
Induced Pluripotent Stem Cells for Modeling Physiological and Pathological Striated Muscle Complexity
Abstract
Neuromuscular disorders (NMDs) are a large group of diseases associated with either alterations of skeletal muscle fibers, motor neurons or neuromuscular junctions. Most of these diseases is characterized with muscle weakness or wasting and greatly alter the life of patients. Animal models do not always recapitulate the phenotype of patients. The development of innovative and representative human preclinical models is thus strongly needed for modeling the wide diversity of NMDs, characterization of disease-associated variants, investigation of novel genes function, or the development of therapies. Over the last decade, the use of patient’s derived induced pluripotent stem cells (hiPSC) has resulted in tremendous progress in biomedical research, including for NMDs. Skeletal muscle is a complex tissue with multinucleated muscle fibers supported by a dense extracellular matrix and multiple cell types including motor neurons required for the contractile activity. Major challenges need now to be tackled by the scientific community to increase maturation of muscle fibers in vitro, in particular for modeling adult-onset diseases affecting this tissue (neuromuscular disorders, cachexia, sarcopenia) and the evaluation of therapeutic strategies. In the near future, rapidly evolving bioengineering approaches applied to hiPSC will undoubtedly become highly instrumental for investigating muscle pathophysiology and the development of therapeutic strategies.
INTRODUCTION
The 2012 Nobel Prize in Medicine awarded to Prs Gurdon and Yamanaka highlighted 60 years of research on pluripotency and pluripotency maintenance together with method for the derivation of induced pluripotent stem cells (iPSC) from any human being [1, 2]. Over the last 15 years, this major through has transformed biomedical research and opened the gate to the elaboration of novel in vitro models of human diseases. The very first consequence of the development of the iPSC technology was to bring pluripotent stem cells to many laboratories worldwide together with the development of increasingly innovative differentiation procedures covering the wide repertoire of human cell types. Most importantly, because they can be derived directly from patients, human induced pluripotent stem cells (hiPSC) offer promising opportunities for modeling and investigating human genetic diseases, which many hoped could prove to be more relevant than animal models that don’t necessarily recapitulate human physiology or diseases. The development of hiPSC-based model also opens a broad range of novel opportunities for enforcement of the 3 R rules to replace or refine the use of animal models. By easing access to relevant human tissues not otherwise accessible, hiPSC slowly became a powerful source of biological material for enhancing our understanding of physiological and pathological processes, disease modeling, drug discovery and optimization of cell-based therapies (Fig. 1A).
Fig. 1
Summary of different in vitro approaches for modeling healthy and diseased skeletal muscle based on the use of induced pluripotent stem cells. A. Schematic representation of possible applications for modeling neuromuscular disorders using hiPSC together with approaches used for the development of therapeutic approaches. HiPSC are derived from NMD patients and healthy controls. The disease mutation can be corrected in patient’s cells, or introduced in healthy control cell lines, using the CRISPR-Cas9 technology. HiPSC are differentiated into skeletal muscle cells (SkMC). The cell phenotype of diseased and healthy cells can be compared by different approaches, including functional testing. HiPSC-SkMC can also be used for screening and development of new therapeutics. B. Schematic representation of different applications and 3D culture systems of hiPSC-derived multilineage progenitors including co-culture in microfluidic devices (2D/3D), 3D culture models for the development of complex mature muscle models. Different physical/chemical stimuli used to induce muscle contractions or maturation are represented.
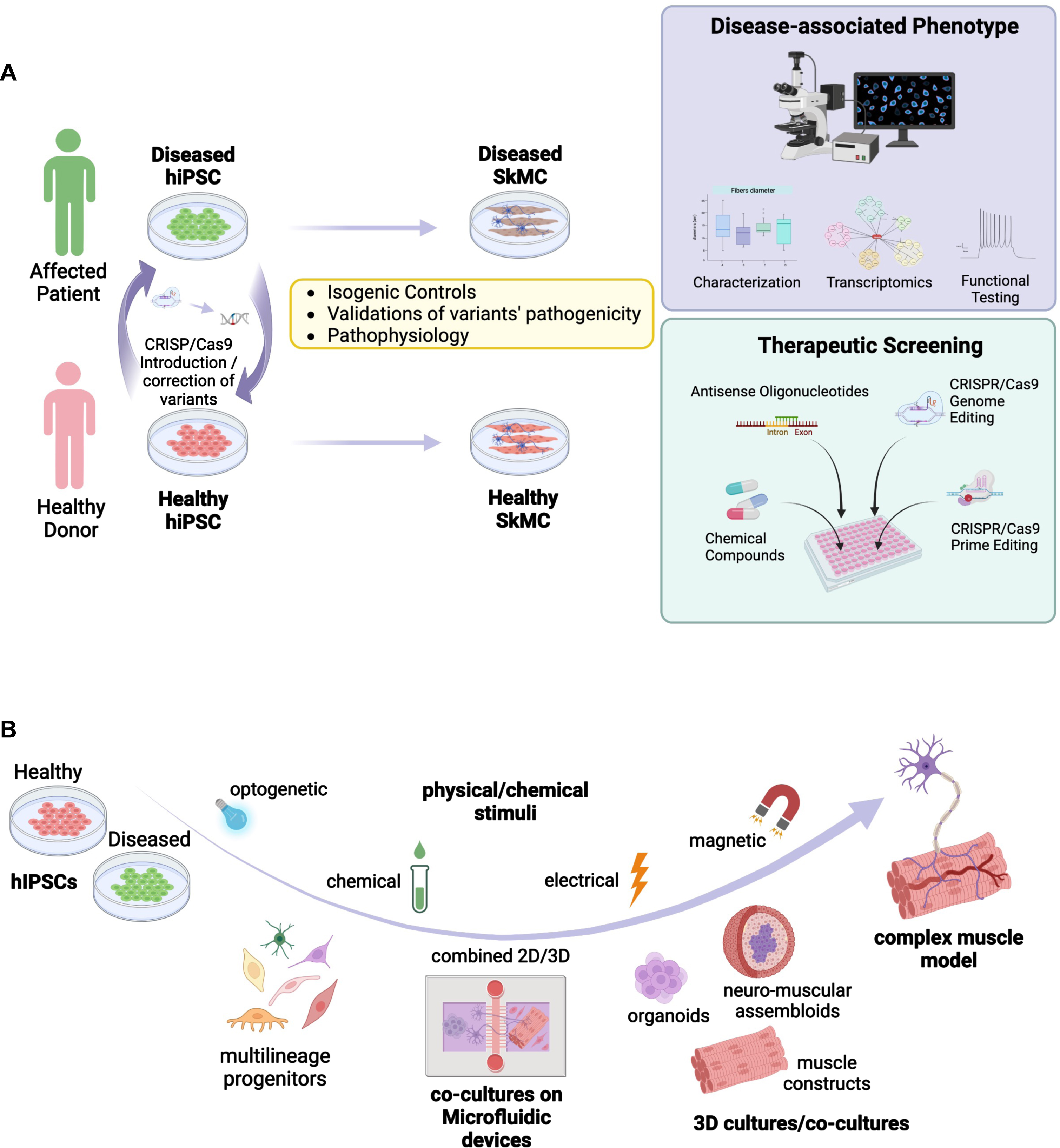
Skeletal muscle is a complex tissue composed of different cell types that all contribute to its function. Mouse models and murine cell lines such as C2C12 cells do not accurately reflect all aspects of human muscle development and biology. Human myoblasts can be obtained by isolation of muscle stem cells from human biopsies [3, 4]. These cells can be readily expanded but may lose their capacity over time with a reduced proliferation potential and cell senescence. Cell immortalization using SV40 T antigen, CDK4 and hTERT overexpression can circumvent these limitations [5, 6]. Alternatively, myoblasts can be obtained from primary fibroblasts after overexpression of PAX7 or MYOD1 [7, 8]. However, these 2-dimensional (2D) primary cell culture systems also have major limitations such as a limited proliferation rate and differentiation capacity, a lack of functionality, the absence of the other cell types present in the muscle such as stromal cells, motor neurons, vascular cells or of the extracellular matrix (ECM) components. They also lack mechanical constraints and 3D organization that characterizes the muscle tissue in vivo. Two-dimensions muscle cells cultures are also poorly amenable for functional assessment such as electro-mechanical coupling, measurement of strength and contraction or calcium handling. Altogether, this implies the need for the development of more elaborate tissue culture models with settings able to mimic more efficiently the in vivo complexity (Fig. 1B). A number of reports describe settings or assemblies of cells of different origin or even species to evaluate myo-innervation, myo-inflammation or myo-vascularization by co-culture between muscle cells together with other cells types. The challenge resides now in building more complex “bio-constructs” with appropriate cell types ratio and optimal ECM for improving cell survival, differentiation and maturation, a challenge that remains to date only partially addressed by available hiPSC-based models. Tackling these various challenges opens up a wide range of opportunities for the development of the next-generation models able to recapitulate physiological and pathological muscle development but also to respond to unmet medical needs with the increasing identification of new disease-causing genes or variants and the emergence of novel non-pharmacological therapies.
FROM DEVELOPMENTAL CUES TO THE MODELING OF MYOGENESIS IN VITRO
Compared to other lineages, hiPSC-based skeletal muscle differentiation is still lagging behind. As a consequence, modeling the large repertoire of NMDs [9] has been hampered by the absence of efficient protocols able to generate fully mature skeletal Muscle Cells (SkMC) in vitro. As for other cell lineages, differentiation of human embryonic stem cells (hESC) or hiPSC toward the skeletal muscle lineage relies on our knowledge of the differentiation of the mesodermal lineage that gives rise to the somites from the anterior to the caudal part of the embryo. During development, the dermomyotome that expresses the PAX3 and PAX7 transcription factors in the dorsal part of the embryo will form skeletal muscles, the dermis, endothelial cells and vascular smooth muscle cells [10, 11]. Under the influence of the neural tube and notochord, the dorsomedial lip of the dermomyotome progressively acquires features specific to the skeletal muscle lineage with expression of the MYOD and MYF5 transcription factors in myoblasts, able to migrate from the myotome and fuse to form embryonic muscle fibers [12, 13]. Based on this sequential activation of myogenic factors, initial protocols for differentiation of hiPSC toward the skeletal muscle lineage made use of induced expression of master myogenic transcription factors such as PAX3 [14] or MYF5 [15] but more efficiently, PAX7 [16–19] or MYOD [20–28] following viral transduction [16–18, 20–22, 24–28], mRNA transfection [29] with eventually co-expression of epigenetic modifiers, BAF60 C [30] or JMJD3 [31]. MYOD induction was more largely employed for modeling muscle diseases, in particular Duchenne Muscular Dystrophy (DMD) and the evaluation of therapeutic approaches (Table 1). However, most of these protocols had the disadvantage of using viral induced expression of myogenic factors with the risk of random transgene insertion that can interfere with the host cells genome activity when considering regenerative medicine approaches.
Table 1
Example of hiPSC- or hESC-based models of Neuromuscular Disorders
| Disease | Differentiation method | Main findings |
| DMD [24] | Dox-inducible MYOD1 transgene; lentiviral vector [24] | Qualitative and transcriptomic characterization of hiPSC-derived muscle fibers for DMD modeling. |
| DMD [115] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | DMD gene correction by TALEN and Crispr/Cas9. |
| DMD [21, 22] | MyoD-ERT; TMX-ERT system, lentiviral vector | Genetic correction of DMD cells. |
| DMD [26] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Exon skipping for DMD correction. |
| DMD [41] | CHIR99021 (day1-4), DAPT (day5–12); 30 days [41] | Comparison of hiPS-derived muscle features revealed concordant phenotypes for patients with different genetic variants. |
| DMD [116] | MyoD-BAF60 C; PiggyBac transposon [30] | Constitutive activation of the TGFβ-SMAD2/3 signaling pathway in DMD iPSC-derived myotubes |
| DMD [117] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23, 28] | Development of collagen gels and electrical field stimulation for maturation of hiPS-derived muscle fibers for drug screening. |
| DMD [118] | 3 steps induction model using small molecules and growth factors [46] | Transcriptome and MiRnome analysis for evidence of early stages of DMD onset. |
| DMD [75] | 3 steps induction model using small molecules and growth factors [41] | High content imaging-based drug screening for identification of drugs; selection of hits and validation in Mdx mice. |
| DMD [72] | 5 steps induction model using small molecules and growth factors [40] | Prednisolone rescues force contraction defects and calcium hyperactivation in hiPSC-derived myogenic cultures. |
| DMD [119] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Method for restoration of Dystrophin by Exon Skipping using CRISPR-Cas9 in myoblasts derived from DMD patient iPSCs. |
| DMD [120] | Dox-inducible MYOD1 transgene; lentiviral vector [24] | Definition of a Crispr/Cas9 approach applicable to 60% of DMD patients. |
| DMD [121] | Dox-inducible MYOD1 transgene; lentiviral vector [24] | Crispr/Cas9 genome editing to induce Utrophin expression in DMD. |
| DM1 [27] | MyoD-ERT; TMX-ERT system, lentiviral vector [21, 22] | RNA-based strategy to excise the GTG-repeat expansion, resulting in the disappearance of ribonuclear foci in DM1 myoblasts. |
| DM1 [90] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Reports changes in repeat expansion length in hiPSC, that remain stable upon differentiation. |
| DM1 [18] | pSAM2-iPAX7-ires-GFP and FUGW-rtTA [16, 19] | Characterization of hiPSC-derived myotubes for patients with DM1, identification of foci sequestrating MBLN1. |
| DM1 [122] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Development of a new model for DM1 phenotype evaluation and testing of antisense oligonucleotides. |
| LGMD2D [21] | MyoD-ERT; TMX-ERT system, lentiviral vector | Derivation of mesangioblasts like cells; generation of muscle fibers upon in vivo transplantation in mouse muscle |
| LGMD2D [22] | MyoD-ERT; TMX-ERT system, lentiviral vector | |
| Carnitine Palmitoyltransferase II deficiency [123] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Phenotypical characterization of Carnitine Palmitoyltransferase II deficiency by qualitative analysis and mass spectrometry. |
| Miyoshi myopathy [23, 76] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Selection of small molecules for degradation and clearance of misfolded Dysferlin in dysferlinopathies. |
| Pompe Disease [124] | CSII-EF1α-MCS-MyoD lentiviral vector [124] | TFEB overexpression promotes glycogen clearance and improves the defective muscle phenotype. |
| Infantile onset Pompe disease [25] | piggyBac-based vector for tetracycline-inducible expression of MyoD [26] | Identification of the mTORC1 pathway as defective in patient’s derived cells; rescue of Glycogen accumulation by dose-dependent rhGAA administration. |
| Nemalin Myopathy [125] | 5 steps induction model using small molecules and growth factors [40] | Characterization of oxidative stress and mitochondrial dysfunction in ACTA1-linked Nemalin myopathy. |
| DMD, LGMD2D, LMNA-related myopathy [21, 22, 98, 99] | PAX7 induction[14] followed by Wnt activation [38] or Wnt activation and BMP inhibition[40] | Detection of disease-specific muscle features. |
| DMD, DM1, FSHD, LGMD2D [48] | 5 steps induction model using small molecules and growth factors [48] | Detection of disease-specific muscle features. |
| FSHD [126] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | DUX4 expression is increased by oxidative stress and activates a DNA damage response in hiPS-derived patient’s myocytes. |
| FSHD [46] | 3 steps induction model using small molecules and growth factors [46] | Characterization of hESC-derived FSHD muscle fibers features, identification of pharmacological hits. |
| FSHD [87] | 3 steps induction model using small molecules and growth factors [46] | Characterization of hiPSC-derived myoblasts (iMyoblasts) for ex vivo and in vivo evaluation of physiological and pathological myogenesis. |
| FSHD [51] | 5 steps induction model using small molecules and growth factors [48] | Transcriptome analysis for identification of sarcomeric defects in FSHD, optimization of live cells assays. |
| Mac Ardle disease [127] | 5 steps induction model using small molecules and growth factors [48] | Characterization of hiPSC-derived myotubes. |
| LGMDR1 (LGMD2A, recessive limb girdle muscle dystrophy linked to Calpain 3 [128] | pSAM2-iPAX7-ires-GFP and FUGW-rtTA [16, 19] | Characterization of hiPSC-derived myotubes |
| LGMDR7 (recessive limb girdle muscle dystrophy linked to Telethonin (TCAP); LGMD2 G) [85] | 3 steps induction model using small molecules and growth factors [46] | Correction of the microduplication using a simple nuclease-induced double strand. |
| Fukutin-related (FKRP) dystroglycanopathies [86] | pSAM2-iPAX7-ires-GFP and FUGW-rtTA [16, 19] | Crispr-Cas9 FKRP gene correction by reversal of α-DG glycosylation. |
| LGMDR9 [129] | 3 steps induction model using small molecules and growth factors [46] | Development of a cell models for identification of molecules able to correct α Dystroglycan for correction of the LGMDR9 phenotype |
| SMA [130] | Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD [23] | Loss of SMN causes mitochondrial bioenergetic failure during myogenesis. |
| C9orf72 hexanucleotide repeat expansions (rGGGGCC), sporadic Fronto temporal dementia [44] | 5 steps induction model using small molecules and growth factors [44] | Accumulation of RNA foci linked to hexanucleotide expansion |
| Myastenia gravis [54] | Neuromuscular trunk organoids | Incubation of organoids with MG patient’s IgG caused a reduction in the number of NMJs together with a reduction in their activity. |
DMD: Duchenne Muscular Dystrophy, DM1: Myotonic Dystrophy; FSHD: FacioScapuloHumeral Dystrophy; LGMD: Limb Girdle Muscular Dystrophy; SMA: Spinal Muscular Atrophy.
A second generation of approaches consisted in the stepwise addition of small molecules to induce mesodermal differentiation, with activation of the Wnt, Sonic hedgehog (Shh) or Notch pathways for the conversion of PAX3 + /PAX7 + progenitors into MYOD+/MYF5 + committed myoblasts [32–34] together with inhibition Bone Morphogenic Proteins (BMPs) that maintains PAX3/PAX7 expression in myogenic progenitors [35–37]. Wnt activation mediated by addition of Wnt3a or more commonly CHIR99021, a GSK3β inhibitors, was efficient in driving muscle differentiation but required Pax3 + /Pax7 + cell sorting, limiting large scale explorations [38, 39]. Alternatively, activation of the Wnt pathway using CHIR99021 together with inhibition of the lateral mesoderm using the LDN193189 BMP inhibitor [40] or followed by inhibition of the Notch signaling using the DAPTγ secretase inhibitor [41] efficiently induced formation of multinucleated and contractile muscle fibers with sarcomeric organization in 50 days and 30 days, respectively. Another GSK3β inhibitor, Bio, was also used in combination with Forskolin, an Adenylyl Cyclase activator that activates cAMP and CREB signaling, to stimulate myoblast proliferation in vitro and led to the production of multinucleated myotubes with Myosin Heavy Chain proteins in 36 days [42]. Treatment of hiPS cell aggregates with a high concentration of bFGF and EGF converted stem cells into 40–50% of myogenic cells expressing MYOD, PAX7 and Myogenin in 6 weeks [43]. Pre-induction of hiPSC in the presence of DMSO followed by the sequential addition of small molecules including CHIR99021, BMP4 and Lys294002 (a PI3K inhibitor) yielded the production of myocytes in 12 days, followed by muscle fusion after 34 days and spontaneous contraction upon addition of 15% of fetal bovine serum [44]. TGFβ inhibitors (SB431542 or A83-01) were also shown as capable of enhancing fusion and maturation of ERBB3 + /NGFR+hiPSC-derived muscle progenitors expressing PAX7 and Myf5 [41, 45]. Alternatively, hiPSC plated on Collagen I in the presence of 5% horse serum and treated with CHIR99021, Ascorbic acid, Alk5 inhibitor, dexamethasone, EGF and insulin, yielded up to 80% of PAX3-positive cells in 10 days and 50–60% of MyoD-positive cells after addition of SB431542 (to inhibit Alk4, 5 and 7), PDGF, EGF, HGF, bFGF, Oncostatin and IGF1 [46] with fusion and terminal differentiation later improved by addition of myokines and anabolic factors [47] (Fig. 2).
Fig. 2
Summary of the different protocols for differentiation of human pluripotent cells toward the skeletal muscle lineage. For the different protocols, the type of coating is indicated (light orange, Matrigel, Collagen I, PLO-Laminin) together with the type of medium (dark grey; HS, Horse Serum; KOSR, Knock-out serum replacement; CDM, chemically defined medium). Growth factors and cytokines are indicated in pink (IGF1, Insulin Growth Factor 1; bFGF, basic Fibroblast growth factor; HGF, hepatocyte growth factor; EGF, Epithelial Growth Factor, PDGF, Platelet derived growth factor; VEGF, Vascular Endothelial Growth Factor; BDNF, Brain Derived Neurotrophic Factor; Il4, Interleukin 4; IL§, Interleukin 6). Cell culture supplements are in yellow (ITS, Insulin-Transferrin-Selenium; AA, non-essential amino acids). Small molecules are indicated in green (CHIR, Chiron 99021, Wnt3a or GSK3β inhibitor; Bio (GSK3β inhibitor); Forskolin (Adenylyl Cyclase activator); Dexamethasone; LDN193189, BMP inhibitor; DAPT, γ secretase inhibitor; SB431542, Ai, RepSox, selective Alk5 inhibitor).
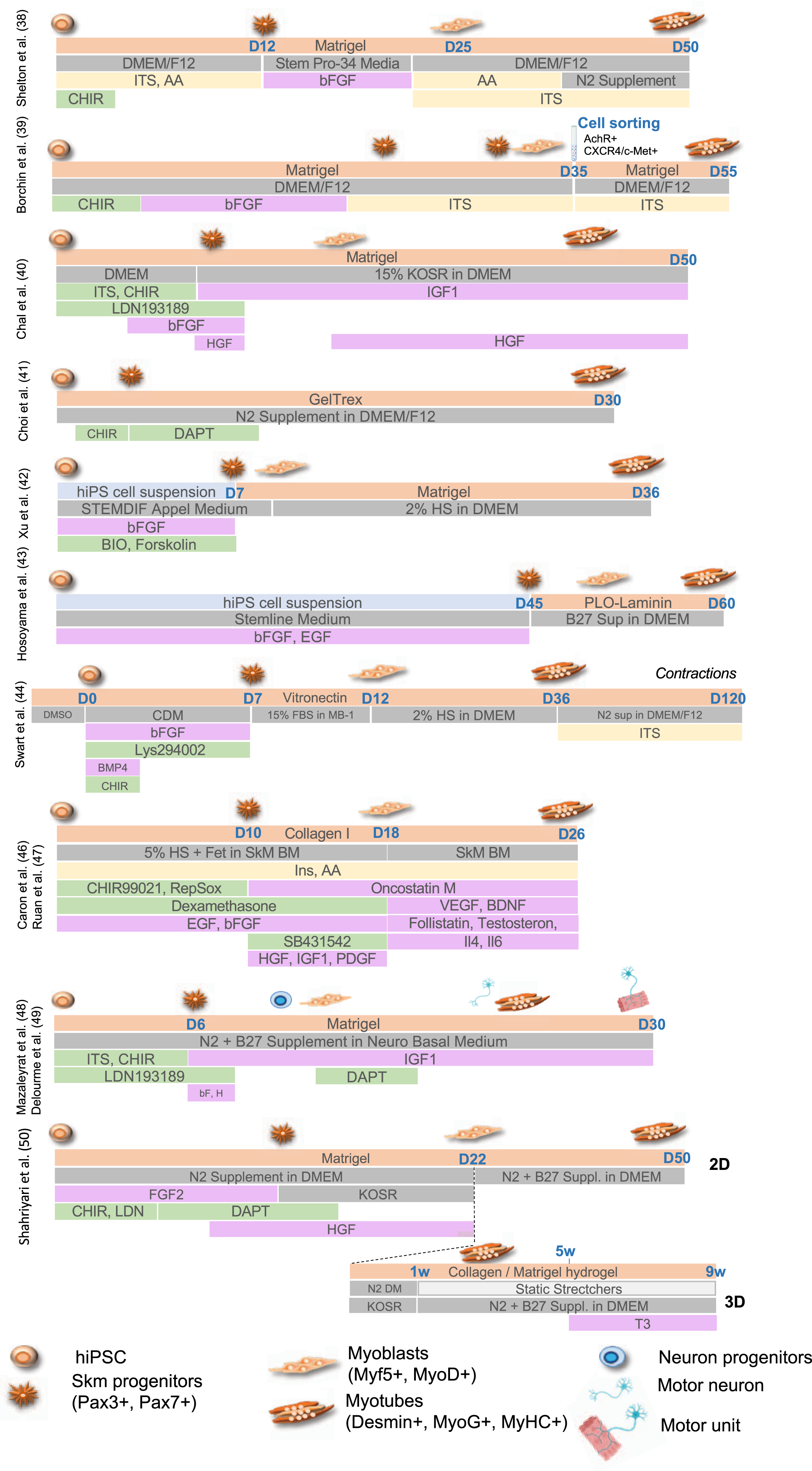
Chal et al. [40] and Hosoyama et al. [43] reported the presence of neural cells differentiating along with muscle cells suggesting an intermediate population of neuro-mesodermal progenitors during the differentiation process. Taking advantage of the transient presence of these two populations of cells, Mazaleyrat et al. [48, 49] proposed a protocol combining activation of the Wnt Pathway (addition of the CHIR99021 GSK3β inhibitor) to induce mesoderm commitment followed by BMP inhibition (LDN193189) for production of muscle and motor neurons progenitors in 8 days. The subsequent addition of DAPT induces the production of motor neurons and formation of contractile millimeter-long multinucleated muscle fibers with sarcolemmal organization in 20 days [48, 49]. Another transgene-free and serum-free protocol and a combination of small molecules to achieve 2D and 3D differentiation was more recently reported [50]. As described by others [40, 48], gene expression profiling at different stages showed that muscle cells differentiation follows developmental trajectories with paraxial mesoderm induction (days 1–4), myogenic specification (days 4–13) and myogenic expansion (days 13–22). At day 22, cells can be plated on Matrigel coated plates for 2D differentiation or into a hydrogel containing Collagen I and Matrigel to achieve 3D differentiation with secondary myogenesis occurring between days 22 to 60. In this setting, only expression of embryonic Myosins (MyH3) or at a lower level, fetal Myosin (MyH8) are detectable but addition of Tricodo-L-Thyronine (T3) at the final maturation step (weeks 5–9 post-differentiation), increase contraction strength together with expression of fast mature MyHC isoforms, sarcomere organization, an important mitochondrial network and a dense ECM [50].
For several of these protocols, cultures can be maintained in the long term thanks to the production of a dense ECM and the presence of PAX7 + satellite cells [23, 35, 46, 48, 51], an advantage in the perspective of regenerative therapy. Furthermore, treatment of myogenic cells [46] with recombinant DLL4 (Delta Like 4, Notch activation) and PDGF-BB (a PDGF receptor β ligand) limits terminal differentiation but favors myogenic progenitors motility and migration, including trans-endothelial migration, opening new perspectives for the systemic delivery of hiPSC-derived muscle progenitors for regenerative therapy [52].
The capacity of self-assembly between muscle and motor neurons was nicely illustrated in human 3D cortico-motor assembloids formed by combining hiPSC-derived cortical and motor neurons together with muscle cells embedded in an extracellular matrix to form 3D spheroids placed on top of a transwell insert. This configuration allows the formation of cortico-spinal connections and neuromuscular junctions (NMJ) between spinal neurons and muscle fibers that triggers muscle contraction up to 10 weeks post-assembly, mimicking the cortico-spinal-muscle circuit that controls muscle activity [53]. In addition, hiPSC are more and more largely used for the production of organoids or mini organs, defined as three-dimensional in vitro cell-derived structures able to self-organize to mimic a given organ. To date, only one report describes the production of neuromuscular organoids composed of spinal cord neurons and skeletal muscle cells that self-organize into 3D trunk neuromuscular organoids that can be maintained for months [54]. These organoids are characterized by the presence of neural and mesodermal cells and formation of NMJ containing pre-synaptic vesicles supported by the presence of S100β-positive Schwann cells, required for NMJ maturation [54].
In agreement with all previous reports, showing that co-culture of muscle cells with fibroblasts, fibro-adipogenic progenitors (FAPs) [55–57], macrophages [58–60] but more importantly motor neurons forming NMJ [61, 62], co-differention and self-organization of different hiPSC-derived cell types increase the differentiation rate and fiber maturation but also permits functional muscle assessments [48, 50, 53, 54] indicating that the presence of non-muscle cells is a key element in a successful in vitro myogenesis.
PLURIPOTENT STEM CELLS FOR MODELING OF NEUROMUSCULAR DISEASES
Neuromuscular diseases (NMDs) form a large and heterogeneous group of genetic diseases that cause progressive degeneration of striated muscles. Most NMDs result in chronic long-term disability imposing a significant burden for patients, families and public healthcare. These pathologies are present in all populations, affecting children as well as adults with a prevalence of 1 in a 1000 people [63, 64]. For many of these diseases, the causative gene is known but the impact of a given mutation on the molecular mechanisms of the disease often remains puzzling and requires further experimental validations for development of targeted and personalized therapies. For a number of hereditary NMDs, well-established animal models contributed to our understanding of the disease process or to the evaluation of therapies. However, many animal models imperfectly recapitulate pathophysiological features of a given disease with more than 95% of all animal-tested therapeutics, inefficiently transferred for the cure of patients.
Relative to the large number of genes associated to NMDs [9], only a few of them have been modelled using hiPSC, with the vast majority of reports focusing on Duchenne Muscular Dystrophy (DMD) for the development or evaluation of therapeutic approaches, including gene therapy (Table 1). In the context of genetic NMDs, hiPS-based models represent unprecedented opportunities to investigate the different aspects of a given neuromuscular disorders, identify new genes, validate the pathogenicity of variants or define early stages of muscle wasting. HiPSC-based models also supports the development of innovative treatments for muscle conditions and may open the door for preclinical screening of a personalized panel of drug candidates to improve these debilitating disorders, as exemplified for instance with the use of neuromuscular organoids to evaluate Myastenia gravis (MG)-associated autoantibodies against Acetylcholine receptor at the NMJ [54] (Fig. 1A).
Regarding the validation of gene variants, the latest advances in next-generation sequencing have uncovered a plethora of genomic variants of unknown significance (VUS) defined as rare or novel genetic variants for which the association with a disease has not been conclusively demonstrated [65]. One of the greatest advances of the twenty-first century is undoubtedly the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas 9 genome editing technology that allows for a precise endogenous genomic correction [66, 67] but also the introduction of single point mutations, insertions, or deletions. The combination of stem cell-based approaches with CRISPR-mediated genomic editing turned hiPSC into powerful tools to study the impact of disease-associated mutations on specific cell types as well as to determine the correlation genotype-phenotype (Fig. 1A). Most importantly, using genome-editing strategies, the pathogenicity of a VUS can be demonstrated by comparing edited hiPSC with their unedited control lines. This approach will likely contribute to the validation of genetic mechanisms underlying NMDs, as previously done for instance in hiPSC-cardiomyocytes (CM) for cardiac diseases [68], whilst maintaining relevance to the human condition and making research findings readily translatable to the clinic.
PLURIPOTENT STEM CELLS FOR THERAPEUTIC DEVELOPMENT
High-throughput drug screening and in vitro preclinical testing
Developing a novel drug to treat a disease is a lengthy and costly process. This is particularly true for muscle disorders, often due to a lack of adequate cellular and animal models that accurately reflect the human muscle pathology. Directly identifying compounds that act on human muscle would facilitate and accelerate drug discovery. HiPSC that provide a plentiful source of patient-derived specialized cells, effective for drug identification and preclinical testing of future therapeutics and captured a growing interest from the pharmaceutical industry and slowly transformed the landscape of drug discovery [69]. As an example, hiPSC-derived neural stem cells from patients with familial dysautonomia, a rare genetic but fatal neurodegenerative disorder, were used a decade ago to screen nearly 7,000 small molecules and led to the identification of a drug hit [70].
However, the optimization of protocols for cell differentiation, maturation and maintenance still represents a significant challenge, in particular for hiPSC-SkMC due to the difficulties in generating and expanding hiPSC-myoblast cultures or obtaining mature differentiated myotubes. As a result, muscle drug discovery approaches remained to date limited to target-based screening in non-muscle cells or rodent myoblasts [71] with testing often focused on known drugs such as androgen receptor modulators (e.g. testosterone), myostatin inhibitors (e.g. Follistatin) or anti-inflammatory agents. The improvement of drug discovery for the broad range of muscle conditions and the development of new therapies for the millions of patients that are currently untreated strongly requires large amount of human skeletal muscle cells that demonstrate a fully mature phenotype.
HiPSC-derived Myoblasts/Myotubes and cardiac myocytes derived from DMD patients have been extensively used to model disease-specific phenotypes that can be reversed by pharmacological compounds (Table 1). DMD hiPSC-myotubes respond to hypertrophic proteins such as IGF-1 and Wnt7a [24], both tested as potential treatments for DMD, and can be pharmacological rescued by “dual-SMAD” inhibition [41] or Prednisolone [72], a corticosteroid and the current standard of care for DMD patients to delay the disease progression by reducing inflammation-induced muscle damage and muscle strength loss. HiPSC-based model demonstrated that Prednisolone has a direct beneficial impact on myofibers and not only on inflammation [72]. Similarly, stress-induced cell death and increased apoptosis can be pharmacologically modulated in DMD hiPSC-CM and improve DMD-associated dilated cardiomyopathy [73, 74]. With the goal of identifying new potential drugs for DMD treatment, a large screen of over 1500 compounds was later carried out on hiPSC-myoblasts carrying a nonsense mutation (c.457 C>T) which abolishes production of all Dystrophin isoforms [75]. Two compounds that restored fusion deficiency of these nonsense DMD hiPSC-myoblasts were identified and further validated in DMD hiPSC–myoblasts carrying different mutations (c.5533 G>C and Ex45del). Fenofibrate and ginsenoside Rd related to TGFβ signaling and FLT3 signaling, also showed efficacy in mdx5cv mice by reducing fibrosis and improving muscle function. Patient-derived hiPSC-myotubes were also used as a drug screening platform for Dysferlinopathy with Nocodazole as a potential drug candidate for clinical applications aimed at increasing Dysferlin protein level [76].
Overall, by efficiently responding to compound testing, hiPSC-derived muscle cells open broad perspectives for drug screening or to evaluates potential therapies on a large-scale in human models of NMDs, maximizing their translational potential, whilst reducing the cost and time of novel therapeutic development.
Development of novel therapeutics
HiPSC-based technologies are also expected to provide platforms for preclinical testing of innovative therapeutics, including gene therapy approaches. Exon skipping by antisense oligonucleotides (ASOs) has gained a lot of interest and proven to be a powerful tool for mRNA splicing modulation to restore open reading frames. ASOs have now extensively been tested as therapeutic for various NMDs. Dystrophin expression was restored in approximately 30% of DMD hiPSC-CM treated with an ASO targeting exon 51 [77], or in DMD hiPSC-myotubes treated with ASO skipping exon 45 [26]. Similarly, an hiPSC-model of DM1 was used to screen ASOs and successfully identified a candidate that effectively abolished RNA foci and rescued mis-splicing in DM1 hiPSC-myotubes [18].
As mentioned earlier, (CRISPR)/Cas 9-genetically corrected patients hiPSC lines provide a powerful source of isogenic control lines for in vitro disease modeling studies. Most importantly, CRISPR/Cas9 genome editing can also be used as therapeutics to restore in vivo deletions or single point mutations. A large number of proof-of-concept studies have already demonstrated the therapeutic use of CRISPR genome editing for NMDs [78, 79], in particular in DMD (Table 1 and reviewed in [80–82]). We do not discuss them all here but will just highlight a few strategies. Myo-editing of the DMD gene mutations was for example elegantly demonstrated in hiPSC-CM corrected for exon 44 deletion, which disrupts the open reading frame of Dystrophin by causing splicing of exons 43 to 45 and introducing a premature termination codon. The reading frame can be restored by using Cas9 with guide RNAs that permits deletion of the splice acceptor or exons 43 and 45 donor sites, allowing splicing between exons 42 and 45 or 43 and 46 respectively [83]. CRISPR/Cas9-mediated excision of exon 51 in DMDΔ52-patient’s hiPSC-derived myoblasts and cardiomyocytes also restores Dystrophin expression and ameliorated skeletal myotube formation as well as cardiac function [84]. CRISPR/cas9 gene correction is not restricted to DMD. As another example, Telethonin (TCAP) expression could be efficiently restored in LGMDR7 (LGMD2 G) hiPSC-derived myoblasts by homology-mediated repair using Cas9 with a guide RNA targeting the microduplication [85]. HiPSC-myoblasts derived from FKRP dystroglycanopathies and Limb-Girdle Muscular Dystrophy (LGMD) R7 patients recapitulated their disease molecular phenotypes responsive to small molecules and gene editing therapeutics [86, 87].
However, CRISPR/Cas9 genome editing is known to create off targets that can be deleterious to the genome. An alternative CRISPR-based technology can now precisely edit individual nucleotides. With the prime editing approach, the therapeutic gene is knocked-in into a safe harbor locus allowing the correction of diseased genes independently of the underlying mutations. This new method was able to successfully restore Dystrophin expression in a DMD-hiPSC model [88] and adenine base editing (ABE) strategy, to restore Dystrophin expression in hiPSC-CM harboring a deletion of exon 51 (ΔEx51) of the Dystrophin gene in DMD patients [89].
Taken together, these different studies show that hiPSC-derived muscle cells represent efficient tools for evaluating the efficacy of drug compounds or CRISPR/Cas9- or exon-skipping-based approaches tailored to individual genetic variants or MD patients. However, except for some specific configurations [90], each of these approaches are mutations specific and therefore have to be re-engineered and adapted for each patient or each variant, a lengthy and costly process. Nonetheless, in the recent years, hiPSC-derived sensory neurons generated from patients with inherited [91] or non-genetic [92] pain disorders highly refractory to all available treatments showed that specific sodium-blocking compounds reduced the hyperexcitability of hiPSC-neurons in vitro while decreasing pain in the same patients. These studies represent proofs of principle of successful clinical translation from patient’s hiPSC in vitro models to individualized treatments. The development of similar assays for NMDs could initiate a new drug discovery paradigm moving us closer toward the era of personalized medicine as valuable tools to predict whether a given patients would respond to a particular drug.
TOWARD THE DEVELOPMENT OF TISSUE ENGINEERING FOR DISEASE MODELING AND REGENERATIVE MEDICINE
Three-dimension (3D) cultures reproducing an extracellular environment which better promotes physiological-like processes allows for the prolonged maintenance together with better level of maturation of the resulting myotubes [93, 94]. The use of hiPSC brings several advantages for the production of 3D models of human skeletal muscle tissue (Fig. 1B). In the last decade, some research groups started to work in this direction, although this field of research remains somewhat unexplored.
One of the first example of 3D engineered muscle tissue from hiPSC dates from 2013, when Tchao and colleagues made a comparative study between muscle derived stem cells and hiPSC-derived cardiac cells. In a collagen-based scaffold, 3D muscle constructs revealed several similarities compared to the development process of skeletal muscle and cardiac progenitors, providing a hybrid model for the study of cardiovascular diseases [95].
In 2018, Rao and colleagues developed a 3D culture model of hiPSC-derived skeletal muscle, starting from induced muscle progenitors embedded in a fibrin-based matrix [96]. With this approach, properly differentiated 3D skeletal muscle bundles were able to respond to electrical or acetylcholine stimulation [96]. The same year, Osaki et al. produced an organ-on-chip 3D model of amyotrophic lateral sclerosis (ALS) using hiPSC-derived motor neurons (MN) and muscle cells. Tedesco and his team were also pioneers in the development of complex skeletal muscle models using fibrin hydrogels loaded with different cellular components (myogenic, endothelial, pericytes and neural hiPSC-derived human progenitors) to generate 3D artificial muscles in normal or pathological contexts [97–99].
The role of the NMJ in muscle disorders was further investigated by exploiting 3D co-cultures of myoblasts-motor neurons and optogenetic technique [100–102]. A 3D culture of healthy muscle cells embedded in a Collagen/Matrigel matrix seeded in one chamber, with light sensitive Channel Rhodopsin-2 (ChR2)-induced ALS-derived MN spheroids in the adjacent and interconnected chamber was used to evaluate the impact of affected MN on muscle functionality and drug screening [101]. Electrophysiological recording and calcium release analyses of 2D vs 3D co-cultures of primary human myoblasts (in a fibrin/Geltrex hydrogel) and hiPSC-derived MN demonstrated a better maturation of NMJs in 3D condition, with a shift of expression from the embryonic to the adult form of nicotinic acetylcholine receptors [102]. A similar approach used a bioreactor for studying the response of 3D muscle constructs to the stimuli transmitted via the NMJ by ChR2-modified MNs in response to a blue light exposure to evaluate the effect of MG auto-antibodies on NMJ functionality [100].
The effect of magnetic fields on 3D muscle construct development and functionality was also investigated by magnetic force-based tissue engineering (Mag-TE). Magnetically labeled cells embedded in a collagen/Matrigel matrix are capable of self-assembly when exposed to the effect of a magnet. The contractile activity of hiPSC-derived “miniaturized skeletal muscle tissue” was increased compared to non-magnetically stimulated constructs in response to stimulating drugs [103].
As demonstrated by the multiple strategies used to obtain a reliable “platform” for studying NMDs, these different developments highlight the growing interest of the scientific community for 3D muscle modeling using hiPSC as the starting material but also the benefit of interdisciplinarity in tissue bioengineering (Fig. 1B). The different techniques cited here such as microfluidic devices, organoids, optogenetic, and magnetic stimulation, represent excellent examples of applying the newest bio-technological throughs to a specific biological application. However, 3D bioprinting, one of the more promising techniques for building a complex 3D artificial muscle tissue [104–106], still remains under exploited. Among the currently available techniques, the extrusion-based bioprinting [107] turned out to be the most suitable for skeletal muscle tissue engineering approaches, thanks to the possibility to produce continuous fibers of cell laden scaffold printed in parallel lines, layer by layer, perfectly reproducing skeletal muscle fibers architecture [108–110]. In the future, 3D bioprinting of hiPSC-derived muscle progenitors to obtain highly organized muscle tissue, combining known proportions of muscle and non-muscle cells will likely open new avenues in modeling NMDs and provide invaluable tools for various medical applications, including regenerative medicine.
CONCLUSION
Together with the undeniable advances provided by cell reprogramming, for the definition of cell lineages trajectories at early developmental stages, in muscle [111, 112], the development of patient-derived hiPSC has resulted in tremendous progress in disease modeling and the design of new therapies. Furthermore, over the recent years, in vivo reprogramming also emerged as a promising approach for tissue regeneration, including muscle [113, 114]. Importantly and interestingly, thanks to their capacity of differentiation toward different cell lineages, hiPSC also offer the advantage of allowing the investigation of the multisystemic aspects of diseases and to cover disease complexity as a whole, including in the course of finding a therapeutic strategy. However, as it is the case for all models, hiPSC-based disease modeling also has several limitations. Among them, one can cite the variability between clones, their respective genomic stability or intrinsic differentiation capacity, but also the need to obtain proper control in a number of cases. To this aim, the most widely used strategy consists at generating isogenic clones either to induce or correct a gene variant using a gene editing technique. However, obtention of truly isogenic clones faces multiple pitfalls as off target effects, copy number variations or point mutations induced by clone selection but also the difficulty in obtaining heterozygous mutations or to induce/correct splice variants. The often-limited efficiency of differentiation protocols in term of yield or maturation also relate to our capacity in recapitulating the in vivo environment largely absent in 2D cultures involving a single cell type. Nevertheless, in a continuously growing field, many of these limitations will be slowly compensated by the fast development of more and more sophisticated cell culture models together with high resolution approach for decoding differentiation steps in physiological and pathological contexts and correcting related defects. The establishment of standardized protocols will also help alleviate the variabilities observed between the different methods and the different batches within the same method.
FUNDING
L.C. received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant, agreement number 101025510. S.T. is the recipient of a post-doctoral fellowship awarded through the ICELARE program (ANR-20-ASTR-004). The work in FM’s laboratory is funded by AFM-Telethon through the MoThARD strategic plan.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
[1] | Takahashi K , Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) ;126: (4):663–76. |
[2] | Takahashi K , Tanabe K , Ohnuki M , Narita M , Ichisaka T , Tomoda K , et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) ;131: (5):861–72. |
[3] | Blau HM , Webster C . Isolation and characterization of human muscle cells. Proc Natl Acad Sci U S A. (1981) ;78: (9):5623–7. |
[4] | Gros J , Manceau M , Thome V , Marcelle C . A common somitic origin for embryonic muscle progenitors and satellite cells. Nature. (2005) ;435: (7044):954–8. |
[5] | Zhu CH , Mouly V , Cooper RN , Mamchaoui K , Bigot A , Shay JW , et al. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell. (2007) ;6: (4):515–23. |
[6] | Simon LV , Beauchamp JR , O’Hare M , Olsen I . Establishment of long-term myogenic cultures from patients with Duchenne muscular dystrophy by retroviral transduction of a temperature-sensitive SV40 large T antigen. Exp Cell Res. (1996) ;224: (2):264–71. |
[7] | Davis RL , Weintraub H , Lassar AB . Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. (1987) ;51: (6):987–1000. |
[8] | Weintraub H , Tapscott SJ , Davis RL , Thayer MJ , Adam MA , Lassar AB , et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A. (1989) ;86: (14):5434–8. |
[9] | Benarroch L , Bonne G , Rivier F , Hamroun D . The version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. (2020) ;30: (12):1008–48. |
[10] | Relaix F , Montarras D , Zaffran S , Gayraud-Morel B , Rocancourt D , Tajbakhsh S , et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol. (2006) ;172: (1):91–102. |
[11] | Buckingham M , Relaix F . The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annual review of cell and developmental biology. (2007) ;23: :645–73. |
[12] | Zammit PS . Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. (2017) ;72: :19–32. |
[13] | Chal J , Pourquie O . Making muscle: skeletal myogenesis in vivo and in vitro. Development. (2017) ;144: (12):2104–22. |
[14] | Darabi R , Gehlbach K , Bachoo RM , Kamath S , Osawa M , Kamm KE , et al. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med. (2008) ;14: (2):134–43. |
[15] | Iacovino M , Bosnakovski D , Fey H , Rux D , Bajwa G , Mahen E , et al. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells. (2011) ;29: (10):1580–8. |
[16] | Darabi R , Arpke RW , Irion S , Dimos JT , Grskovic M , Kyba M , et al. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. (2012) ;10: (5):610–9. |
[17] | Skoglund G , Laine J , Darabi R , Fournier E , Perlingeiro R , Tabti N . Physiological and ultrastructural features of human induced pluripotent and embryonic stem cell-derived skeletal myocytes in vitro. Proc Natl Acad Sci U S A. (2014) ;111: (22):8275–80. |
[18] | Mondragon-Gonzalez R , Perlingeiro RCR . Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: a tool for disease modeling and drug discovery. Dis Model Mech. (2018) ;11: (7). |
[19] | Selvaraj S , Kyba M , Perlingeiro RCR . Pluripotent Stem Cell-Based Therapeutics for Muscular Dystrophies. Trends Mol Med. (2019) ;25: (9):803–16. |
[20] | Goudenege S , Lebel C , Huot NB , Dufour C , Fujii I , Gekas J , et al. Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol Ther. (2012) ;20: (11):2153–67. |
[21] | Tedesco FS , Gerli MF , Perani L , Benedetti S , Ungaro F , Cassano M , et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med. (2012) ;4: (140):140ra89. |
[22] | Maffioletti SM , Gerli MF , Ragazzi M , Dastidar S , Benedetti S , Loperfido M , et al. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat Protoc. (2015) ;10: (7):941–58. |
[23] | Tanaka A , Woltjen K , Miyake K , Hotta A , Ikeya M , Yamamoto T , et al. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi Myopathy in vitro . PLoS One. (2013) ;8: (4):e61540. |
[24] | Abujarour R , Bennett M , Valamehr B , Lee TT , Robinson M , Robbins D , et al. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl Med. (2014) ;3: (2):149–60. |
[25] | Yoshida T , Awaya T , Jonouchi T , Kimura R , Kimura S , Era T , et al. A Skeletal Muscle Model of Infantile-onset Pompe Disease with Patient-specific iPS Cells. Sci Rep. (2017) ;7: (1):13473. |
[26] | Shoji E , Sakurai H , Nishino T , Nakahata T , Heike T , Awaya T , et al. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci Rep. (2015) ;5: :12831. |
[27] | Dastidar S , Ardui S , Singh K , Majumdar D , Nair N , Fu Y , et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. (2018) ;46: (16):8275–98. |
[28] | Uchimura T , Otomo J , Sato M , Sakurai H . A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. (2017) ;25: :98–106. |
[29] | Warren L , Manos PD , Ahfeldt T , Loh YH , Li H , Lau F , et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. (2010) ;7: (5):618–30. |
[30] | Albini S , Coutinho P , Malecova B , Giordani L , Savchenko A , Forcales SV , et al. Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep. (2013) ;3: (3):661–70. |
[31] | Akiyama T , Wakabayashi S , Soma A , Sato S , Nakatake Y , Oda M , et al. Transient ectopic expression of the histone demethylase JMJD3 accelerates the differentiation of human pluripotent stem cells. Development. (2016) ;143: (20):3674–85. |
[32] | Rios AC , Serralbo O , Salgado D , Marcelle C . Neural crest regulates myogenesis through the transient activation of NOTCH. Nature. (2011) ;473: (7348):532–5. |
[33] | Dale JK , Malapert P , Chal J , Vilhais-Neto G , Maroto M , Johnson T , et al. Oscillations of the snail genes in the presomitic mesoderm coordinate segmental patterning and morphogenesis in vertebrate somitogenesis. Dev Cell. (2006) ;10: (3):355–66. |
[34] | Conboy IM , Rando TA . The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. (2002) ;3: (3):397–409. |
[35] | Pourquie O , Coltey M , Breant C , Le Douarin NM . Control of somite patterning by signals from the lateral plate. Proc Natl Acad Sci U S A. (1995) ;92: (8):3219–23. |
[36] | Tajbakhsh S , Borello U , Vivarelli E , Kelly R , Papkoff J , Duprez D , et al. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development. (1998) ;125: (21):4155–62. |
[37] | Kuroda K , Kuang S , Taketo MM , Rudnicki MA . Canonical Wnt signaling induces BMP-4 to specify slow myofibrogenesis of fetal myoblasts. Skelet Muscle. (2013) ;3: (1):5. |
[38] | Shelton M , Metz J , Liu J , Carpenedo RL , Demers SP , Stanford WL , et al. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Reports. (2014) ;3: (3):516–29. |
[39] | Borchin B , Chen J , Barberi T . Derivation and FACS-mediated purification of PAX3+/PAX7+skeletal muscle precursors from human pluripotent stem cells. Stem Cell Reports. (2013) ;1: (6):620–31. |
[40] | Chal J , Oginuma M , Al Tanoury Z , Gobert B , Sumara O , Hick A , et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol. (2015) ;33: (9):962–9. |
[41] | Choi IY , Lim H , Estrellas K , Mula J , Cohen TV , Zhang Y , et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. (2016) ;15: (10):2301–12. |
[42] | Xu C , Tabebordbar M , Iovino S , Ciarlo C , Liu J , Castiglioni A , et al. A Zebrafish Embryo Culture System Defines Factors that Promote Vertebrate Myogenesis across Species. Cell. (2013) ;155: (4):909–21. |
[43] | Hosoyama T , McGivern JV , Van Dyke JM , Ebert AD , Suzuki M . Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl Med. (2014) ;3: (5):564–74. |
[44] | Swartz EW , Baek J , Pribadi M , Wojta KJ , Almeida S , Karydas A , et al. A Novel Protocol for Directed Differentiation of C9orf72-Associated Human Induced Pluripotent Stem Cells Into Contractile Skeletal Myotubes. Stem Cells Transl Med. (2016) ;5: (11):1461–72. |
[45] | Hicks MR , Hiserodt J , Paras K , Fujiwara W , Eskin A , Jan M , et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat Cell Biol. (2018) ;20: (1):46–57. |
[46] | Caron L , Kher D , Lee KL , McKernan R , Dumevska B , Hidalgo A , et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl Med. (2016) ;5: (9):1145–61. |
[47] | Ruan T , Harney D , Koay YC , Loo L , Larance M , Caron L . Anabolic Factors and Myokines Improve Differentiation of Human Embryonic Stem Cell Derived Skeletal Muscle Cells. Cells. (2022) ;11: (6) |
[48] | Mazaleyrat K , Badja C , Broucqsault N , Chevalier R , Laberthonniere C , Dion C , et al. Multilineage Differentiation for Formation of Innervated Skeletal Muscle Fibers from Healthy and Diseased Human Pluripotent Stem Cells. Cells. (2020) ;9: (6) |
[49] | Delourme M , Broucqsault N , Mazaleyrat K , Magdinier F . Production of Innervated Skeletal Muscle Fibers Using Human Induced Pluripotent Stem Cells. Methods Mol 82 Biol. 2020. |
[50] | Shahriyari M , Islam MR , Sakib SM , Rinn M , Rika A , Kruger D , et al. Engineered skeletal muscle recapitulates human muscle development, regeneration and dystrophy. J Cachexia Sarcopenia Muscle. (2022) ;13: (6):3106–21. |
[51] | Laberthonniere C , Novoa-Del-Toro EM , Delourme M , Chevalier R , Broucqsault N , Mazaleyrat K , et al. Facioscapulohumeral dystrophy weakened sarcomeric contractility is mimicked in induced pluripotent stem cells-derived innervated muscle fibres. J Cachexia Sarcopenia Muscle. (2022) ;13: (1):621–35. |
[52] | Choi S , Ferrari G , Moyle LA , Mackinlay K , Naouar N , Jalal S , et al. Assessing and enhancing migration of human myogenic progenitors using directed iPS cell differentiation and advanced tissue modelling. EMBO Mol Med. (2022) ;14: (10):e14526. |
[53] | Andersen J , Revah O , Miura Y , Thom N , Amin ND , Kelley KW , et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell. (1913) ;183: (7):26–29 e. |
[54] | Faustino Martins JM , Fischer C , Urzi A , Vidal R , Kunz S , Ruffault PL , et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell. 2020. |
[55] | Cooper ST , Maxwell AL , Kizana E , Ghoddusi M , Hardeman EC , Alexander IE , et al. C2C12 co-culture on a fibroblast substratum enables sustained survival of contractile, highly differentiated myotubes with peripheral nuclei and adult fast myosin expression. Cell Motil Cytoskeleton. (2004) ;58: (3):200–11. |
[56] | Hicks MR , Cao TV , Standley PR . Biomechanical strain vehicles for fibroblast-directed skeletal myoblast differentiation and myotube functionality in a novel coculture. Am J Physiol Cell Physiol. (2014) ;307: (8):C671–83. |
[57] | Rao N , Evans S , Stewart D , Spencer KH , Sheikh F , Hui EE , et al. Fibroblasts influence muscle progenitor differentiation and alignment in contact independent and dependent manners in organized co-culture devices. Biomed Microdevices. (2013) ;15: (1):161–9. |
[58] | Venter C , Niesler CU . Cellular alignment and fusion: Quantifying the effect of macrophages and fibroblasts on myoblast terminal differentiation. Exp Cell Res. (2018) ;370: (2):542–50. |
[59] | Venter C , Niesler C . A triple co-culture method to investigate the effect of macrophages and fibroblasts on myoblast proliferation and migration. Biotechniques. (2018) ;64: (2):52–8. |
[60] | Juhas M , Abutaleb N , Wang JT , Ye J , Shaikh Z , Sriworarat C , et al. Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat Biomed Eng. (2018) ;2: (12):942–54. |
[61] | Robbins N , Yonezawa T . Developing neuromuscular junctions: first signs of chemical transmission during formation in tissue culture. Science. (1971) ;172: (3981):395–8. |
[62] | Kimura M , Shikada K , Nojima H , Kimura I . Acetylcholine sensitivity in myotubes of nerve-muscle co-culture cultured with anti-muscle antibodies, alpha-bungarotoxin and D-tubocurarine. Int J Dev Neurosci. (1986) ;4: (1):61–7. |
[63] | Carter JC , Sheehan DW , Prochoroff A , Birnkrant DJ . Muscular Dystrophies. Clin Chest Med. (2018) ;39: (2):377–89. |
[64] | Mary P , Servais L , Vialle R . Neuromuscular diseases: Diagnosis and management. Orthop Traumatol Surg Res. (2018) ;104: (1S):S89–S95. |
[65] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. |
[66] | Jinek M , Chylinski K , Fonfara I , Hauer M , Doudna JA , Charpentier E . A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. (2012) ;337: (6096):816–21. |
[67] | Charpentier E , Doudna JA . Biotechnology: Rewriting a genome. Nature. (2013) ;495: (7439):50–1. |
[68] | Garg P , Oikonomopoulos A , Chen H , Li Y , Lam CK , Sallam K , et al. Genome Editing of Induced Pluripotent Stem Cells to Decipher Cardiac Channelopathy Variant. J Am Coll Cardiol. (2018) ;72: (1):62–75. |
[69] | Yang YM , Gupta SK , Kim KJ , Powers BE , Cerqueira A , Wainger BJ , et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. (2013) ;12: (6):713–26. |
[70] | Lee G , Ramirez CN , Kim H , Zeltner N , Liu B , Radu C , et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. (2012) ;30: (12):1244–8. |
[71] | Young J , Margaron Y , Fernandes M , Duchemin-Pelletier E , Michaud J , Flaender M , et al. MyoScreen, a High-Throughput Phenotypic Screening Platform Enabling Muscle Drug Discovery. SLAS Discov. (2018) ;23: (8):790–806. |
[72] | Al Tanoury Z , Zimmerman JF , Rao J , Sieiro D , McNamara HM , Cherrier T , et al. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell-derived skeletal muscle in vitro . Proc Natl Acad Sci U S A. (2021) ;118: (28) |
[73] | Lin B , Li Y , Han L , Kaplan AD , Ao Y , Kalra S , et al. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis Model Mech. (2015) ;8: (5):457–66. |
[74] | Afzal MZ , Reiter M , Gastonguay C , McGivern JV , Guan X , Ge ZD , et al. Nicorandil, a Nitric Oxide Donor and ATP-Sensitive Potassium Channel Opener, Protects Against Dystrophin-Deficient Cardiomyopathy. J Cardiovasc Pharmacol Ther. (2016) ;21: (6):549–62. |
[75] | Sun C , Choi IY , Rovira Gonzalez YI , Andersen P , Talbot CC Jr, Iyer SR et al. Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight. (2020) ;5: (11). |
[76] | Kokubu Y , Nagino T , Sasa K , Oikawa T , Miyake K , Kume A , et al. Phenotypic Drug Screening for Dysferlinopathy Using Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl Med. (2019) ;8: (10):1017–29. |
[77] | Dick E , Kalra S , Anderson D , George V , Ritso M , Laval SH , et al. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. (2013) ;22: (20):2714–24. |
[78] | Chemello F , Bassel-Duby R , Olson EN . Correction of muscular dystrophies by CRISPR gene editing. J Clin Invest. (2020) ;130: (6):2766–76. |
[79] | Olson EN . Toward the correction of muscular dystrophy by gene editing. Proc Natl Acad Sci U S A. (2021) ;118: (22). |
[80] | Happi Mbakam C , Lamothe G , Tremblay G , Tremblay JP . CRISPR-Cas9 Gene Therapy for Duchenne Muscular Dystrophy. Neurotherapeutics. (2022) ;19: (3):931–41. |
[81] | Aslesh T , Erkut E , Yokota T . Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing. Expert Opin Biol Ther. (2021) ;21: (8):1049–61. |
[82] | Kupatt C , Windisch A , Moretti A , Wolf E , Wurst W , Walter MC . Genome editing for Duchenne muscular dystrophy: a glimpse of the future? Gene Ther, (2021) ;28: (9):542–8. |
[83] | Min YL , Li H , Rodriguez-Caycedo C , Mireault AA , Huang J , Shelton JM , et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv.eaav. (2019) ;5: (3):4324. |
[84] | Moretti A , Fonteyne L , Giesert F , Hoppmann P , Meier AB , Bozoglu T , et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. (2020) ;26: (2):207–14. |
[85] | Iyer S , Suresh S , Guo D , Daman K , Chen JCJ , Liu P , et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded. Nature. (2019) ;568: (7753):561–5. |
[86] | Dhoke NR , Kim H , Selvaraj S , Azzag K , Zhou H , Oliveira NAJ , et al. A universal gene correction approach for FKRP-associated dystroglycanopathies to enable autologous cell therapy. Cell Rep. (2021) ;36: (2):109360. |
[87] | Guo D , Daman K , Chen JJ , Shi MJ , Yan J , Matijasevic Z , et al. iMyoblasts for ex vivo and in vivo investigations of human myogenesis and disease modeling. Elife. (2022) ;11: . |
[88] | Tasca F , Brescia M , Wang Q , Liu J , Janssen JM , Szuhai K , et al. Large-scale genome editing based on high-capacity adenovectors and CRISPR-Cas9 nucleases rescues full-length dystrophin synthesis in DMD muscle cells. Nucleic Acids Res. (2022) ;50: (13):7761–82. |
[89] | Chemello F , Chai AC , Li H , Rodriguez-Caycedo C , Sanchez-Ortiz E , Atmanli A , et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci Adv. (2021) ;7: (18). |
[90] | Ueki J , Nakamori M , Nakamura M , Nishikawa M , Yoshida Y , Tanaka A , et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci Rep. (2017) ;7: :42522. |
[91] | Cao L , McDonnell A , Nitzsche A , Alexandrou A , Saintot PP , Loucif AJ , et al. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. (2016) ;8: (335):.335ra56. |
[92] | Namer B , Schmidt D , Eberhardt E , Maroni M , Dorfmeister E , Kleggetveit IP , et al. Pain relief in a neuropathy patient by lacosamide: Proof of principle of clinical translation from patient-specific iPS cell-derived nociceptors. EBioMedicine. (2019) ;39: :401–8. |
[93] | Del Carmen Ortuno-Costela M , Garcia-Lopez M , Cerrada V , Gallardo ME . iPSCs: A powerful tool for skeletal muscle tissue engineering. J Cell Mol Med. (2019) ;23: (6):3784–94. |
[94] | Jalal S , Dastidar S , Tedesco FS . Advanced models of human skeletal muscle differentiation, development and disease: Three-dimensional cultures, organoids and beyond. Curr Opin Cell Biol. (2021) ;73: :92–104. |
[95] | Tchao J , Kim JJ , Lin B , Salama G , Lo CW , Yang L , et al. Engineered Human Muscle Tissue from Skeletal Muscle Derived Stem Cells and Induced Pluripotent Stem Cell Derived Cardiac Cells. Int J Tissue Eng. (2013) ;2013: :198762. |
[96] | Rao L , Qian Y , Khodabukus A , Ribar T , Bursac N . Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat Commun. (2018) ;9: (1):126. |
[97] | Steele-Stallard HB , Pinton L , Sarcar S , Ozdemir T , Maffioletti SM , Zammit PS , et al. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front Physiol. (2018) ;9: :1332. |
[98] | Maffioletti SM , Sarcar S , Henderson ABH , Mannhardt I , Pinton L , Moyle LA , et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. (2018) ;23: (3):899–908. |
[99] | Pinton L , Khedr M , Lionello VM , Sarcar S , Maffioletti SM , Dastidar S , et al. 3D human induced pluripotent stem cell-derived bioengineered skeletal muscles for tissue, disease and therapy modeling. Nat Protoc 2023. |
[100] | Vila OF , Chavez M , Ma SP , Yeager K , Zholudeva LV , Colon-Mercado JM , et al. Bioengineered optogenetic model of human neuromuscular junction. Biomaterials. (2021) ;276: :121033. |
[101] | Osaki T , Uzel SGM , Kamm RD . Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv. (2018) ;4: (10):eaat5847. |
[102] | Afshar ME , Abraha HY , Bakooshli MA , Davoudi S , Thavandiran N , Tung K , et al. A 96-well culture platform enables longitudinal analyses of engineered human skeletal muscle microtissue strength. Sci Rep. (2020) ;10: (1):6918. |
[103] | Yoshioka K , Ito A , Arifuzzaman M , Yoshigai T , Fan F , Sato KI , et al. Miniaturized skeletal muscle tissue fabrication for measuring contractile activity. J Biosci Bioeng. (2021) ;131: (4):434–41. |
[104] | Ostrovidov S , Salehi S , Costantini M , Suthiwanich K , Ebrahimi M , Sadeghian RB , et al. 3D Bioprinting in Skeletal Muscle Tissue Engineering. Small. (2019) ;15: (24):e1805530. |
[105] | Ebrahimi M , Ostrovidov S , Salehi S , Kim SB , Bae H , Khademhosseini A . Enhanced skeletal muscle formation on microfluidic spun gelatin methacryloyl (GelMA) fibres using surface patterning and agrin treatment. J Tissue Eng Regen Med. (2018) ;12: (11):2151–63. |
[106] | Potyondy T , Uquillas JA , Tebon PJ , Byambaa B , Hasan A , Tavafoghi M , et al. Recent advances in 3D bioprinting of musculoskeletal tissues. Biofabrication. (2021) ;13: (2). |
[107] | Cui X , Li J , Hartanto Y , Durham M , Tang J , Zhang H , et al. Advances in Extrusion 3D Bioprinting: A Focus on Multicomponent Hydrogel-Based Bioinks. Adv Healthc Mater. (2020) ;9: (15):e1901648. |
[108] | Costantini M , Testa S , Fornetti E , Fuoco C , Sanchez Riera C , Nie M , et al. Biofabricating murine and human myo-substitutes for rapid volumetric muscle loss restoration. EMBO Mol Med. (2021) ;13: (3):e12778. |
[109] | Fornetti E , De Paolis F , Fuoco C , Bernardini S , Giannitelli SM , Rainer A , et al. A novel extrusion-based 3D bioprinting system for skeletal muscle tissue engineering. Biofabrication. (2023) );15: (2). |
[110] | Kong JS , Huang X , Choi YJ , Yi HG , Kang J , Kim S , et al. Promoting Long-Term Cultivation of Motor Neurons for 3D Neuromuscular Junction Formation of 3D In Vitro Using Central-Nervous-Tissue-Derived Bioink. Adv Healthc Mater. (2021) ;10: (18):e2100581. |
[111] | Xi H , Langerman J , Sabri S , Chien P , Young CS , Younesi S , et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell. (2020) ;27: (1):158–76 e10. |
[112] | Miao Y , Djeffal Y , De Simone A , Zhu K , Lee JG , Lu Z , et al. Reconstruction and deconstruction of human somitogenesis in vitro . Nature. (2023) ;27: (1):500–614–8. |
[113] | Chiche A , Le Roux I , von Joest M , Sakai H , Aguin SB , Cazin C , et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell. (2017) ;20: (3):407–14 e4. |
[114] | Wang C , Rabadan Ros R , Martinez-Redondo P , Ma Z , Shi L , Xue Y , et al. In vivo partial reprogramming of myofibers promotes muscle regeneration by remodeling the stem cell niche. Nat Commun. (2021) ;12: (1):3094. |
[115] | Li HL , Fujimoto N , Sasakawa N , Shirai S , Ohkame T , Sakuma T , et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. (2015) ;4: (1):143–54. |
[116] | Caputo L , Granados A , Lenzi J , Rosa A , Ait-Si-Ali S , Puri PL , et al. Acute conversion of patient-derived Duchenne muscular dystrophy iPSC into myotubes reveals constitutive and inducible over-activation of TGFbeta-dependent pro-fibrotic signaling. Skelet Muscle. (2020) ;10: (1):13. |
[117] | Uchimura T , Asano T , Nakata T , Hotta A , Sakurai H . A muscle fatigue-like contractile decline was recapitulated using skeletal myotubes from Duchenne muscular dystrophy patient-derived iPSCs. Cell Rep Med. (2021) ;2: (6):100298. |
[118] | Mournetas V , Massourides E , Dupont JB , Kornobis E , Polveche H , Jarrige M , et al. Myogenesis modelled by human pluripotent stem cells: a multi-omic study of Duchenne myopathy early onset. J Cachexia Sarcopenia Muscle. (2021) ;12: (1):209–32. |
[119] | Ifuku M , Iwabuchi KA , Tanaka M , Lung MSY , Hotta A . Restoration of Dystrophin Protein Expression by Exon Skipping Utilizing CRISPR-Cas9 in Myoblasts Derived from DMD Patient iPS Cells. Methods Mol Biol. (1828) ;1828: :191–217. |
[120] | Young CS , Hicks MR , Ermolova NV , Nakano H , Jan M , Younesi S , et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell. (2016) ;18: (4):533–40. |
[121] | Sengupta K , Mishra MK , Loro E , Spencer MJ , Pyle AD , Khurana TS . Genome Editing-Mediated Utrophin Upregulation in Duchenne Muscular Dystrophy Stem Cells. Mol Ther Nucleic Acids. (2020) ;22: :500–9. |
[122] | Kawada R , Jonouchi T , Kagita A , Sato M , Hotta A , Sakurai H . Establishment of quantitative and consistent in vitro skeletal muscle pathological models of myotonic dystrophy type 1 using patient-derived iPSCs. Sci Rep. (2023) ;13: (1):94. |
[123] | Yasuno T , Osafune K , Sakurai H , Asaka I , Tanaka A , Yamaguchi S , et al. Functional analysis of iPSC-derived myocytes from a patient with carnitine palmitoyltransferase II deficiency. Biochem Biophys Res Commun. (2014) ;448: (2):175–81. |
[124] | Sato Y , Kobayashi H , Higuchi T , Shimada Y , Ida H , Ohashi T . TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle. Mol Ther Methods Clin Dev. (2016) ;3: :16054. |
[125] | Gartz M , Haberman M , Sutton J , Slick RA , Luttrell SM , Mack DL , et al. ACTA1 H40Y mutant iPSC-derived skeletal myocytes display mitochondrial defects in an in vitro model of nemaline myopathy. Exp Cell Res. (2023) ;424: (2):113507. |
[126] | Sasaki-Honda M , Jonouchi T , Arai M , Hotta A , Mitsuhashi S , Nishino I , et al. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum Mol Genet. (2018) ;27: (23):4024–35. |
[127] | Ortuno-Costela MDC , Cerrada V , Moreno-Izquierdo A , Garcia-Consuegra I , Laberthonniere C , Delourme M , et al. Generation of the First Human In Vitro Model for McArdle Disease Based on iPSC Technology. Int J Mol Sci. (2022) ;23: (22). |
[128] | Mateos-Aierdi AJ , Dehesa-Etxebeste M , Goicoechea M , Aiastui A , Richaud-Patin Y , Jimenez-Delgado S , et al. Patient-specific iPSC-derived cellular models of LGMDR1. Stem Cell Res. (2021) ;53: :102333. |
[129] | Bruge C , Geoffroy M , Benabides M , Pellier E , Gicquel E , Dhiab J , et al. Skeletal Muscle Cells Derived from Induced Pluripotent Stem Cells: A Platform for Limb Girdle Muscular Dystrophies. Biomedicines. (2022) ;10: (6). |
[130] | Ikenaka A , Kitagawa Y , Yoshida M , Lin CY , Niwa A , Nakahata T , et al. SMN promotes mitochondrial metabolic maturation during myogenesis by regulating the MYOD-miRNA axis. Life Sci Alliance. (2023) ;6: (3). |

