
Self-Reported Health-Related Quality of Life of Children with Spinal Muscular Atrophy: Preliminary Insights from a Nationwide Patient Registry in Germany
Abstract
Background:
Spinal muscular atrophy (SMA) is a rare, severely debilitating neuromuscular disease characterized by a wide spectrum of progressive muscular atrophy and weakness.
Objectives:
The objective of this pilot study was to estimate self-assessed health-related quality of life (HRQoL) of children with SMA.
Methods:
Children with SMA were recruited via the German national TREAT-NMD SMA patient registry and asked to self-complete the following rating-scales: KIDSCREEN-27, KINDL, the PedsQL 3.0 Neuromuscular Module (PedsQL 3.0 NMM), EQ-5D-5L, and the Health Utilities Index (HUI). Estimates were stratified by current best motor function of the lower limb and trunk (i.e., non-sitter, sitter, and walker) and SMA type (i.e., type I, II, and III).
Results:
In total, 17 children with SMA (mean age: 9.88 years, SD: 4.33 years, range: 5–16 years; 59% female) participated in the study. Across examined strata, the mean KIDSCREEN-27 total score was estimated at between 48.24 and 83.81; the mean KINDL total score at between 60.42 and 76.73; the mean PedsQL 3.0 NMM total score at between 58.00 and 83.83; the mean EQ-5D-5L utility at between 0.31 and 0.99; and the mean HUI-derived utility at between –0.02 and 0.96.
Conclusions:
The results from this pilot study show that German children with SMA, despite significant physical disability, have surprisingly good HRQoL as assessed using KIDSCREEN-27. Yet, many reside in health states associated with low utility. The disease burden was generally higher among non-sitters compared with walkers, and SMA type I compared with type III, but more research is needed to further delineate this variability. Our preliminary findings contribute to the understanding of HRQoL in pediatric patients with SMA and should be helpful to inform the design of future studies of this patient population.
INTRODUCTION
Spinal muscular atrophy (SMA) is a rare, autosomal-recessive neuromuscular disease characterized by progressive muscular atrophy and weakness. Traditionally, SMA has been classified into subtypes based on age at onset of symptoms and motor milestones achieved [1]. Specifically, children with SMA type I, the most severe form of the disease, experience onset of symptoms during their first six months of life, never learn to sit independently, and are subject to progressive impairment of bulbar function resulting in difficulties with breathing, coughing, and swallowing [2]. Without treatment, the mean life-expectancy at birth for SMA type I is less than 12 months [3]. Children with SMA type II experience disease onset between 6 and 18 months after birth, learn to sit but never walk independently, and over time suffer from progressive functional impairment. However, depending on the disease trajectory, some patients with SMA type II lose the ability to sit independently and may have difficulties coughing, breathing, and swallowing. In the absence of clinical intervention, life-expectancy in SMA II has been estimated at about 60 years [2, 3]. Finally, in SMA type III, disease onset typically occurs after 18 months of age and patients usually learn to walk independently; however, without specific therapy, some patients may lose this ability later in life [4]. Life expectancy of patients with SMA type III is assumed to be normal [2].
Recently, three highly efficacious and potentially curative therapies have been approved for SMA in the European Union, namely nusinersen, onasemnogene abeparvovec, and risdiplam. These drugs have been shown to have the potential to transform the natural disease evolution, thereby challenging the traditional classification of patients into static subtypes [5–7]. Indeed, we have shown that improvements to the medical management, including the introduction of novel therapies, appears to have altered the disease trajectory among children and adults with SMA in Germany [8]. As a result, in line with current standards of care, a complementary disease classification, made in terms of current best motor function of the lower limb and trunk, has been proposed for SMA in response to the observed changes in patterns of morbidity and disability [9, 10]. The system contains three major functional categories: non-sitters, sitters, and walkers. Non-sitters mainly have SMA type I or II, are not able to breathe or eat on their own (thus requiring ventilatory and feeding support), commonly experience respiratory infections due to saliva aspirations and gastroesophageal reflux, have impaired verbal communication skills, and can develop painful contractures. In contrast, sitters mainly have SMA type II or III (but recently, with specific therapy, also type I), they may develop respiratory insufficiency due to muscle impairment, scoliosis (requiring surgery), and overweight, can experience painful contractures, and have a high risk of developing swallowing and chewing problems, as well as bowel dysmotility that may lead to painful gastroesophageal reflux or obstipation. The last category, walkers, mainly have SMA type III (but recently, with specific therapy, also type I and II), experience reduced endurance due to atrophy, scoliosis, weight gain, and mild respiratory insufficiency, may require the use of aids and devices to maintain and promote functional independence, and some experience varying degree of problems with swallowing and chewing [9, 10].
Across the past decade, a substantial body of literature has accumulated with respect to health-related quality of life (HRQoL) in SMA [11–13]. However, few studies have investigated self-assessed HRQoL in pediatric cohorts. Moreover, little is known of the HRQoL in SMA in the era of disease-modifying therapies. To help bridge this evidence gap, the objective of this pilot study was to estimate and describe self-reported HRQoL in a contemporary cohort of German children with SMA exposed to nusinersen and risdiplam. Our aim was to conduct an in-depth analysis of HRQoL, encompassing five commonly utilized rating-scales, to help improve our understanding of this latent trait in SMA in general and inform the selection of measures for future research.
MATERIALS AND METHODS
The data reported as part of this work was collected in a cross-sectional, observational cohort study comprising of children with SMA and one of their caregivers in Germany.
Measuring health-related quality of life
HRQoL is a multidimensional construct referring specifically to the individual’s perception of the impact of health and illness on three aspects (also known as “domains”) of life [14]: (1) physical, (2) mental; and (3) social. As a latent trait, HRQoL must be elicited indirectly through its observable manifestations using rating-scales [15]. Rating-scales consists of one or several questions (also known as “items”), each described in two or more response categories (or “levels”). Two main categories of rating-scales (also referred to as “measures”, “instruments”, or “tools”) of HRQoL exists: (1) generic, and (2) disease-specific. The former is designed to be applicable regardless of the distribution of disease and treatment, whereas disease-specific rating-scales seek to measure HRQoL in patients with specific diseases. Scores of most measures of HRQoL are constructed by counting responses to items using Likert’s method of summated ratings, by which levels are ordered and allocated sequential integers and item scores are subsequently summed to produce total scores [15, 16]. However, an increasing number of tools has been subject to modern psychometric analysis (e.g., Rasch analysis) in which ordinal scales are re-calibrated (upon meeting a set of criteria) into interval continuums [17, 18]. Additionally, some instruments are linked to utilities. Utilities have different meanings in different contexts, but usually reflect preferences for specific health states (where a higher utility is associated with a more preferred health state) [19]. A health state may refer to, for example, the health status associated with a specific disease, one or several disease complications, or general aspects of life (e.g., physical, mental, and social). Moreover, health states can also be classified by HRQoL instruments via their questions and response categories. Utilities are in most cases elicited from members of the general population (i.e., healthy individuals ex ante experiencing the health state) and less frequently from individuals who currently reside in the health state (so called “experience-based utilities”). Irrespective of their source population, utilities typically range between 0 (interpreted as being in a health state that is equal to being dead) and 1 (interpreted as being in a health state of perfect health) [20]. However, health states for some instruments are associated with negative utilities, which are interpreted as being in a health state rated worse than being dead.
Study population
Children with SMA were identified via the German national TREAT-NMD SMA patient registry (www.sma-register.de). The following criteria were imposed for study eligibility: (1) Genetically confirmed diagnosis of SMA, (2) ≥5 years of age, and (3) Currently residing in Germany.
Study procedures and collected data
The SMA patient registry invited patients together with one of their caregivers (e.g., a parent) to complete a questionnaire administered via a dedicated study website online. The questionnaire contained information about the study and age-specific informed consent forms, as well as questions capturing patients’ demographic and clinical characteristics (e.g., age, sex, age at onset of symptoms, age at diagnosis, SMA type, best motor skills of lower and upper extremities, and SMA specific medication). In addition, the children with SMA were asked to self-complete (i.e., without help from their caregivers) a set of age-specific measures of HRQoL, namely KIDSCREEN-27 [21], KINDL [22], the Pediatric Quality of Life Inventory (PedsQL) 3.0 Neuromuscular Module (PedsQL 3.0 NMM) [23], EQ-5D-5L [24], and the Health Utilities Index (HUI) [25] (Table 1). The study was approved by the regional Ethics Committee from the Saarland Medical Association (protocol number 09/20), registered in the German clinical trial registry (DRKS00022876), and conducted in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Table 1
Employed measures of HRQoL
| KIDSCREEN-27 [21] | KINDL (KIDDY and KID) [22] | PedsQL 3.0 NMM [23] | EQ-5D-5L [24] | HUI (Mark III) [25] | |
| Type | Generic | Generic | Disease-specifica | Generic | Generic |
| Age group | 8–17 years | 5–7 years | 5–17 years | 12–17 years | 8–17 years |
| Domains | •Physical well-being•Psychological well-being•Autonomy &parents•Peers &social support•School environment | •Physical well-being•Emotional well-being•Self-esteem•Family•Friends•School | •About my neuromuscular disorder•Communication•About my family resources | •Mobility•Self-care•Usual activities•Pain/discomfort•Anxiety/ depression | •Vision•Hearing•Speech•Ambulation•Dexterity•Emotion•Cognition•Pain |
| No. of items | 27 | 12 or 24b | 25 | 5 | 8 |
| No. of levels | 5 | 3 or 5b | 5 | 3 | 5–6 |
| Recall period | 4 weeks | 4 weeks | 4 weeks | 4 weeks | 4 weeks |
| Scale type | Rasch scale (interval) | Likert scale (ordinal) | Likert scale (ordinal) | Utility scale (interval) | Utility scale (interval) |
| Scale range | 0 to 100c | 0 to 100c | 0 to 100c | –0.661 to 1d | –0.36 to 1d |
Note: EuroQol EQ-5D-5L (EQ-5D-5L). Health Utilities Index Questionnaire (HUI). Pediatric Quality of Life Inventory (PedsQL) 3.0 Neuromuscular Module (PedsQL 3.0 NMM). aSpecific to neuromuscular diseases. bThe KIDDY version contains 12 questions described in three levels, and the KID version contains 24 questions described in five levels. Additionally, a module comprising of six questions pertaining to the chronic illness of participants were included in the KINDL scales. cHigher score = better health status/higher HRQoL. d0 = health state equal to being dead; 1 = health state of perfect health; and < 0 = health state worse than being dead.
Statistical analysis
Continuous demographic and clinical characteristics were summarized using means, standard deviations (SDs), and ranges (i.e., minimum value–maximum value) and categorial characteristics using frequencies and percentages. All measures of HRQoL (described in Table 1) were scored according to their respective manual. For KIDSCREEN-27, we estimated domain and total scores (higher score = better health status/higher HRQoL; scores <40 or >60 are considered significantly different from general population reference data [21]), and also reported results from the patients’ subjective rating of their current health (single-item, described in five levels [“Poor” to “Excellent”]). For KINDL (KID and KIDDY versions), we estimated domain and total scores (higher score = better health status/higher HRQoL). For the PedsQL 3.0 NMM, we estimated domain and total scores (higher score = better health status/higher HRQoL), and also examined the distribution of replies across items and levels. For the EQ-5D-5L, we estimated utilities using the German value set by Ludwig et al. (ranging between –0.661 and 1) [26], and also reported visual analogue scale (VAS) scores (ranging from 0, indicating the worst imageable health status, to 100, indicating the best imageable health status). Finally, for the HUI, we estimated utilities using the HUI Mark III algorithm (ranging between –0.36 and 1) [25]. We stratified results by current best motor function of the lower limb and trunk (i.e., non-sitter, sitter, and walker) and SMA type (i.e., type I, II, and III), respectively. Data analysis was performed using Stata 15 (StataCorp, College Station, TX, USA).
Results
A total of 17 children with SMA met all study criteria and self-completed the study questionnaire between June and September 2021. Demographic and clinical characteristics of the final sample, stratified by current motor function, are summarized in Table 2. The mean age in the total cohort was 9.88 years (SD: 4.33, range 5–16) and 59% were female. All but two patients (88%) were currently on disease-modifying therapy (73% received nusinersen and 27% risdiplam).
Table 2
Demographic and clinical characteristics of the patient sample with SMA
| Non-sitter | Sitter | Walker | Total sample | |
| n (%) | 4 (24%) | 7 (41%) | 6 (35%) | 17 (100%) |
| Age, in years | 11.00 (3.46) (8–14) | 10.14 (5.24) (5–16) | 8.83 (4.17) (5–15) | 9.88 (4.33) (5–16) |
| Female sex, n (%) | 3 (75%) | 5 (71%) | 2 (33%) | 10 (59%) |
| Age at first symptoms, in years | 1.25 (0.50) (1–2) | 0.43 (0.79) (0–2) | 1.83 (0.98) (1–3) | 1.12 (0.99) (0–3) |
| Age at SMA diagnosis, in years | 1.50 (0.58) (1–2) | 0.57 (0.79) (0–2) | 3.00 (2.00) (1–6) | 1.65 (1.66) (0–6) |
| Type of SMA, n (%) | ||||
| Type I | 0 (0%) | 3 (43%) | 0 (0%) | 3 (18%) |
| Type II | 3 (75%) | 4 (57%) | 1 (17%) | 8 (47%) |
| Type III | 1 (25%) | 0 (0%) | 5 (83%) | 6 (35%) |
Note: Data reported as mean (SD) (range) unless otherwise stated. Because of rounding, percentages might not add up to exactly 100%. Spinal muscular atrophy (SMA).
KIDSCREEN-27
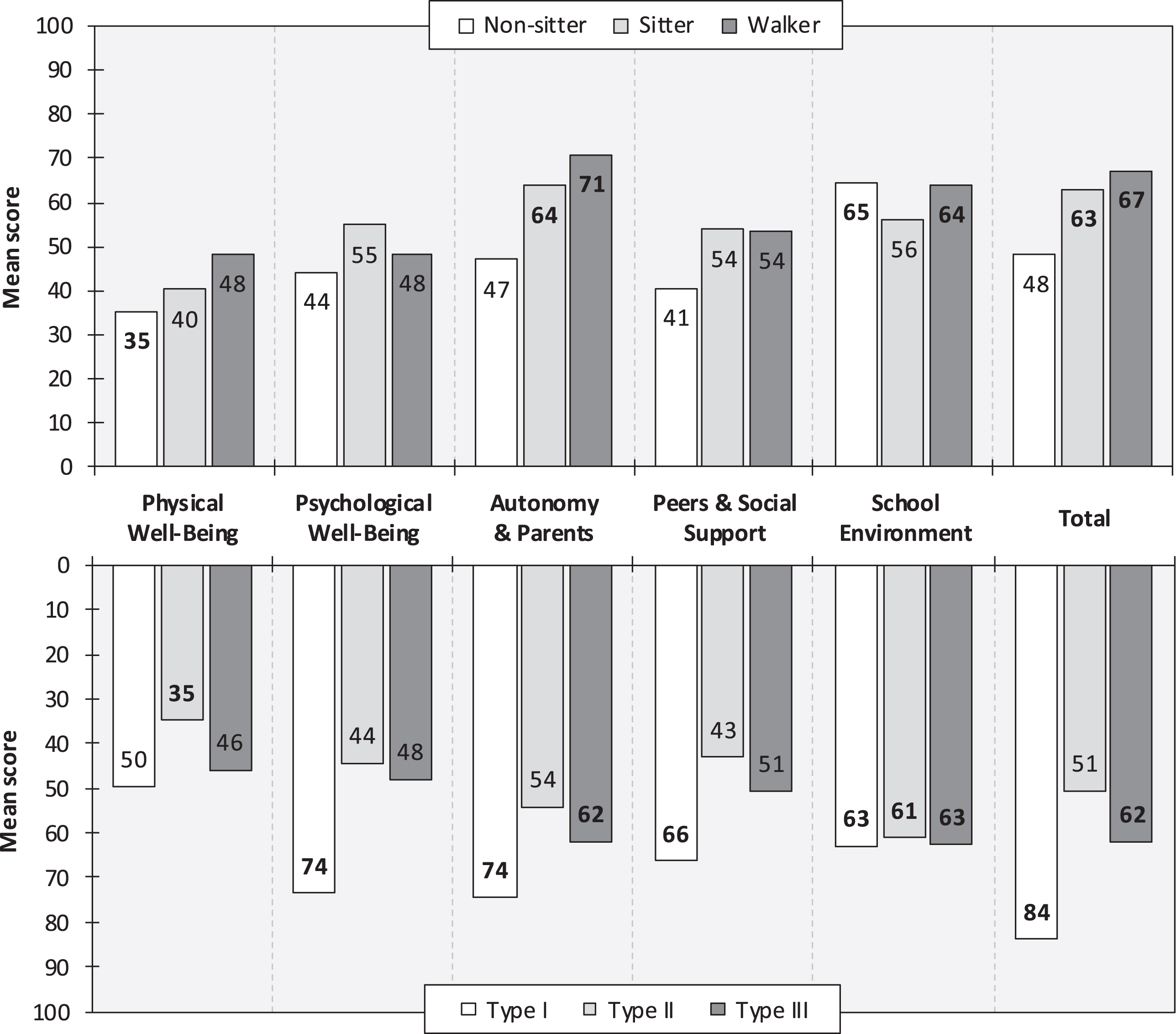
A total of 10 children (mean age: 12.80 years, SD: 3.12 years, range: 8–16 years; 60% female) self-completed the KIDSCREEN-27. In this sample subset, 40% were non-sitters, 30% sitters, and 30% were walkers; 10% had SMA type I, 50% type II, and 40% type III. All but two patients (80%) were currently on disease-modifying therapy (63% received nusinersen and 38% risdiplam). Estimated KIDSCREEN-27 scores (higher score = better health status/higher HRQoL), stratified by current motor function and SMA type, are presented in Fig. 1. The mean total score in all patients was 58.43 (SD: 15.44, range: 40.24–83.81). The proportion of patients rating their current health as “Good”, “Very good”, or “Excellent” was 75% among non-sitters, 67% among sitters, and 100% among walkers. Corresponding estimates for SMA type I, type II, and type III were 100%, 60%, and 100%, respectively. Additional KIDSCREEN-27 results are available as supplemental material (online) (eTable 1).
Fig. 1
KIDSCREEN scores, by highest function (upper panel) and SMA type (lower panel). Note: KIDSCREEN scores range from 0 to 100 (higher score = better health status/higher HRQoL). Bold numbers are significantly different from general population KIDSCREEN reference scores (mean: 50, SD: 10). Spinal muscular atrophy (SMA).
KINDL
A total of 7 children (mean age: 5.71 years, SD: 0.95 years, range: 5–7 years; 57% female) self-completed the KINDL instrument. In this sample subset, 57% were sitters and 43% were walkers; 29% had SMA type I, 43% type II, and 29% type III. All patients were currently on disease-modifying therapy (86% received nusinersen and 14% risdiplam). The mean KINDL total score (higher score = better health status/higher HRQoL) among sitters was 75.00 (SD: 3.07, range: 70.83–78.13) and among walkers 68.05 (SD: 19.69, range: 45.83–83.33). Corresponding estimates for SMA type I were 76.57 (SD: 2.21, range: 75.00–78.13), SMA type II 76.73 (SD: 6.28, range: 70.83–83.33), and SMA type III 60.42 (SD: 20.63, range: 45.83–75.00). The mean total score in all patients was 72.02 (SD: 12.16, range: 45.83–83.33). Additional KINDL results are available as supplemental material (online) (eTable 2).
3.3The pediatric quality of life inventory 3.0 neuromuscular module
All 17 children self-completed the PedsQL 3.0 NMM (Table 1). Estimated PedsQL 3.0 NMM scores (higher score = better health status/higher HRQoL), stratified by current motor function and SMA type, are presented in Fig. 2. The mean total score in all patients was 73.88 (SD: 19.52, range: 29–99). The questions with the lowest mean score (indicative of the largest impairment in health status/loss in HRQoL) for non-sitters were “It is hard to turn myself during the night”, “It takes me a long time to bathe or shower”, and “It is hard for my family to plan activities like vacations”; for sitters “It is hard to turn myself during the night”, “It is hard for my family to get enough rest”, and “It is hard for my family to plan activities like vacations”; and for walkers “I wake up tired”, “It is hard to gain or lose weight when I want to”, and “It is hard for my family to plan activities like vacations”. The questions with the lowest mean score for patients with SMA type I were “It is hard for my family to plan activities like vacations”, “It is hard for my family to get enough rest”, and “It is hard to turn myself during the night”; SMA type II “It is hard to turn myself during the night”, “I take a long time to eat”, and “It is hard for my family to get enough rest”; and SMA type III “I wake up tired”, “My back feels stiff”, and “It is hard to gain or lose weight when I want to”.
Fig. 2
PedsQL 3.0 NMM scores, by highest function (upper panel) and SMA type (lower panel). Note: PedsQL 3.0 NMM scores range from 0 to 100 (higher score = better health status/higher HRQoL). Pediatric Quality of Life Inventory 3.0 Neuromuscular Module (PedsQL 3.0 NMM). Spinal muscular atrophy (SMA).
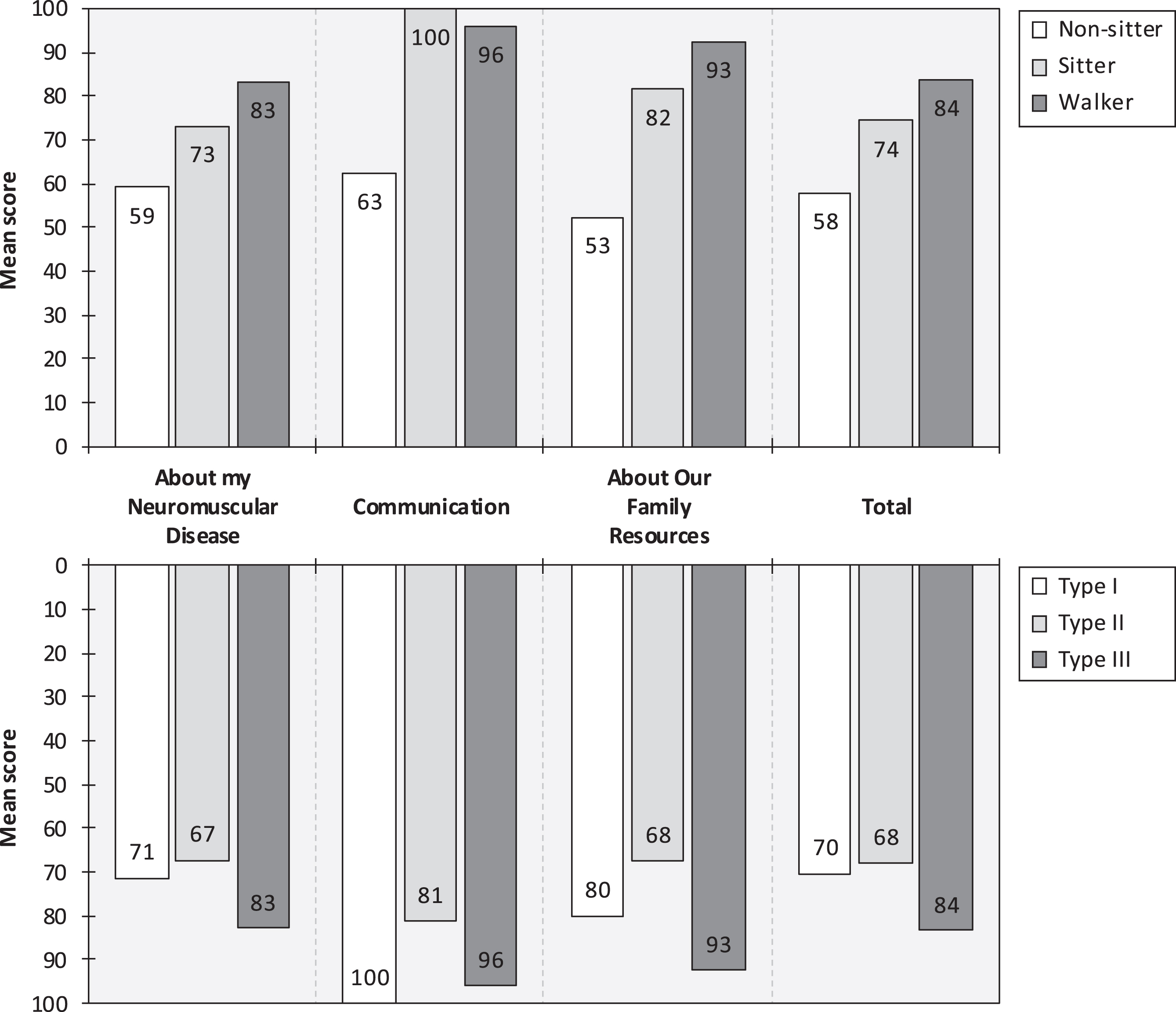
Among non-sitters, at the group level, some problems were noted across all PedsQL 3.0 NMM questions. In contrast, among sitters, all three questions relating to the “Communication” domain (i.e., “It is hard for me to tell the doctors and nurses how I feel”, “It is hard for me to ask the doctors and nurses questions”, and “It is hard for me to explain my illness to other people”), as well as “I get sick easily” and “I think money is a problem in our family”, received the highest score (indicative of no problems/impairment). The highest score among walkers was noted for a total of nine questions (i.e., “It is hard to breathe”, “I get sick easily”, “It is hard to use the bathroom”, “It is hard to swallow food”, “It is hard for me to tell the doctors and nurses how I feel”, “It is hard for me to ask the doctors and nurses questions”, “I think money is a problem in our family”, “I think my family has a lot of problems”, and “I do not have the equipment I need”). All children reported that they “Almost never” or “Never” lack they equipment that they need. Additional PedsQL 3.0 NMM results are available as supplemental material (online) (eTable 3).
EQ-5D-5L
A total of 7 children (mean age: 15 years, SD: 1.40 years, range: 12.00–16.00 years; 57% female) self-completed the EQ-5D-5L. In this sample subset, 29% were non-sitters, 43% sitters, and 29% were walkers; 14% had SMA type I, 57% type II, and 29% type III. All but one patient (86%) were currently on disease-modifying therapy (67% received nusinersen and 33% risdiplam). Estimated EQ-5D-5L utilities, stratified by SMA type and current motor function, are summarized in Fig. 3. The mean total utility in all patients was 0.54 (SD: 0.31, range: 0.30–1.00).
Fig. 3
EQ-5D-5L and HUI (Mark III) utilities, by highest function (left panel) and SMA type (right panel). Note: EQ-5D-5L utilities were derived using the value set by Ludwig et al. [26] and range from –0.661 to 1 (higher value = better health status/higher HRQoL). HUI (Mark III) utilities range from –0.36 to 1 (higher value = better health status/higher HRQoL) [25]. Health Utilities Index (HUI). Spinal muscular atrophy (SMA).
![EQ-5D-5L and HUI (Mark III) utilities, by highest function (left panel) and SMA type (right panel). Note: EQ-5D-5L utilities were derived using the value set by Ludwig et al. [26] and range from –0.661 to 1 (higher value = better health status/higher HRQoL). HUI (Mark III) utilities range from –0.36 to 1 (higher value = better health status/higher HRQoL) [25]. Health Utilities Index (HUI). Spinal muscular atrophy (SMA).](https://ip.ios.semcs.net:443/media/jnd/2024/11-1/jnd-11-1-jnd230071/jnd-11-jnd230071-g003.jpg)
The mean VAS score (ranging from 0, indicating the worst imageable health status, to 100, indicating the best imageable health status) in non-sitters was 87.50 (SD: 3.54, range: 85.00–90.00), sitters 64.67 (SD: 25.01, range: 40.00–90.00), and walkers 90.00 (SD: NA, range: 90.00–90.00). The mean VAS score in patients with SMA type I, type II, and type III were 90.00 (SD: NA, range: NA), 69.75 (SD: 22.81, range: 40.00–90.00), and 90.00 (SD: 0.00, range: 90.00–90.00) respectively. Additional EQ-5D-5L results are available as supplemental material (online) (eTable 4).
The Health Utilities Index
A total of 10 children (mean age: 12.80 years, SD: 3.12 years, range: 8–16 years; 60% female) self-completed the HUI instrument. In this sample subset, 40% were non-sitters, 30% sitters, and 30% were walkers; 10% had SMA type I, 50% type II, and 40% type III. All but two patients (80%) were currently on disease-modifying therapy (63% received nusinersen and 38% risdiplam). Estimated HUI Mark III utilities, stratified by SMA type and current motor function, are summarized in Fig. 3. The mean total utility in all patients was 0.44 (SD: 0.40, range: –0.02–1.00). Additional HUI results are available as supplemental material (online) (eTable 5).
Discussion
In this pilot study, we recorded measurements of self-reported HRQoL from 17 children with SMA in Germany using a battery of commonly employed rating-scales, some administered in this disease population for the first time (i.e., KIDSCREEN-27 and KINDL). Taken together, our preliminary findings show that self-assessed HRQoL in SMA vary substantially depending on the employed rating-scale, as well as across categories of current best motor function and SMA type.
Surprisingly, self-reported estimates from the KIDSCREEN-27 instrument show that our sample of children with SMA has good overall HRQoL (Fig. 1). Indeed, mean scores were significantly higher than general population reference scores, or within the normal range (i.e., 40–60), across all subscales, with two exceptions, namely “Physical Well-Being” among non-sitters (mean score: 35.41) and patients with SMA type II (mean score: 34.83). Particularly high scores were noted for the “Autonomy and Parents” and “School environment” scale domains. Aside the physical disability, these results indicate that most patients in our cohort have subjective HRQoL similar to that of other children. This is also reflected in the fact that the vast majority of patients, >80%, rated their current health as “Good”, “Very good”, or “Excellent”. Possible explanations for these findings include coping mechanisms, in which patients may learn to cope with their illness, adjust their perception and expectations of HRQoL, and adapt to their health state (a phenomenon sometimes referred to as the well-being or disability paradox) [14, 27]. Moreover, the awareness of benefiting from new therapeutic approaches could also have a positive effect on subjective well-being. Similar findings have been observed in patients with other chronic, progressive neuromuscular diseases, such as Duchenne muscular dystrophy [28]. In two KIDSCREEN-27 domains (i.e., “Physical Well-Being” and “Autonomy and Parents”), there was a trend of increasing HRQoL across categories of current best motor function (Fig. 1, upper panel). This was also reflected in the estimated KIDSCREEN-27 total score, which was 40% higher among walkers than non-sitters (the score difference between sitters and walkers was only 6%). On the other hand, estimates were higher for sitters than walkers in two domains, namely “Psychological Well-Being” and “Peers & Social Support” (although very similar for the latter domain). Reasons for these findings warrant further study. In line with expectations, considering that most non-sitters have SMA type II, scores were generally similar for non-sitters and those with SMA type II, in particular “Physical Well-Being” (mean score: 35 vs. 35) and “Physiological Well-Being” (mean score: 44 vs. 44), but also the total score (mean score: 48 vs. 51). Looking at results by SMA type (Fig. 1, lower panel), the single patient with type I (8 years of age; sitter) performed better across all domains than those with type II and type III (but results should be interpreted with caution given the sample size). Moreover, scores for SMA type II were consistently lower than those for type III (mean score difference across domains: 6.27), although comparable for some KIDSCREEN-27 domains (e.g., “Psychological Well-Being” and “School Environment”).
We estimated the mean self-assessed KINDL total score at 75.00 for sitters and 68.05 for walkers (no non-sitters completed the scale). Corresponding scores for SMA type I, type II, and type III were 76.57, 76.73, and 60.42, respectively. Compared with general population KINDL reference data for Germany, estimated at 74.6 for boys and girls 11–13 years of age [29], our findings thus indicate that mainly walkers and patients with SMA type III in our sample had impaired HRQoL (i.e., a score < 74.6) as assessed using the KINDL rating-scale. These differences may be due to different coping mechanisms: SMA children with inferior motor skills are more confident with their disability, while SMA children with better motor function (yet inferior to that of healthy children) have more acceptance problems. However, considering the sample size and age of our study population, more research is needed to delineate the impact on self-reported HRQoL as measured using KINDL across clinical subgroups.
Across scale domains, mean PedsQL 3.0 NMM scores were consistently lower for non-sitters compared with sitters (mean score difference: 26.90), and sitters compared with walkers (mean score difference: 5.63), with one exception, namely “Communication” among sitters (Fig. 2, upper panel). Indeed, all seven patients in this subgroup responded that they “Never” have any problems telling doctors and nurses how they feel, ask doctors and nurses questions, or explain their illness to other people, which is very encouraging. In contrast, 50% of non-sitters and walkers, respectively, reported having problems with these communicative tasks. Moreover, compared with the other strata, non-sitters had particularly low scores also for the “About Our Family Resources” domain. In detail, this mainly concerned “It is hard for my family to plan activities like vacations” and “It is hard for my family to get enough rest”. These findings are consistent with our clinical observation, where non-sitters usually are bedridden and frequently require extensive support (e.g., suction, ventilation, and feeding assistance via gastrostomy tube) which makes it challenging for families to go on vacations and carry out similar activities. Comparing our results with previous research, the mean “About my Neuromuscular Disease” score for non-sitters was almost identical to the self-assessed estimate reported by Iannaccone et al. [23] in their study of 104 US children with SMA (mean age: 9 years, 52% female [total sample; details not reported for patients self-reporting their HRQoL]) (59.25 vs. 58.36). However, both sitters and walkers in our sample had higher scores in this domain than their US counterparts (73.29 vs. 62.20, and 83.33 vs. 72.85, respectively). One obvious explanation for these differences is the fact that almost all patients in our cohort (80%) were treated with nusinersen or risdiplam (no patients were treated in the study by Iannaccone et al. [23]). Similarly, “Communication scores” were lower in our sample compared with the US cohort among non-sitters (62.50 vs. 70.01), but not sitters (100.00 vs. 73.57) or walkers (96.00 vs. 60.97). The same pattern was observed for the “About our Family Resources” domain, for which the mean score among non-sitters in our study was lower than the corresponding US estimate (52.50 vs. 61.00), but higher among sitters (81.67 vs. 72.58) and in particular walkers (92.50 vs. 78.86). Compared with the US data [23], the mean total score in our sample was 9% lower for non-sitters, 9% higher for sitters, and 14% higher for walkers. As noted, reasons for this variability include differences in exposure to disease-modifying drugs but warrant further study. Looking into previously published self-assessed PedsQL 3.0 NMM scores by SMA type, in their study comprising of children and adults with SMA (mean age, sex, and sample size not reported for the subset of patients providing self-assessments), Klug et al. [30] estimated the total score at 61 for SMA type II (11% lower than our estimate of 68.00) and at 69 for SMA type III (22% lower than our estimate of 84).
We estimated the mean self-reported EQ-5D-5L utility at 0.35 among non-sitters, 0.37 among sitters, and 0.99 among walkers. Across SMA types, the mean utility was 0.31 for type I, 0.37 for type II, and 0.99 for type III. Three main observations can be made with regards to these results. First, members of the general population exhibit low preferences for many health states associated with SMA. Compared with the mean EQ-5D-5L utility among male and female members of the general population in Germany 18–24 years of age (the youngest age group for which reference data is available) of 0.94 [31], our findings correspond to a disutility (i.e., loss in utility) of 0.60 among non-sitters, 0.57 for sitters, –0.05 for walkers; and 0.63 for SMA type I, 0.57 for type II, and –0.05 for type III. These data may be compared with the minimally important difference –that is, the smallest difference perceived as important, either beneficial or harmful, and which would lead the patient or clinician to consider a change in the management –in EQ-5D-5L utility estimated at 0.083 (German value set) [32]. Second, compared with HUI-derived utilities (discussed below), the relatively high utility among non-sitters (and to lesser extent also sitters) indicates that the EQ-5D-5L might lack sensitivity in measuring HRQoL in this patient group. Third, and last, the very high utility estimate among walkers suggests that the EQ-5D-5L might lack sensitivity also with regards to the disease impact of SMA in this patient group.
Looking into previous research, no study has assessed HRQoL in children with SMA based on self-reported data from the EQ-5D-5L. However, McMillan et al. [33] reported EQ-5D-5L utilities based on a combination of patient self- and caregiver (mainly parent) proxy reports in a cohort comprising of 965 Canadian patients with SMA (mean age: 11.71 years, 40% female). The mean utility was reported at 0.32 for SMA type I, 0.46 for type II, and 0.65 for type III. Although it is not possible to draw any definite conclusion due to the limited sample size and study differences in patient characteristics, these findings suggests that children with SMA, in particular walkers, might have higher HRQoL than their adult counterparts. Finally, for comparison, published utility estimates (combination of self- and proxy-reports by current best motor function or SMA type) derived using other version of the EQ-5D scales include –0.012 for SMA type II based on the EQ-5D-3L (Spanish sample, mean age: 7.22 years, 58% female) [34]; 0.104 for SMA type I, 0.067 for type II, and 0.252 for type III based on EQ-5D-Y, a child-friendly version of the EQ-5D (Australian sample, mean age: 9.38 years, 53% female) [35]; and between 0.34 and 0.70 depending on the number of SMN2 copy numbers based on the EQ-5D-5L in adults and EQ-5D-Y in children (all treated with nusinersen, onasemnogene abeparvovec, or risdiplam) (Belgian, French, and German sample, median age: 6.25 years, proportion female not reported) [36].
Concerning self-assessed HRQoL measured using the EQ-5D VAS, in line with the EQ-5D-5L utility findings, sitters rated their health status markedly lower than both non-sitters and walkers (64.67 vs. 90.00 vs. 90.00, respectively) and those with SMA type II lower than type I and type III (69.75 vs. 90.00 vs. 90.00, respectively). For comparison, estimates from previous research (combination of self- and proxy-assessments) include 53.03 (SMA type II; Spanish sample) [34]; 59.25 for SMA type I, 67.46 for type II, and 66.11 for type III (Australian sample) [35]; and 46.6 for SMA type I, 66.0 for type II, and 71.5 for type II (Canadian sample) [33].
We estimated the mean self-reported HUI-derived utility at 0.23 among non-sitters, 0.21 among sitters, and 0.96 among walkers. Across SMA types, the mean utility was –0.02 for type I, 0.19 for type II, and 0.88 for type III. Accordingly, the HUI seems relatively insensitive to changes between non-sitters and sitters, but able to differentiate amongst health states characterizing the different SMA types. Compared with HUI general population reference utilities from Canadian children 12–16 years of age of 0.89 [37], the mean disutility in our cohort was estimated at 0.66 for non-sitters, 0.68 for sitters, –0.07 for walkers; and 0.91 for SMA type I, 0.70 for type II, and 0.01 for type III. Only one study, Love et al. [38], has previously estimated self-reported HRQoL in patients with SMA using the HUI. In this study, encompassing 14 Canadian children (mean age and proportion female not reported), the mean self-assessed HUI-derived utility was estimated at 0.29 for SMA type I, 0.23 for type II, and 0.41 for type III. Moreover, two studies have previously estimated HRQoL in patients with SMA based on caregiver proxy- and patient self-reports using the HUI. In their retrospective cohort study of 478 US patients with SMA (mean age: 17.1 years, 59% female), Belter et al. [39] estimated the mean HUI-derived utility (across SMA types, excluding patients receiving permanent ventilation) at 0.12 for non-sitters, 0.24 for sitters, and 0.56 for walkers (independent walking, or with support). Compared with our results, these are lower for non-sitters (potentially [in part] due to the exclusion of ventilated patients, which had a mean utility of 0.00 across SMA types), and in particular for walkers. In the second study, Dangouloff et al. [36] estimated the mean HUI-derived utility at 0.19 (2 SMN2 copy numbers), 0.19 (3 SMN2 copy numbers), and 0.74 (4 SMN2 copy numbers) using the HUI (Mark III) among 42 Canadian children and adults with SMA (median age: 6.25 years, proportion female not reported; all treated with nusinersen, onasemnogene abeparvovec, or risdiplam) and their caregivers.
The main limitation of our pilot study concerns the sample size. Indeed, similarly to most research of children with rare diseases, our cohort was limited to 17 boys and girls with SMA. Accordingly, derived estimates should be interpreted and generalized with some caution. Moreover, due to the self-reported nature of the data, our study is also subject to potential bias from measurement error as a result of incorrect reporting. This also concerns SMA type, which was reported by the patient and/or caregiver. To alleviate this problem, we also stratified our sample by categories of current best motor function (derived based on an analysis of available clinical data). Moreover, due to the sample size, we were unable to formally analyze the impact of specific therapies on estimates of HRQoL, nor the psychometric performance of individual scales, which undoubtedly are important topics for future research. Finally, when interpreting our findings, it is important to keep in mind that Germany is a country characterized by a comparatively very high level of healthcare, as well as relatively low stigmatization of the disabled. For these reasons, our findings would not be expected to be fully generalizable to other settings with less favorable conditions for patients with SMA.
In conclusion, the results from this pilot study show that German children with SMA, despite significant physical disability, have surprisingly good HRQoL as assessed using KIDSCREEN-27. At the same time, many patients were found to reside in health states associated with low preference utilities as rated by members of the general population. Overall, the disease burden was generally higher among non-sitters compared with walkers, and SMA type I compared with type III, but more research is needed to further delineate this variability. Our preliminary findings contribute to the understanding of HRQoL in pediatric patients with SMA and should be helpful to inform the design of future studies in this patient population.
STATEMENTS AND DECLARATIONS
Funding
The non-profit patient association “Initiative SMA –Gemeinsam für eine Therapie” within the “Deutsche Gesellschaft für Muskelkranke e.V.” provided financial support for the license fees of the proprietary HRQoL instruments, as well as the allowance for the SMA patient registry. We acknowledge the financial support of University of Saarland for the publication of this article.
Conflicts of interest
Dr Marina Flotats-Bastardas has received consultant fees from Roche and Biogen.
Dr Landfeldt and Ms. Abner are employees of IQVIA, a contract research organization.
Dr Maggie C. Walter has served on advisory boards for Avexis, Biogen, Novartis, Pfizer, Roche, Santhera, Sarepta, Pharnext, PTC Therapeutics, Ultragenyx, Wave Sciences, received funding for Travel or Speaker Honoraria from Avexis, Biogen, PTC Therapeutics, Ultragenyx, Santhera, Sarepta, and worked as an ad-hoc consultant for AskBio, Audentes Therapeutics, Avexis, Biogen Pharma GmbH, Fulcrum Therapeutics, GLG Consult, Guidepoint Global, Gruenenthal Pharma, Novartis, Pharnext, PTC Therapeutics, Roche.
Simone Thiele has received financial support for advisory services from PTC Therapeutics.
The other authors declare that there is no conflict of interest.
Acknowledgements
The authors would like to sincerely thank all the members of the German SMA community who supported this research with their participation, taking time to answer the questionnaire and sharing information about their physical and mental health and daily lives. We are also very grateful for the financial support provided by the German non-profit patient association “Initiative SMA e.V.”.
Data availability
The data supporting the findings of this study are not publicly available due to ethical restrictions.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230071.
REFERENCES
[1] | Munsat TL , Davies KE . International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul Disord. (1992) ;2: (5-6):423–8. |
[2] | Lunn MR , Wang CH . Spinal muscular atrophy. Lancet. (2008) ;371: (9630):2120–33. |
[3] | Belter L , Cook SF , Crawford TO , Jarecki J , Jones CC , Kissel JT et al., An overview of the Cure SMA membership database: Highlights of key demographic and clinical characteristics of SMA members. J Neuromuscul Dis. (2018) ;5: (2):167–76. |
[4] | Russman BS , Buncher CR , White M , Samaha FJ , Iannaccone ST . Function changes in spinal muscular atrophy II and III. The DCN/SMA Group. Neurology. (1996) ;47: (4):973–6. |
[5] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J et al., Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: (18):1723–32. |
[6] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW et al., Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: (18):1713–22. |
[7] | Baranello G , Servais L , Day J , Deconinck N , Mercuri E , Klein A et al., PP.353FIREFISH Part 1:16-month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with type 1 spinal muscular atrophy. Neuromuscul Disord. (2019) ;29: , S184. |
[8] | Leibrock B , Landfeldt E , Hussong J , Huelle T , Mattheus H , Thiele S et al., Areas of improvement in the medical care of SMA: evidence from a nationwide patient registry in Germany. Orphanet J Rare Dis. 2023; Accepted. |
[9] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al., Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) ;28: (2):103–15. |
[10] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ et al., Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2018) ;28: (3):197–207. |
[11] | Landfeldt E , Edström J , Sejersen T , Tulinius M , Lochmüller H , Kirschner J . Quality of life of patients with spinal muscular atrophy: A systematic review. Eur J Paediatr Neurol. (2019) ;23: (3):347–56. |
[12] | Wan HWY , Carey KA , D’Silva A , Vucic S , Kiernan MC , Kasparian NA , et al., Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. (2020) ;15: (1):70. |
[13] | Yang M , Awano H , Tanaka S , Toro W , Zhang S , Dabbous O , et al., Systematic Literature Review of Clinical and Economic Evidence for Spinal Muscular Atrophy. Adv Ther. (2022) ;39: (5):1915–58. |
[14] | Eiser C , Morse R . Quality-of-life measures in chronic diseases of childhood. Health Technol Assess. (2001) ;5: (4). |
[15] | Wright B . A history of social science and measurement. Educ Meas. (1997) ;52: , 33–52.7. |
[16] | Likert RA . A technique for the measurement of attitudes. Arch Psychol. (1932) ;140: , 5–55. |
[17] | RaschG Probabilistic models for some intelligence and attainment tests (1st Edition). Copenhagen: Danish Institute for Education Research; 1960. |
[18] | Hobart J , Cano S Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess. 2009;13. |
[19] | Drummond MF , Sculpher MJ , Claxton K , Stoddart GL , Torrance GW Methods for Economic Evaluation of Health Care Programmes (4th Edition). Oxford: Oxford University Press; 2015. |
[20] | Neumann PJ , Goldie SJ , Weinstein MC . Preference-based measures in economic evaluation in health care. Annu Rev Public Health. (2000) ;21: :587–611. |
[21] | Ravens-Sieberer U , Herdman M , Devine J , Otto C , Bullinger M , Rose M , et al., The European KIDSCREEN approach to measure quality of life and well-being in children: development, current application, and future advances. Qual Life Res. (2014) ;23: (3):791–803. |
[22] | Ravens-Sieberer U , Bullinger M . Assessing health-related quality of life in chronically ill children with the German KINDL: first psychometric and content analytical results. Qual Life Res. (1998) ;7: (5):399–407. |
[23] | Iannaccone ST , Hynan LS , Morton A , Buchanan R , Limbers CA , Varni JW . The PedsQL in pediatric patients with Spinal Muscular Atrophy: feasibility, reliability, and validity of the Pediatric Quality of Life Inventory Generic Core Scales and Neuromuscular Module. Neuromuscul Disord. (2009) ;19: (12):805–12. |
[24] | Herdman M , Gudex C , Lloyd A , Janssen MF , Kind P , Parkin D , et al., Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. (2011) ;20: :1727–36. |
[25] | Horsman J , Furlong W , Feeny D , Torrance G . The Health Utilities Index (HUI): concepts, measurement properties and applications. Health Qual Life Outcomes. (2003) ;1: :54. |
[26] | Ludwig K , Graf von der Schulenburg JM , Greiner W . German Value Set for the EQ-5D-5L. Pharmacoeconomics. (2018) ;36: (6):663–74. |
[27] | Pangalila R . Quality of life in Duchenne muscular dystrophy: the disability paradox. Dev Med Child Neurol. (2016) ;58: (5):435–6. |
[28] | Landfeldt E , Lindgren P , Bell CF , Guglieri M , Straub V , Lochmüller H , Bushby K . Health-related quality of life in patients with Duchenne muscular dystrophy: a multinational, cross-sectional study. Dev Med Child Neurol. (2016) ;58: (5):508–15. |
[29] | Ravens-Sieberer U , Ellert U , Erhart M . Gesundheitsbezogene Lebensqualität von Kindern und Jugendlichen in Deutschland. Bundesgesundheitsbl. (2007) ;50: :810–8. https://doi.org/10.1007/s00103-007-0244-4 |
[30] | Klug C , Schreiber-Katz O , Thiele S , Schorling E , Zowe J , Reilich P , et al., Disease burden of spinal muscular atrophy in Germany. Orphanet J Rare Dis. (2016) ;11: (1):58. |
[31] | Grochtdreis T , Dams J , König HH , Konnopka A . Health-related quality of life measured with the EQ-5D-5L: estimation of normative index values based on a representative German population sample and value set. Eur J Health Econ. (2019) ;20: (6):933–44. |
[32] | Henry EB , Barry LE , Hobbins AP , McClure NS , O’Neill C . Estimation of an Instrument-Defined Minimally Important Difference in EQ-5D-5L Index Scores Based on Scoring Algorithms Derived Using the EQ-VT Version 2 Valuation Protocols. Value Health. (2020) ;23: (7):936–44. |
[33] | McMillan HJ , Gerber B , Cowling T , Khuu W , Mayer M , Wu JW , et al., Burden of Spinal Muscular Atrophy (SMA) on Patients and Caregivers in Canada. J Neuromuscul Dis. (2021) ;8: (4):553–68. |
[34] | López-Bastida J , Peña-Longobardo LM , Aranda-Reneo I , Tizzano E , Sefton M , Oliva-Moreno J . Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis. (2017) ;12: (1):141. |
[35] | Chambers GM , Settumba SN , Carey KA , Cairns A , Menezes MP , Ryan M , et al., Prenusinersen economic and health-related quality of life burden of spinal muscular atrophy. Neurology. (2020) ;95: (1):e1–e10. |
[36] | Dangouloff T , Hiligsmann M , Deconinck N , D’Amico A , Seferian AM , Boemer F , et al., Financial cost and quality of life of patients with spinal muscular atrophy identified by symptoms or newborn screening. Dev Med Child Neurol. (2023) ;65: (1):67–77. |
[37] | Feeny D , Furlong W , Saigal S , Sun J . Comparing directly measured standard gamble scores to HUI2 and HUI3 utility scores: group- and individual-level comparisons. Soc Sci Med. (2004) ;58: (4):799–809. |
[38] | Love D , Hicks R , Wei Y , Aldana EZ , Almobarak S , Campbell C . P.218Utility based health related quality of life in children and adolescents with spinal muscular atrophy. Neuromuscul Disord. (2019) ;29: , S130. |
[39] | Belter L , Cruz R , Jarecki J . Quality of life data for individuals affected by spinal muscular atrophy: a baseline dataset from the Cure SMA Community Update Survey. Orphanet J Rare Dis. (2020) ;15: (1):217. |


