
Sex Difference in Spinal Muscular Atrophy Patients – are Males More Vulnerable?1
Abstract
Background:
Sex is a significant risk factor in many neurodegenerative disorders. A better understanding of the molecular mechanisms behind sex differences could help develop more targeted therapies that would lead to better outcomes. Untreated spinal muscular atrophy (SMA) is the leading genetic motor disorder causing infant mortality. SMA has a broad spectrum of severity ranging from prenatal death to infant mortality to normal lifespan with some disability. Scattered evidence points to a sex-specific vulnerability in SMA. However, the role of sex as a risk factor in SMA pathology and treatment has received limited attention.
Objective:
Systematically investigate sex differences in the incidence, symptom severity, motor function of patients with different types of SMA, and in the development of SMA1 patients.
Methods:
Aggregated data of SMA patients were obtained from the TREAT-NMD Global SMA Registry and the Cure SMA membership database by data enquiries. Data were analyzed and compared with publicly available standard data and data from published literature.
Results:
The analysis of the aggregated results from the TREAT-NMD dataset revealed that the male/female ratio was correlated to the incidence and prevalence of SMA from different countries; and for SMA patients, more of their male family members were affected by SMA. However, there was no significant difference of sex ratio in the Cure SMA membership dataset. As quantified by the clinician severity scores, symptoms were more severe in males than females in SMA types 2 and 3b. Motor function scores measured higher in females than males in SMA types 1, 3a and 3b. The head circumference was more strongly affected in male SMA type 1 patients.
Conclusions:
The data in certain registry datasets suggest that males may be more vulnerable to SMA than females. The variability observed indicates that more investigation is necessary to fully understand the role of sex differences in SMA epidemiology, and to guide development of more targeted treatments.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disorder, and the most common genetic cause of infant mortality [1]. The majority of SMA cases are caused by the mutation or deletion of the survival motor neuron 1 (SMN1) gene [2], which leads to progressive motor neuron degeneration resulting in progressive muscle weakness [3]. The characteristic symptoms of SMA include hypotonia, muscle atrophy, and weakness of proximal muscles, predominantly affecting the lower extremities [4]. SMA has a broad disease spectrum. It has been classified into five types: 0 –4, according to age of symptom onset and maximum motor milestones achieved. SMA1 and SMA3 have been further divided into subtypes of 1a, 1b, 1c, and 3a, 3b respectively [5]. However, the current demarcation of SMA types is not very strict [6]. The human genome contains the SMN2 gene, an almost identical duplicate of SMN1. A single nucleotide difference in SMN2 alters its splicing pattern causing exclusion of exon 7 in most of the transcripts and resulting in low levels of SMN protein [7]. The severity of the SMA phenotype is correlated with SMN2 copy number, with more copies resulting in a milder phenotype. However, this correlation is not absolute [4].
Recently, the therapeutic landscape for SMA has changed dramatically with the approval by the U.S. Food and Drug Administration of three very costly treatments. Even though all three therapies have an established short-term efficacy in SMA, not all patients respond to the treatments, and the long-term effects among responders are not clear [4, 5]. While promising, these treatments do not represent a cure for SMA [1].
SMN2 is the main modifier for the severity of SMA. But there are also other SMN-dependent and -independent modifiers that influence the phenotype of SMA. SMN-independent modifiers facilitate or counteract disease progression via SMN-downstream pathways. A growing number of proteins have been suggested to modify SMA severity including: plastin (PLS3) [8, 9], neurocalcin delta (NCALD) [10], TIA1 cytotoxic granule associated RNA binding protein (TIA1) [11], ubiquitin like modifier activating enzyme 1 (UBA1) [12], ubiquitin specific peptidase 9 X-linked (USP9X) [13], stathmin-1 (STMN1) [14], myostatin (MSTN) [15] and ZPR1 zinc protein (ZPR1) [16]. Of these modifiers, PLS3, USP9X and UBA1 are encoded by genes on the X chromosome and affect SMA severity in a sex-specific manner [8, 12, 13], indicating that sex may play a role in SMA incidence and severity.
There have been reports of sex difference in SMA patients, but these have not been consistent, nor complete. Pearn (1978) reported a male to female ratio of 2.0 in SMA type 1 patients from Britain, but the sex ratio of cases among family members did not differ significantly from 1.0, and no sex difference was reported in age of onset, or life expectancy [17]. Some studies have indicated the infantile form of SMA is more severe in males [18]. However, other studies have reported no sex differences in sex ratio or symptom severity in SMA types 1 or 2 [18–22]. For the less severe forms of SMA, a male predominance has been reported in most studies, with symptoms reported as milder and the clinical progression course slower in females than males [18, 19, 21, 23–25]. However, a few other studies have not supported this finding [20, 22]. There is a strong correlation between SMN2 copy number and the severity of the SMA phenotype, however no significant difference has been found between the sexes in the number of SMN2 copies [26]. In family case studies, the unaffected or more mildly affected siblings have been reported to be mostly female [25, 26]. These studies indicate a role for sex in the development and severity of SMA pathology.
Sex differences have been demonstrated in brain physiology and behavior during development and adulthood [27–30], and sex is also a significant variable affecting the prevalence and incidence of some neurological disorders [28, 31–33]. Furthermore, sex affects the age-of-onset, progression, disease severity, neuropathology and response to treatment of a number of neurological diseases [28, 30, 34, 35]. Among neurodegenerative disorders, males are more susceptible to Parkinson’s disease [36–38] and motor neuron disease such as amyotrophic lateral sclerosis [39, 40], while females are more susceptible to Alzheimer’s disease [41–43] and multiple sclerosis [44, 45]. A better understanding of the mechanisms leading to these sex difference could help develop more targeted therapies with better outcomes in diseases where sex differences are prominent [46].
In SMA, accumulating evidence of the sexually dimorphic nature of the pathology in many affected tissues suggests the presence of sex-specific vulnerabilities [47]. The ISS-N2-targeting antisense oligonucleotide (ASO) treatment has also shown some sex-specific outcomes in a mouse model [11]. Studying sex difference in SMA is critical both for understanding disease pathogenesis and in developing more effective treatments. The contradictory findings related to the role of sex in SMA may be the result of studies carried out with limited data and small patient populations. To systematically investigate sex difference across different SMA types from large patient population, we obtained aggregated data from two of the biggest SMA registry databases in the world, TREAT-NMD and Cure SMA. While the picture is complex, our data suggest that there are sex differences across different SMA types, in the sex ratio of the patients, in symptom severity, in motor function and in the sex of affected family members. In addition, sex differences in SMA may be more prominent in certain registry datasets. This study highlights the potential value of more in-depth studies of the role of sex in SMA pathogenesis and the consideration of sex difference in SMA treatments.
MATERIALS AND METHODS
Delaware State University’s Institutional Review Board (IRB)-Human Subjects Protection Committee has granted an exemption from the IRB as this project meets Category 4 of Exempt Research specified in 45 CFR 46.101(b).
TREAT-NMD Global SMA Patient Registry
TREAT-NMD (https://treat-nmd.org) is a network for the neuromuscular disease field focused on providing an infrastructure to ensure that the most promising new therapies reach patients as quickly as possible. Since its launch in January 2007 the network’s focus has been on the development of tools for industry, clinicians, and scientists to bring novel therapeutic approaches through preclinical development and into the clinic, and on establishing best practices for treating neuromuscular patients worldwide.
The Global SMA Registry of TREAT-NMD links national SMA patient registries from all over the world. It collects data from genetically confirmed SMA patients. The data can be self-reported and/or provided by professionals. More than 5,000 genetically confirmed SMA patients worldwide have been enrolled in the Global SMA Registry database. The TREAT-NMD Global Registry Network (also known as TGDOC) is governed by a Charter and managed by the TGDOC Executive Board. The Board is responsible for reviewing the requests for data from the Global Registry Network and managing the enquiry governance and contracting. Our enquiry was approved by the TGDOC, and the contract was signed in January 2022. The requested data and data report were sent back in March 2022.
Our enquiry consisted of four primary questions which had been further stratified by the sub-group data items listed below:
Q1: Are there sex-specific difference between different types of SMA, and patients with different SMN2 copy numbers? Sub-group items included: number of SMN2 copies, SMA type (by age of first symptoms), and sex assigned at birth.
Q2: Is the disease severity different between the sexes within the same type of SMA based on motor measures? Sub-group items included: SMA type (by age of first symptoms), clinician global impression of severity total score – at diagnosis/symptom onset, and one motor measure score– at diagnosis/symptom onset. Motor measure scores included were Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), Hammersmith Functional Motor Scale (HFMS) and Expanded (HFMS-E), and Revised Upper Limb Module (RULM) scores. CHOP-INTEND is a clinician-reported outcome measure designed to assess motor function in very weak individuals with neuromuscular disorders and respiratory compromise [48]. It contains 16 items scored from 0 to 4, the total possible score is 64. It is a reliable measure of motor skills in patients with SMA type 1 and neuromuscular disorders presenting in infancy [49]. The HFMS is an assessment of the physical abilities of SMA type 2 and type 3 patients with limited ambulation. It is an ordinal scale consisting of twenty items with individual item scoring as 2 for unaided, 1 for performed with modification an adaption and 0 for unable [50]. To enable its use in the ambulant types 3 population, the HFMS was expanded to include 13 items from the Gross Motor Function Measure to form the HFMSE [51]. The RULM is a revised version of the Upper Limb Module, specifically designed to assess upper limb function in SMA type 2 and type 3 patients. It consists of 19 scorable items, graded on a three-point system with a maximum total score of 37 points [52].
Q3: What is the SMA type and sex of affected family member (s)? Sub-group items included: SMA type (by age of first symptoms), sex assigned at birth (patient), affected genetically related family member sex assigned at birth, and affected genetically related family member SMA type.
Q4: Is the brain development affected differently between the sexes of type 1 patients? Sub-group items included: sex assigned at birth, current weight, and current head circumference.
Twenty-six patient registries representing twenty-five countries were contacted by the TGDOC and asked to provide the requested data within the timeframe of three weeks. The countries contacted including Australia, Belgium, Canada, China, Columbia, Croatia, Czech Republic/Slovakia#, Egypt*, Georgia, Germany/Austria#, Hungary, India, South Korea, Latvia, Lebanon, New Zealand, Poland, Slovenia, Spain, Switzerland, Turkey*, Ukraine, and the United Kingdom (# Represents a registry with dual country coverage, * This country has more than one patient registry). TREAT-NMD Data Enquiries include only data that has been updated in the last 12 months, however given the disruptions caused by the global Coronavirus pandemic this has been extended to 24 months for this enquiry.
Cure SMA membership database
The Cure SMA database constitutes one of the largest patient-reported data repositories on SMA patients worldwide [53]. Self-identified SMA patients were automatically assigned a household and a personal contact ID number in the database. Aggregated data from the membership of affected individuals included personal contact ID number, birthdate, gender, deceased date, date of first contact to Cure SMA, type of SMA, and diagnosis date [53]. Great thanks to the Chief Medical Officer, Mary Schroth MD, FAAP, FCCP and the Director of Research Data Analytics, Lisa Belter, MPH for voluntarily providing the aggregated data from Cure SMA database according the Q1 query for the TREAT-NMD enquiry. The data included 9542 SMA patients and it was provided on May 2nd of 2022.
Analysis
Sex ratio difference in SMA patients of different types and patients with different SMN2 copy numbers
The proportion (percentage) of male and female patients of total SMA patients, within different types of SMA based on symptoms onset and with different SMN2 copy numbers were calculated. Significances were tested with a one proportion Z-test online (https://www.statology.org/one-proportion-z-test-calculator/). The test statistic is calculated as: z = (p-p0)/√(p0(1-p0)/n), Where: P = observed sample proportion, p0 = hypothesized population proportion, n = sample size. P0 was 0.5 based on the estimated overall sex ratio of the world by the United Nations Population Division, Department of Economic and Social Affairs (https://population.un.org/wpp/Download/Standard/Population/). The SMA incidence and prevalence ratio, and carrier frequency were adopted from published literature for the analysis of their correlation with the sex ratio of different countries. To analyze the correlation between the sex ratios of total population and SMA in the same country, the average sex ratio from 1950-2020 was cited from a file downloaded from the Population Division at United Nations (https://population.un.org/wpp/Download/Standard/Population/).
Sex difference in severity and motor scores
To quantify the score of severity, the clinician global impression of severity score was assigned back to individual patients based on the result of query. The severity score was then compared between male and female patients with different types of SMA with a student’s t-test. This score was also compared across SMA types among male and female patients with one-way ANOVA. As the motor scores of CHOP-INTEND, HFMS-E and RULM total score were returned with different ranges, the exact score of individual patients could not be determined. To quantify the motor measurements, the estimated scores of the median value of each range were assigned to individual patients based on the aggregated query result. For example, if there were 17 males who had CHOP-INTEND scores in the range of 1–10, the estimated score of 5.5 [(1 + 10)/2] was assigned to these 17 male patients, and so for other score range and motor measurements (Table 6). Motor scores were compared between male and female with different types of SMA, it was also compared across SMA types among male and female patients. To minimize the error of the midpoint motor score estimation described above, motor score ranges between male and female patients were ranked and compared with Mann Whitney U Test (Wilcoxon Rank Sum Test).
SMA1 head circumference and body weight estimation
The 50th percentiles of head circumference and body weight of control males and females at different ages were obtained from the “head circumference-for-age” and “weight-for-age” charts of the World Health Organization (WHO) child growth standards (https://www.who.int/tools/child-growth-standards/standards), respectively. For SMA1 patients, the median values of each range for head circumference and body weight were assigned to the aggregated patient number from the database query to estimate the values for each patient. Similarly, each patient in an age range was assigned the age of the median value of each range. The cumulative data was plotted for each age group and fitted with “Fit Spline/LOWESS” function with Prism 9 (GraphPad Software, San Diego CA), the 50th percentile value was then read out from the fitted curve for each age group. The median values of the age ranges were chosen to get the control value from the WHO standard charts and plotted together with the estimated SMA patients’ data. Please see data conversion in Tables 8, 9.
Statistics
Student’s t-test (two-tailed), Mann Whitney U test, Pearson correlation, and one-way ANOVA with Tukey post-hoc test were applied with Prism 9 (GraphPad Software, San Diego CA) unless otherwise noted. Statistical significance was set at P < 0.05. The data shown represent mean ± SEM.
RESULT
Sex difference in different types of SMA patients
TREAT-NMD reported a total of 3,838 genetically confirmed 5q SMA patients from 17 registries representing 19 counties, which consists of more than two-thirds (17/26) of the TREAT-NMD Global SMA Registry Network. Of the reports, eight were clinician reported, four were patients and clinician reported, three were patient reported and clinician verified, and two were patient reported. Details of responding registries and data entry are included in Table 1. Cure SMA reported data from 9,542 SMApatients.
Table 1
TREAT-NMD responding registries and data entry. “Name” is the name of patient registry, “Patients (n)” is the number of patients reported in the registry, and “Patient (%)” is the percent of the total number of patients represented in the entry
| Country | Registry | Data Entry type | Patient (n) | Patient (%) |
| Australia | Australian Neuromuscular Disease Registry | Patient and Clinician reported | 152 | 4.0 |
| Belgium | Belgian Neuromuscular Diseases Registry (BNMDR) | Clinician reported | 229 | 6.0 |
| Canada | Canadian Neuromuscular Disease Registry | Clinician reported | 191 | 5.0 |
| Croatia | Croatian Registry for Pediatric Neuromuscular disorders | Clinician reported | 41 | 1.1 |
| Czech Republic and Slovakia | REaDY – DMD &BMD | Patient and Clinician reported | 312 | 8.1 |
| Georgia | Georgian Pediatric NMD Registry | Clinician reported | 47 | 1.2 |
| Germany | German NMD Registry (Munich) | Patient reported / clinician verified | 548 | 14.3 |
| Hungary | Hungarian SMA Registry | Patient reported/ Clinician verified | 161 | 4.2 |
| India | MDA India – NMD registry | Clinician reported | 8 | 0.2 |
| Latvia | NMS datu kolekcija | Clinician reported | 39 | 1.0 |
| Poland | Polish Registry of Patients with NMDs | Patient reported/ Clinician verified | 848 | 22.1 |
| Slovenia | Registry of Slovenian Children with NMD | Clinician reported | 40 | 1.0 |
| Spain | RegistrAME – Registro Nacional de Pacientes FundAME | Patient reported | 290 | 7.6 |
| Switzerland | Swiss Registry for Neuromuscular Disorders | Clinician reported | 130 | 3.4 |
| Turkey | KUKAS | Patient and Clinician reported | 341 | 8.9 |
| Ukraine | Ukrainian Registry of SMA | Patient and Clinician reported | 314 | 8.2 |
| United Kingdom | UK SMA Patient Registry | Patient reported | 147 | 3.8 |
SMA types were classified by symptom onset as follow: SMA1, between birth and 6 months; SMA2, between 7 and 18 months; SMA3a, between 18 and 36 months; SMA3b, between 3 and 18 years; SMA4, over 18 years old. Of the 17 responding registries, Turkey and the United Kingdom did not distinguish SMA3 subtypes, so patients of this type from these countries were included in SMA3a. For the Cure SMA data, the SMA type of patients was not verified with age at symptom onset or proof of genetic testing and the SMA3 type was collected as a single category. The percentages of patients classified into different types of SMA in TREAT-NMD data were as follows: SMA1 23%; SMA2 38%; SMA3a 24%; SMA3b 10%; SMA4 1%; and age of symptoms onset unknown 3%. The percentages of patient classified into different types of SMA in the Cure SMA data were as follows: SMA1 39%; SMA2 31%; SMA3 19%, SMA4 3%; and age of symptoms onset unknown 9%. Of all the patients from TREAT-NMD, only 1 patient was in the intersex/unspecified/unknown category, and about 5% of Cure SMA patients were in this category. Please see the sex distribution in SMA patients of different types in Table 2.
Table 2
Sex distribution in different types of SMA patients, “unknown” is for the group of SMA patients with unknow age of symptoms onset
| SMA types | SMA1 | SMA2 | SMA3a | SMA3b | SMA4 | unknown | Total | |
| Treat-NMD | Male (n) | 474 | 728 | 488 | 232 | 36 | 64 | 2,022 |
| Female (n) | 407 | 744 | 430 | 153 | 12 | 69 | 1,815 | |
| IUU (n)* | 1 | 1 | ||||||
| Cure SMA | Male (n) | 1,789 | 1,381 | 818 | 138 | 402 | 4,528 | |
| Female (n) | 1,746 | 1,449 | 876 | 123 | 332 | 4,526 | ||
| IUU (n)* | 149 | 129 | 89 | 18 | 103 | 488 | ||
*Intersex/Unspecified/Unknown.
The percentages of males and females were calculated with the numbers of patients for whom sex was known from the two databases. Total male and female patients from TREAT-NMD database were 2,022 and 1,815 respectively (Table 2). The percentage of males (52.7%) was significantly higher than females (47.3%) with one-proportional two-tailed Z-test (Fig. 1A). The total number of male (n = 4,528) and female (n = 4,526) patients was about same for the Cure-SMA data (Table 2; Fig. 1B). The Cure SMA membership included patients from United States and from over 100 countries internationally, and U.S. members make up 76% of the total membership database. There was no significant difference in the total number of males (n = 3,423) and females (n = 3,548) among SMA patients from U.S. However, the percentage of males (52%) trended higher than females (48%) in the Cure SMA clinical data registry (Fig. 1C, n = 689, P = 0.05 one-tailed Z-test).
Fig. 1
Male/female ratio of different types of SMA patients. A– C, the percentage of total male and female patients from TREAT-NMD (A), Cure SMA membership (B) and Cure SMA clinical data registry (C) databases; D– F, the male and female percentage of different types of SMA patients from TREAT-NMD (D), Cure SMA membership total (E) and U.S. only (F). *P < 0.05, **P < 0.01 two-tailed Z-test.
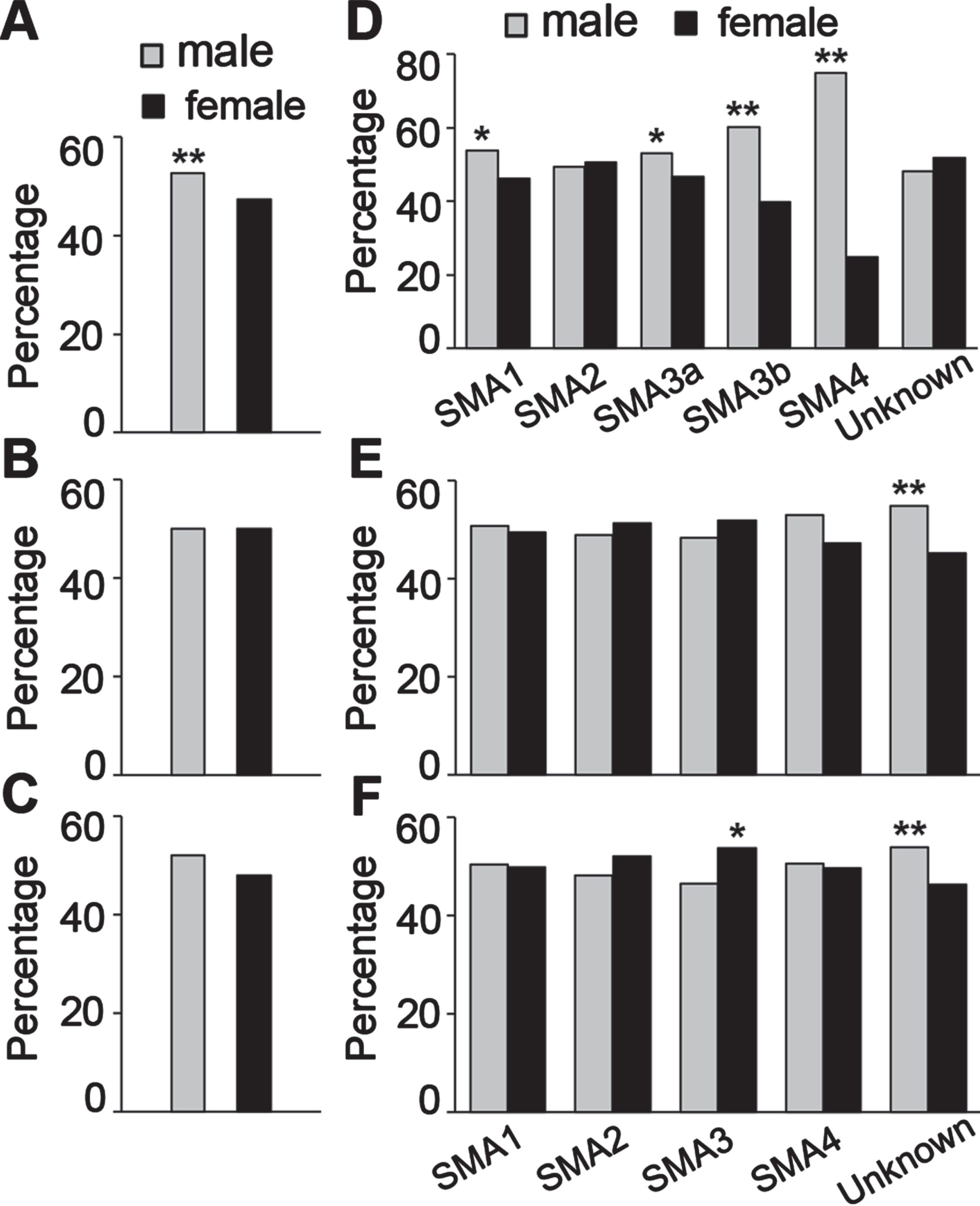
In the TREAT-NMD data, the percentage of males was significantly higher than females in most SMA types except SMA2, and the differences were larger in the milder SMA types (Fig. 1D). For the Cure SMA data, there was no significant difference in sex ratio for any types, but the percentage of male patients (54.8%) was higher than females (45.2%) for which the SMA type was unknown (Fig. 1E). The percentages of females of SMA2 (51.2%) and SMA3 (51.7%) were higher than males, although there was no statistical difference. As the patient numbers in these two types were much larger than the unknown SMA type group. It is likely that patients in the SMA type unknown group have milder forms of SMA, so the skewed sex ratio in the SMA type unknown group data may not represent a true difference. The lack of a sex difference overall in the Cure SMA data is likely due to the skewed sex ratio in the SMA type unknown group (Fig. 1B). The contribution of the SMA type unknown group to the overall difference was even clearer in the data for American patients in the Cure SMA database, where the percentage of females (53.8%) was significantly higher in SMA3 patients, but males (53.5%) were a higher percentage of the SMA type unknown group, so overall there was no difference(Fig. 1F).
Sex difference in SMA patients with different copy number of SMN2
The SMN2 copy number is known to corelate with the severity of SMA symptoms [4]. The percentages of patients for whom the SMN2 copy number was known were 22.8% of the Cure SMA dataset, and 70.1% of the TREAT-NMD dataset (Table 3). Both sets of data showed that with increased SMN2 copy number, the severity of symptoms gets milder (Fig. 2A, C). The percentage of males (62.9%) was significantly higher for patients with 4 + SMN2 copies in the TREAT-NMD data (Fig. 2B). For the Cure SMA data (Fig. 2D), the percentage of females was higher in patients with two copies (56.4%) and three copies (56.2%) of SMN2, but the percentage of males was higher for patients with unknown SMN2 copy number (51.4%). The U.S. data for SMN2 copy number showed the same difference as in the total Cure SMA data (not shown). As the majority of patients in the Cure SMA data (77.2%) had an unknown SMN2 copy number (Table 3), the sex ratio observed in patients with two and three SMN2 copies may not represent a true difference.
Fig. 2
Male/female ratio of SMA patients with different SMN2 copies. A and C, the percentage of patients with different SMN2 copies in different types of SMA from TREAT-NMD (A) and Cure SMA membership (C) datasets; B and D, the percentage of male and female patients with different SMN2 copies from TREAT-NMD (B) and Cure SMA membership (D) databases. **P < 0.01 two-tailed Z-test.
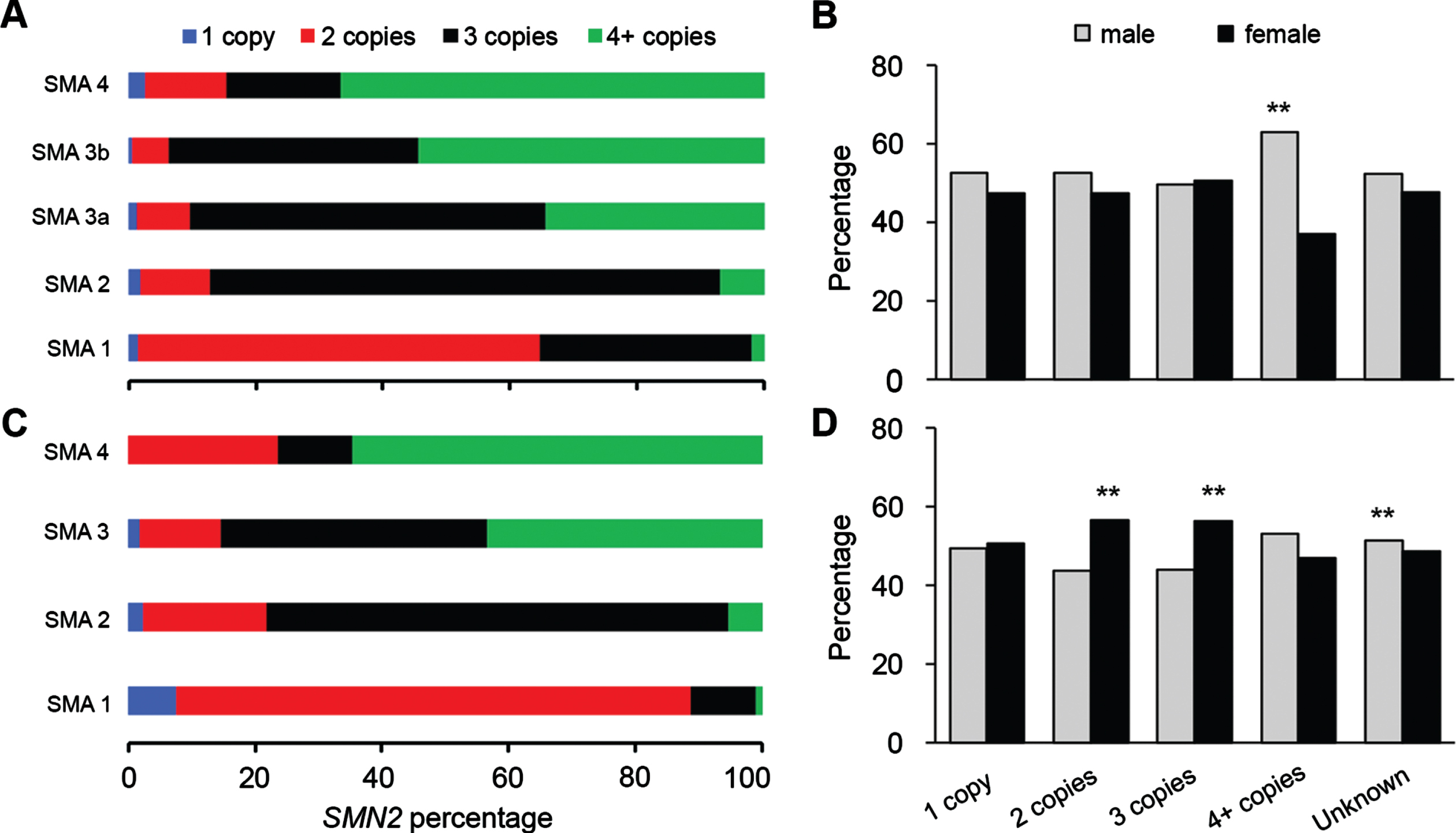
Table 3
Sex distribution in SMA patients with difference SMN2 copy numbers, “unknown” is for the group of SMA patients with unknow age of symptoms onset
| SMN2 copy number | 1 copy | 2 copies | 3 copies | 4 + copies | unknown | Total | |
| Treat-NMD | Male (n) | 20 | 308 | 759 | 334 | 601 | 2,022 |
| Female (n) | 18 | 278 | 774 | 197 | 548 | 1,815 | |
| IUU (n)* | 1 | 1 | |||||
| Cure SMA | Male (n) | 41 | 363 | 384 | 143 | 3,597 | 4,528 |
| Female (n) | 42 | 470 | 492 | 126 | 3,396 | 4,526 | |
| IUU (n)* | 0 | 10 | 14 | 2 | 462 | 488 |
*Intersex/Unspecified/Unknown.
Correlation between sex ratio of SMA patients in a region and the incidence and prevalence of SMA
It has been reported that the rate of incidence of SMA varies in different racial and ethnic populations [53, 54]. The differences we have observed in the sex ratios of patients in our two datasets may be related to demographic differences in the populations from which the data were collected. To test this hypothesis, the available incidence and prevalence rates for SMA, and the carrier frequency of SMA from different counties were determined from published literature [54–62]. The U.S. data from Cure SMA, and the TREAT-NMD data were stratified by country of origin of the patients and the sex ratio among patients was plotted against the male/female ratio in the country, SMA incidence, prevalence, and carrier frequency (Table 4). The male/female ratio was significantly positively correlated with SMA incidence (Fig. 3A) and prevalence (Fig. 3B), but not with the carrier frequency (Fig. 3C). This data suggests that higher male/female sex ratio among SMA patients is associated with higher SMA incidence and prevalence rates. The sex ratio among SMA patients was negatively correlated with the population sex ratio of individual countries averaged from data from the United Nations Population Division for 1950 to 2020 (Fig. 3D). These results indicate that regional demographic characteristics may play a role in sex differences in SMA patients.
Fig. 3
Male/female ratio of SMA patients correlates with incidence and prevalence ratio by country. A– D, male/female ratio plotted with (A) SMA incidence (n = 12, r = 0.67, P = 0.02 two-tailed), (B) SMA prevalence (n = 10, r = 0.68, P = 0.03 two-tailed), (C) SMA carrier frequency (n = 8, r = 0.27, P = 0.26 one-tailed), (D) the average male/female sex ratio of total population (1950– 2020) from United Nations Population Division (n = 17, r = – 0.54, P = 0.03 two-tailed).
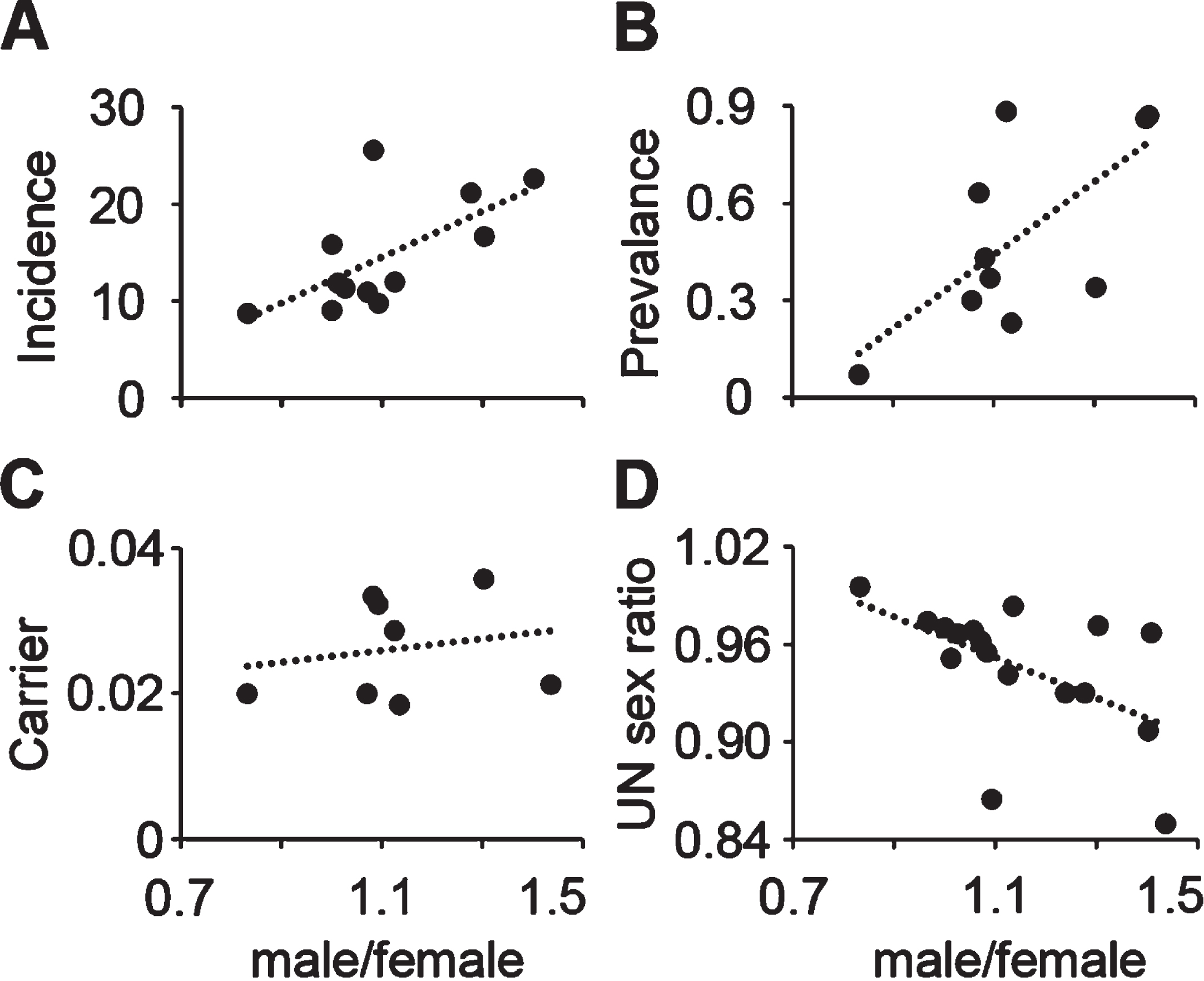
Table 4
Male/female ratio, SMA carrier frequency, incidence, and prevalence from different countries
| Country | Male (M) | Female (F) | M/F | UN M/F | Carrier frequency | Incidence* | Prevalence* |
| Australia | 69 | 83 | 0.831 | 0.995 | 1/50[56] | 8.7[57] | 0.07[55] |
| Belgium | 116 | 113 | 1.027 | 0.966 | 11.3[55] | ||
| Canada | 101 | 89 | 1.135 | 0.983 | 1/54[57] | 0.23[55] | |
| Croatia | 23 | 18 | 1.278 | 0.930 | 21.1[55] | ||
| Czech+Slovakia | 157 | 155 | 1.013 | 0.951 | 11.9[55] | ||
| Germany+Austria | 285 | 263 | 1.084 | 0.955 | 1/30[57] | 25.5[55] | 0.43[55] |
| Hungary | 94 | 67 | 1.403 | 0.907 | 22.6[55] | 0.86[55] | |
| Latvia | 23 | 16 | 1.438 | 0.850 | 1/47[58] | ||
| Poland | 449 | 399 | 1.125 | 0.941 | 1/35[59] | 12[55] | 0.88[55] |
| Slovenia | 20 | 20 | 1.000 | 0.970 | 15.8[55] | ||
| Spain | 149 | 141 | 1.057 | 0.968 | 0.30[55] | ||
| Switzerland | 76 | 54 | 1.407 | 0.967 | 0.87[55] | ||
| Turkey | 193 | 148 | 1.304 | 0.971 | 1/28[60] | 0.34[55] | |
| Ukraine | 164 | 150 | 1.093 | 0.865 | 1/31[61] | 9.8[55] | 0.37[55] |
| UK | 76 | 71 | 1.070 | 0.962 | 1/50[62] | 10.9[55] | 0.63[55] |
| USA | 3423 | 3548 | 1.000 | 0.974 | 1/54[54] | 9.1[54] |
UN, United Nations; *Per 100,000 population.
3.4Sex difference in symptom severity and motor function among SMA patients of different types
The above data address whether there are more males affected than females among SMA patients. The next question we investigated was whether males are more strongly affected by the disease than females. We compared the severity of symptoms between male and female SMA patients with the clinician global impression of severity total score (Table 5), and patient motor function with the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), Hammersmith Functional Motor Scale Expanded (HFMS-E) and Revised Upper Limb Module (RULM) scores from the TREAT-NMD data enquiry (Table 6). To estimate and quantify differences in the scores of symptom severity and motor function, the aggregated data were converted to individual data as described in methods (Tables 5, 6), then the scores were compared between male and female patients.
Table 5
Clinician global impression of severity total score of SMA patients from TREAT-NMD SMA registry
| SMA types | SMA1 | SMA2 | SMA3a | SMA3b | SMA4 | |||||
| Clinician severity score | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) |
| 1 – Not at all affected | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 – Borderline affected | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 3 – Mildly affected | 5 | 7 | 3 | 6 | 7 | 3 | 7 | 9 | 1 | 2 |
| 4 – Moderately affected | 5 | 8 | 12 | 13 | 3 | 3 | 6 | 0 | 0 | 1 |
| 5 – Markedly affected | 15 | 13 | 11 | 14 | 4 | 2 | 0 | 0 | 0 | 1 |
| 6 – Severely affected | 22 | 23 | 7 | 2 | 4 | 1 | 1 | 0 | 0 | 0 |
| 7 – The most extremely affected | 3 | 3 | 5 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| Total patients | 52 | 57 | 38 | 36 | 19 | 9 | 14 | 10 | 2 | 5 |
Table 6
Motor scores of SMA patients from TREAT-NMD SMA registry
| HFMS-E | SMA1 | SMA2 | SMA3a | SMA3b | SMA4 | ||||||
| range | median | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) |
| 1–11 | 6 | 9 | 6 | 44 | 45 | 15 | 8 | 5 | 3 | 0 | 0 |
| 12–22 | 17 | 1 | 1 | 21 | 30 | 10 | 5 | 4 | 4 | 1 | 1 |
| 23–33 | 28 | 2 | 0 | 21 | 19 | 5 | 4 | 10 | 2 | 0 | 1 |
| 34–44 | 39 | 0 | 1 | 6 | 11 | 9 | 12 | 17 | 11 | 1 | 0 |
| 45–55 | 50 | 1 | 0 | 6 | 14 | 21 | 7 | 17 | 13 | 3 | 2 |
| 56–66 | 61 | 0 | 1 | 3 | 3 | 15 | 17 | 17 | 16 | 4 | 1 |
| Total patients | 13 | 9 | 101 | 122 | 75 | 53 | 70 | 49 | 9 | 5 | |
| RULM | SMA2 | SMA3a | SMA3b | SMA4 | |||||
| Range | median | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) | male (n) | female (n) |
| 0–5 | 2.5 | 8 | 7 | 3 | 0 | 0 | 0 | 1 | 0 |
| 6–12 | 9 | 11 | 7 | 4 | 1 | 0 | 1 | 0 | 0 |
| 13–19 | 16 | 11 | 16 | 4 | 1 | 2 | 2 | 0 | 0 |
| 20–25 | 22.5 | 4 | 6 | 5 | 4 | 3 | 2 | 0 | 0 |
| 26–31 | 28.5 | 0 | 3 | 1 | 4 | 1 | 2 | 1 | 0 |
| 31–37 | 34 | 1 | 1 | 15 | 11 | 13 | 10 | 2 | 1 |
| Total patients | 35 | 40 | 32 | 21 | 19 | 17 | 4 | 1 | |
| CHOP-INTEND | SMA1 | ||
| range | median | male (n) | female (n) |
| 1–10 | 5.5 | 17 | 15 |
| 11–20 | 10.5 | 21 | 13 |
| 21–30 | 25.5 | 24 | 26 |
| 31–40 | 35.5 | 17 | 19 |
| 41–50 | 45.5 | 16 | 15 |
| 51–64 | 57.5 | 6 | 14 |
| Total patients | 101 | 102 | |
For severity scores, the dataset did not have enough samples for SMA4 for comparison, so that type was excluded (Table 5). Overall, severity scores were lower in patients with milder SMA. A one-way ANOVA test identified the severity score of male SMA3a patients was significantly lower than of male SMA1 patients, the severity scores for SMA3b patients were significantly lower than SMA1 and SMA2 in both male and female patients; With a two-tailed t-test, the severity scores of SMA2 and SMA3b males were shown to be significantly higher than females in the same SMA type (Fig. 4A). This difference was also confirmed by Mann Whitney U test (not shown) in SMA2 (P < 0.05) and SMA3 (P = 0.05), which suggests that for SMA2 and SMA3, male patients are more severely affected by the disease.
Fig. 4
Sex difference in severity and motor function among different types of SMA patients. A, the clinician global impression of severity total score of different types SMA patients; B, the CHOP-INTEND and HFMS-E total score of SMA1 patients; C and D, the HFMS-E (C) and RULM (D) total scores of milder types of SMA patients. *P < 0.05, **P < 0.01 two-tailed t-test, males were compared with females; #P < 0.05, # #P < 0.01, one-way ANOVA to compare different types of SMA males and females, respectively.
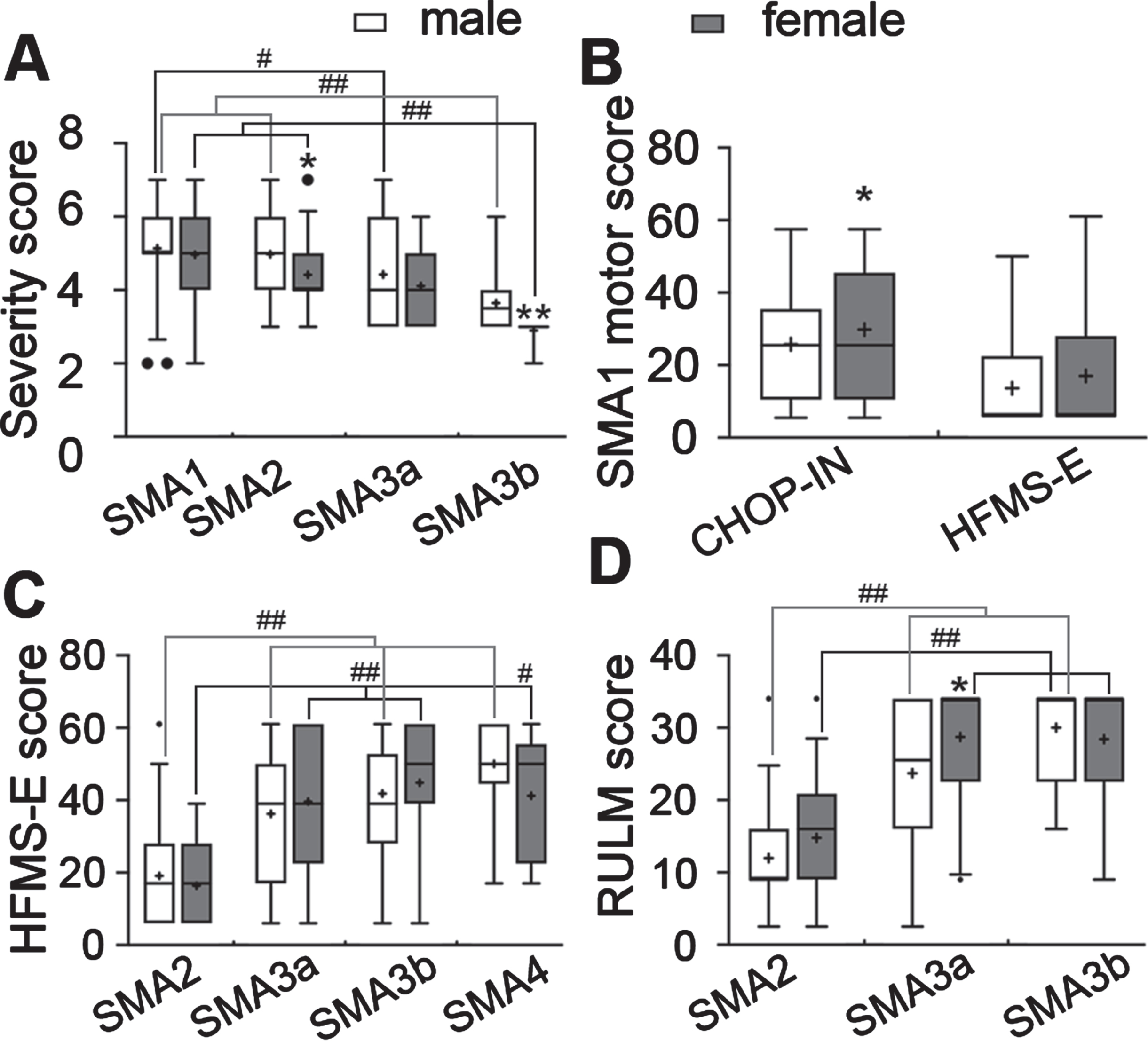
For motor measurements, the TREAT-NMD dataset reported aggregated patient numbers for both CHOP-INTND and HFMS-E scores for SMA1 patients. The estimated CHOP-INTEND score of female SMA1 patients trended higher than males (P < 0.05, one-tailed t-test), but no difference was found in HFMS-E scores between male and female SMA1 patients (Fig. 4B). For the milder types of SMA, both estimated HFMS-E and RLUM scores were reported. Overall, the HFMS-E scores were higher in milder types of SMA, as the scores of SMA3a, 3b and SMA4 were significantly higher than SMA2 in both male and female patients. However, no difference was found in HFMS-E scores between male and female patients in the same type of SMA (Fig. 4C). The RLUM scores of SMA3a and SMA3b were higher than SMA2 in both male and female patients, and the RULM score of females was higher than SMA3a male patients (Fig. 4D). These data suggest that the motor function of female patients in both severe and mild SMA trends better. This is consistent with a recent study which showed a relevant gender effect on motor function in SMA patients, with higher disease severity reported in males especially in the young adult age and in SMA3 patients [63].
To confirm our midpoint motor score estimation above, motor score ranges were ranked and analyzed with the Mann Whitney U test. We found no significant sex differences in motor score ranks in any of the motor measurements for any type of SMA. This may be because of the limitation of converting the aggregated scores over such a big range of motor scores. It also points out the necessity of analyzing individual data from a large population for a more accurate comparison of motor measurements.
Affected family members of SMA patients tend to be male
To study whether SMA equally affects males and females in the same family, the TREAT-NMD database was queried by males only and females only separately (Table 7a), the percentage of affected family members who were male and female was compared. The percentages of patients with no affected family member were not different between the queries for male patients only and female patients only (Fig. 5A). The male/female ratio of affected family members was significantly higher for the male patients only query, while being nearly 50 : 50 in the female patients only query (Fig. 5B). Breaking down the male patients only query by the SMA type of the affected family members, the percentages of males among the SMA1, SMA3a, SMA3b, and symptom onset unknown groups were significantly higher than for females (Table 7b, Fig. 5C). This is similar to the overall male/female ratio for SMA patients of different types shown in Fig. 1D. The non-significance in the SMA4 group among affected family members may be because the number of SMA4 family members was low (n = 10).
Fig. 5
Male/female ratio of family member of SMA patients diagnosed with SMA. A, the percentage of male and female patients with no SMA affected family member (Chi-Square test, P = 0.09); B, the percentage of male and female family member diagnosed with SMA queried with male patients only and female patients only respectively; C, the percentage of males and females in affected family members with different types of SMA queried with male patients only. **P < 0.01 two-tailed Z-test.
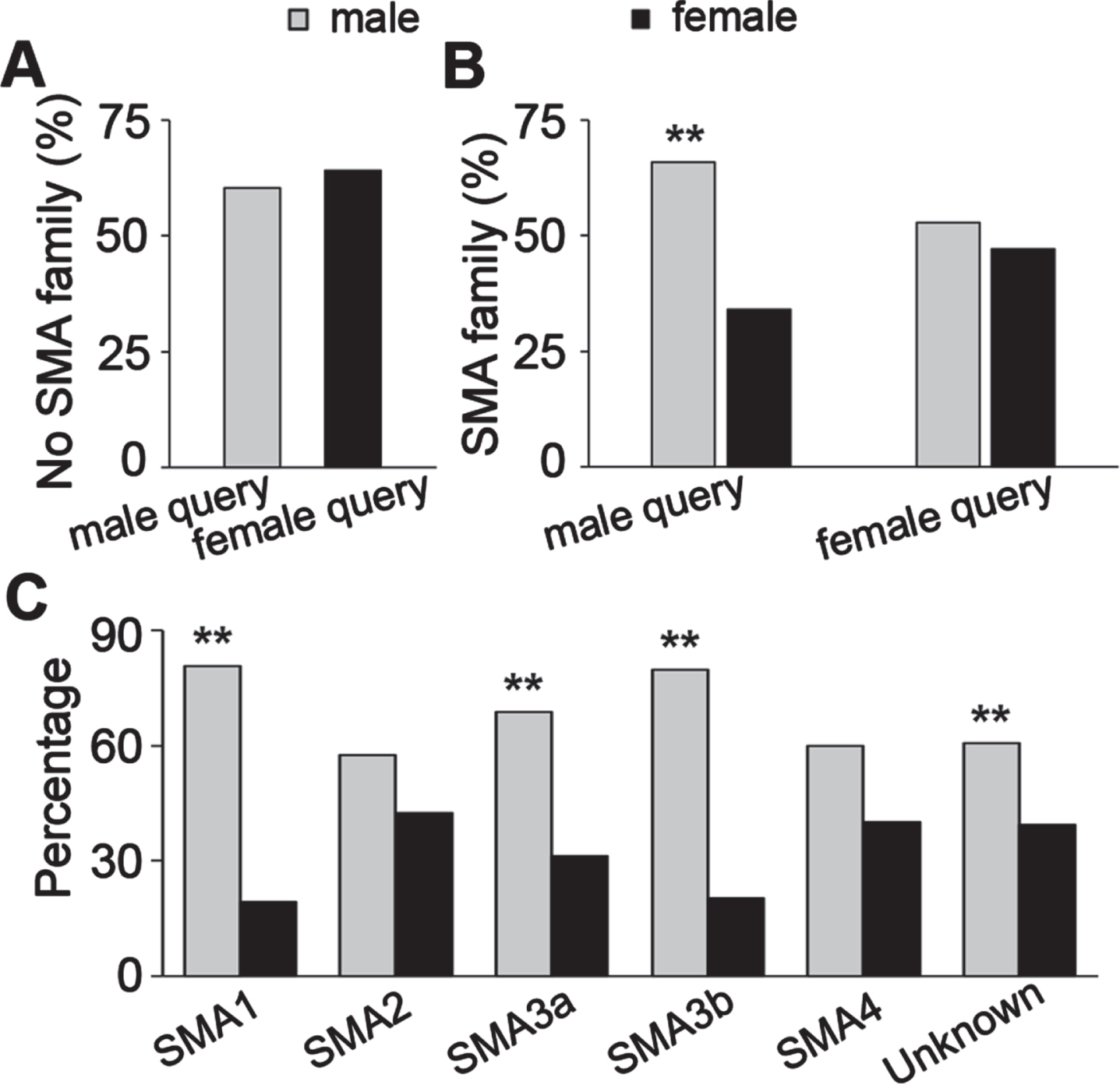
Table 7a
The number of SMA affected family members queried with male and female patients respectively from TREAT-NMD SMA registry
| Query seed | No affected family | Male family | Female family | IUU family | Total |
| Male patients only | 668 | 249 | 129 | 61 | 1107 |
| Female patients only | 600 | 149 | 133 | 55 | 937 |
Table 7b
The patient number of different SMA types of affected family members with male patients only query from TREAT-NMD SMA registry
| Male patients only query | SMA1 | SMA2 | SMA3a | SMA3b | SMA4 | Unknown |
| Male family member | 29 | 27 | 33 | 43 | 6 | 111 |
| Female family member | 7 | 20 | 15 | 11 | 4 | 72 |
Differences in the developmental trajectories of male and female SMA1 patients
Our current study and other studies [25, 26] suggest that males appear to be more strongly affected by SMA, so we looked in more detail at the development of patients with SMA1 by comparing the body weight and head circumference of males versus females. As described in the methods, to quantify the data, the aggregate numbers of patients in the different ranges of body weight and head circumferences in the TREAT-NMD dataset were assigned the median value of the range in which they fell. Cumulative distributions of the data were fitted with Spline to determine the 50th percentile values. To compare the development of body weight and head circumference to the WHO standards, the median values for the age ranges in the dataset were used to find the body weight and head circumference values from the WHO standard charts and compared with SMA patients (Tables 8, 9; Fig. 6).
Table 8
Conversion of the aggregated body weight data to midpoint data in SMA1 patients for growth trend plot in Fig. 6 from TREAT-NMD SMA registry. The n numbers for males and females are the number in that age range across all body weight ranges
| Age range | Body weight ranges (Kg) | Male (n) | Female (n) | ||||||||
| (month) | |||||||||||
| 0–2 | <2 | 2 < 3 | 3 < 4 | 4 < 5 | 5 < 6 | 6 < 7 | 7+ | 3 | 1 | ||
| 2–6 | <4 | 4 < 5 | 5 < 6 | 6 < 7 | 7 < 8 | 8 < 9 | 9+ | 15 | 33 | ||
| 6–12 | <6 | 6 < 7 | 7 < 8 | 8 < 9 | 9 < 10 | 10 < 11 | 11+ | 34 | 41 | ||
| 12–18 | <7 | 7 < 8 | 8 < 9 | 9 < 10 | 10 < 11 | 11 < 12 | 12 < 13 | 13+ | 28 | 23 | |
| 18–24 | <8 | 8 < 9 | 9 < 10 | 10 < 11 | 11 < 12 | 12 < 13 | 13 < 14 | 14 < 15 | 15+ | 18 | 23 |
 | |||||||||||
| Age (month) | Body weight (kg) | Male (n) | Female (n) | ||||||||
| 1 | 1.5 | 2.5 | 3.5 | 4.5 | 5.5 | 6.5 | 7.5 | 3 | 1 | ||
| 4 | 3.5 | 4.5 | 5.5 | 6.5 | 7.5 | 8.5 | 9.5 | 15 | 33 | ||
| 9 | 5.5 | 6.5 | 7.5 | 8.5 | 9.5 | 10.5 | 11.5 | 34 | 41 | ||
| 15 | 6.5 | 7.5 | 8.5 | 9.5 | 10.5 | 11.5 | 12.5 | 13.5 | 28 | 23 | |
| 21 | 7.5 | 8.5 | 9.5 | 10.5 | 11.5 | 12.5 | 13.5 | 14.5 | 15.5 | 18 | 23 |
Table 9
Conversion of the aggregated head circumference data to midpoint data in SMA1 patients for growth trend plot in Fig. 6 from TREAT-NMD SMA registry. The n numbers for males and females are the number in that age range across all head circumference ranges.
| Age range (month) | Head circumference ranges (cm) | Male (n) | Female (n) | ||||||
| 0–2 | <32 | 32 < 34 | 34 < 36 | 36 < 38 | 38 < 40 | 40 < 42 | 42+ | 3 | 1 |
| 2–6 | <36 | 36 < 38 | 38 < 40 | 40 < 42 | 42 < 44 | 44 < 46 | 46+ | 12 | 25 |
| 6–12 | <40 | 40 < 42 | 42 < 44 | 44 < 46 | 46 < 48 | 48 < 50 | 50+ | 22 | 35 |
| 12–18 | <42 | 42 < 44 | 44 < 46 | 46 < 48 | 48 < 50 | 50 < 52 | 52+ | 17 | 20 |
| 18–24 | <44 | 44 < 46 | 46 < 48 | 48 < 50 | 50 < 52 | 52 < 54 | 54+ | 9 | 17 |
| |||||||||
| Age (month) | Head circumference (cm) | Male (n) | Female (n) | ||||||
| 1 | 31 | 33 | 35 | 37 | 39 | 41 | 43 | 3 | 1 |
| 4 | 35 | 37 | 39 | 41 | 43 | 45 | 47 | 12 | 25 |
| 9 | 39 | 41 | 43 | 45 | 47 | 49 | 51 | 22 | 35 |
| 15 | 41 | 43 | 45 | 47 | 49 | 51 | 53 | 17 | 20 |
| 21 | 43 | 45 | 47 | 49 | 51 | 53 | 55 | 9 | 17 |
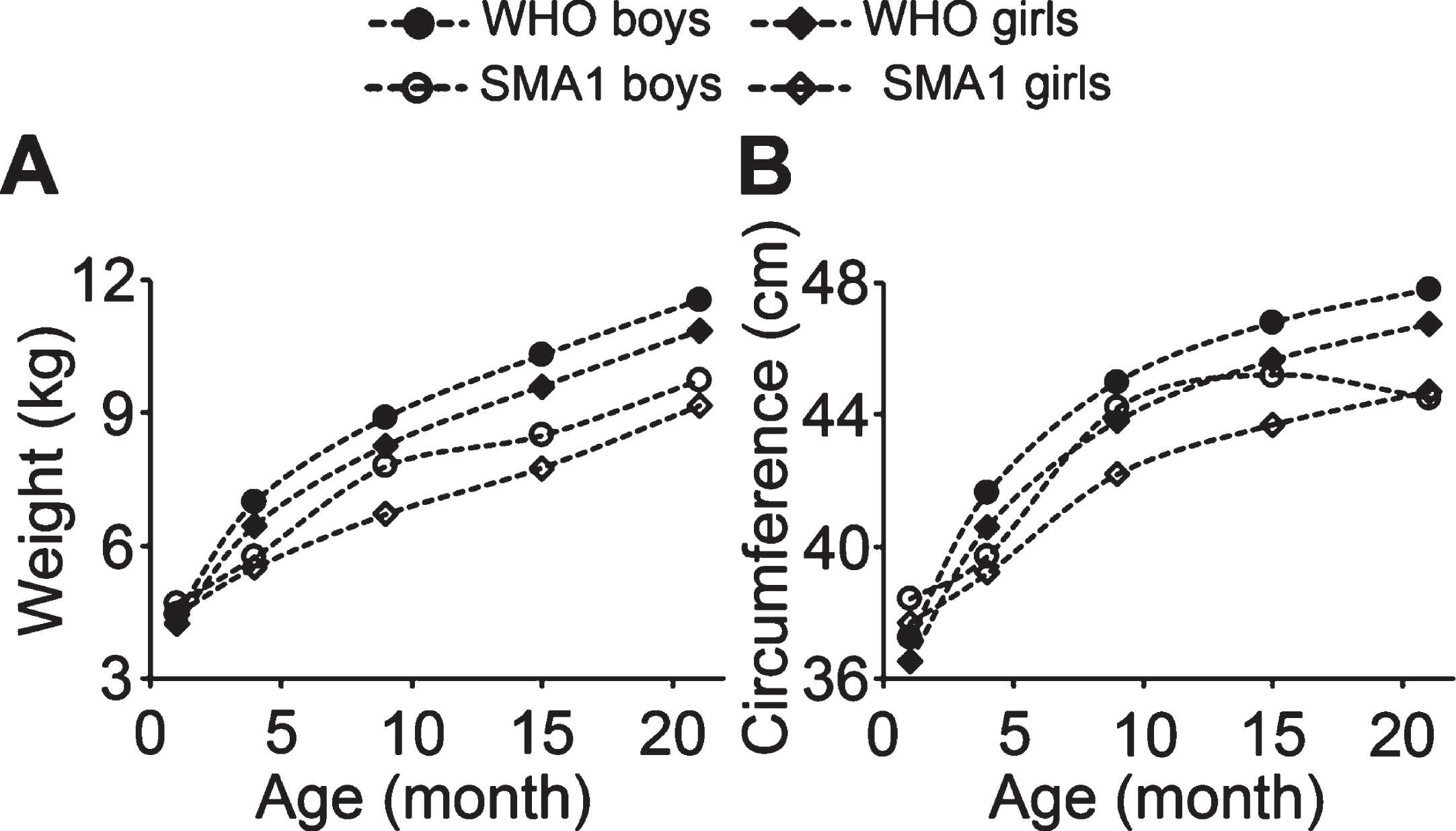
The body weight and head circumference of both male and female SMA1 patients were lower than the WHO standards at all ages reported, and both values were lower in SMA1 female patients than males as compared to the WHO standards. The inconsistency of values for age 1 month old is likely caused by the very small patient number in this age group (Tables 8, 9; Fig. 6). In female SMA1 patients, both body weight and head circumference showed the same developmental trend as the WHO standard but with lower values. In SMA1 males, there was no significant difference in growth pattern of body weight from WHO standard and from female SMA1 patients (Fig. 6A). However, the growth pattern of head circumference in SMA1 males was significantly slowed by age of 15 months, so that the average value of age 21 months was lower than 15 months (Fig. 6B). These results suggest that both body and the head circumference development are slower in SMA1 patients, and the slower growth of head circumference in male patients indicates that brain development may be more strongly affected in male than femalepatients.
Fig. 6
Developmental sex differences in the 50th percentile body weight and head circumference in SMA1 patients. A and B, the growth curve of body weight (A) and head circumference (B) of SMA1 patients plotted with WHO standards.
DISCUSSION
Sex differences have been reported in many neurodegenerative disorders, however sex differences in SMA have received limited attention. It is not clear whether, or how, sex plays a role in SMA pathogenesis and responses to treatment. The current study systematically investigated sex differences in different types of SMA patients with data enquiries from the two largest databases of SMA patients in the world: TREAT-NMD Global SMA Registry and Cure SMA membership database. Our data show that more males are affected by SMA, male SMA patients have more male family members affected by SMA, and male SMA patients have more severe symptoms. In addition, head circumference data suggest that male patients may have greater developmental deficiencies in the brain. Taken together, these results from certain registry datasets indicate that males appear to be more vulnerable to SMA thanfemales.
Modifiers influence the phenotype of SMA
The three officially approved treatments for SMA (Spinrasa, Zolgensma, and Evrysdi) are based on an SMN-restoring strategy [64]. They have had impressive results in extending patient lives and improving motor function. However, they are not curative [1]. The overall limitations of these treatments, including the number of non-responders remain a significant problem, as do adverse effect complications, and the unknown long-term efficacy – especially the newly developed treatment Evrysdi [65]. Optimizing current treatments and developing combinatorial and alternative approaches are needed to improve outcomes for SMA patients. Modifiers that could influence the phenotype of SMA patients open novel therapeutic possibilities for SMA treatment [66]. These modifiers act in both SMN-dependent and SMN-independent ways. SMN-dependent factors include the genomic integrity of SMN gene copies, cis- and trans-regulatory factors, and factors influencing RNA transcription, translation, or protein stability. SMN-independent factors facilitate or counteract disease progression via SMN-downstream pathways. They are involved in major cellular pathways including cytoskeletal dynamics, synaptic vesicle recycling and neurotransmission, axonal transport, control of gene expression and transcription [67].
The SMN complex is involved in many cellular processes, including DNA replication and repair, transcription and splicing [68]. Many different processes have been reported to be modulators of SMN complex activities. Recent evidence has identified post-translational modifications (PTMs) including phosphorylation, methylation, ubiquitination, acetylation, and SUMOylation etc., as ways that the pleiotropic functions of the SMN complex are regulated [69]. Moreover, epigenomic profiles in the early embryo and developing germ cells have revealed dramatic differences in the distribution of the histone PTMs and DNA methylation in female and male mice (for review see [70]). These studies indicate the possibility that sexual dimorphism in these chromatin marks, could interact with SMN-related epigenetic modifications to lead to sex differences in SMA.
Of the SMN-independent modifiers, three – PLS3, USP9X and UBA1, are known to be encoded on the X chromosome and affect SMA severity in a sex-specific manner [47, 71]. For example, it has been reported that asymptomatic SMN1-deleted females exhibit significantly higher expression of PLS3 than their SMA-affect siblings [8]. PLS3 expression has been found to be significantly higher in female patients older than three years, and higher in SMA3 patients than SMA2 patients. In SMA3 patients, those who could walk had higher PLS3 expression than patients who were unable to walk [72, 73]. UBA1 loss was observed in induced pluripotent stem cell-derived motor neurons from SMA patient, and systemic restoration of UBA1 ameliorates SMA phenotype in a mouse model [12]. In addition, mutations of UBA1 cause a very rare, non-SMN-dependent form of SMA in human patients [74]. These studies suggest that modifier proteins with sex-influenced expression can play a role in SMA incidence and severity.
Non-X chromosome genes coding for sex specific SMN-independent SMA modifiers have also been reported. For example, TIA1 was reported as a sex-specific disease modifier in a mild mouse model of SMA. Knockout of TIA1 showed exacerbated developmental defects in male but not in female reproductive organs, promoted body weight only in male mice, and early tail necrosis in female but not in male mice [75]. Myostatin inhibition promoted stronger body weight in male than female in SMA mouse models [15]. Overexpression of ZPR1 was associated with better improvement in females, with higher body weight and lifespan increases compared to male mice [16].
Sex is a risk factor for SMA
Sex differences in SMA have been noticed in earlier case studies, but they did not attract much attention and have not been systematically studied. Before the genetic cause of SMA was revealed, there were reports of sex differences in both Werdnig-Hoffmann and Kugelber and Welander diseases. In 1958, Schmid reported two sisters showed milder manifestations than their brother with infantile SMA Werdnig-Hoffmann disease (SMA1) in a German family with five children [76]. Pearn reported two times more males than females in 78 SMA1 patients in U.K., however no sex differences in familial cases, age of onset or life expectancy were reported [17]. Sprira reported that a male patient with Kugelber and Welander SMA was the most severely affected in a family of 7 in Israel [77]. Dubowitz reported on 12 cases in London with symptom onset in infancy and survival into childhood and adolescence, in which affected males had much more severe symptoms than their female siblings [78]. The predominance of male patients in the milder forms of SMA were reported afterwards in Japanese [19, 25] and Polish [18, 21, 24] and other [26] populations. However, there was no sex difference in sex ratio and severity reported for either Werdnig-Hoffman or Kugelberg-Welander forms of SMA in 91 Canadian cases [22]. A report from an Australian group found no significant sex differences among patients with SMA1 or SMA2, but patients with SMA3b demonstrated a female predominance in 70 genetic diagnosed patients [20].
This inconsistency is present in our study too. In our TREAT-NMD dataset, the percentage of males was significant higher in all types of SMA except SMA2. Males were predominantly and more strongly affected than females, and more male family members were affected as well. This is consistent with other studies discussed above. However, in SMA patients in the Cure SMA membership dataset we found no overall sex differences. In our datasets more than 90% of TREAT-NMD patients were from European countries, while the Cure SMA membership data included patients from over one hundred countries with 75.7% patients from the U.S. Our study and others suggest sex is a risk factor in SMA epidemiology and pathology. However, the inconsistency between the two datasets we queried, and in the other studies discussed above indicate that sex differences may be more common in certain populations. Indeed, we found that the sex ratio of SMA patients within a country was correlated with overall SMA incidence and prevalence rates for that country. Higher male/female ratios among SMA patients were correlated with higher SMA incidence and prevalence rates, and inversely correlated to the total population male/female ratio. This result suggests that genetic background may play a role in sex differences in SMA patients from different registry datasets [54, 55].
SMN2 copy number is strongly correlated with SMA severity [4]. However, we found significantly more males only in patients with more than four SMN2 copies. Our results for fewer than four copies of SMN2 are consistent with a study reporting no overall difference in the distribution of SMN2 copies between sexes [26]. If SMA symptoms tend to be more severe in males, the higher incidence of males in the population of patients with high numbers of SMN2 copies suggests that females with high copy numbers of SMN2 are more likely to appear normal and therefore less likely to be diagnosed with SMA.
Interestingly, a recent study reported that weight and supine length growth are more linear in females with SMA1 and SMA2, while SMA1 males showed a decrease in weight, length, and body mass index velocity at about 3 years of age, and males with SMA2 showed a decrease in both weight and length growth velocity after about 6 years of age [79]. This is consistent with our results showing a decline in average head circumference at age of 21 months in male SMA1 patients. Growth in head circumference in female SMA1 patients was more linear, even though it was lower than the WHO growth standard. These data suggest the presence of sex differences in the development of SMA children.
Male infertility may also be a concern among mild SMA patients, as male reproductive defects including testicular hypofunction and infertility have been reported in mild SMA patients [80]. However, the fertility of woman does not appear to be affected by SMA [81]. This difference is supported by studies in a mild SMA mouse model. Impaired testis development, degenerated seminiferous tubules, reduced sperm count and low fertility in males SMA mice were reported, but no overt sex organ phenotype in female SMA mice [82].
Overall, these studies indicate sex is a risk factor in SMA patients, which needs further investigation.
Potential mechanisms of SMA sex difference: genetic background, sex hormones and X-chromosome inactivation escape genes
Human SMN1 and SMN2 gene copies lie within a repeat unit of a copy number variant on chromosome 5q13. A large genetic variability has been found in the number of repeat units (null to four) per chromosome, and the size, and orientation of this copy number variant [67]. Heterozygous carriers of SMA include four main groups of genotypes due to different SMN1 copy numbers and point mutation or microdeletion of SMN1. Generally, the estimation of the incidence of SMA is around 1 in 10,000 live births, and a prevalence of 1-2 per 100,000 persons. However, significant regional differences exist. This could be because of the method of calculation and/or population size. It could also indicate regional genetic pool difference in the incidence of SMA [55]. The strong correlation of sex ratio with SMA incidence and prevalence in our study supports this regional/ethnical difference. Additional support comes from the inverse correlation of SMA sex ratio with the overall population sex ratio in the countries analyzed. Higher male/female ratio in SMA correlated to lower male/female ratio of the total population in the same country. The mechanism underlying this correlation is not known.
It Is known that sex steroid hormones exert significant effects on neural development and that manipulation of sex hormones during critical periods results in permanent neural changes. Sex-related differences in vulnerability to neurobiological disorders might be due to sexual dimorphisms established during development, as well as to levels of steroid hormones [83]. The brain is particularly vulnerable to age-related androgen loss [84] and the diminishment of estrogen in postmenopausal woman [85]. A neuroprotective role for sex hormones in the brain against neurobiological disorders and injury has been reported [86]. The influence of sex hormones in skeletal muscle quality and health may also contribute to dimorphic responses between sexes during muscle pathologies [87]. It is well established that androgens and estrogens and their respective receptors have specific actions on the different bone envelopes during bone growth and bone maintenance in both sexes [88]. Sex hormones also affect immune responses which could lead to variability in disease phenotypes [89]. Studies also suggest that sex hormones ameliorate the regressive changes in motor neuron and muscle morphology after spinal cord injury [90].
ALS is a motor neuron disease which shows sexual dimorphism in its incidence, age of onset and progression rate [91]. In ALS mouse models, androgen and estrogen receptors are differently dysregulated in motor neurons and astrocytes [92]. Androgen receptor gene mutation of the proximal long arm of the X chromosome is the genetic cause of a rare form of spinal muscular atrophy, spinal and bulbar muscular atrophy (SBMA) [93]. Gene therapy with the androgen receptor isoform for SBMA is under study [94]. In SMA, so far, there has been no report of sex hormone and reproductive system changes in SMA patients, but severe impairment of male reproductive organ development was reported in a mild SMA mouse model [82]. These studies on the role of sex hormones in neurological disorders and motor neuron diseases, and the sex differences in SMA patients observed in our study indicate that sex hormones may play a role in the sexual dimorphism of SMA, something which requires more attention and investigation.
X-chromosome inactivation refers to the fact that in female mammals, for most genes on the X chromosome, one allele on one of the X-chromosomes is silenced. In humans, about 15% of X-linked genes escape inactivation to some degree [95]. Differences in the identity and distribution of escaped genes between species and tissues suggest a role for these genes in the evolution of sex differences in specific phenotypes. Escaped genes have higher expression levels in females than in males implying that they may play a role in sexual dimorphism in complex diseases [96]. Among these escape X-linked genes, PLS3, USP9X, and UBA1 have been recognized as positive SMA modifiers [47]. The female-biased expression of PLS3, USP9X and UBA1 may contribute to sex differences of SMA [8, 97]. Other X-linked genes that may have sex-specific effects on SMA phenotypes include the androgen receptor gene, cell-signaling receptor genes and microRNA genes [95]. The protein and microRNAs coded by some of these genes have direct or indirect interactions with SMN gene products [98, 99]. More study of X-chromosome linked modifiers will advance understanding of sex differences in SMA symptoms and aid in developing novel treatments.
Conclusion and recommendations
Our study suggests that there are sex differences in SMA incidence, phenotypes, and development. However, the limitations of the aggregated data in our datasets prevented determination of details in the differences in the phenotypes and development deficiencies in SMA patients. In the TREAT-NMD dataset there was a high variability in the size of the samples from different countries, and the lack of knowledge of genetic background was a limitation. The total patient numbers were rather small for SMA4 patients with reported severity and motor scores, and for SMA1 patients with reported developmental measurements. These limitations prevent our conclusions from being definitive. Additional studies with data from patients with same genetic background, and who can be age- and sex-matched would make the results more conclusive.
This study strongly indicates the need for more systematic study of sex differences among different populations of SMA patients. Future studies require more careful data management and broader availability of detailed patient information including age, gender, ethnicity, and family history, the severity and disease progression, and the treatment regimens. Thoroughly investigating the mechanisms of sexual dimorphisms in SMA will not only lead to better understanding of the sex-specific pathologies of SMA, it is also of utmost importance for developing effective therapies. This study also strongly supports considering sex in selecting clinical therapies and evaluating potentially sex-specific outcomes, which will allow more complete understanding of the effectiveness of current therapies and potential avenues for developing alternative treatments.
ACKNOWLEDGMENTS
TREAT-NMD thanks all the registry curators, clinical experts of the neuromuscular diseases for their efforts in entering the data, and all patients for their willingness to be part of the neuromuscular disease registries. The following SMA working group of TREAT-NMD is acknowledged for SMA data contribution:
Miriam Rodrigues (TGDOC SMA Subgroup Lead, TREAT-NMD, Newcastle upon Tyne, United Kingdom), Victoria Hodgkinson (Canadian Neuromuscular Disease Registry; TGDOC SMA Subgroup Lead, TREAT-NMD, Newcastle upon Tyne, United Kingdom & The University of Calgary, Canada), Lawrence Korngut (Canadian Neuromuscular Disease Registry; The University of Calgary, Canada), Robin Forbes and Eppie Yiu (Australian Neuromuscular Disease Registry; Murdoch Children’s Research Institute, Australia), Marlène Jagut and Marjan Cosyns (Belgian Neuromuscular Diseases Registry; Sciensano, Belgium), Nina Barisic (Croatian Registry for Paediatric Neuromuscular Disorders; Croatian Child Neurology Society & Croatian Medical Association, Croatia), Vana Vukic (Croatian Registry for Paediatric Neuromuscular Disorders; University Hospital Zagreb, Croatia), Jana Haberlová and Lenka Mokrá (ReaDy – SMA; Czech Neurological Society, Society of Paediatric Neurology, Czech Republic), Nana Tatishvili and Ana Bedoshvili (Georgian Paediatric NMD Registry; M. Iashvili CCH, Georgia), Maggie C. Walter and Simone Thiele (German NMD Registry; Friedrich-Baur-Institute, Dept. of Neurology, Ludwig-Maximilians-University of Munich, Germany), Beatrix Pálmafy (Hungarian SMA Registry; National Institute of Mental Health, Neurology and Neurosurgery, Budapest, Hungary), Veronika Karcagi (Hungarian SMA Registry; Istenhegyi Genetic Diagnostic Centre, Budapest, Hungary), Venkataraman Viswanathan (Muscular Dystrophy Association, India), Ieva Micule and Dace Pretkalnina (NMS datu kolekcija; Children’s Clinical University Hospital, Latvia), Anna Lusakowska and Anna Kostera-Pruszczyk (Polish Registry of Patients with NMDs; Medical University of Warsaw, Poland), Damjan Osredkar and Tanja Golli (Registry of Slovenian Children with NMD; Department of Pediatric Neurology, University Children’s Hospital, University Medical Centre Ljubljana, Slovenia), Maria Grazia Cattinari and Eduardo Tizzano (RegistrAME: Registro de pacientes atrofia muscular espinal España; FundAME, Spain), Andrea Klein (Swiss Registry for Neuromuscular Disorders; Division of Neuropediatrics, Development and Rehabilitation, Department of Pediatrics, Inselspital, Bern University Hospital, University of Bern, Switzerland), Anne Tscherter (Swiss Registry for Neuromuscular Disorders; Institute of Social and Preventive Medicine (ISPM), University of Bern, Switzerland), İpek Gürbüz and Numan Bulut (KUKAS; Hacettepe University, Turkey), Vitaliy Matyushenko and Andriy Shatillo (Ukrainian Registry of SMA; Kharkiv Charitable Foundation “Children with spinal muscular atrophy”, Ukraine), Lindsay Murphy and Chiara Marini-Bettolo (UK SMA Patient Registry; The John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom).
We appreciate the kindness of the Chief Medical Officer, Mary Schroth MD, FAAP, FCCP and the Director of Research Data Analytics, Lisa Belter, MPH for voluntarily providing the aggregated data from Cure SMA database.
We are grateful for statistic consultation from Claudine Jurkovitz, MD, MPH, Lead of the Biostatistics Epidemiology and Research Design Core at Delaware-CTR ACCEL which is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Science of the National Institutes of Health under grant number U54GM104941 (PI: Hicks); and from Dr. Xuanren Goodman in the Department of Sociology and Criminal Justice at Delaware State University.
FUNDING
This study was supported by a Center of Biomedical Research Excellence (COBRE) award from the National Institute of General Medical Science of the National Institutes of Health under grant number P20GM103563 (PI: Harrington).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Day JW , Howell K , Place A , Long K , Rossello J , Kertesz N , Nomikos G . Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatrics. (2022) ;22: :632. |
[2] | Lefebvre S , Bürglen L , Reboullet S , Clermont O , Burlet P , Viollet L , Benichou B , Cruaud C , Millasseau P , Zeviani M , Le Paslier D , Frézal J , Cohen D , Weissenbach J , Munnich A , Melki J . Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: :155–65. |
[3] | Swoboda KJ , Prior TW , Scott CB , McNaught TP , Wride MC , Reyna SP , Bromberg MB . Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Annals of Neurology. (2005) ;57: :704–12. |
[4] | Wirth B , Karakaya M , Kye MJ , Mendoza-Ferreira N . Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annual Review of Genomics and Human Genetics. (2020) ;21: :231–61. |
[5] | Hjartarson H.T.N-BK , Sejersen T. . Disease Modifying Therapies for the Management of Children with Spinal Muscular Atrophy (5q SMA): An Update on the Emerging Evidence. Drug Des Devel Ther. (2022) ;16: :1865–83. |
[6] | Mercuri E . Spinal muscular atrophy: From rags to riches. Neuromuscular Disorders. (2021) ;31: :998–1003. |
[7] | Monani UR , Lorson CL , Parsons DW , Prior TW , Androphy EJ , Burghes AHM , McPherson JD . A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 From the Copy Gene SMN2. Human Molecular Genetics. (1999) ;8: :1177–83. |
[8] | Oprea GE , Kröber S , McWhorter ML , Rossoll W , Müller S , Krawczak M , Bassell GJ , Beattie CE , Wirth B . Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy. Science. (2008) ;320: :524–7. |
[9] | Yener İH , Topaloglu H , Erdem-Özdamar S , Dayangac-Erden D . Transcript levels of plastin 3 and neuritin 1 modifier genes in spinal muscular atrophy siblings. Pediatrics International. (2017) ;59: :53–6. |
[10] | Riessland M , Kaczmarek A , Schneider S , Swoboda KJ , Löhr H , Bradler C , Grysko V , Dimitriadi M , Hosseinibarkooie S , Torres-Benito L , Peters M , Upadhyay A , Biglari N , Kröber S , Hölker I , Garbes L , Gilissen C , Hoischen A , Nürnberg G , Nürnberg P , Walter M , Rigo F , Bennett CF , Kye MJ , Hart AC , Hammerschmidt M , Kloppenburg P , Wirth B . Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. The American Journal of Human Genetics. (2017) ;100: :297–315. |
[11] | Howell MD , Ottesen EW , Singh NN , Anderson RL , Singh RN . Gender-Specific Amelioration of SMA Phenotype upon Disruption of a Deep Intronic Structure by an Oligonucleotide. Molecular Therapy. (2017) ;25: :1328–41. |
[12] | Powis RA , Karyka E , Boyd P , Côme J , Jones RA , Zheng Y , Szunyogova E , Groen EJN , Hunter G , Thomson D , Wishart TM , Becker CG , Parson SH , Martinat C , Azzouz M , Gillingwater TH . Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight. (2016) ;1. |
[13] | Han K-J , Foster DG , Zhang N-Y , Kanisha K , Dzieciatkowska M , Sclafani RA , Hansen KC , Peng J , Liu C-W . Ubiquitin-specific Protease 9x Deubiquitinates and Stabilizes the Spinal Muscular Atrophy Protein-Survival Motor Neuron*. Journal of Biological Chemistry. (2012) ;287: :43741–52. |
[14] | Villalón E , Kline RA , Smith CE , Lorson ZC , Osman EY , O’Day S , Murray LM , Lorson CL . AAV9-Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of spinal muscular atrophy. Human Molecular Genetics. (2019) ;28: :3742–54. |
[15] | Zhou H , Meng J , Malerba A , Catapano F , Sintusek P , Jarmin S , Feng L , Lu-Nguyen N , Sun L , Mariot V , Dumonceaux J , Morgan JE , Gissen P , Dickson G , Muntoni F . Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. Journal of Cachexia, Sarcopenia and Muscle. (2020) ;11: :768–82. |
[16] | Kannan A , Jiang X , He L , Ahmad S , Gangwani L . ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain. (2020) ;143: :69–93. |
[17] | Pearn J . Genetic studies of acute infantile spinal muscular atrophy (SMA type I). An analysis of sex ratios, segregation ratios, and sex influence. Journal of Medical Genetics. (1978) ;15: :414. |
[18] | Hausmanowa-Petrusewicz I , Zaremba J , Borkowska J , Szirkowiec W . Chronic proximal spinal muscular atrophy of childhood and adolescence: Sex influence. Journal of Medical Genetics. (1984) ;21: :447. |
[19] | Ar Rochmah M , Shima A , Harahap NIF , Niba ETE , Morisada N , Yanagisawa S , Saito T , Kaneko K , Saito K , Morioka I , Iijima K , Lai PS , Bouike Y , Nishio H , Shinohara M . Gender Effects on the Clinical Phenotype in Japanese Patients with Spinal Muscular Atrophy. Kobe J Med Sci. (2017) ;63: :E41–e44. |
[20] | Farrar MA , Vucic S , Johnston HM , du Sart D , Kiernan MC . Pathophysiological Insights Derived by Natural History and Motor Function of Spinal Muscular Atrophy. The Journal of Pediatrics. (2013) ;162: :155–9. |
[21] | Lusakowska A , Jedrzejowska M , Kaminska A , Janiszewska K , Grochowski P , Zimowski J , Sierdzinski J , Kostera-Pruszczyk A . Observation of the natural course of type 3 spinal muscular atrophy: Data from the polish registry of spinal muscular atrophy. Orphanet Journal of Rare Diseases. (2021) ;16: :150. |
[22] | Winsor EJ , Murphy EG , Thompson MW , Reed TE . Genetics of childhood spinal muscular atrophy. Journal of Medical Genetics. (1971) ;8: :143. |
[23] | Smith JB , Patel A . The Wohlfart-Kugelberg-Welander disease. Neurology. (1965) ;15: :469. |
[24] | Rudnik-Schöneborn S , Lützenrath S , Borkowska J , Karwanska A , Hausmanowa-Petrusewicz I , Zerres K . Analysis of Creatine Kinase Activity in 504 Patients with Proximal Spinal Muscular Atrophy Types I–III from the Point of View of Progression and Severity. European Neurology. (1998) ;39: :154–62. |
[25] | Furukawa T , Nakao K , Sugita H , Tsukagoshi H . Kugelberg-Welander Disease: With Particular Reference to Sex-Influenced Manifestations. Archives of Neurology. (1968) ;19: :156–62. |
[26] | Feldkötter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. The American Journal of Human Genetics. (2002) ;70: :358–68. |
[27] | Ingalhalikar M , Smith A , Parker D , Satterthwaite TD , Elliott MA , Ruparel K , Hakonarson H , Gur RE , Gur RC , Verma R . Sex differences in the structural connectome of the human brain. Proceedings of the National Academy of Sciences. (2014) ;111: :823–8. |
[28] | Loke H , Harley V , Lee J . Biological factors underlying sex differences in neurological disorders. The International Journal of Biochemistry & Cell Biology. (2015) ;65: :139–50. |
[29] | McCarthy MM , Arnold AP , Ball GF , Blaustein JD , De Vries GJ . Sex Differences in the Brain: The Not So Inconvenient Truth. The Journal of Neuroscience. (2012) ;32: :2241. |
[30] | Ngun TC , Ghahramani N , Sánchez FJ , Bocklandt S , Vilain E . The genetics of sex differences in brain and behavior. Frontiers in Neuroendocrinology. (2011) ;32: :227–46. |
[31] | Lopez-Lee C , Kodama L , Gan L . Sex Differences in Neurodegeneration: The Role of the Immune System in Humans. Biological Psychiatry. (2022) ;91: :72–80. |
[32] | Vegeto E , Villa A , Della Torre S , Crippa V , Rusmini P , Cristofani R , Galbiati M , Maggi A , Poletti A . The Role of Sex and Sex Hormones in Neurodegenerative Diseases. Endocrine Reviews. (2020) ;41: :273–319. |
[33] | Young LJ , Pfaff DW . Sex differences in neurological and psychiatric disorders. Frontiers in Neuroendocrinology. (2014) ;35: :253–4. |
[34] | Bao A-M , Swaab DF . Sex Differences in the Brain, Behavior, and Neuropsychiatric Disorders. The Neuroscientist. (2010) ;16: :550–65. |
[35] | Gillies GE , McArthur S . Estrogen Actions in the Brain and the Basis for Differential Action in Men and Women: A Case for Sex-Specific Medicines. Pharmacological Reviews. (2010) ;62: :155. |
[36] | Gillies GE , Pienaar IS , Vohra S , Qamhawi Z . Sex differences in Parkinson’s disease. Frontiers in Neuroendocrinology. (2014) ;35: :370–84. |
[37] | Haaxma CA , Bloem BR , Borm GF , Oyen WJG , Leenders KL , Eshuis S , Booij J , Dluzen DE , Horstink MWIM . Gender differences in Parkinson’s disease. Journal of Neurology, Neurosurgery & Psychiatry. (2007) ;78: :819. |
[38] | Van Den Eeden SK , Tanner CM , Bernstein AL , Fross RD , Leimpeter A , Bloch DA , Nelson LM . Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. American Journal of Epidemiology. (2003) ;157: :1015–22. |
[39] | Logroscino G , Traynor BJ , Hardiman O , Chiò A , Mitchell D , Swingler RJ , Millul A , Benn E , Beghi E . Incidence of amyotrophic lateral sclerosis in Europe. Journal of Neurology, Neurosurgery & Psychiatry. (2010) ;81: :385. |
[40] | McCombe PA , Henderson RD . Effects of gender in amyotrophic lateral sclerosis. Gender Medicine. (2010) ;7: :557–70. |
[41] | Eikelboom WS , Pan M , Ossenkoppele R , Coesmans M , Gatchel JR , Ismail Z , Lanctôt KL , Fischer CE , Mortby ME , van den Berg E , Papma JM . Sex differences in neuropsychiatric symptoms in Alzheimer’s disease dementia: A meta-analysis. Alzheimer’s Research & Therapy. (2022) ;14: :48. |
[42] | Hebert LE , Weuve J , Scherr PA , Evans DA . Alzheimer disease in the United States – estimated using the census. Neurology. (2013) ;80: :1778. |
[43] | Li R , Singh M . Sex differences in cognitive impairment and Alzheimer’s disease. Frontiers in Neuroendocrinology. (2014) ;35: :385–403. |
[44] | Alonso A , Hernán MA . Temporal trends in the incidence of multiple sclerosis. Neurology. (2008) ;71: :129. |
[45] | Angeloni B , Bigi R , Bellucci G , Mechelli R , Ballerini C , Romano C , Morena E , Pellicciari G , Reniè R , Rinaldi V , Buscarinu MC , Romano S , Ristori G , Salvetti M . A Case of Double Standard: Sex Differences in Multiple Sclerosis Risk Factors. International Journal of Molecular Sciences. (2021) ;22. |
[46] | Pinares-Garcia P , Stratikopoulos M , Zagato A , Loke H , Lee J . Sex: A Significant Risk Factor for Neurodevelopmental and Neurodegenerative Disorders. Brain Sciences. (2018) ;8. |
[47] | Singh NN , Hoffman S , Reddi PP , Singh RN . Spinal muscular atrophy: Broad disease spectrum and sex-specific phenotypes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. (1867) ;166063. |
[48] | Glanzman AM , McDermott MP , Montes J , Martens WB , Flickinger J , Riley S , Quigley J , Dunaway S , O’Hagen J , Deng L , Chung WK , Tawil R , Darras BT , Yang M , Sproule D , De Vivo DC , Kaufmann P , Finkel RS , The Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy The Muscle Study G. Validation of the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatric Physical Therapy. (2011) ;23. |
[49] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , Scott C , Pane M , Messina S , Bertini E , Mercuri E , Finkel RS . The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscular Disorders. (2010) ;20: :155–61. |
[50] | Main M , Kairon H , Mercuri E , Muntoni F . The Hammersmith Functional Motor Scale for Children with Spinal Muscular Atrophy: A Scale to Test Ability and Monitor Progress in Children with Limited Ambulation. European Journal of Paediatric Neurology. (2003) ;7: :155–9. |
[51] | O’Hagen JM , Glanzman AM , McDermott MP , Ryan PA , Flickinger J , Quigley J , Riley S , Sanborn E , Irvine C , Martens WB , Annis C , Tawil R , Oskoui M , Darras BT , Finkel RS , De Vivo DC . An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscular Disorders. (2007) ;17: :693–7. |
[52] | Mazzone ES , Mayhew A , Montes J , Ramsey D , Fanelli L , Young SD , Salazar R , De Sanctis R , Pasternak A , Glanzman A , Coratti G , Civitello M , Forcina N , Gee R , Duong T , Pane M , Scoto M , Pera MC , Messina S , Tennekoon G , Day JW , Darras BT , De Vivo DC , Finkel R , Muntoni F , Mercuri E . Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle & Nerve. (2017) ;55: :869–74. |
[53] | Belter L , Cook SF , Crawford TO , Jarecki J , Jones CC , Kissel JT , Schroth M , Hobby K . An overview of the Cure SMA membership database: Highlights of key demographic and clinical characteristics of SMA members. Journal of Neuromuscular Diseases. (2018) ;5: :167–76. |
[54] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , Flynn K , Hendrickson BC , Scholl T , Sirko-Osadsa DA , Allitto BA . Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72 400 specimens. European Journal of Human Genetics. (2012) ;20: :27–32. |
[55] | Verhaart IEC , Robertson A , Leary R , McMacken G , König K , Kirschner J , Jones CC , Cook SF , Lochmüller H . A multi-source approach to determine SMA incidence and research ready population. Journal of Neurology. (2017) ;264: :1465–73. |
[56] | Archibald AD , Smith MJ , Burgess T , Scarff KL , Elliott J , Hunt CE , Barns-Jenkins C , Holt C , Sandoval K , Siva Kumar V , Ward L , Allen EC , Collis SV , Cowie S , Francis D , Delatycki MB , Yiu EM , Massie RJ , Pertile MD , du Sart D , Bruno D , Amor DJ . Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: Outcomes of 12,000 tests. Genetics in Medicine. (2018) ;20: :513–23. |
[57] | Smith M , Calabro V , Chong B , Gardiner N , Cowie S , du Sart D . Population screening and cascade testing for carriers of SMA. European Journal of Human Genetics. (2007) ;15: :759–66. |
[58] | Lace B , Micule I , Kenina V , Setlere S , Strautmanis J , Kazaine I , Taurina G , Murmane D , Grinfelde I , Kornejeva L , Krumina Z , Sterna O , Radovica-Spalvina I , Vasiljeva I , Gailite L , Stavusis J , Livcane D , Kidere D , Malniece I , Inashkina I . Overview of Neuromuscular Disorder Molecular Diagnostic Experience for the Population of Latvia. Neurology Genetics. (2022) ;8: :e685. |
[59] | Jedrzejowska M , Milewski M , Zimowski J , Zagozdzon P , Kostera-Pruszczyk A , Borkowska J , Sielska D , Jurek M , Hausmanowa-Petrusewicz I . Incidence of Spinal Muscular Atrophy in Poland – More Frequent than Predicted? Neuroepidemiology. (2010) ;34: :152–7. |
[60] | Özdemir Y , Arısoy R , Semiz A , Şanlıkan F , Akar G , Çağ M . Carrier frequency of spinal muscular atrophy in Turkish population. Perinatal Journal. (2022) ;30: :57–60. |
[61] | Soloviov O , Hryschenko N , Livshits L . Spinal muscular atrophy carrier frequency in Ukraine. Russian Journal of Genetics. (2013) ;49: :982–3. |
[62] | Chen X , Sanchis-Juan A , French CE , Connell AJ , Delon I , Kingsbury Z , Chawla A , Halpern AL , Taft RJ , Bentley DR , Butchbach MER , Raymond FL , Eberle MA , BioResource N . Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genetics in Medicine. (2020) ;22: :945–53. |
[63] | Maggi L , Bello L , Bonanno S , Govoni A , Caponnetto C , Passamano L , Grandis M , Trojsi F , Cerri F , Gardani A , Ferraro M , Gadaleta G , Zangaro V , Caumo L , Maioli M , Tanel R , Saccani E , Meneri M , Vacchiano V , Ricci G , Sorarù G , Errico E , Bortolani S , Pavesi G , Gellera C , Zanin R , Corti S , Silvestrini M , Politano L , Schenone A , Previtali SC , Berardinelli A , Turri M , Verriello L , Coccia M , Mantegazza R , Liguori R , Filosto M , Marrosu G , Tiziano FD , Siciliano G , Simone IL , Mongini T , Comi G , Pegoraro E . Adults with spinal muscular atrophy: A large-scale natural history study shows gender effect on disease. Journal of Neurology, Neurosurgery & Psychiatry. (2022) ; 10.1136/jnnp-2022-329320. |
[64] | Hjartarson H , Nathorst-Böös K , Sejersen T . Therapies for the Management of Children with Spinal Muscular Atrophy (5q SMA): An Update on the Emerging Evidence. Drug Design, Development and Therapy. (2022) ;16. |
[65] | Reilly A , Chehade L , Kothary R . Curing SMA: Are we there yet? Gene Therapy. 2022. |
[66] | Wirth B , Garbes L , Riessland M . How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Current Opinion in Genetics & Development. (2013) ;23: :330–8. |
[67] | Wirth B , Mendoza-Ferreira N , Torres-Benito L . Chapter 12 – Spinal Muscular Atrophy Disease Modifiers In Spinal Muscular Atrophy, SumnerCJ, PaushkinS, KoC-P, eds. Academic Press. (2017) ;191–210. |
[68] | Singh RN , Howell MD , Ottesen EW , Singh NN . Diverse role of survival motor neuron protein. Biochimica et Biophysica Acta (BBA) – Gene Regulatory Mechanisms. (1860) ;299–315. |
[69] | Riboldi GM , Faravelli I , Rinchetti P , Lotti F . SMN post-translational modifications in spinal muscular atrophy. Frontiers in Cellular Neuroscience. (2023) ;17. |
[70] | Shirane K , Lorincz M . Epigenetic mechanisms governing female and male germline development in mammals. Sexual Development. (2023) ;16: :1–23. |
[71] | Nishio H . PLS3 expression and SMA phenotype: A commentary on correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. Journal of Human Genetics. (2014) ;59: :64–5. |
[72] | Cao Y , Qu Y , Bai J , Jin Y , Wang H , Song F . Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. Journal of Human Genetics. (2014) ;59: :24–7. |
[73] | Stratigopoulos G , Lanzano P , Deng L , Guo J , Kaufmann P , Darras B , Finkel R , Tawil R , McDermott MP , Martens W , DeVivo DC , Chung WK . Association of Plastin 3 Expression With Disease Severity in Spinal Muscular Atrophy Only in Postpubertal Females. Archives of Neurology. (2010) ;67: :1252–6. |
[74] | Dlamini N , Josifova DJ , Paine SML , Wraige E , Pitt M , Murphy AJ , King A , Buk S , Smith F , Abbs S , Sewry C , Jacques TS , Jungbluth H . Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscular Disorders. (2013) ;23: :391–8. |
[75] | Howell MD , Ottesen EW , Singh NN , Anderson RL , Seo J , Sivanesan S , Whitley EM , Singh RN TIA1 is a gender-specific disease modifier of a mild mouse model of spinal muscular atrophy. Scientific Reports. (2017) ;7. |
[76] | Schmid PC . Beitrag zum Krankheitsbild der infantilen progressiven spinalen Muskelatrophie nach Werdnig-Hoffmann. Zeitschrift für Kinderheilkunde. (1958) ;81: :13–25. |
[77] | Spira R . Neurogenic, Familial, Girdle Type Muscular Atrophy. Stereotactic and Functional Neurosurgery. (1963) ;23: :245–55. |
[78] | Dubowitz V . Infantile muscular atrophy a prospective study with particular reference to a slowly progressive variety. Brain. (1964) ;87: :707–18. |
[79] | De Amicis R , Baranello G , Foppiani A , Leone A , Battezzati A , Bedogni G , Ravella S , Giaquinto E , Mastella C , Agosto C , Bertini E , D’Amico A , Pedemonte M , Bruno C , Wells JC , Fewtrell M , Bertoli S . Growth patterns in children with spinal muscular atrophy. Orphanet Journal of Rare Diseases. (2021) ;16: :375. |
[80] | Lipnick SL , Agniel DM , Aggarwal R , Makhortova NR , Finlayson SG , Brocato A , Palmer N , Darras BT , Kohane I , Rubin LL . Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PLOS ONE. (2019) ;14: :e0213680. |
[81] | Abati E , Corti S . Pregnancy outcomes in women with spinal muscular atrophy: A review. Journal of the Neurological Sciences. (2018) ;388: :50–60. |
[82] | Ottesen EW , Howell MD , Singh NN , Seo J , Whitley EM , Singh RN . Severe impairment of male reproductive organ development in a low SMN expressing mouse model of spinal muscular atrophy. Scientific Reports. (2016) ;6: :20193. |
[83] | Vest RS , Pike CJ . Gender, sex steroid hormones, and Alzheimer’s disease. Hormones and Behavior. (2013) ;63: :301–7. |
[84] | Rosario ER , Chang L , Head EH , Stanczyk FZ , Pike CJ . Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiology of Aging. (2011) ;32: :604–13. |
[85] | Bonomo SM , Rigamonti AE , Giunta M , Galimberti D , Guaita A , Gagliano MG , Müller EE , Cella SG . Menopausal transition: A possible risk factor for brain pathologic events. Neurobiology of Aging. (2009) ;30: :71–80. |
[86] | Siddiqui AN , Siddiqui N , Khan RA , Kalam A , Jabir NR , Kamal MA , Firoz CK , Tabrez S . Neuroprotective Role of Steroidal Sex Hormones: An Overview. CNS Neuroscience & Therapeutics. (2016) ;22: :342–50. |
[87] | Rosa-Caldwell ME , Greene NP . Muscle metabolism and atrophy: Let’s talk about sex. Biology of Sex Differences. (2019) ;10: :43. |
[88] | Venken K , Callewaert F , Boonen S , Vanderschueren D . Sex hormones, their receptors and bone health. Osteoporosis International. (2008) ;19: :1517–25. |
[89] | Bouman A , Heineman MJ , Faas MM . Sex hormones and the immune response in humans. Human Reproduction Update. (2005) ;11: :411–23. |
[90] | Sengelaub DR , Xu X-M . Protective effects of gonadal hormones on spinal motoneurons following spinal cord injury. Neural Regeneration Research. (2018) ;13. |
[91] | Palmieri A , Mento G , Calvo V , Querin G , Ascenzo C , Volpato C , Kleinbub JR , Bisiacchi PS , Sorarù G . Female gender doubles executive dysfunction risk in ALS: A case-control study in 165 patients. Journal of Neurology, Neurosurgery & Psychiatry. (2015) ;86: :574. |
[92] | McLeod VM , Chiam MDF , Lau CL , Rupasinghe TW , Boon WC , Turner BJ . Dysregulation of Steroid Hormone Receptors in Motor Neurons and Glia Associates with Disease Progression in ALS Mice. Endocrinology. (2020) ;161: :bqaa113. |
[93] | Spada ARL , Wilson EM , Lubahn DB , Harding AE , Fischbeck KH . Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. (1991) ;352: :77–9. |
[94] | Lim WF , Forouhan M , Roberts TC , Dabney J , Ellerington R , Speciale AA , Manzano R , Lieto M , Sangha G , Banerjee S , Conceição M , Cravo L , Biscans A , Roux L , Pourshafie N , Grunseich C , Duguez S , Khvorova A , Pennuto M , Cortes CJ , La Spada AR , Fischbeck KH , Wood MJA , Rinaldi C . Gene therapy with AR isoform 2 rescues spinal and bulbar muscular atrophy phenotype by modulating AR transcriptional activity. Science Advances. (2021) ;7: :eabi6896. |
[95] | Carrel L , Willard HF . X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. (2005) ;434: :400–4. |
[96] | Berletch JB , Yang F , Xu J , Carrel L , Disteche CM . Genes that escape from X inactivation. Human Genetics. (2011) ;130: :237–45. |
[97] | Tower J , Pomatto LCD , Davies KJA . Sex differences in the response to oxidative and proteolytic stress. Redox Biology. (2020) ;31: :101488. |
[98] | Wishart TM , Mutsaers CA , Riessland M , Reimer MM , Hunter G , Hannam ML , Eaton SL , Fuller HR , Roche SL , Somers E , Morse R , Young PJ , Lamont DJ , Hammerschmidt M , Joshi A , Hohenstein P , Morris GE , Parson SH , Skehel PA , Becker T , Robinson IM , Becker CG , Wirth B , Gillingwater TH . Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. The Journal of Clinical Investigation. (2014) ;124: :1821–34. |
[99] | Ottesen EW , Singh RN . Characteristics of circular RNAs generated by human Survival Motor Neuron genes. Cellular Signalling. (2020) ;73: :109696. |


