a Mouse Model of ALS
Abstract
Background:
Amyotrophic lateral sclerosis (ALS) is characterized by progressive loss of muscle mass and muscle function. Previous work from our lab demonstrated that skeletal muscles from a mouse model of ALS show elevated intracellular calcium (Ca2+) levels and heightened endoplasmic reticulum (ER) stress.
Objective:
To investigate whether overexpression of sarcoplasmic reticulum (SR) Ca2+ ATPase 1 (SERCA1) in skeletal muscle would improve intracellular Ca2+ handling, attenuate ER stress, and improve motor function ALS transgenic mice.
Methods:
B6SJL-Tg (SOD1*G93A)1Gur/J (ALS-Tg) mice were bred with skeletal muscle α-actinin SERCA1 overexpressing mice to generate wild type (WT), SERCA1 overexpression (WT/+SERCA1), ALS-Tg, and SERCA1 overexpressing ALS-Tg (ALS-Tg/+SERCA1) mice. Motor function (grip test) was assessed weekly and skeletal muscles were harvested at 16 weeks of age to evaluate muscle mass, SR-Ca2+ ATPase activity, levels of SERCA1 and ER stress proteins - protein disulfide isomerase (PDI), Grp78/BiP, and C/EBP homologous protein (CHOP). Single muscle fibers were also isolated from the flexor digitorum brevis muscle to assess changes in resting and peak Fura-2 ratios.
Results:
ALS-Tg/+SERCA1 mice showed improved motor function, delayed onset of disease, and improved muscle mass compared to ALS-Tg. Further, ALS-Tg/+SERCA1 mice returned levels of SERCA1 protein and SR-Ca2+ ATPase activity back to levels in WT mice. Unexpectedly, SERCA-1 overexpression increased levels of the ER stress maker Grp78/BiP in both WT and ALS-Tg mice, while not altering protein levels of PDI or CHOP. Lastly, single muscle fibers from ALS-Tg/+SERCA1 had similar resting but lower peak Fura-2 levels (at 30 Hz and 100 Hz) compared to ALS-Tg mice.
Conclusions:
These data indicate that SERCA1 overexpression attenuates the progressive loss of muscle mass and maintains motor function in ALS-Tg mice while not lowering resting Ca2+ levels or ER stress.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a devastating neurodegenerative disease with an incidence rate of 3 per 100,000 in the United States [1]. The clinical features of ALS include progressive muscle atrophy, weakness and paralysis leading to respiratory failure and death on average 3-5 years after disease onset [1]. The progressive loss of muscle function is due to upper and lower motor neuron degeneration. Among the genetic causes of ALS, mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene contribute to ∼20% of all familial ALS cases. Transgenic mouse models representing SOD1 mutations have been developed [2], well characterized, and greatly increased our understanding of the pathophysiological mechanisms of ALS [3–5]. The Glycine to Alanine at codon 93 (G93A) mutation of SOD1 results in a “gain-of-function” toxicity from increased production of reactive oxygen species (i.e., H2O2) with several proposed downstream pathological mechanisms, such as glutamate excitotoxicity, mitochondrial dysfunction, and intracellular calcium (Ca2+) dysregulation [6].
In skeletal muscle, there are rapid fluctuations in intracellular Ca2+ during muscle contraction, with levels increasing 40 to 100-fold during contractile activity [7]. Consequently, both resting Ca2+, Ca2+ release during contraction and Ca2+ reuptake are mechanisms tightly regulated. The primary regulators in skeletal muscle are the Ca2+ release channels (ryanodine receptor; RyR), Ca2+ pump proteins (sarco/endoplasmic reticulum Ca2+-ATPase; SERCA) and cytosolic Ca2+ buffering proteins (parvalbumin; PV). Redox modifications and impaired function of these Ca2+-handling proteins is detrimental to their ability to bind or sequester Ca2+ [8–10]. Thus, redox-modified proteins such as RyR and SERCA may contribute to elevations in Ca2+ and muscle pathology in various muscle diseases including ALS [11] and Duchenne muscular dystrophy (DMD) [12, 13]. Forced overexpression of Ca2+ handling proteins that increase Ca2+ influx into muscle can induce a pathology that mimics DMD [14–16] while improvements in Ca2+ clearance by overexpression of the fast-twitch SERCA isoform (SERCA1) has been shown to rescue the muscle pathology and improve function in different mouse models of DMD [12, 13]. Thus, improving intracellular Ca2+ homeostasis may be beneficial for neuromuscular diseases where Ca2+ overload is part of the downstream muscle pathology.
Our lab has previously reported elevations in intracellular Ca2+ in single muscle fibers from G93A*SOD1 mice [11]. This was associated with decreased levels of SERCA1, SERCA2 and PV protein in G93A*SOD1 muscles. We have also reported an increase in the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress markers in skeletal muscles of G93A*SOD1 mice which are thought to be due to impaired sarcoplasmic reticulum (SR)/ER intracellular Ca2+ regulation. Based on these findings we hypothesized that improving intracellular Ca2+ clearance capacity in skeletal muscle of ALS mice would ameliorate muscle atrophy, improve motor function, and attenuate the ALS phenotype. We also hypothesized that it would mitigate the ER stress response in skeletal muscle. Thus, the purpose of this study was to evaluate whether disease phenotypic changes would be improved in ALS-Tg mice by overexpression of SERCA1.
MATERIALS AND METHODS
Ethical approval
All procedures were conducted under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Maryland, College Park.
Animals
Control (B6SJL-Tg(SOD1)2Gur/J) female (strain number: 002297) and B6SJL-Tg(SOD1*G93A)1Gur/J (ALS-Tg) (strain number: 002726) male mice were obtained from Jax Laboratories and bred to established a colony at the animal facility of University of Maryland, College Park as previously described [11]. Male and female skeletal muscle SERCA1 overexpression mice (+SERCA1) were obtained from Cincinnati Children’s Hospital Medical Center. SERCA1 overexpression is restricted to skeletal muscle by a modified human skeletal muscle α-actinin promoter [17]. SERCA1 Tg breeders were then used to establish a colony of +SERCA1 mice at the University of Maryland Central Animal Research Facility as previously described [13]. Male wild type (WT)/+SERCA1 mice were bred with female ALS-Tg mice to obtain 4 genotypes: i) WT; ii) SERCA1 overexpression (WT/+SERCA1); iii) G93A*SOD1 (ALS-Tg), and iv) SERCA1 overexpressing G93A*SOD1 (ALS-Tg/+SERCA1). This study was designed to obtain 8-10 mice per genotype. All mice were weaned at 21 days and genotyped for expression of the G93A SOD1 mutation and for SERCA1 using primer sequences and methods previously described [11, 13]. All mice were kept in the same room condition (typical ambient conditions 20.9% O2 and 22±1°C) with equal access to food and water, bedding, and light cycles (12 h light/12 h dark). Mice were used at 16 weeks of age.
Assessment of motor function and disease onset
Motor function of the mice was evaluated weekly from 9 to 16 weeks of age with a grip test. Briefly, the grip test is the time the mice were able to hold their body mass while suspended from a wire mesh lid as previously described [11]. In this study, each mouse was subjected to three trials with the maximum test time being 180 s and a 10 min rest period given between trials. The average of three trials was used for reporting grip test performance. The test was conducted and analyzed in a blinded fashion. Disease onset was defined as the time corresponding to the first signs of myotonic symptoms such as muscle tremor or hindlimb stiffness. The reduction in grip test performance was used as another criterion to evaluate disease progression.
Experimental procedures
At 16 weeks of age mice were euthanized by CO2 inhalation followed by cervical dislocation. Skeletal muscles including gastrocnemius (GAS), tibialis anterior (TA) and quadriceps (QUAD) were removed and weighed by investigators who were blinded to the genotype of the mice. Muscle samples were quick frozen in liquid nitrogen and stored at –80°C for subsequent analyses. At the time of sacrifice, the flexor digitorum brevis (FDB) muscle was removed from a subset of mice (n = 4 for each genotype; 3 females, 1 male each) and single muscle fibers isolated for assessing [Ca2+]i levels.
Single muscle fiber isolation and free [Ca2+]i measurements
Detailed methods for single muscle fiber isolation and [Ca2+]i measurements have been previously described [18]. Briefly, single muscle fibers were obtained from the FDB muscle by collagenase digestion with type 2 collagenase (Worthington) in minimal essential medium (MEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen). After incubation at 37°C in 95% O2-5% CO2, single muscle fibers were obtained by trituration. Subsequently, fibers were maintained in MEM solution with 10% FBS at 37°C, 95% O2-5% CO2 until used for [Ca2+]i assessment.
One day after dissection, fibers were loaded with Fura-2AM for 15 min. The Fura-2 ratio was measured in response to varying stimuli (see protocol below) as an index of [Ca2+]i. Fibers loaded with Fura-2AM were placed in a stimulation chamber containing parallel electrodes and the chamber was positioned on a Nikon TiU microscope stage. Muscle fibers were continuously perfused with a Tyrode solution (121.0 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24.0 mM NaHCO3, and 5.5 mM glucose) with 0.2% FBS [19]. The solution was bubbled with 95% O2-5% CO2 to maintain a pH of 7.3 [19]. Levels of [Ca2+]i were assessed by the Fura-2 fluorescence ratio using an IonOptix Hyperswitch system with dual excitation, single emission filter set for Fura-2 (excitation 340 nm and 380 nm; emission 510 nm). Signals were captured and analyzed using the IonWizard software (IonOptix). Global Fura-2 ratio was measured in muscle fibers using trains of stimuli at 10, 30, 50, 70, 100, 120, and 150 Hz for 350 ms with fibers resting 1 min between frequencies. Peak Fura-2 ratios at each frequency were determined by the average ratio in the last 100 ms of the 350 ms tetanus, when Ca2+ Fura-2 should be at a steady state. All single muscle fibers were evaluated at room temperature. Fura-2 signals were measured in 7-9 fibers per mouse for a total of 28-33 fibers per genotype: WT (n = 33); WT/+SERCA1 (n = 33); ALS-Tg (n = 28); ALS-Tg/+SERCA1 (n = 33).
Muscle protein expression
The QUAD was used to confirm SERCA1 overexpression and to assess UPR and ER stress responses in skeletal muscle as previously described [11, 20]. The ER stress response was assessed by measuring protein expression of the ER stress sensors protein disulfide isomerase (PDI) and Grp78/BiP, and the ER stress-specific cell death signaling protein C/EBP homologous protein (CHOP). Briefly, QUAD was homogenized in lysis buffer containing 20 mM Hepes buffer (pH = 7.4), 150 mM NaCl, 1.5 mM MgCl2, 0.1% Triton X-100, 20 % Glycerol, 1 mM DTT and protease inhibitors (cOmplete mini EDTA-free protease inhibitor cocktail, Roche Diagnostics, Indianapolis, IN, USA). After tissue homogenization, samples were kept at 4°C for 20 min and then centrifuged at 20,000 g. Supernatant was collected and frozen at –80°C until used for protein expression analyses. Sample protein concentration was determined using a BCA assay (Thermo Fisher Scientific Inc., Rockford, IL, USA). For assessing SERCA1, PDI, Grp78/BiP and CHOP protein levels, 30μg of total protein was used. Protein samples were solubilized in 5 x loading buffer and denatured by incubation at 100°C for 5 min. Denatured protein samples were loaded on 8% bis-acrylamide gels and separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After gel electrophoresis, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (EMD Millipore, Billerica, MA, USA) and then blocked with 5% non-fat milk at room temperature for 1 h. The following antibodies and dilutions were used to determine protein expression levels: SERCA1 (1 : 2500, Thermo Fisher Scientific Inc., Rockford, IL, USA) and PDI, Grp78/BiP and CHOP (1 : 1000, Cell Signaling Technology, Danvers, MA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primary antibody (1 : 2000, Thermo Fisher Scientific Inc., Rockford, IL, USA) was used as a total protein loading control. Protein levels were quantified by band densitometry (Bio-Rad Laboratories Inc., Hercules CA) and protein expression levels were expressed in arbitrary units (AU).
Determination of maximal SERCA1 ATPase activity
The QUAD muscle was also used to determine maximal SERCA1 ATPase activity using the method described in Chin et al. 1994 [21] modified to a 96 well plate format [13]. Briefly, QUAD muscles were homogenized in buffer containing 200 mM Sucrose, 10 mM NaN3, 1 mM EDTA, and 40 mM L-histidine (pH 7.8) with a 1 : 20 ratio (w/v, 0.1 g tissue: 2 mL buffer). The Ca2+-ATPase reaction was measured in a crude homogenate in a reaction buffer containing 20 mM Hepes buffer (pH 7.5), 200 mM KCl, 15 mM MgCl2, 10 mM NaN3, 1 mM EGTA, 5 mM ATP, 10 mM phosphoenolpyruvate, 18 U/mL lactate dehydrogenase, 18 U/mL pyruvate kinase, 4μM calcium ionophore A23187 and 0.3 mM NADH. Total ATPase activity was assessed by adding 1 mM CaCl2 and basal ATPase was measured by adding 1 mM CaCl2 and 5 mM cyclopiazonic acid, a selective SERCA inhibitor. Samples were analyzed in triplicate in a 96-well plate at 37°C by measuring NADH absorbance at 340 nm. Maximal SR Ca2+ ATPase activity was defined as the difference between total ATPase and basal ATPase activity.
Statistical analysis
Values are expressed as mean±standard error of the mean (SEM). To evaluate differences between WT, WT/+SERCA1, ALS-Tg and ALS-Tg/+SERCA1 mice, data were analyzed using Student’s T-tests to compare between genotypes, with p < 0.05 used to determine statistical significance.
RESULTS
SERCA1 expression and maximal SR Ca2+-ATPase activity in skeletal muscle of ALS-Tg and +SERCA1 Tg mice
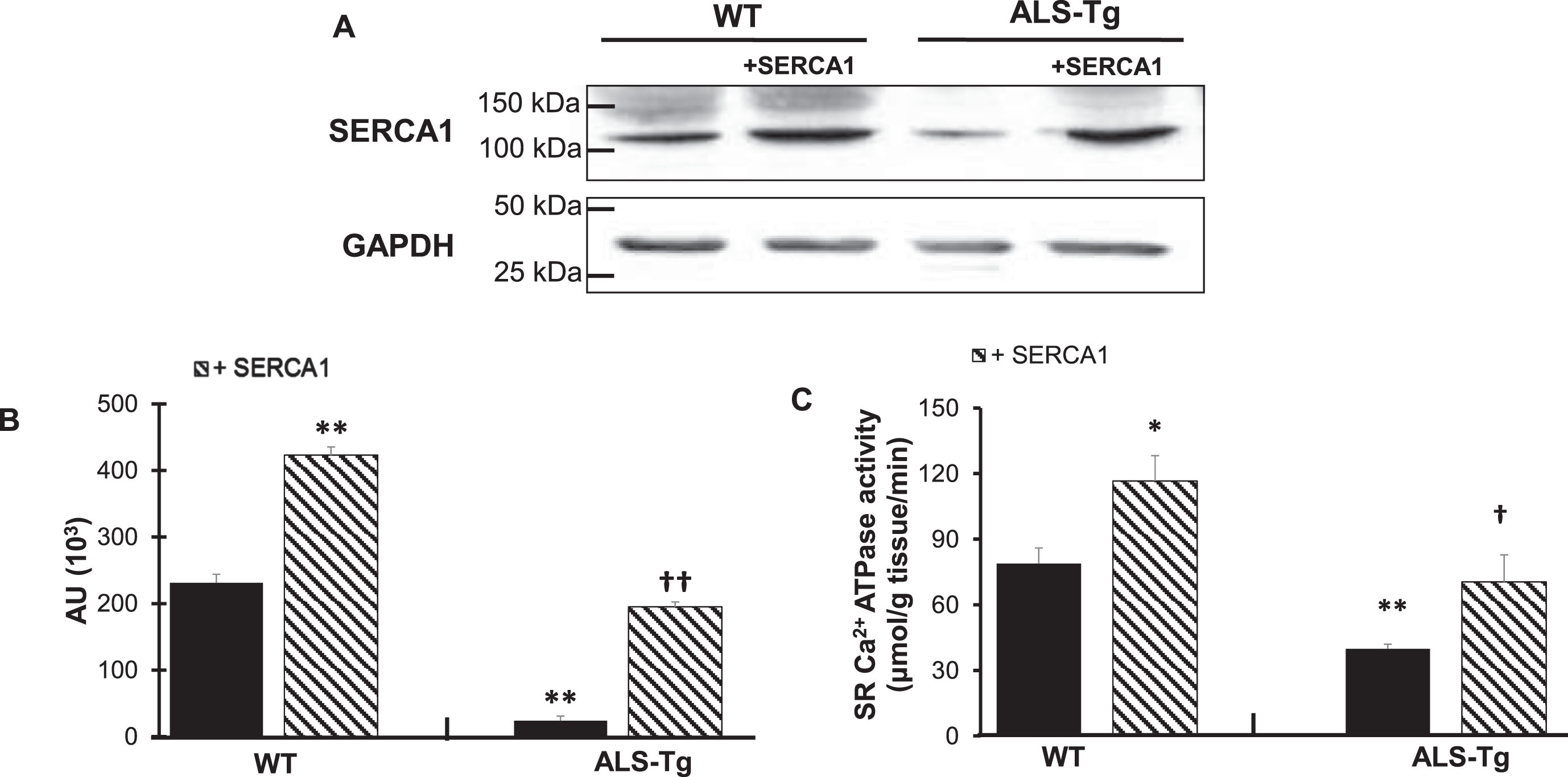
Western blot analysis was used to confirm SERCA1 overexpression in the QUAD muscle of the WT/+SERCA1 and ALS-Tg/+SERCA1 mice (Fig. 1A and 1B). SERCA1 protein levels were on average 1.8-fold higher in QUAD of WT/+SERCA1 compared to WT mice (p < 0.01). Consistent with our previous report, SERCA1 protein levels were lower in QUAD of ALS-Tg mice compared to WT (p < 0.01). SERCA1 overexpression resulted in 8.1-fold higher SERCA1 protein levels in QUAD from ALS-Tg/+SERCA1 mice compared to ALS-Tg mice (p < 0.01). More importantly, SERCA1 overexpression in muscles from ALS-Tg increased SERCA1 protein to similar levels as in WT mice. To determine whether this increase in SERCA1 protein levels resulted in increased function of the Ca2+-ATPase pump protein, we measured muscle SR Ca2+-ATPase activity (Fig. 1C). In QUAD muscles of ALS-Tg mice, maximal Ca2+-ATPase activity was 50% of WT levels (p < 0.01). SERCA1 overexpression increased maximal Ca2+-ATPase activity in QUAD of both WT/+SERCA1 (1.5-fold vs. WT; p < 0.05) and ALS-Tg/+SERCA1 (1.8-fold vs. ALS-Tg; p < 0.05) mice. In ALS-Tg/+SERCA1, the maximal Ca2+-ATPase activity was similar from WT levels. These data confirm that SERCA1 protein levels as well as maximal SR Ca2+-ATPase activity were increased in skeletal muscle of WT/+SERCA and ALS-Tg/+SERCA mice and that both returned to normal WT levels in ALS-Tg/+SERCA1 mice.
Fig. 1
SERCA1 overexpression increases SERCA1 protein levels and maximal SR Ca2+-ATPase activity in quadriceps muscles in WT and ALS-Tg mice. A) Representative western blot image for SERCA1 protein level in quadriceps (QUAD) muscle from WT and ALS-Tg mice with and without SERCA1 overexpression. B) Quantitative analysis of western blot images by densitometry. Data shown are in arbitrary units (AU) for WT (n = 4; 1 male and 3 female), WT/+SERCA1 (n = 4; 1 male and 3 female), ALS-Tg (n = 4; 1 male and 3 female) and ALS-Tg/+SERCA1 (n = 4; 1 male and 3 female). C) Maximal SR Ca2+-ATPase activity of QUAD muscle of WT (n = 4), WT/+SERCA1 (n = 4), ALS-Tg (n = 4) and ALS-Tg/+SERCA1 (n = 4). Average data (B, C) represent mean±SEM. *p < 0.05 vs. WT, **p < 0.01 vs. WT, †p < 0.05 vs. ALS-Tg, ††p < 0.01 vs. ALS-Tg.
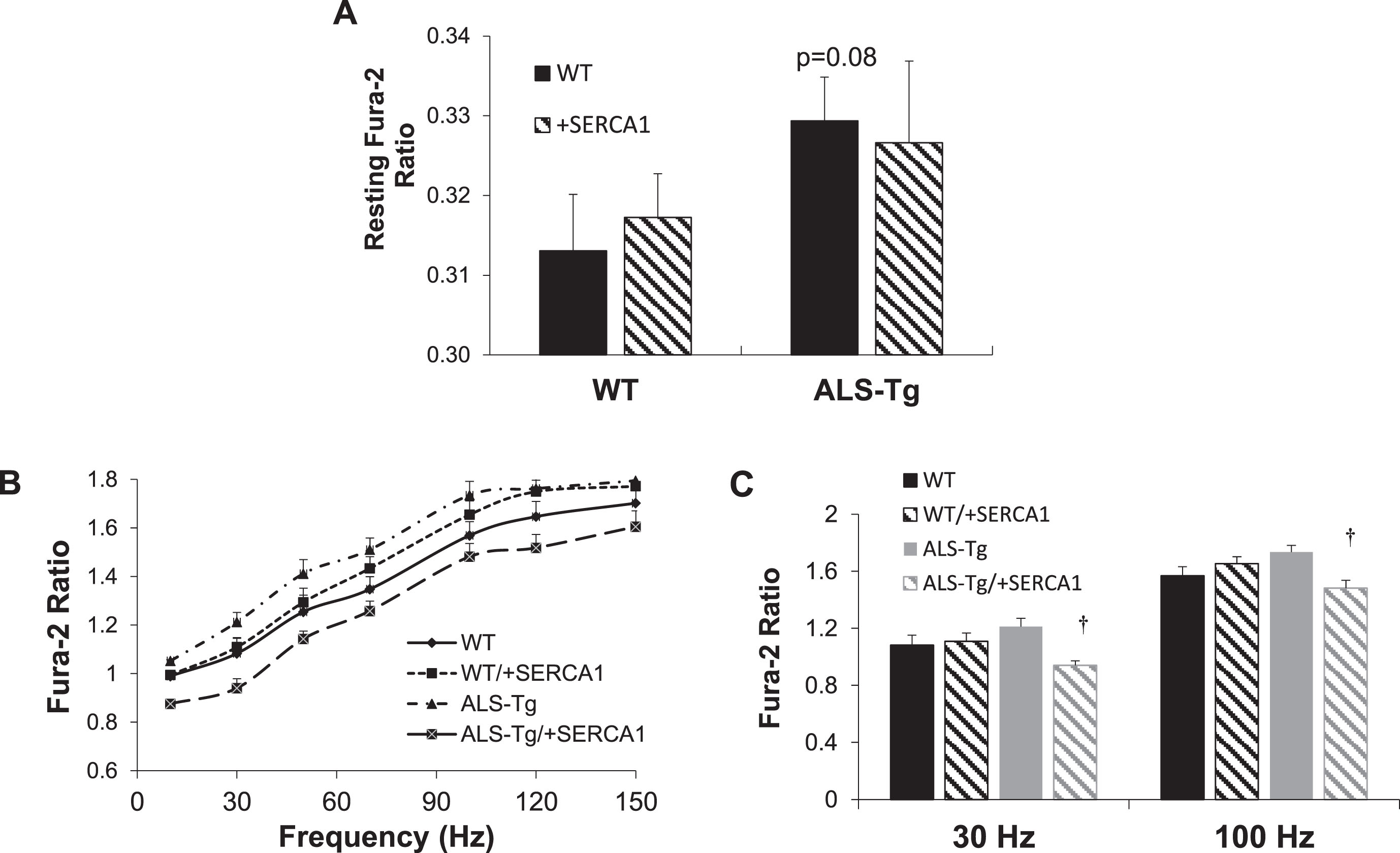
SERCA1 overexpression alters stimulation-induced Ca2+ transients in ALS-Tg mice
In order to assess the effects of SERCA1 overexpression on skeletal muscle Ca2+ cycling during contraction and relaxation, we assessed [Ca2+]i using Fura-2 in single fibers during electrical stimulation. There was no significant difference in resting Fura-2 ratio between any of the groups, although there was a trend (p = 0.08) for single fibers from ALS-Tg to be higher than that in WT fibers (Fig. 2A), consistent with previous findings. There was a significant reduction in peak Fura-2 ratios at all stimulation frequencies in fibers from ALS-Tg/+SERCA1 compared to fibers from ALS-Tg mice, with peak ratios being 16% (10 Hz), 22% (30 Hz), 19% (50 Hz), 17% (70 Hz), 15% (100 Hz), 14% (120 Hz) and 11% (150 Hz) lower in fibers from SERCA1 overexpressing ALS-Tg fibers (Fig. 2B and C). Peak Fura-2 was not different in single fibers from WT and WT/+SERCA1 or between WT and ALS-Tg.
Fig. 2
SERCA1 overexpression reduces electrical stimulation evoked Ca2+ transients in ALS-Tg mice. A) Resting Fura-2 ratios in intact single fibers from the flexor digitorum brevis (FDB) of WT and ALS-Tg mice with and without SERCA1 overexpression. B) Peak Fura-2 ratios in intact single fibers from FDB shown as a function of stimulation frequency. C) Peak Fura-2 ratios in intact single fibers from FDB at 30 and 100 Hz stimulation. Data shown are from WT (n = 33), WT/+SERCA1 (n = 33), ALS-Tg (n = 28) and ALS-Tg/+SERCA1 (n = 33). Values shown represent mean±SEM. †p < 0.05 vs. ALS-Tg.
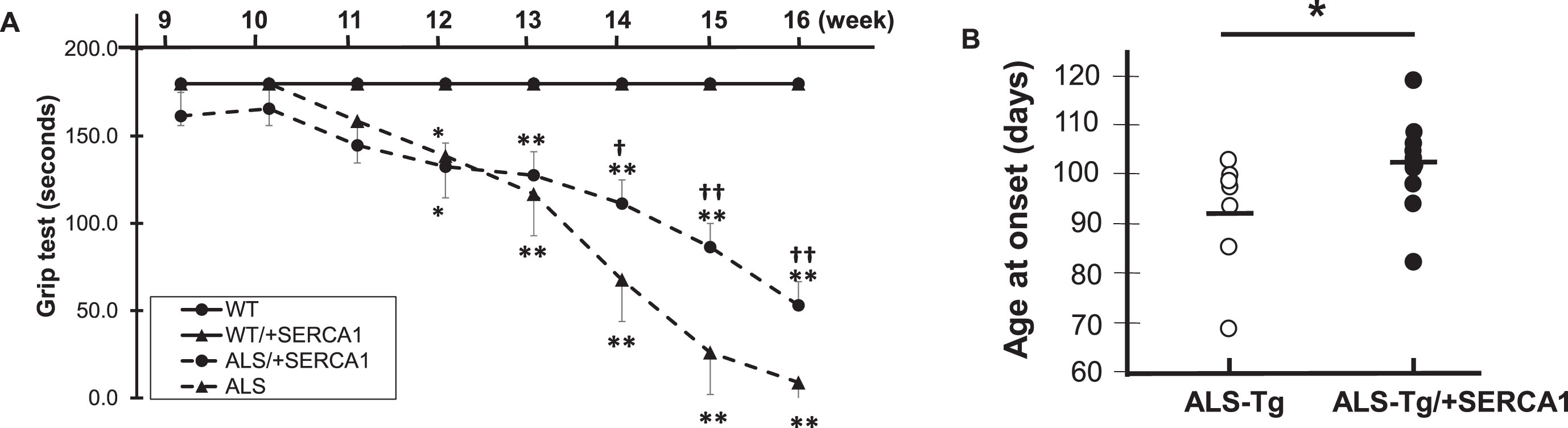
SERCA1 overexpression preserves motor function and delays disease onset in ALS-Tg mice
Deficits in motor function have been consistently observed with disease progression across the lifespan in G93A*SOD1 ALS-Tg mice [3, 19]. To determine whether skeletal muscle-specific increases in SERCA1 would improve motor function, we assessed whole body motor performance using a grip test in mice from 9 - 16 weeks of age (Fig. 3A). WT/+SERCA1 mice showed no signs of decreased motor function, with all mice able to grip for the maximum 180 sec, similar to WT mice. ALS-Tg and ALS-Tg/+SERCA1 mice showed normal motor function up to 11 weeks of age. At 12 weeks, both ALS-Tg and ALS-Tg/+SERCA1 mice showed lower grip time compared to WT mice and this reduction progressively increased until study termination (16 weeks). Despite the rapid deterioration of motor function, SERCA1 overexpression was able to preserve motor function in ALS-Tg mice. Starting at 14 weeks, grip time was longer for ALS-Tg/+SERCA1 mice compared to ALS-Tg mice: ALS-Tg/+SERCA1 grip time was 62% of WT vs. ALS-Tg which was 38% of WT levels (p < 0.05). At 16 weeks ALS-Tg/+SERCA1 mice had grip times that were 29% of WT, compared to ALS-Tg at 5% of WT levels (p < 0.01). These data indicate that SERCA1 overexpression improved grip time 24%, indicating a positive rescue in the ALS phenotype by preserving motor function.
The G93A*SOD1 mice have a shortened life span of 120d [2] with mice surviving 2 weeks after the first signs of disease onset (i.e. muscle tremor and hindlimb stiffness). To determine whether skeletal muscle specific SERCA1 overexpression could delay disease onset, we recorded the age at which disease symptoms were first observed. In ALS-Tg mice, disease onset was observed at 91±4.7 d. In contrast, ALS-Tg/+SERCA1 mice had a significant delay of disease onset (102±2.6 d; p < 0.05) (Fig. 3B).
Fig. 3
SERCA1 overexpression in skeletal muscle preserves motor function and delays disease onset in ALS-Tg mice. A) Grip test time in sec is shown over the time course of the study. B) Age of disease onset (in days) was based on first signs of muscle tremor or hindlimb stiffness. Data shown are combined for male and female mice. Values are means±SEM. *p < 0.05 or **p < 0.01 vs. WT; †p < 0.05 or †† p < 0.01 vs. ALS.
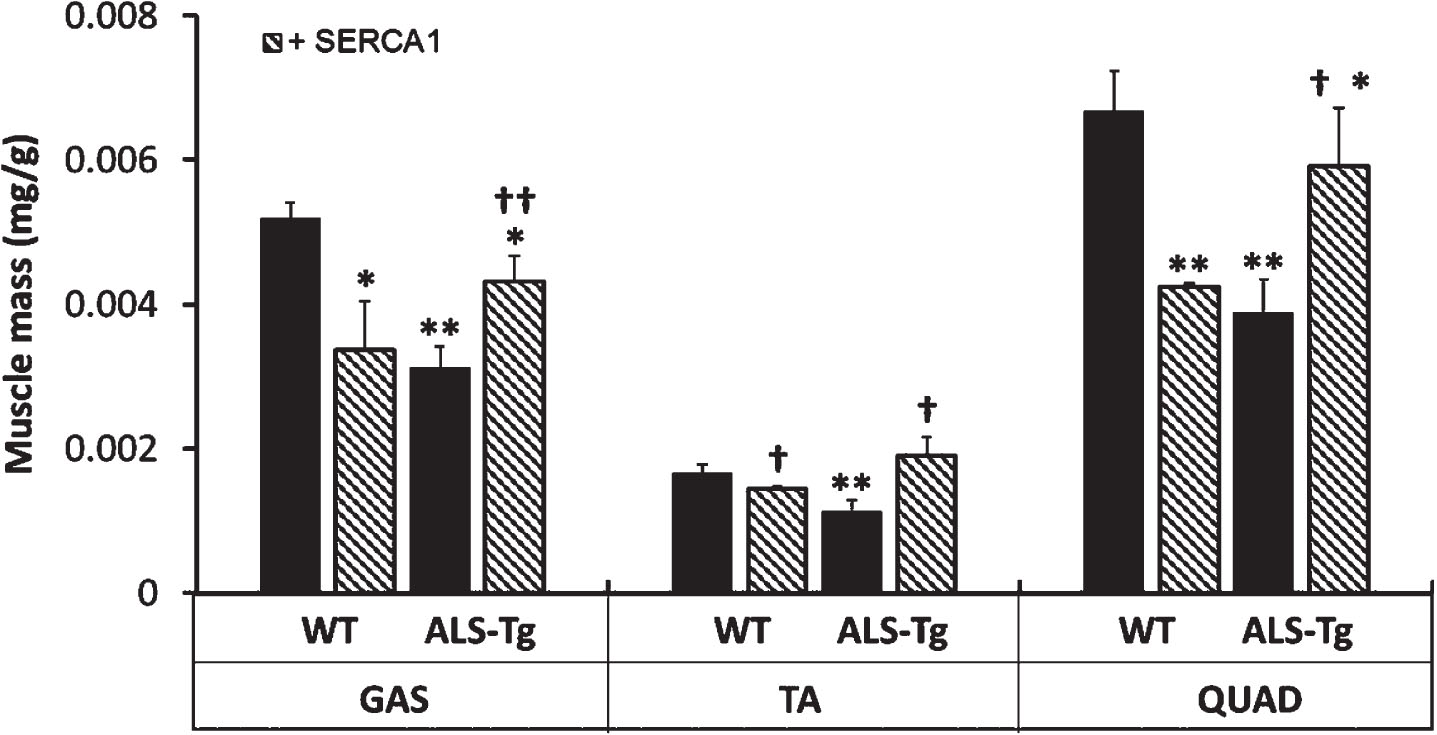
SERCA1 overexpression attenuates skeletal muscle atrophy in ALS-Tg mice
The body mass of mice used in this study are shown in Table 1. At 16 weeks of age, the average body mass of male mice was not different between any of the genotypes. However, for female mice, body mass of WT/+SERCA1 and ALS-Tg were significantly reduced compared to WT (p < 0.05 and p < 0.01, respectively). Body mass of female ALS-Tg/+SERCA1 mice was similar to ALS-Tg mice. Due to the small number of male ALS-Tg mice, the data for both sexes were combined for subsequent data analyses. Combined, the body mass of WT and WT/+SERCA1 were not different, but ALS-Tg and ALS-Tg/+SERCA1 mice were significantly lower compared to WT mice (p < 0.05). All other data reported are for male and female mice combined unless otherwise specified.
Table 1
Effects of SERCA1 overexpression on body mass
| Male | Female | Male + Female Combined | |
| WT | 30.0±1.2 | 25.9±1.1 | 28.0±1.0 |
| (n = 7) | (n = 4) | (n = 11) | |
| WT/+SERCA1 | 30.3±1.2 | 22.0±1.8*† | 26.5±1.2 |
| (n = 7) | (n = 6) | (n = 13) | |
| ALS-Tg | 29.4±11.5 | 17.7±0.9** | 21±3.5 * |
| (n = 2) | (n = 5) | (n = 7) | |
| ALS-Tg/+SERCA1 | 26.4±0.3 | 18.5±0.6** | 21.7±1.5 ** |
| (n = 5) | (n = 6) | (n = 11) |
Body mass is shown in grams (g) with number of mice (n) per group are shown in parentheses. Data represent mean±SEM. *p < 0.05 vs. WT; **p < 0.01 vs. WT; †p < 0.05 vs. ALS-Tg; ††p < 0.01 vs ALS-Tg.
A decrease in muscle mass has been documented by magnetic resonance imaging (MRI) in G93A*SOD1 mice, starting as early as 60 d [19]. In this study we assessed muscle atrophy by measuring muscle mass at 16 weeks (Fig. 4). Muscle atrophy was consistently observed in all muscles from ALS-Tg mice, with muscle mass being 43% (GAS), 50% (TA) and 42% (QUAD) of WT levels (p < 0.01). However, ALS-Tg/+SERCA1 GAS, TA, and QUAD were 40%, 67%, and 53% greater than ALS-Tg (p < 0.01 for GAS; p < 0.05 for TA and QUAD). Muscle mass in ALS-Tg/+SERCA1 was still significantly lower compared to WT muscle mass for the GAS and QUAD; however, the TA muscle mass returned to WT control levels. Interestingly, WT/+SERCA1 mice also showed lower muscle masses compared to WT, being 58% (GAS), 78% (TA) and 58% (QUAD) of WT levels (p < 0.01), which has been previously observed [13].
Fig. 4
SERCA1 overexpression in skeletal muscle attenuates muscle atrophy in ALS-Tg mice. Muscle mass is shown as relative mass (mg/g body mass) for gastrocnemius (GAS), tibialis anterior (TA), and QUAD. Data shown are combined for male and female mice. *p < 0.05 vs. WT, **p < 0.01 vs. WT; †p < 0.05 or ††p < 0.01 vs. ALS-Tg. Values shown are mean±SEM.
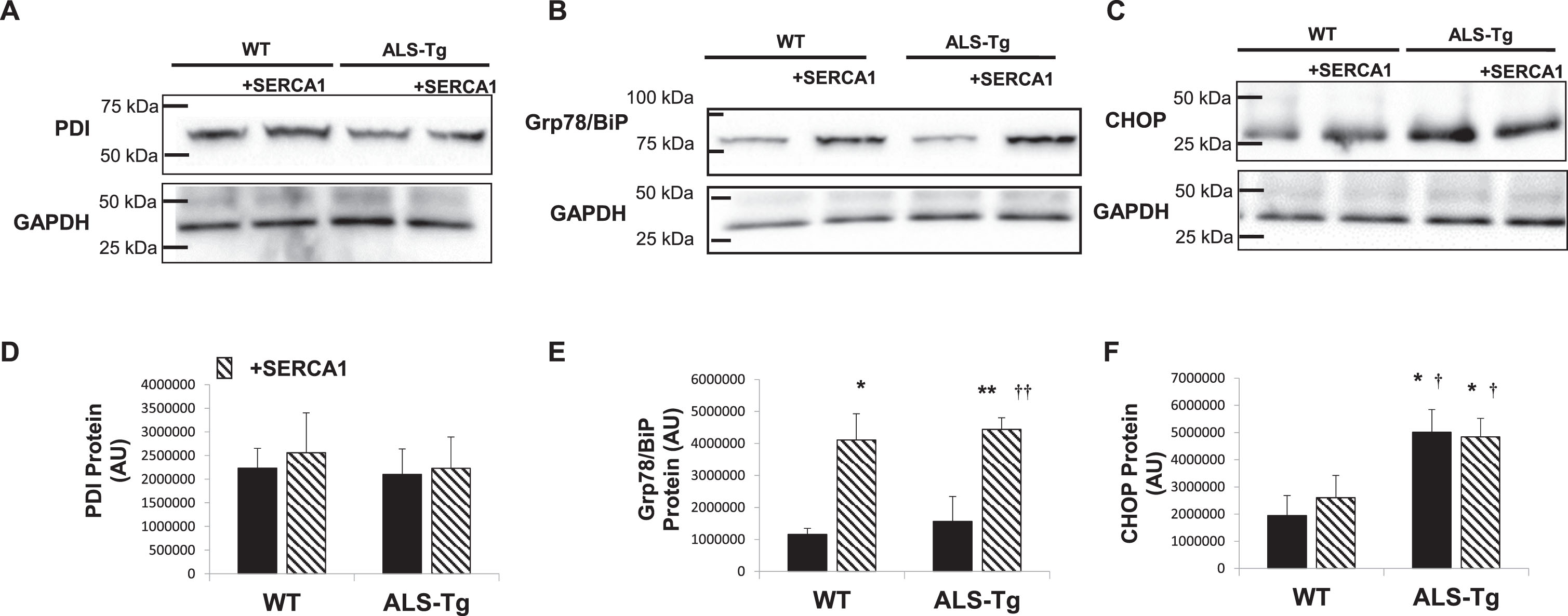
SERCA1 overexpression does not attenuate activation of the ER stress response in ALS-Tg mice
We previously reported an increase in ER stress sensors PDI and Grp78/BiP as well as ER apoptotic signaling protein CHOP in skeletal muscles (GAS, QUAD and diaphragm) of ALS-Tg mice [20]. In the current study, we did not detect an increase in PDI in the QUAD muscle of ALS-Tg or any other group, with expression levels being similar to WT in all genotypes (Fig. 5A). Levels of Grp78/BiP were elevated 3.5-fold in WT/+SERCA1 mice compared to WT (p < 0.05; Fig. 5B). SERCA1 overexpression increased Grp78/BiP levels by 3.8-fold in ALS-Tg/+SERCA1 vs. WT (p < 0.01) and by 2.8-fold compared to the ALS-Tg genotype control (p < 0.01). The apoptosis-signaling protein CHOP was not different between WT and WT/+SERCA1 QUAD muscles. Levels of CHOP protein were 2.6-fold and 2.5-fold higher in muscles from ALS-Tg and ALS-Tg/+SERCA1 mice (p < 0.01), compared to WT mice, respectively. No differences in CHOP levels were reported between ALS-Tg and ALS-Tg/+SERCA1 mice (p > 0.05).
Fig. 5
Proteins of the unfolded protein and ER stress response pathways in in skeletal muscle of WT, ALS-Tg and SERCA1 overexpressing mice. A-C) Representative western blot images for protein disulfide isomerase (PDI) (A), Grp78/BiP (B) and ER stress-specific cell death signaling protein C/EBP homologous protein (CHOP) (C) in QUAD muscle from WT and ALS-Tg mice with and without SERCA1 overexpression. D–F) Quantitative analysis of western blot images by densitometry for PDI (D), Grp78/BiP (E) and CHOP (F). Data shown are in arbitrary units (AU) for WT (n = 4; 1 male and 3 female), WT/+SERCA1 (n = 4; 1 male and 3 female), ALS-Tg (n = 4; 1 male and 3 female) and ALS-Tg/+SERCA1 (n = 4; 1 male and 3 female). Average data (D–F) represent mean±SEM. *p < 0.05 vs. WT, **p < 0.01 vs. WT; †p < 0.05 or ††p < 0.01 vs. ALS-Tg.
DISCUSSION
Our previous study showed elevations in intracellular Ca2+ in single muscle fibers associated with decreases in SERCA1, SERCA2 and PV protein levels in skeletal muscle of G93A*SOD1 mice [11]. Based on these findings, we hypothesized that decreased SERCA1 function could induce skeletal muscle pathology in ALS and that increasing SERCA1 function would improve disease pathology in these mice. To directly assess the effects of increased SERCA1 activity in attenuating the ALS phenotype in skeletal muscle, in the present study we crossed skeletal muscle-specific overexpressing SERCA1 mice with the G93A*SOD1 ALS-Tg mice. SERCA1 overexpression in ALS-Tg mice brought SERCA1 protein and SR Ca2+-ATPase activity back to that of WT control levels. This was associated with a decrease in peak Fura-2 levels during muscle contraction in isolated single muscle fibers in vitro from ALS-Tg/+SERCA1 mice. SERCA1 overexpression was also associated with an overall improvement in motor function, delay in symptom onset and attenuation of muscle atrophy. Overall, we report improvements in several different phenotypic changes in ALS-Tg mice with increased skeletal muscle specific SERCA1 overexpression.
Intracellular Ca2+ plays an essential role in skeletal muscle contractile function, maintaining cellular integrity and, as a second messenger, in regulating muscle gene expression. Ca2+ overload, defined as an elevation in resting intracellular Ca2+ level, leads to severe skeletal muscle damage via activation of Ca2+-dependent proteases such as calpains, release of phospholipase A2, over production of reactive oxygen species, and mitochondrial Ca2+ overload [16, 18, 22]. Abnormal intracellular Ca2+ levels are known to contribute to skeletal muscle damage in pathological conditions such muscular dystrophies and following intense exercise involving eccentric contractions [23, 24]. Skeletal muscle fibers have various ways of regulating intracellular Ca2+ concentration including plasma membrane Ca2+ pumps, cytosolic Ca2+ buffering proteins and SR Ca2+ regulatory proteins. The most significant of these is SERCA1, the SR/ER membrane Ca2+-ATPase responsible for the removal of Ca2+ into the SR during muscle relaxation. SERCA can also reduce intracellular Ca2+ levels after pathological overload. Overexpression of SERCA1 was shown to increase Ca2+ removal and reduce mitochondrial swelling in response to Ca2+ exposure [12]. Increased SERCA1 has also been shown to mitigate skeletal muscle disease pathology in dystrophic mice [12, 13]. Briefly, SERCA1 overexpression attenuated the pathological features such as central nuclei and muscle fibrosis in skeletal muscle and reduced creatine kinase, a serum marker of muscle damage [13, 16]. In both mdx and mdx/Utr-/- mice, SERCA1 overexpression improved force production in response to damage-inducing eccentric contractions [13]. Thus, increased SERCA1 expression and improved intracellular Ca2+ clearance have profound effects on improving pathological and functional consequences in skeletal muscle. These data support the hypothesis that Ca2+ dysregulation contributes to pathological events in skeletal muscle disease and that upregulation of SERCA1 protein level or increasing SERCA1 function is an attractive therapeutic strategy to treating these diseases.
Skeletal muscle atrophy is one of the main disease manifestations in ALS. It has been noted that skeletal muscle atrophy precedes changes in motor neurons [25]. Analysis of muscle volume of G93A*SOD1 ALS-Tg by MRI showed a significant decrease in muscle mass as early as 8 weeks of age, 4 weeks before the mice become symptomatic. The number of functional motor units is reduced by 60% as early as 60d (∼8 weeks) in G93A*SOD1 mice, with this early loss of motor units primarily affecting the high force producing type IIb fast fibers [26]. It is not known whether the motor unit loss and fiber type shift account entirely for the 80% reduction in tetanic force [26]. The role of intrinsic changes in skeletal muscle in the ALS phenotype has also been addressed in mice where mutant SOD1 protein was only expressed in skeletal muscle [27, 28]. Mice with skeletal muscle-specific overexpression of mutant SOD1 also demonstrated severe muscle atrophy and progressive muscle paralysis, although disease progression was delayed [27]. Most importantly, distal degeneration was observed, supporting the hypothesis that skeletal muscle pathology plays an important role in ALS pathogenesis, possibly via a dying-back phenomenon. It is generally accepted that motor neuron dysfunction is the primary pathological event preceding disease progression including skeletal muscle weakness. However, more recent evidence suggests that skeletal muscle atrophy and weakness may precede motor neuron loss. When primary motor neurons degenerate, there is compensatory reinnervation early in disease progression. This reinnervation is critically dependent on healthy myofibers to form stable neuromuscular junctions. Decreased muscle force will not be observed until a large number of motor neurons are lost, and it is hypothesized that improving myocyte health will improve muscle regeneration, neuromuscular junction stability and force production [27, 28]. According to this retrograde signaling hypothesis, disease progression in ALS is due, at least in part, to muscle pathology and neuromuscular junction destabilization. Further, by overexpressing insulin-like growth factor- 1 (IGF1) in a muscle-restricted fashion, Musaro and colleagues found evidence for reduced muscle wasting, increased muscle regeneration, improved neuromuscular junction integrity and delayed disease progression in G93A*SOD1 mice [29, 30]. This was also associated with attenuated skeletal muscle expression of ubiquitin and caspase activity as well as reduced levels of toxic p25 protein accumulation [30]. In the present study, we show that skeletal muscle-specific overexpression of SERCA1 rescued skeletal muscle atrophy and improved motor function in ALS-Tg mice. Collectively these data support the importance of skeletal muscle in ALS disease progression and implicate its contributory role in the ALS pathophysiology. Further, these studies support the notion that muscle-targeted therapies that improve muscle health can improve functional outcomes in ALS.
The attenuation of muscle atrophy in ALS-Tg mice with SERCA1 overexpression represents an improvement in the developmentally regulated gain in muscle mass with as it was observed at 16 weeks of age. Whether this is due to an improvement in muscle protein synthesis and/or a reduction in protein degradation cannot be discerned in the current study. However, we believe that SERCA1 overexpression enhanced muscle health during growth and development, enabling improved motor function. Further, while there are significant increases in muscle mass in ALS-Tg/+SERCA1 mice, the failure to show an overall increase in body mass in ALS-Tg/+SERCA1 mice is likely due to the phenotype of the WT/+SERCA1 mice. Although the mechanisms are not understood, it has been shown in several other studies that the body mass and muscle mass of SERCA1 overexpressing mice is reduced [12, 13]. Thus, whether due to a metabolic adaptation (i.e., reduced adipose mass) of SERCA1 overexpression or insertion of other modifier genes from the α–actinin SERCA1 mouse strain, the gain in muscle mass does not lead to overall gains in body mass.
We reported a reduction in peak tetanic Ca2+ during muscle activation in isolated single fibers from ALS-Tg/+SERCA mice. These data suggest adaptations in excitation-contraction coupling with SERCA1 overexpression, which agree with some of our previous findings [13]. While there may be decreased peak tetanic Ca2+ with muscle activation, this may be beneficial at tetanic levels of activation where muscle is functioning on the plateau of the force-pCa curve [7]. More importantly, the faster clearance of Ca2+ following activation would reduce the possible activation of Ca2+ degratory pathways following bouts of contractile activity. In the present study, we reported no differences in resting Fura-2 levels between WT and ALS-Tg single muscle fibers (p = 0.08). Recent findings showed that specific force and tetanic [Ca2+]i in G93A*SOD1 mice were similar to WT mice at both early (postnatal day 50) and late disease onset (between postnatal day 125–150) [31]. Additionally, resting [Ca2+]i was almost unchanged during repeated muscle contractions in single muscle fibers from G93A*SOD1 mice compared to WT mice, indicating the SERCA function is preserved in intact “healthy” muscle fibers from these mice [31]. This suggests that SERCA function is improved in surviving intact single muscle fibers from late stage G93A*SOD1 mice; yet a similar compensatory mechanism is probably unlikely in other “unhealthy” muscle fibers from G93A*SOD1 [31].
Our previous study showed that ER stress activation was present in skeletal muscle and could contribute, along with altered intracellular Ca2+ regulation, to myocyte death in ALS-Tg mice [20]. We previously showed that ER stress-specific cell death signal CHOP was upregulated by 70 d, prior to symptom onset, and to a greater extent in the later symptomatic stages of disease progression, suggesting that CHOP protein expression is important in ALS disease progression. In the current study, we tested the hypothesis that improving the skeletal muscle phenotype with SERCA1 overexpression would reduce ER stress activation. In contrast to our hypothesis, SERCA1 overexpression failed to suppress the upregulation of CHOP in skeletal muscle of ALS-Tg/+SERCA1 mice, indicating that ER stress per se was not involved in the improvement in the skeletal muscle phenotype. While this result was unexpected, since Ca2+ handling is tightly associated with SR/ER function and protein folding [32], other cell stress mechanisms that might increase the UPR response, such as the mitochondrial specific stress response which increase CHOP expression, could explain the lack of attenuation with SERCA1 overexpression. Surprisingly, the UPR marker Grp78/BiP was upregulated in skeletal muscle of the SERCA1 Tg mice. These data suggest that Grp78/BiP expression is regulated in a Ca2+-dependent manner. Considering that SERCA1 is the most abundant protein in the SR, we cannot rule out the possibility that overexpression of SERCA1 itself induced a protein-overload in the ER lumen and thus activated the UPR/ER stress pathway in the WT/+SERCA1 mice. Nonetheless, with improved Ca2+ clearance capacity, there was no mitigation of ER stress in skeletal muscle of the ALS-Tg mice suggesting that the mutant SOD1-induced oxidative stress activates ER stress upstream of the perturbations in intracellular Ca2+ regulation. Further studies are needed to address the relationship between improved Ca2+ clearance capacity and ER homeostasis and skeletal muscle atrophy and weakness during ALS disease progression.
In summary, this study demonstrates that skeletal muscle-specific overexpression of SERCA1 in ALS-Tg mice improved motor function, delayed disease onset and attenuated muscle atrophy. Our results support the emerging notion that impaired Ca2+ regulation and skeletal muscle pathology is a contributing factor in the ALS disease phenotype. However, while the SOD1-G93A mouse model has been extensively used in preclinical studies for ALS, this model only represents a small percentage of total cases of ALS. This has implications for the ability to translate effective treatments in this model to human clinical trials, and is similar to limitations of mouse models for other diseases such as DMD. While these data support targeting SERCA1 in skeletal muscle as a novel therapeutic strategy to improving muscle function in ALS, future studies need to evaluate whether the altering SERCA1 activity in patients with ALS will provide functional benefits.
ACKNOWLEDGMENTS
The authors thank Pai Chen for his assistance with data collection and analysis and Dr. Jeffery Molkentin for his support in obtaining the +SERCA1 Tg mice. This work was supported by the University of Maryland, College Park new investigator funds to ERC. DAGM was supported by NIH Pre-doctoral Institutional Training Grant T32-AG000268 to J.M. Hagberg.
CONFLICT Of INTEREST
DC and DAGM have no conflict of interests.
ERC is currently a paid employee of Solve FSHD Holdings Inc.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
REFERENCES
[1] | Rowland LP , Shneider NA . Amyotrophic lateral sclerosis. N Engl J Med (2001) ;344: (22):1688–700. |
[2] | Gurney ME , Pu H , Chiu AY , Da Canto MC , Polchow CY , Alexander DD et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 264 (1994) ;264: (5166):1772–5. |
[3] | Turner BJ , Talbot K Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Progress in Neurobiology (2008) ;85: (1):94–134. |
[4] | Patel BP , Hamadeh MJ Nutritional and exercise-based interventions in the treatment of amyotrophic lateral sclerosis. Clin Nutr (2009) ;28: (6):604–17. |
[5] | Bunton-Stasyshyn RK , Saccon RA , Fratta P , Fisher EM SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist. 2014. |
[6] | Blasco H , Mavel S , Corcia P , Gordon PH The glutamate hypothesis in ALS: pathophysiology and drug development. Curr Med Chem (2014) ;21: (31):3551–75. |
[7] | Westerblad H , Allen DG Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol (1991) ;98: (3):615–35. |
[8] | Zaidi A , Barron L , Sharov VS , Schoneich C , Michaelis EK , Michaelis ML Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry (2003) ;42: (41):12001–10. |
[9] | Sharov VS , Dremina ES , Galeva NA , Williams TD , Schoneich C . Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J (2006) ;394: (Pt 3):605–15. |
[10] | Dremina ES , Sharov VS , Davies MJ , Schoneich C Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem Res Toxicol (2007) ;20: (10):1462–9. |
[11] | Chin ER , Chen D , Bobyk KD , Mazala DA Perturbations in intracellular Ca2+handling in skeletal muscle in the G93A*SOD1 mouse model of amyotrophic lateral sclerosis. Am J Physiol Cell Physiol.C (2014) ;307: (11):1031–8. |
[12] | Goonasekera SA , Lam CK , Millay DP , Sargent MA , Hajjar RJ , Kranias EG et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. Journal of Clinical Investigation (2011) ;121: (3):1044–52. |
[13] | Mazala DA , Pratt SJ , Chen D , Molkentin JD , Lovering RM , Chin ER SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am J Physiol Cell Physiol. 308. United States: 2015 the American Physiological Society. 2015. p. C699-709. |
[14] | Parsons SA , Millay DP , Sargent MA , McNally EM , Molkentin JD Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. American Journal of Pathology (2006) ;168: (6):1975–85. |
[15] | Millay DP , Goonasekera SA , Sargent MA , Maillet M , Aronow BJ , Molkentin JD Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America (2009) ;106: (45):19023–8. |
[16] | Goonasekera SA , Davis J , Kwong JQ , Accornero F , Wei-Lapierre L , Sargent MA et al. Enhanced Ca2+influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum Mol Genet. 2014. |
[17] | Paz CAO , Delbono O , Muchnik S Potassium-induced contraction of denervated skeletal-muscle. Medicina-Buenos Aires (1985) ;45: (4):327–8. |
[18] | Mancuso R , Olivan S , Osta R , Navarro X Evolution of gait abnormalities in SOD1(G93A) transgenic mice. Brain Research (2011) ;1406: :65–73. |
[19] | Mead RJ , Bennett EJ , Kennerley AJ , Sharp P , Sunyach C , Kasher P et al. Optimised and Rapid Pre-clinical Screening in the SOD1(G93A) Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). Plos One (2011) ;6: (8). |
[20] | Chen D , Wang Y , Chin ER Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front Cell Neurosci (2015) ;9: :170. |
[21] | Chin ER , Green HJ , Grange F , Mercer JD , Obrien PJ Technical considerations for assessing alterations in skeletal-muscle sarcoplasmic-reticulum Ca++-sequestration function in-vitro. Molecular and Cellular Biochemistry (1994) ;139: (1):41–52. |
[22] | Millay DP , Sargent MA , Osinska H , Baines CP , Barton ER , Vuagniaux G et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nature Medicine (2008) ;14: (4):442–7. |
[23] | Zhang BT , Whitehead NP , Gervasio OL , Reardon TF , Vale M , Fatkin D et al. Pathways of Ca(2)(+) entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol (2012) ;112: (12):2077–86. |
[24] | Allen DG , Whitehead NP , Froehner SC Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol Rev (2016) ;96: (1):253–305. |
[25] | Marcuzzo S , Zucca I , Mastropietro A , de Rosbo NK , Cavalcante P , Tartari S et al. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: A longitudinal MRI study. Experimental Neurology (2011) ;231: (1):30–7. |
[26] | Hegedus J , Putman CT , Tyreman N , Gordon T Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol (2008) ;586: (14):3337–51. |
[27] | Wong M , Martin LJ Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Human Molecular Genetics (2010) ;19: (11):2284–302. |
[28] | Dobrowolny G , Aucello M , Rizzuto E , Beccafico S , Mammucari C , Boncompagni S et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab (2008) ;8: (5):425–36. |
[29] | Dobrowolny G , Giacinti C , Pelosi L , Nicoletti C , Winn N , Barberi L et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. Journal of Cell Biology (2005) ;168: (2):193–9. |
[30] | Dobrowolny G , Aucello M , Molinaro M , Musaro A . Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol Res (2008) ;30: (2):131–6. |
[31] | Cheng AJ , Allodi I , Chaillou T , Schlittler M , Ivarsson N , Lanner JT et al. Intact single muscle fibres from SOD1. J Physiol (2019) ;597: (12):3133–46. |
[32] | Glembotski CC . Roles for the sarco-/endoplasmic reticulum in cardiac myocyte contraction, protein synthesis, and protein quality control. Physiology (Bethesda) (2012) ;27: (6):343–50. |


