
‘A novel TRIP4 Variant Associated with Peripheral Neuropathy: Expanding the Clinical and Genetic Spectrum of ASC1-Related Myopathy’
Abstract
Activating Signal Cointegrator 1 complex (ASC-1 complex) is a ribonucleoprotein tetramer participating in transcriptional coactivation and RNA processing, consisting of four subunits: ASCC1-ASCC3 and ASC-1. Pathogenic variants in the TRIP4 and ASCC1 genes, encoding the ASC-1 and ASCC1 subunits, were recently described in congenital myopathic conditions without signs of motor neuron involvement, and Spinal Muscular Atrophy-like (SMA-like) phenotype with prenatal bone fractures. We present a novel pathogenic TRIP4 variant in two siblings with severe phenotype and mixed sensory-motor polyneuropathy. The reviewed phenotypic spectrum is broad, but sensory-motor polyneuropathy is so-far unreported. We thus expand ASC-1 related myopathy phenotype.
BACKGROUND
Activating Signal Cointegrator 1 complex (ASC-1 complex) is an emerging key modulator of nuclear receptors and transcription factors, acting as a transcriptional coactivator and cell-cycle regulator. It consists of four subunits: ASCC1, ASCC2, ASCC3 and ASC-1 [1–]. Transcriptional and cell-cycle regulation defects are emerging as novel pathogenic mechanisms for congenital neuromuscular diseases following recent identification of pathogenic variants in the Thyroid Hormone Receptor Interactor 4 (TRIP4) and Activating Signal Cointegrator 1 Complex Subunit 1 (ASCC1) genes, encoding ASC-1 and ASCC1 subunits. TRIP4 and ASCC1 variants have been associated with congenital myopathy and Spinal Muscular Atrophy-like (SMA-like) phenotype with prenatal bone fractures [1, 2, 4, 5]. The phenotypic spectrum is broad [1].
We describe a pathogenic TRIP4 variant in two siblings with so far unreported mixed sensory-motor polyneuropathy.
PATIENTS
Patient #1
The proband was the second child of healthy consanguineous Pakistani parents. The older sister, 4 years old, is healthy. The couple’s first pregnancy was terminated due to growth arrest. In this third pregnancy, reduced fetal movements were reported. Delivery, at 41 + 2 weeks of gestation (wGA), was complicated by right shoulder dystocia resulting in brachial plexus injury. She received Apgar scores of 2, 1, and 1 at 1, 5 and 10 minutes.
This female newborn presented with severe hypotonia, poor antigravity movements, bilateral congenital vertical talus, respiratory distress and life-threatening respiratory failure. X-rays showed left humerus fracture (Fig. 1). She required assisted ventilation with tracheostomy since birth, and a percutaneous endoscopic gastrostomy (PEG) was placed aged two months. Right diaphragm hemiparesis and atrial septal defect were also present. 24 hours after birth, she began to suffer from seizures, controlled by phenytoin and levetiracetam. Electroencephalogram (EEG) showed an occipital focus while brain magnetic resonance imaging (MRI) performed aged one week showed left temporal and occipital cortico-subcortical hematoma. Follow-up EEG showed abnormal background without epileptiform discharges, while control brain MRI at three months did not reveal any basal ganglia damage (Fig. 2). At birth, nerve conduction studies (NCS) revealed absence of compound muscle and sensory nerve action potentials (CMAP and SNAP) exclusively in the right upper limb, indicative of complete brachial plexus damage. At six months, repeat NCS showed absent SNAP and reduced motor nerve conduction velocities (NCV) in the lower limbs, in keeping with mixed axonal and demyelinating sensory-motor polyneuropathy (Table 1). Neurological examination was unchanged (diffuse hypotonia, predominating axially, absent head control). She was able to smile and follow visual stimuli. She had no valid cough or gag reflex. She developed chronic mechanical ventilation-dependent respiratory failure. She received early palliative care and died at six months secondary to pneumonia.
Fig. 1
Chest X-ray of patient #1 at 3 months of life showing a diaphyseal fracture on the left humerus, with signs of bone callus apposition (blue arrow). L = left.
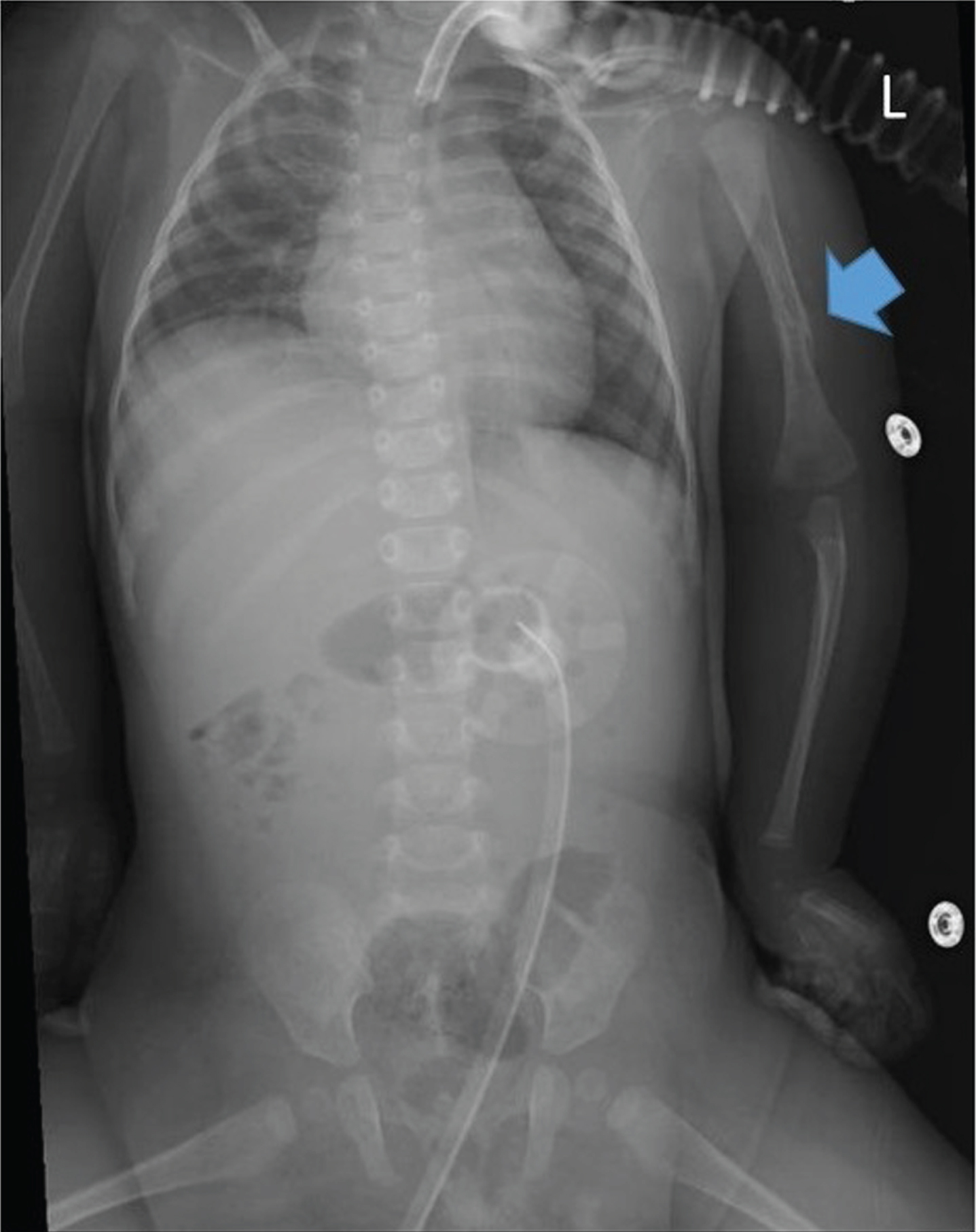
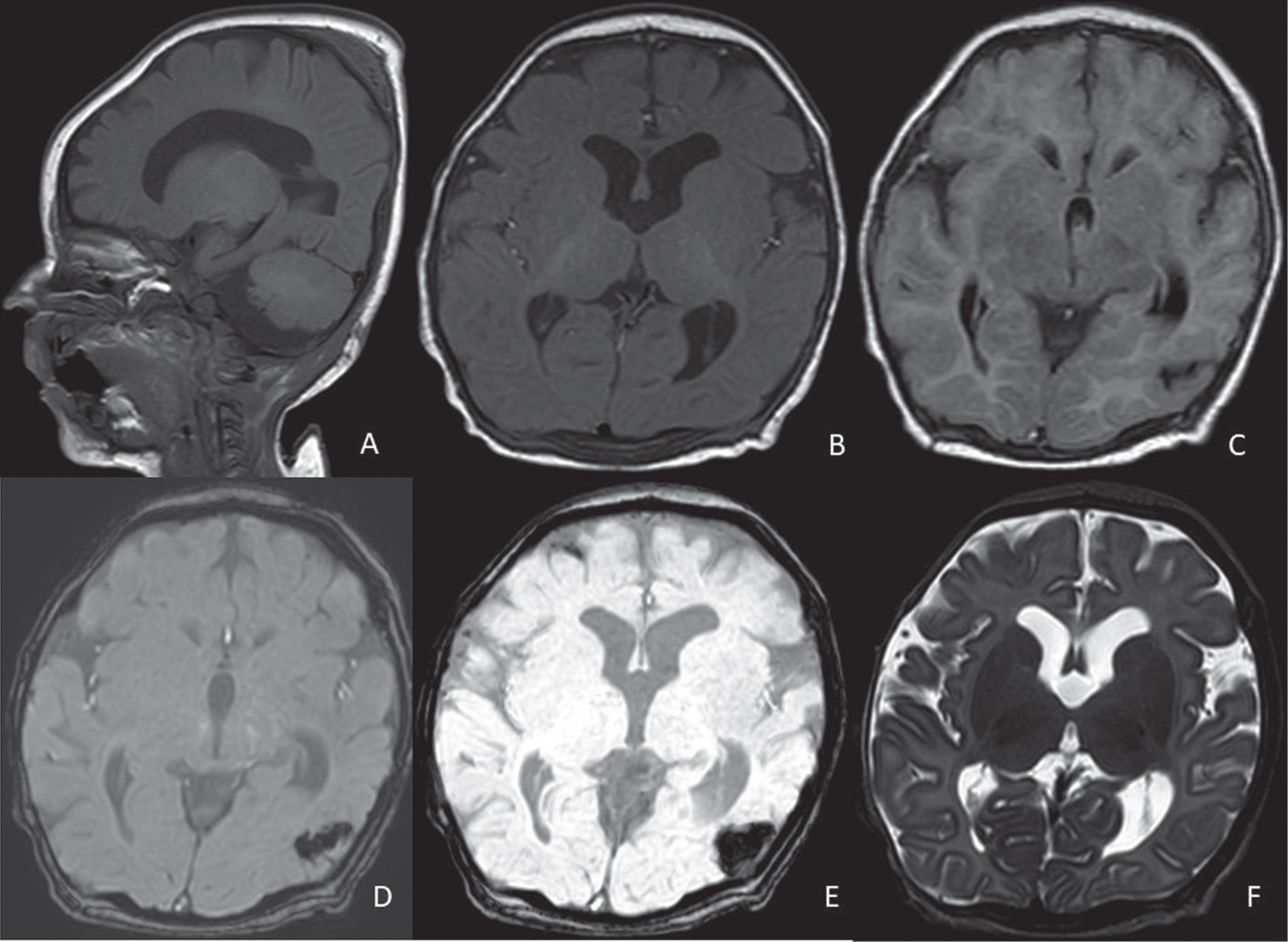
Fig. 2
Control brain MRI of patient #1 at 3 months of age. Dilated ventricualr system (especially at the lateral ventricles level, A-F), increased CSF spaces in the rolandic areas bilaterally, chronic evolution of a focal haemorrhage at the left temporo-occipital carrefour. A: sagittal T1, B: axial T1, C: axial FLAIR, D: axial susceptibility-weighted phase (SWIp) fast, E: axial minimum intensity projection (miniP), F: axial T2-weighted sequence.
Neurometabolic investigations were unremarkable. Echocardiography and eye examination were normal. Muscle biopsy (from the quadriceps muscle) showed mixed neuromyogenic damage. Perimisial connective tissue, fibre size variability, some nuclear centralization, rare regenerative features, and focal PAS-positive fibers were noted. Oxydative enzimes activity was normally distributed, however, some fibers showed COX staining subsarcolemmal enhancement. No alkaline phosphatase positivity was observed. Acid phosphatase was slightly positive in rare fibers. Some hypotrophic fibers were esterase-positive. Myophosphorilase reactivity was normal.
SNP array (GenetiSure Dx Postnatal Array Kit 4x180, Agilent Technologies) showed no unbalanced chromosomal rearrangements, and in particular no deletions/duplications were found in the PMP22 gene (30 probes). Since MLPA of SMN1 gene, DMPK gene analysis for myotonic dystrophy, and a NGS panel investigating RASopathy-associated genes were non-contributory, trio whole exome sequencing (WES) was performed. Phenotype driven analysis coupled with the employment of in silico multigene panels specific for different neuromuscular diseases was used to filter, select and interpret genetic variants, according to both autosomal recessive, autosomal dominant (de novo variants) and X-linked inheritance models. Variant annotation included occurrence in public databases (gnomAD v2.1.1) and analysis with bioinformatic prediction tools (Polyphen2, SIFT, MutationTaster, PhyloP, CADD-Phred). Variant classification was conducted according to ACMG/AMP guidelines [6]. WES identified the homozygous c.136 C > T, p.(Arg46*) variant in the TRIP4 gene (NM_016213.4), detected in heterozygosity in both parents, which was classified as likely pathogenic (ACMG/AMP criteria PVS1 and PM2: nonsense substitution, consistent with variants observed in other patients, generating a premature stop codon in exon 2 expected to lead to nonsense-mediated mRNA decay; at extremely low allele frequency in the reference population database gnomAD v2.1.1, accessed on 2023/07/20, with no reported homozygous individuals).
Table 1
Sensory and motor nerve conduction findings at birth and at 6 months in our proband (A and B, respectively) and in her younger sibling at birth (C); n.r.: no response; (normal values)
| Sensory nerve conduction findings at birth (A) | Sensory nerve conduction findings | Sensory nerve conduction findings at birth (C) | ||||||||
| at 6 months of age (B) | ||||||||||
| Conduction Velocity | Latency | Response Amplitude | Conduction Velocity | Latency | Response Amplitude | Conduction Velocity | Latency | Response Amplitude | ||
| Right Sural | 24 m/s (>22) | 2.0 (<2.2) | 3.1μV (>3) | n.r. | Right Sural | n.r. | n.r. | n.r. | ||
| Left Sural | 23 m/s (>22) | 1.9 (<2.2) | 3.1μV (>3) | n.r. | Left Sural | n.r. | n.r. | n.r. | ||
| Right Median | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | Right Median | n.r. | n.r. | n.r. |
| Left Median | 28 m/s (>21) | 2 (<2.9) | 18μV (>10) | 31 m/s (>21) | 2.2 (<2.8) | 20μV (>15) | Left Median | 18.2 m/s | 1.9 | 10.2 uV |
| Right Ulnar | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | Right Ulnar | n.r. | n.r. | n.r. |
| Motor nerve conduction findings at birth (A) | Motor nerve conduction findings | Motor nerve conduction findings at birth (C) | ||||||||
| at 6 months of age (B) | ||||||||||
| Conduction Velocity | Latency | Response Amplitude | Conduction Velocity | Latency | Response Amplitude | Conduction Velocity | Latency | Response Amplitude | ||
| Right Posterior Tibial | 25 m/s (>24) | 1.3 (<2.5) | 5 mV (>3.5) | 21 m/s (>27) | 4 (<3.7) | 9.5 mV (>5) | Right Tibialis | 19.2 m/s | 6.3 | 3.7 mV |
| Left Posterior Tibial | 25 m/s (>24) | 1.6 (<2.5) | 4.5 mV (>3.5) | 23 m/s (>27) | 4 (<3.7) | 8.9 mV (>5) | Left Tibialis | 19.4 m/s | 6.6 | 3.2 mV |
| Right Peroneal | 26 m/s (>25) | 1.7 (<2.2) | 1.4 mV (>1.2) | 22 m/s (>27) | 4 (<2.5) | 2.5 mV (>2) | Right Peroneal | n.r. | n.r. | n.r. |
| Left Peroneal | 26 m/s (>25) | 1.8 (<2.2) | 1.4 mV (>2) | 21 m/s (>27) | 4.1 (<2.5) | 2.6 mV (>2) | Left Peroneal | n.r. | n.r. | n.r. |
| Right Median | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | Right Median | n.r. | n.r. | n.r. |
| Left Median | 24 m/s (>21) | 2 (<2.9) | 2.4 mV (>1.7) | 32 m/s (>30) | 2.4 (<3.2) | 3.1 mV (>2) | Left Median | 26.4 m/s (>21) | 5.4 (<2.9) | 0.5 mV (>1.7) |
| Right Ulnar | n.r. | n.r. | n.r. | n.r. | n.r. | n.r. | Right Ulnar | n.r. | n.r. | n.r. |
As secondary findings, the heterozygous c.2043_2058del, p.(Leu682Ilefs*24) paternal variant in the DPYD gene (NM_000110.3) and the heterozygous c.1006 C > T, p.(Arg336Cys) paternal variant in the CBS gene (NM_000071.2) were found, conferring carrier status for two recessive conditions, likely not involved in our patient’s phenotype.
SNP array and WES revealed 12 runs of homozygosity spanning about 205 Mb across 7 chromosomes, and containing 1999 genes. Based on current knowledge (OMIM, PubMed), only 34 of these genes have potential relevance to neuromuscular diseases, and none contained any candidate variant that might contribute to the patient’s phenotype.
Patient #2
The third child of the couple is 5 months old. Fetal movements and prenatal ultrasound scans were normal. Genetic analysis on chorionic villus sampling disclosed a normal male karyotype (46, XY) and the same homozygous variant on TRIP4 gene as in patient #1.
He was born by vaginal delivery at 39 wGA with Apgar scores of 3, 6 and 7 at 1, 5 and 10 minutes. As a newborn, NCS showed mixed axonal and demyelinating sensory-motor polyneuropathy (Table 1). Electromyography was normal.
He was discharged with nasal Continuous Positive Airway Pressure (nCPAP) with positive end-expiratory pressure (PEEP) of 4 cmH20. He is PEG-fed.
At 2 months of life he showed microcephaly (OFC 36.5 cm, < 3rd percentile, –2.2 SD), low weight (3,680 g, < 3rd percentile, –2.5SD) and normal length (57 cm, 15–50th percentile). A sacral dimple with hypertrichosis was noticed. He was severely hypotonic with hypoactive patellar deep tendon reflexes.
Chest X-ray and ultrasound scan found diaphragmatic eventration. Echocardiogram found patent ductus arteriosus. No fractures were documented. Cranial and sacral ultrasound scans and ophthalmological evaluation were unremarkable.
DISCUSSION
Recessive TRIP4 variants were described in sixteen families (22 live-born patients, one aborted fetus). Most TRIP4-mutated patients have congenital axial and proximal weakness, reported in twelve families as congenital myopathy (“ASC-1-related myopathy”) and in four as “SMA-like” phenotype with congenital bone fractures, associated with respiratory failure, cardiac and/or cutaneous involvement [2, 4].
Excluding one patient [5] without clinical data, among the 21 described live-born patients (Table S2), the first symptom was neonatal hypotonia in 17, motor delay in three and running difficulties in one. Notably, 13 had perinatal-onset symptoms (asphyxia, respiratory distress). Eight severely affected patients died by 16 months. Four of the remainder never acquired independent ambulation, one lost independent walking in adolescence and six (8–35 years) could walk outdoors despite fatigue/dyspnoea [2, 4, 5, 7]. Four patients needed non-invasive and eight invasive ventilation. Nearly half (9/21) showed myopathic muscle biopsy or electromyography without signs of motor-neuron involvement and normal NCS (when performed). In five, NCS revealed axonal neuropathy [4].
We describe two floppy siblings with severe neonatal hypotonia, poor swallowing, respiratory impairment and a bone fracture in one. The proband presented at birth with congenital vertical talus, but without the most severe reported deformities (arthrogryposis multiplex congenita, scoliosis with rigid spine, joint contractures) [1, 2, 4]. Her younger brother, although less severely affected, has a similar phenotype, with hypotonia, feeding and respiratory difficulties.
Defects in ASC-1 complex components lead to a large spectrum of neuromuscular diseases. 15/21 patients presented clinically, electrophysiologically and/or histopathologically with primary skeletal muscle phenotype characterized by severe depletion or absence of the ASC-1 protein, resulting in congenital myopathy. Six patients from three families [4] had a more severe antenatal phenotype marked by arthrogryposis multiplex congenita, bone fractures and neonatal respiratory distress, caused by TRIP4 variants leading to upregulation of a shorter ASC-1 isoform. In these cases, muscles did not activate upon electrical nerve stimulation, neurography revealed axonal neuropathy, and muscle biopsy (when performed) showed reduced and variable fibre size [4], thus a primary muscle component could not be excluded. However, type I muscle fibres clustering and the presence of apoptotic alpha-motor neurons in the anterior horn of the spinal cord in one muscle biopsy prompted diagnosis of primary motor neuron disease (“SMA-like”) [4, 8].
ASC-1 complex was proposed to play a relevant role in congenital and degenerative motorneuron disease, representing a link between SMA and amyotrophic lateral sclerosis [9]. One patient had cerebellar hypoplasia in addition to “SMA-like” phenotype and congenital bone fractures [10]. We did not observe any signs of first motor neuron involvement nor central nervous system malformations in our two siblings, but growing evidence supports strong association of TRIP4 defects with neuronal involvement [4, 9, 10]. Even if the proband suffered from cerebral haematoma, in our opinion and based on previous clinical cases, at this stage it should be interpreted in the context of dystocic delivery and neonatal distress rather than attributed to a disease-specific mechanism. The association between central and peripheral nervous system involvement in patient #1 might have contributed to her more severe neurological phenotype compared to her sibling. However, phenotypic variability between the two siblings might relate to variable expressivity, or other genetic/epigenetic factors, which should be further investigated in clinical and experimental settings.
Our patients had early-onset mixed sensory-motor polyneuropathy. We highlight, for the first time, a sensory-motor mixed demyelinating and axonal neuropathy. In the proband, CMAP and SNAP were normal soon after birth, while at six months SNAP were absent and motor NCV reduced, indicating progressive polyneuropathy. To our knowledge, TRIP4 gene variants were exclusively associated with axonal neuropathy. Therefore, our cases broaden the phenotypic spectrum. WES analysis was filtered according to a large list of genes involved in neurological conditions, including most of the known causes of neuropathy. Based on our experience, longitudinal NCS is advisable, because polyneuropathy may appear during follow-up.
In our two siblings, we identified the pathogenic homozygous p.Arg46* (c.136 C > T) TRIP4 gene variant, which was described once in the homozygous state in a two-months old patient with a clinical diagnosis of congenital muscular dystrophy Davignon–Chauveau type who presented in the neonatal period with respiratory failure, hypotonia, arthrogryposis, dysphagia and multiple skeletal malformations [11].
Ex-vivo and in-vitro experiments performed on murine myoblastic C2C12 cell lines and on primary muscle cultures from patients with TRIP4 variants revealed ASC-1 depletion and dysregulation of late myogenic differentiation and/or myotube growth [1]. Moreover, TRIP4 variants disrupt motor units and bony structure development. Knockdown ascc1 and trip4 zebrafish revealed severe interference in motor unit development, with damaged axonal outgrowth, neuromuscular junction density and myotome [4].
In conclusion, this work adds to current literature by adding a novel TRIP4 pathogenic variant, and documenting mixed axonal and demyelinating peripheral neuropathy as a possible clinical and neurophysiologic finding.
ACKNOWLEDGMENTS
This work was generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA) (EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516). The authors also wish to thank the parents of the siblings for their cooperation in providing the medical data necessary for this publication. The authors would like to sincerely thank Progetto Pulcino Onlus, as their support made this publication possible.
ETHICS STATEMENTS
The work described in this paper was performed in accord with the Helsinki Declaration of 1975.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230110.
REFERENCES
[1] | Davignon L , Chauveau C , Julien C , Dill C , Duband-Goulet I , Cabet E , et al. The transcription coactivator ASC-1 is a regulator of skeletal myogenesis, and its deficiency causes a novel form of congenital muscle disease, Hum Mol Genet (2016) ;25: :1559–73. |
[2] | Villar-Quiles RN , Catervi F , Cabet E , Juntas-Morales R , Genetti CA , Gidaro T , et al. ASC-1 Is a Cell Cycle Regulator Associated with Severe and Mild Forms of Myopathy, Ann Neurol (2020) ;87: :217–32. |
[3] | Meunier J , Villar-Quiles RN , Duband-Goulet I , Ferreiro A . Inherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases, Int J Mol Sci (2021) ;22: :6039. |
[4] | Knierim E , Hirata H , Wolf NI , Morales-Gonzalez S , Schottmann G , Tanaka Y , et al. Mutations in subunits of the activating signal cointegrator 1 complex are associated with prenatal spinal muscular atrophy and congenital bone fractures, Am J Hum Genet (2016) ;98: :473–89. |
[5] | Marais A , Bertoli-Avella AM , Beetz C , Altunoglu U , Alhashem A , Mohamed S , et al. Further clinical and genetic evidence of ASC-1 complex dysfunction in congenital neuromuscular disease, Eur J Med Genet (2022) ;65: :104537. |
[6] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet Med (2015) ;17: :405–24. |
[7] | Dembour A , Destrée A , Deprez M , Kadhim H , Karadurmus D , Froment O , et al. ASC1 complex related conditions: Two novel paediatric patients with TRIP4 pathogenic variants and review of literature, Eur J Med Genet (2022) ;65: :104469. |
[8] | Oliveira J , Martins M , Pinto Leite R , Sousa M , Santos R . The new neuromuscular disease related with defects in the ASC-1 complex: Report of a second case confirms ASCC1 involvement, Clin Genet (2017) ;92: :434–39. |
[9] | Chi B , O’Connell JD , Iocolano AD , Coady JA , Yu Y , Gangopadhyay J , et al. The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex, Nucleic Acids Res (2018) ;46: :11939–51. |
[10] | Töpf A , Pyle A , Griffin H , Matalonga L , Schon K Solve-RD SNV-indel working Group et al. Exome reanalysis and proteomic profiling identified TRIP4 as a novel cause of cerebellar hypoplasia and spinal muscular atrophy (PCH1), Eur J Hum Genet (2021) ;29: :1348–53. |
[11] | (Kozhanova et al., 2021) |


