
A Novel Mutation in Frabin (FGD4) Causing a Mild Phenotype of CMT4H in an Indian Patient
Abstract
Charcot-Marie-Tooth disease 4H(CMT4H) is an autosomal recessive demyelinating form of CMT caused by FGD4/FRABIN mutations. CMT4H is characterized by early onset and slowly progressing motor and sensory deficits in the distal extremities, along with foot deformities. We describe a patient with CMT4H who presented with rapidly progressing flaccid quadriparesis during the postpartum period, which improved significantly with steroid therapy. Magnetic resonance imaging and ultrasonography demonstrated considerable nerve thickening with increased cross-sectional area in the peripheral nerves. A nerve biopsy revealed significant demyelination and myelin outfolding. This is the first report of an Indian patient with a novel homozygous nonsense c.1672C>T (p.Arg558Ter) mutation in the FGD4 gene, expanding the mutational and phenotypic spectrum of this disease.
Introduction
Charcot-Marie-Tooth disease (CMT) also known as hereditary motor and sensory neuropathy (HMSN)is one of the most common inherited neurological disorders comprising a group of genetically and phenotypically diverse conditions. It is caused by mutations in genes that encode proteins involved in the structure and function of either the peripheral nerve axon or the myelin sheath of nerves [1]. It occurs approximately with a frequency of 1 in every 2500 individuals [2]. It is clinically characterized by progressive motor and sensory deficits arising at distal extremities causing weakness, atrophy, depressed tendon reflexes, and hypoesthesia of feet and legs along with foot deformities such as pes cavus and pes planus [3]. Based on electrophysiological (nerve conduction studies) and histopathological findings, it is classified into demyelinating forms (NCV <38 m/s) such as CMT1 (autosomal dominant) and axonal forms (NCV >38 m/s) such as CMT2 (autosomal dominant or recessive) [3].
CMT4H is an autosomal recessive demyelinating form of CMT [4]. This type of CMT is characterized by an early age of onset, and slow progression, but is more severe than other phenotypes described. It is caused by mutations in the FGD4 gene that encodes a protein called frabin, which serves as a GDP/GTP exchange protein for Rho GTPase Cdc42 (cell-division cycle 42), a small GTP binding protein family member that is involved in regulating the actin cytoskeleton-dependent cellular activities in Schwann cells [5, 6]. In 2005, Giovannoli et al. mapped this disease to a novel locus on chromosome 12p11.2- p13.1 in Lebanese and Algerian families affected with severe autosomal recessive CMT [7]. Later, in 2006, Delague et al., found mutations in FGD4, encoding frabin (FGD1-related F-actin binding protein) in the same patients [8].
This subtype of CMT is relatively uncommon, and there have been no reports of this type in an Indian patient [3, 4]. Here, we describe the first case of an Indian woman with CMT4H with a novel FGD4 mutation, who presented to our tertiary care center with a mild phenotype and postpartum exacerbations by highlighting the clinical, radiological, histopathological and genetic findings.
Materials and methods
Case description
This report is a description of the clinical, radiological, histopathological and genetic mutation findings in a case of FGD4. Institutional ethics committee approval was obtained to collect all data from the medical records maintained at the hospital. The clinical details are written meticulously in the case records and all images were retrieved from the PACS system. The patient provided consent for the unmasking of the face.
Radioimaging
Ultrasonography was performed using Philips Diagnostic scanner EPIQ 7 using an 8–15 MHz linear transducer. CSA (cross-sectional area) was measured at the median (wrist crease, mid-forearm- at the midpoint between the wrist crease and elbow crease, elbow -medial to the brachial artery), ulnar (wrist crease, mid-forearm - at the midpoint between the medial epicondyle and ulnar styloid, elbow-at the ulnar groove). Upper limb nerves were studied with the patient in the supine position, and the arm abducted to 60° at the shoulder and forearm in the supine position. Power and color doppler modes were used. The inner border of the thin hyperechoic epineural rim was traced using the trace function.
MRI (Magnetic Resonance Imaging) of the Brachial plexus was performed on 3T PHILIPS INGENIA scanner with the addition of a 3D NERVE VIEW sequence (TR/TE:2200/170 ms, TI:250 ms, acquisition voxelsize: 0.6×0.5×1.1 mm, Recon Voxel: 0.6×0.6×1.00 mm, Slice thickness: 1.1 mm). It employs a Motion-sensitive driven equilibrium (MSDE) PREPPED 3D sampling perfection with application optimized contrasts using different flip angle evolution (SPACE) scheme for short tau inversion recovery (STIR) contrast (3D NERVE VIEW SEQUENCE, PHILIPS) to achieve optimal MR neurography (MRN). 8-mm-thick slabs of Maximal Intensity Projection (MIP) were reconstructed from the 3D data set.
Mutational analysis
Genetic testing was performed by whole exome sequencing (WES) using Exome Research Panel (Integrated DNA technologies, USA). DNA was extracted from whole blood samples collected from proband, unaffected sister and mother. Sequencing was performed on Illumina sequencing platform with a mean coverage of >50×. Raw sequences were aligned to human reference genome (GRCh37/hg19) using BWA program followed by variant identification using Picard and GATK version 3.6 [9, 10]. Variant annotation was performed using VEP program against the Ensembl release 87 human gene model [11]. Pathogenic / likely pathogenic variants are classified as per American College of Medical Genetics and Genomics (ACMG) criteria [12].
Histopathological examination
The left superficial radial nerve biopsy was fixed in 2.5% glutaraldehyde. Paraffin embedded sections stained with hematoxylin and eosin, and resin embedded semithin sections stained with 1% toluidine blue were studied by light microscopy. Uranyl acetate and lead citrate stained ultrathin sections were subjected to electron microscopy.
Clinical presentation
The patient is a 23-year-old lady evaluated in February 2017 during her postpartum period. She presented at the emergency services with rapidly progressive flaccid quadriparesis. She was born by normal delivery as the first child of non-consanguineous parents. She had bilateral foot deformities (pes cavus and hammer toes) since early childhood. She first noticed bilateral limb weakness during the 7th month of gestation which continued during the postpartum period of the 1st pregnancy at 21 years of age. The weakness improved spontaneously over 5 months. During her second pregnancy at the age of 23 years, she similarly developed worsening of the existing limb weakness and could not rise from the floor. Post-delivery the weakness worsened rapidly and she had flaccid quadriparesis and was evaluated at the emergency services at the end of two months after delivery. She required one person’s support to walk. At the end of 2 months of the onset, she developed distal weakness in all limbs in the form of finger and toe grip weakness. The patient was started on oral steroids (Prednisolone of 30 mg per day) and there was a significant improvement in distal muscle power and she attained independent ambulation. After six months of initial presentation, she complained of squeezing pain in the right shoulder radiating to the hand which subsequently progressed to the other limb as well. The pain was worsened by shoulder movements and relieved by steroids and pain management with prednisolone (30 mg/day) and a combination of gabapentin and amitriptyline (300 mg/day).
The pedigree of the patient is shown in Fig. 1.1. The patient’s mother was born deaf and dumb, and has bilateral profound hearing loss and speech impairment and further examination revealed short stubby fingers, high-arched feet, and coarse facies. A family history of deafness and speech impairment is present in three maternal aunts (Fig. 1.1). The patient’s father has hand and foot deformities since adolescence, but there is no definite or minimal progressive illness and capable of doing all activities of daily living (Fig. 1.2). The cranial nerves and proximal muscles were normal. Severe weakness of the small muscles of hands and feet present. Tendon reflexes were absent. Pain and touch and temperature sensations were impaired in glove and stocking distribution. He refused further evaluation. The younger sister of the proband has normal development and showed no signs of neuropathy.
Clinical examination at 23 years of age revealed a short-statured woman with bilateral hammer toes, ankle contractures, and mild weakness of facial muscles. Weakness of proximal upper and lower limbs (MRC [Medical Research Council] scale = 3) along with mild weakness in the intrinsic hand and foot muscles (MRC scale = 4), global areflexia, loss of superficial sensation (touch, pain, and temperature) in lower limbs in stocking distribution; impaired vibration sense in hands and toes were noted. At the last follow-up, i.e. six years (2023) after the initial presentation, her symptoms remain stable except for occasional pain and moderate proximal limb girdle weakness.
Fig. 1.1
The pedigree of the present proband with the FGD4 gene mutation is illustrated.
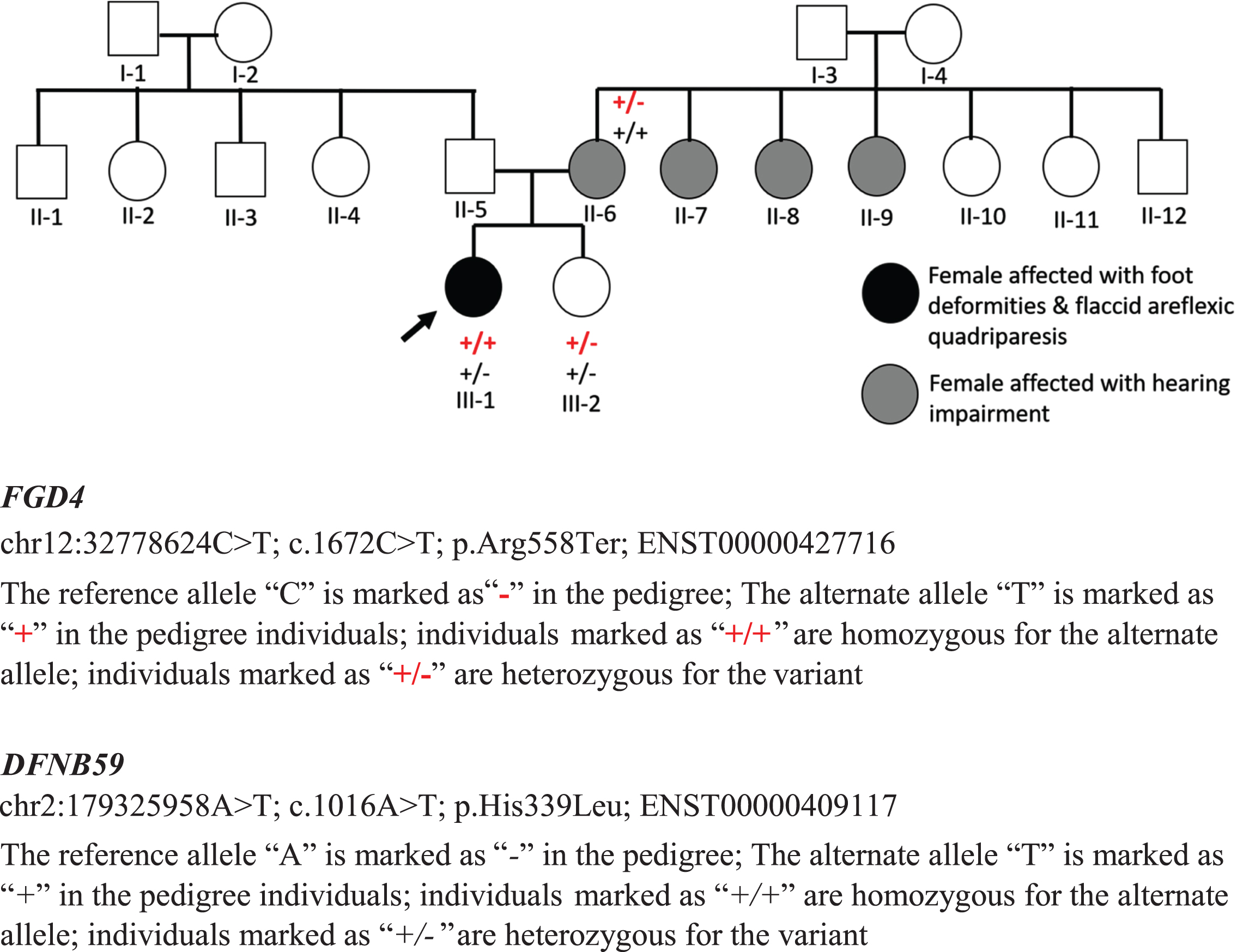
Fig. 1.2
Father aged 64 years developed hand and foot deformities from adolescence which has been minimally to non-progressive. The figures show evidence of severe wasting of the hands and feet with deformities.
Investigations showed normal creatine kinase level (CK). Nerve conduction studies demonstrated absent CMAPs (compound muscle action potentials) in bilateral ulnar and common peroneal nerves; increased distal latencies and amplitudes in the right median nerve (Table 1). SNAPs (sensory nerve action potentials) could not be elicited in upper and lower limb nerves.
Table 1
Nerve conduction studies
| CMAP | ||||
| Nerve | DL (ms) | Amplitude (μV) | Distance (distal/proximal) (mm) | NCV (m/s) |
| Rt. Median at wrist | 17.10 | 273 | 60/220 | 7.0 |
| Rt. Median at elbow | 45.10 | 90 | 200 | NR |
| Ulnar | NR | NR | NR | NR |
| Common peroneal | NR | NR | NR | NR |
CMAP, Compound motor action potential; DL, Distal latency; ms, milliseconds; μV, microvolts; mm, millimeter; NCV, Nerve conduction velocity; m/s, meters per second; NR, Non-recordable. SNAPS were not elicitable in the median, ulnar, and sural nerves.
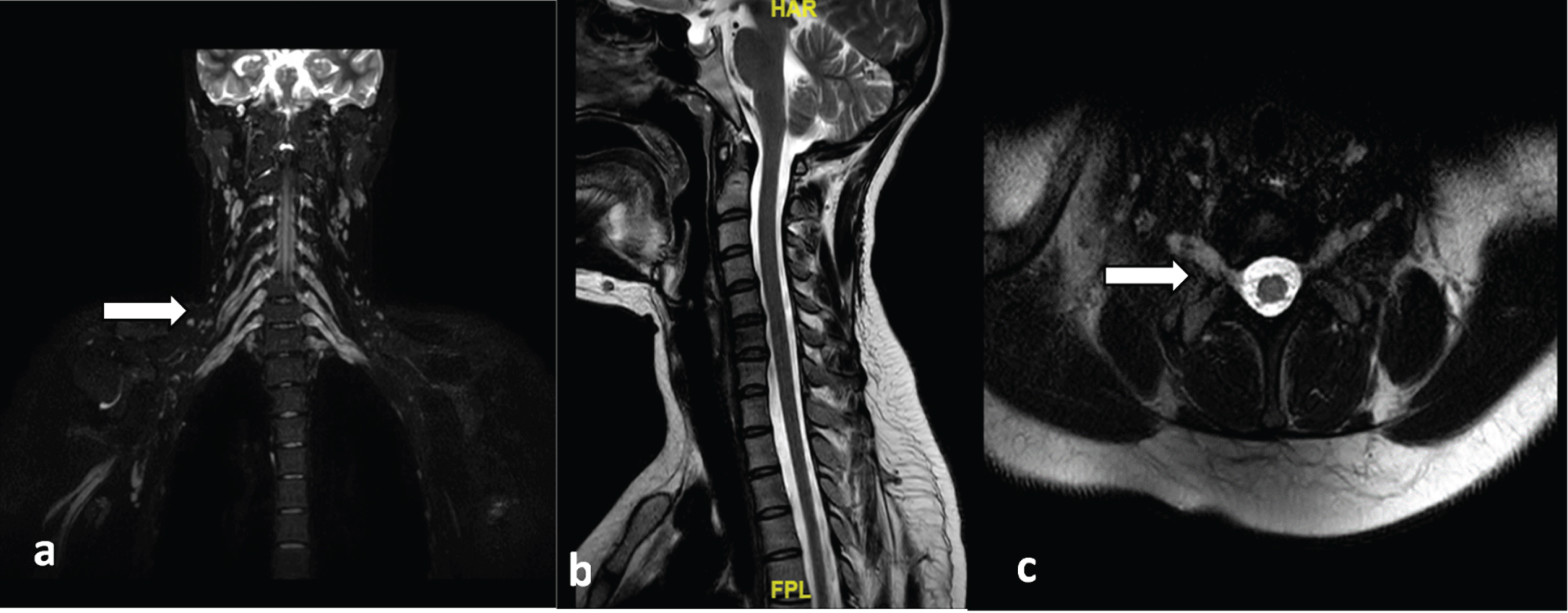
Ultrasound of peripheral nerves showed the CSA of median and ulnar nerves were increased throughout their course (Fig. 2). MRI of the Brachial plexus showed symmetric thickening with T2 hyperintense signal of the roots, trunks, divisions, and cords of bilateral brachial plexus (Fig. 3).
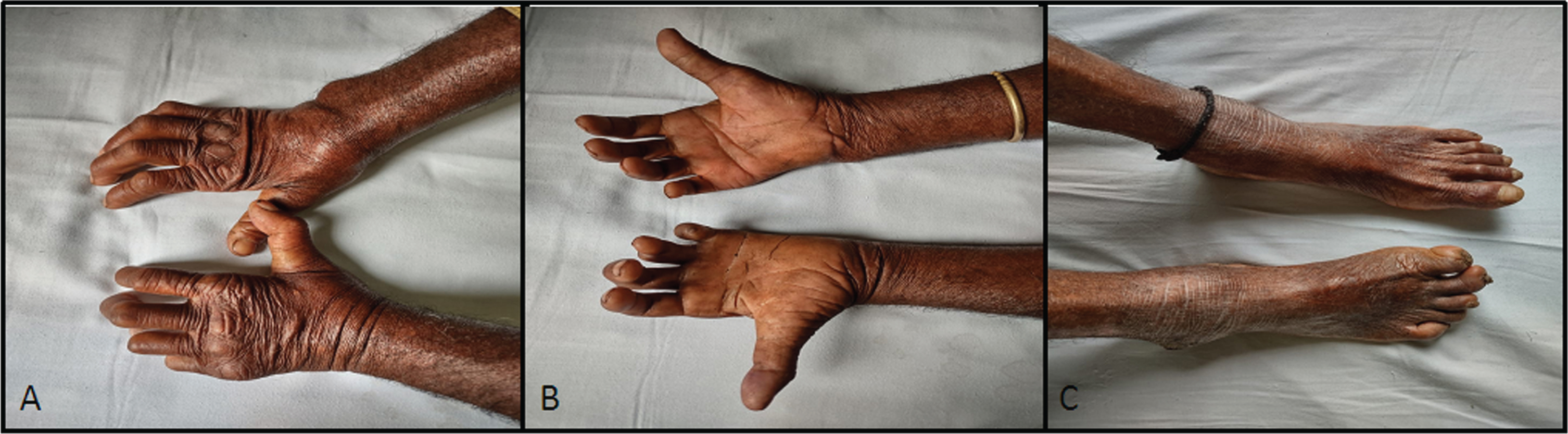
Fig. 2
Ultrasound cross-section images of the median and ulnar nerves. Fig 2.1 (a) Right Median nerve at the wrist (area = 5.9 mm2), (b) Right Median nerve at mid-forearm between the flexor digitorum superficialis and flexor digitorum profundus (area = 17.8 mm2), (c) Right Median nerve at the elbow medial to the brachial artery (area = 15.6 mm2), (d) Right Brachial plexus upper (area = 14.2 mm2), middle (area = 16.9 mm2), and lower truck (area = 25.7 mm2).
Exome sequencing revealed a homozygous nonsense variation c.1672C>T (p.Arg558Ter) affecting exon 14 of the FGD4 gene (Refseq transcript ID: NM_139241.3) in the proband resulting in premature truncation of the protein with loss of distal FYVE and 2nd Pleckstrin homology (PH2) domains. The p.Arg558Ter has been reported as pathogenic in Clinvar (Clinvar ID: 1402656). On segregation analysis, the observed variant was detected in a heterozygous state in the mother and sister who did not have a CMT phenotype. A novel homozygous missense variant, c.1016A>T (p.His339Leu) in exon 7 of the DFNB59 (PJVK) gene (Refseq ID: NM_001042702.5) was found in the proband’s mother affected with hearing loss, which segregated as, heterozygous in the proband and the unaffected sister.
Histopathology showed significant nerve fiber loss, several thinly myelinated fibers, and poorly formed onion bulbs (concentric Schwann cell hyperplasia) indicating a chronic demyelinating process. Several fibers showed abnormal focally folded myelin consisting of thick myelin folds forming redundant outfoldings and inpouchings (Fig. 4).
Discussion
CMT4 is an autosomal recessive form of CMT and mutation in the FGD4 gene is implicated as the cause of this disease. FGD4 encodes a protein called frabin, which belongs to the family of Rho guanine nucleotide exchange factors (RhoGEFs) that activate Rho GTPases, such as Cdc42 and Rac1, by catalyzing the exchange of GDP for GTP [5, 7, 8, 13]. RhoGT-Pases are key regulators of the actin cytoskeleton, which is essential for various cellular processes, such as cell shape, migration, polarity, and adhesion [14]. Frabin is mainly expressed in the nervous system and plays a role in neuronal development and function, such as axon guidance, dendritic spine formation, synaptic plasticity, and neurotransmitter release [15]. Mutations in FGD4 cause CMT4H by disrupting the normal function of frabin and impairing the signaling pathways that regulate the actin cytoskeleton and myelination in Schwann cells [16].
Table 2 presents a summary of Clinical, genetic and histopathological details of previously reported cases. Horn et al., found that frabin plays an important role in myelin development as well as maintenance [6]. They found that disruption in this gene and the subsequent protein cascade can cause myelin abnormalities as soon as the fifth day of postnatal life in animal models. This explains the early onset of neurological symptoms in our patient similar to the other reported studies [16–19]. Our patient had foot deformities since childhood but the other typical neurological features didn’t develop until later in adulthood. The proband had both motor and sensory involvement which was demonstrated by the electrophysiological studies. Usually, CMT4H is characterized by distal weakness at the onset of symptoms, but in contrast, our patient presented with proximo-distal weakness in the postpartum period mimicking an inflammatory demyelinating process [17, 20]. Interestingly, our patient developed these symptoms in the postpartum period of both of her pregnancies. The reason for this phenomenon is unclear, but it may be related to hormonal or immunological changes that occur during pregnancy and postpartum. Previous studies have suggested that estrogen may have a protective effect on peripheral nerve function and myelination, while progesterone may have a detrimental effect [21, 22]. Moreover, pregnancy and postpartum may alter the immune system and trigger autoimmune reactions that could affect nerve integrity [23]. Further investigations are needed to elucidate the role of hormonal and immunological factors in modulating the disease course of CMT4H. Moreover, the present proband had a mild to moderate clinical course as opposed to the first reported cases where they had severe neurological features with deformities, and for this reason, it was thought to be a more severe form compared to other autosomal recessive CMTs [7, 8, 24]. Therefore, our report further widens the clinical spectrum of CMT4H.
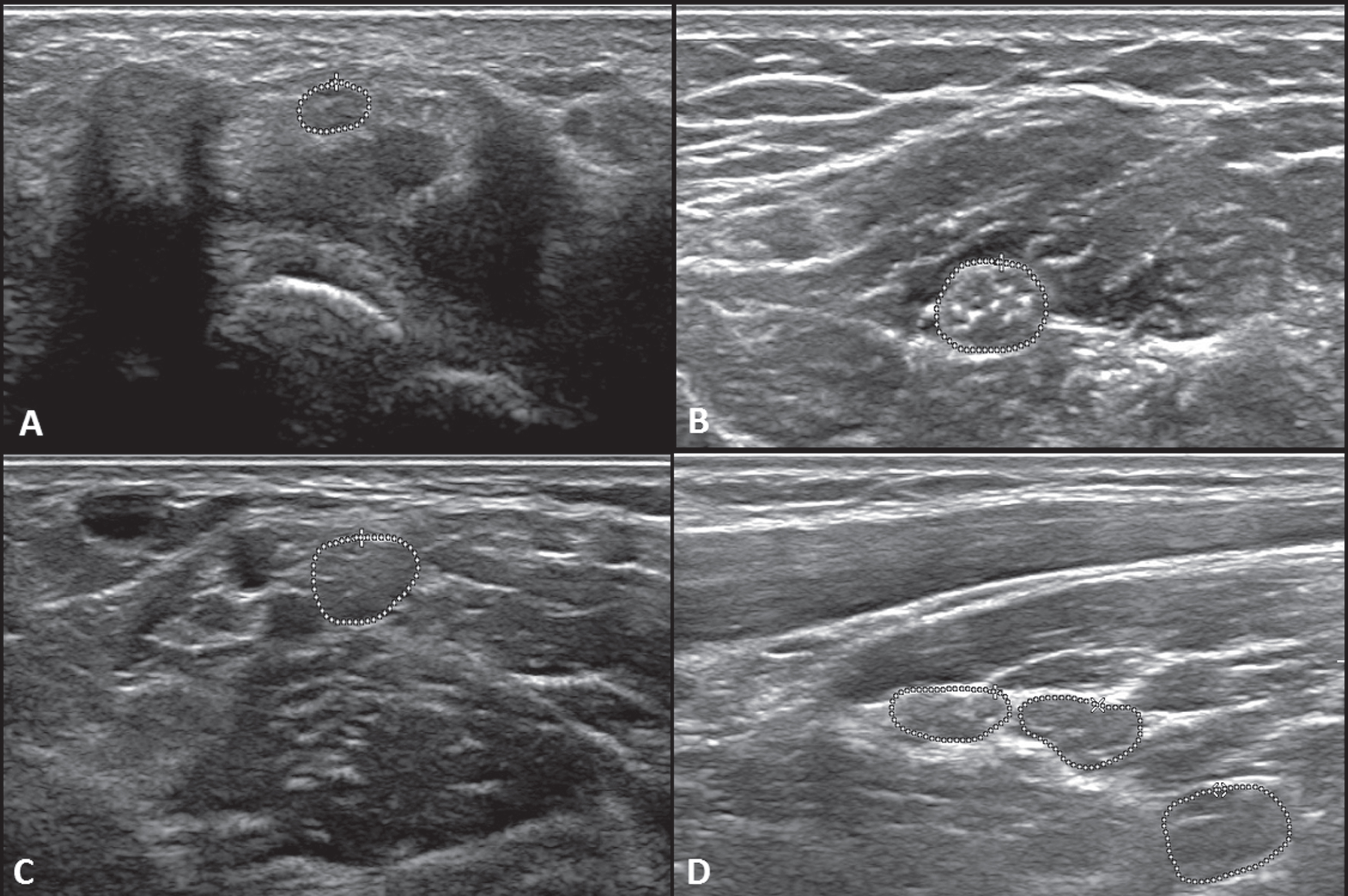
Fig. 2.2
(a) Left Median nerve at the wrist(area = 8.6 mm2), (b) left Median nerve at mid-forearm between the flexor digitorum superficialis and flexor digitorum profundus (area = 18.2 mm2), (c) Left Median nerve at the elbow medial to the brachial artery (area = 20.8 mm2), (d) Left Brachial plexus upper (area = 16.1 mm2), middle (area = 6.8 mm2), and lower trunk (area = 8.0 mm2).
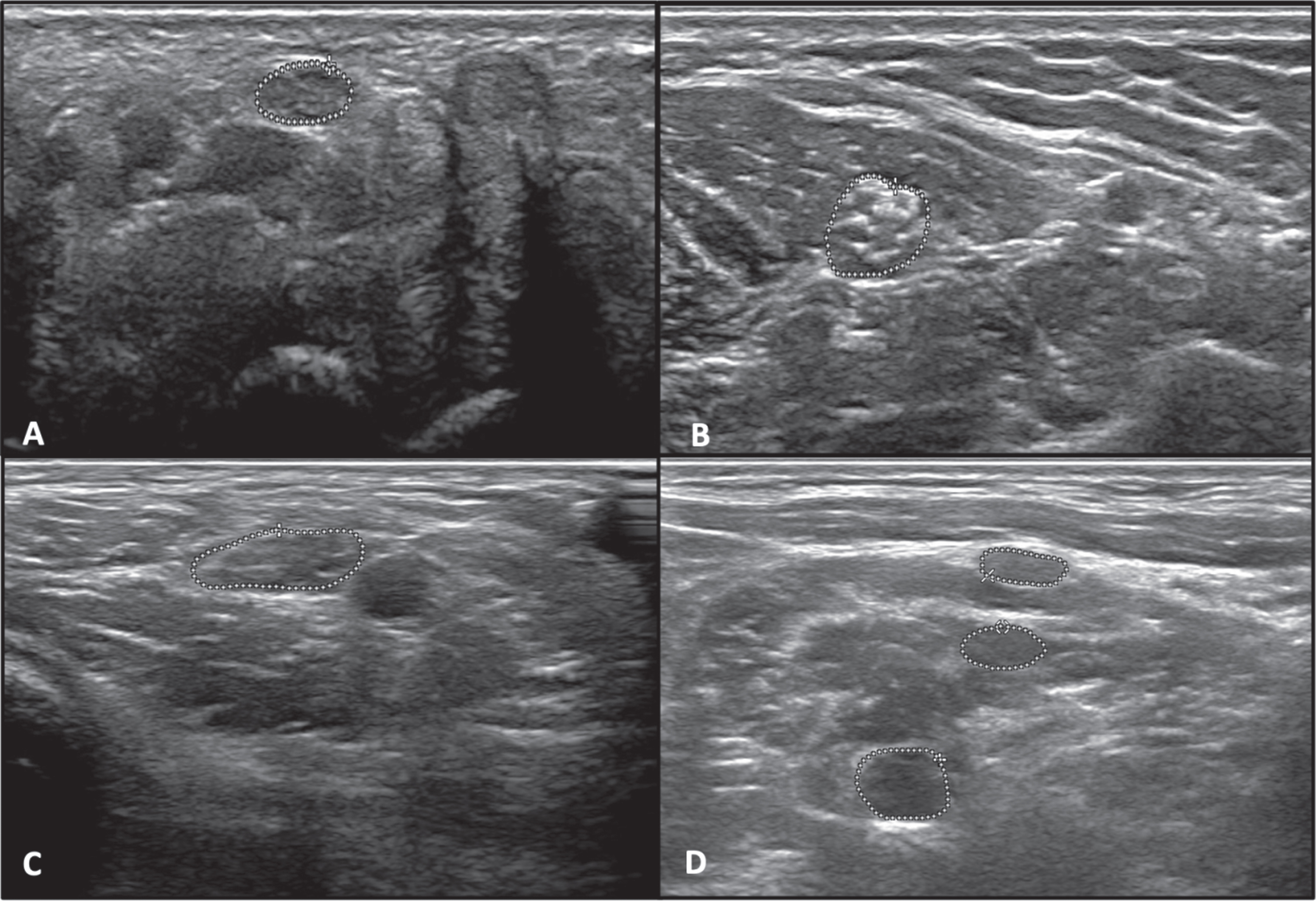
Fig. 2.3
(a) Right Ulnar nerve at mid arm (area = 13.9 mm2), (b) Right Ulnar nerve at the elbow medial to the brachial artery (area = 17.4 mm2).
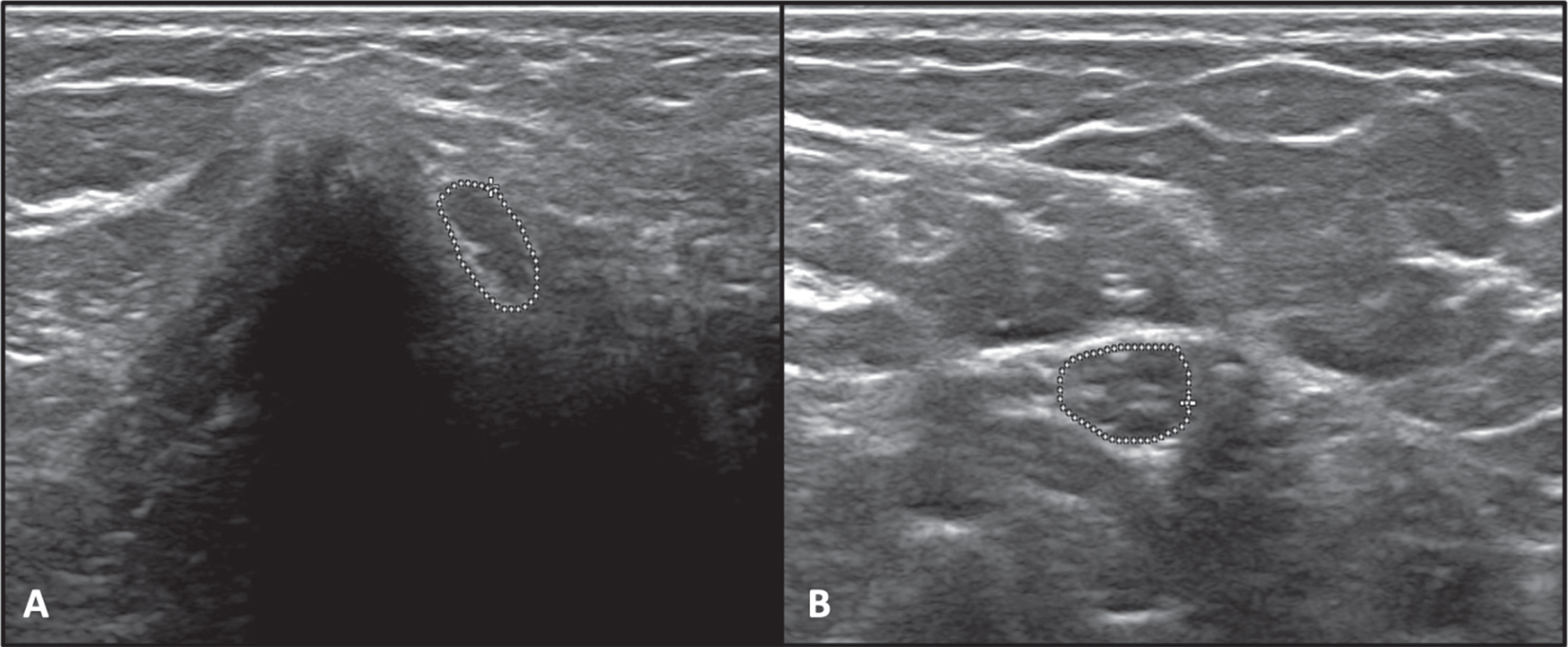
Limited studies are available on nerve ultrasound in inherited neuropathies. In CMT1A, and MPZ- MPZ-associated CMT, uniformly increased CSAs were found whereas, it was not increased in CMT due to NEFL-associated CMT [25]. Focal enlargements have been described in HNPP (Hereditary neuropathy with pressure palsies) [26]. The ultrasonography of our patient showed that the CSAs of median and ulnar nerves were increased throughout their course, suggesting diffuse nerve enlargement. The normal values for median and ulnar nerve CSAs vary depending on the anatomical site, age, sex, height, and weight of the subjects [27]. According to a recent study by Sindhu et al., the mean CSAs of median and ulnar nerves in the Indian population are 7.42±1.39 mm and 4.43±0.79 mm at the wrist, 5.1±0.91 mm and 4.52±0.83 mm at the forearm, 7.77±1.6 mm and 5.64±1.26 mm at the elbow, and 5.9±0.9 mm and 4.3±0.5 mm at the upper arm, respectively [27]. We could not find any previous studies on the USG of nerves in CMT4H.
Fig. 3
MRI of the Brachial plexus. (a,c) Near symmetric thickening with T2 hyperintense signal of the roots, trunks, divisions, and cords of the bilateral brachial plexii (arrows), (b) T2W image of the sagittal section showing normal spinal cord. (MRI, Magnetic resonance imaging).
Whole-exome analysis by next-generation sequencing has been successful in determining the genes causing various CMT types [28, 29]. Over 30 FGD4 mutations causing CMT4H have been reported until now that resulted in truncated or absent protein (missense, nonsense, frameshift, or splicing mutations) [8, 17, 20, 30]. We identified a homozygous nonsense mutation in FGD4, c.1672C>T (p.Arg558Ter) that has not been published in literature before but classified in Clinvar as pathogenic based on a single submission (https://www.ncbi.nlm.nih.gov/clinvar/variation/1402656/). Recessive loss of function mutations including nonsense and frameshift variants in FGD4 have been previously reported in CMT4H patients [16, 30]. Such nonsense and frameshift variants are often expected to result in nonsense mediated decay (NMD), a cellular surveillance mechanism that degrades mRNAs leading to complete loss of protein especially when premature termination codon (PTC) is located significantly upstream to the last exon [31]. The current variant p.Arg558Ter introducing PTC in exon 14 out of 17 exons of FGD4 is thus expected to lead to complete loss of protein due to NMD. Truncating mutations distal to the current variant in FGD4 have been previously reported to cause a severe phenotype [16]. However, Houlden et al reported a milder CMT4H phenotype in a patient with a proximal nonsense mutation p.Arg275Ter which was shown to escape NMD as evidenced by the presence of residual mRNA expression indicating a non-correlation between location of truncation and severity of phenotype in FGD4 patients [20]. Our findings further support this hypothesis and indicate that NMD efficiency may be influenced by factors other than the location of the mutation relative to the last exon-exon junction. For instance, NMD can be modulated by cellular stress, microenvironmental cues, and alternative splicing events [31, 32]. Further studies are needed to elucidate the exact molecular mechanisms underlying NMD regulation and its impact on FGD4 expression and function.
On segregation analysis, we found that the patient’s mother and sister were heterozygous carriers of the mutation. Since the father was not tested further, it is difficult to determine whether his hand and foot deformities can be attributed to FGD4 or if other causative variants were present. The clinical manifestations of CMT4H are highly variable, even among individuals with the same mutation [17]. Some factors that may contribute to this variability are the quality of the genotype, such as the presence of modifier genes or the epigenetic modification, such as the methylation status or alternative splicing of FGD4; the environmental influence, such as exposure to toxins or infections that may affect nerve function; and the mosaicism, such as the presence of somatic or germline mutations that may alter the expression or activity of FGD4 [7, 33].
Table 2
Clinical, genetic and histopathological features of previously reported cases
| Parameter | Current report | Delague et al. 2007 [8] | Houlden et al. 2009 [20] | Fabrizi et al. 2009 [17] | Baudot et al. 2012 [38] | Boubaker et al. 2013 [24] | Arai et al. [18] | Hyun et al. 2015 [39] | Zis et al. 2017 [40] | Kondo et al. [19] | Argente et al. [30] | Ouyang et al. [41] | Aoki et al. 2021 [42] | Zhan et al. 2021 [43] |
| Origin | Indian | Lebanese and Algerian | Irish | Italian | Lebanese and Algerian | Tunisian | Japanese | Korean | Caucasian | Japanese | Spanish | Chinese | Japanese | Chinese |
| Age at onset (years) | Childhood | 1 to 2 | Childhood | <1 year | 3 to 5 | 3 | 4 | 6 to 8 | 8 | 15 | 11 to 19 | 8 | 7 | 11 |
| Sex | F | M | F | F | F and M | F:M = 2:1 | F | M | F | M | M, F | F | F, M (siblings) | M |
| Consan-guinity | Sporadic | Consan-guineous | Consan-guineous | Consan-guineous | Consan-guineous | Consan-guineous | Sporadic | Sporadic | Sporadic | Consan-guineous | Sporadic | Consan-guineous | Consangui-neous | Sporadic |
| Motor symptoms | B/L proximal upper and lower limb weakness | Delayed motor milestones and gait unsteadiness with scoliosis | Distal muscle weakness and wasting | Distal muscle weakness and wasting in lower limbs | Distal muscle weakness and wasting | Wasting of upper and lower limb | Distal left leg weakness | Distal muscle weakness; proximal lower limbweakness | Distal muscle weakness of upper limbs | Distal muscle weakness and wasting | Reduced heel walking | Foot deformities, facial weakness | Distal muscle weakness and wasting | Toe walking, gait disturbance, falls. |
| Sensory symptoms | Sensory loss in lower limbs | Stocking type hypoesthesia | Distal hypoesthesia | Distal hypoesthesia | – | Distal hypoesthesia | Glove and stocking-type sensory loss in both legs | Sensory loss in lower limbs | Severe sensory loss in upper and lower limbs | Sensory loss in upper and lower limbs | None | Numbness of limbs | Distal hypoesthesia | None |
| Scoliosis | No | Yes | No | Yes | No | Yes | No | Yes | Yes | Yes | No | No | Yes | No |
| Foot deformities | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes |
| Mutation | c.1672C >T p.Arg558Ter Nonsense | c.893 T >G p.Met298fs*8; c.893 T >C p.Met298Thr Missense, Splicing | c.823 C>T p.Arg275 Nonsense | c.1762–2 A>G p.Tyr587fs*14 Splicing | c.1698 G >A p.Met566Ile; c.1325 G >A p.Arg442 His Missense | c.514_515insG p.Ala172 Glyfs*7 insertion | c.8371G >A p.Glu280 Lysfs*23 Splicing | c.1512-2A >C (p.Arg468Gln); c.2043 + 1G >A (p.Met345Thr) Missense | c.1192-48_1233del p.(?); c.1304_1305 delins AA p(Arg435Gln) Frameshift, Missense | c.724C>T (p.Arg242X) Splicing | c.514delG p.Ala172Glnfs*28; c.2211dupA p.Ala738Serfs*5 Frameshift | c.1303C>T (p.R435W) Missense | c.1730G >A (p.Arg577Gln) Missense | c.1688C >A (p.T563K) and c.1951C>T (p.Q651X) Nonsense |
| Nerve biopsy | Demyeli-nation with onion bulbs and myelin outfoldings | Demyelination with onion bulbs and myelin out foldings | Not done | Demyelination with onion bulbs and myelin out foldings | Not done | Demyelination with onion bulbs and myelin outfoldings | Demyelination with onion bulbs and myelin out foldings | Demyelination with onion bulbs and myelin out foldings | Not done | Not done | Not done | Not done | Demyelination with myelin outfoldings | Demyelination with myelin outfoldings |
(M, Male; F, Female).
Fig. 4
Nerve biopsy: (A) and (B) Semithin sections of resin embedded tissue stained with toluidine blue show severe and fairly uniform depletion of myelinated fibers, thin myelin sheaths, occasional onion bulbs, and fibers with abnormal myelin folds (A ×200, B ×400). (C) and (D) Electron micrography- Ultrathin sections stained with uranyl acetate and lead citrate reveal thinly myelinated fibers with Schwann cell lamellae (arrow) indicating early onion bulb formation [C ×2000; D, ×4000]. (E) and (F) Fibers with numerous thick abnormal myelin folds forming redundant outpouchings and infoldings [E & F, ×2000].
![Nerve biopsy: (A) and (B) Semithin sections of resin embedded tissue stained with toluidine blue show severe and fairly uniform depletion of myelinated fibers, thin myelin sheaths, occasional onion bulbs, and fibers with abnormal myelin folds (A ×200, B ×400). (C) and (D) Electron micrography- Ultrathin sections stained with uranyl acetate and lead citrate reveal thinly myelinated fibers with Schwann cell lamellae (arrow) indicating early onion bulb formation [C ×2000; D, ×4000]. (E) and (F) Fibers with numerous thick abnormal myelin folds forming redundant outpouchings and infoldings [E & F, ×2000].](https://ip.ios.semcs.net:443/media/jnd/2024/11-1/jnd-11-1-jnd230042/jnd-11-jnd230042-g004.jpg)
The exome analysis also showed a homozygous DFNB59 variant c.1016A>T (p.His339Leu) in the proband’s mother who has profound hearing loss with speech impairment since childhood, but further testing was not done. Biallelic mutations in DFNB59 or pejvakin, have been reported in patients with autosomal recessive non-syndromic hearing loss [34]. The onset of DFNB59-associated hearing loss is often prelingual in childhood with severity ranging from moderate to profound impairment [35]. In the present family, a history of hearing loss has been reported in maternal family (Fig. 1.1). However, the proband and unaffected sister carried only a heterozygous DFNB59 c.1016A>T variant and did not have any hearing impairment. Furthermore, other maternal family members with hearing loss were unavailable for evaluation and performing a segregation study. Based on the genotypes observed and segregation of both variants, we presume that the phenotypes in the proband and mother are unrelated and can be explained by FGD4 and DFNB59 mutations respectively.
Superficial Radial nerve biopsy in our patient displayed prominent demyelination and abnormal focally folded myelin. Myelin outfolding is the characteristic feature of this disease and has been reported previously in other studies of CMT4H [7, 8, 16, 17]. The formation and preservation of myelin structure are dependent on cdc42 signaling in the Schwann cells of peripheral nerves. As FGD4 gene mutations disrupt this signaling pathway, these abnormal myelin outfoldings develop [36]. This pathological finding is also detected in other CMT4 subtypes such as CMT4B1 and CMT4B2 caused by MTMR and SBF2 mutations respectively [36, 37].
To conclude, we report a novel mutation in the FDG4 gene for the first time in an Indian patient. Our patient had milder features compared to previously reported studies broadening the CMT4 phenotype. The postpartum worsening of weakness in the form of flaccid areflexic quadriparesis significantly improved with steroid therapy, which is an interesting observation. The current report expands the phenotypic and genotypic spectrum of CMT4H caused by FGD4 mutations.
ACKNOWLEDGMENTS
We would like to thank the patient and their family for participating in this study.
FUNDING
The authors received no financial support for this article’s research, authorship, and/or publication.
Institutional research support from National Institute of Mental Health And Neurosciences (NIMHANS), Ministry of Health and Family Welfare, Government of India.
CONFLICTS OF INTEREST
Authors do not have any conflict of interests.
DISCLOSURES
Kiran Polavarapu is supported by Canadian Institutes of Health Research (CIHR) postdoctoral fellowship award grant no: 202210MFE-491707-FPP-CECC-404816.
Mainak Bardhan was supported by Indian Council of Medical Research’s Grant in Aid towards the National Registry project entitled “National Registry for Rare and other Inherited Disorders(NRROID)”, Ministry of Health and Family Welfare,Government of India.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
[1] | Bird TD . Charcot-Marie-Tooth (CMT) hereditary neuropathy overview 1. Clinical characteristics of Charcot-Marie-Tooth (CMT) hereditary neuropathy. Gene Reviews. ((2020) )1–22. |
[2] | Barreto LCLS , Oliveira FS , Nunes PS , de França Costa IMP , Garcez CA , Goes GM , et al. Epidemiologic study of Charcot-Marie-Tooth disease: A systematic review. Neuroepidemiology. ((2016) ) 46: (3), 157–65. |
[3] | Pareyson D , Marchesi C . Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. ((2009) ) 8: (7), 654–67. |
[4] | Vallat JM , Mathis S , Funalot B . The various Charcot– Marie– Tooth diseases. Curr Opin Neurol. ((2013) ) 26: (5), 473–80. |
[5] | Nakanishi H , Takai Y . Frabin and other related Cdc42-specific guanine nucleotide exchange factors couple the actin cytoskeleton with the plasma membrane. J Cell Mol Med. ((2008) ) 12: (4), 1169–76. |
[6] | Horn M , Baumann R , Pereira JA , Sidiropoulos PNM , Somandin C , Welzl H , et al. Myelin is dependent on the Charcot-Marie-Tooth Type 4H disease culprit protein FRABIN/FGD4 in Schwann cells. Brain. ((2012) ) 135: (Pt 12), 3567–83. |
[7] | De Sandre-Giovannoli A , Delague V , Hamadouche T , Chaouch M , Krahn M , Boccaccio I , et al. Homozygosity mapping of autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (CMT4H) to a novel locus on chromosome 12p11.21-q13.11. Journal of Medical Genetics. ((2005) ) 42: , 260–5. |
[8] | Delague V , Jacquier A , Hamadouche T , Poitelon Y , Baudot C , Boccaccio I , et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet. ((2007) ) 81: (1), 1–16. |
[9] | Li H , Handsaker B , Wysoker A , Fennell T , Ruan J , Homer N , et al. The sequence alignment/Map format and SAMtools. Bioinformatics. ((2009) ) 25: (16), 2078–9. |
[10] | McKenna A , Hanna M , Banks E , Sivachenko A , Cibulskis K , Kernytsky A , et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. ((2010) ) 20: (9), 1297–303. |
[11] | McLaren W , Pritchard B , Rios D , Chen Y , Flicek P , Cunningham F . Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. ((2010) ) 26: (16), 2069–70. |
[12] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. ((2015) ) 17: (5), 405–24. |
[13] | Obaishi H , Nakanishi H , Mandai K , Satoh K , Satoh A , Takahashi K , et al. Frabin, a novel FGD1-related actin filament-binding protein capable of changing cell shape and activating c-Jun N-terminal kinase. Journal of Biological Chemistry. ((1998) ) 273: (30), 18697–700. |
[14] | Ridley AJ . Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. ((2006) ) 16: (10), 522–9. |
[15] | Ikeda W , Nakanishi H , Miyoshi J , Mandai K , Ishizaki H , Tanaka M , et al. Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. Journal of Cell Biology. ((1999) ) 146: (5), 1117–32. |
[16] | Stendel C , Roos A , Deconinck T , Pereira J , Castagner F , Niemann A , et al. Peripheral nerve demyelination caused by a mutant rho GTPase guanine nucleotide exchange factor, frabin/FGD4. The American Journal of Human Genetics. ((2007) ) 81: (1), 158–64. |
[17] | Fabrizi GM , Taioli F , Cavallaro T , Ferrari S , Bertolasi L , Casarotto M , et al. Further evidence that mutations in FGD4/frabin cause Charcot-Marie-Tooth disease type 4H. Neurology. ((2009) ) 72: (13), 1160–4. |
[18] | Arai H , Hayashi M , Hayasaka K , Kanda T , Tanabe Y . The first Japanese case of Charcot-Marie-Tooth disease type 4H with a novel FGD4 c. 837-1G>A mutation. Neuromuscul Disord. ((2013) ) 23: (8), 652–5. |
[19] | Kondo D , Shinoda K , Yamashita KI , Yamasaki R , Hashiguchi A , Takashima H , et al. A novel mutation in FGD4 causes Charcot-Marie-Tooth disease type 4H with cranial nerve involvement. Neuromuscul Disord. ((2017) ) 27: (10), 959–61. |
[20] | Houlden H , Hammans S , Katifi H , Reilly MM . A novel Frabin (FGD4) nonsense mutation R275X associated with phenotypic variability in CMT4H. Neurology. ((2009) ) . Feb;72: (7), 617–20. |
[21] | Koenig HL , Schumacher M , Ferzaz B , Thi AN Do , Ressouches A , Guennoun R , et al. Progesterone synthesis and myelin formation by schwann cells. Science. ((1979) ).1995;268: (5216);1500–3. |
[22] | Schumacher M , Hussain R , Gago N , Oudinet JP , Mattern C , Ghoumari AM . Progesterone synthesis in the nervous system: Implications for myelination and myelin repair. Front Neurosci. ((2012) )6. |
[23] | Hughes GC . Progesterone and autoimmune disease. Autoimmun Rev. ((2012) ) 11: (6–7), A502–14. |
[24] | Boubaker C , Hsairi-Guidara I , Castro C , Ayadi I , Boyer A , Kerkeni E , et al. A novel mutation in FGD4/FRABIN causes Charcot Marie Tooth disease type 4H in patients from a consanguineous Tunisian family. Ann Hum Genet. ((2013) ) 77: (4), 336–43. |
[25] | Niu J , Cui L , Liu M . Multiple sites ultrasonography of peripheral nerves in differentiating Charcot– Marie– Tooth type 1A from chronic inflammatory demyelinating polyradiculoneuropathy. Front Neurol. ((2017) )8. |
[26] | Noto Y ichi , Shiga K , Tsuji Y , Mizuta I , Higuchi Y , Hashiguchi A , et al. Nerve ultrasound depicts peripheral nerve enlargement in patients with genetically distinct Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry [Internet]. ((2015) ) 86: (4) 378–84. Available from: https://jnnbmj.com/content/86/4/378. |
[27] | Sindhu D , Huddar A , Saini J , Vengalil S , Nashi S , Bardhan M , et al. Cross-sectional area reference values of nerves in the upper and lower extremities using ultrasonography in the Indian population. Ann Indian Acad Neurol. ((2022) ) 25: (3), 449. |
[28] | Choi BO , Koo SK , Park MH , Rhee H , Yang SJ , Choi KG , et al. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum Mutat. ((2012) ) 33: (11), 1610–5. |
[29] | Pipis M , Rossor AM , Laura M , Reilly MM . Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat Rev Neurol. ((2019) ) 15: (11), 644–56. |
[30] | Argente-Escrig H , Sánchez-Monteagudo A , Frasquet M , Millet-Sancho E , Martínez-Rubio MD , Pitarch I , et al. A very mild phenotype of Charcot-Marie-Tooth disease type 4H caused by two novel mutations in FGD4. J Neurol Sci. ((2019) ) 402: , 156–61. |
[31] | Kurosaki T , Maquat LE . Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci [Internet]. ((2016) ) 129: (3)461–7. Available from: https://doi.org/10.1242/jcs.181008. |
[32] | Lewis BP , Green RE , Brenner SE . Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proceedings of the National Academy of Sciences [Internet]. ((2003) ) 100: (1)189–92. Available from: https://doi.org/10.1073/pnas.0136770100. |
[33] | Bongiorno R , Colombo MP , Lecis D . Deciphering the nonsense-mediated mRNA decay pathway to identify cancer cell vulnerabilities for effective cancer therapy. Journal of Experimental & Clinical Cancer Research [Internet]. ((2021) ) 40: (1)376. Available from: https://org/10.1186/s13046-021-02192-2. |
[34] | Delmaghani S , del Castillo FJ , Michel V , Leibovici M , Aghaie A , Ron U , et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. ((2006) ) 38: (7), 770–8. |
[35] | Domínguez-Ruiz M , Rodríguez-Ballesteros M , Gandía M , Gómez-Rosas E , Villamar M , Scimemi P , et al. Novel pathogenic variants in PJVK, the gene encoding pejvakin, in subjects with autosomal recessive non-syndromic hearing impairment and auditory neuropathy spectrum disorder. Genes (Basel). ((2022) ) 13: (1), 149. |
[36] | Laˇsˇsuthov´a P , Vill K , Erdem-Ozdamar S , Schröder JM , Topaloglu H , Horvath R , et al. Novel SBF2 mutations and clinical spectrum of Charcot-Marie-Tooth neuropathy type 4B2. Clin Genet. ((2018) ) 94: (5), 467–72. |
[37] | Pareyson D , Stojkovic T , Reilly MM , Leonard-Louis S , Laurà M , Blake J , et al. A multicenter retrospective study of charcot-marie-tooth disease type 4B (CMT4B) associated with mutations in myotubularin-related proteins (MTMRs). Ann Neurol. ((2019) ) 86: (1), 55–67. |
[38] | Baudot C , Esteve C , Castro C , Poitelon Y , Mas C , Hamadouche T , et al. Two novel missense mutations in FGD4/FRABIN cause Charcot-Marie-Tooth type 4H (CMT4H). J Peripher Nerv Syst. ((2012) ) 17: (2)141–6. |
[39] | Hyun YS , Lee J , Kim HJ , Hong Y Bin , Koo H , Smith AST , et al. Charcot-Marie-Tooth Disease Type 4H Resulting from Compound Heterozygous Mutations in FGD4 from Nonconsanguineous Korean Families. Ann Hum Genet. ((2015) ) 79: (6), 460–9. |
[40] | Zis P , Reilly MM , Rao DG , Tomaselli P , Rossor AM , Hadjivassiliou M . A novel mutation in the FGD4 gene causing Charcot-Marie-Tooth disease. Vol. 22, Journal of the peripheral nervous system: JPNS. ((2017) )224–5. |
[41] | Zhi-Yuan O , You C , Da-Qiang Q , Zhi-Dong C , Xiao-Sheng Z , Fei X , et al. Genetic profile of Chinese patients with Charcot-Marie-Tooth disease. Chin Med J (Engl) [Internet]. ((2020) ) 133: (21)2633–4. Available from: https://doi.org/10.1097/CM9.0000000000001095. |
[42] | Aoki S , Nagashima K , Shibata M , Kasahara H , Fujita Y , Hashiguchi A , et al . Sibling Cases of Charcot-Marie-Tooth Disease Type 4H with a Homozygous FGD4 Mutation and Cauda Equina Thickening. Internal Medicine. ((2021) ) 60: (24), 3975–81. |
[43] | Zhan F , Ni R , Liu T , Tian W , Liu X , Jiang Q . Novel FGD4 Variants and Literature Review of Charcot-Marie-Tooth Disease Type 4H. Ann Clin Case Rep. ((2021) ) 6: , 2052. |


