
Clinical and Genetic Spectrum of Myotonia Congenita in Turkish Children
Abstract
Background:
Myotonia congenita is the most common form of nondystrophic myotonia and is caused by Mendelian inherited mutations in the CLCN1 gene encoding the voltage-gated chloride channel of skeletal muscle.
Objective:
The study aimed to describe the clinical and genetic spectrum of Myotonia congenita in a large pediatric cohort.
Methods:
Demographic, genetic, and clinical data of the patients aged under 18 years at time of first clinical attendance from 11 centers in different geographical regions of Türkiye were retrospectively investigated.
Results:
Fifty-four patients (mean age:15.2 years (±5.5), 76% males, with 85% Becker, 15% Thomsen form) from 40 families were included. Consanguineous marriage rate was 67%. 70.5% of patients had a family member with Myotonia congenita. The mean age of disease onset was 5.7 (±4.9) years. Overall 23 different mutations (2/23 were novel) were detected in 52 patients, and large exon deletions were identified in two siblings. Thomsen and Becker forms were observed concomitantly in one family. Carbamazepine (46.3%), mexiletine (27.8%), phenytoin (9.3%) were preferred for treatment.
Conclusions:
The clinical and genetic heterogeneity, as well as the limited response to current treatment options, constitutes an ongoing challenge. In our cohort, recessive Myotonia congenita was more frequent and novel mutations will contribute to the literature.
INTRODUCTION
Myotonia congenita (MC) is the most common form of non-dystrophic myotonia, and is due to mutations causing loss of function of the voltage-gated chloride channel gene (CLCN1) coding for the ClC-1 voltage-dependent chloride channel [1]. These mutations cause reduction in sarcolemmal-stabilizing chloride conductivity and lead to clinical myotonia [2, 3]. The disease can be inherited autosomal dominant as first identified (Thomsen disease, OMIM:160800) or autosomal recessive (Becker disease, OMIM: 255700). CLCN1 is localized to 7q35 and is 35 kb with 23 exons. More than 275 pathogenic variants have been identified so far in this gene, of which 95% are point mutations [4]. Mutations causing complete loss of function cause the Becker phenotype with more severe clinical progression, while mutations causing Thomsen disease are generally missense mutations with dominant-negative effect on channel function. Additionally, more than ten mutations act in both patterns[5, 6].
For the two types, childhood and adult-onset forms are estimated to have a total prevalence of 1/100.000 [7]. However, prevalence, hereditary pattern and genetic variants display differences between countries due to geographical factors [8–10]. The prevalence of the disease in Türkiye is not known, while data on the clinical and genetic spectrum specific to our country is also limited [11, 12].
Information about the genotype and phenotype of the disease would guide physicians for diagnosis and treatment and assist developing individualized treatment strategies. In this study, we aimed to determine the clinical and genetic features of genetically-proven MC cases in the pediatric age group with larger case numbers.
MATERIALS AND METHODS
Patient selection and clinical characteristics
Detailed research forms questioning patient demographic, clinical, genetic, laboratory and electrophysiological data were sent to participating centers and regional records were retrospectively investigated and anonymously recorded. Cases with genetic confirmation of diagnosis, without missing data, and aged under 18 years at time of first clinical attendance were included in the study. A semi-quantitative scale was used to determine the severity of myotonia. Severe myotonia was classified as myotonia which prevented daily life activities and was easily noticed by others; moderate myotonia was classified as causing some small problems in daily life and mentioned by patients; while mild myotonia was classified as not having any visible negative effect on daily life and only identified by examination [13].
Genetic diagnosis and characteristics
Genetic diagnoses of patients were performed by several different protocols. Sequencing of the CLCN1 gene was performed through Sanger sequencing or next-generation sequencing (NGS). Variants reported to be causative in HGMD Public® / ClinVar and variants predicted to be pathogenic, likely pathogenic, and variant of uncertain significance (VUS) according to dedicated software, such as mutation taster, Polyphen2, Provean, Franklin, and Varsome were enrolled. Analysis of large deletion and duplication was performed through multiplex ligation-dependent probe amplification.
Statistical analyzes
Analysis of data used SPSS version 24. To assess distribution of data, the Kolmogorov-Smirnov test was used. Quantitative data are shown as mean and standard deviation, while qualitative data are shown as case number (n) and percentage. Frequencies were calculated for categorical variables.
Ethical approval
Ethical permission for the study was obtained from Ondokuz Mayıs University Clinical Research Ethics Committee (OMÜ KAEK 2021/582).
RESULTS
Patient demographic and clinical characteristics
Data from 69 patients reported from eleven centers in seven different regions were screened. Fifteen patients without genetic confirmation or without adequate clinical data were excluded from the study. This cohort consists of cases whose diagnosis of myotonic dystrophy type 1 and SCN4A-related sodium channel myotonia, paramyotonia congenita, and periodic paralysis has been excluded. A total of 54 patients, from 40 unrelated pedigrees, were included. The geographical distribution of patients is shown in Fig. 1. Nine patients included in the current study were described in a previously-published study by Özgün et al. [11]. Actual mean age of patients including 13 girls (24%) and 41 boys (76%) was 15.2 years (±5.5) (min=3/max=25). Mean age at diagnosis was 11 (±4.9) (min = 1/max=20) years, with follow-up duration of 3.5 (±2.5) (min = 1/max=10)years.
Fig. 1
Distribution of cases according to geographic region in Türkiye.

Admission complaints included difficulty starting movement (when standing, beginning to walk, going up stairs) (64.8%), difficulty opening hands (29.6%), weakness (9.3%), stiffness (9.3%), leg pain (7.5%), falls (7.5%) and slow movements (1.9%). Complaints began at mean 5.7 (±4.9) (min = 1/max=17) years. For 23 patients (42.5%), complaints increased with cold. When patients were questioned 20 (37%) had episodic weakness attacks, while 49 (91%) described the warmup phenomenon. Myotonia was classified by clinicians as mild for 4 patients (7.5%), moderate for 31 patients (57.5%) and severe for 19 patients (35%). The complaints of 18 patients (Becker; 16/ Thomsen; 2) (33%) progressed during treatment.
Parents of 36 patients (67%) were consanguineous. Thirty-eight patients (70.5%) had individuals with diagnosis of MC in their families.
During examination, 44 patients (81.5%) had hypertrophic appearance of muscles, while 52 (96%) had grip myotonia and 43 patients (80%) had percussion myotonia. Muscle weakness was present in 15 patients (28%).
Creatinine kinase (CK) levels of 48 patients were measured. At attendance, mean serum CK value was 266 (±270) (min=53/max=1814) U/L. No relationship could be established between the CK value and the genotype. Even among individuals carrying the same mutation, CK values differed.
Electromyography (EMG) was performed for a total of 31 patients (57.5%) and 24 had myotonia (44.5%), six had myotonia and myopathy (11%) and one had only myopathy (2%).
Twenty-one of the patients with “only myotonia” and five of the patients with “myotonia and myopathy” showed Becker phenotype. The case with “only myopathy” had difficulty in walking since the age of two and he was diagnosed with Thomsen type MC at the age of ten. The patient’s clinical myotonia was severe and his symptoms were partially relieved with carbamazepine treatment.
The number of participants with Becker phenotype was 46 (85%), and eight (15%) with Thomsen phenotype. One family included individuals with both Thomsen and Becker types. Parental consanguinity was present in 35 patients (76%) with Becker and only for one patient with Thomsen disease (12.5%). The demographic and clinical features of patients according to phenotype are summarized in Table 1.
Table 1
Demographic and clinical features of patients with Becker and Thomsen forms
| Becker form | Thomsen form | |
| n = 46 (100%) | n = 8 (100%) | |
| Demographic characteristics | ||
| Actual age | 15.4±5.5 years | 14.1±6.3 years |
| Age of complaint onset | 5.6±4.2 years | 6.5±4.4 years |
| Diagnosis age | 11.2±4.9 years | 11.9±5.5 years |
| Female/male | 13 (28%) / 33 (72%) | -/ 8(100%) |
| Attendance complaint | ||
| Slow movement | 1 (2.2%) | - |
| Weakness | 4 (8.6%) | 1 (12.5%) |
| Difficulty beginning movement | 30 (65.2%) | 5 (62.5%) |
| Leg pain | 3 (6.5%) | 1 (12.5%) |
| Stiffness | 2 (4.3%) | 3 (37.5%) |
| Fall | 4 (8.6%) | - |
| Difficulty opening hands | 15 (32.6%) | 1 (12.5%) |
| Clinical features | ||
| Symptom increase with cold | 21 (46%) | 2 (25%) |
| Episodic weakness attacks | 18 (39%) | 2 (25%) |
| Warmup phenomenon | 43 (93.5%) | 6 (75%) |
| Myotonia severity | ||
| Mild | 4 (8.5%) | - |
| Moderate | 25 (54.5%) | 6 (75%) |
| Severe | 17 (37%) | 2 (25%) |
| Family history | ||
| Consanguinity | 35 (76%) | 1 (12.5%) |
| MC case in family | 33 (72%) | 5 (62.5%) |
| Examination findings | ||
| Herculoid appearance | 37 (80.5%) | 7 (87.5%) |
| Grip myotonia | 44 (96%) | 8 (100%) |
| Myotonia with percussion | 36 (78%) | 7 (87.5%) |
Genetic characteristics
Eight of the patients (15%) had heterozygous, 36 (66.5%) had homozygous and 8 (15%) had combined heterozygous mutation identified. Two siblings (3.5%), whose healthy parents were cousins, had large homozygous deletion present including exons 1-16 (Table 2). Twenty-three different mutations including ten missense (43.5%), six nonsense (26%), three splice site (13%), two frameshift (8.7%), one indel (4.4%) and one silent (4.4%) were identified (Fig. 2). The mutation distribution according to the EMG findings is summarized inTable 3.
Table 2
Homozygous, combined heterozygous and heterozygous mutations
| Homozygous n = 38 | Combined heterozygous n = 8 | Heterozygous n = 8 | |||
| c.568_569delinsTC (p.Gly190Ser) [14] | 6 | c.830 G>A (p.Cys277Tyr)/ c.1580T>C (p.Ile527Thr) | 5 | c.127 C>T (p.Gln43Ter) | 2 |
| c.697 G>A (p.Gly233Ser) [15,16] | 3 | c.2680 C>T (p.Arg894Ter)/ c.1129 C>T (p.Arg377Ter) | 1 | c.2551 G>A (p.Val851Met) [18] | 1 |
| c.1064 + 1 G>A | 3 | c.1282-1285delTTTG (p.Phe428Thrfs6*)/c.1167-10 T > C | 1 | c.1282-1285delTTTG (p.Phe428Thrfs6*) | 1 |
| c.1129 C>T (p.Arg377Ter) [17] | 3 | c.501 C>G (p.Phe167Leu)/ c.374 G>A (p.Gly125Glu) | 1 | c.2680 C>T (p.Arg894Ter) | 1 |
| c.1012 C>T (p.Arg338Ter) | 2 | c.568_569delinsTC (p.Gly190Ser) | 1 | ||
| c.1063 G>C (p.Gly355Arg) | 2 | c.697 G>A (p.Gly233Ser) | 1 | ||
| c.1167-10T>C | 2 | c.1701 C>A (p.Asn567Lys) | 1 | ||
| c.127 C>T (p.Gln43Ter) | 2 | ||||
| c.1471 + 1 G>A [15] | 2 | ||||
| c.1886T>C (p.Leu629Pro) | 2 | ||||
| c.2680 C>T (p.Arg894Ter) [15] | 2 | ||||
| c.450 C>A (p.Tyr150Ter) | 2 | ||||
| c.774 G>A (p.Glu258Glu) | 2 | ||||
| c.1579A>G (p.Ile527Val) | 1 | ||||
| c.989dupC (p.Ala331Cysfs14*) | 1 | ||||
| c.2305 > T (p.Gln769Ter) | 1 | ||||
| rsa07q34(CLCN1exons1-16) | 2 |
Bold text indicates novel mutations. Superscripts refer to studies about the functional characterization of mutations.
Fig. 2
Two-dimensional representation of the structure of a CLCN1 gene showing the locations of mutations identified in this study.
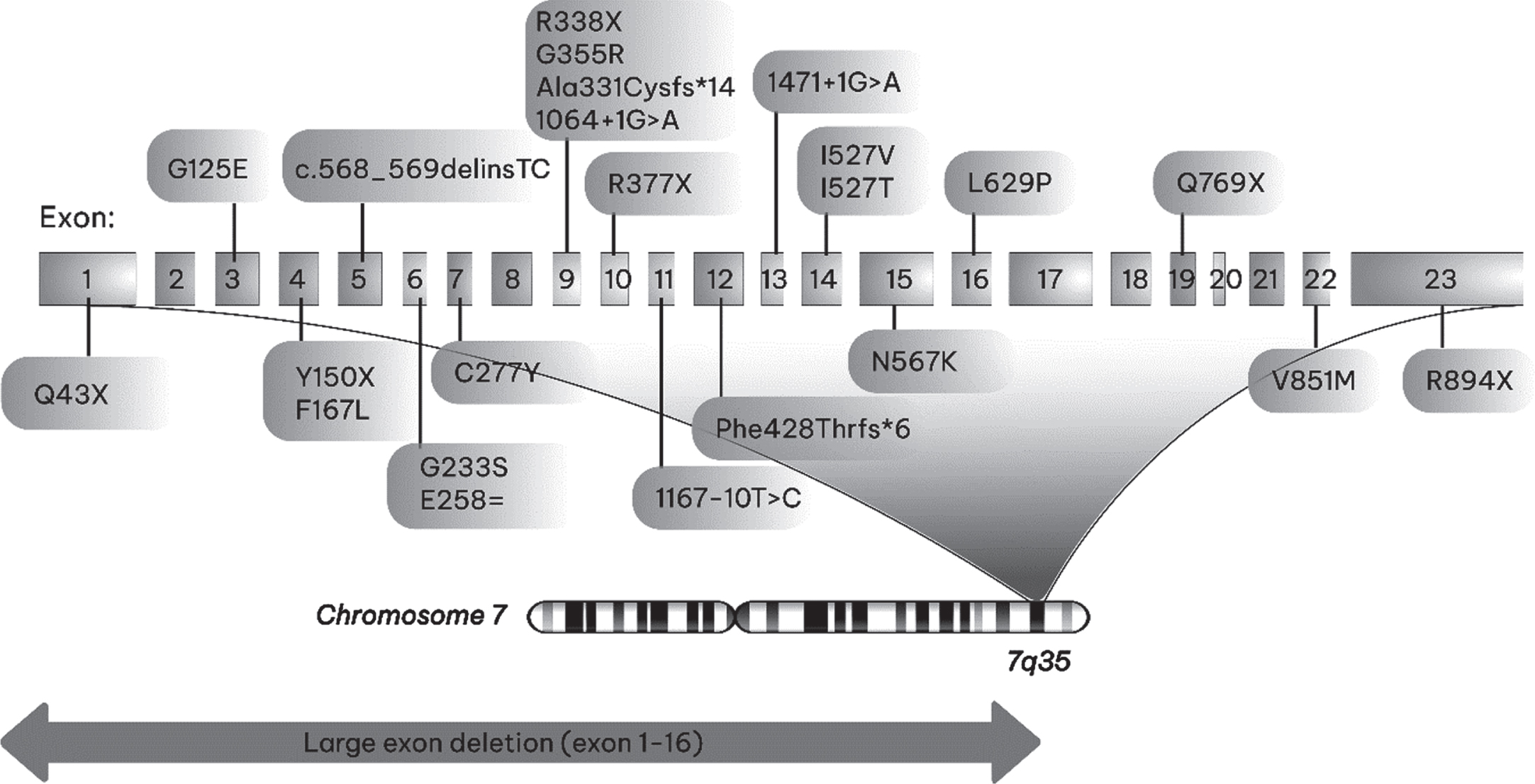
Table 3
Mutation distribution according to electromyography findings
| Myotonia n = 24 | n | Myotonia &Myopathy n = 6 | N | Myopathy n = 1 | n | |||
| c.1129 C>T (p.Arg377Ter) | Ho | 3 | c.1471 + 1 G>A | Ho | 1 | c.2680 C>T (p.Arg894Ter) | He | 1 |
| c.697 G>A (p.Gly233Ser) | Ho | 2 | c.568_569delinsTC (p.Gly190Ser) | Ho | 1 | |||
| c.450 C>A (p.Tyr150Ter) | Ho | 2 | c.1012 C>T (p.Arg338Ter) | Ho | 1 | |||
| c.1063 G>C (p.Gly355Arg) | Ho | 2 | c.1064 + 1 G>A | Ho | 1 | |||
| c.127 C>T (p.Gln43Ter) | Ho | 1 | c.2680 C>T (p.Arg894Ter)/ | CH | 1 | |||
| c.2305 > T (p.Gln769Ter) | Ho | 1 | c.1129 C>T (p.Arg377Ter) | |||||
| c.568_569delinsTC (p.Gly190Ser) | Ho | 1 | c.1701 C>A (p.Asn567Lys) | HE | 1 | |||
| c.1886T>C (p.Leu629Pro) | Ho | 1 | ||||||
| c.1167-10T>C | Ho | 1 | ||||||
| c.1064 + 1 G>A | Ho | 1 | ||||||
| c.1471 + 1 G>A | Ho | 1 | ||||||
| rsa07q34(CLCN1exons1-16) | Ho | 1 | ||||||
| c.830 G>A (p.Cys277Tyr)/ | CH | 2 | ||||||
| c.1580T>C (p.Ile527Thr) | CH | 1 | ||||||
| c.1282-1285delTTTG (p.Phe428Thrfs6*)/ c.1167-10 T > C | 1 | |||||||
| c.501 C>G (p.Phe167Leu)/ c.374 G>A (p.Gly125Glu) | CH | 1 | ||||||
| c.2551 G>A (p.Val851Met) | He | 1 | ||||||
| c.697 G>A (p.Gly233Ser) | He | 1 | ||||||
| c.1282-1285delTTTG | He |
Ho: Homozygous, He: Heterozygous, CH: Combined heterozygous
Among these variants, three were not associated with the disease before including c.2305 C>T (p.Gln769Ter) and c.1579A>G (p.Ile527Val) mutations and the large gene deletion encompassing exons 1 to 16. The demographic and clinical features of patients with these mutations are summarized in Table 4.
Table 4
Demographic and clinical features of patients with mutations not previously associated with disease. All of the patients carried these mutations homozygously
| Mutations | c.2305 > T (p.Gln769Ter) (n = 1) (Nonsense) | c.1579A>G (p.Ile527Val) (n = 1) (Missense) | rsa07q34 (CLCN1 exons1-16) (n = 1) Large exonic deletion | rsa07q34 (CLCN1exons1-16) (n = 1) Large exonic deletion |
| Actual age (years) | 16 | 12 | 11 | 5 |
| Age at complaint onset (years) | 2 | 3 | 3 | 3 |
| Diagnosis age (years) | 6 | 5 | 8 | 4 |
| Sex | Male | Male | Female | Female |
| Attendance complaint | Difficulty beginning movement | Leg pain | Difficulty beginning movement | Difficulty beginning movement |
| Symptom increase with cold | + | - | + | + |
| Episodic weakness attacks | + | - | - | - |
| Warmup phenomenon | + | + | + | + |
| Myotonia severity | Severe | Mild | Moderate | Mild |
| Treatment | Mexiletine | Carbamazepine | Mexiletine | Carbamazepine |
| Treatment response | Partial | Partial | Partial | Partial |
Two variants (c.2551 G>A (p.Val851Met), c.1701 C>A (p.Asn567Lys)) were only found in the heterozygous state. Four variants were observed as both heterozygous and homozygous or combined heterozygous (c.127 C>T (p.Gln43Ter), c.568_569delinsTC (p.Gly190Ser), c.697 G>A (p.Gly233Ser), c.1282-1285delTTTG (p.Phe428Thrfs6*)). One variant was observed as both homozygous and heterozygous and combined heterozygous c.2680 C>T (p.Arg894Ter). The most frequently identified mutations were c.568_569delinsTC (p.Gly190Ser), c.830 G>A (p.Cys277Tyr), and c.1579A>G (p.Ile527Val). Of these variants, c.568_569delinsTC (p.Gly190Ser) caused both Becker and Thomsen phenotypes in one family.
Treatment preferences and responses
Nine patients (16.7%) did not receive any treatment. Although the medication was prescribed for five of them, they did not prefer to use any medication. The treatment was not considered necessary by their physicians for three patients with mild and one with moderate myotonia. Carbamazepine was preferred for 25 (46.3%) patients, mexiletine for 15 (27.8%) and phenytoin for five (9.3%) patients as the first choice for treatment. The first medication of fourteen patients (25.9%) was changed during follow-up due to inadequate treatment response and all of these patients had Becker phenotype. Of those who were receiving treatment, seven out of 45 patients (15.5%) had full response, 36 (80%) had partial response. Two patients (4.5%) had no response to clinical treatment. No correlation was found between treatment response and mutationtypes.
DISCUSSION
Our study included the largest cohort of patients with genetically defined MC in Türkiye. There are differences in the genetic alterations in MC cases from our country with the series reported from other countries. 85% of our cases had Becker phenotype, and this rate is higher than the literature (Table 5). This situation may be explained by the high consanguinity rate of our cohort. In fact, globally parental consanguinity is 67%, with 76% for the Becker type and 12.5% for the Thomsen type when assessed separately. Additionally, 82.5% of patients with Becker disease were homozygote, while only 17.5% were combined homozygote, which supports our argument. On the other hand in Northern Scandinavian and Finnish cohorts, combined heterozygous individuals were mostly observed [19, 20]. The family history rate of MC in our study was 70.5%, while it was 50% in the German cohort [21].
Table 5
Comparison of this study with publications in the literature
| Present study | Canada [22] | Japan [10] | Italy [5] | Germany [21] | |
| 54 | 36 | 30 | 19 | 48 | |
| Thomsen type | 8 (15%) | 9 (25%) | 20 (67%) | 7 (36.8%) | 12 (25%) |
| Becker type | 46 (85%) | 27 (75%) | 6 (6%) | 12 (63.2%) | 28 (58.5%) |
| N/A | 8 (16.5%) |
The mean onset age for complaints was 5.7 (±4.9) years, while diagnosis was made at mean age of 11 (±4.9) years. Our diagnosis latency is very short compared to other studies [8, 21, 23]. This difference may be due to the fact that our cohort included only cases diagnosed in the pediatric period or that the high family history rate providing clinicians diagnostic clues.
Clinical features were not statistically comparable between the Becker and Thomsen phenotype because of the disproportionate number of patients. However, all of the patients who had changes in the treatment regimen due to inadequate response and the majority of patients with progressive course under treatment had Becker phenotype. The higher rate of severe myotonia in Becker patients also suggested that the clinical presentation of this phenotype may be more severe. Dupré et al. similarly observed that recessive CLCN1 mutations seemed particularly resistant to medications since most affected patients had tried more than two medications before improving [22]. Similar to our results Orsini et al. showed the cold effect predominates in Becker patients (75%) compared to Thomsen patients (50%) [5]. No significant difference between these two groups was reported in clinical presentations in the Chinese cohort[24].
“Myotonia and myopathy” were found in six of 31 patients who underwent EMG, and “only myopathy” in one patient. Myopathic electromyographic and histopathological changes have been reported in one-fourth to one-third of cases, and this percentage is 22.6% (7/31) in our study [25]. It is well established that myopathy may develop in some patients with MC. The lower percentage may relate to the younger age of our cohort. In the study of Nojszewska et al., in which they examined needle EMG findings in patients with MC, myotonia was found in 95.8% of the examined muscles. This rate is almost 100% in Becker and 80 % in Thomsen phenotype [25]. Our patient with “only myopathy” had also Thomsen phenotype and concomitant myotonia may not have been detected in the muscle for which needle EMG study was performed.
The current therapy of MC is based on the use of drugs such as Mexiletine, Carbamazepine, Phenytoin, and Lamotrigine. Mexiletine is unavailable in Türkiye. However, Carbamazepine and Phenytoin are readily available and well-known. The possible side effects of Mexiletine and Lamotrigine may also cause the treatment preference to favor carbamazepine.
The Human Gene Mutation Database (HGMD) reported 361 mutations [26]. For the CLCN1 gene, more than 275 pathogenic variants were identified [4]. Most of them are missense or nonsense point mutations, similar to our study [4]. In our study, 54 patients from 40 non-related families were identified to have 24 different mutations. A four-case series from two different families previously reported from Türkiye identified two different novel mutations not found in our cohort (c.450 C>A (p.Tyr150Ter), c.475delC (p.Leu159Cysfs*11)) [12]. One of the publications with largest participation was a prevalence study from England including 312 patients, which identified 104 different mutations, while 15 of these were observed in 83% of the cohort [9]. In the recently published British cohort of 223 probands, 95 of the 115 distinct mutations were also identified as missense. In this cohort a single heterozygous variant was found in 116, homozygous variant in 27, compound heterozygous variants in 75 and more than two variants in five probands [27]. In the Northern Scandinavia study, where the disease is more frequently observed, in 45 index cases from 15 Norwegian families and 70 first-degree relatives of these patients and in five index cases from three Swedish families and two of their first-degree relatives, eight different mutations were identified. c.1238T>G (p.Phe413Cys), c.1592 C>T (p.Ala531Val), and c.2680 C>T (p.Arg894Ter) mutations were the most common [19]. The most frequently identified mutations in the German cohort were c.501 C>G (p.Phe167Leu), c.854 G>A (p.Gly285Glu), c.1238T>G (p.Phe413Cys) and c.2680 C>T (p.Arg894Ter), and it was reported 50% of cases carried the c.2680 C>T (p.Arg894Ter) mutation [21]. A Japanese cohort comprising 30 probands found the Thomsen form was dominant in 63% with 19 different mutations corresponding to the most frequent c.892 G>A (p.Ala298Thr), c.1438 C>A (p.Pro480Thr), c.1615A>G (p.Thr539Ala), and c.1679T>C (p.Met560Thr) mutations [10]. In Europe, the c.2680 C>T (p.Arg894Ter) and c.1238T>G (p.Phe413Cys) mutations were more frequently identified. While c.2680 C>T (p.Arg894Ter) was only found in four patients in our study (7.4%), c.1238T>G (p.Phe413Cys) was not encountered in a similar way to the Japanese cohort [10]. In Italy, the c.501 C>G (p.Phe167Leu) mutation was frequently reported, while it was only present in one of our patients (1.9%) [28]. In another study conducted in Italy, a single mutant allele was found in 49 of 106 probands and two mutant alleles were found in 57 of 106 probands. The three most common mutations in this study were c.180 + 3 A > T, c.501 C>G (p.Phe167Leu) and c.568_569delinsTC (p.Gly190Ser) [29]. The most frequently reported mutations for a recently published Chinese cohort were c.892 G>A (p.Ala298Thr), c.1679T>C (p.Met560Thr) and c.1657A>T (p.Ile553Phe) similar to the Japanese study. As discussed, the frequency of dominant and recessive forms and identified mutations display geographical differences [30].
The c.2305 C>T (p.Gln769Ter) and c.1579A>G (p.Ile527Val) mutations that we identified in our cohort have not been associated with the disease before. c.2305 C>T (p.Gln769Ter) mutation was not found in the gnomAD genome database. According to ACMG classification, c.2305 C>T (p.Gln769Ter) mutation was predicted to be likely pathogenic (PVS1, PM2). It is a null variant, and loss of function is a known disease mechanism. This variant is extremely rare in general population. According to ACMG classification, c.1579A>G (p.Ile527Val) was predicted as likely pathogenic. c.1579A>G (p.Ile527Val) is rare in the general population (gnomAD Total Allele Frequency (TAF): 0.0000159%, 4 heterozygous, 0 homozygous) (PM5). Alternative variant at the same position T > G (Ile527Ser) is classified as pathogenic (PM2). Seven pathogenic and likely pathogenic reported variants were found in a 103bp region surrounding this variant, without any benign variant (PM1). It is located in the exonic hotspot. High number of pathogenic missense variants were reported for the CLCN1 gene rather than missense benign variants (116 to 26 respectively) (PP2). With the combined information for the clinical phenotype of the patients, the absence or scarcity of variations in population databases, and in silico predictions, we suspect that these variants are causative for MC. c.989dupC (p.Ala331Cysfs14*) found in our cohort has been very soon reported in a case with MC. It is a null variant and predicted as pathogenic according to ACMG [31].
In Clinvar, in total 73 whole or partial CLCN1 gene deletions were reported. Of them, eight were contiguous gene deletions including CLCN1 gene. The majority were short variants including deletions of less than 50 base pairs. Exon 2, exon 3 and part of exon 1 were deleted only in one report [15, 32, 33]. In our cases, exons 1-16 were deleted and this is the largest intragenic deletion of CLCN1 to our knowledge.
While mutations causing complete loss of function cause the Becker phenotype with more severe clinical progression, mutations causing the Thomsen disease are generally missense mutations causing a dominant-negative effect on channel functions. Additionally, more than ten mutations do not comply with this distinction due to dominant negative effects, decreased penetrance, differences in helix expression, and they behave both dominantly and recessively, like some variants detected in this study (Table 2) [5, 6]. In our cohort, the same mutation (c.568_569delinsTC (p.Gly190Ser) in one family caused heterozygous and homozygous disease. The intrafamilial variability of this variant was previously reported in a tribe with Bedouin-Arabic origin [34].
According to the Human Gene Mutation database, large-scale deletion and duplications comprise 7-10% of mutations reported in the human genome [35]. A cohort presented by Raja Rajan et al. identified 6% of patients had exon deletion or duplication [4]. In our patients, this rate was 3.7%. All these observations show the importance of checking exon deletion and duplications as a part of the genetic analysis of MC patients considered to have recessive heredity, especially when sequencing defines no mutation or only mutation on one allele.
The diversity of mutations identified make our study valuable, and though the mutations partly overlap with those identified in east Asian and European cohorts, they are notably different. This diversity may be due to the genetic heterogeneity in the Anatolian geography. The Greater Middle East 1111 genome project showed that the Anatolian peninsula shares European nuclear DNA to a large percentage, while a small-scale study in our country showed that genetic variations overlap with Europe, while also carrying traces of Africa/East Asia [36, 37]. These results show that Anatolia is between Europe and Asia, similar to its geographical location. As there is no large-scale study about genotyping MC in the Middle East, our study is important to partly represent this region. In fact, a study reported from our neighbor Iran found that individuals carried the homozygous c.1886T>C, p.Leu629Pro mutation and this variant was present in two of our cases [38].
CONCLUSION
We believe our study enriches the literature due to broad information about the phenotype and genotype of patients with MC in our country and close geography, and the novel mutations identified. The broad genetic spectrum of our cases supports the genetic diversity of the Anatolian geography [39]. Another unique aspect of our study is that it included only cases receiving diagnosis in the childhood period. This study also supports the presence of genetic overlap and differences between countries.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
DISCLOSURE OF INTEREST
None of the authors has any conflict of interest to disclose.
ACKNOWLEDGMENTS
The authors declared no no conflict of interest to report. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
REFERENCES
[1] | Suetterlin K , Männikkö R , Hanna MG . Muscle channelopathies: Recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol. (2014) ;27: :583–90. 10.1097/WCO.0000000000000127. |
[2] | Lipicky RJ, Bryant SH , Salmon JH . Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. J Clin Invest. (1971) ;50: :2091–103. 10.1172/JCI106703. |
[3] | Jentsch TJ , Poët M , Fuhrmann JC , Zdebik AA . Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. (2005) ;67: :779–807. 10.1146/annurev.physiol.67.032003.153245. |
[4] | Raja Rayan DL , Haworth A , Sud R , Matthews E , Fialho D , Burge J , Portaro S , et al. A new explanation for recessive myotonia congenita: Exon deletions and duplications in CLCN1 . Neurology. (2012) ;78: :1953–8. 10.1212/WNL.0b013e318259e19c. |
[5] | Orsini C , Petillo R , D’Ambrosio P , Ergoli M , Picillo E , Scutifero M , et al. CLCN1 Molecular Characterization in 19 South-Italian Patients With Dominant and Recessive Type of Myotonia Congenita. Front Neurol. (2020) ;11: :63. 10.3389/fneur.2020.00063. |
[6] | Mazón MJ , Barros F , De la Peña P , Quesada JF , Escudero A , Cobo AM , et al. Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1gene. Neuromuscul Disord. (2012) ;22: :231–43. 10.1016/j.nmd.2011.10.013. |
[7] | Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) ;1: :19–29. 10.1016/0960-8966(91)90039-u. |
[8] | Baumann P , Myllylä VV , Leisti J . Myotonia congenita in northern Finland: An epidemiological and genetic study. J Med Genet. (1998) ;35: :293–6. 10.1136/jmg.35.4.293. |
[9] | Horga A , Raja Rayan DL , Matthews E , Sud R , Fialho D , Durran SC , et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. (2013) ;80: :1472–5. 10.1212/WNL.0b013e31828cf8d0. |
[10] | Sasaki R , Nakaza M , Furuta M , Fujino H , Kubota T , Takahashi MP . Mutation spectrum and health status in skeletal muscle channelopathies in Japan. Neuromuscul Disord. (2020) ;30: :546–553. 10.1016/j.nmd.2020.06.001. |
[11] | Özgün N , Taşlıdere H . Congenital myotonia: A reviewof twenty cases and a new splice-site mutation in the CLCN1gene. Turk J Pediatr. (2020) ;62: :450–460. 10.24953/turkjped.2020.03.012. |
[12] | Sahin I , Erdem HB , Tan H , Tatar A . Becker’s myotonia: Novel mutations and clinical variability in patients born to consanguineous parents. Acta Neurol Belg. (2018) ;118: :567–572. 10.1007/s13760-018-0893-0. |
[13] | Becker PE . Myotonia congenita and syndromes associated with myotonia. In: BeckerPE, LenzW, VogelF, WendtGG (eds). Vol. 3: , Stuttgard, Germany: Georg Thieme Verlag, 1977. |
[14] | Ulzi G , Lecchi M , Sansone V , Redaelli E , Corti E , Saccomanno D , et al. Myotonia congenita: Novel mutations in CLCN1 gene and functional characterizations in Italian patients. Neurol Sci. (2012) ;318: :65–71. 10.1016/j.jns.2012.03.024. |
[15] | Skálová D , Zídková J , Voháňka S , Mazanec R , Mušová Z , Vondráček P , et al. CLCN1 mutationsin Czech patients with myotonia congenita, in silico analysis ofnovel and known mutations in the human dimeric skeletal musclechloride channel. PLoS One. (2013) ;8: :e82549. 10.1371/journal.pone.0082549. |
[16] | Richman DP , Yu Y , Lee TT , Tseng PY , Yu WP , Maselli RA , et al. Dominantly inherited myotonia congenita resulting from a mutation that increases open probability of the muscle chloride channel CLC-1. Neuromolecular Med. (2012) ;14: (4):328–37. 10.1007/s12017-012-8190-1. |
[17] | Olave-Rodriguez JA , Bonilla-Escobar FJ , Candelo E , Rodriguez-Rojas LX . First Two Case Reports of Becker’s Type Myotonia Congenita in Colombia: Clinical and Genetic Features. Appl Clin Genet. (2021) ;14: :473–479. 10.2147/TACG.S323559. |
[18] | Altamura C , Lucchiari S , Sahbani D , Ulzi G , Comi GP , D’Ambrosio , et al. The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel. Hum Mutat. (2018) ;39: (9):1273–1283. 10.1002/humu.23581. |
[19] | Sun C , Tranebjaerg L , Torbergsen T , Holmgren G , Van Ghelue M . Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur J Hum Genet. (2001) ;9: :903–9. 10.1038/sj.ejhg.5200736. |
[20] | Papponen H , Toppinen T , Baumann P , Myllylä V , Leisti J , Kuivaniemi H , et al. Founder mutations and the high prevalence of myotonia congenita in northern Finland. Neurology. (1999) ;53: :297–302. 10.1212/wnl.53.2.297. |
[21] | Vereb N , Montagnese F , Gläser D , Schoser B . Non-dystrophic myotonias: clinical and mutation spectrum of 70 German patients. J Neurol. (2021) ;268: :1708–1720. 10.1007/s00415-020-10328-1. |
[22] | Dupré N , Chrestian N , Bouchard JP , Rossignol E , Brunet D , Sternberg D , et al. Clinical, electrophysiologic, and genetic studyof non-dystrophic myotonia in French-Canadians. Neuromuscul Disord. (2009) ;19: :330–4. 10.1016/j.nmd.2008.01.007. |
[23] | Trip J , Drost G , Ginjaar HB , Nieman FH , van der Kooi AJ , de Visser M , et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry. (2009) ;80: :647–52. 10.1136/jnn2008.162396. |
[24] | Hu C , Shi Y , Zhao L , Zhou S , Li X . Myotonia Congenita: Clinical Characteristic and Mutation Spectrum of CLCN1 in Chinese Patients. Front Pediatr. (2021) Nov 1;9: :759505. |
[25] | Nojszewska M , Lusakowska A , Gawel M , Sierdzinski J , Sulek A , Krysa W , et al. The needle EMG findings in myotonia congenita. J Electromyogr Kinesiol. (2019) ;49: :102362. |
[26] | Stenson PD , Ball EV , Mort M , Phillips AD , Shiel JA , Thomas NS , Abeysinghe S , et al. Human Gene Mutation Database (HGMD): update. Hum Mutat. (2003) ;21: :577–81. 10.1002/humu.10212. |
[27] | Suetterlin K , Matthews E , Sud R , McCall S , Fialho D , Burge J , et al. Translating genetic and functional data into clinical practice: A series of 223 families with myotonia. Brain. (2022) ;145: :607–620. 10.1093/brain/awab344. |
[28] | Modoni A , D’Amico A , Dallapiccola B , Mereu ML , Merlini L , Pagliarani S , et al. Low-rate repetitive nerve stimulation protocol in an Italian cohort of patients affected by recessive myotonia congenita. J Clin Neurophysiol. (2011) ;28: :39–44. 10.1097/WNP.0b013e31820510d7. |
[29] | Brugnoni R , Kapetis D , Imbrici P , Pessia M , Canioni E , Colleoni L , et al. A large cohort of myotonia congenita probands: Novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet. (2013) ;58: :581–7. 10.1038/jhg.2013.58. |
[30] | Li Y , Li M , Wang Z , Yang F , Wang H , Bai X , Sun B , et al. Clinical and molecular characteristics of myotonia congenita in China: Case series and a literature review. Channels. (2022) ;16: :35–46. 10.1080/19336950.2022.2041292. |
[31] | National Center for Biotechnology Information. ClinVar; [VCV001403542.2], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001403542.2 (accessed April 5, (2023). |
[32] | Nykamp K , Anderson M , Powers M , Garcia J , Herrera B , Ho YY , Kobayashi Y , et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. (2017) ;19: :1105–1117. 10.1038/gim.2017.37. |
[33] | Ronstedt K , Sternberg D , Detro-Dassen S , Gramkow T , Begemann B , Becher T , Kilian P , et al. Impaired surface membrane insertion of homo- and heterodimeric human muscle chloride channels carrying amino-terminal myotonia-causing mutations. Sci Rep. (2015) ;5: :15382. 10.1038/srep15382. |
[34] | Shalata A , Furman H , Adir V , Adir N , Hujeirat Y , Shalev SA , Borochowitz ZU . Myotonia congenita in a large consanguineous Arab family: insight into the clinical spectrum of carriers and double heterozygotes of a novel mutation in the chloride channel CLCN1 gene. Muscle Nerve. (2010) ;41: :464–9. 10.1002/mus.21525. |
[35] | Stenson PD , Mort M , Ball EV , Howells K , Phillips AD , Thomas NS , Cooper DN . The Human Gene Mutation Database: update. Genome Med. (2009) ;1: :13. 10.1186/gm13. |
[36] | Scott EM , Halees A , Itan Y , et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. (2016) ;48: :1071–6. 10.1038/ng.3592. |
[37] | Alkan C , Kavak P , Somel M , Gokcumen O , Ugurlu S , Saygi C , et al. Whole genome sequencing of Turkish genomes reveals functional private alleles and impact of genetic interactions with Europe, Asia and Africa. BMC Genomics. (2014) ;15: :963. 10.1186/1471-2164-15-963. |
[38] | Miryounesi M Md PhD , Ghafouri-Fard S Md PhD , Fardaei M PhD . A Novel Missense Mutation in CLCN1 Gene in a Family with Autosomal Recessive Congenital Myotonia. Iran J Med Sci. (2016) ;41: :456–8. |
[39] | Topaloğlu H . The complex genetic inheritance of Anatolia, Turkey. Dev Med Child Neurol. (2022) ;64: :1181. 10.1111/dmcn.15312. |


