
Expert Insights from a Delphi-driven Neurologists’ Panel: Real-world Mexiletine use in Patients with Myotonic Disorders in Italy
Abstract
Background:
Myotonic disorders, such as non-dystrophic myotonias (NDMs) and myotonic dystrophies (DMs) are characterized by a delay in muscle relaxation after a contraction stimulus. There is general consensus that protocols to treat myotonia need to be implemented.
Objective:
Mexiletine is the only pharmacological agent approved for the symptomatic treatment of myotonia in adult patients with NDM and is considered to be the first-line treatment for DMs; however, its production in Italy was halted in 2022 making its availability to patients problematic.
Methods:
A panel of 8 Italian neurologists took part in a two-round Delphi panel between June and October 2022, analyzing the current use of mexiletine in Italian clinical practice.
Results:
The panelists assist 1126 patients (69% DM type1, 18% NDM and 13% DM type2). Adult NDM patients receive, on average, 400–600 mg of mexiletine hydrochloride (HCl) while adult DM patients receive 100–600 mg, per day in the long-term. The severity of symptoms is considered the main reason to start mexiletine treatment for both NDM and DM patients. Mexiletine is reckoned to have a clinical impact for both NDM and DM patients, but currently drug access is problematic.
Conclusions:
Mexiletine treatment is recognized to have a role in the reduction of the symptomatic burden for NDM and DM patients. Patient management could be improved by facilitating access to therapy and developing new drug formulations.
INTRODUCTION
Myotonic disorders are a heterogeneous group of diseases characterized by a delay in muscle relaxation after a contraction stimulus [1]. They include non-dystrophic myotonias (NDMs) and myotonic dystrophies (DMs) [2, 3]. Both groups of disorders have a genetic origin. NDMs are caused by mutations in the genes encoding for the ion channels of the skeletal muscle membrane (chloride channel, CLCN1, and sodium channel, SCN4A) [4], while DMs, further classified in type 1 (DM1) and type 2 (DM2), are due to the unstable expansion of CTG repetitions at the 3’ untranslated region of the DMPK gene, or CCTG nucleotides within the first intron of the CNBP gene, respectively [5–7].
NDM, DM1 and DM2 are classified as rare diseases, and while NDM is reported to have a prevalence of less than 1 case out of 100,000 [8, 9], DMs are more common: DM1 has a prevalence of 1 in 8,000, and DM2 is reported in 1–9 out of 100,000 [10].
NDM onset is usually at a young age, and patients report muscle stiffness, weakness, fatigue, and pain with a different magnitude and frequency depending on the ion channel carrying the genetic mutation [4]. For DM1, clinical symptoms may also begin in childhood and are multisystemic, manifesting in heart defects, cognitive impairment, respiratory and gastrointestinal disorders, and myotonia, and are characterized by progressive muscle wasting and weakness [11]. DM2 symptoms are similar to DM1, with muscle pain being a prominent feature. Interestingly, DM2 does not have a congenital onset, and symptoms in children usually occur in late childhood and are less pronounced [12]. In all subtypes of myotonic disorders, muscle impairment accompanies the patients throughout their whole life, sometimes worsening and affecting the quality of life (QoL) [13–16] of the patients and their families.
Currently, no cure or genetic treatments are available for the myotonic disorders. Available care recommendations describe the diagnostic and management protocols in the myotonic dystrophies [17, 18] and in the non-dystrophic myotonias [4]. The management of myotonia, one of the most burdensome symptoms for patients, still relies primarily on symptomatic treatment. Specifically, clinicians often resort to off-label use of antiepileptic drugs, anesthetics, and antiarrhythmics to address skeletal muscle myotonia, a practice largely based on anecdotal evidence [1, 19]. Mexiletine (brand name NaMuscla®) is the only pharmacological agent approved in Europe since 2018 with an orphan status for the symptomatic treatment of myotonia in adult patients with NDMs [20]. Mexiletine is mentioned as the first-line treatment by local and national recommendations also for DMs [21–23]. Although these recommendations are supported by patient advocacy groups and experts’ opinions [5, 24–28], and clinicians who consider mexiletine as the standard of care for myotonia symptoms in patients with myotonic disorders [29], a significant under-treatment rate was observed in a multinational prospective study on NDM patients [13, 30]. Mexiletine main characteristics are reported in Table 1. Mexiletine efficacy and safety have been shown previously by multiple studies conducted by Statland et al., Kwiecinski et al., and Suetterlin et al. [31–34], and in the recent short-term trial in adult NDM patients, MYOMEX [35], where it significantly reduced stiffness compared to placebo and significantly ameliorated the patients’ QoL. Indeed, upon mexiletine treatment, patients reported improved stiffness, weakness, fatigue, and pain. Interestingly, the majority of patients showed a preference for the treatment, and the lack of serious adverse events and electrocardiogram (ECG) events make mexiletine a reliable therapy for NDM patients [35].
Table 1
Mexiletine characteristics
| Mexiletine main characteristics | |
| Active substance | Mexiletine hydrochloride |
| Formulation | Capsules (each containing 167 mg of mexiletine, equivalent to 200 mg of mexiletine hydrochloride) |
| Administration | Oral intake |
| Standard regimens | 150 to 200 mg, 2 to 3 times a day |
| Mechanism of action | Sodium channel blocker in cardiac myocytes and nerve cells, classified as a Class 1B antiarrhythmic |
| Side effects | May involve: |
| •the cardiovascular system | |
| •the central nervous system | |
| •gastrointestinal, musculoskeletal, and dermatologic systems | |
In addition, mexiletine efficacy was also demonstrated in patients with adult onset of DM1, with an improvement in hand-grip force relaxation time at 6 months, although no significant effect on the 6-minutes’ walk test was recorded [36]. The positive outcome on myotonia symptoms in DM patients was confirmed by an observational study including both DM1 and DM2 patients [37], and a randomized controlled study testing mexiletine in these patients is currently planned (NCT04700046) [38].
Despite the growing evidence from clinical trials and real-world data, the under-treatment rate of patients is at least in part justified by the difficult access to the drug. Mexiletine, a drug with a peculiar history, was often provided to patients through alternative channels: after its withdrawal from the European market [39], patients had to rely on named patient imports from other countries, including Canada and Japan.
The aim of this study was to describe the experience of a panel of experts in the treatment of myotonic disorders to characterize the current use of mexiletine in Italian clinical practice and potentially identify a common strategy to improve care and standardize treatment approach.
METHODS
Delphi method
The current clinical management of patients with myotonic disorders and the use of mexiletine in the Italian clinical practice were investigated by a modified Delphi panel [40, 41]. The Delphi method is a structured iterative survey that allows the collection of information about a problem from a panel of experts. In each round of the Delphi a series of questions developed by a steering committee are submitted to the experts that can express their opinion anonymously, to allow for group conformity. The opinions are then collected, analyzed and re-proposed to the panel for the next round.
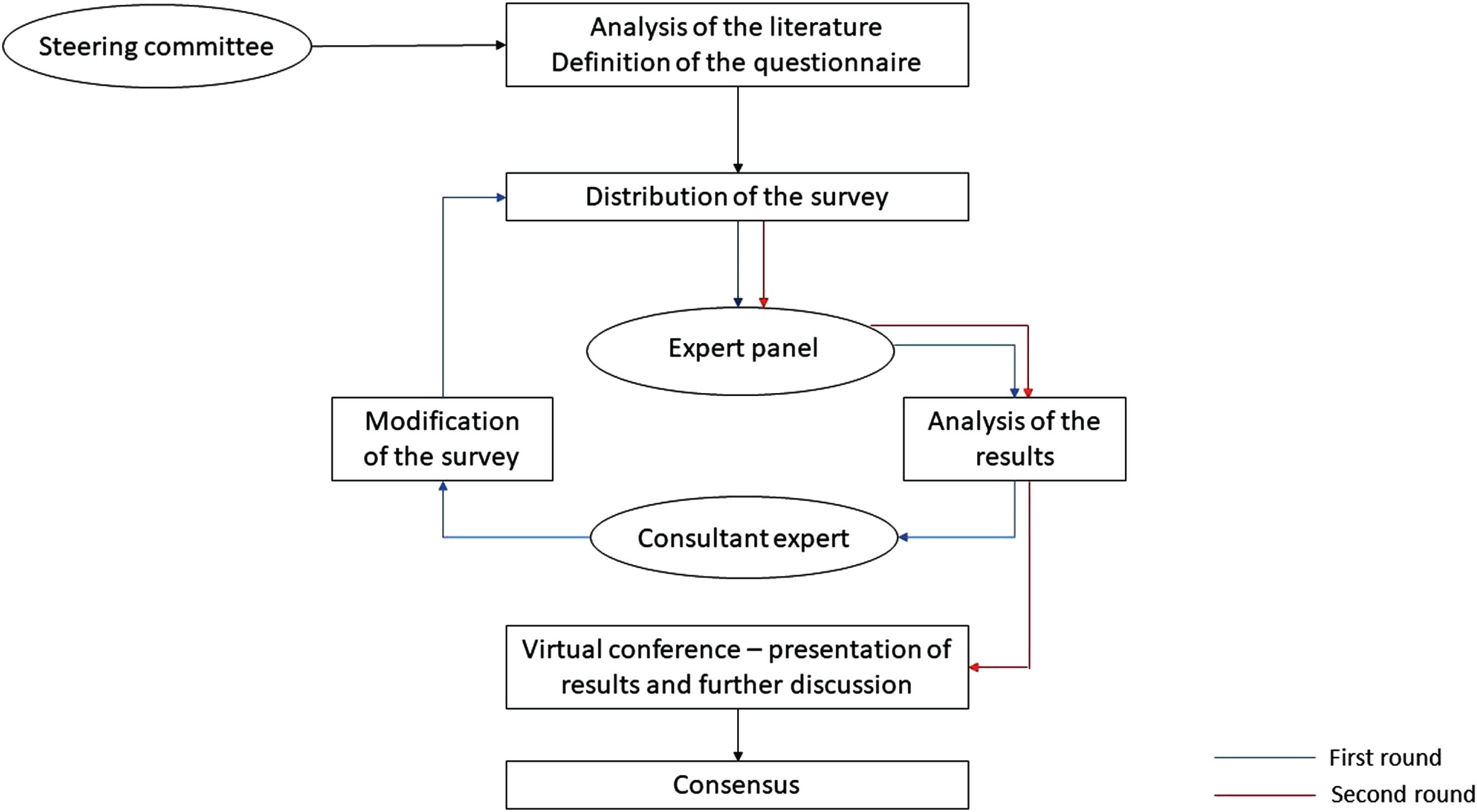
In this study, the two-rounds questionnaire was shared among the expert panelists between June and October 2022, minimizing the time between them to reduce the drop-outs and ensuring that all the expert panelists could provide insights. After the evaluation of the results obtained in the first round by the consultant expert, specific questions were proposed again to the expert panel in the second round. These questions were chosen on the basis of the great divergence in responses, which likely derived from a potential misunderstanding. The results were then collected and shared in aggregated form in a final virtual meeting for further discussion, and the eventual production of a consensus (Fig. 1).
Fig. 1
Flowchart of the modified Delphi method followed in the study.
Selection of the expert panelists
Eight Italian neurologists working in specialized neuromuscular care centers were selected according to their expertise in the diseases’ treatment and the number of patients treated in their centers. The criteria applied by researchers from Pharmalex (expert opinion leader in the field) and the Lupin Neurosciences team for the identification of the experts were the following: panelists must have been working in one of the main neuroscience specialized centers in Italy; the center should treat at least five NDM and/or 15 DM patients (quantifiable expertise); each panelist must have been recognized as a neurology expert in the treatment of myotonia patients (qualitative expertise); the participants must have been previously responsive to Pharmalex team invitation to collaborate.
The small number of expert panelists is a consequence of the rarity of the diseases and the few tertiary care structures dedicated to these pathologies in Italy. All the expert panelists declared to have experience in dealing with NDM and/or DM1/DM2 patients (adults and/or pediatric) for over 5 years.
Questionnaire design
The main points to be evaluated and discussed by the expert panelists through a questionnaire were defined by a steering committee. The questionnaire, including the pre-reading material, was prepared by Pharmalex researchers (DL, PB, and RR) and the Lupin Neurosciences team (AB, RO, MvA, and AZW) and was then validated by one opinion leader expert in the field of myotonic disorders (VAS). The questionnaire proposed spanned the following topics: general experience in the clinical management of patients, a snap-shot of mexiletine prescription, the impact of mexiletine use on clinical effects and patients’ QoL, considerations regarding drug access, and the current status of its use in the Italian clinical practice.
Data analysis
Given the project’s aim, the results are expressed as percentages of responses for all the closed questions and are analyzed according to the different topics. In addition, written explanations to some questions were provided anonymously by the expert panelists. Responses were mandatory for all the questions.
RESULTS
All the expert panelists answered both rounds of questionnaires, with no drop-outs between the first and the second round.
Experience in the clinical use of mexiletine (questions 1–8)
The eight expert panelists declared to assist in total 1,126 patients, mainly DM1, which represented 69% of the total; DM2 patients accounted for 13%, while NDM for 18% (the chloride channel mutation was more represented compared to the sodium channel mutation, 12% vs 6%, respectively) (Fig. 2A). As a general indication, one of the major centers reported to follow roughly 50 patients with NDM sodium or chloride channelopathies, all of them treated with mexiletine; patients with DM1 and DM2 were reported to be 250 and 30, respectively, of whom 30% being treated with mexiletine. On average, an adult NDM patient is expected to receive, with similar likelihood, long-term treatment of either 400 mg or 600 mg of mexiletine hydrochloride (HCl) per day (200 mg of mexiletine hydrochloride [HCl] are equivalent to 167 mg of mexiletine [NaMuscla®], and 400 mg and 600 mg of mexiletine hydrochloride [HCl] are the equivalent to 333 mg and 500 mg mexiletine [NaMuscla®], respectively) [42] (Fig. 2B). A different distribution is instead reported for adult DM patients, depending on which capsule dose was prescribed: when using 200 mg capsules, 75% of panelists indicated prescribing 400 mg (two capsules of 200 mg), while the 50 mg formulation was used by 50% of the panelists who prescribed a daily dose of 200 mg (four capsules), by 25% who prescribed a 400 mg daily dose (i.e., eight capsules) and by 12.5% who prescribed a dose of either 100 mg or 300 mg (Fig. 2C).
Fig. 2
(A) Percentage distribution of the patients assisted by the participants to the Delphi panel, according to the different myotonic disorder. NDM [Na+]: non-dystrophic myotonia due to sodium channel mutation; NDM [Cl-]: non-dystrophic myotonia due to chloride channel mutation; DM1: myotonic dystrophy type 1; DM2: myotonic dystrophy type 2. (B) Expected dosage of mexiletine hydrochloride [HCl] (200 mg capsule) received on average by a NDM adult patient in the long-term in the opinion of the expert panelists. (C) Expected dosage of mexiletine hydrochloride [HCl] (200 mg and 50 mg capsules) received on average by a DM adult patient in the long-term in the opinion of the expert panelists. Note: 200 mg of mexiletine hydrochloride [HCl] are equivalent to 167 mg of mexiletine [NaMuscla®].
![(A) Percentage distribution of the patients assisted by the participants to the Delphi panel, according to the different myotonic disorder. NDM [Na+]: non-dystrophic myotonia due to sodium channel mutation; NDM [Cl-]: non-dystrophic myotonia due to chloride channel mutation; DM1: myotonic dystrophy type 1; DM2: myotonic dystrophy type 2. (B) Expected dosage of mexiletine hydrochloride [HCl] (200 mg capsule) received on average by a NDM adult patient in the long-term in the opinion of the expert panelists. (C) Expected dosage of mexiletine hydrochloride [HCl] (200 mg and 50 mg capsules) received on average by a DM adult patient in the long-term in the opinion of the expert panelists. Note: 200 mg of mexiletine hydrochloride [HCl] are equivalent to 167 mg of mexiletine [NaMuscla®].](https://ip.ios.semcs.net:443/media/jnd/2024/11-2/jnd-11-2-jnd230115/jnd-11-jnd230115-g002.jpg)
Snap-shot of mexiletine prescription (questions 9–11)
In this section of the questionnaire, the experts reported, in their experience, the current situation regarding mexiletine prescription. Eighty-eight percent of the panelists would consider treating an adult NDM patient with mexiletine if the symptoms were considered severe enough to have an impact on the patient’s daily living; all of panelists (100%) would do the same for DM1 and DM2 patients (Table 2). The genetic confirmation of the disorders is considered a reason to treat for 65% of the expert panelists for NDM and 88% for DM, in case of symptoms for which the experts would otherwise not treat. Whether a patient was already receiving mexiletine or not did not influence the choice of treatment, as 75% of panelists stated they would still suggest the drug for NDM and DMs. The absence of potential cardiological finding during a cardiological examination plays a role in the decision process for 75% of panelists in NDM and 63% in DMs. The use of another off-licensed treatment (dyntoin, carbamazepine, lamotrigine, gabapentin for NDM, diphenylhydantoin for DM1 and DM2, and myorelaxants) may influence the decision to prescribe mexiletine, although 38% would proceed independently for the management of NDM and 50% for DM1 and DM2.
Of relevance is that the experts would be more prone to prescribe mexiletine only if the drug was available and reimbursed by NHS (88% of preferences for all the diseases) (Table 2).
Table 2
Real-life prescription and drug access for mexiletine use. Percentage of agreement to prescription according to the reported features
| Feature | % Panelists agreeing | % Panelists agreeing |
| (NDM patients) | (DM1 and DM2 patients) | |
| Factors affecting treatment choice in favor of | ||
| mexiletine | ||
| Symptoms severe enough to impact the patient’s daily living | 88 | 100 |
| Genetically confirmed disease | 65 | 88 |
| Patient already receiving mexiletine | 75 | 75 |
| Symptom judged to be relevant for a patient of any age | 75 | 63 |
| Drug naive patient | 75 | 75 |
| Normal ECG findings and cardiology approval | 75 | 63 |
| Normal ECG findings, 24-hour ECG Holter monitoring, 2D echocardiograms and cardiology approval | 50 | 63 |
| Patient already under another off-licensed treatment | 38 | 50 |
| Other | 13 | 25 |
| Factors affecting drug prescription | ||
| Availability and deliverability of prescribed mexiletine regardless of reimbursement by NHS | 13 | 13 |
| Only if availability and reimbursement of mexiletine occurs through the NHS | 88 | 88 |
| Other | 25 | 25 |
In real-world experience, after the start of mexiletine treatment, the expert panelists estimate seeing an adult patient twice a year regardless of their disease and age.
Evaluation of mexiletine clinical effectiveness and its influence on QoL (questions 12–26)
Overall, 75% of the expert panelists considered the absolute mean change in the overall QoL score of the Individualized Neuromuscular Quality of Life questionnaire (INQoL) of the mexiletine arm of the MYOMEX study [35] as a clinically significant difference; 25% of the expert panelists stated to be unsure.
The locking of the patient’s muscles, the ability to perform general daily activities, and the reduction of pain are the top three domains of the INQoL v1.2 that the panelists believed to be the most impactful for the management of an NDM patient with mexiletine (Table 3).
Table 3
Domains of the INQoL v1.2 that are considered to be the most impactful for the management of NDM patients with mexiletine ranked according to expert panelists’ opinions
| Ranking | Items |
| 1 | Locking of patient’s muscles |
| 2 | The things patient does-daily activities |
| 3 | Patient’s pain |
| 4 | The things patient does-leisure and work activities |
| 5 | Patient’s independence |
| 6 | How tired does patient feel/fatigue |
| 7 | Patient’s muscle weakness |
| 8 | How does patient feel/emotions |
| 9 | Patient’s relationships |
| 10 | The way patient looks/body image |
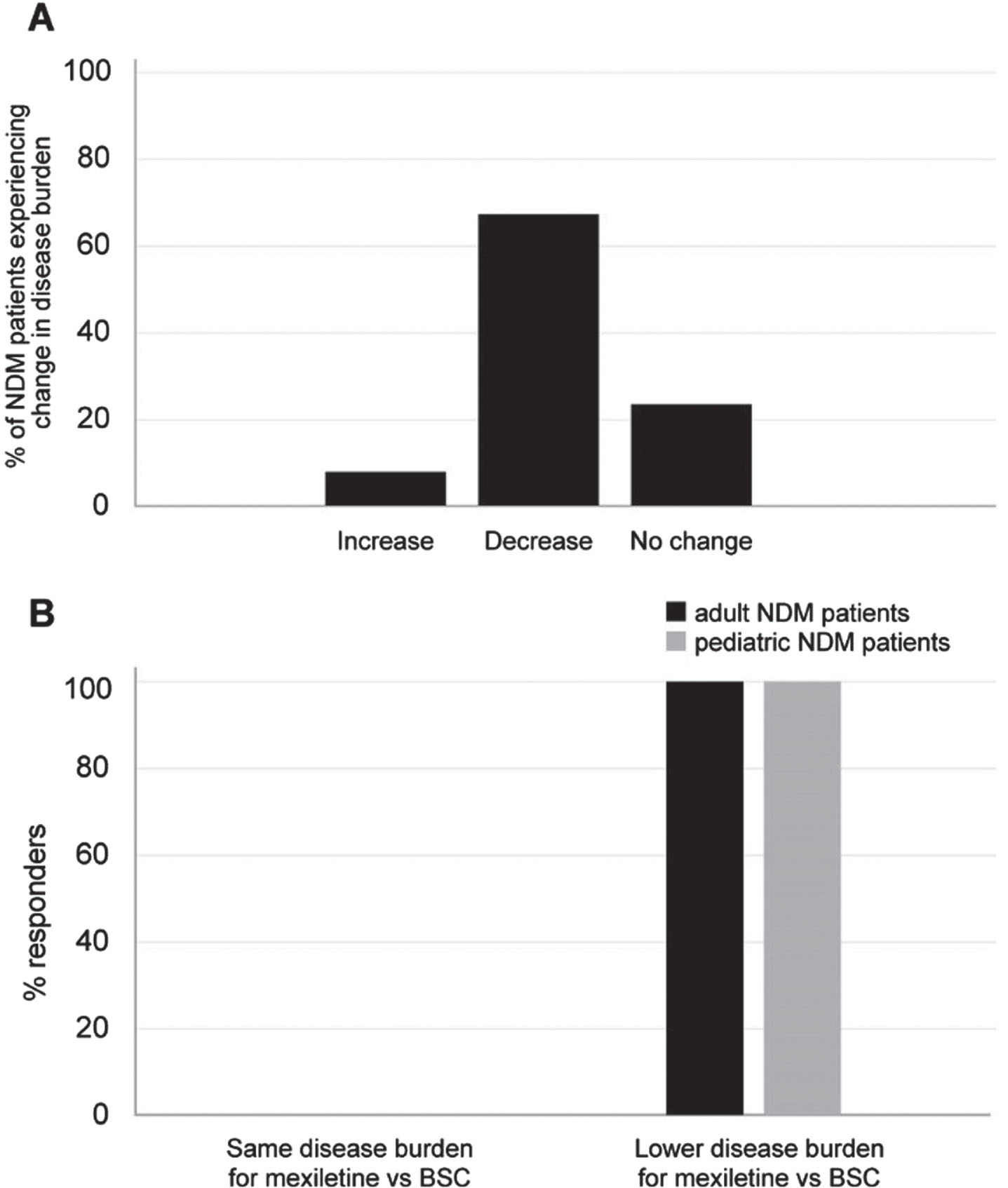
Experts reported they believed that 69% of cases treated with mexiletine would experience a decrease in the myotonia-related burden of the disease over time, compared to the best supportive care (BSC), without mexiletine (Fig. 3A). Overall, all the experts (100%) agreed that a lower disease burden would be perceived by adult and pediatric patients upon mexiletine use compared to BSC (Fig. 3B).
Fig. 3
(A) Percentage of NDM patients expected to experience a change in myotonia-related disease burden upon use of mexiletine vs BSC in the opinion of the expert panelists. (B) Percentage of panelists foreseeing a change in the disease burden for NDM adult (black bar) and pediatric (gray bar) patients upon use of mexiletine vs BSC. BSC: best supportive care.
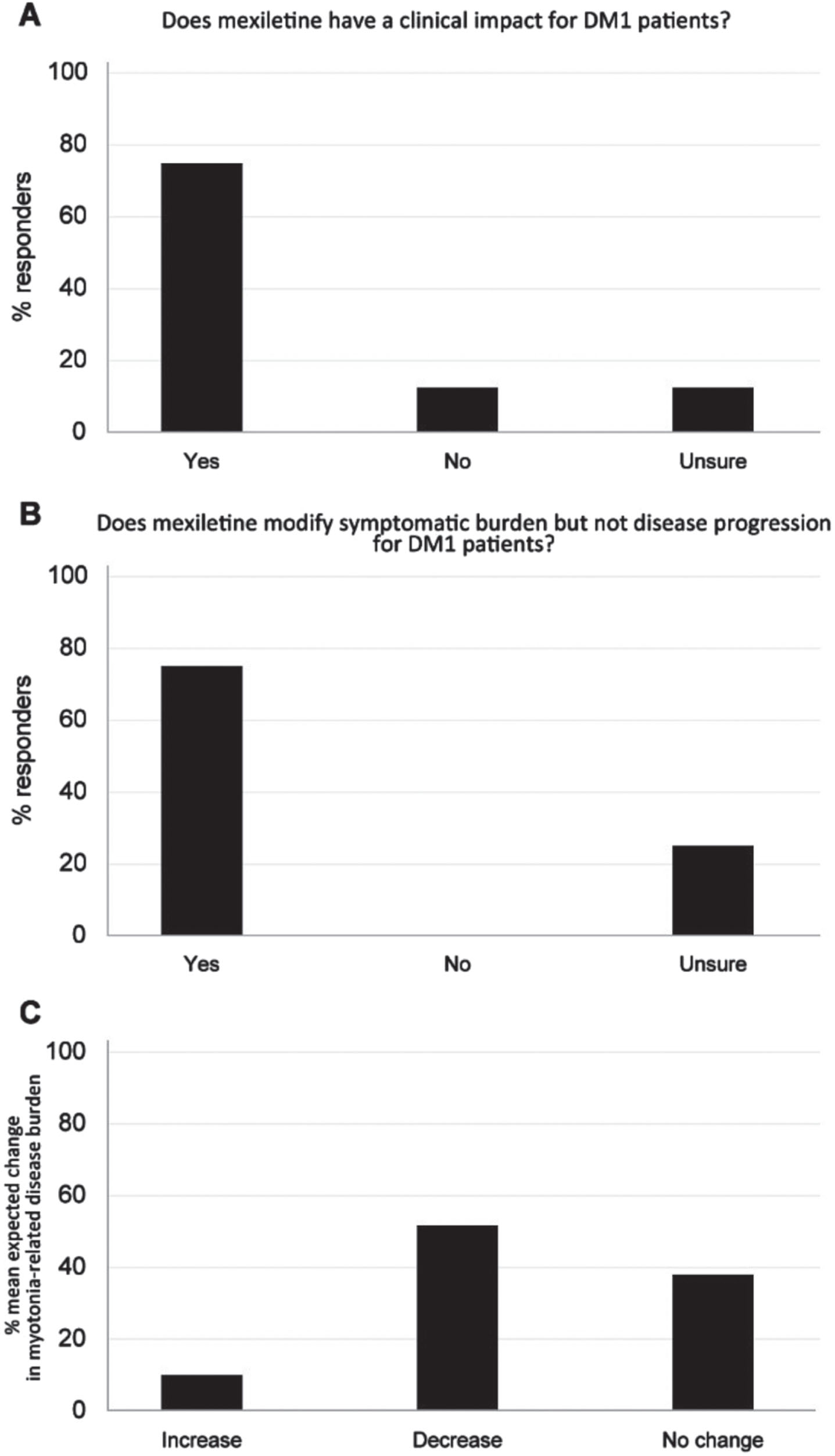
The results on handgrip myotonia presented in the clinical studies of Logigian et al. and Heatwole et al. [36, 43] are defined of clinical importance for DM1 patients by 75% of the expert panelists (Fig. 4A), in spite of the unchanged INQoL and hand/finger myotonia visual analog scale score showed in the study [36]. Seventy-five percent of the experts believed in the benefit of mexiletine as a medication designed to modify symptomatic burden but not disease progression for DM1 patients (Fig. 4B). Experts thought that 53% of DM1 patients would be expected to show a decrease in myotonia-related disease burden upon mexiletine treatment when compared to BSC (Fig. 4C).
Fig. 4
(A) Clinical impact of mexiletine treatment on handgrip myotonia of DM1 patients in the opinion of the expert panelists. (B) Benefit of mexiletine in the modification of the disease burden and disease progression in DM1 patients in the opinion of the expert panelists. (C) Percentage of DM1 patients expected to experience a change in myotonia-related disease burden upon use of mexiletine vs BSC. BSC: best supportive care.
Considering DM2 patients, 75% of the expert panelists declared that mexiletine treatment would be clinically relevant, while 12.5% of the responders believed that the treatment would have no clinical importance, and 12.5% were unsure. VAS for stiffness/myotonia, pain and fatigue were identified as the most impactful measures for the treatment’s benefit assessment on these patients (Table 4).
Table 4
Most impactful measures relevant to assess treatment’s benefit in DM2 patients, ranked according to panelists’ opinion
| Ranking | Items |
| 1 | VAS for stiffness/myotonia |
| 2 | VAS for pain |
| 3 | VAS for fatigue |
| 5 | Handgrip |
| 5 | INQoL |
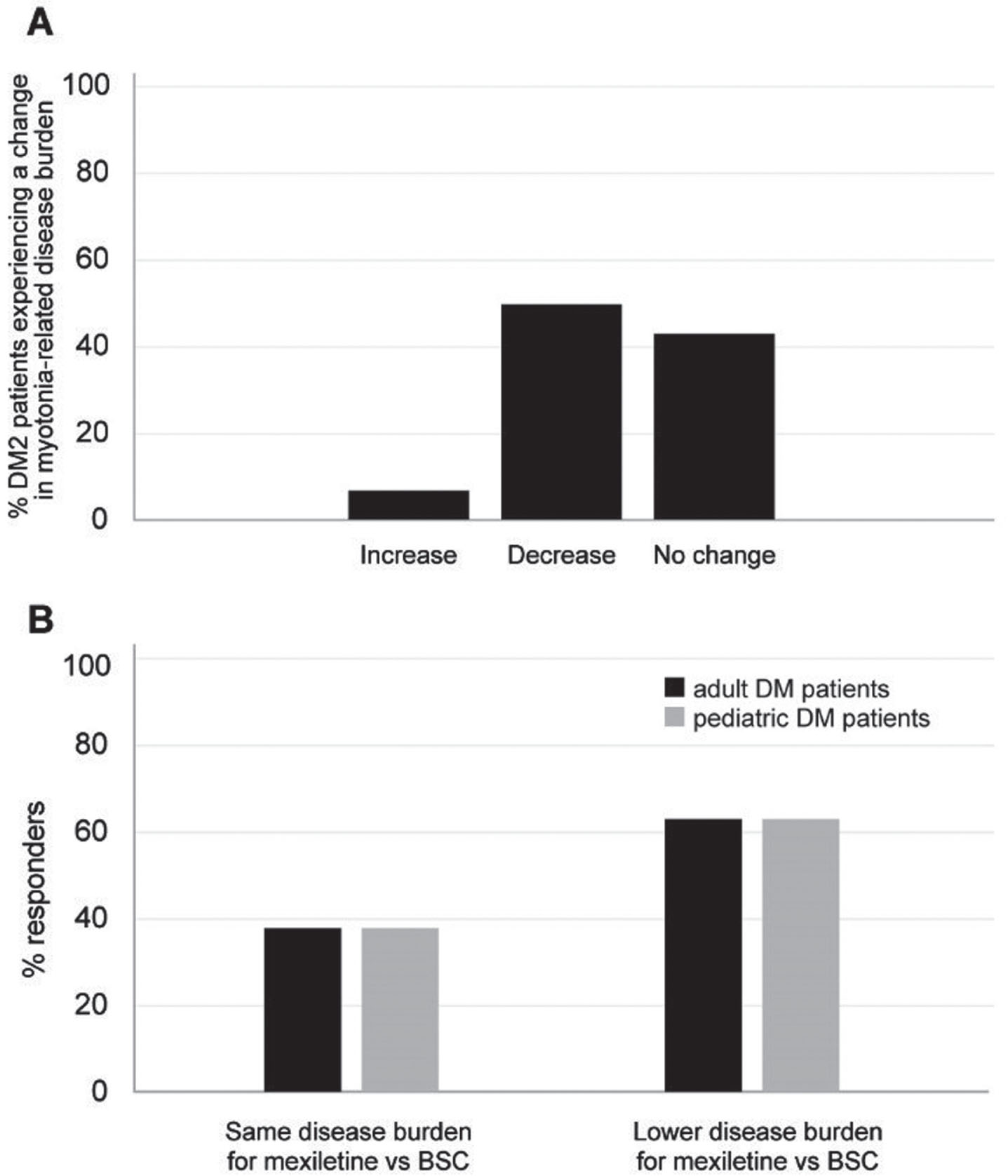
For this particular group of patients, specifically regarding myotonia symptoms, the expert panelists believed that the treatment could alleviate the disease burden related to myotonia in 50% of patients. However, they indicated that it would likely have no effect in 43% of cases. (Fig. 5A). These results are very similar to those obtained for DM1 patients.
When experts were asked to estimate the proportion of DM1 and DM2 patients perceiving a lower disease burden with mexiletine use vs BSC, 63% of them reported they would expect that both adults and pediatric patients would experience a possible decrease in disease burden (Fig. 5B).
Fig. 5
(A) Percentage of DM2 patients expected to experience a change in myotonia-related disease burden upon use of mexiletine vs BSC in the opinion of the expert panelists. (B) Percentage of panelists foreseeing a change in the disease burden for DM adult (black bar) and pediatric (gray bar) patients upon use of mexiletine vs BSC. BSC: best supportive care.
Drug prescription and drug access through Military supply within the 648 law (questions 27–39)
In Italy, the Military Chemical Pharmaceutical Plant, a public institution, started producing and commercializing mexiletine in 2010 for reimbursed off-label use in NDMs and DMs [44]. This production was halted in 2022 [45], and NaMuscla® is not available in Italy as of today, which raises issues for patients as the peculiarity of the situation does not always mirror the inclusion/exclusion criteria and all the clinical evaluations performed in the registration studies.
Military mexiletine was usually prescribed through the Italian 648 law [46] by neurologists (100%), and pharmacists were also indicated by 75% of the expert panelists as the NHS professionals potentially qualified to complete drug prescriptions.
Concerning the length of military mexiletine prescription, the expert panelists reported being allowed to prescribe it, according to the Italian law [46], for an average of 6 months in adults with NDM, DM1, and DM2 and 6.5 months for pediatric patients.
If there is a requirement to report adverse events to pharmacovigilance services, 62.5% of the expert panelists stated that they have a designated contact person either at the Italian Medicines Agency (AIFA) or within their affiliated hospital. The remaining 37.5%, in case of need, usually contacted the pharmacy at their institution.
Concerning the time needed to obtain military mexiletine, 50% of the experts reported that, on average, 7–14 days were needed between the prescription and the pharmacy’s distribution of the drug for patients with NDM, DM1, and DM2 (either adults or children); 25% indicated that 3–7 days were needed and 25% reported the need of more than 14 days, for all types of patients.
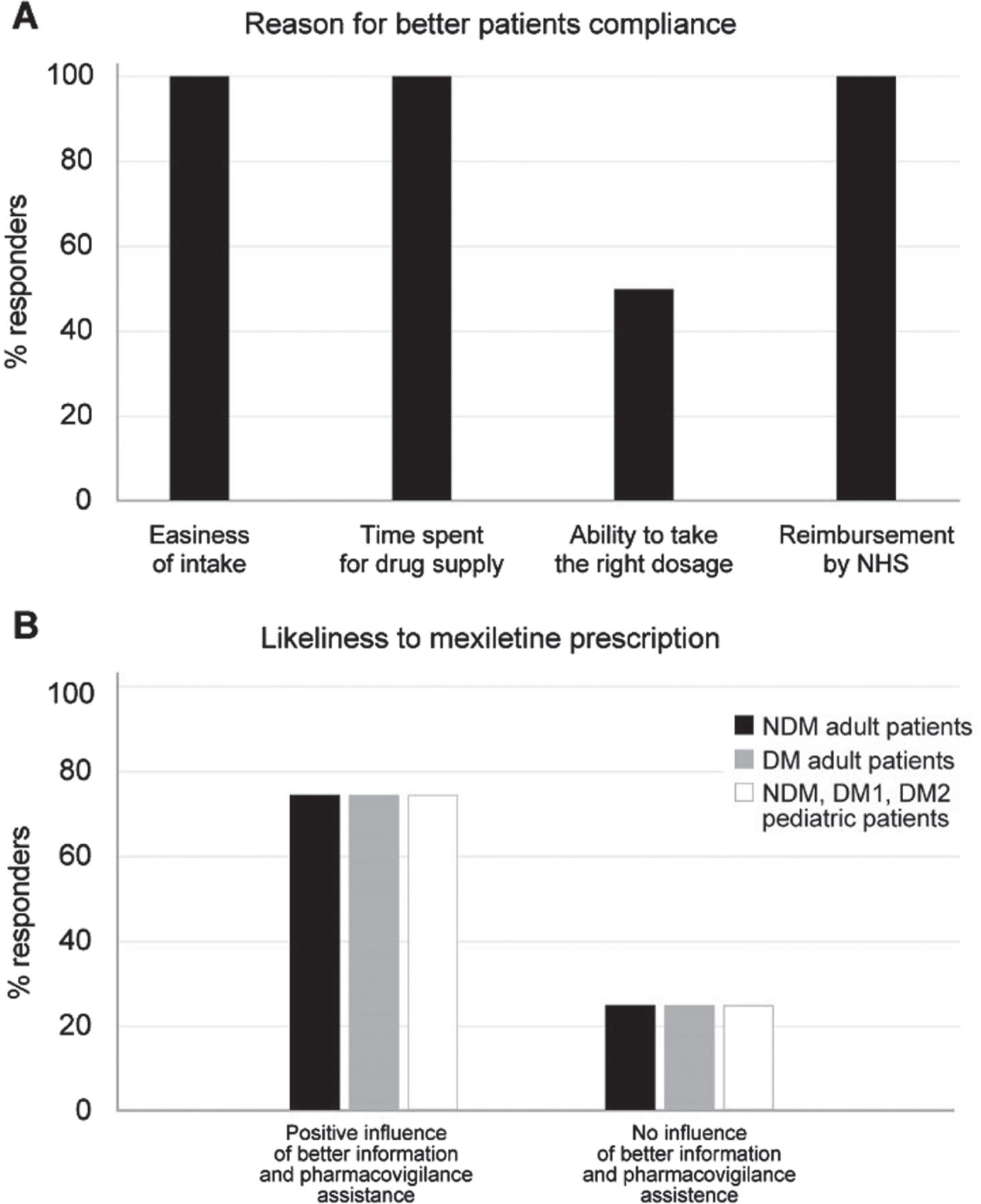
The main reason influencing the timing between the patient’s request and the delivery of military mexiletine, was indicated to be the difficulty of the pharmacy in obtaining the drug (75% of the responders), while the difficulty or delay in the prescription process by the physician was indicated by 25% of panelists. The possibility of having a commercially available product (either reimbursed or not) for patients with myotonic disorders was unanimously believed to make the management of the disease easier. In fact, commercially available mexiletine would positively influence patients’ compliance to the experts’ opinion: again unanimously, the expert panelists agreed that simplicity in obtaining the drug, meaning the reduction in the time spent procuring it, and above all, the possibility of reimbursement by the NHS would greatly impact the compliance (Fig. 6A). The possibility of better access to information (both patient and health care professional education) and pharmacovigilance assistance would positively influence the prescription of a commercial product vs military mexiletine for 75% of the expert panelists when considering adult and pediatric prescriptions (Fig. 6B).
Fig. 6
(A) Reason for improved patient compliance to mexiletine in the opinion of the expert panelists. (B) Influence of better information to patients and presence of pharmacovigilance assistance on the likeliness to mexiletine prescription to adult NDM (black bars), adult DM (gray bars) and pediatric NDM, DM1 and DM2 (white bars) patients according to experts’ opinion.
Finally, in the case of commercialization of non-reimbursed mexiletine provided with hospital’s budgets, 75% of the expert panelists would preferentially prescribe it over military mexiletine, which would remain the choice for 25% of the panelists. All experts agreed that a potential new formulation would contribute to the optimization of patient adherence to treatment and better patient management. Suggested new formulations include drops, syrups, or low-dosage formulations (less than 200 mg), which would be useful for children, soluble or injectable forms to simplify administration to dysphagic DM1 patients, and long-release formulations that would increase the length of the drug action.
DISCUSSION
All myotonic patients experience significant lifetime morbidity due to pain related to their muscle symptoms: there is an urgent need to develop and improve research to better understand and address the hurdles in the clinical management of these patients, including symptomatic treatment for myotonia in NDM, DM1, and DM2, aiming at clear recommendations for the clinical practice [47]. This work was designed to provide a better understanding of the current management of NDM and DMs in Italy, extending what was already reported in the UK [48], and gain more knowledge, as the information related to the treatment of these patients results overall scarce [49]. The opinion of eight neurologists with expertise on the management of NDM, DM1, and DM2 in real-life in Italy collected by a Delphi panel highlighted the benefit of mexiletine treatment and suggested that an improvement in patient management could be reached through a better access to the therapy. Considering the number of patients tracked through the analysis, all the expert panelists agreed that the results of the present questionnaire are representative of the Italian scenario. Indeed, the heterogeneous composition of the population described here, such as the higher proportion of DM1, compared to NDM or DM2 patients, is consistent with what is observed in the clinical practice.
The experts affirm the clinical importance of mexiletine based on evidence emerged from registration studies [35, 43] and their real-world experience: the drug is therefore considered the preferred treatment for NDM, as it is reckoned to reduce the disease burden, and it could be considered also in DM1 and DM2 patients, although the perceived effect on disease burden is less pronounced. The panelists’ opinion is in favor of treatment administration to DM patients, which may benefit from symptomatic burden reduction (DM1 patients) and a possible general improvement on disease burden in both DM1 and DM2 adults and children. A previous analysis reported that overall, dystrophic patients are less treated with anti-myotonia medications despite a similar prevalence of myotonic symptoms in NDM and DM, hinting at a possible undertreatment of this group of patients [49]. The undertreatment is possibly due to the cardiac contraindications in DM patients. Nevertheless, published data using mexiletine [31, 36] confirm the safety profile of the drug. Panelist experience is of good tolerability and drop outs reported are due to gastrointestinal issues but not cardiac arrhythmias. In fact, after discontinuation of mexiletine for lack of drug availability some patients may experience subjective tachycardia and feel better when they resume the drug. To increase and ameliorate mexiletine use in different types of patients, new formulations such as droplets or syrups, together with low dosage capsules usage, are suggested by the expert panelists, especially in the case of administration to pediatric patients or to those with specific impairment (dysphagia, common in DM1 patients), to fit the needs of patients with disease-related disabilities.
Moreover, the average mexiletine hydrochloride (HCl) dosage identified by the panelists (400-600 mg of mexiletine hydrochloride (HCl)/day, equivalent to 333-500 mg mexiletine/day) mirrors the real-life clinical practice described elsewhere [33] and is in line with the dosage suggested by UK experts for NDM adult patients (on average two capsules of mexiletine [NaMuscla®]) [48]. The variability in dosage prescription may depend on the type of myotonia (NDM [chloride or sodium channelopathy], or DM). Indeed, DM patients may experience trouble with full dosage intake, given the possible gastrointestinal symptoms and cardiac effects (the abovementioned tachycardia experienced in rare cases), and thus a slightly lower dosage can be found for such patients; possible malabsorption of the drug, individual variability in drug metabolism, and low compliance have been also hypothesized in these patients during the phase 3 trial [36]. The 50 mg pharmaceutical form is preferred for pediatric and adult patients suffering from gastrointestinal symptoms, as it allows fine-tuning of the dosing regimen.
The experts would prescribe mexiletine in patients with severe symptoms that impact their daily living similar to what was reported by the UK panel [48] and were also in agreement to indicate the severity of the disease symptoms as a relevant indication to prescribe mexiletine, regardless of the age of the patient, especially when considering DM1 and DM2 patients. Considering the potential cardiological implications for DM1 patients [50, 51], the expert panelists reported the unlikeliness of this group to show a complete absence of such events (i.e., branch block) [52]. Therefore, the option “no findings” within the questionnaire as an indication for the therapeutic use of mexiletine is not easily fulfilled in clinical practice. However, it is common to see patients presenting with cardiologic evidence that is compatible with mexiletine administration. Therefore, regardless of the outcomes of the instrumental examinations, a consultation with the cardiologist is always recommended, if not essential, for the eligibility of the treatment [53].
Should a patient start the treatment with mexiletine, twice-a-year follow-up visits are recognized as the standard of care in clinical practice. In the case of pediatric patients, this frequency ensures proper disease attention and reassures parents/caregivers. In the case of reimbursement from the NHS, NDM patients could access the drug more easily, thus potentially improving the overall treatment management and likely reducing the frequency of follow-up visits [48].
Seventy-five percent of the expert panelists reckon that mexiletine use can improve patients’ QoL, as observed by the change in median stiffness VAS score and the overall QoL score compared to baseline reported in the MYOMEX study [35]. The effect of mexiletine is expected to be different between NMD and DM patients: specifically, the experts think that a greater reduction in the disease burden will be observed in NDM rather than in DM patients, because myotonia is the predominant or exclusive disabling symptom in NDM patients. The panel pointed out that people with DM1 possess a peculiar approach to the pathology: these individuals display clinically relevant myotonia which is often minimized by the patients themselves, because of the warm-up phenomenon and the severity and burden of many other symptoms connected to DM1. These patients experience also muscle weakness. Usually, severe weakness is associated with less myotonia. The moderately weak patients may anyway underestimate myotonia because weakness is their main concern [36, 49]. Indeed, in DM1 patients myotonia becomes marginal as the disease progresses, and therefore mexiletine prescription is much more questionable in light of the potential cardiac issues and major muscle weakness in the majority of patients.
On the other hand, the expected slightly lower impact of mexiletine on DM2 with respect to DM1 may be explained by the fact that myotonia is less severe in DM2 patients than in DM1. There is general consensus that DM2 patients experience greater muscle pain, and whether mexiletine could significantly reduce the latter is still to be defined. Overall, the reduction in QoL is to be expected as mainly related to weakness and systemic complications in DM1 and DM2 patients, and less dependent on myotonia.
Access to mexiletine may be a limiting factor for patients with myotonic disorders, as the difficulty in obtaining the drug, due to its peculiar market history [39], may have excluded some patients who could benefit from the treatment. In case of shortages due to restrains in the military pharmacological institute production the drug needs to be imported. This process is currently managed by the hospital pharmacies in agreement with clinicians. Bureaucracy and paperwork, which are required at hospital sites to prescribe and make sure mexiletine gets to the patients is a hindrance to consider in the assessment of patient management. A potentially easy-to-obtain therapy could overcome the difficulties in product availability.
A further step of this project may include an implementation of disease awareness at the territorial level, and an extension of the Delphi project about the real-life use of mexiletine to territorial neurologists that can prescribe the drug. Moreover, other stakeholders to be included could be the patients and caregivers, which can be involved by the MIA (Miotonici In Associazione) and UILDM (Unione Italiana Lotta alla Distrofia Muscolare), the patient associations dedicated to NDM and DM patients, respectively. These stakeholders bring different expertise on the diseases, with different points of view on therapeutic and accessibility issues compared to clinicians and may help acquire extensive real-world data for a complete picture of the situation of NDM, DM1, and DM2 patient management.
The main limitation of this study is the low number of specialists included, which is a consequence of the rarity of the disease. As with any other Delphi panel addressing rare diseases, the number of expert panelists was within the range recommended by the Guidance for this type of study [54], which is between 7 and 12. In addition, the responses are based on subjective perceptions of the specialists rather than on an objective assessment of scales, data sources, and clinical documentation. Moreover, the panelists believe that more emphasis should be given to the paediatric population, give the even more heterogeneous approach to the treatment of muscle symptoms in the paediatric-onset subtypes and the lack of standardized measures of assessment and QoL in this setting. These issues require further attention and need to be addressed in the near future.
Nevertheless, the solid methodology applied, the absence of drop-outs between the rounds of consultancy, and the proven expertise of the participants with real-life NDM, DM1, and DM2 give a precise and reliable picture of the current unmet needs related to the management of myotonic disorders in real-life in Italy.
CONCLUSIONS
In conclusion, a randomized study on the efficacy and especially on the clinical tolerance of mexiletine is necessary to position this treatment in the symptomatic management of myotonic dystrophies.
Based on the current clinical experience in Italy, experts would prescribe mexiletine to NDM, DM1, and DM2 patients of all ages, expecting a reduction of the disease burden with a positive effect on the patient’s QoL. A cardiologist’s opinion is requested before the start of the treatment; as the average daily dosage is identified in 2/3 mexiletine hydrochloride (HCl) capsules, patients experiencing issues with multiple capsule administration would benefit from the introduction of new formulations. An easier-to-obtain drug would likely impact its use by the patients, which may lead to a better control of their disease.
ACKNOWLEDGMENTS
Editorial assistance, funded by Lupin Atlantis Holdings SA, was provided by Health Publishing and Services, srl, according to the Good Publication Practice.
We would like to extend special thanks to the expert panelists who completed the questionnaire and participated in a discussion on the results of the study: Adele D’Amico (Neuromuscular and Neurodegenerative Unit, Department of Neurosciences. Bambino Gesù Paediatric Hospital, Rome, Italy); Massimiliano Filosto (Department of Clinical and Experimental Sciences, University of Brescia, NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy); Lorenzo Maggi (Neuroimmunology and Neuromuscular Disease Unit, Fondazione IRCCS Istituto Neurologico “Carlo Besta”, Milan, Italy); Tiziana Enrica Mongini (Neuromuscular Unit, Department of Neurosciences RLM, University of Turin, Turin, Italy); Elena Pegoraro (Department of Neurosciences, University of Padova, Padova, Italy); Gabriele Siciliano (Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy); Gabriella Silvestri (Neurology Unit, Fondazione Policlinico Universitario “Agostino Gemelli” IRCCS, Department of Neuroscience, Università Cattolica del Sacro Cuore, Roma, Italy); Antonio Toscano (Department of Clinical and Experimental Medicine, University Hospital of Messina, Messina, Italy).
CONFLICT OF INTEREST
DL, PB, RR are employees of Pharmalex Italy SpA. Pharmalex Italy SpA was commissioned and received funding from Lupin Atlantis Holdings SA to conduct this study.
AZW, AB, MvA, RO are employees of subsidiaries of Lupin Atlantis Holdings SA.
VAS worked as an independent clinical advisor on this study and received a financial compensation from Lupin Atlantis Holdings SA for intellectual and scientific advice.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of this study are available within the article and/or its supplementary material.
AUTHORS’ CONTRIBUTIONS
DL, PB, RR conceptualized, supported the methodology, planned and organized the panels. DL, PB, RR, AZW, AB, MvA, RO and VAS drafted the manuscript and critically revised it. All authors read and approved the final version for submission.
FUNDING
This study and the development of the manuscript were sponsored by Lupin Atlantis Holdings SA, Zug Switzerland. The study sponsor was not involved in any form of data collection, analysis or interpretation of the results.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
N.A.
REFERENCES
[1] | Miller F . Differential diagnosis of myotonic disorders. Muscle Nerve. (2008) ;37: (3):293–9. |
[2] | Morales F , Pusch M . An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front Neurol. (2019) ;10: :1404. |
[3] | Heatwole CR , Statland JM , Logigian EL . The diagnosis and treatment of myotonic disorders. Muscle Nerve. (2013) ;47: (5):632–48. |
[4] | Stunnenberg BC , LoRusso S , Arnold WD , et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. (2020) ;62: (4):430–44. |
[5] | Sansone VA . The Dystrophic and Nondystrophic Myotonias. Continuum (Minneap Minn). (2016) ;22: (6, Muscle and Neuromuscular Junction Disorders):1889–915. |
[6] | Brook JD , McCurrach ME , Harley HG , et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. (1992) ;68: (4):799–808. |
[7] | Liquori CL , Ricker K , Moseley ML , et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. (2001) ;293: (5531):864–7. |
[8] | Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) ;1: (1):19–29. |
[9] | Pinessi L , Bergamini L , Cantello R , et al. Myotonia congenita and myotonic dystrophy: descriptive epidemiological investigation in Turin, Italy (1955-1979). Ital J Neurol Sci. (1982) ;3: (3):207–10. |
[10] | Phillips L , Trivedi JR . Skeletal Muscle Channelopathies. Neurotherapeutics. (2018) ;15: (4):954–65. |
[11] | Timchenko L . Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2. Int J Mol Sci. (2022) ;23: (18). |
[12] | Day JW , Ricker K , Jacobsen JF , et al. Myotonic dystrophy type molecular, diagnostic and clinical spectrum. Neurology. (2003) ;60: (4):657–64. |
[13] | Sansone VA , Ricci C , Montanari M , et al. Measuring quality of life impairment in skeletal muscle channelopathies. Eur J Neurol. (2012) ;19: (11):1470–6. |
[14] | Trip J , Drost G , Ginjaar HB , et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry. (2009) ;80: (6):647–52. |
[15] | Landfeldt E , Edstrom J , Jimenez-Moreno C , et al. Health-Related Quality of Life in Patients with Adult-Onset Myotonic Dystrophy Type 1: A Systematic Review. Patient. (2019) ;12: (4):365–73. |
[16] | Rakocevic Stojanovic V , Peric S , Paunic T , et al. Quality of life in patients with myotonic dystrophy type 2. J Neurol Sci. (2016) ;365: :158–61. |
[17] | Johnson NE , Aldana EZ , Angeard N , et al. Consensus-based care recommendations for congenital and childhood-onset myotonic dystrophy type 1. Neurol Clin Pract. (2019) ;9: (5):443–54. |
[18] | Schoser B , Montagnese F , Bassez G , et al. Consensus-based care recommendations for adults with myotonic dystrophy type 2. Neurol Clin Pract. (2019) ;9: (4):343–53. |
[19] | Meola G , Hanna MG , Fontaine B Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry. (2009) ;80: (4):360–5. |
[20] | Agencies EM Mexiletine- Summary of Product Characteristics 2018 [Available from: https://www.ema.europa.eu/en/documents/product-information/namusclaepar-product-information_en.pdf]. |
[21] | Deutsche Gesellschaft fur Neurologie.Myotone Dystrophien, nicht dystrophe Myotonien und periodische Paralysen 2017 [Available from: https://www.dgn.org/leitlinien/3457-030-055-myotone-dystrophien-nichtdystrophe-myotonien-und-periodische-paralysen-2017]. |
[22] | Lehmann-Horn F , Jurkat-Rott K , Rudel R , et al. Diagnostics and therapy of muscle channelopathies–Guidelines of the Ulm Muscle Centre. Acta Myol. (2008) ;27: (3):98–113. |
[23] | England N Standard contract for diagnostic service for rare neuromuscular disorders 2013 [Available from: https://www.england.nhs.uk/wp-content/uploads/2013/06/d04-diagn-serv-rare-neuromusc.pdf]. |
[24] | Matthews E , Fialho D , Tan SV , et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. (2010) ;133: (Pt 1):9–22. |
[25] | Heatwole CR , Moxley RT 3rd The nondystrophic myotonias. Neurotherapeutics. (2007) ;4: (2):238–51. |
[26] | Fondazione Malattie Miotoniche. Canalopatie Muscolari [Available from: http://www.fondazionemalattiemiotoniche.org/malattie-miotoniche/canalopatie-muscolari/]. |
[27] | Miotonici in Associazione. Cos’e la miotonia -Approfondimento [Available from: http://www.miotoniciinassociazioneit/cose-la-miotonia/cose-la-miotoniaapprofondimento]. |
[28] | Mankodi A Myotonic disorders. Neurol India. (2008) ;56: (3):298–304. |
[29] | D’Mello S , Shum L . A review of the use of mexiletine in patients with myotonic dystrophy and non-dystrophic myotonia. Eur J Hosp Pharm. (2016) ;23: (6):359–63. |
[30] | Trivedi JR , Bundy B , Statland J , et al. Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain. (2013) ;136: (Pt 7):2189–200. |
[31] | Statland JM , Bundy BN , Wang Y , et al. Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA. (2012) ;308: (13):1357–65. |
[32] | Kwiecinski H , Ryniewicz B , Ostrzycki A . Treatment of myotonia with antiarrhythmic drugs. Acta Neurol Scand. (1992) ;86: (4):371–5. |
[33] | Suetterlin KJ , Bugiardini E , Kaski JP , et al. Long-term Safety and Efficacy of Mexiletine for Patients With Skeletal Muscle Channelopathies. JAMA Neurol. (2015) ;72: (12):1531–3. |
[34] | Fullam TR , Chandrashekhar S , Farmakidis C , et al. Non-dystrophic myotonia: 2-year clinical and patient reported outcomes. Muscle Nerve. (2022) ;66: (2):148–58. |
[35] | Vicart S , Franques J , Bouhour F , et al. Efficacy and safety of mexiletine in non-dystrophic myotonias: A randomised, double-blind, placebo-controlled, cross-over study. Neuromuscul Disord. (2021) ;31: (11):1124–35. |
[36] | Heatwole C , Luebbe E , Rosero S , et al. Mexiletine in MyotonicDystrophy Type 1: A Randomized, Double-Blind, Placebo-Controlled Trial. Neurology. (2021) ;96: (2):e228–e40. |
[37] | Mousele C , Matthews E , Pitceathly RDS , et al. Long-term Safety andEfficacy of Mexiletine in Myotonic Dystrophy Types 1 and 2. Neurol Clin Pract. (2021) ;11: (5):e682–e5. |
[38] | ClinicalTrials.gov. Study to Investigate the Efficacy and Safety of Mexiletine in Patients With Myotonic Dystrophy Type 1 and Type 2 (MIND) [Available from: https://clinicaltrials.gov/ct2/show/NCT04700046#contacts. |
[39] | EMA. Orphan Maintenance Assessment Report 2018 [Available from: https://www.ema.europa.eu/en/documents/orphan-maintenance-report/namuscla-orphan-maintenance-assessment-report-initial-authorisation_en.pdf]. |
[40] | Chalmers J , Armour M The Delphi Technique. In: Liamputtong P, editor. Handbook of Research Methods in Health Social Sciences. Singapore: Springer; 2019. |
[41] | Niederberger M , Spranger J . Delphi Technique in Health Sciences: A Map Front Public Health. (2020) ;8: :457. |
[42] | EMA. Assessment report NaMuscla 2018 [Available from: https://www.ema.europa.eu/en/documents/assessmentreport/namuscla-epar-public-assessment-report_en.pdf]. |
[43] | Logigian EL , Martens WB , Moxley RTt , et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. (2010) ;74: (18):1441–8. |
[44] | Italiana R Gazzetta Ufficiale, G.U. n. 201 del 28-08-2010 2010 [Available from: https://www.gazzettaufficiale.it/eli/gu/2010/08/28/201/sg/pdf]. |
[45] | Ministero della Difesa. Comunicazione sulla disponibilita di medicinali prodotti dallo SCFM 2022 [Available from: https://portalefarmaci.agenziaindustriedifesa.it/?p=36043]. |
[46] | AIFA. Legge 648/1996 [Available from: https://www.aifa.gov.it/en/legge-648-96. |
[47] | Desaphy JF , Altamura C , Vicart S , et al. Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine. J Neuromuscul Dis. (2021) ;8: (3):357–81. |
[48] | Chapman AM , Schurer M , Weijers L , et al. Improving the understanding of how patients with non-dystrophic myotonia are selected for myotonia treatment with mexiletine (NaMuscla): outcomes of treatment impact using a European Delphi panel. BMC Neurol. (2021) ;21: (1):467. |
[49] | Meyer AP , Roggenbuck J , LoRusso S , et al. Genotype-Phenotype Correlations and Characterization of Medication Use in Inherited Myotonic Disorders. Front Neurol. (2020) ;11: :593. |
[50] | Frommeyer G , Garthmann J , Ellermann C , et al. Broad antiarrhythmic effect of mexiletine in different arrhythmia models. Europace. (2018) ;20: (8):1375–81. |
[51] | Lei M , Wu L , Terrar DA , et al. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation. (2018) ;138: (17):1879–96. |
[52] | Wahbi K , Furling D . Cardiovascular manifestations of myotonic dystrophy. Trends Cardiovasc Med. (2020) ;30: (4):232–8. |
[53] | Agenzia Italiana del Farmaco. DETERMINAZIONE 17 Agosto 2010. [Available from: bit.ly/3KIXkoF] |
[54] | Hasson F , Keeney S , McKenna H . Research guidelines for the Delphi survey technique. J Adv Nurs. (2000) ;32: (4):1008–15. |


