
Abstracts of the 17th International Congress on Neuromuscular Diseases (ICNMD 2022)
Contents
| Hands On Courses [HO]: | 3 |
| Teaching Courses [TC]: | 7 |
| Plenary Sessions [PL]: | 20 |
| Overarching Sessions [OS]: | 24 |
| Workshops [WS]: | 42 |
| Regional Sessions [RG]: | 56 |
| Scientific Sessions [SS]: | 58 |
| Oral Presentations [PS]: | 87 |
| ePosters [eP]: | 133 |
| Author Index | 307 |
Hands On Courses
HO01.02Neuromuscular Imaging in Myopathies
Wijntjes J1
1Radboudumc, Netherlands
Muscle ultrasound is a valuable neuromuscular imaging method in both the clinic and research settings, with proven value and reliability. Ultrasound enables the detection of pathological changes in neuromuscular disease that reflect the fatty replacement and fibrosis of affected muscles. These changes in muscle will lead to increased grayscale levels, as can be seen on muscle ultrasound and can be used to differentiate healthy muscle tissue from diseased.
Simple visual analysis will provide a lot of information about the overall muscle echogenicity and texture, and anatomical context. Taking the subcutaneous fat layer as a reference, clearly abnormal muscles are easy to spot. However, sometimes changes are more difficult to interpret because the skeletal muscles in our body all have different architectures and hence different grayscale levels. Visual interpretation thus strongly depends on observer experience. In practice, this limits the sensitivity for making a visual distinction between normal and diseased muscle to around 70%.
To make visual analysis more objective, the semi-quantitative Heckmatt grading scale can be used. This is a four-point visual grading scale, that classifies images based on the muscle grayscale compared to the overlying subcutaneous fat layer, the presence or absence of a distinct muscle architecture, and the amount of attenuation leading to decreased visibility of the underlying bone reflection.
The most sensitive and validated approach for using muscle ultrasound to discriminate between healthy and diseased muscle is offered by quantitative muscle ultrasound grayscale analysis. This quantitative technique calculates the overall mean grayscale level within a manually selected region of interest in the muscle ultrasound image and compares this to a reference value for that specific muscle that is corrected for the influence of age, length, sex and weight. This makes the technique observer independent. The reported sensitivity for detecting neuromuscular disease varies between 83 and 92%. The technique is device dependent because every ultrasound system will produce different grayscale images due to different software and hardware settings. This means that new reference values are required for each type of device and setting used. This limits the widespread use of this technique.
More and more is known about the promising possibilities of muscle ultrasound as a non-invasive biomarker. There is ongoing research to improve the semi-quantitative assessment method and a lot of energy is being put into the development of a device-independent quantitative measurement method. Alle these efforts aim to tackle the current challenges in the use of muscle ultrasound that hamper its widespread implementation and its benefits for the neuromuscular community.
HO01.04Imaging of the Respiratory Muscles in Neuromuscular Diseases
Doorduin J1
1Donders Institute for Brain, Cognition and Behaviour, Dept of Neurology, Radboud University Medical Center, Nijmegen, The Netherlands
Respiratory muscle weakness is a common feature in many neuromuscular diseases that leads to significant respiratory difficulties, such as dyspnoea, sleep disturbances, lung infections and is often the primary cause of death. Therefore, assessment of respiratory muscle structure and function in neuromuscular diseases is crucial. In the last decade, ultrasound and MRI emerged as promising imaging techniques to assess respiratory muscle structure and function. Respiratory muscle imaging directly measures the respiratory muscles and, in contrast to pulmonary function testing, is independent of patient effort and cognitive function. This makes respiratory muscle imaging suitable to use as tool in clinical respiratory management and as outcome parameter in upcoming drug trials for neuromuscular disorders, particularly in children. Different ultrasound and MRI techniques have been demonstrated useful to detect impaired respiratory muscle structure and function in patients with a neuromuscular disease. Most of these techniques focus on the most important respiratory muscle: the diaphragm. Thickness, thickening, (3D) excursion and fat infiltration of diaphragm can be measured. Using these measurements impaired diaphragm structure and function has been demonstrated in many neuromuscular diseases. Furthermore, new technological advances in ultrasound and MRI are under development that may further increase the value of respiratory muscle imaging. However, to date, a lack of standardization in measurement procedures, evaluation of extra-diaphragmatic respiratory muscles and clinical data from natural history studies impede its widespread clinical use.
HO02.02What’s in a Nerve? Neuropathology Analysis of Frozen Tissue and a New Treatable Neuropathy
Pestronk A1
1Washington University St Louis, United States
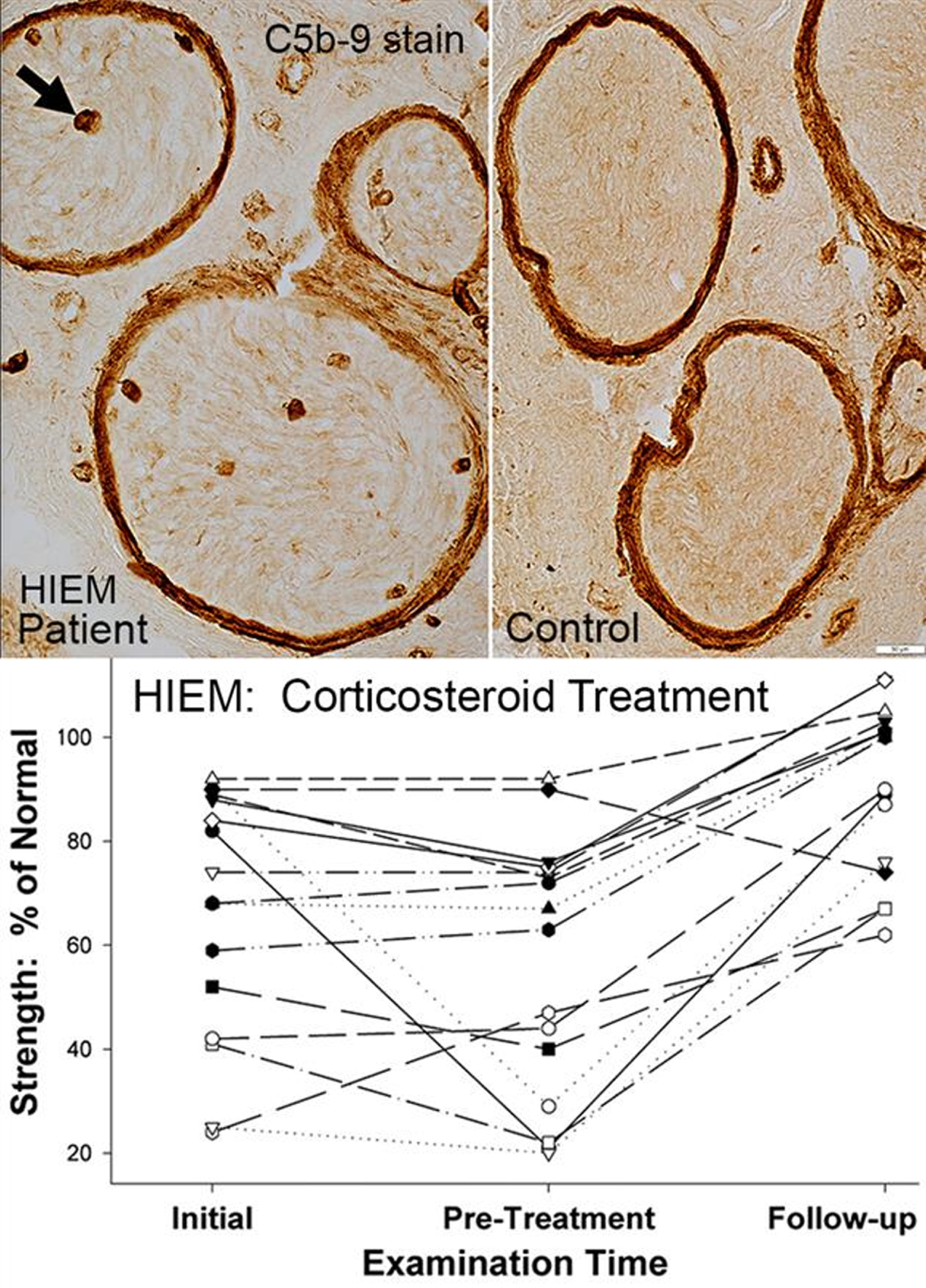
Stained sections of frozen muscle have been the standard of analysis for many years and provide a wealth of information that cannot be gleaned from fixed tissue. Our pathology laboratories routinely evaluate frozen sections of nerve biopsies, in addition to standard studies of fixed paraffin and plastic embedded specimens. Frozen sections of nerve allow for a range of histochemical and immunohistochemical analysis that is not easily performed on fixed tissue. We will describe results of staining of sections of frozen sural nerve with: Hematoxylin & Eosin, Gomori trichrome, VvG, Acid and Alkaline phosphatase, Congo red, ATPase 4.3, and antibodies to Immune features, Neurofilaments (Axons) and Schwann cell proteins. Analysis of nerve pathology using frozen sections allows rapid characterization of the states of large and small axons, Schwann cells, myelin proteins, and humoral and cellular immune disorders. We will describe the clinical and pathology features of Humoral Immune Endoneurial Microvasculopathy (HIEM), a new, treatable axonal neuropathy. HIEM is an adult onset (4th to 9th decade), motor-sensory axonal polyneuropathy with non-inflammatory, humoral immune pathology that includes C5b-9 staining of endoneurial microvessels. HIEM is manifest clinically by slowly progressive asymmetric, distal, lower extremity ± upper extremity weakness, and distal panmodal sensory loss but little pain. Diabetes is present in over 50% of HIEM patients. Weakness improves rapidly during high-dose corticosteroid treatment. HIEM may represent a new class of non-inflammatory, immune, vasculopathic, treatable axonal motor-sensory neuropathies, some masquerading as diabetic neuropathies.
HO02.03Myopathology in Congenital Myopathies
Evangelista T1
1Functional unit of neuromuscular pathology; Institute of Myology; GHU Pitié-Salpêtrière; Sorbonne University, Paris, France
Congenital myopathies (CM) are a heterogeneous group of diseases, typically presenting in the neonatal period or in early infancy with hypotonia and muscle weakness and a slow or non-progressive course. From a genetic point of view, they may be inherited in a dominant, recessive or X-linked manner, or they may arise de novo. The traditional classification of CM was based on the characteristic abnormalities observed in muscle biopsies. Four classical sub-types have been defined: nemaline myopathies, core myopathies, centronuclear myopathies, and congenital fibre type disproportion. With the advances in molecular genetics, as with other muscle diseases, a strict genotype-phenotype-histologic correlation no longer applies. Mutations in different genes can cause the same pathology and mutations in the same gene can cause multiple different phenotypes. However, even if the use of whole exome sequencing has enlarged the clinical and pathological spectrum of congenital myopathies, pathology still has a role in clarifying the pathogenicity of gene variants as well as directing molecular analysis. During this presentation, we will try to summarize the histopathologic characteristics of the “classical” subtypes of CM as well as some of the novel diseases with a CM presentation.
HO02.04Myopathology in Vacuolar and Protein Aggregate Myopathies
Olivé M1
1Neuromuscular Disorders Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau/Center for Biomedical Network Research On Rare Diseases (CIBERER)., Barcelona, España
Protein aggregate myopathies (PAMs) are a growing group of muscle disorders characterized morphologically by the presence of protein aggregates in muscle cells. Protein aggregates, observed as inclusions within muscle fibers, may occur in patients suffering from certain congenital myopathies such as actinopathies, or myosin storage myopathy, but also in late onset hereditary myopathies and even in non-genetic conditions. Structurally and functionally diverse proteins have been implicated in the causation of PAM, allowing for classification of these disorders on the basis of the causative gene.
Myofibrillar myopathies (MFMs) are the largest and best-known group of PAM. They are pathologically defined by focal disintegration of myofibrils and accumulation of degradation products into inclusions containing desmin and many other proteins. MFMs are associated with mutations in more than 15 genes. The accumulation of defective, misfolded, proteins in these conditions overloads the capacity of the ubiquitin-proteasome-system (UPS) to remove them and as a consequence, autophagy pathways are activated leading to protein degradation in lysosomes, resulting in the formation of autophagic vacuoles. The combination of protein aggregates with rimmed vacuoles is a typical feature of MFM. However, rimmed vacuoles are not restricted to MFM. On the contrary, they are also typically observed in sIBM, and are a characteristic feature of large numbers of hereditary muscle conditions including GNE-myopathy, tibial muscular dystrophy, or oculopharyngeal muscular dystrophy among many others. Rimmed vacuoles are typically surrounded by basophilic material that stains red with the modified Gomori trichrome. They usually react with antibodies against p62, ubiquitin and other proteins including TDP-43. Under EM they contain myeloid bodies, degraded organelles, and sometimes tubulofilamentous inclusions.
Besides the rimmed vacuolar myopathies mentioned above, autophagic vacuoles are the major pathological feature of a group of disorders caused by mutations in genes encoding lysosomal enzymes. Pompe’s disease, Danon disease and X-linked myopathy with excessive autophagy (XMEA) caused by mutation in GAA, LAMP2 and VMA21 respectively are classified within this group. Moreover, autophagic vacuoles are also a feature in some toxic myopathies. Immunohistochemical analysis allows to demonstrate sarcolemmal features in most of these conditions, and EM shows the lysosomal origin and contents of the vacuoles.
Finally, prominent vacuolar changes are often found in some glycogenosis. Subsarcolemmal vacuoles with glycogen accumulation are observed in type III, V and VII glycogenosis. In contrast to glycogenosis type II (Pompe’s disease) where glycogen excess is surrounded by a membrane, glycogen deposition in type III, V and VII is not membrane-bound. Histochemical reactions demonstrate lack of phosphorylase and phosphofructokinase activity in glycogenosis type V and VII respectively.
Recognition of protein aggregate myopathies and vacuolar myopathies requires a careful and extensive histochemical, immunohistochemical and electron microscopic workup of muscle biopsy to characterize the composition of the protein aggregates and the nature of the contents of the vacuoles.
In spite of the spectacular advances in the molecular characterization of patients at the genomic level, we still need to identify and characterize the muscle pathology in many patients particularly when there is uncertainty about the pathogenicity of the identified variants.
TC01.03Old and New Antibodies in IIM
Christopher L1
1Johns Hopkins University, United States
This presentation will review myositis- related autoantibodies, their discovery, and the important clinical correlations that relate to each myositis autoantibodies subtype.
We will look back at the timeline of autoantigen discovery from the 1970s until day, exploring well-known autoantibodies and the evidence for newer autoantibodies.
Detection of myositis-specific autoantibodies has undergone a metamorphosis from availability only in highly specialized laboratories to now readily available autoantibodies by line blot technology. But there are pitfalls to these detection systems and the practicing clinician must be able to interpret the results in context and with caution in some circumstances.
Phenotype-autoantibody associations will be elucidated. Specifically, autoantibodies as they relate to the myositis subtypes of dermatomyositis, immune mediated necrotizing myopathy, inclusion body myositis and overlap myositis subtypes will be discussed.
Finally, the presenter will review the evidence for pathogenicity of autoantibodies, the concept of autoantibody epiphenomena, and the intriguing possibility of virally-mediated autoantibody generation.
TC01.04Treatment Options for IIM
Benveniste O1
1Sorbonne Université, Paris, France
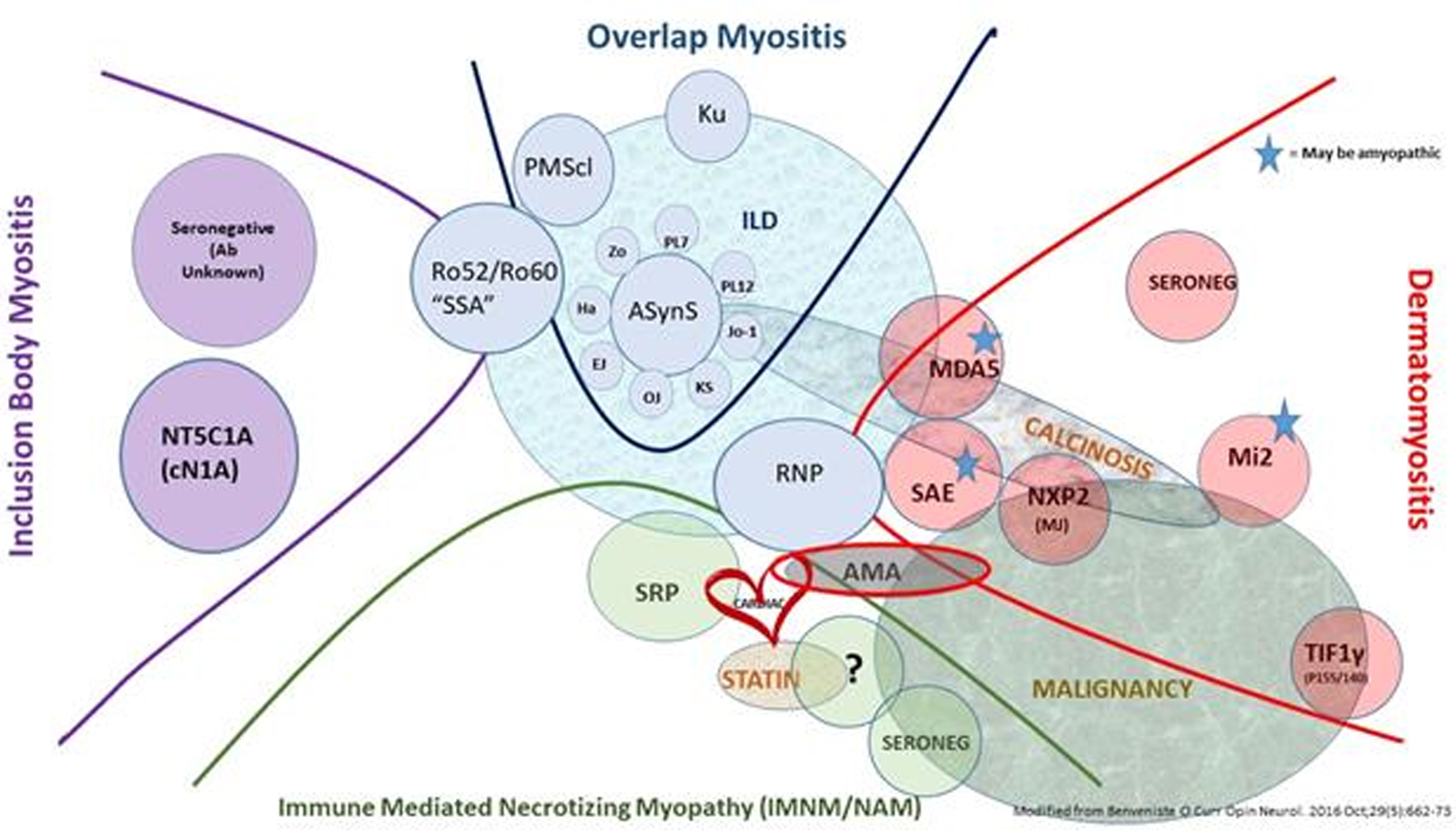
Idiopathic inflammatory myopathies (IIMs), generally known as myositis, are a heterogeneous group of different diseases having muscle weakness and inflammation within muscle tissue in common. Classification has greatly evolved over time. In the 1970s, only two diseases were recognized: polymyositis (PM) and dermatomyosites (DM). My team and I participated extensively in the advancement of IIMs classification by studying our cohort of myositis patients using an unsupervised statistical method, principal component analysis (PCA). And indeed this PCA showed the existence of, not only two, but four distinct subgroups: inclusion body myositis (IBM), anti-synthetase syndrome (ASyS), immune mediated necrotizing myopathy (IMNM) and DM, no longer simply PM. Another team also described different transcriptomic signatures in the muscle that were specific to each of the same subgroups, confirming their existence and different physiopathogenesis among the subgroups.
Therapeutic management today still focuses on high-dose and prolonged corticosteroid usage plus immunosuppressants for patients with DM, IMNM and ASyS, which improves vital prognosis, but does not prevent numerous side effects, relapses and the frequent persistence of disability due to sarcopenia. Furthermore, some DM remain rapidly lethal, and as for IBM, no processing has ever proven to be effective. These unmet needs necessitate a much more concise understanding of the physiological mechanisms to be able to finally develop specific treatments.
In DM, several studies show the presence of an interferon type I (IFN-I) signature in the blood, muscle and skin of patients, which correlates with disease activity. This correlation points to IFN-I as inducing muscle damage and to target this pathway. Based on this and further off label already treated DM patients, different trials of JAK-inhibs will soon be launched.
In IMNM, convergent data demonstrating that anti-HMGCR and anti-SRP autoantibodies (aAbs) are pathogenic. A phase II trial testing an anti-complement strategy have just be completed, but the results are negative. Further trials aiming to decrease the aAb titers will soon be launched.
In ASyS, there is evidence that disruption of tolerance to histidyl-tRNA synthetase, the antigenic target of the anti-Jo-1 aAb (which is by far the most frequent in ASyS) could occur in the lung. And the lung involvement makes the prognosis of this disease. Different immunosuppressant strategies to fight the interstitial pneumonia are ongoing.
The presence of microscopic cellular inflammation (endomysial with the invasion of muscle fibres) is the hallmark of the inflammatory nature of IBM. However, the presence of rimmed vacuoles and other ‘degenerative’ features has led to controversy regarding the pathogenesis of IBM. Nevertheless, it has been shown that IBM is driven by highly differentiated cytotoxic T cells: effector memory (TEM) and terminally differentiated (TEMRA) cells. We did a monocentric phase II trial of rapamycin (sirolimus) with enough evidence of benefit in certain secondary outcomes to pursue a multicentre phase III trial which will start soon. Finally, a Phase I study of a monoclonal antibody against KLRG1 (a marker of TEM/TEMRA cells) designed to deplete KLRG1+ T cells is ongoing, preparing a multicentric phase II/III.
TC02.012022 EAN/PNS GBS Guideline
Hadden R1
1King’s College Hospital, United Kingdom
Guillain-Barré syndrome (GBS) is an acute polyradiculoneuropathy. This is the first systematic clinical guideline, developed by an international task force using formal GRADE methodology.
The diagnostic criteria remain primarily clinical, based on history and examination findings of acute progressive limb weakness and areflexia. Variants of GBS may include motor GBS, Miller Fisher Syndrome, and regional variants with weakness predominantly in lower limbs, face, or pharynx/neck/arms.
The differential diagnosis is wide. When uncertain, diagnosis may be assisted by nerve conduction tests, raised cerebrospinal fluid protein, and less often by MRI spine with contrast, or serum antibodies to gangliosides (especially for variants) or nodal-paranodal antibodies (especially if not improving). Axonal versus demyelinating subtyping does not affect clinical management.
A history of recent gastrointestinal or respiratory infection or of surgery may support the diagnosis. The risk of GBS is only very slightly increased after Covid-19 infection and after the adenovirus-vector vaccines to SARS-CoV2 (AstraZeneca or Johnson & Johnson) but not mRNA vaccines.
Immune treatment is recommended with intravenous immunoglobulin or plasma exchange, for most patients except those mildly affected or after four weeks from onset. A repeat course is reasonable after a treatment-related fluctuation. Corticosteroids are not recommended. There is no evidence of benefit from any other disease-modifying treatment.
Respiratory function should be monitored by forced vital capacity and single breath count to assess the risk of needing mechanical ventilation, guided by the mEGRIS scale.
Pain is very common. It may be musculoskeletal or neuropathic, and treated with gabapentin, tricyclic antidepressants or carbamazepine.
Patients who fail to improve should be reassessed for the correct diagnosis and for axonal degeneration. Around 5% of patients with GBS may later develop CIDP but no test can reliably indicate this within the first eight weeks. Nodal-paranodal antibodies should be tested if CIDP is suspected or if the patient is not recovering well.
The long-term outcome is less good in patients of older age, with preceding diarrhoea, or more severe weakness, as quantified by the mEGOS scale, and also in patients with smaller motor potential amplitudes or raised serum neurofilament light chain level.
TC02.022021 EAN/PNS CIDP Guideline
Van den Bergh P1
1Cliniques universitaires Saint-Luc, Belgium
The 2021 EAN/PNS guideline on diagnosis and treatment of CIDP is a revision of the 2010 EFNS/PNS CIDP guideline. It was carried out by an international task force of disease experts according to modern GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology. Clinical and electrodiagnostic criteria as well as management recommendations and good practice points will be presented. Investigations to discover possible other diseases that may mimic CIDP should be considered and the diagnostic strategy will be addressed. We distinguished typical CIDP and CIDP variants. The previously used term ‘atypical CIDP’ was replaced by ‘CIDP variants’. CIDP variants (multifocal, focal, distal, motor, or sensory CIDP) are well characterized entities with specific clinical and electrodiagnostic phenotypes. Because of insufficient distinction between criteria for probable and definite CIDP, we reduced the levels of diagnostic certainty from three (definite, probable, possible CIDP) to only two: CIDP and possible CIDP. Electrodiagnosis is strongly recommended to confirm the clinical diagnosis of CIDP. If only possible CIDP can be diagnosed, fulfilment of 2 supportive criteria (CSF, imaging, nerve biopsy, objective response to treatment) upgrades the diagnosis to CIDP. In clinically typical CIDP not meeting minimal electrodiagnostic criteria, objective treatment response and fulfilment of 1 other supportive criterion supports the diagnosis of possible typical CIDP. Chronic immune sensory polyradiculopathy (CISP) and autoimmune nodopathies are not classified as CIDP variants. Treatment is recommended only if there is significant disability and impairment and if there is active disease (objective worsening). Recommendations for induction and maintenance treatment will be discussed.
TC02.03Autoimmune Nodopathies
Querol Gutiérrez L1
1LFB BIOMEDICAMENTS, France
Autoimmune neuropathies (AiN) are a heterogeneous group of disorders including, among others, Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). The recent discovery of antibodies targeting cell-adhesion molecules at the node of Ranvier in patients that may present as acute or chronic aggressive AiN, has challenged traditional classifications and provided novel insights into AiN pathogenesis. Antibodies targeting contactin-1, neurofascin 155, contactin-associated protein 1 and pan-neurofascin identify AiN patients with specific clinical and therapeutic features that have recently been considered a differentiated category within the AiN group: the autoimmune nodopathies (ANo). ANo are a rare subset of AiN that may fulfil GBS or CIDP diagnostic criteria, including electrophysiological features that are indistinguishable from those of other acquired demyelinating AiN but that display a type of presentation, associated clinical features, electrophysiological, pathological and genetic profiles and, most importantly, therapeutic response profile that differ from typical GBS or CIDP. These disorders, frequently (though not exclusively) mediated by IgG4 antibodies respond poorly to IVIg but respond well to B-cell depleting therapies.
The presentation at the ICNMD will provide a general overview on the involvement of node of Ranvier in AiN in general and then will review up to date literature on the clinical, pathophysiological and therapeutic features of the different.
TC02.04Biomarkers and Outcome Measures in Immune-mediated Neuropathies
Allen J1
1University of Minnesota, United States
The immune-mediated neuropathies are a collection of diseases that are generally without diagnostic or disease activity biomarkers. As our treatment paradigms get more complex and costly, it is paramount to develop objective ways to assess treatment responses that can justify ongoing immunotherapy. Ideally these tools will reflect not just clinical improvement but also help us understand changes at the tissue and pathobiologic level. To be practical they should also be easy to perform and provide rapid results that are reliable, responsive, valid and cost effective. Clinical outcomes commonly used include measures of disability and strength impairment. For some the minimally important clinical difference (MCID) has been defined, and these measures are critically important to define treatment response groups in clinical trials. In clinical practice we are learning that the strictly defined MCID may be less important than finding a profile of outcomes that move in a similar direction, even if the change is less than the MCID. We also are learning that even our best clinical outcomes are heavily influenced by placebo responses. For example, in one clinical trial that required patients to suspend open-label IVIG and show deterioration and then restabalize with open-label IVIG before randomization, still 37% of subjects who were randomized to placebo remained stable despite a recent deterioration with open-label IVIG suspension. Tissue status biomarkers are poised to supplement clinical outcomes by helping to objectify changes occurring at the nerve level. While no tissue status biomarker is ready for routine use, serum neurofilament light chain (NFL) holds promise as a disease activity biomarker in subset of patients with increased NfL before the start of treatment. Calcitonin and glial fibrillary acidic protein also have have some early data that suggest that they may have a role in understanding axonal status in inflammatory neuropathies, while sphingomyelin, neural adhesion molecule and transmembrane protease serine 5 are currently being explored as myelin status biomarkers. The immunobiologic underpinnings of CIDP have long been elusive. The discovery of the nodal and paranodal antibodies neurofascin155, neurofascin 140/186 and contactin 1 have shed insight into a subset of acquired immune mediated chronic neuropathies. These antibodies may play a role not only as diagnostic biomarkers, but as disease activity biomarkers as well. Serum contactin-1 level, independent of the autoantibody, may be a disease activity marker in some patients. Cytokine/chemokine profiles and complement profiles have been explored in small cohorts of patients. An improved understanding of changes in these profiles may provide influential data on how the immunology of the inflammatory neuropathies changes over time, and by extension how the activity of disease can be measured. Biologic outcomes require study in larger cohorts at various stages of disease to understand their utility. Much is to be learned on how clinical, tissue status, and immuobiologic disease activity outcomes can co-exist. While the search for a uniform biomarker that captures outcome in all with inflammatory neuropathy is unlikely to be realistic, biomarker profiles are poised to make great improvements in our understanding of inflammatory neuropathy disease activity status.
TC03.01Towards Targeted Therapies in Myasthenia Gravis-Pathogenic Mechanisms Translated to Treatments
Narayanaswami P1
1Harvard Medical School/ Beth Israel Deaconess Medical Center, United States
Myasthenia Gravis (MG) is caused by autoantibodies that target various proteins at the neuromuscular junction (NMJ). Various antibodies have been described in MG. The best known are antibodies directed to the AChR, present in 85% of patients with generalized MG. They effect NMJ blockade through a variety of mechanisms including binding the AChR followed by activation of complement resulting in complement mediated lysis of the post-synaptic membrane and loss of AChR, modulation, cross linking and internalization of the AChR with subsequent degradation, or direct blockade of the AChR.
Muscle-specific kinase (MuSK) is a protein that is responsible for clustering of AChRs and maintenance of the NMJ. Antibodies to MuSK are detected in about 5-8% of MG patients. Unlike AChR antibodies, they do not activate complement. These antibodies are thought to interfere with the binding of the protein Low density lipoprotein-related protein-4 (LRP-4) to MuSK and affecting normal AChR clustering. MuSK also participates in the anchoring of the collagen tail of acetylcholine esterase (Col-Q) in the synaptic cleft to the extracellular matrix at the clustering sites and this function is also affected by MuSK antibodies.
Antibodies to LRP-4 have been identified in 2-4% of all MG patients. Other antibodies against cortactin, rapsyn, titin, ryanodine receptor etc. have been described but their role in the pathogenesis of MG is uncertain at this time.
The AChR antibodies belong to the IgG1 and IgG3 subclasses, fixing complement. MuSK antibodies are of the IgG4 subclass. With advances in immunology, there are now attempts at targeting specific antibodies or their effects at the NMJ to treat MG. The first such targeted therapy is complemented inhibition. These drugs are directed to the C5 component of complement and inhibit the cleavage of C5 into C5a and C5b, preventing the formation of the terminal complement component or membrane attack complex (MAC, C5b-C9).
Another targeted therapy involves the neonatal Fc receptor (FcRN) pathway. In addition to its role in the transplacental transfer of maternal antibodies to the fetus, it is also present in endothelial and other cells where it plays a role in the maintenance of IgG and albumin levels. IgG that is internalized into the endothelial cells in the endosome binds to FcRN receptors. The non-bound IgG undergoes lysosomal degradation, while bound IgG moves to the cell surface, where it is released from FcRN and recycled into the circulation. FcRN antagonists are a new class of medications which, by binding and saturating FcRN receptors, inhibit binding of endogenous IgG, causing their degradation. Thus, levels of pathogenic antibodies (in addition to total IgG) are reduced.
Other new therapies in the pipeline include CAR-T- cell therapy, using T cells that are engineered to possess a specialized receptor, the chimeric antigen receptor, directed against B-cell maturation antigen (BCMA). BCMA is found on the plasma cell surface. The rationale behind CAR-T cell therapy in MG is to destroy antibody producing plasma cells. This talk will discuss these mechanisms, setting the stage for the other talks discussing clinical trials of these therapies.
TC03.03The role of FcRn Antagonists in MG Treatment
Gilhus N1
1University Of Bergen, Norway
Myasthenia gravis (MG) is a chronic disease with typical fluctuations. Most patients have only mild or moderate symptoms. However, in 10-15% of the patients the MG is difficult to treat with standard treatments. Even in patients with mild weakness, daily life activities are impaired and quality of life reduced.
Neonatal Fc receptors (FcRn) are instrumental in the recycling of IgG and their activity prolongs IgG half-life. FcRn antagonists prevent this recycling and reduce selectively IgG levels in the body. Efgartigimod, rozanolizumab, nipocalimab, and batoclimab are FcRn antagonists with ongoing therapeutic trials in MG. Efgartigimod is already approved for MG treatment in USA and Japan, and further approvals are expected very soon.
In the efgartigimod phase 3 study of 26 weeks including 167 patients, the primary endpoint was a marked improvement in a MG-specific scale for activities of daily living (MG-ADL responders). This was achieved in 68% of the treated patients and 30% of the placebo controls. Also, other outcome measures, and including physician-assessed muscle weakness (QMG score), showed a significant improvement a few weeks after one treatment cycle. The improvement was regarded as clinically meaningful. Among those not responding after the first treatment cycle, one third improved after a second cycle. In some patients, the improvement lasted for more than 12 weeks. Both patients with acetylcholine receptor and MuSK antibodies responded. The rate of respiratory and urinary infections increased. Phase 2 studies of rozanolizumab and nipocalimab have shown similar results.
FcRn antagonists are expected to be expensive. Cost-benefit considerations are needed before treatment of individual patients. Short-term treatment during an exacerbation may be an alternative to reduce costs. Funding policies for treatment expenses vary between countries.
FcRn antagonists have a proven and clinically meaningful therapeutic effect in MG. The drugs seem safe. Optimal dose has not yet been established. The infection risk when on treatment is probably increased, as for all immunosuppressive treatments. FcRn antagonists can be combined with other immunosuppressive drugs. Studies comparing the various FcRn antagonists are lacking, as are comparisons between FcRn antagonists and standard immunosuppressive therapies and with alternative new treatment options. Efgartigimod represents a new therapeutic option in severe, generalized MG.
TC03.04Peripheral Nervous System Complications of Immune Checkpoint inhibitors
De Bleecker J1
1Ghent University Hospital, Gent, Belgium
Immune checkpoint inhibitors (ICI) are therapeutic monoclonal antibodies that block negative regulators of T cell activation that are used by cancer cells to evade immune surveillance. Targets include cytotoxic T lymphocyte associated antigen 4 (CTLA-4 - ipilumab) and the programmed cell death protein 1 receptor (PD-1 – nivolumab, pembrolizumab, cemiplimab) and its ligand PD-L1 (atezolimab, avelumab, durvalumab). They significantly improve outcome in metaststatic melanoma, non small-cell lung carcinoma, Hodgkin lymphoma and other cancers. Releasing the brakes on immune control not unexpectedly comes with immune-related adverse events (ir-AEs).
Neurologic ir-AEs occur in about 1-2% of patients and are peripheral more than central (encephalitis, meningitis, demyelinating diseases, myelitis). Peripheral neuromuscular syndromes include in decreasing order of frequency inflammatory myopathies (IM), myasthenia gravis (MG), peripheral and cranial nerve neuropathies.
IM patients most often develop subacute pain and weakness in proximal arm and shoulder girdle more than lower limb muscles, often associated with bulbar and respiratory muscle involvement. Most patients have myositis on MRI and irritable myopathy on EMG, and about one third have one of a large variety of myositis-specific antibodies. Biopsies most often show a necrotizing myopathy. Two-thirds have a favorable response to IV or oral steroid therapy. MG manifests as a generalized, often bulbar myasthenia, with more frequent myasthenic crises than expected. About 60% have AChR antibodies; Musk antibodies are very rare. A subset has preexisting AChR antibodies. About half of the MG patients also have subtle to major signs of IM, ranging from myopathic EMG, high CK to clinical manifestations. IM and MG are mainly caused by PD-1/PD-L1 blocker treatment. Combined IM, MG and myocarditis is not uncommon and has a severe prognosis with death from bulbo-respiratory MG or cardiac rhythm and conduction disturbances in more than 25%. LEMS is rare.
Neuropathies present a monophasic or polyphasic course, are diffuse or focal, and more demyelinating than axonal. In contrast with non-irAE patients, typical Guillain-Barré syndromes often do respond well to steroids alone, although most patient have been treated with multiple immunosuppressive therapies, PLEX or IVIg in various combinations. About a fourth have various anti-ganglioside or other antibodies, and most have increased protein and mild cellular reactions in CSF. Cranial neuropathies, single or in combination, are less frequent. Facial, vestibulocochlear, optic and abducens nerve, in decreasing order, have mostly been affected. In some, there was hypophysitis.
Management depends on severity of the ir-AE (graded I-IV). Most symptomatic neurological ir-AEs are grade III-IV and will require ICI treatment be withheld indefinitely and starting high-dose steroids, along with the classic management of the given disease. Many questions remain unanswered. How to differentiate between paraneoplastic syndromes and ir-AEs in some instances, requiring opposite therapeutic decisions towards ICI use? How to approach cancer treatment in patients with underlying disorders such as MG? In daily clinical setting, the neuromuscular neurologist is often confronted with weak cancer patients, having signs of chemotherapy-induced neuropathy, steroid myopathy or vague fatigue. Use of the complete diagnostic armamentarium fortunately allows to make the correct diagnosis in the large majority of cases.
TC04.02Asymptomatic Very High Creatine Kinase
Schoser B1
1Friedrich-Baur-Institute LMU, München, Germany
Persisting high creatine kinase levels over 5000 U/L (normal <200 U/L) without any evidence of a neuromuscular metabolic, dystrophic, or neurogenic disorder are very rare. Nevertheless, on the acquired side, one need to exclude massive muscle injury or ischemia with muscle compartment syndrome, epileptic seizure, heart or barotrauma, rare e.g. snail bit intoxications, intramuscular injections, vaccination, and most commonly bacterial or viral infections and drugs. Persisting very high creatine kinase levels over 20.000 U/L without any evidence of a neuromuscular metabolic, dystrophic, or neurogenic disorder are ultrarare. Here, mostly acute intoxications, status epilepticus seizure, and viral infections like influence virus may be found. In all here mentioned conditions, a muscle mri of the thigh is highly recommended. Clinically monitoring of muscle compartment syndrome is warrant. High frequent monitoring of electrolytes, ECG, and renal function is required and normally ICU submission is recommended. Some curious cases will be presented.
TC04.03Drug Induced Weakness
Argov Z1
1Hadassah-Hebrew University, Israel
Drug induced neuromuscular disorders are more frequently encountered than the common belief. These are potentially reversible conditions if identified early but could be associated with serious clinical syndrome and are rarely even lethal. Drug induced myopathies may appear as myalgia only or as chronic weakness (with or without pain). Acute rhabdomyolysis can also be induced by medications. Emphasis will be made on the more common drugs that may cause various myopathies or other neuromuscular disorder leading to weakness. The two main groups are: statins and immune check point inhibitors.
Drug induced impairment of neuromuscular transmission may present itself as unmasking myasthenia or aggravating it but also as acute episodes in the operative and post operative period. The potential drugs that may aggravate myasthenia and their recognition will be discussed.
The lecture will present the issues with clinical cases and their dilemmas to enhance the relevance between the theory and clinical practice.
TC04.04What Do We Need to Know When Our NMD Patients Get Pregnant?
Rudnik Schoeneborn S1
1Institute of Human Genetics, Medical University, Innsbruck, Austria
Obstetric care and genetic counseling are important issues for women with neuromuscular disorders (NMD) who are contemplating pregnancy. While there is enough information available on pregnancy outcome of prevalent disorders such as Charcot Marie Tooth disease, proximal spinal muscular atrophy and myotonic dystrophy, data are sparse as regards more rare and genetically heterogeneous myopathies and neuropathies. This is a specific concern in limb girdle muscular dystrophy and structural myopathies where genetic identification of clinically distinct subgroups has been implemented in routine work-up in the past few years.
Maternity care has to consider possible cardiopulmonary limitations in pregnancy, an increased risk for thrombosis and urinary tract infections in pregnant and immobile patients. Patients with scoliosis may require individual analgesia for delivery, and specific risks for anesthesia have to be taken into account for several myopathies. In most NMDs the number of miscarriages and of hypertensive diseases in pregnancy is not increased, but deliveries occur more frequently by vaginal operations and by cesarean births. The risk for preterm deliveries and abnormal fetal presentation is increased depending on diagnosis and mobility of patients.
Despite considerable handicap in many women, pregnancy outcome is favorable in most NMDs. This applies to the large number of disorders where the neonatal outcome is not influenced by the fetal or infantile genetic defect, such as in myotonic dystrophy type 1. An exacerbation of muscle weakness in pregnancy is reported in a large proportion of women with early onset progressive proximal weakness. However, the effect is difficult to evaluate in retrospect as many disorders progress unrelated to pregnancy. Prospective studies with neurological scoring systems before, during, and after pregnancy would be required to overcome this problem.
Women with genetic NMD and an increased recurrence risk to offspring should receive genetic counselling, also to discuss their possible reproductive options. High risk couples may decide to pursue a variety of preventive measures to decrease the risk of an affected child. These include prenatal or preimplantation genetic testing, the use of donor gametes, adoption, or remaining childless. Following the decision to have own children, a multidisciplinary team should be formed to develop a care plan. Best practice would include obstetrician, neurologist, geneticist, anesthetist, respiratory and general medicine, and midwifes as appropriate. After delivery an intact social network and sufficient family support is important to make sure that the young family has a good start.
TC05.01What Can we Learn From Recent Advances in ALS Genetics?
Van Damme P1
1KU Leuven, Belgium
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder primarily affecting the motor system. It is characterized by progressive muscle weakness and wasting, limiting survival to 2-5 years after disease onset, mostly due to involvement of respiratory muscles. In about 50% of patients the disease spreads to extra-motor regions in the frontal and anterior temporal cortex, giving rise to problems in behavior, language and other cognitive domains.
Effective therapies to halt disease progression are lacking. ALS has a strong genetic component. In about 10% of patients there is a positive family history pointing towards a monogenetic cause for the disease. But also in the remaining 90% of sporadic cases, genetic factors play a role. More than 50 genes have been associated with ALS, the most commonly mutated genes are C9orf72, SOD1, TARDBP and FUS. Some of these genes cluster in pathways such as protein degradation pathways, RNA metabolism and axonal integrity/function, suggesting that these are implicated in ALS. Understanding the downstream effects of gene mutations in diverse ALS genes will be key to identify therapeutic targets for specific subtypes of ALS. Patient-derived induced pluripotent stem cell models can help to understand the consequences of gene mutations, both in motor neurons and non-neuronal cells. The first attempts to block the negative consequences of gene mutations by gene silencing using antisense oligonucleotides are underway and will hopefully change the treatment landscape of ALS in the near future.
TC05.04PLS and ALS – Part of a Continuum or Two Different Diseases?
Drory V1
1Tel-Aviv Sourasky Medical Center, Tel-Aviv, Israel
Primary lateral sclerosis (PLS) is a very rare, usually non-hereditary motor neuron disease (MND) with a prevalence is of 1-5% of all MND or 1-5/million population. It is characterized by progressive spasticity, related to the selective loss of precentral pyramidal neurons, with secondary pyramidal tract degeneration and preservation of anterior horn motor neurons. The diagnosis is based exclusively on the clinical picture, therefore is challenging and many times delayed.
Historically PLS was considered a MND separate from amyotrophic lateral sclerosis (ALS), due the loss of lower motor neuron (LMN) involvement and much slower progression over decades. But during the last 20 years the dichotomy between the syndromes became blurred, as patients are seen who convert from PLS to ALS many years after disease onset. Interestingly, some PLS patients develop also features of cognitive and behavioral impairment, compatible with a diagnosis of fronto-temporal dementia, as seen in ALS. More similarities between the syndromes relate to existence of TDP-43 cytoplasmic inclusions in frontal cortex neurons, as well as increased levels of neurofilaments in blood and cerebrospinal fluid.
In 2019 an expert meeting convened and published consensus diagnostic criteria for PLS with a classification into probable and definite disease, based on the time elapsed after symptom onset without significant LMN degeneration. Scales have been developed in order to better quantify spasticity and disease burden in PLS and reports regarding the natural history of the disease and its management were published.
Another intriguing observation is the occurrence of PLS cases in families with the need to differentiate this syndrome from hereditary spastic paraparesis (HSP) and for oriented genetic testing.
During this teaching course we will try to build a better understanding of the PLS syndrome, draw its boundaries vs ALS and HSP, and learn about its present and near future research and clinical needs.
TC06.01Clinical Approach to Diagnosis of CMT
Pareyson D1
1Fondazione IRCCS Istituto Neurologico C.Besta, Milan, Italy
Charcot-Marie-Tooth disease (CMT) and related neuropathies are a genetically highly heterogeneous group of neurodegenerative disorders. CMT affects both the sensory and motor nerves, distal Hereditary Motor Neuropathies (dHMN) are phenotypically similar disorders involving only motor nerves, while Hereditary Sensory (and Autonomic) Neuropathies (HSN/HSAN) are rare distinct disorders affecting sensory and sometimes autonomic nerves. Primary myelin involvement is commonly the basis of the demyelinating CMT which represents up to two thirds of all CMT cases, by including autosomal dominant CMT1 and autosomal recessive CMT4 types. There is an area of overlap between CMT1/4 and the primary axonal CMT2, which is filled by the less common “Intermediate CMT”, the main type being the X-linked CMTX1. Hereditary Neuropathy with liability to Pressure Palsies (HNPP) is characterized by recurrent focal neuropathies with autosomal dominant inheritance. About 100 genes have been identified as responsible for these disorders. The diagnostic approach is becoming more complicated as the associated gene number increases and the clinical characteristics of the different CMT types greatly overlap. The availability of next generation sequencing (NGS) techniques has definitely improved the diagnostic yield and at the same time makes it fundamental to well characterize the CMT patients’ phenotype to interpret NGS results. Careful clinical evaluation of patients is therefore still fundamental and need to be completed by the assessment of inheritance pattern and of nerve conduction studies (NCS). NCS and EMG are important to define presence, degree, and pattern of nerve conduction slowing, involvement of motor and/or sensory nerves, presence of spontaneous activity such as in active denervation and neuromyotonia, and to detect or rule out myopathic signs. It is also important to look for peculiar clinical features that may be specific for certain CMT subtypes, such as optic atrophy, glaucoma, cataract, hearing loss, vocal cord palsy, pyramidal tract signs/spastic paraplegia, foot and hand ulcers, autonomic dysfunction, learning difficulties, predominant upper limb involvement, differences in hand musculature involvement (split hand). Vestibular involvement and cough in chronic sensory ataxic neuropathies point to CANVAS. High serum sorbitol levels are typical of SORD-related dHMN/CMT2. Nerve biopsy, now limited to selected cases, may reveal specific myelin (i.e., myelin outfoldings) or axonal (e.g., giant axons) changes. The differential diagnosis may be challenging particularly with dysimmune neuropathies, other hereditary neuropathies, some of which are treatable, distal myopathies, slowly progressive motor neuron diseases.
TC06.03Diagnosis of TTR Amyloid Polyneuropathy for a Curable Disease
Adams D1
1Aphp University Paris Saclay, France
Hereditary transthyretin polyneuropathy (ATTRv-polyneuropathy) are the most disabling hereditary neuropathy of adult onset and life-threatening disease and considered as a worldwide disease with few endemic areas (Portugal, Sweden, Japan, Mallorca, Cyprus). They are associated with systemic manifestations including cardiac, ocular weight loss, and sometimes renal manifestations. They have an autosomal dominant transmission due to point mutation of TTR gene with about 100 TTR variants, the most common one is Val30Met. Age of onset is variable ranging from 20 to 90 yo.
ATTRv-PN belongs to the 5% of rare diseases for which disease modifying therapies (DMT) are available including 3 with marketing authorization: oral tafamidis 20 mg, IV RNAi therapy patisiran, SC ASO inotersen, with 3 other medicines in the pipeline. In this context, early diagnosis of ATTRv-PN is crucial to stop or slow progression of the disease.
Misleading diagnosis are many due to various manifestations sensory as a progressive length dependent polyneuropathy, sensorymotor or autonomic (including erectile dysfunction, gastrointestinal disorders), cardiac (arrhythmias, ventricular blocks), ocular or unexplained weight loss. Main misleading diagnosis are chronic idiopathic axonal polyneuropathy, CIDP, chronic digestive disorder, lumbar spinal stenosis, carpal tunnel syndrome, paraneoplastic neuropathy.
Redflags for diagnosis depends of age of onset and genotype. In early onset Val30Met : progressive painful polyneuropathy or autonomic dysfunction “PLUS” one of the following : positive family story, vitreous opacities, renal abnormalities or unexplained weight loss. In late onset Val30Met and other variants, progressive idiopathic polyneuropathy, or atypical CIDP “PLUS” one of the following: walking difficulties, cardiac (cardiac hypertrophy, arrhythmias, ventricular blocks, or cardiomyopathy), bilateral carpal tunnel syndrome, autonomic dysfunction, vitreous opacities or unexplained weight loss knowing that positive family story is uncommon in 50% of cases.
Tools for diagnosis are simple: TTR gene sequencing looking for one amyloidogenic TTR variant (knowing that there are a dozen of TTR polymorphism) “PLUS” amyloid deposit finding after biopsy. For biopsy, mini invasive 3 mm punch skin biopsy alone or in combination with mini-invasive 4 mm labial salivary gland biopsy allow usually to detect amyloid deposits. Both are required to confirm the disease. In late onset cases, if amyloid deposits are negative on biopsies, DPD scintigraphy may replace cardiac biopsy if myocardial radiotracer uptake.
Challenge for early diagnosis requires also a strong genetic counselling to detect variant TTR carriers at risk to develop the disease and periodic consultations including standardized interview on emerging neurological manifestations, abnormal (Neuropathy impairment score) NIS finding, change in nerve conduction study (NCS), and mini-invasive biopsy when in doubt.
Research are ongoing to validate sensitive biomarkers to detect early onset of the disease in TTR carriers.
TC06.04Approach to Complex Neuropathies
Rossor A1
1UCL Queen Square Institute of Neurology, London, United Kingdom
Peripheral neuropathy is a common finding in patients with complex inherited neurological disease and may be subclinical or a major component of the phenotype. In this talk I will provide an approach to diagnosis for this complex group of patients by addressing key questions including the predominant neurological syndrome, the type of neuropathy, and the other neurological and non-neurological features of the syndrome. The presented approach will give priority to the diagnosis of treatable diseases as well as presenting a format for investigating this group of patients.
TC07.01Morphological Aspects and Introduction
Selcen D1
1Mayo Clinic, United States
Congenital myasthenic syndromes (CMS) are genetically and clinically heterogenous syndromes caused by the impaired neuromuscular transmission. The morphologic and electrophysiological investigations are important for full characterization of the CMS. These results give clues for the appropriate treatment options. In this course, the overview of the CMS with special emphasis of the morphological and electrophysiological aspects will be discussed.
TC07.02Clinical aspects of Congenital Myasthenic Syndromes in Adulthood
Eymard B1
1Sorbonne University, France
If most CMS occur within the first 2 years, diagnosis in adulthood may be observed in two situations: 1) true late onset of the disease, more particularly in Slow-channel syndrome, but also, in our experience, in a small amount of patients with DOK7, COLQ, GFPT1, RAPSN, MUSK, LRP4,TOR1AIP1 CMS; 2) CMS beginning in infancy/childhood but undiagnosed due to minimal expression and /or short duration, or misdiagnosis (eg congenital myopathy). If onset or diagnosis in adults, the risk of misdiagnosis is particularly high: seronegative autoimmune myasthenia gravis was the main mistake (in our series DOK7,7 cases, COLQ, 6 cases, CHRNE low expressor,2 cases, Slow-channel, 1 case, GFPT1, 1 case). In our experience, other erroneous diagnoses were initially made (congenital myopathy, muscular dystrophy, distal myopathy, mitochondrial myopathy, channelopathy, motoneuron disease).
We have retrospectively studied long term course and prognosis including a large cohort of around 100 cases of patients, most of them lastly seen in adulthood, presenting with different CMS: Slow channel, AChR loss (CHRNE), DOK7, COLQ, RAPSN, MUSK, AGRN, GFPT1, DAPGT, LRP4, SLC5A7. Variable successive evolution pattern - progressive worsening, rapid exacerbations with or without regression, stability, improvement- could be present along the life in one single patient (in our series for several DOK7 and COLQ patients). Late-onset deterioration may affect patients with an initially mild disease. DOK7 CMS were particularly prone to worsen in adulthood for limb/axial/ respiratory muscles. For the patients with a Slow-channel syndrome, we observed in most of them a late progressive worsening of respiratory insufficiency, requiring often assisted ventilation. Improvement occurs for most early onset RAPSN patients, even after a very severe condition in infancy. For the AChR low expressor patients, (oldest patient of our series,78 y), recurrent exacerbations were common all along the life, but none had a long-term progressive worsening, all were ambulatory at last visit. Severity and course may differ within the same family, indicating an intrafamilial variability. Pregnancy is a risk period, whatever the gene involved. Impact of the therapy on the course of CMS is positive on long term in most DOK7, COLQ CMS with b2adrenergic (Salbutamol, Ephedrine). However, benefit of therapy was more disappointing for several Slow channel patients due to poor response to Fluoxetine and or Quinidine and for a few DOK7 with severe involvement. Three patients of our series died during, the first with agrin gene mutations, after a three decades progressive worsening from age of 20y resulting despite combined therapies in tetraplegia, severe bulbar and respiratory involvement, the second with severe (tracheostomized) but stable DOK7 CMS, after falling down the stairs, and the last one due to laryngeal spasm after use of AChE inhibitor, DOK7 CMS, diagnosed post mortem. All these features of CMS in adulthood will be illustrated by case reports.
TC07.03Genetic Aspects of Congenital Myasthenic Syndrome
Lochmüller H1,2,3
1University of Ottawa, Ottawa, Canada, 2Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada, 3The Ottawa Hospital, Ottawa, Canada
Neuromuscular junction disorders are a heterogeneous group of acquired (Myasthenia Gravis, MG) and inherited (Congenital Myasthenic Syndromes, CMS) disorders associated with distinctive clinical, electrophysiological, laboratory and ultrastructural abnormalities. The genetic defects in CMS either impair neuromuscular transmission directly or result in secondary impairments, which eventually compromise the safety margin of neuromuscular transmission. The number of genetic defects reported as causative of CMS continues to increase, with over 30 genes now implicated. In addition to early-onset severe phenotypes, we have identified two genes (DOK7, GFPT1) that cause fatigable weakness of muscles in a limb-girdle distribution, but rarely affecting facial or eye muscles. Next-generation sequencing techniques and deep phenotyping, in combination with international data sharing, have revealed not only new genetic causes of CMS, but also unusual, overlapping clinical phenotypes which blur the boundaries with primary myopathies and motor neuropathies. An increasing number of genes linked to mitochondrial function (SLC25A1, TEFM) have been found to cause both neuromuscular transmission defects as well as more severe childhood mitochondrial diseases. This highlights the importance of sharing genomics data for diagnosis and research through a secure platform such as RD-Connect. We will cover the significant progress made in understanding the molecular pathogenesis of CMS, which is important for both patients and clinicians in terms of reaching a definite diagnosis and selecting the most appropriate treatment.
TC07.04Treatment Aspects
Schara-Schmidt U1
1University Of Duisburg-essen, Children’s Hospital, Germany
Congenital myasthenic syndromes (CMS) are a genetically and phenotypically heterogeneous group of disorders caused by impaired neuromuscular transmission and characterized by the leading symptoms of muscle weakness and exercise intolerance. The severity can vary enormously and depends on the underlying genetic cause; it ranges from mild impairment to life-threatening situations in the neonatal period or in the context of crisis-like deterioration in older children and adolescents. Rarely, initial manifestations in later adulthood are also possible. Currently, at least more than 30 genes have been detected as causative for CMS. CMS are rare overall, with an estimated prevalence of 1-9 / 106 depending on the literature, which may be variable in individual countries.
In this lecture the mentioned aspects will be worked on and illustrated by case reports.
Keywords: congenital myasthenic syndromes – genetic diagnosis – treatment - long–term follow-up.
TC08.01Cognitive Impairment
Reimann J1
1Department of Neurology, University Hospital Bonn, Bonn, Germany
Cognition defines processes of knowing, attending, remembering and reasoning. It enables humans to understand themselves, communicate with and understand others, and act accordingly. Impairment of cognition is seen if a sufficient level is not reached in development or if these abilities are acquired but then lost again in maturity. In some myopathies, the frequent occurrence of such CNS involvement is well known and often structural, e.g. Duchenne muscular dystrophy, the Myotonic dystrophies and the severe, early manifestations of the alpha-dystroglycanopathies. In other disorders, changes detected by brain MRI may not be associated invariably with clinical signs (e.g. LAMA2-related muscular dystrophies). CNS involvement may be a disease component in principle, but only manifest rarely as a clinical complaint (e.g. Oculopharyngeal muscular dystrophy, OPMD) or manifest prominently in some cases but not in others due to varying penetrance (e.g. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia, IBMPFD).
Cognitive impairment can be the largest everyday challenge for patients and care-givers. But more frequently the challenge is for the clinician to recognise its presence and relevance to the patient. It is often far from easy to differentiate behaviour reactive to receiving the diagnosis of a chronic disease or to the weakness, pain, and fatigue of a muscle disorder from signs indicating an independent impairment of the CNS. Yet failing to do so will not only leave part of the disease unidentified, it may also sabotage shared decision-making and the patient’s further adaptation to the muscle symptoms in unexpected ways.
TC08.02Swallowing Difficulty in Myopathies
de Visser M1
1Amsterdam University Medical Centres, Location Academic Medical Centre, Department of Neurology, Amsterdam, The Netherlands
Swallowing is the process of clearing food and fluids from the oral cavity into the stomach. It is traditionally separated into the oral phase, the pharyngeal phase and the esophageal phase. Impaired swallowing (dysphagia) in myopathies affects usually the oropharyngeal phases. These phases rely mostly on voluntary (skeletal) muscle activity of the mouth, pharynx and upper esophageal sphincter.
In some myopathies impaired swallowing is a characteristic feature, hence involvement of the pharyngeal muscles is part of the name of the disease, e.g., oculopharyngeal muscular dystrophy. However, there are numerous myopathies in which dysphagia is also prominent, albeit often not recognized or late in the course of the disease when aspiration pneumonia occurs. Myotonic dystrophy type 1 is one of the diseases in which dysphagia may long go unnoticed, yet impaired eating and swallowing is a common and well-known problem. In Duchenne muscular dystrophy and Pompe disease management is mostly focused on alleviating disability caused by limb weakness and on treating respiratory insufficiency, yet swallowing difficulty does occur and may have a marked impact on enjoyment and quality of life. Dysphagia has been reported in all primary mitochondrial myopathies, albeit evaluation of swallowing dysfunction by performing clinical or bedside swallow assessment is not part of the patient care standard. Dysphagia occurs in all types of idiopathic inflammatory myopathies due to inflammation of the swallowing muscles. It is not uncommonly a presenting symptom, and in those patients establishing the diagnosis takes often a longer time as compared to patients with a more diffuse presentation.
There are several reasons why this is the case. First, patients often do not complain about swallowing difficulty until specifically asked for. Second, there are only few validated assessment measures which can be used in daily clinical practice to support the treating physician in establishing whether there is dysphagia and if so, its severity and cause. We can distinguish patient-related outcomes, bedside tests (for fluids and solids) and instrumental tests (imaging, manometry, electromyography), but these measures are mostly not part of the routine evaluation in daily clinical practice.
Why is it important to timely recognize swallowing difficulty? Oropharyngeal dysphagia affects respiratory safety due to aspiration pneumonia. A lack of swallowing efficacy may lead to insufficient nutrition and hydration. Patients may experience a reduced quality of life as a result of these complications and due to psychological and social aspects of dysphagia.
Therefore, it is of utmost importance that the existing swallowing assessment tools undergo disease-specific validation. The availability of clinically meaningful outcome measures is a prerequisite for the development of interventional therapies.
Plenary Sessions
PL01.01An Update on the Limb Girdle Muscular Dystrophies
Straub V1
1Institute of Translational and Clinical Research, Newcastle University, United Kingdom
The limb girdle muscular dystrophies (LGMD) have traditionally been a group of heterogeneous diseases that were defined by an autosomal recessive or dominant mode of inheritance and progressive weakness predominantly affecting the shoulder and pelvic girdle muscles. Over the past 25 years more than 30 diseases have been included in the group, as the definition of LGMD was rather generic. More recently, there have been efforts to review and revise the definition and classification of LGMD and to collect natural history data and establish care standards. The Clinical Genome Resource (ClinGen), funded by the National Institutes of Health (NIH), has established a LGMD Gene Curation Expert Panel, and the TREAT-NMD Alliance a LGMD Task Force. These and other international initiatives, including the GRASP-LGMD consortium and the Jain Foundation’s Clinical Outcome Study for patients with dysferlinopathy, support the development of new treatment strategies for LGMD and first interventional trials have been initiated to address the high unmet needs of the diverse LGMD patient group. Patient advocacy groups are proactively involved in the various activities and have tremendously helped to raise awareness for these rare genetic diseases worldwide. The talk attempts to provide a brief overview of the current forms of LGMD and will focus on genetic and phenotypic features, translational research activities, care aspects and therapy developments. The main objective is for neuromuscular healthcare professionals to get an idea of the differential diagnostic approach to LGMD, the general diagnostic workup of a patient with limb girdle weakness, the relevant care implications and the upcoming clinical trial and treatment options.
PL02.01Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: Overlap Syndromes
Kiernan M1
1Brain and Mind Centre, University of Sydney, Australia
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), once considered distinct disorders, may now be considered to form a continuum, with pure ALS at one end of the spectrum and pure FTD at the other. Traditionally, ALS was characterized by motor dysfunction with intact cognition, while the hallmark of FTD was behavioral and cognitive deficits with little or no motor disability. Such delineation now seems implausible with the mounting clinical, pathological and genetic evidence for the ALS–FTD disease continuum. Early reports of personality and behavioral changes consistent with FTD in ALS patients date back to 1889. As most of these reports were non-English publications, the concept of ALS as a multisystem disorder remained relatively unexplored until the 1980s. The concept, which was initially met with skepticism, has progressively gained acceptance. In addition to clinically established dementia, the concept of a disease continuum has emerged with evidence of subtle changes in frontal executive function, language, social cognition and behavior evident in ALS patients. Conversely, subclinical manifestations, imaging and neurophysiological abnormalities indicative of motor dysfunction, have been reported in FTD. The discovery of a trans-activating responsive (Tar) sequence DNA-binding protein (TDP-43) contributed pathological evidence to support the notion of an overlap. TDP-43 protein deposition has been reported in a subgroup of FTD patients without tau pathology, as well as the majority of familial and sporadic MND cases. A further significant piece of the overlap puzzle emerged in 2011 with identification of the C9orf72 genetic expansion linking FTD and ALS on chromosome 9p21.1. The C9orf72 repeat expansion provides important clues to disease pathogenesis and suggests potential therapeutic targets. Variable diagnostic criteria identify motor, cognitive, and behavioral deficits, but further refinement is needed to define the clinical syndromes encountered in ALS-FTD. In parallel with scientific discovery, major advances have been made in the management of patients with these neurodegenerative conditions and in understanding the molecular pathways contributing to their initial development. With the potential for disease-modifying treatments, there is a clear need to identify and classify patients appropriately, including the provision of genetic testing in sporadic patients; and as early as possible, including pre-symptomatic individuals, in terms of offering neuroprotective therapies.
PL02.03ALS/FTD Genetic Landscape and Therapies
Feldman E1
1Univeristy of Michigan Medical School, Ann Arbor, USA
ALS and FTD genetic architecture is complex, with global effort focused on identifying mutations to single genes causing disease. Forty ALS-associated genes which vary in frequency, penetrance, and mode of inheritance are known, with the most common being C9orf72, TARDBP, SOD1, and FUS. FTD is commonly associated with one of three genes: C9orf72, MAPT, or GRN, and less commonly with TARDBP, VCP, CHMP2B, SQSTM1, UBQLN1, or TBK1. While only 15% of ALS patients meet the formal criteria for FTD, over 50% of ALS patients experience cognitive and behavioral impairment, and ALS is now recognized to be a disease of widespread neurodegeneration with a continuum of signs and symptoms between ALS and FTD. The broad overlap between ALS and FTD is further supported by the fact that 30% of FTD patients develop motor symptoms during the course of their disease. In the same family, one member of the family may present initially with ALS, while another affected member presents with FTD. Investigations of genetic data from large cohorts of ALS/FTD patients have led to an increased understanding of oligogenic inheritance in ALS and FTD, where disease inheritance is affected by several genes that collectively increase disease risk. In addition, polygenic inheritance, where small changes (single nucleotide polymorphisms, a.k.a. SNPs) occur in a large number of genes, is now known to contribute to disease risk. Recent analysis of genetic profiles of ALS/FTD patients also reveals shared polygenic risk of ALS/FTD with multiple other traits, including physical activity and dyslipidemia. New therapeutic efforts have focused on monogenic disease primarily in ALS patients, with the development of antisense oligonucleotides (ASOs) to silence toxic gain-of-function genes including SOD1 and FUS. A recent clinical trial targeting C9orf72 via intrathecal administration of ASOs demonstrated safety among trial participants but has been discontinued due to lack of efficacy. The medical community eagerly awaits future therapeutic trials employing not only ASOs but a variety of other gene therapy approaches targeted against ALS and FTD.
PL03.02Predicting the Clinical Course of GBS
Jacobs B1
1Erasmus MC, Netherlands
Guillain-Barré syndrome (GBS) is an immune-mediated polyradiculoneuropathy with a highly variable clinical presentation, course and outcome. Factors that may contribute to this diversity are the various types of preceding infection and immune responses that may trigger GBS, demographic factors, GBS subtypes and response to treatment, usually with intravenous immunoglobulins (IVIg). The uncertainty about the clinical course is problematic for patients/relatives and treating physicians, especially regarding medical decisions about monitoring, admission/transfer to medium-, high-, or intensive-care beds, treatment, and planning of rehabilitation. The variation in clinical course also hampers the conduct of therapeutic trials in GBS. Numerous clinical and biological predictors of outcome have been reported in GBS, but most studies have limited power, lack multivariate analysis and external validity, have a short follow-up time and are therefore hardly applicable in practice.
Two key clinical endpoints in the course of GBS are respiratory insufficiency and regaining the ability to walk independently. Previously we have conducted extensive prognostic studies in GBS and developed several models for the prediction of these clinical endpoints in individual patients with GBS. The Erasmus GBS Respiratory Insufficiency Score (EGRIS) predicts the risk of developing respiratory failure in the first week of admission and is based on three clinical characteristics that can be obtained at the bedside at hospital entry: number of days from onset of weakness, presence of facial/bulbar palsy and extent of limb weakness (MRC sum score). The modified Erasmus GBS Outcome Score (mEGOS) predicts the risk for patients being unable to regain the capacity to walk independently at 4 weeks, 3 and 6 months after diagnosis and is based on three clinical features available at admission (or at 1 week): age, preceding diarrhea and MRC sum score. These models originated from a population of Dutch patients but have recent been validated in the cohort of the International GBS Outcome Study (IGOS) showing that the EGRIS and mEGOS are also accurate in predicting outcome in patients from other countries. The models can be used online via QxMD platform or https://gbstools.erasmusmc.nl.
Further studies in IGOS have shown that adding further clinical information to the current prognostic models will probably not increase the accuracy. Improving these prognostic models may therefore come from adding prognostic biomarkers. Potential candidates are the findings in nerve conduction studies and biomarkers in serum. These biomarkers may reflect the immune status (anti-ganglioside antibodies, albumin, complement factors and cytokines), nerve degeneration (neurofilament light chain and various myelin markers) or pharmacokinetics of IVIg (IgG). Apart from predicting outcome and monitoring of the disease activity and treatment efficacy in individual patients, the prognostic models may be used to improve the design of treatment trials. An example is the recently finalized second IVIg dose trial in patients with poor expected outcome. The progress made in prognostic modelling, in combination with the new treatments for GBS that are currently evaluated, should result in a more personalized and effective treatment and care of patients.
PL04.02Genetic Causes of Congenital Myasthenic Syndromes
Beeson D1
1Oxford University, United Kingdom
The congenital myasthenic syndromes (CMS) are rare inherited disorders of neuromuscular transmission characterised by fatiguable muscle weakness. Their overall prevalence is uncertain but is thought to be in the order of 1 in 100 000 of the population in the UK. They are genetically determined, usually autosomal recessive, so a history of consanguinity is common. Although impairment of neuromuscular transmission may often give rise to a similar clinical presentation many distinct molecular and cellular mechanisms can be involved. It has become evident over the last ten years that impaired synaptic structure and stability are disease features of equal importance as defects in proteins directly involved in signal transmission. Novel genes that harbour mutations causative for CMS include not only genes encoding proteins specifically expressed at the neuromuscular junction but also those that are more widely, or even ubiquitously, expressed. The list continues to expand and is now over 30. With many of the newly identified genes it is evident that abnormal neuromuscular transmission is only one component of a multifaceted phenotype in which muscle, the central nervous system, and other organs may also be affected. Treatment can be tailored to the underlying molecular mechanism for impaired neuromuscular transmission but treating the more complex multifaceted disorders will require development of new therapies.
PL04.03The Emerging Treatment Landscape for Myasthenia Gravis
Evoli A1
1Università Cattolica del S. Cuore, Roma, Italy
Myasthenia gravis (MG) is generally considered one of best treatable neuromuscular diseases as, with adequate therapy, most patients achieve good control of their symptoms with mild or no disability. However, clinical benefit is often hampered by the burden of treatment-related adverse effects particularly in elderly subjects with comorbidities. In addition, patients refractory to or intolerant of conventional immunosuppression (10-15% of the whole MG population) suffer from chronic disabling weakness and/or disease relapses with significant detriment of their quality-of life. Therefore, the renewed interest in MG management witnessed by the recent proliferation of clinical trials was welcomed by patients and physicians.
New therapeutic options mostly consist of monoclonal antibodies (mAbs) or synthetic peptides that target different levels of the immune system. Randomized controlled trials (RCTs) investigating agents which act at the final steps of MG immune pathogenesis, such as inhibitors of complement activation and antagonists of the Fragment crystallizable neonatal Receptor (FcRn), were generally successful. Although not truly disease-modifying, these agents proved effective in relieving MG weakness and were associated with a good safety profile. On the other hand, RCTs assessing T cell and B cell-directed therapies have failed to show significant benefit, so far. The heterogeneity of both MG itself and the study cohorts, including patients in different disease stages, may have contributed to these disappointing results. B cell depletion with rituximab is largely used in difficult-to-treat MG patients, with clinical responses influenced by disease subtype immunobiology and developmental stage. Currently, treatments targeting plasma cells through anti-CD38 mAb and B cell lineage expressing the B cell maturation antigen (BCMA) with CD8+ CAR-T cells are being investigated in phase II and phase I/II RCTs, respectively.
The possibility of selective immunotherapies is rapidly changing MG treatment, and new options will be available in the near future. In the absence of definite biomarkers of disease activity, clinicians’ responsibility is to decide the best treatment strategy in individual patients.
Overarching Sessions
OS01.01The Emerging Phenotype in Classic-infantile Pompe Disease: Challenges for the Future
van der Beek N1
1Department of Neurology / Center for Lysosomal and Metabolic Diseases, Rotterdam, Netherlands
Pompe disease is a relentlessly progressive, rare, inheritable neuromuscular disease that affects patients of all ages, both men and women, leading to severe functional limitations in daily living. Patients with the most severe ‘classic infantile’ phenotype die within 1 year when untreated due to cardiorespiratory failure, while the phenotype in older children and adults (‘late-onset’ phenotype) is dominated by a progressive limb-girdle myopathy with respiratory failure.
Successful translational research has resulted in the first effective disease-specific treatment for any genetic neuromuscular disease, which has now been used for over 15 years: enzyme-replacement therapy (ERT). This has led to a huge improvement in survival – mainly due to the good response of the heart to ERT – of patients with the classic infantile phenotype: the oldest patients are now young adults. Also, many patients now gain substantial motor skills and learn to walk, which was not possible before. However over the years we have seen a new phenotype emerge in these long-term treated classic infantile patients: 1] neuroimaging of the brain revealed progressive white matter abnormalities, while neuropsychological assessment may show a decrease in processing speed and sometimes more generalized cognitive decline, and 2] development of serious distal muscle weakness of the feet and to a lesser extent of the hands, often even more pronounced then the proximal muscle weakness.
Now that new treatment options are on the horizon, it is of great importance to delineate the variability of this ‘new phenotype’ in great(er) detail, in order to create a new point of reference for these next-generation therapies.
This presentation will provide an overview and specific example of recent successes and challenges in the field.
OS01.02The Changing Phenotype of DMD Patients
De Waele L1
1University Hospitals Leuven, Belgium
Duchenne muscular dystrophy (DMD) is a severe X-linked recessive neuromuscular disorder caused by mutations in the dystrophin gene. Patients most often present in early childhood with progressive muscle weakness resulting in loss of ambulation in early adolescence, and other life-threatening complications such as cardiac and respiratory failure resulting in early death. Life expectancy of these patients used to be shorter than 20 years, but the introduction of chronic treatment with corticosteroids and the continuous optimization of the multidisciplinary standards of care for these patients have resulted in a significant longer survival up to more than 30 years. Promising therapeutic options such as exon skipping, nonsense mutation ribosomal read-through and gene therapy may have an additional impact on life expectancy in DMD patients in the future.
This treatment evolution results in a changing phenotype of DMD patients becoming older with aspects of the disease that are less well studied such as nephrological, gastro-intestinal and cardiovascular complications, in addition to the well-known cardiac, respiratory, orthopedic and endocrinological complications. Moreover, it becomes more important to psychosocially prepare patients for their lives as adults living with DMD, and timely attend the transition process into adulthood.
DMD is no longer a paediatric disease, and a proactive and comprehensive approach is necessary to optimally prepare these patients for the highly complex healthcare needs of their adult DMD life and maximize their quality of life in the future.
OS01.03Spinal Muscular Atrophy: The Paradigm of a Disease with Changing Phenotypes
Servais L1
1University Of Oxford, United Kingdom
Three disease-modifying therapies have recently been approved in spinal muscular atrophy (SMA). In addition, the progressive initiation of newborn screening has led to further change in SMA natural history.
Classically, SMA has been classified according to the maximal motor milestone achieved by patients: SMA type 1 patients have never acquired independent sitting, SMA type 2 have never acquired independent walking, and SMA type 3 have acquired- but may have further lost independent walking. The large utilization of treatment has led to a progressive change in the understanding of this classification. Over 50% of patients with SMA1 acquire sitting position following treatment, and some patients with type 2 may acquire ambulation. Nevertheless, they present specific motor, respiratory and bulbar challenges that were not previously encountered in untreated patients. Specific examples include hip management in SMA1 patients who may become standers, and even walkers, scoliosis management in SMA2 patients who may become walkers, and persistent bulbar difficulties in patients with SMA1 even after treatment.
Patients treated before the onset of symptoms cannot be classified according to their maximal motor ability and are therefore classified on the only information available at the time of treatment, which is the number of SMN2 copies. Several patients identified by newborn screening present with symptoms at the time of treatment initiation and belong therefore to the group of “SMA type Ia” treated very early. Patients identified by newborn screening present with specific phenotypes, including normally developing children, children with motor delay and proximal deficit but with normal development, and patients with bulbar issues disconnected with the level of motor deficit.
Altogether, we observe several new phenotypes that carry on their specific challenges and should certainly prompt new standard of care.
OS02.01Facial and Vocal Recognition as a Decision Support Tool for Neuromuscular Diseases: The FACE-NMD Project
Sacconi S1,2,3
1Université Côte d’Azur, Centre Hospitalier Universitaire de Nice, Système Nerveux Périphérique & Muscle, Hôpital Pasteur 2, 30 voie Romaine CS, Nice, France, 2Université Côte d’Azur, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Institute for Research on Cancer and Aging of Nice, 28 Avenue de Valombrose, Nice, France, 3Fédération Hospitalo-Universitaire Oncoage, CHU Nice, Université Côte d’Azur (UCA), Nice, France
The vast majority of neuromuscular diseases (NMDs) are rare and characterized by complex and extremely heterogeneous phenotypes. They’re awareness among general practitioners and even general neurologists is quite low and patients often experience a long diagnostic odyssey until they are evaluated in one of the few specialized neuromuscular centers. All of this leads to frustrating diagnostic delay hampering the possibility of early treatment or access to specific therapeutic trials.
Furthermore, the clinical and genetic diagnosis of NMDs classically requires the patient presence in the clinic, and time-consuming examinations involving a multidisciplinary team of experts in rare diseases. Thus, it is costly and logistically inconvenient for patients living in isolated rural environments or in underdeveloped countries or having lost their physical autonomy.
Automated computer-assisted facial and voice/speech analysis is a rapidly evolving field, offering clinicians innovative solutions that may make their work more efficient and overcome patient’s unmet medical needs. Recently, automated computer-assisted 2D or 3D facial and voice/speech analysis have been demonstrated effective in the diagnosis of several genetic, neurological and psychiatric diseases but it has never been tested in neuromuscular diseases.
FACE-NMD project will focus on four distinct rare NMDs characterized by a typical pattern of facial and/or voice/speech involvement with a variability of clinical expression, like for example Facio-Scapulo-Humeral Muscular Dystrophy type 1 (FSHD1), Myotonic Dystrophy type 1 and 2 (DM1 and DM2), and Oculo Pharyngeal Muscular Dystrophy (OPMD). These diseases present genetic heterogeneity associated to marked variability in terms of age at onset, clinical features, disease progression and severity.
The typical pattern of facial involvement is one of the highly recognizable and characteristic features of these diseases. Hardly any study has focused on the dysfunction of the voice apparatus, speech and voice quality deriving from a specific combination of progressive facial and/or bulbar muscles impairment, the presence of dysmorphic features (including the teethes), or impact of central nervous system alterations. Facial and voice/speech involvement may be subtle in mildly affected patients or early disease stages and may appear only when the patient closes their eyes or smiles, blows, mimics a kiss, or while talking.
FACE NMD study 0 is aimed in developping facial and vocal recognition algorithms to improve the diagnosis of NMDs. Based on our preliminary results, a combined approach of static and dynamic facial, vocal and speech computer-assisted analysis is being developed to generate algorithms that can address issues related to diagnosis and follow-up of NMD patients with facial involvement.
We would like to set up a FACE-NMD Consortium in order to validate at larger scale the algorithms developed in the abovementioned study in patients affected by FSHD1, DM1, DM2 and OPMD for ther NMD.
We believe that this approach will enable the development of a disease-specific “digital signature” that might be used for early disease diagnosis, follow-up and outcome measure in clinical trials and in real-life studies through widely available mobile technologies.
OS02.02Why and How to Digitalize Bulbar Motor Dysfunction Patterns in NMD
Siciliano G1, Schirinzi E1, Ricci G1, Bechini A2, Donati M2, Marini M2, Vanello N2, Pelagatti S5, Fattori B3, Tavosanis M4, Fanucci L2
1Neurological Clinic, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy, 2Department of Information Engineering, University of Pisa, Pisa, Italia, 3Ear-Nose-Throat Unit, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italia, 4Department of Philology, Literature and Linguistics, University of Pisa, Pisa, Italy, 5Department of Informatics, University of Pisa, Pisa, Italy
Bulbar motor dysfunction in neuromuscular disorders can be related to different grade of dysphagia, respiratory and speech processing deficits. Very few studies have investigated the prosodic characteristic of speech in this type of patients, considering the difficulty to analyze the intersection between the different acoustic properties, such as pitch, speech and pause segment duration, speech rate.
In last years Artificial intelligence (AI)applied in healthcare, changes many aspects of patient care, from diagnosis to follow-up, improving the quality of life of people suffering from physical disabilities. For example, AI can improve automatic speech recognition facilitating the interaction with smart devices through the simple use of voice. In particular, AI has been able to adapt and “translate” the speech of patients with mild to medium levels of dysarthria.
Given the promising results, the DESIRE (improving Daily living for sEverely dySarthric people through enhanced automatIc speech Recognition tEchnology ) project aims to create a clinical useful tool for prognostic and rehabilitation outcome monitoring purposes in dysarthric patients. Through several reading sessions the patients pronounce predetermined selected words aloud. At the end of each session, the tool offers a measure of how much the patient’s pronunciation deviates from the one of the previous sessions, allowing the clinical specialists to work on phonemes that are more difficult for the patient to pronounce. The possibility to monitor the quality of the patient’s verbal production over time allows to tailor the speech therapy treatment based on specific parameters (eg fatigue, reduced intelligibility, difficulty in producing some phonemes / clusters / specific words).
This technological system has been tested in ALS patients and a pivotal study in Myothonic Dystrophy type 1 patients is ongoing. The recognition of non-standard natural language thanks to the collaboration among multidisciplinary competences represents a turning point for the production of solutions addressing communication needs in people with speech impairment, with high impact on the everyday quality of their life.
OS02.03Body Posture Recognition in NMDs
Mongini T1, Gadaleta G1
1Neuromuscular Unit, Department of Neuroscience, University of Torino, Torino, Italy
Postural alterations are a common sign in neuromuscular disorders (NMDs), mainly due to weakness of axial muscles, variably associated with pelvic-femoral and/or scapular and/or distal muscles. An altered posture influences deambulation and limits patients’ autonomy and represents a classic major target for rehabilitation. Moreover, in the last decades, many new phenotypes of NMDs have been genetically characterized, with subtle differences in muscle districts involvement and postural performance; therefore, postural control analysis is a helpful adjunctive tool for diagnostic pattern recognition and the follow-up of NMD patients.
In NMD clinical settings, postural control is variably evaluated by muscle strength testing, specific scales administration, video recording and gait analysis. Muscle MRI is a valuable tool to evidence specific patterns of regional involvement, but has limitations and must be compared to functional scales.
The new human body posture and gesture recognition advanced research involves many various techniques such as computer vision, sensor technology, image processing, and pattern recognition, utilizing wearable sensors located on the body or clothes of a patient to measure definite values, such as the positions of limbs and the slope degree of the body. Other systems utilize acceleration sensors to recognize different exercise postures and may be utilized to classify normal and pathological movement, mainly applied to sports and general rehabilitation fields.
In the new era of Artificial Intelligence (AI), new mathematical algorithms are developed to solve the many problems in the recognition and interpretation of human posture through computer vision, thanks to new methods for image processing, both static and dynamic. It is now also possible to analyze human behaviour automatically by recognizing the posture of the human body. In such context, Artificial Neural Networks (ANNs) and Convolutional Neural Networks (CNNs) are two examples of machine learning processes, able to compute and interpret subsets of data; when combined with video analysis and/or simpler wearable remote devices they may help in the assessment of complex kinematics of posture and gait. Based on the example of the stroke rehabilitation field, ANN - CNN technology and biosensors may supply a qualitative picture of the present physical setting (suggesting even a pattern-based diagnosis of the condition, e.g. axial and gate features in specific hereditary myopathies) and estimate prognosis predicting overall future disability and potential rehabilitation outcomes.
Only a few studies on body posture patterns have considered NMDs, such as Pompe disease or some subtypes of muscular dystrophies. When applied to the neuromuscular field, the accurate recognition of specific postural syndromes might have a differential diagnostic value, as well as a prognostic significance, similarly to the observations in other neurological diseases (i.e., extrapyramidal disorders). Moreover, the advanced programs for body posture analysis could be utilized and validated as outcome measures for the increasing number of clinical trials addressing new treatments for NMDs.
OS02.04The Role of Digital PROMs in the Data Collection for Real World Evidence Evaluation
Balicza P1, Molnar V, Palasti A, Grosz Z, Molnar M
1Semmelweis University Institute of Genomic Medicine and Rare Disorders, Hungary
With the advent of innovative therapies in many neuromuscular disorder, measuring outcomes became a crucial question. Since clinical trials include small number of patients and the development procedure is short due to the huge unmet medical needs we need further information after approval the innovative treatments. It is important for both the patients and for treating physicians, as well as for the funders to know how efficient a therapy is in real world setting. Information collected during routine clinical practice are the real world data (RWD). The result of the analysis of these data is the real world evidence.
In randomized controlled trials objective, easily measurable physical outcomes (such as 6MWT, MMT, RULM, PFTs etc.) are preferred. It is however a question what is a clinically meaningful way of measuring outcomes, what is a real-world value of a drug, how we can measure symptoms not directly in relationship with motor strength, and how we can assess outcomes in patients, who cannot perform well in these tests. Important aspects of a disease sometimes are subjective, not easily quantifiable (such as fatigue, dysphagia) or are not observable (such as sleep problems or sexual dysfunction), or are more complex than measurable in a trial context (such as everyday activities). In these situations, patient reported outcomes may give unparalleled information. General and disease specific patient reported outcome measures help us to identify important aspects of the disease, which cannot be measured in trial settings, and very importantly can serve longitudinal data, when assessed regularly. However gaining the patient for reporting symptoms is not always easy. As most of the neuromuscular diseases need long lasting cooperation with medical staff, it is also relevant to measure patient care experiences (PREMS patient care experience measures). PROMs ad PREMs then can be used in research, quality improvement, controlling and economic evaluation in order to improve patient centered care.
The aim of the lecture is to review the role of PROMs in the field of neuromuscular disorders, as well as to present our experience with adult Pompe disease and spinal muscular atrophy patients.
OS03.02Transition to Adult Services for Young People with Neuromuscular Disease: A Neurologist’s Perspective
Quinlivan R1
1MRC Centre for Neuromuscular Diseases, Queen Square, United Kingdom
Improvements in patient care, including the use of non-invasive ventilation, have led to a rapid growth of young people with neuromuscular disorders transitioning to adult care. Many adult neurology services are not yet ready for this influx of young people with highly complex medical needs. Furthermore, often, the young person and their family have not been adequately prepared for the sizemic shift from a family centred paradigm of care to a patient centred model.
Transition programs should begin in adolescence and focus on preparing the young person to become knowledgable about their condition and its management. The goal should be to coach the young person to become competent in communicating their needs so as to let their voice be heard. They should be helped to develop aspirations for the future to ensure a happy adult life, this may include planning for higher education and or living independently with support. The young person needs to learn how to manage their finances and how to employ and manage carers. Developing interests and hobbies out of school, together with a network of friends, is important to prevent social isolation once they leave full-time education.
The move from paediatric to adult services is stressful for young people and their families, psychological support should be available. Adult and Paediatric teams should work together to ensure a seamless handover of care. The adult team should check the young person’s knowledge about their condition and recognise that the young person may need support in decision making and the importance of family and friends in this process should be recognised. Adult teams should communicate in ways that a young person can understand and avoid complex jargon. To improve compliance, complex multi-disciplinary care should be co-ordinated as much as possible.
OS03.03Transition to Adult Neuromuscular Care: the Pneumologist’s Perspective
Onofri A1, Cutrera R
1Bambino Gesù Children Hospital, Italy
Improvements in management of respiratory disorders coupled with improvements in standards of medical care are increasingly allowing young people with chronic respiratory diseases to survive into adulthood. The process of transition from the pediatric to the adult healthcare system is challenging and requires special attention. Particularly, patients on long-term ventilation tend to be patients with complex needs.
Recent researches explored the transition experience of patients who had undergone the home mechanical ventilation (HMV) transition program of a tertiary children’s hospital. Identified factors that aided transition included early transition discussion, joint pediatric - adult HMV clinic visits, written information about adult services, and communication training for the adolescents to improve their capacity to give accurate medical histories and discussions of their needs with the clinical team. The barriers identified included lack of referral to other medical specialists, difficulty co-ordinating appointments across multiple adult specialists and health care settings, inadequate information on adult community funding structures and limited involvement of family doctors.
Similar to the situation in CF a few decades ago, only a limited number of adult physicians have experience in looking after these patients. Moreover, the multidisciplinary team set up which patients and their families are used to in paediatric care may only exist in a few tertiary centres. Some paediatric teams are therefore having to continue to look after these patients well into adulthood.
As mentioned above, to being under the care of the pediatric pulmonologist who manages the respiratory and LTV aspects of his/her care, patients are often also under the care of several other specialties. For example, a neuromuscular patient will also typically be under the care of the neuromuscular pediatrician, spinal surgeons for scoliosis and orthopaedic surgeons if they have dislocated hips etc. The transfer to the respective adult clinicians ideally should be done sequentially rather than all at the same time and thus requires careful advance planning.
In conclusion, transition is a process, not a single event of transference of care. The insights gained from examination of other chronic diseases highlight the absolute requirements, where possible, for better education and communication. For although all patients, whether manifesting complex needs or not, require some degree of individualized planning, this can only happen if there is a systemic recognition of the need for greater collaborative care partnerships between pediatric and adult clinicians.
OS04.01Overview on Palliative Care in Neuromuscular Disorders
de Visser M1
1Amsterdam University Medical Centres, Location Academic Medical Centre, Department of Neurology, Amsterdam, The Netherlands
Over the last decades there has been increasing awareness of the role palliative care (PC) can play in the care of people suffering from a progressive chronic neurological disease associated with a reduced life expectancy, e.g. amyotrophic lateral sclerosis (ALS). PC aims to improve the quality of life of patients and their families and promote patient autonomy. Advance care planning (ACP) is a substantial part of PC and is an iterative process of discussing and choosing future health care and medical treatment options. ACP is about listening to the patient and their significant others understanding their values, beliefs, and goals of care, and identifying their wishes about treatment options which might also include discontinuation or withholding interventions. ACP should be initiated early in the disease trajectory.
Although ALS is often considered the paradigmatic disease of PC, there are many more neuromuscular diseases (NMDs) which are associated with a severe burden both for the patient and his carer and which have a reduced life expectancy. NMDs include genetic and acquired conditions and there is a wide onset of the disease, ranging from the neonatal period to late adulthood. There is also considerable variability as regards the severity, ranging from death in infancy and early childhood to mild signs and symptoms in elderly patients. Most NMDs in which PC may be offered at some point during the disease course, are hereditary by nature, including Duchenne/Becker muscular dystrophy, congenital myopathy/muscular dystrophy, mitochondrial myopathies, myotonic dystrophy, and spinal muscular atrophy, albeit this list is not comprehensive. These NMDs are characterized by progressive muscle weakness, and there is often also cardiac and respiratory involvement which may lead to premature death. In some NMDs, treatment is currently available changing the perspective of life expectancy and disabilities which makes ACP even more challenging. Amongst acquired NMDs inclusion body myositis and myositis subtypes associated with rapidly progressive interstitial lung disease or progressive dysphagia leading to aspiration pneumonia may cause severe incapacities and not seldom an increased mortality.
Literature on PC in NMDs other than ALS is sparse, albeit there is a tendency to establish standards of care in various NMDs in which PC gets a place. Yet, PC is underutilized in neuromuscular disease. The causes are manifold. First, PC is often considered to be synonymous with end-of-life care, both by health care professionals and patients. Second, there are challenges regarding PC of patients with chronic progressive life-limiting NMDs given the broad spectrum of symptom progression, the lack of reliable and valid condition-specific outcome measures of quality of life, and the scarcity of evidence for efficacy of symptomatic treatments. Third, since patients with NMDs have different symptom profiles, they also have different needs, preferences and psychosocial issues. Fourth, health care professionals in general are found to be not very familiar with communication skills needed to deliver bad news and to discuss ACP.
Optimal palliation requires various skills and should be provided by a multidisciplinary team of health care professionals. Education, directed towards improving communication strategies, is crucial.
OS04.02The Role of the Neurologist and Palliative Care Specialist in ALS
Oliver D
1University Of Kent, United Kingdom
Palliative care services have been closely involved in the care of people with amyotrophic lateral sclerosis (ALS) from the 1960s and a survey in the UK in 2000 showed that over 75% of specialist palliative care services were involved in ALS care, but often only in the later stages (1). A survey in 2019 across Europe showed that the collaboration between palliative care and neurology was greatest for ALS/MND and cerebral tumour (2).
The collaboration varies across and within countries, often according to local expertise and interest of the specialists involved. Surveys have shown that the wider multidisciplinary approaching the care of people with ALS are developing and have a positive effect on people with ALS – in the management of symptoms, quality of life and maybe length of life. However, there may be barriers to collaboration, often depending on the education of all involved – professionals, people with ALS, their families and society.
Palliative care is now recommended according to need rather than prognosis and this challenges both professionals and people with ALS, as often palliative care is associated with end-of-life care. There is a role for palliative care throughout disease progression, based on the regular monitoring and recognition of needs – physical, psycho-social or spiritual / existential. This has been recognised in guidelines, with the close involvement of palliative care services within the multidisciplinary team.
The increased collaboration is a challenge and there is the need for greater education and mutual understanding between services – with training in palliative care for neurologists and in neurology and ALS care for specialist palliative care specialists and teams.
The development of clear pathways of care, with collaboration between all involved, would help to provide a coordinated approach to care throughout the progression of ALS, allowing people with ALS to have as good a quality of life as possible and prepare for deterioration and end of life.
References:
1. Oliver D. Webb S. The involvement of specialist palliative care in the care of people with motor neurone disease Palliat Med 2000; 14: 47-8
2. Oliver D et al. Current collaboration between palliative care and neurology: a survey of clinicians in Europe BMJ Support Palliat Care 2020; doi 10.1136/bmjspcare-2020-002322
OS04.03Approach to Palliative Care in Pediatric NMD’
von der Hagen M, Nolte-Buchholtz S, Janisch M
1Abteilung Neuropädiatrie, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany, 2Sächsisches Kinderpalliativzentrum, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany, 3Sächsisches Kinderpalliativzentrum, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
Introduction: In the last decades, advances in pediatric medicine, the implementation of standards of care and targeted therapies have led to significantly higher survival rates in children with chronic life-limiting neuromuscular (NMD) and neurodegenerative disorders. These advances in care and individualized treatment result in an increasing group of young adult patients with residua or sequelae of childhood diseases. The change introduces new challenges since increased survival results in new morbidities that may significantly affect quality of life, concepts of care and psychosocial requirements. According to the standards of pediatric palliative care by the task force on palliative care for children and adolescents of the European Association of Palliative Care (EAPC), (pediatric) palliative care approaches and principles of care should be integrated from the point of diagnosis in life limiting diseases. Duchenne muscular dystrophy (DMD) is a common NMD in childhood with well-characterized disease trajectories. Simultaneously the clinical course in DMD has been in the focus of disease modifying therapies within the last years. The standards of care in DMD imply palliative care throughout the lifespan
Objective: Algorithms, task areas and interfaces of palliative care in pediatric NMD are exemplary analyzed in DMD in Germany.
Methods: Prospective German nationwide cross-sectional survey based on a mixed-method-design of qualitative and quantitative research.
Results: Although over the last decades and in the era of disease-modifying therapies many aspects of care for children and adolescents with DMD have dramatically improved resulting in survival into adulthood and better quality of life, cardiorespiratory morbidity, pain, the maintenance of nutritional balance, emergent and elective hospitalizations and barriers in transitioning into adult care remain a significant burden for the families. Specialized palliative care levels were rarely integrated within the course of the disease. The triangulated data analyses revealed unmet needs in symptom management, psychosocial care and communication.
Conclusion: The current challenges are to define the task areas and interfaces together with all stakeholders in DMD care and to establish close cooperation. Specialized palliative care can be used as an “add-on” approach in time of need in chronic, progressive diseases rather than as a “prognosis or disease stage”. Treatment algorithms and communication strategies between care givers and the families have to be redefined in the era of novel targeted therapies.
OS05.01The Changing Scope of Newborn Screening: Wilson and Jungner, Duchenne Muscular Dystrophy and Beyond
Clarke A
1Cardiff University, Cardiff, UK
The purpose of newborn screening has usually been the early identification of infants who would benefit directly from early intervention before the recognition of an illness (effectively the Wilson and Jungner criteria of 1968). However, some newborn screening programmes have identified infants unlikely to benefit from early diagnosis but where the purpose underlying screening is different. A valuable example is that of newborn screening for Duchenne muscular dystrophy (DMD), where the clinical diagnosis is often delayed until 4-5 years. In Wales, we operated a pilot programme with an evaluation from 1990 until 1998, and where screening continued until 2011 as a clinical service. The aim of the programme was to reduce diagnostic delays, and to allow informed reproductive decisions and the active planning of care for affected boys.
Screening was offered by opt-in parental choice. More than 343,000 male infants were screened (uptake 92.8%). Of these, screening found a raised bloodspot creatine kinase (CK) in 145 cases, which remained high in 66 cases when a repeat sample was tested at 6–8 weeks, with a positive predictive value of a screen-positive result of 45.5%. Of the 66 cases with a sustained elevation of serum CK, DMD was confirmed in 56 by genotyping and/or muscle biopsy. Becker muscular dystrophy was diagnosed in 5 cases and other rarer forms of muscular dystrophy in 5 cases. Later follow-up identified 13 false-negative cases, with the sensitivity for diagnosing DMD of ~80%.
One concern that arose was about the possible routinisation of consent. This led the programme team to establish small pilot studies, in both Wales and in England, to assess modifications to the ‘information-and-consent’ process to improve the quality of decisions made. Small adjustments have the potential to improve these decisions, leading to lower uptake but greater satisfaction and less decision regret. Another concern was about the level of support for families between diagnosis and the onset of symptoms. The provision of support became problematic after cutbacks, once the programme had moved to regular health service funding.
Lessons learned from this programme can be applied to future screening programmes, whether specifically for DMD or more generally for the identification of infants who will not immediately benefit from medical interventions. The justification of such screening will be framed in terms of benefit to the wider family rather than the direct medical benefit to the affected child. Care needs to be taken to ensure that the ‘information-and-consent’ process helps families to self-identify as likely or unlikely to benefit from early diagnosis. In addition, the continuing provision of emotional and practical support to the families of identified infants and the offer of monitoring the ‘patients in waiting’ for early signs of disease, must be seen as an integral part of the programme.
The discussion of such learning points from previous research will become especially important for the protection and support of families if whole genome sequencing is introduced into programmes of newborn screening.
OS05.02Newborn Screening of Spinal Muscular Atrophy. What Have We Learned?
Servais L1
1University Of Oxford, United Kingdom
Newborn screening pilots and official programs have been progressively implemented since 2018. It was estimated in early 2021 that about 2% of the world population was screened for SMA, and that this number could reach 20% in 2026. Over the last 4 years, there was several quantitative and qualitative learnings from these different pilots
1. The data gathered in these different programs converge to confirm extremely high sensitivity and specificity of SMA NBS. In comparison with any metabolic or endocrine screening, SMA NBS is by far the most accurate.
2. Real world evidence confirmed the data obtained in pre-symptomatic trials, such as Nurture or SPR1NT, which assessed in pre-symptomatic patients the efficacy of nusinersen and gene therapy, respectively.
3. Nevertheless, about 40% of patients with 2 copies of SMN2 appear to be symptomatic at the time of diagnosis, which should mitigate the excellent prognosis obtained in pre-symptomatic trials from which such patients were excluded.
4. Health economic analysis conducted in Australia and in Belgium quantified a major and positive health economic impact of NBS.
5. Despite a published revised consensus for the treatment of patients with 4 copies of SMN2, there are still major discrepancies from one to another country in the management of these patients. No evidence could be gathered so far outside anecdotical cases to immediately treat or to closely observe these patients.
6. The mindset of parents of patients identified by NBS present some specificity, as they have not been confronted to the burden of the disease itself. Aversion for treatment burden and risk is frequently noticed, as well as anxiety for the future.
Altogether, the increasing amount of evidence that converge to demonstrate a high accuracy and cost effectiveness of SMA NBS should facilitate its integration in national newborn screening programs in the coming years.
OS05.03The Role of Preconception and Early Pregnancy Carrier Screening in Neuromuscular Disorders
Laing N1,2
1University of Western Australia, Nedlands, Australia, 2Harry Perkins Institute of Medical Research, Nedlands, Australia
There are many recessive genetic neuromuscular diseases that have early onset. This may be in utero, at birth, shortly after birth, or in infancy. These diseases are often severe, causing early death or life-long challenges for the patient and their family. These diseases include congenital muscular dystrophies, congenital myopathies, spinal muscular atrophy, Duchenne muscular dystrophy. There are no specific treatments for most of these diseases and the treatments that have been developed for some of them can be burdensome for the patient and their family and can be extremely expensive for health systems. Given the choice, couples might not wish to cause their children to have these diseases. The relevant professional bodies in many countries including the USA and Australia (the American College of Obstetricians and Gynaecologists, Royal Australian and New Zealand College of Obstetricians and Gynaecologists, Royal Australian College of General Practitioners), recommend that information about reproductive genetic carrier screening should be made available to all prospective parents. Only government-supported carrier screening can make carrier screening equitably available to all couples. Very few countries have nationwide government-funded genetic carrier screening programs in place. Israel is one. In Australia, in 2018, the Federal Health Department funded Mackenzie’s Mission, the Australian Reproductive Carrier Screening Project, to research how to provide genetic carrier screening free to any couple in this very large country that wanted to use carrier screening. In 2022, recruitment into Mackenzie’s Mission has finished, with over 9,000 couples, pre-pregnancy or in early pregnancy, enrolled from all geographic regions of Australia. The couples were tested for 1,300 genes using next generation sequencing as well as Fragile X syndrome and spinal muscular atrophy. These genes are associated with 750 diseases. Data analysis is ongoing. To date more than 150 couples have been identified as couples who did not know they were at high chance of having a child affected with one of the diseases. This includes couples at risk of having a child with ARSACS, congenital myasthenic syndrome Duchenne muscular dystrophy, Pompe disease, spinal muscular atrophy and other neurological conditions such as mucopolysaccharidoses, Tay-Sachs disease, Usher syndrome. In this year’s budget, 2022, the Australian Federal Government has announced that from September 2023 it will fund carrier testing for three common recessive genetic conditions: cystic fibrosis, Fragile X syndrome and spinal muscular atrophy, for any couple that wishes to use the testing. The Australian Federal Government had previously put in place funding for preimplantation genetic testing for couples known to be at high chance of having a child with a recessive disease. Giving couples information about their carrier status, especially pre-pregnancy, allows couples to make informed choices about their multiple reproductive options.
OS06.01Serum Biomarkers for Myotonic Dystrophy Type 1 (DM1)
Lochmüller H1,2,3
1University of Ottawa, Ottawa, Canada, 2Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada, 3The Ottawa Hospital, Ottawa, Canada
Myotonic dystrophy type 1 (DM1) is the most common adult-onset muscular dystrophy and is primarily characterized by muscle wasting and weakness. Although many biomarkers have been revealed in the rapidly developing biomarker research field that provide information on disease state and progression, limited work has been performed in rare diseases including DM1. We analysed miRNA and protein biomarkers from fibroblasts, skeletal muscle and blood samples of DM1 patients as well as DM1 mouse models. Specific miRNAs that can distinguish DM1 patients from healthy individuals and can be used as potential biomarkers for DM1 progression. We identified dysregulation of the periostin protein as a novel biomarker for DM1 that correlates with disease severity, presence of cardiac dysfunction, and presence of fibrosis. Myostatin in serum of DM1 patients is reduced, which may reflect the reduction of muscle mass. Other protein biomarkers are under investigation. We hypothesize that – while miRNA or protein biomarkers are often unspecific and can be found in other muscle conditions – combinations of biomarkers or biomarker panels can be tailored for specific purposes including the monitoring of disease progression and the effects of therapies.
OS06.02Metabolic and Mitochondrial Biomarkers
Angelini C1
1University Of Padova, Italy
In a series of metabolic lipid myopathies (i.e. carnitine deficiency, CPT2, ETF dehydrogenase defects, NLSD-M) the suggested panel of diagnostic laboratory exams consists of a smear to detect vacuolated leukocytes, the study of lactate, ammonia, acyl-carnitines, Creatine-phospho-Kinase (CK) in plasma, genomic DNA, and is useful performing a muscle with both biochemical and histochemical investigations (Fig.1 ORO stain) and an ultrastructural study or a skin biopsy to culture fibroblasts. Some exams will require the use of complex technology such as the Gas Chromatography-Mass Spectrometry (GC-MS) apparatus for organic acid profile and mass spectrometry for the acyl-carnitines profile. Nowadays a diagnostic genetic analysis is also needed by conventional Sanger technique or Next Generation Sequencing. A history of intermittent myoglobinuric attacks triggered by fasting or some stress factor may suggest a disorder either of the Glycogen (myophosphorylase)/glycolytic pathway or fatty acid oxidation pathway. In glycolytic disorders, the study of lactate during muscle anaerobic or aerobic effort is recommended and demonstrates a flat profile.
In mitochondrial disorders, the use of serum lactate at rest and after effort during aerobic exercise test is a classical biomarker, serum fibroblast growth factor 21 (FGF 21) appears both sensitive and specific for mitochondrial myopathies correlating with disease severity and respiratory deficient muscle fibers in the biopsy, indicating high potential as a serum biomarker for mitochondrial disease: FGF 21 is a specific biomarker for muscle-manifesting defects of mitochondrial translation, including mitochondrial transfer-RNA mutations and primary and secondary mtDNA deletions, the most common causes of mitochondrial diseases.
MicroRNAs (miRNAs) are small non-coding RNAs that have been shown to modulate a wide range of biological functions under various pathophysiological conditions. MiRNAs are 17-27 nucleotide long molecules that regulate posttranscriptional mRNA expression, typically by binding to the 3’-untranslated region of the complementary mRNA sequence and resulting in translational repression and gene silencing. The circulating microRNAs (miRNA) are stable and resist to RNAse activities. They can be actively released by muscle, carried by exosomes, microparticles, and apoptotic bodies, or can be passively released after sarcolemmal damage or injury. We have particularly studied “Canonical myomiRs” (miR-1; miR-133a and miR-133b, miR-206), in serum since they are enriched in muscle and are considered biomarkers of muscle regeneration, myogenesis, fiber type differentiation, degeneration, atrophy and might represent indicators of residual muscle mass consequent to atrophy of muscle lipid storage myopathies, other micro-RNA such as miR-34a and miR-122 are involved in lipid metabolism. A significant increase of myomiRNAs expression levels was detected in the serum of NLSD-M patients: muscle MRI/ CT scan demonstrated that muscle mass correlated with miR-206 levels. A marked elevation of some miRNAs involved in lipid metabolism was observed in the serum of NLSD-M patients. Both miR-34a, which plays a key role in the inhibition of expression of browning genes in obesity, and miR-122, a critical regulator of cholesterol and triglycerides synthesis in the liver, were strongly up-regulated in a case of NLSD-M with hepatosteatosis. An increase in these miRNAs correlated with high levels of hepatic enzymes, but not with CK levels.
Keywords: Creatine-Kinase, Micro-RNA, Acyl-carnitines, lactate, FGF21
OS07.01Overview of COVID-19 Infection and NM Disease Associated with COVID-19 Infection (e.g., GBS/neuritis, Myositis)
Amato A1
1Harvard Medical School / Brigham And Women’s Hospital, United States
The COVID-19 pandemic caused by infection by SARS-CoV-2 has been associated with several neuromuscular disorders including various inflammatory neuropathies (e.g., Guillain-Barre syndrome) and myopathies (e.g., rhabdomyolysis, myositis). Temporal associations however do not imply causality. In this lecture, Dr. Amato will review the literature regarding to neuromuscular complications associated with SARS-CoV-2 infection. There are conflicting studies but if SARS-CoV-2 infection indeed causes GBS it is likely quite rare (probably < 1 case per 100000 infection). Less is known about other inflammatory neuropathies (mononeuritis, plexitis, or CIDP). Larger epidemiological studies are needed to better define the causality. Elevated serum creatine kinase and myalgias are common in hospitalized patients with COVID-19 and in some myositis has been demonstrated on muscle biopsy. Furthermore, autopsy series have shown myositis and neuritis in patients who died from COVID-19 though virus has not been demonstrated in muscle or peripheral nerve. The inflammation is primarily felt to be due to cytokine release and not direct viral invasion of muscle or nerve. Finally, many affected patients have lingering symptoms (Long COVID) or Post-Acute Sequelae of COVID-19 (PASC) that often include neuromuscular symptoms (fatigue, weakness, lightheadedness, sensory loss and pain) that are of unclear nature.
OS07.03Neuromuscular Diseases Associated with COVID-19 Vaccines
Narayanaswami P1
1Harvard Medical School/ Beth Israel Deaconess Medical Center, United States
Several vaccine strategies are in use against the SARS-CoV2 virus which causes COVID-19. There are three major approaches to vaccine development: using the whole virus (live virus, inactivated virus, or viral vector vaccines), using immunogenic parts of the virus, or using the genetic material of the virus. As of April 29, 2022, 10 vaccines with different mechanisms of action have received EUL by WHO.1 In the United States, 3 vaccines have received either emergency use authorization or FDA approval.2 The timing and number of injections in the primary series and timing and need for booster doses of these vaccines depend on the age of the person, underlying immunocompromised status.4
The vaccines have overall been demonstrated to be safe and effective in preventing COVID-19 infection and also in preventing serious COVID-19 infection. However they are rarely associated with some serious side effects. Thrombosis with thrombocytopenia syndrome (TTS) after Johnson & Johnson/Janssen adenoviral vector vaccine is a rare complication that appears to affect women between the ages of 30 and 49 years. It has also been described with the Oxford/Astra Zeneca vaccine. Based on the available data there is no increased risk for TTS after mRNA vaccination, although cases of immune thrombocytopenia have bene reported after the Pfizer/BioNTech and Moderna vaccines.3,4 An increased risk of Guillain-Barré syndrome has also been associated with this the Janssen vaccine.3 Myocarditis and pericarditis, particularly in male adolescents and young adults have been associated with the Pfizer and Moderna mRNA vaccines.5
Currently, antibody testing either post infection or post vaccination are not recommended for various reasons.6, 7
Preexposure prophylaxis with a combination of 2 long-acting antibodies, tixagevimab-cilgavimab target the receptor binding domain of the SARS-CoV-2 spike protein and inhibit virus attachment. In December 2021, the US FDA provided emergency use authorization for preexposure prophylaxis of COVID-19 in patients 12 years or older weighing at least 40 kg, not currently infected with COVID-19 or have a known recent exposure and either have moderate to severe immunocompromise due to a medical condition or receipt of immunosuppressive treatments and may not mount an adequate immune response to COVID-19 vaccines, or in whom available vaccines are not recommended due to prior severe adverse reaction.8 This consists of 2 consecutive intramuscular injections at the same visit and the efficacy appears to last for up to 6 months. It was granted marketing authorization in the EU in March 2022.
References:
1. https://covid19.trackvaccines.org/agency/who/
2. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines.html
3. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html
4. Lee, E et al., Am J Hematol.2021;96:534–537
5. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html
6. Chemaitelly H et al, NEJM 2021; 385:2585
7. Bergwerk M et al. NEJM 2021; 385: 1474
OS08.01Rhabdomyolysis and Acute Myopathies
Argov Z1
1Hadassah-Hebrew University, Israel
The clinical syndrome of rhabdomyolysis is based on the ‘classical’ triad of myalgia, weakness and pigmenturia. For this lecture it will be defined as an acute such clinical syndrome with markedly elevated serum CK. The common general triggers of rhabdomyolysis are: exertion, heat & fever, fasting and dehydration, drugs and anesthesia and muscle trauma. The causes of rhabdomyolysis are divided to two general groups: genetic (metabolic myopathies and dystrophies) and acquired. The main complications of rhabdomyolysis are acute kidney failure and electrolyte imbalance. There is no consensus on treatment guidelines but the main therapeutic modes are high fluid load (if renal status allows) and alkalization of urine. Initial observations on rhabdomyolysis and COVID-19 will be discussed.
The lecture will present the issues with clinical cases and their dilemmas to enhance the relevance between the theory and clinical practice.
OS08.02Myasthenic Crisis
de Visser M1
1Amsterdam University Medical Centres, Location Academic Medical Centre, Department of Neurology, Amsterdam, The Netherlands
Myasthenia gravis (MG) is an acquired autoimmune disorder of the neuromuscular junction, caused by antibodies that target the post-synaptic membrane. These antibodies most commonly bind to the nicotinic acetylcholine receptor (AChR), but in a smaller proportion of cases, antibodies to muscle specific tyrosine kinase (MuSK)(1-10%) or to lipoprotein receptor-related protein 4 (Lrp-4)(1-3%) can be present instead. These antibodies act on the receptors, prevent neuromuscular transmission and induce weakness of skeletal muscles. In 10-15% of MG patients, no antibodies are detected and these patients are designated as “seronegative.”
Weakness can be generalized or localized, is usually more proximal than distal, and nearly always includes eye muscles, causing diplopia and/or ptosis. The pattern of involvement is usually symmetric, except for the eye involvement, which is mostly markedly asymmetric. The muscle weakness typically increases with exercise and repetitive muscle use (fatigue) and varies over the course of a day and from day to day.
Patients commonly present first with ocular manifestations, however, the majority develop generalized muscle weakness, involving the facial and bulbar muscles (dysarthria, dysphagia), the limbs, the neck and axial muscles (dropped head, bent spine), and in severe cases the diaphragmatic and intercostal muscles.
MuSK-MG predominantly appears in women, who show weakness in mostly cranial and bulbar muscles, commonly with an acute onset and a tendency to rapid progression in comparison to AchR-MG.
Myasthenic crisis (MC), the severe end of the disease spectrum, can occur at any age and is potentially life-threatening. This is a clinical emergency that requires management in an intensive care setting. MC is mostly provoked by infections or inadequate treatment. In 15-40% of the reported patients with COVID-19 infection a MC occurred. MC appears in around 15–20% of MG patients in the first 2 years after diagnosis. MC can be the first manifestation of MG. Up to a half of MuSK-MG patients develop a MC in their disease course and it is also common in patients with thymoma-associated disease, or AChR-positive late-onset disease; after surgery (including thymectomy); during or after childbirth; in patients taking a contraindicated medication; at the start of corticosteroid treatment or during the tapering of immunosuppression. In approximately 20% the cause of an exacerbation remains unknown. Characteristic symptoms for the impending MC include rapidly progressive muscle weakness, ‘inverse aspiration’, dysphagia with choking, and dyspnoea associated with orthopnoea and/or tachypnoea which can result in respiratory insufficiency. The clinical management of MC with mechanical ventilation, extended intensive care management and intravenous immunoglobulins (IVIg) or plasmapheresis (PLEX) or in case of persistent MC escalation with rituximab has led to a significant decline in mortality from around 40% in the early 1960s to 5-22% in recent studies with negative prognostic factors including older age at onset, prolonged intubation, and associated comorbidities. At present IVIg and PLEX are considered the gold standard treating MC. However, it may be conceivable that newly developed monoclonal antibody therapy (eculizumab, efgartigimod), could be used as rescue therapy to achieve a significant and rapid clinical improvement.
OS08.03ICU in NMD
Damian M1
1Essex Cardiothoracic Centre, Mid and South Essex NHS Foundation Trust, Basildon, United Kingdom, 2Division of Anaesthetics, University of Cambridge, Cambridge, United Kingdom
Neuromuscular disorders constitute a small proportion of patients treated in the intensive care unit (ICU), but an important one, not least because of their good chance of satisfactory recovery. ICU treatment may be necessary for a newly manifesting neuromuscular disorder; for an exacerbation of a longstanding disease; or neuromuscular disease may arise as a complication of ICU treatment. The general ICU treatment of a neuromuscular disorder includes monitoring and management of respiratory failure; cardiac management; and early neurorehabilitation. These are often successful even when there is no specific treatment. Treatment specific to the underlying disorder may include immunomodulatory treatment in autoimmune disorders, or treatment of an underlying metabolic defect. Precise diagnosis at the earliest possible is essential but may present with difficulties in the ICU setting. This talk focuses on those myopathies which may present to the ICU; their emergency assessment, differential diagnosis in the acute phase; and treatment and pitfalls in their management are discussed.
OS09.01Use of Telemedicine and Home Infusion
Siciliano G1, Bianchi F1, Torri F1, Merico E1, Ricci G1
1Neurological Clinic, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
The COVID-19 pandemic has further highlighted the management difficulties of NMDs patients and the necessity to continue the program of implementation of standard of care yet started in Europe with the definition of the ERNs and with different applications in the various European countries. It will be increasingly necessary to favor and further develop smart management care through the implementation of IT platforms, telemedicine services and other eHealth technologies. In the near future we are moving towards a health system that supports scientific research, strengthens prevention, and brings “medicine home”.
The outbreak of COVID-19 has forced the health care system to undergo profound rearrangements in its services and facilities. In this setting, inpatient and outpatient services had to rethink and reorganize their activities to meet the needs of patients during the “lockdown”.
The rapid and abrupt COVID-19 shutdowns and stay-at-home orders imposed challenges to routine clinical management and clinical trials. The opportunity for real-world evaluation and reduced patient burden are clear benefits to remote assessment and may provide a more robust understanding and characterization of disease impact in NMD.
The quality of patient-physician relationship, the modality of remote clinical assessment and monitoring, and the administration of therapies are the key elements to be provided in neuromuscular Telemedicine. Telemedicine permits the maintenance of a close patient-physician relationship. Other important aspect is digital therapies: i.e., access in hospital settings to perform medical therapies that can be performed only and exclusively in protected settings (such as antisense oligonucleotides for SMA or Patisiran for genetic amyloidosis), but some treatments can be home-based, such as subcutaneous immunoglobulins, or enzyme replacement therapy for Pompe disease.
OS09.03Quick Clinical Outcome Assessments
Angelini C1
1University Of Padova, Italy
The Covid-19 pandemic has highlighted the difficulty in the management of neuromuscular patients and the need for continued implementation of the standard of care.
With the new pandemia psychometrically robust but quick outcome measures are needed to monitor patients’ clinical status. The slow progressive nature of several muscle disorders and the wide pattern of involvement in muscular dystrophies and myopathies make it difficult to establish the prognosis, predict clinical evolution and perform trials and define the impact of natural history, and use new therapies that are becoming available. We constructed a motor function test that is easy and quick to use. This quick Motor Function test: Gait, Stair, Gower’s, Chair (GSGC) was constructed based on the clinical expertise of several physicians involved in the care of DMD; LGMD, and Pompe patients. The GSGC score can be integrated by the use of the motor function of the upper motor limbs with the arm function test (GSGCA). It consists of a simple standardized functional test which grades the ability of the patient to raise their upper arms over the head. Grade 0 corresponds to a full circle of arm abduction, while with grade 6 the patients cannot raise their arms to their mouth and effectively use their hands. The Gardner-Medwin Walton (GMW) scale even modified appears in comparison rather insensitive. The GSGC test includes 4 items. The test provides a detailed picture of motor function by including a quantitative measure of four performances i.e. time to perform four activities: Gait =walking for 10 meters, S=climbing 4 steps on a Stair, G= Gower’s maneuver, C= rising from a Chair (Figure) The GSGC final score is obtained by adding the grades of the four functional tests and ranges from a minimum of 4 (normal performance) to a maximum of 27 (worst performance).GSGCA test includes 5 tests (total score from 5 to 32). Validity and test reliability were determined in a cohort of 9 adult Pompe patients (15 to 54 years of age) and then validated in 40 LOPD cases by a collaborative group. The responsiveness of the GSGCA scale to changes in clinical course over time was examined in a subgroup of 13 LGMD 2B/R2 untreated patients. Interrater and intrarater reliabilities were most usually confirmatory. The motor outcomes are different in various myopathies and depend on a correct diagnosis, while exercise in myopathy patients should be moderate, but not necessarily discouraged. The muscle MRI imaging might be helpful for follow-up of the proximal or distal muscle involvement, to detect fat, and connective tissue replacement, which might be usually absent in metabolic myopathies, except for LOPD. Diet and exercise in LOPD might be an additional therapeutic option synergistic to ERT.
In this presentation, we examine the use of the GSGC scale in LOPD, DMD, and GSGCA scales in the natural history of LGMD R2. The development of “smart care” using telemedicine and eHealth technologies to share images, clinical data, reports, and video meetings of collaborative groups should be implemented.
Keywords: GSGC scale, Covid-19, DMD, LGMD, Pompe
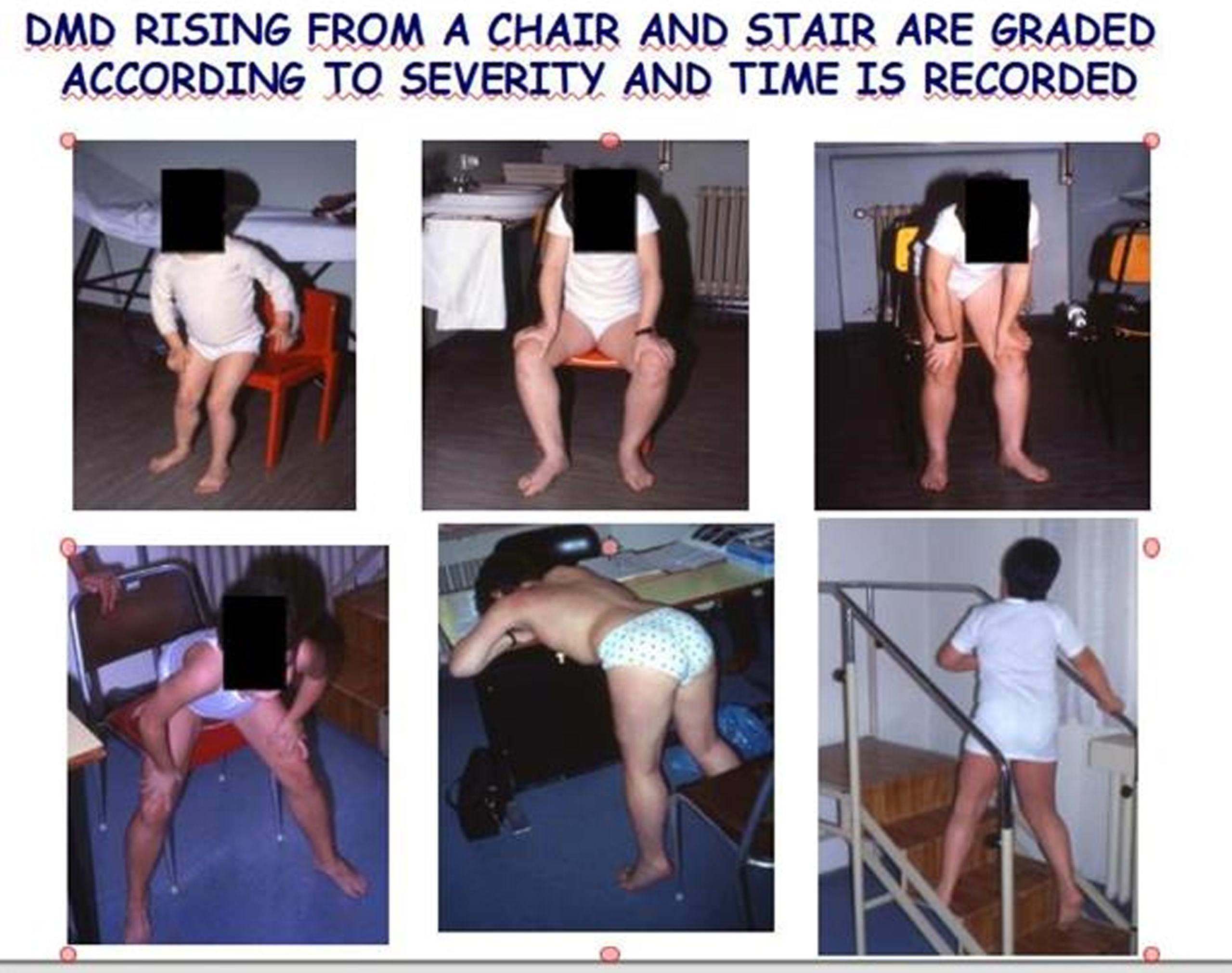
OS10.02Sleep-related Breathing Disorders in Motor Neuron Diseases and Post-poliio Syndrome
Leonardis L1
1University Medical Centre Ljubljana, Ljubljana, Slovenia
Motor neuron diseases (NMD) are progressive neurodegenerative disorders affecting central and/or peripheral motor nervous system leading to progressive muscle weakness. In amyotrophic lateral sclerosis (ALS) nocturnal hypoventilation is frequently reported due to diaphragm, intercostal, pharyngeal, laryngeal and neck muscle weakness. Sleep–related breathing disorders precede daytime symptoms and chronic respiratory failure. The prevalence of sleep apnea is higher compared to healthy controls. Patients with ALS have reduced total sleep time and sleep efficiency, increased sleep fragmentation, increased duration of stage N1 and reduced duration of stage N3 and REM sleep. Disruption of sleep can be caused also by muscle cramps and pain, reduced mobility, depression, hypersalivation and choking.
In patients with spinal muscle atrophy (SMA) weakness of expiratory and intercostal muscles is prominent, while the diaphragm is mostly well preserved. Scoliosis and chest deformities lead to breathing restriction. Thoracoabdominal asynchrony is present during sleep in children with SMA types 1 and 2. Patients with SMA type 1 have severe respiratory abnormalities. They have weak cough; they have increased risk of respiratory infections and aspiration. Due to hypoventilation, especially during sleep they need ventilator support. In young children of SMA types 2 and 3 the decline of respiratory function is prominent, while in early adulthood respiratory functions stabilise. Some of them need due to nocturnal hypoventilation mechanical ventilatory support. In patients with late SMA type 3 and with type 4 respiratory functions decline little, or they are stable. Adult patients with SMA have more common sleep apnea than healthy controls. Analyses of sleep architecture reveal decreased total sleep time, increased duration of stage N1 and increased fragmentation.
Post-polio syndrome includes signs and symptoms that appear between 30 to 40 years after the initial polio illness. Some patients received ventilatory support during polio infection and continue with it afterwards. Some of them have been weaned from ventilator but they may require ventilation after a prolonged latent period. A small proportion of patients require ventilatory support in post-polio period, even if they were not ventilated during the acute illness. The prevalence of sleep apnea (obstructive, central or both) syndrome, nocturnal hypoventilation and restless legs syndrome is higher than in healthy population.
Early detection and treatment of sleep-disordered breathing in patients with motor neuron diseases are especially important for improving their survival and quality of life. Patients with chronic respiratory impairment due to muscle weakness need mechanical ventilation, either non-invasive or invasive. At the early-stage non-invasive ventilation should be used during the night, later also during the day. About invasive ventilation should be discussed with patient in advance. Insufflation/exsufflation devices and secretion management are needed for airway clearance. Patients with sleep apnea need CPAP or other form of non-invasive ventilation.
OS10.03Sleep Apneas in Polyneuropathies and Late-onset Pompe
Young P1
1Medical Park, Germany
Respiratory disturbance and sleep apneas due to neuromuscular disorders is a common aspect in the clinical management of theses patients. The more the proximal muscle wasting is located the more involvement of the diaphragm and other breathing muscle is likely. Patients suffering from Pompe disease are of high risk to develop respiratory involvement during the course of the disease. Clinical approach is to check carefully for signs of respiratory disturbance by clinical examination and by implementing measurement of FVC, maximal respiratory pressure, trancutane capnometry, polygraphy and polysomnography that the diagnosis of hypoventilation, or in much rarer cases sleep apnea, will not be missed to initiate appropriate ventilation therapy and respiratory training. For the vast group of polyneuropathies recent data indicate that respiratory disturbance may also occur. Much more common is obstructive apnea syndrome which correlates more with severity of the neuropathy than with other known risk factors like obesity, gender or age which was shown for the group of Charcot-Marie-Tooth disease (CMT). Therapeutic options are the same as for non-neuromuscular diseased obstructive apnea patients.
In conclusion sleep apneas and other forms of sleep related breathing disorders must be an integrative part or treating patients with Pompe disease and polyneuropathies.
OS11.02Symptomatology of Carriers of X-linked NMD: Duchenne and XL-MTM
Voermans N1, Houwen S, Sarkozy A
1Radboudumc, Netherlands
Duchenne and Becker muscular dystrophies (DMD/BMD) and myotubular myopathy (MTM) are X-linked inherited muscular disorders caused by mutations in the dystrophin gene. At least two-thirds of male cases are thought to be caused by inheritance from carrier mothers.
Symptomatic carriership in DMD and BMD has been recognized for a long time, with a focus on muscle cramps, hyperCKemia and the risk of developing a cardiomyopathy. A minority presents with progressive muscular weakness manifesting in childhood. We have recently performed a longitudinal follow-up study in 12 females with DMD. The results show the wide clinical and participatory spectrum in female patients with childhood onset DMD. Respiratory and cardiac screening, as well as rehabilitation care are recommended to improve health status and to support daily activities and participation for these patients. In May 2022, an ENMC workshop on female carriership of DMD takes place of which the main findings will be summarized.
Symptomatic carriership in XL-MTM has more recently been recognized. We have recently performed an international cross-sectional online questionnaire study among XL-MTM carriers, and a national cross-sectional clinical study. Both studies showed that the prevalence of manifesting XL-MTM carriers may be higher than currently assumed, most having a mild phenotype and a wide variety of symptoms. Manifesting carriers are particularly affected by fatigue, limitations of daily activities, pain, and reduced quality of life. Our findings should increase awareness and provide useful information for health care providers and future clinical trials.
Workshops
WS01.01Peripheral Neuropathy and Brain Abnormalities in the Pathogenesis of LAMA2 Disease
Previtali S1
1Institute of Experimental Neurology (InSpe) and Division of Neuroscience. IRCCS Ospedale San Raffaele, Milano, Italy
Autosomal recessive mutations in the LAMA2 gene affect the production of the alpha2 subunit of the heterotrimer laminin-211 (also called merosin) and are responsible for the development of Merosin deficient Congenital Muscular Dystrophy (MDC1A) or LAMA2-related muscular dystrophy (LAMA2-RD). The disease includes severe early onset forms, characterized by hypotonia, weakness and joint contractures, due to complete laminin-211 deficiency, and milder forms in which only partial loss of laminin-211 is present. Loss or reduction of laminin-211 causes tissue degeneration.
While the disease is predominantly characterized by progressive muscular dystrophy, a dysmyelinating neuropathy is also present and participates to affect patient motricity. Finally, LAMA2-RD is also characterized by brain abnormalities, which mainly manifest with epilepsy.
Our knowledge on pathology and mechanisms underlying the development of peripheral neuropathy and brain abnormalities are mainly due to the use of mouse models.
Aim of this communication is to integrate data from human and mouse studies to discuss consequences and pathogenesis of peripheral neuropathy and brain abnormalities in the course of the disease, which may constitute the basis for future therapeutic strategies.
WS01.02Muscular Dystrophy and Clinical Trial Readiness in LAMA2 Disease
Sarkozy A
1Dubowitz Neuromuscular Centre, Institute of Child Health, United Kingdom
Recessive, pathogenic variants in the LAMA2 gene cause one of the most common genetic form of congenital muscular dystrophies, known as LAMA2 related muscular dystrophy (LAMA2-RD). Patients with LAMA2-RD typically present with hypotonia and weakness at birth, progressive contractures of large joints and respiratory insufficiency. Due to severe muscle weakness and atrophy, independent ambulation is usually not achieved. In addition to skeletal muscle involvement, patients with LAMA2-RD also present central and peripheral nervous system involvement, with white matter abnormalities on brain imaging. Creatine kinase is usually elevated over 1000 IU/L and muscle pathology is dystrophic. Pathogenic variants affect production of the α2 subunit of laminin-211 (or merosin) leading to its deficiency. Complete merosin deficiency is typically observed in patients with congenital onset LAMA2-RD, while partial deficiency mostly associates with later onset forms, with variable but overall milder clinical course compared to congenital forms, often with achievement of independent ambulation. Correlations of clinical severity with amount of residual merosin, location and mechanism of action of the LAMA2 variants have been described, with bi-allelic, loss-of-function LAMA2 variants preferentially leading to severe presentations and complete merosin deficiency. Nevertheless, genotype-phenotype correlations remain challenging, with reports of same variant/s leading to variable clinical presentations, even within the same family. Despite its high prevalence worldwide, there is no cure for LAMA2-RD yet. Still, ongoing preclinical research efforts are promising, and translational efforts are in progress to fully characterise natural history of the disease and identify appropriate outcome measures and improve trial readiness. A first, phase 1 clinical trial of the anti-apoptotic compound omigapil allowed collection of pulmonary and motor outcome measures, and to evaluate exploratory outcome measures and possible biomarkers. Despite some intrinsic limitations, a retrospective, longitudinal natural history study in the UK was able to describe key milestones of disease progression and identify linear decline in percentage predicted forced vital capacity. Currently, multiple, international, coordinated efforts are ongoing towards prospective natural history studies on paediatric patients (in particular children younger than 5 years) as well as adults with LAMA2-RD, aiming to achieve trial readiness.
WS01.03Towards a New Potential Therapy of LAMA2-Related Muscular Dystrophy
Reinhard J, Lin S, Ruegg M1
1Biozentrum, University of Basel, Switzerland
LAMA2-related muscular dystrophy (LAMA2 MD or MDC1A) is the most frequent form of congenital muscular dystrophies. It is caused by mutations in LAMA2, the gene encoding laminin-α2, one chain of the heterotrimeric extracellular matrix protein laminin-211 (α2β1γ1). Most patients lack laminin-α2 due to bi-allelic loss-of-function mutations in LAMA2. The large size of the cDNA encoding laminin-α2 and the heterotrimeric structure of laminin-211 present a challenge for gene replacement or gene editing strategies. Here, we describe the development of an AAV-based gene therapy to functionally replace laminin-211 by two small linker proteins. Prior work in transgenic mice has demonstrated that this Simultaneous Expression of Artificial Linkers (SEAL) in skeletal muscle of LAMA2 MD mice has a tremendous ameliorative effect on disease progression (Reinhard et al., 2017. Sci Transl Med. 9). The two linkers, mini-agrin (mag) and αLNNd, are small enough to be efficiently packed into AAV and are predicted to be well tolerated as they are designed from domains of proteins that are expressed in LAMA2 MD patients. Additionally, the linker proteins are secreted and act in the extracellular matrix, which allows for high targeting and expression efficiency by AAV. Using intravenous injections of two AAV9 vectors expressing either mag and αLNNd at postnatal day 1, we demonstrate high expression of both linkers in skeletal muscle and a significant improvement of the disease phenotype in LAMA2 MD mice. Starting at the age of 4 weeks, AAV9-injected mice gained significantly more weight; at 8 weeks of age, the body and muscle weights were 2 times higher than in vehicle-injected LAMA2 MD mice. Further improvements included a significant increase in grip strength, a substantial increase in myofiber size and a strong reduction of fibrosis. In summary, our study serves as a proof-of-concept in mice and establishes systemic delivery of AAVs that express the two linkers (SEAL technology) as a promising gene therapy for treating LAMA2 MD.
WS02.01Overview of Metabolic Myopathies
Vissing J1
1Rigshospitalet, Denmark
Metabolic myopathies classically encompass inborne errors of muscle metabolism affecting fat and carbohydrate metabolism. Further downstream metabolic blocks affecting the respiratory chain are traditionally classified as mitochondrial myopathies and are not covered in this oversight. Blocks in the breakdown of fat and carbohydrates, which both are important sources of energy supply to contracting muscle, result in a hallmark symptom of exercise intolerance. This intolerance is prominent early in exercise for carbohydrate disorder, because glycogen breakdown and combustion are important early on in exercise, while the exercise intolerance comes later on in disorders of fat metabolism, as fat combustion becomes increasingly important later in exercise. Another common symptom is destabilization of the muscle membrane, due to insufficiency in providing enough energy to support the ion pumps at the sarcolemma. This results in rhabdomyolysis and myoglobinuria. However, it has become clear in the last decades that a number of other inherited muscle diseases can present with a “metabolic myopathy mimic”, in terms of exercise intolerance, rhabdomyolysis and myoglobinuria. This is true of a number of muscular dystrophies (limb girdle muscular dystrophies type 2I/R9, 2B/R2, 2L/R12) and patients with variants in the ryanodine receptor 1 gene, amongst others. In this overview of metabolic myopathies, the about 14 disorders of muscle carbohydrate metabolism and about 8 disorders of muscle fat metabolism will be reviewed, and the two main phenotypic groups of chronic symptoms with permanent muscle weakness and wasting versus episodic symptoms related to the energy deficiency, but without permanent weakness, will be described. Differential diagnoses, which differs in these two main groups will be detailed and lastly current treatment options will be described.
WS02.02Diagnostic Tools and Strategy for Diagnosis of Metabolic Myopathies
Laforêt P1
1Raymond-poincaré Hospital, Garches, France
Metabolic myopathies (MM) are an important group of potentially treatable inherited muscle disorders affecting children and adults. Major advances have been achieved in the diagnosis of these disorders over the past years thanks to improvements in biochemical and molecular techniques. However, exercise testing and muscle biopsy still have an important place in the diagnostic strategy.
We will present in this session the various techniques currently available for the diagnosis of metabolic myopathies, discussing the respective contributions of biochemical analysis, exercise tests (forearm exercise test and cycling test), muscle biopsy and molecular analysis.
We will also discuss the usefulness of these different tests according to the categories of metabolic myopathies (lipid metabolism disorders, glycogenosis, or mitochondrial myopathies), and the current place of targeted gene panels studies.
Diagnostic strategies will be proposed for patients presenting with exercise intolerance or rhabdomyolysis episodes.
WS02.03Current Treatments of Metabolic Myopathies
van der Ploeg A1
1Erasmus MC University Medical Center, Netherlands
Metabolic myopathies are genetic diseases caused by defects in metabolic pathways that lead to accumulation of (toxic) substrates or energy deficits. This may lead to acute episodes cramps, pain and rhabdomyolysis and/or muscle weakness
The main groups of metabolic myopathies are the glycogen storage disorders (GSD II, III, IV, V, VII, IX and Danon disease), fatty acid oxidation disorders and mitochondrial myopathies.
For many of these disorders there are no causative treatments. An exception is GSD II or Pompe disease. Pompe disease is caused by a deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA) needed for the degradation of glycogen. Deficiency of GAA leads to accumulation of glycogen in many tissues. Muscles are affected most. The disease presents as a spectrum. Patients with the classic infantile with complete lack of GAA present shortly after birth with generalized hypotonia and a hypertrophic cardiomyopathy and die without therapy within the first year of life. Patients with late onset (childhood onset and adult) Pompe disease may present at any age with a proximal myopathy. In these patients the heart is (mostly) not involved. Due to progressive muscle weakness patients finally become wheelchair dependent. Importantly respiratory muscles are also affected and pulmonary function becomes increasingly reduced especially in supine position due to involvement of the diaphragm. Many patients eventually need (night time) ventilation. Since 2006 intravenous enzyme replacement therapy consisting of recombinant human GAA has been registered. This has improved prospects of patients. There are still unmet needs. This has led to the exploration of next generation therapies such as next generation ERTs and gene therapies (AAV and lentiviral gene therapy). Recently the first next generation ERT was approved.
For some other metabolic myopathies such as Danon disease gene therapy is explored, but this has not led to market authorization yet. Danon disease is an X-linked disorder caused by LAMP2 deficiency. Importantly the disease presents also in women. A progressive cardiomyopathy and arrhythmias are life threatening to patients and many patients need a heart transplantation at young age
Metabolic myopathies such as fatty acid oxidation disorders may benefit from dietary treatments and/or adequate exercise management. Also these disorders present as a spectrum of disease. For example Very long chain acyl coA dehydrogenase deficiency (VLCAD) may present in infants with hypoglycemia and a cardiomyopathy, while in older children and adults episodes of rhabdomyolysis and muscle pain are most debilitating. Adequate dietary management with sufficient carbohydrates, avoiding of fasting and use of low-fat diet with supplemental medium-chain triglycerides may help these patients.
During the workshop examples of clinical patient presentations and the practical aspects of patient management and treatment will be highlighted.
WS03.02Brain in DM1 Subtypes and DM2 has Differential Involvement
Angelini C, Siciliano G
1University of Padova, Padova, Italy
To establish the degree of the brain and cognitive involvement, studies have addressed neuropsychological features and compared them with gray and white matte involvement detected by brain MRI found in myotonic dystrophy type 1(DM1) subtypes and Myotonic Dystrophy type 2 (DM2). Such studies were done both by a cross-sectional study, but also by longitudinal changes in a series of molecularly defined patients.
Cognitive and brain abnormalities described in DM1, are variably associated with muscle weakness.
We performed neuropsychological studies that revealed a decline in frontal and temporal ability in DM1 and an additional test was done for anosognosia. The expansion size of CTG repeat was classified into 4 groups as E1, E2, E3, or E4., and correlation with (CTG)n DNA was found in frontal tests, anosognosia. The expansion size of CTG repeat was classified into 4 groups E1, E2, E3, and E4.
A typical frontotemporal abnormality was often found by MRI in the white matter it might correlate to a congenital developmental disorder rather than to a progressive lesion.
We clinically followed by Muscle Impairment Rating Scale (MIRS) a series of 30 DM1 patients, in the brain we studied white matter abnormalities in MRI, evaluated according to the AGE Related White Matter Changes (ARWMC) score. The neuropsychological battery included MMSE,memory, linguistic, attentional frontal tests, anosognosia.
White matter abnormalities by MRI were frequent in our DM1 series with juvenile or adult-onset. In the brain the dorsal attention network is affected, with disease progression the temporo-parietal junction and the frontoparietal circuits appeared to be progressively involved, these changes could explain our clinical findings: in juvenile cases when we found anosognosia and compromised linguistic and executive tasks, besides white matter changes a ventriculomegaly could be observed, in adult cases since besides the characteristic temporo-polar abnormalities other lesions were white matter abnormalities and gray matter atrophy.DM1 brain abnormalities contribute to account for social cognitive deficits observed in patients.
DM2 patients present a reduction in cerebral blood flow and occasionally there are alterations in the white matter of the brain, these more specific vascular CNS abnormalities might be related to aging or diabetes mellitus. Other CNS abnormalities are found in other neuromuscular disorders such as LGMD 2I/R9, oculopharingeal muscular dystrophy and glycogenosis type 2, infantile type.
Conclusions: A specific fronto-tempo-polar abnormality (Fig 1) is seen early in the white matter of the brain in DM1. Major degrees of abnormalities did not correlate with disease duration or MIRS score. Global cognitive performance worsened in all patients, particularly in cognitive or linguistic ability, but their evolution probably represents an aging-related decline or a congenital brain abnormality.
Keywords: Myotonic dystrophy, Brain MRI, CNS,LGMD 2I/R9
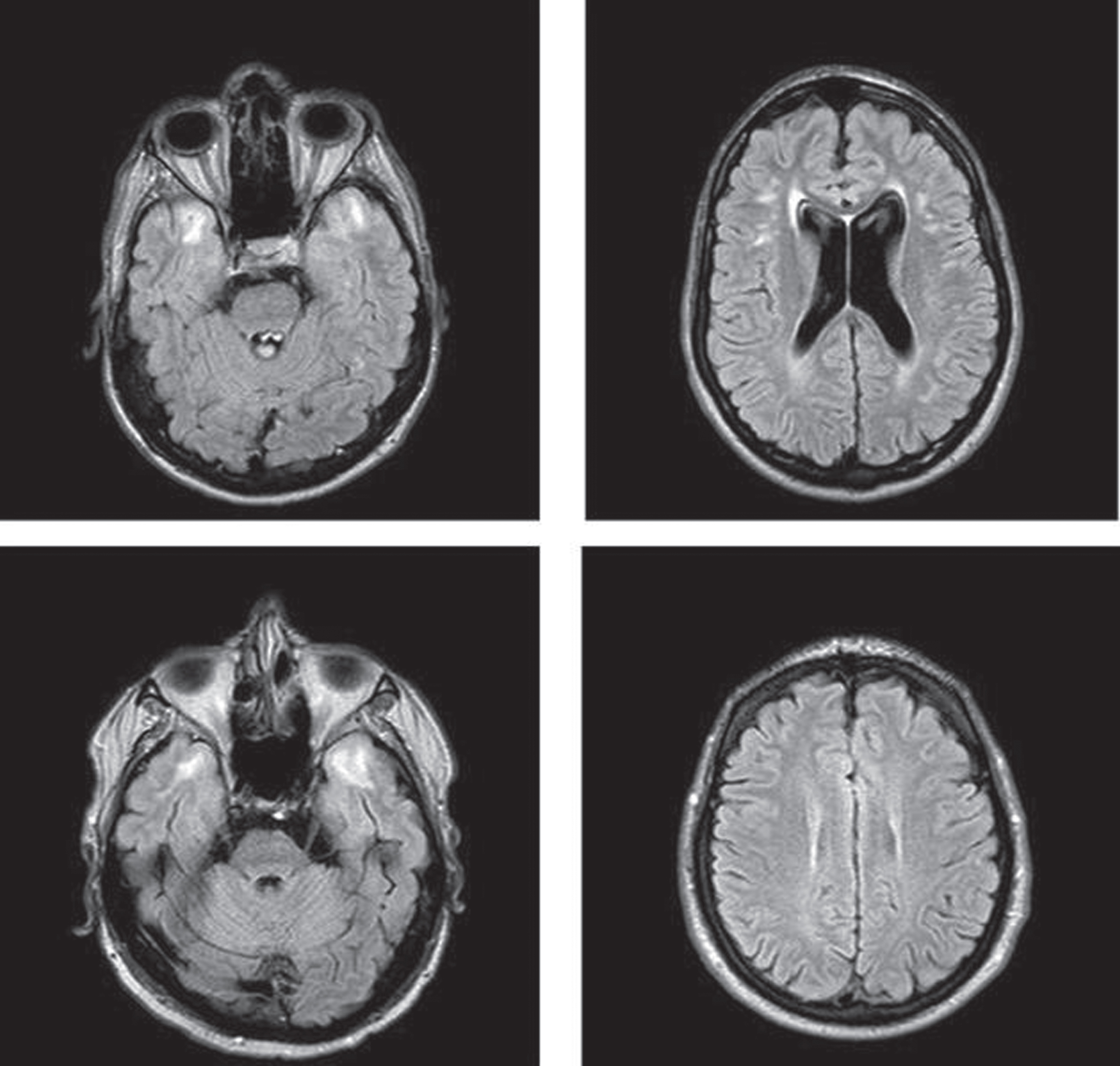
WS04.01Polymyositis, Dermatomyositis and Immune Necrotising Myopathy
De Bleecker J1
1Ghent University Hospital, Gent, Belgium
Myositis represents a heterogeneous group of autoimmune disorders, and polymyositis (PM), dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), anti-synthetase syndrome associated myositis and sporadic inclusion body myositis (IBM) are distinctive clinical and immunopathologic entities. Diagnosis of myositis is based upon clinical signs, creatine kinase (CK) levels and serology, and in most cases requires a muscle biopsy for histopathological evaluation. PM patients are rare, present with subacute predominantly proximal muscle weakness and elevated serum CK levels. A muscle biopsy reveals endomysial inflammation and invasion of non-necrotic muscle fibers by autoaggressive CD8+ T-cells, yet without the degenerative features suggestive of IBM. DM manifests as symmetric proximal muscle weakness and characteristic cutaneous features, with perifascicular muscle fiber atrophy and perimysial B-cell and CD4+ T-cell accumulation on biopsy. Most DM patients can be sub-grouped according to one of five muscle specific antibodies. Microvascular complement deposition is an early pathologic sign. IMNM present as relatively severe proximal muscle weakness with prominent muscle fiber necrosis in the muscle biopsy, yet with absent or minimal inflammatory infiltration. Anti-HMGR and -SRP antibodies occur in about each a third, the remaining third being seronegative. Sarcolemmal complement deposition with the directly toxic antibodies is part of the pathogenesis. Therapy development for PM, DM and IMNM has been mostly founded on empirical data and published case reports, with limited randomized controlled clinical trials (RCT) conducted to date, yet most patients can be treated successfully. First-line choice is steroid therapy, started in severe cases at IV high doses, with gradual tapering and addition of steroid-sparing agents such as methotrexate, azathioprine or mycophenolate to reduce side effects. In steroid-resistant patients, other second-line immunosuppressive agents (cyclophosphamide) or intravenous immunoglobulins (IVIg) may be administered, or plasmapheresis may be attempted. IVIg is an established therapy for DM and has been useful in the acute treatment of IMNM. In addition, the progress made in our understanding of disease pathogenesis has led to the development of more selective anti-inflammatory strategies for treating refractory patients. Patients may be successfully treated with therapeutic antibodies depleting B-cells (rituximab). Monoclonal antibody therapy directed against pro-inflammatory cytokines (TNFα inhibitors infliximab, adhalimumab, etanercept; IL-6: toculizumab; IFNα: sifalimumab), activated T-cells (abatacept, basiliximab), JAK inhibitors (tofacitinib, baricitinib) have been negative or are still studied; some even caused myositis as side effect (TNFα inhibitors). Agents depleting complement (eculizumab) or targeting the FcRN receptor (efgartigimod) are trialed.
To tailor the treatment regimen to the individual patient, precise subtyping of PM, DM and IMNM into subgroups that may predict which treatment options have the most chances of success, is required. Inadequate sub-classification of patients is one of the reasons that previous RCT failed to confirm efficacy, even after multiple case reports with very favorable response. The definition of sensitive outcome measures is an important issue, and has much improved through the IMACS consortium.
Along with medication, physical therapy is now widely accepted to be of some benefit. Prevention of disease damage by the myositis process itself (calcinosis, tendon retraction,..) or its treatment (osteoporosis, steroid myopathy, infections,..) requires thoughtful considerationt.
WS04.02Myasthenic Syndromes
Sacconi S1,2,3
1Université Côte d’Azur, Centre Hospitalier Universitaire de Nice, Système Nerveux Périphérique & Muscle, Hôpital Pasteur 2, 30 voie Romaine CS, Nice, France, 2Université Côte d’Azur, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Institute for Research on Cancer and Aging of Nice, 28 Avenue de Valombrose, Nice, France, 3Fédération Hospitalo-Universitaire Oncoage, CHU Nice, Université Côte d’Azur (UCA), Nice, France
Myasthenia gravis (MG) and Lambert-Eaton myasthenic syndrome (LEMS) are antibody-mediated autoimmune diseases of the neuromuscular junction. Previous works and recently published study on European LEMS registry demonstrate that Lambert-Eaton syndrome prognosis can be influenced by symptomatic drugs and survival by the treatment of overlying cancer or other autoimmune diseases when present. It is very important to early recognise this disease and to detect and treat efficiently associated comorbidities.
Autoimmune myasthénia associated with anti-RAch and Anti-MUSK antibodies is associated with fluctuating weakness in ocular muscles and in muscles of the limb and trunk.
Many first-line treatments for MG, including corticosteroids, acetylcholinesterase inhibitors, and non-steroidal immunosuppressive drugs, target inhibition of acetylcholine breakdown or T-cell function. Even though they allow MG patients to maintain an acceptable level of muscle strenght, functinal ability and quality of life, these treatments are non-specific and may be associated with important side effects. Moreover, MG patients may be or become refractary to these traitements during time or develop contre-indication to the treatement do to coexisting comorbidities.
Over the last few years, new biological agents against complement, the FcRn receptor, or B-cell antigens have been successfully tested in clinical trials. These new therapies with a relatively rapid mode of action and few side effects extend the possibilities for targeted immunotherapies and open new venues to better manage MG. Nevertheless, several challenges may occurs concerning the choice of the drugs in different situation, the frequancy of the administration and the potential long term side effects in the recent context of COVID-19 pandemic.
WS04.03Intensive Care Unit Weakness
Damian M1
1Essex Cardiothoracic Centre, Mid and South Essex NHS Foundation Trust, Basildon, United Kingdom, 2Division of Anaesthetics, University of Cambridge, Cambridge, United Kingdom
ICU treatment initially focuses on life support and cardiorespiratory monitoring, rather than neurological function, and sedation and/or paralysis make muscle weakness hard to appreciate. In cases without a clear history of neuromuscular disease, weakness may be recognised with some delay when the patient fails to wean from the ventilator. The question is then whether a previously unrecognised neuromuscular disorder caused the ICU stay, for instance an inflammatory neuropathy or myopathy, or a hitherto unrecognised metabolic disorder, such as a metabolic myopathy manifesting by acute rhabdomyolysis, or whether the patient suffers from “ICU-acquired weakness” (ICU-AW). This lecture discusses the differential diagnosis of weakness in the ICU, current concepts of ICU-AW, and the management of neuromuscular disease arising in the ICU.
WS05.01Benefits and Limits of ERT in LOPD
Remiche G1
1Centre De Réference Neuromusculaire, Dept. Of Neurology, Hôpital Erasme, Université Libre De Bruxelles, Brussels, Belgium
Glycogen storage disease type II (Pompe disease) is an autosomal recessive mutisystemic disorder due to the deficiency of acid alpha-glucosidase (GAA) lysosomal enzyme. Its prevalence varies following geographic area from 1:40000 to 1:156000.
The pathophysiological process conducts to glycogen accumulation in many tissues with main consequences in skeletal and cardiac muscles associated with cardiomyopathy and proximal/axial myopathy.
The classical infantile Pompe disease manifesting in the first year of life and including severe heart involvement is distinguished from late-onset Pompe disease (LOPD) occurring after the age of one year without cardiac involvement. In LOPD, the clinical picture largely varies and correlates with residual enzyme activity levels. Life prognosis in LOPD is largely determined by a diaphragm weakness inducing restrictive syndrome.
In 2006, recombinant human GAA (rhGAA) was approved as enzyme replacement therapy (ERT) for Pompe disease. In 2010 the unique randomized controlled trial in LOPD with alglucosidase alfa ERT was published and concluded to an improved walking distance and pulmonary stabilization function over an 18-month period (LOTS study).
Conduction of clinical studies in LOPD is limited by several factors including its low prevalence and the large phenotypical spectrum limiting homogenous cohort constitution.
Further studies were all uncontrolled and provided variable results about ERT efficiency especially regarding the long-term benefit. Beside six-minute walking test (6MWT) and seated forced vital capacity, other outcomes have been studied such as medical research council strength measurement, Walton and Gardner-Medwin scale, time on ventilatory support (TOV), and quality of life measured by 36-items Short Form Health Survey (SF-36) questionnaire. For most of these outcomes, no statistically significant benefits were currently demonstrated.
Recent reviews and meta-analyses supported the benefit of rhGAA ERT in LOPD on the 6MWT, the physical part of the SF-36 and TOV.
Whether some predictive factors could help to anticipate the magnitude of the therapeutic response remains controversial. Plausible predictor’s candidates for ERT response include the disease stage at the introduction of ERT, the genetic background and the presence of antibodies targeting rhGAA.
The question of the ERT benefit durability in LOPD merits to be addressed since some published data suggest a trend to its decline after three to five years.
In another hand, recent uncontrolled LOPD cohort’s studies related to interrupted long-term ERT therapy (mean ≈ 7-8 years), concluded to a clear rapid worsening of several outcomes during interrupted treatment phases, raise awareness related to the ERT interruption risk.
In conclusion, rhGAA ERT in LOPD provides clear statistically and meaningful benefit in several outcomes from an intermediate timing exposure perspective. The demonstration of strong statistically and clinically meaningful evidence after longer exposure periods remains uncertain and large interindividual variability is observed.
In order to confirm the long-term cost-effectiveness ERT benefit in LOPD, it is now needed to collect prospective data in well-designed studies performed on large LOPD populations.
These further studies should include stratification strategy integrating potential factors that could discriminate good and poor responders and integrate as main outcomes more meaningful clinical measurements such as quality of life or autonomy.
WS05.02Next-generation Clinical Outcomes and Biomarkers in LOPD
Laforêt P1
1Raymond-poincaré Hospital, Garches, France
Since nearly 15 years, a large number of studies has been dedicated to Late Onset Pompe Disease (LOPD), and among them several multicentric international clinical trials having involved hundreds of patients. Knowing that the main clinical manifestations of LOPD are pelvic-girdle weakness and respiratory involvement, major assessments of disease evolutivity are based on pulmonary function testing (PFT) and 6 minutes walking test (6MWT) since the initial clinical trials.
The 6MWT has several limitations and doesn’t accurately reflect the global severity of limbs and axial muscles involvement.
Various methods of muscle strength assessment have been used in association with 6MWT, such as manual muscle testing or hand held dynamometry, but they don’t present enough interater reliability allowing its use in large multicentric studies. Thus, improving assessment of motor function, including neuromuscular performance, locomotion and postural control in adults with LOPD is an important goal. Usefulness of muscle MRI has also been clearly established in recent years, allowing in particular to detect infraclinical muscle involvement, and analyzing muscles groups that are more difficult to test, such as paravertebral muscles.
Regarding respiratory function, MRI imaging of diaphragm and optoelectronic plethysmography are non invasive methods that have been developped in order to assess lung volume variations and anatomical compartments of chest wall, beside PFT.
We will discuss the contributions and current limitations of these new techniques for the follow-up of patients with LOPD and their potential interest as clinical outcomes for the future clinical trials.
WS05.03The Future of Treatments for Pompe Disease
van der Beek N1
1Department of Neurology / Center for Lysosomal and Metabolic Diseases, Rotterdam, Netherlands
Pompe disease (glycogen storage disease type II) is a lysosomal storage disorder caused by a deficiency of the enzyme acid alpha-glucosidase. A severe enzyme deficiency results in the classic infantile phenotype, which leads to death within the first year(s) of life if left untreated, while a partial lack of enzyme activity is associated with the ‘late-onset’ phenotype, characterized by more slowly progressive limb girdle muscle weakness and respiratory involvement.
Over 15 years ago, enzyme-replacement therapy (ERT) has been successfully implemented as the first disease-specific treatment for Pompe disease. Despite the fact that this has changed the prospects for patients enormously, it has now become evident that the search for a cure does not end here. Two key issues that need to be addressed in the future are: 1] patients with the classic infantile phenotype may develop white matter abnormalities in the brain, with an accompanying cognitive decline, and 2] the inter-individual variation in treatment benefit in patients with the late-onset phenotype is considerable, both in size and duration of the effect.
Fortunately, in recent years a lot has been invested in the development of new therapies, such as next-generation enzyme therapy, AAV gene therapy, RNA-based therapy and lentiviral gene therapy. Several phase 3 studies with new enzyme therapies have since been completed, while AAV gene therapy is now being investigated in a clinical trial. Hopefully, many of these new treatments will reach the clinic within the next 5 years. The availability of multiple treatment options will allow us to treat our patients in a much more individualized way.
This presentation will provide an update on the status of the development of these new innovative therapies.
WS06.02Utilising Novel Technologies to Improve the Diagnostic Yield of Whole Exome Sequencing in Patients with Neurogenetic Disease
Horvath R1
1Department of Clinical Neurosciences, University of Cambridge, United Kingdom
Mutations in either the mitochondrial or nuclear genomes are associated with a diverse group of human disorders characterized by impaired mitochondrial respiration caused by pathogenic variants in >400 different genes. The clinical presentation of mitochondrial diseases is very variable, and there are only very few effective treatments to date.
Whole-exome sequencing has led to a revolution in the diagnosis of mitochondrial diseases, leading to an exponential increase in the number of associated disease genes and the diagnostic yield. However, there are still some patients with a likely mitochondrial disease, where the genetic etiology of a mitochondrial disorder remains unknown, highlighting limitations in variant detection and interpretation. There is a need for improved methods including transcriptomic, proteomic and metabolomic approaches, which may improve the diagnostic yield.
The presentation will cover the current status of the genetic basis of mitochondrial diseases and will discuss current challenges and perspectives and explores the contribution of factors beyond the protein-coding regions and monogenic inheritance expanding the genetic spectrum of mitochondrial disease.
WS07.01The Gestalt Approach to Neuromuscular Disorders of Adulthood - a General Introduction
Schoser B1
1Friedrich-Baur-Institute LMU,Germany
Decryption Gestalt characteristics of neuromuscular disorders is currently based on long-term clinical training by obtaining complex cognitive skills. New technologies will help overcome this tranquil eminence-based method to an amplified precision Gestalt. Current face-to-gene studies in hereditary syndromic disorders were pioneers for this. We are in the decade of transition as neuromuscular disorders become a hotspot of treatable inherited disorders. Thus, we must precisely stratify our neuromuscular disease patient cohorts for modern individualized therapy optimization. Tools like an amplified precision Gestalt approach will help to guide us better. Beyond our growing evidence for treating as early as possible, most of our adult patients are already in advanced stages of the burden of their disease course, for example, previously with moderate to severe muscle wasting and weakness and/or scoliosis and contractures. Therefore, we need to find ways to define more precise subgroups for the best Gestalt treatment outcome. Currently, our decision-making is mainly based on our eye, hand, and brain interaction perception. We need to implement computational decision-making and machine-learning processes incorporate the use of ontologies and algorithms. They permit sophisticated recognition of precision Gestalt patterns and correlate them to genetic variants beyond all clinician’s performative perceptions. However, these modern methods require careful construction of the precision Gestalt to provide the best read-out of the analysis. As principal aim, an individual holistic precision Gestalt of a patient for a specific disease will be defined by a physiological monitoring of data from wearables by continuous real-world data, imaging data from whole body MRI scans or magnetic resonance in combination with surface three-dimensional imaging data sets, neurophysiological mapping, muscle morphological data, and the genetic and molecular information, as genomes, transcriptomes, proteomes, and metabolomes. This amalgamation of various human data sources will entail the full support of machine learning and artificial intelligence.
WS07.03Gait Patterns in Neuromuscular Diseases
Vissing J1
1Rigshospitalet, Denmark
Pattern recognition is key to a neuromuscular specialist. One thing is recognizing the distinct pattern of muscle involvement in each of the several hundred diseases, but another is recognizing the features of gait pattern among different diseases. Thus, distal lower involvement, as seen in many neuropathies, distal SMAs and distal myopathies, such as myotonic dystrophy, is characterized by a high lift of the foot to avoid a drop foot from getting caught by the underlying terrain. More proximal weakness evokes a waddling, hyperlordotic gait as seen in the numerous muscle diseases with limb girdle weakness. Paraspinal muscle weakness produces a backward leaning gait with arms swinging mostly behind the trunk’s mid-axis to prevent falling forward. The talk will review these gait forms and how to distinguish them. Ways of objectively document the gait patterns will be reviewed, including the use of sensors and video assessments of limb and joint movements.
WS08.03Genomic Rearrangements in FSHD
van der Maarel S1
1Leiden University Medical Center, Netherlands
Facioscapulohumeral dystrophy (FSHD) is the third most common muscular dystrophy in adults and is clinically characterized by progressive and irreversible weakness and wasting of the facial and upper extremity muscles. FSHD is caused by incomplete repression of the cleavage stage transcription factor DUX4 in skeletal muscle. The DUX4 gene is located in the D4Z4 macrosatellite repeat in the subtelomere of chromosome 4q which adopts a repressive chromatin structure preventing DUX4 expression in somatic cells. The D4Z4 repeat normally varies between 8-100 units and most often, DUX4 expression in skeletal muscle of FSHD patients is due to D4Z4 chromatin relaxation as a result of a contraction of the repeat to a size of 1-10 units (FSHD type 1 - FSHD1). However, with the advances in genome technologies, new genetic causes for FSHD have been uncovered over the past decade. In addition to genomic rearrangements to D4Z4 other than repeat contractions such as translocations and duplications, also mutations in chromatin factors that are necessary to establish or maintain a repressive D4Z4 chromatin structure (SMCHD1, DNMT3B and LRIF1) have been recognized to cause FSHD (FSHD type 2 - FSHD2). However, rather than a dichotomy between FSHD1 and FSHD2, both forms of the disease should rather be considered a continuum in which the reduced D4Z4 repeat size and the failure in D4Z4 chromatin repression differently contribute to the expression of DUX4 in skeletal muscle. Molecular therapies should therefore be directed at targeting the D4Z4 chromatin structure or DUX4 itself.
WS09.02Advances in Treatment of MAG Neuropathy
Steck A1
1University of Basel, Switzerland
IgM monoclonal gammopathy is frequently associated with a specific phenotype of demyelinating polyneuropathy, particularly with anti-MAG antibodies. The diagnostic utility of anti-MAG antibodies, their specificity, the homogeneity of the clinical syndrome and the consistent pathological findings, do suggest a pathogenetic role for anti-MAG antibodies. Mechanisms of nerve injury consist of antibodies binding to MAG in paranodal myelin and Schmidt Lanterman incisures with subsequent unraveling of myelin lamellae. Since the anti-MAG antibodies appear to exert a direct pathogenic effect on myelin structure and function, B cells depleting therapies have been the main therapeutical mode of treatment. Initially chemotherapy regimens with chlorambucil, cyclophosphamide or fludarabine have been used, but because of toxicity have been replaced by anti-CD20 antibodies. Cumulative data from several studies show that rituximab helps 30–50% of the patients. A retrospective analysis of 50 clinical trials in anti-MAG neuropathy demonstrated that a relative reduction of ≥50% anti-MAG IgM levels was associated with clinical improvement in the responder group. New generation of humanized anti-CD20 monoclonal antibodies that cause more profound or sustained B cell depletion are available and warrant the development of new studies. The discovery of the MYD88 mutation in Waldenström Macrogobulinemia (WM) has led to the use of inhibitors of BTK such as Ibrutinib in the treatment of WM, resulting in efficient tumor cell killing. This mutation is also present in a majority of anti-MAG neuropathy patients. Preliminary data from 2 studies point to a possible efficacy of Ibrutinib in anti-MAG neuropathy. Recently an antigen-specific immuno-therapy has been developed. It consists of a glycopolymer acting as a scavenger for the anti-MAG autoantibodies. Preliminary data provide evidence for the use of this targeted therapy for the removal of anti-MAG antibodies in an extracorporeal approach coupled to an immunoaffinity column. Because complete elimination of the clonal anti-MAG IgM is not possible with current treatments, novel approaches or combination of treatments are needed.
Reference: Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. https://doi.org/10.1016/j.jneuroim.2021.577725
WS10.01Pathology of GBS Focused on its Early Clinical Stage
Berciano J A1
1 University Of Cantabria, Spain
Guillain-Barré syndrome (GBS) is an acute-onset, immune-mediated disorder of the peripheral nervous system, basically comprising demyelinating (AIDP) and axonal forms. The aim of this presentation will be to analyze the pathologic background of early GBS (≤ 10 days after onset). For this purpose, I have carried out a comprehensive literature review of autopsy proven early GBS cases, and an update of our five AIDP patients with autopsy study on whom clinical-electrophysiogical study was done within 10 days after onset. Pathological changes predominated in proximal nerves, in some studies being more prominent at the sides where the spinal roots unite to form the spinal nerves. Particularly in the first few days of the clinical course, endoneurial inflammatory edema was the outstanding feature, sometimes accompanied by centrofascicular or wedge-shaped areas of endoneurial ischemia. Moreover, these changes correlated well with our further prospective nerve sonographic studies in early GBS showing that main changes rely on ventral rami of the investigated cervical nerves (C5-C7). Where appropriate, a comparison of early GBS pathological results with those of experimental autoimmune neuritis was carried out. I will provide new insights into the pathophysiology of early GBS.
Key points:
• Endoneurial inflammatory oedema is the earliest GBS pathogenic event.
• Oedema predominates in proximal nerve trunks.
• It helps to explain the pathophysiology of ascending weakness when conventional electrophysiology shows no consistent changes.
WS10.02Insights in GBS Pathophysiology Gained by Electrodiagnostic Studies
Van den Bergh P1
1Cliniques Universitaires Saint-Luc, Belgium
Although GBS is a clinical diagnosis, the diagnostic certainty can be increased by cerebrospinal fluid and electrodiagnostic studies according to the GBS Brighton Collaboration criteria. Electrodiagnostic studies can show evidence of peripheral neuropathy and can exclude disorders mimicking GBS. GBS is a container term, including typical GGS and GBS variants, such as Fisher syndrome, cervico-pharyngeal-brachial GBS and paraparetic GBS. Moreover, based on pathological studies, axonal (acute motor axonal neuropathy, AMAN, and acute sensorimotor axonal neuropathy, AMSAN) and demyelinating (acute inflammatory demyelinating polyradiculoneuropathy, AIDP) GBS subtypes have been described. Until recently, it was thought that these subtypes could be confidently distinguished by nerve conduction studies. Electrodiagnostic subtype criteria were established by Ho et al. (1995) and Hadden et al. (1998) to this effect. However, the discovery of reversible conduction failure (RCF) in axonal GBS led to the realisation that transient conduction slowing and conduction block can occur due to antibody-mediated immune attack at the nodal/paranodal axolemma. There is no actual demyelination as pathologically defined and if the immune attack continues, conduction failure may not reverse and axonal degeneration ensues. Serial nerve conduction studies can detect RCF and were deemed necessary to correctly diagnose AMAN and AIDP. Recent electrodiagnostic criteria sets by Rajabally et al. (2015) and Uncini et al. (2017) have taken into account this pathophysiological mechanism, which is called nodopathy/paranodopathy. However, the dichotomous distinction between axonal GBS and AIDP, based on correlation of published electrodiagnostic criteria sets with serial studies to detect RCF, and ganglioside antibodies, appears to be difficult as indicated by recent electrodiagnostic studies and by pathological studies. Reasons are that this distinction is partly nerve conduction criteria-specific and that there gold standard electrodiagnostic criteria are not available.
WS10.03Understanding GBS Immune Pathophysiology as it Relates to GM1
Willison H1
1University Of Glasgow, United Kingdom
The autoimmune peripheral neuropathy Guillain-Barré syndrome (GBS) is in part mediated by anti-GM1 ganglioside antibodies induced by preceding bacterial or viral infections. Anti-GM1 antibodies then target plasma membrane GM1 that is extensively distributed in both glial and axonal membranes at the node of Ranvier and motor nerve terminal, in addition to extraneural sites. The wide distribution of the GM1 ganglioside target leads to unwanted complexity is ascribing pathological outcomes to injury of cell-specific membranes, in particular unravelling the consequence of paranodal or terminal Schwann cell membrane injury on axonal function, and vice versa. To overcome this impasse, we have generated transgenic mice through glycosyltransferase manipulation that solely express GM1 in either neurons or glia, thus allowing us to very specifically target and injure axonal or glial membranes with a single anti-GM1 ganglioside antibody. Through this route we have created mouse models of both the axonal and glial forms GBS, induced by one anti-GM1 antibody, thus creating otherwise highly comparable conditions. Herein we describe the axonal injury paradigm in GalNAcT-/--Tg(neuronal) mice receiving an injection of anti-GM1 ganglioside antibody. These mice develop catastrophic weakness, respiratory dysfunction and associated anti-GM1 antibody and complement deposition, and degenerative pathology in nerve axons. Conversely, GalNAcT-/--Tg(glial) mice only deposit anti-GM1 antibody and complement in targeted Schwann cell membranes including paranodal loops. Whilst the acute injury these latter mice sustain is less severe than seen in GalNAcT-/--Tg(neuronal) mice, the GalNAcT-/--Tg(glial) mice develop conduction block and secondary axonal degeneration over time. The new opportunities provided by these novel models are profound - it is now possible to directly and specifically target the axolemmal or glial membranes at the node of Ranvier and other peripheral nerve sites, study associated nodal pathology, and determine the downstream consequences on function and axon fate, currently a major area in GBS clinical research, The effective stages of therapeutic intervention and repair can also be scrutinised using these models, and one such example will be illustrated.
WS11.01Novel Autoantibodies in MG: Do They Matter?
Pasnoor M1
1The University Of Kansas Medical Center, Kansas City, United States
Myasthenia gravis (MG) is an autoimmune condition characterized by muscle weakness and fatigability. It can affect the ocular muscles, bulbar muscles or muscles in the rest of the body including and not limited to the extremities and the neck muscles. It is antibody mediated and patients have autoantibodies targeting various neuromuscular junction proteins. About 85% of patients with generalized MG have acetylcholine receptor antibodies (AChR). These include the binding, blocking and modulating antibodies. About 5-10% of patients have muscle specific tyrosine kinase antibody (MuSK). Autoantibodies against low density lipoprotein receptor–related protein 4 (LRP4) were identified around 2011 in Japanese people with MG and later reported in Germany and USA. LRP4 antibody caused MG symptoms by inhibiting agrin-induced aggregation of acetylcholine receptors. Approximately about 10% of patients with MG have no autoantibodies with the routine diagnostic testing, making it important for discovery of novel antigenic targets. These MG patients are categorized into seronegative group. Antibodies against other targets, such as titin, the ryanodine receptor, agrin, Kv1.4 potassium channel, collagen Q and cortactin have been found in some patients with MG. Anti-Kv1.4 antibodies have been seen frequently in MG patients with thymoma. Identification of these autoantibodies may provide important information for their clinical manifestation and prognosis. Understanding and development of novel autoantibody assays to identify the newer antibodies will aid and support the development of more advanced therapeutics and ultimately improve management of the disease and patient quality of life.
WS11.02Emerging Therapies and Controversies in MG
Dimachkie M
1The University of Kansas Medical Center, Kansas City, USA
Immunosuppressive drugs therapies are the cornerstone of management in myasthenia gravis (MG). Since their introduction in the 1950’s, corticosteroids (CS) were found to be highly effective in improving the lives of people with MG and well tolerated chronically at low dosages. Since then, two issues became apparent. First, some MG patients do not respond well to CS or require moderate to high dosages of CS to maintain treatment response. Second, some MG patients experience intolerable side effects and other may have comorbidities that render treatment with CS undesirable or even impossible. The class of drugs to address CS shortcomings expanded based on case series and some controlled studies. This led to the use a group of MG drugs referred to as adjuvant immunosuppressants which MG neuromuscular medicine specialists borrowed from autoimmune and transplant literature. In MG, an autoantibody mediated disease, therapeutic plasma exchange and pooled immunoglobulin G (intravenously and subcutaneous) have also been shown to be effective. Despite these advances, some MG cases remain refractory to therapies due to lack of efficacy or intolerable adverse events on therapies. To address this challenging group of patients, other approaches were tested in MG. B cell depletion therapy has shown a strong signal in MuSK antibody positive MG but a futility design study did not demonstrate a promise for CS sparing effect in AChR antibody positive cases. More recently, paradigm-shifting therapies have been developed based on knowledge acquired in the last 50 years about the pathogenesis and pathology of MG. This led to the development, successful testing and practical clinical application of complement inhibition and neonatal Fc receptor blockers as novel effective therapeutics for MG. The objectives of this presentation are threefold: first, to discuss established therapies for MG; second, to overview of the definition of refractory MG; and third, to describe promising therapies for MG.
WS11.03The 2020 Update of the International Consensus-based Treatment Recommendations for Myasthenia Gravis
Wolfe G1, Sanders D, Narayanaswami P
1Univ at Buffalo SUNY, United States
Introduction: Consensus-built treatment guidelines are commonly used to support quality care standards and to help guide public health care policy. In the last decade, formalized treatment recommendations for myasthenia gravis (MG) were first developed, initially at a country-wide level. In 2016, the first international treatment guidance for MG based on consensus among a panel of experts was developed and published. This multinational effort was supported by the MG Foundation of America (MGFA). Management categories in the 2016 statement were symptomatic and immunotherapies, intravenous immunoglobulin and plasma exchange, anti-MuSK MG, thymectomy, myasthenic crisis, childhood MG, and pregnancy.
Objective: The MGFA provided support to reconvene the international panel of experts in 2020 to update the 2016 statement. The panel was expanded to include an expert from South America to complement those from North America, Europe and Asia.
Method: The International MG Treatment Task Force, co-chaired by Donald Sanders and Gil Wolfe, was comprised of 16 physicians with expertise in treating adult and childhood MG. The task force also included an observer representing patient interests. A ”modified Delphi” process using RAND-UCLA appropriateness methodology was again used to obtain consensus, with Pushpa Narayanaswami serving as the consensus process facilitator.
Results/Discussion: The task force reinforced recommendations for rituximab and updated those related to thymectomy. New guidance was established for ocular MG, methotrexate, and eculizumab, with cautions developed for immune checkpoint inhibitors. The task force’s consensus statements were published in a major medical journal in 2021. Further updates are planned every 3-5 years based on advances in the field of MG therapeutics.
Keywords: myasthenia gravis, treatment guidelines, UCLA-RAND appropriateness method
WS12.01Muscle and Neuromuscular Junction Autoimmune Complications of Immune Checkpoint Inhibitor Cancer Immunotherapy
Zekeridou A1
1Mayo Clinic, United States
Immune checkpoint inhibitor (ICI) cancer immunotherapy has drastically improved survival in patients with metastatic malignancies that previously had very poor prognosis. By targeting negative regulatory steps of T-cell activation they enhance endogenous ant-tumor immune responses but may lead to autoimmunity that can target any organ. These autoimmune complications are referred to as immune-related adverse events (irAEs). Even tough neurological irAEs are rare, they can affect any level of the neuraxis, including the neuromuscular junction and muscle. Immune-related myositis and myasthenia can coexist, and they are often accompanied by myocarditis that may contribute to morbidity and mortality. There are also cases of Lambert-Eaton myasthenic syndrome described. Treatment is based on ICI withdrawal and corticosteroids; other treatments like plasma-exchange, intravenous immune-globulins or others are also used depending on disease severity at presentation and evolution.
WS12.03Mechanistic Insights Into the Loss of Muscle Mass and Function at High Age
Ham D, Lin S, Ham A, Thürkauf M, Ruegg M1
1Biozentrum, University of Basel, Switzerland
Sarcopenia is defined as the loss of muscle mass and function and is considered a major contributor to loss of life quality at high age. Structural and functional changes at the neuromuscular junction (NMJ), including some denervated skeletal muscle fibers are hallmarks of sarcopenia. Here, we addressed the role of the mammalian target of rapamycin complex 1 (mTORC1) in this phenomenon. mTORC1 is well known to regulate cell size by controlling cap-dependent protein translation in response to growth factors and nutrients. In skeletal muscle, the mTORC1 inhibitor rapamycin prevents load-induced hypertrophy. However, its role in age-induced sarcopenia is not well established. As aging muscle is in a catabolic state, one might expect activity of the growth-promoting mTORC1 to be low, however, mTORC1 activity is actually high in sarcopenic muscle. Moreover, constant mTORC1 activation promotes atrophy in most muscles and prolonged mTORC1 inhibition, via rapamycin treatment, increases lifespan in many species. Based on these observations, we addressed the role of mTORC1 signaling in age-related sarcopenia. We find that (1) mice depleted of the mTORC1 inhibitor TSC1 in skeletal muscle show a phenotype reminiscent of sarcopenia and (2) inhibition of mTORC1 by rapamycin is overwhelmingly anti-sarcopenic. This effect is, at least in part, based on an mTORC1-dependent destabilization of the NMJ. Furthermore, we show that caloric restriction (CR), the best-studied anti-aging intervention thought to act by dampening nutrient-sensing mTORC1, also has an anti-sarcopenia effect. Molecular analysis revealed a strong overlap in the gene signatures of aging and mTORC1 activation, while rapamycin normalized part of the aging signature. Interestingly, CR and rapamycin effects on gene expression were largely distinct, and combining these treatments provided additive muscle function benefits in geriatric mice. We currently also study the gene expression program of myonuclei underneath the NMJ by single nucleus RNA-seq (snRNA-Seq) and investigate the function of candidate genes by adeno-associated virus (AAV)-based methods that allow overexpression and CRISPR-mediated knock-down. Preliminary results from such function screens will be presented. In summary, our experiments clearly show that sustained mTORC1 activation in skeletal muscle causes a gene expression signature and a phenotype similar to sarcopenia that strongly impairs NMJ stability. The use of this model has allowed us to identify candidate genes that may have a function in the maintenance of the NMJ in aging muscle.
Regional Sessions
RG.02Neuromuscular Diseases in Latin America
Verdugo R1
1Department of Neurology and Psychiatry, Faculty of Medicine, Clinica Alemana-Universidad del Desarrollo, Santiago, Chile
In varying degrees, depending on the country, Latin America is facing a combination of neurological health problems characteristic of underdevelopment, along with the diseases of aging and development. In some parts of the region, leprosy is still a significant cause of peripheral neuropathy and -to a lesser degree- Chagas. In most countries, we are seeing a growing incidence of diabetic neuropathy, while there is an increasing recognition of diseases such as amyloidosis and paraneoplastic neuropathies. We are beginning to know with more precision the incidence and characteristics of ALS in our region. We have also faced the epidemic spread of specific neuromuscular diseases, such as the increase of Guillain Barré Syndrome some years ago, as the result of the zika epidemic. On the other hand, we have to define the more prevalent mutations causing hereditary sensory motor neuropathies, and other neuromuscular disorders. Latin America is a challenging region of the world for the Neurologist advocated to Neuromuscular Diseases.
RG.03Neuromuscular Disease in Africa
Imen K1,2, Abida Y1, Gouider R1,2
1Department of Neurology, LR18SP03, Clinical Investigation Centre Neurosciences and Mental health, Razi Universitary Hospital, la Manouba, Tunisia, 2Faculty of Medicine of Tunis, University Tunis El Manar, Tunis, Tunisia
People in low- and middle-income countries (LMIC) tend to have less access to healthcare services and less social security coverage. Moreover, when health care is needed, this one might be delayed or not obtained. Inherited neuromuscular diseases are one of the conditions that may suffer from these limitations. However, it is interesting to note that it is among these regions that the prevalence of these diseases is higher. In fact, LMIC are characterized by a higher prevalence of consanguineous marriages, and autosomal-recessive inheritance in some diseases may account for more than 50% of cases with some phenotype characteristics and particularities. Hence, diagnostic approach of inherited neuromuscular diseases requires careful assessment of mode of inheritance, clinical presentation, neurophysiological studies, and DNA testing. During the past decades, identification of neurological disease genes has expanded and transformed the neurologist’s nosology of this particular condition. However, in some countries, the genetic testing for patients with these diseases especially the access to the high-throughput approaches such as microarray analysis and next generation sequencing (NGS) still limited. Muscle biopsy, which is a cornerstone of the diagnostic approach of neuromuscular diseases, might also be difficult to access in LMIC and specialized physicians are lacking as well. Interest in muscle MRI has been largely stimulated in the last few years by the recognition of an increasing number of genetic defects in the field of inherited neuromuscular disorders. However, in LMIC, the number of MRIs is limited as well as the number of neuroradiologists specialized in this field. Access to new etiopathogenic treatment such as gene therapies is still limited also. In this context, public health and clinical health services, along with specialized neurologists, advanced diagnostic investigations and appropriate treatments can be considered necessary material conditions for good health in LMIC.
RG.04Neuromuscular Disease in Thailand
Witoonpanich R1,2,3
1Division of Neurology, Department of Medicine, 2Faculty of Medicine, Ramathibodi Hospital, 3Mahidol University, Bangkok, Thailand
Neuromuscular diseases in Thailand include most of the common diseases seen worldwide except for diseases caused by some local toxins or infections, or those determined by ethnic and genetic predisposition. Motor neuron disease is common. Spinobulbar muscular atrophy (Kennedy’s disease) is rare. Infection of the anterior horn cell which leads to acute flaccid paralysis is occasionally seen. This is mostly caused by non-polio enterovirus (e.g. echovirus, coxsackievirus and EV71, etc.), and rarely by flavivirus (e.g. dengue virus and Japanese encephalitis virus, etc.). A few cases were infected with vaccine-derived poliovirus, There are polyneuropathy of various causes with diabetic and immune-mediated ones being the most common. The rest includes hereditary, toxic, drug-induced and nutritional polyneuropathy together with POEMS syndrome.
Among diseases of neuromuscular junction, myasthenia gravis is most common. Lambert-Eaton myasthenic syndrome and congenital myasthenic syndrome (CMS) are rare. In the latter, COLQ gene mutation which is associated with acetylcholinesterase deficiency is the most common genetic finding. The first case series of CMS to be reported in Thailand was a large family of slow-channel CMS which is a very rare entity. There were a number of outbreaks of botulism which is caused by botulinum toxin produced by Clostridium botulinum with home-canned bamboo shoots being implicated as the source of the infection. Another toxin which can cause peripheral and central nervous system dysfunction is tetrodotoxin from puffer fish and horseshoe crab.
As regards muscle diseases, all types of muscular dystrophy are seen. In adult, limb-girdle muscular dystrophy type 2B with DYSF gene mutation (dysferlinopathy) is most common. Apart from limb-girdle weakness, dysferlinopathy may also present clinically as distal myopathy. Most patients with distal myopathy have signs of preferential weakness and wasting of posterior leg compartment muscles (Miyoshi myopathy, MM). Some patients may present with initial weakness and wasting of anterior tibial muscle (distal myopathy of anterior tibial onset, DMAT). Distal myopathy with rimmed vacuoles (DMRV) is another type of distal myopathy with clinical presentation similar to DMAT. DMRV is caused by a mutation of the UDP-N-acetylglucosamine 2 epimerase/N-acetylmannosamine kinase (GNE) gene. Regarding inflammatory myopathy, inclusion body myositis which used to be a rarity has now become more common. Dermatomyositis and necrotizing autoimmune myopathy (NAM) are occasionally seen. Among patients with NAM, antibody to the signal recognition particle (anti-SRP) were detected in a large proportion of the patients. Antibody to 3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) were present in a number of patients who either had or had not taken statin medication.
Mitochondrial diseases seen in Thailand include mitochondrial encephalomyopathy, lactic acid and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibers (MERRF), Leber hereditary optic neuropathy (LHON), Kearns-Sayre syndrome (KSS) and chronic progressive external ophthalmoplegia (CPEO). Metabolic myopathy is rather rare. The documented cases include acid α-glucosidase deficiency (Pompe disease), myophosphorylase deficiency (McArdle disease), polyglucosan body myopathy and multiple acyl-CoA dehydrogenase deficiency (MADD). Lastly, hypokalemia with paralysis is a common syndrome in Thailand. This may result from hypokalemic periodic paralysis, thyrotoxic periodic paralysis or distal renal tubular acidosis.
Scientific Sessions
SS01.01Personalized Therapy in Skeletal Muscle Channelopathies
Desaphy J-F1
1Dept. Biomedical Sciences and Human Oncology, School of Medicine, University of Bari Aldo Moro, Bari, Italy
Skeletal muscle ion channelopathies are a genetically heterogeneous family of skeletal muscle diseases due to mutations in ion channel genes. Only recently, a few randomized controlled trials have been performed eventually leading to the designation of orphan drugs and marketing authorization. These drugs however only address the symptoms, without considering the genetic background of the disease. Nondystrophic myotonias (NDM) are rare diseases characterized by skeletal muscle stiffness due to sarcolemma hyperexcitability, which may affect quality of life and trigger life-threatening events in babies. NDM are caused by gain-of-function mutations of the Nav1.4 sodium channel or loss-of-function mutations of the ClC-1 chloride channel. Antiarrhythmic and antiepileptic drugs blocking sodium channels have been empirically used in myotonia because they reduce abnormal action potential firing in myofibers. Although randomized clinical trials confirmed effectiveness of the antiarrhythmic mexiletine in myotonic patients, about 30% of patients remain unsatisfied, due to intolerance or sub-optimal response to the drug. Alternative drugs are required to address the unmet needs of myotonic patients.
Drugs able to correct selectively the molecular defect of channel mutants are expected to be greatly beneficial. Regarding sodium channel myotonia, myotonic Nav1.4 mutations can modify channel sensitivity to mexiletine, due to alteration of binding site or channel gating. For instance, the Nav1.4 mutants presenting a positive shift of fast inactivation voltage dependence are less sensitive to mexiletine but they conserve their sensitivity to the antiarrhythmic flecainide. Patients carrying such mutations and refractory to mexiletine were successfully treated with flecainide, which demonstrates the translatability from bench to bedside. In chloride channel myotonia, ClC-1 loss of function stems from gating alteration or intracellular trafficking impairment. No direct ClC-1 channel activator is currently available. We demonstrated that carbonic anhydrase inhibitors, used empirically in myotonia, can shift the voltage dependence of ClC-1 activation and increase chloride currents and we are evaluating drug effects on myotonic ClC-1 mutants with gating defect. In parallel, we performed proof-of-concept studies to verify the ability of reversible ClC-1 blockers to restore sarcolemma expression of trafficking-deficient ClC-1 mutants, acting as pharmacological chaperones. These studies aim at defining a pharmacogenetics strategy to address precision medicine in myotonic individuals. Supported by Italian-Telethon, Association Française contre les Myopathies, and University of Bari (project Medineuropa).
SS01.02Pathophysiology of Periodic Paralysis
Wu F1, Quinonez M1, Cannon S1,2
1Department of Physiology, David Geffen School of Medicine at UCLA, Los Angeles, USA, 2Department of Neurology, David Geffen School of Medicine at UCLA, Los Angeles, USA
Periodic paralyses (PP) are rare, dominantly inherited disorders of skeletal muscle, in which intermittent loss of muscle fiber excitability and subsequent failure of contraction are caused by missense mutations of voltage-gated sodium channels (NaV1.4 encoded by SCN4A) or calcium channels (CaV1.1 encoded by CACNA1S). Over 70 mutations have been identified, with about 5 accounting for most cases. We and other groups have demonstrated the primary functional consequences of channel mutations in PP are gain-of-function defects that manifest as: (i) disrupted voltage-dependent gaiting of NaV1.4 or (ii) “leaky” channels produced by anomalous ion conduction in the “gating pore” for NaV1.4 or CaV1.1.
These insights on the pathogenesis of periodic paralysis have led to strategies to prevent or abrogate the acute attacks of weakness. For example, stabilizing the normally low intracellular [Cl] by inhibition of the Na-K-2Cl cotransporter with bumetanide prevents the anomalous depolarization of the resting potential and loss of excitability with a low-K challenge in hypokalemic periodic paralysis (HypoPP). Alternatively, K channel openers hyperpolarize and stabilize the resting potential to prevent all forms of PP.
While pharmacologic interventions to suppress acute episodes of weakness in mouse models of PP are convincing, the clinical efficacy in patients remains to be established, and the potential impact on late-onset permanent weakness has not been tested. To develop a more effective and durable intervention that may prevent late myopathy, we explored the feasibility of gene editing technologies to prevent periodic paralysis. Two approaches were used: (1) ablation of the gain-of-function mutant allele using CRISPR/Cas9 or (2) repair of the mutant allele with base editors.
Myoblasts were established from WT and HypoPP mutant mice (homozygous CaV1.1-R528H) and used as a cell-based platform to test the efficiency of mutant allele-specific gene editing. Gene editing constructs were transiently transfected and expression-positive cells were isolated by FACS. Disruption of the R528H allele by CRISPR/Cas9 mediated insertions/deletions (indels) was achieved with 50% - 60% efficiency, while no editing occurred in WT myoblasts. In a second approach, adenine base editing (ABEmax) was used to correct the c.1673G>A p.R528H mutation from A -> G. The mutation was repaired in 34% of alleles, while off-target bystander editing (c.1678A>G) was 6%.
The gene disruption strategy (CRISPR/Cas9) was tested in vivo by electroporation of muscles in the hind foot. Diffuse, but patchy, expression of the CRISPR construct was verified by GFP-positive fibers for which an editing efficiency of 50% indels was detected. The FDB muscle was harvested 4-6weeks after electroporation in other mice, and in vitro contraction studies showed rescue of the HypoPP phenotype. (i) The 40% reduction in baseline isometric force was attenuated to a 15% reduction. (ii) The 35% loss of peak tetanic force in response to a 2 mM K challenge in R528H EDL was absent for R528H + CRISPR/Cas9 muscle. We show feasibility for allele-specific gene editing with meaningful improvement of contractile function, even from partial correction of the HypoPP allele.
SS01.03Treatments of Channelpathies
Fontaine B1
1Institute of Myology, Pitié-Salpêtrière University Hospital, Paris, France
Muscle channelopathies regroup a number of diseases characterized by either muscle weakness and/or stiffness. They include different types of periodic paralysis and non-dystrophic myotonic syndromes. They are mostly due in mutations in the genes encoding muscle ion channels. The better understanding of the dysfunction induced by mutants as well as therapeutic trials has led to a global more accurate treatment of channelopathies. Both scientific evidence and practical problems will be discussed during the presentation.
SS02.01Genetics, Epigenetics and Downstream Consequences
Ganassi M, Heher P, Zammit P1
1King’s College London, United Kingdom
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common inherited myopathy, characterised by a descending dystrophy, initially involving facial and proximal upper limb musculature, that is often left/right asymmetric (Banerji and Zammit, 2021). FSHD1 is autosomal dominant, linked to deletion of a critical number of tandem 3.3kb D4Z4 repeat elements at chromosome 4q35. An open reading frame within each D4Z4 unit encodes a double homeodomain transcription factor - DUX4. Contraction to 10 - 1 D4Z4 repeats results in epigenetic derepression, allowing transcription from the last D4Z4 unit, while certain allelic variants of 4q35 provide a poly(A) signal. Thus, aberrant DUX4 expression underlies pathogenesis in FSHD.
We have recently described how altered mitochondrial ROS metabolism and impaired mitochondrial function cause oxidative stress in FSHD myogenic cells (Heher et al., 2022). We found that elevated mitochondrial ROS levels correlate with increases in steady-state mitochondrial membrane potential. DUX4 triggers mitochondrial membrane polarization prior to oxidative stress generation and apoptosis through mitochondrial ROS, affecting mitochondrial function. Complex I is the main target for DUX4-induced mitochondrial dysfunction. Importantly, mitochondria-targeted antioxidants rescue FSHD pathology more effectively than conventional antioxidants, highlighting involvement of disturbed mitochondrial ROS metabolism (Heher et al., 2022).
DUX4 also compromises myoblast viability, culminating in apoptosis. Such cytotoxicity is rescued by the DUX4 homolog DUX4c through molecular antagonism, also revealing pathomechanisms converging on WNT/β-CATENIN signalling in FSHD.
• Banerji, C.R.S. and Zammit, P.S. (2021). Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: roles of DUX4 and PAX7. EMBO Molecular Medicine 13: e13695.
• Heher, P., Ganassi, M., Weidinger, A., Engquist, E.N., Pruller, J., Nguyen, T.H., Tassin, A., Declèves, A.E., Mamchaoui, K., Banerji, C.R.S., Grillari, J., Kozlov, A.V., and Zammit, P.S. (2022). Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: metabolic stress as potential therapeutic target. Redox Biol. 51, 102251
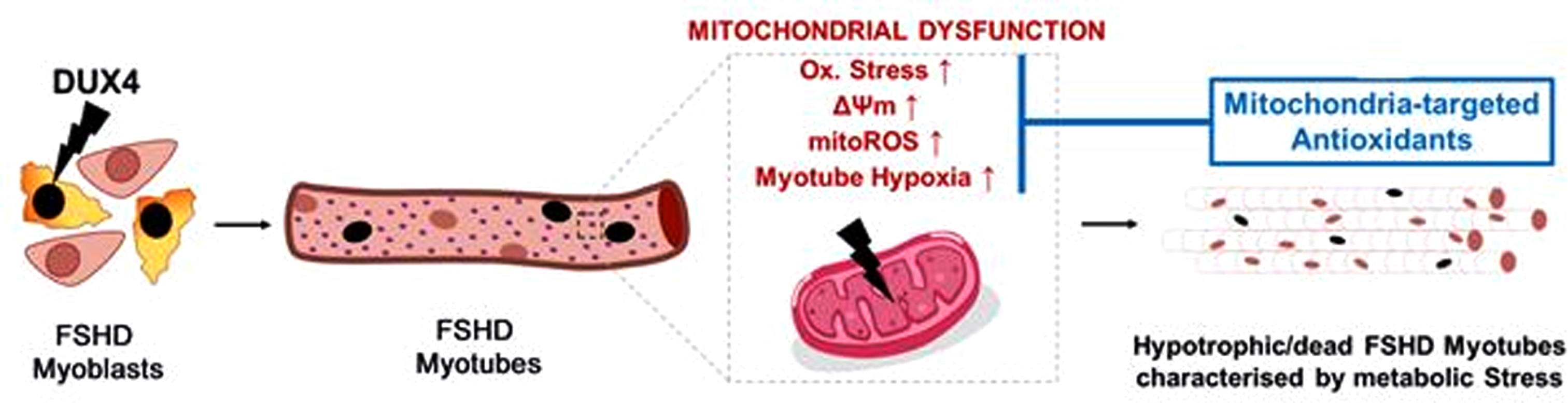
SS02.02Muscle MRI, Echo or EIM: Which is Best for FSHD?
Tasca G1
1Fondazione Policlinico Universitario A. Gemelli IRCCS, Italy
Muscle MRI has become a standard technique to derive secondary outcome measures in clinical trials in several myopathies including FSHD. It is a multimodal technique that allows accurate estimation of fat content in virtually all muscles of the body, besides providing diagnostic clues. Moreover, it is able to identify the only established biomarker of disease activity so far, which is the presence of hyperintense intramuscular signal on STIR sequences. Ultrasound has the advantages of being fast and flexible, non-expensive, and at variance with MRI has the potential to detect fibrosis. However, it is an operator dependent technique, only superficial muscles can be explored and its applications should still be validated in multicentric studies in FSHD. Finally, electrical impedance myography is a non-invasive method to assess changes in muscle structure and composition. It has been successfully applied to derive biomarkers of disease progression in some neuromuscular disorders, although there is a possible concern of low sensitivity in FSHD.
SS02.03Trials, Trial Readiness and the Requirements
Voermans N1, van Engelen B, Kools J
1Radboudumc, Netherlands
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies. Over the last decade, important steps have been made in understanding the underlying cause of FSHD allowing-for the first time-a targeted approach to treatment. FSHD is the result of a toxic gain-of-function from de-repression of the DUX4 gene, a gene not normally expressed in skeletal muscle. With a clear therapeutic target, there is increasing interest in drug development for FSHD, and the first clinical trials have taken place in the USA and Europe (Phase 2 trial with losmapimod).
Overall, treatment approaches target the expression of DUX4 consist by 1) epigenetic silencing of the D4Z4 repeats; 2) blocking DUX4 mRNA production; or 3) targeting one of several identified downstream pathologic pathways triggered by DUX4 expression. These approached are being developed in parallel and are expected to reach to clinical trials in the next few years
Guidelines for clinical trials, pharma regulation and participation, and health care provision in the different European countries differ in various subtle ways. For this reason, the FSHD European trial network (ETN) has been launched in 2021. The research field is expected to benefit from an overall strategy specifically catered to the European context.
The current and future trials call for trial readiness of the expert centers. The four pillars of trial readiness are: 1) clinical and genetic diagnosis; 2) clinical outcome measures; 3) biomarkers and 4) imaging outcome measures. These topics are currently being discussed in four working groups of the FSHD ETN. The recent ENMC workshop on imaging outcome measures by working group 4 (April 2022) has focused on reaching a consensus on MRI sequences, and on the supplementary roles of muscle ultrasound and muscle MRI. Furthermore, the importance of making imaging data of placebo arm participants openly available was stressed. The second workshop will take place in September 2022 and is expected to provide consensus on the related topic. Finally, the network has evolved into a strong European network of physicians and researchers.
At the same time, FSHD patients should become fit for trials. They should be aware of the inclusion criteria of the first trials (genetically confirmed FSHD, mildly to moderately affected, no other neuromuscular diseases) and consider participation if in this range of severity. Participation in a national registry might be the basis for recruitment. Furthermore, symptomatic management should be optimized in order to reached physical fitness. Finally, patients might participate from proper education about the role of trial participants, which is very different from being a clinical patient. The patient advocacy groups play an important role in this.
SS03.01Adult Dominant Distal Actininopathy
Udd B1,2, Savarese M2, Poza J3, Weinberg J4, Olive M5
1Tampere Neuromuscular Center, Tampere, Finland, 2Folkhalsan Research Center, Helsinki, Finland, 3Department of neurology. Hospital Universitario Donostia, San Sebastian, Spain, 4Department of Neurology, Karolinska University Hospital, Stockholm, Sweden, 5Neuromuscular Pathology, Hospital Saint Pau, Barcelona, Spain
The ACTN2 gene encoding the corresponding Z-disc linker protein alpha-actitinin2 was until recently not known to cause muscle disease. As part of our research concept to clarify the genetic cause of unsolved muscular dystrophy patients we first studied four families (three from Spain and one from Sweden) suffering from an autosomal dominant distal myopathy. The affected members showed adult onset asymmetric distal muscle weakness with initial involvement of ankle dorsiflexion and later progressing also to considerable proximal limb muscle weakness.
Neuromuscular examinations showed highly variable CK levels from mildly elevated to 10x the normal value. Muscle biopsy histopathology revealed general myopathic findings, rimmed vacuolated fibers and a few protein aggregates some of them cytoplasmic bodies on EM. Muscle MRI studies were very significant in showing asymmetric fatty degeneration the anterior and lateral compartments and soleus muscle on the lower legs. In the thigh muscles both quads, hamstrings and large adductors were affected with relative sparing of rectus femoris, sartorius, gracilis, semitendinosus and long biceps femoris.
In all the three Spanish families, we identified a unique missense variant in the ACTN2 gene p.C487R, co-segregating with the disease. In the affected members of the Swedish family, we found a different ACTN2 missense variant p.L131P.
Later studies have shown a range of actininopathy phenotypes inherited by both dominant and recessive gene mutations.
SS03.03FSHD-like Dominant Cctininopathy
Savarese M1, Jokela M, Udd B
1Folkhalsan Research Center, Finland
Alpha-actinin-2, encoded by ACTN2, is a sarcomeric protein expressed in cardiac and skeletal muscles and located in the Z-disc where it plays a crucial structural and functional role contributing to sarcomere stability.
In the last few years, mutations in ACTN2 have been identified in patients with different dominant and recessive forms of skeletal muscle disorders with or without cardiomyopathy.
We still lack a clear understanding of the pathomechanisms underlying the different forms of actininopathies and there is a limited correlation between the type of variants identified and the specific clinical picture observed. So far, the only clear genotype-phenotype correlation emerges between indels in the last two exons and a specific phenotype with a combined weakness of the distal lower leg and facial muscles.
Three out-of-frame indels (p.Phe835Serfs*66, p.Glu853Glyfs*48, and p.Pro856Argfs*45 and), predicted to produce a similar extended protein, have been identified in three dominant families from China, Italy and Finland, respectively. Slowly progressive weakness of the distal lower limb and facial muscles was observed in all patients. At a later stage, weakness of proximal lower limb muscles and cardiomyopathy were noticed in some patients. Muscle pathology included internalized nuclei, myofibrillar disorganization positive for alpha-crystallin and myotilin, and rimmed vacuoles. A complete fatty replacement of anterolateral compartment muscles of the lower legs was typical.
ACTN2 should be considered as a possible causative gene in patients with a distal myopathy combined with facial weakness mimicking FSHD.
SS04.01Pathophysiological Mechanisms in the Dominant Centronuclear Myopathy Due to Dynamin 2 Mutations
Bitoun M1
1Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, Paris, France
The autosomal dominant centronuclear myopathy is a rare congenital myopathy associated with a wide clinical spectrum ranging from severe neonatal to milder adult forms. Characteristic histopathological features in muscle biopsies include abnormal centralization of the myonuclei in absence of regeneration, predominance and atrophy of the type I fibres and radiating sarcoplasmic strands. Heterozygous mutations in the DNM2 gene were associated with the entire clinical spectrum. DNM2 gene encodes dynamin 2 (DNM2), a large GTPase, ubiquitously expressed, involved in endocytosis, intracellular membrane trafficking, and regulation of the actin and microtubules cytoskeletons. A Knock-In mouse model (KI-Dnm2R465W/+) expressing the most frequent mutation found in patients has been developed in the laboratory. The heterozygous mice progressively develop a muscle phenotype which recapitulates many aspects of the human condition including muscle atrophy, impairment of the contractile properties and morphological abnormalities mainly affecting oxidative compartments of the muscle fibres. This KI-Dnm2 mouse model was instrumental for identifying several pathophysiological mechanisms at the origin of the muscle dysfunction associated with the DNM2 mutation. Using isolated muscle fibres, we showed a reduction of nuclear number from 20 weeks of age in Tibialis anterior muscle from the heterozygous mice and signs of impaired spatial nuclear distribution including alteration of distance from myonuclei to their nearest neighbours and change in orientation of the nuclei. This study also highlighted a decrease in the satellite cells, the main source for muscle growth and regeneration of mature tissue. In agreement, we found a reduced number of Pax7-positive satellite cells, which were also less activated after induced muscle injury, and a less efficient regeneration in muscles of the KI-Dnm2 mice. Several other defects have been identified in the muscle from the KI-Dnm2 mice including a defect in actin organization and polymerization, the disturbance of the large and flat plaques formed by clathrin at the costamere, a disorganization of the desmin network organized by these clathrin plaques, an impaired calcium homeostasis associating increased cytosol calcium concentration and increased sarcolemmal permeability to the calcium, and an impairment of the excitation-contraction coupling. Signs of defect of autophagy have been also evidenced in the mouse model and this defect was further characterized in patient-derived fibroblasts showing a defect in the early events of autophagosome formation through impairment of the release of autophagosome precursors from the tubular-vesicular recycling endosome membranes. In the same patient-derived cells, clathrin-mediated endocytosis was shown to be altered by DNM2 mutation which can disrupts clathrin-coated pit structure, preventing its maturation and internalization, or slows down the formation of the pits without affecting their internalization. In the absence of arguments in favor of the predominance of one of these mechanisms over the others, we have to consider that the dominant centronuclear myopathy is caused by the combination of all these defects, pointing therapeutic strategies towards approaches targeting the DNM2 mutations rather than one of their consequences.
SS04.02Allele-specific therapy for the Dynamin 2-linked Dominant Centronuclear Myopathy
Trochet D1
1Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, 75013 Paris, France
Dominant centronuclear myopathy (CNM) is a rare form of congenital myopathy associated with a wide clinical spectrum, from severe neonatal to milder adult forms. There is no available treatment for this disease due to heterozygous mutations in the DNM2 gene encoding Dynamin 2 (DNM2). Dominant DNM2 mutations also cause rare forms of Charcot-Mary-Tooth disease, hereditary spastic paraplegia. In addition, a deleterious DNM2 overexpression was reported in several cancers and in X-linked recessive CNM, highlighting a large DNM2 involvement in human diseases. We established the proof of concept for therapy by allele-specific RNA interference devoted to silence the mutated mRNA without affecting the normal allele in a mouse model and patient-derived cells, both expressing the most frequent DNM2 mutation in AD-CNM (R465W, 30% of the AD-CNM patients). We also demonstrated maintenance of the complete rescue of the muscle phenotype one year after a single injection of adeno-associated virus expressing the mutant specific shRNA associated with maintenance of the specific reduction of the mutant Dnm2 transcript. In addition, the long-term study uncovers a pathological accumulation of DNM2 protein occurring with age in the muscle which is prevented by the treatment. Altogether, these results make the allele-specific silencing approach as a robust, safe, and efficient therapy for AD-CNM. However, more than 30 DNM2 mutations have been reported in AD-CNM patients. Therefore, we have developed an approach allowing to silence the mutated allele regardless of the mutation by developing allele-specific siRNAs against 2 DNM2 non-pathogenic single nucleotide polymorphisms (SNP) frequently heterozygous in the population. In addition, allele-specific siRNA against the p.S619L DNM2 mutation, a mutation frequently associated with severe neonatal cases, were developed. All these second-generation AS-siRNAs are efficient for rescuing a panel of defects occurring in patient-derived cell lines. The development of these new molecules allows the extension of the promising AS-RNAi therapy to the large majority of the patients harbouring DNM2 mutations or overexpression using a limited number of allele-specific siRNA.
SS05.02Congenital Myasthenic Syndrome
Selcen D1
1Mayo Clinic, United States
Congenital myasthenic syndromes (CMS) are genetically and clinically heterogenous syndromes. The main presenting symptoms are fatigable weakness and exercise intolerance. The fatigable eyelid ptosis and ophthalmoparesis are important clinical clues for the diagnosis, but some patients present only with limb-girdle muscle weakness without or minimal eyelid ptosis and are frequently misdiagnosed to have congenital myopathy or muscular dystrophy. The correct diagnosis is important to initiate the appropriate treatment of these patients. In this course, CMS presenting as muscular dystrophy will be discussed with illustrated case reports.
SS06.01Drug Repurposing and Biomarker Candidates Discovered Through Molecular Profiling of Myotonic Dystrophy Type 1 Patients on Cognitive Behavioural Therapy
’t Hoen P1, van Engelen B1, the OPTIMISTIC consortium, the ReCognitION consortium
1Radboud university medical center, Nijmegen, Netherlands
Drug repurposing is a key strategy in the development of therapies for rare disease patients with large unmet medical needs. Drug repurposing lowers the costs for drug development and potentially shortens the time from discovery to clinical trials, regulatory approval and patient access. The EU-funded ReCognitION project aims for the identification of drug repurposing candidates for patients with Myotonic Dystrophy type 1. ReCognitION stands for ‘Recognition and Validation of Druggable Targets from the response to Cognitive Behaviour Therapy in Myotonic Dystrophy type 1 patients from Integrated -Omics Networks’. The ReCognitION project is a follow-up project on the successful OPTIMISTIC clinical trial, which has demonstrated clinical benefit of Cognitive Behaviour Therapy (CBT). ReCognitION’s lead hypothesis is that new drug repurposing candidates can be identified from pathways associated with the positive response to CBT and that these drugs can be consolidated or reinforced by conventional drug therapies targeting these pathways. In ReCognitION, we have taken a multi-omics approach to identify the molecular signatures associated with the response to CBT. We have generated RNA-seq profiles from blood samples from 27 participants at 0 and 10 months of intervention and we have generated untargeted serum proteomics for all 255 participants at 0 and 10 months. Specific protein targets were validated in relevant cell and animal models by Western blotting and their interaction partners were identified through co-immunoprecipitation followed by mass spectrometry. We have identified several druggable pathways with support from the large-scale transcriptomics and proteomics datasets. These include the histone (de)acetylation and retinoid acid signalling pathways. Compounds affecting these pathways are currently being evaluated in induced pluripotent stem cell-derived myogenic and neuronal cell cultures and in the HSA-LR and DMSXL mouse models. With these examples, I will illustrate how the drug repurposing strategy based on reverse engineering of a positive response to a behavioural intervention may constitute a novel paradigm for drug repurposing in rare diseases. I will also highlight potential hurdles in the drug repurposing process and discuss ideas to accelerate the drug repurposing pipeline.
ReCognitION is funded by the European Union’s Horizon 2020 research and innovation porgramme ”ERA-NET rare disease research implementing IRDiRC objectives – No 643578” through the E-Rare Joint Translational Call JTC 2018.
SS06.03Drug Repurposing Strategies for Congenital Myasthenic Syndromes
Spendiff S1, Ho K1, McMacken G2, O’Neil D1, O’Connor E1, Grey M3, Lochmüller H4
1Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada, 2Department of Neurosciences, Royal Victoria Hospital, Belfast, United Kingdom, 3Faculty of Health Sciences and Wellbeing, University of Sunderland, Sunderland, United Kingdom, 4Division of Neurology, Department of Medicine, The Ottawa Hospital; Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada. Department of Neuropediatrics and Muscle Disorders, Medical Center – University of Freiburg, Faculty of Medicine, Freiburg, Germany. Centro Nacional de Análisis Genómico (CNAG-CRG), Center for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona, Catalonia, Spain
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by inherited defects of the neuromuscular junction (NMJ). Over 30 mutations have been reported in genes encoding proteins located in the presynaptic, synaptic, and post-synaptic compartments, along with some proteins involved with glycosylation. Onset is often early with patients demonstrating fatigable muscle weakness, delayed motor milestones, and ptosis. Currently, there is no licenced drug for CMS, despite some forms of the condition being controllable through repurposed medications. Treatment relies on a correct genetic diagnosis, as medications that are beneficial for some patients with CMS will be harmful to others with a different genetic mutation. Current treatments include those that increase the amount of acetylcholine (ACh) in the synaptic cleft and β-adrenoreceptor agonists. ACh in the synaptic cleft can be increased using acetylcholinesterase inhibitors, and 3,4-Diaminopyridine which blocks potassium channels, thus prolonging the motor nerve action potential and increasing the quantal release of ACh. These treatments tend to work well for CMS caused by mutations in proteins responsible for ACh recycling, the ACh receptor (AChR) subunits, and the fast channel syndrome. The β-adrenoreceptor agonists are helpful in CMS caused by mutations to COLQ and members of the Agrin/LRP4/Musk AChR clustering pathway. Early on ephedrine was shown to be beneficial, and this was later replaced by salbutamol/albutarol. However, unlike medications that increase synaptic ACh, they often take many months for full benefits to be realised, and their mechanism of action is unknown. We have previously shown that salbutamol treatment improves motility and synaptogenesis in Dok-7 knockdown (KD) zebrafish and ameliorates axon pathfinding, and AChR clustering defects in zMuSK KD zebrafish. Treatment of zebrafish embryos with ICI-118, a selective β2 antagonist, prevents the effects of salbutamol, indicating that the rescue is mediated by β2 receptors. zMuSK KD zebrafish exposed to forskolin had more marked axon pathfinding and AChR clustering than salbutamol treated fish, as well as increased number of prepatterned AChR clusters. The ameliorating effects of forskolin show that the cAMP/PKA pathway is involved in the rescue of NMJ morphology and is a therapeutic target for patients that benefit from salbutamol. Forskolin is already used for asthma, congestive cardiomyopathy and obesity, and does not have the cardiac side effects of salbutamol. We are currently testing both salbutamol and forskolin in mouse models of agrin CMS. Animals are being treated with 3x weekly injections of forskolin, salbutamol, or vehicle control for 35 days. Given our previous results demonstrating beneficial effects of salbutamol in mouse models of CMS caused by mutations to the COLQ gene, we are also testing forskolin as a potential therapeutic in this model. While forskolin offers a potential therapeutic, its low solubility makes it a difficult compound to work with. We have therefore partnered with an organic chemist to synthesise analogues of forskolin with increased solubility and are currently examining their effects on cAMP activity and AChR clustering in C2C12 cells.
SS07.01Muscle Biopsies in the Era of NGS
Claeys K1
1University Hospitals Leuven, Belgium
In the diagnostic workup of patients with hereditary muscle diseases, genetics and in particular recent next-generation sequencing (NGS) technologies have gained an important role. We will tackle the question on the role of muscle biopsy in this new era of NGS, or in other words: do we still need a muscle biopsy? The answer to this question is without any doubt affirmative. Muscle biopsy is a necessary diagnostic tool for differentiating hereditary muscle disorders with treatable diseases such as inflammatory myopathies. Also in cases of double trouble (such as in patients with both an acquired and a hereditary myopathy) or digenic inheritance where mutations in two different genes both cause the phenotype, muscle biopsy will have an important role in the diagnosis. In myopathies with a large phenotype-genotype overlap, such as congenital myopathies for example in ACTA1-related congenital myopathies, the muscle biopsy will provide the exact phenotype of the patient, which is crucial for adequate patient care and management as well as prognosis and counseling. Moreover, muscle biopsy can be used for biochemical analyses and mtDNA genetics in affected tissue. Following NGS often a large number of variants of unknown significance (VUS) result from the analysis. Muscle biopsy can help to determine pathogenicity of the variants in known genes or to establish pathogenicity of variants in novel genes, by performing functional analyses and assessments of proteins on muscle tissue. Transcriptomics (RNA-seq) on muscle tissue can be applied to diagnose deep intronic mutations or mosaicism. Furthermore, biomarkers in muscle tissue can be used for the evaluation of the effect of (novel) treatments and longitudinal follow up. Muscle tissue is also very important to study and understand the underlying pathomechanisms of muscle disorders, which in turn can lead to the development of new therapeutic agents. We conclude that muscle biopsy still has a very important role in the diagnostic workup of patients with hereditary muscle disorders.
SS07.02Nerve Biopsies in the Era of NGS
Weis J1
1institute of Neuropathology, RWTH Aachen University Hospital, Germany
With a prevalence of 1:200, peripheral neuropathies (PNP) encompass one of the largest disease groups among the neurological disorders. The causes of PNP include metabolic, inflammatory, degenerative, toxic, hereditary, vascular, malnutritive, paraneoplastic and other processes. Even though clinical history and examination combined with electrophysiological and laboratory methods often uncover the cause of PNP, many cases remain unsolved. In such situations, nerve biopsy has been a method of choice for decades to classify PNPs and to find clues to uncover their aetiology. However, surgical removal of a piece of nerve causes a sensory deficit and - in some cases - chronic pain. Therefore, a nerve biopsy is usually performed only when other clinical, laboratory and electrophysiological methods have failed to clarify the cause of disease. It must be performed, processed and read by experienced physicians and technicians. Minimal requirements of the workup include paraffin histology as well as resin semithin section histology; cryostat sections, teased fibre preparations and electron microscopy are potentially useful in a subset of cases (1).
On a general note, nerve biopsy analysis provides information about the extent and progression of nerve fibre loss, the type of neuropathy (axonal, demyelinating, combined) and the extent of nerve fibre regeneration. Moreover, “interstitial” pathology (microangiopathy, inflammation/granulomas, abnormal deposits such as amyloid, etc.) is unveiled. The major rationale to perform a (usually sural) nerve biopsy is to gain information about therapeutic options when inflammatory/vasculitic neuropathy is considered. E.g., immunosuppressive drugs can present a risk due to their side effects, and intravenous immunoglobulins are expensive. Nerve biopsies are also useful to detect pathological immunoglobulin and amyloid deposits. In addition, they can provide guidance in the differential diagnosis of hereditary neuropathies with atypical presentation or ambiguous genetic testing results, identify pathological features in the context of an unidentified or ambiguous genetic condition, or detect an inflammatory component in hereditary neuropathies. Often, combined aetiologies are uncovered, including microangiopathic/diabetic and inflammatory or hereditary and inflammatory, or combinations of gene defects (1, 2).
Finally, the PNS gives us an accessible and important window to our nervous system. Scientific nerve biopsy analysis has contributed greatly to our understanding of peripheral neuropathies and nerve fibre pathology in general. Archival specimens, especially paraffin and resin blocks, can be retrieved virtually unaltered after many years and can be a valuable source of information, for example, in cases of hereditary neuropathy. Combined with new molecular methods and in conjunction with the examination of animal models nerve biopsy will continue to contribute informative findings in the future (2).
(1) Weis J, Katona I, Nikolin S, Nobbio L, Prada V, Grandis M, Schenone A. Techniques for the standard histological and ultrastructural assessment of nerve biopsies. J Peripher Nerv Syst. Suppl 2:S3-S10, 2021
(2) Katona I, Weis J. Diseases of the peripheral nerves. Handb Clin Neurol. 145: 453-474, 2017
SS08.02The Distal Hereditary Motor Neuropathies
Rossor A1
1UCL Queen Square Institute of Neurology, London, United Kingdom
The distal hereditary motor neuropathies (dHMN), also referred to as distal spinal muscular atrophy, are a heterogenous group of motor predominant genetic peripheral neuropathies. There is considerable overlap with Charcot-Marie-Tooth disease type 2 and autosomal dominant, recessive and X-linked forms of inheritance exist.
Characteristic features such as upper limb predominant weakness, vocal cord palsy, spasticity and myopathy are specific to certain genes and can help in the interpretation of variants. Furthermore, genes implicated in dHMN preferentially involve heat shock proteins, aminoacyl tRNA synthetases and components of the dynein-dynactin retrograde transport complex.
The recent discovery of recessive loss of function variants in sorbitol dehydrogenase (SORD) as one of the commonest causes of dHMN has generated interest into the use of aldose reductase inhibitors as a potential treatment.
SS08.03The Role of Sphingolipid Synthesis Regulation in Human Motorneuron Diseases
Mohassel P1,2, Dunn T3, Bonnemann C1
1Neuromuscular and Neurogenetic Disorders of Childhood Section, NINDS, NIH, Bethesda, USA, 2Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, USA, 3Department of Biochemistry and Molecular Biology, Uniformed Services University of Health Sciences, Bethesda, USA
Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative disease of the lower and upper motor neurons. Age of onset, pattern of motor neuron degeneration and disease progression vary widely among individuals with ALS and include the more common, adult-onset, sporadic ALS and monogenic forms that typically underlie familial and early-onset cases. We recently reported specific SPTLC1 variants that cause a monogenic form of childhood-onset ALS. These SPTLC1 variants act dominantly and disrupt the normal homeostatic regulation of the enzyme serine palmitoyltransferase (SPT) by its natural inhibitors, ceramide and ORMDL proteins, resulting in unregulated SPT activity and elevated biosynthesis of canonical SPT products. We have also identified a specific pathogenic variant in the related gene SPTLC2 (p.E260K) in five families with ALS, which similarly results in unrestrained SPT activity and overproduction of canonical SPT products. Notably, this is in contrast with SPTLC1 and SPTLC2 variants that shift SPT amino acid usage from serine to alanine or glycine, result in elevated levels of deoxysphingolipids and manifest with the alternate phenotype of hereditary sensory and autonomic neuropathy (HSAN). In addition, genotype-phenotype correlations in patients with SPTLC1 p.S331F/Y variants, who typically manifest with a mixed phenotype of early-onset sensory neuropathy and motor neuronopathy, show elevated levels of dexoysphingolipids concurrent with overproduction of canonical SPT products. Taken together, these findings suggest that sphingolipid biosynthesis is a disease relevant metabolic pathway for motor neuron disease. In addition, these findings directly inform rational therapeutic designs, arguing against serine supplementation (which is used in HSAN) and suggesting SPT inhibition by allele specific silencing, RNA directed therapies, or small molecules as a therapeutic strategy for the associated motor neuron disease phenotype.
SS09.01The Immune System: Friend or Foe in ALS
Feldman E1, Murdock B, Zhao L, Goutman S
1University Of Michigan Medical School, United States
The immune system contributes to the progression of amyotrophic lateral sclerosis (ALS). Despite recent insights into ALS immune mechanisms, however, clinical trials using immunotherapy have failed to improve disease outcomes in human subjects. This may be due to the heterogeneous nature of ALS, as numerous factors contribute to ALS development and the rate of progression. Two of these factors are age and sex: the incidence of ALS increases with age, and men develop ALS at a higher rate than women, particularly among younger individuals. Given these age- and sex-base discrepancies, we hypothesized that immune factors may play a more critical role in ALS progression within specific demographic groups. To test this, we examined immune levels and cellular activation within ALS and control subjects using flow cytometry, correlated these metrics with survival or disease progression rates, and stratified these associations by age and sex. First, we examined whether peripheral neutrophil levels associate with ALS survival in a sex-specific manner. Consistent with previous reports, reduced neutrophil levels was associated with increased survival in ALS subjects. However, this association was driven primarily by female subjects, as females with low peripheral neutrophil levels had an expected median survival that was significantly greater than any other group. In parallel, we examined the activation state of peripheral natural killer (NK) cells in ALS subjects and their association with disease progression. Cytotoxicity markers were significantly upregulated on NK cells in ALS subjects compared to controls, and changes in these markers were significantly associated with disease progression as measured by the revised ALS functional rating scale (ALSFRS-R). Finally, we examined inflammation within the postmortem CNS tissue of ALS subjects using flow cytometry. We found that microglia levels were significantly higher in the spinal cord of female ALS subjects than males. Together, these data suggest that immune mechanisms contributing to ALS progression differ based on age and sex and that these factors should be considered when designing and interpreting clinical trials involving immunotherapy.
Funding: National Institutes of Health R01ES030049, R01NS127188
SS09.02The Promise of Biomarkers in ALS becomes a Reality
Van Damme P1
1KU Leuven, Belgium
The neurodegenerative disorder amyotrophic lateral sclerosis (ALS) is characterized by progressive loss of motor neurons in the motor cortex, the brainstem and the spinal cord. This gives rise to progressive muscle weakness and wasting, limiting the activities of daily living. The survival of people with ALS is often limited to 2-5 years after disease onset, mostly due to respiratory failure. There are no effective disease-modifying therapies for ALS. The cornerstone of the management of ALS is multidisciplinairy care with nutritional and respiratory support.
Over the last 2 decades many clinical trials of new treatments in ALS have failed. There are many possible reasons for this lack of success, but the absence of good biomarkers for ALS is one of them. ALS is a complex disorder, that can manifest in different ways and has proven to be difficult to measure. Biomarkers can help to capture different aspects of the disease such as the aggressiveness of the disease and the underlying disease mechanisms. In addition, they can show target engagement of the drug under study and help to track the treatment responses. Over the last years, many biomarkers for ALS have been developed and they start to find their way into clinical trials. More research is needed to establish which biomarkers are most reliable and easy to use. However, recent studies suggest that biomarkers can help to stratify patients and to measure treatment responses. An overview of recent biomarker developments will be given and future directions will be discussed. The use of biomarkers will hopefully help to derisk clinical trials and to find effective therapies for ALS in the near future.
SS10.01What do we Finally Know About SMN Biology?
Sumner C1
1Johns Hopkins University School of Medicine, United States
The motor neuron disease spinal muscular atrophy (SMA) is caused by reduced expression of the ubiquitously expressed survival motor neuron protein (SMN). Although three new therapeutics each increase SMN expression and improve disease outcomes, there remains variability in response between motor neuron pools within an individual as well as in outcomes between individuals. Recent studies of human autopsy tissues and SMA mouse models indicate that SMN protein levels decline developmentally in the CNS suggesting that SMN may be particularly important for motor neuron maturation during gestation. Indeed, motor neuron axons in both human and SMA mouse ventral roots show impairments of motor axon radial growth and Schwann ensheathment beginning in utero followed by precipitous degeneration postnatally. These observations provide some insight into the fulminant worsening observed in many infantile onset SMA patients and the requirement for neonatal treatment to achieve optimal outcomes. However, there are many pressing biological questions that must be answered in order to continue to advance treatment for patients. The latest data on the transcriptional and posttranscriptional regulators of SMN expression, the molecular and cellular functions of SMN in motor neurons and other organs, and the molecular effects of each of the SMA drugs will be reviewed. Continued basic and translational studies are essential for optimizing therapeutic interventions in SMA patients as well as advancing similar DNA/RNA targeting therapies for other neurogenetic disorders.
SS10.02Challenges in Optimizing SMN Restoration in SMA
Finkel R1
1St. Jude Children’s Research Hospital, United States
Three drugs have now received regulatory approval for treatment of individuals with spinal muscular atrophy (SMA): nusinersen, onasemnogene abeparvovec (OA) and risdiplam. They each increase survival motor neuron (SMN) protein expression in target motor neurons and have demonstrated significant modification of the course of disease. Younger patients respond best, even those with the more severe form, SMA type 1. Population based newborn screening offers the opportunity to identify individuals prior to emergence of symptoms of SMA, and with initiation of treatment generating the most favorable response when started shortly after diagnosis. Three topics of current interest will be discussed here, relevant to considerations for optimal treatment of the individual with SMA. First, time is the enemy. Motor neuron loss begins during fetal development and remains active in early post-natal life, then appears to slow later in childhood and in the adult. The need to restore SMN, therefore, is urgent in the neonate and infant, to rescue faltering motor neurons and sustain them during future growth and development. A substantial fraction of neonates with genetically defined SMA and two copies of the SMN2 gene already demonstrate early features of the disease upon initial assessment. If the pool of motor neurons is reduced at birth, it may be too late to procure a sustained response to these drugs later in life. Fetal therapy then becomes a topic for consideration. Second, target engagement varies. It is important to identify if these drugs reach all the vulnerable motor neurons in the brainstem and spinal cord, if optimal doses are reached and over what time course. Autopsy studies and biomarker data provide some clues. Therapeutic responses would benefit from a better understanding of how intrathecally delivered nusinersen or OA compares to vascular delivery of OA or risdiplam at the motor neuron level and if SMN expression optimized in each cell. Mouse experiments with an OA analogue demonstrated a risk of over-expression of SMN in sensory neurons causing a delayed neurotoxic effect and raises the question if humans treated with OA are at a similar risk. Patients and their caregivers are now requesting combination or sequential therapy with two SMN-enhancing drugs, based upon the unproven hypothesis that a synergistic effect could be achieved that would optimize function of the surviving motor neuron pool. It is challenging, however, to demonstrate that a patient with SMA treated with one SMN-enhancing drug has the capacity to respond further upon the addition of a second drug. Three clinical trials in progress attempt to address these questions. Third, SMA is a non-cell autonomous disease of motor neurons. Increasing SMN expression only in peripheral tissues rescues motor neuron survival in murine studies. The role of increased SMN expression in muscle and other tissues in humans requires further study. Non-SMN directed treatments are in clinical development, e.g., anti-myostatin clinical trials. Addressing these questions will better enable the clinician to discuss a personalized approach as to who, when and how to treat a patient with SMA.
Keywords: fetal therapy, combination therapy, muscle-directed therapy.
SS10.03SMA and New Treatments: Making Sure We Capture All Disease Aspects
Deconinck N
1Neuromuscular reference Center; Hôpital Universitaire des Enfants Reine Fabiola (HUDERF), Brussels, Belgium, 2Neuromuscular Reference Center UZ Gent, Ghent, Belgium
SMA and new treatments: making sure we capture all disease aspects
Over the past decade, new treatment options have been developed such as SMN2 splicing modulation and SMN1 gene replacement. All drugs have resulted in both important survival and functional abilities improvement for patients, changing the disease course. Pivotal trials have indeed focused on survival and motor or respiratory outcomes. Moreover, we now observe new disease trajectories, significantly different from the known natural history and crossing the traditional subtypes of SMA. Clinical approaches have therefore to be adapted.
In the light of such new treatments and disease trajectories, three topics will be discussed: First, the objectives of SMA treatment are not anymore focused only on survival, limb, truncal and respiratory weakness. Emerging evidence and clinical experience have been gained regarding the evaluation and the management of orthopedic and swallowing in SMA. For example, treated symptomatic infants with early onset SMA exhibit a higher rate of early onset scoliosis. This fact highlights the need for early reactions aiming at spine stabilization. However, we have only limited long term experience about the risk/benefits balance of the possible interventions (adapted orthoses, surgical interventions with growing rods...). Moreover, despite feeding being one of the most important aspects in the type 1 SMA care, we are still missing data about possible changes in oral and swallowing abilities in treated patients. We may need new tools for such evaluations although some new tool specifically designed to record oral abilities, swallowing and, more generally, feeding in young type 1 –type 2 SMA patients have been recently developed. Second, as life expectancy has increased, careful monitoring of predictors of life quality are becoming crucial such as growth, communication and cognition. Growth patterns in SMA are now better characterized. Efforts are being made to adequately evaluate cognition and communication and promote its development throughout life. Finally, SMN protein is known to be highly expressed prenatally in most organs. A significant role in organogenesis has therefore been discussed and additional organ involvement, including occurrence of cardiac defects, autonomic dysregulation or abnormal fatty acid metabolism has been reported in SMA. For example, increasing evidence suggest that SMA may also be a vascular disease: necrosis at the tip of toes and fingers have been observed in patients, and thrombotic occlusion of small vessels associated with a significant reductions in vascular density has been demonstrated in severe SMA mouse models.
Altogether, emphasis will be placed on the need to monitor and treat SMA in an inclusive and systemic perspective.
SS11.01Imaging Biomarkers in SMA and ALS
Querin G1, Smeriglio P
1Institut de Myologie, I-Motion Clinical Trials Platform, France
Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are motor-neuron diseases characterized by progressive degeneration of motor neurons in the brain, brainstem, and spinal cord (SC). Patterns of degeneration may be very heterogeneous from one disease to the other and reflect different pathogenic mechanisms underlying variable clinical phenotypes. Neuroimaging techniques have seen a huge development in these last few years, starting from brain and SC anatomical MRI and extending to functional MRI (fMRI) and nuclear imaging. Brain and SC-MRI are the most used and studied techniques that can be applied as diagnostic and prognostic biomarkers, but also as outcome measures in clinical trials. Moreover, and especially after the identification of the C9Orf72 gene mutation as the origin of almost half of familial genetic forms of ALS, brain and SC MRI have revealed their relevance also as pre-symptomatic markers of degeneration useful to detect early pathology before the onset of the symptoms.
Several studies have demonstrated that, in ALS, quantitative MRI methods can detect changes related to the degeneration of upper and lower motor neurons in the motor system, both at the brain and SC. level They also have shown the participation of other systems such as the sensory system, the thalamus, or the basal ganglia, demonstrating in vivo that ALS is a multisystem disorder involving several brain regions. Structural and functional imaging also allows studying dysfunction of brain areas associated with cognitive involvement, namely the frontal and temporal regions. Diffusion imaging and mainly DTI shows a decrease of fractional anisotropy (FA) in the cortico-spinal tracts (CST) as well as in several other white matter bundles in the brain and SC.
When considering C9orf72-ALS asymptomatic and symptomatic patients, diffuse white and grey matter degeneration can be detected, which is already identified up to 20 years before symptom onset and seems slowly progressive over time. In parallel, fMRI shows regions of altered connectivity and [18F] FDG-PET describes several area of brain hypometabolism.
When considering SC-MRI, both sporadic and familial ALS patients present with progressive SC atrophy involving both white and grey matter, which is associated to progressive degeneration of CST. In asymptomatic C9orf72-subjects, SC MRI has also shown progressive decrease of FA in the CST over time.
SMA is a lower motor neuron disease of genetic origin. Recent imaging studies have demonstrated that no evident involvement of the motor system or of other regions is detectable at the brain level, supporting the idea the SMA is a pure lower motor neuron disease with specific spinal involvement. On the other side, SC MRI showed a significant atrophy of grey matter, indicating a progressive degeneration of lower motor neurons without involvement of the white matter.
Globally, a multiparametric approach combining both brain and SC imaging will be required in the diagnostic workup and in the development of biomarkers in ALS and SMA. New methodological developments allowing to investigate the motor system components as well as extra-motor regions, are a promising venue.
SS11.02NatHis for Identification of Biomarkers in Spinal Muscular Atrophy
Attarian S1
1Aix-Marseille Hospital Univ, France
The advent of approved disease-modifying therapies in spinal muscular atrophy (SMA) has transformed the disease field and sparked increased interest in understanding the natural history of SMA. However, there is still an unmet need to identify sensitive biomarkers to capture disease progression and treatment response in this pathology.
Moreover, SMA has several types with onset at different ages and severities and even within each type. The adult forms (>18 years) discussed in this review progress slowly. These characteristics further highlight the above-mentioned crucial need for biomarkers.
Clinical functional scores are the primary tools for assessment. However, they are prone to variation in measurement and are highly dependent on patients’ cooperation and evaluation of the raters.
Several studies have been performed to identify significant sensitive biomarkers for SMA, including electrophysiology, quantified magnetic resonance imaging (qMRI), proteins, and molecular readouts.
Of note, longitudinal studies have reported the relevance of neurophysiological biomarkers such as CMAPS and MUNIX to detect a change at 12 months. In contrast, pulmonary functional tests have not shown sensitivity in detecting significant change at 12 months, nor have serum biomarkers. Muscle qMRI and SMN2 transcript levels could be potential biomarkers. The relevance of serum creatinine, a simple measure, deserves investigation. Further longitudinal studies under real-world conditions are needed to evaluate these potential biomarkers of SMA progression and therapeutic responses.
SS12.1Why Newborn Screening and Early Treatment are Game Changing
Servais L1
1University Of Oxford, United Kingdom
Spinal muscular atrophy (SMA) is the most common genetic cause of childhood death and has long been considered an incurable disease.
In the absence of disease-modifying treatment, patients initially show a loss of muscle strength that progresses to loss of function. In the most severe and frequent cases, patients present with respiratory muscles weakness and respiratory failure and bulbar dysfunction. The clinical phenotypes are divided into five types according to the maximum motor function achieved and the age of onset. Untreated SMA is a cause of severe disability in children and adults, representing an extremely high social cost.
After years of unsuccessful therapeutic trials, three drugs have been successively approved since 2017, having established a clear benefit on survival and functional capacity of patients.
Two of the available treatments Spinraza® and Evrysdi® target the SMN2 gene to prevent splicing of exon 7 during post-transcriptional maturation. The third, Zolgensma®, provides another intact copy of the SMN1 gene.
Regardless of which treatment has been initiated, better efficacy has been demonstrated when treatment is initiated early. Indeed, the daily number of motoneurons that die in absence of treatment is massive. In an infant with 2 copies of the SMN2 gene, it has been estimated that 90% of the motor neurons are lost by the age of six months. This pathological finding is confirmed by the high level of phosphorylated neurofilaments that constitute a marker of neuronal death.
Pre-symptomatic trials converge to show the better efficacy obtained in pre-symptomatic patients. Indeed, whatever is the treatment, patients with three copies of SMN2 treated before 42 days follow today a normal developmental trajectory. Patients with two copies present with a more heterogenous course, with about 50% of patients who present with a normal trajectory and 50% with a mild to moderate motor delay and deficit.
The children included in the pre-symptomatic trials were initially mostly identified through their siblings. The dramatically positive results of these trials have quickly prompted several newborn screenings pilots in which these different data were confirmed. It was estimated in 2021 that about 2% of the world population is screened for SMA, and that this number could reach 20% in 2026. Recently published health economic data points out a major positive health economic impact of NBS strategy.
SS12.02Health Economic Consideration of Newborn Screening of SMA
Dangouloff T1, Thokala P, Deconinck N, D’Amico A, Daron A, Delstanche S, Servais L, Hiligsmann M
1University Of Liège, France
Three new treatments have recently been approved for spinal muscular atrophy (SMA). They reduce mortality in the infantile form, but also provide significant motor benefits in all types of SMA. This benefit is inversely correlated with the duration of the disease at the initiation of treatment. In this context, we set up a newborn screening program (NBS) for SMA in southern Belgium in 2018.
Given the increased importance of economic considerations, studying the health economic impact of this intervention is essential.
We first studied the annual medical costs of patients with SMA. We compared those who did not receive treatment (n=93), those who were treated after being identified clinically after symptoms onset, (n= 42) and those identified by sibling testing or newborn screening (n=14). Ten of these patients were non symptomatic at treatment initiation, four were already symptomatic when treatment was initiated.
Given the high cost of disease modifying treatments, treated patients presented higher medical costs that untreated patients.
The annual medical costs of untreated SMA patients were estimated at €50,798, €24,230 and €3,525 for subjects with 2, 3 or 4 SMN2 copies respectively. For symptomatic patients with 2, 3 or 4 SMN2 copies, medical costs (excluding the cost of treatment) were €30,580, €18,059 and €8,045, respectively. The medical costs of patients identified by screening with 2, 3 or 4 SMN2 copies was €3,913, €1807, and €1,884 respectively.
These data demonstrate the positive impact of the treatments on medical cost, especially in patients identified by screening. Nevertheless, the prohibitive costs of treatment result in a much higher overall cost for treated patients compared to untreated patients, regardless of how they were identified. Since treated patients survive longer, the total cost of living, and thus the budgetary impact of these patients could be even higher.
We then performed a medico-economic analysis to assess the cost-effectiveness of neonatal SMA screening followed by disease-modifying treatment compared to disease-modifying treatment without NBS SMA.
We adapted a previously published Markov model to compare NBS SMA and NBS without SMA, with a lifetime horizon, from the perspective of the Belgian payer. Real-life data, including quality of life, cost and motor development data, collected from 55 SMA patients (43 patients identified by symptoms and treated at least after 2.5 months of life and 12 patients identified by NBS, treated and followed for at least 18 months) were used to populate the model. We studied for each scenario the cost of QALY (1 year of life with full quality of life) and we compared this cost for the two strategies (NBS and absence of NBS).
NBS is associated with a significant gain in QALYs and a significantly favorable cost per QALY gained (€5,820), when only medical costs are considered. When we populate the model with the literature-reported parental choice of treatment and consider the global cost, the total gain per patient identified is 20 QALYS and €2,765,172.
In conclusion, the medico-economic analysis confirmed that NBS is highly beneficial not only for the patients but also for the payers.
SS12.03Organisational, Ethical, and Regulatory Considerations When Setting up an NBS Program
Betts C1, Dangouloff T2, Montague-Johnson C1, Servais L1,2
1Department of Paediatrics, MDUK Oxford Neuromuscular Centre, University of Oxford, Oxford, United Kingdom, 2Neuromuscular Reference Center, CHU de Liège, Hôpital de La Citadelle, Belgium
There is unequivocal evidence demonstrating that treating Spinal Muscular Atrophy (SMA) before the onset of symptoms, profoundly improves the clinical outcome of patients suffering with the debilitating disease. Following the advent of several transformative medicines to treat SMA, it is imperative that a system for early detection of the disease is implemented, which has led to the foundation of newborn screening (NBS) for SMA in several countries.
In the following talk we will compare the organisational, ethical and regulatory experiences and challenges presented to the University of Oxford team, based within the Paediatric Department and the MDUK Oxford Neuromuscular Centre in the UK, alongside the Liège team based at the Neuromuscular Reference Center, in the Hôpital de La Citadelle in Belgium, in the newborn screening programs instituted in both countries.
We will first map the bloodspot screening workflow for both teams, exploring the technical and logistical challenges that were encountered along the pilot study journey. We will discuss each stage in detail from the consenting of the expectant mother, right through to the result reporting phase. Both the Oxford and Liège teams are uniquely placed to capitalise on the surrounding local resources to expand the study beyond their region. We have investigated multiple avenues in an attempt to streamline the screening workflow in an effort to reduce the burden on parents and newborns, and on the NBS programme itself.
There are understandably several ethical and regulatory considerations that must be faced when navigating two highly sensitive topics, specifically paediatrics and neuromuscular diseases.
From an ethical perspective, there is a range of popular opinion. On the one end of the spectrum we explore the impact on parental well-being when they are informed that their seemingly-healthy child carries a life-threatening disease, and whether we should be ‘stealing’ their new-found happiness. On the other side of the spectrum, we consider our societal obligation to offer every child the best opportunity in life and access to transformative drugs which will save their lives, or drastically improve their quality of life.
Finally, we will discuss the regulatory bodies encountered whilst building the programme, specifically the ethical review boards and national screening committees. We will compare the ethical review processes in both countries and the various points raised during their assessment of our screening studies. We also highlight pertinent questions raised during conversations with the national screening committees, and the body of evidence they require in order to implement screening of SMA at a national level.
SS13.03CISP and CISP-plus: a CIDP Variant or Separate Diseases?
Dyck P1
1Mayo Clinic, United States
Objective: Sensory loss with normal nerve conduction studies (NCS) from focal sensory root inflammatory neuropathy is characteristic of chronic immune sensory polyradiculopathy (CISP). However, nonpure cases involving motor and distal sensory nerves exist (CISP-plus). We hypothesize that CISP-plus and CISP are fundamentally part of the same syndrome through comparison of clinical, neurophysiologic, and pathologic features. Methods: CISP-plus cases (primary dorsal root involvement with lesser motor and sensory nerve involvement) and CISP cases were retrospectively analyzed (1986-2019). Results: We identified 44 CISP-plus and 28 CISP cases (n=72) with 86% (38/44) of cases with CISP-plus and 79% of CISP cases (22/28) experiencing imbalance. On examination, large fiber sensory loss was present in 98% (43/44) of patients with CISP-plus and 96% (27/28) of patients with CISP. Gait ataxia was evident in 93% (41/44) of patients with CISP-plus and 79% (22/28) of patients with CISP. Mild distal weakness was common in CISP-plus (75%, 33/44) and did not occur (by definition) in CISP. NCS showed mild abnormalities in all patients with CISP-plus and were normal (by definition) in all patients with CISP. Elevated CSF protein, slowing of somatosensory evoked potentials, and MRI root enhancement occurred in most CISP-plus and CISP cases. Eleven CISP-plus nerve biopsies (3 dorsal lumbar rootlet, 2 fascicular sciatic and 6 distal cutaneous) showed large-myelinated nerve fiber loss, increased frequencies of segmental demyelination, scattered endoneurial inflammation and onion-bulb formations; these findings were most prominent in the rootlet biopsies. Immunotherapy resulted in marked improvement of gait ataxia in 84% (27/32) of patients with CISP-plus and in 93% (13/14) of patients with CISP with return to a normal neurological examination in half of the cases overall (25/46). Conclusion: The recognition of CISP-plus expands the spectrum of CIDP by combining CISP-plus (predominant sensory polyradiculopathy with mild motor and sensory nerve involvement distal to the dorsal root ganglion) with pure-CISP (focal sensory polyradiculopathy) together as proximal sensory CIDP. Both CISP-plus and CISP should be classified as subtypes of CIDP because the disease mechanism is one of inflammatory demyelination with pathological confirmation of segmental demyelination and onion-bulb formation (typical of CIDP) and because of the marked response to immunotherapy. Not classifying these conditions as CIDP because they don’t have the typical electrophysiological characteristics of CIDP poses a large risk to CISP-plus and CISP patients of not being treated with immunotherapy due to denial of coverage from medical insurance carriers.
Keywords: chronic immune sensory polyradiculopathy, chronic immune sensory polyradiculopathy-plus, chronic inflammatory demyelinating polyradiculoneuropathy, peripheral neuropathy, inflammatory neuropathy, sensory neuropathy
SS14.01Genomics and DNA Methylation in Diabetic Neuropathy
Feldman E, Eid S, Noureldein M, Guo K, Hur J1
1University Of Michigan Medical School, United States
Genome wide association studies (GWAS) of large diabetic neuropathy (DN) cohorts have identified multiple genes where the minor polymorphic allele is associated with DN. The identified genes are involved in regulating several important physiological processes and include, among others, ACE (angiotensin-converting enzyme), APOE (apolipoprotein E), ENOS (endothelial nitric oxide synthase), and VEGF (vascular endothelial growth factor). The breadth of these genes emphasizes the complex pathophysiology underlying the onset and progression of DN. The epigenome is the product of modifications to the genome that occur throughout an individual’s lifespan and result in differential patterns of gene expression. Common modifications include DNA methylation, which usually decreases gene expression, and chromatin remodeling, secondary to histone modifications, which usually increases gene expression. To understand the role of the epigenome in DN, we identified and analyzed RNAseq and DNA methylation profiles of sural nerve biopsies from 78 patients with DN, classifying patients by glycemic control, and identified 998 differentially expressed genes (DEGs) and 929 differentially methylated genes (DMGs) between patients with the highest and lowest HbA1c levels. Functional enrichment analysis revealed that DEGs and DMGs were enriched in immune system, extracellular matrix (ECM), and axon guidance pathways. An integrated analysis of the overlapping genes between DEGs and DMGs identified genes and pathways modulating functions such as immune response, ECM regulation, and PI3K-Akt signaling. Our studies reveal that the epigenome joins the genome as a contributor to the pathogenesis of DN, and further suggest that optimal glycemic control is an important component for the prevention of DN.
Funding: National Institutes of Health (R24DK082841, U24DK115255), Nathan and Rose Milstein Emerging Scholar Research Fund, NeuroNetwork for Emerging Therapies
SS15.01Demyelinating Neuropathies: Inherited or Acquired?
Pareyson D1
1Fondazione IRCCS Istituto Neurologico C.Besta, Milan, Italy
The diagnosis of demyelinating neuropathies is often straightforward but may be challenging in other instances. The most important differential diagnosis occurs between demyelinating CMT and acquired dysimmune neuropathies. Diagnosis of CMT is easy when there is a clear-cut family history, disease course is very slowly progressive, onset occurs early in life, skeletal deformities are present, distal involvement predominates, conduction velocities are diffusely and homogeneously slowed, there are neither conduction blocks nor excessive temporal dispersion, and nerve biopsy shows widespread onion-bulb formations with no inflammatory infiltrates. On the other hand, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is characterized by fluctuating course, weakness of distal as well as proximal muscle groups, high protein levels in CSF, non-homogeneous conduction slowing (with asymmetries, excessive temporal dispersion, conduction blocks), edema and inflammatory infiltrates at nerve biopsy. Nerve ultrasounds and magnetic resonance imaging (MRI) of lumbar roots are also helpful in distinguishing CMT from CIDP. However, the differential diagnosis may be difficult in sporadic cases or whenever the familial nature of the disease is not evident; relapses are described in CMT; nonhomogeneous conduction slowing is seen in CMTX1 and occasionally in other CMT types; inflammatory cells may be found at nerve biopsy in some cases. CSF examination, search for anti-nerve antibodies, root MRI, nerve ultrasound, and if needed nerve biopsy may prove useful; response to treatment and results of genetic investigations will solve the issue in most cases. Notably, it is well established that the immune system plays a role in hereditary neuropathies and that there is an immuno-mediated damage in inherited neuropathies; cases of definite CMT with superimposed CIDP which is responsive to treatment have been described, particularly for MPZ-related CMT. The differential diagnosis between hereditary and acquired demyelinating neuropathies includes also anti-MAG and paraproteinemic neuropathies, POEMS syndrome and lymphoma on one side and HNPP, Refsum disease, mitochondrial neuropathies (especially MNGIE), Tangier disease, ARSACS, leukodystrophies on the other; a careful and comprehensive approach, taking into consideration also very rare neuropathies, is needed in such cases.
SS15.02How to Avoid Misdiagnosis of Hereditary Amyloid Neuropathy (ATTRv)
Briani C1
1University of Padova, Italy
Hereditary transthyretin amyloidosis (ATTRv, v for variant) is a rare autosomal dominantly inherited and fatal disease caused by mutations in the transthyretin (TTR) gene, that destabilizes the native TTR eventually leading to the formation and deposition of amyloid fibrils that damage several organs.
More than 130 pathogenic mutations have so far been identified, with the most frequent worldwide being the V30M.
Peripheral nerves (both somatic and autonomic) and heart are the most commonly damaged organs, but also renal, gastrointestinal and ocular involvement may occur.
Some mutations are more associated with cardiac phenotype, other with neuropathy phenotype, and a few with the oculo-leptomeningeal disease form. Mixed phenotypes are also common.
Recently, innovative pharmacological therapies have emerged able to significantly modify the natural course of the disease and improve survival.
An early diagnosis is crucial for a prompt therapy initiation. Unfortunately, the diagnosis is often delayed because of heterogeneous clinical presentations.
In endemic areas (e.g Portugal and Brazil) the neuropathy has both an early and a late onset, whereas in non endemic areas it is generally an adult onset progressive length-dependent axonal polyneuropathy involving both small and large nerve fibres. Misdiagnoses are common, especially in the latter form, the most frequent being with inflammatory demyelinating polyradiculopathy (CIDP). The other crucial differential diagnosis is with light chain amyloidosis (AL).
In a patient with axonal polyneuropathy and history of carpal tunnel syndrome, autonomic symtoms or signs, biochemical markers or instrumental findings (e.g. cardiac uptake at bisphosphonate bone scintigraphy) may help identify involvement of other organs, and help the diagnostic workup. Recently red flags for a correct diagnosis have emerged from neuroimaging, especially nerve ultrasound or plexi MRI. Also skin biopsy seems to allow early detection of amyloid. The multisystemic nature of the disease requires a multidisciplinary approach.
SS15.03Acute Recurrent Focal or Generalized Neuropathy: Inherited or Acquired?
Peric S1
1University of Belgrade - Faculty of Medicine, University Clinical Center of Serbia - Neurology Clinic, Belgrade, Serbia
Neuropathies are very heterogeneous group of disorders based on their course and causes. Majority of them are chronic, but some may be acute. Acute neuropathies can be disabling and even life treatening so their recognition is of great importance. Among patients with acute neuropathies, some may have recurrent episodes and in these cases we should consider specific diagnoses. In this lecture we will focus on upper and lower limb acute recurrent neuropathies, excluding cranial neuropathies.
When we see a patient with focal recurrent neuropathy, we should make differentiation between hereditary vs. acquired forms, i.e. between hereditary neuropathy with liability to pressure palsies (HNPP) and even some other rare hereditary forms vs. entrapment neuropathies, mononeuritis multiplex (MMx) and multifocal motor neuropathy (MMN). Clinical and electrophysiological clues that may direct us toward right diagnosis of HNPP will be specifically discussed. If high suspicion of HNPP is considered and patient is negative for deletion in PMP22 gene, we should consider analysis of point mutations in the same gene and then whole exome or genome sequencing in order to find new genes. In a case of acute recurrent plexopathy, one also must consider hereditary vs. acquired disorders, i.e., hereditary neuralgic amyoptrophy (HNA) vs. Parsonage-Turner syndrome and diabetic plexopathy. During the lecture, we will try to select the most specific clinical features that may help in etiological diagnosis of plexopathies. Both typical and extended phenotypes of HNA will be discussed. Different causes and clinical presentations of Parsonage-Turner syndrome will also be assessed. Finally, we will also look at diabetic plexopathy and its specificities.
Besides focal presentation, acute recurrent neuropathies may be generalized. In this case main differential diagnosis is between acute polyradiculoneuritis vs. porphyria. Acute polyradiculoneuritis was considered as once-in-life disease for decades, however in recent twenty years number of recurrent cases and case series have been described. We will discuss which patients with acute polyradiculoneruitis are in a specific risk of a recurrent episode of the same disease. One must also consider acute-onset chronic inflammatory demyelinating polyradiculoneuropathy (A-CIDP) and some rare metabolic neuropathies.
Timely and adequate diagnosis of acute reccurent neuropathies and their causes have significant impact on patients’ treatment, outcome, further disease course and on genetic counselling in family.
SS16.01Update on Chemotherapy Neuropathy
Cavaletti G1
1Università di Milano-Bicocca, Italy
Chemotherapy-induced peripheral neurotoxicity (CIPN) is a frequent, potentially severe, and dose-limiting side effect of cancer treatment. However, in several instances, the real impact of CIPN on patients’ daily life seems to be not yet completely understood, and it is sometimes considered an unavoidable event in the course of life-saving chemotherapy. CIPN can be induced by several different classes of drugs that are widely used in the treatment of solid as well as hematological malignancies, such as platinum drugs, antitubulins, and proteasome inhibitors. Also, the most recent treatments (e.g. checkpoint inhibitors) and targeted drugs (e.g. vedotins) are not free from neurotoxic side effects, which may be very severe.
Despite our incomplete knowledge of the mechanism at the basis of CIPN, it is conceivable that structural properties of the various neurotoxic compounds contribute to variations in the pathogenetic mechanisms of the damage as well as in the type, severity of the clinical picture, and incidence of CIPN.
Another still unsettled issue is represented by the optimal tool to be used to detect and grade CIPN and its severity. Patient-reported outcome (PRO) measures, composite scales (e.g. the Total Neuropathy Score), instrumental and pathological assessments, and severity surrogate biomarkers, such as neurofilament light serum/plasma levels have all been considered, but a general consensus has not yet been achieved. This unmet need has a relevant impact on the possibility to design reliable clinical trials, particularly when aimed at finding effective preventive/therapeutic treatments for CIPN.
In fact, so far no drugs capable of preventing the occurrence of CIPN or ameliorating its long-term course are available, and chemotherapy schedule modification (including drug withdrawal) is often required to limit its severity, thus preventing patients from receiving the most effective treatment of cancer. Moreover, also symptomatic therapy is often largely ineffective in reducing CIPN symptoms.
SS16.02Checkpoint Inhibitors and Neuropathy
Dubey D1
1Mayo Clinic, United States
Immune-checkpoint inhibitors (ICIs) are increasingly being utilized for the management of advanced or metastatic malignancies. Effect of ICIs on immunosurveillance and interference with self-tolerance contributes to immune related adverse events (irAEs) affecting multiple organ system including central and peripheral nervous systems. Several studies have highlighted varied presentations of ICI associated neuropathies, their management, and long-term outcomes. Furthermore, our understanding of mechanisms of neurological irAEs has also improved, potentially helping with formulation of patient specific management strategies.
SS17.01Early Diagnosis of TTR Amyloidosis
Planté-Bordeneuve V1
1Neurology-Amyloid network, Hospital Henri Mondor-APHP, East-Paris-Créteil University, Créteil, France
Hereditary transthyretin amyloidosis (ATTRv) is characterized by a severe progressive sensorimotor and autonomic polyneuropathy associated or not with a cardiomyopathy. Renal or ocular manifestation are less frequent. The disease is due to amyloid depositions in tissues, caused by a spectrum of autosomal dominant mutations in the transthyretin (TTR) gene. Age at first manifestations varies from the 3rd to 8th decade depending on the TTR variant and on the origin of patients. In the past decade, considerable advances have occurred in the treatment of ATTRv amyloidosis. Oral treatment with TTR stabilizers and more recently TTR gene silencing therapies have proven to stabilize the polyneuropathy in about 70% of patients, along with a survival improvement.
Such non-invasive treatments should be administered from inaugural manifestations to avoid further amyloid deposition and preserve the neurological function. In this context, early recognition of ATTRv is a real challenge.
Recent works on the disease risks give insights on the appropriate time to initiate the monitoring of asymptomatic TTR mutation carriers. Such timeline varies according the type of the TTR variant, the AO in family members as well as the geographical origin for the ATTR-Val30Met carriers, most frequent in Europe.
Given the variable disease expression, one has to investigate the different facets of the disease, involving a multidisciplinary team e.g. neurologist, cardiologist, nephrologist, gastroenterologist and ophthalmologist. Also, the investigations should focus on the expected phenotype according to the specific mutation. A careful review of symptoms, clinical examination and neurophysiological tests will be used to detect the neuropathy, including nerve conduction studies, a battery of small nerve fiber tests and test of autonomic function e.g. heart rate variability, laser evoked potentials, sudomotor testing. Relevant cardiac investigations include electrocardiography, echocardiography, cardiac MR imaging and biomarkers. Identification of amyloid deposition via non-invasive biopsy or cardiac fixation in scintigraphy with bone tracers are also key in the diagnosis. Baseline investigations will be important to serve as reference in the follow up. A consensus emerges on minimum diagnosis criteria before initiating treatment.
SS17.02Update on Therapies for TTR Amyloidosis
Coelho T1
1Centro Hospitalar Universitário do Porto, Portugal
Hereditary ATTR amyloidosis is a multisystemic progressive and fatal disorder caused by the extracellular deposition of variant transthyretin (TTR) as amyloid. A progressive axonal sensory and motor neuropathy with autonomic involvement and/or a severe cardiomyopathy are the most common clinical presentations.
Liver transplant was the first disease-modifying treatment available, but its benefits were limited. Fortunately, In the last decade, three drugs were approved for the treatment of ATTR amyloidosis with polyneuropathy and for cardiomyopathy.
Tafamidis is a TTR stabilizer available as 20mg and 61mg capsules, for daily oral intake. The lowest dose was approved in Europe in 2011 for neuropathy treatment and the higher dose was recently approved for cardiomyopathy patients. For patients with neuropathy the best response was seen in female patients with early disease. No safety issues were so far detected.
Patisiran is an interference RNA preventing the synthesis of wild and variant TTR in the liver. A dose of 300 µg/Kg is infused every three weeks after pre-medication with a low steroid dose. The phase 3 clinical trial showed a highly significant difference between treatment arms, with some degree of improvement in a large percentage of patients. Some mild infusion reactions may occur, but they are easily controlled.
Inotersen is an antisense oligonucleotide that also prevents TTR synthesis. It is formulated for weekly subcutaneous injection. The pilot trial showed a highly statistically significant difference between treatment arms. The drug may cause severe thrombocytopenia and renal disease. Monitoring rules with frequent blood and urine analysis are needed.
Recently a drug based on CRISP/Cas9 technology was tested in a phase 1 study with ATTR patients and showed a significant reduction of TTR plasma concentration after a single dose. Phase 2 study is moving forward.
There are other drugs under development, including antibodies aiming to remove amyloid deposits from tissues and others addressing specific unmet needs, namely the late occurrence of disease in the CNS and the eyes.
Anyway, the approval of these three new drugs is changing a devastating and life-threatening disease into a chronic condition with better quality of life and prolonged survival.
SS17.03Retinal and CNS TTR Amyloidosis – an Emerging Problem
Sousa L1,2,3
1Unidade Corino de Andrade, Centro Hospitalar Universitário do Porto, Porto, Portugal, 2Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, 3Centro Hospitalar de Entre o Douro e Vouga
Hereditary transthyretin amyloidosis is caused by the accumulation of abnormally configured transthyretin (TTR). TTR is produced predominantly by the liver but also, in smaller amounts, by the choroid plexus and the ciliary and retinal pigment epithelia of the eye.
In the central nervous system (CNS), TTR amyloid accumulates in the leptomeningeal membranes and arteries, causing cerebral amyloid angiopathy. In patients with the V30M mutation, this deposition seems to start very early in the disease course. CNS symptoms appear in late phases of the disease, after more than 14 years of systemic involvement. The most common manifestations are transient focal neurological episodes (TFNEs). These are characterized by transitory impairment of language, visual, sensory, or motor functions. They are usually brief, stereotyped, and recurrent. Other reported manifestations include ischemic stroke, cerebral hemorrhages, and cognitive decline. There is also anecdotal evidence of cranial nerve dysfunction.
In the eye, locally produced TTR causes lens and vitreous opacities, glaucoma, and vascular changes, including retinal and choroidal amyloid angiopathy. Symptoms include floaters, loss of contrast sensitivity, and progressive visual loss.
Disease modifying therapies such as liver transplant, TTR stabilizers, and gene silencing therapies reduce plasma TTR but have no significant impact in the CSF and retinal TTR production.
Better systemic treatments extend disease duration, which allows for the symptoms of CNS and ocular dysfunction to become more prevalent. More research is needed to better define the pathophysiology of CNS and ocular dysfunction in ATTR amyloidosis; and to explore new treatment targets or CNS penetrability strategies.
SS18.01Classical Pharmacological Therapy
Sereda M1
1University Medical Centre Göttingen, Germany
Novel treatment approaches in Charcot-Marie-Tooth Disease (CMT) using small molecules
There is no therapy available for Charcot-Marie-Tooth Disease. This may be due to the huge genetic heterogeneity; to the fact that animal models not always reproduce human phenotypes, particularly for axonal CMT2; to the limited numbers of natural history studies, and finally to the lack of informative and robust outcome measures for many of the CMT subtypes. Nevertheless, in the past years proof-of-concept of efficacy has been provided at the preclinical level for several therapeutical strategies. Some of them are already translated to clinical trials.
In this session, I will describe the potential of the most promising approaches applying classical pharmacology (e.g. Progesterone Antagonists, P2X7 Inhibitors, IFB-088, Lipids, Neuregulin, PXT-3003) and describe their mode of action in the diseased Schwann cell. My focus will be CMT1A.
SS18.02Development of a Targeted Therapy by siRNA for Charcot-Marie-Tooth 1A Neuropathy
Massade L1
1U1195 INSERM, France
Charcot-Marie Tooth disease 1A (CMT1A) is the most common inherited neuropathy in the world. It affects the peripheral nerves and causes progressive paralysis of the legs and hands. No treatment is currently available to fight this disease, which is due to the overexpression of a specific protein called PMP22 which is made twice as much as normal.
The challenge was to standardize the expression of this protein in patients with CMT1A. French scientists have developed a patented therapy based on reducing by twice, the RNA coding for the PMP22 protein by small interfering RNA (siRNA).
The difficulty in developing this therapy has been to stabilise these siRNA, which degrade very rapidly in biological environments. Researchers have coupled them with another molecule called squalene, which is typically used in cosmetology and pharmacology. Biocompatible, biodegradable and forming nanoparticles in water, squalene protects siRNA from degradation. It also controls the size of the particles formed and the amount of siRNA injected via intravenous route.
Then they showed, in CMT1A mice models, that injecting these siRNAs, completely and rapidly restored of mouse locomotor activity and strength. The siRNAs penetrate the peripheral nerves, strengthen the myelin sheath around those nerves, and normalize the nerve signal velocity. The effect of treatment lasts for three weeks for severe forms and more than ten weeks for milder forms of the disease.
In line of these data we are now attempting to transition into a preclinical study, bearing in mind the concentration of the effective injectable product without risk of toxicity. Our aim is to define the minimal efficient dose showing functional and histological recovery without any adverse side effects. In case of success, this approach could be also be applied to other hereditary neuropathies.
SS18.03Gene Therapy and Gene Editing
Kleopa K1
1The Cyprus Institute Of Neurology And Genetics, Cyprus
Charcot-Marie-Tooth (CMT) inherited neuropathies result from variable molecular-genetic alterations in neurons and their axons, or in Schwann cells and the myelin sheath they form, leading to either loss of function or toxic gain of function cellular mechanisms. In order to treat the cause of the most common demyelinating CMT neuropathies resulting from either cell-autonomous loss of function or toxic mechanisms in myelinating Schwann cells, we have developed cell-targeted gene replacement or gene silencing approaches. Using clinically translatable intrathecal injection of AAV vectors we have demonstrated widespread biodistribution and expression throughout the PNS. Delivery of the GJB1 gene associated with CMT1X resulted in restoration of Cx32 expression in Schwann cells. Pre- and post-onset gene replacement therapy provided therapeutic benefit including improved motor function, nerve conduction velocities, and nerve pathology in different knockout and transgenic mouse models of the disease. Likewise, replacement of the SH3TC2 gene associated with CMT4C resulted in functional and morphological improvement in a model of CMT4C neuropathy. To treat CMT1A, the commonest CMT type, caused by PMP22 gene duplication, we developed a microRNA-based gene silencing approach. Delivery of microRNA by AAV9 both at early as well as late stages of the disease efficiently silenced PMP22 expression in PNS tissues, leading to functional and morphological phenotypic improvement in a CMT1A model overexpressing the human PMP22 gene. Clinically relevant treatment-responsive blood biomarkers, including neurofilament light (NF-L), neural cell adhesion molecule 1 (NCAM1) and growth differentiation factor 15 (Gdf15) have also been validated in these neuropathy models. Our studies provide the proof of concept for the therapeutic potential of gene replacement or gene silencing therapies to treat patients suffering from inherited demyelinating neuropathies.
SS19.2Advances, Controversies, and Care of Treatment Induced Diabetic Neuropathy
Freeman R1,2,3
1Director, Center for Autonomic and Peripheral Nerve Disorders, 2Harvard Medical School, 3Beth Israel Deaconess Medical Center
Treatment-induced neuropathy was first described in 1933 in a woman with diabetes who reported tingling and shooting pains in the lower extremities that appeared four weeks after the initiation of insulin. The pain increased despite the use of analgesics and sedatives but resolved within 3 days of stopping insulin concurrent with severe hyperglycemia. Recent reports have described treatment-induced neuropathy in individuals with type 1 and type 2 diabetes treated with oral hypoglycemic agents or with insulin. Pre-treatment glycosylated hemoglobin (A1C) is typically high and glycemic control rapid.
Pain and autonomic dysfunction are the most prominent features of this neuropathy. In our report all individuals with treatment induced neuropathy exhibited symptoms of autonomic impairment and had evidence of autonomic dysfunction on testing. Autonomic symptoms of treatment induced neuropathy are more prevalent and more severe than in patients with generalized diabetic peripheral neuropathy. Sixty-nine percent of our cohort had systolic blood pressure falls > 20mmHg. Autonomic symptoms and test results tend to improve over time, particularly in those with type 1 diabetes. The autonomic symptoms are accompanied by pain that may occur distally in a length-dependent fashion or be more generalized. Unlike the pain associated with a generalized polyneuropathy, the pain in treatment-induced neuropathy may involve proximal sites including the trunk. Evoked pain – hyperalgesia and allodynia – is more prevalent than in generalized polyneuropathy. In our series evoked pain was present in 60% of subjects (80% of type 1 subjects and 40% of type 2 patients). Similar to the autonomic dysfunction associated with treatment induced neuropathy, all subjects reported an improvement in pain after many months of continued glucose control.
Most individuals with treatment-induced neuropathy have a substantial worsening of nephropathy and retinopathy within 1 year of rigorous control. The underlying pathophysiology is not known.
SS19.03Advances, Controversies, and Treatment in Diabetic Lumbosacral Radiculoplexus Neuropathy
Dyck P1
1Mayo Clinic, United States
Lumbosacral radiculoplexus neuropathy (LRPN) is a condition that involve nerve roots, plexus and peripheral nerves of the lower limb. It can occur in diabetic patients (DLRPN) and in non-diabetic patients (non-DLRPN). When the condition affects people with diabetes mellitus (DM), the diabetes is usually mild type 2 and the neuropathy may be diagnosed simultaneously with the onset of DM. LSRPN usually starts focally with pain in the upper lumbar plexus (hip/thigh) more commonly than in the lower lumbosacral (foot/leg) plexus and evolves into a more widespread and often bilateral illnesses with weakness. It typically has associated weight loss and extreme pain (burning, lancinating and allodynia), although a painless variety can occur. The painless form is more symmetrical and has more upper limb involvement than the painful form and so can resemble chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Both DLRPN and non-DLRPN are due to ischemic injury (multifocal fiber loss, perineurial thickening, neovascularization and injury neuroma) from an inflammatory attack on nerve and microvasculitis. Because microvasculitis is the putative mechanism, DLRPN and non-DLRPN have been classified as forms of non-systemic vasculitic neuropathy by a PNS vasculitic taskforce. A controlled trial of intravenous corticosteroids in DLRPN did not achieve the primary endpoint (time to improvement of the neuropathy impairment score lower limb by 4 points) but many secondary endpoints measuring pain and positive neuropathic symptoms showed significant improvement. In an epidemiological study of Olmsted County MN, the incidence of LRPN was found to be 4.16/100,000/year which makes LRPN a more common inflammatory neuropathy than CIDP, Guillain Barre Syndrome (GBS) or inflammatory brachial plexopathy in the same population. People with DM were 8 times more likely to develop LRPN than non-diabetic people. Risk factors for developing LPRPN included DM and other autoimmune conditions. The clinical features of the epidemiological LRPN cohort were essentially the same as the referral-based cohorts and both groups usually had spontaneous improvement although the community cohort had less severe disease. Long-term survival is reduced in LRPN patients and in multivariate analysis DM, age and stroke were mortality risk factors but having an episode of LRPN was not. LRPN is often triggered by rapid changes in glycemic control, change in diet, change in exercise and recent surgery. LRPN is a monophasic illness and patients generally have neurological recovery although it is incomplete.
Keywords: diabetic lumbosacral radiculoplexus neuropathy, non-diabetic lumbosacral radiculoplexus neuropathy, vasculitic neuropathy, peripheral neuropathy, diabetic neuropathy, lumbosacral plexopathy.
SS20.02Correlation Between Muscle MRI and Genotype in Myasthenic Syndromes
Nalini A1, Polavarapu K, Preethish-Kumar V, Vengalil S, Nashi S, Kulanthaivelu K, Siddiqui S, Ranjan S
1National Institute of Mental Health and Neurosciences, India
Correlation Between Muscle MRI and Genotype in Myasthenic Syndromes
Congenital myasthenic syndromes (CMS) characterized by fatigable weakness are potentially treatable and majorly autosomal recessive disorders resulting from gene mutations affecting the neuromuscular junction structure and function. Drugs that help in certain forms may worsen others underscoring the importance of establishing a specific genetic diagnosis. As genetic screening can be time consuming, Muscle magnetic resonance imaging plays an important role in the diagnosis and identifies distinct patterns of muscle involvement of many muscle disorders including CMS- CDG (Congenital disorders of Glycosylation) (GMPPB, GFPT1, DPAGT1, ALG12), which may help target genetic analysis and muscle selection for biopsy as well. This group of CMS-CDG usually has elevated creatine kinase levels. The MRI findings relate to hyperintensity on T1w images, reflecting fatty infiltration, and hyperintensity on fat-suppressed T2w sequences (STIR), reflecting inflammation or increased blood flow. We share our experience on muscle MRI findings on a group of genetically confirmed CMS-CGD disorders. Our biggest group had mutations in GMPPB gene. Various clinical phenotypes are described in GMPPB gene defects. All had fatigable limb girdle weakness and all underwent muscle MRI of lower limbs. In our cases of GMPPB related CMS-LGMD phenotype with identical homozygous mutation, c.1000G > A in the GMPPB gene, we identified distinct patterns of muscle involvement. Qualitative assessment by Mercuri staging and Borsato score revealed early and severe involvement of paraspinal muscles, gluteus minimus, and relatively less severe involvement of the short head of the biceps femoris. Moderately affected muscles were gluteus maximus and medius, hip adductors, obturatus, pyriformis, rectus femoris, vastus intermedius, gracilis, lateral head of the gastrocnemius, and medial part of soleus. The mildly affected muscles were tensor fascialata, pectineus, the short head of biceps femoris and Sartorius. The muscles spared were: posterolateral part of Vastus Lateralis (VL), Tibialis posterior (TA), Flexor Hallucis Longus (FHL) and Flexor digitorum Longus. Myoedema was prominently noted in adductor longus, Vastus medialis, VL, gracilis, medial gastrocnemius, TA, peroneii, and FHL. A distinct proximo-distal gradient of affliction was identified in the glutei, vasti, TA and peronei. Also, a postero-anterior gradient was observed in the gracilis muscle. One patient with GFPT1 homozygous mutation [c.44C>T; p.(Thr15Met)] showed fatty atrophy of gastrocnemii with asymmetric myoedema involving the right gastrocnemius and negligible fatty atrophy of bilateral gluteus maximus and thigh muscles. The other patient with GFPT1 showed mild fatty infiltration of gluteal muscles, moderate symmetrical fatty infiltration involving gluteus maximus, quadriceps, adductor brevis and magnus. Two siblings with DPAGT1(homozygous, c.652C>T) showed mild fatty infiltration of gluteal muscles, moderate symmetrical fatty infiltration gluteus maximus. The distinctive MR imaging pattern, coupled with relatively slowly progressive fatigable limb-girdle weakness, would facilitate an early diagnosis of the milder form of CMS-CDG disorders that are more benign in nature. Thus MRI could aid in directing the accurate approach to genetic diagnosis as well as selection of therapy.
Keywords: Congenital myasthenic syndromes; Muscle magnetic resonance imaging, CMS-CDG disorders; GMPPB; DPAGT1; GFPT1
SS20.03Efficacy of Efgartigimod in Generalized Myasthenia Gravis: Myasthenia Gravis Composite Score Analysis From ADAPT
De Bleecker J1, Meisel A2, Howard Jr J3, Vu T4, Bril V5,6, Ulrichts P7, Guglietta A7, T’joen C7, Murai H8, Utsugisawa K9, Mantegazza R10
1Ghent University Hospital, Ghent, Belgium, 2Department of Neurology and NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, Berlin, Germany, 3Department of Neurology, The University of North Carolina, Chapel Hill, USA, 4Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 5Ellen & Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, Toronto, Canada, 6University of Toronto, Toronto, Canada, 7argenx, Ghent, Belgium, 8Department of Neurology, School of Medicine, International University of Health and Welfare, Tokyo, Japan, 9Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 10Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Introduction: Efgartigimod is a human IgG1 antibody Fc-fragment that blocks the neonatal Fc receptor and reduces IgG autoantibody levels. The safety and efficacy of efgartigimod was assessed in ADAPT, a 26-week, global, multicenter, phase 3, randomized, double-blind, placebo-controlled trial in patients with generalized myasthenia gravis (gMG). The Myasthenia Gravis Activities of Daily Living (MG-ADL) scale and Quantitative Myasthenia Gravis (QMG) scores were utilized as primary and secondary outcome measures, while the Myasthenia Gravis Composite (MGC) score was evaluated as an exploratory endpoint. The MGC comprises both physician-reported (muscle strength and ocular function) and patient-reported (talking, chewing, swallowing, and breathing) outcomes. MGC response options are weighted and were developed using the highest performing items from preexisting MG-specific scales. Total scores range from 0-50, with higher scores indicating more severe symptoms. Based on published literature, a 3-point improvement in score is considered clinically meaningful. Previously reported data have shown that a significantly greater proportion of efgartigimod-treated patients achieved clinically meaningful improvement (CMI) in MG-ADL and QMG scores compared to placebo-treated patients. The goal of the current analysis is to evaluate the efficacy of efgartigimod using the MGC, which was assessed during the ADAPT study.
Methods: Patients were randomized to receive efgartigimod (n=84) or placebo (n=83). A subset of patients were anti-acetylcholine receptor (AChR) antibody positive (n=65 efgartigimod versus n=64 placebo). Efgartigimod 10 mg/kg was administered intravenously in cycles of once-weekly infusions for 4 weeks, with subsequent cycles initiated based on clinical evaluation. Mean MGC change from baseline was a predefined exploratory endpoint.
Results: The mean (SE) reduction in MGC score at week 4 was -8.913 (0.974) for efgartigimod-treated anti-AChR antibody positive patients during cycle 1 compared with -2.871 (1.007) for placebo-treated patients (95% CI, -8.181 to -3.904; P<.0001). A similar improvement was seen in the overall population, with a mean (SE) change of -9.231 (0.878) in the efgartigimod group and -4.497 (0.885) in the placebo group (95% CI, -6.668 to -2.800; P<.0001). Similar results occurred during subsequent treatment cycles in both populations. A CMI of ≥3 points was observed in 81.0% of anti-AChR antibody positive patients in the efgartigimod group compared to 60.0% in the placebo group at week 4, with improvements reaching ≥10 points in 47.6% of patients in the efgartigimod group and 8.3% in the placebo group. In the overall population at week 4, 82.5% (efgartigimod) versus 63.3% (placebo) achieved a ≥3-point improvement, with improvements reaching ≥10 points in 46.3% (efgartigimod) and 12.7% (placebo) of patients. The most frequently observed adverse events are described below. Headache occurred with similar frequency between treatment groups. Nasopharyngitis, nausea, and diarrhea were more frequent in placebo patients. Upper respiratory tract infections and urinary tract infections occurred more in efgartigimod-treated patients. Adverse events were predominantly mild or moderate.
Conclusions: In the ADAPT study, efgartigimod treatment resulted in greater mean reductions of MGC scores (a composite measure incorporating both physician- and patient-reported outcomes) compared to placebo. The current analysis adds to the existing data that show efgartigimod is a promising new treatment for patients with gMG.
SS21.01The Interface of Neuromuscular Transmission and Mitochondrial Diseases
Horvath R1
1Department of Clinical Neurosciences, University of Cambridge, United Kingdom
Mutations in either the mitochondrial or nuclear genomes are associated with a diverse group of human disorders characterized by impaired mitochondrial respiration. The clinical presentation of mitochondrial diseases is very variable, and there are only very few effective treatments to date.
Within this group of diseases, an increasing number of mutations have been identified to cause a defect of neuromuscular junction which can be treated with drugs improving neuromuscular transmission. The fatigable weakness in these conditions may improve on therapy.
The overall prevalence of neuromuscular transmission defects in mitochondrial diseases is about 25%. Patients with some selected gene defects have been shown more frequently to present with a defect of the neuromuscular transmission, such as pathogenic dominant RRM2B variants or specific recessive variants in SLC25A1 and TEFM. The use of drugs improving the neuromuscular transmission may be beneficial in some of these patients.
The presentation will give an overview on mitochondrial diseases with neuromuscular transmission defects and will provide some guide to the diagnosis and treatment of these diseases.
SS21.02New Synaptic and Presynaptic Defects of the Neuromuscular Junction
Rodriguez Cruz P1
1Centro Nacional de Análisis Genómico - Centre For Genomic Regulation, Spain
Congenital myasthenic syndromes (CMS) are a heterogenous group of disorders caused by my mutations in genes encoding proteins that are essential for neuromuscular transmission. Next-generation sequencing (NGS) technology has helped to expand the genetic landscape of CMS with more than 30 subtypes described to date. The most common classification of CMS relies on the location of the mutated protein into presynaptic, synaptic or postsynaptic CMS and those thar are ubiquitously expressed. Postsynaptic CMS is by far the most common subtype of CMS accounting for approximately 70-80% of cases, but presynaptic and synaptic CMS are gaining attention as we improve our understanding on the function and molecular organization of the neuromuscular synapse. Until recently, mutations in the choline acetyltransferase gene (CHAT) were the only cause of presynaptic CMS described, but in recent years the landscape of presynaptic CMS genes has greatly expanded with the discovery of CMS genes involved in axonal transport (MYO9), synthesis and recycling of acetylcholine (SLC5A7, SLC18A3, PREPL) and vesicle exocytosis (SYT2, VAMP1, SNAP25B, UNC13A). Presynaptic CMS are generally rare and may present with central manifestations such as intellectual disability or learning difficulties derived from the expression of the mutated protein beyond the neuromuscular junction (NMJ), challenging the original definition of CMS. Other key clinical features include early onset, severe disease, respiratory apnoeas and modest response to cholinesterase inhibitors. Synaptic CMS is also rare and, until recently, endplate acetylcholinesterase deficiency caused by mutations in COLQ, which encodes the collagen-like tail subunit of asymmetric acetylcholinesterase, was the only subtype described. The spectrum has broadened with the discovery of novel synaptic CMS genes including COL13A1, LAMB2 and LAMA5. This has contributed to our understanding of the synaptic basal lamina, which is now considered a complex network in which key signal transduction processes for the development, maintenance and regeneration of pre- and post-synaptic NMJ structures take place, rather than a structure exclusively required for mechanical support. Major molecules at the synaptic basal lamina include collagens, laminins, nidogens and heparan sulfate proteoglycans such as Agrin, which is released into the synaptic cleft from the presynaptic terminal and plays an essential role in the AChR clustering pathway. COL13A1 encodes the α-chain of an atypical collagen, which is concentrated at the NMJ and is involved in AChR clustering and endplate maturation. Patients harboring COL13A1 mutations often suffer from a severe subtype of CMS early in life, with breathing and feeding difficulties and greater axial than appendicular weakness. Interestingly, disease severity appears to improve over time, which could be related the predominant role of COL13A1 in endplate maturation. Laminin-β2 deficiency and laminin-α5 deficiency are extremely rare diseases with only single CMS cases reported and manifestations extending beyond the boundaries of the NMJ, such us renal and ocular abnormalities. Cholinesterase inhibitors are usually not effective in synaptic CMS but 3,4-diaminopyridine and salbutamol can be beneficial in some subtypes, which supports the increasing role of β2-adrenergic agonists in the treatment of disorders of the neuromuscular junction.
SS21.03Genetic Epidemiology of Congenital Myasthenic Syndromes
Lochmüller H1,2,3
1University of Ottawa, Ottawa, Canada, 2Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada, 3The Ottawa Hospital, Ottawa, Canada
Congenital myasthenic syndromes (CMS) are a heterogeneous group of rare inherited neuromuscular disorders characterized by fatigable weakness owing to compromised function of the neuromuscular junction, either through direct impairment of neuromuscular transmission or through secondary defects that eventually compromise the safety margin of neuromuscular transmission. There is substantial clinical heterogeneity among the CMS subtypes, from childhood onset with episodic apnea and intellectual disability, to later onset with limb-girdle distribution of weakness and less facial and eye involvement, but all are associated with distinctive clinical, electrophysiological, laboratory and ultrastructural abnormalities.
Early gene discovery work in CMS included the discovery of a number of founder mutations (including in the CHRNE, DOK7 and RAPSN genes) and larger international studies with larger cohorts from Europe, Brazil, India and Turkey have revealed epidemiological differences between populations. In recent years, next-generation sequencing has dramatically increased the number of genetic defects reported as causative of CMS, with over 30 genes now implicated. Nevertheless, the clinical and genetic heterogeneity of CMS, as well as referral and ascertainment biases in diagnostic practice, have made phenotype-based epidemiological studies and prevalence estimations difficult, with publications in the last decade providing estimates ranging from 1.8 cases per million total population to 22.2 cases per million children. This lack of accurate data hampers diagnosis, healthcare provision and therapy development.
Efforts to better understand CMS epidemiology have resulted in the development of patient registries collecting clinical and genetic data on affected individuals and establishing trial-ready patient cohorts. In parallel, the new large-scale population databases of genomic data that have come online in recent years provide the opportunity to calculate the genetic prevalence or lifetime risk of autosomal recessive CMS based on the allele frequency of individual variants in an unaffected population, an approach made possible by the fact that most CMS subtypes are recessive, meaning that carriers of single variants that are pathogenic when present in homozygosity or compound heterozygosity can be found in the healthy general population. This data may be used to assign diagnostic probabilities, provide insights into numbers of patients amenable to clinical research and novel therapies, and aid resource allocation in therapy development and healthcare provision.
SS22.01Clinical Trial Update for Myasthenia Gravis
Bril V1
1University Health Network, University of Toronto, Toronto, Canada
Traditional myasthenia gravis (MG) therapies include corticosteroids and other immunosuppressant therapies (IST). These are limited by troublesome side-effects and delayed onset of action. As a result, many MG patients suffer ongoing impairments with odds of 5.76 that they will be unemployed at 2 years after diagnosis. Many patients prioritize avoiding both MG crises and rescue treatments, and fear that their treatments are not targeting the underlying causes of their disease. An explosion in novel therapies for myasthenia gravis (MG) promises to improve the treatment landscape. Recently approved drugs such as eculizumab and efgartigimod are leading the way to expanded treatment choices for the MG patient. These new therapies aim at different targets of the immune system: eculizumab (complement inhibitor) at the downstream complement deposition and membrane attack complex stage, and efgartigimod (Fc receptor [FcR] inhibitor) at pathogenic antibody recycling upstream in the immune system. Eculizumab has shown benefit for AChR antibody positive, refractory MG and efgartigimod for AChR antibody positive, generalized MG with some benefits in seronegative patients. Both drugs have been generally well tolerated in phase 3 clinical trials. Rozanolixizumab, another FcR inhibitor, has shown benefits in AChR antibody positive patients and also MuSK antibody positive patients. Additional FcR inhibitors (nipocalimab and batoclimab) and complement inhibitors (zilucoplan and ravilizumab) are in development. Future potential therapies include anti-B cell therapies such as inebilizumab, mezagitamab (anti-CD 38 antibody), satralizumab (anti-interleukin 6 antibody) and chimeric anti-T cell therapy.
SS22.02Pregnancy and Treatment Considerations in Myasthenia Gravis and Congenital Myasthenic Syndrome
Palace J1
1Oxford University Hospitals Trust, United Kingdom
The outcomes of pregnancy in both the congenital myasthenic syndromes (CMS) and acquired myasthenia gravis (MG) are generally favourable. Good pre-pregnancy control of MG appears to be a prognostic factor for stability during pregnancy, and although rare and so there is a paucity of evidence, the subtype of CMS may play a role in the likelihood of stable symptoms during pregnancy.
Planning should be instituted well in advance of conception particularly in regards to therapeutic strategies to ensure teratogenic immunosuppressive drugs can be substituted. Other drugs where the effects are less clear should be discussed, including the strength of evidence, and combined decision making is encouraged, and where possible treatment should be reduced to a minimum effective dose. In those with CMS prior formal genetic counselling should have taken place.
A multidisciplinary approach involving the relevant specialists throughout pregnancy, delivery and in the neonatal period is important to ensure good foetal outcomes. Awareness and prompt appropriate treatment is crucial of transient neonatal myasthenia in those with MG and of the rare cases of inherited CMS from the affected mother.
SS22.03Role of Autoantibodies in Diagnosis, Treatment Choice and Monitoring of Myasthenia Gravis
Vincent A1, Leite M2
1University of Oxford, Oxford, United Kingdom, 2Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
Myasthenia gravis (MG) is the best-known antibody-mediated neurological disease. Acetylcholine receptor antibodies (AChR-Abs) are present in around 85% of patients with generalised disease (GMG) and muscle specific kinase (MuSK) antibodies in up to 50% of those without AChR antibodies. Around 50% of ocular MG patients have AChR-Abs and only a small and variable proportion have MuSK-Abs. LRP4 interacts with MuSK and is important at the neuromuscular junction, but LRP4-Abs are rare in our experience.
AChR antibodies can be measured by radioimmunoprecipitation but increasingly an ELISA is used. ELISA is a little inferior in specificity and sensitivity but both can occasionally detect antibodies in patients without clinical evidence of MG (but who might be at risk). If they are both negative, AChR-Abs may be detected using a cell-based assay, unfortunately not available in many centres, where the AChR is clustered by the intracellular protein rapsyn and the antibodies are able to bind divalently to adjacent AChRs as they would at the neuromuscular junction. MuSK-Abs and LRP4-Abs can also be detected by the appropriate cell-based assays. Antibodies with strong preference for foetal (rather than adult) AChR are importantly found in the very rare mothers whose babies have loss of foetal movement in utero leading to life-threatening arthrogryposis multiplex congenital or a less severe, persistent facial myopathy. Cortactin and titin antibodies are associated with MG but not frequently used for diagnosis.
In general, immunotherapies are needed to reduce antibody levels and it can be helpful to know which antibodies you are dealing with. Thymectomy is highly relevant to AChR-Ab MG but is not usually performed in MuSK-Ab MG where the thymus pathology is normal. Drugs that target the FcRn that recycles IgG in vivo maintaining circulating IgG levels, can reduce total IgG within a few days, and offer an alternative to plasma exchange; they have been shown effective in AChR-Ab MG and could be used in MuSK-Ab MG. Most of the antibodies to AChR are IgG1, and complement-mediated lysis is an important mechanism in causing AChR loss; anti-complement therapeutic antibodies have been shown to be effective in MG. Conversely, MuSK-Abs are IgG4 that does not activate complement and this form of MG is unlikely to respond to anti-complement treatment but appears to respond better to anti-CD20 therapies than AChR-Ab MG.
Finally, how useful are serial antibody estimations in monitoring MG? In practice, not very. This is because the routine diagnostic laboratories use assays designed, principally, for diagnosis and rather than to measure accurately the titres. High titres in particular are often underestimated. Monitoring the patient’s symptoms is much more informative. On the other hand, if a patient is not improving, despite immunotherapies, it may be worth checking whether the antibodies have fallen (ie if treatment has failed), but for this to be informative, serial dilutions of the post-treatment serum to identify the true titre in comparison with a pre-treatment serum would be required. This is only likely to be available at specialist research centres.
Oral Presentations
PS01.01Analysis of the Longitudinal CINRG Becker Natural History Study Dataset
Dang U1, Gordish-Dressman H, Niizawa G, Gorni K, Guglieri M, Connolly A, Wicklund M, Bertorini T, Mah J, Thangarajh M, Smith E, Kuntz N, McDonald C, Henricson E, Upadhyayula S, Byrne B, Manousakis G, Harper A, Iannaccone S, Clemens P, CINRG-BNHS Investigators
1Carleton University, Ottawa, Canada
Background: The phenotype of Becker muscular dystrophy (BMD) is extremely variable, ranging from Duchenne-like severity (loss of ambulation in second decade) to much milder disease with ambulation retained well into adulthood. We performed a prospective, natural history study (BNHS) of males with in-frame dystrophin gene mutations causing BMD to better characterize the clinical course and outcomes crucial to rigorous clinical treatment trials.
Objective: To present natural history of clinical outcomes in BMD from a longitudinal dataset.
Approach: The BNHS followed 83 ambulatory and non-ambulatory males with BMD aged 5.5 to 75.5 years at enrollment for up to three years with annual assessments. Baseline characteristics of these participants have been published. In this post-baseline analysis, we studied binned repeated cross-sectional clinical outcome data, shift-based analyses of NSAA, longitudinal mixed-effect modeling of clinical outcomes, and survival analysis of time to stand. Analyses were stratified by age (<18 or ≥18 years old).
Results: Deletion exons del 45-47 and 45-48 were most common. In those <18 years, the NSAA showed a ceiling effect not observed with other outcomes. In longitudinal modeling of percentage predicted FVC, 3 timed function tests, 6-minute walk distance, and NSAA, age was found to be significantly associated with outcome performance in adulthood but not in those <18 years. Mutation status (del 45-47, 45-48, vs. others) was significantly associated with some outcomes. The median age at which it took 10 seconds or longer to stand was estimated as 51 years (95% CI: 45 years, infinity) by time to event analysis.
Conclusions: While the sample size was small, our data demonstrated variable progression of different outcomes based on age groups, and mutation groups, and found that disease progression seems to largely manifest in adulthood for BMD. Our study has clinical trial design implications for enrollment criteria, efficacy determination, and sample size calculations.
PS01.02Results of a Double-Blind Cross-over Trial of Vamorolone in DMD: A Safer Alternative to Corticosteroids
Hoffman E1, Guglieri M2, Clemens P3, Perlman S4, Smith E5, Horrocks I6, Finkel R7, Mah J8, Deconinck N9, Goemans N10, Haberlova J11, Straub V2, Harper A12, Sheih P13, De Waele L14, Castro D15, Yang M16, Ryan M17, McDonald C18, Tulinius M19, Webster R20, McMillan H21, Kuntz N22, Rao V22, Baranello G23, Spinty S24, Childs A25, Sbrocchi A26, Selby K27, Monduy M28, Nevo Y29, Vilchez-Padilla J30, Nascimiento-Osorio A31, Niks E32, de Groot I33, Katsalouli M34, James M2, van den Anker J1, Damsker J1, Ahmet A21, Ward L21, Dang U35, Leinonen M36
1Reveragen, Rockville, United States, 2Newcastle University, Newcastle, UK, 3University of Pittsburgh School of Medicine, Pittsburgh, USA, 4Seattle Children’s Hospital, Seattle, USA, 5Duke University Medical Center, Durham, USA, 6Royal Hospital for Children, Glasgow, UK, 7St. Jude Children’s Research Hospital, Memphis, USA, 8University of Calgary, Cumming School of Medicine, Calgary, Canada, 9Hôpital Universitaire des Enfants Reine Fabiola, Brussels, Belgium, 10University Hospitals Leuven, Leuven, Belgium, 112nd School of Medicine Charles University in Prague, Prague, Czech Republic, 12Children’s Hospital of Richmond at Virginia Commonwealth University, Richmond, USA, 13UCLA Life Sciences, Los Angeles, USA, 14Catholic University of Leuven, Leuven, Belgium, 15University of Texas Southwestern Medical Center at Dallas, Dallas, USA, 16Children’s Hospital Colorado, Denver, USA, 17Royal Children’s Hospital, Melbourne, Australia, 18University of California-Davis, Irvine, USA, 19Gothenburg University, The Queen Silvia Children’s Hospital, Gothenburg, Sweden, 20The Institute for Neuroscience and Muscle Research, The Children’s Hospital at Westmead, Sydney, Australia, 21Children’s Hospital of Eastern Ontario, Ottawa, Canada, 22Ann & Robert H. Lurie Children’s Hospital, Chicago, USA, 23Great Ormond Street Hospital NHS Trust, London, UK, 24Alder Hey Children’s NHS Foundation Trust, Liverpool, UK, 25Leeds Teaching Hospitals NHS Trust, Leeds, UK, 26Montreal Children’s Hospital, Montreal, Canada, 27University of British Columbia, Vancouver, Canada, 28Nemours Children’s Hospital, Orlando, USA, 29Schneider Children’s Medical Center of Israel, Tel Aviv, Israel, 30Neuromuscular Research Unit, IIS La Fe; CIBERER, Valencia, Spain, 31Hospital Sant Joan de Deu, Barcelona, Spain, 32Leiden University Medical Center, Leiden, Netherlands, 33Radboud University Nijmegen Faculty of Medical Sciences, Nijmegen, Netherlands, 34Aghia Sophia Children’s Hospital, Athens, Greece, 35Carleton University, Ottawa, Canada, 36Santhera, Basel, Switzerland
Vamorolone is a dissociative steroidal anti-inflammatory drug that changes structure/activity relationships with the glucocorticoid receptor. Steroid-naïve boys with genetically confirmed DMD (5.4±0.9 years) were randomized (n=121) in a double-blind, placebo- and prednisone-controlled efficacy and safety trial of vamorolone. The trial had a 24-week Period 1 with participants randomized to 4 groups (placebo, prednisone, vamorolone 2 mg/kg/day, vamorolone 6 mg/kg/day), and a 24-week Period 2 where the placebo and prednisone groups crossed over to vamorolone. Both vamorolone-treated groups showed significant improvements in motor function tests over the 24-week treatment Period 1. The trial met the primary and first four sequential secondary end points for change from baseline to Week 24 (time to stand velocity: vamorolone 6 mg/kg/day vs. placebo p=0.002, 2 mg/kg/day vs. placebo p=0.02; 6-minute walk test 6 mg/kg/day vs. placebo p=0.003, 2 mg/kg/day vs. placebo p=0.009; time to run/walk 10 meters velocity 6 vs. placebo p=0.002). Height percentile declined in prednisone-treated, but not vamorolone-treated participants (least square means [standard error] prednisone -1.58%tile [se=1.44] vs. vamorolone 6 mg/kg/day +3.40%tile [se=1.55]; p=0.02). Bone turnover markers declined with prednisone treatment but not vamorolone (p<0.001 for all comparisons). All 3 treatment groups led to adrenal insufficiency, but vamorolone 2 mg/kg/day less than prednisone (p<0.002).
Of the 56 participants receiving vamorolone during both Period 1 and 2, 2 of 56 patients discontinued treatment during Period 2 (1 adverse event, 1 consent withdrawn). For vamorolone 6 mg/kg/day, efficacy was maintained or further improved from Week 24 to Week 48, whilst for vamorolone 2 mg/kg/day, efficacy was maintained in some efficacy endpoints. Comparing the 2 vamorolone dose groups over a 48-week treatment period, vamorolone 6 mg/kg/day showed greater improvements in motor outcomes compared to 2 mg/kg/day for some outcomes (TTSTAND, 6MWT and TTCLIMB), but similar efficacy in others (NSAA, TTRW). Three serious adverse events were reported during the 48 weeks: perforated appendicitis (2 mg/kg/day), asthma (6 mg/kg/day) and viral gastroenteritis (6 mg/kg/day), all considered unrelated to vamorolone. Stunting of growth was not seen at either dose group over the 48-week treatment period, and body mass index stabilized after an initial increase during first 24 weeks.
Prednisone-treated participants (Period 1) that crossed over to vamorolone showed maintenance of efficacy across all efficacy endpoints for vamorolone 6 mg/kg/day. No serious adverse events were reported after the switch. Annualized rates of adverse events were reduced after the switch from prednisone to vamorolone (all events: 20% reduction, steroid-related events: 40% reduction). Stunting of growth observed with prednisone during Period 1 was reversed during treatment with vamorolone during Period 2.
Placebo-treated participants in Period 1 that crossed over to vamorolone in Period 2 (delayed starters) showed an improvement in multiple efficacy outcomes after the switch to vamorolone. Early-starters in the vamorolone 6 mg/kg/day group (treatment continued throughout 48 weeks) had increased outcome means compared with placebo->vamorolone 6 mg/kg/day delayed-starters for 4/5 outcomes at four post-baseline timepoints, suggesting continued efficacy beyond 24-weeks treatment.
In summary, the trial showed compelling evidence of efficacy of vamorolone with improved safety compared to prednisone.
PS01.3Early Effect of Steroids on Functional Outcomes in Young Boys with Duchenne Muscular Dystrophy
Schiava M1, Broomfield J2, Abrams K2, McDermott M3,4, Martens W4, Gregory S4, Mayhew A1, McDonald C5, Griggs R4, Guglieri M1
1Newcastle University, John Walton Muscular dystrophy Research Centre, Newcastle Upon Tyne, United Kingdom, 2Department of Health Sciences, University of Leicester, Leicester, United Kingdom, 3Department. of Biostatistics and Computational Biology, University of Rochester Medical Centre, Rochester, USA, 4Department. of Neurology, University of Rochester Medical Centre, Rochester, USA, 5Department of Physical Medicine and Rehabilitation, University of California Davis Health, Sacramento, USA
Introduction: Glucocorticoids (GC) are part of the standard of care in Duchenne Muscular Dystrophy (DMD) and have changed the disease natural history. Previous studies reported a favourable effect of early treatment with GC.
However, the effect of GC on motor function outcomes (FO) at the time of treatment initiation remain to be characterized. This study aims to describe motor FO in young boys with DMD after starting GC treatment and identify the factors that influence FO trajectories
Methods: Functional outcome data from the first 18 months of the Finding the Optimum Regimen for Duchenne Muscular Dystrophy (FOR-DMD) study were analysed. The FOR-DMD study was an international, multi-centre randomized, double-blind trial including 196 GC-naïve boys with DMD age 4 - <8 years. Boys were randomized 1:1:1 to receive daily prednisone (0.75 mg/kg/day), daily deflazacort (0.9 mg/kg/day), or intermittent prednisone (0.75 mg/kg 10 days on/10 days off). Boys were assessed at screening, baseline and at Months 3, 6, 12 and 18 after initiation of treatment; motor FO included rise from floor velocity (TRF-vel), 10-meter walk/run velocity (10MWR-vel), North Star Ambulatory Assessment (NSAA) total score and the 6-minute walking test distance (6MWT).
A linear mixed model was performed; fixed effects included: age at baseline, GC regime, mutation type; baseline height and dichotomized FO cut-off values at baseline based on the literature.
Results: One hundred ninety-six boys were included in the study. The distribution by age group was: 20% (n=40) 4 – 4.9 y, 40% (n=78) 5 – 5.9 y, 24% (n=47) 6 – 6.9 y, and 16% (n=31) 7 – 7.9 y. Sixty-five boys were allocated to daily prednisone and daily deflazacort and 66 boys to intermittent prednisone regime. The predominant mutation type was out of frame deletions (67.4%, 130/193) of which 68.5% (89/130) were amenable to exon skipping. The distribution by dichotomized FO cut-off values at baseline was: 60% (n=118) boys had a 6MWT > 330 mts; 73% (n=143) TRF-vel > 0.138 units/second; 69% (n=136) 10MWR-vel > 0.142 units/second and 42% (n=83) NSAA > 22 points.
A significant effect of GC regimen and dichotomized FO cut-off values at baseline was observed in all FO at month 18. Boys on intermittent, 10-days on/10-days off, prednisone had lower trajectories in the TRF-vel,10MWR-vel, 6MWT compared to daily prednisone. Boys with a baseline FO below the cut-off values showed lower trajectories than boys above them.
Boys in the 7 – 7.9 y age group at baseline showed lower performances in all the FO up to 18 months compared to boys in the 5 – 5.9 y group. Mutation type had no effect on FO at 18-months.
Discussion: This study describes the factors that affects FO trajectories in DMD at the time of GC treatment initiation. This data can be useful for evaluation of the effect of novel therapies targeting young boys with DMD and inform clinical trial design.
PS01.04Retrospective, Longitudinal Clinical Analysis in a Large UK Cohort of Patients with Nemaline Myopathies
Perry L1,4, Steel D1, Brusa C1, McCauley J2, Scoto M1, Manzur A1, Quinlivan R1, Munot P1, Robb S1, Baranello G1, Muntoni F1,3,4, Sarkozy A1,4
1The Dubowitz Neuromuscular Centre, University College London, Great Ormond Street, Institute of Child Health and MRC Centre for Neuromuscular Diseases, Neurosciences Unit, Great Ormond Street Hospital, London, United Kingdom, 2Genetics Department, Molecular Genetics Laboratory Viapath, Guy’s Hospital, London, United Kingdom, 3NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Institute of Child Health, University College London, London, United Kingdom, 4MRC UCL International Centre for Genomic Medicine in Neuromuscular Diseases, London, United Kingdom
Introduction: Nemaline myopathies (NM) are rare, clinically, and genetically heterogeneous conditions with relevant co-morbidity and mortality. Limited knowledge on long term natural history and outcome predictors are available to guide management of patients.
Aims: To describe genotype phenotype correlations, long term clinical outcomes and prognostic factors in NM.
Methods: We completed a retrospective cross-sectional phenotypic and genotypic analysis of patients with a genetic diagnosis of NM referred to the highly specialised national service for congenital myopathies/muscular dystrophies at Great Ormond Street Hospital, between January 2001 and March 2021.
Results: We identified 134 NM patients with the following genotypes: 60 NEB, 39 ACTA1, 11 TPM3, 9 KLHL40, 8 TPM2, 3 LMOD3, 2 TNNT1 and 2 CFL2-NM. Overall mortality was 19% (23/120) with a median age at death of 3 months (IQR 16.3, range 1 day to 19 years). CFL2 and KLHL40 genotypes had the highest mortality rates and lowest median age at death of 2 months (IQR 2.4, range 12 days to 4.5 months). Commencement of respiratory support in the neonatal period was associated with an increased mortality risk (P = 0.0003). Median living age at last assessment was 17.4 years (IQR 15.03, range 3-79.5 years). Symptom onset was ante/neonatal in 65% (72/110) of patients, with later onset, including adulthood, observed in 35% (38/110) of patients with NEB, ACTA1 and TPM3-NM. Ambulation was attained in 86% (79/92) of patients older than 18 months, and this was most common in NEB, ACTA1, TPM2 and TPM3-NM, and absent in TNNT1-NM patients. Motor deterioration was observed in 45% (35/77) of ambulant patients with 17% (13/77) losing independent ambulation. 17% (13/77) of patients exhibited a degree of motor improvement during the first decade of life. Loss of ambulation was more common in patients with a history of gross motor delay (23%, 10/43) compared to those with normal early gross-motor milestones (7%, 2/28). Respiratory and feeding support were required by 44% (47/107) and 46% (47/102) of patients respectively, and were most common in CFL2, KLHL40 and LMOD3-NM, and least frequent in TPM2 and TPM3-NM.
Conclusions: Our study confirms NEB and ACTA1-NM as the most common genotypes. We report motor deterioration in 2/3 of patients, which appears more prevalent in patients with a history of gross motor delay. Our data also highlights higher respiratory and/or feeding co-morbidities in CFL2, KLHL40 and LMOD3-NM, with CFL2 and KLHL40-NM having the highest rates of infantile mortality. Our data also showed that early respiratory insufficiency was associated with a significantly increased mortality risk. Overall, TPM2-NM appears to have the best motor, co-morbidity, and mortality outcomes. International collaborations and large prospective natural history studies are needed to confirm these findings and to build trial readiness for these conditions.
PS01.05GNE Myopathy – Phenotype, Genotype Characteristics and Disease Progression in Large Cohort of Indian Patients
Baskar D1, Vengalil S1, Nashi S1, Arunachal G1, Narayanappa G1, Kumar P1, Huddar A1, U G1, Thomas A1, Atchayaram N1
1National Institute Of Mental Health And Neurosciences (nimhans), Bangalore, India
Background: GNE myopathy (GNEM) which is also known as hereditary inclusion body myopathy or distal myopathy with rimmed vacuoles is a rare and unique adult-onset inherited myopathy. It mostly presents with distal lower limb weakness with conspicuous quadriceps sparing. GNEM is autosomal recessive disorder due to mutation of GNE gene in chromosome 19p13.3 which encodes a bifunctional enzyme involved in sialic acid biosynthesis. Here we present clinical and genetic features of genetically proven GNEM patients from India.
Methods and materials: This was an observational follow-up study done at NIMHANS, India of GNEM genetically diagnosed by Sanger and Next generation sequencing. Clinical characteristics along with functional disability assessment using IBMFRS and MDFRS was recorded.
Results: The study cohort consisted of 152 patients with 65.8% being males. The mean age of symptom onset was 26.6 ± 6.2 years with majority of patients in the 3rd decade of life. The most common presenting symptom was foot drop (44.1%). About 18% of patients had proximal lower limb weakness masquerading as limb girdle syndrome. Bifacial weakness was noted in 9 patients and Beevor’s sign was seen in 87 patients (57.2%) denoting lower abdominal muscle weakness. Most of the patients had predominant hamstring and tibialis anterior weakness with only 5 patients having quadriceps weakness. The founder Indian mutation (c.2179G>A, V727M) was found in 126 patients (84%) and Roma Gypsy founder mutation (c.1853T>C, I618T) in 7 patients (4.6%). All compound heterozygotes had V727M in one allele. 56.3% of patients with V727M were unable to walk at presentation with loss of ambulation on follow-up noted in 21.8% patients. Most of the patients had involvement of kinase (44.7%) or kinase and epimerase domains (39.4%). Missense mutations were seen in 59.8% with 36.8% having novel mutations. 18 patients had rapid progression of symptoms with most of them having V727M mutation with involvement of either kinase or epimerase and kinase domains. Muscle biopsy was done in 36 patients with 75% of patients showing rimmed vacuoles. Among the 78 patients who were followed up majority had only mild to moderate disability with IBMFRS scale (92%), whereas 76.3% had a mobility disability score of <50% with MDFRS scale.
Discussion and conclusion: This is one of the few studies with large cohort of patients analysed prospectively. This study highlights important phenotypic and genotypic characteristics and their relationship in patients with GNEM.
PS01.06Multimodal Assessment of Dysphagia in Patients with Inclusion Body Myositis and Oculopharyngeal Muscular Dystrophy
Zeng R1, Rietveld A2, Al-Bourini O3, Olthoff A4, Weidenmüller M4, Kroon R2, Carstens P1, Kommerell I1, Hofer S5, Frahm J5, Uecker M3, Seif A3, Friede T6, Saris C2, Schmidt J1
1Department of Neurology, University Medical Center, Goettingen, Goettingen, Germany, 2Department of Neurology, Radboud University Medical Center, Nijmegen, The Netherlands, 3Department of Diagnostic and Interventional Radiology, University Medical Center Goettingen, Goettingen, Germany, 4Department of Otorhinolaryngology, Phoniatrics and Pedaudiology, University Medical Center Goettingen, Goettingen, Germany, 5Biomedical NMR, Max Planck Institute for Multidisciplinary Sciences, Goettingen, Germany, 6Department of Medical Statistics, University Medical Center Goettingen, Goettingen, Germany
Background: Swallowing dysfunction (dysphagia) is associated with higher morbidity and mortality in patients with muscle diseases, however the underlying pathophysiology is only poorly understood. In addition to standard instrumental assessments of dysphagia such as fiberoptic endoscopic evaluation of swallowing (FEES) and videofluoroscopy (VFSS), new diagnostic tools may enable a more precise evaluation of dysphagia. Aim of this project is the characterization of myogenic dysphagia in a multimodal approach using real-time MRI, quantitative T1 mapping, quantitative muscle ultrasound, FEES and logopaedic assessments. For this purpose, two different muscle diseases were analysed: Oculopharyngeal muscular dystrophy (OPMD), a genetic disorder characterized by ptosis, dysphagia and proximal muscle weakness, and inclusion body myositis (IBM), an inflammatory myopathy with typically asymmetric limb weakness and often associated with severe dysphagia.
Methods: 16 patients with IBM and 13 patients with OPMD were included in the study. Swallowing function was studied in each participant using real-time MRI of bolus passage times, FEES and logopaedic assessments. Tissue analysis of swallowing muscles was performed using quantitative T1 mapping and quantitative muscle ultrasound. The results of the two cohorts were compared with each other and to data of a healthy cohort.
Results: Tissue analysis using quantitative T1 mapping and quantitative muscle ultrasound demonstrated characteristic patterns of fibrosis and fatty infiltration in the tongue and masseter muscle in OPMD. Functional analysis with real-time MRI revealed significantly prolonged oral transit times in patients with OPMD, as well as prolonged pharyngeal transit times in IBM and OPMD. FEES and logopaedic assessments reliably identified dysphagia in affected patients, but failed to differentiate the underlying disease.
Conclusion: OPMD and IBM show distinct disease-specific patterns of oropharyngeal muscle involvement and swallowing dysfunction, which are revealed by novel MRI techniques and ultrasound. These innovative diagnostic parameters widen the spectrum for diagnostics, follow-up examination and further research on the development of treatments of dysphagia.
PS02.01Comparison of Inflammation and Neurodegeneration Markers in CSF as Predictors of Survival in ALS Patients
Masrori P2,3,4, De Shaepdryver M1, Poesen K1,5, Van Damme P2,3,4
1Laboratory for Molecular Neurobiomarker Research, Department of Neurosciences, Leuven Brain Institute, KU Leuven, Leuven, Belgium, 2Department of Neurology, University Hospitals Leuven, Leuven, Belgium, 3Experimental Neurology, Department of Neurosciences, Leuven Brain Institute, KU Leuven, Leuven, Belgium, 4Laboratory of Neurobiology, Center for Brain & Disease Research, Leuven, Belgium, 5Laboratory Medicine, University Hospitals Leuven, Leuven, Belgium
Objective: To compare markers of neurodegeneration [neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain (pNfH)] and markers of inflammation [chitotriosidase-1 (CHIT1), chitinase-3-like protein 1 (YKL-40) and monocyte chemoattractant protein-1 (MCP-1)] as biomarkers of survival in cerebrospinal fluid (CSF) of patients with amyotrophic lateral sclerosis (ALS).
Methods: Protein biomarkers were determined in CSF of 94 patients with ALS using enzyme-linked immunoassays. Univariate survival analyses were performed using the Kaplan-Meier analysis, and the log-rank test was conducted to determine differences between the survival curves (n = 94, censored: 14%). A Cox regression survival model was calculated including 8 established clinical predictors of survival in ALS (n = 84, censored: 13%). For this purpose patients were stratified as low and high biomarker level based upon the median concentration of the biomarker in the total ALS cohort.
Results: Univariate survival analyses revealed a significantly shorter survival in patients with ALS having high levels of pNfH (χ2 = 12.69, p < 0.001), NfL (χ2 = 12.34, p < 0.001), CHIT1 (χ2 = 7.62, p < 0.01), YKL-40 (χ2 = 14.05, p < 0.001) and MCP-1 (χ2 = 8.45, p < 0.01). In a multivariate Cox regression model, high levels of pNfH (HR: 3.36, 95% CI: 1.87 – 6.04, p < 0.001), NfL (HR: 2.06, 95% CI: 1.23 – 3.46, p < 0.01), CHIT1 (HR: 2.13, 95% CI: 1.22 – 3.73, p < 0.01), YKL-40 (HR: 2.17, 95% CI: 1.21 – 3.90, p = 0.01) and MCP-1 (HR: 2.49, 95% CI: 1.47 – 4.21, p = 0.001) were independently associated with a shorter survival. Patients with both high NfL and YKL-40 levels harbored a significantly shorter survival (median: 15.3 months, range: 1.43 – 49.0 months) compared to those patients with both low NfL and YKL-40 levels (median: 45.5 months, range: 2.03 – 74.5 months; p < 0.0001), but did not when compared to those patients with low NfL yet high YKL-40 levels (median: 20.2 months, range: 0.47 – 77.3 months; p = 0.74).
Conclusion: This study highlights the importance of CSF biomarkers to predict survival in patients with ALS. Furthermore, it demonstrates that patients with ALS without pronounced neurodegeneration have a short survival in the presence of neuroinflammation, e.g. reflected by low NfL and high YKL-40 levels. These findings may have implications for future stratification of patients in clinical trials.
PS02.2Spinal Cord MRI for Tracking of Early Degeneration in C9orf72 Asymptomatic Carriers: A Longitudinal Study
Querin G1,2, Bede P3, Péllegrini-Issac M4, Rinaldi D5,6, Saracino D5,6, Salachas F7, Camuzat A5, Marchand-Pauvert V4, Cohen-Adad J8, Colliot O5, Isabelle L5,6, Pradat P4,7
1Institut de Myologie, I-Motion Clinical Trials Platform, Paris, France, 2APHP, Pitie-Salpetriere Hospital, Service de Neuromyologie, Paris, France, 3Computational Neuroimaging Group, Academic Unit of Neurology, Trinity College Dublin, Dublin, Ireland, 4Laboratoire d’Imagerie Biomédicale, CNRS, INSERM, Sorbonne Université, Paris, France, 5Institut du Cerveau et de la Moelle Épinière, Sorbonne Université, INSERM U1127, CNRS UMR 7225, Hôpital Pitié-Salpêtrière, Paris, France, 6Institute of Memory and Alzheimer’s Disease, Center of Excellence of Neurodegenerative Disease, Sorbonne University, Hôpital Pitié-Salpêtrière, Paris, France, 7APHP, Département de Neurologie, Centre référent SLA, Hôpital Pitié-Salpêtrière, Sorbonne Université, CNRS UMR7622, INSERM ERL 1156, IBPS, Paris, France, 8NeuroPoly Lab, Institute of Biomedical Engineering, Polytechnique Montreal, Montreal, QC, Canada; Functional Neuroimaging Unit, CRIUGM, Université de Montreal, Montreal, Canada
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal dementia (FTD) share genetic susceptibility and a large portion of familial cases are due to C9orf72 gene mutations. Brain and spinal cord (SC) imaging studies in asymptomatic C9orf72 carriers have demonstrated white (WM) and grey matter (GM) degeneration up to 20 years before the expected symptom onset.
The objective of this study is to longitudinally describe, using quantitative MRI, the evolution of spinal cord (SC) degeneration over 36-months observation time in a cohort of asymptomatic C9orf72 mutation carriers.
Methods: 40 asymptomatic C9orf72 carriers (C9+) were enrolled in the study. Each subject underwent a 3T cervical SC MRI at baseline, after 18 and 36 months. Quantitative measures of GM and WM atrophy and DTI parameters (FA, AD, MD, RD) in the cortico-spinal tracts (CST) and dorsal columns were computed.
We used linear mixed-effects models (LMEMs) to test for significant differences in MIR parameters between time points. We employed the following terms as fixed effects: time point (from T0 up to T3), baseline age, gender, genetic status and interaction terms between time point and age, and between time point and genetic status. Random intercept terms for participants were included in the model.
Results: Mean age at inclusion was 41.18 years +/- 11.46 (range 25.42 – 72.79). 18 subjects were male and 22 female, 3 subjects came from ALS-only families, while 17 of them originated from FTD-only families and 20 from ALS/FTD families. Clinical examination was normal for all the participants. No significant difference was identified in MRI parameters between subjects coming from ALS and FTD families. No significant difference in GM and WM cross-sectional area was observed over the three time points (p > 0.05). A significant progressive reduction of fractional anisotropy (FA) in the pyramidal tracts was observed over time with a significant difference between the baseline and the 36-months evaluation (p = 0.04). No other significant modifications in DTI parameters in the cortico-spinal tract were detected. When considering only patients coming from families having at least one ALS member, no significant evolution of CST degeneration was identified (p > 0.05).
Discussion: Cervical SC imaging of C9orf72 hexanucleotide carriers detect a progressive pyramidal tract FA reduction which seems to be continuous but not linear. Subjects coming from ALS families, do not seem to have faster pyramidal tract degeneration compared to the global cohort.
PS02.03Analysis of Muscle Resonance Imaging of Cohort of Chronic Motor Neuropathy/Neuronopathy Patients Reveals Characteristic Features
Diaz-manera J1, Esteller D2, Morrow J3, Reyes D4, Tasca G5, Alangary A3, Bisogni G6, Monforte M5, Marini-Bettolo C1, Sabatelli M6, Ramdharry G3, Alonso-Pérez J4, Bolaño-Díaz C1, Schiava M1, Turon-Sans J4, Guglieri M1, Rossor A3, Vesperinas A4, Collet R4, Olive M4, Straub V1, Reilly M3, Rojas-Garcia R4
1Newcastle University, Newcastle Upon Tyne, United Kingdom, 2Neurology Department. Hospital Clinic de Barcelona. Universitat de Barcelona., Barcelona, Spain, 3Department of Neuromuscular Disease, Queen Square, UCL Institute of Neurology and the National Hospital of Neurology and Neurosurgery, London, UK., London, United Kingdom, 4Neuromuscular Disorders Unit. Neurology department. Hospital de la Santa Creu i Sant Pau. Universitat Autònoma de Barcelona. Barcelona. Spain., Barcelona, Spain, 5UOC di Neurologia, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy., Rome, Italy, 6Centro Clinico NeMO Adulti, Rome, Italy., Rome, Italy
Background: Chronic motor neuropathy/neuronopathy (CMNs) encompass a group of rare diseases characterized by muscle weakness mainly affecting distal muscles of the limbs. Muscle magnetic resonance imaging (MRI) can be a useful tool for the diagnostic workup but has not been explored systematically in these patients.
Methods: This is a retrospective study collecting clinical, genetic and imaging data. Muscle MRI included T1-weighted and Short Tau Inversion Recovery (STIR) sequences. Fat replacement was quantified using the Mercuri score. Identification of patterns was performed using hierarchical clustering.
Results: We included 76 patients with a clinical and electrophysiological diagnosis of CMN. A pathogenic gene variant was identified in 29 subjects, a VUS was identified in 15 and 32 subjects remained without molecular diagnosis. We collected 25 whole-body and 51 lower limb (LL) MRIs. All patients but one had pathological findings. We identified features common to all patients regardless of the genetic diagnosis. Muscle fat replacement was predominant in the LL distal muscles (75/76 cases), but also affected the thigh in 59, the pelvis in 18 and the trunk/upper limb muscles in seven. The posterior compartment of the leg, including soleus and gastrocnemius, was the most affected (71/76) followed by the peronei (63/76). Asymmetric involvement was found in 19 patients. A distal to proximal gradient of fat replacement was observed in 39 patients (52%). Texture analysis showed a reticular pattern or “muscle islets” in 63 patients (83%). Hyperintensities on STIR were observed in 49/59 and were predominantly located in the distal LL muscles. A distal to proximal gradient in STIR was observed in 25 subjects. Besides features common to all individuals, we identified a pattern of muscle fat replacement characteristic of BICD2 and HSPB1 patients. We did not identify a correlation between fat content and age, disease duration or motor functionality.
Conclusion: Muscle MRI of CMN patients reveals common features that could be helpful for the diagnosis of patients. We have observed a pattern of MRI involvement characteristic of patients with mutations in specific genes. There was no correlation between fat content and functional status or time of disease progression.
PS02.04Deep Learning-Powered Hybrid Optoacoustic Imaging for Characterization of Pediatric Spinal Muscular Atrophy
Wagner A1, Regensburger A1, Danko V1, Jüngert J1, Schlereth M2, Stromer D2, Breininger K2, Tan L1, Maier A2, Federle A3, Klett D3, Schuessler S1, Neurath M3, Roos A4, Lochmüller H5, Woelfle J1, Trollmann R1, Waldner M3, Knieling F1
1 Department of Pediatrics And Adolescent Medicine, University Hospital Erlangen,Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg, Erlangen, Erlangen, Germany, 2Department of Artificial Intelligence in Biomedical Engineering, Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg, Erlangen, Erlangen, Germany, 3 Department of Medicine 1, University Hospital Erlangen,Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg, Erlangen, Erlangen, Germany, 4University of Duisburg-Essen, Department of Pediatric Neurology, Developmental Neurology and Social Pediatrics, Essen, Germany, Essen, Germany, 5Children’s Hospital of Eastern Ontario Research Institute, Division of Neurology, Department of Medicine, Ottawa, Canada, Ottawa, Canada
Introduction: Spinal muscular atrophy (SMA) is a severe neuromuscular disease caused by a homozygous deletion or mutation in the survival motor neuron 1 gene resulting in insufficient expression of the survival motor neuron protein. This leads to degeneration of motor neurons in the spinal cord and brain stem with consecutive muscular atrophy and weakness. While promising causal therapies are now available, prognostic biomarkers are missing. We investigate the ability of hybrid optoacoustic imaging to visualize and quantify muscle degeneration in SMA patients.
Methods: In this case-control proof-of-concept trial (clinicaltrails.gov ID NCT04115475) n=10 SMA patients n=10 gender/age-matched healthy volunteers (HV) were included. Optoacoustic imaging of muscles of upper and lower extremity of participants was performed using Multispectral Optoacoustic Tomography (MSOT) combined with Reflected-Ultrasound Computed Tomography (RUCT) for anatomical guidance. In MSOT, laser light transmitted through a handheld probe is used to excite tissue chromophores like hemoglobin or lipid and subsequently generates specific ultrasonic signals. The optoacoustic spectrum was obtained at 13 single wavelengths (SWL) including 680nm, 715nm, 730nm, 760nm, 800nm, 850nm, 930nm, 950nm, 980nm, 1000nm, 1030nm, 1064nm and 1100nm. SWLs and spectrally unmixed MSOT parameters (hemoglobin, collagen, lipid) were compared between groups and correlated with standard motor outcome tests and standard B-mode ultrasound imaging (US). In addition, a deep learning (DL) approach using convolutional neuronal networks was evaluated for automatic classification of optoacoustic imaging data.
Results: While MSOT signals were homogeneous in muscles of HV, SMA patients showed a moth-eaten optoacoustic pattern. The single wavelength 800nm showed the greatest difference of MSOT signal intensities between HV and SMA patients. A correlation between MSOT SWL 800nm and quantitative motor outcomes was found. In addition, MSOT parameter total hemoglobin showed lower signal levels according to the severity of the SMA type. The evaluation of quantitative agreement of RUCT and standard US using greyscale analysis (GSL) revealed significantly higher GSL in SMA patients in both techniques with good correlation with motor outcome tests. Qualitative agreement between RUCT and US was excellent for the differentiation of pathological vs. healthy muscles, echogenicity and distribution pattern, moderate for Heckmatt scale, and poor for muscle texture. DL approaches showed promising results with high accuracies to distinguish SMA from a other neuromuscular disorders and healthy controls.
Conclusions: Handheld MSOT imaging is a promising technology for non-invasive visualization and quantification of individual disease burden in pediatric SMA patients. The results suggest the potential for use for therapeutic monitoring in this rare genetic disease. Integrated RUCT may allow the assessment of basic qualitative and quantitative measures for muscular diseases with comparable results to conventional US. In addition, imaging-based differentiation and classification of SMA from other neuromuscular disorders and healthy muscles by DL approaches is feasible, underlining the translational potential for this technology for clinical applications.
PS02.05Description of a Spanish Cohort with Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS)
Santirso D1, Alvarez M2, Menendez M1, Suarez V3, Moris G1
1Neurology Department, Central Universitary Hospital of Asturias, Oviedo, Spain, 2Genetics Department, Central Universitary Hospital of Asturias, Oviedo, Spain, 3ENT Department, Central Universitary Hospital of Asturias, Oviedo, Spain
Background: Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS) was first described in 1990s as a late-onset progressive neurological disease with a triad of cerebellar ataxia, bilateral vestibular areflexia, and sensory neuropathy. Thus, diagnosis was based on clinical and electrophysiological findings. In 2019, an intronic biallelic repeat expansion in the gene encoding the Replication Factor Complex Subunit 1 (RFC-1) was reported in 100% of the familial cases and 92% of the sporadic cases. Since then, a growing number of patients has been diagnosed with CANVAS. Few studies have described the phenotypical spectrum of this entity in Spanish population, and exceptionally after genetic diagnosis.
Methods: A cross-sectional study was developed in a cohort of patients with genetic diagnosis of CANVAS in the Central University Hospital of Asturias in the period 2019-2021. Clinical, electrophysiological, and vestibular test characteristics were assessed.
Results: Thirty-one patients were identified. 71% were women with a median age of 53.6 years (range 35-75) at onset of symptoms and 69.6 years (range 46-83) at the time of diagnosis. Four patients reported a familial history of CANVAS. Gait instability (87%), dysesthesia of the limbs (74%), and spasmodic chronic cough (77%) were the most common complaints. Diminished vibratory sense (90%), impaired tandem (74%), abnormal Romberg’s test (68%), and ataxic gait (68%) were frequent findings in the neurological examination. Deep tendon reflexes were commonly diminished (58%), however, 13% presented with hyperreflexia. Vestibular testing with video Head Impulse Test (vHIT) showed bilateral vestibular dysfunction in 71% of the patients. Nerve conduction studies (NCS) showed reduced or absent sensory nerve action potentials in 97% of the patients and Somatosensorial Evoked Potentials (SSEP) were commonly impaired (90%). Additionally, Quantitative Sensory Tests (QST) were performed in 6 patients showing abnormalities in Ad and type C fibers in every subject.
Conclusion: Our investigation showed that the phenotypical characteristics of this cohort of Spanish patients are similar to studies of other populations. Gait instability with diminished vibration sense are present in the majority of the cases, whereas spasmodic cough is a common symptom.
Electrophysiological studies showed evidence of sensory neuronopathy in virtually every patient as previously reported. In spite of the small number of QST performed, it is noteworthy that every patient showed concomitant alteration of the small fiber nerves reflecting that autonomic dysfunction may be a frequent finding in CANVAS.
PS02.06Contribution of Magnetic Resonance Spectroscopy in the Study of Hereditary Spastic Paraplegia
Martinuzzi A1, Montanaro D2, Frijia F3, Vavla M4, BAratto A5, Coi A6
1Irccs E. Medea Scientific Institute, Conegliano, Italy, 2U.O.S.D. Servizio Autonomo di Risonanza Magnetica, Dipartimento di Neuroscienze Cliniche dell’Età Evolutiva, IRCCS Fondazione Stella Maris, Pisa, Italy, 3U.O.C Bioengineering and Clinical Technology, Fondazione CNR/Regione Toscana G. Monasterio, Pisa, Italy, 4University of Padova, Padova, Italy, 5Imaging Unit, Conegliano hospital, AULSS2 Veneto Region, Conegliano, Italy, 6Unit of Epidemiology of Rare Diseases and Congenital Anomalies, Institute of Clinical Physiology, National Research Council, Pisa, Italy
Hereditary Spastic Paraplegias (HSP) is a genetic neurodegenerative disorder affecting the corticospinal tract. No established neuroimaging biomarker is associated with the condition.
45 patients affected by HSP (18 SPG4, 6 SPG3a, 5 SPG5, 3 SPG30, 3 SPG31, 2 each of SPG7, 8, 11, 72, 1 SPG10 and 1 undetermined) and 46 healthy control (HC) matched by age and gender were recruited at the Medea Institute.
All subjects underwent an MRI study with a 1.5T equipment (Philips Achieva 2.5 XR, Royal Philips Healthcare, Eindhoven, NL) at baseline (T0) and follow-up (T1), the mean interval between the two examinations was 2.40±0.70 years. The MRI protocol included single-voxel spectroscopy sampling bilaterally the Rolandic (motor functions) and pre-motor regions (TR 2000msec, TE 35msec, voxel size 15x15x15mm).
The major brain metabolites detectable with 1H-MRS were analyzed: N-Acetyl Aspartate (NAA) Choline (Cho), Creatine-phosphocreatine complex (Cr), myo-Inositol (mI) and lipids (Li). All me-tabolites were processed with Spectro View Software (Philips Healthcare) measuring the peaks and evaluated as ratio to Cr. MRI data were analyzed at baseline (T0) and longitudinally (T0 vs T1).
MRS data were analyzed with Student’s t-test or the nonparametric unpaired Wilcoxon test for continuous variables, evaluating differences in concentration of metabolites mean values with confidence interval of 95%. Statistical analyses were performed using STATA software (StataCorp. 2019. Stata Statistical Software: Release 16. College Station, TX: StataCorp LLC).
A strong statistically significant difference was observed at T0 in the concentration of mI/Cr (Rolandic left p<0.001, Rolandic Right p<0.038) and in NAA/Cr (Rolandic left p<0.025) between HSP subjects and HC. In the left premotor area only NAA/Cr show a significant difference (p<0.022) be-tween the two group. mI and NAA levels were significantly correlated with clinical severity as as-sessed by SPRS.
The longitudinal analysis involved fewer patients (30). Statistically significant differences were ev-idenced in ml/Cr concentration (p<0.014) in the Rolandic left area among HSP subjects, with high-er measured levels of ml in the follow up (mean±SD = 0.72±0.03) than at baseline (mean ± SD = 0.61 ±0.04).
The “ctree” method that was applied in this study, allowed the development of decision trees able to classify HSP subjects and controls with an overall accuracy (total number of subjects cor-rectly predicted as HSP or control) of 79%, a sensitivity (number of HSP subjects correctly predict-ed as HSP) of 85% and a specificity (number of control subjects correctly predicted as control) of 73% assessing just two metabolites (mI and NAA).
The results are to be taken with caution given the very small number of subject and the still missing statistical validation step (blinded assignment to HSP vs HC group). Nevertheless, this pilot study indicates that brain MRS in HSP is a valuable approach, potentially exploitable as objective biomarker in this condition.
PS03.01Phase 1/2a Trial of Delandistrogene Moxeparvovec in Patients with DMD: 4-year Update
Mendell J R1,2, Sahenk Z1,2, Lehman K J1,Lowes L P1,2, Reash N F1, Iammarino M A1, Alfano L N1, Lewis S1,3, Church K1, Shell R1, Potter R A1,3, Griffin D A1,3, Pozsgai E R1,3, Hogan M1, Hu L3, Mason S3, Darton E3, Rodino-Klapac L R3
1Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, USA, 2The Ohio State University, Columbus, USA, 3Sarepta Therapeutics, Inc, Cambridge, USA
Delandistrogene moxeparvovec (SRP-9001) is an investigational gene transfer therapy developed for targeted skeletal and cardiac muscle expression of micro-dystrophin (a shortened, functional dystrophin protein) that is being studied in patients with Duchenne muscular dystrophy (DMD).
The objective of this Phase 1/2a, single-dose, open-label clinical trial (NCT03375164) is to evaluate the safety of systemic delivery of delandistrogene moxeparvovec in patients with DMD.
Four ambulatory patients with DMD (4–7 years old) were enrolled. Patients were given an intravenous infusion of delandistrogene moxeparvovec at a dose of 2.0x1014 vg/kg (supercoiled qPCR, linear plasmid standard equivalent of 1.33x1014 vg/kg) and prednisone (1 mg/kg/day) 1 day pre- to 30 days post-gene delivery. The primary outcome measure is safety. The secondary outcome measures include micro-dystrophin expression quantified by western blot and immunofluorescence (sarcolemmal micro-dystrophin expression) in pre- and post-muscle biopsies (Week 12 post-infusion). Key efficacy outcome measures include change in the North Star Ambulatory Assessment (NSAA) and timed function tests (10-metre Walk/Run and 100-metre Walk/Run, 4-stair Climb and Time to Rise).
Previously, delandistrogene moxeparvovec demonstrated an acceptable long-term safety profile 3 years post-treatment. Treatment-related adverse events (TRAEs) were mild to moderate, occurred mostly in the first 90 days of treatment, and all resolved. No serious adverse events (AEs), study discontinuations, or AEs associated with clinically relevant complement activation were reported. The most common TRAE was vomiting (9 of 18 TRAEs). All patients demonstrated a clinically meaningful improvement on NSAA (mean change [standard deviation] from baseline to Year 3: +7.5 points [3.42]). Patients treated with delandistrogene moxeparvovec generally maintained muscle strength (Time to Rise and 4-stair Climb) and showed improvement in ambulation ability (10-metre and 100-metre Walk/Run) from baseline to Year 3.
The observed safety profile and the enduring response following gene transfer provide proof of concept for the continuation of clinical trials assessing delandistrogene moxeparvovec using single-dose gene transfer therapy in patients with DMD. We present the latest long-term (4-year) safety and functional data from this study.
This study is funded by Sarepta Therapeutics.
PS03.02IGNITE DMD Phase I/II Study of SGT-001 Microdystrophin Gene Therapy for DMD: 2-Year Outcomes Update
Donisa Dreghici R1, Redican S1, Lawrence J1, Brown K1, Wang F1, Houston P1, Gonzalez P1, Schneider J1, Morris C1, Clary C1, Shieh P2, Byrne B3
1Solid Biosciences, Cambridge, United States, 2University of California, Neurology and Pediatrics, Los Angeles, United States, 3University of Florida College of Medicine, Powell Gene Therapy Center, Gainsville, United States
Duchenne muscular dystrophy (DMD) is a progressive, lethal neuromuscular disease caused by the absence of functional dystrophin protein in skeletal and cardiac muscle. SGT-001 is a systemically administered AAV9 microdystrophin gene therapy developed to deliver and express a surrogate functional dystrophin protein that uniquely includes the neuronal nitric oxide synthase (nNOS) binding domain. It is being evaluated for the treatment of DMD in the ongoing IGNITE DMD Phase I/II clinical trial. Following the administration of a low dose of 5E13 vg/kg to 3 subjects the dose was escalated, and all subsequent subjects have received SGT-001 at 2E14 vg/kg (n=6).
To date the most common treatment emergent adverse events are nausea, emesis, pyrexia, thrombocytopenia, and headache. Three subjects experienced SAEs associated with complement activation within the first weeks following dosing, which have all resolved without sequelae. Patients continue to undergo long-term follow up and are doing well up to approximately 4 years post-dosing.
Data previously presented up to 1.5 years post-dosing showed continued benefit in motor function, pulmonary function, and patient reported outcome measures (PROMs) in 2E14 vg/kg cohort subjects, compared to untreated control patients in IGNITE DMD and natural history. In addition, biopsies collected from these subjects at 12-24 months post-dosing showed continued expression of microdystrophin, restored membrane localization of β-sarcoglycan and nNOS, and only very mild active dystrophic pathology.
Additional long-term data collected at 2 years post-dosing will be presented, suggesting the durability of SGT-001 treatment effect. Subjects receiving SGT-001 at 2E14 vg/kg show stabilization or improvement profiles in motor function (North Star Ambulatory Assessment, 6-Minute Walk Test), pulmonary function (Forced Vital Capacity [FVC] % predicted [%p], peak expiratory flow [PEF %p], and PROMs (Pediatric Outcomes Data Collection Instrument [PODCI]) compared against expected natural history decline profiles. An update on more recent patients administered SGT-001 will also be provided.
These preliminary data continue to suggest a positive benefit-risk profile for SGT-001 warranting continued evaluation for the treatment of DMD.
PS03.03Minimizing Immune Responses Against Micro-Dystrophin
Chamberlain J1,2, Crudele J1,2, Vohra R1,2, Hauschka S1,2, Lee D1,2, Odom G1,2
1University Of Washington, Seattle, United States, 2Wellstone Muscular Dystrophy Specialized Research Center, Seattle, United States
Duchenne muscular dystrophy (DMD) is caused by dystrophin mutations. A potential hurdle for DMD gene therapy is the possibility of an immune response against dystrophin, which has now been observed in several patients treated with AAV-microdystrophin (µDys) vectors. This response has led to several reported serious adverse events in patients with deletions that overlap exons 8-11. To circumvent such T cell responses we have been using Rosetta-based computational protein deimmunization & redesign protocols with integration of experimental immunogenic epitope data, MHC epitope prediction tools, and host genomic data. Our initial studies have explored whether µDys can be redesigned to remove immunodominant epitope(s) within the actin-binding domain and beginning of hinge 1 (encoded by exons 6-8) that displayed T-cell reactivity in the first AAV-µDys human clinical trial (Mendell et al; NCT00428935). Importantly, the algorithm used in our studies predicted T-cell responses to the same epitopes, independently of their detection in the clinical trial. We used Rosetta to develop 3 redesigns of the first epitope (P17, exon 6), 5 for a second (P19, exon 7), and 10 for a third (P23 in exon 8). The first 2 redesigns (P17 and P19) displayed loss of immune recognition when assayed via interferon-γ ELISpot against patient T-cell lines; such T-cells were not available for P23. While some redesigned proteins displayed reduced stability, several appeared as stable and functional as the original µDys in both myogenic cultures and in skeletal muscles of mdx4cv mice. We further evaluated cardiac function of the two leading P17/P19 redesigns. For this we infused 2E14 vg/kg of redesigned (R) AAV6-CK8-µDys vectors into mdx4cv mice at 2 weeks of age, with an evaluation end point at ~1.5 years of age. Cardiac function was evaluated using cardiac magnetic resonance imaging. Significant differences were noted between the ejection fractions of C57Bl/6 control and mdx4cv mice, with a near complete return to normal using redesigned µDys. Furthermore, we quantified fibrosis via the collagen fraction (extracellular volume, ECV) using gadolinium-enhanced MRI. The amount of ECV was significantly higher in untreated mdx4cv mice compared to control mice, while treated mice displayed values close to controls. We have also observed that driving µDys expression from muscle-restricted expression cassettes (MSEC) is also important to limit T cell immunity. These studies demonstrate that multiple dystrophin T cell epitopes can be deimmunized and redesigned without loss of function or stability in both skeletal and cardiac muscles.
PS03.04Safety, β-Sarcoglycan Expression, and Functional Outcomes From Systemic Gene Transfer of rAAVrh74.MHCK7.hSGCB in LGMD2E/R4
Rodino-Klapac L1, Pozsgai E1,2, Lewis S1,2, Griffin D1,2, Meadows A1,3, Lehman K2, Church K2, Reash N2, Iammarino M2, Sabo B2, Alfano L2, Lowes L2, Neuhaus S1, Li X1, Mendell J2,4, Müller-York A
1Sarepta Therapeutics, Inc., Cambridge, USA, 2Center for Gene Therapy, The Research Institute at Nationwide Children’s Hospital, Columbus, USA, 3Wexner Medical Center, The Ohio State University, Columbus, USA, 4Department of Pediatrics and Neurology, The Ohio State University, Columbus, USA
Background: Limb-girdle muscular dystrophy type 2E/R4 (LGMD2E/R4) is caused by mutations in the β-sarcoglycan gene (SGCB), resulting in loss of SGCB protein and subsequent absence of the dystrophin-associated protein complex (DAPC) at the sarcolemma. LGMD 2E/R4 manifests as progressive hip/shoulder muscle weakness. This first-in-human, phase 1/2 trial (NCT03652259) evaluated SRP-9003, a self-complementary rAAVrh74.MHCK7.hSGCB construct designed to restore SGCB protein production in patients with LGMD2E.
Methods: Patients aged 4–15 years with SGCB mutation (both alleles) received 1 SRP-9003 IV infusion: Cohort 1 (n=3), 1.85×1013 vg/kg; Cohort 2 (n=3), 7.41×1013 vg/kg. Endpoints included safety (primary), SGCB protein expression (secondary), and function (North Star Assessment for Limb-girdle Type Muscular Dystrophies [NSAD], time to rise [TTR], 4-stair climb [4-sc], 100-meter timed test [100m], 10-meter timed test [10m]).
Results: Previously reported results showed that SRP-9003 was well tolerated; adverse events occurred early and were manageable. In Cohort 1, robust SGCB protein expression and correct sarcolemmal localization post treatment was seen, accompanied by durable DAPC reconstitution up to year 2 (Y2). Patients treated with SRP-9003 showed durable functional improvements as well, which were maintained through year 2 evaluations (NSAD, +5.7 points; TTR, -0.6 sec; 4-stair climb, -0.3 sec; 100m, -2.8 sec; 10m, -0.2 sec.). In Cohort 2, followed for 1 year, SRP-9003 gene transfer also showed functional improvements (NSAD, +4 points; TTR, -1.1 sec; 4-stair climb, -0.4 sec; 100m, -7.9 sec; 10m, -0.6 sec). Post hoc analysis over 24 months showed improved NSAD outcomes (Cohort 1) versus untreated natural history cohort (+9.2-point difference, Y2; 95% CI, 3.2-15.1). An update with 3-year functional data for Cohort 1 and 2-year protein expression and functional data for Cohort 2 will be presented.
Conclusions: These data suggest sustained efficacy of SRP-9003 therapy, supporting advancement of the clinical development program.
PS03.5AAV Vector-mediated RNAi of Mutant LDB3 Expression as a Therapeutic Strategy for Myofibrillar Myopathy
Pathak P1, Hale J1, Palmeri J1, Sabu-Kurian A1, Mankodi A1
1National Institute of Neurological Disorders and Stroke, National Institutes Of Health (NIH), Bethesda, United States
Background: Myofibrillar myopathies are characterized by protein aggregation and the Z-disc disintegration in muscle fibers. There are currently no curative treatments for patients. Previously we showed that the p.Ala165Val mutation in LIM Domain Binding 3 (LDB3) causes myopathy by impairing the chaperone-assisted selective autophagy, and downregulating PKCα and TSC2-mTOR through gain-of-function effects in the lab-generated Ldb3A165V/+ knock-in mice. Mutant mice develop filamin C and chaperone aggregation in muscle fibers at the age of 4 months followed by myofibrillar disintegration in same fibers. Here, we aimed to prevent or reverse the disease phenotype by AAV vector mediated RNAi of the mutant allele expression. AAV2/9 serotype efficiently transduces skeletal muscle.
Materials and methods: Allele-specificity and efficacy of siRNA oligonucleotides to downregulate the mutant Ldb3 transcript were demonstrated in transfected cell culture systems to inform in vivo approach in mutant mice. We injected a single dose of AAV2/9-shRNA-miR-Ldb3 and AAV2/9-shRNA-miR-ctrl (5×10e12 vg/kg) in opposite tibialis anterior (TA) muscles of the 3 month (n=27; before the onset of muscle pathology) and 5 month (n=22; after the onset of muscle pathology) old mutant mice. Muscle tissue was harvested at 1 month and 3 months from the intramuscular injection for efficacy assays such as quantitative PCR, immunoblotting, and immunofluorescence.
Results: Quantitative PCR and immunoblotting assays showed a significant decrease of mutant Ldb3 mRNA and LDB3 protein levels with two siRNA (si16 and si10) out of five tested in transfected HEK293 cells (p < 0.01). Four weeks after injecting AAV 2/9-shRNA-miR-Ldb3s16 and AAV-shRNA ctrl into the opposite tibialis anterior muscles of the mutant mice, GFP expression was visualized throughout muscle, suggesting efficient viral transduction. Quantitative PCR and immunoblotting results confirmed the Ldb3 knockdown at RNA and protein levels in AAV 2/9-shRNA-miR-Ldb3s16 or s10-injected muscle compared to scramble shRNA at 1 month and 3 months. The s16 shRNA preferentially downregulated the mutant allele, whereas s10 shRNA demonstrated mutant allele specific knockdown in the transfected cells and in mouse muscle. Importantly, we observed reversal of PKCα levels to wildtype levels in treated muscle of the mutant mice at both timepoints. Quantitative phosphoproteomics assays using tandem mass spectroscopy are ongoing to characterize cell signaling network signature of the mutation effects and therapeutic response in muscle tissue. Early studies indicate a reversal in myopathology features of increased internal myonuclei and filamin C aggregation in the muscle fibers of the treated muscle at 3 months after the injection in 5 month old mice.
Conclusions: Our results demonstrate a robust mutant LDB3 knockdown, identify PKCα as a treatment response biomarker, and provide an effective strategy to treat myofibrillar myopathy based on reduction of toxic mutant protein.
PS03.06ASPIRO Gene Replacement Therapy (Resamirigene Bilparvovec) Trial in XLMTM: Pathologic Findings in Four Deceased Participants
Lawlor M1, B. Shieh P2, G. Bönnemann C3, Müller-Felber W4, L. Kuntz N5, J. Dowling J6, Schoser B7, Margeta M8, Meng H1, M. Hopp A1, Wozniak L2, Foley A3, N. Saade D9, E. Kleiner D10, Dikoglu E10, Jones C3, Lopes Abath Neto O10, Blaschek A4, Lurz E11, Mueller S4, R. Wadhwani N5, Mohammad S5, A. Chapin C5, C. Reed R12, Hsu E13, Prasad S14, Rico S14, Murtagh M14, Bachtell N14, Miller W15
1Medical College of Wisconsin, Milwaukee, USA, 2University of California, Los Angeles, USA, 3Neuromuscular and Neurogenetic Disorders of Childhood Section, NINDS, NIH, Bethesda, USA, 4Klinikum der Universität München, Munich, Germany, 5Ann & Robert H Lurie Children’s Hospital of Chicago, Chicago, USA, 6Hospital for Sick Children, Toronto, Canada, 7Friedrich-Baur-Institute, Dep. of Neurology Ludwig-Maximilians-University Munich, Munich, Germany, 8University of California San Francisco, San Francisco, USA, 9University of Iowa Hospitals and Clinics, Iowa City, USA, 10Laboratory of Pathology, NCI, Bethesda, USA, 11von Hauner Children’s hospital, Ludwig Maximilian University, Munich, Germany, 12Department of Laboratories, Seattle Children’s Hospital, Seattle, USA, 13Department of Pediatrics, University of Washington, Seattle, USA, 14Formerly of Astellas Gene Therapies (formerly Audentes Therapeutics), San Francisco, USA, 15Astellas Gene Therapies (formerly Audentes Therapeutics), San Francisco, USA
Introduction: X-linked myotubular myopathy (XLMTM) is a life-threatening congenital myopathy caused by mutations in the MTM1 gene, leading to absent or dysfunctional myotubularin and resulting in respiratory failure at birth, profound muscle weakness, and early death.
Method: ASPIRO (NCT03199469) is an open-label, Phase 1/2/3 randomized trial where young boys with genetically confirmed XLMTM and chronic ventilator dependence received a single intravenous dose of adeno-associated viral (AAV) vector delivering human MTM1, resamirigene bilparvovec (AT132). As of 08/2021, 24 study participants had received AT132 (AT132): seven at the lower dose, 1.3x1014 vg/kg and 17 at the higher dose, 3.5x1014 vg/kg. Preliminary safety and efficacy results have been presented previously. Three participants in the higher dose and one recently dosed participant in the lower dose cohort died.
Results: All four deceased participants had ongoing hepatobiliary cholestasis with decompensated liver disease at time of death. Resulting immediate causes of death included sepsis in three participants and gastrointestinal hemorrhage in one participant. While severe liver pathology was observed in all four participants at time of death, two serial liver biopsies obtained from one (participant 12) demonstrated progression to liver fibrosis over the course of ~7 months. Histological similarities among all four participants indicate that a similar process was responsible for the liver disease observed in the fatalities. This progressive, severe cholestatic liver disease appears to be associated with 1) a previously unrecognized cholestatic tendency that has now been described in untreated XLMTM patients, 2) exposure to AT132, with mechanism of exacerbation of cholestatic disease not presently understood, and 3) evidence of decreased expression of bile salt export protein (BSEP) in liver tissue, the cause of which is yet to be elucidated.
Conclusion: While sepsis and gastrointestinal hemorrhage were the immediate causes of death in four participants on the ASPIRO trial, all were attributable to AT132 triggered severe exacerbation of cholestatic liver disease as an underlying cause. Retrospective analyses of preclinical mouse and canine XLMTM models treated with AAV8 gene transfer did not reveal evidence of cholestatic disease in the absence or presence of treatment. Similarly, AT132 in healthy non-human primates was not associated with liver toxicity, despite doses higher than those used in the clinical trial. The factors that would specifically help predict this susceptibility in treated patients remain under investigation. The ASPIRO study is currently on hold while investigations continue. The pathological findings from the four deceased participants will be presented.
PS04.1Useful and Cost-effective Workup in Chronic Polyneuropathy (the EXPRESS Study)
Wiersma M1, Vrancken A1, van Doorn P2, Eftimov F3, van Eijk R4, Frederix G5, Notermans N1, Eurelings M6, Verstraete E7, van de Wijdeven G8, Piepers S9, van der Star G1, Teunissen L10, van der Meulen M G F10, van Doormaal P11,1
1Department of Neurology, UMC Utrecht Brain Center, University Medical Centre Utrecht, Utrecht, Netherlands, 2Department of Neurology, Erasmus MC, University Medical Center Rotterdam, Rotterdam, Netherlands, 3Department of Neurology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands, 4Biostatistics & Research Support, Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht, Netherlands, 5Department of Public Health, Healthcare Innovation & Evaluation and Medical Humanities, University Medical Center Utrecht, Utrecht, Netherlands, 6Department of Neurology, Spaarne Gasthuis, Haarlem/Hoofddorp, Netherlands, 7Department of Neurology, Rijnstate, Arnhem, Netherlands, 8Spierziekten Nederland, Baarn, Netherlands, 9Department of Neurology, Meander MC, Amersfoort, Netherlands, 10Department of Neurology, St. Antonius, Nieuwegein, Netherlands, 11Department of Neurology, Tergooi MC, Hilversum, Netherlands
Background: polyneuropathy is a common disease. An estimated 300.000 persons have polyneuropathy and more than 13.000 patients are newly diagnosed each year in the Netherlands. Polyneuropathy has many causes and risk factors, some common (diabetes, excessive alcohol consumption) and some not. There are knowledge gaps about the usefulness and extent of blood tests and nerve conduction study (NCS) to diagnose and search for a cause. The recently updated Dutch guideline Polyneuropathy therefore recommends to conduct a complete workup. Our hypothesis is that in many patients with a clinical diagnosis chronic polyneuropathy, a limited or even no further workup improves cost-effectiveness without loss of diagnostic reliability or disadvantageous effect on treatment choice.
Methods: the EXPRESS study is a prospective observational multi-center study carried out in five large general hospitals and three neuromuscular expertise centers. Adult patients 18 years or older with symptoms suspect for polyneuropathy, who are referred to a neurologists for an outpatient workup are eligible. Patients’ electronic medical records (EMR) are used to gather all relevant data pertaining to the workup of polyneuropathy and the outcomes measures. Direct medical costs and other health care costs are determined from these data and questionnaires, as well as time to diagnosis. Real-time workup by patients’ neurologists will be compared to a limited or no further workup according to consensus by a panel of neuromuscular specialists and experienced neurologists. Primary outcome is effectiveness of a limited or no further workup expressed as concordance between panel diagnosis and patients’ neurologists’ diagnosis (i.e., percentage overlooked diagnoses). This will be related to differences in costs and impact on treatment or patient management otherwise. Other outcomes are burden/gain for the patient in terms of number of investigations, time to diagnosis, hospital visits, sick-leave, loss of productivity, expenses, experienced quality of care. Furthermore, a clinical prediction model will be developed to objectively guide the decision-making if nerve conduction study (NCS) is required. Each patient is his own control and follow-up time is 6 months. The total sample size will be 1200 patients, of whom 200 will be enrolled as validation cohort.
Discussion: this study is a quality in health care evaluation to ascertain if and which blood tests and when NCS are necessary and cost-effective to establish the diagnosis and identify the cause of polyneuropathy.
Funding: ZonMw Evaluatieonderzoek ZE&GG grant
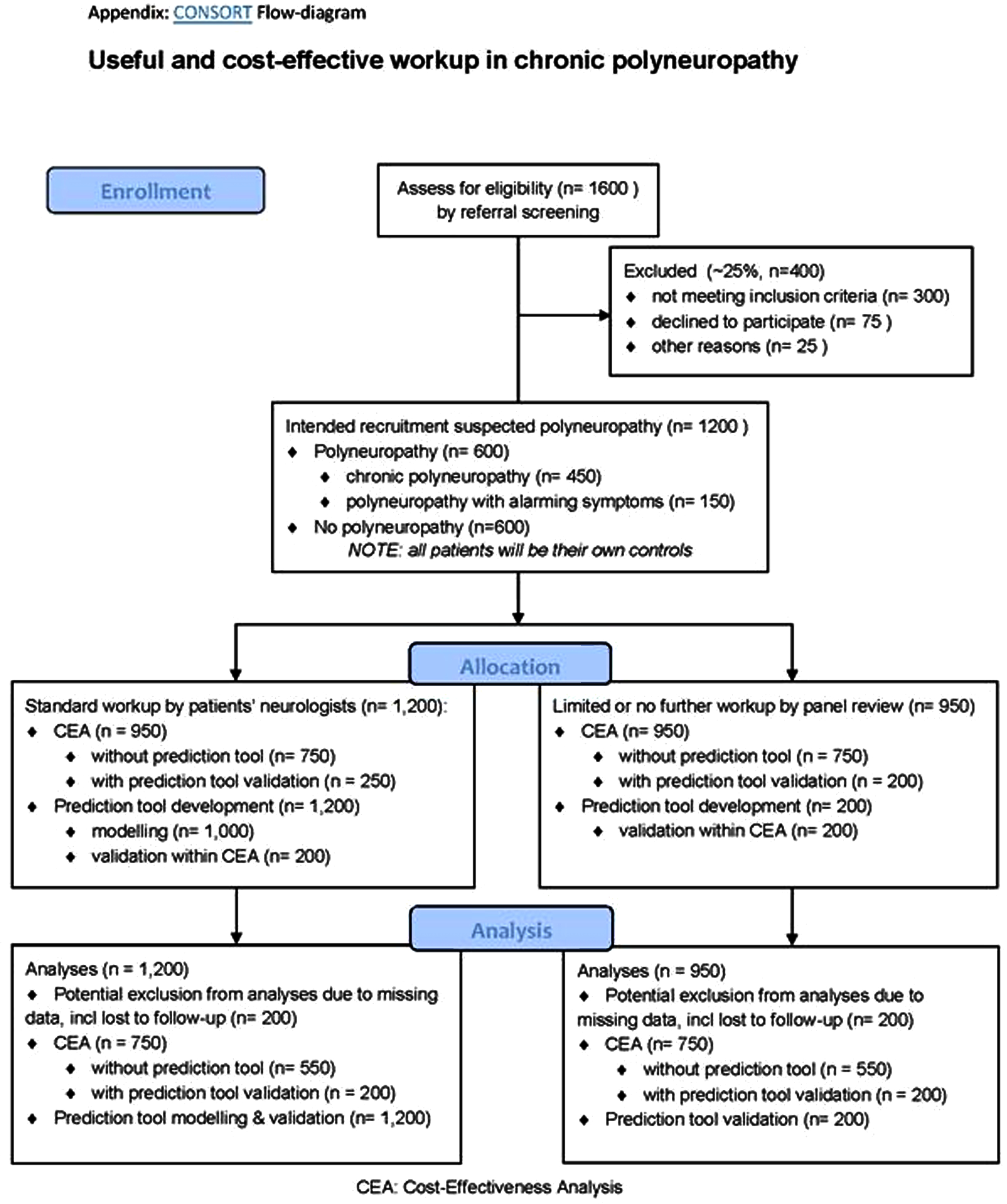
PS04.02The Prevalence of and Risk Factors for Distal Symmetric Polyneuropathy in a Low-income, U.S. Population
Feldman E1, Gagne A1, Marcus H2,3, Dawood T2,3, Bachuwa G2,3, Kvalsund M4,5, Skolarus L1, Callaghan B1, Elafros M1
1University Of Michigan, Ann Arbor, United States, 2Hurley Medical Center, Flint, United States, 3Michigan State University, East Lansing, United States, 4University of Rochester, Rochester, United States, 5University of Zambia, Lusaka, Zambia
Introduction: Distal symmetric polyneuropathy (DSP) is a disabling, painful condition associated with falls and reduced quality of life. While DSP risk factors, such as glucose intolerance, are more common among Black people and people with low-income, these populations are underrepresented in existing DSP studies. Therefore, the Flint Neuropathy Study is an ongoing study assessing DSP prevalence and associated risk factors in a predominantly Black, low-income setting.
Methods: Patients >40 years of age presenting to the Hurley Medical Center Outpatient Internal Medicine Residency Clinic predominantly serving Medicaid patients in Flint, Michigan were enrolled. Demographics, clinical characteristics including medication use, anthropomorphic measurements, fasting lipids, fasting glucose, and Hemoglobin A1C, were collected. Glucose intolerance was defined using the 2021 ADA diagnosis and classification of diabetes mellitus criteria, whereas metabolic syndrome was defined using the harmonized criteria from the IDF, NHLBI, AHA, WHF, IAS, and IASO societies. DSP was defined using the Michigan Neuropathy Screening Instrument Questionnaire Index (MNSIQI). Descriptive statistics were performed using means and frequencies. Analysis of variance was used to examine the association between DSP and metabolic syndrome and glycemic status.
Results: 81 participants (62% female, 57.5 years. 67% Black, 53% Medicaid, 22% <grade 12 education) have enrolled to date. At enrollment, 28 (35%) reported a history of T2DM whereas 9 (11%) were pre-diabetic. 4 (5%) denied a history of glucose intolerance despite a prior diagnosis. Criteria for metabolic syndrome was met in 39 (48%). 7 (9%) were newly diagnosed with glucose intolerance during the study. Mean MNSIQI score was 2.65 (SD 1.56) with 48/81 (56%) meeting criteria for DSP. Among participants with a history of glucose intolerance and DSP (n=21), 16 (76%) were unaware they had DSP and 5 (24%) were aware. 7/11 (64%) participants who did not know they had glucose intolerance prior to participation had DSP. DSP was associated with a history of glucose intolerance (prediabetes: MNSIQI score 2.74, T2DM: 3.21 vs no history: 2.03, p<0.01) as well as metabolic syndrome (2.95 vs 2.11 p=0.028).
Conclusions: DSP is extremely common and underrecognized in this patient population with low incomes. Characterizing the DSP burden and risk factors in these individuals is essential to improve our understanding of DSP and to ensure equal representation in implementation efforts to address DSP symptoms and associated outcomes.
PS04.03Assessment Timing and Choice of Outcome Measure in Determining Treatment Response in CIDP: Post-hoc PRISM
Ouaja R1, Rajabally Y2, Pujol S1, Kasiborski F1, Nobie-Orazio E3
1LFB, Les Ulis, France, 2Aston University, Birmingham, Royaume-Uni, 3Milan University, Milan, Italie
Introduction: Treatment response and its timing is variable in chronic inflammatory demyelinating polyneuropathy (CIDP). We here aimed to study this variability with multiple outcome measures.
Methods: We performed a post-hoc analysis of the PRISM trial, a 24-week prospective, multicentre, single-arm, open-label phase 3 study of IqYmune, a 10% intravenous immunoglobulin preparation, for CIDP. We ascertained the timing of response with primary/secondary outcome measures.
Results: At 6 weeks post-treatment initiation, 13/40 subjects (32.5%) were defined as responders on the primary outcome measure, the adjusted Inflammatory Neuropathy Cause, and Treatment (INCAT)scale. This increased to 20/41 (48.8%) at 12 weeks and to 32/42 (76.2%) at 24 weeks. Use of minimal important difference (MID)-determined amelioration of the inflammatory Rasch-built Overall Disability Scale (I-RODS), or of the Medical Research Council Sum Score (MRCSS), or of dominant handgrip strength, in addition to the adjusted INCAT, offered a sensitivity of 41.7% in identifying adjusted INCAT non-responders at week 12 who subsequently responded at week 24. Specificity was of 60% versus INCAT non-responders at week 24.
Consideration of amelioration of any amplitude on any secondary outcome measure offered a75% sensitivity but only 30% specificity versus adjusted INCAT non-responders at week 24.
Conclusions: Immunoglobulin treatment continuation may be justified for up to 24 weeks in CIDP. Additional outcome measures may help in early treatment stages to predict delayed response on the adjusted INCAT. However, their use is limited by high false-positive rates. More robust, reliable, and relevant outcome measures are needed to detect early improvement in CIDP.
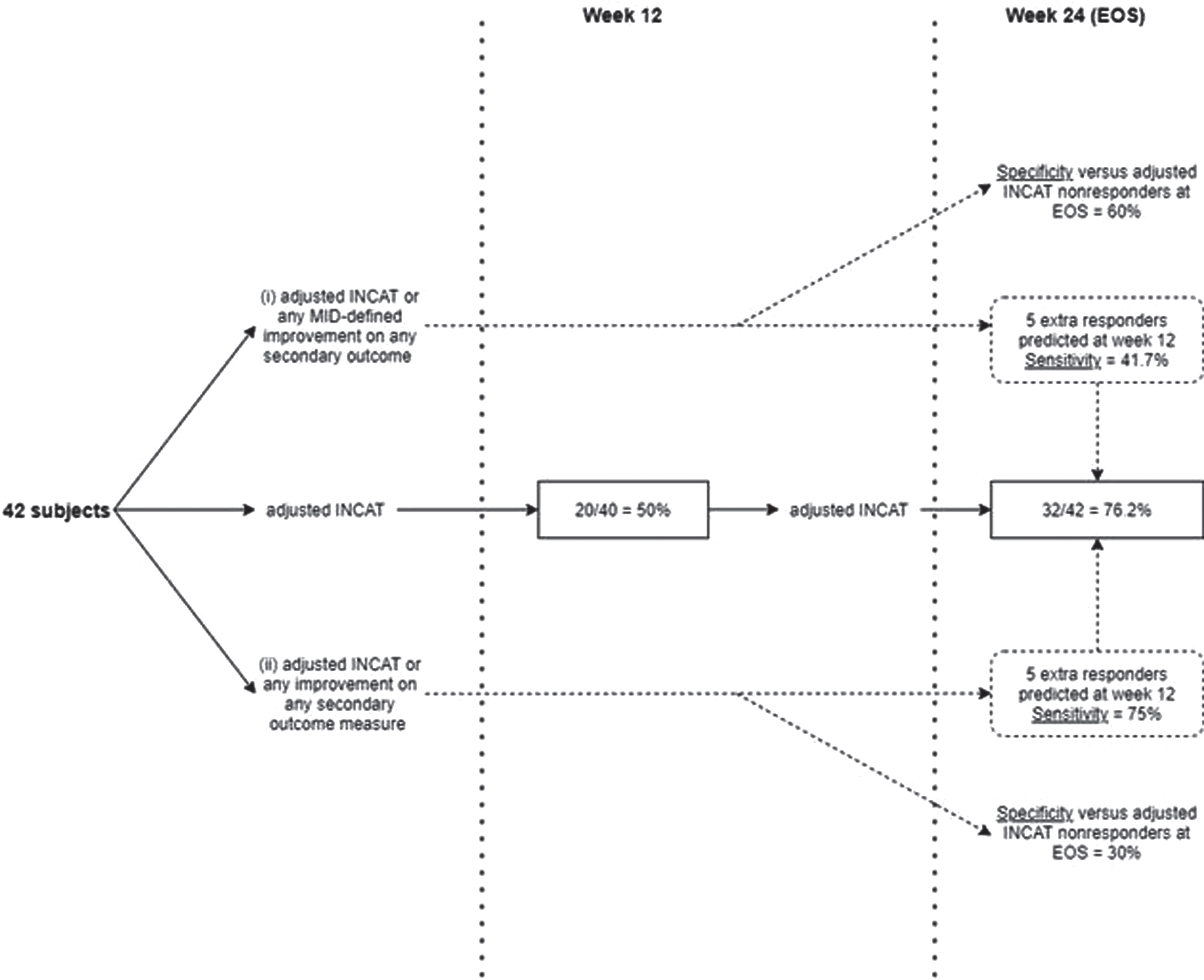
PS04.04Peripheral Neuropathies Associated with Systemic Autoimmune Disorders: A Single-Centre Retrospective Study
Moura J1, Neves Cardoso M2, Samões R1,3, Santos E1,3,4, Martins da Silva A1,3,4, Sousa A2,4
1Department of Neurology, Hospital de Santo António (HSA), Centro Hospitalar Universitário do Porto (CHUP), Porto, Portugal, Porto, Portugal, 2Department of Neurophysiology, Hospital de Santo António (HSA), Centro Hospitalar Universitário do Porto (CHUP), Porto, Portugal, Porto, Portugal, 3Multidisciplinary Unit for Biomedical Research, Instituto de Ciências Biomédicas Abel Salazar, University of Porto, Porto, Porto, Portugal, 4Clinical Immunology Unit, Centro Hospitalar Universitário do Porto, Porto, Portugal, Porto, Portugal
Introduction: Peripheral nervous system involvement is frequent in connective tissue diseases and systemic immune-mediated disorders, particularly vasculitis. The peripheral nerve may be damaged by several pathophysiological mechanisms, translating in various clinical presentations.
Objectives: To describe a group of patients with peripheral neuropathies secondary to systemic autoimmune disorders (SAD) from a Portuguese tertiary hospital.
Methods: Retrospective analysis through an institutional registry of patients followed in the Neuroimmunology outpatient clinic. Only neuropathies with neurophysiological confirmation were included. When available, biopsy results were also collected.
Results: A total of 39 patients were identified, 84.6% female, with a mean age of onset of neuropathy symptoms of 56.1 years (SD 13.1). The most common SAD was sarcoidosis (20.5%), followed by eosinophilic granulomatosis with polyangiitis (15.4%), Sjögren’s syndrome (10.3%), systemic sclerosis (10.3%), rheumatoid arthritis (10.3%), Behçet’s disease (7.7%), IgG4-related disease (5.1) and systemic lupus erythematosus (2.6%). Neuropathy was the presenting feature of SAD in 51.3%, with the most common symptoms being paresthesia (30.8%) and hypoesthesia (23.1%).
Small fibre polyneuropathies were present in 15 patients (38.5%), five with additional neuropathies. Mononeuropathies were present in 20.5% (including three rheumatoid arthritis cases), sensory-motor axonal polyneuropathies in 15.4% (with 3 cases of eosinophilic granulomatosis with polyangiitis), multiple mononeuropathies in 12.8% (2 cases of eosinophilic granulomatosis with polyangiitis and 1 case of rheumatoid vasculitis), sensory axonal polyneuropathies in 10.3%, inflammatory radiculopathy in 7.7% (all being cases of sarcoidosis) and ganglionopathy in 1 IgG4-related disease case. Tissue biopsy was used to diagnose 10 cases, including 3 multiple mononeuropathies. Approximately 49.0% improved after immunosuppressive therapy, which consisted in methylprednisolone in 5 patients and intravenous immunoglobulin in 4. Approximately 35.9% of patients were on maintenance immunosuppressive therapy, most commonly azathioprine (n=5).
Conclusion: We demonstrated what was previously recognised by several larger cohorts regarding the variety of neuropathies associated with different SAD. While rheumatoid arthritis is mainly associated with entrapment mononeuropathies, Sjögren syndrome was found primarily on small fiber polyneuropathies and vasculitis with multiple mononeuropathies. Nerve biopsy appears to be particularly useful for diagnosing multiple mononeuropathies (60.0%).
Disclosures: The authors have nothing to disclose
PS04.05Autoantibody Screening in Idiopathic Small-Fiber Neuropathy
Pascual-Goñi E1, Lleixà C, Caballero-Ávila M, Martín-Aguilar L, Geerts M2, Hoeijmakers J2, de Greef B2, Merkies I2, Faber C2, Querol L
1Neuromuscular Diseases Unit, Hospital De La Santa Creu I Sant Pau, Barcelona, Spain, 2Department of Neurology, Maastricht University Medical Centre, Maastricht, The Netherlands.
Introduction: Small-fiber neuropathies (SFN) are a heterogeneous group of disorders of thinly myelinated Ad-fibers and unmyelinated C-fibers. Multiple causes of SFN have been reported, including genetic mutations and immune-mediated systemic disorders, but the cause remains unknown in up to 50% of cases (idiopathic SFN=iSFN). Recently, autoantibodies to plexin-D1 were detected by ELISA in SFN patients. ELISA had 75% sensitivity and 100% specificity using DRG sections as the gold standard for anti-Plexin-D1 detection. We aimed to screen for autoantibodies reacting against neural structures and Plexin-D1 in a well-defined cohort of iSFN.
Methods: We tested baseline sera from 60 skin biopsy-proven iSFN patients enrolled in a clinical trial assessing IVIG effectiveness for iSFN. Sera from HC, ALS, CMT and MS were used as controls. IgG against neural structures were tested by (1) ICC using primary cultures of rat DRG neurons; (2) ICC using human neuroblastoma-derived neurons; (3) IHC using macaque peripheral nerve sections; (4) ELISA using RH Plexin-D1 Protein (R&D).
Results: iSFN sera reacted against DRG neurons significantly more frequently than controls [16/60 (27%) vs 6/90 (7%) (p <0.001)]. No differences were observed in the proportion of patients reacting against neuroblastoma-derived neurons [4/60 (7%) vs 10/90 (11%) (p=0.36)]. In macaque peripheral nerve sections we identified a significantly higher frequency of IgG reactivity to NCAM+ unmyelinated Schwann cells in iSFN patients vs controls [13/60 (22%) vs 3/56 (5%) (p=0.01)]. Anti-Plexin-D1 antibodies were detected by ELISA in 3/60 (5%) of patients but not in controls. All 3 anti-PlexinD1+ sera showed IgG reactivity to DRG neurons.
Conclusion: iSFN patients showed a heterogeneous repertoire of autoantibodies against neural structures. Anti-Plexin-D1 IgG were detected in 5% of iSFN. Another subset of patients showed IgG reactivity to unmyelinated Schwann cells. The specific antigens and clinical implications of these autoantibodies are yet to be determined.
PS04.06Diagnostic Value of Standardized Nerve Ultrasound of the Plexus Brachialis in Chronic Inflammatory Neuropathies
Dubuisson N1, van de Scheur J, Lemmen D, Van den Bergh P, van der Pol W, van den Berg L, Goedee H
1Cliniques Universitaires St-luc, Brussels, Belgium
Background: Nerve ultrasound (US) is a practical and widely available, innocuous technique with low costs that is now increasingly used as an important complementary diagnostic tool in immune mediated neuropathies. Although brachial plexus was often included in the published sonographic studies, there is still considerable variability with regards to which elements of the brachial plexus should be evaluated. We therefore aimed to evaluate the diagnostic accuracy of an extensive US protocol of the brachial plexus in chronic inflammatory neuropathy (CIN).
Methods: All consecutive patients with suspected CIN, seen at our neuromuscular outpatient between March 2018 to March 2020 were eligible for inclusion. All patients underwent a standardized set of extensive electrodiagnostic testing and US protocol, including (nerve roots, trunks and supraclavicular part of plexus), and appropriate laboratory testing. We used logistic regression and ROC analysis to determine the most optimal plexus US protocol and compared the test characteristics with that of most recent diagnostic consensus criteria.
Results: We included 98 patients (.. CIDP, .. MMN) and 98 disease controls (axonal neuropathies, lower motor neuron syndromes...) We found that combination of cross-sectional measurements (CSA) of the plexus trunks and C5 to C7 nerve roots had the highest diagnostic yield. In contrast, longitudinal and supraclavicular measurements appear to have no added diagnostic value. In addition, our shortened protocol with improved cut-off values yielded a sensitivity of 90.2% and an enhanced specificity of 92.3%. Importantly, our US protocol identified 22% of patients with a CIN diagnosis that responded to treatment who were otherwise missed with routine electrodiagnostic studies and other supportive criteria.
Conclusion: Our study shows that plexus US protocol of plexus for CIN can be limited to a practical protocol, improving detecting with minimal burden. This shortened US protocol is relatively easy to implement in routine practice, but still warrants consideration of relevant imaging mimics.
PS05.01Rozanolixizumab in Generalized Myasthenia Gravis: Responder Analyses From the Phase 3 MycarinG Study
Bril V1, Druzdz A2, Grosskreutz J3, Habib A4, Mantegazza R5, Sacconi S6, Utsugisawa K7, Vissing J8, Vu T9, Boehnlein M10, Bozorg A11, Gayfieva M12, Woltering F13, Kaminski H14
1University Health Network, Toronto, Canada, 2Department of Neurology, Municipal Hospital Poznan, Poland, 3Precision Neurology, Department of Neurology, University of Luebeck, Germany, 4MDA ALS and Neuromuscular Center, University of California, Irvine, Orange, USA, 5Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Italy, 6Université Côte d’Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, France, 7Department of Neurology, Hanamaki General Hospital, Japan, 8Department of Neurology, Rigshospitalet, University of Copenhagen, Denmark, 9Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 10UCB Pharma, Germany, 11UCB Pharma, Morrisville, USA, 12UCB Pharma, Slough, UK, 13UCB Pharma, Germany, 14George Washington University, USA
Background: Generalised myasthenia gravis (gMG) is a rare neuromuscular disease caused by pathogenic immunoglobulin G (IgG) autoantibodies that reduce signal transmission by disrupting neuromuscular junction components such as acetylcholine receptors (AChR) and muscle-specific kinase (MuSK) receptors. Conventional treatments (immunosuppressants and immunomodulators) often do not provide adequate disease control, with patients continuing to experience burdensome symptoms or potentially life-threatening exacerbations. The Phase 3 MycarinG study evaluated the efficacy and safety of rozanolixizumab, a fully humanised IgG4 monoclonal antibody that can rapidly and specifically reduce circulating IgG, including pathogenic autoantibodies, as a treatment for gMG.
Methods: The MycarinG study (MG0003/NCT03971422) was a Phase 3, multicentre, randomised, double-blind, placebo-controlled, three-arm study. Patients were aged ≥18 years, had Myasthenia Gravis Foundation of America (MGFA) Class II–IVa gMG and were positive for anti-AChR or anti-MuSK autoantibodies at first visit. After a screening period of up to 4 weeks, participants were randomised 1:1:1 to weekly subcutaneous rozanolixizumab 7mg/kg, 10mg/kg or placebo for a 6-week double-blind treatment period, followed by an 8-week observation period. The primary efficacy endpoint of MycarinG was the change from baseline (CFB) to Day 43 (one week after final dose) in MG Activities of Daily Living (MG-ADL) score. Responder analyses at Day 43 included the proportion of MG-ADL responders (≥2.0-point improvement from baseline) (secondary variable), proportion of responders for Quantitative Myasthenia Gravis (QMG) score (≥3.0-point improvement from baseline) and Myasthenia Gravis Composite (MGC) score (≥3.0-point improvement from baseline). Additional endpoints included Minimal Symptom Expression (MSE, defined as having an MG-ADL score of 0 or 1). Safety endpoints included treatment-emergent adverse events (TEAEs) and TEAEs leading to treatment discontinuation.
Results: In total, 200 participants were randomised to rozanolixizumab 7mg/kg (n=66), 10mg/kg (n=67) or placebo (n=67). At Day 43, least squares mean CFBs in MG-ADL (difference vs placebo [95% CI]) were: -3.370 (-2.586 [-4.091, -1.249]; p<0.001) for 7mg/kg; -3.403 (-2.619 [-3.994, -1.163]; p<0.001) for 10mg/kg; versus -0.784 for placebo. More patients in the rozanolixizumab arms than the placebo arm achieved response in MG-ADL (p<0.001), QMG, and MGC scores (Table). MSE was achieved in 25.8%, 28.4% and 3.0% of patients in the 7mg/kg, 10mg/kg and placebo arms, respectively. TEAEs occurred in 81.3%, 82.6% and 67.2% of patients receiving 7mg/kg, 10mg/kg and placebo, respectively. Serious TEAEs occurred in 7.8%, 10.1%, and 9.0% of patients, respectively, and 3.1%, 7.2% and 3.0% patients, respectively, discontinued due to TEAEs.
Conclusions: Clinical efficacy of rozanolixizumab was demonstrated by improvement in responder rates over placebo, in multiple disease-specific outcome measures, with no new safety concerns. Funded by UCB Pharma.
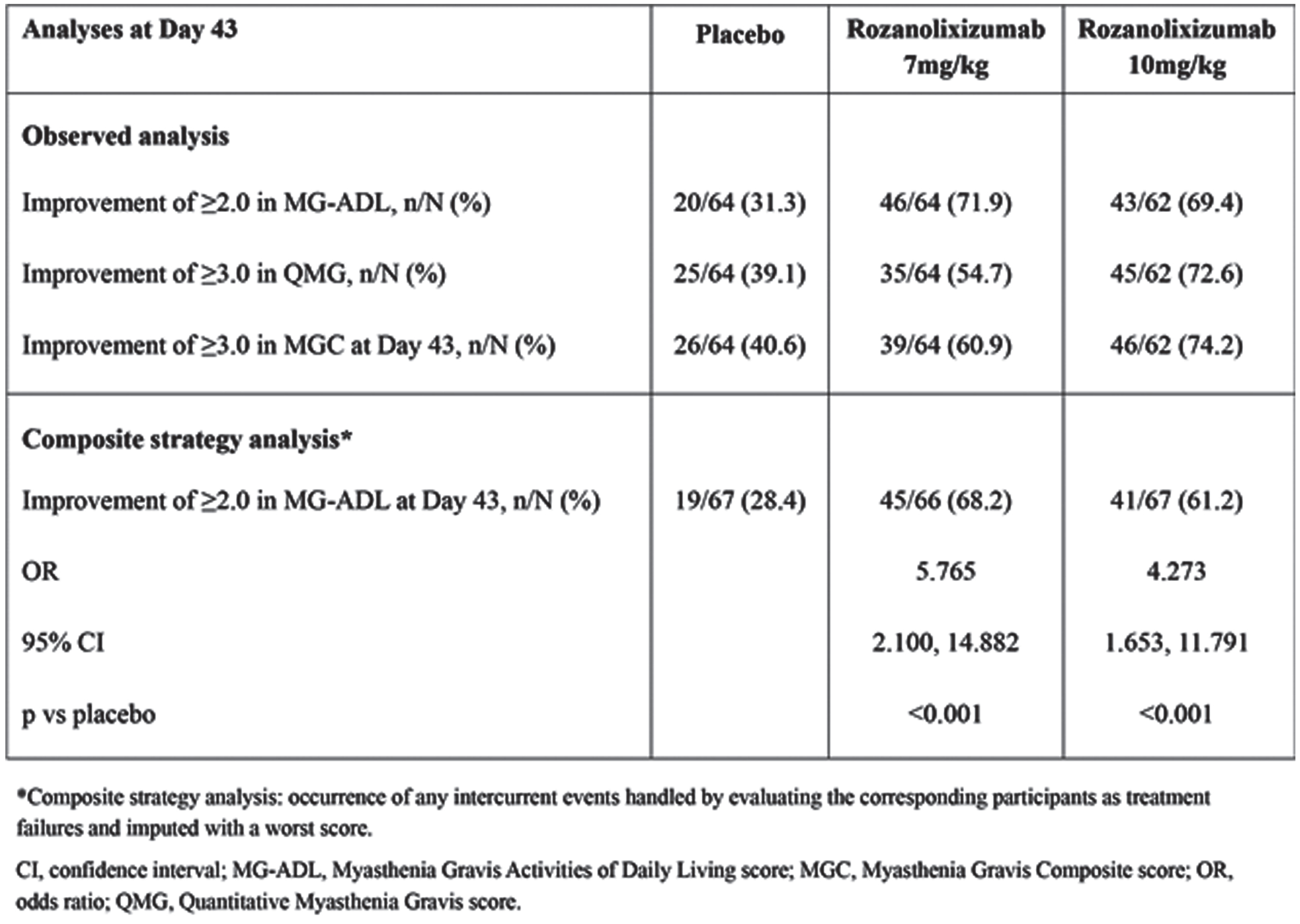
PS05.02Combining Clinical Trial and Real-world Data to Model the Benefit of Efgartigimod on Productivity Losses
Saccà F1, Dewilde S2, Nivelle E3, Qi C4, Jacob S5, Meisel A6, Palace J7, Claeys K8, Mantegazza R9,10, Paci S11, Phillips G4
1University of Naples Federico II, Naples, Italy, 2SHE, Brussels, Belgium, 3Independent HEOR Consultant, Melbourne, Australia, 4argenx, Boston, USA, 5University Hospitals Birmingham and Institute of Immunology and Immunotherapy, Birmingham, UK, 6Charité Universitätsmedizin Berlin, Department of Neurology, Neuroscience Clinical Research Center, Berlin, Germany, 7John Radcliffe Hospital, Department of Clinical Neurology, Oxford, UK, 8University Hospitals Leuven, Department of Neurology, Leuven, Belgium, 9Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy, 10Associazione Italiana Miastenia e Malattie Immunodegenerative, Milan, Italy, 11argenx, Ghent, Belgium
OBJECTIVE: Generalized Myasthenia Gravis (gMG) is a rare chronic autoimmune neuromuscular disease resulting in generalized muscle weakness affecting ocular motility, swallowing, speech, mobility and respiratory function considerably impacting a patient’s independence. Efgartigimod is a novel therapy recently approved by the FDA with demonstrated efficacy per ADAPT trial to improve the daily function of gMG patients. An economic model was developed to quantify the impact of efgartigimod in reducing the economic burden of productivity losses among gMG patients and their caregivers, combining data from ADAPT and a real-world evidence study.
METHODS: The ADAPT study is a multicenter, randomized, placebo-controlled, phase 3 study in 167 adult gMG patients comparing efgartigimod add-on treatment to conventional therapy (CT) alone. The distribution of Myasthenia Gravis Activities of Daily Living (MG-ADL) scores after 4 weeks was used to quantify the clinical benefit of efgartigimod. The MyRealWorld-MG study is a digital, observational, multi-country survey of gMG patients collecting monthly data on MG-ADL, days of sick leave, number of hours of caregiver help needed, and caregiver worktime lost. All data were self-reported by the patients and were not cross-checked with other sources. Combining data from ADAPT and MyRealWorld-MG enabled the calculation of changes in productivity losses associated with efgartigimod. The need for caregiver support and patient and caregiver worktime lost were analyzed and stratified by MG-ADL score. Total annual costs of productivity losses per patient were calculated for the UK and compared between efgartigimod and CT patients.
RESULTS: In ADAPT, the mean MG-ADL score of efgartigimod patients after 4 weeks of treatment was 4.4 versus 6.7 for CT, with 23.3% and 3.3% of patients having “Minimum Symptom Expression” (MG-ADL 0-1); 45.0% and 31.7% having mild MG (MG-ADL 2-5), 30.0% and 58.3% having moderate MG (MG-ADL 6-13); and 6.7% and 6.7% having severe MG (MG-ADL 14-24), respectively. In MyRealWorld-MG, the number of self-reported patient sick days, the proportion of patients needing help from caregivers and caregiver worktime lost was significantly positively associated with increasing MG-ADL scores (all p<0.001). Median UK annual income in 2021 for full-time employment (1786 hours worked) was £31,881.
The model calculation showed that the worktime hours lost due to sick leave was reduced by 21% with efgartigimod treatment versus CT, and caregiver worktime losses were cut by -16%, as presented in Table 1. The resulting total productivity losses per gMG patient were estimated to be 27% lower for efgartigimod than for CT patients, totaling £8,740 versus £11,906 per year. Productivity losses were mainly borne by patients (72% of total productivity losses), representing the economic cost of sick days taken. The remainder 39% of productivity costs was due to caregiver job loss (77%) or reduced working hours (33%). Total productivity savings in patients and caregivers amounted to £3,165 per patient per year.
CONCLUSION: gMG is associated with significant economic burden in terms of patient and caregiver productivity losses. By improving daily functioning, efgartigimod has the potential to reduce this burden. The findings need to be further validated once real-world data on efgartigimod becomes available.
PS05.03Humoral Responses Following SARS-CoV-2 Vaccination in Patients with Commonly Used Immunosuppressants in Neuromuscular Disorders
van Dam P1, Wieske L1, Steenhuis M2, Stalman E1, Kummer L1,2, van der Kooi A1, Raaphorst J1, Verschuuren J3, Ruiter A3, Brusse E4, van Doorn P4, Baars M4, van der Pol L5, Goedee S5, van Ham M2,6, Kuijpers T7, Rispens T2, Eftimov F1
1Department of Neurology and Neurophysiology, Amsterdam Neuroscience, Amsterdam UMC, location AMC, University of Amsterdam, Amsterdam, the Netherlands, 2Department of immunopathology, Sanquin Research and Landsteiner Laboratory, Amsterdam UMC, Amsterdam, the Netherlands, 3Department of Neurology, Leiden University Medical Center, Leiden, the Netherlands, 4Department of Neurology, Erasmus MC University Medical Center, Rotterdam, the Netherlands, 5Brain center UMC Utrecht, department of neurology and neurosurgery, Utrecht, the Netherlands, 6Swammerdam Institute for Life Sciences, University of Amsterdam, the Netherlands, 7Department of Pediatric Immunology, Rheumatology and Infectious Disease, Amsterdam UMC, location AMC, University of Amsterdam, Amsterdam, the Netherlands
Background: Disease-specific studies have reported impaired humoral responses after SARS-CoV-2 vaccination in patients with immune-mediated inflammatory disorders (IMIDs) treated with specific immunosuppressants and immunomodulating agents. The objective of this study is to investigate the humoral immune response after SARS-CoV-2 vaccination in patients using immunosuppressive and immunomodulating mono- and combination therapies, focussing on frequently prescribed therapies for inflammatory neuromuscular diseases.
Methods: National prospective observational cohort study in selected patients with prevalent IMIDs including neuromuscular disease, and immunosuppressive or immunomodulating monotherapy (n=1273), combination therapies (n=419), patients without immunosuppressants (n=473), and healthy controls (n=174). Anti-RBD IgG responses and neutralisation capacity were monitored following standard vaccination regimens and a three-vaccination regimen in subgroups. Hybrid immune responses, i.e. vaccination after previous SARS-CoV-2 infection, were studied as a proxy for recall responses.
Findings: Sera from 1869 participants without and 470 participants with previous SARS-CoV-2 infection were analysed. We included 168 (7·2%) patients with inflammatory neuropathies and myopathies, and 127 (5·4%) patients with myasthenia gravis. Humoral responses did not differ between disorders. Anti-CD20 therapy and mycophenolate mofetil combined with corticosteroids were associated with lower relative risks (RR) for reaching seroconversion following standard vaccination (RR: 0·32 and 0·61 respectively). The monotherapies corticosteroids, purine antagonists, methotrexate, mycophenolate mofetil and IVIg were not associated with a lower RR for reaching seroconversion (RR: 0·97, 0·98, 1·01, 0·86, and 0·99, respectively). Similarly, corticosteroids combined with either methotrexate or purine antagonists was not associated with a lower RR for reaching seroconversion (RR 0·89). A third vaccination increased seroconversion for mycophenolate mofetil combination treatments but not for anti-CD20 therapies. Most immunosuppressant groups showed moderately reduced antibody titres after standard vaccination that, in subgroups, did not increase after a third vaccination, although seroconversion rates and neutralisation capacity were unaffected. In participants with previous SARS-CoV-2 infection, SARS-CoV-2 antibodies were boosted after vaccination, regardless of immunosuppressive treatment.
Interpretation: Humoral responses following vaccination are impaired by specific immunosuppressants, most relevant for neuromuscular diseases being anti-CD20 and mycophenolate mofetil combination treatments. After standard vaccination regimens most immunosuppressants show equal seroconversion to controls although antibody titres may be moderately reduced. As neutralisation capacity and recall responses are also preserved in these patients, this is not likely to translate in loss of (short term) protection. Alternatively, in immunosuppressants showing poor humoral responses after standard vaccination regimens such as, a third vaccination resulted in additional seroconversion in mycophenolate mofetil combination treatments whereas the effect for anti-CD20 therapy was limited.
Funding: ZonMw (The Netherlands Organization for Health Research and Development)
PS05.04Disease Activity after SARS-CoV-2 Vaccination and Infection in Patients with Immune-Mediated Neuromuscular Diseases
Stalman E1, van Dam P1, Roosen J1, Wieske L1, Kummer L1,2, van der Kooi A1, Raaphorst J1, Verschuuren J3, Ruiter A3, Brusse E4, van Doorn P4, Baars A4, van der Pol W5, Goedee S5, Steenhuis M2, van Ham S2,6, Kuijpers T7, Rispens T2, Eftimov F1
1Department of Neurology and Neurophysiology, Amsterdam Neuroscience, Amsterdam UMC, location AMC, University of Amsterdam, Amsterdam, Netherlands, 2Department of immunopathology, Sanquin Research and Landsteiner Laboratory, Amsterdam, Netherlands, 3Department of Neurology, Leiden University Medical Center, Leiden, Netherlands, 4Department of Neurology, Erasmus MC University Medical Center, Rotterdam, Netherlands, 5Brain center UMC Utrecht, department of neurology and neurosurgery, Utrecht, Netherlands, 6Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, Netherlands, 7Department of Pediatric Immunology, Rheumatology and Infectious Disease, Amsterdam UMC, location AMC, University of Amsterdam, Amsterdam, Netherlands
Importance: Viral infection or vaccination has the potential to increase disease activity in immune-mediated neuromuscular diseases.
Objective: We aimed to evaluate whether SARS-CoV-2 vaccination and infection leads to increase of disease activity in patients with immune-mediated neuromuscular diseases.
Methods: This is an interim analysis of a subset of patients from an ongoing prospective multi-center cohort study on SARS-CoV-2 vaccination in patients with various immune mediated inflammatory diseases in the Netherlands, the Target to-B!-COVID study (T2B!). Patients received digital questionnaires every two months from study entry to assess disease activity compared to previous visit using a 5-point Likert scale. In addition, in case of SARS-CoV-2 infection (prior to vaccination) patients received an extra questionnaire to assess disease activity in the four weeks after infection. In cases of self-reported increase of disease activity, medical files were used to assess whether disease activity was reported by the treating physician, and whether changes were made in type or dose of immunosuppressive or immunomodulating treatment.
Results: In total, we included 303 patients with immune-mediated neuromuscular disease of which 127 patients with inflammatory neuropathies, 133 patients with myasthenia gravis, and 43 patients with myositis. In the four months after completed vaccination, 67 (22.1%) patients indicated an increase in disease activity, of which 62 (93%) was reported as “worse” and 5 (7%) as “much worse”. In 10 (3.3%) of the cases with self-reported increase, disease activity was also reported by the treating physician in the medical chart. In 4 (1.3%) of patients with self-reported increase disease activity treatment was adjusted because of the increase in disease activity. A SARS-CoV-2 infection prior to vaccination occurred in 24 (8%) patients, from which 3 (12.5%) indicated an increase in disease activity, not leading to change in treatment.
Conclusion: Increase of disease activity after SARS-CoV-2 vaccination or infection was reported infrequently, and was self-limiting in most cases. Findings from our cohort may help physicians in neuromuscular disease to adequately inform patients on the risk of increased disease activity due to SARS-CoV-2 vaccination or infection. Full and verified results will be reported at the ICNMD 2022.
Funding ZonMw (The Netherlands Organization for Health Research and Development)
PS05.05Immunosuppressive Therapy as Risk Factor for Severe SARS-CoV-2 Infection in Myasthenia Gravis
Stascheit F, Grittner U, Hoffmann S, Mergenthaler P, Schroeter M, Ruck T, Blaes F, Kaiser J, Schara U, Della Marina A, Thieme A, Hagenacker T, Jacobi C, Berger B, Urban P, Knop K, Schalke B, Kalischewski P, Pawlitzki M, Wiendl H, Meisel A
1Charité- Universitätsmedizin Berlin, Berlin, Germany
Importance: Patients with myasthenia gravis (MG) and IST are potentially at increased risk for poor COVID-19 outcome.
Objective: To determine whether immunosuppressive therapy (IST) compared to no IST is associated with a higher risk for, first, a symptomatic SARS-CoV-2 infection and, second, a more severe COVID-19 disease course as measured by hospitalization rate and death.
Design, setting, and participants: The present study included all available MG patients from the German myasthenia gravis registry, which is a nationwide registry conducted by expert centers since February 2019 (German Clinical Trials Registry DRKS-ID 00024099).
Main outcomes and measures: Between May 2020 and June 2021, data were collected on demographics, disease duration, comorbidities, preexistent IST including standard (corticosteroids, azathioprine, mycophenolate mofetil, methotrexate, cyclosporine) and escalation (rituximab, eculizumab) IST, thymectomy, COVID-19 characteristics, and outcomes. COVID-19 was diagnosed with a nasopharyngeal swab by polymerase-chain-reaction. Multiple binary logistic regression models and generalized estimation equation regression models based on matched SARS-CoV-2 infected to non-infected patients were used to estimate the association of IST with SARS-CoV-2 infection. Multiple binary logistic regression models were used to assess the association of IST with outcome of COVID-19 in MG patients.
Results: Of 1388 MG patients, 95 (7%) MG patients with a mean age of 58 (SD 18) and median disease duration of 65 months (IQR 27-126) presented with COVID-19. Among them, 39 patients (41%) were male, and 76 (80%) received IST at the time of infection. There were 32 patients (34%) admitted to hospital due to COVID-19, 12 (13%) to the intensive care unit, and a total of 11 patients (12%) died. IST was a risk factor for hospitalization and death in the group of COVID-19 affected MG patients (adjusted odds ratio [OR] 3.04, 95% confidence interval [CI] = 1.02-9.06, p=0.046), but not for symptomatic SARS-CoV-2 infection itself in the whole group of MG patients.
Conclusions and relevance: In MG patients, preexistent IST was a factor for a severe disease course of COVID-19 but not for the risk for SARS-CoV-2 infection. These data support the consequent implementation of effective strategies to prevent COVID-19 in this high-risk group.
PS05.06Pathogenic Effects of IgG1-MuSK Antibodies on the Agrin-induced AChR Clustering Pathway in C2C12 Myotubes
Cao M1,2, Cossins J1, Liu W1, Maxwell S1, Webster R1, Beeson D1, Vincent A1
1Nuffield Department of Clinical Neurosciences University Of Oxford, Oxford, United Kingdom, 2Department of Neurology, Norfolk and Norwich University Hospital, Norwich, UK
Antibodies to muscle specific kinase (MuSK) are found in a variable proportion of patients with generalised myasthenia gravis (MG). The agrin/MuSK pathway is essential for maintenance of the acetylcholine receptors (AChRs) at the neuromuscular junction. In MG with MuSK-antibodies, predominant monovalent IgG4 antibodies inhibit MuSK function, leading to reduced MuSK phosphorylation and reduced AChR clusters on the surface of C2C12 myotubes. Immunisation of mice against MuSK leads to defective neuromuscular transmission with reduced acetylcholine receptors. Enhancement of MuSK phosphorylation with a SRC homology 2 domain-containing phosphotyrosine phosphatase 2 (SHP2) inhibitor protected AChR clusters from the pathogenic effect of IgG4 MuSK antibodies. Interestingly, divalent IgG1,2 or 3 MuSK antibodies also reduced agrin-induced AChR clusters but did not appear to inhibit MuSK function directly.
To understand better the pathogenic mechanisms of IgG1-3 MuSK antibodies, C2C12 mouse myotubes were incubated with IgG1-3 or IgG4 antibodies purified from five MuSK-MG patients. 12-20% of the MuSK antibodies were IgG1-3 (80 - 88% IgG4). Expression and phosphorylation of MuSK, DOK7 and AChR-beta subunit were measured by immunoprecipitation and western blotting. AChR clusters were described as fully formed, ≥3 µm, or microclusters, <3 µm.
IgG1-3, in contrast to IgG4, did not reduce MuSK or DOK7 phosphorylation but reduced AChR clusters as previously reported. These were restored by NSC-87877, the SHP2 inhibitor (as previously reported for IgG4 MuSK antibodies). Independent application of IgG1-3 or agrin stimulated the phosphorylation of MuSK, DOK7 and AChR-beta subunit with a similar time-course peaking at around 1-2 hours and partially decreasing thereafter. These observations failed to explain the inhibitory effect of IgG1-3 on AChR clusters. With MuSK IgG4, however, microclusters were increased but failed to form full clusters, whereas with MuSK IgG1-3 both types of clusters were reduced suggesting an effect on microcluster formation. Results are consistent with a pathogenic effect of IgG1-3 in MuSK-MG, likely by inhibiting the formation of microclusters that precede full cluster formation, but the molecular mechanisms, and how NSC-87877 restores the clusters, remain unexplained.
PS06.01SUNFISH: 3-year Efficacy and Safety of Risdiplam in Types 2 and 3 Spinal Muscular Atrophy
Deconinck N1,2, Day J3, Mazzone E4, Nascimento A5, Oskoui M6, Saito K7, Vuillerot C8,9, Baranello G10,11, Boespflug-Tanguy O12,13, Goemans N14, Kirschner J15,16, Kostera-Pruszczyk A17, Servais L2,18,19, Braid J20, Gerber M21, Gorni K22, Martin C20, Scalco R23, Yeung W20, Mercuri E4, on behalf of the SUNFISH Working Group
1Neuromuscular Reference Center, UZ Gent, Ghent, Belgium, 2Neuromuscular Reference Center and Paediatric Neurology Department, Queen Fabiola Children’s University Hospital, Université Libre de Bruxelles, Brussels, Belgium, 3Department of Neurology, Stanford University, Palo Alto, USA, 4Pediatric Neurology Institute, Catholic University and Nemo Pediatrico, Fondazione Policlinico Gemelli IRCCS, Rome, Italy, 5Neuromuscular Unit, Neuropaediatrics Department, Hospital Sant Joan de Déu, Fundacion Sant Joan de Déu, CIBERER – ISC III, Barcelona, Spain, 6Departments of Pediatrics and Neurology Neurosurgery, McGill University, Montreal, Canada, 7Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan, 8Service de Rééducation Pédiatrique Infantile “L’Escale”, Hôpital Femme Mère Enfant, CHU-Lyon, Bron, France, 9Neuromyogen Institute, CNRS UMR 5310 - INSERM U1217, Université de Lyon, Lyon, France, 10The Dubowitz Neuromuscular Centre, NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Institute of Child Health University College London, & Great Ormond Street Hospital Trust, London, UK, 11Developmental Neurology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, 12I-Motion - Hôpital Armand Trousseau, Paris, France, 13Université de Paris, UMR 1141, NeuroDiderot, Paris, France, 14Neuromuscular Reference Centre, Department of Paediatrics and Child Neurology, University Hospitals Leuven, Leuven, Belgium, 15Department of Neuropediatrics and Muscle Disorders, Faculty of Medicine, Medical Center-University of Freiburg, Freiburg, Germany, 16Department of Neuropediatrics, University Hospital Bonn, Faculty of Medicine, Bonn, Germany, 17Department of Neurology, Medical University of Warsaw, Warsaw, Poland, 18MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, UK, 19Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Liège, Belgium, 20Roche Products Ltd, Welwyn Garden City, UK, 21Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 22PDMA Neuroscience and Rare Disease, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 23Pharma Development Neurology, F. Hoffmann-La Roche Ltd, Basel, Switzerland
Objective: To assess the efficacy and safety of risdiplam in patients with Types 2 and 3 spinal muscular atrophy (SMA) who have received treatment for 3 years (36 months).
Background: Risdiplam is a centrally and peripherally distributed, oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier that has been approved by the US Food and Drug Administration for the treatment of patients with SMA aged ≥2 months, and by the European Commission for the treatment of patients aged ≥2 months with a clinical diagnosis of Type 1, 2 or 3 SMA or 1–4 SMN2 copies.
Design/Methods: SUNFISH (NCT02908685) is a multicenter, two-part, randomized, placebo-controlled, double-blind study of risdiplam in patients with Types 2 and 3 SMA (inclusion criteria 2–25 years at enrollment). Part 1 (N=51) assessed the safety, tolerability and pharmacokinetics/pharmacodynamics of different risdiplam dose levels in patients with Types 2 and 3 SMA (ambulant and non-ambulant). Part 2 (N=180) assesses the efficacy and safety of the Part 1-selected dose of risdiplam versus placebo in a broad population of patients with Type 2 and non-ambulant Type 3 SMA. In Part 2, patients were treated with risdiplam or placebo for 12 months; all participants then received risdiplam until Month 24. At Month 24, patients were offered the opportunity to enter the open-label extension.
Results: In SUNFISH Part 2, total scores on the 32-item Motor Function Measure increased from baseline to Month 12 in patients treated with risdiplam (n=120, data cut-off: 6 September 2019); these increases were maintained between Months 12 and 36. At Month 36, no treatment-related safety findings leading to withdrawal had been reported in any patients in SUNFISH.
Conclusions: SUNFISH is ongoing and is providing long-term efficacy and safety data of risdiplam in a broad population of children, teenagers, and adults with Types 2 and 3 SMA.
PS06.02Impact of Nusinersen on Caregiver Experience and HRQoL in Presymptomatic SMA: NURTURE Study Results
Kirschner J1,2, Crawford T3, Ryan M4, Finkel R5, Swoboda K6, De Vivo D7, Bertini E8, Hwu W9, Sansone V10, Pechmann A11, Montes J12, Krasinski D12, Chin R13, Berger Z13, Zhu C13, Raynaud S13, Paradis A13, Johnson N13
1University Hospital Bonn, Bonn, Germany, 2Medical Centre, University of Freiburg, Freiburg, Germany, 3Deptartments of Neurology and Pediatrics, Johns Hopkins University School of Medicine, Baltimore, United States, 4Murdoch Children’s Research Institute and University of Melbourne, Parkville, Australia, 5Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, United States, 6Department of Neurology, Massachusetts General Hospital, Boston, United States, 7Departments of Neurology and Pediatrics, Columbia University Irving Medical Center, New York, United States, 8Unit of Neuromuscular and Neurodegenerative Disorders, Post-Graduate Bambino Gesù Children’s Research Hospital, Rome, Italy, 9Department of Medical Genetics and Pediatrics, National Taiwan University Hospital, Taipei, Taiwan, 10Neuromuscular Omniservice Clinical Center, Neurorehabilitation Unit, Univ. of Milan, Milan, Italy, 11Department of Neuropediatrics and Muscle Disorders, Medical Ctr. Univ. of Freiburg, Freiburg, Germany, 12Department of Rehabilitation and Regenerative Medicine, Columbia University Irving Medical Center, New York, United States, 13Biogen, Cambridge, United States
Background: Spinal muscular atrophy (SMA) is associated with substantial disease burden and impacts health-related quality of life (HRQoL) of patients and their caregivers. NURTURE is an ongoing, Phase 2, open-label, multinational, single-arm study evaluating nusinersen in participants who initiated treatment in the presymptomatic stage. The interim results from ~5 years of follow-up show that all participants treated with nusinersen were alive and none required permanent ventilation; the majority exceeded motor milestones predicted by SMA natural history. The aim of this study was to evaluate the impact of nusinersen treatment on caregiver experience and HRQoL of presymptomatically treated participants over time.
Methods: Interim data from NURTURE (n=25, February 2021 datacut) were evaluated by SMN2 copy number: 2 (n=15) and 3 (n=10) SMN2 copies. Assessment of Caregiver Experience with Neuromuscular Disease (ACEND) and Pediatric Quality of Life Inventory (PedsQL) Generic Core Scale (GCS) and Neuromuscular Module (NM) were administered to caregivers of participants. ACEND includes 7 total subdomains related to physical impact on caregivers (feeding/grooming/dressing, sitting/play, transfer, mobility) and general caregiver impact (time, emotion, finance). ACEND and PedsQL scales are scored from 0-100 with higher scores indicating a reduced caregiver impact and improved HRQoL, respectively. Mean scores from first assessment of ACEND or PedsQL scales in NURTURE to Day 1440 were evaluated and descriptive analyses were conducted.
Results: ACEND was administered to caregivers of participants at a median (min, max) of 2.4 (1.9, 3.2) years after initiation of nusinersen. PedsQL-GCS and NM were administered to caregivers of participants at a median (min, max) of 3.1 (1.9, 4.4) years after initiation of nusinersen. There was an overall pattern of observed increases in ACEND mean scores among caregivers of participants with 2 (n=13) and 3 (n=7) SMN2 copies from first assessment in NURTURE to Day 1440 in the physical impact subdomains: feeding/grooming/dressing, transfer, and mobility. Near-maximum mean scores (signifying reduced caregiver impact) were observed at first assessment and maintained over time to Day 1440 for the sitting/play physical impact subdomain, regardless of SMN2 copy number. ACEND mean scores in all seven domains were generally higher among caregivers of participants who had 3 versus 2 SMN2 copies at both timepoints. In addition, PedsQL-GCS and NM mean scores were higher among caregivers of participants who had 3 (n=7 or 8) versus 2 (n=14) SMN2 copies at first assessment and at Day 1440. Although small decreases were observed over time, PedsQL-GCS mean scores observed at first assessment and at Day 1440 remained high, especially among caregivers of participants with 3 SMN2 copies. The mean (SD) scores of the PedsQL-GCS reported among caregivers of participants with 3 SMN2 copies at both first assessment: 93.7 (5.9) and at Day 1440: 88.3 (10.8) were comparable to those reported by caregivers in healthy toddlers aged 2-4: 87.4 (12.5) as reported in a validation study by Varni et al 2003.
Conclusions: Nusinersen was associated with sustained reduced impact on caregiver experience and high levels of HRQoL over time among caregivers of participants with presymptomatic SMA who had been treated for several years.
PS06.03Combination of Antisense Oligonucleotide Therapy with BIO101 Demonstrates synergistic Beneficial Effects in Severe SMA-like Mice
Bezier C1,2, Nazari Hashemi P1, Cottin S2, Lafont R1,3, Veillet S1, Charbonnier F2, Dilda P1, Latil M1, Biondi O2,4
1Biophytis, Sorbonne Université, Paris, France, 2Université de Paris, Faculté des Sciences Fondamentales et Biomédicales, INSERM UMR-S 1124, Paris, France, 3Sorbonne Université, Paris-Seine Biology Institute (BIOSIPE), CNRS, Paris, France, 4Laboratoire de Biologie de l’Exercice pour la Performance et la Santé (LBEPS), UMR, Université d’Evry, IRBA, Université de Paris Saclay, Evry-Courcouronnes, France
Spinal Muscular Atrophy (SMA) is a neurodegenerative disease characterized by the loss of spinal motor neurons and progressive muscular atrophy, due to insufficient level of survival of motor neuron protein (SMN). SMA is defined as a non-cell autonomous disease, involving numerous tissues and cell-types, including skeletal muscles. In this context, strategies to overexpress SMN protein and approaches to maintain neuromuscular functions are currently developed and appear as promising long-term prospect for therapy in SMA.
We evaluated the efficacy of Sarconeos (API BIO101), a Mas receptor activator, as monotherapy and in combination with ASO therapy in severe SMA-like mice (SmnΔ7/Δ7; 2 copies huSMN2+/-). We showed beneficial effects of BIO101 as monotherapy on the entire motor unit with a complete protection of lumbar motor neurons, a limited muscular atrophy (-17% in the tibialis, -40% in the plantaris, and -12% in the soleus), an increased vascularization (+10% in the tibialis and the plantaris, +3% in the soleus), and an accelerated maturation of both muscular fibers and neuromuscular junctions. Interestingly, these benefits were independent of SMN expression. In combination with ASO therapy, BIO101 demonstrated synergistic effects on body weight (+1% with BIO101, +11% with ASO, +32% with ASO + BIO101 at 10 days after birth compared to vehicle) and increased survival (+5 days of median survival). Most importantly, the co-treated SMA-like mice improved their moving capacity (+40%) and their fatigue resistance (4,2-fold) compared to SMA-like mice treated only with ASO.
These results provide strong evidences that BIO101, with beneficial effects on the entire motor unit, constitutes an efficient SMN-expression-independent therapy for improving muscle function and should be considered as a potential combinatorial option for a new therapeutic strategy in SMA patient.
PS06.04RAINBOWFISH: Preliminary Efficacy and Safety Data in Risdiplam-Treated Infants with Presymptomatic Spinal Muscular Atrophy
Servais L1,2,3, Farrar M4, Vlodavets D5, Zanoteli E6, Al-Muhaizea M7, Finkel R8, Nelson L9, Prufer A10, Wang Y11, Fisher C12, Gerber M13, Gorni K14, Kletzl H15, Palfreeman L12, Scalco R16, Bertini E17, on behalf of the RAINBOWFISH Study Group
1MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, UK, 2Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Liège, Belgium, 3I-Motion - Hôpital Armand Trousseau, Paris, France, 4Sydney Children’s Hospital Network and UNSW Medicine, UNSW Sydney, Sydney, Australia, 5Russian Children Neuromuscular Center, Veltischev Clinical Pediatric Research Institute of Pirogov Russian National Research Medical University, Moscow, Russia, 6Department of Neurology, Faculdade de Medicina, Universidade de São Paulo (FMUSP), São Paulo, Brazil, 7Department of Neurosciences, King Faisal Specialist Hospital & Research Center-Riyadh, Riyadh, Kingdom of Saudi Arabia, 8Center for Experimental Neurotherapeutics, St Jude Children’s Research Hospital, Memphis, USA, 9UT Southwestern Medical Center, Dallas, USA, 10Federal Uni Rio de Janeiro, Rio de Janeiro, Brazil, 11Children’s Hospital of Fudan University, Shanghai, China, 12Roche Products Ltd, Welwyn Garden City, UK, 13Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 14PDMA Neuroscience and Rare Disease, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 15Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland, 16Pharma Development Neurology, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 17Department of Neurosciences and Neurorehabilitation, Bambino Gesù Children’s Research Hospital IRCCS, Rome, Italy
Objective: To determine the efficacy, safety and pharmacokinetics (PK)/pharmacodynamics (PD) of risdiplam in presymptomatic infants with genetically diagnosed spinal muscular atrophy (SMA).
Background: Risdiplam is a centrally and peripherally distributed, oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier that has been approved by the US Food and Drug Administration for the treatment of patients with SMA aged ≥2 months, and by the European Commission for the treatment of patients aged ≥2 months with a clinical diagnosis of Type 1, 2 or 3 SMA or 1–4 copies of SMN2.
Design/Methods: RAINBOWFISH (NCT03779334) is an open-label, single-arm, multicenter study of risdiplam in presymptomatic infants with genetically diagnosed SMA. RAINBOWFISH is actively enrolling infants from birth–6 weeks of age (at first dose), regardless of SMN2 copy number. The primary analysis will be conducted at 12 months in infants with two SMN2 copies and compound muscle action potential (CMAP) amplitude ≥1.5mV at baseline.
The primary endpoint is the proportion of infants sitting without support for ≥5 seconds (assessed by Item 22 of the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development, Third Edition). Secondary endpoints include the development of clinically manifested SMA; survival and permanent ventilation; achievement of motor milestones; motor function; growth measures; nutritional status; CMAP; PK/PD; and safety monitoring.
Results: As of the data cut-off (1 July 2021), the median age at first dose was 26.5 days (range: 16–40 days) for the first 18 enrolled infants. No serious adverse events were reported in infants treated for up to 22.8 months. As of the data cut-off, seven infants have been treated with risdiplam for ≥12 months (4/7 infants have 2 SMN2 copies, of which two had CMAP amplitude <1.5mV and two had ≥1.5mV at baseline; 3/7 infants have >2 SMN2 copies, of which three had CMAP amplitude ≥1.5mV at baseline). Efficacy data from these seven infants demonstrated that most infants reached near maximum scores on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders scale by 4–5 months of age and achieved motor milestones within the World Health Organization windows for healthy children. At the data cut-off, all infants treated with risdiplam for at least 12 months were alive without permanent ventilation, maintained swallowing and feeding abilities, and had not required hospitalization. We will report updated baseline demographics and safety data in enrolled infants, and the latest efficacy data in infants who have received risdiplam for at least 12 months.
Conclusions: RAINBOWFISH will provide valuable information about outcomes following presymptomatic administration of risdiplam and will help determine the dose for infants <2 months of age. Recruitment is ongoing worldwide.
PS06.05Risdiplam: Pharmacokinetic, Pharmacodynamic, Safety and Efficacy Exposure Response Analyses
Kletzl H1, Cleary Y1, Grimsey P2, Gerber M3, Scalco R4
1Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland, 2Roche Products Ltd, Welwyn Garden City, UK, 3Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 4Pharma Development Neurology, F. Hoffmann-La Roche Ltd, Basel, Switzerland
Objective: To determine the pharmacokinetic (PK), pharmacodynamic (PD), safety and efficacy exposure response of risdiplam in patients with spinal muscular atrophy (SMA).
Background: Risdiplam is a centrally and peripherally distributed, oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier that has been approved by the European Commission for the treatment of patients aged ≥2 months, with a clinical diagnosis of Type 1, 2 or 3 SMA or 1–4 copies of SMN2.
Methods: PK and PD data were obtained in all risdiplam clinical studies in patients with SMA, from infants to adults. Risdiplam plasma concentrations were measured, and the PD markers SMN protein and mRNA were assessed in blood.
Results: PK and PD data were used to select the dosing regimen of 0.2 mg/kg for infants aged between 2 months and 2 years, 0.25 mg/kg for children aged >2 years with a body weight ≤20 kg, and 5 mg for patients with a body weight ≥20 kg. This dosing regimen was assessed in the pivotal studies in patients with Type 1 (FIREFISH Part 2; NCT02913482) and Type 2/3 SMA (SUNFISH Part 2; NCT02908685), which led to the approval of risdiplam. PK and PD data were obtained in all patients, with a body weight ≤109 kg and aged ≤61 years of age. Body weight and age were found to have a significant effect on the PK of risdiplam, and the chosen dosing regimen based on age and body weight accounts for that observation. Risdiplam’s mode of action was confirmed by measuring the shift from SMN2Δ7 mRNA to full-length mRNA.
Conclusion: The selected dosing regimen led to a ≥2-fold median increase in SMN protein within 4 weeks after treatment start in all studies, which was maintained throughout treatment. Individual exposure was generally within the same range, and exposure response analyses showed no relevant differences for efficacy or safety outcomes within the observed exposure range for all patients.
PS06.06Matching-adjusted Indirect Comparison of Risdiplam Versus Nusinersen in Type 1 Spinal Muscular Atrophy: 2-year Update
Hawkins N1,2, Aponte Ribero V3, Gorni K4, Daigl M3, Martí Y3, Evans R2, Scott D2, Mahajan A5
1Institute of Health & Wellbeing, University Of Glasgow, Glasgow, UK, 2Visible Analytics, Oxford, UK, 3Global Access, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 4PDMA Neuroscience and Rare Disease, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 5Bridge Medical Consulting Ltd., London, UK
Objective: To conduct an updated matching-adjusted indirect comparison (MAIC) to evaluate relative efficacy and safety of risdiplam versus nusinersen in Type 1 spinal muscular atrophy (SMA) using longer-term data covering at least 24 months of treatment.
Background: Disease-modifying therapies approved for the treatment of SMA have not been directly compared in head-to-head trials. Indirect treatment comparisons are needed to provide information on the relative efficacy and safety of available treatments for healthcare decision-making. However, cross-study heterogeneity may bias relative effect estimates from naïve indirect comparisons. Population-adjusted indirect comparison methodologies, such as MAIC, can reduce biases resulting from heterogeneity in population characteristics.
Design/Methods: Pooled 2-year risdiplam data from 58 infants in FIREFISH Part 1 (NCT02913482; pivotal dose cohort, n=17) and Part 2 (n=41) were compared with nusinersen data from 81 patients in the ENDEAR cohort (NCT02193074) of the open-label extension study SHINE (NCT02594124; treated for approximately 3.5 years). MAIC methodology was used to compare individual patient-level risdiplam data with nusinersen data extracted from publicly available sources. Populations were matched based on known baseline prognostic factors and effect modifiers in Type 1 SMA: age at first dose, symptom duration and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) score. Comparisons were conducted on time-to-event outcomes; hazard ratios (HRs) of risdiplam versus nusinersen were estimated using Cox proportional-hazards models, which handle differences in follow-up time across studies and allow comparisons over time.
Results: FIREFISH and ENDEAR enrolled similar populations with comparable baseline characteristics in terms of age and disease duration (within 1 week). Differences in baseline CHOP-INTEND scores across the two studies were reduced after matching. The effective sample size for risdiplam after matching was 36.5. MAIC results suggest that patients treated with risdiplam may be 79% less likely to die (overall survival HR 0.21; 95% confidence interval [CI] 0.03–0.53) and 80% less likely to die or require permanent ventilation (event-free survival HR 0.20; 95% CI 0.06–0.37) compared with patients treated with nusinersen. Results were statistically significant. Treatment with risdiplam may also lead to a statistically significant delay in the time to occurrence of the first serious adverse event (HR 0.42; 95% CI 0.24–0.64) versus treatment with nusinersen. Similar results were seen with unadjusted indirect comparison analyses. These results are consistent with previously published findings from an indirect treatment comparison based on 12-month data.
Conclusions: Updated results from a MAIC based on longer-term data from the FIREFISH and SHINE (ENDEAR cohort) studies suggest that in patients with Type 1 SMA, risdiplam may significantly reduce the number of deaths and permanent ventilation events, and delay the occurrence of the first serious adverse event, as compared with nusinersen over at least 2 years of follow-up.
PS07.01Dysregulation of ER Import Proteins in IMNM with Particular Differences Between SRP54+ and HMGCR+ Patients
Preusse C1,2, Lang S3, Marteau T4, Morales-Gonzales S5, Hentschel A6, Sickmann A6, Allenbach Y7, Benveniste O7, Schülke-Gerstenfeld M8, Stenzel W1, Dittmayer C1, Roos A4,9,10
1Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neuropathology, Berlin, Germany, 2Department of Neurology with Institute for Translational Neurology, University Hospital Münster, Münster, Germany, 3Medizinische Biochemie und Molekularbiologie, Medizinische Fakultät der Universität des Saarlandes, Saarbrücken, Germany, 4Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Essen, Germany, 5Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, CharitéCrossOver, Berlin, Germany, 6Biomedical Research Department, Leibniz-Institut für Analytische Wissenschaften - ISAS - e.V., Dortmund, Germany, 7Department of Internal Medicine and Clinical Immunology, Pitié-Salpêtrière University Hospital, Paris, France, 8Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Paediatrics, Berlin, Germany, 9Pediatric Neurology, University Children’s Hospital, University of Duisburg-Essen, Faculty of Medicine, Essen, Germany, 10Department of Neurology, Heimer Institute for Muscle Research, University Hospital Bergmannsheil, Ruhr-University Bochum, Bochum, Germany
Immune mediated necrotizing myopathies (IMNM) are part of the idiopathic inflammatory myopathies (IIM), precisely defined by international consensus recently. Criteria were based on clinical, morphological as well as serological features. The main characteristics include proximal lower limb-predominant muscle weakness, substantially increased serum CK levels and detection of the pathognomonic myositis-specific auto-antibodies anti-SPR54 or -HMGCR in many patients, while 1/3 of IMNM patients remain without a positive serostatus for these antibodies. Notably, both auto-antibodies target proteins of the endoplasmic reticulum (ER) / sarcoplasmic reticulum (SR) which are involved in protein processing. Impaired protein processing may lead to ER/SR-stress. The ER/SR-stress is in turn accompanied by the activation of the unfolded protein response (UPR), which is modulated by proteins belonging to the three UPR-branches. In a previous study on muscles derived from IMNM-patients’ we confirmed activation of the protein clearance machinery by increased abundances of UPR-related proteins, demonstrating activation of all three UPR-branches in IMNM, independent of the serostatus.
Here, we expanded the analyses by focussing on proteins crucial for the import of nascent polypeptides into the ER/SR and thus for protein synthesis. Using proteomics, histology, transcriptional analyses and immunoblotting, we detected interesting changes in the pathophysiology of IMNM with differences among the different auto-antibody conditions. Thus, our studies unravel myopathological differences between SRP54+ and HMGCR+ IMNM patients providing new starting points for patient stratification. Our transcript studies, among others, revealed an up-regulation of Calumenin in HMGCR+ patients, encoding a calcium-binding protein localized within the ER/SR, which is involved in protein folding and sorting. Additionally, we detected SEC61B as up-regulated, encoding for a central component of the protein translocation apparatus resident within the ER/SR-membrane. Interestingly this increase was less pronounced in SRP54+ patients. Our protein studies using histological staining and immunoblotting confirmed these findings.
These combined studies - for the first time - show a pathophysiological difference between the two IMNM subgroups, not only increasing our understanding of the underlying pathophysiology but also providing new aspects in the stratification of IMNM patients.
PS07.02Autoantibodies Against TRIM72/MG53 in Dysferlinopathy Patients and Mouse Models Decrease Sarcolemmal Membrane Repair Capacity
Weisleder N1, Bulgart H1, Banford K1, McElhanon K1
1Ohio State University, Columbus, United States
Limb Girdle Muscular Dystrophy Autosomal Recessive 2 (LGMDR2, previously known as LGMD2B) results from mutations in the dysferlin gene that lead to a range of different clinical presentations. Variation in both the age of onset and the degree of pathologic involvement led to multiple designations of the dysferlinopathies, including Miyoshi Myopathy, Distal Myopathy with Anterior Tibial Onset, Proximodistal weakness, Pseudometabolic myopathy, and HyperCKemia. Dysferlin acts in the sarcolemmal membrane repair response, a conserved cellular pathway where disruptions of the lipid bilayers are resealed by exocytoic trafficking of intracellular vesicle to the injury site where they fuse with each other and the membrane fuse to form a repair patch topped by a protein enriched cap containing dysferlin. The loss of dysferlin expression or function during dysferlinopathy leads to compromised membrane repair and decreased sarcolemma integrity that leads to myocyte death and progression of muscular dystrophy. There have been extensive efforts to better understand the disease mechanisms contributing to the progression of dysferlinopathies, there are many aspects of the pathophysiology that lead to muscle dysfunction that are not clearly resolved. Understanding these mechanisms is essential for effective management of LGMDR2 and development of new potential therapies. We recently observed that autoantibodies against TRIM72/MG53, a dysferlin binding partner protein that also contributes to membrane repair, can decrease membrane repair capacity in skeletal muscle. These autoantibodies appear in the serum of human immune-mediated myopathy (IMM) patients (McElhanon, et al., JCI, 2020). Since IMM and LGMDR2 share several pathologic hallmarks that lead to occasional misdiagnosis, we tested if autoantibodies against TRIM72/MG53 appear in the LGMDR2 patient serum and if these antibodies could compromised membrane repair in skeletal muscle. To determine if such antibodies are present in LGMDR2 patients we conducted ELISA testing of 106 LGMDR2 patient serum samples. These studies revealed 17.9% of patients had highly elevated levels of MG53/TRIM72 autoantibodies. Similar results were seen in multiple mouse models of LGMDR2. Serum enriched in these autoantibodies from LGMDR2 patients or mouse models, as well as exogenous MG53/TRIM72 antibodies, compromise sarcolemmal membrane repair in laser wounding repair assays. Compromised repair is dependent on TRIM72/MG53 antibodies as depletion of these antibodies from serum samples removes these effects on membrane repair. This antibody mediated compromised membrane repair could represent a therapeutic target for LGMDR2 as we find poloxamer compounds known to increase sarcolemma integrity can reduce the impact of TRIM72/MG53 antibodies skeletal muscle. Our results reveal the autoantibodies against TRIM72/MG53 that compromised sarcolemma repair could represent a novel disease mechanism in LGMDR2. We find targeting membrane integrity minimizes these effects, indicating a new potential therapeutic intervention.
PS07.03A Structural Variant of the C-Terminal Prion-like Domain of TDP-43 Causes Vacuolar Muscle Degeneration
Ervilha Pereira P1,2, Schuermans N1,2, Meylemans A3,4, LeBlanc P1,2, Versluys L1,2, Debackere E1,2, Vanakker O1,2, Janssens S1,2, Baets J5,6,7, Verhoeven K8, Lammens M9, Symoens S1,2, De Paepe B3,4, De Bleecker J3,4, Bogaert E1,2, Dermaut B1,2
1Center for Medical Genetices, Ghent University, Gent, Belgium, 2Department of Biomolecular Medicine, Faculty of Medicine and Health Sciences, Ghent University, Gent, Belgium, 3Department of Neurology, Ghent University Hospital, Gent, Belgium, 4Department of Head and Skin, Faculty of Medicine and Health Sciences, Ghent University, Gent, Belgium, 5Neuromuscular Reference Centre, Department of Neurology, Antwerp University Hospital, Antwerp, Belgium, 6Translational Neurosciences, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium, 7Laboratory of Neuromuscular Pathology, Institute Born-Bunge, University of Antwerp, Antwerp, Belgium, 8Department of Neurology, Bruges Amyloidosis Centre, Sint-Jan Hospital Bruges, Bruges, Belgium, 9Department of Pathology, Antwerp University Hospital, Edegem, Belgium
Neurodegenerative diseases are often associated with the accumulation of protein aggregates in neuronal tissues, the nature of which varies from condition to condition and plays a crucial role in the distinction and categorization of the individual pathologies. For one, neuronal TDP-43-positive inclusions are a pathological hallmark lesion found in frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), accounting for 45% and 97% of cases, respectively.
Remarkably, missense mutations in the C-terminal prion-like domain (PrLD) of TDP-43 are the main type of disease-causing mutations reported for ALS. This domain of the TDP-43 protein is mainly responsible for mediating self-interaction, and mutations of this region are thought to have an impact on the aggregation dynamics of the protein, ultimately leading to the formation of the hallmark pathological aggregates.
However, the scope of TDP-43 proteinopathies is not limited to neural tissues, with a wealth of reports showing TDP-43 pathology in vacuolar myopathies including sporadic inclusion body myositis (sIBM), oculo-pharyngeal dystrophy, rimmed vacuole myopathies and other distal myopathies. Nevertheless, genetic evidence indicating a direct pathogenic role for TDP-43 in myopathies is still lacking.
Here we identified a multigenerational family with an autosomal dominant rimmed vacuole myopathy. Whole exome sequencing and genome-wide linkage analysis conclusively mapped the disease to an 11bp deletion in TDP-43 (maximum multipoint LOD-score of 3.6). The deletion leads to a frameshift mutation in the C-terminal region of TDP-43, resulting in an altered and shortened PrLD (TDP-43 p.Trp385IlefsTer10). Notably, this novel mutation is unlike any mutation reported for TDP-43 so far, which are predominantly single-base missense mutations.
Patient-derived muscle biopsies revealed the presence of p62/TDP-43-positive sarcoplasmic inclusions along with nuclear depletion of TDP-43. Additionally, we also verified an increased number of autophagosomes and a transcriptomic signature indicative of reduced mitochondrial and lipid metabolism, concomitant with a switch in sarcomeric protein isoforms pointing towards increased muscle regeneration. Together with these pathological observations in patient samples, functional assays in D. melanogaster showed that TDP-43 p.Trp385IlefsTer10 retains normal function but results in a reduction in toxic gain-of-function properties for the protein.
With the study of this unique variant of TDP-43 it is our goal to shed some light on the importance of the C-terminal domain of the protein and how its profound remodelling can impact the dynamics of TDP-43 aggregate formation. Furthermore, these results genetically link TDP-43 to vacuolar muscle degeneration for the first time. This not only highlights the crucial role of the PrLD in the development of pathological outcomes in a tissue-specific manner, but it also expands the implications of TDP-43 proteinopathies, from being almost exclusively associated to a neurodegenerative context into a broader spectrum encompassing myopathies as well.
PS07.04A Randomized, Double-Blind, Placebo-Controlled Study of Arimoclomol in Patients with Inclusion Body Myositis
Dimachkie M1, Machado P2, Barohn R3, McDermott M4, Blaettler T5, Lloyd T6, Shaibani A7, Freimer M8, Amato A9, Ciafaloni E4, Jones S10, Mozaffar T11, Gibson S12, Wicklund M13, Levine T14, Sundgreen C5, Carstensen T5, Bonefeld K5, Jørgensen A5, Phonekeo K5, Heim A1, Herbelin L1, Hanna M2
1University Of Kansas Medical Center, Kansas City, United States, 2University College London, London, England, 3University of Missouri, Columbia, United States, 4University of Rochester, Rochester, United States, 5Orphazyme A/S, Coppenhagen, Denmark, 6Johns Hopkins University, Baltimore, United States, 7Nerve & Muscle Center of Texas, Houston, United States, 8Ohio State University, Columbus, United States, 9Brigham & Women’s Hospital, Boston, United States, 10University of Virginia, Charlottesville, United States, 11University of California - Irvine, Orange, United States, 12University of Utah, Salt Lake City, United States, 13University of Colorado, Denver, United States, 14HonorHealth, Phoenix, United States
Background: Inclusion body myositis (IBM) is the most common idiopathic inflammatory myopathy occurring in patients over the age of 45 years. Since immune suppression has not been effective, modulating the cytoprotective “heat shock response” (HSR) represents a candidate therapeutic approach targeting both inflammation and degeneration. In a pilot study, arimoclomol, an amplifier of the HSR, was safe and well tolerated with some trends suggesting efficacy at 8 months in subjects with IBM.
Objectives: To present the efficacy and safety/tolerability data from a phase 2/3 randomized controlled trial of arimoclomol in IBM (NCT02753530).
Methods: In this international multicenter, double-blind, placebo-controlled trial, subjects were randomized (1:1) to receive either arimoclomol citrate 400 mg or matching placebo capsules three times a day (1,200 mg/day) for 20 months. The primary outcome measure was the change from baseline to Month 20 in the IBM Functional Rating Scale (IBMFRS) total score. Hierarchically ordered key secondary outcome measures included hand grip strength (strongest hand), Modified Time Up and Go, Manual Muscle Testing (24 muscles), 6-minute walk test distance, and the Short-Form 36 health survey. Other outcome measures included patient and clinician impressions, and other measures of muscle strength and function. Drug safety and tolerability were evaluated.
Results: One hundred fifty-two IBM subjects fulfilling ENMC 2011 criteria were randomized with mean age 67.2 years (SD 8.1), mostly men (76%), mean disease duration 98 months (SD 58), and mean baseline IBMFRS of 27.4 (SD 4.6). The IBMFRS declined by a mean of 3.25 points with arimoclomol vs. 2.26 points with placebo over 20 months (p=0.11). Secondary efficacy outcome measures did not show any statistically significant treatment group differences. Most frequently reported AEs observed with higher incidence in arimoclomol group were gastrointestinal disorders (54.8% vs. 39.7%). Patients receiving arimoclomol were more likely to discontinue treatment due to AEs (17.8% vs. 5.1%). The relative frequency of serious AEs was comparable in the two treatment arms (arimoclomol 15.1% vs. placebo 23.1%). Elevated transaminases were reported in the first three months and were more frequently observed with arimoclomol than with placebo (15.4% vs. 6.4%).
Conclusions: This trial did not demonstrate a benefit of arimoclomol in IBM with respect to its primary and secondary efficacy endpoints.
PS07.05Clinical and Magnetic Resonance Imaging of Muscles in anti-Mi2B Inflammatory Myositis
Huddar A1, Nashi S1, Kulanthaivelu K2, Vengalil S1, Unnikrishnan G1, Thomas A1, Baskar D1, Nalini A1
1Department of Neurology, Nimhans, Bangalore, India, 2Department of Neurointervention and Neuroradiology, NIMHANS, Bangalore, India
Background and Aims: Mi2b antibody is associated with classical dermatomyositis (DM) and is found in upto 10-20% of DM. In this study we aimed to determine clinical features and Magnetic resonance imaging (MRI) characteristics of muscles in Anti- Mi2b Inflammatory Myositis, and look for correlation between imaging findings with clinical parameters.
Methods: We retrospectively reviewed case files of patients with anti-Mi2b positive inflammatory myositis who underwent lower limb MRI with axial STIR, T1 sequences (1.5 T Aera; SIEMENS healthcare MR scanner) from 2017-2020. Manual Muscle Testing-8 (MMT 8) was used for clinical severity, modified Stramare scoring to grade muscle edema and modified Mercuri scale for muscle atrophy on MRI.
Results: Among a total of 110 patients with inflammatory myositis, there were 25 patients with Mi2b antibody positive inflammatory myositis. In this cohort, 16 patients underwent lower limb MRI and were included in current study. There were 8 men and 8 women (F:M=1:1). The mean age at diagnosis was 44.3±11.6 years (Range: 24 to 60 years) and mean duration of illness was 21.3±23.8 months (Range: 1 to 72 months). Presenting symptoms included muscle pain and weakness. Proximal lower limb and upper limb weakness was seen in 15 and 13 patients respectively with truncal weakness in nine and neck weakness in 11 patients. Five patients were wheelchair bound at time of diagnosis. Skin changes were seen in 15 (photosensitivity-4, heliotrope rash-6, shawl sign, V sign and Gottrons papules in 2 each). Other features included: joint pains (n=10), loss of weight (n=10) and respiratory symptoms (n=7). None had cardiac involvement. Mean creatine kinase (CK) was 6044.1±7673 (Range: 100 to 30013). CT chest revealed non-specific interstitial changes in one patient. Muscle biopsy done in one patient revealed polygonal to round fibers, internal nuclei, variation in fiber size, myophagocytes, necrosis, endomysial inflammation and moth eaten fibers. Lower limb MRI revealed fatty infiltration (n=11) and edema (n=16) in thigh muscles. None of the muscles had atrophy of grade >3. Fatty infiltration was maximum in hamstrings (1.36±0.915) followed by iliopsoas (1.3±1.04), glutei muscles (1.3±1), and least in quadriceps (1.26±0.9). Obvious muscle edema (grade >2) was seen in 13/16 patients. Most severely affected muscles were quadriceps (vastus medialis, intermedius, lateralis and rectus femoris equally affected with grade 3.8±1.6) followed by hip adductors (3.53±1.4), iliacus, iliopsoas (3.4±1.64) and glutei muscles (3.4±1.5). Among hamstrings, semitendinosus (3.53±1.6) was more involved than semimembranosus (2.66±1.95) and biceps femoris (2.66±1.75). All showed fascial edema in thigh muscles. Gluteus medius and minimus muscle edema showed significant correlation with CK levels, however, there was no significant correlation between muscle edema of most muscles and MMT-8, CK. MMT-8 did not correlate with CK values by Spearman ‘s rho test. There was a trend towards correlation of muscle edema in rectus femoris, semitendinosus and soleus with CK.
Conclusions: This study showed preferential severe affection of the hip and thigh muscles with uniform fascial edema in patients with anti Mi2b antibody positive inflammatory myositis. However, most of these muscles on Stramare scoring did not show significant correlation with MMT.
PS07.06Key Features for Morphological Classification of Idiopathic Inflammatory Myopathies in Children
Schänzer A1, Rager L1, Dahlhaus I2, Dittmayer C3, Preusse C3,4, Della Marina A5, Goebel H3, Hahn A6, Stenzel W3
1Institute of Neuropathology, Justus Liebig University, Giessen, Germany, Giessen, Germany, 2Institute of Medical Informatics, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin und Humboldt-Universität zu Berlin, Berlin, Germany, 3Department of Neuropathology, Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany, 4Department of Neurology with Institute of Translational Neurology, University Hospital, Münster, Münster, Germany, 5Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Hospital Essen, University Duisburg-Essen, Essen, Germany, 6Department of Child Neurology, Justus Liebig University, Giessen, Giessen, Germany
Background: In adult patients, the classification of idiopathic inflammatory myopathies (IIM) has been well described, and distinct morphological features have been associated with clinical subtypes and myositis specific autoantibodies (MSA). Although there are certain clinical and serological similarities, there may be important differences in pathogenesis of myositis between adults and children. In juvenile IIM, the classification and morphological characteristic features of distinct subgroups are not well-defined. New treatment strategies require a precise diagnosis of the subgroups in IIM, and, therefore knowledge about pathomorphology of juvenile IIM is warranted.
Methods: Muscle biopsies from 15 patients (mean age 8.9±4.6, range 3-19 years, 73% female) with IIM and 7 controls were analysed by standard methods, immunohistochemistry and transmission electron microscopy (TEM). Detailed clinical and laboratory data were accessed retrospectively.
Results: The leading clinical symptom was proximal muscle weakness and skin symptoms. Dermatomyositis (DM) was diagnosed in 9/15, antisynthetase syndrome (ASyS) in 4/15 and overlap-myositis (OM) in 2/15. MSA were positive in 6/12 patients, with 2 positive anti-NXP2+, 2 anti-Jo-1+ and 2 anti-PL7+ patients. In two patients, anti-SLE antibodies were detected. Analysis of skeletal muscle tissues showed inflammatory cells and upregulation of MHC class I in all IIM-subtypes. Morphological key findings were COX-deficient fibres as a striking pathology in DM and perimysial alkaline phosphatase positivity in anti-Jo-1-ASyS. Vascular staining of the type 1 IFN-surrogate marker MxA correlated with endothelial tubuloreticular inclusions in both groups. None of these specific morphological findings were present in anti-PL7-ASyS or OM.
Conclusions: Using a comprehensive panel of stains for muscle biopsies is helpful to classify juvenile IIM subtypes even if MSA as serologic markers are missing. Morphological key features are helpful to discriminate IIM subtypes underlining differences in their aetiopathogenesis and therefore individual and targeted therapeutic strategies may be applied to these children.
PS08.01A Recurrent Missense Variant In ITPR3 Is Associated With Demyelinating CMT
Beijer D1, Dohrn M1,2, Rebelo A1, Feely S3, Reilly M4, Scherer S5, Lee Y6, Shy M7, Zuchner S1
1John P. Hussman Institute for Human Genomics, University Of Miami, Miami, United States, 2Department of Neurology, Medical Faculty, RWTH Aachen University, Aachen, Germany, 3University of Iowa, Iowa City, United States, 4Centre for Neuromuscular Diseases, UCL Queen Square Institute of Neurology, Queen Square, London, United Kingdom, 5Department of Neurology, The Perelman School of Medicine at the University of Pennsylvania, Philadelphia, United States, 6Department of Neurology, Taipei Veterans General Hospital, Taipei, Taiwan, 7Department of Neurology, University of Iowa Carver College of Medicine, Iowa City, United States
The diagnostic yield in demyelinating Charcot-Marie-Tooth disease (CMT1) is typically ~80-95%, of which at least 60% is due to the PMP22 gene duplication. The remainder of CMT1 is more genetically heterogeneous. We used whole exome sequencing (WES) and whole genome sequencing (WGS) data to investigate novel causal genes and mutations in a cohort of ~2,000 individuals with CMT disease submitted to the Genesis project. We identified a recurrent missense variant in ITPR3, a recently described CMT gene, in more than 16 individuals from seven different families. All families presented with slow nerve conduction velocities and an autosomal dominant or de novo inheritance, matching the diagnostic category of CMT1. Sanger sequencing confirmed the co-segregation of the CMT phenotype with the presence of the variant, including a four-generation family with multiple affected second-degree cousins, and a de novo inheritance in an isolated patient. ITPR3 encodes IP3R3 (inositol 1,4,5-trisphosphate receptor 3), which, like its paralogs ITPR1and ITPR2, is highly expressed in the nervous system. Based on protein modeling, this residue is located in the dimerization interface and could interfere with the dimerization process. We are currently testing this hypothesis using in vitro modeling. Here we show that a recurrent ITPR3 missense mutation is associated specifically with a demyelinating phenotype and could account for a relatively large proportion of unsolved CMT1 patients.
PS08.02SCREEN4PN; a Novel iPSC Testing Platform for Efficient Evaluation of Compounds for CMT Neuropathies
Van Wermeskerken T1, van Lent J1,2, Da Silva Authier T1,2, Lanens D3, Timmerman V1,2,3
1Peripheral Neuropathy Research Group, Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium,2Laboratory of Neuromuscular Pathology, Institute Born Bunge, Antwerp, Belgium, 3iMARK consortium, University of Antwerp, Antwerp, Belgium.
SCREEN4PN is an induced pluripotent stem cell (iPSC) testing platform being developed to efficiently test new therapeutic compounds for Charcot-Marie-Tooth (CMT) neuropathies. Over 2,5 million people suffer from CMT neuropathy, but most CMT therapeutic studies are in a laboratory or pre-clinical phase, with only one clinical study reaching phase III for the most common form of CMT, type 1A.
Due to the tremendous clinical and genetic heterogeneity characteristic of CMT neuropathy, the development of efficient therapies is a convoluted process. So far, all therapeutic studies have been performed in specific small animal models mimicking one specific gene mutation causing a CMT disease subtype. With over a thousand mutations resulting in CMT neuropathy, creating an animal model for every gene mutation would be unrealistic, due to high costs and ethical objections (3R principle for the use of laboratory animals). In addition, a long lead time must be taken in consideration since the progressive neuromuscular symptoms typically only appear in (young) adulthood, and complications can arise during the development of transgenic laboratory animals. Moreover, the metabolism between humans and small laboratory animals differs significantly, which can have implications for determining the correct dose and the long-term effects of a drug.
The SCREEN4PN platform overcomes these problems by using iPSC technology. We recently demonstrated that iPSC-derived nerve cells from CMT type 2 patients, caused by different gene mutations, share common features. Targeting these common features could allow for the development of a uniform therapy for CMT. We showed that we could partially restore progressive mitochondrial dysfunction in these iPSC neurons by means of a therapeutic molecule. The test platform currently includes: measuring nerve outgrowth, determining the axonal transport, characterizing the mitochondrial dysfunction, and phenotyping by means of microscopy techniques.
SCREEN4PN will is a testing platform focusing on CMT-targeted therapies, which can be applied to other peripheral neuropathies and neuromuscular disorders by expanding it from a 2D to a 3D cell model by introducing neuromuscular organoids (NMOs). The platform significantly shortens the process for testing drug candidates and biomarkers by a factor of 5 compared to research using animal models, taking only four months compared to over more than a year. The cost of screening candidate therapeutic molecules should decrease by a factor of 4. The number of experiments and the experimental variability should also decrease in comparison to animal model research.
The SCREEN4PN platform, consisting of patient and control-derived iPSC neurons and NMOs (2D and 3D cultures), combined with standardized assays, will benefit the pharmaceutical industry and clinical research organizations in evaluating and/or validating their therapies or biomarkers in a relevant model; not only for CMT but for other related neuromuscular and neurodegenerative disorders as well.
PS08.03Biomarkers and Outcome Parameters: A Global Natural History Study on SORD Neuropathy
Dohrn M1,2, Cortese A3,4, Curro R3,4, Seeman P5, Stojkovic T6, Hammans S7, Previtali S8, Bolino A9, Schenone A10, Kennerson M11, Rojas Garcia R12,13, Sevilla T14, Manganelli F15, Zhang R16, Hahn K17, Schlotter-Weigel B18, Claeys K19, Brunkhorst R2, Parman Y20, Cavalcanti F21, Obermaier C22, Houlden H3,4, Scherer S23, Hermann D24, Pareyson D26, Reilly M3,4, Shy M25, Züchner S1
1Hussman Institute For Human Genomics, Miller School Of Medicine, University Of Miami, Miami, United States, 2Medical Faculty of the RWTH Aachen University, Department of Neurology, Aachen, Germany, 3Centre for Neuromuscular Diseases, UCL Queen Square, London, United Kingdom, 4Institute of Neurology, Queen Square, London, United Kingdom, 5Neurogenetic laboratory, Department of Paediatric Neurology, 2nd Faculty of Medicine, Charles University and University Hospital Motol, Prague, Czech Republic, 6Institut de Myologie, Paris, France, 7Wessex Neurological Centre, University Hospital Southampton NHS Foundation Trust, Southampton, United Kingdom, 8Neuromuscular Repair Unit, InSpe (Institute of Experimental Neurology) and Division of Neuroscience, IRCCS San Raffaele Scientific Institute, Milan, Italy, 9Human Inherited Neuropathies Unit, Institute of Experimental Neurology (INSPE) and Division of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ospedale San Raffaele, Milan, Italy, 10Department of Neurosciences, Rehabilitation, Ophthalmology, Genetic and Maternal Infantile Sciences, IRCCS Policlinico San Martino, University of Genoa, Genoa, Italy, 11Northcott Neuroscience Laboratory, ANZAC Research Institute, Sydney, Australia, 12Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, 13Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain, 14Neuromuscular Diseases Unit, Department of Neurology, HospitalUniversitari i Politècnic La Fe, Valencia, Spain, 15Dipartimento di Neuroscienze, Scienze della Riproduzione ed Odontostomatologiche, Naples, Italy, 16Department of Neurology, The Third Xiangya Hospital, Central SouthUniversity, Changsha, China, 17Department of Neurology, Charité University Medicine, Berlin, Germany, 18Department of Neurology, Ludwig Maximilian University Munich, Munich, Germany, 19Department of Neurology, University Hospitals Leuven, Leuven, Belgium, 20Department of Neurology, Istanbul Faculty of Medicine, Istanbul, Turkey, 21Institute for Biomedical Research and Innovation, National Research Council, Mangone, Italy, 22Center for Genomics and Transcriptomics, Tübingen, Germany, 23Department of Neurology, The University of Pennsylvania, Philadelphia, United States, 24Department of Neurology, University of Rochester, Rochester, United States, 25Department of Neurology, University of Iowa Hospitals and Clinics, Iowa City, United States, 26Department of Clinical Neurosciences, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Biallelic loss-of-function mutations in SORD lead to the accumulation of sorbitol and to the development of neuropathy. The underlying polyol-pathway involving the enzymes aldose reductase and sorbitol dehydrogenase, the latter encoded by SORD, has been studied already in the context of diabetes mellitus and diabetic neuropathies. Therapeutically, this can be used to specifically reduce the production of sorbitol. To define valid outcome measures for clinical trials, we have been conducting a global natural history study on SORD neuropathy, which will help us to understand phenotype patterns and disease dynamics. At the time of the ICNMD meeting, our study will be closed, and we will be able to present the first statistically significant results. Through an international network of collaborators, we have so far enrolled 150 patients carrying biallelic SORD mutations. Detailed clinical data, collected following a standardised protocol, are so far available from 101 patients. Out of these, 73 carry the most common c.753delG; p.Ala253GlnfsTer27 variant in a homozygous state. Further 27 patients have the c.753delG variant in compound-heterozygosity with a second missense or nonsense variant, including c.458C>A; p.Ala153Asp, which appears to be the second most common mutation in this cohort (n=18). Patient phenotypes were classified as axonal Charcot-Marie-Tooth disease (CMT2) in 64%, as distal hereditary motor neuropathy (dHMN) in 31%, and intermediate CMT in 5%. The mean age of symptom onset, mostly manifested through difficulty walking and running, was 17±10 years (range 3-51 years). Foot dorsal and plantar flexion strengths were reduced in 96% and 79% of patients, respectively, while sensation was preserved in over 60% of the cases. According to the CMTES score, the neuropathy was mild in two thirds of cases. MRC scores of foot dorsiflexion correlated inversely with the subjects’ age and declined significantly over 1 year, while CMTES did not (n=23). Analyzing fasting sorbitol levels in patient serum, we found a significant elevation (14.2±2.7 gr/L, n.v.<0.25) without differences between genotypes.
With these preliminary data at hand, we conclude that foot dorsi- and plantar flexion are prominently involved in patients with SORD neuropathy. Especially the former seems to be a promising outcome measure to be considered for future clinical trials. Sensory symptoms were less severe and less frequently reported, so that the leading phenotype was motor predominant axonal CMT. Fasting serum sorbitol is a reliable biomarker that can functionally validate a variant’s pathogenicity within the expanding genotype spectrum of the disease. SORD neuropathy is probably (one of) the most common autosomal recessive inherited neuropathy(/ies), and with clinical trials in preparation, it might become the first treatable CMT subform in the near future.
PS08.04Response Predictors to Patisiran Treatment in Non-endemic ATTRv Patients
Martínez-Vicente L1, Gajate-García V1, Gutiérrez-Gutiérrez G2, Guerrero-Peral Á3, Horga A1, Guerrero-Sola A1, Matías-Guiu J1, Galán-Dávila L1
1Hospital Universitario Clínico San Carlos, Madrid, Spain, 2Hospital Universitario Infanta Sofía, San Sebastián de los Reyes, Spain, 3Hospital Universitario de Valladolid, Valladolid, Spain
INTRODUCTION: Hereditary amyloid transthyretin (ATTRv) amyloidosis is a rare disease with different clinical manifestations in endemic and non-endemic areas. Patisiran, a small interference RNA that inhibits hepatic synthesis of TTR, was recently approved for the treatment of ATTRv. Our objective was to study predictive factors of response to Patisiran in a cohort of non-endemic ATTRv patients.
METHODS: Retrospective study with prospective data collection of patients with ATTRv on Patisiran treatment for ≥6 months. The patients were classified into Good-Responders, Partial-Responders or Non-Responders according to their response to treatment. Demographic, clinical, and laboratory data were collected and correlated with response to Patisiran.
RESULTS: 22 patients with ATTRv on Patisiran treatment for ≥6 months were evaluated. 8 patients were women (36.4%) and 14 patients were men (63.6%). All patients had neuropathy, and 20 patients also had cardiomyopathy (91%). Mean age of disease onset was 62 years (SD 10.7). Val50Met mutation was found in 13 patients (59.1%, of which 12 patients were late-onset), followed by Ser97Tyr and Glu109Lys in 2 patients respectively (9.1% and 9.1%). Mean time of treatment with Patisiran was 27.6 months (SD 18.4). At last follow-up, 19 patients were on Patisiran treatment (86.4%), 1 patient underwent a change in treatment (4.5%), and 2 patients died (9.1%). 13 patients (59.1%) were previously treated with Tafamidis.
11 patients were Good-Responders (50%), 8 patients were Partial-Responders (13.6%) and 3 patients were Non-Responders (36.4%). Predictive factors of good response to treatment with Patisiran were male gender (p=0.006: Good-Responders=90.9% vs Partial and Non-Responders=36.4%), initial PND stage ≤IIIA (p=0.002: Good-Responders=100% vs Partial and Non-Responders=50%) and sustained NTproBNP<300 (p=0.027: Good-Responders 40% vs Partial and Non-Responders 0%).
CONCLUSIONS: In this study, male gender, initial PND stage ≤IIIA, and sustained NTproBNP<300 were predictors of good response to Patisiran.
PS08.05Muscle Ultrasound as a Potential Progression Marker in Hereditary Neuropathies
Winter N1, Vittore D1, Grimm A1, Dohrn M3
1Department of Neurology and Epileptology, Hertie Institute for Clinical Brain Research, University of Tuebingen, 72076 Tübingen, Germany, 2Department of Neurology, Medical Faculty, RWTH Aachen University, Aachen, Germany, 3Dr. John T. Macdonald Foundation, Department of Human Genetics and John P. Hussman Institute for Human Genomics, Miami, United States of America
Background: Hereditary neuropathies are progressively disabling diseases with limited treatment options only. High resolution ultrasound (HRUS) of peripheral nerves and muscles is a new method for the examination of neuromuscular disorders. While the analysis of nerve alterations focuses more on differential diagnosis of different polyneuropathies (Grimm et al. 2016), muscle changes, on the other hand, may primarily reflect the extent of nerve damage and thus might enable a characterization of disease progression.
Objective: Characterization of muscle ultrasound patterns in hereditary neuropathy patients to establish a biomarker for disease follow-up.
Methods: We analyzed HRUS data in a cohort of 150 patients with different, genetically confirmed hereditary neuropathies and of 71 healthy volunteers and compared them to clinical and paraclinical results.
We measured the muscle echo intensity of the tibial anterior muscle, the gastrocnemius muscle including both the medial and lateral heads, the brachioradial, the dorsal interosseous 1, and the sternocleidomastoid muscle, each on the non-dominant side. For semiquantitative classification, we used the grading scale by Heckmatt (ranging from 1 to 4) and for an objective analysis, we performed a grey scale histogram analysis with the software ImageJ. For comparison with clinical and paraclinical parameters, we summarized all muscle echo intensities (EI sum), using complete muscle data sets only.
We further analyzed whether the muscle changes occurred homogeneously or inhomogeneously.
The study was funded by Pfizer (ASPIRE 2018).
Results: The most frequent mutation was the heterozygous PMP22 duplication accounting for 36.7% (n=55), followed by mutations in MPZ (n=16, 10,7%), TTR (n=14, 9.3%), GJB1 (n=15, 10%), and GLA (n=7, 4.7%). We analyzed the muscle echo intensity (EI) in 651 muscles, examined in 124 patients, and 417 muscles of 71 healthy controls.
Tibial anterior muscles revealed the highest echo intensity in comparison to all other muscles (EI TA mean=98.24±17.52), while the sternocleidomastoid muscle had the lowest EI values (mean =60.08±15.02), suggesting a rather late or lower clinical manifestation of cranial and proximal spinal nerves. When muscle changes occurred, they were more often inhomogeneously distributed (214 vs 104 cases, 333 normal muscles), consistent with a typical neuropathic pattern of damage.
Regarding clinical parameters, the patients’ age and estimated disease duration correlated with the EI sum (age r=0.46 p<0.001; estimated disease duration r=0.35, p=0.0007).
EI values of all patient muscles were significantly higher than in control muscles (p<0.0001 for all muscles, mean EI healthy muscles IOD=40.84±23.85; BR=41.59±14.73; SCM=48.67±12.98; TA=59.24±18.14; GCNM=52.27±16.62; GCNL=55.05±18.3). CMTNS-2 and CMTES showed a positive correlation and MRC sum an inverse correlation with the overall muscle echointensity (CMTNS-2 p<0.0001; CMTES p<0.001; MRC sum p<0.0001).
Discussion: Muscle HRUS is a useful tool for monitoring of disease progression in hereditary polyneuropathies.
PS08.06Administration of AICAR, an AMPK Activator, Prevents and Reverses Diabetic Polyneuropathy (DPN) by Regulating Mitophagy
Russell J1, Chandrasekaran K1, Choi J1, Salimian M1, Hedayat A1
1University Of Maryland, Baltimore, United States
Purpose: Exercise interventions have been shown to slow progression of diabetic neuropathy (DN). AICAR (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside or Acadesine), which importantly mimics the effect of exercise, is an Adenosine Mono Phosphate Kinase (AMPK) activator. AMPK is a master activator of the SIRT1-PGC1a mitochondrial (Mt) biogenesis pathway. This study determined if administration of AICAR rescued mice from DPN induced by a high fat diet (HFD) or by Streptozotocin (STZ) administration.
Methods: WT C57BL6 mice were fed with Control Diet (CD) or HFD for 4 months. Neuropathy was determined by mechanical allodynia (MA), nerve conduction velocity (NCV) at 0, 2 and 4 months and Intraepidermal nerve fiber density (IENFD) at 4 months. AICAR was administered at 2 months (reversal) and at the start of HFD administration (prevention). AICAR was administered subcutaneously (500 mg/kg). In further experiments, AICAR was administered to STZ treated mice with early neuropathy after 2 months. Presence of neuropathy was measured 4 months post STZ.
Results: Administration of AICAR prevented and reversed the development of neuropathy in HFD-fed mice and reversed neuropathy in STZ mice. AICAR levels in blood and neurons were increased in treated mice. Western blot of neuronal protein extracts showed decreased levels of phosphorylated AMPK in HFD and STZ mice. AICAR treatment increased phosphorylation of AMPK, increased the mitochondrial fission factor (Mff), and recruit dynamin-related protein 1 (DRP1) to promote mitochondrial fission. AMPK also phosphorylated autophagy activating kinase 1 (ULK1) at Serine-555 to mitochondria to promote mitophagy.
Conclusions: AICAR mediated AMPK phosphorylation prevented and reversed DPN and is associated with increased mitochondrial turnover that is critical for axonal regeneration. AICAR, unlike metformin, is a direct activator of AMPK, and may be more effective in preventing diabetic complications.
PS09.01Contractile Skeletal Muscle Organoids for Modelling Duchenne Muscular Dystrophy and Evaluating Potential Therapies
Tejedera-Villafranca A1, Ramón-Azcón J1,2, Fernández-Costa J1
1Institute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology (BIST), Barcelona, Spain, 2Institució Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain
Neuromuscular disorders present a high prevalence worldwide and their outcomes are usually life-threatening. They span a range of heterogeneous and rare conditions, as well as complex pathophysiology. This challenges the modelling of neuromuscular disorders, usually performed using traditional cell cultures and animals. Because of this, preclinical research is not efficiently performed, so few curative treatments reach the patients. New approaches, such as human bioengineered organoids, are emerging as new tools to overcome these limitations. These advanced models would allow the study of both pathological processes and the discovery of new potential drugs. Its development is usually reached by culturing cells on a hydrogel scaffold that provides structural support and integrity for the tissue. However, skeletal muscle represents a complex and challenging tissue for its in vitro generation, given the importance of the structure of myofibers for their correct functionality. In this work, we focused on the paediatric neuromuscular disease of Duchenne muscular dystrophy (DMD). Although there are several molecules in drug development for DMD, there is no treatment available for patients to date. Intending to accelerate the testing of anti-DMD drugs, we developed patient-derived 3D functional skeletal muscle organoids. By using a 3D-printed casting mold, we encapsulated patient-derived muscle satellite cells in a fibrin-composite matrix. This platform incorporates two flexible T-shaped pillars that provide continuous tension to the tissue, thus allowing the orientation of the muscle fibers. These posts served as anchoring points and allowed an easy evaluation of the contractibility of the muscles. After seven days of differentiation, DMD organoids expressed mature myogenic markers and showed functional phenotypes as they responded to electrical pulse stimulation (EPS) by contracting. Using this strategy, we identified DMD-related functional phenotypes that are similarly present in patients. Moreover, we analysed the presence of muscle damage markers after the performance of EPS. Using several strategies, we observed that DMD muscle organoids reproduced the loss of myotube integrity that is observed in dystrophinopathies due to the sarcolemmal instability. Finally, the applicability of this dystrophic skeletal muscle model in evaluating therapeutic compounds was explored using anti-DMD drug candidates, such as Ezutromid. Taking all these considerations together, our results show that muscle organoids technology has great potential to be especially valuable in the context of current and future discovery and development of drugs to treat DMD and other neuromuscular disorders.
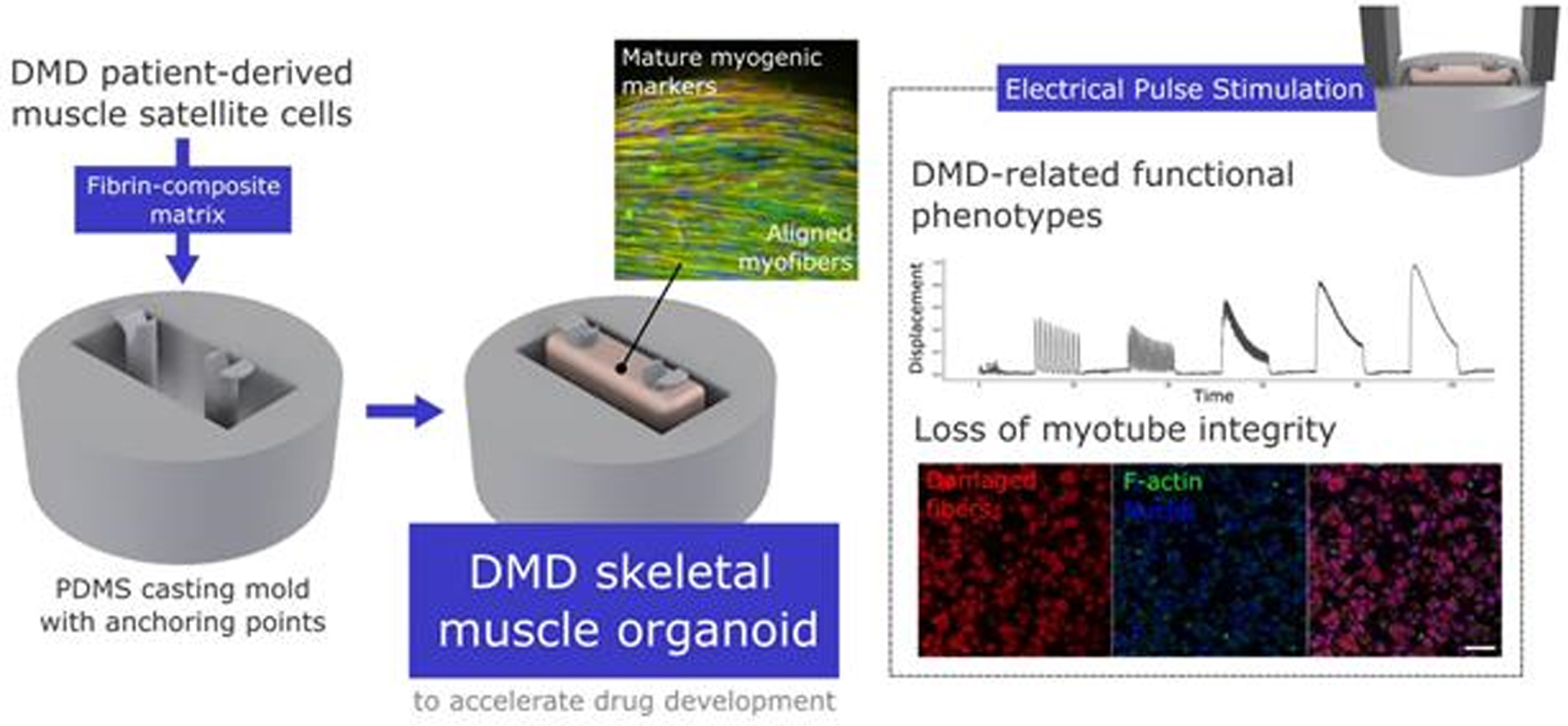
PS09.02Brody Disease: A Novel Potential Therapeutic Approach for This Rare Human Disease
Akyurek E1, Jacinto J2, Bolcato M2, Bianchini E3, Sandonà D3, Gentile A2, Sacchetto R1
1Department of Comparative Biomedicine and Food Science, University of Padova, Legnaro, Italy, 2Department of Veterinary Medical Sciences, University of Bologna, Ozzano Emilia, Italy, 3Department of Biomedical Science, University of Padova, Italy
Brody disease is a “rare” genetic disorder due to defects in SERCA1 gene and it is characterized by exercise-induced muscle stiffness and impairment of relaxation (Brody 1996). Bovine “congenital pseudomyotonia” (PMT) is a genetic muscular disorder very similar to human Brody myopathy for clinical signs. A missense mutation in the ATP2A1 gene (Drögemüller et al 2008), encoding sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1), causes congenital PMT in cattle and Brody disease in humans.
The mouse is the preferred animal model for preclinical evaluation of new therapeutic molecules. Unfortunately, it is not available for Brody disease (due to the prevalence of type II fibers in diaphragm muscle). Clinical symptoms genetic and biochemical findings clearly demonstrated that congenital PMT in Chianina cattle is the real analogue of Brody myopathy (Sacchetto et al 2009).
Our findings show that a SERCA1 mutation in bovine PMT results in a protein that is most likely misfolded, ubiquitinated, and destroyed prematurely by the ubiquitin-proteasome system, albeit retaining its catalytic capabilities (Bianchini et al. 2014).
The treatment with proteasome inhibitors restores SERCA levels and Ca2+ homeostasis in a cellular model and in muscle fibres from PMT affected animals (ex vivo experiments) (collected in conformance with the institutional guidelines for the care and use of animals).
At present, neither specific therapy nor mouse model for Brody myopathy exists.
However, a novel pharmacological approach based on the use of protein folding correctors known as CFTR (Cystic Fibrosis Transmembrane Regulator) explored in Cystic Fibrosis has recently been created and demonstrated in vitro. In a HEK293 cell model, treatment with CFTR correctors restored the expression level of mutated SERCA1.
These data have been confirmed in vivo by local treatments of bovine PMT muscle with the most effective in vitro CFTR corrector. (Pharmacological treatments and biopsies were authorized by the Italian Ministry of Health-authorization ID 1251/2021.) Besides, treatments with the same CFTR corrector on SERCA1 mutated zebrafish line Accordion, are ongoing.
Brody, I. A. (1996) Muscle contracture induced by exercise: a syndrome attributable to decreased relaxing factor. New Eng. J. Med. 281, 187-192.
Drögemüller C., Drögemüller M., Leeb T., Mascarello F., Testoni S., Rossi M., Gentile A., Damiani E., Sacchetto R. (2008) Identification of a missense mutation in the bovine ATP2A1 gene in congenital pseudomyotonia of Chianina cattle: An animal model of human Brody disease. Genomics 92:482-491
Sacchetto R., Testoni S., Gentile A., Damiani E., Rossi M., Liguori R., Drögemüller C., Mascarello F. (2009) A defective SERCA1 protein is responsible for congenital pseudomyotonia in Chianina cattle. Am. J. Pathol. 174:565-573.
Bianchini E., Testoni S., Gentile A., Calì T., Ottolini D., Villa A., Brini M., Betto R., Mascarello F., Nissen P., Sandonà D., Sacchetto R. (2014) Inhibition of Ubiquitin Proteasome System Rescues the Defective Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA1) Protein Causing Chianina Cattle Pseudomyotonia. J. Biol. Chem. 289:33073-33082.
PS09.03In Vivo Validation of the Efficacy of the CFTR Corrector C17 in Sarcoglycanopathy
Scano M1, Benetollo A1, Nogara L1,2, Bondì M1, Dalla Barba F1, Caccin P1, Carotti M1, Blaauw B1,2, Sacchetto R3, Sandonà D1
1Department of Biomedical Sciences, University of Padova, Padova, Italy, 2Venetian Institute of Molecular Medicine, University of Padova, Padova, Italy, 3Department of Comparative Biomedicine and Food Science, University of Padova, Legnaro, Italy
Sarcoglycanopathies or LGMDR3-6 are rare autosomal recessive disease still incurable, affecting mainly the skeletal muscle of the shoulder and pelvic girdle. The onset of symptoms occurs in childhood, the disease is progressive, very often forcing affected subjects to the wheelchair, even though mild forms present late onset and slow progression. Most of the reported cases are due to missense mutations originating a folding-defective but potentially functional sarcoglycan (SG), which is eliminated by the cells’ quality control system. The consequent loss of function, leads to progressive muscle degeneration. In the recent years, thanks to the elucidation of sarcoglycanopathy molecular mechanism, novel therapeutic strategies have been developed.
With the intent to recover the mutants, avoiding SG-complex disruption, we exploited the use of protein folding correctors belonging to the CFTR modulators family. The effective rescue of different SG-mutants has been proved for a panel of such molecules by using cell models and, importantly, myogenic cells from sarcoglycanopathy patients.
To validate the pharmacological strategy in vivo, there is the need of an organism expressing a SG carrying a missense mutation. However, the available SG-KI mice bearing a pathogenic missense mutation in either sgca or sgcb genes did not develop the expected phenotype on the basis of the equivalent human mutations. The SG-complex properly localized at the sarcolemma and no sign of myopathy developed, even in mice undergoing overload exercise. To bypass this bottleneck, we adopted an alternative approach to generate a valuable model of LGMDR3.
In the background of the sgca-null mouse, we delivered the human α-SG carrying either the V247M or the R98H missense mutation by exploiting the transduction via AAV1. Virus injection, restricted to hind-limbs, was carried out in 1-2-day-old pups to induce tolerance toward the human protein. Transduction with human wild type α-SG sequence, as positive control of humanization, resulted in hind-limb muscles with a nearly healthy phenotype with the SG-complex containing the exogenous and the murine subunits correctly localized at the sarcolemma. Conversely, the transduction of the R98H-α-SG sequence led to a dystrophic muscle with only traces of α-SG at the fiber surface and clear myopathic features at the histological level. Mice with the “mutated humanized” hind-limbs, well mimicking the human pathology, allowed proving the efficacy of C17, the most promising CFTR corrector identified in vitro. Histological and molecular analyses revealed an increase of the SG complex at the sarcolemma and a clear reduction of myopathic signs in comparison to vehicle treated animals. Notably, at the functional level, the muscle force generated by treated animals was nearly identical to that of healthy mice. All C17 treated animals reached the experimental endpoint; they showed no behavioural difference in comparison to vehicle treated mice; they grew at similar rate and no sign of toxicity was observed in liver and kidneys.
The findings collected in vivo, together with in vitro data, are the proof of concept of C17 corrector efficacy in sarcoglycanopathies, opening the way for the preclinical studies of a novel therapeutic intervention for this neglected disease.
PS09.04REN001, PPARd Agonist, Preserves Muscle Strength and Promotes Recovery of Muscle Atrophy After Leg Immobilization
Davies M1, Dorenbaum A2, Wang S3, Mittendorfer B4
1Reneo Pharma Ltd, Sandwich, United Kingdom, 2Reneo Pharmaceuticals Inc, Irvine, United States, 3Shenzhen Xbiome Biotech Co. Ltd, Shenzhen, China, 4Washington University School of Medicine, St Louis, United States
Introduction: REN001 is an oral, once-daily peroxisome proliferator-activated receptor delta (PPARd) agonist being investigated for the treatment of PMM.
Methods: A randomized, investigator and subject blind, placebo-controlled phase 1 study in healthy volunteers was conducted to evaluate the safety of oral REN001 (formerly known as HPP593), 100 mg twice daily, administered during and after limb immobilization. All subjects had the left leg immobilized with a knee brace (30° flexion) and used crutches from Day 1 to Day 14. Changes in muscle strength, gene expression from muscle biopsies, and cross section area (CSA) in the immobilized leg was evaluated. After 14 days of dosing the brace was removed and the subjects continued to take REN001 for an additional 14 days and gradually resumed regular physical activity of their previously immobilized limb.
Results: A total of 24 subjects (all men) were randomized to receive 28 days treatment with REN001 (n=12, mean [SD] age 42 [8.9] years) or placebo (n=12, mean [SD] age 39 [8.2] years).
After removal of the knee brace at Day14, subjects receiving REN001 had significantly less loss of knee strength compared to those receiving placebo (Mean change: REN001 = -5.8 lb, Placebo = -36.2 lb; p=0.01). The primary pharmacodynamic endpoint was change in single knee extension strength at Day 21 from baseline. REN001 treated subjects had a significant increase in knee extension strength compared to placebo (Mean change: REN001 = 32.8 lb, Placebo = 2.7 lb; p<0.001, mixed model repeated measures analysis; p=0.004, with baseline as a covariate). At day 29, change in mean knee extension strength was greater for the REN001 group compared to placebo (Mean change: REN001 = 25.5 lb, Placebo = 13.7 lb; p<0.2.). No differences in CSA were noted.
Muscle biopsies were analyzed for changes in mRNA expression of PPARd-regulated genes involved in mitochondrial biogenesis and function. Compared to placebo, REN001-treated individuals had significant increases (>4-fold) in pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4), angiopoietin-like 4 (ANGPTL4) and solute carrier family 25 member 34 (SLC25A34).
REN001 was safe and well tolerated in this study. No serious adverse events were reported. Most treatment-emergent adverse events (TEAEs) were mild in severity and similar in placebo or REN001 treated subjects. The most common TEAEs were headache, post-procedural hematoma and rash. Mild elevation in CPK, not related to study drug, were observed. All TEAEs resolved with no sequelae.
Conclusions: REN001, 100 mg given orally twice daily for 28 days to healthy volunteers was safe and well tolerated. Compared to placebo, treatment with REN001 increased expression of genes involved in mitochondrial biogenesis and oxidative phosphorylation, prevented muscle wasting and promoted recovery of muscle atrophy after prolonged leg immobilization. This is the first demonstration of the impact of REN001 directly on human muscle and these results offer a rationale to evaluate REN001 in patients with mitochondrial myopathies.
PS09.05Impact of Mechanical Stretch on Nuclear Shape and Chromatin Organization in Skeletal Muscle
Jabre S1,2, Hleihel W1, Coirault C1
1Sorbonne Université, INSERM UMRS_974, Centre for Research in Myology, Paris, France,2 Université de Saint Esprit, Kaslik, Liban
Background. An integrated network of nuclear envelope proteins defines the mechanical properties of the nucleus and regulates cell mechanical signaling in response to mechanical challenges. Whereas the lamina and specifically A-type lamins have been recognized for many years as a major contributor to nuclear stiffness and deformations, chromatin and its histone modification state also contribute to nuclear mechanics independently of A-type lamins. However, how A-type lamins and chromatin-mediated mechanoresponse contribute to mechanical load-mediated adaptation in normal and pathological skeletal muscle remains unknown.
Aims. We first sought to determine how muscle differentiation impacts nuclear characteristics in muscle cell precursors (MuSCs) and myotubes. Then, we investigated the respective roles of nuclear envelope proteins (lamin A/C, SUN1 and SUN2) and chromatin compaction on the mechanical load-mediated nuclear response in myonuclei.
Methods. Immortalized MuSCs and myotubes (72h of differentiation) obtained from patients without neuro-muscular disorders were analyzed with different experimental setups to modulate chromatin compaction, nuclear envelope protein expression and mechanical stretch. Cells were treated with Trichostatin A (TCA0.1µM for 48h), a histone deacetylase inhibitor known to decrease chromatin compaction, or silenced for LMNA, SUN1 or SUN2 expression with silencing mRNA strategies. Cyclic stretch (10%, 4hours) was performed in myotubes. Nuclear characteristics were assessed on nuclear volume and nuclear sphericity. Histone post-translational modifications including chromatin repression markers (H3K27me3 and H3K9me3) and chromatin active marker (H3K4me3) were analyzed by immunofluorescence.
Results. Muscle cell differentiation into myotubes was associated with a significant elongation of nuclear long axis and a decrease in nuclear volume (each p<0.05). In myotubes, the nuclear volume was significantly higher after TCA treatment and after SUN2 mRNA silencing. Additionally, siLMNA induced important deformations of the myonucleus shape and increased nuclear thickness but did not significantly modify the nuclear volume. Cyclic stretch increased the nuclear volume in untreated and TCA-treated myotubes, whereas silencing of LMNA, SUN1 or SUN2 abolished the stretch-induced nuclear volume response.
The relative intensity of H3K27me3 labelling was significantly lower in myotubes compared with MuSCs whereas H3K9me3 and H3K4me3 intensities were higher in myotubes compared with MuSCs, thereby showing that myogenic differentiation is modulating the accessibility of the transcriptional machinery. In stretched and unstreched myotubes, the intensity of the H3K27me3 and H3K9me3methylation markers was significantly modified by treatment with siLMNA, siSUN1 and siSUN2 compared to controls. This demonstrates that proteins of the nuclear envelope are required for proper response to mechanical stress.
Conclusion. Overall, our study highlights crucial changes in nuclear shape and histone post-translational markers during muscle differentiation and upon mechanical challenge. Moreover, our results uncover that the nuclear mechano-response is tightly regulated by nuclear envelope proteins in skeletal muscle.
PS09.06Pilot project of Home Mechanical Ventilation in Ukrainian Patients with Duchenne Muscular Dystrophy
Toussaint M1, Tsarenko A
1Ulb Erasme University Hospital, Brussels, Bel, Braine L’alleud, Belgium
Background. The mean survival of DMD boys affected by Duchenne Muscular Dystrophy (DMD) is estimated between 18-21 years of age.[1] Fortunately, HMV prolongs survival in this patient group until ± 35 years of age. [2,3] There is increasing number of programs of HMV across the World. So far, the Ukrainian Parliament has not approved a State Program for the care of DMD patients. We conducted a pilot project of Home Mechanical Ventilation in Ukrainian patients with Duchenne Muscular Dystrophy
Methods. Children with DMD were invited to Kirovograd Regional Children’s Clinical Hospital, Kropyvnytskyi, Ukraine, for 5-days training with noninvasive ventilation (NIV). Donated equipment comprised second-hand Covidien PB560 ventilators from Belgium. Due to the absence of Carbon Dioxide Pressure (PCO2) and Pulse Oximetry (SpO2) monitoring, indications of HMV included: sleep-related symptoms, restrictive lung function test, loss of ambulation for more than 1 year, or age higher than 17 years. Master Class lectures on HMV were conducted for Ukrainian doctors in conjunction with patient training.
Results. Twelve and fifty Ukrainian physicians took part in face-to-face and online Master Classes, respectively. Simultaneously, 8 Duchenne inpatients, mean age 15.4 (SD: 1.8) years and body mass index 25.8 (SD: 4.0), were included in the study. All patients chose nasal masks and volume-pressure assisted control mode. After 6 weeks, one patient stopped HMV, two others used HMV partially during sleep, and 5/8 used nocturnal HMV increasingly with few complaints. Follow-up via phone call was organized after hospitalization. Results are reported in table 1.
Conclusions. This study reports the first experience of implementation of HMV in Ukraine. NIV is feasible in DMD inpatients in Ukraine. In the short term, the Ukrainian Parliament should recognize official Centers for HMV, and, primarily, define the conditions for the reimbursement of equipment for HMV and ongoing maintenance. In the future, distributors of equipment should support these specialized Centers for HMV by delivering: NIV and adjunct devices for monitoring PCO2/ SpO2, in addition to cough augmentation apparatus.
References
1. Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002 Dec;12(10):926-9. doi: 10.1016/s0960-8966(02)00140-2.
2. Ishikawa Y, Miura T, Ishikawa Y, Aoyagi T, Ogata H, Hamada S, Minami R. Duchenne muscular dystrophy: survival by cardio-respiratory interventions. Neuromuscul Disord. 2011 Jan;21(1):47-51. doi: 10.1016/j.nmd.2010.09.006
3. Kohler M, Clarenbach CF, Bahler C, Brack T, Russi EW, Bloch KE. Disability and survival in Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2009 Mar;80(3):320-5. doi: 10.1136/jnnp.2007.141721.
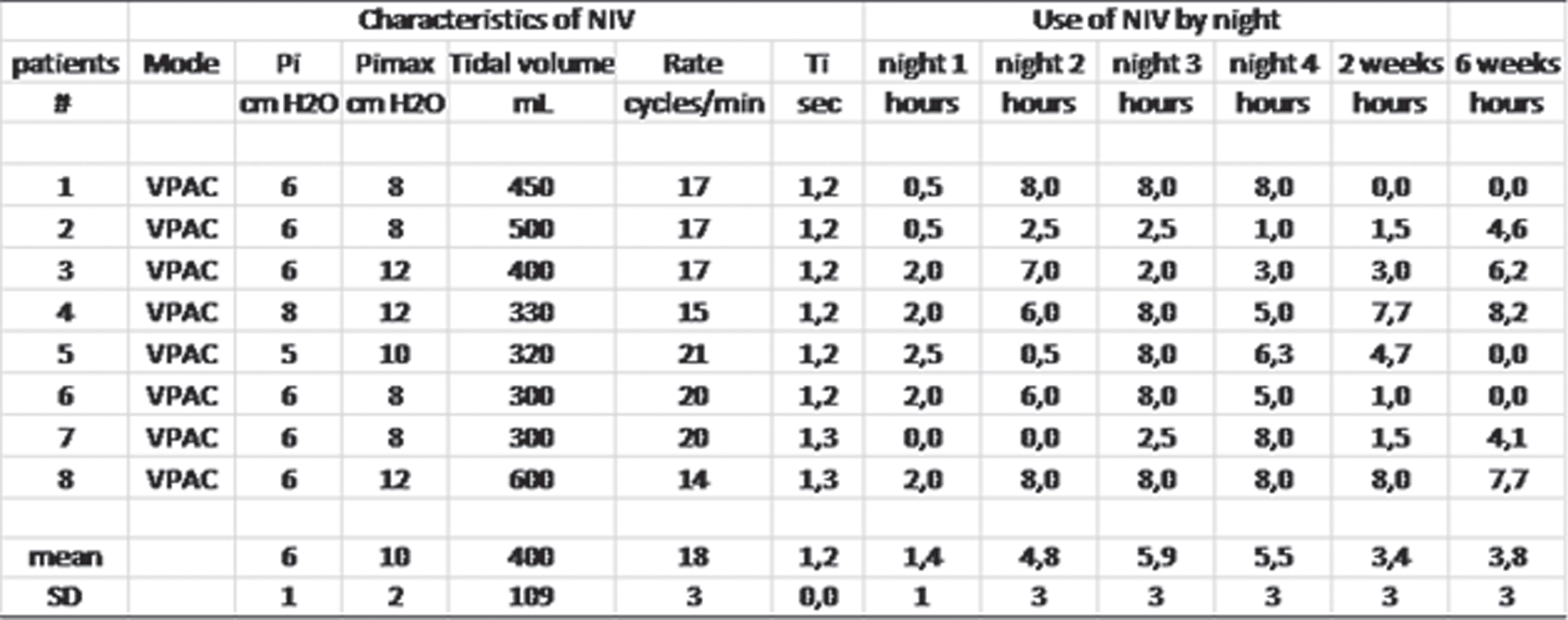
ePosters
eP01.01.01Genetic, Proteomic and Morphological Characterisation of Two Children With Novel Nonsense Mutations of BVES (POPDC1)
Schänzer A1, Gangfuß A2, Hentschel A3, Heil L4, Gonzales M5, Schönecker A6, Depienne C7, Nishimura A1, Kohlschmidt N8, Sickmann A3, Schara-Schmidt U2, Fürst D4, van der Ven P4, Hahn A9, Roos A1,10
1Institute of Neuropathology, Justus Liebig University, Giessen, Giessen, Germany, 2Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Essen, Germany, 3Leibniz-Institut für Analytische Wissenschaften – ISAS – e.V. Dortmund, Germany, Dortmund, Germany, 4Institute for Cell Biology, Department of Molecular Cell, University of Bonn, Bonn, Germany, 5Pediatric Heart Center, Justus Liebig University Giessen, Giessen, Germany, 6University Hospital Essen, University Duisburg-Essen, Department of Pediatric Cardiology, Essen, Germany, 7Institute of Human Genetics, University Hospital Essen, University of Duisburg-Essen, Essen, Germany, 8Institute of Clinical Genetics and Tumour Genetics, Bonn, Bonn, Germany, 9Department of Child Neurology, Justus Liebig University, Giessen, Germany, Giessen, Germany, 10Children’s Hospital of Eastern Ontario Research Institute; Division of Neurology, Department of Medicine, The Ottawa Hospital; and Brain and Mind Research Institute, University of Ottawa , Ottawa, Canada
Background: Popeye domain containing protein 1 (POPDC1) is a highly conserved transmembrane protein essential for striated muscle function and homeostasis. Recessive pathogenic variants in the gene encoding POPDC1 (BVES, Blood vessel epicardial substance) are causative for an ultra-rare limb-girdle muscular dystrophy (LGMDR25), associated with cardiac arrhythmia and myopathy. Only ten patients have been described in literature so far.
Methods: We describe the phenotype of four affected children of two consanguineous families, each with a novel variant in BVES (c.457C>T;p.Gln153Ter and c.578T>G;p.Ile193Ser). Detailed analyses including immunofluorescence and electron microscopy and proteomic profiling of muscle biopsies from an affected patient of each family were performed.
Results: Creatine kinase values (300-16.000 U/l) and cardiac involvement were variable symptoms in affected family members. Detailed morphological analysis of skeletal muscle showed a myopathy with reduced expression of POPDC1 accompanied by altered sarcolemmal dysferlin and XinA abundance. At the electron microscopic level, the muscle fiber sarcolemm was focally disrupted. The proteomic signature showed statistically significant dysregulation of 191 proteins of which 173 were increased and 18 were decreased. Focusing on proteins presenting with decreased abundance in BVES-patient derived muscle, GO-term analysis of affected biological processes revealed - among others - perturbation of muscle fibril assembly mo- filament sliding and contraction as well as transition between fast and slow fibers. Decreased proteins are also highly indicative for a vulnerability of myocardium by indicating altered regulation of the force of heart contraction, ventricular cardiac muscle tissue morphogenesis and development, regulation of the heart rate, as well as cardiac hypertrophy in response to stress.
Conclusion: We identified two novel recessive pathogenic BVES variants associated with early onset muscle pathology complicated by cardiac conduction defects. Biochemical studies provided insights into the molecular etiology of the muscular phenotype and might even explain the cardiac manifestation upon loss of functional POPDC1 in muscle cells. We conclude that in young patients with cardiac conduction disturbance and elevated CK, a neuromuscular disorder should be ruled out by genetic analysis for providing a good therapy strategy.
eP01.01.02Cardiac Analysis Reveals Morphological Alterations in an Intermediate Mouse Model of Spinal Muscular Atrophy
Nair N1, Gray G2, Murray L1
1Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, United Kingdom, 2Centre for Cardiovascular Science, University of Edinburgh, Edinburgh, United Kingdom
Traditionally considered a motor neuron disease, spinal muscular atrophy (SMA) is now known as a multi-system disorder. Effective new treatments are improving life expectancy, however since they are frequently targeted to the central nervous system, defects in non-neuronal tissues like the heart could become prominent. Existing literature on severe mouse models of SMA reports cardiac defects including thinning of left ventricular wall (LV) and interventricular septum (IVS), fibrosis and increased apoptosis. However, since morphological measurements are frequently not presented relative to body size, it is unclear how the decreased size of SMA mice confound these results.
The aim of our study was to use a milder mouse model of SMA to explore the underlying basis of cardiac pathology. We have therefore examined the cardiac structure and function at P18 in the Smn2B/- mice. Initial histological analysis revealed thinning of LV (p≤0.01) and IVS (p≤0.05) in the Smn2B/- mice, which is consistent with the literature. However, when these were normalised to the heart size, there was a relative increase in the thickness of both LV and IVS (p≤0.01 each) when compared to controls, which was confirmed by in vivo high-resolution LV echocardiography (p≤0.01). Systolic and diastolic cardiac function were preserved; however, there were changes (p≤0.05) in longitudinal strain that may indicate subtle cardiac stress in the Smn2B/- mice.
Together these data suggest that cardiac function is preserved in the Smn2B/- mice, but at the cost of cardiac remodelling which can increase the risk of subsequent heart failure. Such defects may underlie and precede the more significant cardiac defects described in severe mouse models of SMA. This work also highlights the importance of using a normalisation factor while measuring the cardiac parameters in a multi-systemic disorder like SMA, where normalisation reversed many of the trends observed in this mouse model.
eP01.01.03Respiratory Follow-Up In Children With Spinal Muscular Atrophy - A Descriptive Study
Pereira I1, Saianda A2, Coelho J3, Moreno T3, Ferreira R2
1Department Of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisboa, Portugal, 2Pulmonology Unit, Department of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisboa, Portugal, 3Neurology Unit, Department of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisboa, Portugal
Introduction and Aim: Respiratory complications such as infections and respiratory failure are the main cause of morbidity and mortality in patients affected by Spinal Muscular Atrophy (SMA). The multidisciplinary approach should include respiratory evaluation and therapy - ventilatory support and secretion management as needed - as it is part of the standards of care of SMA.
The aim of this study was to characterize a group of children with SMA, regarding respiratory evaluation and monitoring (including sleep and respiratory function tests) and respiratory support. Data on its basic features and specific therapy received (nusinersen, risdiplan or onasemnogene abeparvovec) is also detailed.
Methods: Clinical data was collected from the clinical electronic records of 15 children with SMA currently (January 2022) being followed at the Pediatric Pulmonology Unit in a tertiary hospital. Descriptive statistics was done.
Results : 7(46.7%) patients have SMA type 1 diagnosis and 8(53.3%) SMA type 2, median age 33(12;40)mths and 141,5(68;203)mths, respectively. All patients received specific treatment. The median age at diagnosis was 4(1;12)mths for SMA 1 and 17(10;72)mths for SMA 2. Median age at start of specific treatment, nusinersen, risdiplan or onasemnogene abeparvovec, was 6(3;14)mths for SMA 1 and 105(22;198)mths for SMA 2.
14(93%) patients are on non-invasive ventilation (NIV), 100% in type 1 and 88% in type 2 SMA patients. The median age at which NIV was started was 4.9(2;9)mths in patients with SMA 1 and 60(18;148)months in SMA 2. The median number of hours on NIV is 14(±5)h in SMA 1 and 8,68(±1,65)h in SMA 2. 80% of all SMA patients use cough assist. No SMA type 2 patients and 1 SMA type 1 needed hospital admissions for respiratory reason in the last year.
Respiratory evaluation and monitoring using oximetry, capnography and polysomnography was undertaken in 9(60%) patients in the last year and respiratory function was assessed in 7(88%) of patients with SMA 2.
Discussion: Children with SMA are a complex group of patients, requiring a comprehensive respiratory follow-up and support. NIV is frequently required, as well as cough aids. The introduction of novel therapies, modifying the prognosis, is changing the disease paradigm, with longer survival and less dependence on NIV.
eP01.01.04Worldwide Prevalence of Home Mechanical Ventilation in Neuromuscular Disorders
Toussaint M1
1Erasme University Hospital, Brussels, Bel, Brussels, Belgium
Background: Hypercapnic respiratory failure due to alveolar hypoventilation is a common complication of severe neuromuscular disorders (NMD). In this patient subgroup, home mechanical ventilation (HMV) improves quality of life and survival. HMV is often offered via noninvasive ventilation (NIV) through a nasal mask.
Methods:
• Search of Medline for publications in English or French from 2005 to 2020.
• Analysis of data from 22 international programs of HMV.
Results: The prevalence of NMD is about 30/100.000 population [1]. The prevalence of HMV in NMD is reported as stable or slowly increasing with years [2, 3]. Patients with NMD account for 39.5% of all HMV users. The prevalence of NMDs using HMV is calculated at 2.9/100.000 in a general population (= ±10% of all NMD patients). Large variations in the prevalence of HMV in NMD, from 0.2 to 8.3/100.000 population, exist among countries (figure 2).
Depending on the time period of ventilator use per 24 hours, three categories of HMV users can be defined: nocturnal (8/24h), discontinuous (8-16/24h), and continuous (>16/24h, life-support) ventilation. [1] In NMD, nocturnal, discontinuous, and continuous HMV is likely to concern about 60, 20 and 20% of patients using HMV.
The following equipment for HMV can be used according to the 3 categories.
• Nocturnal HMV: bi-level devices can be sufficient.
• Discontinuous HMV: a battery is desirable but not mandatory.
• Continuous HMV: two life-support devices with disconnection alarms and batteries are recommended. If no internal battery is available, external supply of electricity is mandatory.
Large differences in the prevalence of HMV are reported across regions within the same country, despite a homogeneous national health care system and similar indications for treatment [4].
There is a positive relationship between national prevalence of HMV and the national GDP per capita. Despite this relationship, HMV recently started to develop in middle and low-income countries. Today, 13 new middle, and low-income countries cheerfully develop HMV programs: Thailand, China, Brazil, Iran, South Africa, Tunisia, Malaysia, Turkey, Argentina, Serbia, India, Pakistan, and Ukraine.
Key messages
• Almost 10% of NMD patients (3/100.000 population) would use Home Mechanical ventilation (HMV), of which 3/5 use it during sleep at night (8/24h)
• The prevalence of HMV is associated with the Gross Domestic Product per capita.
• Large variations in the prevalence of HMV in NMD, from 0.2 to 8.3/100.000 in a general population, exist among countries
References
1. HAS 2012, Ventilation mécanique au domicile. ISBN 978-2-11-128563-7. www.has-sante.fr https://www.has-sante.fr/upload/docs/application/pdf/2013-01/rapport_ventilation_cnedimts_2013.pdf
2. Gajdoš O, Rožánek M, Donin G, et al. Cost-Utility Analysis of Home Mechanical Ventilation in Patients with Amyotrophic Lateral Sclerosis. Healthcare (Basel) 2021;9:142. doi: 10.3390/healthcare9020142.
3. Rose L, McKim D, Leasa D, et al. Trends in incidence, prevalence, and mortality of neuromuscular disease in Ontario, Canada: A population-based retrospective cohort study (2003-2014). PLoS One 2019;14:e0210574. doi: 10.1371/journal.pone.0210574.
4. Laub M, Berg S, Midgren B, et al. Home mechanical ventilation in Sweden--inequalities within a homogenous health care system. Respir Med 2004;98:38-42. doi: 10.1016/j.rmed.2003.08.005.
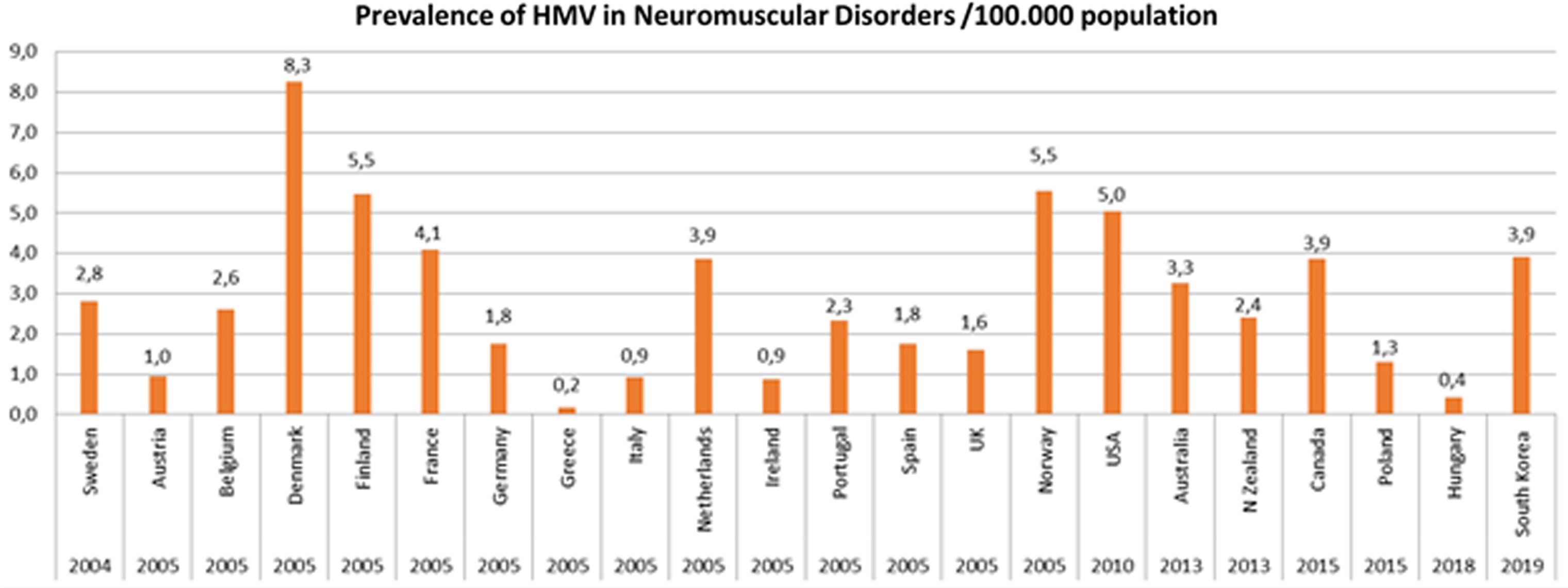
eP01.01.05De Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia
De Winter J1,2,3, Van de Vondel L1,2, Beijer D1,2,4, Coarelli G5, Wayand M6,7, Palvadeau R8, Pauly M9,10, Klein K11, Rautenberg M11, Guillot-Noël L5, Deconinck T12, Vural A13, Ertan S13, Dogu O14, Uysal H15, Brankovic V16, Herzog R9, Brice A5, Durr A5, Klebe S17, Stock F18, Bischoff A19, Rattay T6,7, Sobrido M20,21, De Michele G22, De Jonghe P2,3, Klopstock T19,23,24, Lohmann K10, Zanni G25, M. Santorelli F26, Timmerman V2,27, Haack T11,28, Züchner S4, PREPARE consortium, Schüle R6,7, Stevanin G5,29, Synofzik M6,7, Basak A8, Baets J1,2,3
1Translational Neurosciences, Faculty of Medicine and Health Sciences, University Of Antwerp, Wilrijk, Belgium, 2Laboratory of Neuromuscular Pathology, Institute Born-Bunge, University of Antwerp, Wilrijk, Belgium, 3Neuromuscular Reference Centre, Department of Neurology, Antwerp University Hospital, Edegem, Belgium, 4Dr John T. Macdonald Foundation Department of Human Genetics, John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, USA, 5Sorbonne University, ICM-Paris Brain Institute, INSERM, CNRS, APHP, Pitié Salpêtrière Hospital, Paris, France, 6Department of Neurodegenerative Diseases, Hertie Institute for Clinical Brain Research (HIH), Center of Neurology, University of Tübingen, Tübingen, Germany, 7German Center for Neurodegenerative Diseases (DZNE), University of Tübingen, Tübingen, Germany, 8Koc University, School of Medicine, Suna and Inan Kirac Foundation, Istanbul, Turkey, 9Department of Neurology, University Hospital Schleswig Holstein, Lübeck, Germany, 10Institute of Neurogenetics, University of Lübeck, Lübeck, Germany, 11Institute of Medical Genetics and Applied Genomics, University of Tuebingen, Tübingen, Germany, 12Center of Medical Genetics, University of Antwerp and Antwerp University Hospital, Edegem, Belgium, 13Koc University, School of Medicine, Department of Neurology, Istanbul, Turkey, 14Department of Neurology, School of Medicine, Mersin University, Mersin, Turkey, 15Department of Neurology, School of Medicine, Akdeniz University, Antalya, Turkey, 16Clinic for Child Neurology and Psychiatry, University of Belgrade, Belgrade, Serbia, 17Department of Neurology, University Hospital Essen, Essen, Germany, 18Institute of Human Genetics, University Hospital Essen, Essen, Germany, 19Department of Neurology, Friedrich-Baur-Institute, LMU Munich, Munich, Germany, 20Centro de Investigación Biomédica en red de Enfermedades Raras (CIBERER), Santiago de Compostela, Spain, 21Neurogenetics Research Group, Instituto de Investigación Sanitaria (IDIS), Hospital Clínico Universitario, SERGAS, Santiago de Compostela, Spain, 22Department of Neurosciences and Reproductive and Odontostomatological Sciences, Federico II Univeristy, Naples, Italy, 23German Center for Neurodegenerative Diseases (DZNE), Munich, Germany, 24Munich Cluster for Systems Neurology (SyNergy), Munich, Germany, 25Unit of Neuromuscular and Neurodegenerative Disorders, Department Neurosciences, Bambino Gesù Children’s Hospital, Rome, Italy, 26Molecular Medicine, IRCCS Fondazione Stella Maris, Pisa, Italy, 27Peripheral Neuropathy Research Group, Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium, 28Centre for Rare Diseases, University of Tübingen, Tübingen, Germany, 29Paris Sciences Lettres Research University, Ecole Pratique des Hautes Etudes, Paris, France
Background: Pathogenic variants in SPTAN1 have been linked to a remarkably broad phenotypical spectrum. Clinical presentations include epileptic syndromes, intellectual disability, and hereditary motor neuropathy.
Objectives: We investigated the role of SPTAN1 variants in rare neurological disorders such as ataxia and spastic paraplegia.
Methods: We screened 10,000 NGS datasets across two international consortia and one local database, indicative of the level of international collaboration currently required to identify genes causative for rare disease. We performed in silico modelling of the identified SPTAN1 variants.
Results: We describe 22 patients from 14 families with five novel SPTAN1 variants. Of six patients with cerebellar ataxia four carry a de novo SPTAN1 variant, two showed a sporadic inheritance. In this group one variant (p.Lys2083del) is recurrent in four patients. Two patients have novel de novo missense mutations (p.Arg1098Cys, p.Arg1624Cys) associated with cerebellar ataxia, in one patient accompanied by intellectual disability and epilepsy. We furthermore report a recurrent missense mutation (p.Arg19Trp) in 15 patients with spastic paraplegia from seven families with a dominant inheritance pattern in four and a de novo origin in one case. One more patient carrying a de novo missense mutation (p.Gln2205Pro) has a complex spastic ataxic phenotype. Through protein modelling we show that mutated amino acids are located at crucial interlinking positions, interconnecting the three-helix bundle of a spectrin repeat.
Conclusions: We show that SPTAN1 is a relevant candidate gene for ataxia and spastic paraplegia. We suggest that for the mutations identified in this study, disruption of the interlinking of spectrin helices could be a key feature of the pathomechanism.
eP01.01.06Use of NGS for Diagnosis of Asymptomatic Hyperckemia in Childhood
Marti P1, Pitarch I2, Muelas N3, Azorin I1, Vilchez J1
1IIS La Fe, Neuromuscular, Valencia, Spain, 2Hospital UiP La Fe, Neuropediatric., Valencia, España, 3Hospital UiP La Fe, Neurology., Valencia, España
Background/aims: Studies of isolated hyperCKemia in pediatric population, particularly applying NGS sequencing are scarce, except for a few systematic new born screenings for DMD. Herein we report data of a study performed in a single Neuromuscular Centre.
Methods: We studied 63 children (0-18 years old) with pauci-symptomatic or asymptomatic hyperCKemia in which rearrangement of the DMD gene was excluded by MLPA. For this purpose, it was designed an extensive NGS Illumina panel with 273 genes involved in neuromuscular diseases.
Results: We obtained a surprisingly high rate of diagnoses: 56%. Fifty-seven percent of the diagnosed group involved mutations in only 5 genes and the remaining cases spanned a many groups of less frequent occurrence genes. The diagnose rate and mutation distributions changes in different ranges of age. Immunodetection analysis in muscle biopsy was performed to corroborate loss-of-function variants in 15 cases. Of patients diagnosed 13% had a CK min below 500 IU/L. Of the point mutations detected in DMD gene 60% of patients shown primary stop codon mutations.
Conclusion: This study proves that the use of NGS can radically change the approach of the HCK study since it clearly increases the diagnostic yield, is non-invasive and less expensive. However, muscle biopsy is still necessary to confirm uncertain variants.
eP01.01.07Prevalence of Titinopathy in India
Dhall A1, Dhiman N1, Jassal B1, Faruq M2, Shamim U2, Bhatia R1, Venugopalan Y V1, Suri V1, Sharma M1
1All India Institute Of Medical Sciences(aiims), New Delhi, India, 2CSIR-Institute of Genomics and Integrative Biology, New Delhi, India
Background: Limb-girdle muscular dystrophies (LGMDs) are a large group of heterogeneous genetic diseases characterised by proximal muscle weakness of limb girdles with variable clinical course. According to the 229th ENMC workshop in Naarden in 2017, 38 different subtypes of LGMDs were identified. The prevalence of LGMD is estimated to range from one in 14,500 to one in 123,000. The age of onset can vary greatly. The relative occurrence of each subtype of LGMD varies among different ethnic populations, but worldwide LGMD2G, 2H and 2J are extremely rare.
In Spain, Italy, England, Turkey, Russia, China, Brazil and Australia, the most common LGMD is calpainopathy (LGMD2A). LGMD2I is more frequent than other forms in the Scandinavian Peninsula. However, dysferlinopathy is the most frequent in US, Japan and Mexico. In India, GNE myopathy has the highest incidence, whereas in Finland, anoctaminopathy ranked the first (25%) amongst other forms.
Titinopathy is caused by pathogenic mutations in TTN gene located on the short arm of chromosome 2 (q31.2). It has the highest number of exons (364) and the longest coding sequence (>100kb) of any human gene. TTN gene encodes for the skeletal protein titin that spans from the Z-disk to the M-band. Phenotypic variants includes several muscle disorders, including cardiomyopathy, recessive congenital myopathies and limb-girdle muscular dystrophy (LGMD) 2J or LGMDR10.
Affected individuals have severe progressive proximal muscle weakness. Eventually the distal muscles become involved and some individuals become wheelchair bound.
Materials and Methods: 96 patients (2018-19) suspected of LGMD from non-related families underwent thorough phenotypic characterization followed by muscle histopathological analysis. These cases were subjected to targeted next generation sequencing using a customized panel of 54 genes. Sequencing data was analysed using CLC Genomics by Qiagen for generating variants
Results: 25 patients (16 male and 9 female) were diagnosed with Titinopathy. Mean age of diagnosis was 23.9 years with onset as early as 6 months. CPK levels ranged from 30 to 18,660U/L. EMG was myopathic in 24% (6 out of 25) patients. Histomorphological analysis showed predominant dystrophic changes. Sequencing results show 10 novel and 32 reported mutations including exonic (frameshift and non-synonymous SNVs) and splice variants.
Conclusion: In our ongoing study Titinopathy is found to be the most common LGMD in Indian cohort. NGS sequencing has enabled a thorough and rapid investigation of genes as huge as TTN leading to identification of a large number of variants. A high proportion of mutations were identified in the TTN gene thus broadening the genetic spectrum of Titinopathy. Some mutations were unique in the Indian population which signifies that the distribution of TTN mutations is associated with ethnic background. Also, identifying TTN mutations is important as unlike most congenital myopathies, there is a significant risk of cardiomyopathy. No genotype-phenotype correlation existed suggesting that the clinical phenotype is determined not only by TTN variants but also likely through a complex interplay of environmental, epigenetic, and genetic factors.
Keywords: LGMD (Limb girdle muscular dystrophy), Titinopathy, Immunohistochemistry, targeted sequencing
eP01.01.08Optimisation of a Cell-Based Strategy for Rapid Evaluation of Compounds in Myotonic Dystrophy Type I
López-Martínez A1, Soblechero-Martín P1,2, Catalli C1,3, Jauregi-Barrutia A1,4, Nogales-Gadea G1,5, Kapetanovic-Garcia S1,5, Arechavala-Gomeza V1,6
1Neuromuscular Disorders, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain, 2Clinical Laboratory Service, Basurto University Hospital, Bilbao-Basurto Integrated Health Organisation, Osakidetza Basque Health Service, Bilbao, Spain, 3Department of Genetics, Cruces University Hospital, Osakidetza Basque Health Service, Barakaldo, Spain, 4Department of Neurology, Cruces University Hospital, Osakidetza Basque Health Service, Barakaldo, Spain, 5Neuromuscular and Neuropediatric Research Group, Institut d’Investigació en Ciències de la Salut Germans Trias i Pujol (IGTP), Campus Can Ruti, Universitat Autònoma de Barcelona, Badalona, Spain, 6ALS and Neuromuscular Unit – Department of Neurology, Bilbao-Basurto Integrated Health Organisation, Osakidetza Basque Health Service, Bilbao, Spain, 7Ikerbasque, Basque Foundation for Science, Bilbao, Spain
Introduction: Recently, many compounds derived from in silico and in vitro high-throughput drug screenings have been proposed to have a therapeutic effect on DM1, but robust in vitro validation methods are not available to the DM1 community. Gold-standard techniques for the evaluation of drug candidates in DM1 are based in visualization of foci in the cell nuclei, evaluation of altered splicing patterns in DM1 tissues, and assessment by western blotting of gross changes in protein expression. These are laborious procedures with poor reproducibility and little consensus on their methodological protocols. An in-depth characterization of DM1 patient-derived cultures is needed to accurately evaluate new drug DM1 candidate therapies.
Objectives: The objective of this project is to develop a strategy to characterize patient-derived cultures both at protein and RNA level, for rapid evaluation of new compounds in DM1.
Methods: Myoblots and fibroblots are assays for protein quantification in microplates based in the In-Cell Western method that we have adapted to the study of DM1-relevant proteins in myoblast and fibroblast cultures, respectively. They allow studying the expression of several proteins with many biological and technical replicates per experiment, resulting in high reproducibility and a throughput similar to ELISA applications. In addition, we have optimised a digital droplet PCR (ddPCR) protocol for the quantification of the mRNA expression of the same proteins to assess the consequences of MBNL1 sequestration in the DMPK mRNA repeats at post-transcriptional level.
Results: Optimisation of myoblots and fibroblots allowed us to accurately quantify MBNL1, DMPK and CELF1 in immortalized differentiated myoblasts cultures and primary human fibroblasts cultures, in addition to CLCN1 and MyHC in myoblasts. Statistically significant differences in protein expression among DM1 and CTRL groups were found. We further validated our approach by treating our cultures with several small molecules that have been proposed as drug candidates for DM1 and quantified their effects on these key proteins’ expression.
Conclusion: A combination of optimised myoblots/fibroblots and ddPCR analysis of DM1 cultures, allows a highly reproducible and less laborious characterization of DM1 patient-derived cultures suitable for evaluation of potentially therapeutic compounds in DM1.
eP01.01.09Spectrum of Muscular Dystrophies in India
Sharma M1, Dhall A1, Faruq M2, Pathak D1,3, Shamim U2, Dhiman N1, Jassal B1, Suri V1, Bhatia R1, Venugopalan Y V1, Gulati S1, Chakrabarty B1
1All India Institute Of Medical Sciences(aiims),new Delhi,india, New Delhi, India, 2CSIR-Institute of Genomics and Integrative Biology, New Delhi, India, 3National Institutes of Health, Bethesda, United States of America
Background: Muscular dystrophy (MD) is a heterogenous group of inherited diseases characterised by muscle degenaration, fibrosis and adipose tissue infiltration leading to weakness of muscles over time. This is due to the lack of various proteins, which are necessary for normal muscle functions. The absence of these proteins can cause problems with walking, swallowing, and muscle coordination. There are more than 30 different types of muscular dystrophies, which vary in symptoms and severity. Among them DMD/BMD and LGMD are most frequently reported subtypes.
Limb-girdle muscular dystrophies (LGMD), a group of disorders, are a //genetically inherited conditions that primarily affects skeletal muscle leading to progressive, predominantly proximal muscle weakness at presentation caused by a loss of muscle fibres. To be considered a form of limb girdle muscular dystrophy the condition must be described in at least two unrelated families with affected individuals achieving independent walking, must have an elevated serum creatine kinase activity, demonstrate degenerative changes on muscle imaging over the course of the disease, and have dystrophic changes on muscle histology, ultimately leading to end-stage pathology for the most affected muscles.
According to 229th ENMC international workshop (Naarden, 2017) there are more than 30 different genetic subtypes of LGMDs transmitted through autosomal recessive (AR) and, less frequently, autosomal dominant (AD) inheritance .Currently there are 25 AR (LGMD2A-2T) and eight AD genes (LGMD1A-1H) already identified. Affected patients can have a mild disease course remaining ambulant until late in life or have a severe phenotype, clinically very similar to X-linked DMD. Among patients diagnosed with AR LGMD, LGMD2A (Calpainopathy) is the most frequent form.
Materials and Methods: 96 patients (2018-19) suspected of MD from non-related families underwent thorough phenotypic characterization followed by muscle histopathological analysis. These cases were subjected to targeted next generation sequencing using a customized panel of 54 genes. Sequencing data was analysed using CLC Genomics by Qiagen for generating variants
Results: Evaluation ofclinico-pathological results and genetic analysis revealed a spectrum of muscle disorders including uncommon and rare subtypes:titinopathy(26%),Congenital Muscular Dystrophy (13.54%), calapinopathy (12.5%), dysferlinopathy (11.45%), Ulrich (5.2%), GNE myopathy (4.16%), Plectinopathy (3.12%), Sarcoglycanopthy (2.08%),DMD (1.04%),Desminopathy (1.04%) and X-linked autophagy (2.08%).
Age of diagnosis varied from 10 months to 54 years. EMG was myopathic in 17.7% (17/96) of cases.Histomorphological analysis showed predominant dystrophic changes. CPK levels ranged from 30 to 18,660U/L. We identified 33 unique variants that were either pathogenic or VOUS.Majority of variants were exonic including frameshift, stopgain and SNVs. An equal proportion of compound heterozygous and homozygous mutations were observed suggesting the need for carrier-testing for autosomal-recessive disorders.
Conclusion:In our ongoing study Titinopathy is found to be most prevalent LGMD after Congenital Muscular Dystrophy in Indian cohort. Next generation sequencing hasexpanded the clinical spectrum and genetic variability of patients with Muscular Dystrophy. A proportion of unique variants were identified. No genotype-phenotype correlation could be established suggesting the interplay of factors other than just genetics contributing to the cause of disease.
Keywords: LGMD (Limb girdle muscular dystrophy), Immunohistochemistry, targeted sequencing, muscular dystrophies.
eP01.01.10Diagnostic Approach after Whole Exome Sequencing: A Single-center Experience in Neuromuscular Disorders
Chen P1, Hsueh H1,2, Tsai L1, Lee N3, Chao C1, Yang C1
1Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan, 2Department of Anatomy and Cell Biology, National Taiwan University College of Medicine, Taipei, Taiwan, 3Department of Medical Genetics, National Taiwan University Hospital, Taiwan
Background: Whole-exome sequencing (WES) had become one of the powerful diagnostic approaches to identify the pathogenic variants in patients with genetic disorders. To achieve an accurate diagnosis for patients with neuromuscular disorders, physician should interpret the genetic report carefully with the information of clinical manifestations, pathology, and electrophysiological studies. However, the judgment became difficult if the WES yielded equivocal or negative results. Herein, we described our experience to integrating the WES into the diagnostic approach in patients with neuromuscular disorders. We focused on (1) further molecular tests in patients with variants found in WES, and (2) the diagnostic approach for those with negative findings in WES.
Methods: WES was performed on patients with the impression of hereditary neuromuscular disorders. The information of clinical manifestations, neurological examination, nerve conduction study, electromyography, nerve and muscle biopsy, and laboratory tests were collected. Further available molecular tests based on the culprit gene found in WES were done to confirm the pathogenicity. In those with negative findings of WES, details in further examinations or plans were also collected. Based on the above examinations, patients were categorized into four scenarios : (1) definite diagnosis by WES, (2) probable diagnosis by WES, (3) negative findings in WES, but final diagnosis in further work-up, and (4) undetermined etiology by whole-exome sequencing and in further work-up.
Results: In total, 38 patients accepted WES, and the diagnostic yield was 68.4% (26/38). Fifteen patients, who all had myopathy, fulfilled the proposed criteria of definite diagnosis and nine patients (myopathy: 4; neuropathy:5) were categorized into the probable diagnosis group. The diverse culprit genes guided further molecular tests to confirm the pathogenicity. Notably, in the twelve patients with equivocal or negative findings in WES, three patients (25%) had a diagnosis in the further workup: tumor-induced osteomalacia, mitochondrial myopathy with pathogenic variant in mitochondrial DNA (m.3243A>G), and vasculitis-related neuropathy. The etiologies remained undetermined in nine patients after WES and further workup. Re-analysis of WES 1-2 years later, whole-genome sequencing, or specific molecular studies were needed for these patients.
Conclusions: WES facilitates the diagnosis of hereditary neuromuscular disorder. However, it is essential to arrange further molecular studies to correlate the identified variants with the clinical features. For patients who had negative findings from WES, secondary causes and mitochondria disease should be considered. Moreover, observation of treatment response, segregation analysis as well as further molecular evaluation should be beneficial to the final diagnosis.
eP01.01.10Median Nerve Ultrasound in Carpal Tunnel Syndrome
Abida Y1,2, Kacem I1,2, Mrabet S1,2, Gharbi A1,2, Atrous A1,2, Souissi A1,2, Gargouri A1,2, Nasri A1,2, Gouider R1,2
1Department Of Neurology, Lr18sp03, Clinical Investigation Centre Neurosciences And Mental Health, Razi Hospital, Manouba, Tunisia, 2Faculty of Medicine of Tunis, University Tunis EL Manar, Tunis, Tunisia
Introduction: Carpal tunnel syndrome (CTS) is the most common compressive neuropath. Its diagnosis is primarily clinical. Although the electroneuromyography (ENMG) is recognized to be the gold standard for the diagnosis, high resolution ultrasound can be helpful. The aim of our study is to describe ultrasonic features of patients with clinical symptoms of CTS and to compare results to ENMG findings.
Methods: This prospective cross-sectional study involved 61 patients (122 hands) with CTS signs. ENMG was applied to all patients as well as an ultrasonographic evaluation of the median nerve using a transducer with 13 MHZ linear array. The evaluation included the study of the nerve surface at the entry of the carpal tunnel, the echogenicity of the median nerve, the flattening of the nerve and the palmar bulge of the retinaculum of the extensors.
Results: Average age of our patients was 50.95 years. Sex ratio was 0.1. Bilateral involvement was present in 37 patients. Average cross-sectional area of the median nerve was 15.66±7.22 mm2. Average flattening ratio was 2.98±0.83. Palmar bulge of retinaculum was present in 42.3% of cases. ENMG was pathological in 95%. Severity of the CTS did not correlate with the cross-sectional area (p=0.58). Overall sensitivity was 73.77% for ultrasound and 80.38% for ENMG.
Conclusion: In our study, ultrasound was a simple and a reliable method in exploring CTS with a good sensitivity. Although it allowed morphological study of the nerve, it couldn’t replace the ENMG which constitute a gold standard examination particularly regarding prognosis.
eP01.02.01Clinical Features and the Novel p.K357E Mutation in a Cohort of Patients With mfn2-Related Neuropathy
Abati E1,2, Manini A1, Velardo D2, Del Bo R2, Napoli L3, Rizzo F1,2, Moggio M3, Bresolin N1,2, Bellone E4, Bassi M5, D’Angelo M6, Comi G1,2, Corti S1,2
1Department of Pathophysiology and Transplantation (DEPT), University Of Milan, Milan, Italy, 2Neurology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy, Milano, Italy, 3Neuromuscular and Rare Diseases Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, MI, 4Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (Dinogmi) – Medical Genetics, University of Genoa, Genoa, Italy, 5Laboratory of Molecular Biology, Scientific Institute IRCCS E. Medea, Bosisio Parini, Italy, 6Neuromuscular Disorder Unit, Scientific Institute IRCCS E. Medea, Bosisio Parini, Italy
Introduction: Charcot-Marie-Tooth disease type2A (CMT2A) is a rare inherited axonal neuropathy caused by mutations in MFN2 gene, which encodes Mitofusin 2, a transmembrane protein of the outer mitochondrial membrane. The aim of this study is to further expand our knowledge about the genetic background and clinical presentation of CMT2A.
Methods: We performed a cross-sectional analysis on thirteen patients carrying mutations in MFN2, from ten families, describing their clinical and genetic characteristics.
Results: Evaluated patients presented a variable age of onset and a wide phenotypic spectrum, with most patients presenting a severe phenotype. Overall, patients with childhood-onset CMT2A (1-20 years of age) presented a more severe phenotype comparing with patients with adult-onset CMT2A (>20 years of age). Clinical sensory involvement was reported only in 46% of patients and did not seem to be associated to specific mutations nor to age of onset. Additional symptoms were described in 61% of patients. Among them, optic atrophy and reduced visual acuity were the most common. A novel heterozygous missense variant was detected, p.K357E. It is located at a highly conserved position and predicted as pathogenic by in silico tools. At a clinical level, the p.K357E carrier shows a severe sensorimotor axonal neuropathy.
Discussion: In conclusion, our work expands both the genetic and phenotypic spectrum of CMT2A by providing a detailed description of clinical features and disclosing a novel mutation and its related clinical effect. Obtaining a precise genetic diagnosis in affected families is crucial both for family planning and prenatal diagnosis, and in a therapeutic perspective, as we are entering the era of personalized therapy for genetic diseases.
eP01.02.02Patient-Reported Symptom Burden of Charcot-Marie-Tooth Disease Type 1A (CMT1A): Findings from a Real-World Digital Study
Thomas F1, Sevilla T2, Sivera R3, Genovese F4, Gray A5, Bull S6, Paoli X7, Sénéchal T7, Boutalbi Y7, Day L8, Llewellyn S8, Larkin M8
1Hackensack Meridian School of Medicine, Hackensack University Medical Center, Hackensack, United States, 2Hospital Universitari i Politècnic La Fe, Universitate de Valencia, CIBERER, Valencia, Spain, 3Hospital Francesc de Borja, Gandia, Spain, 4ACMT-Rete per la malattia di Charcot-Marie-Tooth, OdV, Bologna, Italy, 5Charcot-Marie-Tooth Association, Glenholden, United States, 6Charcot-Marie-Tooth UK, Christchurch, United Kingdom, 7Pharnext, Paris, France, 8Vitaccess, Oxford, United Kingdom
INTRODUCTION: This analysis aimed to examine patient-reported symptom burden for Charcot-Marie-Tooth disease type 1A (CMT1A) in European and US real-world practice.
METHODS: Adults with CMT1A were recruited to an ongoing international observational study exploring the real-world impact of CMT. Data were collected via CMT&Me, a bespoke digital app developed for this study, through which participants were asked questions via patient-reported outcome measures. This interim analysis examined participants (n=937) from France, Germany, Italy, Spain, the UK and the US.
RESULTS: The CMT1A symptoms ranked with highest importance by participants (n=826 patients who responded to this question) were weakness in hands and fingers (most important, 32%), difficulty walking (15.5%), weakness in the feet (13.0%), fatigue (8.7%), weakness in the legs (5.7%), and problems with balance (sixth most important, 4.2%). The majority of participants reported the severity of their symptoms to be moderate (58.5%, n=400/684) or severe (24.4%, n=167/684), with almost half of participants (48%, n=310/662) experiencing a worsening of CMT1A symptom severity from initial diagnosis.
Anxiety and depression were each reported by over a third of participants (39.3%, n=247/628 and 37.9%, n=238/628 respectively), higher than the prevalence in the general global population.
There was a high recorded use of rehabilitative interventions (of 445 respondents, 86.5% reported using physical therapy), medications (of 404 respondents, 72.5% reported using painkillers), and orthotics/walking aids (of 477 respondents, 48.6% reported using off-the-shelf insoles). There was a similar reported use of these across all countries.
CONCLUSIONS: Patients with CMT1A experience a high level of symptom burden, aligned with clinical observations and literature describing a wide variety of symptoms. Patient first suffer in using limbs, fatigue, pain symptoms, and depression, conducing to impairment to quality of life. It is apparent that there remains a high unmet need in CMT1A caused by the burden on patients.
eP01.02.03One Cause, Many Courses: Leveraging Whole-Genome Sequencing for Comprehensive Modifier Studies in CMT1A
Dohrn M1,2, Xu I1, Ruiz A1, Danzi M1, Reilly M3, Shy M4, Scherer S5, Inherited Neuropathy Consortium4, Züchner S1
1Hussman Institute For Human Genomics, Miller School Of Medicine, University Of Miami, Miami, United States, 2Medical Faculty of the RWTH Aachen University, Department of Neurology, Aachen, Germany, 3Centre for Neuromuscular Diseases, UCL Queen Square, Institute of Neurology, London, United Kingdom, 4Department of Neurology, University of Iowa Hospitals and Clinics, Iowa City, United States, 5University of Pennsylvania, Department of Neurology, Philadelphia, United States
Heterozygous duplications of PMP22 are the most common cause of Charcot-Marie-Tooth disease. Why some patients are more severely affected than others is still not fully understood. In this study, we aim at identifying new genetic disease modifiers using whole-genome sequencing and genotype-phenotype correlations.
The study design encompasses four steps: 1. statistical analyses to define mild vs. severe phenotypes and conduct sample size estimations; 2. phenotype-based patient selection; 3. whole-genome sequencing; and 4. bioinformatic analysis of genetic variant clusters in defined phenotype subsets. For the first step, we have analyzed previously collected CMT1A clinical datasets retrieved from the RDCRN-INC database. Phenotype information was available from 2,190 patients out of 1,317 families. Including 1 to 10 years of follow-up, we assessed 12,441 visits in total. To understand not only the disease severity at baseline, but phenotype dynamics, we compared the 5-year follow-up data for foot dorsiflexion strength (MRC scale) and CMT examination score version 2 (CMTES-2), the two most complete data sets overall.
Our results confirmed that foot dorsiflexion strength and CMTES-2 correlate with age and disease duration, reflecting on the natural disease course. As a limitation, foot dorsiflexion was normal in 27% of the patients, which makes it difficult to define statistical outliers. CMTES, on the other hand, only reached a significant increase after a follow-up time span of seven years, therefore being less representative for disease dynamics in this cohort. We decided to combine those two parameters in a curated minimal data set specific for this CMT1A modifier study, together with information on age at onset, ancestry, and sex. In order to avoid potential confounders, we defined diabetes mellitus, previous chemotherapies, or other known acquired or genetic neuropathy causes as exclusion criteria for this study.
Conclusions: Based on power analyses, we aim at sequencing whole-genome data from 500 mildly and 500 severely affected patients, ideally coming from different families. Interested colleagues and patients are being invited to contact us.
eP01.02.04Work Impacts in Charcot-Marie-Tooth Disease Type 1A (CMT1A): Findings from a Real-World Digital Study
Thomas F1, Sevilla T2, Sivera R3, Genovese F4, Gray A5, Bull S6, Paoli X7, Sénéchal T7, Boutalbi Y7, Day L8, Llewellyn S8, Larkin M8
1Hackensack Meridian School of Medicine, Hackensack University Medical Center, Hackensack, United States, 2Hospital Universitari i Politècnic La Fe, Universitate de Valencia, CIBERER, Valencia, Spain, 3Hospital Francesc de Borja, Gandia, Spain, 4ACMT-Rete per la malattia di Charcot-Marie-Tooth, OdV, Bologna, Italy, 5Charcot-Marie-Tooth Association, Glenholden, United States, 6Charcot-Marie-Tooth UK, Christchurch, United Kingdom, 7Pharnext, Paris, France, 8Vitaccess, Oxford, United Kingdom
INTRODUCTION: This analysis aimed to examine patient-reported impacts to working life for Charcot-Marie-Tooth disease type 1A (CMT1A) in European and US real-world practice.
METHODS: Adults with CMT1A were recruited to an ongoing international observational study exploring the real-world impact of CMT. Data were collected via CMT&Me, a bespoke digital app developed for this study, through which participants were asked questions on demographic and employment variables. This interim analysis examined participants (n=937) from France, Germany, Italy, Spain, the UK and the US.
RESULTS: Of participants who responded to this question (n=607, mean age: 45), 54% (n=328/607) reported working for pay; this was similar across countries with the lowest being France (46%, n=33/72). Twenty percent (n=122/607) reported not working due to disability; this was highest in the US (27%, n=46/172) and lowest in Italy (10%, n=9/91).
Of those working for pay, 74% (n=241/328) reported their work life was affected by CMT1A. Highest rates were in Spain (96%, n=23/24), while lowest were in Italy (65%, n=35/54). Frequently reported ways that CMT1A affected work life were type of job (54%, n=131/241 who specified ways in which work life was affected), number of sick days (30%, n=73/218) and working part-time (30%, n=73/218). Participants reported missing a mean 1.4 workdays in the past two weeks due to CMT1A, equivalent to approximately 36 days per year.
Of those unemployed (7%, n=42/607), 71% (n=30/42) reported that CMT1A was a contributing factor. Highest rates were in Italy (91%, n=10/11), while lowest were in Germany (0%).
CONCLUSIONS: CMT1A has a substantial impact on patients’ ability to work, which is comparable across European countries and the US. Patients are absent from work approximately 36 days per year due to CMT1A. Further research is needed to explore indirect costs associated with these losses, and to better manage impact on patients’ work lives.
eP01.02.05R298C LMNA Mutation Can Cause either Peripheral Neuropathy, Cardiomyopathy or Both: A Case Series Study
Tamaoui L1, Kably B1, Bouhouch R2, Bouhouch A3, Birouk N1
1Clinical neurophysiology department, Hôpital des Spécialités RABAT, Ibn Sina University Hospital, Rabat, Morocco, 2Private cardiology clinic, Rabat, Morocco, 3Neurology B and Neurogenetic department, Hôpital des Spécialités RABAT, Ibn Sina University Hospital, Morocco
Background: LMNA mutations can lead to variety of disorders, collectively termed laminopathies, involving heart, adipose, nerve, bone, and skin tissues, and some premature ageing syndromes. The phenotype associated with the homozygous R298C LMNA mutation is characterized by isolated axonal sensorimotor neuropathy or Charcot-Marie-Tooth disease (CMT) without any features suggestive of cardiac disease. Still, to our knowledge, no case of combined cardiac and neurogenic features has been reported so far in relation with this mutation.
Methods: The follow up of 2 female patients presenting with autosomal recessive axonal neuropathy allowed the identification of two families bearing homozygote lamine A/C mutations with different combinations of axonal neuropathy and cardiac involvement among the same family.
Results: Here we report 5 patients from 2 unrelated families with a homozygous R289C mutation in the LMNA gene and marked intra-familial variability of the disease severity and the coexistence of the cardiac involvement. 3 patients had both axonal neuropathy and cardiac manifestations, mainly conduction disturbances and 1 patient had only axonal neuropathy. Interestingly, one patient had an isolated cardiomyopathy with heterozygote defect of the R289C LMNA gene.
Conclusion: An increased number of overlapping syndromes manifest as a result of LMNA gene mutation. Here we report, for the first time, that R289C mutation in the LMNA gene can cause cardiac manifestations in addition to the well-known phenotype of axonal neuropathy. This highlights the relevance of the screening of heart involvement in individuals who present with an axonal peripheral neuropathy related to an LMNA gene defect.
eP01.02.06Overlap Between Hereditary Sensory-Motor Neuropathy (HSMN) And Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP).
Hustinx M1, Poleur M1, Daron A1, Maertens A1, Servais L1,2, Delstanche S1
1Neuromuscular Reference Centre, CHR Citadelle Hospital, Liège, Belgium, 2Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, United Kingdom
OVERLAP BETWEEN HEREDITARY SENSORY-MOTOR NEUROPATHY (HSMN) AND CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY (CIDP).
Background: Hereditary sensory-motor neuropathy (HSMN) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) have distinct clinical and neurophysiological features. However, the distinction may be challenging : HSMN may be complicated by superimposed CIDP and CIDP may sometimes closely mimic the disease course of inherited neuropathy.
Case series We report 3 cases with significant phenotypic overlap.
Cases 1 (25 years) and 2 (37 years) were initially considered consistent with definite CIDP according to EFNS/PNS 2010 criteria. After demonstrating an objective and robust response to intravenous immunoglobulin therapy (IVIG), a slightly progressive clinical and neurophysiological deterioration appeared in both patients, leading to re-evaluation and confirmation of a genetic neuropathy (PMP22 and MPZ respectively). The coexistence of CMT1 and CIDP is supported by favorable clinical (GRIP, PINCH and 6-minutes-walk tests) and neurophysiological responses to immunoglobulin therapy, still currently observed.
Case 3 was at first presented as a subclinical HSMN. This patient had indeed morphological features (scoliosis, hollow feet) consistent with long-term evolution, when she was initially assessed at the age of 12 years for running disabilities. Cerebrospinal fluid (CSF) was normal. Neurophysiological examination showed slow nerve conduction velocities around 13m/s, but also motor conduction blocks in multiple nerves. However, no mutation was found among a panel exploring more than 4000 genes. The patient then developed a rapidly progressive quadriparesis at the age of 16, which responded favorably to IVIG (GRIP test + 2kg bilaterally and a reduction of 50% of the time needed to walk 10 meters in a 4-week period). She is currently dependent on a regular immunoglobulin course and present with significant weakness when delay between infusion is increased.
Conclusion: This description adds evidence indicating an overlap between genetic neuropathies and CIDP. Indeed, defective structural myelin protein may predispose peripheral nerves to immune attacks, while neuropathies mimicking HSMN may sometimes respond to IVIG. These cases underline the challenge in confirming the diagnosis, especially in the context of expensive intravenous treatment and potential genetic counselling implication.
eP01.02.07Plasma and Skin Biomarkers for Charcot Marie Tooth Disease
Shy M1, Nicolaisen E1, Wu X1, Prada V1, Stork S1, Villapando N1, Zuccarino R1, Bai Y1, Moran J2, Won S2, Duong P3, Rossor A4, D’Antonio M3, Svaren J2
1University Of Iowa, Iowa City, United States, 2University of Wisconsin-Madison, Madison, USA, 3Centre for Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, England, 4Division of Genetics and Cell Biology San Raffaele Scientific Institute, Milan, Italy
BACKGROUND: Building upon previous studies of CMT1A, we have initiated biomarker studies to identify biomarkers in other common forms of Charcot Marie Tooth (CMT) disease, including CMT1B, CMT1X, and CMT2A. Recent studies had identified elevated levels of microRNA’s in plasma from individuals with CMT1A. The goal of this study was to determine if microRNA’s (miR) are elevated in the plasma of individuals with other common types of CMT, and to determine if Schwann cell-derived transcripts in skin can serve as biomarkers of CMT.
METHODS: We have collected plasma and skin samples from individuals with genetically confirmed cases of CMT1B, CMT1X, and CMT2A, and are evaluating plasma samples using microRNA profiling. We also have screened for plasma nerve-enriched transcripts using Nanostring analysis of skin biopsies with a custom gene Codeset based on bioinformatic analysis of peripheral nerve data sets from CMT models. Biomarker levels are correlated with other patient data to test if biomarkers levels correlate with disease severity.
RESULTS: Screening plasma miRNA revealed that muscle-derived microRNA’s (myomirs) are elevated in CMT1B, 1X, and 2A compared to controls. While the myomiRs likely reflect the progressive muscular atrophy in CMT1A, some of the elevated miRs may reflect Schwann cell processes that underlie the pathogenesis of the disease. In addition, we have further developed our nanostring assay to apply to CMT1B, CMT1X, and CMT2A, and several candidate biomarkers have emerged from pilot studies of these forms of CMT.
CONCLUSION: Several candidate biomarkers that have been developed to assess not only CMT disease status, which may provide useful treatment-responsive biomarkers for clinical trials. Some biomarkers identified so far appear to be elevated in several major forms of CMT, whereas others are more specific to a given subtype. The elevation of muscle-derived myomiR’s likely reflects ongoing muscular atrophy in individuals with CMT, and it is possible that this could provide a complementary biomarker in clinical trial design for CMT.
eP01.02.08Depression in Patients with Charcot-Marie-Tooth Disease Type 1A (CMT1A): Findings from a Real-World Digital Study
Thomas F1, Sevilla T2, Sivera R3, Genovese F4, Gray A5, Bull S6, Paoli X7, Sénéchal T7, Boutalbi Y7, Day L8, Llewellyn S8, Larkin M8
1Hackensack Meridian School of Medicine, Hackensack University Medical Center, Hackensack, United States, 2Hospital Universitari i Politècnic La Fe, Universitate de Valencia, CIBERER, Valencia, Spain, 3Hospital Francesc de Borja, Gandia, Spain, 4ACMT-Rete per la malattia di Charcot-Marie-Tooth, OdV, Bologna, Italy, 5Charcot-Marie-Tooth Association, Glenholden, United States, 6Charcot-Marie-Tooth UK, Christchurch, United Kingdom, 7Pharnext, Paris, France, 8Vitaccess, Oxford, United Kingdom
INTRODUCTION: This analysis aimed to examine patient-reported diagnosis of, and consequences associated with, depression among Charcot-Marie-Tooth disease type 1A (CMT1A) patients in European and US real-world practice.
METHODS: Adults with CMT1A were recruited to an ongoing international observational study exploring the real-world impact of CMT. Data were collected via CMT&Me, a bespoke digital app developed for this study, through which participants were asked questions on demographic and employment variables. This interim analysis examined participants (n=937) from France, Germany, Italy, Spain, the UK and the US.
RESULTS: Thirty-eight percent of participants (n=238/628 who reported other medical conditions) reported having been diagnosed with depression in addition to CMT1A; higher than in the general population. Of these, 54% (n=112/208 who also reported symptom severity) and 35% (n=72/208) reported moderate or severe CMT1A symptom severity respectively. Forty-three percent of participants diagnosed with depression (n=102/238) reported that they used, or had previously used, antidepressants.
Reported diagnosis of depression varied considerably by country. Highest rates were among participants in the US and UK (48% and 40% respectively – of which 17% (n=17/103) and 38% (n=25/66) reported severe symptom severity respectively), while lowest rates were among participants in France and Italy (29% and 18% respectively – of which 53% (n=9/17) and 31% (n=5/16) reported severe symptom severity respectively).
Of participants who responded to the EuroQol 5 Dimensions 5 Levels (EQ-5D-5L) instrument (n=685), 62% reported concerns with anxiety/depression.
CONCLUSIONS: Over a third of participants reported diagnosis of depression in addition to CMT1A. This is not surprising for a disease with this symptom burden; however, depression itself as a comorbid condition represents significant disease burden and can affect treatment and outcomes for CMT1A – this warrants further analysis and exploration.
eP01.02.09Novel Variation in the Stalk Domain of KIF5A in a Patient With CMT2-Like Phenotype
Liouta E1, Poulidou V1, Frontistis A1, Moschou M1, Spilioti M1, Kimiskidis V1, Arnaoutoglou M1
11st Neurology Department, AHEPA University Hospital, Thessaloniki, Greece
Introduction: Mutations in the kinesin family member 5A (KIF5A) gene, encoding the heavy chain of kinesin, can cause variable phenotypes ranging from Hereditary Spastic Paraplegia (HSP) and Amyotrophic Lateral Sclerosis (ALS) to Charcot-Marie-Tooth disease type 2 (CMT2). The heavy chain of kinesin-1, a protein with important implication in axonal transport, consists of a motor, a stalk and a tail domain. In the majority of previous reports, most pathogenic mutations are located in the kinesin motor domain whereas variants within the stalk domain are relatively rare. Herein we describe a man with axonal neuropathy consistent with CMT2-like phenotype, in whom a novel mutation in the stalk domain of KIF5A gene has been identified.
Case presentation: We describe the case of a 15-year-old man presenting with progressive walking difficulties. The parents describe a toe-walking gait since childhood. Neurological examination revealed distal leg weakness and foot drop, normal knee-jerks and diminished ankle reflexes. There was also a mild sensory impairment with slight loss of vibration distally. From the upper limbs, no muscle weakness was detected and the tendon reflexes were normal. There were neither pyramidal signs nor bladder dysfunction. Nerve conduction studies showed symmetrically absent compound muscle action potentials (CMAPs) and sensory action potentials (SNAPs) in lower limbs with normal findings from the upper limbs, consistent with sensorimotor axonal polyneuropathy. Needle electromyography showed chronic neurogenic abnormalities in muscles of the lower limbs. MRI of cervical and thoracic spine revealed mild focal dilation of the central canal in C1-C4 and in the middle of the thoracic spine. Clinical exome sequencing identified a heterozygous missense mutation in the stalk domain of KIF5A gene (p.Arg707Gln), which is a single nucleotide variant (SNV), following an autosomal dominant inheritance. The same heterozygote variant was also identified in the mother of the patient who does not experience any symptoms yet. To the best of our knowledge, this nucleotide change has not been previously described, but a different amino acid change (p.Arg707Trp) has been interpreted as a variant of uncertain significance for HSP in the database ClinVar.
Conclusions: It has been proposed from previous reports that KIF5A mutations disrupt axonal transport in different ways, ultimately leading to axonal degeneration. Taking into consideration that the stalk domain allows the dimerization with another kinesin heavy chain, we could assume a potential destabilizing effect of the p.Arg707Gln mutation. The expression of KIF5A in all neurons partially explains the variety of clinical spectrum associated with KIF5A mutations, but how mutations in the same gene affect both central and peripheral motor pathways is a question that requires further research. The identification of novel mutations of this gene could potentially shed light on pathogenic mechanisms of the associated diseases.
eP01.03.01Deacetylation of E3 Ubiquitin Ligase NEDD4-1 by Sirtuin1 Regulates Axonal Growth and Treats Diabetic Neuropathy
Russell J1, Chandrasekaran K1, Hedayat A1, Choi J1
1University Of Maryland, Baltimore, United States
Purpose: Neuronal over expression of Sirtuin 1 (SIRT1) protein, a NAD+-dependent deacetylase, prevented and reversed high fat diet (HFD)-induced diabetic peripheral neuropathy (DPN) [Brain. 2019;142(12): 3737-3752]. NeuroSIRT1 deacetylated NEDD4-1 protein, which is a modulator of axonal and dendritic growth. The purpose of this study was to determine, if knockout of SIRT1 protein in neurons exacerbates DPN, prevents NEDD4-1 deacetylation and decreases neurite length in cultured DRG neurons.
Methods: Neuron-specific CamK2a-CRE/ERT2 mice was mated with floxed SIRT1 mice to generate an inducible nSIRT1 fl/fl mouse. Adult nSIRT1KO phenotype was induced with tamoxifen administration. nSIRT1 fl/fl and nSIRT1KO mice were fed with HFD for 4 months. A second group of mice were fed with HFD for 2 months and then an allosteric activator of SIRT1, SRT2104 (1.33 mg/kg), was added to the HFD for additional 2 months. Neuropathy end points (Sciatic Motor Nerve Conduction Velocity (SMNCV), Tail Sensory Conduction Velocity (TSNCV), Tail Motor Latency (TML) and Mechanical Allodynia (MA-Von Frey) were measured at 2 month and 4 months. Intraepidermal nerve fiber density (IENFD) was measured in hind-paw.
Results: There was a progressive decrease in SIRT1 protein in DRG neurons in nSIRT1KO mice. MA, NCV and IENFD were abnormal in HFD-fed mice compared to CD-fed in both nSIRT1fl/fl and nSIRT1 KO mice at 4 months. Addition of SRT2104 to the HFD after 2 months on HFD reversed changes in MA, NCV and IENFD at 4 months only in nSIRT1 fl/fl mice but not in nSIRT1 KO mice. Dorsal root ganglion (DRG) protein extracts showed increased acetylation of NEDD4-1 protein in HFD mice and addition of SRT2104 prevented NEDD4-1 acetylation only in nSIRT1 fl/fl mice but not in nSIRT1 KO mice. NEDD4-1 siRNA decreased neurite growth in DRG cultures.
Conclusions: SIRT1 mediated ubiquitination is important in prevention of DPN and failure of SIRT1 activation increases the severity of DPN.
eP01.03.02CuidAME Registry: Using Process Automation and Machine Learning Technology to Build SMA Data Analytics Repository
Benavides L1, Segovia S1,2, Exposito J1, Nascimento A1
1Neuromuscular Unit, Neuropediatrics Department, Institut de Recerca Sant Joan de Déu, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain, 2The John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle, United Kingdom
CuidAME is a nationwide registry established in 2019 with the aim of collecting longitudinal data on patients with Spinal Muscular Atrophy (SMA) in Spain. CuidAME uses the SMArtCARE software developed by Open App, which guarantees the systematisation of data collection on a highly secure web platform. Currently, 11 hospitals are participating in the project and data from 240 patients have been collected. Data reporting requires prior data management to facilitate data cleaning and analysis. To date, the preparation of data has involved a manual process which has increased the workload with the consequent risk of compromising data quality. In response of these challenges, it has been decided to develop a set of IT services to automate data management, improving response times and the quality of data processing for analytics purpose.
We have used a process of automation and machine learning technology approach developed with python to improve data quality and data model structure. We have used a stepwise approach to build the data analytics repository: Step 1 - data profiling to identify data errors such as data type errors, missing and duplicated data; Step 2 - data integration from different data sources to create and unique master file; Step 3 - data quality to improve accuracy and enhance reliability; Step 4 - data approval; Step 5 - data reporting with a customized query builder. The automation process supports the data profiling, data integration, and data quality, and machine learning technology support the data quality and data approval.
The automation of processes will allow us to transform the past experiences of manual procedures into an IT service that improves results in terms of efficiency and data quality.
eP01.03.03Spinal Muscular Atrophy Disease Registries: Overview and Recent Progress
Raynaud S1, Viscidi E1, Youn B1, Wang N1, Eaton S1, Tiar F2, Kangas N3, Gooch J4, Makepeace C4, Johnson N1, Paradis A1
1Biogen, Cambridge, United States, 2Biogen, Paris, France, 3Biogen, Baar, Switzerland, 4Biogen, Maidenhead, United Kingdom
Background: Patient registries in rare diseases such as spinal muscular atrophy (SMA) can play a critical role in furthering scientific understanding of the natural history of the disease, patterns of treatment use, and the effectiveness and safety of disease modifying therapies, especially in patient populations outside those included in clinical trials. Biogen supports a disease registry approach for SMA with multiple partners to enable the collection of high quality, robust real-world data to meet key data needs of the SMA community, healthcare providers, researchers, regulators, payors, and industry. An overview of these partnerships and the progress made to date is provided.
Methods: Based on guidance from the European Medicines Agency (EMA), Biogen established collaborations with multiple SMA disease registries to gather robust information on disease outcomes, to characterize the natural course of SMA and evolving phenotypes, and to improve our understanding of nusinersen and other emerging treatments in a real-world setting. Over the past 6 years, work has been undertaken with registry partners to improve the capacity and capability of registries to collect reliable patient-level data, standardize data across registries to an international aligned core data set (TREAT-NMD), and provide financial support to implement data collection and ensure sustainability. The aligned data set includes information on demographics, clinical characteristics, medical history, functional outcomes, hospitalizations, and treatments for SMA.
Results: Biogen is currently partnering with 24 SMA disease registries across 22 countries, which have enrolled over 6,000 individuals living with SMA across the globe as of December 2021. As both the number of patients and the longitudinal data are continuing to grow, the data have been increasingly used to provide insights for understanding the natural history of SMA and the effectiveness and safety of nusinersen treatment. The SMA registries supported by Biogen have led to more than 20 independent publications (manuscripts and congress presentations) through 2021. Findings from registries in several European countries (Italy, Spain, and Germany) have also been included in regulatory submissions and as part of reimbursement submissions to inform treatment value in specific patient populations including adults. In 2021, registry data were used to support the inclusion of real-world evidence on nusinersen safety and effectiveness in adults with SMA into the EMA Summary of Product Characteristics (SmPC). These registry data, which demonstrated the benefits of nusinersen treatment in adult SMA patients, played a role in expanding access and reimbursement for Type III SMA patients in the UK who had lost the ability to walk.
Conclusions: Registry data will continue to play a critical role in addressing key questions relevant for management of SMA in patients, such as predictors of treatment response, and inform treatment decision making in a multi-treatment era of SMA. Answering these research questions will help healthcare professionals optimize outcomes for the heterogenous real-world patient population. Registry data can also help in broadening our disease understanding. Real-world evidence will continue to be used to support access and reimbursement decisions, ultimately helping more patients gain access to effective treatments.
eP01.03.04Cognitive Assessment of Spinal Muscular Atrophy
Akodad S1
1Université Libre De Bruxelles, Brussels, Belgium
Introduction: Spinal Muscular Atrophy (SMA) is a rare neuromuscular disease that has a heavy burden on patients. It destroys the nerve cells in the brainstem and spinal cord that control essential skeletal muscle activity such as walking, speaking, breathing, and swallowing. According to arecent study little is known about the impact of the disease across different types of the disease (SMA type 1-4) and clinical interventions. Fortunately, the progression of SMA was shown to be halted with the help of new treatments. Patients have a prolonged life expectancy (mainly for SMA type 1), enabling patients to set longer-term life goals. Improving our knowledge on the cognitive profile is becoming mandatory however current evidence on cognitive aspects of SMA type 1 and 2 is limited and contradictory.
Indeed, the commonly used diagnostic tests usually cannot be used due to the motor and verbal limitations associated with this pathology. We are therefore conducting a clinical study to investigate cognition in SMA patients. To this end, we recommend removing physical limitations (intrinsically related to SMA) by introducing cognitive tests into the practice of “eye” targeting. The objective is to develop and validate of a new tool for the evaluation of cognitive functions of patients with SMA type 1 and 2 based on oculometric.
Methods: We developed an innovative set of cognitive tests and implemented it through an eye tracking system: Matrix subtest of Weschler non-verbal scale (WNV) of ability studying fluid reasoning and perceptual reasoning (4-13 years), Recognition subtest of WNV of ability studying visuo-spatial memory (4-8 years), the chimeric animal Stroop test studying inhibition (4-12 years),Face recognition subtest of NEPSY-II studying the face encoding, discrimination and recognition (5-13 years), Picture complement test studying the spatial reasoning (4-8 years) and Matching pair test (4-8 years). In order to validate our method, we performed the equivalent of these tests in their paper-and-pencil version from the neuropsychology routine.
Tests were performed by children aged from 4 years to 13 years.
Results: SMA children results did not significantly differ from healthy controls in each task.
Nevertheless, results must be interpreted with caution since validation of their implementation on an eye tracking system is still in progress. Furthermore, the number of SMA children needs to be increased to power the study.
Conclusions: This study investigated for the first time a set of multiple cognitive tasks in SMA children using an eye tracker device. It reinforces the hypothesis of a normal cognition in SMA children without currently confirming it. Validation of the newly implemented tests (for example with comparison to the paper version) and inclusion of new patients are still in progress to answer to the key question of the cognitive impairments of SMA, especially in type 1.
Keywords: Spinal muscular atrophy, cognition, eye tracking.
eP01.03.05SMA-REACH-UK, Adult-SMA-REACH and UK SMA-Patient-Registry an Integrated Model: Transition of Data and Longitudinal Data Collection.
Segovia S1,2, Fitzsimmons S1, Samsuddin S3, Anifowoshe D3, Murphy L1, Cavalcante E3, Madden M3, Blewitt C1, Mayhew A1, Muni-Lofra R1, Rohwer A3, Moss C3, Scoto M3, Baranello G3, Muntoni F3, Marini-Bettolo C1
1The John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK., Newcastle, United Kingdom, 2Neuromuscular Unit, Neuropediatrics Department, Institut de Recerca Sant Joan de Déu, Hospital Sant Joan de Déu, Spain, Esplugues de Llobregat, Spain, 3The Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health., London, United Kingdom
Spinal muscular atrophy (SMA) is a form of motor neuron disease caused by a mutation in the survival motor neuron 1 gene (SMN1) which results in a wide disease spectrum affecting infants and adults. The recent advances in drug development and European Medicines Agency approval of three drugs for SMA highlight an urgent need to establish stronger clinical networks to monitor and gain a better understanding of the impact of new drugs on the natural history of the disease. The generation of real-world evidence requires the standardisation and systematic collection of data generated during routine clinical visits and data reported directly from patients.
The objective of SMA REACH UK, Adult SMA REACH and the UK SMA Patient Registry is to systematise the collection of paediatric and adult patient data to enable integrated and longitudinal analysis of clinical and patient-reported data.
In the UK there are three databases currently collecting data on SMA patients. SMA REACH UK is led by Great Ormond Street Hospital (GOSH) and is a paediatric SMA clinical network and data collection study already established and actively collecting both natural history and treatment-related data. Adult SMA REACH is led by the John Walton Muscular Dystrophy Research Centre (JWMDRC) and is a more recently established adult SMA clinical network and data collection study. The UK SMA Patient Registry, also led by the JWMDRC, is a long-established web-based patient registry that collects patient-reported data across all ages.
SMA REACH UK and Adult SMA REACH work collaboratively and use the same IT platform for data collection. The datasets of both databases have been harmonised to enable easy data integration and transition. In addition, the UK SMA Patient Registry has established a process of data sharing of Patient Reported Outcome Measures (PROMs) to allow this data to be integrated with SMA REACH’s clinician-reported data for analysis. Patients included in the three projects consent to the integration and sharing of their data.
The integration of the three UK SMA databases will advance our understanding of the impact of standards of care and new therapies on the natural history of the disease and ensure continuity of data collection throughout transition. The inclusion of patient perception of the impact of the disease is ensured by integrating data directly reported by patients.
eP01.03.06Proposal of New Functional Motor Scale to Evaluate Muscle Fatigue in Adult SMA Patients
Torri F1, Ricci G1, Govoni A1, Chiappini R1, Fontanelli L1, Manca L1, Siciliano G1
1Department of Clinical and Experimental medicine, University of Pisa, Pisa, Italy
Spinal Muscular Atrophy (SMA) is a potentially devastating disease affecting the lower motor neuron, carrying a significant burden on patients’ general motor skills and quality of life, characterized by a great variability in phenotypic expression, only partially due to the genetic defect. As new therapeutic options make their appearance on the scene, clinical tools aiming at defining any changes in the patients’ performance are needed along the already used RUHLM and Hammersmith scales for the upper and lower limbs, especially in adult patients undergoing treatment with Nusinersen and Risdiplam, in which the expected clinical response is a stabilization of disease progression rather than frank improvement. Here we propose a new functional motor scale evaluating the fatigue dimension for the upper and lower limbs in adult SMA patients, in order to possibly objectify variations in a quite invalidating symptom that is often reported as improving with treatment by adult SMA patients. The scale is composed by twelve timed motor tasks evaluating both the upper and the lower limbs, including sitting, standing and walking for ambulant patients. The tasks and the order in which they are proposed to the patient have been decided in order to obtain the maximum patient’s compliance and endurance and target both the proximal and the distal segments of the limbs. Moreover, many of the included tests are similar to daily life gestures and tasks that the patient may be required to achieve (i.e. fine movements of the fingers, postural changes, personal hygiene and dressing). The scale was validated in adult SMA patients and proved good intra- and inter-observer reliability. We believe that the evaluation of fatigue and endurance as an objective parameter should be an essential part in the management and follow-up of SMA adult patients.
eP01.03.07CuidAME: A New Registry for Longitudinal Data Collection of Spanish SMA Patients
Segovia S1,2, Exposito J1, Nungo C3, Vazquez J3, Pitarch I3, Caballero J4, Rodriguez-Sanchez C4, Pascual S4, Marco C5, Dominguez R5, Povedano M5, Pareja A6, Lopez-Lobato M6, Alvarez M7, Costa L7, Gomez-Andres D7, Munell F7, Moreno A8, Martinez-Salcedo E8, Nascimento A1
1Neuromuscular Unit, Neuropediatrics Department, Institut de Recerca Sant Joan de Déu, Hospital Sant Joan de Déu, Spain, Esplugues de Llobregat, Spain, 2The John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle, United Kingdom, 3Pediatric Neurology of Neuromuscular Disease Unit.Hospital Universitario y Politécnico La Fe, Valencia, Spain, 4Department of Pediatric Neurology, Hospital la Paz, Madrid, Spain, Madrid, Spain, 5Department of Neurology, Hospital Universitari de Bellvitge, Hospitalet de Llobregat, Spain, 6Servicio de Neuropediatría, Hospitales Universitarios Virgen del Rocío, Sevilla, Spain, 7Pediatric Neurology, Vall d’Hebron Institut de Recerca (VHIR), Hospital Universitari Vall d’Hebron, Barcelona, Spain, 8Neuromuscular Unit, Neurology Department, Hospital ClínicoUniversitario Virgen de la Arrixaca, Murcia, Spain
Spinal muscular atrophy (SMA) is a genetic disorder characterized by loss of motor neurons in the spinal cord, leading to progressive muscle atrophy, weakness, and disability. Natural history of SMA has changed due to improvements in treatment and technological advances. However, the real-world evidence of the outcome of new treatments is unknown and registries can be a useful tool for this purpose. The aim of CuidAME Registry is to collect Longitudinal Data of Spanish SMA patients.
CuidAME uses Smartcare platform to collect retrospective and prospective data on all SMA patients, regardless of their treatment regimen, collected during routine clinical visits and updated every eight months.
Herein we analysed the baseline data collected for 104 seen at 7 different hospitals. We included 104 patients: 20% were SMA type 1, 60% type 2, 18% type 3, and 2% type 4. 71 patients (68%) were treated, all received Nursinersen except two who received Zolgensma and one who received Risdiplam. Mean age at baseline visit was 5.98 (0-39). 57 % of patients were female. For SMA type 1, the mean age was 4y.o. (0-9), 81% were treated, 82% were able to sit, 70,6% used Non-invasive ventilation (NIV) and 41.1% used a feeding tube. For SMA type 2, the mean age was 11y.o. (2-37), 64,5% were treated, 82,5% were able to sit, 45% used NIV, 2.5% used a feeding tube, and 30% had a scoliosis surgery. The mean age for the SMA type 3A was 9 y.o (3-16). and 11 y.o. (7-65) for 3B. 68,42% (19) were treated, 69% (13) were able to walk. None of the SMA type 3 patients had scoliosis surgery nor ventilatory support.
These are the results of the baseline visit. In the final poster the first year data with longitudinal data analysis will be presented.
CuidAME is providing a platform for harmonizing and systematising data collection across centers in Spain and it will promote national and international collaboration between centers and registries expanding the knowledge on SMA.
eP01.03.08Swallowing Evaluation in Treated SMA Patients – A Pilot Prospective Study
Benmechri S1, Tahon V2, De Vos E2, Christiaens A1, Deconinck N1,2
1 Paeditric neurology department and Neuromuscular Reference Center ULB ,Hôpital Universitaire des Enfants Reine Fabiola (HUDERF), Université Libre de Bruxelles, Brussels, Belgium, 2Neuromuscular Reference Center (NMRC), UZ Ghent, Ghent, Belgium
Background: Spinal muscular atrophy (SMA) is a group of autosomal recessive disorders associated with degeneration of motor neurons in the spinal cord and, in the most severely affected patients, brain stem. Recent new therapies have demonstrated clear progress on survival, and general motor function. However, swallowing problems is a common complication in SMA type 1 but also in SMA type 2 that still requires better evaluation methods.
Objective: To evaluate the feasibility and reliability of a swallowing questionnaire (Qdeglut) and the Iowa Oral Performance Instrument (IOPI, kPa) as a marker of the oral phase of swallowing in a cohort of treated SMA patients.
Methods: We prospectively included (n= 25) normally developing (ND) children aged between 3 and 15 years old and 20 age matched children with a confirmed genetic diagnosis of SMA. All SMA patients were receiving a SMN2 splicing modifying therapy at inclusion. All patients underwent at least one IOPI pressure measurement and Qdeglut. Repeated measures are ongoing. The CHOP INTEND and HMFSE was used as a measure of motor function.
Results: ND children showed consistent lip, tongue, and masticatory pressures that were expected for their ages with tight standard deviations, and a positive correlation with age. Taken as group SMA patients showed lower pressure values than age matched ND children. Pathological scores on Qdeglut were correlated with lower pressures in SMA patients. Finally, a clear correlation between pressure and SMA type was observed.
Conclusion: From this pilot project, we could show that IOPI measurement system and Qdeglut can be applied reliably and easily in the context of a simple outpatient consultation. Early results indicate that SMA patients, although benefitting from a disease modifying therapy, demonstrate clear weaker inter-lips, palate lingual and masticatory pressures. Impairment appears to be more severe along the SMA spectrum (SMA1>SMA2>SMA3). Additional longitudinal data will be presented.
eP01.03.09Safety and Effectiveness of Onasemnogene Abeparvovec Alone or with Other Disease-Modifying Therapies: Findings from RESTORE
Servais L1, Benguerba K2, De Vivo D3, Kirschner J4, Muntoni F5, Proud C6, Tizzano E7, Saito K8, Raju D9, LaMarca N9, Sun R9, Anderson F10, Faulkner E9, Finkel R11
1MDUK Oxford Neuromuscular Centre, University Of Oxford, Oxford, United Kingdom, 2Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 3Departments of Neurology and Pediatrics, Columbia University Irving Medical Center, New York, USA, 4Clinic for Neuropediatrics and Muscle Disease, University Medical Center Freiburg, Freiburg, Germany, 5The Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health & Great Ormond Street Hospital, London, UK, 6Children’s Hospital of The King’s Daughters, Norfolk, USA, 7Department of Clinical and Molecular Genetics, Hospital Valle Hebron, Barcelona, Spain, 8Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan, 9Novartis Gene Therapies, Inc., Bannockburn, USA, 10Center for Outcomes Research, University of Massachusetts Medical School, Worcester, USA, 11Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, USA
Objective: We sought to describe the safety and effectiveness of onasemnogene abeparvovec (OA) alone or in association with other disease-modifying therapies (DMTs) for patients with spinal muscular atrophy (SMA) in a real-world setting.
Methods: We evaluated patients with two or three copies of the survival motor neuron 2 (SMN2) gene receiving OA alone or in combination with any other DMTs in RESTORE (an ongoing, prospective, multicenter, multinational, observational SMA patient registry).
Results: As of May 23, 2021, data were available for 197 patients who had received OA. Of these, 97 (49.2%) received OA alone (Group 1), while 40 (20.3%) and 60 (30.5%) received OA before (Group 2) or after (Group 3) another DMT, respectively. All patients in Group 3 received nusinersen before OA. No patients received risdiplam before OA. Clinical characteristics of the study patients are presented by treatment regimen in Table 1. For patients with two or more milestone assessments (one or more occurring after OA administration; n=72), 47 (65.3%) achieved additional milestones, including 27/47 (57.4%) in Group 1, 11/47 (23.4%) in Group 2, and 9/47 (19.1%) in Group 3. There was no significant difference in time to first milestone achievement after OA administration between the three groups (p>0.05). Changes in motor function scores were also similar between treatment groups (p>0.05). Median changes in Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders scores for patients in Groups 1, 2, and 3 were 7 (IQR, 5–11.5, n=24 with 19 patients [79.2%] achieving an increase of ≥4 points), 9 (IQR, 5–21, n=15 with 12 patients [80%] achieving an increase of =4 points), and 4.5 (IQR, 0.75–13, n=14 with eight patients [57.1%] achieving an increase of ≥4 points), respectively. Median changes in Hammersmith Functional Motor Scale Expanded scores for Groups 1, 2, and 3 were 6 (IQR, 1.5–25.5, n=5 with four patients [80%] achieving an increase of ≥3 points), 13 (IQR, 0–16.5, n=5 with three patients [60%] achieving an increase of =3 points), and 4 (IQR, 0.5–12, n=5 with three patients [60%] achieving an increase of ≥3 points), respectively. Median changes in Hammersmith Infant Neurological Examination scores for Groups 1, 2, and 3 were 3.5 (IQR 2–11.25, n=10), 4 (IQR 0.5–5.5, n=5), and 3 (IQR 0–12.5, n=5), respectively. Treatment-emergent adverse events (TEAEs) of any grade were recorded for 33 (34.0%) patients in Group 1. Of these, 12 (12.4%) patients reported TEAEs of Grade ≥3. In Group 2, TEAEs were reported in 18 (45%) patients, with 10 (25%%) reporting TEAEs of Grade ≥3. For patients in Group 3, TEAEs were reported for 38 (63.3%) patients, with 19 (31.7%) reporting TEAEs of Grade ≥3. No new safety signals were identified during the study.
Conclusions: Based on the data available from the RESTORE registry, similar real-world effectiveness was observed in patients receiving OA monotherapy compared with patients who switched from nusinersen or patients who received subsequent DMTs. Patients who received nusinersen before OA appear to have experienced greater frequency and severity of adverse events.
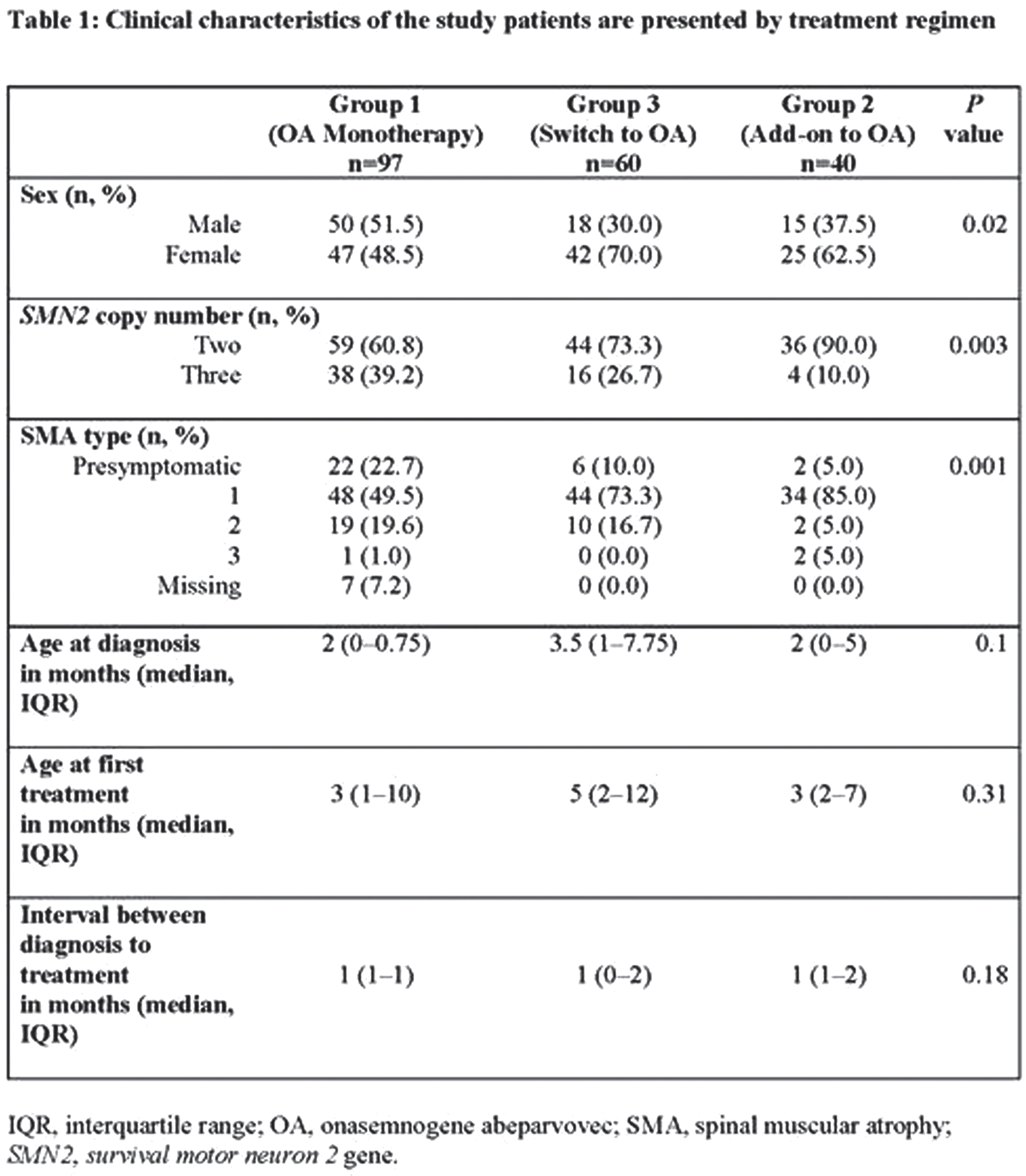
eP01.03.10Brown-Vialetto-Van Laere Syndrome, Temporary Clinical Stabilization with Intravenous Immunoglobulin (IVIg) and MRI Abnormalities
Martínez-Esteban P1, Alvarez-Toledo K2, Mattos-Castillo J2, Méndez-Dávalos C1, Bobadilla E3
1Instituto Nacional De Salud Del Niño San Borja, Lima, Peru, 2Instituto Nacional de Ciencias Neurológicas, Lima, Peru, 3Hospital Universitario San Ignacio, Bogota, Colombia
INTRODUCTION: Riboflavin transporter deficiency(RTD) is a rare neurological disease that includes Brown Vialetto Van Laere syndrome(BVVL) and Fazio Londe syndrome(FL), as a result from heterozygous or homozygous pathogenic variants in SLC52A2 and ALC52A3 genes, producing neurodegeneration.(1,2)
BVVL syndrome is characterized by motor-sensory neuropathy, sensorineural hearing loss, cranial nerve disorders, optic atrophy, upper and lower motor neuron involvement, ataxia and respiratory failure requiring mechanical ventilation.(1,2)
CLINICAL CASE: Male 1 year 10 months, without relevant medical history, was admitted having with 3 months of disease; beginning with right peripheral facial paralysis, dysarthria and dysphagia, progressing to facial diparesis, respiratory distress and difficulty walking.
On admission, the patient was awake, alert, preserved eye movements, photoreactive pupils, ophthalmoscopy showed bitemporal papillary pallor, facial diplegia, decreased gag reflex. Proximal quadriparesis, preserved sensation and preserved osteotendinous reflexes.
The Brain tomography, cerebrospinal fluid study(CSF) and heavy metal dosage were negative.
Four days after admission he developed acute respiratory failure, requiring mechanical ventilation. Electrodiagnostic(EDX) studies showed decreased amplitudes of compound muscle action potentials. Magnetic resonance imaging(MRI) of the brain was normal, spinal MRI in T1w with gadolinium showed enhancement of the lumbo-sacral roots (Figure-1), for which an autoimmune pathology was suspected, starting intravenous immunoglobulin(IVIg).
Two weeks after admission, he presented global hiperreflexia and bilateral Babinski, a control brain/spinal MRI was performed with similar findings on admission, starting a 2nd cycle of IVIg, showing improvement in muscle strength, disappearance of hiperreflexia and Babinski.
After 1 month of admission, EDX studies showed active denervation in upper extremity muscles suggestive of lower motor neuron pathology. The CSF study remained normal.
Two months after admission, he became encephalopathic, whit reappearing global hiperreflexia and bilateral Babinski. The electroencephalogram recorded slow generalized activity. Due to clinical worsening and response observed previously to IVIg, plasmapheresis was started, with no improvement.
Due to the torpid evolution and the lack of response to treatment, RTD was suspected. Brainstem auditory potentials showed bilateral neurosensorial hearing los, with this finding BVVL syndrome was suspected, and treatment with riboflavin was immediately started, with a favorable clinical response.
The genetic study found an alteration in the SLC52A3 gene, variant c.1156T>C (p.Cys386Arg) in homozygosis, classified as of uncertain significance(VUS).
DISCUSSION: Due to the clinical presentation, the involvement of cranial nerves, upper and lower motor neuron involvement, the need for mechanical ventilation and hearing loss and MRI abnormalities of nerve roots, SBVVL was suspected as the best diagnostic alternative. (1-3)
SBVVL is potentially treatable disease, therefore riboflavin should be instituted when clinically suspected. (4-6) During the evolution, we observed a partial response and temporary stabilization of symptoms with IVIg, which has been previously reported in isolated cases. (7) Although the mechanism by which IVIg has a favorable and transitory effect is still unknown.
Although the genetic mutation found was classified as VUS, this mutation was described in a patient 2 years 06 months, with similar clinical characteristics (6). In addition, the segregation study showed the same variant in each of the heterozygous parents, so we assume that the variant found should be reclassified.
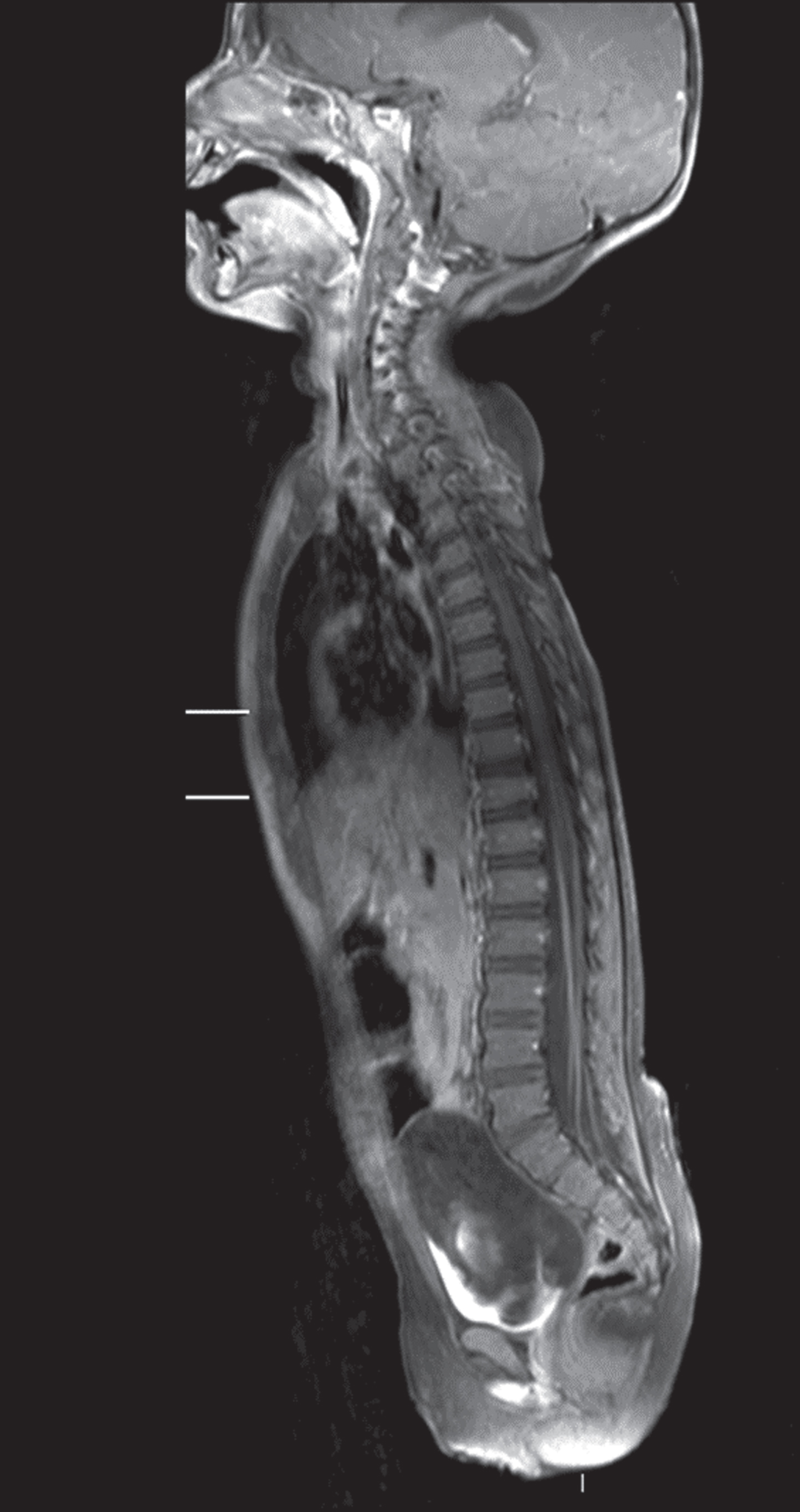
eP01.03.11A Compound Heterozygous Mutationin Calpain-1 Identifies a New Gene for Spinal Muscular Atrophytype-4
Perez Siles G1,2, Ellis M1, Grosz B1,2, Vucic S4, Kiernan M3,5, Morris K4, Reddel S3, Kennerson M1,2,6
1Northcott Neuroscience Laboratory, ANZAC Research Institute, Sydney, Australia, 2Sydney Medical School, University of Sydney., Sydney, Australia, 3Brain and Mind Centre, The University of Sydney, Sydney, Australia, 4Concord Clinical School, University of Sydney and Royal North Shore Hospital, Sydney, Australia, 5Department of Neurology, Royal Prince Alfred Hospital., Sydney, Australia, 6Molecular Medicine Laboratory, Concord Repatriation General Hospital, Sydney, Australia
Spinal Muscular Atrophy (SMA) is a heterogeneous group of neuromuscular diseases characterized by degeneration of anterior horn cells of the spinal cord, leading to muscular atrophy and weakness. Although the major cause of SMA is autosomal recessive exon deletions or loss-of-function mutations of survival motor neuron 1 (SMN1) gene, next generation sequencing technologies are increasing the genetic heterogeneity of SMA. SMA type 4 (SMA4) is an adult onset, less severe form of SMA for which genetic and pathogenic causes remain elusive.Whole exome sequencing in a 30-year-old brother and sister with SMA4 identified a compound heterozygous mutation (p. G492R/p. F610C) in calpain-1 (CAPN1). Mutations in CAPN1 have been previously associated with cerebellar ataxia and hereditary spastic paraplegia. Using skin fibroblasts from a patient bearing the p. G492R/p. F610C mutation, we demonstrate reduced levels of CAPN1 protein and protease activity. Functional characterization of the SMA4 fibroblasts revealed no changes in SMN protein levels and subcellular distribution. Additional cellular pathways associated with SMA remain unaffected in the patient fibroblasts, highlighting the tissue specificity of CAPN1 dysfunction in SMA4 pathophysiology. This study provides genetic and functional evidence of CAPN1 as a novel gene for the SMA4 phenotype and expands the phenotype of CAPN1 mutation disorders.
eP01.04.01COVID-19: Retrospective Analysis in Neuromuscular Disease Patient’s Impact on Healthcare, Quality of Life and Anxiety
Buscemi L1, Delstanche S2, Demortier F1, Dubois C1, Duclos M1, Medard L1, Servais L3
1Department of Paediatrics, CHU Liège/University of Liège, Belgium, 2University department of neurology, CHR Citadelle de Liège, Belgium, 3MDUK Neuromuscular Center, Department of Paediatrics, University of Oxford, UK
In March 2020, Belgium followed the world sanitary regulations related to covid-19, and instaured a general lockdown. To prevent health care saturation, only urgent consultations were allowed to happen face-to-face. This situation has largly impacted the activity of chronic patients care, including in the NeuroMuscular disease Reference Center (NMRC) of Liège.
In order to measure the impact of lockdown on patients’ health, we set up a retrospective analysis with data collected in 680 patients, adults and children followed at the NMRC Liège. We collected demographic, medical, social, quality of life and anxiety data. We found that 25.1% of neuromuscular adults patients presented with a significative change (< 5%) of weight. 22.53% of adults and 21.26% of children experienced change regarding medical support, 26.0% of adults and 25.5% of children regarding social aids support, 32.2% of adults and 32.3% of children regarding paramedical support and, finally, 37.7% of adults and 46.9% of children regarding their family environement. Anxiety was reported by 36.3% and 41.8% of children and adults, respectively. Anxiety was mostly reported to be caused by loneliness and fear for relatives. Comparing in pre and post lockdown the standardized ActiVlim questionnaire that measures patient autonomy, we found that 35.4% of adults (n=147) were overall stable whilst 31.3% reported progress and 33.3% reported decline. Similar comparison in children (n= 35) demonstrated stability in 17.1%, progress in 45.7% and decline in 37.1%.
A clear disease specificity was observed in Activlim data when we compare pre and post surveys. For example, children with SMA disease tended to stabilize in quality of life questionnaire and even progressed for some. This is consistent with the fact that these patients are treated with newly available disease modifying treatments. On the other hand, ALS patients tended to lose a very significant part of their autonomy during this lockdown, which is of course related to the course of the disease itself.
Our study helps to better appreciate the impact of lockdown on neuromuscular patients, and could add further evidence in the benefit/risk balance of lockdown on the health of vulnerable population.
eP01.04.02COVID-19-Related Neuropathy in Colombia: The Experience During the First 23 Months Of Pandemic
Peña Guzmán L1,2, Rodríguez Hernández L1,2, Gómez Mazuera A1,2
1Fundación Santa Fé De Bogotá, Bogotá, Colombia, 2Universidad del Rosario, Bogotá, Colombia
COVID-19-related neuropathy in Colombia: the experience during the first 23 months of pandemic
Introduction: The SARS-CoV-2 virus has a high neuroinvasive capacity due to the increased expression of angiotensin-converting enzyme receptor 2 (ACE-2) in neurons (1) and it is believed that the mechanism by which it can cause injury to the nervous system peripheral nervous system is immune-mediated, although a direct cytotoxic effect of the virus cannot be ruled out (2). Multiple types of neuropathy associated with SARS-CoV-2 infection have been described, the most frequent being Guillain-Barré syndrome, pre-existing diabetes, compression neuropathies and drugs used to treat symptoms of COVID-19 (3).
Objetives: To characterize the patients who were referred to the electromyography laboratory at the Fundacion Santa Fé de Bogotá, Colombia due to suspected COVID-19-related neuropathy
Methods: Descriptive observational study, case series type. The electrodiagnostic studies carried out between January 2020 and December 2022 in the electromyography laboratory at the Fundacion Santa Fé de Bogotá, Colombia with suspected COVID-19-related neuropathy were reviewed.
Results: 94 patients were evaluated in the electromyography laboratory with suspected COVID-19-related neuropathy between January 2020 and December 2021, of which 53% (50/94) were men. The average age was 54.8 years. 32% (30/94) had severe COVID and 31% (29/94) were hospitalized in the ICU.
Most of the studies were normal: 35% (33/94). Of the abnormal findings, it was found in order of frequency: symmetric motor and sensory axonal polyneuropathy in 21.2%, and of this group of patients, 55% were in the ICU, 35% had no data and 20% were hospitalized-not ICU. 18% presented compression neuropathy of the median nerve in the carpal tunnel, 6.3% asymmetric motor and sensory axonal neuropathy, 6.3% suggestive findings of cervical and/or lumbosacral root involvement, 4.2% Guillain Barré syndrome, 4.2% compression neuropathy of the peroneal nerve, 2.1% brachial plexus axonal injury, 2.1% peroneal nerve axonal injury, 2.1% radial axonal injury, 2.1% myopathic changes, 1% hypoglossal nerve axonal injury, 1% symmetric axonal and demyelinating polyneuropathy, 1% hereditary neuropathy, 1% asymmetric demyelinating neuropathy, 1% axonal injury of the sciatic nerve, 1% axonal injury of the median nerve in the forearm, 1% axonal injury of the lumbosacral plexus, 1% compression neuropathy of the ulnar nerve in the elbow and 1% axonal injury from a sensory branch of the median nerve.
Conclusions: The most frequent abnormality in the study was symmetric motor and sensory axonal polyneuropathy, which can be explained by the prolonged ICU stay, which increases the risk of Critical illnes Neuropathy.
References
1. Natoli S, Oliveira V, Calabresi P, Maia L, Pisani A. Does SARS-Cov-2 invade the brain? Translational lessons from animal models. Eur J Neurol. 2020, 0: 1–10
2. Paliwal V, Garg R, Gupta A, Tejan N. Neuromuscular presentations in patients with COVID-19. Neurol Sci. 2020 Nov;41(11):3039-3056
3. Finsterer, Scorza F, Scorza C, Fiorini A. Peripheral neuropathy in COVID-19 is due to immune-mechanisms, pre-existing risk factors, anti-viral drugs, or bedding in the Intensive Care Unit. Arq Neuropsiquiatr. 2021 Oct;79(10):924-928.
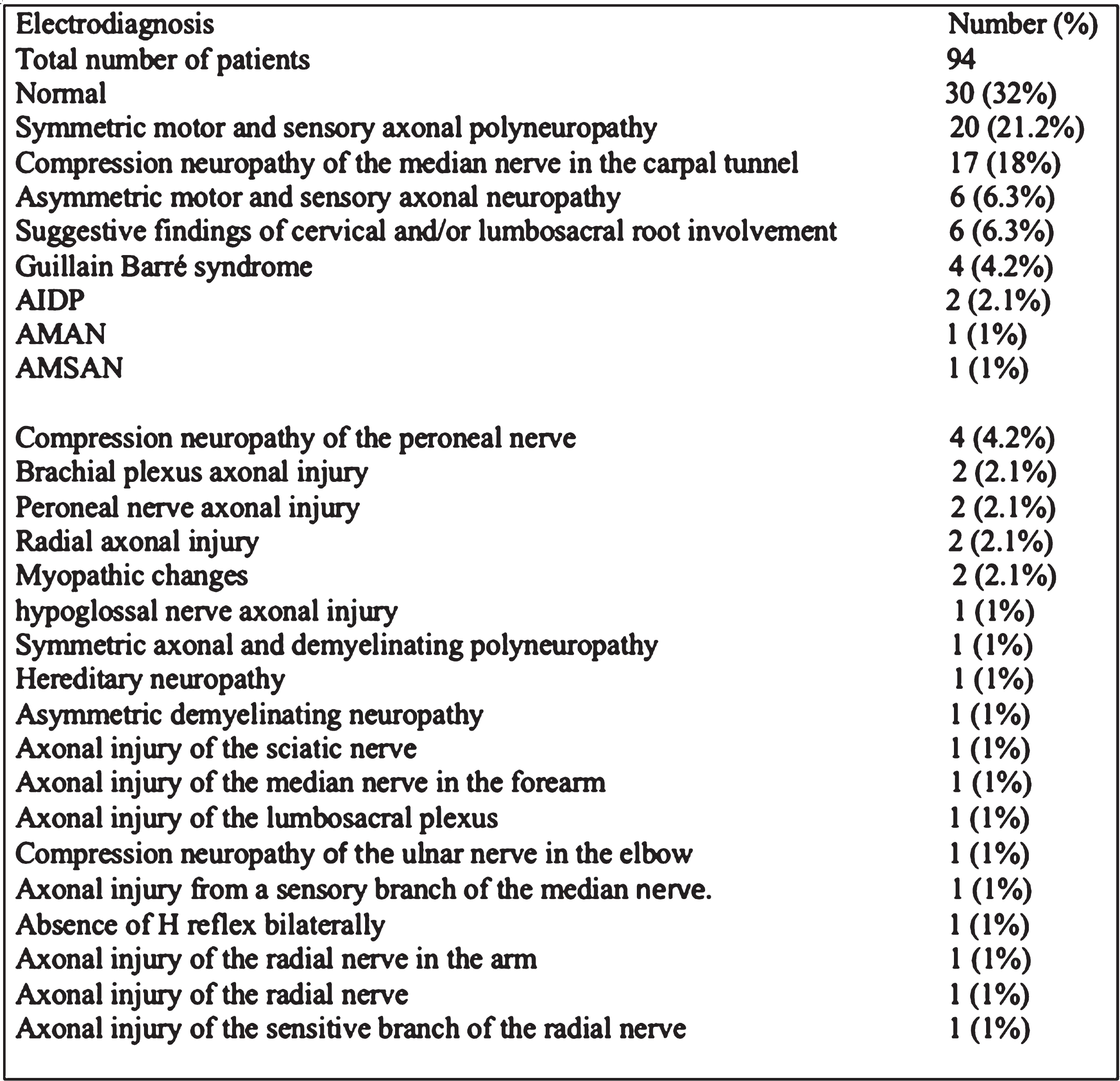
eP01.04.03Myositis And Myocarditis With Anti-Jo-1 Antibodies Following SARS-CoV-2 mRNA Vaccination Or COVID-19 Infection
Raaphorst J1, Willems M1, Kamperman R1, Walter H1, Smithuis F2, Aronica E3, Kok W4, Boekholdt M4, de Groot L5, van Leeuwen E6, van Onna M7, Wieske L1, Eftimov F1, Gelderman K8, Hamann D9, van der Molen R10, van Lochem E11, Meek B12, Schreurs M13, van der Kooi A1
1Department of Neurology, Amsterdam Neuroscience, Amsterdam UMC, location AMC, Amsterdam, The Netherlands, 2Department of Radiology and Nuclear Medicine, Amsterdam University Medical Centre, Amsterdam, The Netherlands, 3Department of (Neuro)Pathology, Amsterdam University Medical Centre, Amsterdam Neuroscience, Amsterdam, The Netherlands, 4Department of Cardiology, Amsterdam University Medical Centre, Amsterdam Neuroscience, Amsterdam, The Netherlands, 5 Department of Rheumatology, Isala Hospital, Zwolle, The Netherlands, 6Department of Experimental Immunology, Amsterdam institute for Infection & Immunity, Amsterdam UMC, Amsterdam, The Netherlands, 7Amsterdam Rheumatology and Immunology Center, Amsterdam UMC, Department of Rheumatology and Clinical Immunology, University of Amsterdam, Amsterdam, The Netherlands, 8Department of Immunopathology, Sanquin Research and Landsteiner Laboratory AMC/UvA, Amsterdam, The Netherlands, 9Department of Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, The Netherlands, 10Department of Laboratory Medicine, Laboratory of Medical Immunology, Radboud University Medical Center, Nijmegen, The Netherlands, 11Department of Microbiology and Immunology, Rijnstate Hospital, Arnhem, The Netherlands, 12Department of Medical Microbiology and Immunology, St Antonius Hospital, Nieuwegein, The Netherlands, 13Department of Immunology, Erasmus Medical Center, Rotterdam, The Netherlands, Rotterdam, The Netherlands
Background: Myocarditis without myositis has been described following mRNA SARS-CoV-2 vaccination. The literature on post-vaccine antibody mediated myositis is limited and to date no case series have been reported with a distinct clinical syndrome and a single myositis specific antibody, related to SARS-CoV-2 mRNA vaccination or COVID-19.
Over a 6-month period in 2021, 54 patients were referred to our tertiary referral centre for suspected myositis. Out of 25 patients with a diagnosis of myositis, we identified three patients with a distinct clinical syndrome with myositis and myocarditis with anti-Jo-1 antibodies, following SARS-CoV-2 mRNA vaccination (BNT162-Pfizer-BioNtech; n=2) or following a mild COVID-19 infection (n=1).
Results: Three patients (one woman, two men; 49, 50 and 58 years old) developed progressive muscle weakness and muscle pain following either vaccination (patient 1 and 2) or mild COVID-19 infection (patient 3). Patients 2 and 3 had a history of anti-CCP positive rheumatoid arthritis (RA), which had been untreated for three years in patient 2.
Both post-vaccine cases had severe pitting edema of the legs, patient 2 also had arthritis. None of the patients had mechanic’s hands, Raynaud’s phenomenon, or interstitial lung disease (ILD). The time interval between the SARS-CoV-2 trigger and the onset of progressive muscle weakness was between 10 and 14 days (patient 1 and 3) and was estimated between three and seven days in patient 2. Laboratory tests showed highly elevated CK levels (17-32 times upper limit of normal (ULN)) and troponine T levels (14-34 times ULN). In patient 2, in addition, troponin I was tested (42 times ULN), which is more specific for myocardial involvement. In patient 1 supraventricular tachycardia, unspecific ST- and T-wave abnormalities and elevated NTproBNP were found. In all patients, testing for myositis specific antibodies (MSAs; EUROline myositis 16 Ag. line-blot assay) showed anti-Jo-1 antibodies (semi-quantitatively in the highest possible range). Muscle MRI showed widespread muscle edema in all patients and extensive fascial and subcutaneous edema in the legs in the post-vaccine cases (figure 1). Muscle biopsies showed inflammatory myopathy. Cardiac MRI showed abnormalities in all patients: pericardial effusion and/or late contrast enhancement of the epicardial myocardium (figure 1). All patients showed major improvement in response to immunosuppressive therapy and could discontinue high-dosed steroids after three and six months.
Discussion: In conclusion, we report three patients with a distinct clinical picture of anti-Jo-1 myositis and myocarditis without ILD, following SARS-CoV-2 mRNA vaccination or COVID-19. Although it is difficult to determine a causal relationship between SARS-CoV-2 and anti-Jo-1 myositis based on these small numbers, we suspect a SARS-CoV-2 trigger of anti-synthetase syndromes given the typical combination of symptoms and previously demonstrated association with antecedent viral infections.
In addition, we have collected nationwide data on myositis specific antibodies (MSAs) in 2019 (pre-COVID-19) and 2021 (during COVID-19) from six medical centers in the Netherlands. We are currently analysing these data to examine whether the proportion of positive MSAs in 2021 is higher as compared to 2019. The results will be presented at the ICNMD.
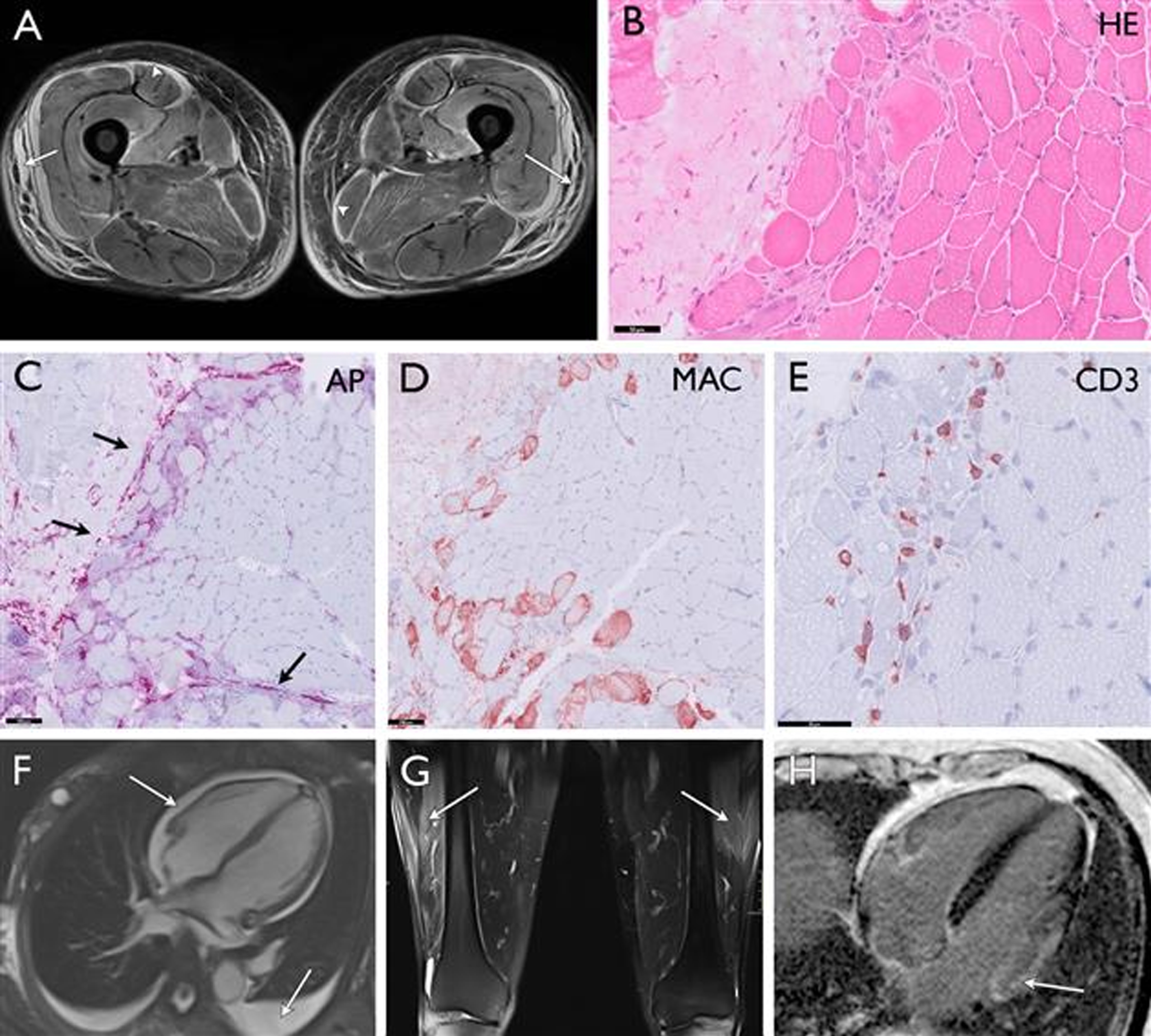
eP01.04.04Guillain-Barre Syndrome in 220 Patients with COVID-19.
Finsterer J1, Scorza F1
1Neurolgy Neurophysiolgy Center, Postfach 20, Austria
Objectives: Guillain-Barre syndrome (GBS) is an established manifestation of neuro-COVID. Here we summarise and discusses recent findings concerning the pathophysiology, clinical presentation, diagnosis, treatment and outcome of SARS-CoV-2-associated GBS (SC2-GBS).
Methods: Literature review
Results: By the end of December 2020, at least 220 patients with SC2-GBS have been published in 95 papers. SC2-GBS is most likely secondary due to an immune reaction against SARS-CoV-2 since the virus has not been found in the CSF of any SC2-GBS patient so far reported. SC2-GBS occurs in each age group, does not differ from non-SC2-GBS regarding clinical presentation and treatment but the outcome of SC2-GBS is worse compared to non-CS2-GBS patients, and the prevalence/incidence of GBS most likely increased since the outbreak of the pandemic.
Conclusions: SC2-GBS is most likely secondary to an immune reaction against SARS-CoV-2 since the virus has not been found in CSF of any SC2-GBS patient reported. SC2-GBS occurs at any age. SC2-GBS does not differ from non-SC2-GBS regarding clinical presentation and treatment but the outcome of SC2-GBS is worse compared to non-CS2-GBS patients. The prevalence/incidence of GBS most likely increased since the outbreak of the pandemic. Since there are no studies about the optimal treatment of SC2-GBS subtypes available, they should be treated empirically in the same way as non-SC2-GBS subtypes. Early diagnosis of SC2-GBS is warranted because if appropriate treatment is applied in due time the overall outcome from the infection may improve.
eP01.04.05IVIg Treatment in Chronic Inflammatory Neuropathies During the COIVD19 Pandemic
Baranska J1, Lipowska M1, Sobieszczuk E1, Kierdaszuk B1, Kostera- Pruszczyk A1
1Department of Neurology, Medical University of Warsaw, Warsaw 02-097, Poland
During the COVID19 pandemic, there were changes in the organization of healthcare together with immunoglobulins shortage, which had influence on cyclic treatment regimens in some patients. In Poland, intravenous immunoglobulin (IVIg) treatment is used only as inpatient treatment, which was challenging during the COVID19 pandemic. The aim of the study was to assess how the COVID19 pandemic influenced the therapy regimen and the course of the disease in patients with chronic inflammatory demyelinating polyneuropathy (CIDP) and multifocal motor neuropathy (MMN) treated with cyclic IVIg.
Materials and methods: 15 patients were cyclically treated with IVIg our center: 8 with MMN (5F and 3M) mean age 54.6 years and 7 with CIDP (3F and 4M) mean age 51 years. Three periods were compared: before the pandemic (Jan.2019-Feb.2020) and two periods during the pandemic: I (Mar-Dec.2020), II (Jan-Dec.2021). The following parameters were assessed: mean doses (g / kg bw / week), mean intervals between drug administrations (weeks) and treatment efficacy assessed using functional scales: for MMN-INCAT, MMN-RODS, for CIDP-INCAT.
Results: During the pandemic, lengthening of the intervals between administrations or reduction of the doses per treatment were observed in the majority of patients. A reduction in mean doses was demonstrated in 4/8 patients with MMN and CIDP in 1/7 compared to period I pandemic and in 3/7 compared to period II pandemic. Due to disease progression, the mean dose was increased in 3/8 patients with MMN. Prolongation of intervals was observed in 3/8 patients with MMN and 3/7 with CIDP. Most patients had a point in time when symptoms worsened (worsening of INCAT by at least 1) due to lack of treatment in MMN in 6/8 and in CIDP in 5/7, reversible with increased dose. The extension of the intervals resulted not only from the pandemic restrictions, but from the limited availability of IVIg as well. No statistically significant differences were found between the mean periods, between consecutive IVIg administrations and the mean IVIg doses in the periods before the pandemic and during the pandemic (I, II) and functional assessments at the end of analyzed period.
Conclusion: The limited availability of IVIg and epidemiological restrictions during the COVID19 pandemic required modification of treatment regimens. Drug dose reduction and prolongation of the intervals between drug administrations, lead in the majority of CIDP patients to temporary deterioration in functional status, reversible by increased dosage of IVIg.
eP01.04.06Quality of Life of Myasthenia Gravis Patients During COVID-19 Pandemic – One Year Follow Up
Stojanov A1, Djordjevic G, Stojanov J, Petrovic S
1Clinic Of Neurology, Unniversity Clinical Center Nis, Nis, Serbia
Purpose: The coronavirus disease 2019 (COVID-19) is the largest pandemic of our times. Pandemics are severe stressors to vulnerable groups (such as patients with chronic diseases) and this highly contagious disease exerts considerable impacts on mental health. We wanted to investigate the possible impact of COVID-19 pandemic on the quality of life (QoL) of myasthenia gravis (MG) patients and potential changes during the period of one year.
Methods: Data on the epidemiological and clinical characteristics of MG was collected. We used a self-designed questionnaire (consisting of 12 questions), a revised 15-item Myasthenia Gravis Quality of Life Questionnaire (MGQOL15r), a 36-item health survey of the Medical Outcomes Study Short Form (SF36), Hamilton scales for the assessment of anxiety (HAM-A) and depression (HAMD) were used. The actual severity of the clinical manifestation was estimated using MG activities of daily life (MGADL). We reassessed patients in April and May 2021, who were tested during April 2020 using the same questionnaires.
Results: The study included 57 adult MG patients. We noticed a statistically significant difference between the results obtained at these two time points regarding scores on MGQOL15r (p<0.05). The obtained scores were significantly better in 2021. Some scores on SF-36 subscales were also better in 2021 than in 2020 (such as social functioning, emotional well-being, role limitation due to emotional problems) (p<0.05). MGQOL15r and SF36 scores correlate with severe clinical manifestation, high scores on HAM-A and HAM-D (p<0.01). Higher scores on HAM-D and fear that MG symptoms will be worse if the patient gets an upper respiratory infection were independent predictors of the lower SF36 scores. Regarding MGQOL15r independent predictors of the higher score were higher scores on HAM-D.
Conclusions: QoL of myasthenia gravis patients improved during the pandemic. It is important for healthcare workers to provide professional therapeutic advice and psychosocial support for this population of patients during the pandemic.
eP01.04.07COVID Spike Antibodies in Neuromuscular Conditions: A KU Experience
Pasnoor M1, Tajuddin A1, Jawdat O1, Farmakidis C1, Jabari D1, Heim A1, Higgs K1, Dimachkie M1
1The University Of Kansas Medical Center, Kansas City, United States
Background: Little is known about the immune response to COVID vaccination in immune suppressed neuromusuclar patients. Multiple studies showed variable data regarding the effect of immunosuppression on the immune response to the vaccination.
Objective: To evaluate the COVID spike antibody levels in patients with various neuromuscular conditions who received vaccination and assess the effect of immunosuppressive therapies on antibody levels.
Method: We performed a retrospective chart review of patients in neuromuscular clinic who had COVID antibody testing. We collected demographic, clinical, diagnostic and treatment information. Descriptive statistics was performed on the data obtained.
Results: The total number of patients enrolled in the study were129, 64 male and 65 female . The mean age of the patients enrolled was 64.82 ±14.50 and the mean duration of antibody acquisition since date of last vaccination dose was 172 days. The number of patients on immunosuppressive therapies was 88(68.2%), 47(53.41%) had high antibody titer (>250), 31(35.23%) had low antibody titer (0.4-250), 9(10.23%) had undetected antibody titer (<0.4) and 1 (1.1%) had detected unmeasured titer. Immunosuppressive therapies included steroids, methotrexate, azathioprine, myophenolate mofetil, eculizumab, efgartigimod, intravenous immunoglobulin and rituximab. The other group of patients who were not on immunosuppressive medication were 41 (31.8%), 28 (68.29%) showed high antibody titer (>250), 9(21.95%) showed low antibody titer (0.4-250), 1(2.44%) showed undetected titer (>0.4) and 3(7.32%) had detected unmeasured titer. 50% of patient who were on rituximab showed undetected antibody titer and 60% of patients who received eculizumab had low antibody titer. 45% of patients on immunosuppressive drugs did not develop adequate spike antibodies levels to vaccination, compared to 24% on no immunosuppressive therapies.
Conclusions: Our study reveals modest impact of immunosuppression on COVID spike antibody titer. While this finding is limited by small number of patients and heterogeneity in therapies, age and interval between vaccination and antibody testing, our finding supports the importance of booster vaccine in this patient population.
eP01.04.08Parsonage-Turner Syndrome after COVID-19 Vaccination
Van Boxstael E1, Maldonado Slootjes S1, Dorban S1
1CHU UCL Namur, Godinne, Yvoir, Belgium
Parsonage-Turner syndrome (PTS) is an inflammatory disorder of the brachial plexus, which typically presents as severe neuropathic pain, followed by rapid multifocal weakness and amyotrophy of the upper limb. Triggering events such as infection, vaccination and trauma have been described.
We present the case of a 53-year-old man who developed PTS twelve days after receiving a second dose of the AZD1222 vaccine against COVID19 (Oxford/AstraZeneca), as confirmed by electroneuromyography and MRI of the brachial plexus. As common triggering events were excluded, we consider the AZD1222 vaccine as possible trigger.
Even though causality cannot be proven, we present this case of PTS following COVID-19 vaccination to raise awareness amongst caregivers to identify this underrecognized plexopathy timely. We will discuss PTS pathophysiology and the possible link between PTS and COVID19 vaccination.
eP01.04.09Multi-Centre Study to Assess the Safety of Alglucosidasi and of Laronidasi in Home Infusion Setting
Scarpa M, Parini R, Sacchini M, Crescimanno G, Fiumara A, Fischetto R, Gasperini S, Siciliano G, Ravaglia S, Sechi A, Piperno A, Cianci V, Maggi L, Musumeci O, Taurisano R, Verrecchia E, Toscano A1
1Sanofi, Milano, Italy
Background – During Coronavirus Disease-19 (COVID-19) pandemic, the temporary and exceptional authorization 341/2020 0f the Italian Drug Agency (AIFA), allowed to guarantee the adherence to treatment for patients with lysosomal storage disorders (LSDs) through home therapy. Stable patients affected with Pompe Disease and Mucopolysaccharidosis type I (MPS I) could then receive regular Enzyme Replacement Therapy (ERT) infusions at home. Indeed, a data collection seemed to us a good opportunity to assess the safety of home infusions and thus fill the existing information gap.
Methods - This is an Italian, multicenter, non-interventional, double cohort study sponsored by Sanofi Genzyme with both retrospective and prospective data collection to obtain safety information on ERT treatment of Pompe Disease and Mucopolysaccharidosis type I (MPS I) patients in a home-care setting. The study will enroll 60 patients at 15 sites. The retrospective observation will start from the first ERT infusions in a homecare setting and the prospective observation will last after 12 months from the enrollment. During the control visits, investigators will administer the questionnaires and will record any documented clinical data occurred during the home infusions.
Objectives - This study aims at obtaining safety information on patients with Pompe Disease treated with Myozyme® (alglucosidase alfa) and of patients with MPS I treated with Aldurazyme® (laronidase) in a home-care setting, as well as evaluating personal satisfaction of both cohorts of patients and documenting infusion compliance.
Conclusions – The outcomes will mirror real-life management of patients in home-care infusion setting, including safety profile, treatment compliance and quality of life.
Keywords: COVID-19, Lysosomal storage disorders, Enzyme Replacement Therapy, Pompe Disease, MPS I, Alglucosidase alfa, Laronidase, Home-care setting, Safety profile, Quality of Life
eP01.04.10Clinical Course of Four Neuromuscular Disease Patients Infected COVID-19
Ishii A1, Tsuji H1, Mamada N1, Ogawa R1, Kurihara Y1, Hitomi S
1University Of Tsukuba, Tsukuba, Japan
Introduction: Information on COVID-19 infection prevention measures and vaccines for patients with neuromuscular diseases has been sufficiently disseminated, but the details of the actual course of infected patients are rarely directly involved by neurologists. We report four cases of COVID-19 with neuromuscular diseases and were able to observe their progress.
Subjects: 1 case of multiple sclerosis (MS), 1 case of chronic inflammatory demyelinating polyradiculoneuropathy / dermatitis (CIDP / DM), 1 case of limb-girdle muscular dystrophy (LGMD) and one Duchenne muscular dystrophy carrier (DMD-C) were examined.
Result: MS: A 32-year-old man who was taking fingolimod, but improved by waiting at home, and he did not relapse. CIDP / DM: 54-year-old female, PSL, taking tacrolimus, using remdesivir for pneumonia. After recovery, peripheral neuropathy worsened, and steroid pulse treatment was added. LGMD: 48-year-old female Although she had pneumonia, she did not need to be ventilated and improved with only oxygen administration and favipiravir without deterioration of% VC. DMD-C: 59-year-old female, improved only by oxygen administration. The DMD (second son, 29 years old) who were cared by her was hospitalized because no one could care him.
Discussion: All cases were affected prior to vaccination. Regarding CIDP, there was a case report of deterioration after illness, and this case also deteriorated and required treatment. LGMD / DMD-C did not show any deterioration in respiratory function. It is a study of a small number of cases, and it is necessary to accumulate future cases.
eP01.05.01Muscle Inflammation Drives Mitochondrial Dysfunction in Inclusion Body Myositis
Meyer S1, Scholle L2, Zechel S3, Kummer S4,5, Kazmaier L1, Götze R1, Kummer K1, Zierz S2, Stadelmann C3, Sehmisch S6,7, Lorenz H6, Hell A6, Zschüntzsch J1, Schmidt J1,8,9
1Department of Neurology, University Medical Center Göttingen, Göttingen, Germany, 2Clinic of Neurology, University Hospital Halle, Halle, Germany, 3Institute of Neuropathology, University Medical Center Göttingen, Göttingen, Germany, 4Department of Infectious Diseases, Virology, Heidelberg University Hospital, Heidelberg, Germany, 5Risk Group 4 Pathogens – Stability and Persistence, Biosafety Level-4 Laboratory, Center for Biological Threats and Special Pathogens, Robert Koch Institute, Berlin, Germany, 6Department of Trauma Surgery, Orthopedics and Plastic Surgery, University Medical Center Göttingen, Göttingen, Germany, 7Department of Trauma Surgery, University Medical Center Hannover, Hannover, Germany, 8Department of Neurology and Pain Treatment, University Hospital of the Brandenburg Medical School Theodor Fontane, Immanuel Klinik Rüdersdorf, Rüdersdorf bei Berlin, Germany, 9Faculty of Health Sciences Brandenburg, Brandenburg Medical School Theodor Fontane, Rüdersdorf bei Berlin, Germany
Background: Inclusion body myositis (IBM) is an inflammatory myopathy in patients above fifty years of age, presenting with progressive muscle weakness and wasting. Histopathological findings include autoreactive inflammation, muscular protein degeneration as well as mitochondrial impairment. Mitochondrial changes characterized by COX-deficiency, mtDNA-mutations and pathological mitochondrial proliferation are abundantly present in IBM compared to other forms of myositis. The role of mitochondria during IBM pathogenesis has yet remained elusive. We hypothesized that chronic muscle inflammation can trigger mitochondrial damage, which would diminish levels of ATP and cause muscle weakness and wasting.
Methods: Expression of genes responsible for mitochondrial fusion and fission (DNM1L, Fis1, Mfn1/2) was assessed by RT-PCR for 14 IBM patients and 10 healthy donors. Muscle tissue samples were obtained from diagnostic biopsies of four patients with IBM and compared to two non-myopathic controls regarding expression of markers of inflammation, protein degeneration and mitochondrial damage. Human primary myotubes were studied in a well-established inflammatory cell culture model of IBM that allowed assessment of mitochondrial morphology and function. Mediators of mitochondrial fission and fusion were examined by Western Blot and immunocytochemistry. Mitochondrial function was assessed via spectrophotometry of COXIV activity as well as seahorse metabolic cell analysis.
Results: Expression of MHCI, αB-crystallin, iNOS, 6E10 and p62 was increased in IBM muscle samples compared to controls. Mitochondrial damage was evidenced by an increased rate of COX-deficiency and increased TOM20 levels. A significant co-upregulation of TOM20 and MHCI (p<0.0001) as well as TOM20 and αB-crystallin (p<0.0001) was observed. Non-myopathic individuals showed a positive correlation between mitochondrial fusion/fission genes and age, whereas this correlation was absent in IBM, suggestive of dysregulated mitochondrial function. In vitro, exposure to pro-inflammatory cytokines IFN-γ and IL-1β led to a shift of the mitochondrial network morphology towards fragmentation. STED microscopy after 72 hours of cytokine exposure revealed a mitochondrial fragmentation rate of 37.9% compared to 16.8% in the control group (p=0.011). Protein level of the fission mediator Drp1 was increased and COX IV activity was significantly reduced after 72hours of cytokine exposure (p=0.013). Seahorse analysis identified a trend towards reduced ATP production upon pro-inflammatory cytokine exposure of muscle cells.
Conclusion: We demonstrate a dysregulation of mitochondrial fusion and fission in IBM, suggesting a disturbance of the mitochondrial quality control in this disorder, which can readily explain the known mitochondria pathology in IBM muscle. Our ex vivo data demonstrate that, in IBM muscle, inflammation and mitochondrial distress act in concert, whereas in vitro findings clearly showed that inflammation can induce mitochondrial fission and disrupt mitochondrial function. The data help to explain how myoinflammation leads to mitochondrial dysfunction with consecutive muscle weakness and wasting. Further studies are necessary to elucidate implications on mitophagy and accumulation of unwanted proteins.
Acknowledgments: We thank Iris Iben for excellent technical support. We thank our patients and donors of muscle specimen for their voluntary participation.
eP01.05.02B Cell Receptor Profiling before and after IVIg Treatment in Idiopathic Inflammatory Myopathies
Anang D1,2,3, Walter H4, Lim J4, Niewold I1,3, van der Weele L1,2, Aronica E5, Eftimov F4, Raaphorst J4, van Schaik B6, van Kampen A6, van der Kooi A4, de Vries N1,2
1Department of Rheumatology and Clinical Immunology, Amsterdam Rheumatology and Immunology Center, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands., Amsterdam, The Netherlands, 2Department of Experimental Immunology, Amsterdam Infection and Immunity Institute, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands., Amsterdam, The Netherlands, 3Department of Genome analysis, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands., Amsterdam, The Netherlands, 4Department of Neurology, Amsterdam UMC, University of Amsterdam, Amsterdam Neuroscience, Amsterdam, The Netherlands., Amsterdam, The Netherlands, 5Department of (Neuro)Pathology, Amsterdam Neuroscience, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands., Amsterdam, The Netherlands, 6Bioinformatics Laboratory, Department of Epidemiology and Data science, Amsterdam Public Health Institute, Amsterdam Infection and Immunity Institute, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands., Amsterdam, The Netherlands
Objective: To gain novel insights into the role of B cells in the pathogenesis of newly diagnosed idiopathic inflammatory myopathies (IIMs), and to explore if the B cell receptor (BcR) repertoire changes over time after treatment with intravenous immunoglobulin (IVIg) monotherapy.
Methods: Next generation sequencing at the RNA level was used to study the BcR repertoire in muscle tissues collected at diagnosis before IVIg treatment as well as in peripheral blood (PB) collected before and nine weeks after IVIg treatment in treatment naïve IIM patients. Results were correlated to patients’ clinical improvement based on the total improvement score (TIS) (score ≥40 responders).
Results: Prior to IVIg treatment, BcR clones found in muscle tissue could be retrieved in peripheral blood. In peripheral blood 9 weeks after IVIg treatment, new patient specific dominant BcR clones were formed and pre-treatment dominant BcR clones disappeared. The cumulative frequency of all dominant BcR clones before treatment was significantly higher in individuals who responded to IVIg compared to those who did not respond to IVIg. Over time at follow up, a decrease in the cumulative frequency of dominant clones correlated with an increased TIS.
Conclusion: In IIMs, BcR clones present in muscle tissue can be detected in peripheral blood. The correlation between a higher cumulative frequency of dominant BcR clones at baseline and eventual treatment response suggests that response to IVIg may depend on the composition of the pre-treatment BcR repertoire.
eP01.05.03Multi-Muscle Pathology Assessment in Inclusion Body Myositis: Post-mortem Study in Two Cases
Glaubitz S1, Kummer K1, Götze R1, Zechel S2, Mittelbronn M3, Harter P4, Heide S5, Flössel U5, Schmidt J1,6,7
1Muscle Immunobiology Group, Department of Neurology, Neuromuscular Center, University Medical Center Göttingen, Göttingen, Germany, 2Institute of Neuropathology, University Medical Center Göttingen, Göttingen, Germany, 3National Center of Pathology & Luxembourg Center of Neuropathology, Dudelange, Luxembourg, 4Institute of Neurology, University Hospital Frankfurt, Frankfurt, Germany, 5Institute of Forensic Medicine, Dresden Technical University, Dresden, Germany, 6Department of Neurology and Pain Treatment, University Hospital of the Brandenburg Medical School Theodor Fontane, Immanuel Klinik Rüdersdorf, Rüdersdorf bei Berlin, Germany, 7Faculty of Health Sciences Brandenburg, Brandenburg Medical School Theodor Fontane, Rüdersdorf bei Berlin, Germany
Introduction: Inclusion body myositis (IBM) is an inflammatory myopathy leading to slowly progressive weakness and atrophy of arms and legs, particularly the finger flexors and knee extensors. The pathology includes inflammatory infiltrates and degenerative muscle damage. Cell stress cascades around nitric oxide and αB-crystallin are thought to be crucial factors of disease progression. However, a better understanding of the temporal and mechanistic interrelationship between inflammation, cell stress and protein accumulation could help to design more effective treatment strategies.
Methods: Two clinically well-characterized IBM patients from our neuromuscular center were studied after autopsy. Informed consent had been obtained. One patient was only mildly affected (IBM-FRS= 26) while the other had severely impaired mobility and suffered from dysphagia (IBM-FRS= 11). All major skeletal muscles from arms and legs were subjected to a histopathological work up that included standard morphology (H&E, trichrome), enzyme histochemistry (COX/SDH etc.) as well as immunochemical assessment for inflammation (MHC-I, CD8), cell-stress (COX, αB-crystallin) and intracellular protein accumulation (p62). Muscle fibers that showing pathological results in those stainings, especially regarding morphological abnormalities such as vacuoles, necrosis, atrophy etc. were quantitatively analyzed. A correlation analysis was performed between histological abnormalities and clinical symptoms such as muscle weakness.
Results: Different patterns of histopathological changes were observed in the various muscle groups and both patients differed as well. Whereas some muscles showed a marked deposition of p62, others displayed much more inflammatory or mitochondrial pathology. A significant correlation was noted between the frequency of αB-crystallin positive fibers and the inflammatory marker MHC-I in the milder affected patient. There was a positive correlation between increased fiber diameters and COX deficient fibers in the severely affected patient. Rimmed vacuoles were detected more frequently in the more severely affected patient. Clinically affected muscles showed a higher proportion of atrophic fibers.
Discussion: The combined analysis of clinical parameters and definition of a histopathological spectrum of post-mortem pathology including atrophy, hypertrophy, p62, CD8+ cells, αB-crystallin, rimmed vacuoles, and MHC class I in skeletal muscles from IBM patients with different disease severities can serve as a valuable contribution to help to differentiate the complex pathogenesis of inclusion body myositis in the future.
eP01.05.04Design of a Global Phase 2/3 Randomized, Placebo-Controlled Trial of Ravulizumab in Adult Dermatomyositis
Aggarwal R1, Cavagna L2, Iikuni N3, Rakhade S3
1University of Pittsburgh Medical School, Pittsburgh, United States, 2IRCCS Policlinico S. Matteo Foundation, University of Pavia, Pavia, Italy, 3Alexion, AstraZeneca Rare Disease, Boston, United States
Introduction: Dermatomyositis (DM), a rare and life-altering chronic immune-mediated disease, is characterized by distinct skin rashes and/or progressive muscle weakness and other systemic manifestations. Established DM medications (e.g., high-dose systemic steroids and immunosuppressive therapies, frequently prescribed off-label) inadequately control symptoms and/or lead to severe adverse effects in many patients,1_3 highlighting the need for new DM treatments with improved risk-benefit profiles.
The classical complement pathway, including endothelial deposition of the C5b-9 membrane attack complex (MAC), is thought to play a key role in DM organ damage.4 Approved for the treatment of several complement-mediated diseases, the long-acting monoclonal antibody ravulizumab targets the terminal complement protein C5, causing its immediate, complete, and sustained inhibition and preventing its cleavage to form MAC and pro-inflammatory mediators.5
Objective: Given the need for new DM therapies and the implication of terminal complement proteins in DM pathophysiology, we designed a global, double-blind, randomized, Phase 2 (Part A)/Phase 3 (Part B) trial to evaluate the efficacy and safety of ravulizumab compared with placebo in adults with DM (ALXN1210-DM-310; NCT04999020; EudraCT2021-001200-15).
Methods: A total of 180 adults with DM6 who have active disease with muscle weakness and inadequate responses or intolerances to two or more DM medications will be randomized to the ravulizumab or placebo arm, with treatment delivered intravenously as a loading dose followed by maintenance dosing once every eight weeks. Parts A and B will contain different participants and each part will consist of a screening period, a randomized, controlled period (Part A; 26 weeks; Part B: 50 weeks), and an open-label extension period. The primary endpoints are the percent of participants with a ≥20-point improvement from baseline on the ACR/EULAR Myositis Response Criteria Total Improvement Score (TIS20) at 26 weeks (Part A) and 50 weeks (Part B). The trial also includes a variety of secondary and exploratory outcome measures, e.g., mean changes from baseline in the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) activity score and a novel participant-reported assessment, the Dermatomyositis-Disease Symptoms Questionnaire (DM-DSQ). Safety evaluations will focus on the incidence rates of treatment-related adverse events (TEAEs), e.g., those classified as serious and/or leading to treatment discontinuation.
Results: The ALXN1210-DM-310 trial is currently enrolling patients in multiple countries.
Conclusions: ALXN1210-DM-310 is the first global, randomized, placebo-controlled Phase 2/3 interventional trial designed to evaluate the efficacy and safety of a C5 inhibitor in adult patients with DM who have active disease despite treatment with standard medications.
References:
1. Joffe MM, Love LA, Leff RL, et al. Am J Med.1993; 94:379.
2. Zieglschmid-Adams ME, Pandya AG, Cohen SB, et al. J Am Acad Dermatol. 1995; 32:754.
3. Kissel JT, Levy RJ, Mendell JR, Griggs RC. Neurology 1986; 36:35.
4. Yang SH, Chang C, Lian Z-X. J Transl Autoimmun. 2019;2:100018.
5. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761108s012lbl.pdf. Accessed January 26, 2022.
6. Lundberg IE, Tjarnlund A, Bottai M, et al. Arthritis Rheumatol. 2017;69(12):2271-2282.
eP01.05.05A Prospective Diagnostic Accuracy Study of Multi-Modality Testing in Patients Suspected of a Treatable Iim
Kamperman R1, Walter H1, Raaphorst J1, Verhamme C1, Koelman J1, Potters W1, Hemke R2, Smithuis F2, Aronica E3, van Leeuwen E4, Baars P4, de Visser M1, van Schaik I1,6, Bossuyt P5, van der Kooi A1
1Department of Neurology and Clinical Neurophysiology, Amsterdam University Medical Centre, University of Amsterdam, Amsterdam Neuroscience, Amsterdam, The Netherlands, 2Department of Radiology and Nuclear Medicine, Amsterdam University Medical Centre, Amsterdam Movement Sciences, Amsterdam, The Netherlands, 3Department of (Neuro)Pathology, Amsterdam University Medical Centre, Amsterdam Neuroscience, Amsterdam, The Netherlands, 4Department of Experimental Immunology, Amsterdam institute for Infection & Immunity, Amsterdam, The Netherlands, 5Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Amsterdam University Medical Centre, Amsterdam Neuroscience, Amsterdam, The Netherlands, 6Spaarne Gasthuis, Haarlem, The Netherlands
Introduction: Idiopathic inflammatory myopathies (IIMs) excluding inclusion body myositis (IBM) are a group of heterogeneous auto-immune disorders characterised by subacute onset and progressive proximal muscle weakness which are frequently part of a multisystem auto-immune disorder. The clinical symptoms and signs differ widely between patients at disease onset and reaching a correct diagnosis in a timely manner can be challenging. There is no gold standard for the diagnosis of IIM. Diagnostic modalities include serum creatine kinase (sCK) activity, muscle imaging (magnetic resonance imaging (MRI) or ultrasound (US)), electromyography (EMG), myositis auto-antibody testing and muscle biopsy. Several diagnostic criteria have been developed for IIMs, varying in reported sensitivity and specificity.
Hypothesis: We hypothesize that an evidence-based diagnostic strategy, using fewer and preferably the least invasive diagnostic modalities, can achieve the accuracy of a complete panel of diagnostic tests, including MRI, US, EMG, myositis-specific auto-antibody testing and muscle biopsy.
Study design: The OptimizAtion of Diagnostic Accuracy in idioPathic inflammaTory myopathies (ADAPT) study is a prospective diagnostic accuracy study with an over-complete study design. This means that all consenting participants undergo standardized history taking, physical examination, standard laboratory testing (including sCK), muscle imaging by whole body muscle MRI and muscle US, EMG, myositis auto-antibody testing, and muscle biopsy. One-hundred patients suspected of an idiopathic inflammatory myopathy excluding IBM will be included. To be eligible, a patient must be suspected of an IIM based on symmetrical proximal muscle weakness causing a functional limitation that justifies treatment with high dose glucocorticoids, with an onset of symptoms ≤24 months before inclusion. A reference diagnosis will be assigned by an expert panel using all clinical information and all results of all ancillary tests available, including 6 months follow-up. Several predefined diagnostic strategies will be compared against the reference diagnosis to find the optimal diagnostic strategy. In addition, the patient burden of the ancillary investigations will be assessed and compared between the diagnostic modalities.
Discussion: Although the diagnostic accuracy of some of the previous mentioned diagnostic modalities have been studied before, to the best of our knowledge, no previous study has examined a complete diagnostic panel for myositis. Our prospective study enables the evaluation of the diagnostic accuracy of individual items and procedures and of the incremental value of multi-test diagnostic strategies, and the assessment of the burden of each diagnostic test.
eP01.05.06Fibroblast Model Unveils New Molecular Insights in Inclusion Body Myositis
Cantó-Santos J1, Valls-Roca L1, Tobías E1, García-García F1, Moreno-Lozano P1, Milisenda J1, Grau-Junyent J1, Garrabou G1
1Muscle Research and Mitochondrial Function Lab - Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) - Faculty of Medicine and Health Sciences, University of Barcelona - Hospital Clinic of Barcelona – CIBERER, Barcelona, Spain
Inclusion body myositis (IBM) is an inflammatory myopathy characterized by proximal and distal muscle weakness, in addition to inflammatory infiltrate, rimmed vacuoles and mitochondrial changes in myofibers. IBM etiology is not well-known, and there are no biomarkers or effective treatments, in part due to the lack of validated disease models. Here we present the use of patients’ derived fibroblasts as a disease model by identifying muscle pathological hallmarks at transcriptomic (778 deregulated expressed genes) and functional level (increased inflammation, impaired autophagy and dysfunctional mitochondria). These findings confirm the presence of molecular alterations in peripheral tissues from IBM patients and propose patients’ derived fibroblasts as a promising disease model, that may be eventually exported to other neuromuscular disorders. We additionally reveal new molecular players in IBM associated with disease progression, prompting to delve into disease etiology, in the discovery of novel biomarkers and in testing new therapeutic strategies.
Funding: FIS PI1800498 and PI2100935 granted by ISCIII and FEDER
eP01.05.07Prevalence and Clinical Correlation of Myositis-Specific and Myositis-Associated Autoantibodies in Indian Myositis Cohort
Jassal B1, Dhall A1, Vishnu V1, Chakrabarty B1, Bhadu D1, Gulati S1, Bhatia R1, Kumar U1, Suri V1, Sharma M1
1All India Institute Of Medical Sciences, New Delhi, India
Abstract: Background: Idiopathic inflammatory myopathies (IIMs), collectively known as myositis, are a heterogeneous group of systemic autoimmune disorders. Approximately two-thirds of the IIM patients carry myositis-specific or myositis-associated autoantibodies which aid in creating homogenous subsets of myositis.
Objective: This study aimed at estimating the prevalence and evaluating the clinical correlations of MSAs and MAAs in Indian IIM patients.
Methods: Patients satisfying Bohan and Peter’s criteria were recruited between 2019- 2021 and their clinical data were obtained from the archive. Sera of 66 IIM patients and 16 healthy controls were tested for the antibodies against Mi-2α/β, TIF1γ, MDA5, NXP2, SAE1, Ku, PM-Scl100, PM-Scl75, Jo-1, SRP, PL-7, PL-12, EJ, OJ, Ro-52 using commercial line blot (Euroimmun, Germany). Serum samples were also screened for Anti-HMGCR autoantibody by QUANTA Lite HMGCR ELISA.
Results: Out of the 66 patients, 39 serum samples (59.09%) had either MSA or MAA whereas 27 patients (40.90%) and 16 healthy controls (100%) were seronegative for all of the tested autoantibodies. Only a single patient showed the presence of more than one MSA. Ten patients revealed the coexistence of MSA with MAA; anti-Ro52 being the most concurrent. Anti-Ro52 (14, 21.21%) was the most prevalent antibody among this cohort, followed by anti-Mi2α/β (13, 19.69%), anti-NXP2 & anti-SRP (5, 7.57%), anti-PL-7 &anti-PM-Scl75 (3, 4.54%) and anti-TIF1γ (2, 3.03%). Anti-Ro52 was associated with dysphagia (5, 35.71) and celiac disease (2, 14.28), anti-Mi2α/β with heliotrope rash (4, 30.76%), and gottron’s papules (3, 23.07%) in dermatomyositis patients. Lung involvement and alopecia were observed in anti-TIF1γ positive patients.
Conclusion: Two-thirds of the IIM patients had MSA/MAA whereas one-third of patients were seronegative. Although in some cases MSA coexisted with MAA, MSAs were almost always mutually exclusive. Clearly, myositis-related autoantibodies are associated with clinical manifestations but to establish a particular clinico-serological correlation large sample size is needed to establish a definitive correlation.
eP01.05.08Immune-Mediated Necrotizing Myopathy: An Emerging Disorder
Portela Sánchez S1, Sánchez Soblechero A1, Catalina Álvarez I1, Lozano Ros A1, López Muñoz S2, Sola Vendrell E1, Sánchez Mateos P1, Muñoz Blanco J1
1Hospital General Universitario Gregorio Marañón, Madrid, Spain, 2Hospital Universitario La Paz, Madrid, Spain
Introduction and objectives: Immune-mediated necrotizing myopathies (IMNMs) comprise a group of idiopathic inflammatory myopathies that are characterized by more severe weakness and higher creatine kinase (CK) levels. It has been related with myositis specific antibodies including the anti-signal recognition particle (SRP) and an autoantibody that targets 3-hydroxy-3-methyl-coenzyme A reductase (HMGCR), which is a key enzyme in the cholesterol biosynthesis pathway that is upregulated in statin-treated patients.
The objective of this study is to describe the main features and the clinical management of a cohort of IMNMs patients.
Methods: We conducted an observational, monocentric, retrospective study of IMNM patients diagnosed in the Neuromuscular Unit of a tertiary hospital in Madrid (Spain) between 2013 and 2020.
Results: Fifteen patients were included. The median age at symptoms onset was 71 (IQR, 62-75) years and nine patients (60%) were female. Previous treatment with statins was documented in 13 patients (86.7%), atorvastatin the most common was used in nine patients followed by simvastatin and rosuvastatin. The most common clinical presentation was subacute in seven patients (46.7%) and the time from symptoms onset to diagnosis was less than six months in ten patients (66.7%). Proximal limb muscle weakness, which was present in each case, was the most common clinical feature followed by myalgias, present in eleven patients (73.3%). Proximal weakness was more pronounced in lower limbs, and it was considered as severe (based on the grade of proximal weakness if MRC ≤ 3/5) in nine patients (60%). Median CK levels at diagnosis was 4,971 units per liter (U/L) (IQR, 3,901-7,984 U/L). Regarding the MSA testing, anti-HMGCR were the only detected in 12 patients (80%). A muscle biopsy was performed in 14 patients and the histological features (Figure) were characterized by the presence of necrosis and regeneration fibers in each case (100%), seven patients (46.7%) exhibited mild inflammatory lymphocyte infiltrates, and immunohistochemical analysis of HLA-I performed in ten cases, was positive in seven patients. The initial treatment approach started with prednisone in fourteen patients (93.3%). Based on the individual clinical response other drugs were required in combination including intravenous immunoglobulins (IVIG) in four patients (26.7%), methotrexate in four patients (26.7%), and azathioprine and mycophenolate used in two cases each (13.3%). After six months, twelve patients (80%) experienced clinical improvement. One patient experienced a relapse during the follow-up and cancer was diagnosed after the IMNM diagnosis in one patient who was positive for anti-HMGCR antibodies.
Discussion: This retrospective study describes the main clinical characteristics, autoantibody profiles, muscle biopsy pathological features, treatment strategies, and outcome data from a cohort of patients with IMNM treated in a neuromuscular unit. The subacute instauration of proximal weakness in the presence of greatly elevated CK levels is crucial to a clear clinical suspicion, while muscle biopsy and MSA are fundamental for a correct diagnosis. In this sample, anti-HMGCR antibodies were the most common. Clear recognition of the disease would facilitate early and aggressive treatment, which should be individualized based on the initial response to corticosteroids, and usually requires a combination of several drugs.
eP01.05.09COVID-19 and Vaccination Against Sars-CoV-2 in Patients With Myasthenia Gravis From Belgrade, Serbia
Peric S2, Rankovic M3, Bozovic I1, Ivanovic V1, Palibrk A1, Basta I2, Stevic Z2, Rakocevic-Stojanovic V2, Lavrnic D2
1Neurology Clinic, University Clinical Centre Of Serbia, Belgrade, Serbia, 2Neurology Clinic, University Clinical Centre Of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia, 3School of Medicine, University of Belgrade, Belgrade, Serbia
Introduction: Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction that causes muscle weakness and fatigability. Majority of MG patients request a long-term immune suppression.
Aim: To analyze frequency and severity of COVID-19 infection in MG patients, as well as frequency of patients vaccinated against SARS-CoV-2.
Patients and Methods: 125 MG patients from the central Belgrade municipalities were included – 60.0% females, age at MG onset 50.1 (19.7) years, age at testing 61.7 (16.8) years, AChR positive 78.4% and MuSK positive 8.6%.
Results: One third of MG patients have had a COVID-19 and they were younger compared to those who did not suffer from a COVID-19. Severe COVID-19 was registered in 27.9% of MG patients, mostly in elder subjects with comorbidities such as cardiac diseases and history of malignancies. Two patients had a lethal outcome. MG worsening was noticed in 21.4% of patients after COVID-19 and 41.8% had COVID-19 sequalae. Majority of our MG patients were vaccinated against SARS-CoV-2 (68.8%). Vaccination was more common among MG patients with diabetes and in those with a milder form of MG. Main reasons not to accept a vaccine were: do not wish to be vaccinated (12 patients), afraid of MG worsening (10), advise of a neurologist (8), and advise of a general practitioner (6). The most common types of vaccine were Sinopharm (42.2%) and Pfizer-BioNTech (25.6%). Adverse events were noticed in 36.0% of vaccinated patients, with mild infection (77.4%) and local reactions (12.9%) being the most common. MG worsening was noticed in 5 (5.8%) vaccinated patients.
Conclusion: COVID-19 has placed a significant burden for MG patients, with severe COVID-19 forms and MG worsening being common. Percentage of vaccinated MG patients was higher than in general Serbian population.
Keywords: myasthenia gravis (MG), COVID-19, SARS-CoV-2, vaccination, comorbidities
eP01.06.01Two-Years Prospective Natural History Study in 24 Adult LGMDR12 Patients: Clinical and Radiological Outcome Measures
De Wel B1,2, Huysmans L3,4, Peeters R5, Goosens V5, Ghysels S5, Byloos K5, Putzeys G5, D’Hondt A1, De Bleecker J6, Dupont P7, Maes F3,4, Claeys K1,2
1Department of Neurology, University Hospitals Leuven, Leuven, Belgium, 2Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium, 3Medical Imaging Research Centre, University Hospitals Leuven, Leuven, Belgium, 4Department ESAT - PSI, Image & Speech processing, KU Leuven, Leuven, Belgium, 5Department of Radiology, University Hospitals Leuven, Leuven, Belgium, 6Department of Neurology, University Hospital Gent, Gent, Belgium, 7Laboratory for Cognitive Neurology, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium
Introduction: Limb Girdle Muscular Dystrophy type R12 (LGMDR12) is a genetic neuromuscular disorder caused by recessive mutations in the Anoctamin-5 (ANO5) gene, leading to a slowly progressive limb-girdle weakness in most patients. Natural history data in LGMDR12 are lacking. With genetic treatments in development, a comprehensive evaluation of natural disease course and outcome measures is highly needed in these patients.
Methods: We prospectively measured the following outcome measures in 24 patients and 24 age/sex-matched healthy controls at baseline and after 12 and 24 months: 6-minute-walk-distance (6MWD), 10-metre-walk-test (10MWT), 60- and 150-points Medical Research Council (MRC) sum scores, isometric dynamometry of quadriceps and hamstrings muscles (Biodex®), serum creatine kinase (CK) and whole-body MRI including 6-point Dixon images of the thighs to calculate intramuscular fat fraction. Thigh muscles were segmented individually and entirely, in order to allow analysis of fat fraction at the level of individual muscles as well as of muscle groups, and to avoid slice selection bias.
Results: Patients with intermediate stage thigh muscle fat replacement at baseline (proton density fat fraction (PDFF) 20-70%) already showed a significant increase in PDFF in 8/14 evaluated thigh muscles after one year. The standardized response mean (SRM) demonstrated a high responsiveness to change in PDFF for 6 individual muscles over 2 years in this group. However, in patients with early (<20%) or end stage (>70%) muscle fat replacement, PDFF did not increase significantly over two years of follow-up. Biodex® isometric dynamometry showed a significant decrease of muscle strength in all patients in the right and left hamstrings (-6.2Nm, p<0.01 and -4.6 Nm, p<0.01, respectively) and right quadriceps muscles (-9 Nm, p=0.04) after 1 year of follow-up, whereas the 6MWD, 10MWT, and MRC sum scores were not able to detect a significant decrease in muscle function/strength even after two years. There was a strong inverse correlation between total thigh PDFF and hamstrings peak torque (Rho=-0.7; p<0.01), and a moderately strong inverse correlation with the other clinical outcome measures at baseline.
Conclusion: Thigh muscle PDFF imaging is a sensitive outcome measure to track progressive muscle fat replacement in selected LGMDR12 patients even after one year of follow-up and correlates with clinical outcome measures. Biodex® isometric dynamometry can reliably capture loss of muscle strength over the course of one year in LGMDR12 patients and should also be included as an outcome measure in future clinical trials.
eP01.06.02Assessing the Relationship of Patient Reported Outcome Measures with Functional Status in Dysferlinopathy
Hilsden H1, Mayhew A1, James M1, Moore U1, Rufibach L2, Spuler S3, Day J4, Jones K5, Goebel D6, Salort-Campana E7, Pestronk A8, C. Walter M9, Paradas C10, Stojkovic T11, Mori-Yoshimura M12, Bravver E13, Díaz-Manera J14, Pegoraro E15, Mendell J16, Straub V1
1The John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Central Parkway, Newcastle Upon Tyne, United Kingdom, 2The Jain Foundation, Seattle, USA, 3Charite Muscle Research Unit, Experimental and Clinical Research Center, a joint cooperation of the Charité Medical Faculty and the Max Delbrück Center for Molecular Medicine, Berlin, Germany, Berlin, Germany, 4Department of Neurology and Neurological Sciences, Stanford University School of Medicine; Stanford, USA, 5The Children’s Hospital at Westmead, and The University of Sydney, Sydney, Australia., 6Department of Neurology Children’s National Health System, Washington, USA, 7Service des maladies neuromusculaire et de la SLA, Hôpital de La Timone, Marseille, France, 8Department of Neurology Washington University School of Medicine, St. Louis, USA, 9Friedrich-Baur-Institute, Dept. of Neurology, Ludwig-Maximilians-University of Munich, Munich, Germany, 10Neuromuscular Unit, Department of Neurology, Hospital U. Virgen del Rocío/Instituto de Biomedicina de Sevilla, Sevilla, Spain, 11Institut de Myologie, AP-HP, GH Pitié-Salpêtrière, Paris, France, 12Department of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, Japan, 13Neuroscience Institute, Carolinas Neuromuscular/ALS-MDA Center, Charlotte, USA, 14Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 15Department of Neuroscience, University of Padova, Padova, Italy, 16The Abigail Wexner Research Institute at Nationwide Children’s Hospital, Columbus, USA
Dysferlinopathy is a muscular dystrophy with a highly variable functional disease progression in which the relationship of function to some patient reported outcome measures (PROMs) has not been previously reported. This analysis aims to identify the suitability of PROMs and their association with motor performance. 204 patients with dysferlinopathy were identified in the Jain Foundation’s Clinical Outcome Study in Dysferlinopathy from 14 sites in 8 countries. All patients completed the following PROMs: Individualised Neuromuscular Quality of Life questionnaire (INQoL), International Physical Activity Questionnaire (IPAQ), and activity limitations for patients with upper and/or lower limb impairments (ACTIVLIM). In addition, non-ambulant patients completed the Egen Klassifikation Scale (EK). Assessments were conducted annually at baseline, year 1, 2, 3 and 4. Data were also collected on the North Star Assessment for Limb Girdle Type Muscular Dystrophies (NSAD) and Performance of Upper Limb (PUL) at these time points from year 2. Data were analysed using descriptive statistics and Rasch analysis was conducted on ACTIVLIM, EK, INQOL. For associations, graphs (NSAD with ACTIVLM, IPAQ and INQOL and EK with PUL) were generated from generalized estimating equations (GEE). The ACTIVLIM appeared robust psychometrically and was strongly associated with the NSAD total score - Pseudo R2 0.68. The INQOL performed less well and was poorly associated with the NSAD total score - Pseudo R2 0.18. EK scores were strongly associated with PUL - Pseudo R2 0.69. IPAQ was poorly associated with NSAD scores - Pseudo R2 0.09. This study showed that several of the chosen PROMs demonstrated change over time and a good association with functional outcomes. An alternative quality of life measure and method of collecting data on physical activity may need to be selected for assessing dysferlinopathy.
eP01.06.03Clinical and Genetic Features in Two Families Carrying Novel and Reported DYSF Variants
Burnyte B1, Vilimiene R2, Morkuniene A1, Petroska D1,3, Utkus A1
1Institute of Biomedical Sciences, Faculty of Medicine, Vilnius University, Vilnius, Lithuania, 2Institute of Clinical Medicine, Faculty of Medicine, Vilnius University, Vilnius, Lithuania, 3National Center of Pathology, Affiliate of Vilnius University Hospital Santaros Klinikos, Vilnius, Lithuania
Background: Pathogenic variants in DYSF have been associated with a spectrum of rare muscle disorders known as dysferlinopathies. Here we report 4 patients from 2 families including one family with novel homozygous multiexonic duplication of DYSF gene and one with previously reported compound heterozygous point mutations.
Family 1: Two brothers (patients 1 and 2, 39 and 37 years old, respectively) were referred for evaluation of progressive leg weakness and gait difficulties. Ages at onset were 29 and 30 years, respectively. Examination showed bilateral distal weakness in both brothers and proximal weakness greater in lower than upper extremities (MRC 0/5) in patient 1. CK level was 5728 U/L in patient 1 and 12552 U/L in patient 2. EMG studies of patient 2 at age of 34 revealed that myopathic changes were more pronounced in the calf muscles with mild spontaneous activity in the left leg. Muscle biopsy from left quadriceps demonstrated non-specific myopathic change without any signs of inflammation. Muscle MRI showed mild oedema in the proximal muscles on the right shoulder girdle. Marked atrophy with fatty replacement in adductor longus, biceps femoris, semimembranosus, gastrocnemius and soleus muscles were observed on both sides. WES analysis in patient 1 identified a homozygous duplication of exon 19 to 31, which was confirmed by MLPA. Segregation analysis was conducted on family members and the same homozygous variant was identified in the affected brother, while both parents were heterozygous carriers. This variant has not been reported previously. Duplication predicted to be truncating and probably results in a loss of function of the protein.
Family 2: Patient 3 developed difficulty climbing stairs and weakness before age of 20. Examination at age of 24 revealed muscle weakness, which was more prominent in the distal parts of the extremities. CK 18905 U/L. EMG showed brief small polyphasic motor unit action potentials without spontaneous muscle activity. A right quadriceps biopsy showed non-inflammatory and non-neurogenic myopathic change. His affected sister (patient 4) noticed weakness in the lower parts of the legs and frequent falls at the age of 18. Later she developed difficulties getting up from a sitting position. Examination showed muscle wasting and weakness of the calves. For both siblings sequencing analysis revealed a compound heterozygous for pathogenic single base substitutions that were previously reported: splice site variant c.2864+1G>A (rs199954546) and missense mutation c.5419C>T (rs746243052) in DYSF.
Conclusion: Mutations in the DYSF gene found to date are point mutations or deletions or insertions distributed all over the coding sequence. This report is about a novel multiexonic out of frame duplication encompassing exons 19 to 31 of DYSF gene affecting the sequence encoding highly conserved the C-terminal part of the ferlin family domain and DYSF domain. Novel variant results in distal myopathy phenotype at the onset and a limb-girdle pattern of muscle weakness predominant in lower extremities as the disease progresses. Reported families contribute to the expanding clinical and molecular data of dysferlinopathies and further expands the pathogenic variants spectrum.
eP01.06.04Description of motor function in Duchenne Muscular Dystrophy in a center of expertise in Colombia
Castellar S1,2, Ortiz F1,2, Ruiz E1,2, Salcedo M1, Soto D1, Suarez F1, Sanchez D1, Garcia A1, Correa C1, Cediel M1, Anzola M1, Quiroga M1
1Instituto Roosevelt, Cra 4 East # 17-50, Colombia, 2Universidad Nacional de Colombia, Cra 45 # 26-85, Colombia
Duchenne muscular dystrophy (DMD) is an inherited, recessive, X-linked myopathy.
The main aims of the current work were to describe the motor function in patients with Duchenne Muscular Dystrophy (DMD), to establish the correlation between the functional tests and the age of the patients and to determine the correlation between the different tests.
We describe the functional characteristics according to the age of a group of patients with DMD attended at the Roosevelt Institute in Bogotá, Colombia.
A retrospective descriptive study was carried out with confirmed DMD patients (onset of symptoms before six years) evaluated between the years 2010 to 2020.
Results from the MFM (Motor Function Measure), North Star Ambulatory Assessment (NSAA) scales, the six-minute, and the box and cubes tests were included. Those scales and tests were previously applied to 85 children diagnosed with DMD.
Most of the patients (67.1%) were at a level 7 on the Vignos scale. 28 patients (32.9%) retained the ability to walk independently. At functional level 7 of the Vignos scale, the maximum MFM score was 56.3%. A child classified in level 4 in Vignos scale obtained a total MFM of 44.8%. Another patient, classified in level 5 of Vignos scale, achieved a score of 52.1%. The other 26 patients (92.8%), with independent walking capacity, obtained scores higher than 57% in the MFM.
The correlation of the MFM-D1 and the NSAA was excellent (r = 0.92; p = 0.000) and MFM-D2 and the NSAA was moderate (r = 0.47; p = 0.009). The correlation between NSAA scale and 6MWT was excellent (r = 0.90; p= 0.001). Correlations above 0.75 were found between domain 1 of the MFM and the NSAA scale with the 6-minute test.
The correlation of MFM-D3 and Brooke Classification was significant (r = -0.68 p = <0.0001) and box and cubes tests with the Brooke classification was significant too (r = -0.76 p = <0.0001).
Patients who received steroids lost walking ability at 11 years of age and those who did not receive steroids lost their ability at 9 years of age.
This study describes the gross motor function and shows the wide variability of motor phenotypes of patients with DMD treated at a reference institution in Colombia. The observed correlation between each of the tests used helps to more accurately characterize gross motor function. The MFM-1D and NSAA scales have an excellent correlation with the 6-minute test.
These findings serve as a reference for the functional evaluation of patients with DMD and help to detect the presence of contextual factors that are affecting their mobility.
eP01.06.05Preliminary Results from MLB-01-003: An Open Label Phase 2 Study of BBP-418 in LGMD2I
Harper A2, Langeslay R2, Rodriguez H1, Hutchaleelaha A1, Kelley K1, Sproule D1
1ML Bio Solutions, Inc., Carlisle, United States, 2Virginia Commonwealth University, Richmond, United States
Introduction: Limb Girdle Muscular Dystrophy (LGMD) Type 2I, also called LGMDR9 FKRP-related, is caused by bi-allelic loss-of-function of the fukutin-related protein (FKRP) gene which results in hypoglycosylation of alpha-dystroglycan (αDG). BBP-418 (ribitol) is an orally administered substrate supplementation intended to saturate the FKRP enzyme driving increased glycosylation of αDG, thus ameliorating the root cause of disease in LGMD2I.
Objectives: The MLB-01-003 Phase 2 study is intended to explore the safety and tolerability, feasibility and usefulness of selected clinical efficacy and pharmacodynamic (PD) assessments in patients with LGMD2I receiving ascending doses of BBP-418.
Methods: This is an open label study in ambulatory and non-ambulatory patients with LGMD2I conducted at Virginia Commonwealth University. Part 1 of the study involved three ascending dose cohorts treated for 3 months with BBP-418: cohort 1 (n=4, dose 6 g daily), cohort 2 (n=4, dose 6 g twice daily) and cohort 3 (n=6, dose 12 g twice daily). During Part 2 of the study, all patients received 12 g twice daily (weight adjusted for lower weight patients) for 3 months.
Results: 14 patients aged 12-53 with LGMD2i were enrolled in the study. 8/14 were homozygous for the common mutation. 11/14 were ambulatory (able to complete the 10 m walk test in <12 seconds). All 14 patients showed declines in creatine kinase (CK) (mean ~70% decrease) from baseline assessment after 3 months of treatment with BBP-418 in Part 1. No drug-related SAEs have occurred. Additional updated safety, laboratory and clinical data will be provided at the time of the meeting.
Conclusions: Preliminary MLB-01-003 data from patients with LGMD2i treated with BBP-418 suggests a positive effect on CK, a widely used biomarker of muscle injury. A larger, global, double-blind placebo-controlled phase 3 study is planned for 2022.
eP01.06.06The Founder Mutation TRAPPC11 c.1287+5G>A is a Frequent Cause of Limb-girdle Muscular Dystrophy in Roma Population
Natera De Benito D1, Justel M1, Sariego A2, Jou C1, Hernandez-Lain A6, Juliá N1, Ortez C1, Poch M3, Hedrera A4, Gómez H5, Carrera-García L1, Expósito-Escudero J1, Domínguez-Carral J1, Codina A1, Vila-Bedmar S6, Zulaica M9, Cancho-Candela R7, de la Osa A8, Pascual I10, Jimenez-Mallebrera C1, Lopez-de Muniain A9, Camacho A6, Domínguez-González C6, García-Cazorla A1, Nascimento A1
1Hospital Sant Joan De Déu, Barcelona, Spain, 2Hospital Marqués de Valdecilla, Santander, Spain, 3Hospital San Pedro, Logroño, Spain, 4Hospital Universitario Central de Asturias, Oviedo, Spain, 5Hospital Universitario de Salamanca, Salamanca, Spain, 6Hospital 12 de Octubre, Madrid, Spain, 7Hospital Rio Hortega, Valladolid, Spain, 8Hospital Peset, Valencia, Spain, 9Hospital Donostia, San Sebastian, Spain, 10Hospital La Paz, Madrid, Spain
Limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of genetically determined muscle disorders. TRAPPC11-related LGMD is an autosomal-recessive condition that is clinically characterised by muscle weakness, intellectual disability and hyperkinetic movements. We present a comprehensive description of the heterogeneous clinical features of the TRAPPC11-related LGMD caused by the homozygous c.1287+5G>A mutation in 21 Roma individuals from 14 different families. The c.1287+5G>A mutation, which we suggest as a founder mutation in the Roma ethnic group, leads to a phenotype characterized by early-onset muscle weakness, movement disorders, intellectual disability and elevated serum CK, which is similar to other series. Nevertheless, our observations indicate a strong prevalence of progressive and highly significant microcephaly. Moreover, an infectious trigger was described in 24% of the individuals, which has not been reported to date. These two interesting findings may be considered reliable signs to distinguish the c.1287+5G>A variant in TRAPPC11 from other variants or, alternatively, they can be underreported features associated to LGMD R18, regardless of which variant causes it.
eP01.06.07A Familial Case with Phenotypic Differences in a CAV3 Pathogenic Variant
Lee S3, Lee H1, Park K2, Choi Y1
1Gangnam Severance Hopspital, Gangnam-gu, Republic of Korea, 2Ewha Womans University Mokdong Hospital, Yangchun-gu, Republic of Korea, 3Ewha Womans University Mokdong Hospital, Seoul, Republic of Korea
Background: Caveolin-3 is one of the muscle cell membrane proteins regulates sarcolemma stability and modulates various signaling pathways. Pathogenic variants in CAV3 lead to different skeletal muscle phenotypes, including limb-girdle muscular dystrophy(LGMD); rippling muscle disease-2(RMD2); distal myopathy; and isolated hyperCKemia. It is known that pathogenic variants in CAV3 can cause different and overlapping clinical phenotypes even within the same family. We report the first Korean family with the c.296A>C (p.His99Pro) variant exhibiting marked phenotypic variabilities. Case report: A 32-year-old man complained of progressive proximal muscle weakness. His family history was unremarkable, and initial development after birth was reportedly normal, but he recalled that he was far behind his peers in running when he was in school. He experienced frequent myalgia after intense physical activities since he was 20 years old. On examination, he displayed proximal muscle weakness and bilateral hypertrophy on posterior calf. However, sensory examination revealed no abnormalities. Laboratory studies revealed a serum creatine kinase (CK) level of 1,908 IU/L (normal, <135 IU/L). Electromyography reveled active generalized myopathy. A biceps brachii muscle biopsy represented marked muscle size variation with some degenerating and regenerating fibers. Immunohistochemical analysis of the muscle specimens showed reduced expression of Caveolin-3. Targeted next-generation sequencing (NGS) of 598 neuromyopathy genes identified a pathogenic variant in CAV3 (NM_033337.2: c.296A>C, p.His99Pro). It was classified as a likely pathogenic variant according to following evidences: 1) Located in a mutational hot spot, 2) absent from the Genome Aggregation Database (gnomAD) full exome and genome databases, 3) multiple lines of computational evidence support a deleterious effect on the gene, 4) Reputable source recently reports variant as pathogenic. Based on the clinical and pathologic features with genetic analysis, our patient was diagnosed with LGMD caused by CAV3 mutation. After identification of the causative gene, all family members underwent neurology exam, laboratory studies, and genetic studies. They all showed normal muscle power. However, among all family members, his mother and brother represented high CK level of 317 IU/L and 819 IU/L, respectively. The same pathogenic variant in CAV3 was detected in those family members, and was not found in his father with normal CK level. Conclusion: Although the patient reported unremarkable family history, we identified the same pathogenic variant in CAV3 in asymptomatic family members with hyperCKemia. This is the first CAV3 pathogenic variant reported in a Korean family with significantly different phenotypes such as LGMD and idiopathic hyperCKemia.
eP01.06.08Clinical and Genetic Spectrum of LGMDR Cases in the East of Algeria
Sifi Y1
1Departement of Neurology, Constantine3 University, Constantine, Algeria, 2Labooratory of Biology and Molecular Genetics, Constantine3 University, Constantine, Algérie, 3Laboratory of Biochemistry Constantine3 University, Constantine, Algérie, 4Institut de Myologie, Hôpital de la Salpêtrière, Constantine, Algérie
Introduction: Autosomal recessive limb-girdle muscular dystrophies (LGMDR) constitute an heterogeneous group of genetic neuromuscular disorders, characterized by progressive muscle weakness involving the shoulder and pelvic girdles. They can mimic many other neuromuscular conditions in particular the late-onset form of Pompe disease (LOPD). Here we report the clinical and genetic spectrum of a cohort of patients diagnosed with LGMDR in the East of Algeria, irrespective of their respiratory status.
Methods: Our study was sponsored by Sanofi-Genzyme as part of a project called “Pompe and LGMD NGS diagnostic project in international regions “Nine autosomal recessive LGMDs and Pompe disease were investigated by Next-Generation Sequencing (NGS) using a 10- gene panel (CAPN3, DYSF, SGCA, SGCB, SGCD, SGCG, FKRP, ANO5, TCAP, GAA). 97 families whose phenotype was consistent with LGMDR, with or without respiratory symptoms, were included in the study.
Results: Thirty nine families out of 97 families showed mutations in theCAPN3, DYSF, SGCA, and SGCG genes. All patients showed progressive weakness predominantly in the lower limbs. Other findings were: scapular winging, joint contractures and calf hypertrophy or atrophy. In total, 39 mutations, seven of which were novel,have been identifiedin SGCG (n= 14), CAPN3 (n = 12), DYSF (n = 9), and SGCA (n = 4) respectively.. The most prevalent forms of LGMDR in our cohort remain sarcoglycan-deficiencies (gamma and alpha) followed by calpainopathy and dysferlinopathy. Such a high prevalence of gamma-sarcoglycanopathy can be explainted by the existence of a founder mutation (c.del525T) in the whole region (Maghreb). No patients have been identified with Pompe disease.
Conclusions: Our results demonstrate the usefulness and effectiveness of a targeted approach by NGS in patients presenting with LGMDR, with or without respiratory problems. Our next goal is to extend the study to a larger groups of families with the hope of diagnosing LOPD patients for which a treatment is available.
eP01.06.09Molecular Diagnosis of Muscular Dystrophy Using the LGMD Gene Panel in Adult Neurology
Choi S1, Lee J1, Han J1, Shin J1, Hong Y2, Sung J1
1Department of Neurology, Seoul National University Hospital, Seoul, Republic of Korea, 2Department of Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, Republic of Korea
Background: Molecular genetic analyses have allowed the identification of more than 40 genes involved in muscular dystrophies, with very rare muscular dystrophy variants being continually identified. While the diagnostic yield of the limb-girdle muscular dystrophy (LGMD) gene panel in Korean pediatric patients (57.1%) was previously reported, its utility in adult neurology remains uncertain.
Objective: The goal of this study was to investigate the diagnostic value of the LGMD gene panel, which consisted of 43 genes, in adult patients with suspected myopathy of genetic etiologies, and to identify clinical features influencing the molecular diagnosis through genotype-phenotype correlations.
Methods: We retrospectively reviewed the medical records of 98 patients older than 20 years old who tested the LGMD gene panel at Seoul National University Hospital from October 2017 to November 2021. Concerning diagnostic certainty, we used the following definition: definite for patients harboring pathogenic or likely pathogenic variants conforming to their inheritance pattern, potential for those harboring pathogenic or likely pathogenic variants and/or variants of uncertain significance.
Results: We identified a definite genetic diagnosis in 29 (29.6%) of the 98 patients tested with the LGMD gene panel. When potential diagnoses were included, the diagnostic yield of the LGMD gene panel increased to 46.9%. Among the genes that accounted for the definite diagnoses, DYSF (n = 9) was the most common, followed by CAPN3 (n = 6) and DMD (n = 6). Other genes included GNE (n = 2), EMD, FHL1, ADSSL1, AHCY, DUX4, and GAA. A family history of muscular dystrophy was found in 34.5% of the patients. Patients with calpainopathy mostly presented with limb-girdle weakness, whereas those with dysferlinopathy had diffuse or distal weakness as well as limb-girdle weakness.
Conclusion: Next generation sequencing-based gene panel can be a helpful tool for the diagnosis of muscular dystrophies in adult neurology.
eP01.06.10Observational Study: The Quality of Life in Patients with Alpha-Sarcoglycan, Beta-Sarcoglycan and Gamma-Sarcoglycan Gene Mutation
VOLA B, Maggi R, Cerletti M, Torrente Y, Sanchez Riera C, Paniga C1
1Gruppo Familiari Beta-sarcoglicanopatie, Talamona, Italia
SUMMARY: Sarcoglycanopathies are underfunded “orphan” diseases and there is no cure for them. Our volunteer organization named Family Group of Beta-sarcoglycanopathy Odv (GFB Odv www.lgmd2e.org) was founded in 2013 with the aim of stimulating scientific research on this disease. Since 2012 families of the GFB Odv are supporting preclinical and clinical projects of gene therapy carried out by Prof. J. Mendell at the Ohio State University (Columbus, Ohio, USA) and Sarepta Therapeutics. In 2018 GFB Odv promoted a natural history study on Italian patients, “Clinical Determinants of Disease Progression in Patients with Beta-sarcoglycan Gene Mutations”. Since 2021 GFB Odv has started an ongoing observational study on the quality of life in patients with Beta-sarcoglycan, Alpha-sarcoglycan, and Gamma-sarcoglycan gene mutation.
ABSTRACT TEXT: LGMDR3, LGMDR4, LGMDR5 are recessive autosomal diseases, there are no specific treatments, except for some physical therapies that prevent worsening of muscle contractures and therapies associated with cardiac and respiratory problems.
In 2013 the volunteer organization named Family Group of Beta-sarcoglycanopathy (GFB ODV; www.lgmd2e.org) was established for stimulating and supporting both basic and clinical research on these diseases.
Since 2012, families of the GFB ODV funded a gene therapy research project for LGMDR4, under the supervision of Prof. J. Mendell, at the Ohio State University (Columbus, Ohio, USA). The first clinical trial, conducted by Sarepta Therapeutics, is currently underway.
In 2018 GFB Odv promoted a natural history study on Italian patients affected by LGMD2E, published on 1st July 2021 in the journal “Frontiers in Neurology”, with the title “Clinical Determinants of Disease Progression in Patients with Beta-sarcoglycan Gene Mutations”, led by Prof. Yvan Torrente, of the University of Milan and Ospedale Maggiore Policlinico in Milan, Italy.
Since 2021 GFB Odv has started an ongoing observational study on the quality of life in patients with Beta-sarcoglycan, Alpha-sarcoglycan, and Gamma-sarcoglycan gene mutation, again in collaboration with prof. Yvan Torrente.
The study is aimed at international GFB patients. At this time 130 patients from 43 countries have joined the Project: in particular, 63 with LGMDR4, 35 with LGMDR5, 32 with LGMDR3. 181 questionnaires have already been filled out.
The ACTIVLIM, ABILHAND, EK, PEDSQL and PROMIS scales are used in the study, both in paper and electronic format. The study will run for 3 years and the scales will be given to patients every 6 months. Patient recruitment is still open.
KEYWORDS: Limb-girdle muscular dystrophy, LGMD, LGMDR4, LGMD2E, beta-sarcoglycanopathy, LGMDR3, LGMD2D, alpha-sarcoglycanopathy, LGMDR5, LGMD2C, gamma-sarcoglycanopathy, orphan disease, website, GFB ODV, Quality of life, observational study
eP02.01.01Value of Muscle Ultrasound in Pediatric Neuromuscular Patients
Kuepper H1, Horber V
1Neuropaediatric Department, University Hospital Tuebingen, Tübingen, 72076, Germany
Introduction: Structural muscle changes, as frequently associated with neuromuscular conditions, can be well depicted by muscle ultrasound. Gray scale analysis of muscle ultrasound imaging has been shown to be a useful adjunctive test, as an estimate of infiltration of fat and fibrous tissue (Pillen et al., Neurological research, 2011).
Methods: Muscle ultrasound of 20 pediatric patients with different muscle diseases, i.e. Duchenne / Becker muscular dystrophy, alpha- sarcoglycanopathy (SGCA gene), myofibrillar myopathy-9 with early respiratory failure (TTN gene), congenital muscle dystrophy with partial merosine deficiency (LAMA2 gene), myotonia congenita (CLCN1 gene), myotonic dystrophy type I and congenital myotonic dystrophy were analyzed and compared to 10 pediatric patients with non- neuromuscular conditions. A Canon Aplio ultrasound system was used with a linear probe (4,5-18 MHz). Uniform imaging settings were standardized (frequency at 18 MHz, dynamic range at 60, gain at 70%). All ultrasound studies were performed by the same investigator (H.K.). Images were acquired in cross-sectional planes with an image depth of 4cm and a single focal point at 1,8 cm while the transducer was positioned perpendicular to the underlying bone. Grayscale analysis was performed using ImageJ software (NIH, Bethesda) by drawing a region of interest underneath the superficial fascia of the muscle and calculating a mean grayscale value.
Results: Muscle echogenicity was markedly higher in patients with Duchenne muscular dystrophy (DMD), alpha- sarcoglycanopathy, myofibrillar myopathy-9 with early respiratory failure (MFM9) and congenital muscle dystrophy with partial merosine deficiency (MDC1a). For DMD patients, this was most pronounced in the vastus lateralis and biceps femoris muscle (mean echogenicity 3,2 times higher). Within the group of DMD patients, those with a higher degree of motor impairment showed a higher muscle echogenicity. A patient with alpha- sarcoglycanopathy showed a similar/milder muscle ultrasound pattern compared to DMD patients, with comparably accentuated hyperechogenicity of the sartorius and deltoid muscle. Notably, muscle echogenicity in an infant aged 9 months with a presymptomatic diagnosis of DMD showed only subtle abnormalities in the vastus lateralis muscle (mean echogenicity 1,6 times higher than age matched control). A patient with MDC1a showed a more pronounced hyperechogenicity of the medial gastrocnemius muscle (mean echogenicity 2,3 times higher than age matched control) than the lateral gastrocnemius muscle. The pattern of muscle involvement in a patient with MFM9 appeared more selective (e.g. marked hyperechogenicity of digastric and paraspinal muscles).
Discussion: The positive value of muscle ultrasound and gray scale imaging analysis within the evaluation of these pediatric neuromuscular patients could be demonstrated. A correlation between the amount of muscle hyperechogenicity and clinical progression of the muscle disease was visible in the DMD group. A tendency of disease specific structural muscle changes could be seen in some of the patients. Further studies with greater patient numbers are needed to further characterize these specific patterns of muscle involvement and the type of progression in the course of the disease. This diagnostic tool is of particular benefit not only within the differential diagnostic work-up but also follow- up examination in the context of new treatment strategies.
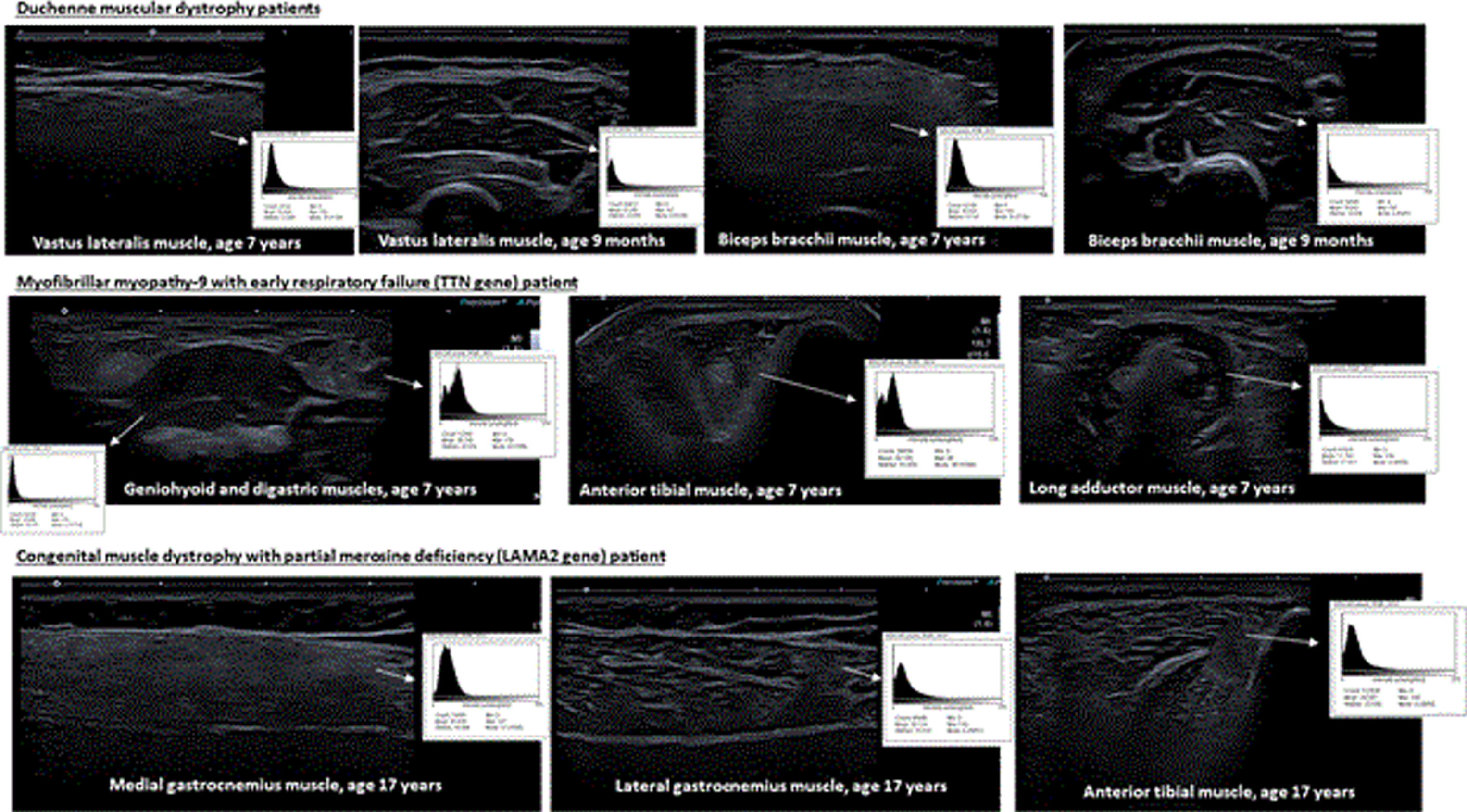
eP02.01.02Place of Muscle Ultrasound in the Diagnosis and Follow-up of Pediatricneuromuscular Diseases
Oulhissane Omar A1, Quijano-Roy S1
1CRMR CHU Raymond Poincaré, GARCHES, France
Background: Imaging techniques are become increasingly important in the diagnostic workup of a suspected neuromuscular disorders. High-resolution ultrasound allows the visualisation of the muscle, nerve and adjacent structures. It even allows dynamic imaging of contracting muscles and visualise pathological muscle activity such as fasciculation. Selective involvement of certain muscles is an indicator for muscle diseases and help to direct genetic testing. It can be used to localize specific muscles to target needle placement of EMG and biopsy.
Objectives: The purpose of this study was to describe our own institutional experience, in order to determine the possibility of using high-resolution ultra-sound as a routine tool in screening, diagnosis and follow-up of muscle diseases in the pediatric population.
Subjects and methods: This is a descriptive study. A total of 30 patients (1 month – 26 years old) were examined over a period of 18 months (May 2020 to January 2022). The ultrasound was performed blind to the results of other biological, physiological and radiological investigations (EEG, MRI, CPK ...) Replacement of healthy muscle with fat and fibrosis, manifested by an increase in echogenicity. It was assessed visually (Qualitative US) and semi-quantitativelly (Hickmat scale). Specific pattern were determined and described if presents. The following muscles were inspected : 1) anterior thigh : rectus femoris, vasts intermedius, lateralis and medialis, sartorius and gracilis, 2) posterior thigh : adductors, biceps femoris, semitendinosus and semimembranosus, 3) leg : tibialis anterior, peronii and gastrocnemius soleus. Other groups such as the axial muscles and muscles of the face can also be examined. The results obtained are then compared with biologic, physiological and radiological data.
Results: The sensibility and specificity of US in the discrimination of myopathies vs neuropathic conditions are high. The concordance ratio of US to other laboratory methods including MRI, EMG, muscle histology, immuno-histochimistry, and mutation analysis is elevated. The specificity of the ultrasound pattern decreases at the very advanced stage of the pathology. This accuracy is earlier in infants compared to other techniques such as MRI.
Summary / Conclusion: The value of muscle ultrasound as a non-invasive, accessible and accurate tool for the diagnosis and follow-up of pediatric neuromuscular disorders is well established. It should be an extension of the physical exam in the work-up of any neuromuscular disease. Quantitative US is well suited to monitor disease progression. It should be developped in our center.
eP02.01.03Machine Learning in Ultrasound-Guided Differentiation of Myopathic From Neurogenic Patterns: A Pilot Study
Bara S1, Vlachopoulos G1, Spiliopoulos K2, Veltsista D2, Chroni E2, Costaridou L1
1Department of Medical Physics, School of Medicine, University of Patras, Patras, Greece, 2Department of Neurology, School of Medicine, University of Patras, Patras, Greece
Background: Muscle ultrasound (MUS) has been proven a reliable examination in the detection of muscle abnormality and the monitoring of progression in neuromuscular disorders. Discriminating myopathic from neurogenic disease via MUS can be a challenging task, especially in early or severely advanced stages of muscular pathology. Image texture analysis and Machine Learning (ML) of MUS images is a novel approach in discrimination of myopathic from neurogenic diseases, which can predict disease characteristics.
Objective of this study is to investigate differentiation among neurogenic, myopathic and healthy biceps brachii (BB), by means of ML analysis of MUS image textural features.
Methods: Nineteen patients diagnosed with Amyotrophic Lateral Sclerosis (ALS) (mean age 63.79 ± 8.7 years, 6 females and 13 males, mean BMI 26.64 ± 5.9 kg/mm2) and 22 patients with muscular dystrophy (mean age 44.05 ± 13.6 years, 12 females and 10 males, mean BMI 25.26 ± 4.82 kg/mm2) underwent B-mode ultrasonography of the BB muscle. Forty-one sex-, age- and body mass-matched healthy subjects constituted the control group. Transverse ultrasound images of BB were analyzed. A total of 46 features were extracted, 14 of which being histogram (first order) derived parameters and 32 texture (second order). A ML approach was utilized to evaluate the discriminating ability of the extracted features, using 3 different classifiers. Specifically, Logistic Regression (LR), Random Forest (RF), Support Vector Machine (SVM) were employed for differentiating between neurogenic-myopathic, healthy-neurogenic, as well as healthy-myopathic groups. The area under curve (AUC) of the receiver operating characteristic (ROC) curve was used in order to evaluate the classification performance of the 3 classifiers.
Results: Analysis between neurogenic and control group revealed 13 features with performance >0.65 AUC. Best performing features were histogram first quartile (Q1) (AUC=0.71) with the use of LR classifier and texture Gray-Level Run Length Matrix (GLRLM) High Gray-level Run Emphasis (HGRE) (AUC=0.79) with the use of RF classifier. Analysis between myopathic and healthy groups demonstrated 29 features with performance >0.80 AUC. Best performing features were histogram Skewness (AUC=0.90) with the use of RF classifier and texture GLRLM Short-Run Low Gray-level Emphasis (SRLGE) (AUC=0.94) with RF classifier. Finally, analysis between neurogenic and myopathic group showed 13 features with performance >0.80 AUC. Best performing features were histogram Q1 (AUC=0.83) with LR classifier and texture GLRLM Long-Run Low Gray-level Emphasis (LRLGE) (AUC=0.85) with RF classifier.
Conclusion: Results demonstrate that both histogram and texture analysis may offer meaningful insights for the discrimination between myopathic and neurogenic disease. Texture analysis is especially promising towards increasing disease classification accuracy.
eP02.01.04Novel Titin Mutation Responsible for Hereditary Myopathy With Early Respiratory Failure in Adult Moroccan Man
Tamaoui L1, Belkhayate A2, Birouk N1
1Clinical neurophysiology department, Hôpital des Spécialités RABAT, Ibn Sina University Hospital, RABAT, Morocco, 2Private genetic laboratory, RABAT, Morocco
Introduction: Respiratory insufficiency is a common feature of many neuromuscular disorders. However, its occurrence as a prominent or sole symptom in adulthood should rise suspicion of an extremely rare condition called hereditary myopathy with early respiratory failure (HMERF). Titinopathies is one of the likely diagnoses. However, the differential diagnosis is broad and might be challenging for diagnosis. Here we describe a first sporadic case of HMERF in Moroccan man with a novel titin mutation.
Methods: case report
Result: A 48-year-old Moroccan man with no family history of muscle disease who complained about slowly progressive respiratory insufficiency since his 40s in addition to a long-standing asthenia since his early 20s with no obvious weakness. Neurologic exam was normal but physical examination revealed dorsal kyphosis with forward tilt of the pelvis. Electro-diagnostic study showed evidence of a subtle signs of myopathy. MRI of the lower extremities revealed characteristic fatty infiltration mostly affecting the posterior muscles of the thigh, adductor and gluteus maximus muscles. Muscle pathology showed dystrophic changes. In the first place, we excluded the most common diseases related to these symptoms; in particular Pompe disease and Steinert disease. Therefore, a clinical exom sequencing was performed and revealed heterozygous mutation C.37101 + 1G>A of the titin gene which is not previously reported. Currently, the patient is stable using continuous noninvasive ventilation at night and few hours a daytime for almost 10 years.
Conclusion: This case illustrates an uncommon condition of chronic muscle diseases presenting as respiratory insufficiency. Although, the myofibrillar myopathy du to Titin mutation is a rare genetic disorder, we aim to stress on its hallmark features, the differential diagnosis, and the positive contribution of supportive care.
eP02.01.05Deep Learning-Based Electrodiagnosis of Needle-Electromyography
Yoo I1
1Nowon Eulji Medical Center, Eulji University School of Medicine, Bundang Gu, Pangyo Yeok Ro, Republic of Korea
Background: It has been demonstrated that deep learning shows good performance in reading a surface electromyography, needle electromyography (nEMG) in resting state. However, it is not well elucidated whether deep learning can be applied to reading the nEMG in contraction state, which plays more important role in differentiating among myopathy, neuropathy, and normal. We investigated whether convolutional neural network (CNN) algorithm can identify the abnormality of nEMG.
Methods and findings: We classified nEMG data (58 patients; 382 muscles) stored in Seoul National University Hospital database from June 2015 to July 2020 among myopathy, neuropathy, normal by using the CNN algorithm.
Based on the classified results by CNN algorithm, the accuracy, sensitivity, specificity, positive predictive value and F1 score were 0.820, 0.820, 0.904, 0.820, and 0.820, respectively; mean values of the results electro-diagnosed by physicians were 0.537, 0.527, 0.770, 0.582, and 0.511, respectively. The performance of CNN algorithm for predicting myopathy, neuropathy, and normal was also evaluated with area under the receiver operating characteristic curve, and the results were 0.898 (95% confidence interval [CI] 0.884–0.912), 0.840 (95% CI 0.838–0.841), and 0.948 (95% CI 0.928–0.968), respectively.
Conclusions: This study demonstrated that the CNN algorithm is valuable in interpreting nEMG of patient with neuropathy or myopathy on behalf of physicians and assisting physician’s decision making in diagnosing patients with suspected neuromuscular disease. Large, prospective cohort studies with more diverse neuromuscular disease are needed in the future.
Keywords: Electromyography, Machine learning, Neuromuscular disease, Convolutional neural network.
eP02.01.06Normative Values for Commonly Used Nerve Conduction Studies in Russian Population
Kovalchuk M1, Kovalchuk M2, Druzhinin D3, Pyatkov A4, Druzhinina E5, Sarycheva A6
1Moscow City Clinical Hospital after V.M. Buyanov, Moscow, Russian Federation, 2Chaika Clinics, Moscow, Russian Federation, 3Yaroslavl State Medical University, Department of Neuropathology, Yaroslavl, Russia, 4Moscow Centre for Research and Practice in Medical Rehabilitation, Restorative and Sports Medicine of Moscow Healthcare Department, Moscow, Russia, 5Pirogov Russian National Research Medical University, Moscow, Russia, 6Central military clinical hospital No3 after A.A. Vishnevsky, Krasnogorsk, Russia
Introduction: Reference values used in routine nerve conduction studies (NCS) depend on the technical and lab settings, as well as on ethnical and physical factors such as subject’s height, age and skin temperature. Each geographical region and even neurophysiology department is preferred to have own NCS normative dataset as it is crucial in suspected peripheral neuropathy. Our aim was to establish limits of standard NCS values in Russian population.
Materials and Methods: 77 healthy volunteers aged between 18 and 76 years were investigated by four neurophysiologists in a standardized technique using extended NCS protocol: motor, sensory and F-waves of median, ulnar, radial, tibial and peroneal nerves both sides. Skin temperature was monitored along the whole study. Following maximal and minimal parameters were analyzed for each nerve, except radial: compound muscle action potential (CMAP) amplitude; sensory nerve action potential (SNAP) amplitude; nerve conduction velocity (CV); distal motor latency (DML); min F-latency. Gender and age differences were assessed.
Results: Out of 77 volunteers 32 were male (41.5%) and 45 were female (58.5%). Mean age was 36.2 years old; mean height 174.4 cm. Complete set of NCS values is presented in Table 1. The spread of CMAP values for tibial and median nerves was larger, than for other nerves.
Conclusion: This study established normative parameters for routine NCS in the Russian cohort. This can be of use in a selected geographic region when peripheral nerve abnormality is suspected.
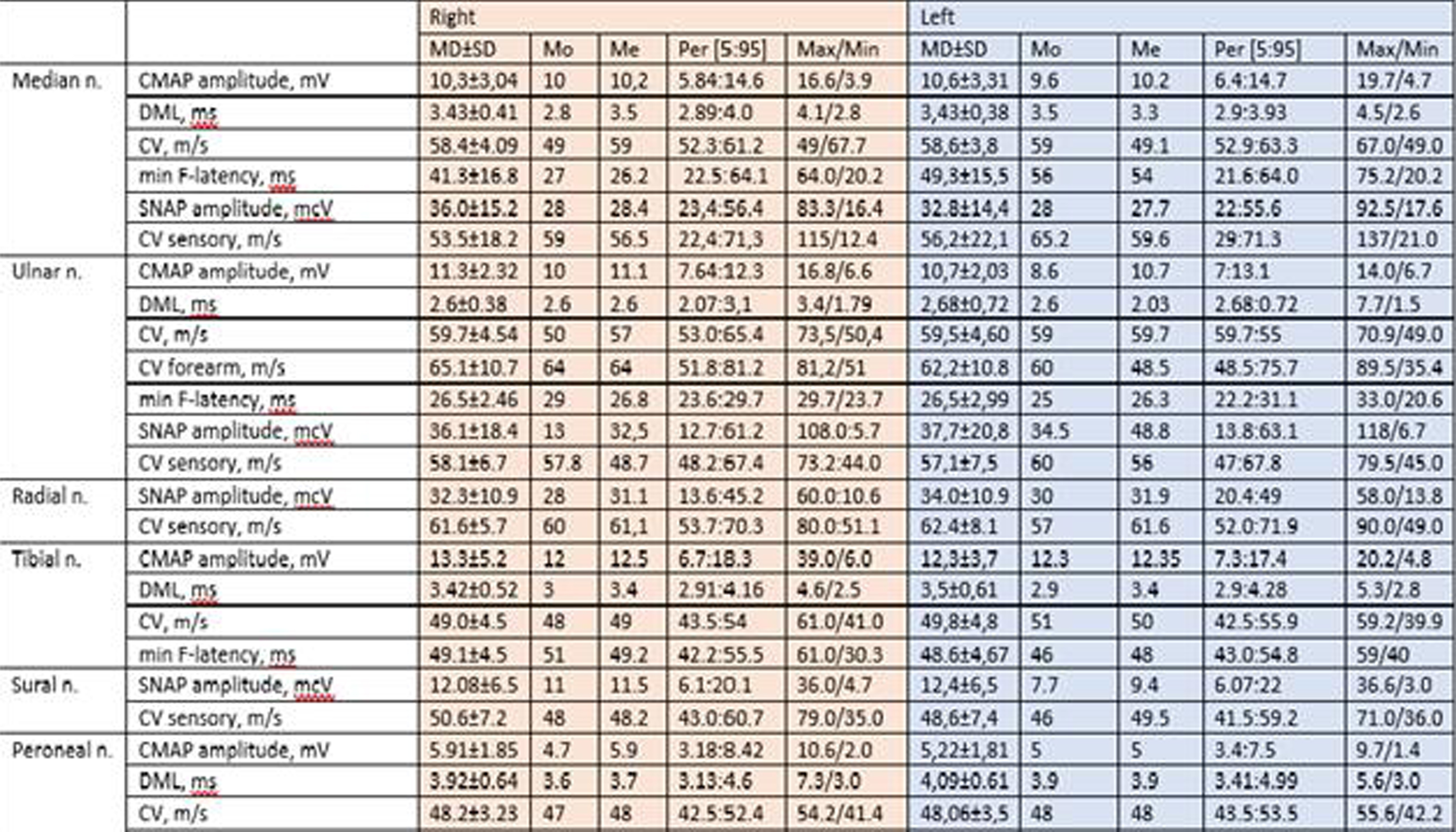
eP02.01.07Cardiac MRI in Duchenne and Becker Muscular Dystrophy
S G M1, Nalini A1, Vengalil S1, Barthur A2, Nashi S1
1NIMHANS, Bangalore, India, 2Sri Jayadeva Institute of Cardiovascular Sciences and Research, Bangalore, India
Objectives – We sought to determine the prevalence of cardiac involvement in Duchenne and Becker Muscular Dystrophy (DMD and BMD) using Cardiovascular MRI (CMRI) and its association with clinical and laboratory parameters.
Background - Cardiac disease is a common manifestation of DMD and BMD. CMRI can be used as a non-invasive technique for detection of early myocardial fibrosis in DMD and BMD.
Methodology – A prospective, observational study was conducted on 30 DMD and 5 BMD patients, genetically confirmed using Multiplex Ligation-dependant Probe Amplification (MLPA) and underwent cardiac MRI between March 2020 and August 2021.
Results –DMD patients had mean age of disease onset– 4.03 ± 1.63 years, and mean disease duration- 7.3 ± 3.42 years; BMD patients had mean age of onset- 13.4 ± 4.56 years and mean disease duration- 6.4 ± 2.41 years. All patients had predominantly proximal lower and upper limb weakness. 83.87% had variable deletion of exons in DMD gene (84.61% confined to regions between exons 45 to 54). CMRI revealed late gadolinium enhancement (LGE) of left ventricular (LV) wall, suggestive of myocardial fibrosis in 51.43% of cases, but only 5.71% was symptomatic. Among the patients with LGE, 55.55% (10 of 18) had reduced LV Ejection Fraction. Myocardial fibrosis in CMRI was associated with disease duration, but not with age, genotype or creatine kinase levels. CMRI changes showed good correlation with ECG – a characteristic descending pattern of voltage in anterior chest leads and low voltage in V6 lead.
Conclusions – There is a high prevalence of subclinical cardiac involvement in DMD/BMD patients which can be detected using Cardiac MRI, which helps in initiating early cardioprotective therapy in this subgroup. Myocardial fibrosis is confined to inferior and lateral LV wall in patients with mild involvement and progress towards LV apex, anterior and septal wall in patients with greater involvement.
eP02.01.08Artificial Intelligence Based Automatic Muscle MRI Segmentation: Towards a Generalized Solution for Quantitative Imaging
Snezhko E1, Carlier P2, Reyngoudt H3, Carlier R4, Bardakov S5, Suslov V6
1United Institute Of Informatics Problems, National Academy Of Sciences Of Belarus, Minsk, Belarus, 2University Paris-Saclay, CEA, SHFJ, Orsay, France, 3Institute of Myology, Pitie-Salpetriere University Hospital, Paris, France, 4Medical Imaging Department, Raymond Poincare University Hospital, Garches, France, 5SM Kirov Military Medical Academy, Russian Ministry of Defense, Saint-Petersburg, Russia, 6Saint-Petersburg State Pediatric Medical University, Saint-Petersburg, Russia
The need to manually segment muscles on NMR images has been the main bottleneck to a wider diffusion of the quantitative imaging protocols and their use as an outcome measure in clinical trials. The motivation for this work was to apply recent techniques based on Convolutional Neural Networks (CNNs) to the segmentation of individual muscle groups and whole segment muscle volumes segmentation on NMR images.
The cohort for individual muscle group segmentation contained 300+ Dixon out-of-phase volumes of legs and thighs volumes. In the thighs, the quadriceps and hamstrings were manually segmented as well as the foot extensors, the peroneal group and the triceps surae in the legs. The cohort for whole segment muscle volume determination contained 700+ Dixon out-of-phase data sets together with water and fat maps from nine different neuromuscular diseases.
Convolutional neural network (CNN) was applied on the data, composed of an encoder part, which is MobileNet v2 without fully-connected layers, and a decoder part consisting of up-sampling layers and a soft maximum classifier at the end. Four layer pairs from encoder and decoder parts are connected in UNet manner.
The median values of CNN vs manual segmentation Dice’s coefficient for muscle groups ‘Quadriceps’, ‘Hamstrings’ were 0.968 and 0.948, respectively. The median values of Dice’s coefficient of segmentation for muscle groups ‘Foot Extensors’, ‘Peroneal group’ and ‘Triceps Surae’ were 0.949, 0.929 and 0.951.
The median values of Dice’s coefficient of segmentation for whole segment muscle volumes at the level of legs and thighs were 0.980 and 0.984, respectively. The median Hausdorff distance for whole segment muscles were 3.390 (legs) and 4.243 (thighs).
These results show the ability of an AI based automatic segmentation software to contour precisely muscle groups and segment global muscle volumes of diseased muscles with variable degrees of fatty replacement.
The experiments on individual muscle groups and whole segment muscle volumes segmentation on NMR images using CNNs showed the acceptable quality for further quantitative analysis of automatically segmented regions of interest.
CNN based automatic segmentation of individual muscle groups and whole segment muscle provides a robust solution for volumes of Dixon NMR images. Additional tests and experiments are being conducted on individual and global muscle segmentation of standard gradient echo and spin echo images collected from platforms of different vendors.
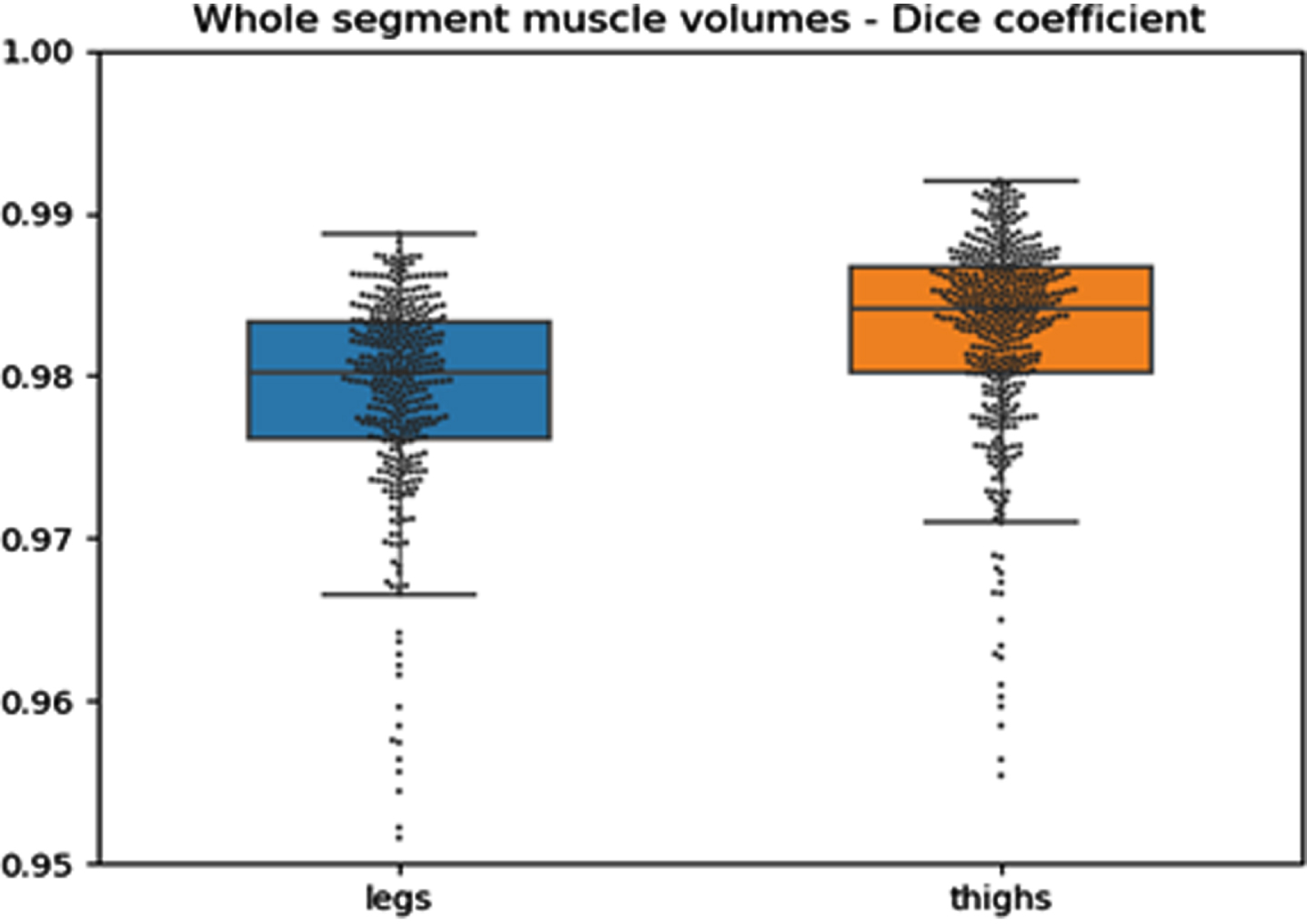
eP02.01.09Neuropathies Amidst the Pandemic: Remote Phenotype Validation and Assessment of Patient Needs
Dohrn M1,2, Broschwitz R2, Dumke C2, Schöne U2, Bähr F2, Peschke G2, Achenbach P2, Brunkhorst R2, Schulz J2, Züchner S1
1Hussman Institute For Human Genomics, Miller School Of Medicine, University Of Miami, Miami, United States, 2Medical Faculty of the RWTH Aachen University, Department of Neurology, Aachen, Germany
The past two years have been significantly overshadowed by the respiratory virus Covid-19. This has shown a relevant impact on health care systems, so that at a specialized neuromuscular center, we experienced a shortness of personnel and time, and in parallel, we were facing many unanswered questions addressed to us by patients with rare diseases. In this study, we developed a new questionnaire to assess patient needs, concerns, and symptoms confronting the global pandemic.
We included individuals with hereditary neuropathies (n=15), autoimmune-inflammatory neuropathies (n=26), or idiopathic small fiber neuropathies (n=45). For validation, we used previous clinical examination reports. Forty-six percent of the included patients were female, 52% male, and one patient “diverse”. The mean age at examination was 52.67±13.37 years (range: 19-79 years).
Most of the patients (59%) reported mild to moderate limitations in their daily life activities due to Covid-19. Severe impairment was reported in 28%. Due to the pandemic, 54% of the patients reported to be concerned about their own and 76% about their relatives’ health. Patients with a positive family history were 2.4x more likely to be seriously worried about other family members. We observed that patients with more wide-spread sensory loss reported higher impairment levels than those with distal sensory loss only. Overall, 37% of the patients said that contracting Covid-19 was their main concern, including the presumed risk of a severe course. Further 34% were worried that their neuropathy might worsen if they ever contracted Covid-19. Thirty-three percent of the patients experienced limitations in their treatment options, e.g. by not being able to continue their physical therapy. Seven percent were concerned about social distancing, as daily care required direct interactions with others. Whereas 65% of the included individuals confirmed that they felt appropriately informed by their treating physicians, 21% wished to receive more information. Sixteen percent, however, said that they did not dare to ask their questions in order to not disturb the health care personnel amidst the crisis. Patients with hereditary, autoimmune, or small fiber neuropathies did not show any differences in their Covid-related daily-life impairment. Previous clinical results correlated with patient-reported sensory levels; and gait unsteadiness was reported significantly more often in patients with afferent ataxia.
We conclude that Covid-19 imposes a relevant daily-life burden on neuropathy patients. Patient-reported outcome measures are a valid remote strategy if in-person visits are not possible.
eP02.01.10Subclinical Status of Dysferlinopathy
Bardakov S1, Deev R, Suslov V, Carlier P, Reyngoudt H, Tsargush V1, Musatova E, Umakhanova Z, Isaev A
1Kirov S.M. Military Medical Acadamy, St.Petersburg, Russian Federation
Dysferlinopathy is a phenotypically heterogeneous, inherited, progressive muscular dystrophy caused by mutations in the DYSF gene. Dysferlinopathy is marked by elevated serum creatine kinase (CK) and can manifest as hyperCKemia in asymptomatic or low-symptom states.
We describe the clinical signs and symptoms and laboratory and imaging results of eight patients at the preclinical stage of dysferlinopathy (aged 3-12 years), with homozygous mutation of the DYSF gene. MRI scanning was performed from the anterior, superior, iliac spine to the lower third of the legs on an Ingenia 1.5T tomograph (Philips Healthcare, Eindhoven, Netherlands) using a body surface receiver coil. The protocol included T2-multi-slice multi-echo (T2 MSME), T1 weighted (T1w), T2 weighted (T2w) and Short Tau Inversion Recovery T2 weighted (STIR T2w) pulse sequences. Water T2 (wT2), an imaging biomarker of oedema and disease activity and fat fraction, a marker of chronic degenerative changes were extracted by a triexponential deconvolution of the MSME signal. The control group consisted of eight healthy children of the corresponding gender and age.
Based on this case series, we propose three stages of dysferlinopathy disease progression. The first stage is asymptomatic hyperCKemia laboratory syndrome of myocytolysis, which is marked by a mild increase in CK (>1.5 times the upper limit of normal (ULN)) and lactic dehydrogenase (LDH). Second stage: moderate increase in CK (2.7-3.2 × ULN) and LDH, minimal signs of fatty muscle infiltration, displacement of the center of support to the back of the foot during plantography, and decrease in Achilles reflexes. Third stage: higher increase in CK (3.2-6.2 × ULN), LDH, alanine aminotransferase (ALT), and myoglobin; moderate fatty infiltration of muscles; slight afterload fatigue of the calf muscles; slight decrease in muscle strength; reduction of Achilles and knee tendon reflexes. Taken together, isolated hyperCKemia and low-symptom patients form a single continuum of pre-symptomatic cases.
Quantitative MRI revealed an t increase in wT2 in the anterior thigh muscles (in particular, in the m. vastus intermedius), the adductor muscles of the thighs, the anterior and peroneal groups of the lower leg, and the medial head of the gastrocnemius muscle. A slight increase in the fat fraction was noted in the gluteal muscles (in particular in the m. gluteus maximus) and the posterior leg muscle group (m. soleus and the lateral head of the gastrocnemius muscle).
Surprisingly, there was an inverse correlation between wT2 in the muscles of the anterior thigh group and the CPK(rp=0.83, p = 0.03), LDH (rp=0.85, p = 0.02), myogoglobin (rp=0.88, p = 0.02) levels and the age of patients (rp=0.89, p = 0.01).
Quantitative MRI methods can detect minimal changes, in the absence of anomalies on the standard images. In particular signs of disease activity were revealed by an increase in water T2.
eP02.02.01Overview of Patients With Chronic Inflammatory Demyelinating Polyneuropathy of the Neuromuscular Reference Center of LIèGE
Poleur M1, Maertens de Noordhout A1, Lievens I2, Delstanche S1
1University department of neurology, Centre Hospitalier Régional de la Citadelle, Liège, Belgium, 2Departement of neurology, Centre Hospitalier Universitaire, Liège, Belgium
Introduction: There is currently a worldwide shortage on intravenous immunoglobulins (IVIG). In 2015, the Belgian authorities limited the prescription of IVIG for Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) to neurologists attached to a neuromuscular reference center (NMRC).
Method: We established a registry to characterize our patients diagnosed with CIDP. We reviewed medical records of all CIDP patients registered in the NMRC of Liège in 2021. We recorded in a table clinical and electrophysiological data such as supportive criteria (cerebrospinal fluid analysis, MRI, biopsy and response to therapy) relative to the previous EFNS/PNS and new EAN/PNS criteria in order to verify if our patients meet them.
Results: We studied 117 patients aged between 7 and 87 years. Mean age at the time of diagnosis was 52 (± 14.1) years old. The ratio male/female is 2/1. Clinically, two-thirds of patients had typical CIDP. Only 41% of patients showed absent deep tendon reflexes. Among EFNS/PNS electrophysiological criteria, significant nerve conduction velocity slowing was most frequently found. Forty-four percent of patients presented at least one conduction block. Some nerves (e.g. tibial) and other criteria (e.g. distal compound muscle action potential duration) were seldom assessed. Among supportive criteria, patients showed less frequent albumin cytologic dissociation (46%) than previously described. In patients with albumin cytologic dissociation, mean proteinorachia was 1053 mg/dL. A nerve biopsy was performed in four patients and influenced the diagnosis in none of these cases. Among 8 plexus magnetic resonance imaging performed, two showed typical signs of CIDP. It influenced the diagnosis for one patient. Eighty-two percent of patients responded to steroids or IVIG. Response to treatment was used as a supportive criterion in 17.8% of cases. Currently, 89 patients are treated. Among them, 93% of patients receive IVIG.
According to 2010 EFNS/PNS criteria, 82% and 7.7% of our population met the criteria of defined and probable CIDP, respectively. Six patients initially included for analysis do not suffer from CIDP and their diagnosis was modified. Seven percent of patients diagnosed with CIDP did not meet the 2010 EFNS/PNS criteria. There was no difference between these patients in terms of supportive criteria except that they tended to present poorer response to standard treatment. For three of these patients, the diagnosis is under consideration. We will also present a comparison between previous EFNS/PNS and new EAN/PNS criteria at the ICNMD.
Discussion: Due to the lack of specific test, the diagnosis of CIDP is based on a combination of clinical, electrophysiological, and laboratory features. Unfortunately, misdiagnosis is common, leading to unjustified prescription of IVIG. Guidelines and diagnosis criteria are designed to avoid diagnostic pitfalls and prevent unnecessary treatment.
This study highlights significant ways for us to improve our clinical practice. In the future, we should create a standardized protocol of electrophysiological studies for CIDP patient, use more widely the supportive criteria and assess objectively the response to treatment as recommended by the guidelines.
This study also highlights the necessity to find new treatments for CIDP patients in an era of IVIG shortage.
eP02.02.02Intravenous Immunoglobulin Therapy in Patients with Chronic Inflammatory Demyelinating Polyneuropathy: A Systematic Literature Review
Anderson-Smits C1, Mitchell S2, Hawe E2, Hartley L2
1Takeda Development Center Americas, Inc., Cambridge, United States, 2RTI Health Solutions, Manchester, United Kingdom
Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is a rare, chronic, progressive or relapsing immune-mediated disorder that typically requires long-term therapy. Intravenous immunoglobulin (IVIG) and systemic corticosteroids are recommended as first-line treatment options for both induction of therapy and maintaining clinical response in CIDP, with plasma exchange (PE) considered if these treatments fail.
Objective: To systematically identify and describe published randomized controlled trials (RCTs) on the use of IVIG (versus placebo, systemic corticosteroids or PE) in patients with CIDP.
Methods: Systematic searches of the MEDLINE, Embase, BioSciences Information Service and Cochrane Library electronic databases were performed (database inception–March 2020) to identify RCTs of IVIG versus placebo, systemic corticosteroids or PE in patients with CIDP. Selected congress, society and key health technology assessment and regulatory websites were also searched (2017–2020). Articles were screened using pre-specified criteria for population, intervention, comparison, outcome and study design. Efficacy, safety and health-related quality of life (HRQoL) outcomes data were extracted and summarized using descriptive statistics.
Results: In total, 10 eligible RCTs were identified for analysis: five placebo-controlled studies and five head-to-head comparison studies (two versus other IVIG therapies, two versus systemic corticosteroids and one versus PE). Across RCTs, IVIG was administered at 0.4–2 g/kg over 1 day to 9 weeks, or as 50 or 100 g/L infusions. Compared with placebo, IVIG was generally associated with statistically significant improvements (p≤0.035) in disability (4 of 5 studies reporting relevant data), muscle-strength (3 of 4 studies) and nerve-conduction outcomes (3 of 5 studies), and in Rotterdam Handicap Scale scores and all domains of the Short Form 36 Health Survey (1 study; p<0.001 and p≤0.044, respectively). The different IVIG therapies compared in the two head-to-head RCTs showed generally equivalent efficacy with respect to disability measures. In the trials comparing IVIG with other treatments for CIDP, no significant differences were identified between IVIG and systemic corticosteroids for disability, most muscle strength or HRQoL outcomes, or between IVIG and PE for disability outcomes (muscle strength and HRQoL not assessed). Across RCTs, the adverse events reported with IVIG were mostly mild, with few serious adverse events observed.
Conclusion: In this systematic literature review of RCTs in patients with CIDP, IVIG use was shown to improve disease symptoms and HRQoL measures significantly compared with placebo and to have a favorable safety profile. Although few head-to-head comparisons of IVIG with other IVIG products/regimens, systemic corticosteroids or PE exist, those available have demonstrated no meaningful differences between therapies in the efficacy or HRQoL outcomes evaluated.
eP02.02.03Chronic Inflammatory Demyelinating Polyradiculoneuropathy in Patients in Diabetic and Non-Diabetic Patients: A Comparative Study
Abida Y1, Mrabet S1,2, Nasri A1,2, Atrous A1,2, Souissi A1,2, Gharbi A1,2, Gargouri A1,2, Kacem I1,2, Gouider R1,2
1Department Of Neurology, Lr18sp03, Clinical Investigation Centre Neurosciences And Mental Health, Razi Hospital, Manouba, Tunisia, 2Faculty of Medicine of Tunis, University Tunis EL Manar, Tunis, Tunisia
Introduction: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an acquired demyelinating neuropathy. The possibility of co-association between diabetes mellitus (DM) and chronic inflammatory demyelinating polyneuropathy (CIDP) has long been a focus of interest as well as of its clinical significance. Our objective is to determine whether the clinical characteristics, electrodiagnostic classification of nerve injury and treatment response differed in diabetes and non-diabetes patients diagnosed with CIDP.
Methods: Retrospective study including all patients with CIDP defined according to the EFNS 2010 criteria (n=56). DM was diagnosed according to the American Association of Diabetes criteria. Subjects with a confirmed diagnosis of type 2 diabetes mellitus (DM) and CIDP were included in this study (n =16). Clinical and electrophysiological data were collected,
Results: Mean age of onset was 43 in CIDP + DM patients and 55 in CIDP without DM (p=0.01). Clinical features including early signs, motor deficit and superficial sensitivity did not differ between the two groups. Deep sensitivity was mostly impaired in CIDP+DM patients (p=0.002). Electrodiagnostic features were similar in the two groups. Treatment response (p=0.824) as well as number of prednisone or IVIG courses were also equivalent between the groups (p=0.134 and p=0.9 respectively)
Conclusion: Our study did not find a major difference between diabetic and non-diabetic patients with CIDP apart from age of onset and deep sensation involvement
This is in contrast with previous studies showing that CIDP+DM patients are found to be proximally weaker, to have more severe motor and sensory impairment in nerve conduction studies and to have less favorable outcome. This may be due to a shorter time since the onset of the diabetes which could lower the effects of the superimposed diabetic distal symmetric sensorimotor polyneuropathy.
eP02.02.04Identical Late Responses in Early Stages of Guillain-Barré Syndrome: A-Waves or Repeater F-Waves
Veltsista D1, Kintos V2, Kefalopoulou Z1, Chroni E1
1University of Patras, Medical School, Rio, Greece, 2Department of Neurology, General Hospital of Elefsina Thriasio, Magoula-Elefsina, Greece
Purpose: Early detection of neurophysiological abnormalities in patients with suspected Guillain-Barré Syndrome (GBS) is clinically valuable. We herein analyzed the frequency and significance of identical late motor responses following the compound muscle action potential (CMAP), in the initial examination of GBS patients.
Methods: We retrospectively reviewed nerve conduction studies (NCS) performed in our institute between January 2020 and December 2021. Twenty-two GBS patients (12 females; mean age 55.3 ± 19.1 years) who underwent NCS within 10 days from symptom onset (median interval 5 days, IQR 5) were included. A total of 86 nerves, including 27 ulnar, 19 median, 25 peroneal and 15 tibial nerves, were studied. Late responses were elicited by a series of 40 supramaximal stimuli in each nerve. A-waves were recognized as identical in latency and configuration responses in ≥16 of 40 traces. Repeater F-waves (Freps) were identified by means of a software program (F Wave Analyzer) and index total Freps (100 x total number of Freps/ total number of traces with F-waves) was calculated.
Results: At the initial NCS, whose late responses were evaluated herein, 9 patients fulfilled the neurophysiological diagnostic criteria for GBS. At later stages, a diagnosis of acute inflammatory demyelinating polyneuropathy (AIDP) was confirmed in 13 and acute motor axonal neuropathy (AMAN) in 9 patients.
A-waves were present in 44 of the 86 studied nerves (13/27 ulnar, 6/19 median, 13/25 peroneal, 12/14 tibial). An abnormal increase in Index Total Freps was calculated in 54 out of the 57 nerves with F-wave recordings (17/18 ulnar, 13/13 median, 13/14 peroneal, 11/12 tibial). Late responses were absent in 15/ 86 nerves. Co-occurrence of A-waves and increased index total Freps was found in 29/ 86 nerves.
In nerves with conduction block (CB), A-waves appeared more frequently compared to increased index total Freps (15.9% vs 5.9%, p<0.05). However a comparison of A-waves presence between AIDP and AMAN (22.1% vs 13.9%, p=0.275) did not reach a significant level. A-waves morphology was polyphasic (>4 phases) in 18 nerves (13 AIDP), and single in 26. Minimum latencies of A-waves were always shorter than that of F-waves.
In nerves with otherwise normal motor conduction studies increased index total Freps was significantly more common compared to A-waves (94.1% vs 84.1%, p<0.05). Disease subtype and CMAP amplitude abnormalities did not correlate significantly with increased index total Freps (p=0.124). Index total Freps correlated negatively with overall F-wave persistence (p= 0.025).
Discussion: A-waves and Freps are late responses frequently observed in the majority of nerves studied in early GBS. Both are characterized by identical potentials occurring in consecutive recordings. A-waves appeared in nerves with motor conduction abnormalities and are associated with CB. Whereas, an increased index total Freps suggests that a reduced number of motor neurons are able to produce F-waves and constitutes an early finding even when standard nerve conduction studies are unaffected.
eP02.02.05An Unusual Clinical Evolution in Anti-contactin-1 Positive CIDP
Vivier Y1, Alonso Jimenez A
1University Hospital of Antwerp, Schoten, Belgium
Specific clinical, electrophysiological and serological features are used to recognise a phenotype fitting the atypical chronic inflammatory demyelinating (CIDP) variant spectrum. We report a 28-year-old male patient, without any significant history apart from a recent asymptomatic COVID-19 infection, presenting at first with bilateral facial nerve palsy, subsequently –three months later- developing an subacute onset symmetric sensory ataxia and areflexia, and thirdly experiencing diffuse rapidly progressive motor deficits. Additional investigations suggested an autoimmune polyneuropathy: Liquor analysis showed cytoalbuminologic dissociation. Cerebrospinal fluid protein elevation was remarkable: 631 mg/dL. Nerve conduction studies showed prominent distal latencies prolongation and dispersion of the potentials, meeting the electrodiagnostic criteria of the European Federation of Neurological Societies/Peripheral Nerve Society for CIDP (2021). Full spine magnetic resonance imaging depicted pathological thickening and enhancement of the roots of the cauda equina as seen in radiculitis. There was no or poor response to conventional treatment, i.e. immunoglobulins (IVIG), corticosteroids and even plasmapheresis. Muscle weakness deteriorated. Presence of serum IgG4 anti- contactin-1 (CNTN1) antibodies was found by ELISA identification and titration, and the patient improved substantially after rituximab treatment. While contributing to the expanding confidence in nodal and paranodal antibodies as valuable biomarkers in clinical practice, our case entails several peculiarities: 1/ SARS-CoV2 positivity as a possible trigger of this auto-immune polyneuropathy 2/ A considerably younger age of onset than in the patients already described (range 33-76 years). 3/ The clinical course progressed in an atypical manner even for atypical CIDP: initial presentation with bilateral asymmetric facial palsy, followed by sensory ataxia, which prompted the initial diagnosis of Miller-Fisher syndrome, and later development of severe motor impairment. 4/ Proteinorachy was so pronounced that we considered neuroborreliosis as a potential associated disorder. Borrelia seroconversion occurred after the first IVIG-treatment, and could be false positive. However, the patient was treated with intravenous ceftriaxone, which had no effect on the clinic. 5/ Antibodies against CNTN1 were undetectable after 2 months of rituximab. Emphasising the both diagnostic and therapeutic importance of recognising a phenotype compatible with atypical CIDP, an underrecognized and consequently undertreated disease where early diagnosis and prevention of axonal damage is crucial in.
eP02.02.07Characteristics and Epidemiology of Patients with Multifocal Motor Neuropathy in Latvia.
Roddate M1,2, Glazere I1,3, Zukova V1,2, Rots D4, Gailite L4, Kurjane N3, Kenina V5
1Pauls Stradins Clinical University Hospital, Department of Neurology, Riga, Latvia, 2Riga Stradins University, Faculty of Continuing Education, Riga, Latvia, 3Riga Stradins University, Department of Biology and Microbiology, Riga, Latvia, 4Riga Stradins University, Scientific Laboratory of Molecular Genetics, Riga, Latvia, 5Riga East Clinical University Hospital, Rare disease centre, Riga, Latvia
Introduction: Multifocal motor neuropathy (MMN) is a chronic immune-mediated disorder characterized by progressive asymmetric weakness with spared sensation and conduction blocks on electrophysiological assessment. The disease has an estimated prevalence of 0.6 to 2 per 100,000 population with male-to-female ratio of 2.7:1 and remains a diagnostic challenge considering wide spectrum of differential diagnosis of lower motor neuron syndromes. This is a first study presenting clinical characteristics and epidemiological data of MMN patients in Latvia.
Objectives: To determine incidence (2015-2021) and prevalence (on November 1, 2021) of MMN in Latvia and describe the main characteristics of MMN patients in Latvia.
Methods: A review of patient’s files diagnosed with MMN in a 6-year period (2015-2021) in Pauls Stradins and Riga East Clinical university hospitals. Repeated neurologic evaluation for all patients, including MRC scoring scale, grip strength, MMN-RODS and 6MWT for clinical assessment.
Results: 8 patients (4 men and 4 women) with confirmed MMN (according to EFNS/PNS clinical and electrophysiological criteria) were enrolled in our study. At the time of enrolment, the median age was 46 years (range 16-76). The median age of symptom onset was 37 years with a median duration of the disease until the diagnosis of 24 months. The prevalence and incidence were 0,39 per 100 000 and 0,40 per 100 000, respectively. 6 patients had monophasic course of disease and 2 female patients reported polyphasic course with few relapses. All patients reported upper limb involvement as the first symptom. Electrophysiological evaluation revealed initial conduction blocks in median, ulnar and tibial nerves in most cases. At the time of the last follow-up all patients were able to walk without assistance. Clinical evaluation revealed significant distal atrophy or hypotrophy, mostly in thenar/hypothenar and palms as well as proximal interphalangeal joint contractures in 3 patients; 3 patients had areflexia in the affected limbs. The median MMN-RODS was 43 in our patient group (summed raw score, range 3-48). All patients received immunoglobulins as the first line of treatment: intravenous administration was chosen in 6 patients, two others received medication subcutaneously. Only one patient reported no improvement after received treatment.
Conclusion: The incidence and prevalence of MMN patients in Latvia are lower to previously reported studies worldwide. Our study also does not show male predominance. All things considered; we suspect that MMN is likely to be under-recognised pathology in our region. Results of our study emphasises importance of careful clinical and electrophysiological evaluation in patients with lower motor neuron syndrome since specific immunomodulating treatment with good response is available for MMN patients.
eP02.02.08Herpes Zoster May Be a Trigger for Lumbosacral Radiculoplexus Neuropathy
Aragon Pinto C1, Pinto M1, Dyck P1
1Mayo Clinic, Rochester, United States
OBJECTIVE: To describe the association between herpes zoster and lumbosacral radiculoplexus neuropathy (LRPN).
BACKGROUND: LRPN is an immune mediated neuropathy that may have a trigger in approximately 1/3 of cases. The most common triggers are surgery, rapid glycemic changes, vaccines and infection. Varicella-zoster virus (VZV) is known to cause central and peripheral nervous system complications but the association of VZV and LRPN has never been systematically studied.
METHODS: We performed an electronic chart review to identify patients who developed LRPN up to 4 weeks after the appearance of VZV infection rash at our institution between 1/1/2000 to 12/31/2017. We only included patients who had classic herpes zoster diagnosed by a physician.
RESULTS: Seven patents were identified. 5 (71.4%) had diabetes. Median age-at-onset was 68 years (range 48-76), 5 (71.4%) were female and median time to LRPN diagnosis was 6 (2-16) months. Median time from rash onset to LRPN onset was 3 (1-4) weeks, and median time from LRPN onset to nadir was 6 (2-12) months. The rash location was in the trunk region in 3 (42.9%) patients, face in 2(28.5%), groin in 1 (14.3%) and arm in 1(14.3%). No patient had diffuse VZV infection. All patients had weakness and pain in roots and nerves distributions outside of the rash location. At diagnosis, 3 (42.8%) patients had more than 10 lbs of weight loss, 5 (71.4%) had bilateral lower extremity involvement, 3 (42.9%) had panplexus involvement, and the median modified Rankin scale was 3 (2-4). In 3 patients, herpes zoster anteceded a recurrence of patient’s previous episode of LRPN. One patient was immunosuppressed (liver transplant).
CONCLUSIONS: Herpes zoster can be associated with LRPN. Even though VZV might cause direct peripheral nerve injury, the protracted progression of LRPN, neuropathic symptoms far away from the rash location and recurrence of LRPN may suggest that VZV is rather a trigger for LRPN.
eP02.02.09Acute Worsening of Anti-mag Neuropathy Following Treatment With Rituximab
Muley S, Kaldawi S
1Barrow Neurologic Institute, Phoenix, United States
Rituximab is efficacious in the treatment of anti-MAG neuropathy. Rare cases of worsening of neuropathy after rituximab have been reported. All cases improved, but rarely back to baseline. We report a case of rituximab related worsening of anti-MAG neuropathy that did improve with plasma exchange.
Overall, 10 cases have been described in the literature with rituximab related worsening of anti-MAG neuropathy. Only 1 of these reports utilizes plasma exchange as treatment and that case reported patient stabilization to pretreatment level. All other cases utilized either IVIg or no treatment. None of these reported improvement beyond pretreatment level.
Patient is a 70 year old female who first noticed tingling in her feet 5-6 years ago that progressed gradually to the lower legs and led to gait incoordination. She was diagnosed with anti-MAG neuropathy by an outside neurologist 1-2 years after symptom onset (antibody titer 1:51200). She did not respond to a one year course of IVIg.
She was seen at our institution 4 years after symptom onset. At this point she had numbness that had progressed to her knees and finger tips with some gait instability. Strength in the legs was normal and she was walking two miles per day (despite the imbalance). Romberg’s test was negative. Serum immunofixation showed IgM kappa gammopathy with total IgM level of 234mg/dL. Repeat EMG/NCS showed conduction slowing with disproportionate severe prolongation of distal motor latencies. Treatment with two induction doses of rituximab 1 gm, 14 days apart was initiated because of progressive numbness and the gait instability.
Two days after the second dose she had significant worsening of numbness in her hands /legs with severe impairment of balance and was no longer able to do her walks as before, even though strength remained normal. Examination showed absent proprioception and severely reduced vibratory perception at the ankles and fingertips with positive Romberg’s test. Sensation to light touch was now reduced up to midcalf and wrist level. Repeat EMG/NCS showed significant worsening of conduction slowing compared to the prior study. Plasma exchange was initiated, 5 cycles (every other day), followed by 3 times per week for a total of 6 weeks. Patient reported subjective improvement in balance about 4 weeks into the plasma exchange, with plateau. She reported being able to walk a total of 3 miles without any assistive devices. A repeat EMG/NCS was done and showed significant improvement in median and ulnar motor amplitudes, and distal latencies have improved.
Our case illustrates that despite the initial worsening with rituximab, initiation of plasma exchange leads to robust improvement beyond pretreatment levels. While overall there is a limited number of cases, it is possible that in some patients worsening with rituximab is expected, transient, and eventually improves to some degree without intervention. The addition of plasma exchange could improve recovery beyond baseline function. Thus, rituximab may be effective even in patients who show initial worsening in cooperation with plasma exchange. Future research is necessary to determine predictors of this outcome.
eP02.02.10Clinical and Prognostic Characteristics of Guillain-Barre Syndrome Associated With COVID-19, Is This Coincidental?
Ansari B1
1University Of Medical Science In Isfahan, Isfahan, Iran (Islamic Republic of)
Introduction: During Covid-19 pandemic periods, various studies have been revealed the coexistence of these two diseases, raising the question of whether SARS-CoV-2 has a role in triggering GBS or it’s just co-incidentally. So far, 255 cases of this concurrence have been reported.
In this study, we publish 45 patients’ demographic, clinical, electro diagnostic study, response to treatment and prognostic features association of Covid-19 and GBS during the 5 corona’s epidemiologic peaks in Isfahan province.
Methods: This cross-sectional, multi-central study was performed during covid-19 pandemic since 2020 February until 2021 October. In this period 5 epidemiologic peaks of corona virus occurred in Isfahan (one of providence of Islamic republic of Iran) and total of 417166 people became infected. 45 patient with definitive Covid-19 (based on positive nasopharynx Reverse transcription polymerase chain reaction (RT-PCR) or highly suggestion of High-resolution computed tomography (HRCT) for covid-19) were referred to one of the 2 referral hospitals (Alzahra and Kashani hospital). All patients whom suspected of peripheral nerve symptoms referred to the neuromuscular fellowship for further examination and performing EDx. Demographic, clinical, therapeutic and prognostic features were collected according to Hospital records.
Results & discussion: 45 patients (60% male, 40% female) were surveyed. The mean age was 54.66±10.021 (max: 84, min:14, range:80). The most EDx pattern was AIDP (57.8%, n=26).42.2%(n=19) of patients had axonal pattern. 8 of them were Acute motor axonal neuropathy(AMAN) and 11 patients were Acute motor-sensory axonal neuropathy(AMSAN). The most (91%) GBS phenotype was classic pattern which defined as acute-sub acute flaccid length dependent paralysis. 2 patients had pure para paretic pattern and 2 had miller-fisher pattern.
The most common symptom of covid-19 was fever (89.7%), Other symptoms included dyspnea (48.7%), cough (46.2%), myalgia (28.2%), headache (28.2%), diarrhea (28.2%) and the less common was anosmia (12.8%). No significant difference was found between any of the covid-19 symptoms with EDx patterns. 7 patients had a history of GBS which were more than 1 year before the onset of new symptoms. 13.6% of patients had no any symptoms of covid-19 on the day of the onset of neurological symptoms, either the symptoms of covid-19 developed after the neurological symptoms or covid-19 was discovered accidentally.
Mean distance between onset of covid-19 and neurological symptoms was 18.05±8.88 which was significantly lower in the axonal injury groups (12.00±800 pvalue: 0.013). Also There was also significant difference between frequency of para/post infectious patient in axonal and demyelinating subtypes (p value: 0.045). So that Para infection was more associated with axonal injuries. Among other prognostic findings, include respiratory equipment (33% no equipment, 44% none-invasive and 22.2% mechanical ventilation), required to ICU admission (46.7%), length of ICU admission (16.66 ±12.03), length of intubation (12.10±6.24), length of hospitalization(23.66±14.13) and mortality(8.9%) no Significant differences were detected among each subgroups of EDx patterns and also between axonal/demyelinating injuries.
There was also significant difference among erythrocyte sedimentation rate and C-reactive protein among axonal patterns that means axonal patterns (AMAN and AMSAN) had more level of ESR and CRP at the first neurological symptom’s day.
eP02.03.01Risdiplam in Children With Spinal Muscular Atrophy: Real-World Experience After One Year of Treatment
Gomez Garcia De La Banda M1, Bouadi A1, Spigarelli M1, Bocassin C1, Dehache L1, Benezit A1, Amthor H1, Dabaj I1, Tirolien S1, Vaugier I1, Villart M1, Quijano-Roy S1
1Paris Saclay University, Raymond Poincare Hospital, Garches, France
Introduction: The evolution of standards of care and the arrival of innovative specific therapies for spinal muscular atrophy (SMA) have changed the natural history of this disease. The first oral molecule approved for SMA is Risdiplam (Evrysdi®), wich modifies SMN2 splicing, leading to an increase of SMN protein levels in blood.
Available in France since 2020, it has been shown to be effective and safe in clinical trials, but data in real-world conditions are scarce. We want to share our initial experience treating children with symptomatic SMA with risdiplam, with focus safety and functional aspects
Patients and Methods: This study retrospectively reviewed all patients treated by risdiplam between November 2020 and January 2022 in a single neuromuscular pediatric reference center. A multidisciplinary assessment was performed at baseline and periodically after, including clinical controls (neurological, orthopedics, nutritional and ophthalmological), blood and urinary tests and motor and respiratory function evaluations. Genetic, tolerability and clinical complementary data were also reviewed.
Results: Up to January 2022, a total of 24 patients with genetic confirmation of SMA were included: 2 patients SMA1; 21 SMA2 and 1 SMA3, with a median age of 14 years (4-20 years). 21 patients (95%) had pre-treatment ventilatory support (19 NIV; 2 tracheostomy) and 16 a minimally invasive fusionless surgery. 19 patients (79%) had been previously treated with nusinersen.
All patients are alive and only 2 patients decided to stop treatment. The remaining 22 patients had a good treatment adherence and did not present severe side effects.
The median treatment and follow-up time was 7 months (1-14 months) and 9 patients had been followed for at least 12 months.
No loss of acquisition was observed and new achievements or motor improvement acquisitions were reported in 15 patients (62%), with a subsequent gain in daily life autonomy. The most frequent reported side effects were nausea and diarrhea.
Two patients reported urinary infection, which was resolved with antibiotics without sequels. Two patients referred photosensitivity reactions triggered by sunlight and light from screens, as well as skin hyper and hypopigmentation. No patient had respiratory problems during this period and 25% of the series (6 patients) reported an improvement in terms of swallowing and speech (better articulation of words and louder voice) as well as an improvement in weight gain.
Conclusion: Preliminary short-term results using daily risdiplam are encouraging. It seems to be easy to administer by families, with a good therapeutic adherence. In our cohort, risdiplam has been a safe molecule, well tolerated without severe side effects. A variable motor improvement has been observed in a proportion of patients from the first months of treatment. No major changes were observed in 25% of the series (6 patients), but sensitivity to change may need longer follow-up. More specific motor and respiratory results and the update of the characteristics of the cohort will be provided at the congress
eP02.03.02Cost-Utility Analysis of Risdiplam Compared with Onasemnogene Abeparvovec in Spinal Muscular Atrophy Type 1
Bischof M1, Dean R2, Toro W3, Weidlich D4, Howells R5, Dabbous O3
1Novartis Gene Therapies, Inc., Rotkreuz, Switzerland, 2Precision HEOR, Boston, USA, 3Novartis Gene Therapies, Inc., Bannnockburn, USA, 4Clarivarte, Frankfurt, Germany, 5Clarivate, Manchester, UK
Objective: We sought to assess the cost utility of daily risdiplam oral therapy for spinal muscular atrophy (SMA) type 1 in the United States in a naïve comparison against onasemnogene abeparvovec single-dose gene replacement therapy.
Methods: We updated and adapted a multi-state survival Markov model developed for SMA type 1 to include data for risdiplam. Health state transitions were based on motor milestone attainment (sitting, walking) from published Phase III trials (FIREFISH [1] for risdiplam; START [2] and STR1VE-US [3] for onasemnogene abeparvovec). The FIREFISH pooled cohort (n=58) included 10 sitters and 0 walkers in Part 1 (n=17, 24 months follow-up from first treatment) and 12 sitters and 0 walkers in Part 2 (n=41, 12 months follow-up from first treatment). The START and STR1VE-US cohorts (n=34) had 22 sitters and three walkers by age 36 months. Survival benefit was extrapolated using long-term data from SMA type 2 (sitting) and type 3 (walking) patients. For non-sitters, survival was extrapolated from a published sham control arm (ENDEAR [4]). Survival on permanent ventilation was from a natural history study from Gregoretti et al. [5]. Risdiplam cost (manufacturer wholesaler price) was sourced from Micromedex (Red Book, Jan. 2022) and long-term usage assumptions were based on commercial insurer policy documents (i.e., treatment was stopped after transition to permanent assisted ventilation). Onasemnogene abeparvovec cost included the price of a single dose, plus costs of IV administration and laboratory monitoring. Lifetime cost of SMA medical care for both arms was based on values from Tan et al. [6] indexed to 2021 US dollars. Utilities were taken from the US Institute for Clinical and Economic Review and values preferred by the UK Evidence Review Group.
Results: Undiscounted per-patient total quality-adjusted life-years (QALYs) were 8.7 for risdiplam and 20.5 for onasemnogene abeparvovec (5.0 and 10.4, respectively, if discounted at 3%). Estimated discounted lifetime costs for risdiplam were $4.324M at the price of $11,170 per pack (weight-based dose). Lifetime discounted costs for onasemnogene abeparvovec at the price of $2.125M per dose was $4.253M per patient. Treatment with risdiplam resulted in greater costs and fewer QALYs gained compared with onasemnogene abeparvovec treatment, resulting in risdiplam being dominated by onasemnogene abeparvovec.
Conclusions: In comparison with onasemnogene abeparvovec in this analysis, risdiplam was not cost-effective and was dominated by onasemnogene abeparvovec for treatment of patients with SMA type 1.
References
1. Baranello G, et al. N Engl J Med 2021;384:915–23.
2. Mendell JR, et al. N Engl J Med 2017;377:1713–22.
3. Day JW, et al. Lancet Neurol 2021;20:284–93.
4. Finkel RS, et al. N Engl J Med 2017;377:1723–32.
5. Gregoretti C, et al. Pediatrics. 2013;131:e1509–14.
6. Tan H, et al. JHEOR. 2019;6:185–195.
eP02.03.03SAPPHIRE: Efficacy and Safety of Apitegromab in Later-Onset SMA; Phase 3 Trial in Progress
Crawford T1, Darras B2, Day J3, SAPPHIRE Study Team4, Barrett D5, Song G5, O’Neil J5, Howard S5, Rossello J5, Osman N5, Patel J5, Nomikos G5, Kertesz N5, Chyung Y5
1Johns Hopkins Medical, Baltimore, United States, 2Boston Children’s Hospital, Boston, USA, 3Stanford Neuroscience Health Center, Palo Alto, USA, 4SAPPHIRE Study Team, includes clinical trial investigators, physical therapists, study coordinators, 5Scholar Rock, Inc., Cambridge, United States
Objective: The SAPPHIRE study is a placebo-controlled phase 3 trial to determine the safety and efficacy of 12 months treatment with apitegromab (SRK-015) nonambulatory patients with Type 2 and Type 3 Spinal Muscular Atrophy (SMA). The primary efficacy objective compares baseline to motor function change on the HFMSE scale.
Background: SMA is a severe, progressive neuromuscular disease caused by reduced levels of survival motor neuron (SMN) protein leading to muscular atrophy. Apitegromab is a fully human investigational monoclonal antibody that selectively inhibits activation of myostatin, a negative regulator of muscle mass and function. This Phase 3 trial, SAPPHIRE (Study SRK-015-003) is being conducted in patients ≥ 2 years old at screening, who were previously diagnosed with later-onset SMA, and are receiving an approved SMN upregulator therapy (either nusinersen or risdiplam). This study will primarily confirm or evaluate the efficacy and safety of apitegromab in combination with SMN upregulator therapies.
Design/Methods: SAPPHIRE (NCT 05156320) is a global, multicenter, two-part, randomized, placebo-controlled, double-blind study in nonambulatory patients, aged 2–21 years, with Type 2 or Type 3 SMA. SAPPHIRE is comprised of two parts: Part 1, the main efficacy population (n=156, randomized 1:1:1, apitegromab 20mg/kg: apitegromab 10mg/kg: placebo) will assess the efficacy, safety, tolerability, pharmacokinetics and target engagement of different apitegromab dose levels in nonambulatory patients with SMA currently taking background SMN upregulator therapy, either nusinersen or risdiplam. Part 2 is an exploratory subpopulation of nonambulatory patients with Type 2 and Type 3 SMA (n≈48; randomized 2:1 to apitegromab 20mg/kg or placebo) that will assess the safety and efficacy of the apitegromab high dose compared with placebo in nonambulatory patients with SMA, aged 13-21, currently taking background SMN upregulator therapy, nusinersen or risdiplam.
Results: This is an overview of the study design of the SAPPHIRE trial currently enrolling Type 2 and Type 3 SMA participants to evaluate the safety and efficacy of a muscle targeted therapy, apitegromab in combination with background SMN upregulator therapy.
Conclusions: SAPPHIRE is an ongoing pivotal phase 3 study. Apitegromab has the potential to be the first muscle-directed therapy in patients with SMA.
eP02.03.04Safety of Onasemnogene Abeparvovec in Patients With SMA in Real Clinical Practice
Nevmerzhitskaya K1, Sapego E1, Morozova D2
1Regional Children Hospital, Yekaterinburg, Russian Federation, 2Ural State Medical University, Ekaterinburg, Russian Federation
Background: Gene replacement therapy with onasemnogene abeparvovec in spinal muscular atrophy was approved in Russia in December 2021 for children weighing less than 21 kg, but safety data in real clinacal practice are still limited. We aimed to evaluate the safety of onasemnogene abeparvovec in a case series in children with spinal muscular atrophy with short-term follow-up in real clinical practice.
Methods: 12 patients (7 male, 5 female, age range 7-43 months) with a body weight between 6.6 and 12.5 kg received standard dose of onasemnogene abeparvovec (1.1 × 10*14 vg/kg) the Regional Children’s Clinical Hospital in the city of Yekaterinburg (Russia). The age of disease onset was 4 (2; 6.5) months, the age at the time of treatment was 19 (12; 22) months. Safety of therapy was assessed based on clinical and laboratory monitoring for at least 60 days after drug administration. All patients received 1 mg/kg/d prednisolone for four weeks starting on the day before gene replacement therapy.
Results: During the first week after drug administration, all patients developed at least one clinical event - hyperthermia, vomiting, lethargy, diarrhea and at least one laboratory abnormality: thrombocytopenia (<150 × 109/L) in 10/12 patients, neutropenia in 6/12 patients, monocytosis in 12/12 patients, increased troponin I in 4/12 patients. The most significant and long-lasting laboratory abnormality was the increase of transaminases (in 12/12 patients), in three cases it required an elevation of the dose of prednisolone to 2 mg / kg and in one case–steroid pulse therapy for five days. All laboratory abnormalities were asymptomatic and returned to normal levels during the post-treatment observation period. There were no patients with identified clinical signs of insufficiency of the functions of body systems. There were no serious clinical adverse events that led to hospitalization and / or death of the patient.
Conclusions: Gene replacement therapy with the onasemnogene abeparvovec in children with spinal muscular atrophy has shown acceptable safety. All clinical manifestations and laboratory abnormalities during the first week of follow-up that were associated with drug administration were reversible and did not lead to serious consequences.
eP02.03.05Treatment of Spinal Muscular Atrophy with Onasemnogene Abeparvovec in Switzerland
Stettner G1, Hasselmann O2, Klein A3
1Neuromuscular Center Zurich and Department of Pediatric Neurology, University Children’s Hospital Zurich, Zurich, Switzerland, 2Department of Neuropediatrics, Children’s Hospital of Eastern Switzerland, St. Gallen, Switzerland, 3Division of Neuropediatrics, Development and Rehabilitation, University Children’s Hospital, Inselspital, Bern, Switzerland
Introduction: Spinal muscular atrophy (SMA) is a rare degenerative neuromuscular disorder characterized by progressive muscle hypotonia and weakness leading to early death due to compromised bulbar and respiratory functions in the majority of affected individuals without treatment. Recently, targeted treatment approaches were introduced for the treatment of SMA including Onasemnogene Abeparvovec (OA), a gene replacement therapy. In Switzerland, OA was approved for the treatment of SMA in July 2021, but the Federal Social Insurance Office granted cost approvals in individual cases already since late 2020. This study describes the first experiences with OA in Switzerland.
Material and Methods: Between September 2020 and end of 2021 nine individuals with SMA were treated with OA in three neuromuscular centers in Switzerland. Motor function, bulbar and respiratory outcome, development of scoliosis, and adverse events focusing on liver function, thrombocytopenia, and cardiotoxicity were analyzed using prospectively collected data from the Swiss Registry for Neuromuscular Disorders (Swiss-Reg-NMD).
Results: 6 patients with SMA type 1 (including 2 with nusinersen pre-treatment), 1 SMA type 2 patient, and 2 pre-symptomatic individuals with 3 SMN2 gene copies and 3 copies of a SMN hybrid gene, respectively, were included. Mean age at OA treatment was 5 months (range 2 weeks – 17 months), mean follow up was 270 days (range 68 – 482 days).
In SMA type 1, mean CHOP Intend score increased from 21 to 47/64 following treatment. 4/6 (67%) SMA type 1 patients were dependent on nutritional support at end of follow up, and 3/6 (50%) patients developed scoliosis. At last follow up, no patient required ventilatory support. HFMS score of the SMA type 2 patient increased from 14 to 25/40. CHOP Intend scores of the pre-symptomatic individuals were 22 (poor compliance) and 52 at baseline, respectively, and increased to 44 and 62.
Concomitant steroid treatment was modified according to recommendations due to increased transaminases in 3/9 (33%) patients (both nusinersen pre-treated and the SMA type 2 patient). Transient thrombocytopenia occurred in 5/7 (71%) patients, but did not require intervention. No patient had significant troponin I and/or T elevations.
Discussion: As demonstrated in clinical trials and first post-marketing observational studies, OA proved to be effective and disease modifying in the treatment of SMA also in this study. All study participants showed significant improvements of motor function. However, the proportion of SMA type 1 patients, who required nutritional support at the end of follow up and developed scoliosis was higher in this cohort compared to previously published reports. Overall, OA treatment was not associated with significant safety issues during the follow up period.
eP02.03.06The Impact of Nusinersen Treatment on Patient-Reported Outcome Measure in Later-Onset Spinal Muscular Atrophy
Bae H1, Kim A2, Lee J3, Shim Y4, Ryu H5, Kwon S1, Chae J5, Lee Y1
1Department of Pediatrics, School of Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, Republic of Korea, 2Department of Rehabilitation Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, Republic of Korea, 3Department of Neurology, School of Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, Republic of Korea, 4Department of Pediatrics, Korea University Ansan Hospital, Seoul, Republic of Korea, 5Department of Pediatrics, Pediatric Clinical Neuroscience Center, Seoul National University Children’s Hospital, Seoul National University College of Medicine, Seoul, Republic of Korea
Background: This study aims to evaluate the impact of nusinersen treatment on health-related QoL in patients with later-onset spinal muscular atrophy (SMA) and their caregivers.
Methods: We assessed the changes of self-and proxy-reported QoL cross overtime during treatment with nusinersen by measuring the pediatric Quality of life inventory (PedQL) 4.0 Generic Core Scale (PedsQL GCS), and the PedQL 3.0 Neuromuscular Module (NMM). We also evaluated the activity of daily living and the caregiver burden using pediatric evaluation of disability inventory-computer adaptive test (PEDI-CAT) and Assessment of Caregiver Experience with Neuromuscular Disease (ACEND), respectively. Factors associated with QoL were also evaluated.
Results: Twenty-three patients with SMA (SMA type 2: n= 19, SMA type 3: n= 4) and their caregivers were included in the analysis. There are significant changes in the self-reported PedQL GCS total score. (p <0.001) “Psychosocial health summary” score of PedQL GCS was significantly changed with a trend toward improvement both on self- (p < 0.001) and proxy-reported assessment (p = 0.003). The score of “physical health” on PedQL GCS and “about my neuromuscular disorder” on PedQL NMM was not significantly changed both on self-and proxy-reported assessment. Patients’ responsibility tended to improve with treatment. Caregiver’s financial burden increased over time (p = 0.018). Self-reported PedsQL GCS total scores were positively correlated with the score of Hammersmith Functional Motor Scale- Expanded (HFMSE) (p < 0.001).
Conclusion: Our study demonstrated that nusinersen might improve the QoL of patients with later-onset SMA, particularly in terms of psychosocial function. PROMs, including QoL, the activity of daily living, and caregiver burden, should be considered the additional outcome measure but needs further evaluation in a larger patient cohort over a more extended period.
eP02.04.01Tocilizumab in Therapy Myasthenia Gravis Patients With COVID-19
Martic V1, Jovin Z2, Udovicic I1, Tausan D1, Begovic V1, Pilcevic D1
1Military Medical Academy, Belgrade, Serbia, 2Clinical Center of Vojvodina, Novi Sad, Serbia
Background: Beneficial effect of IL6 inhibitors in patients with Covid19 can be explained by production proinflammatory cytokines- IL-6 by viral stimulation. Developed renal insufficiency and decrease in saturation accompanied by an increase IL-6 was indication for tocilizumab therapy in these patients with Covid-19 infection as a part of cytokine storm therapy.
Patients and methods: We present clinical outcome in 3 our patients with generalized autoimmune myasthenia gravis (MG) and COVID-19 infection treated with tocilizumab. All patients had severe clinical condition and required hospitalization. Clinical course of the disease was commented in this group of patients.
Results: Long-term clinical remission of MG was noted in 2 of 3 patients treated with tocilizumab, one patient died.
Conclusion: Our experience confirms experience from literature about usefulness tocilizumab therapy in treatment of refractory MG.
Keywords: myasthenia gravis, Covid-19, therapy, tocilizumab, outcome
eP02.04.02Myasthenia Gravis Demographics Re-Visited
Witting N1, Kjaer Andersen R1, Holst Axelsen K1, Vissing J1
1Neuromuscular Research Center And Dept Neurol, Rigshospitalet, Copenhagen, Denmark
Myasthenia gravis is a common autoimmune neuromuscular disease with failure of neuromuscular transmission. Much research has been done but issues such as diagnostic delay and its causes and symptom evolution are still insufficiently elucidated. We conduct an ongoing cohort study with chart review of more than 400 patients. At present 178 patients fulfilling 2 of 3 diagnostic criteria (elevated MG-antibodies (acetylcholine receptor, MuSK or LRP4), positive pyridostigmine response or abnormal electrophysiology pointing to failure of neuromuscular transmission) were analyzed. Average age at first symptom was 54 years (range 9-94), younger for women (54 years) than for men (64 years), p= 0.004. The classic bimodal pattern of onset with a peak for women in young adulthood and a second peak for both genders in old adulthood was not observed.
Onset symptoms were ocular (diplopia or ptosis) in 105, bulbar in 34, axial/drophead in 3, respiratory failure in 4 and in limbs in 12. Many with ocular onset, however, experienced evolution to a generalized type, hence the final phenotype was generalized in 114, purely ocular in 32 and ocular with discrete other symptoms in 23. Average diagnostic delay was 319 days (range 7-3862). 73, 68, 16 and 2 had visited 1, 2, 3 or 4 non-neuromuscular experts before MG diagnosis. The experts were mostly ophthalmologists or neurologists. 87, 50, 20 and 38 had a brain MRI, brain CT, lumbar puncture or other not MG-related exams carried out pre-MG diagnosis. More data will be presented at the poster.
eP02.04.03Incidence of Skin Changes in Patients with Myasthenia Gravis Prescribed Mycophenolate or Azathioprine
Mehrabyan A1, Brooks C1, Dark E1, Hwang S2, Iyer S1
1University Of North Carolina Medical Center, Chapel Hill, United States, 2University Of North Carolina Eshelman School Of Pharmacy, Chapel Hill, United States
Objective: To examine the impact of mycophenolate mofetil (MMF) and azathioprine (AZA) on the incidence of development of skin lesions in patients with myasthenia gravis (MG).
Background: International consensus guidelines for the management of MG recommend the use of nonsteroidal immunosuppressive agents, such as MMF and AZA. It has been reported that AZA increases the risk of malignancies. Limited information exists on the risk of skin cancer in MG patients treated with MMF.
Methods: This is a single center, cohort study of patients with MG. Retrospective chart review was conducted to gather demographic data, MG history and management. Patients were contacted via EHR patient message, telephone, and/or during clinic appointment to request participation in a standardized survey to assess skin lesion history. Patients were assigned to either non-exposed or exposed groups, determined by treatment with MMF or AZA for ≥ 12 months. Statistical analysis was performed using chi-square for categorical data and logistic regression to analyze the relationship between age and MMF/AZA exposure on incidence of skin lesions.
Results: Of 194 total patients in MG clinic population, 142 patients met inclusion criteria and were contacted to request participation. Majority (67%) were white, with median age of 67 years (IQR 48-74), and 56% identifying as female. A total of 103 patients (72%) responded to the survey, with 63 patients (61%) in the exposed group and the remaining 39% either naïve to these agents or exposed for < 12 months. Of those surveyed, 51 and 14 patients had been exposed to MMF or AZA, respectively, including 2 patients who had been exposed to both agents. Per the patient-reported survey, we found that 8 (16%) and 5 patients (36%) developed skin lesions after initiation of MMF and AZA, respectively. Of those exposed, only 6 patients (10%) reported a malignant lesion. Of the non-exposed patients, 16 patients (40%) reported historical or current skin lesions, including 10 (25%) who reported a malignant lesion. The incidence of skin lesions was significantly lower in the exposed group compared with the non-exposed cohort (X2=4.536, p=0.033). When adjusting for current age, it was found that the odds of experiencing skin lesions was lower in the patients exposed to MMF or AZA compared with the non-exposed group (OR=0.383, 95% CI 0.149 - 0.959).
Conclusions: Based on this large MG cohort survey study, we found that the incidence of reported skin lesions in patients exposed to MMF or AZA was statistically lower than the incidence of reported lesions in patients not exposed to these medications. After adjusting for current patient age, the incidence of skin lesions reported in the exposed group compared with the non-exposed group remains significantly lower. Two potential factors could explain these findings: variable sun exposure in MG populations due to risk of MG exacerbation and/or refused treatment with AZA/MMF after being counseled on potential risk of skin malignancies. Prospective studies with detailed dermatological evaluation may be essential for further investigation of the skin cancer risk in MG patients treated with AZA and MMF.
eP02.04.04Quality of Life in Myasthenia Gravis and Correlation of MG-QOL15 With Other Functional Scales
Porras L1, Homedes Pedret C1, Vélez Santamaria V2, Alberti M2, Casasnovas Pons C2
1Institut d’Investigació Biomèdica de Bellvitge, L’Hospitalet del Llobregat, Barcelona, Spain, 2Hospital Universitaride Bellvitge, L’Hospitalet del Llobregat, Barcelona, España
Health related quality of life (HRQOL) in myasthenia gravis (MG) is frequently decreased. Some studies showed that optimizing medical treatment may improve HRQOL. Further, there are many validated clinical scales and questionnaires to evaluate the clinical status and HRQOL in MG. The objetives of ou study were to determine if there was a significant improvement in HRQOL following an intensive treatment for MG, to identify which demographic and clinical features influenced patients’ HRQOL and to investigate if the questionnaire MG-QOL15 correlated with other clinical evaluation scales.
We conducted a post-authorization, experimental study with prospective follow-up. In line with the Declaration of Helsinki, the study protocol was approved by the institutional review board of the Clinical Research and Clinical Trials Unit. We included 45 patients with generalized MG who were starting immunomodulatory treatment with intravenous immunoglobulins (IVIG) and prednisone for the first time. At each visit we administered several validates scales and questionnaires for MG. We analysed changes in HRQOL from the basal visit to the visits at 4 and 6 weeks and calculated if the questionnaire MG-QOL15 correlated with other clinical measures (MG-ADL, MGC, QMG and Neuro-QOL Fatigue). Also, we studied if there was a correlation between HRQOL and some demographic and clinical or laboratory variables.
The mean MG-QOL15 score improved significantly 4 and 6 weeks after starting intensive immunomodulatory treatment. We found that QMG score prior to study enrolment was significantly associated with HRQOL in our patients. However, the remaining demographic and clinical factors studied did not affect HRQOL. Also, the MG-QOL15 score correlated significantly with the other clinical evaluation scales. The strongest correlation was with the MG-ADL and the Neuro-QOL Fatigue scales and the weakest with the QMG scale.
HRQOL of our patients improved after receiving an intensive medical treatment and achieving a better control of the symptoms of the disease. The questionnaire MG-QOL15 had a positive correlation with other measures of clinical assessment. As MG is a fluctuating condition and some symptoms are difficult to examine, we aim physicists to use scales and questionnaires that include items perceived by the patient.
eP02.04.05Barriers and Facilitators to Exercise in Auto-Immune Myasthenia Gravis
Birnbaum S1, Archer A, Hogrel J, Stalens C
1Association Institute Of Myology, Paris, France
Exercise in myasthenia gravis has been shown to be safe and effective in improving certain health components such as strength and functional capacity. However, recent quantitative data shows that not all individuals with myasthenia partake in regular exercise. The reasons for this are unclear due to a lack of data and mixed results in the literature with respect to, for example, disease severity as a possible barrier as well as age and body mass index. Current research has not explored whether other possible non-disease related factors such as motivation, fear, practical aspects such as access to resources or time could contribute to limiting exercise participation. It is essential to identify these factors in order to provide adequate programs to individuals to meet current needs. Therefore, we aim to identify extrinsic and intrinsic factors which contribute to limiting physical activity, or on the contrary facilitate exercise participation for adults living with myasthenia gravis and explore relationships between these factors and self-perceived quality of life (MG-QOL 15), fatigue (NeuroQoL fatigue), disease duration, type of MG and sociodemographic factors.
Method: Ethics approval was obtained. A purpose-built online survey was created and diffused nationwide via patient associations and healthcare networks. The questionnaire will remain online for six months. Individuals are required to confirm their diagnosis of myasthenia and provide informed consent prior to completing the questionnaire. Participants complete the questionnaire one-time only. Response time is approximately 30 minutes.
Results: We have currently received 372 responses since November 2021 of which 360 are eligible for analysis. The study population includes 73% femmes, 28% hommes (age min 19, age max 88). Study inclusion will terminate in May and full results will be available in June.
eP02.04.06Clinical Characteristics of Patients With Seronegative Myasthenia Gravis
Andersen R1, Axelsen K1, Vissing J1, Witting N1
1Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, Denmark
Myasthenia gravis (MG) is an autoimmune disease characterized by antibodies against proteins in the postsynaptic membrane of the neuromuscular junction causing weakness in skeletal muscle. Around 10% of patients diagnosed with MG do, however, not have detectible levels of any antibodies associated with MG and are categorized as seronegative. The objective of this study is to compare the clinical features of seronegative MG patients to the much larger group of seropositive patients who have measurable levels of antibodies against at least one protein associated with the acetylcholine receptor in the muscular membrane.
We will compare the two patient groups on parameters concerning sex, age at disease onset, thymus pathology, severity of symptoms and treatment response. Furthermore, we wish to investigate possible differences in the symptom profile of seronegative versus seropositive patients. Our hypothesis is that seronegative MG patients differ from seropositive MG patients, especially in regards to symptom profile and severity and response to treatment.
We aim to include all approximately 400 patients with a verified diagnosis of MG currently being treated at Copenhagen Neuromuscular Center, and expect 5-10% to be in the seronegative group. We will collect data on these patients from their medical records in a REDCap database.
This is an ongoing study; results will be presented at the poster presentation.
eP02.04.07TREAT-NMD Global Registry Network: Facilitating 12 Years of Neuromuscular Drug Development
Turner C2, Porter B1, Walker H1, Bennett N1, TGDOC Subgroup Leads and Patient Representatives1, Straub V2, Allison D1, Guglieri M2, Campbell C3, Ambrosini A4
1TREAT-NMD Services Ltd, Newcastle upon Tyne, United Kingdom, 2John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom, 3Department of Paediatrics, Clinical Neurological Sciences & Epidemiology, Western University, London, Canada, 4Fondazione Telethon, Milan, Italy
Background: TREAT-NMD is an international network of excellence facilitating collaborative research in neuromuscular disease (NMD) that was established through an EU-funded project in 2007. Having developed a range of infrastructures, the network aims to accelerate drug development, provide new therapies to patients swiftly and improve access to relevant information on standards of diagnosis and care. A key infrastructure is the Global Registry Network (GRN), created in 2009 in collaboration with clinicians and patient organisations. The GRN is a federated network of individual, independent patient registries that collect information on consenting NMD patients.
To join the GRN, registries must agree to share data to the GRN in response to specific enquiries and collect the relevant TREAT-NMD Core Dataset(s) for their disease area(s). These standardised datasets include information required for clinical trial feasibility purposes or to establish clinical trial eligibility. Aggregated data reports and other information can be requested securely through the Global Registry Enquiries process.
Aims: The GRN aims to provide an environment where registries can discuss challenges, share best practice, network, and collaborate with colleagues from around the globe.
Through Global Registry Enquiries, the GRN aims to support stakeholders with data provision or information distribution to primarily facilitate clinical trial planning or recruitment, and to further academic research.
Methods: Disease-specific subgroups have been established for DMD, SMA, DM, FSHD, LGMD and Ultra-Rare NMDs. These groups allow registry curators, Principal Investigators, and patient representatives to meet, with regular updates provided via newsletters and online workshops.
Global Registry Enquiries are reviewed by the TREAT-NMD Global Data systems Oversight Committee (TGDOC) who govern the GRN. Enquiries can be categorised as data enquiries, providing stakeholders with de-identified, aggregated data from the network, or recruitment enquiries, where clinical trial information is distributed to eligible NMD patients via those registries.
Results: The GRN now comprises more than 60 registries, in 30 countries, collecting data in more than 10 NMDs. Collectively, registries in GRN hold data on approximately 80,000 patients, including more than 50,000 with a genetically-confirmed NMD. Since 2009, there have been approximately 86 initial enquiries made from academics, clinicians, industry, contract research organisations, regulatory agencies, and patient organisations as part of the Global Registry Enquiries process. Of these, 41 (48%) have resulted in successful registry enquiries that have been conducted. The majority (78%) of which have supported data or recruitment enquiries specifically for industry.
Conclusion: TREAT-NMD is still positioned to support high quality registry enquiries from a wide range of stakeholders. As new therapies are approved there will be an increasing demand for provision of de-identified patient-level data to align with Post Marketing Surveillance commitments from pharmaceutical regulatory agencies. The development of the TREAT-NMD Global Registries Platform and the adoption of expanded TREAT-NMD core datasets created through a consensus-building process with clinicians, academics and patient organisations will help to support this new stream of work.
eP02.04.08Immunofluorescence Signal Intensity Measurements as a Semi-quantitative Tool to Assess Sarcoglycan Expression in Muscle Biopsy
Zanotti S1, Magri F1, Poggetti F1, Ripolone M1, Ciscato P1, Comi G1, Sciacco M1
1Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Diseases Unit, Milan, Italy
Quantitative immunoanalysis of sarcoglycan complex in skeletal muscle biopsy is an important part of the diagnostic path in patients with limb-girdle muscular dystrophy. Sarcoglycanopathy diagnosis is genetic, but muscle analysis - by both immunohistochemistry and Western blot - is still mandatory to establish the correct diagnostic process. Unfortunately, however, this analysis cannot be performed if the bioptic specimen is scarce.
The aim of our study is to provide a sensitive tool for semi-quantification of residual amount of sarcoglycans in patients affected by sarcoglycanopathies, based on immunofluorescence staining of muscle sections, image acquisition and software elaboration.
To develop and validate this method, skeletal muscle cryostatic sections from 11 sarcoglycanopathy, seven Becker muscular dystrophy and four age-matched control samples were analyzed for the expression of sarcoglycan complex and laminin-α2.
The staining of laminin-α2 was used as internal standard to evaluate the efficiency of the immunofluorescence reaction, as sarcolemmal reference to prove muscle fiber integrity, and to allow fiber counts in each analyzed image.
Sarcoglycanopathies are highly heterogeneous in terms of disease progression, muscular weakness, loss of ambulation and cardiac/respiratory involvement, and clinical severity usually correlates with the residual protein amount, which makes protein quantification extremely relevant.
Sarcoglycan fluorescence normalized data were analyzed in LGMDR3, LGMDR4 and LGMDR5 patients compared to age-matched controls showing a significant reduction of mutated sarcoglycan expression and a variable reduction of the other sarcoglycans, in agreement with literature reports.
Fluorescence normalized data were also analysed in relation to the age of onset of the disease. To this purpose patients were subdivided into three groups: early, intermediate and late age of onset respectively. Our results showed a negative correlation between the residual sarcoglycan expression and the fibro-adipose tissue deposition. In particular, data showed a negative correlation of sarcoglycan-α fluorescent signal versus fibrosis in patients with an early age of onset and a negative correlation between sarcoglycan-d signal and fibrosis in both intermediate and late age of onset groups.
Bland-Altman plot of difference between data obtained by this method and Western blot show agreement between two quantitative measurements even if the analysis was limited to a small number of samples.
The availability of a method that allows objective quantification of the sarcolemmal proteins, faster and less consuming than Western blot analysis and able to detect low residual sarcoglycan expression with great sensitivity, proves useful to better define both patient prognosis and expected disease evolution.
To date, there is no curative treatment for sarcoglycanopathies, but some clinical trials aimed at restoring protein expression and correct localization are underway. A quantitative evaluation of protein expression and the visualization of their correct membrane localization are fundamental targets for therapeutic strategies based on the partial or complete restoration of protein expression. The proposed method could find application also when monitoring the efficacy of therapeutic interventions and during clinical trials.
eP02.04.09Three Cases of Congenital Fibre Disproportion With Etiology Other Than Congenital Myopathy
Farhat E1, Miladi N, Marrak S, Chaabouni M1
1Clinique Les Jasmins, Centre Urbain Nord, Tunisia
Introduction: Congenital fibre type disproportion (CFTD) is a histological entity characterized by type 1 muscle fibres smaller for at least 12% than type 2 fibres, and absence of other pathological features. CFD has been recognized for decades as a subtype of congenital myopathy (CM). We report phenotype and genotype description of three patients who had shown muscle weakness from early childhood, typical CFD on muscle biopsy, and final diagnosis different than CM.
Patient 1: Is an 8-year-old girl with a history of neonatal hypotonia, and delayed motor development. At examination she had weakness in the proximal extremities and a positive Gower’s sign. CK plasma level was elevated and an electromyogram (EMG) showed a myopathic pattern. Brain MRI was normal. Muscle biopsy concluded to a CFD. The Whole Exome Sequencing (WES) revealed a missense homozygous mutation in POMK gene, confirming the diagnosis of congenital muscular dystrophy.
Patient 2: was born of normal pregnancy and delivery, had delayed speech development, major learning problems and excessive slowness of movement. On first examination she was12 years-old, showed a slim phenotype, lordotic stance, facial involvement with ptosis, dysphonia and moderate proximal-predominant limb muscle weakness. On muscle biopsy we found non-specific myogenic changes with no dystrophic signs, and a selective atrophy of type 1 fibres. CK levels were slightly increased. EMG revealed myotonic discharges. This particular sign prompted the analysis of the DMPK gene allowing the diagnosis of type 1 myotonic dystrophy, with 61 expanded CTG repeats.
Patient 3: had a history of congenital hypotonia with delayed motor development. He underwent scoliosis surgery at the age of 14, and a non-invasive nocturnal ventilation was recently indicated due to a severe sleep apnea. At time of first examination he was 34 years old, was still able to walk and had severe weakness of facial, axial and proximal limbs. EMG was myogenic, CK levels were normal. The WES allowed the identification of a homozygous mutation in COL13A gene, confirming the diagnosis of the autosomal recessive congenital myasthenia syndrome type 19. He was treated with pyridostigmine and salbutamol.
Discussion and conclusion: CFD is a histochemical pattern encountered mainly in CM but appearing to represent a larger spectrum of a diseases. Mutations in the ACTA, SEPN1, and TPM3 genes are known genetic causes of CM with histological pattern of CFD. Other disorders in which histological aspect of CFD is reported included muscular dystrophy, myotonic dystrophy, congenital myasthenic syndromes, Low’s syndrome, rigid spine syndrome, and Cockayne’s syndrome. In our patients the clinical presentations differ, reflecting the phenotype typically associated with the respective gene. The neonatal onset of symptoms, myogenic EMG and isolated pattern of CFD lead us to retain initially the diagnosis of CM. However, the results of genetic analysis excluded this diagnosis. We insist through this observation that we should consider the possibility of other diagnosis beyond the spectrum of CM whenever a diagnosis of CFD is made.
eP02.05.01One-year ENDEAVOR Data (Ambulatory, ≥4 to <8-year-olds): Phase 1b Trial of Delandistrogene Moxeparvovec in DMD
Lehman K, Zaidman C1, Proud C2, McDonald C3, Mason S4, Guridi M5, Wang S4, Reid C6, Darton E4, Wandel C5, Lewis S4, Malhotra J4, Griffin D A4,7, Potter R A4, Rodino-Klapac L R4, Mendell J R7,8
1Department of Neurology, WUSTL, Washington, USA, 2Children’s Hospital of the King’s Daughters, Norfolk, USA, 3UC Davis Health, Sacramento, USA, 4Sarepta Therapeutics, Inc., Cambridge, USA, 5F. Hoffmann-La Roche Ltd, Switzerland, 6Roche Products Ltd, Welwyn Garden City, UK, 7Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, USA, 8The Ohio State University, Columbus, USA
Delandistrogene moxeparvovec (SRP-9001) is an investigational gene transfer therapy developed for targeted skeletal and cardiac muscle expression of micro-dystrophin (a shortened, functional dystrophin protein) that is being studied in patients with Duchenne muscular dystrophy (DMD).
Findings from ongoing Phase 1 and Phase 2 trials have demonstrated micro-dystrophin expression in patients treated with delandistrogene moxeparvovec clinical process material, suggesting the possibility for clinical benefit in people with DMD.
ENDEAVOR (NCT04626674) is a two-part, open-label, Phase 1b study assessing the expression and safety (over 260 weeks) of commercially representative delandistrogene moxeparvovec material in four cohorts of boys with DMD.
Study inclusion requires a diagnosis of DMD, with a confirmed mutation in the DMD gene, and stable steroid dosing (Cohorts 1-3 only, ≥12 weeks prior to screening). Cohort-specific criteria include: ≥4 to <8-year-old ambulatory boys (Cohort 1), ≥8 to <18-year-old ambulatory boys (Cohort 2), non-ambulatory boys (Cohort 3), and ≥3 to <4-year-old ambulatory boys (Cohort 4).
Participants weighing ≤70 kg received a single intravenous dose (1.33x1014 vg/kg, linear standard qPCR) of commercially representative delandistrogene moxeparvovec material. The follow-up period consists of two parts: Part 1, from post-infusion through to Week 12, and Part 2, from Week 12 through to Week 260.
The primary outcome measure is the change in micro-dystrophin protein expression from baseline to Week 12 (Part 1), as measured by western blot analysis. Key secondary outcome measures include change in micro-dystrophin expression at the sarcolemma from baseline to Week 12 (Part 1), as measured by immunofluorescence (muscle fibre intensity and percentage of dystrophin-positive fibres), and safety (over 260 weeks). Exploratory functional endpoints for ambulatory boys include: the North Star Ambulatory Assessment and timed function tests (100-metre Walk/Run, 4-stair Climb, Time to Rise, and 10-metre Walk/Run).
Previously presented expression data from the first 11 patients in Cohort 1 demonstrated micro-dystrophin protein expression at Week 12 following treatment with delandistrogene moxeparvovec. This expression corresponded with vector genome copy numbers in the nuclei of target cells, confirming successful delivery. These data from ≥4 to <8-year-old ambulatory boys suggest that commercially representative delandistrogene moxeparvovec material demonstrates safety and expression consistent with previous studies that used clinical process material. We present 1-year safety and functional data and 12-week expression data from all patients in Cohort 1 (n=20) following treatment with commercially representative delandistrogene moxeparvovec material.
This study is sponsored by Sarepta Therapeutics and funded by Sarepta Therapeutics and F. Hoffmann-La Roche.
eP02.05.02Whole Genome Sequencing in a Pair of Duchenne Muscular Dystrophy Siblings with Discordant Cognitive Phenotype
Bello L1, Sabbatini D1, Alexander M2
1University of Padova, Padova, Italy, 2University of Alabama, Birmingham, USA
Background: Central nervous system (CNS) involvement in dystrophinopathies is variable, although loosely associated with mutations in the C-terminal domains of dystrophin. We aimed to identify candidate variants involved in the modulation of CNS phenotype in dystrophinopathies, by performing whole genome sequencing (WGS) in a pair of Duchenne muscular dystrophy (DMD) siblings with discordant cognitive phenotype and filtering variants with a dedicated bioinformatic algorithm.
Materials and Methods: Patient 1 was born of healthy, non-consanguineous parents. He was diagnosed with DMD at the age of 4 years (deletion DMD exons 45-52), and lost ambulation at the age of 11 (currently 29 y.o.). He had no speech delay nor intellectual disabilities. Patient 2, his younger brother, was diagnosed with DMD due to the same mutation, and lost ambulation at the age of 12 (currently 14 y.o.). However, he presented severe speech delay and an autism-spectrum disorder (ASD) with behavioral issues requiring neuroleptics. We performed WGS using DNA extracted from peripheral blood of Patient 1 and 2 and their parents. Structural Variants were analyzed with Breakdancer algorithm, while indel and SNPs (single nucleotide polymorphisms) were filtered exploiting our own Python scripts in according to the following criteria: not shared between the two siblings; frequency < 5% in European population and considering function of genes involved.
Results: WGS confirmed identical DMD deletion breakpoints in both brothers and their mother. The bioinformatic algorithm filtered a “short list” of 41 variants situated in 36 loci, reviewed individually for potential pathogenetic mechanisms and previous reports of involvement in neurodevelopmental phenotypes. The most notable variants were rs112339619, a G>A single SNP upstream of the ASD risk gene ANK3, previously reported in a WGS study of ASD twins; and rs117696080, an intronic variant in NRXN3, encoding neurexin 3, a dystrophin-associated glycoprotein (DAPG), and also identified in the same ASD twin study. Patient 2 was heterozygous for both these variants, while patient 1 had a wild-type genotype at both loci. Both of these variants are in regions of strong conservation: vertebrate PhastCons of 0.96 e 1.00 respectively. BreakDancer shows an interesting homozygous deletion (2166 bp) in an intronic region of ASD risk gene NALCN. Moreover, a co-expression with DMD gene has been shown in literature.
Conclusions: Identified variants represent putative modifiers of the cognitive phenotype of DMD. We plan to validate these findings by sequencing a larger cohort of dystrophinopathy patients with and without cognitive issues.
eP02.05.03Growth Patterns and Loss of Ambulation in Boys with Duchenne Muscular Dystrophy (DMD)
Stimpson G1, Ridout D2, Manzur A1,3, Sarkozy A1,3, Muntoni F1,3, Baranello G1,3
1Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, Faculty of Population Health Sciences, London, United Kingdom, 2Population, Policy & Practice Department, UCL Great Ormond Street Institute of Child Health, Faculty of Population Health Sciences, London, United Kingdom, 3NIHR Great Ormond Street Hospital Biomedical Research Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Glucocorticoids (GC) are the only recommended treatment for boys with DMD, as they have been shown to prolong ambulation. However, chronic use is associated with several side-effects, particularly weight gain and stunted growth. This is of interest currently as DMD boys are tending to initiate GC at very young ages. As clinicians can consider adverse growth side-effects when making decisions on GC use and discontinuation for patients, it is crucial to understand the role of growth in functional decline in DMD.
An analysis of 2,602 assessments of height and weight on 599 UK NorthStar patients was completed. We considered only patients in one of five treatment subgroups: “GC naive”, “deflazacort daily” (DD), “deflazacort intermittent” (DI), “prednisolone daily” (PD) and “prednisolone intermittent” (PI), which were assigned based on majority regime between starting GC and loss of ambulation. We used SD scores for weight and height calculated using the UK90 reference population. Shared random-effects joint modelling was used with the rate of change of the longitudinal growth outcomes also included in the model.
Height trajectory was associated with risk of loss of ambulation, with an increment of 19% for each SD of height increase at a given time (hazard ratio of 1.19 (95 % CI: 1.04,1.38)). The steepness of the relative height gain also played a major role. For every 0.25 SD increase in yearly height gain, the risk of loss of ambulation doubled (hazard ratio of 2.00 (95% CI: 1.44, 2.77)). The GC naïve subgroup had the highest risk of loss of ambulation over time, however between the subgroups receiving GC the risk was driven by the height profiles, with the tallest subgroup (PI) being the most at risk of loss of ambulation and the shortest subgroup (DD) being the least.
Weight trajectory was also associated with risk of loss of ambulation, with an increase in risk of loss of ambulation of 27% for each SD of weight gain at a given time (hazard ratio of 1.27 (95% CI: 1.12,1.43)). The relative weight gain was also significantly associated with loss of ambulation risk. For a 0.25 SD increase in yearly weight gain, the risk of loss of ambulation increased 70% (95% CI: 34%, 115%). The subgroup most at risk of loss of ambulation over time was the GC naive group. The PI subgroup, who had the heaviest weight trajectories across the four GC groups after the age of 10 had the highest risk of loss of ambulation over time.
In conclusion, patients not on steroids are those more at risk of losing ambulation. However, increased weight and/or height significantly affect the risk of loss of ambulation. This data provides further insights into the long-term clinical management of patients with DMD.
eP02.05.04An Alu-Mediated Insertion in the DMD Gene Canceled Out by Exon 15 Splicing
Budillon A1, Farina A1, Cesarone E1, Tartaglione I2, Torella A1,3, Nigro V1,3
1University of Campania Luigi Vanvitelli, Naples, Italy, 2Department of General and Specialized Surgery for Women and Children, Università degli Studi della Campania “Luigi Vanvitelli”, Naples, Italy, 3Telethon Institute of Genetics and Medicine (TIGEM), Pozzuoli, Naples, Italy, Naples, Italy
The DMD gene provides instruction for making muscle protein dystrophin. Pathogenic mutations affecting this gene cause X-linked dystrophinopathy. Its spectrum is broad since different variants can lead to mild forms of Becker Muscular Dystrophy (BMD) or severe forms of Duchenne Muscular Dystrophy (DMD). In the early stages, the discernment between these two clinical forms is not always clear. Therefore, early molecular findings should always be integrated with a clinical prediction which is mainly based on reading frame studies of the transcript.
Herein we describe the complex case of a 3-year-old adopted boy with elevated serum CK levels and positive Gower’s sign. Following our diagnostic workflow, we first performed an 80-plex PCR method, known as Log-PCR. It showed the absence of the band corresponding to exons 14 and 15. To validate this result, we performed a second independent method (MLPA - multiplex ligation-dependent probe amplification) which, unexpectedly, ended up being negative. With the aim of successfully combining the discrepancy between these two methods, we performed a PCR of exon 14 and 15 and a Sanger sequencing of exon 15. Within exon 15, we identified a transposon element corresponding to an Alu sequence.
Among the rarer explanations of an altered gene expression there is transposon insertion. Being the DMD gene the largest one, some cases of transposon-induced dystrophinopathy have been described.
Transposons are selfish genetic elements that jump from one site of the DNA to another. One of them is Alu sequence which is an RNA transposable element (TE) of 300 bp length and it is a non-autonomous and non-coding SINE. Depending on the target region of the insertion, it can either be silent or affect gene expression in different ways.
Since no similar cases have been reported in the literature, we sequenced the cDNA obtained from a muscle biopsy. By observing a partially functioning in-frame transcript produced by exon 15 skipping, we predicted that the phenotype would have been mild in the long term and comparable to BMD. Seven years after the molecular diagnosis, the phenotype prediction was found to be accurate.
Here we present the first documented case of exon 15 skipping in the DMD gene. So far, there has been no clinical evidence to support our prediction of a mild phenotype. Today, thanks to a random insertion of an Alu sequence within exon 15, we are able to empirically affirm that it leads to Becker Muscular Dystrophy.
eP02.05.05FU-5Cv: Inducible Muscle-Specific Downregulation of Utrophin in Dystrophic Mice to Better Mimic Duchenne Muscular Dystrophy
Neff L1, Gayi E1, Scapozza L1, Dorchies O1
1Pharmaceutical biochemistry, School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland
The mdx and mdx5Cv mice are commonly used animal models for Duchenne muscular dystrophy (DMD). However, they do not properly replicate disease severity and progression experienced by DMD patients. The mild phenotype of mdx/mdx5Cv mice is probably due to upregulation of utrophin, which compensates partly the functions of the missing dystrophin. The dko mouse (dystrophin-utrophin double KO) has been proposed as an alternative model. However, dko mice are very severely affected, breed poorly, and the constitutive absence of utrophin from all tissues is not relevant for DMD.
We aimed at creating a strain of dystrophic mice that would be a better phenocopy of DMD. We used a CRISPR-Cas9 strategy for generating FU-5Cv, an mdx5Cv dystrophic mouse bearing a floxed exon 7 in Utrn, the utrophin gene. Excision of Utrn exon 7 causes a frameshift and premature termination of utrophin translation. FU-5Cv also carries a tetracycline-dependent muscle-Cre recombinase transgene. Administration of doxycycline allows conditional invalidation of Utrn in skeletal muscles only. A constitutive KO was also obtained (KU-5Cv), which is genetically similar to dko.
Here we show preliminary characterization of FU-5Cv and comparison to wildtype (WT), dystrophic (mdx5Cv) and KU-5Cv. Small groups (N = 5-6) of male mice were used. At weaning (P28), mice were given a doxycycline-supplemented diet for 1 week. This triggered muscle-specific recombinase expression and deletion of Utrn exon 7, giving rise to DEL-FU-5Cv mice. For consistency, all groups were exposed to doxycycline, irrespective of their Utrn alleles. Longitudinal monitoring started at 1.5 months, including body weight recording and horizontal grid hanging test. At 6 or 12 months of age, isometric muscle contraction features (phasic tension, tetanic tension, fatigability) were recorded. Mice were euthanized and muscles were weighed and prepared for histology and molecular analyses. Sciatic nerves were collected for examination of axon morphology. Additional groups were sacrificed at 1 and 3-months as reference for subsequent measurement of utrophin levels decay.
WT and mdx5Cv mice had similar body weights during the whole study. The body weight of DEL-FU-5Cv mice was reduced from 4-months of age. KU-5Cv mice (similar to dko) were markedly smaller throughout the study. At the horizontal grid hanging test, mdx5Cv mice had low scores compared to WT. Motor performance was impaired in DEL-FU-5Cv mice compared to mdx5Cv with further decline beyond 9 months of age. KU-5Cv mice had very low scores from start and rapidly declined. Phasic and tetanic tensions developed by the triceps surae were significantly reduced in DEL-FU-5Cv compared to mdx5Cv mice but higher than in KU-5Cv mice. By contrast to mdx5Cv mice that showed paradoxical hypertrophy of most muscles, DEL-FU-5Cv mice showed widespread muscular atrophy (sparing only TA and EDL). Histological examination revealed marked myopathic changes with enhanced fibrosis.
In conclusion, using the FU-5Cv line, we showed that post-natal downregulation of utrophin caused a progressive dystrophy, characterized by reduced body weight, muscle wasting, impaired motor function, decreased muscle force and early accumulation of fibrosis. The phenotype of FU-5Cv mice resembles DMD without the shortcomings of regular mdx/mdx5Cv and dko mice.
eP02.05.06Preclinical Assessment of Therapeutic Cocktails in Dystrophic Mice: Tamoxifen Combined to Metformin, Citrulline and Steroids
Neff L1, Scapozza L1, Dorchies O1
1Pharmaceutical biochemistry, School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland
We published earlier (1,2) that tamoxifen (Tam), a non-cytotoxic drug used for nearly 40 years to treat hormone-dependent breast cancers, had remarkable protective actions in mdx5Cv mice, an animal model of Duchenne muscular dystrophy (DMD). Subsequently, Dr Talya Dor (Hadassah Medical Center, Jerusalem, Israel) offered tamoxifen to DMD boys as a compassionate treatment. Encouraged by the absence of safety concern and by apparent stabilization of the boys over 24-36 months, she launched a larger open-label trial (3), the results of which has been published in 2021 (4): based on CK levels and motor assessments (NSAA, 6-minute walking distance) their study suggests safety and efficacy of tamoxifen in ambulant DMD boys. Since mid-2018, almost 100 DMD boys (80 ambulant on steroids, 16 non-ambulant) have been enrolled in TAMDMD (5,6), a double-blind international, placebo-controlled phase 3 clinical trial led by Prof. Dirk Fischer (UKBB, Basel, Switzerland).
The British charity Duchenne UK is supporting TAMDMD massively and is eager to make funding decisions about other drug candidates for DMD, including citrulline (Cit) and metformin (Met). These compounds are believe to improve muscle perfusion and enhance mitochondrial biogenesis. Cit+Met was found promising in a pilot trial in patients with Becker muscular dystrophy. Because clinical trials are very expensive and the number of patients to be recruited is limited, Duchenne UK requested that we evaluate the efficacy of Tam/Cit/Met cocktails in dystrophic mice, using a comprehensive panel of functional, biochemical, molecular and histological investigations.
Groups of 10-12 male dystrophic mice aged 8-9 months were treated for 3 months with drug-supplemented diets. Groups of untreated wildtype and dystrophic mice were used as controls. First, we compared Tam alone at the optimal dose (3 mg/kg/day) with Tam+Cit or Tam+Met (3 doses at half-log intervals). The highest doses caused minor toxicity. Based on improvement of motor function and muscle force, we determined that the intermediate doses of Cit (3 g/kg/day) and of Met (0.3 g/kg/day) produced the best benefits when combined to Tam. Next, we combined the 3 drugs at the optimal dosing, with or without prednisolone (Pred; 1 mg/kg/day) and compared their efficacy to Tam alone, Tam+Pred and Met+Cit. Addition of Cit+Met to Tam marginally increased the efficacy of Tam alone. Addition of Pred to either Tam or Tam+Cit+Met caused a delay in functional improvement. Unexpectedly, Cit+Met slightly worsened the condition.
Molecular and histological analyses are ongoing. Our findings so far suggest that (i) Cit+Met is not a valid option for DMD, (ii) the therapeutic benefit of adding Cit+Met to Tam is limited, (iii) Pred attenuates the improvements afforded by Tam. Further investigations are warranted to confirm and extend these findings which may have important consequences for developing efficient therapies for DMD patients.
(1) Dorchies et al., Am J Pathol (2013), 182(2):485-504.
(2) Gayi et al., Chimia (Aarau) (2018), 72(4):238-240.
(3) https://clinicaltrials.gov/ct2/show/NCT02835079
(4) Tsabari et al., Neuromuscul Disord (2021), 31(9):803-813.
(5) https://clinicaltrials.gov/ct2/show/NCT03354039
(6) Nagy et al., Trials (2019), 20(1):637.
eP02.05.07Roles of Ltbp4 and Abcc6 on the Phenotype of Mouse Models of Duchenne Muscular Dystrophy
Neff L1, Gayi E1, Scapozza L1, Dorchies O1
1Pharmaceutical Biochemistry, School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland
The original mdx mouse (C57BL/10ScSn background) and mdx5Cv mouse (C57BL/6J background) are widely used for decades as models for Duchenne muscular dystrophy (DMD). However, they do not properly replicate DMD severity and progression. Dystrophic mice show a mild disease, compensatory muscle hypertrophy instead of atrophy, late-onset fibrosis that affects muscles to various degrees, and normal lifespan. Recently, the D2.mdx mouse emerged as an attractive DMD model: the mdx allele was transferred into the DBA/2J background, which naturally harbours mutations in Ltbp4 (Ltbp4DEL), a gene encoding a TGF-binding protein, and in Abcc6 (Abbc6MUT), a gene that causes ectopic calcification in muscle cells. D2.mdx mice show early-onset frailty, muscle atrophy and enhanced fibrosis resembling the human condition, presumably due to Ltbp4DEL. However, Abbc6MUT causes widespread ectopic calcification in myofibres, a feature that is not relevant for DMD.
We aimed to decipher the contribution of Ltbp4DEL and Abbc6MUT in aggravating the phenotype of dystrophic mice. First, we generated the D2-5Cv mouse line (>97% DBA/2J background), similar to the D2.mdx line but bearing the mdx5Cv allele instead of the mdx allele. The phenotype of D2-5Cv mice was similar to that of D2.mdx, including frailty, substantial drop in body weight, marked atrophy of tibialis anterior (TA) and gastrocnemius muscles, enhanced muscular fibrosis, and unwanted ectopic calcification in myofibres. Next, we performed genetic background swapping from DBA/2J to C57BL/6J: D2-5Cv males were mated with C57BL/6 females and pertinent individuals in the progeny selected over 5 generations until we obtained the following lines of mdx5Cv mice: 5CV/0 (bearing wildtype versions of both Ltbp4 and Abcc6), 5Cv/A (bearing Abbc6MUT), 5Cv/L (bearing Ltbp4DEL), and 5Cv/LA (bearing both Ltbp4DEL and Abbc6MUT). These lines were characterized at 1, 3, 6 and 12 months of age and compared to wildtype (WT) mice.
Preliminary phenotyping confirmed that Abbc6MUT caused ectopic calcification in muscles of 5Cv/A and 5Cv/LA mice. The extent of calcification seems not to correlate with a specific muscle type or with age: for a given muscle at a given age, some specimens were free of calcification whereas others were heavily loaded with abnormal white deposits. This supports our view that Abbc6MUT is not a relevant genetic factor for aggravating the phenotype of dystrophic mice and would act as a confounding factor instead. Clinically relevant features of dystrophic mice in the DBA/2J background (D2.mdx and D2-5Cv) were not replicated in the C57BL/6J background: at all ages examined 5Cv/A, 5Cv/L and 5Cv/LA mice had similar body weight as WT and 5Cv/0. Muscle size and isometric force was similar in all groups of dystrophic mice, independently of Ltbp4DEL and Abbc6MUT alleles. Fibrosis was similar in 5Cv/0, 5Cv/L and 5Cv/LA mice, suggesting that, in the C57BL/6J background, Ltbp4DEL did not promote fibrosis.
The lines 5Cv/LA and D2/5Cv show highly divergent phenotypes. They share the alleles mdx5Cv, Ltbp4DEL and Abbc6MUT but differ for the rest of their genetic backgrounds. Our findings question the roles of Ltbp4DEL and Abbc6MUT in aggravating the phenotype of dystrophic mice and strongly suggest that other genetic factors are involved.
eP02.05.08Integrated Analyses of Data from Clinical Trials of Delandistrogene Moxeparvovec in DMD
Zaidman C1, Shieh P B2, Proud C3, McDonald C4, Day J W5, Mason S6, Guridi M7, Hu L6, Yu L6, Reid C7, Darton E6, Wandel C8, Richardson J6, Malhotra J6, Singh T6, Rodino-Klapac L R6, Mendell J R9,10
1Department of Neurology, WUSTL, Washington, USA, 2UCLA Medical Center, Los Angeles, USA, 3Children’s Hospital of the King’s Daughters, Norfolk, USA, 4UC Davis Health, Sacramento, USA, 5Department of Neurology, Stanford University, Palo Alto, USA, 6Sarepta Therapeutics, Inc., Cambridge, USA, 7Roche Products Ltd, Welwyn Garden City, UK, 8F. Hoffmann-La Roche Ltd, Basel, Switzerland, 9Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, USA, 10The Ohio State University, Columbus, USA
Delandistrogene moxeparvovec (SRP-9001) is an investigational gene transfer therapy developed for targeted skeletal and cardiac muscle expression of micro-dystrophin (a shortened, functional dystrophin protein) that is being studied in patients with Duchenne muscular dystrophy (DMD).
The objective of this integrated analysis is to evaluate functional data from patients (≥4 to ≤8 years old) with DMD who have participated in the delandistrogene moxeparvovec studies and compare these data with a control cohort of propensity-matched external control patients. Functional data were pooled from three studies:
• Study 101 (NCT03375164) is a Phase 1/2a, single-dose, open-label clinical trial evaluating the safety of systemic delivery of delandistrogene moxeparvovec clinical process material in patients with DMD (N=4), aged 4-7 years at enrolment
• Study 102 (NCT03769116) is a three-part, Phase 2, double-blind, placebo-controlled clinical trial evaluating the safety and efficacy of systemic delivery of delandistrogene moxeparvovec clinical process material in patients with DMD (N=41), aged 4-7 years at screening
• ENDEAVOR (NCT04626674) is an open-label, Phase 1b study assessing the expression and safety of commercially representative delandistrogene moxeparvovec material in patients with DMD (N=38). Cohort-specific criteria include age at screening and ambulatory/non-ambulatory status: ≥4 to <8-year-old ambulatory boys (Cohort 1), ≥8 to <18-year-old ambulatory boys (Cohort 2), non-ambulatory boys (Cohort 3), and ≥3 to <4-year-old ambulatory boys (Cohort 4).
Expression and safety data from Studies 101 and 102 and Cohort 1 of ENDEAVOR (n=11) have been previously presented, along with functional data from Studies 101 and 102.
Here we present an integrated analysis of functional data from the 53 patients assessed to date from Study 101 (N=4), Parts 1 and 2 of Study 102 (n=29) who received the target dose (1.33x1014 vg/kg by linear qPCR) of delandistrogene moxeparvovec over 1 year, and patients from Cohort 1 of ENDEAVOR (n=20). The primary endpoint of this integrated analysis is 1-year change from baseline in the North Star Ambulatory Assessment (NSAA) total score. Exploratory endpoints include 1-year change from baseline in timed function tests, including Time to Rise and the 10-metre Walk/Run (10MWR).
The control cohort includes natural history and external clinical trial data from several studies. The following criteria were used to identify the external control patients to ensure they were similar to patients enrolled in the delandistrogene moxeparvovec studies: age (≥4 to ≤8 years old), NSAA score (≥13 and ≤30), Time to Rise (≤10.4 seconds), and 10MWR (≤9.1 seconds) at baseline and receiving a stable dose or dose equivalent of oral corticosteroids for ≥12 weeks before baseline. Functional data from all patients who received the target dose from the delandistrogene moxeparvovec studies will be analysed and compared with the propensity-matched external control cohort to contextualise the results.
Collective safety data (as of the latest data-cut) from all patients (all cohorts in Studies 101 and 102 and ENDEAVOR) will be presented.
Studies 101 and 102 and ENDEAVOR are sponsored and funded by Sarepta Therapeutics. ENDEAVOR is also funded by F. Hoffmann-La Roche.
eP02.05.09Phenotypic Variability of Becker Muscular Dystrophy: A Detailed Clinical Characterization Protocol Towards Trial Readiness
Torri F1, Ricci G1, Govoni A1,4, Astrea G2, Castiglione V3, Giannoni A3, Emdin M3, Siciliano G1
1Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy, 2Neurology, IRCCS Fondazione Stella Maris, Pisa, Italy, 3Cardiology Department, Fondazione Toscana Gabriele Monasterio, Pisa, Italy, 4Neuromuscular and Rare Disease Unit, La Fondazione IRCCS Ca’ Granda Ospedale Maggiore di Milano Policlinico, Milano, Italy
Although consisting of a less severe, slower form of dystrophinopathy, Becker Muscular Dystrophy (BMD) carries a variable amount of disability burden and various clinical trials are ongoing, prompting the need for detailed phenotypic definition of patients and identification of outcome measures.
We present a detailed phenotypic characterization of 32 adult patients with BMD, providing a description of follow-up data over the course of 24 months for 13 patients; moreover, in a subgroup of 10 patients clinical data have been compared with muscle magnetic resonance acquisitions over the same period.
Each patient has been genetically characterized. Baseline, and 1-year and 2 years assessments included North Star Ambulatory Assessment (NSAA), timed function tests (time to climb and descend four stairs), 6-minute walk test (6MWT), Walton and Gardner-Medwin Scale and manual force measurement (MRC scale). Muscle magnetic resonance imaging (MRI) was acquired at baseline and in a subgroup of patients after an average period of 24 months. Data on cardiac function (electrocardiogram, echocardiogram and cardiac MRI) were also collected. Patients motor tests and muscle MRI have been registered on the developing InGene2.0 (Bando Ricerca e Salute 2018 Programme, Tuscany Region) informatic platform, designed by clinicians, biomedical engineers and bio-IT technicians to collect multimodal and multiparametric data (clinical, molecular, imaging, muscle biopsy) from patients affected by neuromuscular diseases, in order to ameliorate diagnosis, follow-up and identification of disease outcome measures.
Baseline evaluation confirmed the broad phenotypic spectrum of BMD and the different disease course based on the disease severity, highlighting the complex genotype-phenotype correlation. As to the follow-up period, taken globally, significant worsening of motor impairment was not detected in our patients, while it was present only when considering the subgroup of subjects carrying deletions starting form exon 45, thus suggesting that standardized motor scales could not represent an optimal tool to evaluate changes in disease progression, especially in the perspective of future clinical trials. With regard to muscle MRI, overall semiquantitative analysis via Mercuri scale did not reveal a significant disease progression, with a mean increase of 1 point, with a certain heterogeneity of the carried mutation among severity groups.
Our data stress the need of precise phenotypic stratification of patients and identification of more suitable and sensible outcome measures, capable of catching little but significant changes in disease progression that could not be obvious if assessed with the classical clinical tools and biomarkers.
eP02.05.10Non-neuromuscular Manifestations in a Colombian Patient with Megaconial Congenital Muscular Dystrophy
Bobadilla E1, Rojas S2, Restrepo D3, Castro S4, Castro D5, López García J6, Giraldo Castro H7
1Hospital Universitario San Ignacio, Bogotá, Colombia, 2Universidad de Antioquia, Medellín, Colombia, 3Universidad CES- Cenpi, Medellín, Colombia, 4Ayudas Diagnósticas SURA, Medellín, Colombia, 5CIGE- Centro de inmunología y Genética, Medellín, Colombia, 6CENPI- Clínica de las Américas, 7CENPI- Private Practice Child & Adolescent Psychiatry
Introduction: Megaconial congenital muscular dystrophy, whose initial description was in 1998 by Nishino, is due to mutations in the CHKB (Cholin Kinase Beta) gene that affect the biosynthesis of phosphatidylcholine, causing mitochondrial structural disturbances. So far there are 35 identified patients, mainly Turkish and Japanese patients, however its incidence in Latin America is unknown.
Case report: We present a Colombian girl was born to non-consanguineous parents with a normal pregnancy. She was referred to pediatric neurology at 17 months of age for presenting motor and language development delay as well as hypotonia. On physical examination, she had severe global hypotonia, with axial and proximal weakness. She also had global hyporeflexia and hypermobility mainly in distal joints. She had no skin lesions or cardiac abnormalities.
She had difficulties in socialization and since the age of 2 years old, she began with hand flapping stereotypies with behavioral symptoms. A diagnosis of severe autism was made. She started walking about 3 years old and apnea-hypopnea syndrome and significant sleep disturbance was identified at that time. She also had seizures with EEG showing right frontotemporal epileptiform activity, however her brain MRI was normal. CPKs were 1013-1095 IU/L. Muscle biopsy showed a dystrophic pattern with mitochondrial abnormalities.
A clinical exome was performed for congenital muscular dystrophies that presented two variants: CHKB, c.582-1G>A, heterozygous, probably pathogenic CHKB, c.101T>C (p.Trp61Arg), heterozygous, VUS. The above findings added to the muscle biopsy findings confirmed CHKB-CMD also called megaconial muscular dystrophy.
Conclusion: This patient is the first patient reported in Latin America. With this case we want to highlight that CHKB-CMD should always be a diagnosis to rule out in patients with central nervous system symptoms such as epilepsy, sleep disorders and neurocognitive symptoms such as autism and severe behavioral disorders in patients with neuromuscular manifestations.
eP02.06.01DYNE-101 Corrects the DM1 Splicing Phenotype of hTfR1/DMSXL Mice and Is Well Tolerated in NHPs
Picariello T1, Russo R1, Schlaefke L1, Hildebrand S1, Najim J1, Qiu Q1, Weeden T1, Davis J1, Zanotti S1, Beskrovnaya O1
1Dyne Therapeutics, Inc., Waltham, United States
Lack of targeted delivery of oligonucleotide therapeutics to muscle tissues remains a challenge in the treatment of muscle diseases. The FORCE™ platform, designed to overcome these limitations, consists of an oligonucleotide payload conjugated to an antigen-binding fragment (Fab) targeting the extracellular region of the human transferrin receptor (TfR)1 which is enriched in muscles. This allows for the rational selection of payloads to target the genetic basis of disease such as myotonic dystrophy type 1 (DM1), a rare neuromuscular disorder caused by the expansion of CUG repeats in the 3’-untranslated region of the dystrophia myotonica protein kinase (DMPK) RNA. The expanded CUG repeats form hairpin-loop structures that sequester splicing regulators into toxic nuclear foci, leading to a spliceopathy that drives DM1 clinical manifestations. Naked antisense oligonucleotides (ASO) designed to knock down DMPK have shown promise in preclinical models; however, clinical application has failed because of insufficient delivery to muscle upon systemic administration. To determine applicability of the FORCE platform as a potential treatment for DM1, hTfR1/DMSXL mice, a novel model expressing human TfR1 and a human DMPK with >1,000 CUG repeats, were dosed with DYNE-101, a Fab-conjugated ASO that targets toxic human nuclear DMPK, on days 0 and 7. On day 28, assessment of DMPK RNA levels and splicing in the heart and skeletal muscles revealed a 40-49% reduction of toxic human DMPK RNA, leading to spicing correction. These effects were accompanied by a 49% DMPK foci reduction in the heart. Analysis of gene expression in nuclear and cytoplasmic fractions provided direct evidence that DYNE-101 reduced accumulation of toxic human DMPK RNA trapped in the cell nucleus. DYNE-101 was well tolerated in a GLP toxicology study in non-human primates (NHPs). Collectively, these data demonstrated that DYNE-101 is well tolerated and targets toxic DMPK in the nucleus, thereby driving splicing correction in cardiac and skeletal muscle with a long-lasting effect. The findings support advancement of DYNE-101 into the clinic.
eP02.06.02Myotonic Dystrophy Type 2 and Autoimmune Diseases
Peric S4, Zlatar J2, Pesovic J3, Nikolic L2, Bozovic I1, Ivanovic V1, Palibrk A1, Basta I4, Savic-Pavicevic D3, Rakocevic-Stojanovic V4
1Neurology Clinic, University Clinical Centre Of Serbia, Belgrade, Serbia, 2School of Medicine, University of Belgrade, Belgrade, Serbia, 3Center for Human Molecular Genetics, Faculty of Biology, University of Belgrade, Belgrade, Serbia, 4Neurology Clinic, University Clinical Centre Of Serbia, School of Medicine, University of Belgrade, Belgrade, Serbia
Introduction: Myotonic dystrophy type 2 (DM2) is a rare autosomal dominant multisystemic disease with highly variable clinical presentation. Several case reports and one cohort study suggested significant association between DM2 and autoimmune diseases (AIDs).
Aim: To analyze frequency and type of AIDs in DM2 patients from the Serbian DM registry.
Patients and Methods: 131 DM2 patients from 108 families were included - 62.6% females, age at DM2 onset 40.4 (13.0) years, age at entering Registry 52.0 (12.8) years, age at analysis 58.4 (12.8) years. Data were obtained from the Akhenaten, Serbian Registry for DM, as well as through the medical electronic data system.
Results: At entering Registry, 35 (26.7%) of 131 DM2 patients had AIDs including Hashimoto thyroiditis (18.1%), rheumatoid arthritis, diabetes mellitus type 1, systemic lupus, Sjogren, localized scleroderma, psoriasis, celiac disease, Graves, neuromyelitis optica, and Guillain-Barre syndrome. At the time of data analysis, two additional patients developed new AIDs, so eventually 37 (29.6%) of 125 DM2 survivors had AIDs. Antinuclear antibodies (ANCA) were found in 14 (10.7%) of 63 tested patients, including 12 patients without defined corresponding AID (all in low titers – 1:40 to 1:160). Antineutrophil cytoplasmic antibodies (ANCA) were negative in all 50 tested cases. The percentage of women was significantly higher among patients with AIDs (82.9% vs. 55.2%, p<0.01). Patients with AIDs were less likely to have myopathy on electromyography (66.7% vs. 84.8%, p<0.05).
Conclusion: AIDs were present in as high as 30% of DM2 patients. Thus, screening of AIDs in DM2 seems reasonable. Presence of AIDs and/or ANA may lead to false diagnosis of AIDs and under-diagnosis of DM2.
Keywords: myotonic dystrophy type 2 (DM2), autoimmune diseases, antinuclear antibodies, antineutrophil cytoplasmic antibodies
eP02.06.03Nuclear Envelope Dysfunction in Myotonic Dystrophy Type 1
Viegas D1, Pereira C1, Martins F1, Mateus T1, da Cruz e Silva O1, Herdeiro M1, Rebelo S1
1Department of Medical Sciences, Institute of Biomedicine (iBiMED), University of Aveiro, 3810-193 Aveiro, Portugal, Aveiro, Portugal
Myotonic Dystrophy type I (DM1) is a rare autosomal dominant genetic disease caused by the abnormal expansion of unstable CTG repeats in the 3’ untranslated region of Myotonic Dystrophy Protein Kinase (DMPK) gene being the most common muscular dystrophy among adults (1). The number of CTG expansions can vary in different organs and tissues, contributing to the great phenotypic variability observed in patients with DM1. The clinical phenotype is wide, even between members of the same family, ranging from severe congenital-onset forms to late-onset forms which present mild symptoms (1–4). DM1 is characterized by myotonia, progressive peripheral muscle weakness and atrophy, involving also multiple organs and systems: cardiovascular, respiratory, endocrine, gastrointestinal, central nervous systems and eye (2–4).
To date, the molecular mechanism underlying this devasting disease is still poorly characterized, although there are some hypotheses that envisage to explain the multisystemic features observed in DM1 (reviewed in 5). An emergent hypothesis is that nuclear envelope (NE) dysfunction may contribute to muscular dystrophies, particularly to DM1. Therefore, the main objective of the present study was to evaluate the nuclear profile of DM1 patient-derived and control fibroblasts to determine the protein levels and subcellular distribution of relevant NE proteins.
Our results clearly demonstrate that nuclear profile and NE proteins are substantially altered in DM1-patient derived fibroblasts. Concerning nuclear profile, a larger nuclear area and a higher number of deformed nuclei and micronuclei are the most prominent alterations observed in DM1 patient-derived fibroblasts (6). The protein levels of several NE proteins were increased namely, lamin A/C, LAP1 and SUN1, while the levels of emerin and nesprin-1/nesprin-2 remained unaltered and decreased, respectively. Further, altered location of these NE proteins were accompanied by the presence of nuclear deformations (blebs, lobes and/or invaginations) and higher number of nuclear inclusions (6).
Our study has strengthened the hypothesis that changes in the NE are important hallmarks of DM1 and supports further studies and the exploitation of NE dysfunction in DM1 as a target for the development of DM1 therapies.
References
1. Darras Basil T. Myotonic dystrophy: Etiology, clinical features, and diagnosis. UpToDate. 2009;
2. Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol. 2014 Oct;27(5):599-606.
3. Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol. 2013 Dec;32(3):154-65.
4. Thornton CA. Myotonic dystrophy. Neurol Clin. 2014 Aug;32(3):705-19, viii.
5. Mateus T, Martins F, Nunes A, Herdeiro MT, Rebelo S. Metabolic Alterations in Myotonic Dystrophy Type 1 and Their Correlation with Lipin. Int J Environ Res Public Health. 2021 Feb 12;18(4):1794.
6. Viegas D, Pereira CD, Martins F, Mateus T, da Cruz E Silva OAB, Herdeiro MT, Rebelo S. Nuclear Envelope Alterations in Myotonic Dystrophy Type 1 Patient-Derived Fibroblasts. Int J Mol Sci. 2022 Jan 4;23(1):522.
Acknowledgments: This research was funded by Fundação para a Ciência e a Tecnologia (FCT) through the Institute of Biomedicine (iBiMED)—UIDB/BIM/04501/2020/UIDP/04501/2020 and by the MEDISIS project (CENTRO-01-0246-FEDER-000018). Image acquisition was performed in the LiM facility of iBiMED, a node of PPBI (Portuguese Platform of BioImaging): POCI-01-0145-FEDER-022122.
eP02.06.04Does Small Fiber Neuropathy Contribute to Chronic Muscle Pain in Patients with Myotonic Dystrophy?
Schmitt V1, Bäumler P2, Irnich D2, Schoser B1, Montagnese F1
1Friedrich Baur Institut, Department of Neurology, Ludwig Maximilians University, Munich, Germany, 2Multidisciplinary Pain Centre, Department of Anaesthesiology, University Hospital LMU Munich, Munich, Germany
Introduction: One of the most common and most disabling symptoms in myotonic dystrophy type 1 (DM1) and 2 (DM2) is chronic muscle pain. Little is known about the pathophysiology of myalgia, and the response to common analgesics is often not satisfactory. Previous studies with quantitative sensory testing (QST) in DM2, identified some similarities with fibromyalgia and suggested the presence of peripheral sensitization mechanisms involved in the origin of chronic pain in DM patients. Our aim was to investigate whether the presence of small fiber neuropathy could be a contributor to chronic muscle pain in myotonic dystrophy patients.
Methods: We included DM1 and DM2 patients between 18 and 65 years old with chronic myalgia. Exclusion criteria were presence of diabetes mellitus or polyneuropathy. Patients completed three pain questionnaires (Brief pain inventory – BPI, PAIN-DETECT and pain disability index – PDI), underwent neurological examination, nerve conduction study, QST and skin biopsy (distal and proximal lower limb) to determine the intraepidermal nerve fiber density (IENFD). QST data were compared with a group of 30 gender and age matched healthy controls. IENFD data were interpreted with existing reference values.
Results: We recruited 32 DM2 (F 59%, mean age 53.4±7.3) and 21 DM1 (F 62%, mean age 40.7±12.6) patients. DM2 patients, in comparison to DM1, showed higher pain related disability (p=0.026) and higher pain interference (p=0.012), took painkillers more frequently (34% vs 14%, p=0.006) and described pain more often as radiating (78% vs 25%, p<0.001). Pain patterns differed (p=0.035) with more DM2 patients suffering chronic constant pain (38% vs 25%) and more DM1 patients with pain attacks (55% vs 25%).
Pain severity and location was similar between DM1 and DM2. In QST, we found multiple differences between DM1 and DM2, as well as between DM and healthy controls. DM2 patients were characterized by a loss in cold, warm, mechanical and vibration sensitivity, while DM1 patients showed signs of mechanical hyperalgesia (lower mechanical pain threshold, higher mechanical pain sensitivity). Both DM patient groups showed increased pressure pain sensitivity. IENFD was reduced in 63% of DM1 patients and in 50% of DM2. This correlated with age (p=0.050) but, interestingly, not with QST data.
Conclusion: This study shows the presence of loss in detection sensitivity and gain in pain sensitivity in patients with myotonic dystrophy, as well as reduced IENFD. Ongoing analyses will further examine how these abnormalities correlate with the severity and quality of pain.
eP02.06.05Frequently Used Outcome Measures to Evaluate Muscle Strength in Patients with Myotonic Dystrophy Type 1
Mateus T1, Costa A1, Viegas D1, Marques A2, Herdeiro T1, Rebelo S1
1Department of Medical Sciences, Institute of Biomedicine (iBiMED), University of Aveiro, Aveiro 3810-193, Portugal, Aveiro, Portugal, 2Respiratory Research and Rehabilitation Laboratory – Lab3R, Institute of Biomedicine (iBiMED), School of Health Sciences (ESSUA), University of Aveiro, Aveiro, Portugal, Aveiro, Portugal
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy in adults with a prevalence of 1 in 3,000 to 8,000 individuals worldwide (1,2). DM1 is an autosomal dominant disease caused by abnormal expansion of unstable repetitions of cytosine-thymine-guanine trinucleotide (CTG) in the 3’ untranslated region of Myotonic Dystrophy Protein Kinase (DMPK) gene. DM1 present different clinical phenotypes according to the age of onset and length of CTG repeat expansion (1–3). DM1 is characterized by myotonia, progressive muscle weakness and other multisystemic alterations (3,4). However, respiratory and cardiac dysfunction are the most common cause of death in DM1 population, leading to a mortality of 51% to 76% and 30%, respectively (2,4). Considering that muscle strength measurements are crucial to manage DM1 skeletal, cardiac and respiratory muscle dysfunction, understanding which outcome measures are the most frequently used to assess muscle strength in DM1 is of paramount importance. Therefore, the aim of this study was to gather and summarise the most frequent outcome measures used to assess muscle strength in adult patients with DM1 to contribute for future clinical practice guidance and research. The search strategy used was based on the extraction and gathering data from Pubmed, Web of Science and Embase databases. All observational studies using measures of muscle strength in adult patients with DM1 were included (80 studies), where 24 measured cardiac, 45 skeletal and 23 respiratory muscle strength (2). Among the studies that indirectly assessed the cardiac muscle strength, the most frequent method and outcome measure used were echocardiography and left ejection fraction, respectively (2). Studies that assessed skeletal muscle strength, the most frequent methods and outcome measure used were quantitative muscle test and manual muscle test, and maximum isometric torque, respectively (2). The most frequent method and outcome measures used to assess the respiratory muscle strength were manometry, and maximal inspiratory and expiratory pressure, respectively (2).
The results of this study regarding muscle strength evaluation of adult patients with DM1 are essential to correctly assess disease severity and response to interventions. Further, clinicians and researchers should consider using the same methodologies and outcome measures described in this study, to contribute to a better understanding of the response in muscle strength in future clinical trials and interventions.
References:
1. Ashizawa, T. et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol. Clin. Pract. 8, 507–520 (2018).
2. Mateus, T. et al. Outcome measures frequently used to assess muscle strength in patients with myotonic dystrophy type 1: a systematic review. Neuromuscul. Disord. (2021) doi:10.1016/j.nmd.2021.09.014.
3. Wenninger, S., Montagnese, F. & Schoser, B. Core Clinical Phenotypes in Myotonic Dystrophies. Front. Neurol. 9, (2018).
4. Mateus, T., Martins, F., Nunes, A., Herdeiro, M. T. & Rebelo, S. Metabolic Alterations in Myotonic Dystrophy Type 1 and Their Correlation with Lipin. Int. J. Environ. Res. Public Health 18, 1794 (2021).
Acknowledgments: This research was funded by Fundação para a Ciência e a Tecnologia (FCT) through the Institute of Biomedicine (iBiMED)—UIDB/BIM/04501/2020/UIDP/04501/2020. Also financed the MEDISIS project (CENTRO-01-0246-FEDER-000018).
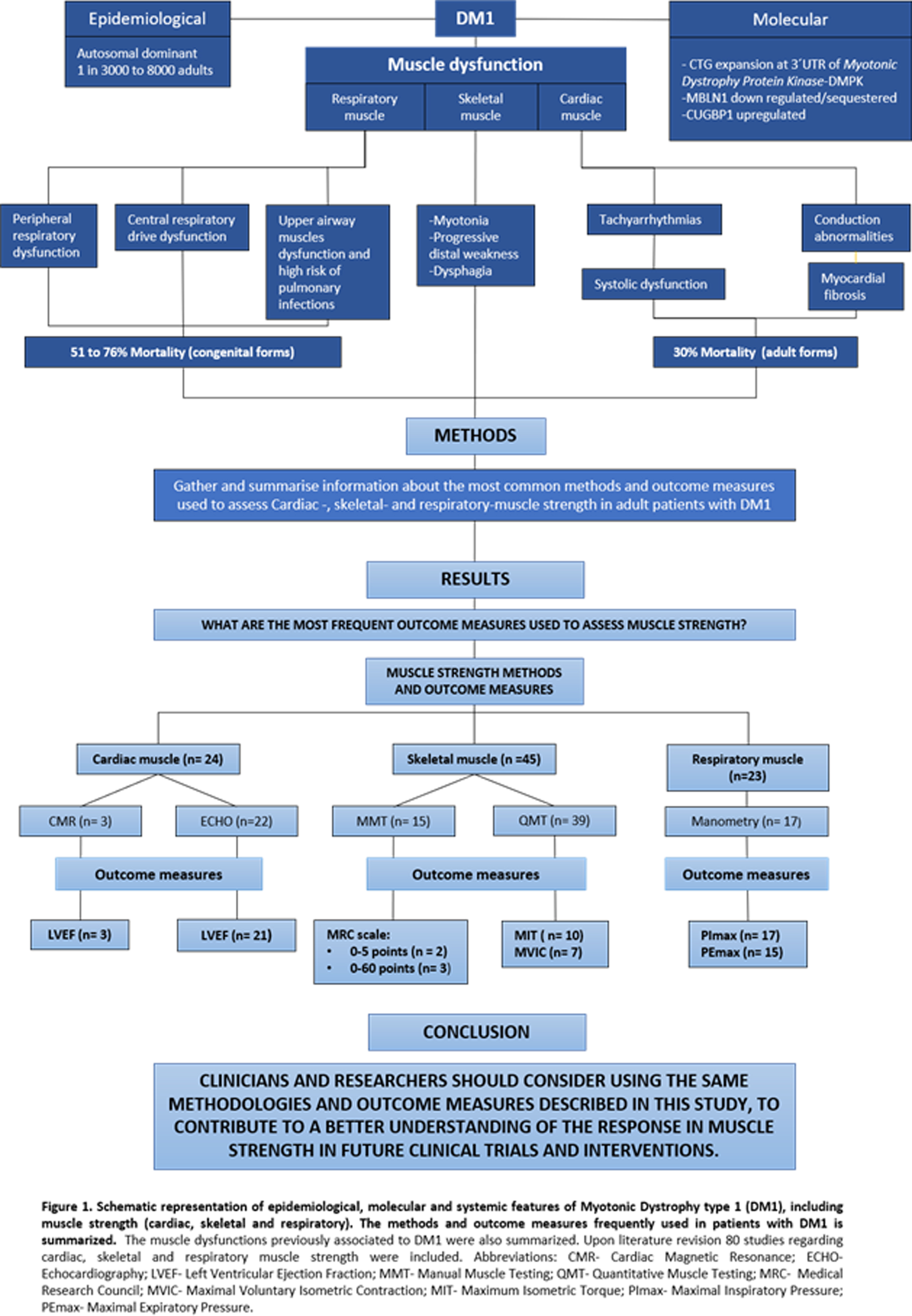
eP02.06.06Clinical Features of the UK Myotonic Dystrophy Patient Registry
Walker H1, Turner C2, Bowler M3, Ashley E4, Rogers M5, Orrell R6, Donachie J7, Williams D8, Hamilton M9, Hewamadduma C10, Sodhi J1,11, Marini-Bettolo C1,11
1The John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom, 2University College Hospital, National Hospital for Neurology and Neurosurgery, London, United Kingdom, 3Myotonic Dystrophy Support Group, Nottingham, United Kingdom, 4Cure Myotonic Dystrophy UK Charity, United Kingdom, 5Institute of Medical Genetics, University Hospital of Wales, Cardiff, United Kingdom, 6UCL Queen Square Institute of Neurology, University College London, London, United Kingdom, 7School of the Arts, English and Drama, Loughborough University, Loughborough, United Kingdom, 8Patient Representative, United Kingdom, 9West of Scotland Clinical Genetics Service, Queen Elizabeth University Hospital, Glasgow, United Kingdom, 10Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, United Kingdom, 11Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
The UK Myotonic Dystrophy Patient Registry is a patient self-enrolling online database collecting clinical and genetic information about myotonic dystrophy type 1 (DM1) and type 2 (DM2). The registry was established in May 2012 with support from Muscular Dystrophy UK and the Myotonic Dystrophy Support Group and is coordinated Newcastle University. The registry aims to facilitate academic and clinical research, better characterise and understand DM, and disseminate information relating to upcoming studies and research advancements.
The registry is used to capture longitudinal, self-reported data through an online portal available to patients and clinicians. Where specialised clinical or genetic information is required, the neuromuscular specialist involved in the patient’s care can be invited to provide some additional information and the patient can select them from a pre-populated list at the registration stage. The registry is a Core Member of the TREAT-NMD Global Registries Network for DM1.
Between May 2012 and January 2022, there were 834 patient registrations. On average there are 5 new registrations per month. For those reporting a clinical diagnosis, 96% have DM1 (of which 14% have a diagnosis of congenital DM) and 4% have DM2. Overall, 40% of patients have had genetic confirmation of their condition provided.
The registry has previously supported almost 30 research enquiries to date. Since 2020, the registry has facilitated 11 enquiries including an industry enquiry, three COVID-19 surveys, and various surveys capturing information on dysphagia, pregnancy, patient preferences for future treatments and the patient/caregiver experience.
The registry continues to be a versatile, cost-effective research tool, helping facilitate and advance a range of DM research. Additional work continues to be done to improve reporting of genetic information on the registry and there are future data linkage plans between the registry and the Newcastle Research Biobank for Rare and Neuromuscular Diseases.
eP02.06.07Myofibrillar Myopathy: Clinico-Genetic Spectrum from an Indian Neuromuscular Center
Oommen A1, Vengalil S, Nashi S, Polavarapu K, Arunachal G, Bardhan M, Narayanappa G, Kumar V, Unnikrishnan G, Huddar A, Baskar D, Nalini A
1National Institute Of Mental Health And Neurosciences (NIMHANS), Bangalore, India
Background: Myofibrillar myopathies (MFM) occur secondary to mutations in DES, CRYAB, MYOT, LDB3, FLNC, BAG3, FHL1, TTN, PYROXD1 and KY genes. The study aims to characterize the clinico-genetic spectrum in the Indian population.
Method: A description of the clinical, radiological and mutation spectrum of 14 genetically confirmed MFM patients.
Results: The male: female ratio is 10:4. Mean age of onset, presentation and illness duration were 16, 27 and 11 years, respectively. Consanguinity (28.6%) and motor developmental delay (28.6%) were present. One-third had ptosis and bulbar symptoms at birth. Clinical features: ptosis and ophthalmoparesis (42.9%), bifacial weakness and flaccid dysarthria (21.4%), neck weakness (50%), proximal weakness of upper and lower limbs (35.8%), only proximal lower limb weakness (28.6%), foot drop (14.3%) and dyspnoea (14.3%). Contractures at ankles (64.3%), neck (14.3%), hips (14.3%) and knees (14.3%) were observed. Cardiac involvement (28.6%) included restrictive, dilated and hypertrophic cardiomyopathy. Mean creatine kinase=1607U/L. Muscle biopsy in two patients revealed reduced/ absent desmin expression. Muscle MRI in three patients showed fatty infiltration predominantly in glutei muscles, adductor muscles of thighs and pancompartmental muscles of legs. Next generation sequencing (NGS) showed DES gene mutation in nine patients. The most common mutation was in intron 5 with variant c.1023+5G>A. All three patients with intronic mutations had ptosis or easy fatigability in childhood and subsequently presented with quadriparesis and oculofacialbulbar weakness. Other DES mutations include [Exon 6, c.1216C>T, pArg406Trp; Exon 5, C958delG, p. E320fs*2; Exon1, c.20C>A, p.Ser7Tyr; Exon 7, c.1280A>T, p.Asn427Ile; Exon 1, c.448C>T, p.Arg150Ter]. TTN [Exon343, c.95134T>C, p.Cys31712Arg], HSPB8 [Exon 3, c.566_567del, p.Asp189GlufsTer26], FLNC [Exon 39, c.6398G>T,p.Arg2133Leu], CRYAB [Exon 3, c.238G>A, p.Asp80Asn], LDB3 [Exon 12, c.1909A>G,p.Thr637Ala] gene mutations were seen in each of the patients. Intronic and exonic mutations in DES gene and exonic mutations in TTN and CRYAB represent novel variants.
Conclusion: The study highlights the diverse clinical manifestations and genetic mutation patterns in Indian patients of MFM. Majority had DES gene mutations. Few patients with congenital ocular or bulbar weakness resembling congenital myasthenic syndrome had intronic DES mutations. The study features the significance of NGS in diagnosis, identifying novel variants and expanding the genotype-phenotype correlation associated with MFM in Indian population.
eP02.06.08Muscular Dystrophies Due to Collagen VI Mutations
Coelho J1, Moldovan O2, Saianda A3, Ferreira R3, Moreno T1
1Neurology Unit, Department of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisbon, Portugal, 2Genetic Department of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisbon, Portugal, 3Pulmonology Unit, Department of Pediatrics, Hospital Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Lisbon, Portugal. Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
Introduction: Mutations in Collagen VI are associated to Congenital Muscular Dystrophies both Ulrich and Bethlem phenotypes. Recessive and dominant forms are possible and more than classical presentations, a broad clinical spectrum is possible. We aimed to analyze the clinical, pathologic, and genetic characteristics of patients with collagen VI-related myopathy followed in our tertiary care hospital from the last 15 years (2006-2021).
Results: We analyzed 5 patients, all females; from different families and origins (4 from Portugal, one from Guiné); the median age is 10,4 years (5-19 years). Four of them presented with neonatal hypotonia and the other patient started symptoms in the first year of life. Those who had neonatal hypotonia showed joint contractures (from arthrogryposis to club feet). One had also a congenital torticollis. All have proximal weakness with distal hyperlaxity and all but one have cutaneous alterations and calcaneus fat pads. Our patients present pathogenic alterations in all collagen VI associated genes (3 were COL6A1, one COL6A2 and one COL6A3). Only one is on non-invasive ventilation. 2 patients lost walk capacity, one at 7 years and the other around 10 years.
Conclusions: Despite neonatal presentation, in most of our patients, the diagnosis came several years after the onset of symptoms. The clues for clinical diagnosis are proximal weakness, distal hyperlaxity and cutaneous alterations. Genetic approaches are becoming easily available and will permit earlier diagnosis and definition of new phenotypes.
eP02.06.09Proteomics Changes in Response Voluntary Exercise in a Murine Model of Myotonic Dystrophy Type 1
Manta A1, Ljubicic V2, Spendiff S3, Roos A3,4, Lochmuller H3,5,6,7,8
1Faculty of Medicine, University of Ottawa, Ottawa, Canada, 2Department of Kinesiology, McMaster University, Hamilton, Canada, 3Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada, 4Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Essen, Germany, 5Department of Neuropediatrics and Muscle Disorders, Medical Center -University of Freiburg, Faculty of Medicine, Freiburg, Germany, 6Centro Nacional de Análisis Genómico (CNAG-CRG), Center for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona, Spain, 7Division of Neurology, Department of Medicine, The Ottawa Hospital, University of Ottawa, Ottawa, Canada, 8Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada
Myotonic dystrophy type 1 (DM1) is a genetic neuromuscular disorder, caused by the expansion of the CUG trinucleotide repeat in the untranslated region of the DMPK gene. Clinical manifestations include skeletal muscle weakness, wasting, myotonia, and insulin resistance. Exercise training studies with DM1 participants demonstrate that exercise is safe, well tolerated and elicits modest clinically improvements in muscle strength and function. The molecular mechanisms underlying the therapeutic benefit of exercise in DM1 skeletal muscle has only recently been investigated. Proteomic surveys of skeletal muscle allow for high-throughput evaluation of the beneficial adaptations to both chronic and acute exercise interventions. More recently, this technique has begun to be employed to study exercise adaptations in various chronic diseases to further our understanding of the pathophysiology and perturbed signalling pathways. The purpose of this study is to analyze the proteomic response in a murine model of DM1 in response to 6-8 weeks of voluntary wheel running. Three groups of mice (3-6 months old) were utilized: i) sedentary human skeletal actin-long repeat (HSA-LR, a known rodent model of DM1) mice (SED-DM1), ii) HSA-LR mice with volitional access to a home cage running wheel for 6-8 weeks (EX-DM1), and iii) sedentary wild-type mice (WT). As previously published, our EX-DM1 mice demonstrated improvements in strength and muscle function compared to the SED-DM1, to the levels of WT mice. This was complimented with improvements in hallmarks of the disease pathogenesis: a reduction in the sequestration of RNA splicing regulator, Muscleblind-1, and the associated missplicing of target pre-mRNA. The goal of the following study is to highlight the particular pathways that augment these beneficial adaptations. Moreover, we are interested in investigating canonical exercise pathways that may not be upregulated in DM1 biology when compared to available proteomic datasets in healthy wildtype mice that underwent chronic training. This study’s preliminary results are part of the first study to comprehensively investigate the molecular responses and adaptations to exercise in DM1 skeletal muscle.
eP03.01.01Validation of the Individualized Neuromuscular Quality of Life Questionnaire in Korean Patients with Neuromuscular Diseases
Lee S1, Han H1, Suh B2, Choi Y1, Rose M3, Park H1
1Gangnam Severance Hopspital, Gangnam-gu, Republic of Korea, 2Kangbuk Samsung Hospital, Jongro-gu, Republic of Korea, 3King’s college Hospital, London, Republic of Korea, 4Ewha Womans University Mokdong Hospital, Seoul, Republic of Korea
Background and Purpose: The Individualized Neuromuscular Quality of Life (INQoL) questionnaire is a widely used measure of quality of life in patients with neuromuscular diseases. The purpose of this study was to translate and validate the Korean version of the INQoL questionnaire in Korean patients with neuromuscular diseases.
Methods: We translated the original version of the INQoL into Korean and applied language adaptations. Internal consistency, known-group validity, and test-retest reliability were also assessed. Validity was measured using the following methods: 1) construct validity using the modified Rankin Score (mRS) and the manual muscle testing (MMT)-sum score, and 2) concurrent validity with the 36-Item Short Form Survey (SF-36) questionnaire.
Results: Because of linguistic and cultural differences between English and Korean, there are several difficulties in translation. Translating ‘myotonia’ was difficult because there is no such terminology in Korean that is familiar to the general public. Moreover, there were some cultural differences in the selection of the terms, such as ‘at the moment’ and ‘in the face of my condition’ in Korean, but these were not practical and did not cause misunderstandings in the meaning of each item. A total of 193 patients (141 males and 52 females) with genetic neuromuscular diseases were enrolled in this study. Among the 193 patients, 175 (91%) and 18 (9%) had genetic myopathy and neuropathy, respectively. The most common disease was facioscapulohumeral muscular dystrophy type I in 27 patients, followed by myotonic dystrophy in 24, dysferlinopathy in 20, Becker muscular dystrophy in 17, and spinal muscular atrophy in 9. Twenty-six patients (14%) were not genetically confirmed. The median age at symptom onset and examination was 18.0 (interquartile range: 8.0–30.0) and 38.0 (interquartile range: 30.5–46.0) years, respectively. Invasive or non-invasive mechanical ventilators were used in 28 patients (15%). The median mRS and MMT-sum scores were 2 (interquartile range: 1.5–3.0) and 230.0 (interquartile range: 176.0–270.0), respectively. Twenty patients (16 males and 4 females) completed the INQoL questionnaire twice within 6 months. All patients had genetic myopathy. The coefficients for internal consistency (Cronbach’s α = 0.871 to 0.933) and test-retest reliability (Spearman’s ρ 0.453 to 0.886) were adequately high in all subscales, except the ‘treatment effects’ dimension. The INQoL subscales, with the exception of locking, droopy eyelids, double vision, and swallowing difficulties, were significantly associated with their relevant SF-36 domains (Spearman’s ρ -0.274 to -0.833). Functional status and muscle strength were most strongly associated with independence (Spearman’s ρ = 0.753; p < 0.001 for mRS, and Spearman’s ρ = -0.741; p < 0.001 for MMT-sum score).
Conclusions: The Korean version of the INQoL questionnaire is a reliable and validated measurement tool for Korean patients with neuromuscular diseases.
eP03.01.02Improving Clinical Trials in Neuromuscular Diseases: The TREAT-NMD Advisory Committee for Therapeutics (TACT)
Turner C1, Straub V1, Robertson L2, De Luca A3, Aartsma-Rus A4
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle Upon Tyne, United Kingdom, 2TREAT-NMD Services, Newcastle Upon Tyne, United Kingdom, 3Dipartimento di Farmacia, University of Bari, Bari, Italy, 4Leiden University Medical Center, Leiden, Netherlands
Established in 2009 as part of TREAT-NMD, TACT is an expert multidisciplinary body that provides the neuromuscular community with independent, confidential and objective guidance on advancing new therapies for rare neuromuscular diseases.
TACT’s aim is to position potential therapies along a realistic and well-informed pathway to or through clinical trials, and eventual registration. This is achieved by a thorough, independent, confidential and expert review resulting in detailed advice. TACT believes that this leads to de-risked, better designed and conducted studies that generate more meaningful data and have increased potential to be funded longer term.
Applications for advice are reviewed by a bespoke panel of world-leading experts drawn from the TACT committee of around 70 members in response to the specific needs of a particular application.
To date TACT has reviewed over 70 applications from both academic investigators and industry across all NMDs. However, the majority have come from those working in Duchenne muscular dystrophy (DMD) therapy development. TACT works closely with and is generously supported by major DMD patient organisations around the world to ensure the process assists the community as a whole.
Feedback shows that TACT has generated recommendations that have greatly helped investigators, including industry, in evaluating their potential compounds and therapy development programmes. It has encouraged applicants to consider these in a methodical fashion with clear go/no-go decisions and with optimal use of funding and resources. Examples of the impact of TACT in DMD are given in this poster as well as details of how organisations can access TACT advice.
eP03.01.03Impact of Independence in Mobility on Independence Outdoors in People With Neuromuscular Conditions
Wong K1, Sodhi J1, Moat D1, McCallum M1, Richardson M1, James M1, Mayhew A1, Mason J1, Straub V1, Marini-Bettolo C1, Muni-Lofra R1
1The John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
Background: Neuromuscular conditions are characterised by progressive muscle weakness, which may result in loss of ambulation. Capturing patient quality of life with an appropriate outcome measure, alongside functional measures can help to gain a better understanding of the impact of mobility status on quality of life. The Quality of Life Measure for people with slowly progressive and genetic neuromuscular disease (QOL-gNMD) is a validated questionnaire specifically designed for people with neuromuscular conditions, which explores different aspects of quality of life. In this study, we focus on the impact of disease on general perception in health, quality of life and outdoor mobility.
Aims: To understand how different levels of mobility may impact on patients’ perceived health, quality of life, falls risk, and ability to leave the house independently.
Methods/Materials: Patients with a neuromuscular condition were asked to fill in the QOL-gNMD questionnaire, prior to attending their routine clinical appointment at the John Walton Muscular Dystrophy Research Centre. Mobility scores were obtained as part of the clinical assessments.
Results: A total of 184 patients were included in the study. Patients who are still ambulant but require the use of walking aids reported the worst perceived general health (70% reported moderate/ poor health), compared to those who can walk unaided or use a wheelchair (47% and 44% reported moderate/ poor health). Reported overall quality of life is also the worst in those who use walking aids (78% reported moderate/ poor quality of life), but better in those who walk unaided or use a wheelchair (44% and 38% reported moderate/ poor quality of life). In terms of outdoor mobility, 70% of patients who use walking aids reported that they are unable, or only occasionally able to leave their home independently. This improves in wheelchair users (62%) and in those who can walk unaided (32%). It was also found that all patients (100%) who use a walking aid avoid certain places due to risk of a fall. Those who can walk unaided or use a wheelchair are less likely to avoid venues (59% and 56% reported avoidance due to falls risk).
Conclusion: Wheelchair users reported better health, quality of life and outdoor mobility, compared to those who use walking aids to walk. The results of this study suggest that timely discussions regarding the introduction of wheelchair use and early implementation of therapy support to assess fall’s risk may help to improve quality of life in people with neuromuscular conditions. Further studies are required to understand the different factors that influence the introduction of supportive care in neuromuscular diseases.
eP03.01.04Neuropsychological and Behavioral Profile in a Cohort of Becker Muscular Dystrophy Pediatric Patients
Tosi M1, Cumbo F1,2, Catteruccia M1, Diodato D1, Nicita F1, Grimaldi Capitello T2, Alfieri P3, Vicari S3,4, Bertini E1, D’Amico A1
1Unit of Muscular and Neurodegenerative Disorders, Department of Neuroscience, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy, 2Unit of Clinical Psychology, Department of Neuroscience, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy, 3Child and Adolescent Psychiatric Unit, Department of Neuroscience, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy, 4Department of Life Sciences and Public Health, Università Cattolica del Sacro Cuore, Rome, Italy
INTRODUCTION: Cognitive and executive function impairment as well as the association between executive functions and dystrophin gene mutation position have been widely investigated in individuals with Duchenne muscular dystrophy (DMD), whereas few studies explored these functions in Becker muscular dystrophy (BMD) patients. The aim of this study was to investigate the neuropsychological and behavioral profile in a cohort of BMD patients and whether there is any correlation with site of DMD gene mutation.
METHODS: This is a single-center, observational, prospective, cross-sectional study in which a full neuropsychological assessment, including intellectual functioning, executive functions, and language abilities, was performed in children and adolescents affected by BMD without cognitive impairment. A comparison between groups based on site of mutation or IQ level was attempted.
RESULTS: 22 patients were enrolled. The mean IQ in the whole cohort was 94 (range 71-122). Overall, the patients in our cohort did not perform well in tests investigating the executive functions. No statistically significant difference was found in groups stratified by site of mutation or IQ level.
DISCUSSION: This study confirms BMD patients have a risk of impairment of the executive functions, despite having a normal IQ in most cases. This is a very important aspect, as it puts them at risk of developing learning disabilities.
eP03.01.05Evolution of Gait Parameters and Influence of Orthotics on Gait in a Patient With Dysferlinopathy
James M1, Alcock L2, Straub V1, Mayhew A1
1The John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom, 2Clinical Ageing Research Unit, Campus for Ageing and Vitality, Newcastle University, Newcastle upon Tyne, United Kingdom
Background: Dysferlinopathy (LGMDR2/2B), caused by mutations in the DYSF gene, presents with a broad spectrum of muscle weakness and variable rates of progression. Patients with LGMDR2 have a distinct gait pattern although this has not been quantified using instrumented gait analysis. Gait analysis is a novel outcome measure introduced into the Clinical Outcome for dysferlinopathy II natural history study (COSII, http://www.jain-foundation.org/dysferlinoutcomestudy) to determine its utility as a clinical biomarker. A reduction in walking speed indicates functional decline. Clinically, lower limb orthoses may be prescribed to improve walking, although limited data exist to examine their impact on gait parameters in patients with LGMDR2.
Aim: This case study aims to quantify the temporospatial characteristics of gait and change in those over one year of one COSII individual with LGMDR2 using instrumented gait analysis; and to compare gait when walking with and without ankle orthoses.
Methods: Using the GAITrite (240Hz) instrumented walkway, we quantified six temporospatial features of gait whilst our COS II patient, a 30 year old male, walked barefoot and then in ankle orthoses with footwear. Temporospatial characteristics including gait velocity, step length, step width, stance and swing duration were extracted in addition to gait asymmetry and variability. The patient was asked to walk at self-selected preferred and fast gait velocities and complete up to 6 trials in each condition. Wilcoxon signed rank tests were used to evaluate statistical differences (p<.05).
Results: Gait characteristics were compared between conditions and over time. At baseline the subject’s preferred walking speed barefoot (1.16m/s)and shod (1.14m/s) was slow compared to an unaffected population, and only when asked to walk as fast as safely possible did he achieve a gait speed (1.4m/s) reported as that required for community ambulation1. The GAITrite was able to capture differences in temporospatial characteristics of walking when the patient was walking with or without lower limb orthoses, particularly step width which improved when using a silicon ankle foot orthoses.
Conclusions: This case study objectively characterises the gait patterns of a patient with LGMDR2 with and without lower limb orthoses. Future analysis aims to describe gait in a wider population of COSII patients and examine the indications for successful lower limb orthotic prescription in LGMDR2.
References
1. Middleton A, Fritz SL, Lusardi M. Walking speed: the functional vital sign. J Aging Phys Act. 2015;23(2):314-22.
eP03.01.06Predictors of Gait in Patients with Late-Onset Pompe Disease
Maulet T1,2,3, Cattagni T5, Dubois F3, Laforet P1,4, Bonnyaud C2,3
1End: Icap Laboratory, Inserm Unit 1179, Uvsq, France, Versailles, France, 2Paris-Saclay University, UVSQ, Research Unit ERPHAN, 78000, Versailles, France, Versailles, France, 3Motion analysis laboratory, Functional Exploration Department, Raymond Poincaré Hospital, APHP, Garches, France, Garches, France, 4Neurology Department, Nord/Est/Ile de France Neuromuscular Center, Raymond-Poincaré Hospital, Garches, France, Garches, France, 5Nantes Université, Movement - Interactions - Performance, MIP, UR 4334, F-44000 Nantes, France, Nantes, France
Background: Gait disorders concern 87% of patients with late-onset Pompe disease (LOPD), impacting their daily activities. Patients report gait instability and, a decrease in walking speed is observed. The relationships between gait disorders and muscle weakness remain poorly explored. The only study assessing jointly walking and muscle strength in LOPD used a therapist-dependent measure for strength (held-hand dynamometer) and a walking description in 4 categories (unable to walk, walking with aids, waddling gait without aids, normal gait). It is essential to explore relationships between gait parameters and lower-limb strength of each muscle group with the gold standard tools to increase knowledge on motor disorders in LOPD, to highlight new gait biomarkers and to guide assessments.
Objectives: We aimed to determine in LOPD patients 1) which muscle groups are the most important contributors to gait speed and stability during gait, 2) which kinematic gait parameters are the most important contributors to gait speed and, 3) whether isokinetic dynamometer measures are most important predictors of gait parameters than manual muscle testing (MMT).
Method: Eighteen LOPD patients (58.3 ± 12.2 years) performed gait at comfortable speed (3D analysis) and, 3 trials of maximal voluntary contraction for 6 lower-limb muscle groups (isokinetic dynamometer and MMT). We first used correlations between muscle strength (hip, knee, and ankle flexors and extensors, hip adductors and abductors) and gait parameters (joint kinematics and spatiotemporal parameters) to select variables for the multiple regressions. Variables significantly correlated were included in the stepwise models to highlight explanatory parameters of 1) gait speed with strength, 2) gait speed with kinematics, 3) single support during gait with strength.
Results: Concerning the muscular contributors to gait speed, hip abductors strength is the main predictor with 64% of variance explained (p=0.001). The variance prediction increased to 73% (p=0.001) by adding ankle dorsiflexors strength. The percentage of single support during gait was the main kinematic predictor of gait speed, with 81% of variance explained (p=0.001), followed by step length and cadence (98% variance explained with the three). Concerning stability, hip abductors strength was the only predictor of percentage of single support with 44% of variance explained (p=0.004). The MMT score was poorly to moderately correlated with the gait parameters (r between 0.49 and 0.62, p <0.05) while the isokinetic dynamometer results were moderately to strongly correlated (r between 0.57 and 0.77, p <0.05). MMT scores were not predictive of gait speed.
Discussion: This is the first study exploring the relationships between muscle weakness and gait biomechanics in LOPD patients. We showed that abductors strength is the major contributor to gait instability, which is known in other populations. Interestingly, the major contribution of the abductors to gait speed appears a specificity of LOPD. To conclude, it is essential in LOPD to systematically assess the hip abductors strength with isokinetic dynamometers and standardized methods, considering its importance in gait.
eP03.01.07Simplified and Non-invasive Optical Motion Tracking of Respiratory Dynamics in the MDX Mouse Model.
Svetlove A1, Albers J2, Alves F2, Dullin C2, Zschüntzsch J2
1Max-Planck-Institute For Multidisciplinary Sciences, Göttingen, Germany, 2University Medical Centre Göttingen, Göttingen, Germany
Duchenne muscular dystrophy (DMD) is the most common x-chromosomal inherited dystrophinopathy which leads to progressive muscle weakness and a premature death due to cardiorespiratory dysfunction. The mdx mouse lacks functional dystrophin protein and has comparatively human-like diaphragm phenotype. Moreover, respiratory muscle weakness is a known consequence of many other neuromuscular diseases which portray abnormal breathing patterns and changes in respiratory dynamics. This observation makes monitoring of abnormal breathing patters an attractive analysis strategy to develop a potential biomarker for neuromuscular diseases such as DMD. We aimed to establish a sensitive, reliable, harmless and easy way to assess the reduction in breathing capability based on observed respiratory muscle weakness and subsequent irregularity in breathing pattern. Optical respiratory dynamics tracking (ORDT) was developed utilising a single view camera facing the chest of the mouse that tracks the movement of paper markers placed on its thoracic-abdominal region. For the standard ORDT the breathing motion of anaesthetised mice was recorded by a camera (Basler acA640-90gc, Pentax Cosmicar TV Lens 25mm 1:1.4) acquiring images with 600 by 400 pixels for over 10 seconds at 100 frames per second (fps) in 8 bit grey value format. Paper markers with a printed black cross-hair pattern (3 mm side length and 1 mm centre diameter) were used. The markers in the video feed were located by a Laplacian of Gaussian particle detection and tracked by Linear Assignment Problem algorithms implemented in the TrackMate plugin for ImageJ. Karhunen-Loève transformation implemented in Python 3 was used to reorient the data in the direction of the strongest displacement. Thus, subsequent analysis was done on a simple 1D oscilation function over time. Parametrization of this function was done using the established XLF software. Wt mouse ORDT traces displayed an active breaking of the lung recoil during expiration which was absent in the mdx animals due to known weakening of the diaphragm. ORDT successfully distinguished diseased mdx phenotype from healthy controls. In conclusion we believe that this easy method has merit in detecting and monitoring of various neuromuscular disorders and has great potential for patient translatability.
eP03.01.08Serum Neurofilament Light Chain as a Marker of Nervous System Damage in Myopathies
Schaefer J1, Benkert P2, Akgün K1, Ziemssen T1, Jackson S1, Saak A1
1Uniklinikum C.G.Carus, Dresden, Germany, 2University Hospital Basel, Basel, Switzerland
Background: Neurofilament light chain in serum (sNfL) has been suggested as a biomarker for the assessment of neuroaxonal damage. Since NfL are not expressed in muscle, elevated sNfL in patients with primary myopathies suggest additional involvement of the nervous system. To verify this hypothesis, we measured sNfL in a series of patients with myopathies.
Methods: sNfL were determined in 62 patients with molecular proven primary myopathies in whom some nervous system involvement may be predicted: myotonic dystrophy type I and II (DM I, II) and mitochondrial disease. In addition, sNfL were measured in 8 patients with facioscapulohumeral muscular dystrophy (FSHD) and in a disease control group caused by genetic defects exclusively expressed in muscle.
Results: sNfL values were significantly elevated in the DM I, the DM II and the mitochondrial group, with FSHD patients showing the lowest sNfL elevations. sNfL levels in the disease control group were not different from the healthy controls. A significant correlation between repeat length and sNfL levels was found in the DM I patients, but not in the DM II patients. Mitochondrial patients with encephalopathy showed significantly higher sNfL concentrations compared to patients with only muscular symptoms.
Conclusions:sNfL levels are elevated in myopathies with, based on the underlying molecular defect or clinical features, established nervous system involvement, i.e. myotonic dystrophies and mitochondrial disorders. sNfL were also raised in FSHD, where involvement of the nervous system is not usually clinically apparent. Thus, the lack of sNfL elevations in patients from the disease control group (myopathies due to mutations in genes exclusively expressed in muscle tissue) supports the concept of sNfL as a simple and non-invasive biomarker to detect to detect and monitor neuronal damage in myopathies, even in those without clinically evident CNS-involvement.
eP03.01.09Troponin T in Spinal and Bulbar Muscular Atrophy
Musso G1, Sorarù G2, Mion M1, Zaninotto M1, Bello L2, Pegoraro E2, Plebani M1,3, Basso D1,3
1Department of Laboratory Medicine, University-Hospital of Padova, Padova, Italy, 2Department of Neurosciences, University of Padova, Padova, Italy, 3Department of Medicine-DIMED, University of Padova, Padova, Italy., Padova, Italy
Introduction: Cardiac isoforms of troponins are essential and highly specific components of cardiac muscle. Cardiac troponin I (cTnI) and troponin T (cTnT) are currently fundamental diagnostic biomarkers of acute myocardial injury, as they are immediately released in blood following tissue damage. Their elevation in blood is promptly detected with high sensitivity methods and correlates with extension of myocardial damage.
Elevation of cTnT has been also reported in a wide range of neuromuscular disorders (Rittoo D, 2014). Recently, a systematic study has been conducted in amyotrophic lateral sclerosis (ALS) (Castro-Gomez S, 2021) showing an increase of cTnT that correlates to disease severity, with cTnI levels always within normal limits.
In spinal and bulbar muscular atrophy (SBMA) a well-established muscular biomarker as creatin kinase (CK) was reported highly increased compared to controls, but not correlating to clinical measures (Lombardi, et al., 2019).
Methods: In this retrospective study we selected 10 male patients (median age 52.5 years) from a cohort of genetically confirmed SBMA and 5 male patients (median age 41 years) with genetically confirmed diagnosis of spinal muscular atrophy (SMA) type 3, each patient attending a regular clinical follow-up at Neuromuscular Clinic of University of Padova. SBMA disease severity and progression was evaluated with Spinal and Bulbar Muscular Atrophy Functional Rating Scale (SBMAFRS).
Serum cTnT and cTnI concentrations were measured using high sensitivity (hs) assays: Roche Diagnostics Troponin T hs STAT (99th percentile upper reference limit 14 ng/L) and Abbott Architect Stat High Sensitive Troponin I assay (99th percentile upper reference limit 16 ng/L for female and 34 ng/L for male). Serum CK was measured with Roche Diagnostics assay (reference range 20-180 U/L).
Correlation of cTnT and cTnI with SBMAFRS scores was evaluated using Spearman correlation coefficients (Analyse-it Software).
Results: cTnT was above the reference range in all SBMA patients (median 43.5 ng/L, measured range 22-167), while elevated values were observed in only one SMA (23 ng/L, median in SMA patients 12 ng/L, measured range 7-23); cTnI was within reference range in each sample tested (measured range <2-25.3 in SBMA and <2-3.7 in SMA). Median CK was 1320 U/L in SBMA and 385 in SMA (measured range 124-2983 versus 128-1032 U/L)
No correlation was found for cTnT nor cTnI with SBMAFRS (p=0.8277 and p=0.3818 respectively).
Conclusion: In this preliminary study we specifically demonstrated for the first time that serum levels of cTnT, but not cTnI, increase in SBMA, its pattern being close to that of CK. Disease severity as measured with SBMAFRS does not correlate to cTnT. Further studies should address the biological re-expression of cTnT in injured adult skeletal muscle and increased levels of cTnT should be cautiously evaluated in these patients when myocardial injury is suspected.
eP03.01.10Urinary Titin as a Biomarker of Myotonic Dystrophy Type 1
Varga D1, Perecz B1, Sipos A1, Fülöp K2, Pál E1,2
1University of Pécs, Department of Neurology, Pécs, Hungary, 2University of Pécs, Department of Pathology, Neuropathology Unit, Pécs, Hungary
Myotonic dystrophy is one of the most common autosomal dominant inherited muscular diseases in adulthood. We tested urinary titin N-fragment/urinary creatinine ratio as a biomarker, in 30 patients with myotonic dystrophy type 1 (DM1) and compared to 30 healthy controls. Clinical data, such as strength, blood creatine kinase level and DM1-related outcome measures were correlated. Titin was significantly elevated in urine samples of DM1 patients compared to controls and was related to clinical stages according to MIRS. Titin was related to the patients’ age, handgrip strength and DM active score. Urinary titin seems to be a sensitive biomarker in different neuromuscular disorders, including DM1.
eP03.01.11InGene 2.0: An Intelligent Technological Approach to Genotype-Phenotype Relationship Study in Rare Neuromuscular Diseases
Tonacci A1, Sansone F1, Conte R1, Roccella S1, Santorelli F1, Siciliano G1, Matà S1, Malandrini A1, Tupler R1, Angelini C1
1Institute Of Clinical Physiology, National Research Council Of Italy (ifc-cnr), Pisa, Italy
Rare Neuromuscular Diseases (NMD) include a wide range of clinical conditions, characterized by common hallmarks and often subtle differences, making their differential diagnosis and characterization often tricky even for the expert clinicians.
In recent years, noteworthy technological developments have allowed the clinician to go into depth of the clinical characteristics in a disease or, more specifically, in an individual, while researchers found benefits in having the chance to investigate the links between the genotype and the clinical phenotype of the conditions studied, retrieving relationships once hard to even be figured out.
In this framework, the InGene 2.0 project, supported by the Tuscany Region (Italy), allowed the implementation of a smart, highly customizable, multimodal platform, composed of software and hardware components, to this end. The platform, named “Health 360” to give an overview of the health status of an individual at 360 degrees, was designed to be used by the clinician to collect and safely store, with GDPR compliance, different kinds of data to characterize the affected individuals. At the same time, the tool is also conceived to be used by data scientists as it includes some basic Machine Learning facilities, expected to be expanded soon at the platform level.
The platform, as reported above, conceived as a modular tool to be easily configurable and scalable upon the needs of different end-users and trials or projects, actually consists of several modules. They include Genostore, devoted to the collection of genetic data, MRIndex, aiming to store and analyze, through Artificial Intelligence (AI), muscular MRI files, NeuroExam, devoted to the administration of neurological examination, PhysioTest, whose accessible user interface allows performing physiotherapy assessments, including the Six-Minute Walk Test and beyond. In addition, the InGene module, actually under upgrading phase, carries on the AI algorithms, mainly based on Machine Learning methodologies, to retrieve basic correlations between genotype and clinical phenotype of groups of individuals. Further modules are actually under construction, including one, named AUTOMA 2.0, to retrieve data from wireless wearable sensors (i.e. force sensors, joints angles, EMG, ECG, respiration), smartphones and Internet-of-Things (IoT), and one to analyze muscular biopsy records through AI, similarly to what is currently used within the MRIndex.
The experimental phase has been carried out in four clinical centers in Tuscany, Italy (IRCCS Stella Maris Foundation, University Hospitals in Pisa, Firenze Careggi and Siena), and Health360 is actually hosting data from more than 230 patients with rare NMDs treated in the four centers, with the target to increase this number to 500 by 2023. This further scaling up could probably help bringing Health360 at the forefront of clinical data collection in rare NMDs, making it a reference tool for genotype/phenotype association in NMDs to the benefit of both clinicians and researchers.
eP03.02.01Coexistence of Charcot-Marie-Tooth Disease and Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Vukojevic Z1
1Clinic of Neurology, University of Banja Luka, University Clinical Centre of Republic of Srpska, Faculty of Medicine, Bosnia and Herzegovina, Starine Novaka 44 78252 Trn, Laktaši, Bosnia and Herzegovina
Introduction: Charcot-Marie-Tooth disease (CMT) is the most common inherited polyneuropathy. Symptoms usually appear between age 5 and 25, and the disease is slowly progressive. The most important symptoms are muscle weakness and atrophy of the feet, lower part of the legs, hands, and forearms. Foot deformities (hammertoes, high arches) and changes in sensation (“stocking-glove” pattern) are common. Over the course of this polyneuropathy, other forms of polyneuropathies can occur and in these cases the symptoms and signs are more pronounced. It is particularly important to diagnose associated polyneuropathy especially the ones that could be treated. One such polyneuropathy is chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) – an acquired chronic immune-mediated demyelinating polyneuropathy. Patients are presented with muscle weaknesses and changes in sensation, but they occur significantly faster than with CMT. CIDP is a treatable polyneuropathy, the most important drugs are intravenous immunoglobulins (IVIg), corticosteroids and plasma exchange.
Case report: A 19-year-old female patient was admitted to hospital due to weaknesses in legs and arms and difficulty walking. When she was 14 years old she experienced mild weaknesses in the feet. Over the next few years, muscle weaknesses progressed to distal parts of the legs and hands. She was diagnosed with CMT type 1 (CMT1). The diagnosis was confirmed by genetic testing. Other possible causes of polyneuropathy were excluded. Six months before the current hospitalization, she was able to walk 3-4 km, and had a slight weakness in the hands. Then, suddenly the weakness begun to worsen. Walking becomes more and more difficult, she was able to walk only 200-300 meters. Neurological examination revealed: moderate weakness in the arms, severe weakness in the legs, more pronounced distally. Mild hypotrophy of the muscles of the hands, and moderate in the feet and lower part of the legs. Areflexia. Changes in sensation (“stocking-glove” pattern). Cerebrospinal fluid analysis: elevated protein levels (1.10 g/l), normal cell counts. Electroneurography (ENG) examination was compared with previous examinations and showed tipycal finding for CIDP (low motor and sensory nerve conduction velocity, abnormal temporal dispersion of the CMAP, motor conduction blocks). Additional tests were used to exclude other polyneuropathies. The patient was diagnosed with CIDP associated with CMT1. She was treated with IVIg for 5 days (0.4 g/kg per day). A significant recovery began, which continuied over following months. Over the next six months, she was treated once a month with IVIg 0.4/kg for one day. After six months, she could walk for 2 km. Strength in the hands is significantly better. In the following period, it is planned to continue treatment with IVIg once a month.
Conclusion: CMT1 is an inherited sensorimotor polyneuropathy that has a mild progressive course. If the course of the disease is unusually rapid, and the weaknesses begin to progress significantly faster, we have to consider the association with other type of polyneuropathy. CIDP is the most common possible associated polyneuropathy. It is very important to diagnose CIDP because it is a treatable polyneuropathy.
Keywords: Charcot-Marie-Tooth Disease, chronic inflammatory demyelinating polyradiculoneuropathy
eP03.02.02HINT1-Related Autosomal Recessive Axonal Neuropathy With Neuromyotonia (ARAN-NM). Presentation of a Greek Pedigree
Moschou M1, Poulidou V1, Liouta E1, Notas K1, Gavriilaki M1, Kimiskidis V1, Arnaoutoglou M1
11st Neurology Department, AHEPA University Hospital, Thessaloniki, Greece
Background: Autosomal recessive axonal neuropathy with neuromyotonia (ARAN-NM) related to mutations in HINT1 gene is a rare form of hereditary neuropathy. Probands presented with motor greater than sensory neuropathy of various onset, accompanied by muscle stiffness and cramps in the limbs. Furthermore, they displayed non-classical symptoms, including pain in the extremities and signs of central nervous system involvement
Case report: We report two patients, siblings, who presented at puberty with myotonia, gait impairment and loss of upper limb dexterity. Disease progression included progressive distal muscle weakness and atrophy, neuromyotonia and foot and hand deformities. Electrophysiological studies showed axonal motor neuropathy and neuromyotonic discharges. Using Next-generation sequencing a homozygous mutations with two pathogenic variants, c.110G>C (p.Arg37Pro) (homozygous), were identified in HINT1 gene. ARAM_NM related to HINT1 mutation as a known cause of hereditary neuropathy is considered to be quite rare in Greece, as only 4 known cases have been reported. Our study also includes a review of the spectrum of clinical and neurophysiological manifestations (frequent and rare) in relations to other HINT1 gene mutations and in comparison to our patients.
eP03.02.03The Importance of Functional Assessment In Hereditary Sensory and Autonomic Neuropathy
Castaño Herrera L1, Salazar Lengua C3, Ruiz Ospina E1, Castellar Leones S1, Correa Arrieta C2, Ortiz Corredor F1, Sánchez D2
1Universidad Nacional De Colombia, Bogotá, Colombia, 2Instituto de ortopedia infantil Roosevelt, Bogotá, Colombia, 3Universidad de la Sabana, Bogotá, Colombia
INTRODUCTION: Hereditary sensory autonomic neuropathy (HSAN) is a clinically heterogeneous group characterized by significant sensory and autonomic involvement. The classification of HSAN is based on inheritance mode, genetic mutation, and phenotype. The diagnosis of these conditions is important to continue advances in research regarding phenotypic characterization and the development of biochemical and genetic advances in the era of precision medicine. Although most hereditary neuropathies do not have disease-modifying treatments, we present below a clinical case of a potentially treatable neuropathy.
CLINICAL CASE: 44-year-old man onset 8 years of bilateral sensory loss in feet and hands, associated with frequent stumbling and ulcer in the first toe joint of the left foot. Physical examination showed hyposensitivity and impaired vibratory and cold thermal sensitivity in stocking-glove pattern, painless ulcer, edema in the first toe of the left foot, pes cavus, and bilateral hammertoes. The electrodiagnostic study reports chronic sensorimotor axonal polyneuropathy with greater involvement in lower limbs. Nuclear magnetic resonance reports osteomyelitis of the large left knuckle. Genetic studies report a heterozygous mutation in the SPTLC1 gene: c.1036G>C (p. Ala346Pro). A hereditary sensory autonomic polyneuropathy type IA was diagnosed. Supplementation with high doses of serine is indicated. Hearing and ophthalmological studies are requested to rule out superimposed sensory compromise. Quantitative sensory testing and neuropathic questionnaries are indicated for follow-up and evaluation of response to treatment.
DISCUSSION: we present a patient with a mutation in the serine palmitoyl transferase long-chain 1 subunit (SPTLC). This mutation is present in most of patients with HSAN. Early diagnosis depends on physical examination, detailed sensory and autonomic evaluations, and genetic studies for hereditary polyneuropathies. The identification of specific mutations has allowed diagnostic confirmation according to the phenotype. As in the clinical case, pupillary anomalies, loss of the corneal reflex, deafness, restless legs, cramps, hyporeflexia, tongue lesions, osteomyelitis, amputation and sepsis have been described, OMIM*605712. This mutation reduces the affinity of the enzyme for L-serine and leads to the accumulation of neurotoxic deoxysphingolipids. Understanding this mechanism has allowed the initiation of biologically rational and potentially disease-modifying therapy with L-serine at 400mg/kg/day. The need to complement the evaluation with neuropathic tests would help characterize and monitor in the context of a variant that has recently been associated with pathogenicity. As mentioned by Fridman, et al. in which they perform follow-up with the Charcot-Marie-Tooth Neuropathy Score version 2 (CMTNS) test, finding continuous improvement after second year of treatment.
CONCLUSION: HSAN represent a diagnostic challenge for clinicians. Lack of evidence on disease-modifying treatments often leads to frustration for patient and treating physician. The case presented represents a type of neuropathy that has a potential treatment with L-serine that could work as a modifier of the disease. It is important to emphasize to the physiatrist the need to initiate therapeutic interventions and perform functional evaluation. This has allowed the characterization of the variability of the different phenotypes and has allowed a better focus on diagnosis, follow-up and measurement of impact on patients.
eP03.02.04Dose-Exposure-Efficacy Response Relationships for Intravenous Immunoglobulin,10% in Patients with Multifocal Motor Neuropathy
Li Z1, Roepcke S2, Franke R2, Yel L1
1Takeda Development Center Americas, Inc., Cambridge, United States, 2Cognigen, a division of Simulations Plus, Buffalo, United States
Background: Despite widespread use of intravenous immunoglobulin (IVIG) to treat patients with multifocal motor neuropathy (MMN), dose-exposure-efficacy response relationships for IVIG in patients with the disease are not well understood.
Objective: To assess relationships between IVIG,10% dose, serum total IgG levels and the efficacy measure of grip strength in patients with MMN using population pharmacokinetic/pharmacodynamic (popPK/PD) modeling.
Methods: A popPK/PD model was developed and validated using data from a phase 3 clinical trial of IVIG,10% in 44 adults with MMN on stable IVIG treatment (NCT00666263), randomized to receive either placebo or IVIG,10% over two 12-week crossover periods (arm 1: IVIG-placebo; arm 2: placebo-IVIG). IVIG,10% dose ranged from 0.4–2.0 g/kg administered every 2–4 weeks (Q2W–Q4W). The linked PK/PD indirect response model was based on observed serum trough total IgG levels, patient dosing history and grip strength data for the more affected hand. Covariates, including baseline grip strength, gender and other patient/treatment characteristics were assessed. The model was used to simulate the following dosing regimens: 0.4, 0.8 and 1 g/kg Q2W; 1 and 2 g/kg Q3W; 0.4, 0.8, 1 and 2 g/kg Q4W; and 2 g/kg Q4W split into four daily infusions per dose. A minimum grip strength improvement of 4 kg was used as a threshold for clinically meaningful change.
Results: Observed grip strength of the more affected hand at first measurement during the trial ranged from 1.15–44.80 kg (median: 14.92 kg). The PK/PD model adequately described observed grip strength and serum total IgG concentrations, capturing the central tendency of the observed data and the extent of variability. Covariate analysis identified a relationship between lean body mass and volume of distribution; no covariates significantly affected grip strength. Model-based simulations showed that average grip strength at steady state increased with increasing IVIG,10% dose, and correspondingly increasing exposure, for the Q2W, Q3W and Q4W dosing intervals (median improvement 1.93–7.21 kg). Doses ≥1.6 g/kg/month resulted in ≥70% of patients achieving clinically meaningful improvements in median grip strength, with more frequent dosing at the same total monthly dose leading to smaller fluctuations in IgG concentrations and grip strength. Splitting the dose over multiple days did not markedly affect the PK concentration–time profiles of IVIG,10%, and had no discernable effect on grip strength.
Conclusion: The popPK/PD model successfully characterized the relationship between IVIG,10% dose, serum IgG levels and efficacy response in patients with MMN. Model-based simulations suggest a dosing regimen of ≥1.6 g/kg/month (e.g. 0.8–1 g/kg Q2W, 2 g/kg Q3W or 2 g/kg Q4W) would achieve clinically meaningful improvements in grip strength in most patients, and that more frequent dosing may maintain a more consistent response. Splitting one dose over multiple days is not expected to affect grip strength, allowing the flexibility to tailor dosing to increase patient comfort and convenience.
eP03.02.05Long-Term Course of Chronic Inflammatory Demyelinating Polyneuropathy: Clinical and Neurophysiological Outcomes
Melnik E1, Suponeva N1, Grishina D1, Arestova A1
1Research Center of Neurology, Moscow, Russian Federation
Introduction: Chronic inflammatory demyelinating polyneuropathy (CIDP) is the most common chronic immune-mediated polyneuropathy. CIDP is characterized by a long-term progressive or relapsing course of the disease, leading to temporal and permanent disability and requiring maintenance of long-term immunotherapy.
Objective: To assess clinical and paraclinical outcomes in patients with CIDP history of more than 5 years.
Methods: We included 46 adult patients that fulfilled the European Academy of Neurological Societies/Peripheral Nerve Society diagnostic criteria for CIDP 2021 and disease duration of more than 5 years. Treatment response, treatment status, remissions (improved and untreated), treatment changes, and residual symptoms or deficits were assessed. Outcome measures included Medical Research Council (MRC) sum score, the Neurological Impairment Scale (NIS), the Inflammatory Neuropathy Cause and Treatment (INCAT) disability scale, the Inflammatory Rasch-built overall disability scale (I-RODS), CIDP Disease Activity Status (CDAS), Questionnaire Short Form 36 Health Survey (SF-36), chronic acquired polyneuropathy patient-reported index (CAP-PRI). Electrophysiological examination and nerve ultrasound (Ultrasound pattern sum score, Grimm et al, 2015) were performed.
Results: Median follow-up period was 10 [7.0; 13.45], average age 48.1 ± 13.4 years. There were 32 (70%) typical CIDP patients and 14 (30%) CIDP variants (multifocal CIDP). The period from the disease onset to initiation of immunotherapy for typical CIDP was up to a year (60%) and for CIDP variants 5 years (50%). Up to 86% of multifocal CIDP required maintenance treatment after 5 years of the disease. 50% typical CIDP had CDAS 1 (cure: ≥5 years off treatment), 64% multifocal CIDP - CDAS 5 (unstable active disease: abnormal examination with progressive or relapsing course). For CIDP variants NIS motor score (p=0.002), INCAT score for arms (p=0.006), CAP-PRI score (p=0.025) were higher compared to typical CIDP. 80% CIDP patients had neurophysiological signs of a demyelination process, fulfilled the criteria of EAN/PNS 2021. UPSS sum score was 7.7 [2.0; 10.75], but most CIDP patients had nerve enlargement (increased mean cross-sectional area) in proximal ulnar and median nerves segments and brachial plexus.
Conclusions: The typical CIDP is characterized by a favorable long-term course, up to 60% of patients are considered clinically cured or are in stable remission. The progressive unstable course of the disease (64% of patients, p<0.03), a more severe neurological deficit with predominant involvement of the hands, and a greater degree of disability are significantly more common in CIDP variants. 80% of patients in the long-term follow-up have neurophysiological and sonographic signs of a chronic demyelinating process, fulfilled the criteria of EAN/PNS 2021.
eP03.02.06Hematological Effects of Intravenous Immunoglobulin Therapy in Patients with Neuromuscular Diseases – A Retrospective Analysis
Olivier P1, De Bleecker J1
1Ghent University Hospital, Gent, Belgium
Introduction: Administration of intravenous immunoglobulins (IVIGs) is a frequently used treatment in Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), multifocal motor neuropathy (MMN) and myasthenia gravis. IVIGs are generally well-tolerated and adverse reactions seldom prohibit its usage. Recently, an increasing number of cases with IVIG-related hemolysis have been reported. Hemolytic anemia is associated with changes in haptoglobin, lactate dehydrogenase (LDH) and bilirubin combined with a positive direct agglutination test (DAT).
Material and methods: A retrospective analysis of patients hospitalized for IVIG administration for neurological disorders at the department of neurology in an academic and in a general hospital was performed. Data collection period was arbitrarily set between 1 September 2017 and 31 August 2018.
Laboratory data was searched for blood samples of patients taken at hospital admission and at hospital discharge. Three different brands of IVIG (Privigen, Sandoglobulin and Multigam) were used. Pre-treated patients received a dose of 1.0 g/kg and de novo treated patients a dose of 2.0 g/kg. Duration of administration varied between 2-5 days. Patients were divided in first-time treated versus pre-treated patients.
Statistical analysis was performed using R software (R Core Team). The paired t-test and the Wilcoxon signed rank test were used for normally and non-normally distributed data, respectively.
Furthermore the frequency of “IVIG-associated hemolysis” was calculated and was defined as a decrease of more than 1g/dL in Hb and a positive DAT and at least one of the following findings: 1) increased reticulocyte count 2) increased LDH 3) low haptoglobin 4) unconjugated hyperbilirubinemia or 5) elevated free Hb in plasma.
Results: Forty patients were selected after exclusion based on comorbidities or incomplete datasets. 26 patients had a diagnosis of CIDP, 12 of MMN and 2 of polymyositis. 25 patients were treated with Sandoglobulin, 7 with Privigen and 8 with Multigam.
Treatment with IVIG showed a significant decrease in RBC (-0.31 x10^6/µL, p<0.001), Hb (- 0.95 g/dL, p<0.001) and increase in reticulocyte count (+ 0.6/10^3RBC, p=0.01). There was a significant decrease in LDH (-37 U/L, p<0.001), haptoglobin (-0.14 g/L, p=0.001) and increase in unconjugated bilirubin (+0.03 mg/dL, p=0.002). White blood cell and platelet counts dropped significantly after IVIG treatment (-1.9 x10^3/µL, p<0.001 and -33.5 x10^3/µL, p<0.001). There were no significant differences between the first-time treated and pre-treated groups.
DAT was negative in 34 patients, slightly positive or positive in 4 and 2 patients, respectively, prior to IVIG. After IVIG administration DAT was negative in 9 patients, slightly positive or positive in 7 and 24 patients, respectively. 5 out of 40 patients met all the criteria for IVIG associated hemolysis and 3 other patients met the 2 main criteria. Blood groups of the patients meeting all criteria were A+, A-, B+ and AB+.
Discussion: Our data shows significant changes in various hematological parameters in both pre-treated as first-time treated patients. A surprisingly high amount of patients met the criteria for IVIG-related hemolysis. These findings confirm the need for monitoring hematology parameters in all patients treated with IVIG, despite lack of consensus or guidelines.
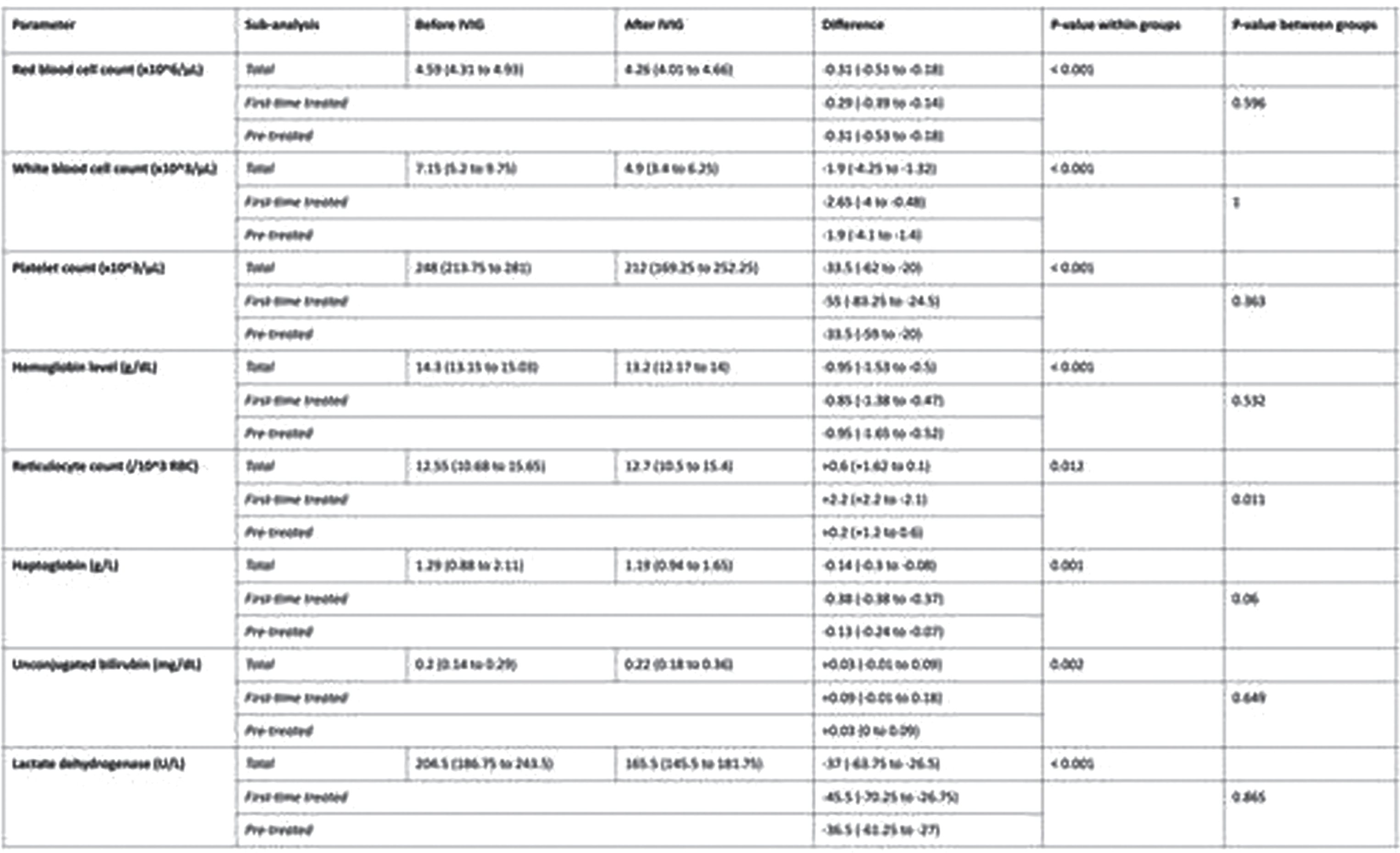
eP03.02.07Man-In-The-Barrel Syndrome as a Manifestation of Multiple Myeloma Relapsed
Vallejo D1,2,3, Millan J1,3, Cardona D1,3, Patiño V1,3, Cuervo J2, Velez J2,4
1Universidad De Antioquia, Medellin, Colombia, 2Ips Universitaria Universidad de Antioquia, medellin, Colombia, 3Neuroclínica, 4Prodiagnóstico
A 47-year-old man with a history of IgG-kappa stage IIA multiple myeloma diagnosed in 2015. He received treatment with CyBorD chemotherapy (CT) and Cyclophosphamide / dexamethasone for five cycles, as well as an autologous bone marrow transplant in 2015. At the time of admission, he was in a complete remission due to negative PET SCAN and continued to receive chronic post transplantation treatment with Thalidomide.
He consulted for a clinical picture of sudden onset consisting of monoparesis of the left upper limb and 15 days later in the right upper limb.
On physical examination, when evaluating strength with the Medical Research Council scale, a predominantly asymmetric proximal weakness was found: left upper limb: proximal 3/5, distal 4- / 5; right upper limb: proximal 3- / 5, distal 2/5; lower limbs 5/5. The myotendinous reflexes were asymmetrically diminished or absent: left radial style ++; brachial and tricipital 0; right: tricipital +, brachial and brachioradialis 0; also, with bilateral hypoesthesia in the C5-C7 territory. The plantar response was bilateral flexor, gait, proprioception and pallesthesia were normal. There were no other significant findings on the general physical or neurological examination. Brachial plexus involvement was suspected bilaterally, and a study was started.
VDRL, HIV, HBsAg, ACS HCV, vitamin B12, blood count, and kidney function were normal. He had elevated LDH (613), elevated IgG (2476). The lumbar puncture showed cerebrospinal fluid cytochemical with slight cytological albumin dissociation (0 leukocytes, 63.9 proteins) and negative microbiological studies for bacteria, fungi, and mycobacteria. CSF flow cytometry showed 61.5% of pathological plasma cells with Kappa light chains. Electromyography and limb neuro conditions showed axonal sensory polyneuropathy in the lower limbs, the involvement of the right C5 root, and neuropathy of the left axillary nerve and radial nerve. Brain MRI without alterations, but contrasted cervical and brachial plexus resonance confirmed infiltrative tumour involvement of roots, trunks, anterior and posterior divisions, predominantly left bilateral, reporting hyperintensity in vertebral bodies C4 and C7, posterior elements of C5, compromise of the epidural space extending from the T3 to T6 level, exerting a mass effect on the medullary cord, displacing it in the left lateral direction, also with thickening and hyperintensity of the signal and enhancement after the administration of contrast medium, compromising the roots of the brachial plexus bilaterally but with left predominance See figure 1
Extramedullary relapse of multiple myeloma with Neuromyelomatosis was diagnosed, so intrathecal triple chemotherapy was started with methotrexate, cytarabine and dexamethasone and cyclophosphamide.
Peripheral neuropathy associated with MM, which is frequently underdiagnosed, is found in 12% of patients at the time of diagnosis and up to 50-60% have subclinical peripheral neuropathy (PN). So far we have only found one case report of a patient who, as the first manifestation of his MM, had infiltration of the brachial plexus and another case of and brachial plexitis due of stem cell transplantation.The cause of PN associated with MM has been associated with perineural or perivascular paraproteinemic deposits, root compression and 21-70% related to treatment and infrequently neuromyelomatosis
eP03.02.08Sensory Polyneuropathy Associated With Vitamin D Deficiency
Kang S1
1College Of Medicine, Jeju National University, Jeju, Republic of Korea
Vitamin D deficiency is closely related with diabetic polyneuropathy. However, there is no report of peripheral neuropathy associated with vitamin D deficiency in non-diabetic patient. We describe a 55-year-old woman with vitamin D deficiency presenting with progressive gait ataxia and paresthesia. Neurological examination showed hypoactive deep tendon reflexes and proprioceptive sense impairment. Nerve conduction studies revealed sensory polyneuropathy. Serum 25(OH)D3 and 1,25(OH)2D3 levels were 1.9 ng/mL (normal range; 9.5~55.5) and 1.4 pg/mL (19.6~54.3), respectively. Serum total calcium and vitamin B12 values were 5.5 mg/dL (8.6~10.0) and 185.9 pg/mL, respectively. Serial nerve conduction studies were performed for 1 year, while she received treatment for vitamin D deficiency. There was progressive clinical improvement, but electrophysiological findings were not improved. To my knowledge, this is the first report of sensory polyneuropathy in association with vitamin D deficiency. It is interesting to note that the sensory polyneuropathy associated with vitamin D deficiency state was developed in non-diabetic patient.
eP03.02.09Diagnosing Small Fiber Neuropathy Remains Challenging in Sarcoidosis - Preliminary Data
Raasing L1, Grutters J, Vogels O, Veltkamp M
1St. Antonius Hospital, Nieuwegein, Netherlands
Introduction: Small fibre neuropathy (SFN) occurs in many sarcoidosis patients.1 A gold standard for diagnosis of SFN general is lacking but the diagnostic criteria using quantitative sensory testing (QST) and intra-epidermal nerve fiber density (IENFD) published by amongst others Devigili et al. are being used in clinical practice. However, determining IENFD according to the guidelines published by Lauria et al. remains challenging in general hospitals, due to its labour intensity.
Aims and objectives This study investigates whether the current diagnostic criteria are applicable for diagnosing SFN specifically in patients with sarcoidosis.
Methods: Clinical evaluation by a blinded neurologist, the SFN screening list (SFNSL), nerve conduction studies (NCS), 10 um skin biopsy and quantitative sensory testing (QST) at both hands and feet are used to diagnose SFN. An intra-epidermal nerve fiber density (IENFD) <40/mm2 is used as cut-off value for SFN. According to Devigili et al. the most sensitive combination of QST parameters is a combination of abnormal warm detection threshold (WDT) and cold detection threshold (CDT) on both feet with method of levels (LEV) and method of limits (LIM). These criteria are compared with the total amount of abnormal QST parameters.
Results: Of 53 sarcoidosis patients with symptoms of sensory neuropathy only 8 patients were diagnosed with definite SFN based on clinical signs, normal NCS, abnormal IENFD and/or abnormal WDT and CDT on both hands and/or feet with method of levels and method of limits. Remarkably, none of the patients had an abnormal QST based on those criteria. However, when we looked at abnormal results in either Thermal Sensory Limen, Paroxsysmal Heat Sensations or Pain Thresholds instead of only looking at warm-cold detection thresholds we found a correlation between the amount of abnormal QST parameters and clinical signs of SFN. In addition, patients with clinical signs of SFN show a higher SFNSL-score. No correlation could be found with IENFD.
Discussion: During this study, it is taken into account that not following the recommended guidelines for IENFD determination, makes it more challenging to interpret the result. It does give a relative indication on IENFD. Moreover, the main focus of this abstract is defining abnormal QST. Much research is performed to QST measurements, resulting in published guidelines for performing the test and well defined normative values. However, a huge heterogeneity is seen between studies in measurement sites and the parameters used to define abnormality. Devigili et al. did a good suggestion by determining sensitivity and specificity of several combinations in QST parameters compared with IENFD.
Conclusion: Using more abnormal QST parameters in combination with higher SFNSL-score, result into a more reliable diagnosis of SFN in sarcoidosis patients.
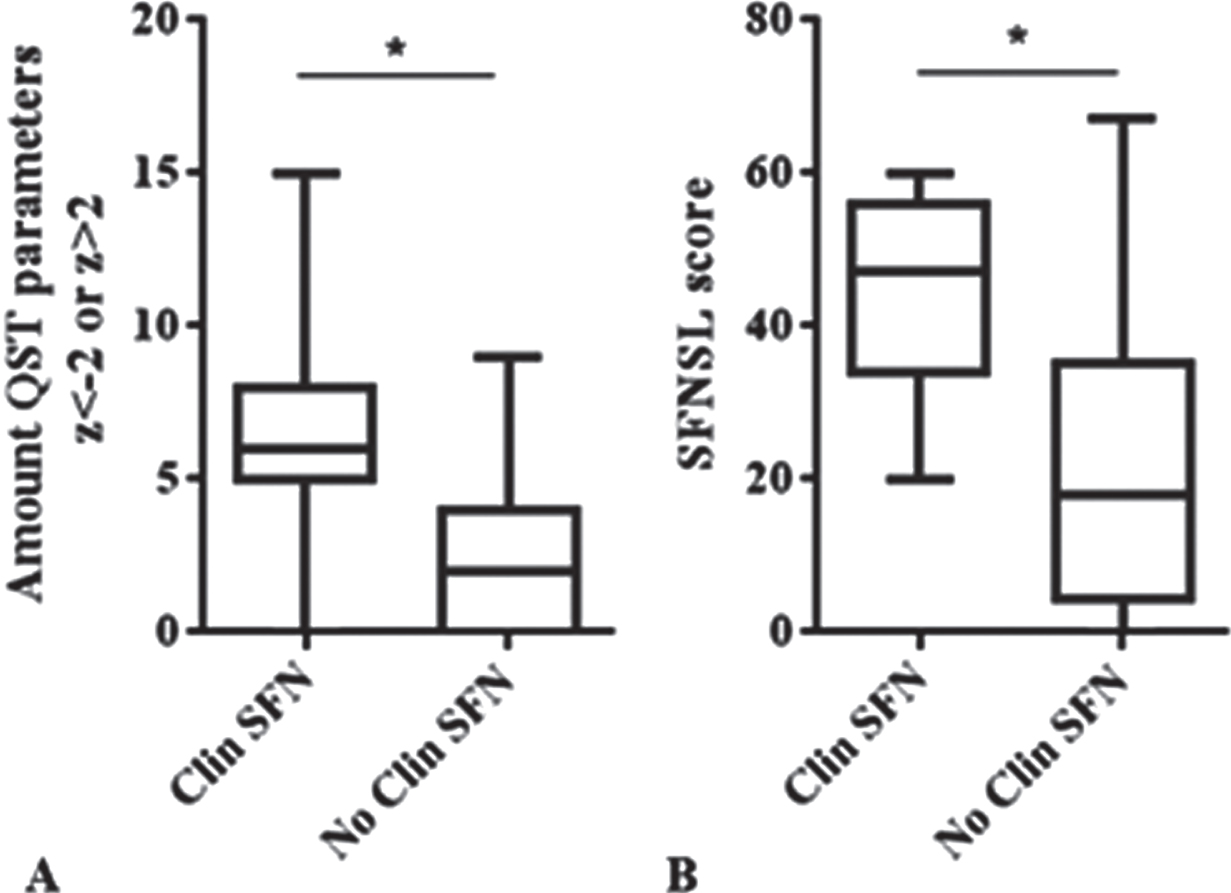
eP03.02.11In Vitro Comparison Between Different 10% Intravenous Immunoglobulin Preparations
Ouaja R1, Boyer S1, de Coupade C1, Chevalier A1, Guillou H1, Burlot L1
1LFB, Les Ulis, France
Introduction: This work aimed at reporting the extent to which 10% IVIg preparations differ in their purity profile and biological characteristics.
Methods: Purity profile and biological characteristics of IqYmune® were compared to other marketed 10% IVIg (Privigen®, Octagam®, Gamunex®, andKiovig®). For biochemical comparisons, IqYmune® batches used were performed with European plasma source, and for activity comparison,IqYmune® batches used were performed with US plasma. For competitors, commercial batches were performed using US plasma and are in compliance with regulatory requirements.
Results: Anticomplementary activity (ACA) Variable levels of ACA were observed among the brands with intermediate levels for IqYmune®. Complement activation is known to induce fever, chills, and hypotension [1].
Functional characterization: All compared batches are issued from US plasma. The results show no significant difference for all antibodies and functional activities.
Purity: Purity assessment shows differences between these preparations: IgA contents are the highest for Octagam® (>1.4 mg/g Prot); IgA are intermediate for Gamunex®, Kiovig®; for Privigen® and IqYmune®, contents in IgA are very low (around 0.1 mg/gProt). (Fig1)
Coagulant factors: IqYmune® exhibits the lowest Factor XI Ag content (102-109mEIU/mL) compared to competitors (151-338mEU/mL). Maximum Factor XI Agcontent was observed with Privigen® (309mEIU/mL).
Conclusions: Although different IVIg preparations essentially consist of polyclonal IgG, the different purification processes lead to variations in other characteristics. The IqYmune® manufacturing process developed with a QbD approach [2] has generated a 10% IVIg having a higher level of quality and enhanced safety profile while maintaining a high level of efficacy.
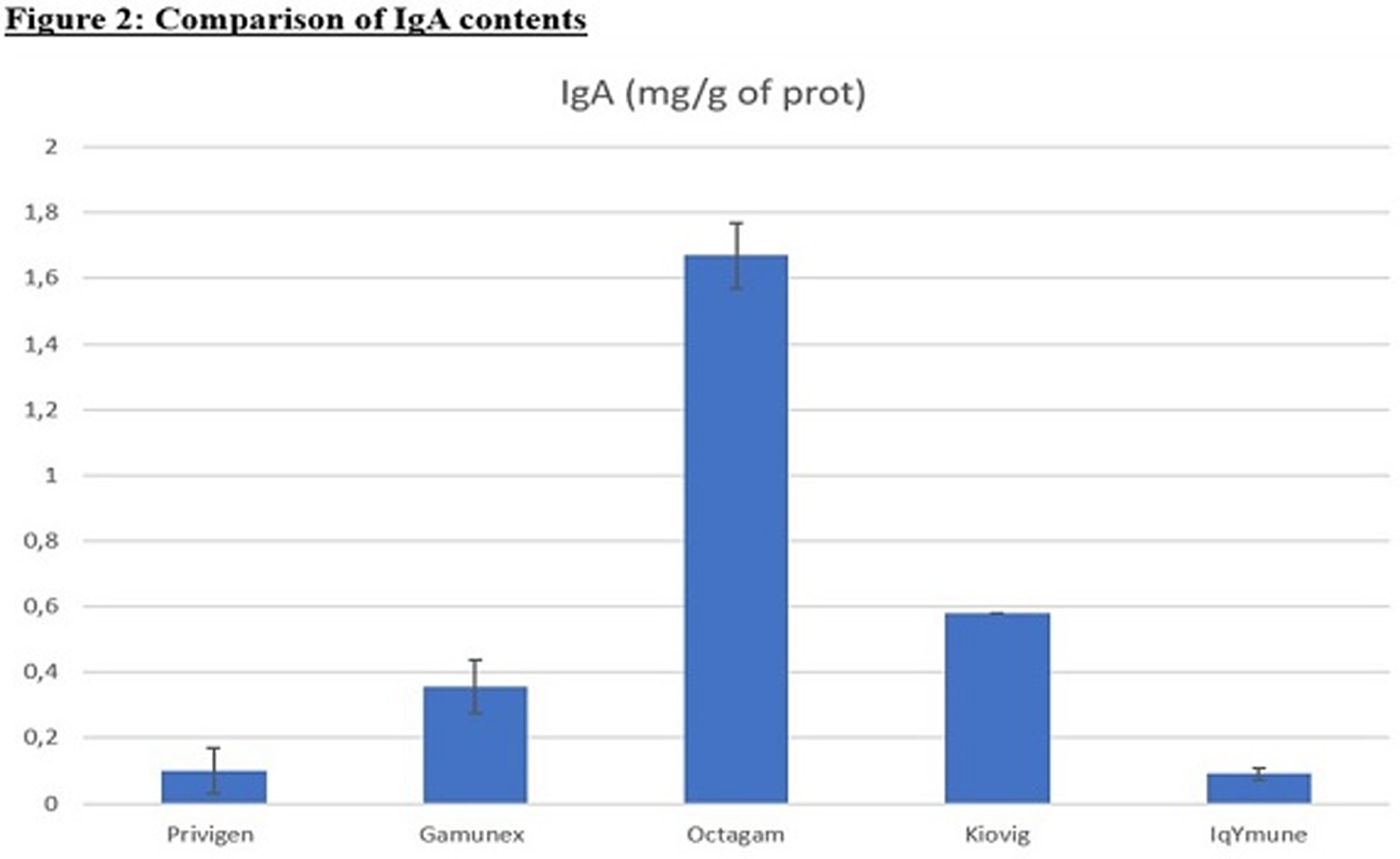
eP03.03.01MANATEE: GYM329 (RO7204239) in Combination with Risdiplam Treatment in Ambulant Children with Spinal Muscular Atrophy
Muntoni F1, Darras B2, Duong T3, Morrow J4, Servais L5,6,7, Carruthers I8, Gerber M9, Kletzl H10, Martin C8, Zhang B11, Scalco R12, Wagner K12, Mercuri E13
1The Dubowitz Neuromuscular Centre, NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Institute of Child Health University College London, & Great Ormond Street Hospital Trust, London, UK, 2Department of Neurology, Boston Children’s Hospital, Harvard Medical School, Boston, USA, 3Department of Neurology, Stanford University, Palo Alto, USA, 4Queen Square Centre for Neuromuscular Diseases, UCL Institute of Neurology, London, UK, 5MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, UK, 6Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Paediatrics, University Hospital Liège & University of Liège, Liège, Belgium, 7I-Motion - Hôpital Armand Trousseau, Paris, France, 8Roche Products Ltd, Welwyn Garden City, UK, 9Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 10Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland, 11PD Neuroscience, Roche Products Ltd, Welwyn Garden City, UK, 12Pharma Development Neurology, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 13Pharma Development Neurology, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 14Pediatric Neurology Institute, Catholic University and Nemo Pediatrico, Fondazione Policlinico Gemelli IRCCS, Rome, Italy
Objective: To assess the safety, tolerability, pharmacokinetics (PK)/pharmacodynamics (PD) and efficacy of GYM329 (RO7204239) in combination with risdiplam in ambulant children with spinal muscular atrophy (SMA).
Background: Disease-modifying treatments for patients with SMA that directly target the deficiency of survival of motor neuron (SMN) protein have been approved in over 50 countries. While these treatments have positively impacted the disease trajectory, additional complementary therapies may further benefit treated patients.
Risdiplam is a centrally and peripherally distributed, oral SMN2 pre-mRNA splicing modifier that has been approved by the US Food and Drug Administration for the treatment of patients with SMA aged ≥2 months, and by the European Commission for the treatment of patients aged ≥2 months with a clinical diagnosis of Type 1, 2 or 3 SMA or 1–4 copies of SMN2. GYM329 is an investigational, recycling and antigen-sweeping monoclonal anti-myostatin antibody (myostatin is a negative regulator of skeletal muscle growth). In a mouse model of SMA, the combination of GYM329 and an SMN2 splicing modifier was found to further improve muscle size and strength compared with SMN2 splicing modifier treatment alone.
Design/Methods: MANATEE (NCT05115110) is a multicenter, two-part, randomized, placebo-controlled, double-blind study that will investigate the effect of GYM329 in combination with risdiplam on treatment-naïve and non-treatment-naïve ambulant patients, aged 2–10 years at screening. Part 1 (target enrollment N~36) will assess the safety, tolerability and PK/PD of different GYM329 doses in combination with risdiplam. Part 2 (target enrollment N~144) will assess the efficacy and safety of the Part 1-selected dose of GYM329 in combination with risdiplam. The primary endpoint of Part 2 will be the change from baseline in the Revised Hammersmith Scale (RHS) total score at Week 72. Secondary efficacy endpoints will include changes from baseline in Motor Function Measure (MFM) score (MFM total score and Domain 1 + Domain 2 score); time taken to rise from the floor as measured by RHS Item 25; time taken to walk/run 10 meters as measured by RHS Item 19; and lean muscle mass as assessed by dual-energy x-ray absorptiometry (patients aged ≥5 years).
Discussion: Risdiplam has demonstrated clinically meaningful benefit in patients with SMA and there have been no treatment-related safety findings leading to treatment withdrawal in the risdiplam studies to date. This combination therapy approach may lead to a complementary effect on improvement of patients’ motor function. Here, we present the design of the MANATEE study with additional information on the objectives and endpoints that will be used.
Conclusions: This study will provide valuable information about the safety and efficacy of GYM329 in combination with risdiplam treatment in ambulant children with SMA.
eP03.03.02Progression of the Revised Hammersmith Scale Items in Patients with Spinal Muscular Atrophy Treated with Nusinersen
Castellar S1,2, Ortiz F1,2, Salcedo M1, Ruiz E1,2, Soto D1
1Instituto Roosevelt, Av 4 East # 17-50, Colombia, 2Universidad Nacional de Colombia, Cra 45 # 26-85, Colombia
Treatment with Nusinersen has modified the natural history of patients with Spinal Muscular Atrophy (SMA). The magnitude of the therapeutic response is variable.
The therapeutic results in children with SMA II and III have been measured with the Expanded Hammersmith Functional Scale. Subsequently, Revised Hammersmith Scale was validated following the RASH methodology, which improved its metrics, properties and score score interpretation.The Revise Upper Limb Module (RULM) has been developed to evaluate the upper limb performance in patients with SMA over time.
In this paper we are going to present the detailed progress of the Hammersmith scale and RULM in four cases of patients who received the Nusinersen treatment in Colombia.
Patient A shows the highest functional changes after the onset of the Nusinersen treatment. The revised Hammersmith scale started from 14 points to 26 score, and the expanded from 20 to 36. Functional gains are represented in items 3, 13, 6,16,7, 14 and 22. These items are from low and median complexity.
By the age of 2, this boy had completely lost the crawl ability. This function has been recovered by the age of 3.1 years. Additionally, the patient achieved a 2 point-score in item 16 after 4 months of treatment (cruise at least 5 steps around furniture). With the RULM scale, this patient improved 3 points, going from 22 to 25 points. The items that have changed are items I, R and L.
In contrast, patient B only shows functional gains in low complexity items (4, 2, 9, and 13). Revised Hammersmith scale beginning with 5 and finishing in 9 points, the expanded scale going from 6 to 12 score. In the RULM scale, this patient improved 3 points, moved from 16 to 19 points. Items that have changed are C, E and M.
Patient C, between 6,5 and 8.3 years, the revised Hammersmith scale moved from 27 to 32 and the expanded scale from 37 to 40. Patient C shows improvement on items 10,17,22,23 and 21. In particular, there were improvements in some items of the median complexity (21: stand to sit on the floor), before minor complexity item (item 3: sitting to lying). In the RULM scale, the patient improved 1 point (item H: tear paper), from 28 to 29 with the loading dose of Nusinersen, after this, in the course of 8 months the scale kept the score, without significant change.
Patient D, between 3,2 and 4,5 years, the score of the revised Hammersmith scale was from 15 to 19 and the expanded from 23 to 28. Functional increase was observed in less complexity items (11,13,6,10 and 12). At the age of 4 years, after 8 months of the beginning of the treatment, item 14 (lie to sit) pass from 0 to 1, which is a big accomplishment for functional gain. Furthermore, the RULM scale improved 3 points from 21 to 24. The enhanced items were items H, K,O and I.
eP03.03.03Critical Review of the Spanish Pharmacoclinic Protocol for the Monitoring of Nusinersen Treatment
Carrera L1, Expósito J1, Natera D1, Medina J1, Moya O1, Marco C2, Roca S1, Jimenez-Mallebrera C1, Trifunov S1, Codina A1, Colomer J1, Povedano M2, Ortez C1, Nascimento A1
1Hospital Sant Joan De Deu, Barcelona, Spain, 2Hospital Universitari de Bellvitge, Barcelona, Spain
The use of nusinersen for the treatment of spinal muscular atrophy (SMA) was approved in Spain in March 2018. The establishment of pharmacoclinical protocols is necessary to guarantee adequate follow-up of patients and a more precise evaluation of the treatment response. The withdrawal criteria in the Spanish protocol vary according to the SMA type and assess exclusively the gain or loss of points in motor function scales. SMA type 1 patients are evaluated prior to administration of the 10-month dose (6th dose) from baseline treatment, and from this moment, in the clinical assessment every four months. Discontinue treatment if there is a worsening in motor function, loss of 1 point in any of the motor milestones on the HINE scale (Hammersmith Infant Neurological Examination); or worsening in lung function (Permanent ventilation defined as more than 16 hours/day). SMA type 2 and type 3 patients are assessed after 2 years of nusinersen treatment. Discontinue treatment if there is no improvement equal or greater than 3 points from baseline in Hammersmith Functional Motor Scale Expanded score (HFMSE) and 2 points in Revised Upper Limb Module (RULM) in non-ambulant patients or more than 30 metres in 6-Minute Walk Test (6MWT) in ambulant SMA type 3. Here, we evaluate critically the Spanish protocol through a review of the patients treated in our hospital.
Methods. We collected demographic, clinical data and outcome measures in SMA patients treated with nusinersen between March 2018 and January 2022. We assessed changes in motor function every 4 months in SMA type 1 using Hammersmith Infant Neurological Examination (HINE) and the Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorder scale (CHOP INTEND) and every 8 months in SMA type 2 and 3, using HFMSE, RULM, 6MWT and Egen Klassifikation 2 scale (EK2).
Results. We included 25 patients: 3 SMA type 1, 19 SMA type 2 and non-ambulant type 3, and 3 ambulant SMA type 3. The age range at the nusinersen treatment onset from 0,4 to 17 years (mean 8,3). Withdrawal criteria were evaluated in 22 patients. Treatment was withdrawn in 63% of patients after 2 years of treatment (12 SMA type 2 and non-ambulant type 3 patients and 2 ambulant SMA type 3). All patients who were withdrawn started treatment with nusinersen at age 5 years or older. Nusinersen treatment was restarted after 8 months off treatment in 3 patients who had marked worsening of their motor function. Treatment was continued in all SMA type 1 patients.
Conclusion. Pharmacoclinical protocols must be updated according to current knowledge. Modifying factors such as the age of treatment onset or the baseline motor functional level are not considered when assessing the treatment withdrawal.
eP03.03.04Experience in the Treatment With Pyridostigmine Monotherapy, in Patients With Spinal Muscular Atrophy in Colombia
Pareja I1, Múnera L2, Giraldo G2, Baquero C2, Mesa S1
1Universidad de Antioquia, Hospital Pablo tobón Uribe, Medellín, Colombia, 2Hospital Pablo Tobón Uribe, Medellin (ANT), Colombia
Spinal muscular atrophy (SMA) is a characterized by progressive muscle weakness and atrophy due to degeneration of anterior horn cells. 95% of cases are due to mutations in SMN1 located on 5q13, this results in the death of motor neurons . SMA affects 1 in 6,000 to 10,000 infants
The therapeutic strategies can be: SMN-targeted mechanisms or Non-SNM-based mechanisms. So they can be at the level of the motor neuron or at the muscular level.
To date, among patients who are or not candidates for targeted therapy approved to date, treatment with pyridostigmine has been offered.
Based on the description that in this disease there is a dysfunction of the neuromuscular junction wich can contribute to symptoms of weakness and fatigability in patients. Pyridostigmine as an acetylcholinesterase inhibitor that improves neuromuscular transmission, could improve this symtoms
Patients admitted to a program for neuromuscular diseases in the city of Medellín were chosen. They were selected those who were not candidates to pharmacological therapy approved to date for the treatment of SMA.
Pyridostigmine therapy was offered with the approval of each child’s parents. Initial response and follow-up based on motor scores after initiation of therapy were documented; as well as the level of satisfaction in distal tremor, lingual fasciculations and final motor functionality with therapy, this was classified as poor, good or very good.
The age at diagnosis had a mean of 7 months of age. 60% of the population were women and 40% were men. Of these, 60% corresponded to classification SMA 3, 30% SMA 2 and 10% SMA 1. 100% of them denied consanguinity, and 100% of them had a deletion mutation in exon 7, with 3 or more copies of SMN2 The earliest diagnosed child was at 0.8 months and the latest was at 18 months. None of our patients had neonatal screening. None of the patients were candidates for alternative therapy and all parents gave consent to initiate pyridostigmine therapy.
The mean age at initiation of therapy was 11,7 years. At the beginning of therapy, the median of the Hammersmith scale was 29 points, with a dose of one milligram per kilo day of pyridostigmine on average, distributed in two doses. In 50% of the children, there was stability of the scale score at 6 months from the start, with an interquartile range of 1 (0-1); which is not considered a statistically significant finding
There was a significant response described for distal tremor (especially for handling cutlery and study instruments) as well as lingual fasciculations; with a level of satisfaction with the medication of 100% as very good.
It is considered that pyridostigmine is not a disease-modifying drug, but its use can be considered to improve the quality of life of patients, on fine motor functionality.
More studies and more patients are needed to assess its impact on gross motor skills.
eP03.03.05Onasemnogene Abeparvovec Treatment Outcomes by Patient Weight at Infusion: Initial Findings from the RESTORE Registry
Servais L1, Benguerba K2, De Vivo D3, Kirschner J4, Mercuri E5, Muntoni F6, Proud C7, Tizzano E8, Quijano-Roy S9, Saito K10, Menier M11, LaMarca N12, Sun R12, Anderson F13, Faulkner E12, Finkel R14
1MDUK Oxford Neuromuscular Centre, University Of Oxford, Oxford, United Kingdom, 2Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 3Departments of Neurology and Pediatrics, Columbia University Irving Medical Center, New York, USA, 4Clinic for Neuropediatrics and Muscle Disease, University Medical Center Freiburg, Freiburg, Germany, 5Department of Paediatric Neurology and Nemo Clinical Centre, Catholic University, Rome, Italy, 6The Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health & Great Ormond Street Hospital, London, UK, 7Children’s Hospital of The King’s Daughters, Norfolk, USA, 8Department of Clinical and Molecular Genetics, Hospital Valle Hebron, Barcelona, Spain, 9Garches Neuromuscular Reference Center (GNMH), APHP Raymond Poincare University Hospital (UVSQ Paris Saclay), Garches, France, 10Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan, 11Atlas Clarity LLC, San Francisco, USA, 12Novartis Gene Therapies, Inc., Bannockburn, USA, 13Center for Outcomes Research, University of Massachusetts Medical School, Worcester, USA, 14Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, USA
Objective: Interventional trials of onasemnogene abeparvovec demonstrated safety and efficacy for infants typically weighing <8.5 kg at infusion. We aimed to describe real-world outcomes for patients with spinal muscular atrophy (SMA) according to weight at the time of onasemnogene abeparvovec infusion.
Methods: We compared baseline characteristics, effectiveness (measured by Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders [CHOP INTEND]), and safety of onasemnogene abeparvovec for patients weighing <8.5 kg (Group 1) or ≥8.5 kg (Group 2) at the time of onasemnogene abeparvovec infusion in RESTORE (a prospective, multinational SMA patient registry).
Results: As of May 23, 2021, RESTORE included 163 patients with available data on weight at onasemnogene abeparvovec infusion. One hundred twenty patients weighed <8.5 kg (Group 1; range, 1.6–8.4 kg), and 43 weighed ≥8.5 kg (Group 2; range, 8.6–12.5 kg). Clinical characteristics and treatment patterns between Group 1 and 2 differed markedly. A greater percentage of patients in Group 1 had two survival motor neuron 2 (SMN2) gene copies (74% vs. 42% in Group 2), while a greater percentage of patients in Group 2 had three SMN2 gene copies (51% vs. 20% in Group 1). Most patients in Group 1 were diagnosed with SMA type 1 (67.8%) or were presymptomatic (23.7%), while most patients in Group 2 were diagnosed with SMA type 1 (43.9%) or type 2 (43.9%). The majority of patients in Group 1 (80/120; 66.7%) were aged <6 months at onasemnogene abeparvovec infusion, while most patients in Group 2 (41/43; 95.3%) were aged ≥6 months at infusion. Monotherapy with onasemnogene abeparvovec was more prevalent in Group 1 (60.8%) versus Group 2 (41.9%). Different polytherapy regimens were observed in both groups. Of 43 patients in Group 1 who were evaluable for CHOP INTEND (two or more assessments, more than one occurring after onasemnogene abeparvovec infusion), 42 (97.7%) improved or maintained score, and 37 (86.0%) achieved increases of ≥4 points. Of five evaluable patients in Group 2, all improved or maintained score, and three (60.0%) achieved ≥4-point increases. In Group 1, 60/119 (50.4%) patients experienced one or more treatment-emergent adverse events (TEAEs) of any grade, and 28 (23.5%) reported one or more serious adverse events. In Group 2, TEAEs of any grade were reported in 25/43 (58.1%) patients, and 10 (23.3%) reported one or more serious adverse events.
Conclusions: Preliminary results suggest that patients weighing ≥8.5 kg at onasemnogene abeparvovec infusion may benefit from treatment, as demonstrated by improved CHOP INTEND scores. We did not observe a difference in the incidence of TEAEs or serious adverse events based on patient weight at onasemnogene abeparvovec infusion.
eP03.03.06Treatments and Outcomes for Patients with Spinal Muscular Atrophy Type 2: Findings from RESTORE Registry
Servais L1, Benguerba K2, De Vivo D3, Kirschner J4, Muntoni F5, Proud C6, Tizzano E7, Quijano-Roy S8, Desguerre I9, Saito K10, Raju D11, LaMarca N11, Sun R11, Anderson F12, Faulkner E11, Finkel R13
1MDUK Oxford Neuromuscular Centre, University Of Oxford, Oxford, United Kingdom, 2Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 3Departments of Neurology and Pediatrics, Columbia University Irving Medical Center, New York, USA, 4Clinic for Neuropediatrics and Muscle Disease, University Medical Center Freiburg, Freiburg, Germany, 5The Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health & Great Ormond Street Hospital, London, UK, 6Children’s Hospital of The King’s Daughters, Norfolk, USA, 7Department of Clinical and Molecular Genetics, Hospital Valle Hebron, Barcelona, Spain, 8Garches Neuromuscular Reference Center (GNMH), APHP Raymond Poincare University Hospital (UVSQ Paris Saclay), Garches, France, 9Hôpital Necker Enfants Malades, APHP, Paris, France, 10Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan, 11Novartis Gene Therapies, Inc., Bannockburn, USA, 12Center for Outcomes Research, University of Massachusetts Medical School, Worcester, USA, 13Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, USA
Objective: We sought to describe real-world treatment patterns and outcomes for patients with spinal muscular atrophy (SMA) type 2.
Methods: Patients with SMA type 2 in the RESTORE registry (an ongoing prospective, multicenter, multinational, observational SMA patient registry) who have received one or more disease-modifying therapies (DMTs; nusinersen, risdiplam, onasemnogene abeparvovec [OA]) were included. Changes in motor milestones and disability scores were analyzed, and treatment-emergent adverse events (TEAEs) were assessed.
Results: As of May 23, 2021, RESTORE included 43 patients with SMA type 2. Twenty-seven (62.8%) were female. Of patients with survival motor neuron 2 (SMN2) gene copy number information, seven (16.3%), 33 (76.7%), two (4.7%), and one (2.3%) had two, three, four, and more than four copies of the SMN2 gene, respectively. The median age at SMA diagnosis was 14.5 months (IQR 10.4–19.4 months), and the median age at first DMT administration was 16.5 months (IQR 11.1–22.0 months), with a median interval between diagnosis and first treatment of 1.5 months (IQR 1–2.8 months). The median interval was 37.5 days for patients receiving OA monotherapy, 84 days for patients receiving nusinersen monotherapy, and 40 days for patients switching from other therapies to OA or for those receiving add-on therapy to OA. A significant difference in age at first DMT administration was observed between groups (p=0.0014). Median (IQR) age at first treatment for OA monotherapy, nusinersen monotherapy, switching from other therapies to OA, and receiving add-on therapy to OA was 16.5 months (10.25–19.75 months, n=20), 27 months (19.0–90.0 months, n=11), 11.5 months (7.5–16.0 months, n=10), and 12.5 months (8.0–17.0 months, n=2), respectively. Of 15 patients who had two or more motor milestone assessments (one or more occurring after DMT administration), all but one patient (OA only) maintained or achieved additional milestones: 11/14 (78.6%) received OA monotherapy; 2/14 (14.3%) switched from nusinersen to OA; 1/14 (7.1%) received nusinersen only. Median change in Hammersmith Functional Motor Scale (HFMSE) score was 8.5 (IQR: 2.3–16.8, n=4, with 3/4 [75%] reporting a clinically important ≥3-point improvement), with median monthly change in score of 0.9 (IQR: 0.5–2.1, n=4). Median change in HFMSE score for patients receiving OA monotherapy was 14 (IQR: 8.5–19.5, n=2, with 100% reporting a clinically important ≥3-point improvement), with median monthly change in score of 0.9 (IQR: 0.8–1.1, n=2). TEAEs of any grade were recorded for 7/22 (31.8%) patients who received OA monotherapy, none of whom experienced a TEAE of Grade ≥3. Following administration of OA, two patients required nasogastric tube insertion for feeding and two patients required BiPAP ventilation. TEAEs were recorded for 5/10 (50%) patients who switched from nusinersen to OA. One (10%) patient experienced an adverse event of Grade ≥3. No new safety signals were identified.
Conclusions: These preliminary data suggest that OA is effective and has an acceptable safety profile for patients with SMA type 2. Future analyses will include additional comparisons between DMT regimens as they become feasible with the ongoing enrollment of patients into the RESTORE registry.
eP03.03.07Characteristics and Epidemiology of Amyotrophic Lateral Sclerosis in a Health-Care Area in Northwestern Spain
Espinosa Trujillo A1, Guijarro Del Amo M, García Pazos O, Santamaría Montero P, Rodriguez Rodriguez M
1University Hospital Lucus Augusti, Lugo, Spain
BACKGROUND AND AIMS: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease affecting motor neurons and other neural cells that eventually leads to severe disability and death. ALS prevalence ranges between 4.1 and 8.4 per 100.000. In Europe the annual incidence ranges from 2.1 to 3.8 per 100.000 people. Since there are no disease-modifying therapies, much of the clinician-patient relation is based on managing their functional impairment, taking care of symptoms and helping anticipate disease progression. Multidisciplinary clinical attention aims to address these needs and improve patient care.
We describe the characteristics of the group of patients with ALS followed within a multidisciplinary setting in our health-care area of 486.237 inhabitants.
METHODS: We have retrospectively reviewed the full medical records of all patients diagnosed with ALS at the University Hospital Lucus Augusti, in Lugo, Spain since January 1st, 2018 until December 31st, 2021. The diagnosis was determined according to the El Escorial-Awaji criteria, using clinical and EMG findings.
RESULTS: We obtained a sample of 58 patients diagnosed with ALS from which we analyzed the following data. ALS prevalence at the time of ending the registry was 7.8 per 100.000 people. The annual incidence was 2.41 per 100.000 people. The mean age at the time of diagnosis was 64 years, with a minimum age of 27 years and a maximum of 84 years. 65.5% of the sample were men and 34.5% were women, which means an affected male-female ratio of 1.89:1. All the patients underwent EMG studies and 15.5% needed a second EMG to be diagnosed. The mean diagnostic delay was 13.8 months, ranging from 0.5 to 57 months. As part of diagnosis, genetic tests were made in 22.41%. In regard to the site of onset, 74.13% had spinal-limb, 20.68% bulbar and 5.17% respiratory onsets. About other clinical features, only 1 patient (1.72%) had a familial history of ALS, 18.97 % were or had been smokers, 19% had cognitive impairment and 6,9% met criteria for fronto-temporal dementia. On follow-up, 34.5% suffered from significant disabling cramps and/or spasticity, 10.34% had pseudobulbar palsy/emotional lability and 55,2% had depressive symptoms. Regarding the treatment, 86% were treated with riluzole, 36.2% used noninvasive ventilation (NIV), 6.9% underwent tracheostomy and subsequent invasive ventilation, 39.6% used cough-assist devices (CAD) and 27.6% used both NIV and CAD. The 20.7% underwent gastrostomy and enteral nutrition (EN) and, within the patients without EN, 25.8% needed dietary supplements.
CONCLUSIONS: We observed that the incidence, prevalence and affected male-to-female ratio in our sample is similar to those described in the literature, however the proportion of patients with family history of ALS is lower. The mean diagnosis delay is in keeping with the data of previous studies; however, in some cases it was greater than two years due to non-specific or atypical symptoms and/or delayed referral to a neurologist or diagnostic tests.
Finally, we have achieved better awareness and management of patients’ symptoms and needs since they are attended in our multidisciplinary unit, and that has improved their overall care.
eP03.03.08Characterization of the ALS Patients’ Population in a Large Italian Centre
Bianchi F1, Becattini L, Fontanelli L, Siciliano G
1Azienda Ospedaliero Universitaria Pisana, Via Sant’antonio 61 Pisa, Italy
Introduction: Amyotrophic Lateral Sclerosis is a fatal neurodegenerative disorder involving upper and lower motor neurons. It is sporadic for 90% of cases and familiar for 10%, with a median age at onset ranging from 58 to 63 years; some phenotypes have a gender prevalence. Here we describe the recent motor neuron disease (MNDs) population of the Santa Chiara Hospital, Pisa, to compare it with what described in Literature
Methods: In the last year, 153 patients (77 men and 76 women) with a median age at onset of 63.4 underwent regular neurological evaluation for MNDs. A database to better characterize our population was developed and analyzed
Results: Fifteen female and eighteen male patients had a predominant involvement of I MN at onset (median age 58.2 years). Fifty men and thirty women (median age 61.8 years) had a prevalent involvement of II MN. Twenty-eight women and five men (median age 68.7 years) had a bulbar picture. Two men (median age 60 years) had a cognitive impairment and two female and one male patients had a respiratory onset (median age 68.3). Eight out of 143 ALS patients had a positive genetic test. Thirty-eight patients had a familiarity for neurodegenerative diseases
Among the 100 patients that underwent a high camp cerebral MRI, sixty-eight had signs of motor cortex degeneration. Seventeen patients had predisposing risk factors
Conclusion: Our database is aligned with previous literature reports, supporting the validity of the current body of knowledge of ALS prevalence. Similar databases are useful to track patients and increase the efficiency of follow-ups
eP03.03.09Descriptive Analysis of Bulbar Amyotrophic Lateral Sclerosis (ALS) In the Northern Area of Tenerife
Pérez Pérez H1, González Toledo G1, Hernández García M1, Hernández Javier C1, Crespo Rodríguez M1, Carrillo Padilla F1
1Neurology Service. Hospital Universitario de Canarias (HUC), La Laguna, Spain
Objective: To describe the basic epidemiological and clinical aspects of bulbar ALS in the northern area of Tenerife.
Methods: We retrospectively reviewed the medical history of all the patients with diagnosis of ALS in follow-up by the Neuromuscular diseases unit of our hospital in the period 1/1/14 to 31/10/21, to do a descriptive analysis of their main epidemiological and clinical aspects. The reference population of our hospital is 396483 people.
Results: 69 cases of ALS were found, 23 (33%) of them with bulbar onset. 19 (83%) patients were females and the mean age at diagnosis was 68 (±10,1) years old. Average time from the first symptom to diagnosis was 12,5 (±9) months. None of them had family history of motor neuron disease.
The most frequent debut were isolated dysarthria (56%) and dysarthria with dysphagia (35%). Just 2 patients had isolated dysphagia as the first symptom. All patients had limb weakness later in the evolution of the disease. Also all patients had dysarthria at some point of the disease, becoming anarthric 83% of them. 4 (18%) showed some degree of cognitive impairment and 2 fullfilled diagnostic criteria for associated frontotemporal dementia: 1 frontal variant and 1 primary progressive aphasia. 61% of patients fulfilled Awaji-Shima criteria of clinically definite ALS in the first electromyographic study performed and 26% fulfilled at least clinically possible ALS criteria. All patients had clinically definite ALS later in the evolution.
Non-invasive ventilation was offered to 62% patients, but 2 didn’t tolerate it and 1 rejected it. Mean time from diagnosis to ventilation was 6,5 (±6,5) months. No patient was tracheostomized. Food thickeners were recommended to 70% patients and gastrostomy to 62%, but 1 rejected it. Mean time from diagnosis to gastrostomy was 14 (±9) months. 74% needed a wheelchair in a mean time from diagnosis of 11 (±7,5) months.
Riluzole -50 mg orally twice daily- was used throughout the disease in 91% of patients. Due to secondary effects, 1 patient needed a dosage reduction. 1 patients stopped it voluntarily. 18 (78%) patients died in this period, with an average surveillance since diagnosis of 20 (±12) months. The most frequent cause of death was respiratory failure in end-stage disease (73%). Respiratory infection was the cause of death of the other 27%.
Conclusions: We report the epidemiological and clinical data about bulbar ALS patients in a tertiary-level hospital specialized ALS consultation of the Canary Islands. Our results globally agree on those reported in other regions of the world. Dysarthria-anarthria was the main symptom in the debut of our bulbar-onset ALS patients, but all patients developed spinal symptoms. In spite of bulbar onset, 61% showed generalized denervation signs in the first electromyography. Riluzole was globally well tolerated. Prognosis was globally poor and most patients required respiratory or nutritional support early in the evolution of the disease
eP03.03.10Atypical Onset of Amyotrophic Lateral Sclerosis: Seven Cases Report
Marzouk B1, Kably B1, Erguig L1, Birouk N1, Tamaoui L
1Hopital Des Spécialité, CHU IBN Sina, Rabat, Morocco
Background and Aims: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects both lower motor neurons (LMN) and upper motor neurons (UMN) with a marked phenotypic heterogeneity. We present atypical onset of this disease.
Methods: This is a retrospective and descriptive study of seven clinical cases of defined ALS with atypical onset.
Results: This report concern seven patients with a diagnosis delay varying from 1 to72 months:
Case 1: 51-years old woman with inaugural neuromyotonia revealed by painful tonic contractures, cramps, fasciculations and tetraparesis, electroneuromyography (ENMG) showed neuromuscular hyperexcitability for which the etiological assessment remained negative. She progressed to typical ALS.
Case 2,3: Two men aged 59 and 57 years old with inaugural acute respiratory failure due to diaphragmatic paresis in whom ALS was diagnosed later.
Case 4: 30-years-old woman, presented with subacute pseudomyositis with proximal weakness of lower limbs and myalgia, the initial ENMG showed myogenic appearance, but follow up ENMG disclosed diffuse denervation patterns. She progressed to ALS features and died 2 years later from respiratory failure.
Case 5: man of 53 years old with isolated pseudobulbar palsy syndrome where dysarthria lasted for more than 4 years before the onset of signs of LMN impairment.
Case 6: 60-years old woman with a Flail Leg (FL), associated with sicca syndrome whose treatment was not effective, rapidly progressing to diaphragmatic involvement that required non invasive ventilation, before apparition of few sign of upper limbs impairment.
Case 7: 42-years-old man with chronic right brachial monoparesis, which remained isolated with focal involvement of LMN in C6-T1 levels for 6 years before progressing to typical ALS.
Discussion: Respiratory muscle weakness rarely presents as the initial symptom of ALS. It accounts for only 1-3% of all ALS cases and only 14% of these patients presented acutely and required emergency intubation. ALS/polymyositis share immunologic features and ALS/inclusion body myositis in her hereditary form, may share mutations in the gene for valosin containing proteinandTDP-43. The occurrence of isolated bulbar symptoms due to UMN damage, for at least 3-4 years is more suggestive of primary lateral sclerosis, in our patient the bulbar palsy remained isolated without signs of LMN impairment for more than 4 years. In general, FL had a relatively good prognosis, with a long median time from onset to the use of NIV and 5-years survivals rates, but our patient progressed rapidly to respiratory failure and death. More than 80% of patients with arm onset develop ipsilateral or contralateral leg symptoms at 60 months after onset. Studies showed that some motor neurons remain resistant to the effects of the disease process to the end. In our last case the focal involvement remained isolated more than 6 years which was a real dilemma for diagnosis of ALS and only the repetition of ENMG test leaded to diagnosis. Finally, in our Knowledge no cases of ALS associated with neuromyotonia have been reported previously.
Conclusion: An unusual onset of ALS necessarily leads to differential diagnoses, but clinical and ENMG monitoring remains the key to diagnosis.
eP03.03.11Cognitive Decline in ALS Patients. MoCA Score and CSF Biomarkers (T-Tau, P-Tau, β 42)
Sanchez Soblechero A1, Portela Sanchez S1, Catalina Alvarez I1, Muñoz Blanco J1
1ALS-Neuromuscular Unit. Hospital General Universitario Gregorio Marañón, Madrid, Spain
Introduction:
Cognitive decline in ALS patients is of utmost recognition as it implies difficulties in treatment adherence and shorter survival. Over 50% of ALS patients presents with cognitive disfunction or behavioral disturbances along Frontotemporal Dementia (FTD) spectrum, even 15% meet FTD criteria. Montreal Cognitive Assessment (MoCA) is considered a reliable test to evaluate cognitive decline in ALS patients in clinical practice.
In last decades biomarkers have been evaluated in ALS in cerebrospinal fluid (CSF). Amyloid- β (β42), Total-Tau (T-tau) and phosphorylated Tau (P-Tau) are the hallmarks of Alzheimer Disease. Due to its clinical accessibility some studies have tried to use them as diagnostic of prognostic biomarkers, although with contradictory results. It is thought that elevated T-tau and low β42 might be related with a shorter survival time.
Only few studies have evaluated cognitive profile and CSF biomarkers simultaneously. The objective of this study is to evaluate β42, T-Tau and P-Tau levels in CSF of ALS patients at diagnosis and correlate them with initial cognitive decline evaluated by MoCA test.
Methods: We enrolled patients fulfilling the El Escorial Criteria for ‘possible’, ‘probable’ or ‘definite’ ALS attended in our ALS-Neuromuscular Unit from June 2021 to November 2021 who underwent a lumbar puncture for routine laboratory analysis. CSF biomarkers were determined by LUMIPULSE® automated platform (a fully automated chemiluminescent enzyme immunoassay (CLEIA)) technique standardized in Alzheimer Disease.
We recorded prospectively demographic (age, sex, educational level (low<14 years)) and clinical data in first visit: initial symptoms (spinal, bulbar, generalized) and scores in clinical scales (MoCA test, ALSFRS-R, MITOS, KINGS).
Percentages, median and standard deviation were used for clinical data comparison. We used lineal regression (ANOVA) to assess correlation between MoCA score and CSF biomarkers, adjusted by age and educational level.
Results: We included 11 patients (median age 65.6±9.5), 7 (63%) were male and 6 (54.6%) presented with spinal phenotype. Median time from initial symptoms to diagnosis were 11 months. CSF was available in 11 patients. Mean values were 353,36(±152,97)pg/mL for T-Tau, 32,37(±9,46)pg/mL for P-Tau and 701,91(±234,93)pg/mL for β42. T-Tau was higher than cut point (404pg/mL) in three patients and β42 was lower than cut point (599pg/mL) in four different patients. β42/β40 ratio was low in only one patient. None of the patients was diagnosed as Alzheimer Disease.
MoCA score was available in 9 patients (81%). Median score was 20.1±5. 6 patients (66.6%) scored lower than cut point (<25). Two patients (18%) fulfilled FTD criteria. There is an inverse correlation between elder age and MoCA score (β=-0.396 (IC:-0.71-0.08);p=0.02).
There is no correlation between any of the CSF biomarkers (T-Tau: β=-0.028 (IC: -0.065-0.009);p=0.119); P-Tau: β=-0.202 (IC:-0.638-0.234);p=0.3; β42: β=-0.007 (IC:-0.024-0.01);p=0.369) and MoCA score. Linear regression (R2) of T-Tau is 0.558. There are no significant changes in these correlations adjusted by age and educational level.
Discussion: Cognitive decline was present in 66% of patients in our sample, two of them fulfilling FTD criteria. Initial MoCA test in ALS might be influenced by age. More studies are warranted to evaluate correlation among CSF biomarkers, especially T-tau, and MoCA score.
eP03.04.01Effectiveness of Eculizumab Treatment for Generalized Myasthenia Gravis in Us Clinical Practice: gMG Registry Data
Muppidi S1, Narayanaswami P2, Cutter G3, Nowak R4, Greene E5, Mozaffar T6, Rodrigues E7, Korideck H7, Howard Jr J8
1Department of Neurology & Neurological Sciences, Stanford University School of Medicine, Palo Alto, USA, 2Department of Neurology, Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, USA, 3Department of Biostatistics, School of Public Health, University of Alabama at Birmingham, Birmingham, USA, 4Department of Neurology, Yale University School of Medicine, New Haven, USA, 5Department of Neurology, Houston Methodist Neurological Institute, Houston, USA, 6Department of Neurology, University of California, Irvine, USA, 7Alexion, AstraZeneca Rare Disease, Boston, USA, 8Department of Neurology, University of North Carolina at Chapel Hill, Chapel Hill, USA
INTRODUCTION: Clinical trial data have demonstrated that eculizumab improves clinical outcomes in individuals with refractory generalized myasthenia gravis (gMG). Data from clinical practice on treatment patterns and effectiveness of eculizumab in gMG are being collected by the Alexion-sponsored gMG Registry.
OBJECTIVE: To describe treatment outcomes and safety (serious adverse events [SAEs]) in current gMG Registry participants during eculizumab therapy in clinical practice in the USA.
METHODS: Starting in December 2019, adults with gMG who had ever received eculizumab enrolled in the gMG Registry (NCT04202341). After obtaining consent, demographic data, myasthenia gravis activities of daily living (MG-ADL) total score and Myasthenia Gravis Foundation of America (MGFA) classification were collected from medical records at two time points: in the 6 months before eculizumab initiation and at first gMG Registry assessment after eculizumab treatment initiation (at Registry enrollment). SAEs in patients receiving eculizumab during Registry participation were recorded.
RESULTS: As of November 29, 2021, in total, 111 adults with gMG had enrolled in the gMG Registry (male, 52.3%; mean [range] age at MG diagnosis, 56.1 [16.0–92.0] years). The mean (range) time from eculizumab initiation to gMG Registry enrollment was 2.0 (0.0–6.7) years. Mean (standard deviation) MG-ADL total score decreased from 8.3 (3.6) before eculizumab initiation to 3.1 (3.6) after eculizumab treatment. MGFA classification improved with eculizumab treatment: class I, 0.0% of patients before eculizumab initiation versus 28.9% after eculizumab treatment; class II, 36.8% versus 55.3%; class III, 52.6% versus 15.8%; class IV, 10.5% versus 0.0%. The median MGFA class was III before eculizumab initiation and II after eculizumab treatment. One SAE (invasive pulmonary aspergillosis) considered by the investigator to be related to eculizumab was reported in a patient who died, and two serious infections considered unrelated to eculizumab were reported (COVID-19/pneumonia and urinary tract infection); there were no meningococcal infections. Two patients died of causes considered to be unrelated to eculizumab (lung adenocarcinoma and acute congestive heart failure/myocardial infarction).
CONCLUSION: These data from the gMG Registry provide evidence of the effectiveness of eculizumab for the treatment of gMG in clinical practice across the USA, demonstrating a benefit/risk profile consistent with that observed in clinical trials.
eP03.04.02Pharmacokinetics and Pharmacodynamics of Nipocalimab in Healthy Participants and Patients with Generalized Myasthenia Gravis
Zhu Y1, Valenzuela B2, Dosne A2, Peréz-Ruixo J2, Leu J1, Karcher K3, Ramchandren S3, Ling L3, Sun H3, Vermeulen A2, Samtani M4
1Janssen Research & Development, LLC, Spring House, United States, 2Janssen Research & Development, Beerse, Belgium, 3Janssen Research & Development, LLC, Titusville, United States, 4Janssen Research & Development, LLC, Bridgewater, United States
Background: Nipocalimab is a fully human IgG1λ monoclonal antibody that blocks the neonatal Fc receptor (FcRn), inhibits IgG recycling, and lowers circulating IgG, including IgG-based pathogenic autoantibodies. The potential therapeutic benefit of nipocalimab for treatment of autoimmune diseases has been demonstrated in a Phase 2 (Ph2) study in patients with generalized myasthenia gravis (gMG). This analysis was conducted to build the quantitative relationship between pharmacokinetics (PK) and pharmacodynamics (PD) of nipocalimab to inform the dose selection for a Ph3 study in adult patients with gMG.
Methods: Nipocalimab PK, PD (serum IgG), and/or efficacy (MG - Activities of Daily Living [MG-ADL]) data were obtained from Ph1 studies in healthy participants and a Ph2 study in patients with gMG (NCT03772587). Population PK/PD/efficacy modeling (NONMEM version 7.4.4) and simulation (Simulo® 8.0 Expert version) analyses were conducted to evaluate the relationship between PK, IgG lowering, and MG-ADL.
Results: Nipocalimab exhibited one-compartment PK with nonlinear target-mediated drug disposition. Asymptomatic, self-limited, recoverable, and dose-dependent decreases in total serum IgG concentrations of up to ~85% from baseline were observed across the Ph1 and Ph2 studies. In contrast, no nipocalimab-related changes in total IgM, IgA, and IgE were observed. The observed PK and IgG lowering profiles of nipocalimab were generally comparable between healthy participants and patients with gMG. For example, the observed mean IgG lowering was 74% from baseline on Day 10 in healthy participants (n=6) versus 72% on Day 15 in patients with gMG (n=13) following a single 30 mg/kg IV infusion. Consistent with Ph1 results, rapid, dose-dependent mean IgG lowering was observed 1 week after the initial dose across all dose groups in the Ph2 study (from 5 mg/kg every 4 weeks [q4w] up to 60 mg/kg q2w). A maximum IgG lowering (~80%) was observed at Week 2 in the highest dose groups (60 mg/kg single-dose and q2w). Similar dose-dependent reductions in all IgG subclasses (IgG1, IgG2, IgG3, IgG4) and in pathogenic autoantibodies against acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) were also observed across nipocalimab treatment groups. The modeling results suggest a quantitative exposure-response relationship between systemic exposure to nipocalimab and IgG lowering. Among all intrinsic and extrinsic factors evaluated, only patient body weight was found to be a clinically relevant covariate that would impact the PK of nipocalimab and the associated IgG lowering. The simulation results demonstrate comparable total IgG-time profiles between healthy participants and patients with gMG for the same dose regimen of nipocalimab. The modeling and simulation results also indicate that the IgG lowering was associated with the improvements in MG-ADL in patients with gMG. This link between IgG lowering and MG-ADL improvement was consistent with the relationship reported for other FcRn inhibitors.
Conclusion: Total serum IgG lowering is a good predictor of efficacy. The PK/PD/efficacy model developed was used in the dose regimen selection for a Ph3 gMG study.
eP03.04.03Zilucoplan in Myasthenia Gravis: Safety and Tolerability Results From the Phase 3 Randomised RAISE Study
Leite M1, Vu T2, Hussain Y3, Kaminski H4, Utsugisawa K5, Genge A6, Mantegazza R7, Brock M8, Boroojerdi B9, Vanderkelen M10, de la Borderie G11, Duda P12, Howard Jr J13
1Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK, 2Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 3Department of Neurology, Dell Medical School, The University of Texas at Austin, Austin, USA, 4George Washington University, Washington DC, USA, 5Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 6Clinical Research Unit, The Montreal Neurological Institute, Montreal, Canada, 7Neuroimmunology and Neuromuscular Diseases Department, Fondazione Istituto Neurologico, Italy, 8UCB Pharma, Raleigh, USA, 9UCB Pharma, Germany, 10UCB Pharma, Belgium, 11UCB Pharma, Belgium, 12UCB Pharma, Cambridge, USA, 13The University College of North Carolina at Chapel Hill, Department of Neurology, Chapel Hill, USA
Background: Patients living with generalised myasthenia gravis (gMG) experience high treatment burden due to the need for chronic immunosuppressant and corticosteroid therapy, which can be associated with significant adverse effects and a slow or incomplete response. There is a clear need for well-tolerated new treatments. In AChR Ab+ gMG, a proportion of pathogenic AChR autoantibodies precipitate the classical complement pathway, which converges at complement C5, leading to complement-mediated damage of the neuromuscular junction and subsequent impaired synaptic transmission. Zilucoplan (ZLP) is a C5 inhibitor that has shown a promising efficacy and safety profile in a Phase 2 study in patients with gMG. RAISE further investigated the efficacy, safety and tolerability of ZLP in subjects with AChR+ gMG.
Methods: RAISE (NCT04115293) was a Phase 3, multicentre, randomised (1:1), double-blind, placebo-controlled study. Participants had a diagnosis of MGFA Class II–IV gMG, confirmed AChR autoantibodies, MG Activities of Daily Living (MG-ADL) score of ≥6 and Quantitative Myasthenia Gravis (QMG) score of ≥12. Randomisation was stratified by MG-ADL and QMG baseline scores and geographical region. Subjects self-administered daily subcutaneous doses of 0.3mg/kg ZLP or placebo over 12 weeks. The primary efficacy endpoint was change from baseline (CFB) at Week 12 in MG-ADL score. Secondary efficacy endpoints included CFB to Week 12 in QMG, Myasthenia Gravis Composite score, and Myasthenia Gravis Quality of Life 15-Item Scale. The key safety and tolerability endpoint was incidence of treatment-emergent adverse events (TEAEs). Additional safety assessments included TEAEs leading to discontinuation and adverse events of special interest, laboratory tests and immunogenicity. Pharmacodynamic endpoints assessed the effect of zilucoplan on the classical complement pathway activation and C5 levels.
Results: The study has completed enrolment with 174 study participants randomized. Data are being analyzed and results will be available for presentation at the congress, with a focus on safety and tolerability, including safety-related secondary endpoints (incidence of treatment-emergent AEs), analysis of other safety endpoints (e.g. TEAEs leading to discontinuation, laboratory tests and immunogenicity) and adverse events of special interest.
Conclusions: The RAISE study will report the efficacy, safety, and tolerability of ZLP as a chronic treatment for AChR+ gMG. Funded by UCB Pharma.
eP03.04.04Baseline Characteristics of Patients with Myasthenia Gravis Enrolled in an Expanded Access Programme for Efgartigimod
De Bleecker J1, Löscher W2, Schneider-Gold C3, Verschuuren J4, Gelinas D5, Rahman O5, Lambert E5, Van Hoorick B5, Beauchamp J5, Mantegazza R6
1University Hospital and St. Lucas General Hospital, Ghent, Belgium, 2Department of Neurology, Medical University Innsbruck, Innsbruck, Austria, 3St. Josef Hospital, Ruhr-University of Bochum, Bochum, Germany, 4Department of Neurology, Leiden University Medical Center, Leiden, Netherlands, 5argenx, Ghent, Belgium, 6Carlo Besta Neurological Institute, Milan, Italy
Background: Generalised myasthenia gravis (gMG) is a rare, immunoglobulin G (IgG) autoantibody-mediated disease that results in disability and reduces patients’ quality of life. Efgartigimod is a human IgG1 antibody Fc-fragment that blocks the neonatal Fc receptor, thereby decreasing IgG recycling and reducing IgG autoantibody levels. In the phase 3 ADAPT study, efgartigimod was shown to be well tolerated and efficacious in patients with gMG. The ongoing, multinational expanded access programme (EAP) for efgartigimod addresses the unmet need for patients with gMG not enrolled in a clinical trial, who are unable to effectively manage their disease with currently approved therapies.
Methods: The EAP is managed according to country-specific or individual clinic protocols, with the purpose of following local clinical practice (US protocol: NCT04777734). Recruitment is ongoing in Austria, Belgium, Germany, Italy, Spain and the Netherlands. Following the approval by the US Food and Drug Administration in December 2021 of efgartigimod for the treatment of gMG in patients who are AChR-Ab+, recruitment into the EAP has ceased in the US.
Adult patients are eligible for the EAP if they have had a confirmed diagnosis of gMG and need treatment, regardless of antibody status, and have had a documented IgG level of ≥4 g/L 1 month prior to screening. Some countries require patients to have a total MG-ADL score of ≥5 at screening (with >50% of the total score due to non-ocular symptoms). Enrolled patients receive intravenous efgartigimod (10 mg/kg) on an individualised treatment cycle dosing pattern.
Results: As of 19 January 2022, 30 patients have been enrolled in the EAP. The majority are female (n=18), with most patients aged between 45 and 64 years (n=14). There are a similar number of patients aged 25–44 years (n=7) and ≥65 years (n=6), with 3 patients in the younger group (18–24 years). Median time from diagnosis was 4 years (n=29). Nineteen patients are AChR-Ab+, 8 are seronegative and 3 are MuSK-AB+. Twenty-six patients had IgG levels >6 g/L. Most patients are MGFA class III (n=12), with 6 patients each included in class II and class IV. Plasma exchange had previously been received by 10 patients, with 11 patients having previously received intravenous immunoglobulin, including 4 patients who had received both. A total of 6 patients had previously received eculizumab and 5 patients had previously received rituximab. A total of 19 patients had experienced MG crisis and/or been hospitalised owing to their gMG in the 12 months prior to screening. Additionally, 20% of patients had a history of thymectomy and 67% of patients had ≥2 comorbidities.
Conclusions: Efgartigimod was well tolerated and efficacious in the phase 3 ADAPT trial. The EAP offers an important treatment option for patients lacking an effective management strategy for their gMG, who are not enrolled in an ongoing clinical trial.
eP03.04.05Promise-MG: Results of a Multicenter Comparative Effectiveness Study of Myasthenia Gravis Treatments
Narayanaswami P1, Sanders D, Guptill J, Bibeau K, Krueger A, Venitz J, Li F, Liu B, Desai R, PROMISE-MG Study Group
1Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, United States
Background: Immunosuppressive treatment of myasthenia gravis (MG) is influenced by disease related and other patient factors, co-morbidities and treatment availability/cost. There are no studies of the comparative effectiveness of immunosuppressive treatments used for MG.
Objectives: To compare:
1. Patient-reported outcomes (PRO) and clinician-reported outcomes (CRO) in MG patients who receive either azathioprine (AZT) or mycophenolate mofetil (MMF)
2. PROs and CROs in MG patients receiving an adequate dose/duration of AZT or MMF vs. patients receiving these agents at less than adequate doses/duration (as defined by the International Consensus Guidance for MG treatments).
Methods: Prospective, observational multicenter cohort study. Patients were recruited when first seen at a study site and followed for up to 36 months. There were no study-imposed interventions - patients received conventional care. The primary outcome was the greatest change from baseline in the Myasthenia Gravis Quality of Life- 15, revised (MG-QOL15r). The co-primary outcome measure was a composite CRO measuring clinical improvement and treatment adverse effects (AEs) and was defined as achieving the Myasthenia Gravis Foundation of America Post-Intervention Status Minimal Manifestation (MM) or better with AEs no greater than Grade 1 CTCAE (Common Terminology Criteria for Adverse Events). Secondary outcomes were the greatest changes from baseline in the MG Composite (MGC), MG-Activities of Daily Living Scale (MG-ADL), MG- manual muscle testing score (MG-MMT), and hospitalizations for MG. Overlap weight propensity scores were used in a weighted generalized linear regression model with appropriate link functions.
Results: Of 167 patients enrolled, 82 received either MMF (48) or AZT (34). Similar proportions in both treatment groups had clinically meaningful changes in the outcome measures. (MMF vs. AZT: MG-QOL15r: 75% vs. 65%, MG-ADL: 90% vs. 82%, MGC: 92% vs. 79%, MG-MMT 93% vs. 74 %). Mean difference in MG-QOL15r score reduction from baseline was 2.8 (95% confidence interval 0.56- 5.05, p=0.01), favoring MMF; this difference was not clinically meaningful. There was no difference in the co-primary outcome measure (2% more participants receiving AZT achieved the composite outcome, CI -19% to 14%). AEs were more frequent with AZT, observed in 11/34 (32%) patients (hepatotoxicity 5, influenza-like reaction 3, hematologic AEs 2, pancreatitis 1). AEs to MMF were seen in 9/48 (19%, gastrointestinal disturbances 7, renal impairment 1, hemoglobin drop 1). A similar proportion of patients who received AZT 1-2 mg/kg/day achieved meaningful improvements in all outcome measures, (AZT ≥2 mg/kg/day vs. AZT ≥1 mg/kg/day: MG-QOL15r: 85% vs. 71%, MG-ADL: 85% vs. 86%, MGC: 71% vs. 64%, MG-MMT: 71% vs. 71%).
Conclusions: Both MMF and AZT produced clinically meaningful improvement in patients with MG, and there was no clinically meaningful difference between them. AEs were more frequent in those receiving AZT. A lower than recommended dose of AZT (1-2 mg/kg/day) may be effective. Exploratory outcomes including time to improvement, demographics and outcomes in the non AZT/MMF treated patients, sub-group analyses of early/late onset MG, acetylcholine receptor antibody-positive vs. negative patients, and correlations between outcome measures over time will be presented.
Narayanaswami and Sanders contributed equally
eP03.04.06The Association Between QMG Scores and Health-related Quality of Life in Myasthenia Gravis Patients
Dewilde S2, Qi C1, Janssen B3, Gelinas D1, Brauer E1, Phillips G1, Ollendorf D4
1Argenx, Boston, United States, 2Services in Health Economics, Brussels, Belgium, 3Erasmus University Rotterdam, Department of Medical Psychology and Psychotherapy, Rotterdam, Netherlands, 4Center for the Evaluation of Value and Risk in Health, Tufts University Medical Center, Boston, United States
Introduction: Generalized myasthenia gravis (gMG) is a neurological condition affecting patients’ muscle strength and often results in problems with energy, vision, swallowing/chewing, limb weakness, and breathing. Patients also frequently suffer from mental health problems such as anxiety and depression. Utility values (a measure of health-related quality of life (HRQoL)) of gMG patients are lower than in the general population; however, a relationship to gMG severity has not yet been established. The aim was to estimate the association between gMG symptom scores and utility.
Methods: The Quantitative Myasthenia Gravis (QMG) score is a quantitative assessment of patient strength in 13 questions, based on the endurance of key muscle groups (eyes, facial muscles, swallowing, speech, arms, hands, neck, legs, vital capacity). Total scores range from 0 to 39, with higher scores indicating more severe disease. The EuroQol-5-Dimension 5-Level (EQ-5D-5L) is a 5-question HRQoL instrument of which responses can be summarized in a utility value using a country-specific value set. Utility values range from 1=full health to 0=dead and values can also be negative (to -1). QMG was measured simultaneously with the EQ-5D-5L on a (bi)weekly basis for up to 26 weeks in ADAPT, a Phase-3, multicenter, randomized, placebo-controlled clinical trial among adult gMG patients who were randomized to efgartigimod in combination with conventional therapy (CT) versus CT alone.
Descriptive statistics were reported for QMG and EQ-5D-5L utilities at baseline and at follow-up. A Generalized Estimating Equation (GEE) statistical model with a normal distribution, identity link and compound symmetry variance-covariance matrix was estimated to predict utility values based on the patient’s QMG score and treatment received.
Results: 167 patients (84 efgartigimod, 83 CT) contributed a total of 3032 simultaneous measurements of QMG and EQ-5D-5L, of which 167 were at baseline and 2867 at follow-up (all time points combined). The mean QMG scores declined from 16.2 at baseline to 11.7 at follow-up for efgartigimod; corresponding values were 15.5 and 14.5 for CT. Mean EQ-5D utility values at baseline were 0.650 for efgartigimod and 0.605 for CT. At follow-up, utility values increased to 0.756 and 0.651, respectively.
A statistical association was evaluated with the GEE model, which showed a significant improvement in utility values per unit improvement in QMG score. Each additional unit increase in QMG total score lead to a utility decline of 0.019 (p<0.001). In addition, for the same QMG score, a statistically significant improvement of 0.062 (p=0.0275) in utility was found for patients taking efgartigimod compared to CT. This finding suggests that efgartigimod provided an additional benefit to gMG patients that was not captured with the QMG score alone, and this benefit was characterized in the comparative proportion of efgartigimod versus CT patients reporting no or mild problems in mobility (75% vs 59%), self-care (88% vs 64%), usual activities (72% vs 53%), pain/discomfort (80% vs 70%) and anxiety/depression (91% vs 83%).
Conclusions: Among gMG patients, QMG was significantly associated with EQ-5D utility scores. QMG scores alone were not sufficient to capture the utility gained from efgartigimod therapy.
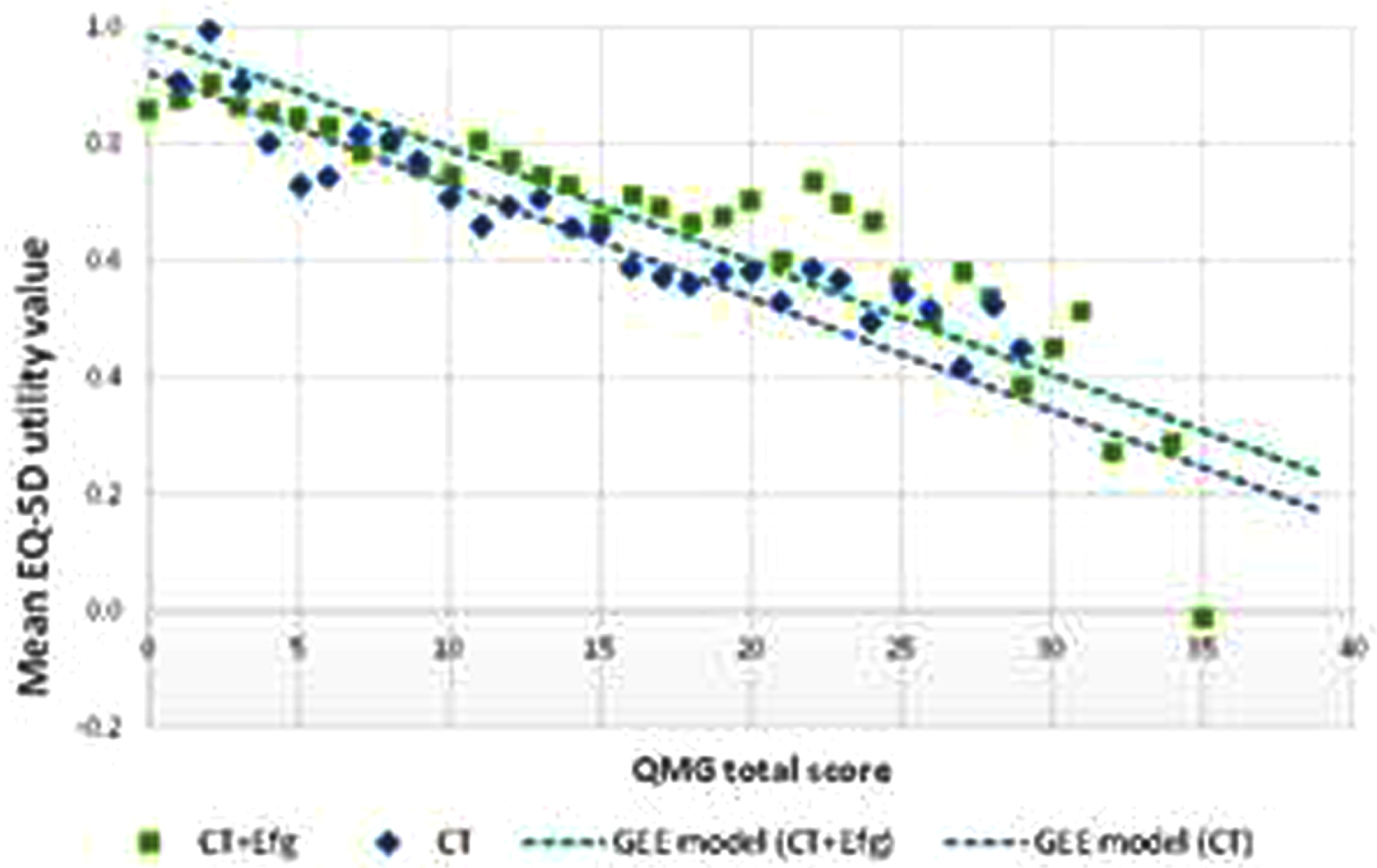
eP03.04.07Oral Tobacco, but Not Smoking, Is Associated With an Increased Risk of Myasthenia Gravis.
Petersson M1, Jons D2, Feresiadou A3, Ilinca A4, Lundin F5, Johansson R6, Budzianowska A7, Roos A8, Kågström V9, Gunnarsson M10, Sundström P11, Klareskog L1, Olsson T1, Kockum I1, Piehl F1,12, Alfredsson L1, Brauner S1,12
1Karolinska Institutet, Stockholm, Sweden, 2Sahlgrenska University Hospital and University of Gothenburg, Gothenburg, Sweden, 3Uppsala University Hospital and Uppsala University, Uppsala, Sweden, 4Skåne University Hospital and Lund University, Malmö, Sweden, 5Linköping University Hospital and Linköping University, Linköping, Sweden, 6Karlstad Central Hospital, Karlstad, Sweden, 7Ryhov Hospital and Linköping University, Jönköping, Sweden, 8Östersund Hospital and Umeå University, Östersund, Sweden, 9Sundsvall Hospital, Sundsvall, Sweden, 10Örebro University Hospital and Örebro University, Örebro, Sweden, 11University Hospital of Umeå and Umeå University, Umeå, Sweden, 12Karolinska University Hospital, Stockholm, Sweden
Introduction: Myasthenia gravis (MG) is a complex disease, where lifestyle and environment are believed to contribute to the majority of the risk. Still, few such factors have been identified. Smoking and nicotine use have been associated to disease risk across several autoimmune diseases. While rate of smoking is comparatively low in Sweden, use of oral tobacco (snuff) is more frequent, containing up to four times higher nicotine concentrations per dose compared to cigarettes. Here, we examine the influence of smoking and snuff on MG disease development.
Methods: In a cross-sectional nation-wide Swedish study conducted 2018-2019, patients with MG were invited to submit an extensive environmental and lifestyle questionnaire. Cases were categorized based on age at inclusion, sex and area of residence, and matched to up to 15 randomly selected population controls per case. Age at disease onset was set as index age. Smoking and snuff use related to disease onset was investigated with multivariate conditional logistic regression, adjusting for sex, area of residence, index age and year. Nicotine exposure in relation to acetylcholine receptor autoantibody status (anti-AChR) was also investigated.
Results: Out of 1485 invited patients, 1077 participated, representing approximately 42% of the total Swedish MG population. Of these, 1062 were matched to 8545 controls. The average (SD) age at inclusion was 64 (16) years, disease duration was 16 (15) years and 53% were female. Comparing smoking habits, we did not observe an association to disease risk for smoking at onset or ever smoking prior to disease onset (OR=0.96, 0.80-1.2, P=0.70 and OR=0.99, 0.86-1.2, P=0.94, respectively). In contrast, use of snuff at onset was associated to increased risk (OR=1.9, 1.4-2.5, P<0.001). Among non-current smokers with available anti-AChR data (n=402), the effect of snuff use was even more prominent (OR=2.5, 1.6-3.9, P<0.001).
Conclusion: In this nationwide, prevalent study we observed that use of oral snuff but not smoking was associated with increased risk of MG disease development. This contrasts with findings in other autoimmune diseases, such as multiple sclerosis and rheumatoid arthritis, where an opposite relation is observed, possibly due to non-nicotine mediated effects. It may therefore be speculated that the association found in MG is related to the higher nicotine exposure with snuff.
eP03.04.08Productivity Losses for Generalized Myasthenia Gravis Patients and their Caregivers: Association with Disease Severity
Jacob S1, Dewilde S2, Qi C3, Saccà F4, Meisel A5, Palace J6, Claeys K7, Mantegazza R8,9, Paci S10, Phillips G3
1University Hospitals Birmingham and Institute of Immunology and Immunotherapy, Birmingham, UK, 2SHE, Brussels, Belgium, 3argenx, Boston, USA, 4University of Naples Federico II, DNSRO Department, Naples, Italy, 5Charité Universitätsmedizin Berlin, Department of Neurology, Neuroscience Clinical Research Center, Berlin, Germany, 6John Radcliffe Hospital, Department of Clinical Neurology, Oxford, UK, 7University Hospitals Leuven, Leuven, Belgium, 8Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy, 9Associazione Italiana Miastenia e Malattie Immunodegenerative, Milan, Italy, 10argenx, Ghent, Belgium
OBJECTIVE: Generalized Myasthenia Gravis (gMG) is a rare chronic autoimmune neuromuscular disease resulting in muscle weakness affecting vision, swallowing, speech, mobility, dexterity and respiratory function. Patients suffer from fatigue and frequently experience anxiety and depression. The disease considerably impacts the patient’s ability to work and his/her independence, which may result in needing help from a caregiver to assist with activities of daily living. The objective of this study was to document the burden of gMG in terms of productivity losses experienced by patients and caregivers, and to investigate whether there is an association with disease severity.
METHODS: The MyRealWorld-MG study is a digital, observational, multi-country survey (US, UK, Canada, Italy, Germany, Spain, Japan) among adult gMG patients. Patients downloaded a mobile application onto their phones and entered background characteristics, self-assessed their disease severity with the Myasthenia Gravis Activities of Daily Living (MG-ADL) instrument; and recorded the number of days of sick leave in the previous month, whether they needed support from a caregiver, the amount of caregiver help needed per week, and whether their caregiver reduced or stopped their own work time. These data were analyzed by categorizations of the total MG-ADL score, which is the sum of the sub-scores on 8 domains and ranges from 0 (no symptoms) to 24 (worst symptoms).
RESULTS: The MyRealWorld-MG survey provided data from 591 MG patients on work productivity losses. Patients had a mean age of 47.4 (SD 14.3), 70% of them were female, 12% has had a thymectomy, 63% suffered from other medical conditions. The number of patient sick days, the proportion of patients needing help from caregivers and caregiver work time lost were all positively associated with increasing MG-ADL scores.
On average, 40.6% of patients had taken sick leave during the past month. This proportion was strongly statistically associated with the MG-ADL score (p<0.001) and is presented in Figure 1. The average duration of sick leave per month was about 2 weeks for patients with MG-ADL<12 and 4 weeks for MG-ADL>=12.
In the survey, 32.4% of the respondents reported needing help from caregivers (family, friend, nurse), and this proportion was also significantly positively associated with increasing MG-ADL total scores (p<0.0001) and is presented in Figure 1. Most caregivers spent about 2 hours per day caring for their patient, regardless of the patient’s disease severity, although the spread in these hours increased with more severe disease. Of these caregivers, 20.8% needed to give up paid employment altogether, and this was notably less frequent among the caregivers of mild patients (14.3% for MG-ADL 0-1 compared to 27.3% for MG-ADL 14-24). In addition, 15.6% of caregivers had to cut down on average 14.4 hours of their worktime.
CONCLUSION: These results suggest that gMG is associated with significant productivity losses from patients and caregivers. Improvements in symptom burden of the disease, as measured with the MG-ADL and covering domains such as talking, chewing, swallowing, breathing, dexterity, mobility, and vision, will lead to reductions in worktime lost, not only by patients but also among their caregivers.
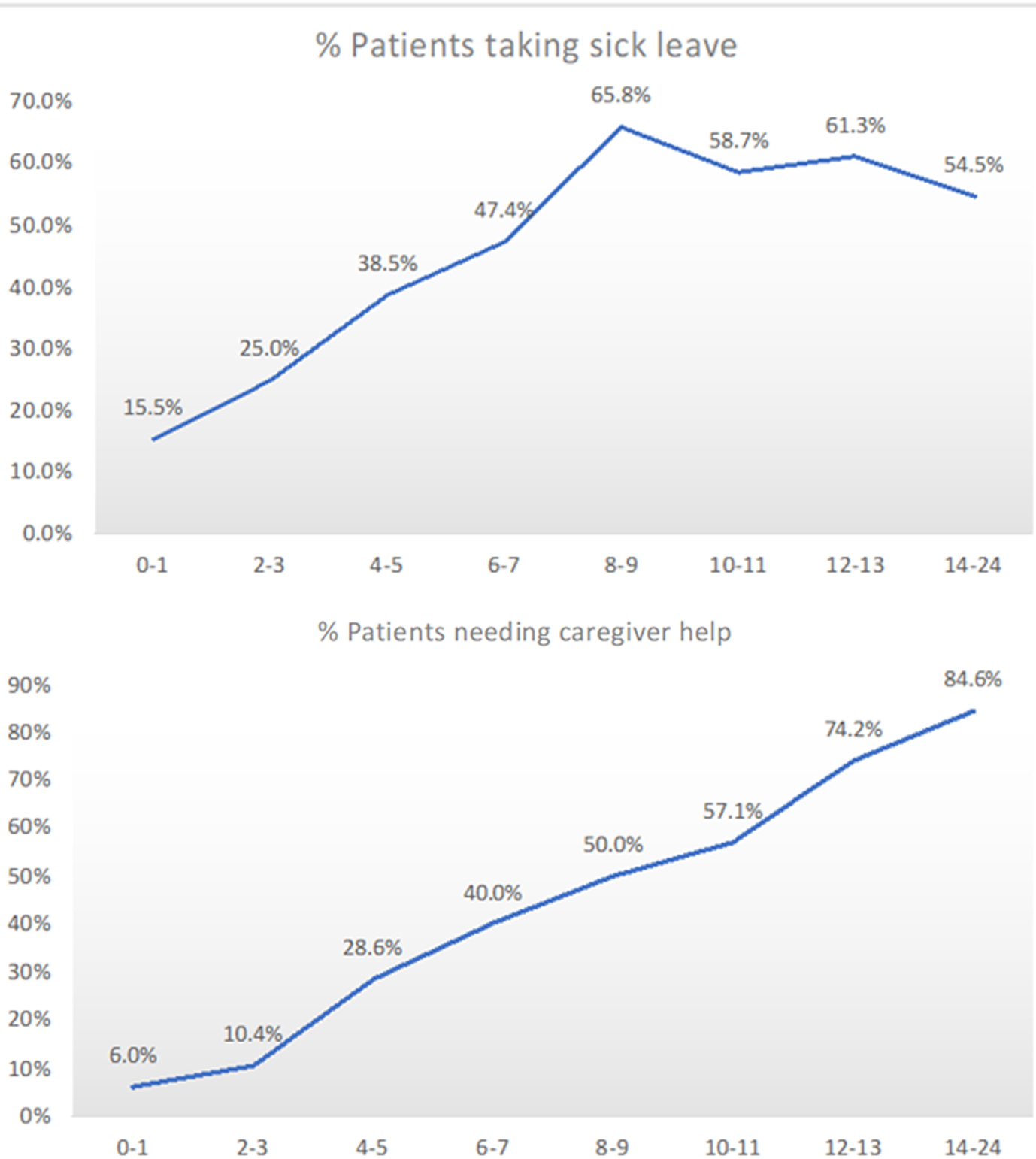
eP03.04.09Efficacy and Safety of Tolebrutinib in Adults with Generalized Myasthenia Gravis: Phase 3 Study Design
Syed S1,2, Kaminski H3, Benoit P4, Chen Y5, Syed S6, Turner T6, Smyth B7, Howard Jr J8
1The Ellen and Martin Prosserman Centre for Neuromuscular Disease, Toronto General Hospital, Toronto, Canada, 2Division of Neurology, University Health Network and University of Toronto, Toronto, Canada, 3Department of Neurology, George Washington University, Washington, USA, 4Sanofi R&D Neuroscience Unit, 1 Avenue Pierre Brossolette, Chilly-Mazarin, France, 5Biostatistics and Programming, Sanofi, 640 Memorial Drive Cambridge, USA, 6Neurology Development, Sanofi, 50 Binney Street Cambridge, USA, 7Global Pharmacovigilance, Sanofi, 55 Corporate Drive Bridgewater, USA, 8Department of Neurology, The University of North Carolina at Chapel Hill, Chapel Hill, USA
Background: Current treatments in generalized myasthenia gravis (gMG) have limited clinical benefit, with slow onset and substantial side-effects. There is an unmet need in gMG for effective treatment options with long-term safety and more feasible modes of administration. Tolebrutinib is an oral, covalent, irreversible inhibitor of Bruton’s tyrosine kinase, an enzyme expressed in multiple cell types implicated in gMG pathogenesis, thereby suggesting it may be an effective treatment option for gMG. Tolebrutinib is currently under development for multiple sclerosis and has shown no safety or tolerability concerns with its oral administration.1 Here, we describe the phase 3 study design (NCT05132569) that will evaluate efficacy and safety of tolebrutinib in participants with gMG receiving standard-of-care (SOC) treatment.
Methods: This multicenter, randomized, double-blind, placebo-controlled study, comprises a screening period (up to 28 days) and a 26-week treatment period; the double-blind period will be followed by a 2-year open-label extension (OLE) (Figure). Key eligibility criteria include age 18-85 years; clinically confirmed diagnosis of gMG with generalized muscle weakness (Myasthenia Gravis Foundation of America [MGFA], Class II-IV disease); seropositivity for anti-acetylcholine receptor (anti-AChR) or anti-muscle-specific kinase (anti-MuSK) autoantibodies, or prior confirmed diagnosis in case of seronegativity (for anti-AChR and anti-MuSK autoantibodies); and Myasthenia Gravis-Activities of Daily Living (MG-ADL) score ≥6 at screening and Day 1. Participants with MGFA Class I/V, history of thymectomy within 6 months before screening, and use of certain treatments without predefined washout periods, will be excluded. Approximately 192 participants will be enrolled to achieve the sample size of 154 randomized participants.
Eligible participants will be randomized (1:1) to receive oral tolebrutinib daily or matching placebo, as an add-on therapy to SOC treatment. The primary endpoint of the double-blind period is the change in MG-ADL score from baseline to Week 26. Secondary endpoints include change from baseline to Week 26 in Quantitative Myasthenia Gravis (QMG) score, Myasthenia Gravis Impairment Index score, and Myasthenia Gravis-Quality of Life 15-item questionnaire score; proportion of participants with ≥2-point reduction in MG-ADL score and ≥3-point reduction in QMG score at Week 26; and safety assessments. All participants who complete the double-blind period will enter OLE to receive tolebrutinib daily while continuing to receive the same SOC treatment. The goal of the OLE is to assess long-term safety, tolerability, and efficacy of tolebrutinib in participants with gMG. An interim analysis, based on the change in QMG score from baseline to Week 12, will be done once 40 randomized participants (only seropositive) complete the Week 12 visit or withdraw early from the treatment/study during the double-blind period.
Discussion: This is a phase 3 randomized, placebo-controlled, double-blind study with a planned interim analysis (using change from baseline in QMG score at Week 12) and an OLE period, designed to generate long-term efficacy and safety data on the use of tolebrutinib as a treatment for gMG. The primary endpoint is the change from baseline in MG-ADL score at Week 26, a validated endpoint for MG management. The study is currently enrolling.
Reference: 1. Reich DS, et al. Lancet Neurol. 2021;20(9):729-738.
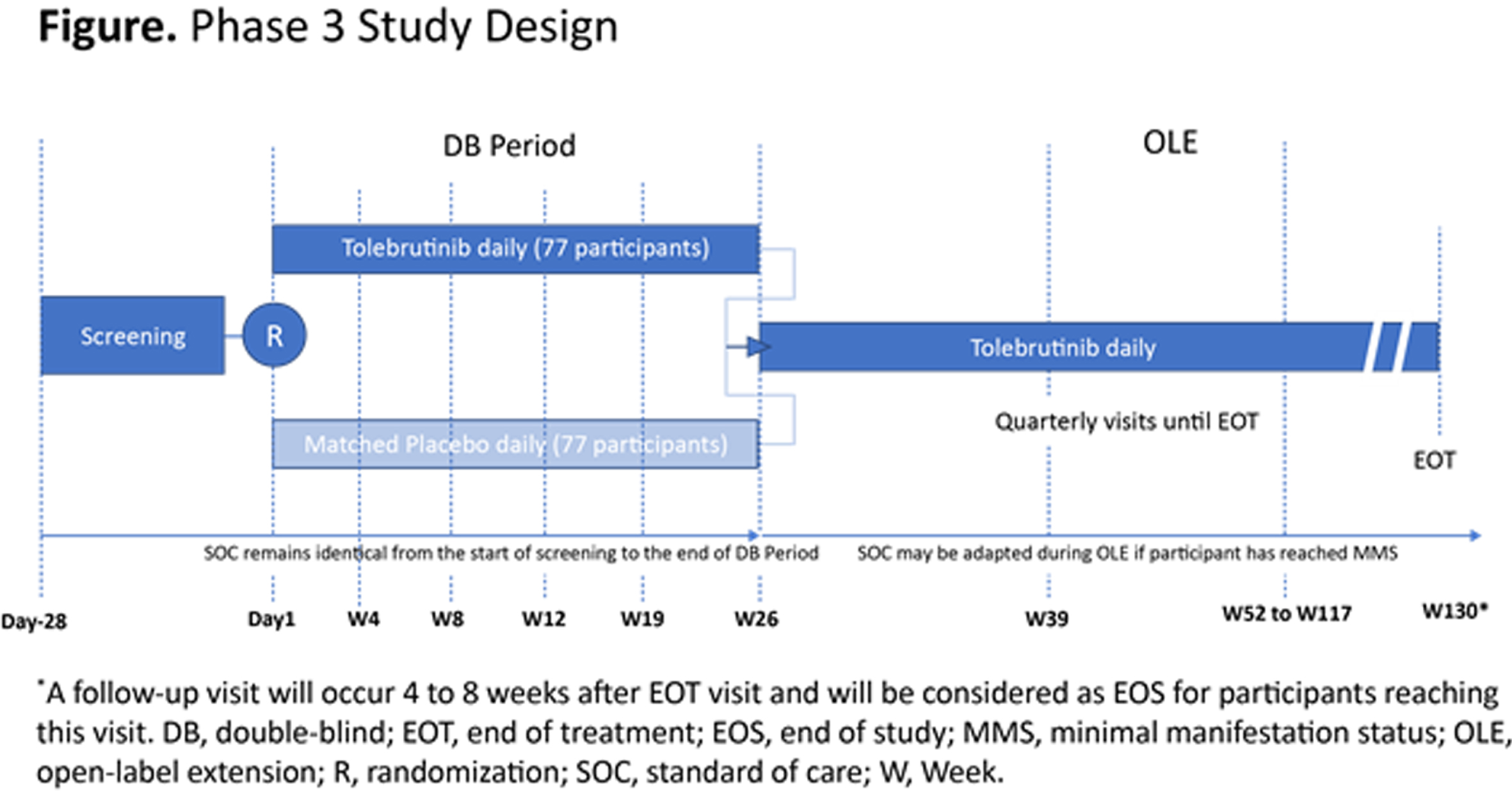
eP03.04.10Serological Diagnostics of MuSK Myasthenia Gravis in South Korea: Comparison of ELISA, RIPA and CBA
Kwon Y1, Woodhall M2, Lee J1, Han J1, Sung J1, Waters P2, Hong Y3
1Department of Neurology, Seoul National University Hospital, 101, Daehak-ro, Jongno-gu, Republic of Korea, 2Neuroimmunology Group, Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, Oxford, UK, 3Department of Neurology, Seoul National University Seoul Metropolitan Government Boramae Medical Center, Republic of Korea
We investigated the diagnostic accuracy of enzyme-linked immunosorbent assay (ELISA) for MuSK antibody in a large cohort of 105 Korean patients with generalized MG who were seronegative for acetylcholine receptor antibody on radioimmunoprecipitation assay (RIPA). We also investigated the clinical context in which the serological test for MuSK antibody is requested in practice. The seropositive rate of ELISA was similar to RIPA (22% and 23%, respectively), but lower than cell-based assay (CBA, 27%). The results of ELISA were in good agreement with those of CBA and RIPA, with the Cohen’s kappa of 0.76 (0.61 – 0.91) and 0.91 (0.78 – 1.0), respectively (95% CI, p < 0.001 for both). There were significant correlations between MuSK antibody concentrations in ELISA and CBA scores (r = 0.44, p < 0.001) and RIPA values (r = 0.44, p < 0.001). Motor neuron disease, particularly bulbar onset, was a major differential diagnosis for MuSK MG with overlapping clinical and electrophysiological features. Combined with a high specificity in other disease controls (0.95), our results support a diagnostic utility of MuSK ELISA as an alternative to CBA and RIPA when the latters are not available.
eP03.04.11Clinical Differences Between Ocular and Generalized Myasthenia Gravis
Axelsen K1, Kjær Andersen R, Witting N, Vissing J
1Copenhagen Neuromuscular Center, Copenhagen, Denmark
Myasthenia Gravis (MG) is an autoimmune disease, that leads to activity induced weakness in skeletal muscle, caused by autoantibodies against acetylcholine receptors (AChR). Some patients present only with ocular symptoms throughout life (ptosis and diplopia) whereas others have more generalized symptoms such as fatigue in extremities, weakness of the neck muscles, dysphagia and dysarthria.
Not many studies have evaluated whether differences regarding demographics and phenotype separate the two groups of MG patients. Such differences may have consequences for the diagnostic process and management of the disease.
This project is an ongoing, retrospective chart review. We aim to include 400 patients who have been diagnosed with MG, and evaluate characteristics such as age, gender, thymoma, disease onset and evolution, antibody profile, medication, treatment response and satisfaction level.
We hypothesize that there will be differences between ocular- and generalized MG patients regarding to the above-mentioned parameters.
So far, our data suggest that there is a larger time delay from symptom onset until diagnosis in the purely ocular group. 13.3% of the ocular patients are diagnosed within two months, where it is 33.3% in the generalized group.
In the ocular group, the proportion of woman seems to be larger as well. Almost 70% women versus 60% in the generalized group. The average number of MG drugs used to treat the ocular patients are 2.5 drugs compared with 3.2 drugs used to treat the generalized patients (p-value = 0.075), which suggest that the treatment is less complicated in the ocular group.
Multiple other comparisons of characteristics will be analyzed and presented when collection of all data is achieved.
eP03.05.01Effects of ERT on Cardiac Function in Classic Infantile Pompe Disease- 19 Years of Follow-up
Scheffers L1, Kok R2, van den Berg L1, van den Hout J1, Boersma E2, Capelle C2, Helbing W2, van der Ploeg A2, Koopman L2
1Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, Rotterdam, Netherlands, 2Department of Pediatric Cardiology, Erasmus MC - Sophia Children’s Hospital, Rotterdam, Netherlands
Objective: Patients with Classic Infantile Pompe disease are born with a hypertrophic cardiomyopathy. Treatment with enzyme replacement therapy (ERT) improves motor function and normalizes cardiac hypertrophy. Despite an initial increase in muscle strength, on the long term most patients develop residual muscle weakness. Potentially, this decrease in function can also be detected within the cardiac muscle. We aimed to assess long-term effects of ERT on cardiac structure and function.
Methods: In total 27 Classic Infantile Pompe patients treated with ERT were included. Cardiac function was assessed at regular time intervals (before and after start of ERT) using conventional echocardiography and a more sensitive and innovative echocardiography technique: myocardial deformation. Separate linear mixed effect models were used to asses temporal changes within the first year and the long-term follow-up period thereafter. Echocardiograms of 103 healthy children served as controls.
Results: A total of 192 echocardiograms were analyzed. Median follow-up was 9.9 years (IQR: 7.5 – 16.3). Mean LVMI before start of ERT was increased, 292.3 g/m2 (95% CI: 202.8 - 381.8, mean Z-score +7.6 ) and normalized after 1 year of ERT 87.3 g/m2 (CI: 67.5 - 107.1, mean Z-score +0.8, p<0.001). Cardiac function measured by shortening fraction was within normal limits before start of ERT, and mean shortening fraction remained within normal limits up to 22 years of follow-up. Cardiac function measured by LV/RV longitudinal and circumferential strain was decreased before start of ERT: mean LV longitudional strain was -12.1% (SD±4.8%, normal = <-16%), mean LV longitudinal strain was -12.2% (SD±5.2, normal = <-16%) and mean RV longitudinal strain was -14.0% (SD±7.0, normal = <-16%), but all views normalized within 1 year after start of ERT. During long-term follow up, LV circumferential strain worsened slightly in Pompe patients (+0.24%/year) compared to controls. LV and RV longitudinal strain did not change significantly over time compared to controls. Despite a statistical differences in Pompe patients compared to controls in the LV circumferential strain, mean LV and RV longitudinal, and LV circumferential strain remained within normal limits up to 19 years of follow-up.
Conclusion: Even with more sensitive echocardiographic myocardial deformation analysis cardiac function normalizes after start of ERT, and seems to remain stable over a median follow-up period of 9.9 years. These results are unique due to the long follow up period, relatively large cohort, and longitudinal use of myocardial deformation analyses to asses cardiac function in more detail. These results suggest that the frequency number of echocardiographic studies can be reduced in the follow-up of Classic Infantile Pompe patients once the cardiac function has been normalized, as this 19 year follow-up period did not show a clinically relevant decline in cardiac function or other structural abnormalities.
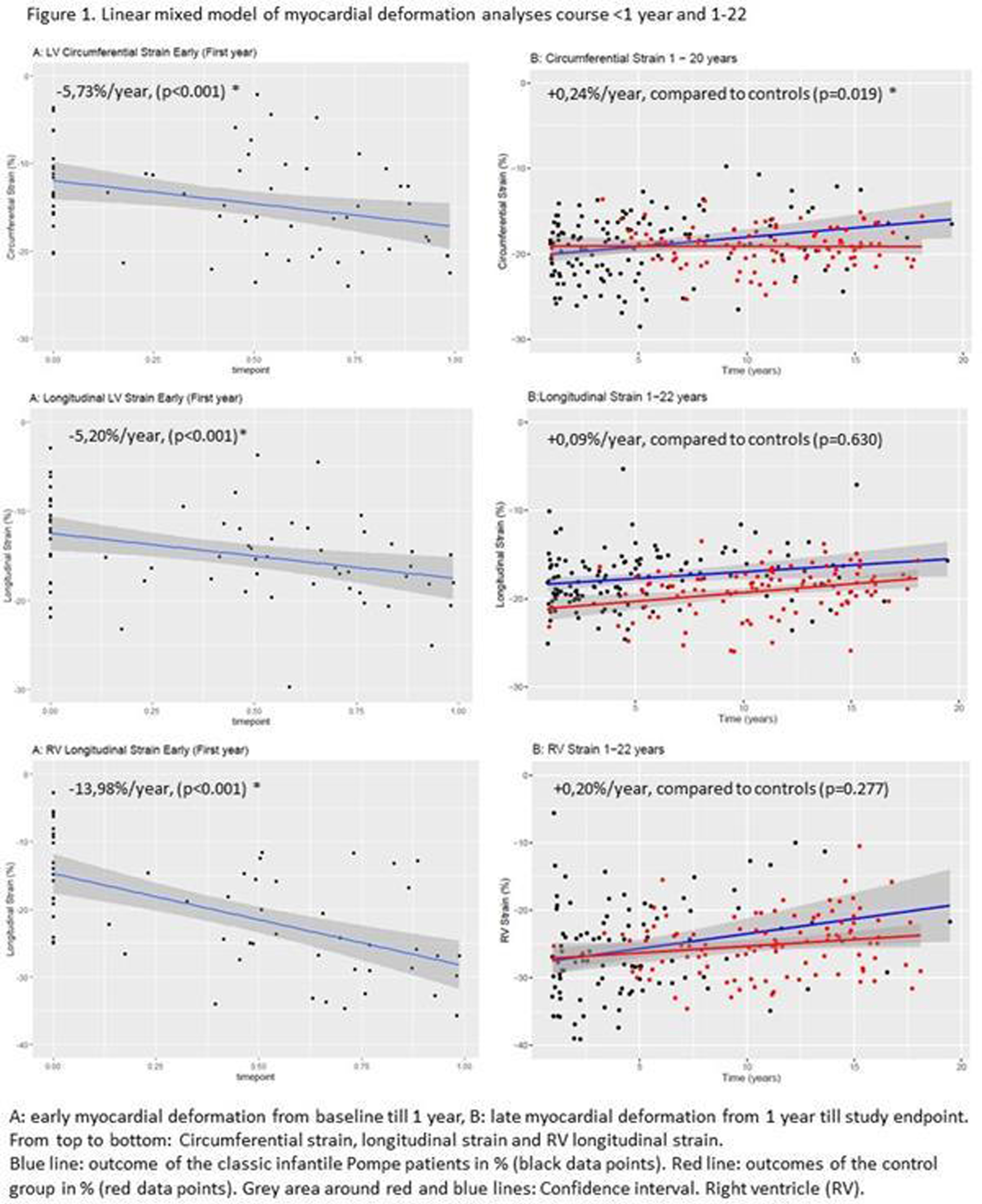
eP03.05.02Safety of Home-Based Infusion of Alglucosidase Alfa in Adults With Late-Onset Pompe Disease
Ditters I1, van Kooten H2, van der Beek N2, van der Ploeg A1, van den Hout H1, Huidekoper H1
1Department of Pediatrics, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, 3015 GD Rotterdam, Netherlands, 2Department of Neurology, Center for Lysosomal and Metabolic Diseases, Erasmus MC, University Medical Center, 3015 GD Rotterdam, Netherlands
Background: Biweekly infusions with alglucosidase alfa (recombinant human alpha-glucosidase), are the cornerstone of treatment in late onset Pompe disease. Home infusion therapy may improve quality of life. In many countries providing enzyme replacement therapy (ERT) at home is not possible due to safety concerns related to the risk of infusion associated reactions (IARs) or logistical constraints. The COVID-19 pandemic has prompted the need to provide ERT for Pompe patients at home. In the Netherlands, currently over 80% of infusions are given at home. Here we present data on the safety of home-based infusions in adult patients with late-onset Pompe disease (LOPD).
Methods: Data on patient descriptives, infusion characteristics and IARs from patients starting ERT between 1999 and 2018 were collected and analyzed. The Dutch infusion schedule for adult late-onset patients differs from the schedule recommended by the pharmaceutical company and is as follows: 0.2, 0.8, 3.5, 10 mg/kg/hour with steps of 30 minutes for the first three steps and 10 mg/kg/hour for the remainder of the infusion. IARs were graded by the healthcare provider. If no classification was available infusions were retrospectively classified using the Common Terminology Criteria for Adverse Events (CTCAE) classification. Descriptive analyses were performed tabulating patient and infusion characteristics as well as types of IARs and actions needed to resolve these.
Results: Data on 18380 infusions with alglucosidase alfa in 121 adult LOPD patients were analysed. 4961 infusions (27.0 %) were given in hospital and 13419 (73.0 %) at home. The majority of infusions (88.4%) was administered using a standard infusion schedule. In 144 (2.9%) of hospital infusions and 113 (0.8%) of home infusions an IAR occurred. Mild IARs occurred in 115 hospital infusions and in 104 infusions at home. Twenty-five moderate IARs were reported in hospital and 8 at home. Very few severe IARs occurred (4 in hospital, 1 at home). The most prevalent symptoms in hospital were itching and chills; at home chills and trembling were most prevalent. The most common most severe interventions taken in hospital in response to an IAR were giving medication (antihistamines), or pausing the infusion and restarting it later. This was also the most common intervention at home. Sixteen infusions (11.1%) in hospital and 6 infusions (5.3%) at home were stopped completely after the IAR occurred. Only one IAR in the home situation required immediate clinical evaluation in hospital. The consecutive infusion after an IAR occurred in hospital, patients most commonly received premedication or the infusion scheme was adapted, whereas at home after the majority of IARs no action was taken. The most common premedication in hospital were an antihistamine and a corticosteroid and at home an antipyretic and antihistamine were most common.
Conclusion: Our data demonstrate that very few IARs occur during alglucosidase alfa infusions in adult patients with LOPD. Very few severe IARs occurred. The majority of IARs at home were mild and did not require additional medical intervention. This demonstrates that alglucosidase alfa can be safely administered in the home situation, provided the appropriate infrastructure is present.
eP03.05.03Minimal Clinical Important Difference for Lung Function and Walking Ability in Adult Pompe Patients.
Lika A1,2,3, Kruijshaar M1, Andrinopoulou E2,3, Van der Beek N4, Rizopoulos D2,3, Van der Ploeg A1
1Department of Pediatrics, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, Rotterdam, The Netherlands, 2Department of Biostatistics, Erasmus MC University Medical Center, Rotterdam, The Netherlands, 3Department of Epidemiology, Erasmus MC University Medical Center, Rotterdam, The Netherlands, 4Department of Neurology, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, Rotterdam, The Netherlands
Introduction: The Minimal Clinical Important Difference (MCID) is the smallest change in a treatment outcome that a patient would consider worthwhile, and usually serves as a benchmark for improvement. Pompe disease, a rare metabolic myopathy, presents with progressive muscle weakness leading to limitations in motor function and respiratory difficulties. Here we estimate the MCID in adult patients with Pompe disease after a year of enzyme replacement therapy (ERT).
Methods: During clinical follow-up, pulmonary function was assessed as Forced Vital Capacity in upright position (FVCup) and walking ability was measured using the six-minute walk test (6MWT). Both outcomes were expressed as the percentage of the predicted normal value.
The MCID was determined using an anchor-based approach. Anchor questions included item 2 of the Medical Outcome Study 36-item Short-Form Health Survey ‘how has your health in general changed compared to one year ago’, and the question ‘how has your physical functioning changed compared to one year ago?’. Answer options ‘much better’ and ‘a bit better’, were combined to increase patient numbers per group, although this will overestimate the MCID (it is no longer just the smallest change). As a third anchor we used the SF-36 physical component summary score categorized into ‘better’ (change >+5), ‘worse (<-5) and ‘same’ (>=-5 and <=+5).
The MCID was calculated as the difference in the mean changes in the clinical outcomes after a year of ERT, comparing patients with anchor values ‘same’ and ‘better’ (the group ‘worse’ is not needed for this calculation). As the data were not collected at fixed time points, the measurements closest to the start of ERT and one year after were chosen. As an alternative a modelling approach was used to impute the data at the specific time point. Last, a distribution approach was also investigated.
Results: Data on at least one clinical outcome and one anchor were available for 109 patients treated with ERT.
The Figure below shows the change in the two clinical outcomes after one year of ERT for the patients who answered ‘better’ (‘a bit’/’much’), ‘the same’, and ‘worse’ (‘a bit’/’much’) to SF-36 item 2. We observed a general trend in which the change in the clinical outcomes was larger in patients who indicated they felt ‘better’ than in those who felt ‘the same’ and ‘worse’, but there was also considerable overlap between these groups.
Depending on the anchor used, the estimated MCID for FVCup ranged from 1.56 to 4.83 percentage points (pp) and from 3.29 to 7.47 pp for the 6MWT. This means that in a study, increases in the percentage predicted of FVCup/6MWT within these ranges are likely to be clinically meaningful.
Conclusion: The estimated MCIDs for adult Pompe patients ranged from 1.56 - 4.83 pp for FVCup and from 3.29 - 7.47 pp for the 6MWT. The MCIDs varied considerably according to the method used, and the combination of “a bit better” and “much better” may have inflated the results above the minimum level.
Keywords: minimal important difference, anchor-based approach, Pompe disease, GSD-II
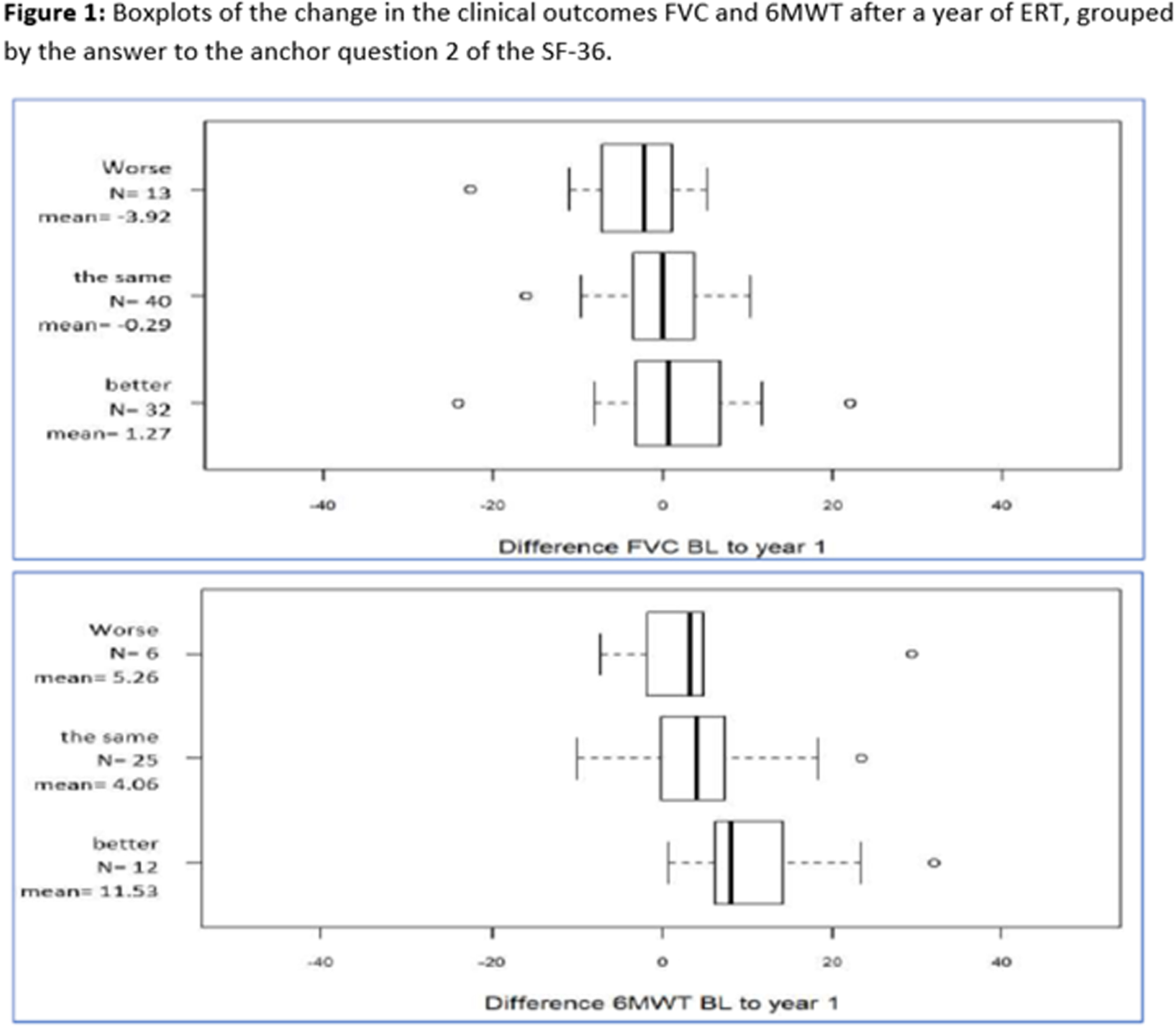
eP03.05.04Effect of Alglucosidase Alfa Dosage on Survival and Walking Ability in Classic Infantile Pompe Disease
Ditters I1, Huidekoper H1, Kruijshaar M1, Rizopoulos D2, Hahn A3, Mongini T4, Labarthe F5, Tardieu M5, Chabrol B6, Brassier A7, Parini R8, Parenti G9,10, van der Beek N11, van der Ploeg A1, van den Hout H1
1Department of Pediatrics, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, Rotterdam, the Netherlands, 2Department of Biostatistics, Erasmus MC University Medical Center, Rotterdam, the Netherlands, 3Department of Child Neurology, University of Giessen, Giessen, Germany, 4Neuromuscular Center, AOU Città della Salute e della Scienza, Turin, Italy, 5Department of Pediatrics, Center for inborn errors of metabolism ToTeM, Tours, France, 6University hospital of Marseille, Marseille, France, 7Hôpital Necker-Enfants Malades, Paris, France, 8Department of Pediatrics, S. Gerardo Hospital, Monza, Italy, 9Department of Translational Medical Sciences, Federico II University, Napels, Italy, 10Telethon Institute of Genetics and Medicine, Napels, Italy, 11Department of Neurology, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, Rotterdam, the Netherlands
Background: Enzyme replacement therapy (ERT) with alglucosidase alfa has been found to improve outcomes in patients with classic infantile Pompe disease, who without treatment typically die before the age of 1 year. Variable responses to the standard recommended dosage have led to alternative dosing strategies. We aimed to assess the effect of real-world ERT regimens on survival and walking ability in these patients.
Methods: In this observational cohort study, we obtained data collected as part of a collaborative study within the European Pompe Consortium on patients with classic infantile Pompe disease from France, Germany, Italy, and the Netherlands diagnosed between Oct 26, 1998 and March 8, 2019. Eligible patients had classic infantile Pompe disease with a disease onset and proven diagnosis before age 12 months, and a hypertrophic cardiomyopathy. A proven diagnosis of classic infantile Pompe disease was defined as a confirmed deficiency of α-glucosidase in leukocytes or lymphocytes, fibroblasts or muscle, or two pathogenic GAA variants in trans, or both. We collected data on demographics, GAA variants, ERT dosage, age at death, and walking ability. We analysed the effects of ERT dosage on survival and walking ability using Cox regression, Kaplan-Meier curves, and log-rank tests.
Findings: We included 124 patients with classic infantile Pompe disease, of whom 116 were treated with ERT (median age at start of treatment 3·3 months [IQR 1·8–5·0, range 0·03–11·8]). During follow-up (mean duration 60·1 months [SD 57·3]; n=115), 36 (31%) of 116 patients died. 39 different ERT dosing regimens were applied. Among the 64 patients who remained on the same dosage, 16 (52%) of 31 patients on the standard dosage (20 mg/kg every other week), 12 (80%) of 15 patients on an intermediate dosage (20 mg/kg per week or 40 mg/kg every other week), and 16 (89%) of 18 patients on the high dosage (40 mg/kg per week) were alive at last follow-up. Survival was significantly improved in the high dosage group compared with the standard dosage group (hazard ratio [HR] 0·17 [95% CI 0·04–0·76], p=0·02). No significant difference in survival was identified between the intermediate dosage group and the standard dosage group (HR 0·44 [0·13–1·51], p=0·19). Of the 86 patients who reached 18 months of age, 44 (51%) learned to walk. Ten (53%) of 19 patients on the standard dosage regimen, six (67%) of nine patients on intermediate dosage regimens, and 14 (93%) of 15 patients on high dosage regimens learnt to walk, but the differences between groups were not statistically significant.
Conclusion: Patients with classic infantile Pompe disease treated with the high ERT dosage of 40 mg/kg per week had significantly improved survival when compared with patients treated with the standard recommended ERT dosage of 20 mg/kg every other week. Based on these results, we suggest that the currently registered dosage should be reconsidered.
Funding: Prinses Beatrix Spierfonds and Wishdom Foundation.
Link to publication: Ditters et al. Lancet Child Adolesc Health. 2022 Jan;6(1):28-37. doi: 10.1016/S2352-4642(21)00308-4. Epub 2021 Nov 22. PMID: 34822769.
eP03.05.05Czech Nationwide Screening of Pompe Disease – Case Reports
Mensova L1, Mazanec R1, Parmová O2, Vohánka S2, Mazurová S3
1Department of Neurology, 2nd Medical Faculty od Medicine, Charles University in Prague and Motol University Hospital, Prague, Czechia, 2Neurological Clinic of University Hospital Brno, Brno, Czechia, 3Department of Paediatrics and Inherited Metabolic Disorders 1. LF UK a VFN, Prague, Czechia
Introduction: Pompe disease (type II glycogenosis) is a rare autosomal recessive hereditary metabolic disorder. The course of the disease and the overall survival time can be influenced by the enzyme replacement therapy. Since 2018 Neuromuscular centres in Prague and in Brno are implementing a project aimed at detecting new Pompe disease cases and providing the newly diagnosed patients with dispensary care and substitution therapy.
Methods: Information campaign by means of addressed letters, presentations at national conferences and journal articles was held among neurologists, paediatric neurologists, internal medicine specialists, pulmonologists and rheumatologists. The aim of the project is to perform the dry blood spot test (DBS) for Pompe disease in patients with muscle weakness of unknown etiology or asymptomatic hyperCKemia or idiopatic restrictive respiratory insufficiency. In patients with positive DBS genetic testing and GAA enzyme activity is performed. Newly found patients are examined and offered enzyme substitution therapy and regular follow ups.
Results: Between 2018 and 2021 1679 DBS tests were received and examined and 2 patients with late onset Pompe disease found. They are brother and sister of retirement age, both have significantly decreased GAA enzyme activity and they both share heterozygous GAA gene mutations (c.-32-13T>G/c.307T>G (p.(Cys103Gly)) however they differ in phenotype.
Patient 1 – 71 year old male, first symptom at 63 – dyspnoea while playing tennis, slowly progressing, at 67 unstable waddling gait, unable to climb a chair, CK 240 IU/l (upper limit 194). DBS sent by patients general practitioner. At 71 – able to walk without support, proximal weakness of lower limbs (4/5 according to MRC), axial weakness, severe hypotrophy of thoracolumbar paraspinal muscles, weakness of the diaphragm - forced vital capacity (FVC) while lying with more than 30 % decrease comparing to the sitting FVC. Whole body muscle MRI corresponds with described pattern of involvement including involvement of the tongue, spinal extensors, diaphragm and tights. Normal cardiologic findings.
Patient 2 – 74 year old female, biologically younger, still physically very active, walks 8-10 km per day, without any symptoms, tested as a relative in risk. Normal muscle strength, no hypotrophy, lying FVC without significant decrease comparing to the sitting FVC. CK 230 IU/l (upper limit 171 IU/l). Whole body MRI not performed for severe claustrophobia. Normal cardiologic findings.
Conclusion/Discussion: High number of indicated DBS tests shows increase in awareness of Pompe disease and good cooperation of clinicians. Despite the project we were not able to achieve the estimated prevalence 1:283 000 (Schosser et. al, 2015), according to literature in Czechia there should be 70-250 patients, but till today only 24 people we diagnosed. Reported cases illustrate that screening has its place even among patients of senior age.
Acknowledgements: The authors of this publication are member of the European Reference Network for Neuromuscular
Diseases - Project ID N° 870177. Univ. Prof. Dr. Berthold Streubel, MD, Medical Laboratory Archimed Life Science GmbH Project is financially supported by Sanofi Genzyme.
eP03.05.06Involvement of Muscle Capillaries in Late Onset Pompe Disease (LOPD) With Childhood Onset
Meznaric M, Angelini C
1University of Ljubljana, Faculty of Medicine, Institute of Anatomy, Ljubljana, Slovenia, 2University of Padova, Department of Neurosciences, Padova, Italy
Pompe disease, OMIM # 232300, is the lysosomal storage disease due to mutations in the GAA gene which encodes acid alpha-1,4-glucosidase (GAA), the enzyme that breaks down glycogen at acidic environment of the lysosome. Traditionally Pompe disease has been considered a muscle disorder (cardiac and/or skeletal). Findings of involvement of multiple organic systems at autopsy studies and signs and symptoms as dysarthria, dysphagia, osteoporosis, vertebral fractures, scoliosis, kyphosis, lumbar hyperlordosis, rigid/bent spine syndromes, sleep apnoea, painful paraesthesia (burning feet i.e. small fibre neuropathy), urethral and anal sphincter impairment, gastrointestinal symptoms, vacuolated lymphocytes, hearing loss, cerebral and extracerebral vascular involvement due to accumulation of glycogen in the smooth muscle cells of media which may contribute to aneurysm formation, blood vessel occlusion, (vertebral) dolichoectasia, aortic stiffness, lacunar encephalopathy, all emphasize the multisystem nature of Pompe disease.
The following case of a 3-and-a-half-year old girl with late onset Pompe disease (LOPD) with childhood onset is of interest because glycogen accumulation was demonstrated by electron microscopy in muscle capillaries in the biopsy of vastus lateralis muscle before enzyme replacement therapy was available and could be considered as an illustration of natural history of LOPD (Figure 1). At the time of the biopsy the patient already required mechanical ventilation.
In conclusion, the concept of diffuse vascular involvement, including small vessels, in LOPD may contribute to multisystem nature of Pompe disease and may influence the effectivity of possible therapeutic interventions in LOPD.
Figure 1. Biopsy of vastus lateralis muscle in a 3-and-a-half-year old girl with LOPD. (a) Scale bar = 5µm; (b) Scale bar = 500nm; Endothelial cells of endomysial capillaries are filled with numerous abnormal lysosomes.
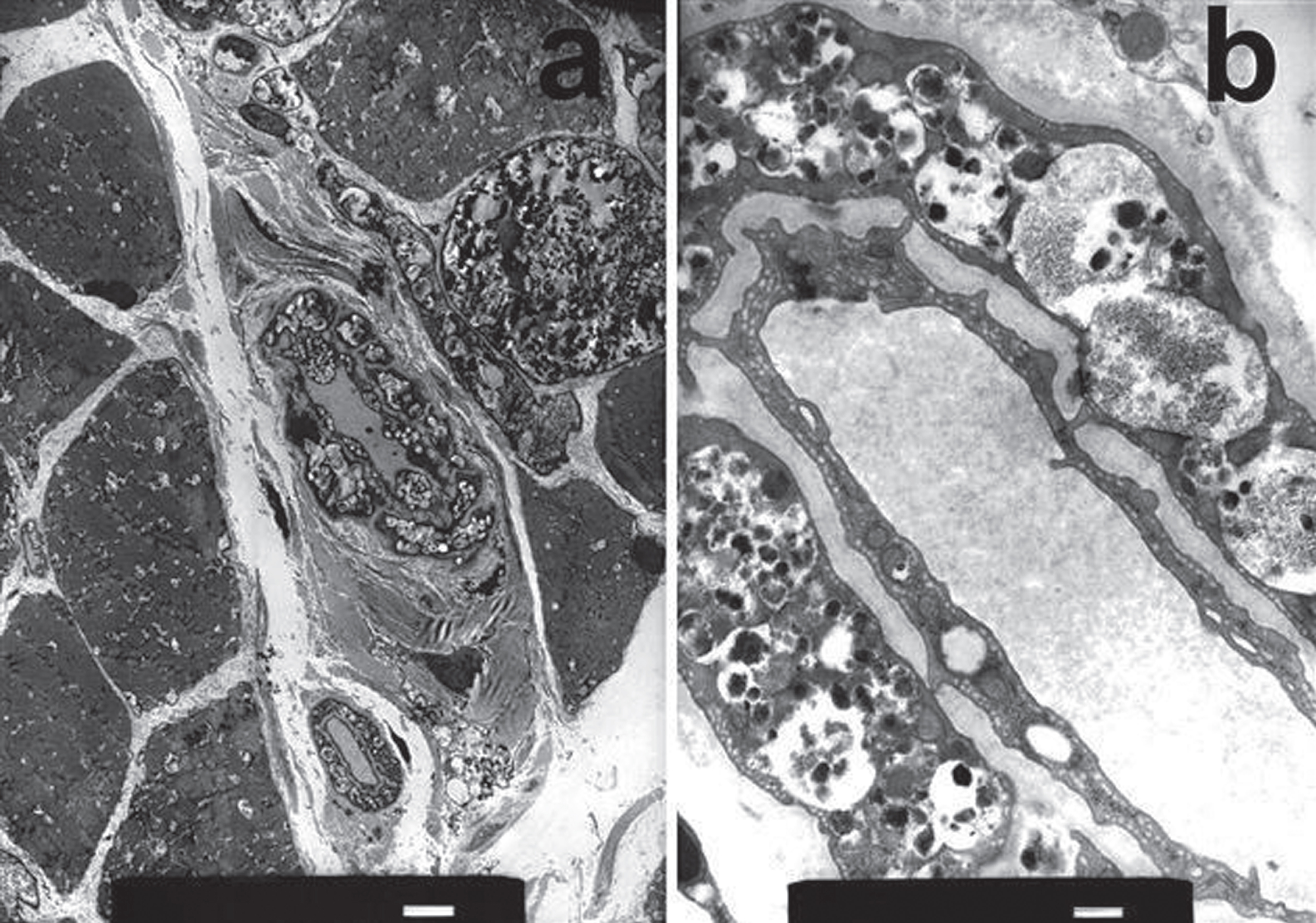
eP03.05.07Neuromuscular Symptoms and Gene Variants From a Long-Chain Fatty Acid Oxidation Disorder Gene Panel Program
Rangel Miller V1, Japalaghi O1, Marsden D1, McLaughlin H2, Simmons K1, Yong J2, Miller N1
1Ultragenyx Pharmaceutical Inc, Novato, United States, 2Invitae Corporation, San Francisco, United States
Introduction: Long-chain fatty acid oxidation disorders (LC-FAOD) are rare, life-threatening, autosomal recessive conditions that impair the utilization of fats for energy production. Undiagnosed LC-FAOD may present with hypoglycemia, cardiomyopathy, cardiac arrhythmias, and neuromuscular symptoms, including rhabdomyolysis and myopathy. For this reason, the differential diagnosis of unexplained rhabdomyolysis includes LC-FAOD along with physical and nonphysical causes, such as drugs, toxins, electrolyte imbalances, endocrine/autoimmune disorders, and other genetic disorders.
Methods: Patients in the US, Canada, and Mexico who have either a clinical diagnosis or suspicion of LC-FAOD with a confirmatory acylcarnitine test either ordered or performed are eligible for this clinician-ordered, sponsored no-charge genetic testing program. The next generation sequencing gene panel with copy number variant (CNV) detection includes the 6 genes associated with LC-FAOD (ACADVL, CPT1A, CPT2, HADHA, HADHB, SLC25A20) plus 18 additional genes associated with disorders that cause a similar acylcarnitine profile. Results for the LC-FAOD genes were classified as positive (positive = 2 P or LP) or potential positive (potential positive = 2 variants, at least 1 VUS).
Results: As of 5 January 2022, 478 patients were tested. Clinical symptoms were reported for 186 (39%) of the 478 patients. The most common neurologic and muscular symptoms were elevated creatine kinase (65), myopathy (59), rhabdomyolysis (40), peripheral neuropathy (14), cardiomyopathy (14) and myoglobinuria (13). One or more LC-FAOD gene variants (pathogenic, likely pathogenic, or uncertain significance) were found in: 38% of those reporting elevated creatine kinase (25), 27% of those reporting myopathy (16), 33% of those reporting rhabdomyolysis (13), 36% of those reporting cardiomyopathy (5), 31% of those reporting myoglobinuria (4), and 7% of those reporting peripheral neuropathy (1). Of 20 LC-FAOD gene variants found in patients with myopathy and 19 variants found in patients with rhabdomyolysis, c.1528G>C in HADHA and c.338C>T in CPT2, were most frequently identified.
While patients <1y accounted for 56% (268) of NGS panel tests, 44% were for older patients: 1-12y (80) or >13y (130). Outcome of confirmatory acylcarnitine testing was reported with panel test orders for 272 (57%) of 478 patients, of which 24% (66) were positive and 65% (178) were inconclusive. Of 66 acylcarnitine-positive patients, 44% (29) had positive/potential positive panel findings. Of 178 acylcarnitine-inconclusive patients, 15% (26) had positive/potential positive panel findings. Of 206 patients with no acylcarnitine data provided, 17% (34) had positive/potential positive panel findings. Overall, 19% (91) of patients tested had a positive/potential positive molecular diagnosis from the gene panel and 9.6% (46) of patients tested had a positive/potential positive LC-FAOD molecular diagnosis specifically (2 had positive/potential positive diagnosis for LC-FAOD plus another panel gene).
Conclusions: Program results demonstrate the diverse composition of gene variants in patients referred for LC-FAOD genetic testing. Patients with myopathy and rhabdomyolysis and a suspicion of LC-FAOD frequently had gene variants identified, suggesting the importance of considering LC-FAOD in patients with neuromuscular symptoms. Rhabdomyolysis, cardiomyopathy and neuromuscular gene panels that include the 6 LC-FAOD genes (ACADVL, CPT1A, CPT2, HADHA, HADHB, SLC25A20) may prove a productive approach to diagnose LC-FAOD.
eP03.05.08Natural History of Muscular Forms of Fatty Acid Beta-Oxidation Disorder: Description of 44 Patients
Rouyer A1, Tard C2, Douillard C3, Nadaj-Pakleza A4, Chanson J4, Spinazzi M5, Bedat-Millet A6, Dimitri-Boulos D7, Laforêt P8
1Neurology Department, Raymond Poincaré Hospital, AP-HP, Garches, France, 2Neurology Department, Salengro Hospital, Lille, France, 3Endocrinology-Diabetologist-Metabolist-Nutrition Department, Reference Center for Hereditary Metabolic Diseases, Huriez Hospital, Lille, France, 4Nord / Est / Ile-de-France Reference Center for Neuromuscular Diseases, Neurology Department, Strasbourg, France, 5Reference Center for Neuromuscular Diseases AOC, Neurology Department, Angers, France, 6Neurology Department, Charles Nicolle Hospital, Rouen, France, 7Neurology Department, Bicêtre Hospital, AP-HP, Le Kremlin Bicêtre, France, 8Nord/Est/Ile de France Neuromuscular Reference Center, Neurology Department, Raymond-Poincaré Hospital, AP-HP, Garches, France
Fatty acid beta-oxidation (FAO) disorders are recessive genetic diseases. The evolution and long-term prognosis of muscular forms are poorly described. We report the clinical and paraclinical data of 44 adult with a muscular form of FAO disorders with the aim of improving knowledge of their natural history, increasing diagnostic performance and optimizing management.
In this retrospective study, we collected the data from carriers of FAO disorder with muscle involvement in 6 French reference centers of neuromuscular or metabolic diseases: demographics, symptoms and clinical examination, biological and molecular analyses, neuromuscular explorations (muscle imaging and biopsy, ECG and echocardiography, EFR), hepatic ultrasound, treatments and long-term clinical course.
The cohort includes 44 adult patients including 14 carriers of CPT II deficiency (32%), 13 carriers of VLCAD deficiency (30%), 9 carriers of MAD deficiency (20%), 5 carriers of SCAD deficiency (11%) and 3 carriers of LCHAD deficiency (7%). The main symptoms are acute muscular manifestations, sometimes associated with a permanent muscular deficit. Episodes of rhabdomyolysis are frequent (84%) with an average CPK level of 68958 [660; 300000] IU/L, complicated by 3 intensive care hospitalizations (7%) and 4 acute renal failures (9%). Exercise intolerance occurred in 52% of cases. General metabolic complications are observed in 58% of patients (vomiting, asthenia, severe weight loss, encephalopathy and coma). Respiratory manifestations are noted in 18% of cases (dyspnea on exertion or at rest, sleep apnea syndrome) and cardiological manifestations in 9% of cases (syncope, heart failure during rhabdomyolysis). The average age at disease onset is 15 [0.5; 35] years and the average diagnosis delay is 13 [0; 58] years. The analysis of fasting acylcarnitine profile allowed for orienting genetic explorations in 65% of cases. A muscle biopsy was performed in 67% of patients, showing lipid overload in 61% of cases. The treatments administered are: Triheptanoin in 25% of carriers of long-chain FAO disorder (LCHAD, VLCAD) and the association Riboflavin + L-Carnitine in 78% of carriers of MAD deficiency. After an average follow-up of 10 years, 33% of patients remained asymptomatic and 56% continued to show symptoms after exercise. After diagnosis and appropriate management, the frequency of rhabdomyolysis decreased in 64% of cases, and only one patient (2%) was hospitalized in intensive care (VLCAD deficiency).
This large cohort of muscular adult patients with FAO disorder shows: 1) The main manifestations of these adult forms of metabolic disease are dominated by muscular symptoms (exercise intolerance and rhabdomyolysis); 2) Main explorations for diagnosis are fasting acylcarnitines profile (for all deficiencies) and muscle biopsy for MAD deficiency; 3) Life-threatening complications are rare, except for acute rhabdomyolysis, with a good long-term prognosis in the majority of cases.
eP03.05.09A Case of VCP Mutation Featuring With Lobulated Myofiber, Motor Neuron Disease and Frontotemporal Dementia
Chae J1, Sung J2
1Jeonbuk National University Hospital, Jeonju, Republic of Korea, 2Seoul National University, Seoul, Republic of Korea
Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD), caused by mutations in the valosin-containing protein (VCP) gene, is an autosomal-dominant, hereditary multisystem proteinopathy. It is considered a unique clinical syndrome with heterogeneous clinical presentation due to its diversity and familial variation. In particular, muscle biopsy findings that may appear in various ways from rimmed vacuole to atrophy, muscle fiber size variation, and VCP/ubiquitin/TDP-43 inclusion. A case with ALS and FTD, showing a biopsy finding of the muscular dystrophy pattern has not been reported.
A 52-year-old female patient came to the hospital with a feeling of gait impairment and proximal weakness that started gradually from 5 years ago. She was complaining of gait disturbance, her right calf muscle gradually began to atrophy, with dysarthria of unknown onset. Neurological examination showed shoulder abduction, hip flexion/extension, knee flexion extension mRC grade 4, presenting with proximal muscle weakness. Toe gait was possible, but heel gait was not. Gowers sign was positive, she had no skin lesions commonly observed in inflammatory myositis. Needle electromyography showed widespread denervation with reinnervations, suggesting motor neuron disease. Cognitive function test was performed because of patient is complaining of decreased memory and cognitive function. Possibility of accompanying frontotemporal dementia considered with 13 points on the frontal assessment battery (FAB), and 8 points on the f/u FA, frontotemporal lobe atrophy in brain MRI was shown. In muscle CT, diffuse atrophy with multifocal fatty change in upper and lower extremity muscles was observed on shoulder, gluteal, and thigh muscle (especially limb-girdle). Spine MRI also shown severe fatty degeneration of paraspinal muscle. Muscle biopsy at vastus lateralis shows marked size variation of myofibers with rimmed vacuole, lobulated myofibers are frequently noted. Ultrastructually, several large myelin figures are seen, which are consistent with rimmed vacuole, TDP-43 and Ubiquitin inclusions are presented in the rimmed vacuole. Genetic testing was performed, heterozygous mutations in the VCP gene were identified as the cause of IBMPFD c.463C>T, p.Arg155Cys, and heterozygote (p.R155C).
IBMPFD, which involves multiple systems, has various clinical presentations and many diseases that can appear overlapping. So, there are cases where incorrectly diagnosed as other neuromuscular diseases or lately diagnosed. Thus, IBMPFD should be suspected through detailed history taking and neurological exam, and process of checking VCP gene mutation is absolutely necessary. However, can be used bisphosphonate when bone pain is accompanied by Paget disease, physical, occupational therapy and treatment that enhances functionality through braces are all of the treatment. Therefore, although the diagnosis of IBMPFD is challenging, if it is clinically highly suspicious, it will be helpful to improve the quality of life of the patient and offer better management through a rapid, accurate diagnosis of IBMPFD. In addition, it should be considered that the biopsy finding of the muscular dystrophy pattern, which was rarely reported in IBMPFD, may appear together with MND and FTD.
eP03.05.10Head Drop and Hyperckemia Associated With a Carnitine Palmitoyltransferase II (CPT2) Deficiency
Tatillo C1, Bisciglia M1, Elands S1, Vandernoot I2, Desmyter L2, Remiche G1
1Centre de Référence Neuromusculaire, Department of Neurology, Hôpital Erasme, Université Libre de Bruxelles, 1070 Brussels, Belgium, 2Department of Genetics, Hôpital Erasme, Université Libre de Bruxelles, 1070 Brussels, Belgium
Introduction: Carnitine palmitoyltransferase II (CPT2) deficiency is an autosomal recessive muscle disorder. It is the most common disorder of long-chain fatty-acid (LCFA) metabolism, as well as the most frequent cause of hereditary rhabdomyolysis. We hereby aim to broaden the phenotypic spectrum of CPT2 by presenting a patient who exhibited a head drop at initial examination.
Materials and Methods: A 26 years-old woman presented to the emergency department with myalgia and cramps. Symptoms had started 48 hours beforehand with muscular pain, inability to stand up and walk. She denied fasting or extreme physical exercise. Neurological examination showed a symmetric proximal weakness of both lower limbs, axial weakness and head drop. Since her first pregnancy five years ago, the patient described having episodes of cramps and leg pain with difficulties in carrying weights and climbing stairs. On two occasions, these episodes were associated with head drop, dysphonia and dark urines. The episodes generally lasted one to two weeks, resolved spontaneously and were concomitant with her menstruation. No medical work-up had ever been made.
Past medical history included pyelonephritis, complicated by rhabdomyolysis. She suffered from gestational diabetes and severe obesity (BMI 42 kg/m2) after pregnancy and underwent a gastric bypass at the age of 25 years.
Results: Acylcarnitine level assay revealed an abnormal profile with increased C14-C18, C18:1, C18:2 and an increased ratio (C1+C18:1)/C2, suggestive of an inborn metabolism error in the long chain fatty acids oxidation.
Clinical exome sequencing identified the presence of 3 heterozygous mutations in CPT2 gene : c.338C>T p. (Ser113Leu), c.1239_1240delGA p.(Lys414Thrfs*7) and c.1342T>C p.(Phe448Leu). The three mutations were all previously reported in the literature at homozygote or compound heterozygote state in individuals affected by CPT2 deficiency. The patient was subsequently advised to avoid fasting and intense physical efforts. A specific low-fat and rich-carbohydrate diet with frequent meals was initiated. Since the introduction of the new regimen she has not relapsed into an episode of rhabdomyolysis.
Conclusions: To the best of our knowledge, this is the first case of head drop as clinical manifestation of the myopathic form of CPT II deficiency, thereby enlarging the phenotypical spectrum of this entity.
eP03.05.11Mitochondrial Network Disruption in Skeletal Muscles of the McArdle Mouse Model
Villarreal-Salazar M1,2, Real-Martinez A1, L. Andreu 3, A. Martín M2,4, Arenas J2,4, Santalla 5, Lucia A6, Vissing J7, O. Krag T7, Pinós T1,2
1Vall D’hebron Research Institute, Barcelona, Spain, 2Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain, 3EATRIS, European Infrastructure for Translational Medicine, Amsterdam, The Netherlands, 4Mitochondrial and Neuromuscular Diseases Laboratory, 12 de Octubre Hospital Research Institute (i+12), Madrid, Spain, 5Department of Sport Sciences, Universidad Pablo de Olavide, Sevilla, Spain, 6Faculty of Sport Sciences, European University, Madrid, Spain, 7Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
Introduction: McArdle disease is an autosomal recessive disorder caused by an inherited deficiency of muscle glycogen phosphorylase. Thus, McArdle disease patients are unable to obtain energy from their muscle glycogen stores leading to abnormal accumulation of subsarcolemmal glycogen. Although the disease is well described, the pathophysiological mechanisms produced by impaired muscle glycogenolysis have not yet been well established. Since 2012, with the knock-in mouse model developed by our group, has been found that the absence of GP-MM in the model leads to progressive muscle degeneration. However, muscle damage varies according to fiber type composition; muscles with a higher proportion of IIa and IIx fibers are the most affected by severe glycogen accumulation. In contrast, oxidative muscles (type I fibers) and highly glycolytic muscles (IIx/IIb and IIb fibers) are less affected. Notably, no major changes in the relative distribution of different muscle fiber types (based on MHC profiles) have been reported in patients with McArdle disease or McArdle mice.
Objective: To determine whether glycogen accumulation in skeletal muscles, particularly in TA, affects the mitochondrial network and cytoskeleton structure, thereby altering aerobic metabolism in the McArdle mouse model and patients.
Methodology: For this purpose, proteins of the cytoskeleton, sarcomere, and mitochondrial membrane, as well as proteins of mitochondrial dynamics and autophagy, were analyzed by immunofluorescence in two different muscle types such as TA (rich in IIa/IIx fibers) and quadriceps (with a higher proportion of IIx/IIb fibers) with airyscan microscopy. The ultrastructure of muscle fibers was analyzed by electron microscopy, while quantification of mitochondria amount and sarcomere proteins was performed by WB. Finally, mtDNA content and oxphos complex activities were determined in four different muscles (soleus, gastrocnemius, EDL, TA, and quadriceps).
Results: Immunofluorescence imaging showed altered integrity of the cytoskeleton, sarcomere structure, and mitochondrial network, both along the A-band and across the Z-line. Furthermore, an abnormally high proportion of isolated and enlarged mitochondria were also observed. The presence of structurally damaged mitochondria was confirmed at the ultrastructural level by electron microscopy, along with an obvious disruption of the myofiber structure. Additionally, abnormal accumulation of autophagic proteins was observed. These results suggest that mitochondrial turnover regulated by mitochondrial biogenesis, dynamics, and degradation mechanisms might be impaired. In this regard, our data shows a decrease in the mitochondrial content in the skeletal muscle (especially in TA) of the McArdle mouse model.
Conclusion: Due to glycogen deposits, the structure throughout the myofiber is compromised by altering the sarcoplasmic structure, the mitochondrial network and dynamics, confirming that there is damage not only to those muscular fibers of glycogen-driven metabolism (mainly anaerobic) but also of aerobic metabolism in skeletal muscle. These results not only contribute to the characterization of the pathophysiological mechanism of McArdle’s disease, but also to specific underlying processes such as fiber type degeneration, mitochondrial dynamics, or autophagy processes that have not yet been revealed.
eP03.06.01Etiology, Genetics and Prevalence of Myopathies in the Population of Alicante (Spain)
Ros-Arlanzón P, Aledo-Sala C, Pelegrín-Durá L, Díaz-Marín C
1Hospital General Universitario de Alicante, Alicante, Spain
Introduction: Myopathies form a group of clinically heterogeneous diseases, produced by different etiologies and affecting skeletal muscle. The diagnosis is based on multiple clinical data and is supported by various complementary tests. Myopathies require multidisciplinary management, and they benefit from early identification of the etiology that produces them to plan adequate treatment and follow-up of the patient. However, there are no studies that clarify the frequency of these diseases from a global perspective, most of them focusing on a specific subtype or subtypes of myopathies.
Methodology: a retrospective observational study of 132 patients with the diagnosis of Myopathy was carried out in a region of Alicante (a southern region of Spain) with 280,535 inhabitants registered in 2021. Demographic, clinical, genetic, and morbi-mortality variables of the selected patients were collected and analyzed using R statistical software. Each case was classified depending on its etiology after meticulous examination of informatic clinical history and at some cases after a clinical visit and new ancillary tests. Prevalence at the date of 31 December 2021 was calculated.
Results: The mean age of the cases was 51.81 (SD ± 17.10) years without preferences for sex (p-value 0.747). We observed 59.09% of the cases with genetic etiology, 9.85% of acquired cause, and 31.06% of undetermined cause. The global prevalence of myopathies in the city of Alicante was 47.05 cases per 100,000 inhabitants (95% CI: 39.52–55.98). The requested molecular diagnostic techniques showed alterations in 70.41% of the cases. Genetic analysis showed a not previously described pathogenic variant in the SMCHD1 gene in two patients with FSHD phenotype and in the FLNC gene in one patient with MFM5 phenotype. Other 67 genetic alterations were identified and described.
Conclusions: the prevalence of myopathies in Alicante is higher than expected. Recent studies showed similar prevalences of genetic myopathies or acquired myopathies in other regions of Spain. There is a lack of studies showing the prevalence of myopathies as a whole, independent of their etiology. The distribution of the etiology of the myopathies in the study population was congruent with medical literature.
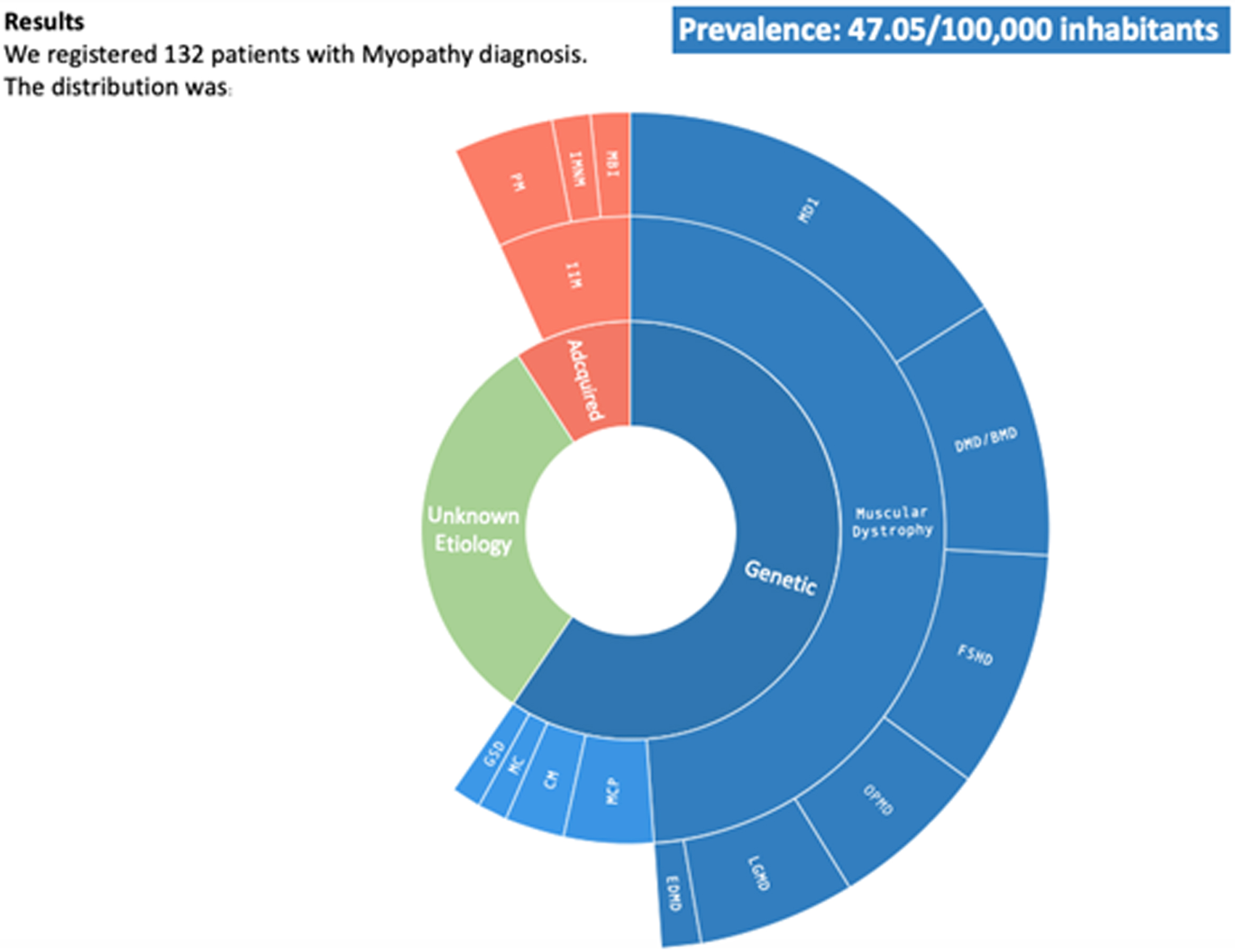
eP03.06.02Far More than Vacuoles? Proteomic Profiling of Danon Disease Reveals a Striking Mitochondrial Phenotype
Kleefeld F1, Hahn K1, Preuße C2, Goebel H2, von Moers A3, Hentschel A4, Roos A5, Stenzel W2
1Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology, Germany, 2Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neuropathology, Germany, 3DRK Kliniken Berlin-Westend, Klinik für Kinder- und Jugendmedizin, Germany, 4Leibniz-Institut für Analytische Wissenschaften -ISAS- e.V., Germany, 5Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Germany
Danon disease is a rare, multisystemic disorder presenting with skeletal myopathy, cardiomyopathy, and mental retardation. This X-linked genetic disorder is caused by mutations in the lysosomal-associated membrane protein-2 (LAMP2) gene. More than 160 pathogenic mutations in the LAMP2 gene have been identified. LAMP2 plays an important role in lysosomal degradation and autophagy and thus in cell homeostasis. Nevertheless, the exact pathophysiological mechanisms underlying the disease remain elusive. So far, no causal treatment is available for Danon disease, resulting in a limited prognosis for patients.
To gain further insights into the pathophysiology, we analyzed skeletal muscle biopsies from two male patients by proteomic profiling, histology and electron microscopy. The two and five years-old boys presented with muscle weakness and mild cardiomyopathy. Analysis of the proteomic profile revealed a significant up-regulation of 117 proteins and down-regulation of 83 proteins. Strikingly, the vast majority of downregulated proteins showed a mitochondrial localization and were involved in mitochondrial response to cellular stress and mitophagy. Subsequent histological and electron microscopic studies focusing on mitochondrial ultrastructure revealed pronounced mitophagy, e.g. lysosomes having engulfed mitochondria and signs of mitochondrial damage with enlarged mitochondria, abnormal and rarified cristae in both biopsy samples. However, Cox-negative-SDH-positive myofibers were not identified.
In sum, the first proteomic data on muscle biopsies in Danon disease indicate an important pathophysiologic role of mitochondrial damage and mitophagy linking lysosomal dysfunction with mitochondria.
eP03.06.03Global FKRP Registry - The Research Database for Limb Girdle Muscular Dystrophyase R9 (2I)
Murphy L1, Alfano L2, Brazzo K3, Johnson N4, Laurent J5, Mathews K6, Thiele S7, Vissing J8, Walter M7, Woods L9, Ørstavik K10, Straub V1
1John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom, 2Center for Gene Therapy at the Research Institute, Nationwide Children’s Hospital, Columbus, USA, 3CureLGMD2i Foundation, Lancaster, USA, 4Virginia Commonwealth University, Department of Neurology, Richmond, USA, 5LGMD2i Research Fund, Bellevue, USA, 6Carver College of Medicine, University of Iowa, Iowa City, USA, 7Friedrich-Baur-Institute, Department of Neurology, Ludwig-Maximilians-University, Munich, Germany, 8Copenhagen Neuromuscular Center, University of Copenhagen, Copenhagen, Denmark, 9Patient representative, USA, 10Section for Rare Neuromuscular disorders, Department of Neurology, Oslo University Hospital, Oslo, Norway
The Global FKRP Registry is an international registry for individuals with conditions caused by mutation of the Fukutin-Related Protein gene (FKRP): limb girdle muscular dystrophy R9 (LGMDR9, formerly LGMD2I) and the congenital muscular dystrophies (MD) MDC1C, Muscle-Eye-Brain Disease and Walker-Warburg Syndrome. The registry seeks to further understanding of the natural history and prevalence of FKRP-related MD.
The purpose of the registry is to aid the rapid identification of eligible patients for clinical studies. It disseminates FKRP-relevant information; provides a source of information to academics, industry and healthcare professionals; and supports the FKRP community.
Registration is patient-initiated through a secure online portal (www.fkrp-registry.org). Participants give their consent and are invited to complete a questionnaire about their condition. Data is reported by both patients and their healthcare professionals and includes: gene mutation, age of onset, presenting symptoms, motor function and muscle strength, respiratory and cardiac function, and medication. In addition, participants are invited to complete validated questionnaires on quality of life (INQoL) and pain (McGill).
Currently, 849 patients are registered in the Global FKRP Registry, with an age range of 2-81 years. Registrations are from 50 countries, with the greatest numbers from the USA (28%), Germany (21%) and UK (10%). More than 200 healthcare professionals participate in the registry. Diagnoses are reported as LGMDR9 (89%), MDC1C (2%), other FKRP-related MD (2%), unspecified (7%). Sixty-nine percent of patients are reported as being ambulant, 25% as non-ambulant and 6% as unspecified. The mutations reported within the registry are: 66% homozygous for the common mutation (c.826C>A), 27% heterozygous for the common mutation, 5% heterozygous with two unique mutations (non-c.826C>A) and 2% homozygous with one unique mutation.
In recent years, the Global FKRP Registry has assisted recruitment to numerous natural history studies and clinical trials in LGMDR9. It has facilitated research by responding to data enquiries and by circulating surveys, demonstrating its effectiveness as a repository of patient data, a tool for data collection and assembly of a trial-ready patient cohort. In the near future, the registry will implement the newly developed TREAT-NMD LGMD Core Dataset. Collection of a shared dataset by individual LGMD patient registries from around the world will enable a larger and potentially more powerful body of data to be collected on this diverse patient cohort.
As knowledge of rare neuromuscular conditions increases and advances in the development of potential therapies are made, the registry is centrally placed to help support the accumulation of natural history and post-marketing surveillance data and facilitate recruitment to clinical trials.
eP03.06.04First Description of a Caveolin-3-Related Rippling Disease in a Peruvian Family
Martínez-Esteban P1, Saavedra Ruiz M2, Bobadilla E5, Malfatti E3, Urtizberea J4
1Instituto Nacional De Salud Del Niño San Borja, Lima, Peru, 2Hospital Nacional Guillermo Almenara Irigoyen, Lima, Peru, 3Henri Mondor University Hospital, Paris, France, 4Institut de Myologie, AFM-Téléthon., Paris, France, 5Hospital Universitario San Ignacio, Bogota, Colombia
Introduction: Caveolinopathies are nosological entities defined by mutations in genes encoding, caveolin-1(CAV1), caveolin-2(CAV2), and caveolin-3(CAV3). Caveolins are expressed in skeletal, smooth, and cardiac muscle. Caveolin-3 (CAV3) is a muscle-specific membrane protein and belongs to the dystrophin glycoprotein complex. (1). The various phenotypes related to CAV3 gene defects are: 1) Rippling muscle disease; 2) Distal myopathy; 3) Isolated hyperCKemia; 4) Hypertrophic familial cardiomyopathy; 5) Long QT syndrome, and 6) Sudden infant death syndrome (2,3). A few cases present a combination of these phenotypes.
Case report: The proband is a 6-year-old male without relevant medical history. He started walking independently at 14 months, and developed a progressive tiptoe walking over the years. Spontaneous undulating movements consistent with a rippling phenomenon, stiffness, and increasing mild pain in the lower limbs upon waking and mild exercise were noted. Examination revealed tiptoe walking, with shortening of both Achilles’ tendons, mild lower limb girdle weakness(4+/5) and bilateral calf hypertrophy(Fig 1). Serum Creatine kinase(CK) were 1,143 IU/l.
His 66-year-old maternal grandfather and 41-year-old mother presented a similar phenotype with childhood-onset stiffness, calf hypertrophy, and pain in the lower limbs upon awakening. Symptoms remain mild and stable even with ageing. NGS study reported pathogenic heterozygous variant in the CAV3 gene, thec.99C>G; p.Asn33Lys. Segregation studies confirmed the presence of the variant in the affected mother, grandfather and in three years old twin brother and sister of the proband . None of these twins have symptoms or rippling phenomenon.
Discussion: Caveolinopathies caused by CAV3 mutations are infrequent entities. In a series of 663 cases of unclassified muscular dystrophies, only 1% of them had mutations in the CAV3 gene (4). Clinical manifestations appear around the first two decades of life and are characterized by: symmetric, proximal and progressive muscle weakness, positive Gowers’ sign, calf hypertrophy, myalgias, cramps and stiffness after exercise and muscle hyperirritability (5).
Rippling muscle disease(RMD) is characterized by signs of increased muscle irritability, such as percussion induced rapid contraction(PIRC), percussion-induced muscle mounding (PIMM) and/or electrically silent muscle contractions (rippling muscle) (5). Here, the proband presented with stiffness and rippling phenomenon, myalgia, calf hypertrophy, tiptoe walking and hyperCKemia, all this matching the characteristics of two CAV3-related phenotypes: rippling muscle disease and hyperCKemia. RMD is dominantly inherited and this pattern was confirmed by segregation studies.
Fulizio L et al. reported two patients with distal weakness and hyperCKemia starting in the third decade of life were described. One of them had calf hypertrophy and her biopsy indicated myopathic changes (4). Sugie K et al. published the observation of a 6-year-old Japanese boy who had fatigability, myalgias since age 3, calf hypertrophy and hyperCKemia. (6). Renard D et al., reported a 16-year-teenager with asymptomatic hyperCKemia. Electromyography and muscle biopsy showed myopathic changes, the muscle MRI showed STIR hyperintensities in the bilateral medial gastrocnemius (7).
Caveolinopathies should be taken into consideration when rippling phenomenon, myalgia, and stiffness are associated with calf hypertrophy, tiptoe walking, hyperCKemia, and autosomal dominant mode of inheritance. Genotype-phenotype correlations are not yet fully understood.
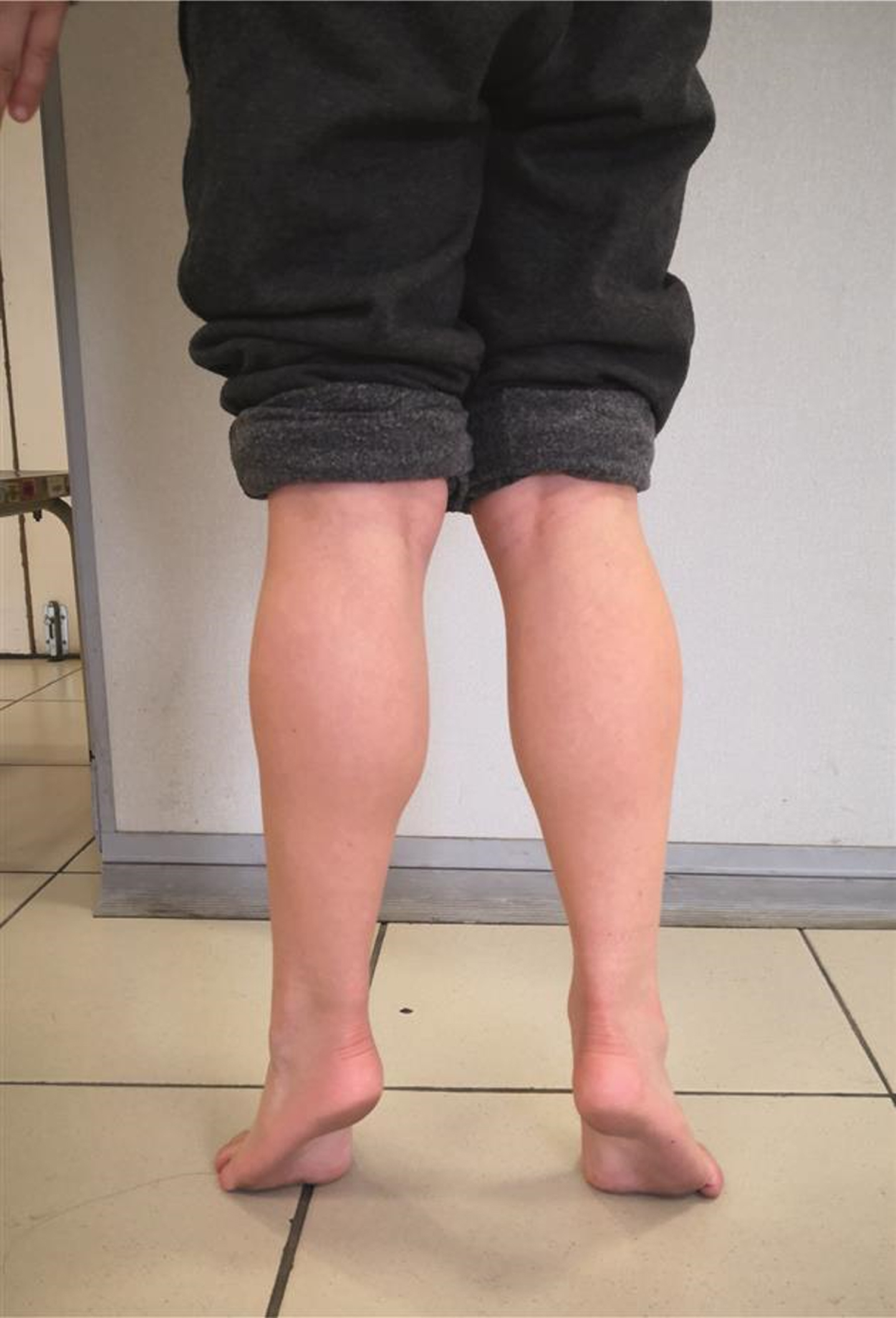
eP03.06.05Paucisymptomatic Hyperckemia as a Phenotype of Myopathy Associated with ano5: Case Report
Ruiz E1, Correa C, Castellar S, Ortiz Corredor F, Sanchez D, Salcedo M, Soto D
1 Universidad Nacional De Colombia Instituto Roosevelt, Bogota, Colombia
Introduction. The ANO5 gene codes for Anotacmine-5, a chloride channel involved in sarcolemma repair. Recessive mutations in ANO5 are associated with myopathies characterized by proximal or distal weakness or isolated hyperCKemia.
Case report. We present the case of a patient with generalized pain, progressive onset, which began in 2006 (age of onset 35 years) with non-specific characteristics, without exacerbations, which partially improved with pregabalin, without weakness or other associated symptoms. For several years she was managed with a diagnosis of fibromyalgia, with little response to analgesic treatment with NSAIDs, opioids and neuromodulators. In 2018, she went to emergency department due to pain exacerbation, finding CPK at 2,750 IU/L, so it was considered that she had inflammatory myopathy, receiving corticosteroids and immunosuppressants without improvement.
The electrodiagnostic study showed signs of myopathy without denervation. Muscle biopsy showed nonspecific myopathic changes, with muscle MRI in the leg with selective and asymmetric atrophy in both medial gastrocnemius, predominantly left. The genetic panel for myopathies showed compound heterozygosity for 2 pathogenic variants in the ANO5 gene: c.2609T>G (p. Leu870*) classified as nonsense and c.1119+1G>T in the non-coding splicing donor site.
Conclusion. We describe the case of a patient compound heterozygous for 2 pathogenic variants for the ANO5 gene, with a paucisymptomatic hyperCKemia phenotype, managed for several years with an initial diagnosis of fibromyalgia, and later as an inflammatory myopathy without response to immunomodulators.
eP03.06.06Artificial Intelligence Electrocardiogram-Derived Age Detects Accelerated Aging in LMNA Patients
Shelly S1,2, Lopez-Jimenez F3, Chacin-Suarez A3, Cohen- Shelly M3,4, Medina-Inojosa J3,5, Kapa S3, Attia Z3, Chahal A3, Somers V3, Friedman P3, Milone M1
1Department of Neurology, Mayo Clinic, Rochester, United States, 2Department of Neurology, Sheba Medical Center, Tel Aviv, Israel, 3Department of Cardiovascular Medicine, Mayo Clinic, Rochester, USA, 4Department of Cardiology, Sheba Medical Center Tel Aviv, Israel, Tel Aviv, Israel, 5Division of Epidemiology, Department of Quantitative Health Science, Mayo Clinic, Rochester, USA
Background: LMNA encodes the nuclear envelope lamin A/C. Lamins are proteins involved in ageing processes. Mutations in LMNA cause a spectrum of phenotypes which include muscular dystrophy, arrhythmogenic dilated cardiomyopathy, peripheral neuropathy, lipodystrophy, bone/skin disorders, progeroid syndrome and overlap syndrome. Early diagnosis is crucial to prevent sudden cardiac death. We hypothesized that LMNA mutations result in an ECG biological age older than chronological age and that this finding could impact patient care.
Methods: We have previously demonstrated that artificial intelligence (AI)-enabled ECG predicted age and its gap with chronological age (AI-ECG age-gap) correlate with total and cardiovascular mortality and may be a measure of biological age. We applied this previously trained convolutional neural network model to predict biological age by ECG in LMNA patients manifesting with a spectrum of phenotypes, and seen between 2003 and 2019. Findings were compared with age/sex matched control ECGs.
Results: Thirty-one LMNA patients from 28 families had 12-lead ECGs (n=271) electronically available for analysis at our institution. Median age at symptoms onset was 22 (range: <1 to 53 years) and was not different in males and females (15). Neurological symptoms at onset included muscle weakness due to skeletal myopathy (n=7), 3 of whom presented with hypotonia at birth and/or delayed motor development, chronic myalgia and fatigue (n=1), rhabdomyolysis (n=1), and stroke (n=1). Three patients who manifested with skeletal myopathy were found to have cardiac involvement at the time of the neurological evaluation or during follow-up. Cardiac symptoms were heterogeneous and preceded the onset of skeletal myopathy in four patients. Patients presenting with neurological symptoms were significantly younger (median age 11 years) than those presenting with cardiac symptoms (median age 36 years). Five LMNA patients were asymptomatic. We included a total of 1532 controls. We found significant differences in positive AI-ECG age-gap (positive age-gap means biologically older than chronological age) in LMNA patients (median of 17.5 years) compared to controls (4.9 years; p<0.001). AI-ECG age with 10 years or higher age-gap was observed in 73% of LMNA patients compared to 27% of controls (p<0.001). Consecutive serial analysis for all ECGs in the study versus predicted age showed faster aging when comparing the rates of age-gap overtime in the LMNA group and controls (p<0.0001). Moreover, a subset analysis of asymptomatic LMNA patients still showed significantly higher AI-ECG age-gap compared to controls (p=0.05).
Conclusion: The study showed that LMNA patients, including asymptomatic mutation carriers, are biologically older than controls with a significantly positive AI-ECG age-Gap and have an accelerated aging. AI-ECG age could function as a biomarker of cardiac disease and could be helpful in detecting early cardiac involvement and assisting in the treatment decision process. It could also be a potential tool to assess response to genetic therapy.
eP03.06.07Design of ‘Time Is Muscle’: RCT on IVIg Add-on to Prednisone in Newly Diagnosed Myositis
Kamperman R1, Walter H1, de Visser M1, Bogaards H2, de Borgie C2, Colen – de Koning J3, Verhamme C1, Maas M4, Eftimov F1, van Schaik I1,5, van der Kooi A1, Raaphorst J1
1Department of Neurology and Clinical Neurophysiology, Amsterdam University Medical Centre, University of Amsterdam, Amsterdam Neuroscience, Amsterdam, The Netherlands, 2Clinical Research Unit, Amsterdam UMC, University of Amsterdam, Amsterdam UMC location AMC, Amsterdam, The Netherlands, 3Department of Clinical Pharmacy, Amsterdam University Medical Centre, University of Amsterdam, Amsterdam UMC location AMC, Amsterdam, The Netherlands, 4Department of Radiology and Nuclear Medicine, Amsterdam University Medical Centre, Amsterdam Movement Sciences, Amsterdam, Netherlands, 5Spaarne Gasthuis, Haarlem, The Netherlands
Introduction: Idiopathic inflammatory myopathies (IIM, also known as myositis), include dermatomyositis, anti-synthetase syndrome, overlap myositis and immune mediated necrotizing myopathy. The standard initial treatment is high dosed glucocorticoids, which results in relatively slow improvement of muscle strength. Early intensive treatment (‘hit-early, hit-hard’) may induce faster reduction of disease activity and prevent chronic disability due to disease damage. Compared to other immunomodulating compounds, intravenous immunoglobulin (IVIg) is more fast-acting, which could lead to an early and sustained suppression of the inflammatory process when administered together with glucocorticoids. Addition of IVIg may be promising in this regard: data from previous studies have shown that add-on IVIg improved symptoms and muscle strength in refractory myositis patients, and that monotherapy IVIg improved outcomes after nine weeks, in about half of treatment naive patients.
Hypothesis: We hypothesize that early add-on IVIg leads to a greater clinical response after twelve weeks in patients with newly diagnosed myositis, in comparison to prednisone monotherapy. Secondary, we expect that early treatment with add-on IVIg leads to a shorter time to improvement and sustained positive effects on health-related quality of life, physical activity, fatigue and reduction of muscle MRI abnormalities on the longer term (26 and 52 weeks).
Study design: The Time Is Muscle trial is a phase-2 double-blind randomized controlled clinical trial. Recruitment of patients will be performed through the Dutch Myositis Network (DMN), a collaboration between myositis centres in the Netherlands, of which the Amsterdam UMC serves as a tertiary referral centre for IIM. Forty-eight patients with IIM will be treated with IVIg or placebo at baseline and after 4 and 8 weeks, in addition to standard therapy with prednisone. The first infusion will be administered within one week after diagnosis. The primary outcome is the Total Improvement Score (TIS) of the myositis response criteria after 12 weeks compared to baseline. At baseline, and after 4, 8, 12, 26 and 52 weeks, secondary outcomes will be assessed, including time to improvement, daily prednisone dosage, physical activity and MRI muscle imaging parameters.
Discussion: Time Is Muscle is the first double-blind randomized controlled trial in early myositis aiming to provide evidence for an effect of IVIg in addition to standard treatment with prednisone. Multiple secondary outcomes will be assessed at several time points, providing more data on the disease course of myositis. The first patients have been included, and we expect to conclude enrolment of patients in the first half of 2024. At the conference the concept of the Dutch Myositis Network will be presented, in addition to the detailed study design.
eP03.06.08The PACE-DM1 tool: An Adapted Home-Based Physical Activity Program for DM1 to Counter Physical Deficiencies
Fortin A1,2, Aubertin-Leheudre M4,5, Gagnon C2,3, Duchesne E2,6
1Department of Health Sciences;UQAC, Saguenay, Canada, 2Groupe de recherche interdisciplinaire sur les maladies neuromusculaires (GRIMN), Jonquière, Canada, 3Faculté de médecine et des sciences de la santé; Université de Sherbrooke (Sherbrooke, Sherbrooke, Canada, 4Département des sciences de l’activité physique, faculté des Sciences, UQAM, Montreal, Canada, 5Centre de recherche de l’Institut universitaire de gériatrie de montréal (CRIUGM), Montreal, Canada, 6Département des sciences de la santé, physiothérapie; UQAC, Chicoutimi, Canada
Introduction: Myotonic dystrophy type 1 (DM1) is related to muscle weakness, impaired balance, and a high risk of falls resulting in decreased quality of life and social participation. Indeed, DM1 is considered as a model of premature aging. However, the Covid-19 pandemic has exacerbated these health problems by decreasing the access to rehabilitation services and significantly decrease volunteer total weekly physical activity (PA) level. A pragmatic tool: PACE (Promoting Autonomy through exerCisE), which has been developed to allow safe and adapted physical activity (APA) practice in older adults at home, could be a solution to counter physical deficiencies and lockdown restrictions in DM1 population.
Objectives:
1. To adapt the PACE tool (decisional tree related to 35 APA programs) for the DM1 population (Pace-DM1 tool);
2. To evaluate if assessing functional and muscular capacity in remote mode is feasible and valid in the DM1 population compared to in person modality;
3. To evaluate the acceptability, feasibility and usability of implementing the PACE-DM1 tool for the health professionals and the patients.
Methods:
O-1: Two patient-partners, 3 participants who will experience the PACE-DM1 APA program, 2 health professionals from the Neuromuscular Disease Clinic of Jonquière (NMC), 1 NMC manager and 3 researchers were recruited to adapt the PACE tools using a co-creation design.
O-2: Participants are assessing functional and muscular capacity [Time Up and Go, 30sec. chair stand test, 5 reps of sit-to-stand, 4-meter gait speed, balance and functional reach test] per and post-intervention (12 weeks) in remote (zoom ©) and in-person methods.
O-3: After co-creation meetings, health professional were asked to evaluate the acceptability (satisfaction assessed via Likert scales; semi-opened questions), usability (System Usability Scale questionnaire) and feasibility (adherence, recruitment rate, etc.) of the PACE-DM1 tool.
Results: First, co-creation meetings allowed us to adapt PACE tool (e.g. decisional tree cut points) for DM1 participants. Health professionals found PACE-DM1 tool exercises safe and adapted to the physical deficiencies of the DM1 population.
Our preliminary data (n=2) using the adapted decisional tree showed similar results between in-person and remote assessment. More precisely, both assessment modality identified the same physical deficiencies (lower limb cardio-muscular, trunk mobility and stability, and balance) and prescribed the same program difficulty (out of a possible 5 levels).
In the pre-intervention phase, implementing the PACE-DM1 tool was considered acceptable, usable and feasible for health professionals.
Conclusion: Preliminary results demonstrate that an APA pragmatic, easy-to-use and adapted tool is implementable to prevent care trajectory of people with DM1. Nevertheless, these promising results need to be confirmed and validated with a larger sample.
eP03.06.09Photosensitive Epilepsy and Polycystic Ovary Syndrome as Manifestations Of MERRF
Finsterer J1
1Neurolgy Neurophysiolgy Center, Postfach 20, Austria
Objectives: Though endocrinologic involvement and epilepsy are frequent features of myoclonic epilepsy with ragged-red fibers (MERRF), polycystic ovary syndrome (PCOS) and photosensitive epilepsy have not been reported.
Case report: A 32yo female was diagnosed with MERRF at age 19y upon presence of the four canonical features and the variant m.8344A>G in MT-TK (tRNA(Lys)) (blood heteroplasmy rate: 50%). She experienced recurrent photosensitive focal and generalised seizures since age 19y, which could be triggered by flickering light or by looking at small stones, leaves, or dirty snow on the ground. Since the last 42 months she was seizure-free upon levetiracetam (4000mg/d), clonazepam (1.5mg/d), and topiramate (25mg/d). Additionally, she suffered from secondary amenorrhoea since adolescence. She was married between ages 19y and 25y but did not get pregnant. PCOS was diagnosed and treated with desogestrel plus estradiol. Nonetheless the course was progressive, particularly with regard to ataxia, myocloni, and myopathy.
Conclusions: The phenotypic spectrum of MERRF is broader than anticipated and may additionally include PCOS and photosensitive epilepsy. PCOS in MERRF may respond to hormone substitution and photosensitive epilepsy to levetiracetam, clonazepam, and topiramate.
eP04.01.01The Burden of Duchenne Muscular Dystrophy in Belgium: A Registry-Based Study
Cosyns M1, De Meulemeester E1, Beysen D2, Deconinck N3, De Braekeleer K4, De Waele L5, Paquay S6, Smeets N7, Vanden Brande L8, Van Lander A9, Devleesschauwer B1
1Sciensano, Brussels, Belgium, 2NMRC UZ Antwerpen, Edegem, Belgium, 3NMRC ULB Erasme HUDERF, Brussels, Belgium, 4NMRC Inkendaal, Vlezenbeek, Belgium, 5NMRC UZ Leuven, Leuven, Belgium, 6NMRC Cliniques Universitaires Saint-Luc, Brussels, Belgium, 7NMRC UZ Brussel, Brussels, Belgium, 8NMRC CHR de la Citadelle, Liège, Belgium, 9NMRC UZ Gent, Ghent, Belgium
Introduction. Duchenne muscular dystrophy (DMD) is a severe, progressive, muscle-wasting disease leading to ambulation loss, the need of ventilation support and premature death. As a rare disease, DMD has a small health impact at population level. However, given its course, the health impact at patient level is significant. The preferred metric for quantifying health impact of rare diseases is the Disability-Adjusted Life Year (DALY) as it allows for comparable estimates of both the population and patient level impact. DALYs reflect the healthy life years lost due to illness and death, and combine both morbidity and mortality, as well as disease occurrence and severity. Quantifying the burden of DMD using the DALY helps identify disease characteristics to target and provides important input into health care planning. It allows to compare DMD to other diseases for which DALYs have been calculated and, on the long-term, to monitor the evolution of DMD, which is particularly interesting in light of emerging disease-modifying therapies. Therefore, the present study aimed to estimate the health burden of DMD in Belgium by using the DALY approach. To our knowledge, this approach has not yet been applied to the neuromuscular field.
Methods. DALYs were calculated as the sum of the adjusted number of years lived with disability (YLDs) and the number of years of life lost due to premature mortality (YLLs) using a hybrid perspective (often called a prevalence approach) in a multi-aspect disease model. Data were drawn from the 2017 to 2019 data collections of the Belgian neuromuscular diseases registry (BNMDR) and international literature (for disease aspects not included in the registry). Disability weights (DWs) for DALY calculation were derived from the Global Burden of Disease Study. If not available, the European DW study, DWs from proximate health states and international literature were consulted, in that order.
Results. In Belgium, DMD resulted in an average of 601 DALYs per year, which represents an average of 2.31 DALYs per patient (composed of 85% YLLs and 15% YLDs). For comparison, a similar study in cystic fibrosis (CF) resulted in 0.75 DALYs per patient (composed of 53% YLLs and 47% YLDs). The highest morbidity was caused by ambulation loss (an average of 38 YLDs per year), followed by cardiomyopathy (12 YLDs) and respiratory dysfunction (11 YLDs). Constipation (9 YLDs) and long bone fracture (9 YLDs) complete the top 5.
Discussion. As expected, with an average of 601 healthy life years lost per year, DMD especially represents an individual health burden. The burden per patient was found to be three times higher than the burden per patient in CF, with the highest contribution coming from the YLLs due to premature mortality. The YLD ranking could be used to guide subsequent data collections. While many DMD registries include motor, cardiac, respiratory and bone data, constipation is often not enquired. Further, with this study we encourage the use of the DALY on other neuromuscular diseases as combining the data might reveal an important health impact at population level.
eP04.01.02Improving and Harmonising Care Standards for Duchenne Muscular Dystrophy in the UK
Turner C1, Childs A2, Johnson A3, Manzur A4, Muntoni F5, Quinlivan R6, Rodney S7, Sarkozy A5, Wong J8, Guglieri M1
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle Upon Tyne, United Kingdom, 2Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom, 3Duchenne UK, London, United Kingdom, 4Great Ormond Street Hospital for Children NHS Trust, London, United Kingdom, 5The Dubowitz Neuromuscular Unit, UCL Great Ormond Street Institute of Child Health, London, United Kingdom, 6MRC Centre for Neuromuscular Disease, UCL Institute of Neurology, London, United Kingdom, 7Duchenne Research Fund, London, United Kingdom, 8Royal Hospital for Children in Glasgow, Glasgow, United Kingdom
DMD Care UK was established in 2020 as a collaboration between Newcastle University, the UK North Star network and Duchenne UK. The project aims to improve care provision across the country for Duchenne muscular dystrophy (DMD) by reaching expert-consensus, facilitating implementation and education of UK-relevant standards of care (SoC) based on international guidelines. The project also identifies gaps in resource, practice, evidence and research, aiming to address these to better facilitate and understand the most effective interventions across the clinical care setting.
Through a series of sub-specialist working groups comprising clinical experts in their fields, neuromuscular specialists and patient representatives, the project has now produced or is in the process of releasing a series of UK-specific guidelines with endorsement from the neuromuscular community and relevant professional bodies. This work continues and includes education of the patient and clinical community and efforts to promote and support implementation.
We report here on progress so far, in particular in bone and endocrine, respiratory, cardiac, physiotherapy, emergency and psychosocial care. We also summarise next steps.
DMD Care UK was conceived by Duchenne UK after feedback from patients about inconsistencies in care across the UK and the reality of delivery compared to International recommendations published in 2018. DMD Care UK is a strong example of how the clinician and patient community can work together to improve standards of care for all those living with DMD.
The project has taken international guidelines and turned them into actions for implementation within the UK healthcare system. This is a model that can be supported by external stakeholders and also adopted in other national healthcare settings.
Acknowledgements: DMD Care UK is funded through grants received from Duchenne UK, Joining Jack and the Duchenne Research Fund
eP04.01.03Illness Perceptions And Quality Of Life In Adolescents With Neuromuscular Disorders And Caregivers
Prikken S1, Geuens S1, Willen J1, Goemans N1, Luyckx K2, De Waele L1
1University Hospitals Leuven, Leuven, Belgium, 2KU Leuven, Leuven, Belgium
Background. Neuromuscular disorders (NMDs) are a heterogeneous group of diseases characterized by their impact on physical functioning. However, these diseases can also have a wide-ranging impact on the psychosocial development of affected children and adolescents. Previous research has demonstrated an impaired quality of life in some subgroups of NMDs, like Duchenne Muscular Dystrophy (DMD) and Spinal Muscular Atrophy (SMA). The ways in which patients perceive their illness and how these perceptions affect their psychosocial well-being, remains to be fully understood. Therefore, this multi-informant study explored associations between adolescents’ illness perceptions and their quality of life as reported by caregivers. In addition, the quality of life of their caregivers was also taken into account.
Methods. A total of 41 adolescent NMD patients (66% male, Mage=14.29) participated along with their parents (N=41, 76% mothers). Patients were subdivided into 6 disease groups: DMD (34.1%), SMA (17.1%), congenital myopathy (19.5%), Charcot Marie Tooth (12.2%), limb-girdle muscular dystrophy (9.8%), and a category labeled as ‘other’ (7.3%). Patients completed the Brief Illness Perception Questionnaire (BIPQ). To assess patients’ quality of life, parents completed the parent-reported PedsQL Neuromuscular Module. Parents also gave an estimation of their own perceived quality of life on a VAS-scale (ranging from 0-10). First, we explored whether disease groups differed on the different quality of life measures (controlling for patient age). Next, correlations between scores on the illness perceptions and the different quality of life measures were calculated.
Results. First, the role of disease groups for explaining quality of life appeared rather limited. Only for the PedsQL subscale of physical functioning, the effect was marginally significant (F(5,34)=2.44; p=.054). Results suggested that particularly the SMA group may be at risk for experiencing lower physical quality of life. Next, the PedsQl subscale of family resources was closely related to illness perceptions. In families with low scores on family resources, patients reported more consequences (r=-.46, p<.01), more complaints (r=-.53, p<.01), lower personal control (r=.38, <.05), more concerns (r=-.32, p<.05) and a higher emotional impact (r=-.41, p<.01) of the illness. Associations between illness perceptions and the PedsQL subscales of physical functioning and communication were less pronounced. In addition, parental quality of life was not significantly correlated with illness perceptions of NMD youth.
Conclusion. These preliminary findings illustrate that the ways in which adolescents with NMDs perceive their illness may be closely related to family functioning. These preliminary findings thus underscore the need for assessing illness perceptions in youth with NMDs along with family functioning indicators to identify the particular support needs for these families.
eP04.01.04A Pilot T1-Weighted MRI Study to Evaluate Chronic Corticosteroid-Use in Duchenne Muscular Dystrophy on Brain
Geuens S1, Sleurs C1, Lemiere J1, Niks E2, Goemans N1, Kan H3, De Waele L1, Doorenweerd N2
1University Hospitals Leuven, Leuven, Belgium, 2Leiden University Medical Center, Leiden, The Netherlands, 3C.J. Gorter Center for High Field MRI, LUMC, Leiden, The Netherlands
Prednisolone and deflazacort have been proven to be effective in slowing down Duchenne Muscular Dystrophy (DMD) disease progression. Previous studies in other pathologies demonstrated corticosteroids can cross the blood-brain barrier and cause brain atrophy. In this pilot study, we assessed the effect of both compounds on brain structure in boys with DMD.
3D T1-weighted images were obtained on a 3T Philips system at two different sites. Scans from 20 DMD patients (n=10 daily deflazacort (13.1±3.1y), n=10 10 days on/10 days off prednisolone (13.0±3.0y)) and 16 age-matched healthy controls (14.0±2.8y) were processed using FSL software to calculate intracranial (ICV), total brain (TBV), grey matter (GMV), white matter (WMV) and CSF volumes. In addition, volumes of the hippocampus, amygdala, thalamus, nucleus accumbens, caudate nucleus, pallidum and putamen were calculated. Volumes in cc were compared between controls and DMD and between DMD groups on different steroid regimens using multivariate analyses of variances in SPSS and considered significant at p<0.05.
Consistent with previous reports the DMD group had lower ICV (1522.5 vs 1701.7), TBV (1256.3 vs 1405.1) and GMV (713.1 vs 803.5) compared to healthy controls. Of the subcortical structures, caudate nucleus (8.1 vs 9.0), hippocampus (7.9 vs 9.7), pallidum (3.5 vs 4.0), putamen (10.7 vs 12.1) and thalamus (15.3 vs 16.6) of DMD patients were smaller than their healthy peers. However, this was partially explained by the TBV. Interestingly, the DMD group on daily deflazacort showed higher CSF volumes (301.6 vs 234.3), but lower WMV (511.0 vs 572.2), TBV (1201.2 vs 1305.9) and volume of the pallidum (3.3 vs 3.8) as compared with DMD patients on an intermittent prednisolone regime.
This pilot study indicates that the different corticosteroid regimes need to be assessed further to determine if it results in different regional volume reductions.
eP04.01.05Immune Function Indices in Patients with Amyotrophic Lateral Sclerosis
Kononets O1
1Shupyk National Healthcare University Of Ukraine 9 Dorohozhytska Str., Kyiv, Ukraine 04112, Borodianka District, Ukraine
The aim of the study is to closely examine and analyze immune function indices in patients with amyotrophic lateral sclerosis.
Material and methods. We analyzed the dynamic clinical and laboratory data of 63 amyotrophic lateral sclerosis patients, aged 54 ± 8. Having done the laboratory examination, we evaluated the patients’ immunogram indices, i.e., B-lymphocytes, T-lymphocytes, T-helpers, T-suppressors, CD4:CD8 ratio, natural killer cells ratio.
Research results. Having analyzed the immunological profile of amyotrophic lateral sclerosis patients, we found that the count of B-lymphocytes ranged from 1 to 22 % (the mean value was 10,9 %); by comparison, the count was within normal limits in only 4 patients (6,3 %). The rest 59 amyotrophic lateral sclerosis patients (93,7 %) were found to have the below normal count.
The T-lymphocytes count ranged from 46,0 to 84,3 % (the mean value was 65,96 %); by comparison, the count was within normal limits in only 18 patients (28, 6 %); the count was beyond the reference interval in the rest 45 patients with amyotrophic lateral sclerosis: it was below the norm in 25 patients (39,7 %) and the count was above the norm in 20 patients (31,7 %).
The T-helpers count was observed to range from 6,0 to 54,0 % (the mean value was 37,54 %); whereas the count was within normal limits in more than half the patients, i. e. in 39 cases (61,9 %); the rest 24 amyotrophic lateral sclerosis patients were observed to have the count beyond the reference interval: the count was below the norm in 6 patients (9,5 %) and it was above the norm in 18 patients (28,6 %).
The T-suppressors count was observed to range from 6,0 to 71,0 % (the mean value was 22,69 %); whereas the count was within normal limits in almost half the patients, i. e. in 29 cases (46 %); the rest 34 amyotrophic lateral sclerosis patients were observed to have the count beyond the reference interval: the count was below the norm in 26 patients (41,3 %) and it was above the norm in 8 patients (12,7 %).
The natural killer cells level was observed to range from 0,95 to 39,4 % (the mean value was 10,82 %); whereas the level was discovered to be within the reference values in more than half the patients, i. e. in 42 cases (66,7 %); the rest 21 amyotrophic lateral sclerosis patients were observed to have the level beyond the reference interval: the level was below the norm in 15 patients (23,8 %) and it was above the norm in 6 patients (9,5 %).
Conclusions. We found significant changes when evaluating the immune status in the patients with amyotrophic lateral sclerosis: almost all the patients (93.7 %) had decreased B-lymphocytes levels; 41.3 percent of those patients had a reduced level of T-lymphocytes – T-suppressors subpopulation. The nature and predictive value of the changes need further investigation.
Keywords: amyotrophic lateral sclerosis, immune function indices.
eP04.01.06Duchenne Muscular Dystrophy Diagnostic Gaps in Primary Medical Chain
Gremiakova T1, Gremiakova O1, Sakbaeva G2, Stepanov A2, Souslov V3, Artemieva S4
1Fund “Gordey”, Moscow, Russian Federation, 2Federal State Budget Institution «Central Clinical Hospital with Ambulance», Moscow, Russian Federation, 3Federal State Budget Educational Institution of Higher Education «Sankt-Petersburg State Pediatric Medical University» RF Ministry of Education, Sankt-Petersburg, Russian Federation, 4Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, Moscow, Russian Federation
Amount of DMD patients in Russia, based on a population of ~146 million people, should be statistically 4000-4500 boys, adolescents and young adults in accordance with global data. Hence, awareness of DMD among pediatricians and pediatric neurologists in primary medical chain is extremely low, so the diagnosis is made after a long sorting of the wrong options, by accidentally detecting transcendental concentrations of serum creatine kinase (CK), or - what happens more often in the regions, the diagnosis is fully incorrect, the child’s condition is attributed to other reasons.
In Russia, there is no national medical register, disparate data in different patient, medical, diagnostic organizations are limited and fragmented. According to Fund “Gordey” information, the list of diagnosed patients is around 1000-1500 patients, what is approximately third part of the total number of sick boys.
As of today, there is no problem in genetic diagnostics of DMD in Russia. Diagnostic medical centers have introduced modern methods for the genetic diagnosis of DMD, which can reliably determine the presence and type of gene mutation. Blood sample with CK level more than 2000 units from any region in Russia currently can be delivered to certified genetic lab for DMD genetic testing.
Research has been conducted by questioning families and doctors on the various issues of diagnostics. A group of 187 families was requested to answer the following questions:
Ø age at diagnosis
Ø whether child was misdiagnosed before being diagnosed with DMD
Ø if yes, what was the first diagnosis made to the boy.
According answers received, average age of diagnostics in Russia is around 7 yo. Adequate formulation of the DMD diagnosis in the primary medical chain was found in less than half of cases - only 47.1% of boys received a diagnosis of DMD as a primary disease. The other most frequent diagnose was hepatitis (28.1%), perinatal lesions of the central nervous system (6.4%), cerebral palsy (2.7%), autism (2.7%), flat feet, hallux valgus and others (13%).
About 500 pediatricians and pediatric neurologists working in the primary medical chain were requested to answer questions concerning symptoms of DMD. As of classical DMD symptoms for 3-6 yo Duchenne boys, over 75% were familiar with such symptoms as Gower’s maneuver, fatigue, problems with stairs. Less than half mentioned often falls, pseudohypertrophy of the calf muscles, toe walking.
For early diagnostics of the boys aged 1-3 yo, 82,7% of doctors mentioned fatigue, 61,8% - clumsiness, 57,8% - motor development delays, 46,2% - increase of ALT, AST. Less than 20% mentioned speech delays and peculiarities in behavior and social development. 76,1% of responders gave correct answers that CK is the only serum marker to confirm/exclude DMD.
In order to bring DMD boy to the therapy that changes the course of the disease, patient needs to be visible. The current situation, when only the tip of the iceberg is visible showing-up 30% of the DMD country population, needs urgent work to identify patients, making genetic diagnosis and targeting them to appropriate therapy.
eP04.02.01Nothing to Laugh About.
Supiot F1, Bisciglia M1
1C.u.b. Erasme, Anderlecht, Belgium
Nitrous oxide alias N2O or laughing gas is usually used in medicine and dentistry as a anesthetic agent. In Belgium, until recently, it was sold in grocery stores as a cooking aid for making whipped cream. It was soon discovered by some people that it had profound anti-anxiety and euphoriant effects. Known side effects of nitrous oxide include dizziness, dissociation, disorientation, loss of balance, impaired memory and cognition. Recreational nitrous oxide use is generally moderate, but heavy and/or sustained use may provoke a functional vitamin B12 deficiency. We describe here three cases of nitrous oxide-induced peripheral neuropathy, one of which also developed acute encephalopathy after a relapse of use, associated with low vitamin B12 count and high homocysteine levels. A remarkable characteristics of the patients was that they were all living in a particular Brussels borough, for two of them knew each other and were all young men. Nerve conduction studies and pathophysiologic issues will be discussed in detail.
eP04.02.02Acute Arsenic Intoxication Presentation Guillain-Barre Syndrome Mimic in Oncohematological Patient
Gutiérrez Martínez A1, Amela Peris R1, Díaz Díaz A1, Ruano Hernández A1
1Complejo Hospitalario Insular-Materno-Infantil, Las Palmas De Gran Canaria, Spain
Introduction: Acute arsenic intoxication is a rare cause of an acute demyelinating polyneuropathy. Systemic manifestations included acute gastroenteritis, variably associated with encephalopathy, pancytopenia, hepatitis, cardiomyopathy and dermatitis. However, chronic low-level arsenic exposure may cause distal axonopathy, predominantly sensory polyneuropathy, that is not preceded by multiorgan involvement. The acute neuropathy is usually initially misdiagnosed as Guillain-Barré syndrome (GBS), with additional tests supportive of GBS.
Case report: A 43-year-old male presented weakness in the upper limbs associated with paresthesia in both hands for 1 month. It progressively associates weakness in the lower limbs that initially required the use of a cane and later a wheelchair. He denied diplopia, visual disorder, bulbar disorder or sphincter incontinence. Nor does he refer to other symptoms by organs and apparatus.
Past medical history: Diabetes mellitus type 2 without apparent metadiabetic complications. Acute myeloid leukemia type PML-RARa positive, receiving chemotherapy treatment for 1 month and a half before the onset of symptoms.
Additional tests show a normal blood count and coagulation. Biochemistry with glucose 221 mg/dl, HbA1c 6.3%, vitamin B1, B6, B12 normal. HIV, Lues, HBV, HCV, Borrelia burgdorferi and Rickettsiosis negative. Negative IgG and IgM antiganglioside antibodies. ANA and negative onconeuronal antibodies. In the lumbar puncture there is an absence of cells with proteins of 94 mg/dl. CSF culture and cytology were normal. The initial electromyogram shows a predominantly demyelinating sensory-motor polyneuropathy with signs of motor block at the proximal level and lengthening of the absent F wave at the median level and lengthening at the ulnar level.
Treatment with intravenous immunoglobulins was started at a dose of 0.4 g/kg/day for 5 days with no evidence of improvement. After speaking with hematology about the chemotherapy treatment (ATO and ATRA), which includes the presence of arsenic, levels are requested, presenting levels of 650 mcg/L in urine and 74 mg/L. After said result, treatment with parenteral dimercaprol for 14 days. After treatment, arsenic levels decrease more rapidly in the control.
Conclusion: Polyneuropathy due to arsenic can appear acutely even with hardly any systemic manifestations. A high index of suspicion should be maintained, even despite the prescribed dose with the absence of severe affectation in clinical trials. Probably some prior individual factor (diabetes, genetics, vitamin B1...) can precipitate the degree of polyneuropathy despite the dose. Depletive treatment can help improve symptoms.
eP04.02.03Six Months of Electroneumyography in Ouagadougou, a Great Experience
Kapto O1, Kyelem J2, Ouedraogo M3, Dabilgou A2, Napon C4, Millogo A5, Kabore B2
1Polyclinique Notre Dame De La Paix, Ouagadougou, Burkina Faso, 2Centre Hospitalier Universitaire Yagado-Ouedraogo,3Centre Hospitalier Régional de KOUDOUGOU, 4Centre Hospitalier Universitaire de Bogodogo, 5Centre Hospitalier Universitaire Sanou Sourou
Introduction: WFN gives opportunity to many neurologists to learn Neurophysiology. We were admitted to WFN’s Fellowship in Morocco in 2020. We learn electroneuromoygraphy and we start it in our hospital in Burkina Faso since August 2021. We present the report of our activities.
Methodology : From August 2021 to January 2022, we collect all EMG done in our Hospital.
Results: We realised 63 tests with a mean age at 43.82 years. Sex ratio was 0.9 with 30 men and 33 women. Tests were done in upper limbs in 30.6%, in lower limbs in 7.93%, both in 60.32% and facial in 1.59%. Results were normal in 53.97%. Carpal Tunnel syndrome was present in 22.22% of which bilateral sensorymotor in 42.86%, right sensorymotor in 21.43%, Left sensory in 21.43%, right sensory in 7.14% and left motor in 7.14%. Root canal involvement was present in 7.94% with L5 (60%) and S1 (40%). Myogenic syndrome was present in 1.59% cases. Traumatic involvement were present in two cases with ulnar sensory axonotmesis and median sensitive axonotmesis. Polyneuropathies were present in 15.87% with axonal sensory (40%), axonal sensorymotor (40%) and demyelinisant sensory (20%). Facial nerve involvement was present in 1.59%.
Conclusion: Our Fellowship in Neurophysiology allow us to improve diagnose in many pathologies. Our activity is still low but our hope is perfom better diagnostic for more people
Keywords: EMG, Carpal canal syndrome, Polyneuropathy.
eP04.02.04A Case Report on Orbital Myositis
Kuqo A1, Reka E1, Xhelili M1, Rroji A2, Kruja J3
1University Hospital Center “Mother Theresa”, Neurology Service, Tirana, Albania, Tirana, Albania, 2University of Medicine, Faculty of Medicine, Neurology Service, Tirana, Albania, Tirana, Albania, 3University of Medicine, Faculty of Medicine, Neurology Service, Tirana, Albania Tirana, ALBANIA
Introduction: Orbital Myositis is a rare inflammatory disorder involving the extraocular muscles of the eye. Recent classifications of orbital myositis categorize this disorder as either a typical acute type of Idiopathic orbital myositis which is responsive to steroids or as atypical type including idiopathic chronic or recurrent orbital myositis. Orbital myositis has many similarities to uveitis and other intracranial disorders manifesting with orbital swelling, redness and altered eye function in the setting of intracranial disease. Other etiological factors to orbital myositis include viral infections, parasitic, associated autoimmune disease, myositis as a result of drug reactions and rarely paraneoplastic disease which may also cause extraocular muscle inflammation.
Case presentation: A 62- year old right handed woman presented to the emergency room with a sudden onset of painful double vision and redness around the left eye. She experienced pain localized in the periorbital region and a left hemicrania along with it. The patient referred a 3 day history of the above signs and symptoms following a two week period of high fever, malaise and flu-like symptoms for which she has been treated with antibiotics. She suffered only from hypertension and overall physical examination was unremarkable. The Neurologic examination revealed painful diplopia, semi-ptosis, partial ophthalmoplegia of the left eye and conjunctival hyperemia. No motor or sensory deficits were found. An increased Intra ocular pressure of the left eye with visual acuity of 7/10 was evidenced from the ophthalmological examination. A normal fundus oculi was found. No proptosis, chemosis, discharge or limbic injection was observed in the left eye. In the emergency room, the patient was presumptively diagnosed with orbital cellulitis and treated empirically with intravenous antibiotics. A CT scan was performed as a routine emergent test in the emergency department showing thickened medial rectus muscle in order to rule out vascular pathologies of the brain with ocular manifestations.
Results: On imaging MRI performed the next day orbital myositis of the left medial rectus muscle with contrast enhancement in a fusiform way with involvement of the muscle tendon was detected. The patient was started on high dose corticosteroids and after initiation of steroid therapy, there was rapid and complete resolution of signs and symptoms. The patient was discharged on hospital day 5 with steroids to be tapered as an outpatient. Follow ups with ophthalmological evaluation and MRI control was recommended.
Conclusion: Orbital myositis occurs in a typical and atypical type with the former one being responsive to steroids, but atypical types related to specific autoimmune and inflammatory conditions are increasingly recognized. Orbital myositis has many similarities to other systemic and neurologic diseases and a further checkup is needed in order to rule out other etiological factors in order to effectively treat the patient and avoid complications.
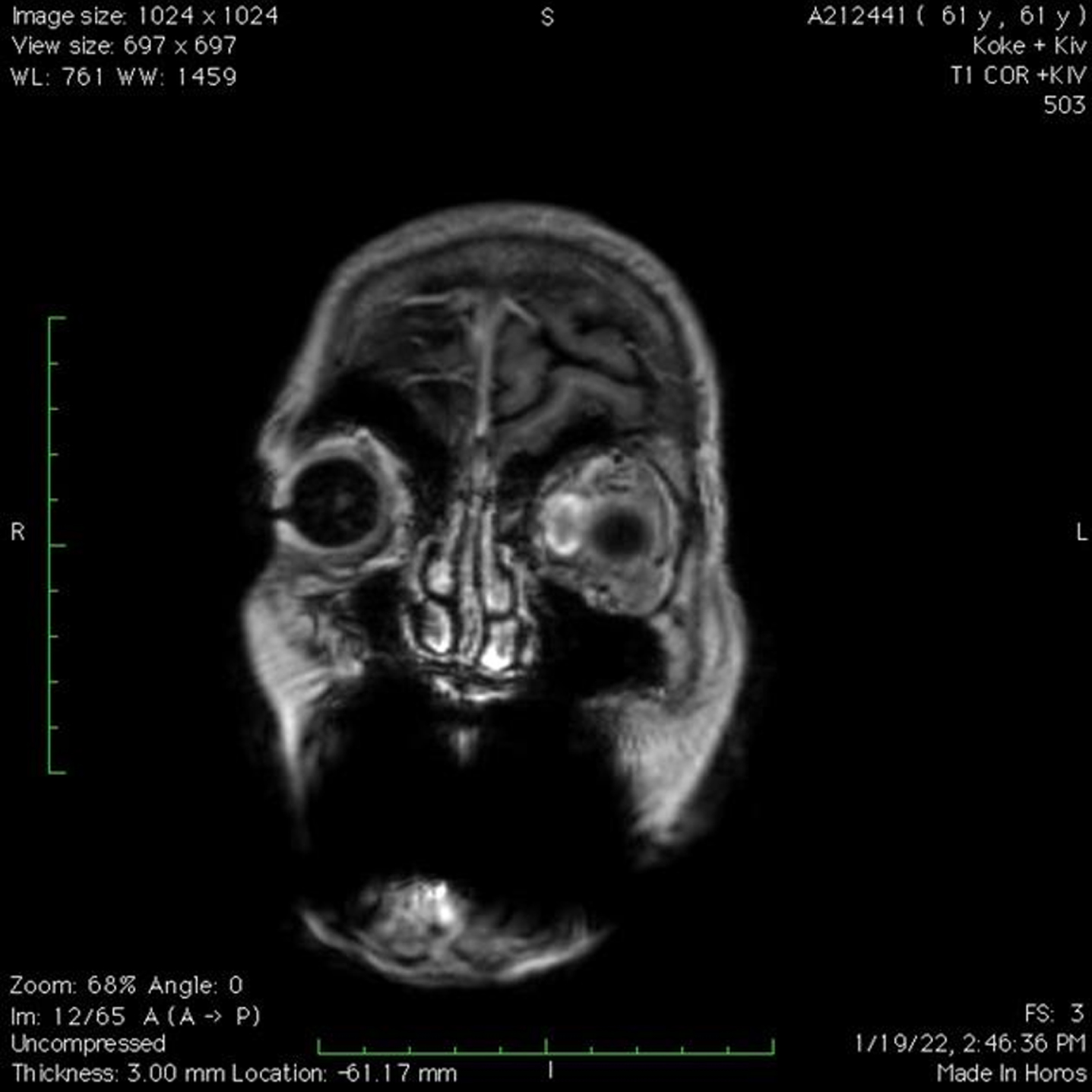
eP04.02.05Belgian Retrospective Survey of Hereditary Transthyretin-Mediated (hATTR) Amyloidosis Patients Treated With Patisiran in Real-World Practice
Tilleux S, De Bleecker J1, Claeys K2, Delstanche S3, Van Parys V4, Baets J5, Remiche G6
1Department of Neurology, University Hospital Ghent, Ghent, Belgium, 2Department of Neurology, University Hospitals Leuven, and Laboratory for Muscle Diseases and Neuropathies, KU Leuven, Leuven, Belgium, 3Department of Neurology, Centre Hospitalier Universitaire de Liège, Liège, Belgium, 4Department of Neurology, Cliniques Universitaires Saint-Luc, Brussels, Belgium, 5Department of Neurology, Antwerp University Hospital, and Translational Neurosciences, Faculty of Medicine and Health Sciences, UAntwerpen, Antwerp, Belgium, 6Department of Neurology, Hôpital Erasme, Université Libre de Bruxelles (ULB), Brussels, Belgium
Introduction: Hereditary transthyretin-mediated (hATTR) amyloidosis is an autosomal dominant, multisystemic disease caused by mutations in the transthyretin (TTR) gene. Having a worldwide prevalence of 1/1,000,000 patients, hATTR amyloidosis leads to a progressive sensorimotor and autonomic neuropathy sometimes associated with cardiac, renal and ophthalmologic manifestations resulting in a poor prognosis with a median survival of 4.7 years following diagnosis. In Belgium, current available therapies are tafamidis, patisiran and inotersen, of which the latter is not reimbursed. To date, few real-world data related to treated Belgian hATTR amyloidosis patients have been published. Objectives: To describe the current Belgian hereditary hATTR amyloidosis population with Coutinho polyneuropathy stage 1 or 2, as well as the real-world clinical impact of patisiran use in adult patients with hATTR amyloidosis, in terms of neurological and cardiac symptoms. Methods: This study was initiated to provide clinical evidence to support the Belgian reimbursement procedure for patisiran and to collect real-world evidence of clinical practice with patisiran in Belgium. It is a retrospective study based on data collected from the medical files/records of patients with hATTR amyloidosis treated with patisiran in the period from 01/07/2018 to 01/02/2021. Results: In Belgium, 31 patients were diagnosed with hATTR amyloidosis with polyneuropathy stage 1 or 2 and eligible for treatment with an active therapeutic during the data collection period. Of all hATTR amyloidosis patients treated with patisiran in Belgian Neuromuscular Reference Centers (n=12) there was an even distribution between patients with polyneuropathy stage 1 (n=7) and stage 2 (n=5). Half of the patients (n=6) had cardiac symptoms with NYHA class 2 or above. For nine patients who started patisiran treatment in the early access program, more extensive information was available. Following patisiran treatment, almost all patients (n=8) showed stable or improved assessments for most of the neurological and/or cardiological parameters. Only one of the 9 patients disclosed/presented worsening statuses (for FIM and NYHA scores) recorded at the end of the data collection period. Conclusion: The patients with hATTR amyloidosis polyneuropathy in Belgian have similar baseline demographics and disease characteristics to those studied in the pivotal patisiran APOLLO study and in real-world show a similar therapeutic response, altering the expected disease progression in most patients.
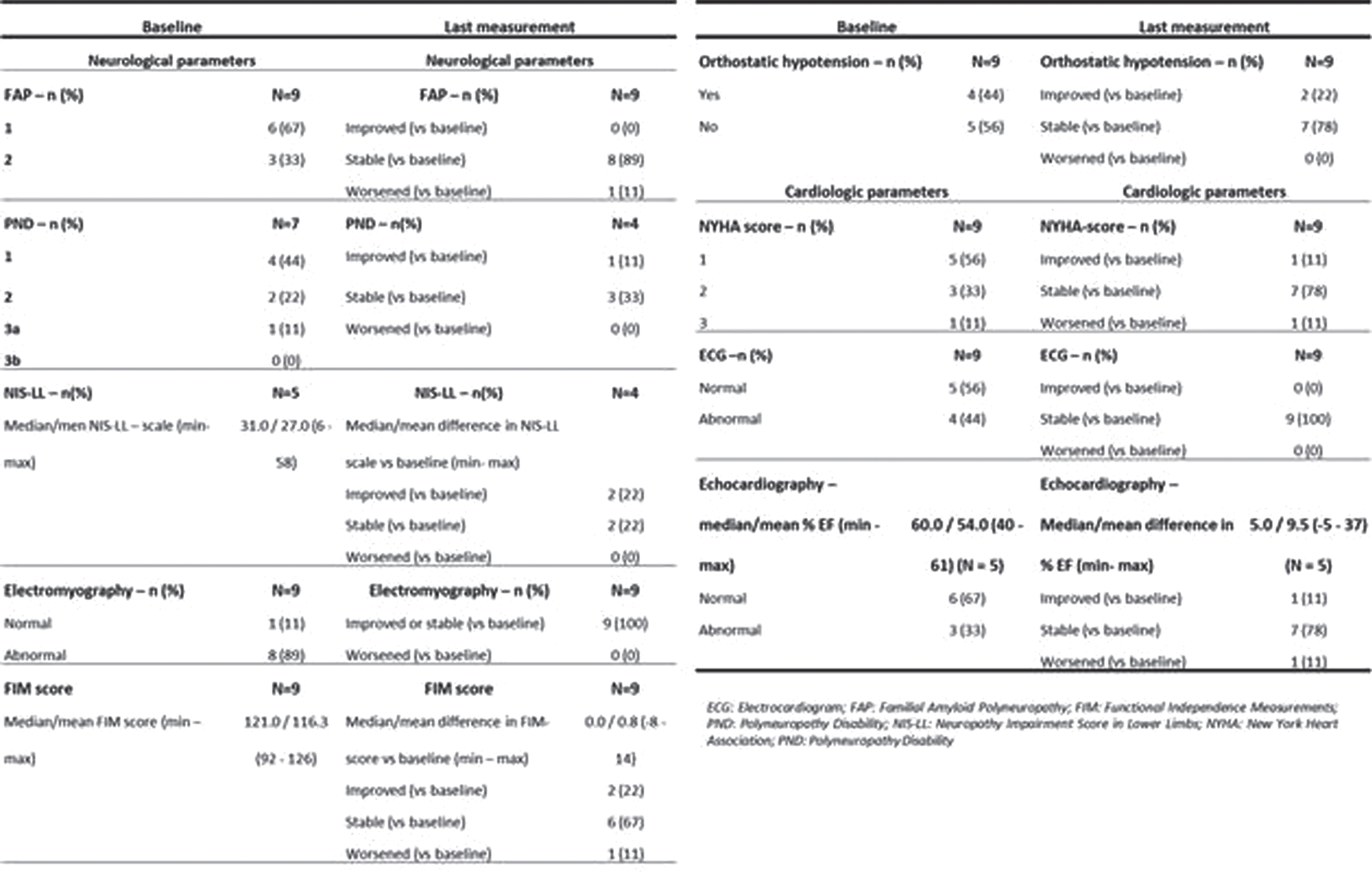
eP04.02.06Predictive Factors of Response to Tafamidis in a Cohort of Non-endemic ATTRv Patients
Martínez-Vicente L1, Gajate-García V1, Gutiérrez-Gutiérrez G2, Guerrero-Peral Á3, Horga A1, Guerrero-Sola A1, Matías-Guiu J1, Galán-Dávila L1
1Hospital Universitario Clínico San Carlos, Madrid, Spain, 2Hospital Universitario Infanta Sofía, San Sebastián de los Reyes, Spain, 3Hospital Universitario de Valladolid, Valladolid, Spain
INTRODUCTION: Hereditary amyloid transthyretin (ATTRv) amyloidosis is a rare disease with a broad clinical spectrum that varies between endemic and non-endemic areas. The first treatment approved for ATTRv was Tafamidis, a TTR stabilizer. Our objective was to study predictive factors of response to Tafamidis in a cohort of ATTRv patients from a non-endemic area.
METHODS: Retrospective study with prospective data collection of patients with ATTRv on Tafamidis treatment for ≥6 months. The patients were divided into three groups based on their response to Tafamidis (Good-Responders, Partial-Responders or Non-Responders). Demographic, clinical, and laboratory data were collected and correlated with response to Tafamidis.
RESULTS: 40 patients with ATTRv on Tafamidis treatment for ≥6 months were evaluated. 19 patients were women (47.5%) and 21 patients were men (53.5%). Mean age of disease onset was 61 years (SD 12.2). Val50Met mutation was found in 17 patients (42.5%, of which 16 patients were late-onset), followed by Ser97Tyr in 7 patients (17.5%), Glu109Lys in 4 patients (10%), and Val142Ile and Ser43Asn in 3 patients respectively (7.5% and 7.5%). Mean time of treatment with Tafamidis was 38 months (SD 25.8). At last follow-up, 20 patients were on Tafamidis treatment (50%), 16 patients underwent a change in treatment (40%), and 4 patients died (10%). 16 patients were Good-Responders (42.1%), 6 patients were Partial-Responders (15.8%) and 16 patients were Non-Responders (42.1%). Predictive factors of non-response to Tafamidis were an initial PND stage II (p=0.021), initial large fiber involvement (p=0.006), significant weight loss (p=0.008) and a history of spinal stenosis (p=0.009). Initial PND stage I (p=0.02), initial NIS≤15 (p=0.001), initial Norfolk ≤25 (p=0.002) and initial RODS>40 (p=0.025) were associated with good response to Tafamidis.
CONCLUSIONS: In this study, initial PND stage I, NIS≤15, Norfolk ≤25 and RODS>40 were associated with a good response to Tafamidis, while the presence of an initial PND stage II, large fiber involvement, weight loss and spinal stenosis were predictive factors of non-response to treatment with Tafamidis.
eP04.02.07Quantitative Sensory Testing in patients diagnosed with ATTR Amyloidosis in Colombia: A Case Series
Ruiz E1,2, Castellar S1,2, Diaz J1,2, Ortiz F1,2, Correa C
1Universidad Nacional de Colombia, Bogota, Colombia, 2Centro de investigación en Fisiatría y electrodiagnóstico CIFEL, Bogota, Colombia
Introduction: Transthyretin amyloidosis (ATTR) is a progressive condition, potentially lethal caused by mutations in TTR gene leading to deposition of amyloid in different tissues. Small fiber neuropathy has been previously described as consequence of amyloid deposit in individuals carrying the pathogenic mutation. We present a descriptive small fiber function profile by Quantitative Sensory Testing (QST) of 4 individuals with molecular diagnosis of ATTR
Cases: 4 patients. 1 male, 3 female mean age 58.5y (Range 75-41y) at time of evaluation. All patients had confirmed diagnosis of ATTR by TTR gene sequencing. Val142Ile Variant was found in 2 individuals and Val50Met variant was found in other 2 individuals. Electrodiagnostic studies were normal except for one patient which revealed bilateral moderate Carpal Tunnel Syndrome. QST was performed in upper and lower extremities with Computed Aided Sensory Evaluator (CASE IV Software) using 4,2,1 stepping algorithm. Vibratory detection thresholds (VDT), Cold Detection Thresholds (CDT) and Heat-Pain Detection Thresholds (HPDT) were calculated. Increased CDT was the main dysfunction observed in 50% of carriers of both mutations with percentiles calculated above 97.5 indicating hyposensitivity to cold stimuli. Increased VDT was found in an individual affected by Val50Met variant in four extremities percentiles calculated 99.9. One patient with Val142Ile showed significant decrease of HPT percentiles calculated 0.01 demonstrating marked hypersensitivity to heat stimuli in both upper and lower limbs. This patient had atrial fibrillation as associated cardiopathy and was the oldest among the individuals observed.
Conclusion: This is the first study of this kind in Colombia. Abnormal findings in QST were revealed in 75% of patients affected with ATTR. Although Val142Ile Variant is associated mainly with cardiac amyloidosis we found neuropathic manifestations in our study. It is important to rule out small fiber neuropathy in patients affected by ATTR to characterize them and correlate it with genotypic, phenotypic, and demographical features. QST is an useful tool to determine sensory deficits caused by small fiber neuropathy. There are new molecules that can be used to modify the natural history of the disease and is imperative to have different biomarkers which quantify and determine neuropathic manifestations in order to evaluate goals, objectives and potential outcomes to pharmacological treatments. This is the first study of this kind in Colombia
eP04.03.01Respiratory-Onset of ALS in a Pregnant Woman With a Novel SOD1 Mutation
Masrori P1,2,3, Ospitalieri S4,5, Forsberg K6,7, Moens T2,3, Poesen K8,9, Race V10,Brännström T6, Andersen P6, Thal D4,5, Van Damme P1,2,3
1Department of Neurology, Neuromuscular Reference Centre, University Hospitals Leuven, Leuven, Belgium, 2Department of Neurosciences, Experimental Neurology, and Leuven Brain Institute (LBI), KU Leuven - University of Leuven, Leuven, Belgium, 3Laboratory of Neurobiology, Center for Brain and Disease Research, VIB, Leuven, Belgium, 4Department of Imaging and Pathology, KU Leuven, Leuven, Belgium, 5Department of Pathology, University Hospitals Leuven, Leuven, Belgium, 6Department of Clinical Sciences, Neurosciences, Umeå University, Umeå, Sweden, 7Department of Medical Biosciences, Units: Pathology, Umeå University, Umeå, Sweden, 8Laboratory for Molecular Neurobiomarker Research, Department of Neurosciences, Leuven Brain Institute, KU Leuven, Leuven, Belgium, 9Laboratory Medicine, University Hospitals Leuven, Leuven, Belgium, 10Laboratory for Molecular Diagnosis, University Hospitals Leuven, Leuven, Belgium
Background: With the advent of gene therapies for amyotrophic lateral sclerosis (ALS) the importance of gene testing in ALS is increasing. This will likely lead to the identification of new variants for which the pathogenicity is not established. We aimed to study the pathogenicity of a newly identified variant in superoxide dismutase 1 (SOD1).
Methods: Gene testing was performed using Sanger sequencing. SOD1 activity in erythrocytes was measured using spectrophotometry. Post-mortem brain and spinal cord sections were stained with antibodies against phosph-TDP-43 and SOD1.
Results: We identified a novel c.416G>T (p.Gly139Val) mutation in SOD1, which caused a rapidly progressive respiratory onset form of ALS. The mutation resulted in a 50% drop of SOD1 activity. Post-mortem examination confirmed the absence of TDP-43 pathology and displayed typical SOD1 inclusions in remaining motor neurons, confirming the pathogenic nature of the mutation.
Discussion: Novel variants of unknown pathogenicity will be identified as a result of a surge in gene testing in people with ALS. An in depth study of a newly identified p.Gly139Val mutation in SOD1 confirmed that pathogenicity of this mutation. Future patients with this particular mutation should qualify for SOD1 silencing or editing therapies.
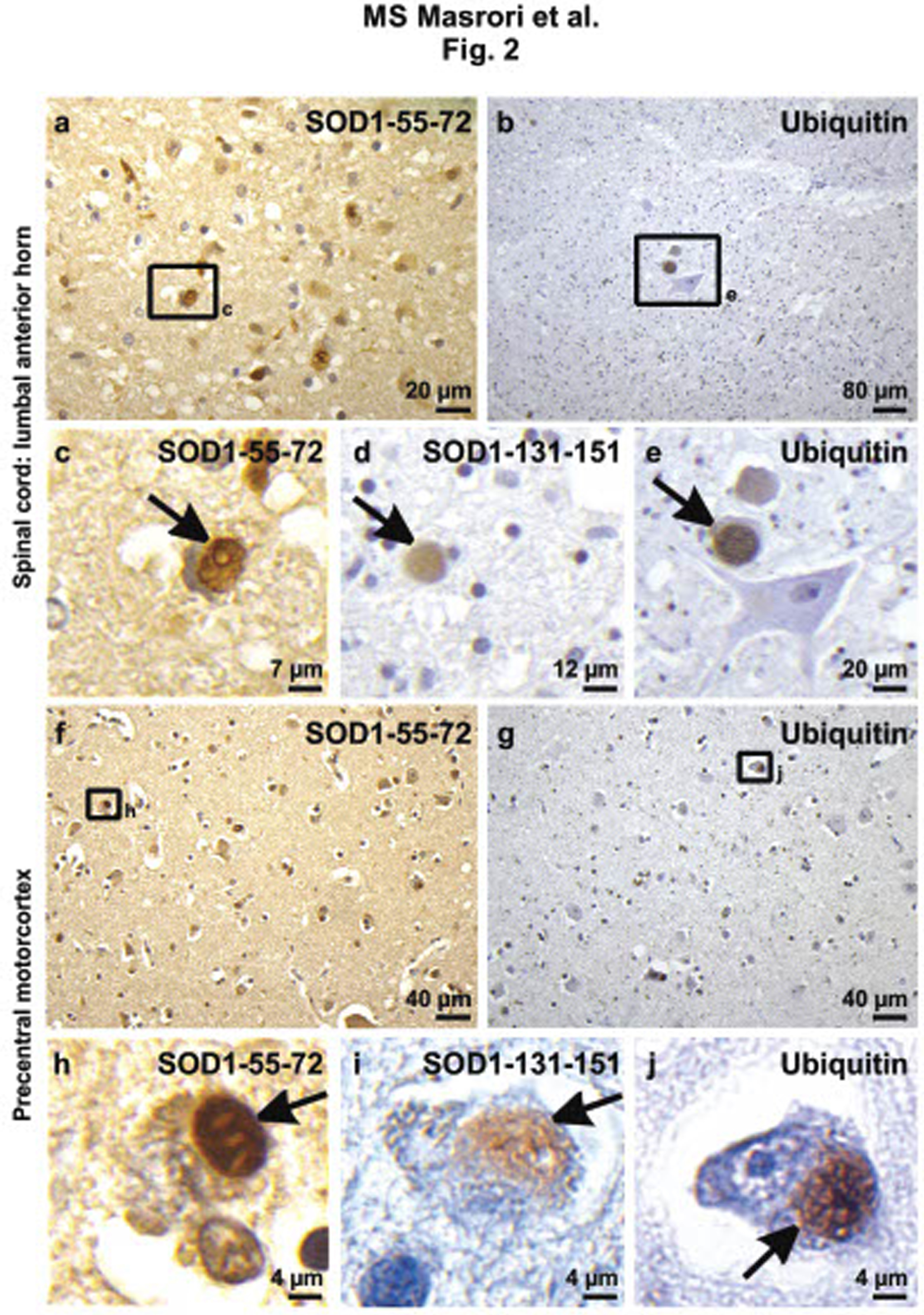
eP04.03.02Association of APOE e4 Allele With Survival in Amyotrophic Lateral Sclerosis Among Tunisian Cases
Kacem I1,2, Seghaier I1, Abida Y1,2, Mrabet S1,2, Jelassi C1, Nasri A1,2, Gouider R1,2
1Department Of Neurology, Lr18sp03, Clinical Investigation Centre Neurosciences And Mental Health, Razi Hospital, Manouba, Tunisia, 2Faculty of Medicine of Tunis, University Tunis EL Manar, Tunis, Tunisia
Introduction: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative motor neuron disorder affecting both upper and lower motor neurons. The pathogenesis of sporadic ALS, the most common form of disease, is unknown and probably multifactorial, including overexposure of motor neurons to glutamate and oxidative stress-induced cell death. Further causative factors, including a genetic predisposition, may have a modulatory influence. APOE e4 allele is associated with poorer outcome in ALS and several studies correlated the carriage of the risk allele e4 with low survival rate.
Objective: The aim of the present study was to analyze the association of APOE e4 allele with progression and survival of ALS as well as evaluate the possibilities of complex formation of APOE e4 with the four main ALS genes (SOD1, TARDBP, C9ROF72 and FUS).
Methodology: We conducted a retrospective study in the Department Neurology of Razi Universitary Hospital, including patients with definite or probable ALS from January 2011 to December 2021. Genetic analysis comprised the genotyping of APOE gene using Restriction fragment length PCR (PCR-RFLP). Additionally, ALS cases were genotyped for the 4 main associated ALS genes; SOD1, TARDBP, C9ORF72 and FUS. Clinical data was analyzed in order to evaluate the correlation of APOE e4 as prognosis factor of survival using Kaplan Meier analysis. The association of APOE and ALS were evaluated using a case control study. Written informed consent was obtained from all patients for the genetic study, which was approved by the appropriate ethics committees.
Results: Two hundred and nine ALS cases were included. The mean age of onset for the total cohort was 54.6+13.7 years and the median of survival was 4 years [1STQu =2 and 3RDQu=6]. The total cohort comprised 167 ALS with spinal onset, 37 bulbar, and 5 generalized. Significant differences were seen in term of e4 allele presence in both groups (patients and controls) regardless of allele copy number (p<0.001). In fact, 30.14% (N=63) of ALS patients were carriers of e4 allele vs. 69.86% Non-carriers (N=146). However, in our healthy control cohort we found only 4 cases carriers of e4 allele (2.51%) and 190 are non-carriers (97.93%). There was a significant shortening of the 50% probability of survival (by 12 months) in patients carrying the APOE e4 allele (p=0.037). Moreover, the effect of APOE e4 allele on survival rate was correlated to the ALS form. In fact, patients with spinal ALS carrier of e4 had poorer survival rate compared to spinal ALS non carriers (p=0.0083). While no significant correlation was observed regarding APOE variants and bulbar ALS onset (p=0.81).
Interestingly, we report two ALS cases carriers of G294A-TDP-43 and e4 APOE allele mutation as well as another two cases with C9orf72 expansion were carriers of APOE-e4 risk allele simultaneously.
Conclusion: Carrying an APOE-e4 allele is a poor prognostic factor in ALS. This is compatible with a role of apolipoprotein on neuronal survival and repair.
eP04.03.03The Thymus in the Pathogenesis/Pathophysiology of Amyotrophic Lateral Sclerosis
P. Lemos J1, Marais T1, Pezet S1, Butler-Browne G1, Mouly V1, Savino W2,3,4, Smeriglio P1
1UMRS974 Sorbonne University, Inserm, Institute of Myology, Myology Research Center, Paris, France, 2Laboratory on Thymus Research, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil, 3Brazilian National Institute of Science and Technology on Neuroimmunomodulation, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil, 4Rio de Janeiro Research Network on Neuroinflammation, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil
Background: Amyotrophic Lateral Sclerosis (ALS) is the most common motor neuron disease in adults, characterized by the loss of motor neurons and consequent muscle atrophy leading to paralysis and death 3 to 5 years after diagnosis. While most cases of ALS are sporadic in nature (sALS), approximately 20% of the familial forms of ALS (fALS) are caused by a mutation in the Cu/Zn superoxide dismutase 1 (SOD1) gene. Neuroinflammation seems to play a critical role in ALS, with significant T lymphocyte inflammatory infiltrates being observed in the central nervous system (CNS) of SOD1 mutated ALS patients. CD4+ and CD8+ T cells are found in close proximity to degenerated neurons in the spinal cord. Interestingly, the regulatory T lymphocytes (Treg), a subset of CD4+ T cells bearing the phenotype CD4+CD25+Foxp3+, have been described to confer neuroprotection. In ALS patients, a lower number of Treg in the peripheral blood correlates with a faster disease progression. The thymus is responsible for the maturation and differentiation of T lymphocytes and evidence suggests that thymic function may be compromised in the mouse SOD1 model (mSOD1). However, whether there is a thymic dysfunction involved in the T-cell imbalance and, notably, in the decrease of peripheral Treg cells observed in both ALS patients and in mSOD1 mice, is not yet known.
Methods: We used male B6SJL-Tg(SOD1*G93A)1Gur/J mice (herein named mSOD1) at four different postnatal time points, i.e. before (30 and 60 days), at the onset (90 days), and at the late-stage of the disease (120 days old), and compared them with B6SJLF1/J control littermates (herein named WT).
Results: We observed that, in addition to a lower body weight gain, mSOD1 mice at 120 days of age have reduced relative thymus weight, compared to the WT controls. Analysis of the cellular compartment by flow cytometry demonstrated that mSOD1 mice present a reduction in the number of CD4+CD25+Foxp3+ Tregs in the thymus at the onset of the disease (90 days old) which persists until the final stage (120 days old). Additionally, using annexin V staining we observed a higher relative number of apoptotic cells in the CD4+CD25+ compartment within the mSOD1 thymus at 120 days old, as compared to controls.
Conclusion: Our results suggest that the decrease in the number of Treg observed in the periphery of ALS patients and in the mSOD1 mouse model is at least partially related to a decrease in the tTreg exit from the thymus, due to an impaired intrathymic Treg cell development.
Funding: This work was supported by the Association Institut de Myologie (AFM) and the Inserm-Fiocruz French-Brazilian Cooperation. W.S. is funded by CNPq, CAPES and FAPERJ (Brazil) and the MercoSur Fund for Structural Convergence (FOCEM).
eP04.03.04Systemic Genetic Screening of Korean Patients With Amyotrophic Lateral Sclerosis
Kim J1, Choi S1, Shin J1, Hong Y2, Sung J1
1Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Republic of Korea, Seoul, Republic of Korea, 2Department of Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, Republic of Korea
Intoduction: To understand pathophysiology of amyotrophic lateral sclerosis (ALS) and improve outcomes in ALS clinical trials, genetic study is a research priority. The genetic spectrum of ALS varies widely between ethnic groups. In 2011, the most common ALS gene in Caucasian populations, C9orf72 hexanucleotide repeat expansion, was identified in the three landmark studies. However, C9orf72 repeat expansion is less frequently observed in Asian populations and has still not been reported in Korean ALS patients.
Method: In this study, we performed targeted sequencing of a 27 gene panel in 154 patients with ALS. 8 patients had familial ALS and the rest of the patients were sporadic.
Result: 14/154 patients carried a pathogenic or likely pathogenic variant of causative genes, of whom 3 patients had family history of ALS, and SOD1 was the most common gene. Notably, 22% of patients harbored a variant of uncertain significance (VUS), of whom three patients carried more than one VUS.
Discussion: We identified genetic heterogeneity in ALS between ethnic groups. Moreover, considering clinical trials in progress targeting specific genetic forms of ALS including SOD1, systemic genetic screening of patients with ALS might be a crucial step in real practice.
eP04.03.05New Mutation (Val31Gly) In PFN1 Gene Responsible for the Development of ALS18 in Bulgarian Pedigree
Angelov T1, Chamova T1, Todorov T2, Ormandzhiev S2, Todorova A2,3, Tournev I1,4
1Clinic of Nervous Diseases, University Hospital “Alexandrovska”, Medical University of Sofia, Bulgaria, 2Genetic Medico-Diagnostic Laboratory “Genica”, Sofia, Bulgaria, 3Department of Medical Chemistry and Biochemistry, Medical University of Sofia, Bulgaria, 4Department of Cognitive Science and Psychology, New Bulgarian University, Sofia, Bulgaria
Introduction: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive loss of motor function, disability, and death. Mutations in PFN1 encoding the protein Profilin-1, are responsible for the development of ALS 18.
Material and methods: We present a pedigree consisting of 3 generations and 4 affected individuals with a genetically confirmed heterozygous mutation 17:g.4851598A>C, c.92T>G, p.Val31Gly in the PFN1 gene. The mean age at onset of the disease is 59.75 (±10.11) years with ranging between the first two generations (females) and the third (male) of 22.33 (±3.4) years. It is more benign, with a relatively high survival rate, as three of the affected are still alive – at least two, four and seven years respectively after the disease onset and only one had deceased three years after the onset. The clinical features encompass predominant involvement of lower motor neuron (LMN) in one limb and gradual impairment of the other limbs, without obvious involvement in the brainstem and the upper motor neuron (UMN).
Discussion: ALS 18 is a rare clinical form of ALS, with only 31 genetically proven patients described up to now. The Bulgarian pedigree is a carrier of a novel mutation V31G (Val31Gly) and its clinical features are very similar to previously reported cases with relatively late onset, slow progression of the disease and predominant impairment of LMN in the limbs.
Conclusion: Our data will help to enrich the conceptions of the clinical-genetic phenotype of ALS and to improve the differential-diagnostic notions of the rare clinical-genetic forms.
Keywords: ALS, genetics, phenotype, pedigree, PFN1
eP04.03.06Clinical Characteristics of c.63C>G (p.Phe21Leu) Variant in SOD1 gene in Colombian Patients with ALS
Ruiz E1, Correa C, Ortiz corredor F, Castellar S, Sanchez D, Salcedo M, Soto D
1Universidad Nacional de Colombia - Instituto Roosevelt, Bogota, Colombia
Introduction. Amyotrophic lateral sclerosis (ALS) is a progressive and irreversible neurodegenerative disorder of motor neurons, which alters walking, speech, swallowing, among other functions. The SOD1 gene codes for Cu/Zn Superoxide dismutase 1, and its mutations are related to 20% of cases of familial ALS.
Number of cases. The c.63C>G variant in the SOD1 gene was found to occur more commonly in men (6/7, 85.7%), with a mean age of onset of 50.1 years (range 40-70 years); the mean time of follow-up was 79.2 months (range 43 – 152 months); phenotypically, 71.4% presented as progressive muscular atrophy, with a predominance of cervical onset (5/7, 71.4%), with signs of upper motor neuron in 2 cases (28.5%), dysphagia, dysarthria and use of non-invasive mechanical ventilation in 6 cases (85.7%), although none have required gastrostomy, tracheostomy or alternative communication systems.
There is a family history in 2 cases (28.5%), and the mean rate of progression on the ALSFRS-R scale is 0.26 points/month (range 0.1-0.5).
Conclusion. The SOD1:c.63C>G variant in Colombian patients predominate in men, with an early age of onset, low rate of progression, cervical onset segment, with progressive muscular atrophy phenotype, and mean follow-up in months above the general average of patients with ALS.
eP04.03.07Serum Creatine Kinase and Creatinine in the Diagnosis and Prognostic Prediction of Amyotrophic Lateral Sclerosis
Seghaier I1, Mrabet S1,2, Abida Y1,2, Saad Y1,2, Riahi S1,2, Nasri A1,2, Kacem I1,2, Gouider R1,2
1Department Of Neurology, Lr18sp03, Clinical Investigation Centre Neurosciences And Mental Health, Razi Hospital, Manouba, Tunisia, 2Faculty of Medicine of Tunis, University Tunis EL Manar, Tunis, Tunisia
Introduction: Several prognostic biomarker candidates have been received increasing attention recently in typical Amyotrophic Lateral Sclerosis (ALS) cases such as muscle enzyme creatine kinase (CK) and creatinine (Cr). Serum CK and creatinine have been considered to be associated with muscle damage. Hence may be considered as potential biomarkers for ALS and survival indicator
Objective: To evaluate serum creatine kinase (CK) levels and creatinine (Cr) of ALS patients and to explore the relationship between CK levels and the clinical characteristics and survival prognosis of ALS patients.
Methodology: We conducted a retrospective observational cohort study that included ALS patients followed in the department of Neurology in Razi University Hospital from October 2021 to December 2021. We collected demographic data, clinical phenotype, initial ALSFRS-R score, disease duration, survival, and CK and Cr levels when it was possible. We studied correlation between serum CK and Cr concentrations and clinical features. A p-value of <0.05 was regarded as statistically significant. Statistical analyses were performed using R studio software.
Results: We included 297 patients. Sex ratio was 1.9, the mean age of onset was 55.14±14.3 (Spinal 53.5 years and Bulbar 61.1 years) and the median disease duration was 2 years (max=46 years and min 0 years). Males had significantly higher baseline serum Cr and CK levels (p=0.021 and p=0.042 respectively). Besides, Cr levels were positively associated with initial ALSFRS-R scores (p=0.046). Serum Cr levels tended to be positively and significantly correlated along with the age of onset (p=0.087), while, the CK level was inversely correlated to the age of onset (p=0.011).
Regarding the ALS form, the CK level was significantly higher (p=0.043) among spinal ALS cases compared to bulbar and generalized form 262.5 vs. 147.4 and 122.4 respectively. Moreover, spinal and bulbar female ALS cases tends to had higher level of initial CK than males (186.8 vs.177 and 151 vs. 100.5 respectively). However, the generalized male ALS form have more increased CK than female (148 vs. 96 respectively). No differences were noted in the creatinine level according to ALS form (p=0.949) even with gender adjustment (p=0.117). Regrading the survival rate, we found a significant association of CPK initial level and survival (p=0.023). In fact, there was a significant shortening of the 50% probability of survival (by 24 months) in patients carrying with abnormal CK compared to ALS cases with normal CK level.
Conclusion: Serum CK levels of ALS patients were correlated to gender, site of onsite, ALSFRS-R score and serum Cr. Serum CK could be reliable and easily accessible prognostic chemical markers for ALS and abnormal CK baseline levels could predict a poor prognosis and short survival in ALS patients. Serum CK as well as Cr could be an independent prognostic factor for ALS progression and/or severity among Tunisian patients.
eP04.03.08Potential Role of Mitochondrial Dysfunction in an Unusual Co-occurrence of ALS and Primary Biliary Cirrhosis
Kim J1, Chavez-Keatts M1, Paul P1
1University Of Illinois At Chicago, Chicago, United States
Objective: To describe a case of co-existing Primary Biliary Cirrhosis (PBC) and Amyotrophic Lateral Sclerosis (ALS) and hypothesize a common disease pathomechanism.
Background: Mitochondrial dysfunction has been proposed to play a role in pathogenesis of ALS. Since PBC is a rare autoimmune disease with antibodies against mitochondrial antigens, co-occurrence of ALS in patients with PBC warrants further scrutiny, especially when both are rare conditions. We present a case of concomitant ALS and PBC; only two such have been previously reported1.
Case report: A 71-year-old woman presented with a 5-year history of progressive weakness. She was diagnosed with PBC about 20 years ago confirmed by a liver biopsy and elevated anti-mitochondrial antibodies and had been on treatment with ursodiol. She first noticed weakness in her left lower extremity, followed by gradual weakness of bilateral upper extremities and finally became wheelchair bound. She was referred to neurology clinic for suspected hepatic myelopathy. On exam, patient had spasticity and diffuse hyperreflexia in all 4 limbs along with flaccid and spastic dysarthria and limb muscle atrophy. MRI brain and spinal cord imaging were unremarkable to explain diffuse motor findings. Needle EMG showed evidence of diffuse active and chronic denervation. Overall findings were consistent with diagnosis of definite ALS per El Escorial criteria. Patient was started on riluzole, which was well tolerated.
Discussion: Hepatic myelopathy presenting as progressive spastic paraparesis has been described in patients with chronic liver disease, particularly after portosystemic shunt placement. However, in such cases exact pathomechanism and basis of predilection for motor neurons remain unknown. Our patient developed weakness before shunt placement and myelopathy would not explain the diffuse lower motor neuron findings.
The literature on ALS is unfolding it into a multi-system disease with evidence of metabolic changes and energy dysregulation. Hepatic derangement in ALS has been noted in both clinical study and murine models. Further, respective rare incidences of ALS and PBC suggest coincidental co-occurrence is statistically very unlikely. It is thus possible that anti-mitochondrial immune response in PBC attacks other systems including motor neurons. Relatedly, bile acids including those used in PBC treatment have been found to have protective potential in many neurodegenerative conditions. The results from recent phase II clinical trial using taurursodiol, a bile acid, demonstrated slowing of disease progression. Consistent with this, both in the previously reported case and our patient, ALS related weakness occurred many years after being on treatment with ursodiol suggesting possible protective role.
In a recent study, reduced mitochondrial respiration has been identified as metabolic hallmark in ALS motor neurons, both familial and sporadic, suggesting defective mitochondrial respiration could be the final common pathway2. However, in the same study, both Riluzole and Edaravone, the only 2 FDA approved medications failed to improve the mitochondrial bioenergetics and perhaps explains their limited benefits. Amidst the growing knowledge of molecular complexity of ALS pathogenesis, our case emphasizes the continuing relevance of unusual clinical co-occurrences.
References
1. Zhang H et al. Front Neurol. 2019;10:890. doi:10.3389/fneur.2019.00890
2. Hor JH et al. Cell Death Differ. 2021;28(4):1379-1397. doi:10.1038/s41418-020-00664-0
eP04.03.09Hereditary Spastic Paraparesis Type 9A Mimicking ALS: A Case Report
Ruiz E1, Correa C, Castellar S, Sanchez D, Ortiz F
1Instituto Roosevelt - Universidad Nacional De Colombia, Bogota, Colombia
Introduction. Hereditary spastic paraparesis (SPG) are a heterogeneous group of rare, hereditary disorders that affect the longest axons of the corticospinal tract, causing weakness and progressive spasticity in the lower extremities. SPG9 is caused by mutations in the ALDH18A1 gene. The inheritance pattern can be autosomal dominant or recessive, and has been described more frequently in British, French, and Italian families.
Case report. We present the case of a male patient of French and Italian descent, with progressive, continuous weakness in the right hand that began in April 2019 (age of onset 43 years), associated with cramps, generalized fasciculations, and gait disturbance, without bulbar symptoms. He has had a slow progression (0.2 monthly points on the ALSFRS-R scale).
The electrodiagnostic study showed signs of denervation and reinnervation in the cervical and lumbosacral segments. The genetic panel for motor neuron disease showed the variant c.1385A>G (p.Lys462Arg) in the ALDH18A1 gene, in heterozygosis, classified as probably pathogenic in VARSOME, which codes for hereditary spastic paraparesis type 9A, described with a phenotype that combines signs of upper motor neuron and motor neuronopathy.
Conclusion. We describe the case of a patient with a probably pathogenic variant in the ALDH18A1 gene, who presented with complicated spastic paraparesis associated with motor neuronopathy, with a clinical phenotype and electrodiagnostic findings that simulate slow-progressing amyotrophic lateral sclerosis.
eP04.03.10International Phase 3 Trial Evaluating Sodium Phenylbutyrate/Taurursodiol in Amyotrophic Lateral Sclerosis (PHOENIX): Enrollment Update
van den Berg L1, van Eijk R1,2, Al-Chalabi A3,4, Andrews J5, Chiò A6,7, Corcia P8, Cudkowicz M9, Ludolph A10, McDermott C11, Manuel M12, Mehta L12, Timmons J12, Whitney E12, Paganoni S9,13
1Department of Neurology, UMC Utrecht Brain Center, University Medical Center Utrecht, Utrecht, the Netherlands, 2Biostatistics & Research Support, Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht, the Netherlands, 3Maurice Wohl Clinical Neuroscience Institute, King’s College London, Department of Basic and Clinical Neuroscience, London SE5 9RX, United Kingdom, 4Department of Neurology, King’s College Hospital, London SE5 9RS, United Kingdom, 5Division of Neuromuscular Medicine, Columbia University, New York, United States, 6’Rita Levi Montalcini’ Department of Neuroscience, University of Turin, Turin, Italy, 7Azienda Ospedaliero-Universitaria Città della Salute e della Scienza of Turin, Turin, Italy, 8ALS Center, CHU Tours, France, 9Sean M. Healey and AMG Center for ALS & the Neurological Clinical Research Institute, Massachusetts General Hospital, Harvard Medical School, Boston, United States, 10Department of Neurology, University of Ulm, Ulm, Germany, 11Sheffield Institute of Translational Neuroscience, University of Sheffield, Sheffield, United Kingdom, 12Amylyx Pharmaceuticals, Inc., Cambridge, United States, 13Spaulding Rehabilitation Hospital, Harvard Medical School, Boston, United States
Background: An oral, fixed-dose coformulation of sodium phenylbutyrate (PB) and taurursodiol (TURSO, also known as ursodoxicoltaurine) was designed to reduce neuronal death by blocking key cell death pathways originating in the endoplasmic reticulum and mitochondria. In the 24-week US multicenter, randomized, placebo-controlled, phase 2 CENTAUR trial (NCT03127514), PB/TURSO was shown to significantly slow functional decline in adults with definite amyotrophic lateral sclerosis (ALS; revised El Escorial criteria), symptom onset ≤18 months, and slow vital capacity (SVC) >60% at screening. Overall survival was significantly longer among those originally randomized to PB/TURSO versus placebo at nearly 3 years after randomization. Overall adverse event (AE) incidence was similar for PB/TURSO and placebo in CENTAUR, but early gastrointestinal events occurred with greater frequency in the PB/TURSO group.
Objective: PHOENIX (NCT05021536) is an ongoing international, phase 3 trial aimed at demonstrating the safety and efficacy of PB/TURSO in a larger, more heterogeneous sample of people with ALS over a longer duration.
Methods: Up to 600 adults (EU, n≈400; US, n≈200) with clinically definite or clinically probable ALS (revised El Escorial criteria), symptom onset <24 months, and SVC ≥55% will be enrolled from >70 Treatment Research Initiative to Cure ALS (TRICALS) and Northeast ALS Consortium (NEALS) sites. Participants will be randomized in a 3:2 ratio to receive PB/TURSO or matching placebo by mouth or feeding tube for 48 weeks. PB/TURSO safety will be assessed via incidence and severity of AEs and trial discontinuations. The primary efficacy objective is to evaluate the impact of PB/TURSO versus placebo on disease progression, using a joint assessment of function (as measured by the ALS Functional Rating Scale–Revised) over 48 weeks and survival. Secondary objectives will assess the effect of PB/TURSO on SVC, quality of life (40-item ALS Assessment Questionnaire), transition time through King’s and MiToS stages, ventilation-free survival, and long-term overall survival beyond the planned 48-week follow-up duration. Caregiver burden and plasma biomarkers of neuron damage and neuroinflammation will be assessed as exploratory outcomes.
Results: Updates on trial enrollment and progress will be provided.
Discussion: By incorporating a larger, more heterogeneous population of people with ALS followed for a longer duration, PHOENIX will build on the findings of the phase 2 CENTAUR trial. PHOENIX applies various telemedicine-friendly interventions, including limited in-person visits, electronic data capture, and home spirometry under guidance of a remote avatar, thereby highlighting a patient-centric ALS trial design.
eP04.04.01Long Term Preliminary Safety and Efficacy Outcomes for X-Linked Myotubular Myopathy with Gene Replacement Therapy
L. Kuntz N1, B. Shieh P2, J. Dowling J3, Müller-Felber W4, Blaschek A4, G. Bönnemann C5, Foley A5, N. Saade D6, M. Seferian A7, Servais L8, Coats J9, Lakshman N9, Han C10, Prasad S11, Rico S11, Miller W9
1Ann & Robert H Lurie Children’s Hospital of Chicago, Chicago, USA, 2University of California, Los Angeles, USA, 3Hospital for Sick Children, Toronto, Cannada, 4Klinikum der Universität München, Munich, Germany, 5Neuromuscular and Neurogenetic Disorders of Childhood Section, NINDS, NIH, Bethesda, USA, 6University of Iowa Hospitals and Clinics, Iowa City, USA, 7I-Motion, Institute of Myology, Paris, France, 8MDUK Oxford Neuromuscular Centre, Oxford, UK, 9Astellas Gene Therapies (formerly Audentes Therapeutics, Inc), San Francisco, USA, 10Astellas Pharma Global Development, Northbrook, USA, 11Formerly Astellas Gene Therapies, San Francisco, USA
Introduction: XLMTM is a rare, life-threatening congenital myopathy caused by mutations in the MTM1 gene. Approximately 80% of XLMTM patients experience profound muscle weakness leading to congenital respiratory failure and chronic ventilator dependency, profound impairment of motor development, and high negative quality of life impact. There is no approved treatment for XLMTM.
Method: ASPIRO (NCT03199469), a Phase 1/2/3 randomized, open-label study is investigating the safety and efficacy of AT132 (resamirigene bilparvovec), a single-dose gene replacement therapy for ventilatory-dependent XLMTM. Participants were young boys with genetically confirmed XLMTM. The primary efficacy outcome was the change in hours of daily ventilator support from baseline through Week 48. The key secondary efficacy outcome was percentage of participants who achieve functionally independent sitting by Week 48. We report long-term safety and key efficacy outcomes (up to 42 months [m]) for the first 6 participants dosed in ASPIRO, all receiving the lower dose of 1.3 x 10^14 vg/kg and compared with 15 untreated controls (including 12 participants from INCEPTUS), as of 29JAN2021.
Results: The mean age at dosing was 20·4m (range: 9·5-49·7m) and 19·6m (5·9-39·3m) at enrollment among dosed participants and controls, respectively. Major developmental milestones achieved by dosed participants over time is shown in Figure 1. All dosed participants were ventilator dependent at first assessment; 5 (83.33%) requiring transtracheal invasive ventilation >22 hours/day and 1 (16.67%) used non-invasive ventilation of 12 hours/day. All dosed participants achieved ventilator independence, 5 remain so (mean durability 25.6m; range 18.3-36.6m) of which 4 have been decannulated. No control participants became ventilator independent or were decannulated.
At baseline, 1/6 dosed participant was able to sit independently without support for 30 seconds; 5/6 participants did not have full head control and were unable to sit independently. Major motor milestones were achieved in all dosed participants (Figure 1); 5/6 remain independently ambulatory without assistive device (achieved mean [SD] time 21.92 [5.57]m); 4 of whom have achieved the ability to ascend stairs. 5/15 (33.3%) control participants achieved independent sitting; none achieved higher milestones.
Among 6 dosed in cohort 1, 4 (67%) participants experienced treatment-emergent severe adverse events; infections in 4 (67%) and respiratory/thoracic/mediastinal disorders in 1(17%). All dosed participants currently have stable liver function test values. As of January 2021, three deaths in the higher dose cohort occurred following severe decompensated liver disease, and three deaths in the control cohort (aspiration pneumonia; cardiopulmonary failure; hepatic hemorrhage with peliosis) were observed. As of September 2021, a newly dosed participant in the lower dose cohort experienced severe liver function test abnormalities and has died.
Conclusion: A rapid improvement in respiratory and motor outcomes was observed among 6 XLMTM participants dosed with AT132 at 1.3 x 10^14 vg/kg vs control participants; these improvements have been maintained and expanded upon over time, indicating improved strength, function, and quality of life for these dosed participants. These substantial improvements must be weighed against the occurrences of fatal serious adverse events, for which the ASPIRO program is on clinical hold while relevant clinical information is being gathered and reviewed.
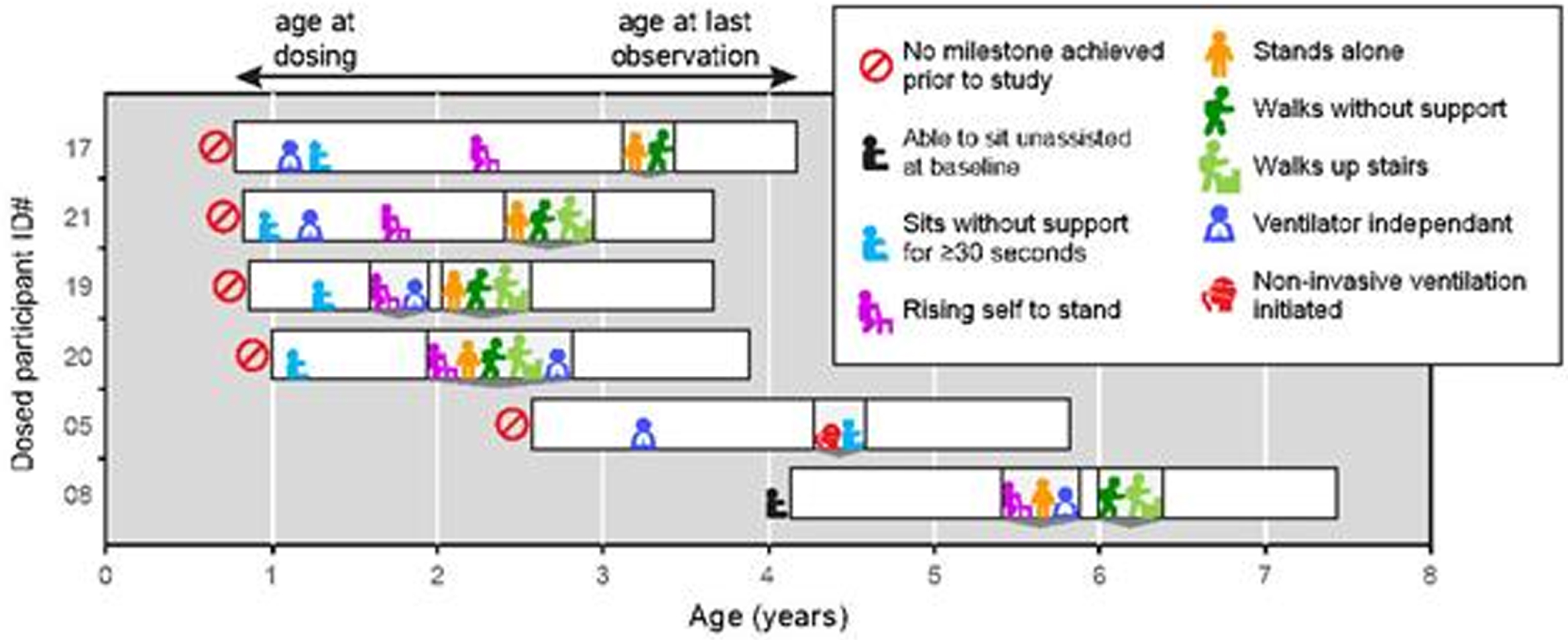
eP04.04.02FORCETM Platform Demonstrates Durable Dystrophin Expression in Mdx Mice and Favorable Safety Profile in NHPs
Desjardins C1, Venkatesan R1, O’Donnell E1, Hall J1, Russo R1, Spring S1, Tang K1, Davis J1, Weeden T1, Zanotti S1, Beskrovnaya O1
1Dyne Therapeutics, Inc., Waltham, United States
Current therapies for Duchenne muscular dystrophy (DMD) use phosphorodiamidate morpholino oligomer (PMO) to induce exon skipping in the dystrophin pre-mRNA thereby restoring the open reading frame and enabling translation of a shortened but functional dystrophin protein. However, success of this strategy has been hampered by insufficient distribution of PMO to cardiac and skeletal muscle. To overcome these limitations, we developed the FORCE™ platform, consisting of an antigen-binding fragment (Fab) that specifically recognizes the transferrin receptor 1, conjugated to an oligonucleotide payload. FORCE-M23D is a mouse-specific Fab-PMO conjugate designed to skip exon 23 of the murine Dmd pre-mRNA. We demonstrated that a single dose of FORCE-M23D delivers substantial levels of exon 23-skipping PMO to cardiac and skeletal muscles in mdx mice, a model of DMD caused by a nonsense mutation in exon 23 of the mouse Dmd transcript. Treatment with FORCE-M23D led to dose-dependent, robust, and durable exon skipping and dystrophin restoration. Dystrophin protein restoration reached peaks of 20%, 11%, and 27% of wild-type (WT) levels in the quadriceps, diaphragm, and heart, respectively, with a single 10 mg/kg PMO-equivalent dose, and peaks of 51%, 90%, and 77%, respectively, with a single 30 mg/kg PMO-equivalent dose. Dystrophin expression was detectable at the sarcolemma at 8 weeks in a dose-dependent manner. Mice administered the higher conjugate dose showed 68% dystrophin-positive fibers in quadriceps and near-complete restoration of sarcolemmal dystrophin localization in heart and diaphragm at 4 weeks post-dose. We also demonstrated that FORCE-M23D treatment resulted in improved functional outcomes compared with administration of the unconjugated PMO payload. Dyne’s clinical candidate, DYNE-251, a Fab-conjugated PMO designed to skip exon 51, was evaluated in non-human primates (NHPs). Five weekly 30 mg/kg doses of DYNE-251 resulted in 43%, 52%, and 18% exon 51 skipping in the heart, diaphragm, and quadriceps, respectively, 8 weeks after the first dose. A GLP toxicology study of DYNE-251 in NHPs demonstrated a favorable safety profile. Collectively, data in mice and NHPs suggest that DYNE-251 may provide a promising approach to addressing DMD.
eP04.04.03Givinostat for the Management of Becker Muscular Dystrophy: A Randomised, Placebo-Controlled, Double-Blind Study
Velardo D1, Magri F2, Kan H4,6, Vandenborne K7, Willcocks R7, Cinnante C5, Ripolone M1, van Benthem J8, van de Velde N3,4, Nava S5, Ambrosoli L9, Cazzaniga S10, Niks E3,4, Bettica P10, Comi G1,2
1Neuromuscular and Rare Diseases Unit, Department of Neuroscience, IRCCS Foundation Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy, 2Neurology Unit, Neuroscience Section, Department of Pathophysiology and Transplantation, Dino Ferrari Centre, IRCCS Foundation Ca’ Granda, Ospedale Maggiore Policlinico, University of Milan, Milan, Italy, 3Department of Neurology, Leiden University Medical Center, Leiden, The Netherlands, 4Duchenne Center Netherlands, The Netherlands, 5Neuroradiology Unit, IRCCS Foundation Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy, 6Department of Radiology, Leiden University Medical Center, C.J. Gorter Center for High Field MRI, Leiden, The Netherlands, 7ImagingDMD and Department of Physical Therapy, University of Florida, Gainesville, USA, 8Department of Orthopedics, Rehabilitation and Physiotherapy, Leiden University Medical Center, Leiden, The Netherlands, 9OPIS srl, Desio, Italy, 10Italfarmaco SpA, Milan, Italy
Becker muscular dystrophy (BMD) is a heterogeneous condition, with substantial variability in age of onset and clinical presentation. There are no treatments specifically approved for the management of BMD, with current therapies aimed at control of symptoms. BMD is caused by loss-of-function mutations in the dystrophin gene, resulting in a reduced amount or a truncated size of the dystrophin protein. Neuronal nitric oxide synthase (nNOS) is an important component of dystrophin-associated protein complex (DAPC). Nitric oxide produced by nNOS specifically inactivates histone deacetylase (HDAC) via S-nitrosylation of a cysteine residue.This mechanism is dysfunctional in dystrophic muscle, leading to aberrantly upregulated HDAC activity. A potential target of therapy for BMD is therefore HDAC inhibition. Here we report the results of a phase II, randomised, double-blind, placebo-controlled study that aimed to evaluate the efficacy and safety of givinostat, a HDAC pan-inhibitor, on micro- and macroscopic muscle morphology and function. Following a four-week screening period, patients were randomised to receive givinostat or placebo for 12 months, attending study visits every two weeks for the first two months, then every 12 weeks for the remainder of the study. Eligible patients were males aged 18–65 years, inclusive, with a diagnosis of BMD confirmed by genetic testing (based on patient records), who were able to walk between 200 and 450 m in a 6 min walk test (6MWT). During the screening period and after 12 months, MRI and MRS of the lower limbs, thigh and pelvic girdle, and an open muscle biopsy of the brachial biceps were performed. In addition, patients undertook a series of functional tests. The primary objective was to demonstrate superiority of givinostat over placebo in terms of the mean change from baseline in total fibrosis after 12 months of treatment. Secondary efficacy endpoints included change from baseline after 12 months of treatment in: other histological parameters (muscle fibre area [MFA], adipose tissue, other histological structures, fibres with nuclear centralizations, total number of fibres on each slide, regenerative fibres, fibres cross-sectional area [CSA], fibre size variability, and dystrophin); MRI fat fraction, CSA, and contractile area of the pelvic girdle, thigh and lower limb muscles; MRS fat fraction of the vastus lateralis and soleus; motor function measure [MFM] (total and component); timed function tests (time to climb four standard steps, time to walk/run 10 m, and time to rise from the floor); 6MWT; and muscle strength evaluated by knee extension and elbow flexion measured by hand-held myometry. The study failed to achieve the primary endpoint. However, there was a potential signal from the MRI assessments that suggests givinostat could prevent (or at least delay) disease progression in BMD, in terms of fat infiltration into the whole thigh and quadriceps muscles. This also provides additional support to the use of MRI as an assessment tool in future BMD studies.
eP04.04.04Post-Authorisation Safety Study of Mexiletine Treatment in Patients with Non-Dystrophic Myotonia: Methodology Overview
Pandey B1, Vicart S2, Tard C3, Schneider-Gold C4, Zozulya-Weidenfeller A5, Sedehizadeh S6, Kaufman G1, Rosenbohm A7, Jayaseelan D8,9
1Lupin Pharmaceuticals, Baltimore, United States, 2Assistance Publique-Hôpitaux de Paris, Sorbonne Université, INSERM, Service of Neuro-Myology, Muscle Channelopathies Reference Center and UMR 974, Institute of Myology, University Hospital Pitié-Salpêtrière, Paris, France, 3U1171, Centre de référence des maladies neuromusculaires Nord Est Ile de France, Hôpital Salengro CHU de Lille, Lille, France, 4Department of Neurology, St. Josef-Hospital, Ruhr-University Bochum, Bochum, Germany, 5Lupin Neurosciences, Lupin Atlantis Holdings SA, Zug, Switzerland, 6Department of Clinical Neurology, Queen’s Medical Centre, Nottingham, United Kingdom, 7Department of Neurology, University of Ulm, Oberer Eselsberg 45, 89081, Ulm, Germany, 8National Hospital for Neurology and Neurosurgery, University College London Hospitals NHS Foundation Trust, London, West Hertfordshire Hospitals NHS Trust, London, United Kingdom, 9West Hertfordshire Hospitals NHS Trust, Watford, United Kingdom
Introduction: Non-dystrophic myotonias (NDMs) are rare genetic disorders that cause hyperexcitability of muscle fibres via ion channel dysfunction. Lifelong stiffness, locked muscles and pain contribute to severe disease burden that can impact daily life. Mexiletine is the only licensed anti-myotonic treatment approved for symptomatic release of myotonia in adult patients with NDM. Previous studies for mexiletine have assessed only short-term efficacy and safety. There is a need for long-term data in a real-world setting, to provide a proven positive risk–benefit profile for effective treatment of myotonia symptoms. This non-interventional, prospective, observational, multicentre study (NCT04616807) aims to provide data demonstrating long-term safety, tolerability and efficacy of mexiletine.
Study design: Up to 50 adult patients diagnosed with NDMs will be enrolled over 2 years in approximately six investigator sites in the UK, Germany and France (additional EU sites TBC). Study population will include patients newly initiated on NaMuscla™ (mexiletine) and patients already on mexiletine at enrolment (only patients who switch to NaMuscla™ will be included).
Baseline for all patients is the enrolment visit. For patients already on mexiletine, cumulative data will be collected for adverse events (AEs) that occurred on treatment.
Data will be captured in electronic case report forms, through physician assessment (handgrip, eyelid myotonia), patient self-assessment tools (visual analogue scale [VAS] and Myotonia Behaviour Scale [MBS]), individualised neuromuscular quality of life (INQoL) questionnaires, and patient diaries. Assessment frequency will align with routine clinical practice and local treatment protocols. Data collection is at 12, 24 and 36 months from enrolment, with follow-up calls at 6, 18 and 30 months. Total study duration is 5 years including enrolment period and data collection, with interim analyses at 1 and 3 years.
Primary endpoints are (i) proportion of patients with treatment-emergent AEs, including serious AEs (SAEs); and (ii) proportion of patients requiring dose reduction or treatment discontinuation due to AEs. Secondary safety endpoints include patients with AEs, SAEs or AEs of special interest; AEs in patients ≥65 years or with abnormal hepatic or renal function; patients presenting with cardiac arrhythmia; severe cutaneous adverse reactions; drug reactions with eosinophilia and systemic symptoms; and Stevens-Johnson syndrome; or seizures. Secondary efficacy endpoints include changes in VAS, MBS and INQoL questionnaire scores from baseline; and clinical myotonia evaluation over time. Summary statistics will be provided for all collected parameters. Limitations include selection bias (minimised by inclusion/exclusion criteria); no control group (outcomes to be discussed in context of MYOMEX study); and clinical practice differences between countries (flexibility provided within study protocol).
This study is being conducted as part of the agreed European Risk Management Plan.
Conclusion: Previous studies assessed efficacy and safety of mexiletine in NDMs over short treatment periods (≤22 days in MYOMEX study; 4-weeks in randomised controlled and aggregated n-of-1 trials). This study will inform on long-term safety and effectiveness of mexiletine in symptomatic management of myotonia in NDM adults in a real-world setting.
eP04.04.05An Open-Label, Non-Comparative Study of Mexiletine in Children and Adolescents with Myotonic Disorders: Methodology Overview
Barnerias C1, Isapof A2, Pandey B3, Zozulya-Weidenfeller A4, Kaufman G3
1Consultations Maladies Neuromusculaires, Service de Neurologie pédiatrique, Hôpital Necker-Enfants Malades, Paris, France, 2Service de Neuropédiatrie - Unité des Pathologies Musculaires Cliniques, CHU Paris Est - Hôpital d’Enfants Armand-Trousseau, Paris, France, 3Lupin Pharmaceuticals, Baltimore, United States, 4Lupin Neurosciences, Lupin Atlantis Holdings SA, Zug, Switzerland
Introduction: Paediatric myotonic dystrophy (DM) and non-dystrophic myotonias (NDM) have a significant impact on patients’ quality of life. Treatment can be challenging for patients due to a historic lack of treatment options coupled with delayed diagnosis as a result of variable disease onset and non-specific symptoms. Mexiletine is the only anti-myotonic treatment approved for symptomatic release of myotonia in adult patients with NDM. A short-term, open-label, non-comparative study (EudraCT 2019-003757-28) will assess the safety and efficacy of mexiletine for the treatment of myotonia in adolescents (cohort 1, aged 12–17 years) and children (cohort 2, aged 6–11 years). Secondary objectives include evaluation of efficacy via patient reported outcomes; pharmacokinetics of mexiletine in children and adolescents; and assessment of the acceptability of the capsule formulation.
Study design: Approximately 7 patients will be enrolled in each cohort, with genetically confirmed NDM/DM (DM1 or DM2), presenting with symptoms of myotonia and without significant cardiac abnormalities on echocardiogram (ECG) 3 months prior to enrolment or history of significant liver disorder. Study duration is ~12 weeks, with a ~4-week screening phase and 8-week treatment phase comprising 4 weeks’ titration (starting at an age-appropriate bodyweight-based dose of 62, 83 or 167 mg once daily as evaluated by investigator, with up-titration every 14 days to a maximum of 3 times per day) followed by 4 weeks’ maintenance at the best-tolerated dose. Blood samples for pharmacokinetic evaluation will be collected pre- and post-dose on Day 42. Following completion, participants will be offered follow-up in an open-label 24-month extension study (EudraCT: 2019-003758-97).
Primary safety endpoints are number and frequency of adverse events (AEs) and serious AEs, incidence of AEs of special interest, and ECG changes from baseline. Efficacy endpoints, as submitted to and accepted by the Paediatric committee of the European Medicines Agency, are adapted from commonly used adult scales, according to patient age and ability to participate in the evaluation. Primary efficacy endpoints are mean change from baseline (to Days 14, 28, 42 and 56) in handgrip myotonia score and visual analogue score (VAS) or faces score for muscle stiffness. Secondary endpoints include mean change in VAS or faces score for muscle pain, weakness and fatigue, clinical myotonia assessment (based on time to eye opening, Timed Up and Go test, and flexor myotonia assessment), paediatric quality of life (PedsQL) scores and Clinical Global Impression scores. Summary statistics will be provided for all collected parameters.
Conclusion: This study will inform on the safety and efficacy of 8-week treatment with mexiletine in patients aged 6–17 with genetically-confirmed NDM/DM (DM1 or DM2). Pharmacokinetic data will be used to establish dosing recommendations for each paediatric age subset from 6 to <18 years old.
eP04.04.06Readily Available Low-Cost Highly Effective Treatment for Inherited Muscle Disorders Diagnosed by Whole Exome Sequencing
Tsiverdis I1, Mathioudakis L, Latsoudis H, Skoula I, Kokosali E, Bitsori M, Vergadi E, Vorgia P, Galanakis E, Evangeliou A, Zaganas I
1Neurology/Neurogenetics Laboratory, Medical School, University of Crete, Heraklion, Greece, 2Information Systems Laboratory, Foundation of Research and Technology, Heraklion, Heraklion, Greece, 3Pediatrics Department, University Hospital of Heraklion, Heraklion, Greece 4Hellenic Mediterranean University, Heraklion, Heraklion, Geece, 5Aistotle University of Thessaloniki, Thessaloniki, Greece
Introduction: Inherited muscle diseases are caused by genetic variants in numerous genes and present high phenotypic heterogeneity. This heterogeneous presentation poses a diagnostic challenge and leads to diagnostic delays, occasionally depriving patients from timely initiation of disease modifying therapies. Our aim here is to describe two patients for whom diagnosis of a treatable muscle disorder by whole exome sequencing (WES) led to initiation of readily available low-cost highly effective treatment.
Methods: After obtaining informed consent, WES was performed for a 12- and a 13-year-old patient with a myopathic phenotype deemed genetic in origin. Sequencing was performed at Otogenetics, Norcross, GA, USA and Macrogen, Seoul, Korea, using the Illumina HiSeq2000/25000 and the HiSeq4000 platform, respectively, aiming at a >50x coverage. Variant annotation was performed using the Ingenuity Clinical Insight (Qiagen, USA) software and workflows developed at the Neurogenetics Laboratory, University of Crete, Greece, based on related genetic databases and bibliography
Patients: The first patient, a 12-year-old female, presented weakness of the upper extremities and easy fatiguability from the age of 2 years old, with subsequent development of scoliosis and weakness of the lower extremities. Muscle biopsy showed non-specific findings, and several other tests were non-diagnostic. Finally, WES suggested the diagnosis of Multiple Acyl-CoA Dehydrogenase Deficiency type II, due to the p.Pro483leu (c.1448C>T) and p.Arg559Ter (c.1675C>T) variants in the ETFDH gene. The compound heterozygosity of the variants was established by testing of the unaffected parents with Sanger sequencing. Administration of riboflavin (150mg/day) for over one year led to clinical benefits for the patient, including increased endurance in long-distance walking and stair climbing and improvement of scoliosis, obviating the need for a brace.
The second patient was a 13-year-old female with recurrent respiratory infections, chronic obstructive pulmonary disease, gaze palsy, myopathy, walking instability, and scoliosis. Extensive investigations, including muscle biopsy and neurophysiological testing, were inconclusive. The patient’s sister had been bedridden for years due to an uncharacterized neuromuscular disorder. For this patient, WES revealed two variants in the DOK7 gene, p.Arg158Trp (c.472C>T) and p.Glu463Ter (c.1387G>T), in compound heterozygosity, as a cause of congenital myasthenia. Administration of oral salbutamol led to impressive clinical improvement, including a 38% increase in the 6min walk test (from 437 to 604 m.).
Discussion: Inherited muscle diseases display high phenotypic and genetic heterogeneity, making their diagnostic classification challenging and often involving tedious and expensive inconclusive tests. Nowadays, next generation sequencing technologies, such as WES, have allowed accurate molecular diagnosis of these diseases, at an affordable cost. Occasionally, this genetic diagnosis can lead to effective treatment approaches tailored to the patient’s genotype. This genome based personalized treatment includes, in addition to costly modalities such as enzyme replacement therapy, low-cost highly effective substances, such as salbutamol and riboflavin.
eP04.04.08Diagnosing Necrotizing Myopathy With Hmgcr Antibodies – A Quest Proving Time is Muscle.
Malfatti E1, Major Z2,3, Urtizberea J4
1Centre de Référence de Maladies Neuromusculaires, Hopital Henri Mondor, Créteil, France, 2Neurophysiology Department, National Center for Spinal Disorders, Budapest, Hungary, 3Neurology Department, Municipal Clinical Hospital, Cluj-Napoca, Romania, 4Institut de Myologie, Pitié-Salpêtrière, Paris, France
We present the case of a 27 year old woman with an assymmetrical myopathy. Symptoms started in 2005-2006 with mild weakness in the upper limbs, accentuated in 2012 when climbing the stairs and raising the left hand over the head became difficult. Muscle pain developed. Physical therapy helped to an almost complete recovery. Between 2016-2020 there were episodes of accutization followed by stationery periodes, and since 2020 the weakness progressed fast, the patient is not able to stand up and the right upper limb is also affected, she’s not able to raise it even to shoulder level. Pain became more intense.
Several investigations were conducted. The first laboratory finding was a persistent elevation of transaminases and LDH, then elevated CK levels. Electromyography identified a myopathic trace, and ruled out the presence of myasthenic decrementum and neuropathies.
Rheumatological investigation followed when even if the symptoms suggested an inflammatory myopathy, the myositis profile (anti Mi-2, Ku, Pm-Scl 100, Pm-Scl 75, Jo-1, SRP, PL-7, EJ, OJ, Ro-52 antibodies), anti nuclear antibodies, anti dcDNA, anti RNP/Sm, anti Ro (SS-A) antibodies were all negative. The patient was treated with methylprednisolon, with improvement, still with reduction of CK levels during the treatment. At a later evaluation, lactic acid levels during ischemia raised the suspicion of a mitochondrial myopathy. Muscle biopsy followed, revealing sarcoplasmic homogenization and loss of striations along with regenerative changes, myocites amphophilic cytoplasm, central nucleus and visible nucleoli, without inflammatory infiltrates. Yet, HLA-I and terminal complement complex which are sensitive markers inflammatory changes could not be analyzed due to lack of frozen tissue. The conclusion was a necrotizing myopathy without a further evidence for a disease specific process.
Imagery revealed diffuse edema at the level of the muscles of both thighs. Azathyoprine and Pregabaline treatment followed, without significant improvement.
Hypothyroidism and bilateral cataract was diagnosed, with debatable significance. Another muscle biopsy excluded mitochondrial myopathies, immunohystochemistry reveale d the presence of distrophine dys 1, 2, 3, merosine, normally expressed DRP, alpha, beta, gamma sarcoglycans and disferline. Western blotting for calpaine 3 and disferline proved the presence. The whole exome sequencing was also negative. Pompe, Fabry and MPS proved also negative, ASMD negative and SMN1 mutations highly unlikely.
We started to evaluate at this point the patient. The whole body MRI revealed adipous infiltrates and edema at the gastrocnemius muscles. The patient refuses immunosuppressive or corticosteroid treatment.
The asymmetric character raised the possibility of a FSHD, and another biopsy was made, which finally proved to be a necrotizing myopathy with HMGCR antibodies.
The above presentation proves that efforts should be made to get a rapid and proper diagnosis of NMDs, in order to help in such disabling clinical presentations, not only to maintain functionality, but also the adherence to treatment, and emphasizes the possibility that immune-mediated necrotizing myopathy (IMNM) might be a necrotizing myopathy with HMGCR antibodies, even in young patients – in this case debute at the age of 11-12 years – without the presence of treatment with statins, and with a peculiar asymmetrical presentation.
eP04.04.09Anti-SRP-Positive Immune-Mediated Necrotizing Myopathy Accompanied by Hashimoto Thyroiditis
Baek S1, Sung J1, Park J1, Choi K2, Oh J2, Kim B1
1Department of Neurology, Korea University Anam Hospital, Korea University College of Medicine, Seoul, Republic of Korea, 2Department of Neurology, Konkuk University Medical Center, Seoul, Republic of Korea
Background: Immune-mediated necrotizing myopathy (IMNM) is one of the categories of idiopathic inflammatory myopathy and is characterized by the necrotic change of muscle fiber with no or little inflammatory cells. Anti-signal recognition particle (SRP) and anti-3-hydroxyl-3-methylglutaryl-coenzyme A reductase autoantibodies play a crucial role in distinguishing IMNM from other inflammatory myopathies. IMNM typically presents severe limb muscle weakness and sometimes presents extra-muscular manifestations. Some cases with IMNM are associated with other autoimmune diseases. Here, we report a patient with anti-SRP-positive IMNM accompanied by Hashimoto thyroiditis.
Case presentation: A previously healthy 61-year-old woman came to the neurologic clinic with a 2-month history of proximal muscle weakness. She had myalgia, fatigue, and mild shortness of breath. She could not walk independently because of limb weakness. On neurologic examination, the muscle strength was MRC grade 4 in arms and grade 3 in legs. Serum creatine kinase (CK) level was elevated to 14441 IU/L (normal range: 38 – 185 IU/L). Electromyography showed myopathic potentials with positive sharp waves on upper and lower extremities and paraspinal muscles. Magnetic resonance imaging revealed diffuse symmetric T2 high signal changes in multifocal limb muscles. Muscle biopsy revealed mild muscle size variation with scattered degeneration and regenerating muscle fibers without inflammatory cell infiltrate. Muscle-specific antibodies study by line immunoassay revealed highly positive anti-SRP antibody. Immunologic examination teste revealed that antinuclear antibody was positive at 1:640 in the cytoplasmic pattern. Anti-SSA was also positive (204 U/mL), but anti-Jo-1, anti-SSB, anti-RNP, anti-Scl-70, anti-centromere, and anti-dsDNA were all negative. In addition, anti-microsomal Ab was 363.3U/mL (normal range: 0 – 60U/mL) and anti-thyroglobulin antibodies was 61.73 IU/mL (normal range: 0 – 4.11 IU/mL). Thyroid function test was revealed that TSH 12.82 µU/mL (normal range: 0.17 – 4.05), free T4 1.01ng/dL (normal range 0.89 – 1.79), and T3 71.1ng/dL (normal range 78 – 182 ng/dL). Thyroid ultrasound revealed the normal size of both thyroid glands with diffuse parenchymal heterogeneity and increased vascularity. She was diagnosed with anti-SRP-positive IMNM accompanied by Hashimoto thyroiditis with subclinical hypothyroidism. She was treated with intravenous steroid pulse therapy (methylprednisolone 1g/day for 5 consecutive days) following a high-dose oral steroid (1mg/kg/day) for IMNM. In addition, she was treated with levothyroxine 0.05mg/day for Hashimoto thyroiditis. After treatment, her symptoms were gradually improved and she could walk independently outdoors 4 months later.
Conclusion: A recent study has reported that approximately 1 – 2 % of idiopathic inflammatory myopathy could be affected by autoimmune thyroid disease. However, the concomitant with autoimmune thyroid disease in IMNM is few reported. Our case may suggest that anti-SRP-positive IMNM is concomitant with autoimmune thyroid disease. Therefore, it would be necessary to consider evaluating thyroid autoantibodies and thyroid function in patients with IMNM.
eP04.04.10Necrotizing Autoimmune Myopathy Anti-HMGCR Antibodies and Demyelinating Polyneuropathy
Vallejo D1,2,3, Robledo S1,3
1Universidad De Antioquia, Medellin, Colombia, 2Ips Universitaria Universidad de Antioquia, Medellin, Colombia, 3Neuroclínica, Medellin, Colombia
A 54 year old man presented to the neuromuscular clinic for evaluation of two years of slowly progressive proximal weakness in all limbs and dysphagia during the last months. During these two years he had several CK measurements, all of them between 6000 and 8000 µmol/l. He had a medical history of dyslipidemia and had been treated with atorvastatin for five years, until two years ago. The general examination was unremarkable and the neurologic examination revealed mild symmetric proximal weakness of all limbs and generalized hyporeflexia. The antibodies against 3-hydroxy-3-methylglutaryl–CoA reductase (antiHMGCR) were positive and the MRI showed diffuse muscle involvement, without a specific pattern, and without severe atrophy or fat replacement. The pectoral muscle histopathology revealed great variability of muscle size, fiber atrophy, myobags, fat replacement areas, degenerative changes with vacuoles, regenerative changes in some fibers, myonecrosis and myophagocytosis, with scarce endomysial inflammatory infiltrate.
Although he was treated with oral steroids for one month, the weakness worsened until he could not walk by himself, and the CK rose from 6000 to 7916 µmol/l. He was then admitted to the hospital where he received intravenous immunoglobulin regaining independent gait. The CK levels decreased to 4500 µmol/l. An electromyography with conductions velocities was performed. Aside from revealing denervation in multiple proximal muscles, it showed absence of sensitive responses in sural and superficial peroneal nerves, and also revealed slow motor conduction velocity in fibular and tibial nerves, with prolonged latency of the fibular nerve. All the studies for polyneuropathy were unrevealing except for a gamma peak in serum protein electrophoresis. The serum immunofixation evidenced a IgG Kappa monoclonal gammopathy; the bone marrow biopsy was normal. PET-CT was normal and Anti-MAG antibodies were negative and there was no albuminocytological dissociation in cerebrospinal fluid, We considered neuropathy associates with MGUS ( Monoclonal gammopathy of undetermined significance). POEMS syndrome was excluded ((polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) With immunoglobulin, methotrexato and steroids he was stable for six months.
Peripheral neuropathy is extremely rare during the course of necrotizing autoimmune myopathy and we did not find published cases of polyneuropathy with necrotizing autoimmune myopathy anti-HMGCR antibodies.
It is not possible to determine whether the co-occurrence of myopathy and polyneuropathy in this patient was due to chance or autoimmune predisposition or whether gammopathy or neuropathy could be part of the spectrum of extramuscular manifestations of myopathy.
eP04.04.11Hereditary Inclusion Body Myopathy (HIBM) as a Rare Clinical Entity: A Case Report
Barbov I1, Kalcev G
1University Clinic For Neurology Skopje, Skopje, North Macedonia
Introduction: Hereditary inclusion body myopathy (HIBM) is a rare slowly progressive muscle disease that frequently develops between age 20 and 40 years with bilateral foot drop induced by anterior tibialis weakness. Involvement of lower-extremity muscle continues from the anterior to the posterior compartment of the lower leg, followed by hamstrings, then hip girdle muscles, with correlative sparing of the quadriceps.
Methods: Description of the case report with hereditary inclusion body myopathy form in North Macedonia.
Results: The authors report female patient that is genetically homozygous of the pathogenic modification in the GNE (Glucosamine (UDP-N-Acetyl)-2-Epimerase/N-Acetylmannosamine Kinase) gene, which means that this condition is inherited from the both parents. Three years ago, the patient noticed instability in walking and peroneal type of walking, accompanied by weakness of the calf muscles. Furthermore, it is also important the fact that the patient’s sister also was clinically diagnosed with this type of distal myopathy 16 years ago. In addition, the evolution of the disease in her is with progressive character matching with the weakness of the lower extremities in the earlier stage, together with the weakness of the upper extremities at a later stage.
Conclusion: A plenty of new facts remains to be learned in the future studies about the other and less common forms of HIBMs beyond the definition of the corresponding causative gene defect.
eP04.05.01Improved Quality of Life in Patients with X-Linked Myotubular Myopathy (XLMTM) Treated with Resamirigene Bilparvovec
Servais L1, Shieh P2, L. Kuntz N3, Dowling J4, Müller-Felber W5, Blaschek A5, Bönnemann C6, Foley A6, N. Saade D7, M. Seferian A8, Haselkorn T9, Bowden A9, Coats J9, Han C10, Prasad S11, Rico S11, Miller W9
1MDUK Oxford Neuromuscular Centre, Oxford, UK, 2University of California, Los Angeles, USA, 3Ann & Robert H Lurie Children’s Hospital of Chicago, Chicago, USA, 4Hospital for Sick Children, Toronto, Canada, 5Klinikum der Universität München, Munich, Germany, 6Neuromuscular and Neurogenetic Disorders of Childhood Section, NINDS, NIH, Bethesda, USA, 7University of Iowa Hospitals and Clinics, Iowa City, USA, 8I-Motion, Institute of Myology, Paris, France, 9Astellas Gene Therapies (formerly Audentes Therapeutics, Inc), San Francisco, USA, 10Astellas Pharma Global Development, Northbrook, USA, 11Formerly Astellas Gene Therapies (formerly Audentes Therapeutics, Inc), San Francisco, USA
Introduction: XLMTM is a rare, life-threatening myopathy caused by pathogenic variants in the MTM1 gene. Approximately 80% of XLMTM patients experience profound muscle weakness with respiratory distress/failure at birth and chronic ventilator dependency. Most children with XLMTM cannot sit without support and 70-85% are non-ambulant. XLMTM poses substantial burden on quality of life for patients and their families due to high medical need, and emotional, time, and financial constraints. No approved treatment exists for XLMTM.
Method: ASPIRO (NCT03199469), a Phase 1/2/3 randomized, open-label study is investigating the safety and efficacy of AT132 (resamirigene bilparvovec), a single-dose gene replacement therapy for XLMTM. Participants were boys (aged <5 years at day 1 and/or enrolled in INCEPTUS, NCT02704273, a prospective run-in study) with genetically confirmed XLMTM. A total of 23 AT132 dosed participants were included (n=6, lower-dose of 1.3 x 10^14 vg/kg; n=17, higher-dose of 3.5 x 10^14 vg/kg) and 15 untreated controls (including 12 participants from INCEPTUS), as of 01/29/2021. We examined quality of life outcomes based on the ACEND (Assessment of Caregiver Experience with Neuromuscular Disease) total score, a questionnaire reported on a scale from 0 to 100, with higher scores indicating caregivers experiencing less intense care-giving impact. Mean differences (SD) in total scores were calculated from baseline to last observation (data cut 29JAN2021) and from baseline to week 48 for each cohort. Data from three patients dosed prior to data cut and who died during ASPIRO from AT132-related liver toxicity were excluded due to limited data points occurring post-dose.
Results: Mean age at dosing (months, range) was 20.4 (9.5, 49.7) in the lower-dose cohort, 39.4 (6.8, 72.7) in the higher-dose cohort, and 19.6 (5.9, 39.3) at enrollment among control participants. Mean length of follow-up (months, range) in the lower-dose, higher-dose, and control cohorts were 36.7 (33.4, 40.4), 16.1 (3.4, 29.8), and 16.1 (5.7, 32.7), respectively. In both the lower-dose and higher-dose cohorts, post-dose total ACEND scores at last observation markedly improved, whereas they remained static over time in untreated controls (Figure). Mean differences (SD) from baseline to week 48 were: lower-dose, 35.4 (10.0); higher-dose, 17.6 (16.5); controls, 0.6 (6.7). As of January 2021, three deaths in the higher-dose cohort (attributed to decompensated, severe cholestatic liver disease) occurred, and three deaths in the control cohort (attributed to respiratory events or hepatic peliosis hemorrhage) were observed. As of September 2021, a newly dosed participant in the lower-dose cohort died following abnormal liver function tests observed in the weeks after dosing; the cause of death is pending.
Conclusion: Preliminary results from ASPIRO indicate substantial improvement in quality of life for both patients with XLMTM dosed with AT132 and their parents. Treated patients decrease their dependence on caregivers to eat, wash, dress, sit, play, and move. This has a marked positive impact on their caregivers from a time, emotional, and financial perspective. These substantial improvements must be weighed against the occurrences of fatal serious adverse events, for which the ASPIRO study is on clinical hold while relevant clinical information is being gathered and reviewed.
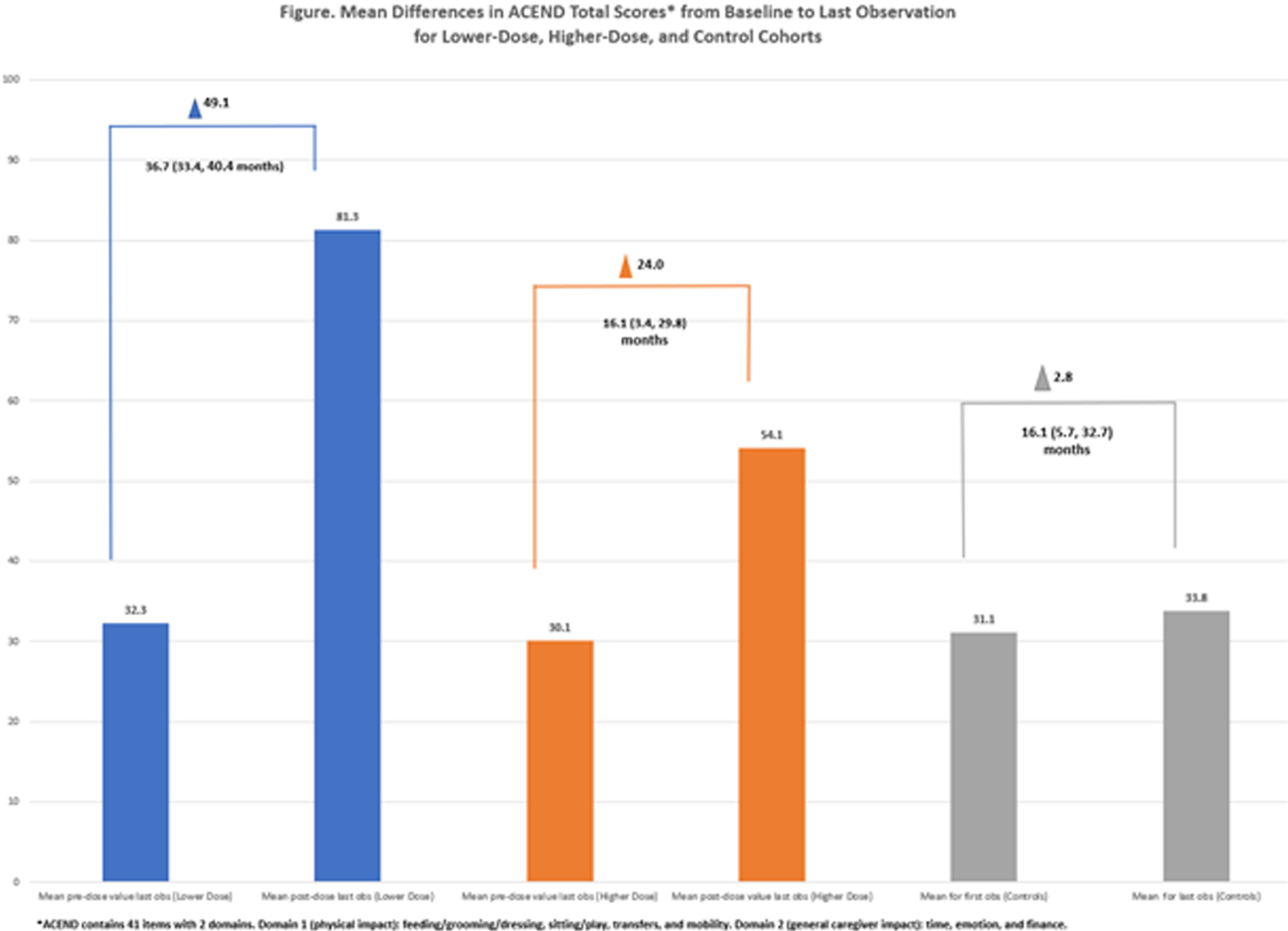
eP04.05.02Clinical and Genetic Features of the Myotubular and Centronuclear Myopathy Patient Registry Cohort
Bullivant J1, Borland H1, Lennox A2, den Hollander A3, Saegert C3, Lynch O, Moat D1,8, Graham R9, Schara-Schmidt U10, Bönnemann C4, Jungbluth H5,11, Buj-Bello A6, Dowling J7, Marini-Bettolo C1,8
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle Upon Tyne, UK, 2Myotubular Trust, London, UK, 3ZNM - Zusammen Stark! e.v., Stuttgart, Germany, 4National Institute of Neurological Disorders & Stroke, Neurogenetics Branch, Bethesda, USA, 5Department of Paediatric Neurology – Neuromuscular Service, Evelina Children’s Hospital, Guy’s & St Thomas’ NHS Foundation Trust, London, UK, 6Genethon, Université Paris-Saclay, Univ Evry, Inserm, Integrare research unit UMR_S951, Evry, France, 7Division of Neurology, Hospital for Sick Children, Toronto, Canada, 8John Walton Muscular Dystrophy Research Centre, Newcastle Hospitals NHS Trust, Newcastle upon Tyne, UK, 9Department of Anesthesiology, Critical Care and Pain Medicine, Boston Children’s Hospital, Harvard Medical School, Boston, USA, 10Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioural Sciences, University Duisburg-Essen, Essen, Germany, 11Randall Centre for Cell and Molecular Biophysics, Muscle Signalling Section, FoLSM, King’s College London, London, UK
The centronuclear myopathies are congenital neuromuscular conditions characterised by the central location of the nucleus in muscle cells and a highly variable clinical picture. The presumably most common form is the ultra-rare X-linked myotubular myopathy (XLMTM) with an estimated incidence of 1 in 50,000-100,000 male births and usually a severe phenotype.
The Myotubular and Centronuclear Myopathy Patient Registry has been collecting demographic, genetic and longitudinal clinical data on affected individuals (living and deceased) and female carriers of XLMTM from all over the world, since 2013.
The registry is funded by patient organisations and industry and coordinated by the John Walton Muscular Dystrophy Research Centre at Newcastle University in the UK. It facilitates translational research by providing data to answer research questions or inform clinical trial feasibility studies, and by supporting recruitment into clinical trials and other research studies. It also serves as an important communication and engagement tool for this patient population.
Patients and caregivers provide longitudinal data via online questionnaires available in English, German, French, Spanish, Italian, Polish, Hindi, and Brazilian Portuguese, with more languages to follow in 2022. Registrations are verified by reviewing the genetic report, and every 6 months the registry platform prompts participants to log in and confirm or update their responses. In March 2022 we will assure greater data accuracy by enabling participants to nominate their doctor via the registry platform who will then be invited to log in, verify their patient’s diagnosis and provide a limited number of data points to the registry record.
This poster will present the reported clinical features of the registry cohort according to genotype. On 26-01-2022 the registry contained 398 participants (259 male and 139 female), of which 40 were reported deceased (39 male and 1 female) and 358 living (220 male and 138 female). Of the 138 living females, 70 were registered as female carriers of XLMTM and 68 as patients. 51 countries were represented, with the largest cohorts being UK & Ireland (103), United States (99), and Germany (34). Causative genes reported include MTM1, DNM2, BIN1, RYR1 and TTN.
In summary; the Myotubular and Centronuclear Myopathy Patient Registry contains important data on a diverse and growing international cohort of individuals affected by this group of rare and ultra-rare conditions. It aims to support all areas of translational research including epidemiology, clinical trial planning and recruitment, outcome measure development, standards of care, and real-world data for regulatory decision-making.
eP04.05.03Novel Splicing Mutation in MTM1 Leading To Two Abnormal Transcripts Causes Severe Myotubular Myopathy
Bosco L1,2, Leone D3, Costa Comellas L4, Monforte M5, Pane M3,6, Mercuri E3,6, Bertini E1, D’Amico A1, Fattori F1
1Unit of Neuromuscular and Neurodegenerative Disorders, IRCCS-Bambino Gesù Children’s Hospital, Rome, Italy, 2Department of Science, University “Roma Tre”, Rome, Italy, 3Centro Clinico Nemo, Fondazione Policlinico Universitario A. Gemelli IRCCS-Università Cattolica del Sacro Cuore, Rome, Italy, 4Pediatric Neurology, Vall d’Hebron Institut de Recerca (VHIR), Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain, 5Unità Operativa Complessa di Neurologia, Fondazione Policlinico Universitario IRCCS-A. Gemelli, Rome, Italy, 6Pediatric Neurology, Department of Woman and Child Health and Public Health, Fondazione Policlinico Universitario A. Gemelli IRCCS-Università Cattolica del Sacro Cuore, Rome, Italy
X-linked myotubular myopathy (XLMTM) is a severe form of centronuclear myopathy characterized by generalized weakness and respiratory insufficiency. XLMTM is associated with pathogenic variants in MTM1 gene encoding the dual-specificity phosphatase named myotubularin. NGS targeted Sequencing on DNA of a three months old child affected by XLMTM identified the novel hemizygous MTM1 c.1261-5T>G intronic variant. Sequencing MTM1 cDNA obtained from total RNA extracted from patient’s muscle biopsy confirmed that the novel intronic variant, located in intron 11, interferes with the normal splicing process generating two different abnormal transcripts simultaneously expressed in patient’s muscular cells. The first aberrant transcript, induced by the activation of a cryptic splice site in intron 11, includes four intronic nucleotides (UCAG) upstream of exon 12 resulting in a shift in the transcript reading frame, introducing a new premature stop codon in the catalytic domain of the protein (p.Arg421Serfs*7). The second aberrant MTM1 transcript, due to the lack of recognition of the 3’ acceptor splice site of intron 11 from the spliceosome complex, leads to the complete skipping of exon 12. We expanded the genotypic spectrum of XLMTM underlying the importance of intron-exons boundaries sequencing in male patients affected by XLMTM.
eP04.05.04Severe Congenital Myopathy With Type II Fibers Atrophy Due to MYL1
Exposito Escudero J1, Ciutad M2, Fernández C2, Sebastiani G2, Coronel O2, Madrigal I2, Jou C1, Codina A1, Ortez C1, Carrera L1, Natera D1, Arca G2, Nascimento A1
1Hospital Sant Joan De Deu, Esplugues de Llobregat, Spain, 2Hospital Clínic, Barcelona, Spain
Introduction: Congenital myopathies are increasingly considered a spectrum of diseases; however, they are usually subclassified by the presence of distinct histopathological features on muscle biopsy. Several new genes have been discovered in recent years thanks to NGS technology. The MYL1 gene encodes two splice isoforms of skeletal muscle fast essential light chain which are well studied in model organisms. They interact with fast myosin heavy chain as either homodimers or heterodimers, recently associated with 2 cases of severe congenital myopathy. Here we describe the clinical and pathological features of a new patient with this ultra-rare disorder with a hall mark in the muscle pathology.
Case Summary: Premature newborn of 34 weeks of low weight for gestational age, without consanguinity or family history of neuromuscular disease. She presented severe polyhydramnios and decreased fetal movements during pregnancy, which led to premature delivery. At birth, mechanical ventilation was required from birth. He presents several weakness and muscle wasting with minimal spontaneous movements, areflexia, myopathic facies and humeral fracture at birth, with other minor signs such as micro retrognathia, kyphoscoliosis, bilateral cryptorchidism. He does not present ptosis, lingual fasciculations, ophthalmoplegia, or arthrogryposis.
An etiological study was started with a normal metabolic study, muscle enzymes, echocardiogram, cerebral and abdominal ultrasound, and ophthalmologic examination. He has negative studies for Steinert and Prader Willi diseases. MLPA for SMN1 was also normal.
The muscle biopsy showed fibers with marked variation in the size (<2-70 µm) randomly distributed without forming groups although the atrophic fibers are distributed surrounding the hypertrophic fibers. The small fibers were type 2 expressed fast myosin and the hypertrophic fibers was type I expressing slow myosin. The small fibers were pale with NADH. No necrosis was seen.
An exome targeting myopathies study was completed in which 2 variants (c.334C>T and c.478+1G>A)in the MYL1 gene were detected.
During his evolution, the patient presented respiratory complications with bronchial aspiration and bilateral necrotizing pneumonia, which finally led to an adaptation of the therapeutic effort with death at 24 days of life.
Conclusions: The mutation in MYL1 causes a new, albeit rare, condition of severe congenital myopathy with unusual muscle pathology of myofiber hypotrophy type II and myofiber hypertrophy type I. Patients with a congenital myopathy and these pathologic features should be screened for MYL1 mutations.
eP04.05.05Clinical and Pathologic Findings of Korean Patients With Selenon-Related Myopathy
Lee S2, Choi Y1, Park H1
1Gangnam Severance Hopspital, Gangnam-gu, Republic of Korea, 2Ewha Womans University Mokdong Hospital, Seoul, Republic of Korea
Introduction: Selenoprotein N-related myopathy (SELENON-RM), caused by pathogenic variants in SELENON, is a rare congenital myopathy characterized by slowly progressive axial muscle weakness, early-onset spinal rigidity, and respiratory insufficiency. The skeletal muscles of SELENON-RM frequently showed multiple-minicores, congenital fiber type disproportion, or Mallory body-like inclusions. These recessive conditions share so many clinical and genetic features that they are considered the same disorder, termed SELENON-RM. We evaluated the characteristics of SELENON-RM in a Korean population by analyzing clinical, pathological, and genetic analysis obtained from seven unrelated patients with pathogenic variants in SELENON.
Methods: We reviewed the medical records of a myopathy database from 2002 to December 2021 at Gangnam Severance Hospital. Results: We identified five different SELENON pathogenic variants in seven patients from unrelated families, which were all classified into pathogenic or likely pathogenic according to ACMG/AMP guidelines. SELENON pathogenic variants include six missense, five frameshift, and one nonsense variants. All variants were previously reports. We found two pathogenic variants in five patients, but one heterozygous variant in two. Among seven patients, five patients (71.4%) were male and two patients (28.6%) were female. The median age of symptom onset was 3 years old [interquartile range: 2.5-7.5 years old]. Early-onset scoliosis and pulmonary insufficiency were found in six patients (85.7%). Delayed motor development, including poor head control and late independent ambulation were reported in five patients (71.4%). The serum CK level was within normal range in five patients, and mildly elevated in two patients (600 IU/L in MF157 and 555 IU/L in MF1481), respectively. Muscle biopsied were performed in five patients (MF27, MF157, MF942, MF1467, and MF1481). H&E staining demonstrated wide variations in fiber size and increased interstitial fibrosis. One muscle specimen showed focal reduction of NADH activity reminiscent of minicores. Muscle MRI scan was performed in two patients (MF942 and MF1467), and showed homogeneous remarkable findings of severe atrophy in semimembranosus muscle. Conclusions: This study demonstrates the genetic and clinical spectra Korean patients with SELENON-RM as the first report in Korea.
eP04.05.07RYR1-Related Congenital Myopathy in a Cohort of Peruvian Patients
Martínez-Esteban P1, Bobadilla E2, Lazarte C1, La Serna J3, Celis L1, Juarez T4, Rendu J5, Sternberg D6, Malfatti E7, Urtizberea A8
1Instituto Nacional De Salud Del Niño San Borja, Lima, Peru, 2Hospital Universitario San Ignacio, Bogotá, Colombia, Bogota, Colombia, 3Instituto Nacional de Ciencias Neurologicas, Lima, Perú, 4Hospital Santa Rosa, Piura, Perú, 5CHU Grenoble, France, GMGF, Marseille, France, 6Assistance Publique-Hôpitaux de Paris, Centre de Référence des Canalopathies Musculaires, Centre de Référence des Maladies Neuromusculaires, Paris, France, 7APHP, Centre de Référence de Pathologie Neuromusculaire Nord-Est-Ile-de-France, Henri Mondor University Hospital, Paris, France, 8Institut de Myologie, AFM-Téléthon, Paris, France
1. BACKGROUND Ryanodine Receptor 1-Related Myopathies(RYR1-RM) are the most frequent congenital myopathies(1) with an incidence of at least 1: 90,000 in the US pediatric population(2). The incidence of congenital myopathies in South America remains unknown due to limited access to molecular diagnosis.
The spectrum of clinical manifestations of RYR1-RM is wide, including congenital hypotonia, delayed motor milestones, proximal, axial and bulbar muscular weakness with swallowing and respiratory disorders, ocular involvement and fetal akinesia in most severe cases (3,4). RYR1-RM can be either autosomal dominant or autosomal recessive inherited (5).
2. PATIENTS AND METHODS: Seven patients from five unrelated families with a clinical and genetic diagnosis of RYR1 related of congenital myopathy were included. P1, P2 and P3 belong the same family. All patients and or their parents gave consent for clinical examination and genetic studies. Clinical and genetic features are reported in Table 1.
The most common initial symptoms were neonatal hypotonia (5/7), poor sucking and motor delay(3/7). The pattern of weakness was mainly axial and proximal. Facial weakness was observed in 5 patients (71%). Ophthalmoplegia associated with ptosis was only found in 1 patient with a recessive mutation (P7).
Normal serum CK levels was a consistent feature in the whole cohort. One patient reported cardiac conduction disorders requiring a pacemaker. Two patients became wheelchair-dependent at 14 years(P1) and at 6 years(P7).
Muscle magnetic resonance imaging (MRI) in P5 an P6 showed diffuse fatty infiltration showing relative preservation of the rectus femoris and gracilis.
3. DISCUSSION: RYR1 have been described as the most frequently genetic etiology diagnosed in congenital myopathy in all worlds (6). The difficulty of accessing diagnostic support such as muscle biopsy and genetic studies limited access to the diagnosis in our country for many years
Proximal and axial muscle weakness and hypotonia were most frequent clinical features in our patients, similar to cohorts reported in other countries (7-9). Different studies that have tried to establish a genotype phenotype correlation have shown that dominant mutations have a milder phenotype and have less functional impairment, unlike recessive mutations that have a more severe phenotype with greater facial involvement and usually present with ophthalmoplegia (8). In our cohort, all patients with recessive mutations had facial weakness and only one had ptosis and ophthalmoplegia. The findings in MRI with classic pattern of selective involvement of the vastus muscles and relative sparing of the rectus femoris, adductor longus and gracilis helps to identify patients with potential RYR1-RM (10,11)
In the described family(P1, P2,P3) with the dominant mutations, the variable expressivity was evident. The 41-year-old mother, although she has weakness, is still able to walk, while her son lost the ability to walk in adolescence and her other daughter at 7 years old is able to run, jump and climb stairs without difficulty.
The adequate clinical characterization of each patient with suspected neuromuscular disease continues to be the fundamental pillar to direct the available diagnostic studies. In our context, the possibility to perform MRI has also become an important tool non-invasive that added to identification of the RYR1-RM patients.
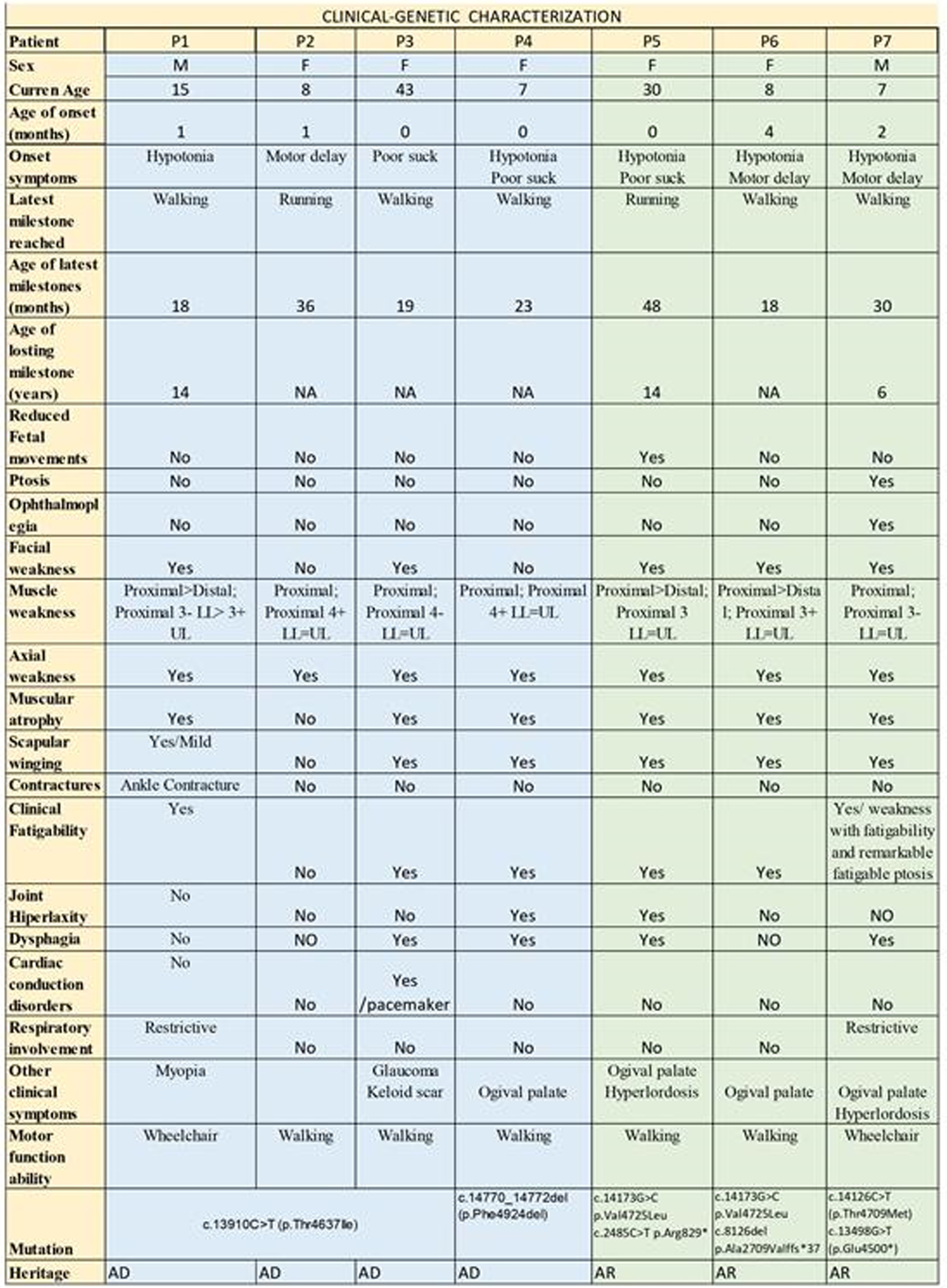
eP04.05.08Risk of Malignant Hyperthermia in Patients Carrying a Variant in the Ryanodine Receptor 1 Gene
Herdewyn S1, Janssens L2, De Puydt J3,4, Symoens S1,2, De Bleecker J1,2
1University Hospital Ghent, Ghent, Belgium, 2Ghent University, Ghent, Belgium, 3University Hospital Antwerp, Antwerp, Belgium, 4Antwerp University, Antwerp, Belgium
Introduction: Malignant hyperthermia (MH) is a life-threatening pharmacogenetic disorder of the skeletal muscle metabolism, which can be prevented by adaptation of the anesthetics used, if the patient is known with the condition. Pathogenic variants in the Ryanodine Receptor 1 (RYR1)-gene are often linked to MH.
Methods: We retrospectively studied 15 patients who presented to our adult neuromuscular clinic between 2016 and 2021 with muscle complaints and who turned out to have a variant in the RYR1 gene. Symptoms at presentation, CK-levels, EMG, muscle biopsy and in vitro contracture test (IVCT) results and pathogenicity prediction were reviewed, alongside previous exposure to general anesthesia.
Results: Most patients presented with (exercise induced) myalgia, some with elevated CK-levels. Six out of the eleven patients with a variant of unknown significance in RYR1 had a positive IVCT, indicating MH-susceptibility. These variants of unknown significance should thus be reclassified as pathogenic variants. The pathogenicity prediction software was not always in line with the IVCT results.
In one patient, with two variants of unknown significance, only the presence of both variants expressed the MH-susceptibility trait. The presence of one of both variants alone resulted in a negative IVCT, as was seen in both parents. Interestingly, one MH-susceptible patient was also diagnosed with facioscapulohumeral muscular dystrophy (FSHD).
Conclusion: Neurologists should think of screening for a variant in the RYR1 gene in patients with muscle complaints, even in the absence of CK-elevation. Additional IVCT can confirm or rule out MH-susceptibility in a reliable matter, which is of uttermost importance given the potentially fatal complications.
eP04.05.09Congenital Myopathy Caused by Mutations in the Neubulin Gene Associated to Schizophrenia: A Case Report.
Portela Sánchez S1, Palacios Mendoza M1, Sánchez Soblechero A1, Catalina Álvarez I1, Lozano Ros A1, Muñoz Blanco J1
1Hospital General Universitario Gregorio Marañón, Madrid, Spain
Introduction: Congenital myopathies are a group of inherited clinically and genetically heterogeneous muscle disorders characterized by the presence of distinctive features on muscle biopsy including nemaline myopathy and central core disease as two of the most common. Mutations in the nebulin (NEB) gene cause autosomal recessive congenital myopathies. Here we report a patient with a congenital myopathy caused by mutations in the nebulin gene associated to schizophrenia.
Case report: The patient is a 36-year-old boy. He presented at birth with generalized hypotonia and macrocephaly. The parents were non-consanguineous with no family history of neuromuscular disorders. Pregnancy and delivery were normal. Motor milestones were delayed since the early childhood with difficulties in walking. Main symptoms were frequent falls and inability to run that had not progressed since adolescence. At the age of 14-years-old, the patient developed behavioral impairment and he was referred to Psychiatry department where paranoid schizophrenia was diagnosed at the time. The neurological examination revealed craniofacial dysmorphia, cavus feet and a mild degree of axial and lower limb girdle weakness. Serum creatin kinase (CK) levels were normal, and electromyography showed a proximal myopathic pattern. A brachial biceps biopsy performed at the age of 34-year-old showed cores in type 1 fibers with NADHTR staining and no nemaline rods were apparent with the Gomori trichrome, at light microscopy. Following the diagnosis of congenital myopathy to identify the genetic cause an exome on genomic DNA of the patient was performed. The patient presented two mutations in heterozygosis in the NEB gene, a missense mutation in exon 141 c.18916G>A (p. Ala6306Th) and a missense mutation in exon 27 c.2573C>T (p. Ala858Val), both of them previous associated to nemaline myopathy.
Discussion: We report a case of a patient with congenital myopathy with cores and schizophrenia associated with nebulin mutations. Recessive pathogenic variants in NEB are the major cause of nemaline myopathy. However, the spectrum of myopathies caused by variants in the gigantic NEB mutations is wide, with different weakness distribution and clinical severity. In addition to clinical heterogenicity, a wide muscle pathological spectrum has been described in patients with congenital myopathies. In this way, at this time, pathogenic variants in NEB have also been identified in distal myopathies and core-rod myopathies. To the best of our knowledge this is the first case of NEB associated congenital myopathy presenting with cores in muscle biopsy and psychiatric disturbances. Currently, it is extremely difficult to stablish genotype-phenotype correlations and new generations sequence (NGS) techniques are a valuable tool for the molecular diagnosis of congenital myopathies. Thus, to perform targeted NGS to study genes as NEB should be strongly considered in patients with congenital myopathies to allow further description of the wide clinical and pathological spectrum associated to NEB gene mutations.
eP04.05.10Clinical Features of the UK FSHD Patient Registry
Walker H1, Orrell R2, Graham A3, Watt S3, Lunt P4, Norwood F5, Roberts M6, Willis T7, Matthews E8, Muni-Lofra R1,9, Marini-Bettolo C1,9
1The John Walton Muscular Dystrophy Research Centre, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom, 2UCL Queen Square Institute of Neurology, University College London, London, United Kingdom, 3Muscular Dystrophy UK, United Kingdom, 4University of Bristol, Bristol, United Kingdom, 5Department of Neurology, Kings College Hospital, London, United Kingdom, 6Department of Neurology, Salford Royal NHS Foundation Trust, Salford, United Kingdom, 7Neuromuscular Service, The Robert Jones and Agnes Hunt Orthopaedic Hospital NHS Foundation Trust, Shropshire, United Kingdom, 8The Atkinson Morley Regional Neurosciences Centre, St George’s University Hospital NHS Foundation Trust, London, United Kingdom, 9Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
The UK Facioscapulohumeral Muscular Dystrophy (FSHD) Patient Registry is a patient self-enrolling online database collecting clinical and genetic information about FSHD type 1 (FSHD1) and type 2 (FSHD2). The registry was established in May 2013 with support from Muscular Dystrophy UK and is coordinated by Newcastle University. The registry aims to facilitate academic and clinical research, better characterise and understand FSHD, and disseminate information relating to upcoming studies and research advancements.
The registry is used to capture longitudinal, self-reported data through an online portal available to patients and clinicians. Where specialised clinical or genetic information is required, the neuromuscular specialist involved in the patient’s care can be invited to provide some additional information and the patient can select them from a pre-populated list at the registration stage. The registry is a Core Member of the TREAT-NMD Global Registries Network for FSHD.
Between May 2013 and January 2022, there were 1,074 patient registrations, with 84% based in the UK. On average, there are 9 new registrations per month. For those reporting a clinical diagnosis, 97% have FSHD or FSHD1, and 3% have FSHD2. Overall, 46% of patients have had genetic confirmation of FSHD1 provided.
The registry has previously supported almost 30 registry enquiries to date. Since 2020, the registry has facilitated 12 enquiries including, three COVID-19 surveys, and various surveys capturing information on dysphagia, pregnancy, sleep and the patient/caregiver experience.
The registry is currently one of the largest national FSHD patient registries and is an example of a versatile, cost-effective research tool, helping facilitate and advance a wide range of FSHD research. Additional work continues to be done to improve reporting of genetic information on the registry and there are future data linkage plans between the registry and the Newcastle Research Biobank for Rare and Neuromuscular Diseases.
eP04.05.11Safety and Tolerability of Losmapimod for the Treatment of FSHD
Shoskes J1, Ramana V1, Mellion M1
1Fulcrum Therapeutics, Inc., Cambridge, United States
Objective: Evaluate the safety and tolerability of losmapimod in the treatment for FSHD.
Background: FSHD is a relentless, variably progressive disease leading to accumulation of disability over decades. Fulcrum is developing losmapimod, a small molecule p38 α/β MAPK inhibitor, to treat FSHD. Losmapimod has been generally well-tolerated in more than 3,600 subjects across multiple clinical studies, including >100 subjects with FSHD. Fulcrum has assessed losmapimod in FSHD in one completed phase 1 study (FIS 001-2018) and two ongoing Phase 2 studies in the open label extension period (FIS 001-2019 and FIS 002-2019).
Methods: Subjects aged 18-65 years with genetically confirmed FSHD1, Clinical Severity Score 2-4, and MRI-eligible muscles for biopsy were exposed to losmapimod 7.5 or 15 mg twice daily PO for 14 days and up to 76 weeks. In study FIS 001-2018, 6 subjects were exposed to 7.5 mg and 11 subjects to 15 mg twice daily dosing for 14 consecutive days. In studies FIS 001-2019 and FIS 002-2019, 14 and 77 subjects respectively, received at least one dose of losmapimod 15 mg twice daily for up to 76 weeks.
Results: A total of 108 subjects with FSHD1 have been exposed to losmapimod, with approximately 131 patient-years of exposure. Fifty-seven subjects have been exposed to losmapimod for 12 to 18 months, and 30 have been exposed for over 18 months. Most adverse events (AEs) observed during the studies were considered of mild to moderate in severity. The most common AEs were alanine aminotransferase (ALT) increase, headache, dizziness, dry skin, eczema and gastrointestinal disorders. The majority of AEs resolved with continued dosing. Dosing has been paused for 14 days in four subjects (3 in FIS 001-2019 and 1 in FIS 002-2019) subjects due to COVID-19 infection. There were no reported drug related SAEs, deaths, discontinuations due to AEs, or clinically significant changes in vital signs, clinical laboratory results, or ECG parameters.
Conclusion: Losmapimod given as up to 15 mg twice daily in >100 subjects with FSHD1 for up to 76 weeks has been generally well-tolerated, consistent with that previously reported in other patient populations. Therefore, the benefit-risk profile of losmapimod for the treatment of FSHD remains favorable.
eP04.06.01Automated Integrative Splicing Predictor Tool: Focus on Deep Intronic Variants Prioritization for the DMD Gene
Zanobio M1, Zeuli R1, Peduto C1, Onore M1, Romano F1, Napoli M1, Torella A1,2, Picillo E1, Del Vecchio Blanco F1, Pinelli M3, De Riso G3, Piluso G1, Nigro V1,2
1University of Campania Luigi Vanvitelli, Naples, Italy, 2Telethon Institute of Genetics and Medicine, Pozzuoli, Italy, 3Dep. of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Napels, Italy
NGS technology allows to detect virtually all DNA variations, nevertheless deep intronic single nucleotide variants (SNVs) remain truly elusive. Indeed, defining their pathogenic role is the most fascinating challenge of the last years. One of their major effects is the partial or full pseudo-exon (PEs) inclusion, mainly due to aberrant splicing, through activation of cryptic intronic acceptor and/or donor splice sites and the alteration of sequence motifs for enhancer or silencer splicing factors. This is a major challenge for large genes with numerous and huge introns, such as the DMD gene
With the aim of generating an automated tool to predict and prioritize deep intronic SNVs detected by NGS analysis, we tested four different prediction tools (SpliceAI, NNSplice Predictor, HSF and SFMap) to evaluate possible effects on splicing: we studied the deep intronic SNVs in the DMD gene annotated in the Leiden Open Variation Database (LOVD). We divided the collected LOVD variants into two groups, TRAINING (variants with known RNA effect) and TESTING (variants with unknown RNA effect). Validating this approach on TRAINING group, we confirmed that 77% of variants is involved in alternative splicing leading to PEs inclusion. Similarly, on TESTING group we predicted the effect on splicing for 72% of thevariants studied.
We are strongly confident that our automated tool is very practical to predict the effect of deep intronic SNVs on alternative splicing and PEs formation, thus providing a good indication for prioritizing NGS variants and transcriptomic studies on muscle biopsy.
eP04.06.02The Epidemiology of Mutations of Dystrophin in the Hungarian Population
Udvari S1, Andó S2, Bessenyei B2, Pikó H3, Koczok K2, Gál A1, Karcagi V4, Balogh I2, Molnár M1
1Institute Of Genomic Medicine And Rare Disorders, Budapest, Hungary, 2Debrecen University, Clinical Genetic Faculty, Debrecen, Hungary, 3PentaCore Laboratory, Budapest, Hungary, 4Istenhegyi Gene Diagnostic Centre, Budapest, Hungary
The genetic diagnostics of Duchenne and Becker muscular dystrophies have a more than 30-year history in Hungary. In distrophinopathies, therapies specific for certain mutations have become part of the clinical practice, so it is highly important to obtain knowledge about the disease causing mutations and the phenotypes connected to them in the Hungarian population. In this presentation we show the diagnostic results of the dystrophin gene analysis. The data summarises the mutations found in Debrecen University, Semmelweis University and the former institute of National Public Health (OKI) and compares those with the internationally published data.
The methodology used to identify the mutations was initially multiplex PCR, later MLPA. In the last 5 years NGS technology made the sequencing of the entire dystrophin gene possible.
Results: Until now in Hungary 356 dystrophinopathies have been confirmed. The ratio of deletions and duplications were similar to published data. Some Becker genotypes were over-represented in the Hungarian cohorts. In general, point mutations were under-represented, as sequencing prior to NGS technology was rarely carried out. Phenotype analysis indicated that autism spectrum disorder and obsessive-compulsive symptoms were sometimes associated with dystrophinopathy. The number of symptomatic carriers is 4 in our cohort. There were at least 16 cases where siblings or close relatives inherited the same mutations, but only unrelated cases are included in the study.
Conclusions: The knowledge of the mutation spectrum of the dystrophin gene is very important in the age of mutation specific treatment, because with the aid of these numbers, the number of patients who would benefit from the innovative therapies or who are waiting for novel, non mutation specific therapies could be accurately predicted. These data are relevant not only for the clinicians, but for those financing the treatments, in view of the planning of the budgeting for rare disorders in the future.
eP04.06.03Targeting NAPDH Oxidases in Duchenne Muscular Dystrophy: diapocynin Therapeutic Effect on Adult mdx Mice
Guedira G1, Petermann O1, Scapozza L1, Ismail H1
1Pharmaceutical Biochemistry/Chemistry Group, Institute of Pharmaceutical Sciences of Western Switzerland, University of Geneva, Geneva, Switzerland
Duchenne muscular dystrophy (DMD) is the most common muscular disease affecting children. It affects nearly 1 male birth over 3500. There is a general consensus that oxidative stress is a pervasive feature in the pathogenesis of DMD. Recent work on NADPH oxidase (NOX) showed that they can be a target of interest in diseases involving oxidative stress like DMD. Previously, we have shown that the putative NOX inhibitor, diapocynin, demonstrated high efficacy in inhibiting reactive oxygen species (ROS) production in dystrophic myotubes and preventing eccentric contraction induced damage in isolated dystrophic mice in situ. Diapocynin in vivo treatment also showed a restoration of spontaneous locomotor activity, enhanced wheel running capabilities, improvement in fatigue and diaphragm structure in young mice treated from 14 days post-natal to 12 weeks of age.
In light of the encouraging results obtained with diapocynin in young mdx5Cv mice, we decided to test it further on the chronic phase of the disease. We report here a comprehensive analysis of diapocynin in adult mice treated between 6 and 9 months of age. Read-outs previously done in young mice were done in older mice. Classical methods such as grid tests, locomotor activity, resistance to fatigue and eccentric contractions among others were performed as well as histological evaluation and biochemical analyses.
The treatment of dystrophic mice in advanced stage of the disease in a curative setting revealed the potential of NOX targeted therapy. Our findings showed that different functional and histological parameters were improved compared to control. These observations correlate with our findings on quantifying NOXes expression at different stages of the disease.
eP04.06.04Eteplirsen Safety, Tolerability, and Pharmacokinetics in Young Patients with DMD Amenable to Exon 51 Skipping
Mercuri E1, Seferian A2, Servais L2,4, Deconinck N5,6, Stevenson H7, East L7, Zhang W7, Upadhyay S7, Muntoni F8,9
1Pediatric Neurology Unit, Università Cattolica del Sacro Cuore Roma, Rome, Italy and Nemo Clinical Centre, Fondazione Policlinico Universitario A Gemelli IRCCS, Rome, Italy, 2I-Motion Institute, Hôpital Armand Trousseau, Paris, France, 3Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Liège, Belgium, 4MDUK Oxford Neuromuscular Centre, University of Oxford, Oxford, UK, 5Centre de Référence Neuromusculaire and Paediatric Neurology Department, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, 1020 Brussels, Belgium, 6Neuromuscular Reference Center, UZ Gent, Ghent, Belgium, 7Sarepta Therapeutics, Inc, Cambridge, USA, 8Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health, London, UK, 9National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, London, UK
Background: Eteplirsen is indicated for treatment of exon 51 skip-amenable patients with Duchenne muscular dystrophy (DMD). Previous studies in patients >4 years of age indicate eteplirsen is well tolerated and attenuates pulmonary and ambulatory declines compared with matched natural history cohorts.
Objective: Here we evaluate the safety, tolerability, and pharmacokinetics of eteplirsen in patients aged 6–48 months, the youngest population of patients with DMD in a clinical trial to date, in Study 4658-102 (NCT03218995).
Methods: In this open-label, multicenter, dose-escalation study, patients who had a confirmed mutation of the DMD gene amenable to exon 51 skipping (Cohort 1: aged 24–48 months, n=9; Cohort 2: aged 6 to <24 months, n=6) received ascending doses (2, 4, 10, 20, 30 mg/kg) of once-weekly eteplirsen intravenously over 10 weeks, continuing at 30 mg/kg up to 96 weeks. Endpoints included incidence of adverse events and clinically significant laboratory abnormalities (primary) and pharmacokinetics (secondary).
Results: All patients completed the study (N=15). Average time since diagnosis was 10.5 months, and most (13/15, 86.7%) were not taking corticosteroids. An implantable venous access device (IVAD) port was placed in 9/15 (60%) patients during the study. Eteplirsen was well tolerated with no treatment-related discontinuations, deaths, or evidence of kidney toxicity. Most treatment-emergent adverse events were mild, and the most common were consistent with those commonly seen in pediatric populations (pyrexia, nasopharyngitis, vomiting, cough, diarrhea). There were no IVAD-related serious bloodstream infections reported. Eteplirsen pharmacokinetics were consistent between both cohorts and aligned with expectations based on previous clinical experience in boys with DMD older than 4 years of age.
Conclusion: These data support the safety and tolerability of eteplirsen at the approved 30 mg/kg dose in patients as young as 6 months old.
eP04.06.05Description of Osmolyte Pathways in Maturing MDX Mice Reveals Altered Taurine and Sodium/Myo-Inositol Co-transporter Levels
Merckx C1, Cosemans G1, Zschüntzsch J2, Schmidt J2,3, De Paepe B1, De Bleecker J1
1Department of Neurology, Ghent University and Ghent University Hospital, Ghent, Belgium, 2Department of Neurology, University Medical Center Göttingen, Göttingen, Germany, 3Department for Neurology and Pain Management, Immanuel University Clinic Rüdersdorf, Rüdersdorf bei Berlin, Germany
BACKGROUND: Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle degeneration and weakness. Osmotic stress participates to DMD pathology and altered levels of individual osmolyte pathway members have been reported. The regulation of osmolyte pathways in dystrophin deficient tissue remains, however, poorly understood. The goal of this study was to gain insight in osmoregulatory changes in the mdx mouse model, by examining the expression of osmolyte pathway members taurine transporter (TauT), sodium myo-inositol co-transporter (SMIT), and aldose reductase (AR) in skeletal muscle and diaphragm in mice aged 4 to 26 weeks.
METHODS: Mdx and C57BL/10SnJ control mice were sacrificed at age 4, 8, 12, and 26 weeks. Muscle damage was evaluated by determining percentages of healthy, regenerating and necrotic fibers as visualized on haematoxylin–eosin stained sections of the tibialis anterior. Expression of osmolyte pathway members TauT, SMIT, and AR was studied in tibialis anterior, gastrocnemius and diaphragm using immunofluorescence, qPCR, and western blot.
RESULTS: Histological analysis of the tibialis anterior indicated necrosis was most extensive in 12-week-old mdx mice (9.2%) and decreased to 3.1% in 26-week-old mice, whereas the number of regenerated fibers reached its peak at week 26 (64.2%). TauT was down regulated in tibialis anterior and gastrocnemius (p<0.01) of 4-,8-, and 12-week-old mdx mice but not in 26-week-old mice, whereas expression remained significantly lower in the diaphragm of 26-week-old mdx mice. By contrast, expression of SMIT was significantly higher in skeletal muscles of mdx mice compared to control mice (p <0.01). No difference in AR protein levels between mdx and age-matched control mice could be observed.
CONCLUSIONS: Our study revealed differential regulation of osmolyte pathway members TauT, SMIT and AR, which points to their complex involvement in mdx pathogenesis going beyond general osmotic stress responses. These results highlight the potential of osmolyte pathway members as a research interest and therapeutic target in dystrophinopathy.
eP04.06.06Non-dystrophic Myotonia; The Patient Journey to Diagnosis
Aldwinckle T2, Jayaseelan D2, Pilling K1, Luckin J3
1The National Hospital of Neurology and Neurosurgery, UK., 2Lupin Healthcare (UK) Ltd, Slough, SL1 2BE, UK, 3Strategic North, Hale, WA14 2EX, UK
Background. Non-dystrophic myotonia (NDM) is a recognised but rare channelopathy with a prevalence of approximately 1:100,000. In the UK, diagnosis and treatment are undertaken by patient referral to specialist centres, however like other rare diseases the patient journey to diagnosis may be lengthy.
Aims. To explore and document the patient referral pathway from the patient’s self-recall of first onset of symptoms to primary care consultation and subsequent specialist diagnosis and treatment.
Methods. A third-party research consultancy (Strategic North) conducted one-hour in-depth telephone interviews with NDM patients referred by either neurologists with expertise in NDM or patient organisations. Patient responses were supplemented by commentary from the expert neurologists participating.
Results. 17 patients with NDM participated in the interviews. While their symptoms occurred early in life, patients initially self-managed these using a range of coping strategies for 5-10 years following onset. With more prominent symptoms developing and coping mechanisms failing, patients then consulted their general practitioner (GP) for diagnosis; this period of seeking a diagnosis ranged from 1 to 10 years with repeated GP consultations. Of the 17 patients interviewed, less than one third were referred by their GP directly to regional neurology centres. The remaining patients were referred initially to a range of non-neurology specialties, including physiotherapy, paediatrics and rheumatology over a period of 2-3 years. Patients who were referred to the above specialists were subsequently discharged back to their GP without an NDM diagnosis, with associated patient frustration. All patients were eventually referred to either general neurologists or regional specialist neurology centres where a diagnosis of NDM was made followed by further referral to the UK national specialist neurology centre for sub-type diagnosis and treatment management plans, representing a period of 0-3 years duration. Patients reported a range of emotions through their diagnostic journey.
Conclusions. The data presented are from a small cohort of patient interviews, with patient responses based on self-recall of historic events. Nevertheless, it appears that initial diagnosis of NDM remains a challenge, with many patients reporting periods of several years from initial consultation with their GP and further non-neurology referrals prior to attending neurology centres for diagnosis and initiating treatment. Availability of further tailored educational resources including informing patients of NDM as a potential diagnosis for their symptoms may support shortening this patient journey.
eP04.06.07Sensory Polyneuropathy in Oculopharyngeal Muscular Dystrophy, it this a novel Phenotypical Findings ?
Remiche G1, Bisciglia M1, Desmyter L2, Vandernoot I2, Mavroudakis N1
1Centre De Réference Neuromusculaire, Dept. Of Neurology, Hôpital Erasme, Université Libre De Bruxelles, Brussels, Belgium, 2Department of Genetics, Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium
Introduction: Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant myopathy typically occurring between the fourth and seventh decade. It involves mainly in early stages pharyngeal and levator palpebrae superioris muscles and will progress with involvement of limb-girdle muscles.
Brais et al. identified in 1998 that OPMD is caused by an expansion of a short (GCN) trinucleotide repeat in the coding sequence of PABPN1 (GCG6 expanded to GCG8–13) that create a short alanine expansion (10 alanines expanded to 12–17 alanines).
Currently co-existing polyneuropathy in OPMD is debated in the literature with highly variable prevalence reported on rare small series. However, to date, no series of genetically confirmed OPMD cases had demonstrated a high prevalence of coexisting polyneuropathy.
Mutated PABN1 protein polymerizes and accumulates in the nucleus forming toxic filaments and nuclear or cytoplasmic inclusions.
Describing a small series of genetically confirmed OPMD patients having an associated unexplained polyneuropathy and making a review of the literature, we address the question of that potential association.
Methods: We retrospectively assessed medical records of genetically confirmed OPMD patients followed in our center in order to identify clinical and neurophysiological data suggesting a polyneuropathy. We also systematically looked for the presence of potential etiologies for polyneuropathy. Clinical and neurophysiological polyneuropathy international diagnostic criteria were applied.
We systematically reviewed the literature published since 1998, in order to identify previously genetically confirmed OPMD patients harboring a coexisting polyneuropathy.
Results: We include three female and two male patients aged between 63 and 85 years.
The age of the OPMD diagnosis ranged between 60 and 68 and ages at the first symptoms were between 50 and 63 years-old.
Four of the five OPMD patients found in our database had clinical and electrodiagnostic evidence for polyneuropathy that was observed between the age of 59 and 75 years. Apart one patient having a past medical history of excessive alcohol intake none had other known common cause of polyneuropathy.
Only two genetically proofed OPMD patients, in the literature, were found to have an associated polyneuropathy.
Conclusion: Our observation seems to confirm some scarce previous data supporting the possibility of an over-representation of mild sensory polyneuropathy in OPMD patients.
A possible explanation for peripheral nerves involvement in OPMD could be a toxicity of PABPN1 protein aggregates on peripheral nerves.
This potential association is of clinical importance since sensory polyneuropathy increases the risk of falls in these osteoporosis-predisposed aged patients having in most of the cases a low body-mass-index due to dysphagia. Polyneuropathy is an actionable condition by physiotherapy and prevention of neurotoxic exposure including many drugs.
Our study includes some limits such as potential confusing factors including the high prevalence of peripheral neuropathies in aged populations and the limited number of patients in our cohort.
Larger well-designed multicentric prospective studies are needed to confirm this possible association.
eP04.06.08Effects of FGF21 Supplementation in Muscle Cells from Mitochondrial Disease Patients
Chudenkova M1, S. Müller J1, Hathazi D1, Horvath R1
1Department of Clinical Neurosciences, School of Clinical Medicine, University of Cambridge, Cambridge, United Kingdom
Background: FGF21 is an endocrine hormone known to regulate energy homeostasis in cells. Due to its increased levels in the serum of patients with skeletal muscle-specific mitochondrial disease, it has been proposed as a biomarker for mitochondrial myopathies. As skeletal muscle is the primary affected tissue in these diseases, studying the effect of FGF21 in muscle is crucial for understanding its roles.
Aims: Our goal is to validate the effects of FGF21 supplementation in primary myoblasts from patients with different mitochondrial diseases.
Methods/Materials: human primary myoblasts from healthy controls and patients with the most common mitochondrial translation defect m.3243A>G with different heteroplasmy levels (20%, 40% and 80%) were grown in Skeletal Muscle Cell Growth Media (Promocell) and supplemented next day after seeding with 50 ng/ml FGF21 for 8 days. We first evaluated the effects of FGF21 supplementation on mitochondrial biogenesis. Western blotting was used to detect the steady-state protein levels of mitochondrial respiratory chain complex subunits. We assessed cell viability and the activation of master regulators of cell metabolism such as AMPK and its downstream targets in mTOR signaling.
Results: Our initial assessment of our myoblasts cohort shows that mitochondrial respiratory chain protein levels were decreased in all m.3243A>G patients and it is correlating with the mutational load, while the FGF21 supplementation did not result in increased steady-state levels of respiratory chain subunits. Moreover, we detected that FGF21 activates AMPK in patient cells, potentially remodeling the metabolism and mitochondrial function.
Conclusion: Our preliminary data on FGF21 treatment in muscle cells of patients with mitochondrial myopathies showed promising results. Further experiments are in progress with different FGF21 concentrations and varying treatment times to better understand the role of FGF21 in mitochondrial myopathies.
eP04.06.09Clinical Characterization of Familial Hyperkalemic Periodic Paralysis with a SCN4A Met1592Val Mutation
Kang S1
1College Of Medicine, Jeju National University, Jeju, Republic of Korea
Background: Hyperkalemic periodic paralysis (HyperPP) is characterized by episodic flaccid paralysis of skeletal muscles that is exacerbated by the consumption of potassium-containing foods, fasting, or rest following exercise. We describe the clinical and electromyographic characteristics in familial HyperPP with the Met1592Val mutation in the SCN4A gene.
Methods: Thirty patients from seven families were assessed by interviews and clinical examinations. Standardized protocols comprising short and long exercise tests were applied to 15 unaffected control subjects and the 30 patients with familial HyperPP. To identify comorbidities prevalent in patients, we surveyed subjects for common medical conditions. Results: Precipitants of attacks were vigorous exercise and hunger in all patients. All patients experienced clinical myotonia at the eyelids or lips. Attack duration varied from less than 1 hour to greater than 3 weeks. Reports of symptom duration varied within and between individuals, but the maximal duration of symptoms was generally prolonged. The mean age of onset was 7.3 years (range 1- 14 years), attacks beginning before 10 years in 86.3% of patients studied. Exercise of short duration induced an immediate increase in the amplitude of the compound motor action potential (CMAP) in the patients, and this was significantly larger and lasted longer than that observed in controls within 50 seconds (p<0.05). A long exercise test induced a large increase in the CMAP amplitude in patients immediately after exercise completion, which decreased to normal values with 1 minute. In contrast, controls showed a decreased CMAP amplitude immediately after exercise, which subsequently also returned to the normal value.
Conclusions: Affected members were phenotypically heterogeneous and showed similar response in exercise test. The exercise tests may be helpful in confirming abnormal excitability of muscle membrane in HyperPP patients.
eP04.06.10Skeletal Muscle Injury by Electroporation: A Model to Study Degeneration/Regeneration Pathways Murine Models For NMD
Vainzof M1, Vasconcelos F1, Almeida C1, Ishiba R1, Bitoun M2, Souza L1, Ribeiro-Jr A1, Souza B1, Zogby I1, Saldys N1
1Human Genome and Stem Cells Research Center, University Of Sao Paulo, Sao Paulo, Brazil, 2Sorbonne Université, INSERM, Institute of Myology, Centre of Research in Myology, France
Skeletal muscle has a remarkable capacity to regenerate after injuries mainly due to a reservoir of precursor cells named satellite cells (SCs), which are responsible for after-birth muscle growth and response to lesions. Upon injury, the regenerative response includes SCs exit of quiescence, activation, proliferation and fusion to repair or form new myofibers. Every phase of regeneration is highly regulated by many molecules and signaling pathways. In murine models for neuromuscular diseases, the pattern of degeneration is variable, and can be different from the observed in the human respective disease.
Here we used a model of muscle injury induced by electroporation, which is an efficient and safe method to induce muscle damage and followed the steps of degeneration and regeneration. In the normal muscle, three days after electroporation, the muscle showed prominent signals of degeneration, with areas of necrosis and infiltration of macrophages, followed by regeneration observed by the presence of centrally nucleated myofibers. After five days, the regeneration was very active, with small dMyHC positive fibers. Fifteen days later, we observed a general regeneration of the muscle, with almost normal fiber size after 30 days.
We applied this fast degeneration/regeneration inducing muscle damage approach in mouse models for neuromuscular disease, such as the SJL/J model for Limb-girdle Muscular Dystrophy Recessive 2 Dysferlin-related (LGMDR2) and the KI-Dnm2 mouse model for Centronuclear Myopathy, both with discrete histopathological alterations and compared to a dystrophic model with muscle degeneration as observed in the mdx mouse model for Duchenne Muscular Dystrophy.
The proportion of Pax7+ fibers was elevated in the mdx model and reduced in the KI-Dnm2 mouse. Upon induced degeneration, the genes related to the regeneration pathway (MYOD, MYF5, MYOG), were activated in the SJL/J and the KI-Dnm2 models in days 3 and 5, similarly to normal controls, but at lower levels in the KI-Dnm2 model. Regeneration, marked by positive dMyHC fibers was observed after days 3 and 5, in the SJL/J and mdx, but was delayed in the KI-Dnm2 model (days 5 and 10). In overall, while the SJL/J model showed a remarkable regenerative capacity, the regeneration was less efficient in the KI-Dnm2 and mdx models by forming fewer new myofibers, or with a smaller diameter.
The elucidation of players and mechanisms involved in muscle degeneration and regeneration in the normal and the myopathic muscles is of extreme importance, especially for therapeutic strategies for muscle diseases. FAPESP-CEPID, CNPq-INCT.
eP04.06.11Evaluation of the Relationship between Genotype and Phenotype of Dystrophinopathy in Iranian Race
Ansari B1
1University Of Medical Science In Isfahan, Isfahan, Iran (Islamic Republic of)
Background: Duchenne Muscular Dystrophy (DMD) and Becker muscular dystrophy (BMD) are X-linled disorders caused by a mutation in the dystrophin gene. Genotype and phenotype matching studies show that phenotype severity depends on the amount of muscle dystrophin or the mutation / deletion site in the dystrophin gene. The purpose of this article is to investigate the relationship between any genetic mutation in the dystrophin gene and the clinical picture associated with that mutation in the Iranian race.
Methods: This is a cross-sectional study that was performed in 2021 in Isfahan on 54 patients with muscle weakness with problems in dystrophin gene. Demographic data of patients were collected using a checklist. These data included age, family history of muscle dystrophies, family history of other medical diseases and type of muscle dystrophy. We also evaluated the number and area of deleted exons divided based on dystrophy types. The gait of patients was also assessed based on using a wheelchair, waddling gait or toe gait. The severity of muscle dystrophy was assessed in each limb based on muscle force. We also evaluated and compared clinical condition of patients, cardio-pulmonary, mental and bulbar conditions.
Results: the type of dystrophin was 40.7% of Becker patients, 55.6% Duchenne and 3.7% intermediate between the deletions. The highest frequency of deleted exons in this area was in Becker patients in exons 47-45 (n = 5) and 48-45 (n = 4) and in Duchenne patients in removed exons 45, 52-48, 55-51 and 53. Patients with deletion in exons of 45-47 had higher ages at the time of wheelchair boand and patients with deletion in exons 51-55 had lower ages at the time of wheelchair boand. The frequencies of proximal and distal muscle weakness in lower ages were significantly higher in patients with DMD . Hotspot area in our study is exons of 45-55 where 63% all of our patients had mutation in this area.
Conclusion: Exon deletion was most common genotype in patients. We found no significant differences between DMD and BMD regarding the number of deleted exons. Patients with deletion in exons of 45-47 had higher ages at the time of wheelchair boand and patients with deletion in exons 51-55 had lower ages at the time of wheelchair boand.
Author Index
Aartsma-Rus A. eP03.01.02, S217
Abati E. eP01.02.01, S142
Abida Y. eP01.01.10, S141, eP02.02.03, S186, eP04.03.02, S276, eP04.03.07, S279, RG.03, S56
Abrams K. PS01.3, S89
Achenbach P. eP02.01.09, S183
Adams D. TC06.03, S15
Aggarwal R. eP01.05.04, S166
Ahmet A. PS01.02, S87
Akgün K. eP03.01.08, S220
Akodad S. eP01.03.04, S149
Akyurek E. PS09.02, S128
Al-Bourini O. PS01.06, S91
Al-Chalabi A. eP04.03.10, S281
Alangary A. PS02.03, S93
Albers J. eP03.01.07, S220
Alberti M eP02.04.04, S197
Alcock L. eP03.01.05, S218
Aldwinckle T. eP04.06.06, S302
Aledo-Sala C. eP03.06.01, S259
Alexander M. eP02.05.02, S202
Alfano L. eP03.06.03, S261, PS03.04, S99, PS03.01, S97
Alfieri P. eP03.01.04, S218
Alfredsson L. eP03.04.07, S244
Allenbach Y. PS07.01, S117
Allen J. TC02.04, S10
Allison D. eP02.04.07, S198
Almeida C. eP04.06.10, S305
Alonso-Pérez J. PS02.03, S93
Alonso Jimenez A. eP02.02.05, S187
Alvarez-Toledo K. eP01.03.10, S154
Alvarez M. eP01.03.07, S151, PS02.05, S95
Alves F. eP03.01.07, S220
Al Muhaizea M. PS06.04, S114
Amato A. OS07.01, S35, PS07.04, S120
Ambrosini A. eP02.04.07, S198
Ambrosoli L. eP04.04.03, S284
Amela Peris R. eP04.02.02, S271
Amthor H. eP02.03.01, S191
Anang D. eP01.05.02, S164
Andersen P. eP04.03.01, S275
Andersen R. eP02.04.06, S198
Anderson-Smits C. eP02.02.02, S185
Anderson F. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Andreu L. eP03.05.11, S258
Andrews J. eP04.03.10, S281
Andrinopoulou E. eP03.05.03, S251
Andó S. eP04.06.02, S300
Angelini C. WS03.02, S45, eP03.01.11, S222, eP03.05.06, S254, OS06.02, S34, OS09.03, S38
Angelov T. eP04.03.05, S278
Anifowoshe D. eP01.03.05, S150
Ansari B. eP02.02.10, S190, eP04.06.11, S305
Anzola M. eP01.06.04, S173
Aponte Ribero V. PS06.06, S116
Aragon Pinto C. eP02.02.08, S189
Arca G. eP04.05.04, S293
Archer A. eP02.04.05, S197
Arechavala-Gomeza V. eP01.01.08, S139
Arenas J. eP03.05.11, S258
Arestova A. eP03.02.05, S226
Argov Z. OS08.01, S36, TC04.03, S13
Arnaoutoglou M. eP01.02.09, S147, eP03.02.02, S224
Aronica E. eP01.04.03, S158, eP01.05.02, S164, eP01.05.05, S167
Artemieva S. eP04.01.06, S270
Arunachal G. eP02.06.07, S214, PS01.05, S90
Ashley E. eP02.06.06, S213
Astrea G. eP02.05.09, S207
Atchayaram N. PS01.05, S90
Atrous A. eP01.01.10, S141, eP02.02.03, S186
Attarian S. SS11.02, S71
Attia Z. eP03.06.06, S263
Aubertin-Leheudre M. eP03.06.08, S265
Axelsen K. eP02.04.06, S198, eP03.04.11, S248
Azorin I. eP01.01.06, S137
Baars A. PS05.04, S110
Baars M. PS05.03, S109
Baars P. eP01.05.05, S167
Bachtell N. PS03.06, S100
Bachuwa G. PS04.02, S103
Bae H. eP02.03.06, S195
Baek S. eP04.04.09, S289
Baets J. eP01.01.05, S136, eP04.02.05, S273, PS07.03, S119
Bai Y. eP01.02.07, S145
Balicza P. OS02.04, S28
Balogh I. eP04.06.02, S300
Banford K. PS07.02, S118
Baquero C. eP03.03.04, S233
Baranello G. eP01.03.05, S150, eP02.05.03, S202, PS01.02, S87, PS01.04, S89, PS06.01, S112
Bara S. eP02.01.03, S179
Baratto A. PS02.06, S96
Barańska J. eP01.04.05, S160
Barbov I. eP04.04.11, S290
Bardakov S. eP02.01.08, S182, eP02.01.10, S184
Bardhan M. eP02.06.07, S214
Barnerias C. eP04.04.05, S286
Barohn R. PS07.04, S120
Barrett D. eP02.03.03, S193
Barthur A. eP02.01.07, S182
Basak A. eP01.01.05, S136
Baskar D. eP02.06.07, S214, PS01.05, S90, PS07.05, S120
Bassi M. eP01.02.01, S142
Basso D. eP03.01.09, S221
Basta I. eP01.05.09, S170, eP02.06.02, S209
Beauchamp J. eP03.04.04, S242
Becattini L. eP03.03.08, S236
Bechini A. OS02.02, S26
Bedat-Millet A. eP03.05.08, S256
Bede P. PS02.2, S92
Beeson D. PL04.02, S22, PS05.06, S111
Begovic V. eP02.04.01, S195
Beijer D. eP01.01.05, S136, PS08.01, S122
Belkhayate A. eP02.01.04, S180
Bello L. eP02.05.02, S202, eP03.01.09, S221
Bellone E. eP01.02.01, S142
Benavides L. eP01.03.02, S148
Benetollo A. PS09.03, S128
Benezit A. eP02.03.01, S191
Benguerba K. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Benkert P. eP03.01.08, S220
Benmechri S. eP01.03.08, S152
Bennett N. eP02.04.07, S198
Benoit P. eP03.04.09, S246
Benveniste O. PS07.01, S117, TC01.04, S8
Berciano J.A. WS10.01, S52
Berger B. PS05.05, S110
Berger Z. PS06.02, S113
Bertini E. eP03.01.04, S218, eP04.05.03, S293, PS06.02, S113, PS06.04, S114
Bertorini T. PS01.01, S87
Beskrovnaya O. eP02.06.01, S208, eP04.04.02, S284
Bessenyei B. eP04.06.02, S300
Bettica P. eP04.04.03, S284
Betts C. SS12.03, S73
Beysen D. eP04.01.01, S266
Bezier C. PS06.03, S114
Bhadu D. eP01.05.07, S168
Bhatia R. eP01.01.07, S138, eP01.01.09, S139, eP01.05.07, S168
Bianchi F. eP03.03.08, S236, OS09.01, S37
Bianchini E. PS09.02, S128
Bibeau K. eP03.04.05, S242
Biondi O. PS06.03, S114
Birnbaum S. eP02.04.05, S197
Birouk N. eP01.02.05, S144, eP02.01.04, S180, eP03.03.10, S238
Bischoff A. eP01.01.05, S136
Bischof M. eP02.03.02, S192
Bisciglia M. eP03.05.10, S257, eP04.02.01, S270, eP04.06.07, S303
Bisogni G. PS02.03, S93
Bitoun M. eP04.06.10, S305, SS04.01, S62, eP04.04.06, S287
Blaauw B. PS09.03, S128
Blaes F. PS05.05, S110
Blaettler T. PS07.04, S120
Blaschek A. eP04.04.01, S282, eP04.05.01, S291, PS03.06, S100
Blewitt C. eP01.03.05, S150
Bobadilla E. eP01.03.10, S154, eP02.05.10, S208, eP03.06.04, S262, eP04.05.07, S295
Bocassin C. eP02.03.01, S191
Boehnlein M. PS05.01, S106
Boekholdt M. eP01.04.03, S158
Boersma E. eP03.05.01, S248
Boespflug-Tanguy O. PS06.01, S112
Bogaards H. eP03.06.07, S264
Bogaert E. PS07.03, S119
Bolaño-Díaz C. PS02.03, S93
Bolcato M. PS09.02, S128
Bolino A. PS08.03, S123
Bondì M. PS09.03, S128
Bonefeld K. PS07.04, S120
Bonnemann C. SS08.03, S67
Bonnyaud C. eP03.01.06, S219
Borland H. eP04.05.02, S291
Boroojerdi B. eP03.04.03, S241
Bosco L. eP04.05.03, S293
Bossuyt P. eP01.05.05, S167
Bouadi A. eP02.03.01, S191
Bouhouch A. eP01.02.05, S144
Bouhouch R. eP01.02.05, S144
Boutalbi Y. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Bowden A. eP04.05.01, S291
Bowler M. eP02.06.06, S213
Boyer S. eP03.02.11, S230
Bozorg A. PS05.01, S106
Bozovic I. eP01.05.09, S170, eP02.06.02, S209
Braid J. PS06.01, S112
Brankovic V. eP01.01.05, S136
Brassier A. eP03.05.04, S252
Brauer E. eP03.04.06, S243
Brauner S. eP03.04.07, S244
Bravver E. eP01.06.02, S171
Brazzo K. eP03.06.03, S261
Breininger K. PS02.04, S94
Bresolin N. eP01.02.01, S142
Briani C. SS15.02, S75
Brice A. eP01.01.05, S136
Bril V. SS20.03, S82, PS05.01, S106, SS22.01, S85
Brock M. eP03.04.03, S241
Brooks C. eP02.04.03, S196
Broomfield J. PS01.3, S89
Broschwitz R. eP02.01.09, S183
Brown K. PS03.02, S97
Brunkhorst R. eP02.01.09, S183, PS08.03, S123
Brusa C. PS01.04, S89
Brusse E. PS05.03, S109, PS05.04, S110
Brännström T. eP04.03.01, S275
Budillon A. eP02.05.04, S203
Budzianowska A. eP03.04.07, S244
Buj-Bello A. eP04.05.02, S291
Bulgart H, PS07.02, S118
Bullivant J. eP04.05.02, S291
Bull S. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Burlot L. eP03.02.11, S230
Burnyte B. eP01.06.03, S172
Buscemi L. eP01.04.01, S156
Butler-Browne G. eP04.03.03, S277
Byloos K. eP01.06.01, S170
Byrne B. PS01.01, S87, PS03.02, S97
Bähr F. eP02.01.09, S183
Bäumler P. eP02.06.04, S211
Bönnemann C. eP04.04.01, S282, eP04.05.01, S291, eP04.05.02, S291, PS03.06, S100
Caballero-Ávila M. PS04.05, S105
Caballero J. eP01.03.07, S151
Caccin P. PS09.03, S128
Callaghan B. PS04.02, S103
Camacho A. eP01.06.06, S174
Campbell C. eP02.04.07, S198
Camuzat A. PS02.2, S92
Cancho-Candela R. eP01.06.06, S174
Cannon S. SS01.02, S58
Cantó-Santos J. eP01.05.06, S168
Cao M. PS05.06, S111
Capelle C. eP03.05.01, S248
Cardona D. eP03.02.07, S228
Carlier P. eP02.01.08, S182, eP02.01.10, S184
Carlier R. eP02.01.08, S182
Carotti M. PS09.03, S128
Carrera-García L. eP01.06.06, S174
Carrera L. eP03.03.03, S232, eP04.05.04, S293
Carrillo Padilla F. eP03.03.09, S237
Carruthers I. eP03.03.01, S230
Carstensen T. PS07.04, S120
Carstens P. PS01.06, S91
Casasnovas Pons C. eP02.04.04, S197
Castaño Herrera L. eP03.02.03, S224
Castellar Leones S. eP03.02.03, S224
Castellar S. eP01.06.04, S173, eP03.03.02, S231eP03.06.05, S263, eP04.02.07, S275, eP04.03.06, S279, eP04.03.09, S281
Castiglione V. eP02.05.09, S207
Castro D. eP02.05.10, S208, PS01.02, S87
Castro S. eP02.05.10, S208
Catalina Alvarez I. eP03.03.11, S238, eP01.05.08, S169, eP04.05.09, S296
Catalli C. eP01.01.08, S139
Cattagni T. eP03.01.06, S219
Catteruccia M. eP03.01.04, S218
Cavagna L. eP01.05.04, S166
Cavalcante E. eP01.03.05, S150
Cavalcanti F. PS08.03, S123
Cavaletti G. SS16.01, S76
Cazzaniga S. eP04.04.03, S284
Cediel M. eP01.06.04, S173
Celis L. eP04.05.07, S295
Cerletti M. eP01.06.10, S176
Cesarone E. eP02.05.04, S203
Chaabouni M. eP02.04.09, S200
Chabrol B. eP03.05.04, S252
Chacin-Suarez A. eP03.06.06, S263
Chae J. eP02.03.06, S195, eP03.05.09, S257
Chahal A. eP03.06.06, S263
Chakrabarty B. eP01.01.09, S139, eP01.05.07, S168
Chamberlain J. PS03.03, S98
Chamova T. eP04.03.05, S278
Chandrasekaran K. eP01.03.01, S147, PS08.06, S126
Chanson J. eP03.05.08, S256
Chao C. eP01.01.10, S140
Chapin C.C. PS03.06, S100
Charbonnier F. PS06.03, S114
Chavez-Keatts M. eP04.03.08, S280
Chen P. eP01.01.10, S140
Chen Y. eP03.04.09, S246
Chevalier A. eP03.02.11, S230
Chiappini R. eP01.03.06, S151
Childs A. eP04.01.02, S267, PS01.02, S87
Chin R. PS06.02, S113
Chiò A. eP04.03.10, S281
Choi J. eP01.03.01, S147, PS08.06, S126
Choi K. eP04.04.09, S289
Choi S. eP01.06.09, S176, eP04.03.04, S278
Choi Y. eP01.06.07, S174, eP03.01.01, S216, eP04.05.05, S294
Christiaens A. eP01.03.08, S152
Christopher L. TC01.03, S7
Chroni E. eP02.01.03, S179, eP02.02.04, S187
Chudenkova M. eP04.06.08, S304
Church K. PS03.01, S97, PS03.04, S99
Chyung Y. eP02.03.03, S193
Ciafaloni E. PS07.04, S120
Cianci V. eP01.04.09, S162
Cinnante C. eP04.04.03, S284
Ciscato P. eP02.04.08, S199
Ciutad M. eP04.05.04, S293
Claeys K. eP01.06.01, S170, eP03.04.08, S245, eP04.02.05, S273, PS05.02, S108, PS08.03, S123, SS07.01, S65
Clarke A. OS05.01, S32
Clary C. PS03.02, S97
Cleary Y. PS06.05, S115
Clemens P. PS01.01, S87, PS01.02, S87
Coarelli G. eP01.01.05, S136
Coats J. eP04.04.01, S282, eP04.05.01, S291
Codina A. eP01.06.06, S174, eP03.03.03, S232, eP04.05.04, S293
Coelho J. eP01.01.03, S134, eP02.06.08, S215
Coelho T. SS17.02, S77
Cohen-Adad J. PS02.2, S92
Cohen- Shelly M. eP03.06.06, S263
Coi A. PS02.06, S96
Coirault C. PS09.05, S130
Colen – de Koning J. eP03.06.07, S264
Collet R. PS02.03, S93
Colliot O. PS02.2, S92
Colomer J. eP03.03.03, S232
Comi G. eP01.02.01, S142, eP02.04.08, S199, eP04.04.03, S284
Connolly A. PS01.01, S87
Conte R. eP03.01.11, S222
Corcia P. eP04.03.10, S281
Coronel O. eP04.05.04, S293
Correa Arrieta C. eP03.02.03, S224
Correa C. eP01.06.04, S173, eP03.06.05, S263, eP04.02.07, S275, eP04.03.06, S279, eP04.03.09, S281
Cortese A. PS08.03, S123
Corti S. eP01.02.01, S142
Cosemans G. eP04.06.05, S302
Cossins J. PS05.06, S111
Costa A. eP02.06.05, S211
Costa Comellas L. eP04.05.03, S293
Costa L. eP01.03.07, S151
Costaridou L. eP02.01.03, S179
Cosyns M. eP04.01.01, S266
Cottin S. PS06.03, S114
Crawford T. eP02.03.03, S193, PS06.02, S113
Crescimanno G. eP01.04.09, S162
Crespo Rodríguez M. eP03.03.09, S237
Crudele J. PS03.03, S98
Cudkowicz M. eP04.03.10, S281
Cuervo J. eP03.02.07, S228
Cumbo F. eP03.01.04, S218
Curro R. PS08.03, S123
Cutrera R. OS03.03, S29
Cutter G. eP03.04.01, S239
Dabaj I. eP02.03.01, S191
Dabbous O. eP02.03.02, S192
Dabilgou A. eP04.02.03, S271
da Cruz e Silva O. eP02.06.03, S210
Dahlhaus I. PS07.06, S121
Daigl M. PS06.06, S116
Dalla Barba F. PS09.03, S128
Damian M. OS08.03, S37, WS04.03, S47
Damsker J. PS01.02, S87
Dangouloff T. SS12.02, S72, SS12.03, S73
Dang U. PS01.01, S87, PS01.02, S87
Danko V. PS02.04, S94
Danzi M. eP01.02.03, S143
Dark E. eP02.04.03, S196
Daron A. eP01.02.06, S145, SS12.02, S72
Darras B. eP02.03.03, S193, eP03.03.01, S230
Darton E. eP02.05.01, S201, eP02.05.08, S206, PS03.01, S97
Da Silva Authier T. PS08.02, S122
Davies M. PS09.04, S129
Davis J. eP02.06.01, S208, eP04.04.02, S284
Dawood T. PS04.02, S103
Day J. eP01.06.02, S171, eP02.03.03, S193, PS06.01, S112
Day J W. eP02.05.08, S206
Day L. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Dean R. eP02.03.02, S192
Debackere E. PS07.03, S119
De Bleecker J. eP01.06.01, S170, eP03.02.06, S226, eP03.04.04, S242, eP04.02.05, S273, eP04.05.08, S296, eP04.06.05, S302, PS07.03, S119, SS20.03, S82, TC03.04, S12, WS04.01, S46
de Borgie C. eP03.06.07, S264
De Braekeleer K. eP04.01.01, S266
Deconinck N. eP01.03.08, S152, eP04.01.01, S266, eP04.06.04, S301, PS01.02, S87, PS06.01, S112, SS10.03, S69, SS12.02, S72
Deconinck T. eP01.01.05, S136
de Coupade C. eP03.02.11, S230
Deev R. eP02.01.10, S184
de Greef B. PS04.05, S105
de Groot I. PS01.02, S87
de Groot L. eP01.04.03, S158
Dehache L. eP02.03.01, S191
De Jonghe P. eP01.01.05, S136
de la Borderie G. eP03.04.03, S241
de la Osa A. eP01.06.06, S174
Del Bo R. eP01.02.01, S142
Della Marina A. PS05.05, S110, PS07.06, S121
Delstanche S. eP01.02.06, S145, eP01.04.01, S156, eP02.02.01, S185, eP04.02.05, S273, SS12.02, S72
De Luca A. eP03.01.02, S217
Del Vecchio Blanco F. eP04.06.01, S299
De Meulemeester E. eP04.01.01, S266
De Michele G. eP01.01.05, S136
Demortier F. eP01.04.01, S156
den Hollander A. eP04.05.02, S291
De Paepe B. eP04.06.05, S302, PS07.03, S119
Depienne C. eP01.01.01, S133
De Puydt J. eP04.05.08, S296
De Riso G. eP04.06.01, S299
Dermaut B. PS07.03, S119
Desai R. eP03.04.05, S242
Desaphy J.-F. SS01.01, S58
Desguerre I. eP03.03.06, S234
De Shaepdryver M. PS02.01, S92
Desjardins C. eP04.04.02, S284
Desmyter L. eP03.05.10, S257, eP04.06.07, S303
de Visser M. eP01.05.05, S167, eP03.06.07, S264, OS04.01, S29, OS08.02, S36, TC08.02, S18
De Vivo D. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234, PS06.02, S113
Devleesschauwer B. eP04.01.01, S266
De Vos E. eP01.03.08, S152
de Vries N. eP01.05.02, S164
De Waele L. eP04.01.01, S266, eP04.01.03, S268, eP04.01.04, S268, OS01.02, S24, PS01.02, S87
De Wel B. eP01.06.01, S170
Dewilde S. eP03.04.06, S243, eP03.04.08, S245, PS05.02, S108
De Winter J. eP01.01.05, S136
Dhall A. eP01.01.07, S138, eP01.01.09, S139, eP01.05.07, S168
Dhiman N. eP01.01.07, S138, eP01.01.09, S139
Diaz-manera J. PS02.03, S93
Diaz J. eP04.02.07, S275
Dikoglu E. PS03.06, S100
Dilda P. PS06.03, S114
Dimachkie M. eP01.04.07, S162, PS07.04, S120, WS11.02, S53
Dimitri-Boulos D. eP03.05.08, S256
Diodato D. eP03.01.04, S218
Ditters I. eP03.05.02, S250, eP03.05.04, S252
Dittmayer C. PS07.01, S117, PS07.06, S121
Djordjevic G. eP01.04.06, S161
Dogu O. eP01.01.05, S136
Dohrn M. eP01.02.03, S143, eP02.01.09, S183, PS08.01, S122, PS08.03, S123, PS08.05, S125
Dominguez R. eP01.03.07, S151
Domínguez-Carral J. eP01.06.06, S174
Domínguez-González C. eP01.06.06, S174
Donachie J. eP02.06.06, S213
Donati M. OS02.02, S26
Donisa Dreghici R. PS03.02, S97
Doorduin J. HO01.04, S3
Doorenweerd N. eP04.01.04, S268
Dorban S. eP01.04.08, S162
Dorchies O. eP02.05.05, S204, eP02.05.06, S204, eP02.05.07, S205
Dorenbaum A. PS09.04, S129
Dosne A. eP03.04.02, S240
Douillard C. eP03.05.08, S256
Dowling J. eP04.05.01, S291, eP04.05.02, S291, PS03.06, S100
Drory V. TC05.04, S14
Druzdz A. PS05.01, S106
Druzhinina E. eP02.01.06, S181
Druzhinin D. eP02.01.06, S181
Dubey D. SS16.02, S77
Dubois C. eP01.04.01, S156
Dubois F. eP03.01.06, S219
Dubuisson N. PS04.06, S106
Duchesne E. eP03.06.08, S265
Duclos M. eP01.04.01, S156
Duda P. eP03.04.03, S241
Dullin C. eP03.01.07, S220, eP02.01.09, S183
Dunn T, SS08.03, S67
Duong P. eP01.02.07, S145
Duong T. eP03.03.01, S230
Dupont P. eP01.06.01, S170
Durr A. eP01.01.05, S136
Dyck P. eP02.02.08, S189, SS13.03, S73, SS19.03, S80
Díaz-Manera J. eP01.06.02, S171
Díaz-Marín C. eP03.06.01, S259
Díaz Díaz A. eP04.02.02, S271
D’Amico A. eP03.01.04, S218, eP04.05.03, S293, SS12.02, S72, eP01.02.01, S142, eP01.02.07, S145
D’Hondt A. eP01.06.01, S170
East L. eP04.06.04, S301
Eaton S. eP01.03.03, S149
Eftimov F. eP01.04.03, S158, eP01.05.02, S164, eP03.06.07, S264, PS04.1, S101, PS05.03, S109, PS05.04, S110
Eid S. SS14.01, S74
Elafros M. PS04.02, S103
Elands S. eP03.05.10, S257
Ellis M. eP01.03.11, S156
Emdin M. eP02.05.09, S207
Erguig L. eP03.03.10, S238
Ertan S. eP01.01.05, S136
Ervilha Pereira P. PS07.03, S119
Espinosa Trujillo A. eP03.03.07, S236
Esteller D. PS02.03, S93
Eurelings M. PS04.1, S101
Evangeliou A. eP04.04.06, S287
Evangelista T. HO02.03, S5
Evans R. PS06.06, S116
Evoli A. PL04.03, S23
Exposito Escudero J. eP04.05.04, S293
Exposito J. eP01.03.02, S148, eP01.03.07, S151
Expósito-Escudero J. eP01.06.06, S174
Expósito J. eP03.03.03, S232
Eymard B. TC07.02, S16
Faber C. PS04.05, S105
Fanucci L. OS02.02, S26
Farhat E. eP02.04.09, S200
Farina A. eP02.05.04, S203
Farmakidis C. eP01.04.07, S162
Farrar M. PS06.04, S114
Faruq M. eP01.01.07, S138, eP01.01.09, S139
Fattori B. OS02.02, S26
Fattori F. eP04.05.03, S293
Faulkner E. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Federle A. PS02.04, S94
Feely S. PS08.01, S122
Feldman E. PL02.03, S21, PS04.02, S103 , SS09.01, S67, SS14.01, S74
Feresiadou A. eP03.04.07, S244
Fernández-Costa J. PS09.01, S127
Fernández C. eP04.05.04, S293
Ferreira R. eP01.01.03, S134, eP02.06.08, S215
Finkel R. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234, PS01.02, S87, PS06.02, S113, PS06.04, S114, SS10.02, S69
Finsterer J. eP01.04.04, S160, eP03.06.09, S266
Fischetto R. eP01.04.09, S162
Fisher C. PS06.04, S114
Fitzsimmons S. eP01.03.05, S150
Fiumara A. eP01.04.09, S162
Flössel U. eP01.05.03, S165
Foley A. eP04.04.01, S282, eP04.05.01, S291
Foley A.N. PS03.06, S100
Fontaine B. SS01.03, S59
Fontanelli L. eP01.03.06, S151, eP03.03.08, S236
Forsberg K. eP04.03.01, S275
Fortin A. eP03.06.08, S265
Frahm J. PS01.06, S91
Franke R. eP03.02.04, S225
Frederix G. PS04.1, S101
Freeman R. SS19.2, S80
Freimer M. PS07.04, S120
Friede T. PS01.06, S91
Friedman P. eP03.06.06, S263
Frijia F. PS02.06, S96
Frontistis A. eP01.02.09, S147
Fülöp K. eP03.01.10, S222
Fürst D. eP01.01.01, S133
Gadaleta G. OS02.03, S27
Gagne A. PS04.02, S103
Gagnon C. eP03.06.08, S265
Gailite L. eP02.02.07, S188
Gajate-García V. eP04.02.06, S274, PS08.04, S125
Galanakis E. eP04.04.06, S287
Galán-Dávila L. eP04.02.06, S274, PS08.04, S125
Ganassi M. SS02.01, S59
Gangfuß A. eP01.01.01, S133
Garcia A. eP01.06.04, S173
García-Cazorla A. eP01.06.06, S174
García-García F. eP01.05.06, S168
García Pazos O. eP03.03.07, S236
Gargouri A. eP01.01.10, S141, eP02.02.03, S186
Garrabou G. eP01.05.06, S168
Gasperini S. eP01.04.09, S162
Gavriilaki M. eP03.02.02, S224
Gayfieva M. PS05.01, S106
Gayi E. eP02.05.05, S204, eP02.05.07, S205
Geerts M. PS04.05, S105
Gelderman K. eP01.04.03, S158
Gelinas D. eP03.04.04, S242, eP03.04.06, S243
Genge A. eP03.04.03, S241
Genovese F. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Gentile A. PS09.02, S128
Gerber M. eP03.03.01, S230, PS06.01, S112, PS06.04, S114, PS06.05, S115
Geuens S. eP04.01.03, S268, eP04.01.04, S268
Gharbi A. eP01.01.10, S141, eP02.02.03, S186
Ghysels S. eP01.06.01, S170
Giannoni A. eP02.05.09, S207
Gibson S. PS07.04, S120
Gilhus N. TC03.03, S11
Giraldo Castro H. eP02.05.10, S208
Giraldo G. eP03.03.04, S233
Glaubitz S. eP01.05.03, S165
Glāzere I. eP02.02.07, S188
Goebel D. eP01.06.02, S171
Goebel H. eP03.06.02, S260, PS07.06, S121, PS04.06, S106
Goedee S. PS05.03, S109, PS05.04, S110
Goemans N. eP04.01.03, S268, eP04.01.04, S268, PS01.02, S87, PS06.01, S112
Gomez-Andres D. eP01.03.07, S151
Gomez Garcia De La Banda M. eP02.03.01, S191
Gonzales M. eP01.01.01, S133
Gonzalez P. PS03.02, S97
González Toledo G. eP03.03.09, S237
Gooch J. eP01.03.03, S149
Goosens V. eP01.06.01, S170
Gordish-Dressman H. PS01.01, S87
Gorni K. PS01.01, S87, PS06.01, S112, PS06.04, S114, PS06.06, S116
Gouider R. eP01.01.10, S141, eP04.03.02, S276, eP04.03.07, S279, RG.03, S56
Goutman S. SS09.01, S67
Govoni A. eP01.03.06, S151, eP02.05.09, S207
Graham A. eP04.05.10, S298
Graham R. eP04.05.02, S291
Grau-Junyent J. eP01.05.06, S168
Gray A. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Gray G. eP01.01.02, S134
Greene E. eP03.04.01, S239
Gregory S. PS01.3, S89
Gremiakova O. eP04.01.06, S270
Gremiakova T. eP04.01.06, S270
Grey M. SS06.03, S64
Griffin D.A. eP02.05.01, S201, PS03.01, S97
Griffin D. PS03.04, S99
Griggs R. PS01.3, S89
Grimaldi Capitello T. eP03.01.04, S218
Grimm A. PS08.05, S125
Grimsey P. PS06.05, S115
Grishina D. eP03.02.05, S226
Grittner U. PS05.05, S110
Grosskreutz J. PS05.01, S106
Grosz B. eP01.03.11, S156
Grosz Z. OS02.04, S28
Grutters J. eP03.02.09, S229
Guedira G. eP04.06.03, S300
Guerrero-Peral Á. eP04.02.06, S274, PS08.04, S125
Guerrero-Sola A. eP04.02.06, S274, PS08.04, S125
Guglieri M. eP02.04.07, S198, eP04.01.02, S267, PS01.01, S87, PS01.02, S87, PS01.3, S89, PS02.03, S93
Guglietta A. SS20.03, S82
Guijarro Del Amo M. eP03.03.07, S236
Guillot-Noël L. eP01.01.05, S136
Guillou H. eP03.02.11, S230
Gulati S. eP01.01.09, S139, eP01.05.07, S168
Gunnarsson M. eP03.04.07, S244
Guo K. SS14.01, S74
Guptill J. eP03.04.05, S242
Guridi M. eP02.05.01, S201, eP02.05.08, S206
Gutiérrez-Gutiérrez G. eP04.02.06, S274, PS08.04, S125
Gutiérrez Martínez A. eP04.02.02, S271
Gál A. eP04.06.02, S300
Gómez H. eP01.06.06, S174
Gómez Mazuera A. eP01.04.02, S157
Götze R. eP01.05.01, S163, eP01.05.03, S165
Haack T. eP01.01.05, S136
Haberlova J. PS01.02, S87
Habib A. PS05.01, S106
Hadden R. TC02.01, S8
Hagenacker T. PS05.05, S110
Hahn A. eP01.01.01, S133, eP03.05.04, S252, PS07.06, S121
Hahn K. eP03.06.02, S260, PS08.03, S123
Hale J. PS03.5, S99
Hall J. eP04.04.02, S284
Ham A. WS12.03, S55
Hamann D. eP01.04.03, S158
Ham D. WS12.03, S55
Hamilton M. eP02.06.06, S213
Hammans S. PS08.03, S123
Han C. eP04.04.01, S282, eP04.05.01, S291
Han H. eP03.01.01, S216
Han J. eP01.06.09, S176, eP03.04.10, S247
Hanna M. PS07.04, S120
Harper A. eP01.06.05, S173, PS01.01, S87, PS01.02, S87
Harter P. eP01.05.03, S165
Hartley L. eP02.02.02, S185
Haselkorn T. eP04.05.01, S291
Hasselmann O. eP02.03.05, S194
Hathazi D. eP04.06.08, S304
Hauschka S. PS03.03, S98
Hawe E. eP02.02.02, S185
Hawkins N. PS06.06, S116
Hedayat A. eP01.03.01, S147, PS08.06, S126, eP01.06.06, S174
Heher P. SS02.01, S59
Heide S. eP01.05.03, S165
Heil L. eP01.01.01, S133
Heim A. eP01.04.07, S162, PS07.04, S120
Helbing W. eP03.05.01, S248
Hell A. eP01.05.01, S163
Hemke R. eP01.05.05, S167
Henricson E. PS01.01, S87
Hentschel A. eP01.01.01, S133, eP03.06.02, S260, PS07.01, S117
Herbelin L. PS07.04, S120
Herdeiro M, eP02.06.03, S210
Herdeiro T. eP02.06.05, S211
Herdewyn S. eP04.05.08, S296
Hermann D. PS08.03, S123
Hernandez-Lain A. eP01.06.06, S174
Hernández García M. eP03.03.09, S237
Hernández Javier C. eP03.03.09, S237
Herzog R. eP01.01.05, S136
Hewamadduma C. eP02.06.06, S213
Higgs K. eP01.04.07, S162
Hildebrand S. eP02.06.01, S208
Hiligsmann M. SS12.02, S72
Hilsden H. eP01.06.02, S171
Hitomi S. eP01.04.10, S163
Hleihel W. PS09.05, S130
Hoeijmakers J. PS04.05, S105
Hofer S. PS01.06, S91
Hoffman E. PS01.02, S87
Hoffmann S. PS05.05, S110
Hogan M. PS03.01, S97
Hogrel J. eP02.04.05, S197
Ho K. SS06.03, S64
Holst Axelsen K. eP02.04.02, S196
Homedes Pedret C. eP02.04.04, S197
Hong Y. eP01.06.09, S176, eP03.04.10, S247, eP04.03.04, S278
Hopp A. PS03.06, S100
Horber V. eP02.01.01, S177
Horga A. eP04.02.06, S274, PS08.04, S125
Horrocks I. PS01.02, S87
Horvath R. eP04.06.08, S304, SS21.01, S83, WS06.02, S49
Houlden H. PS08.03, S123
Houston P. PS03.02, S97
Houwen S. OS11.02, S40
Howard Jr J. eP03.04.01, S239, eP03.04.03, S241, eP03.04.09, S246, SS20.03, S82
Howard S. eP02.03.03, S193
Howells R. eP02.03.02, S192
Hsu E. PS03.06, S100
Hsueh H. eP01.01.10, S140
Huddar A. eP02.06.07, S214, PS01.05, S90, PS07.05, S120
Huidekoper H. eP03.05.02, S250, eP03.05.04, S252
Hu L. eP02.05.08, S206, PS03.01, S97
Hur J. SS14.01, S74
Hussain Y. eP03.04.03, S241
Hustinx M. eP01.02.06, S145
Hutchaleelaha A. eP01.06.05, S173
Huysmans L. eP01.06.01, S170
Hwang S. eP02.04.03, S196
Hwu W. PS06.02, S113
Iammarino M.A. PS03.01, S97, PS03.04, S99
Iannaccone S. PS01.01, S87
Iikuni N. eP01.05.04, S166
Ilinca A. eP03.04.07, S244
Imen K. RG.03, S56
Irnich D. eP02.06.04, S211
Isabelle L. PS02.2, S92
Isaev A. eP02.01.10, S184
Isapof A. eP04.04.05, S286
Ishiba R. eP04.06.10, S305
Ishii A. eP01.04.10, S163
Ismail H. eP04.06.03, S300
Ivanovic V. eP01.05.09, S170, eP02.06.02, S209
Iyer S. eP02.04.03, S196
J. Dowling J. eP04.04.01, S282
Jabari D. eP01.04.07, S162
Jabre S. PS09.05, S130
Jacinto J. PS09.02, S128
Jackson S. eP03.01.08, S220
Jacobi C. PS05.05, S110
Jacob S. eP03.04.08, S245, PS05.02, S108
Jacobs B. PL03.02, S21
James M. eP01.06.02, S171, eP03.01.03, S217, eP03.01.05, S218, PS01.02, S87
Janisch M. OS04.03, S31
Janssen B. eP03.04.06, S243
Janssens L. eP04.05.08, S296
Janssens S. PS07.03, S119
Japalaghi O. eP03.05.07, S255
Jassal B. eP01.01.07, S138, eP01.01.09, S139, eP01.05.07, S168
Jauregi-Barrutia A. eP01.01.08, S139
Jawdat O. eP01.04.07, S162
Jayaseelan D. eP04.04.04, S285, eP04.06.06, S302
Jelassi C. eP04.03.02, S276
Jimenez-Mallebrera C. eP01.06.06, S174, eP03.03.03, S232
Johansson R. eP03.04.07, S244
Johnson A. eP04.01.02, S267
Johnson N. eP01.03.03, S149, eP03.06.03, S261, PS06.02, S113
Jokela M. SS03.03, S62
Jones C. PS03.06, S100
Jones K. eP01.06.02, S171
Jones S. PS07.04, S120
Jons D. eP03.04.07, S244
Jou C. eP01.06.06, S174, eP04.05.04, S293
Jovin Z. eP02.04.01, S195
Juarez T. eP04.05.07, S295
Juliá N. eP01.06.06, S174
Jungbluth H. eP04.05.02, S291
Justel M. eP01.06.06, S174
Jørgensen A. PS07.04, S120
Jüngert J. PS02.04, S94
Kably B. eP01.02.05, S144, eP03.03.10, S238
Kabore B. eP04.02.03, S271
Kacem I. eP01.01.10, S141, eP04.03.02, S276, eP04.03.07, S279
Kacem I Gouider R. eP02.02.03, S186
Kaiser J. PS05.05, S110
Kalcev G. eP04.04.11, S290
Kaldawi S. eP02.02.09, S189
Kalischewski P. PS05.05, S110
Kaminski H. eP03.04.03, S241, eP03.04.09, S246, PS05.01, S106
Kamperman R. eP01.04.03, S158, eP01.05.05, S167, eP03.06.07, S264
Kangas N. eP01.03.03, S149
Kang S. eP03.02.08, S228, eP04.06.09, S304
Kan H. eP04.01.04, S268, eP04.04.03, S284
Kapa S. eP03.06.06, S263
Kapetanovic-Garcia S. eP01.01.08, S139
Kapto O. eP04.02.03, S271
Karcagi V. eP04.06.02, S300
Karcher K. eP03.04.02, S240
Kasiborski F. PS04.03, S103
Katsalouli M. PS01.02, S87
Kaufman G. eP04.04.04, S285, eP04.04.05, S286
Kazmaier L. eP01.05.01, S163
Kefalopoulou Z. eP02.02.04, S187
Kelley K. eP01.06.05, S173
Kenina V. eP02.02.07, S188
Kennerson M. eP01.03.11, S156, PS08.03, S123
Kertesz N. eP02.03.03, S193
Kierdaszuk B. eP01.04.05, S160
Kiernan M. eP01.03.11, S156, PL02.01, S20
Kim A. eP02.03.06, S195
Kim B. eP04.04.09, S289
Kimiskidis V. eP01.02.09, S147, eP03.02.02, S224
Kim J. eP04.03.04, S278, eP04.03.08, S280
Kintos V. eP02.02.04, S187
Kirschner J. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234, PS06.01, S112, PS06.02, S113
Kjaer Andersen R. eP02.04.02, S196, eP03.04.11, S248
Klareskog L. eP03.04.07, S244
Klebe S. eP01.01.05, S136
Kleefeld F. eP03.06.02, S260
Klein A. eP02.03.05, S194
Kleiner D. PS03.06, S100
Klein K. eP01.01.05, S136
Kleopa K. SS18.03, S79
Klett D. PS02.04, S94
Kletzl H. eP03.03.01, S230, PS06.04, S114, PS06.05, S115
Klopstock T. eP01.01.05, S136
Knieling F. PS02.04, S94
Knop K. PS05.05, S110
Kockum I. eP03.04.07, S244
Koczok K. eP04.06.02, S300
Koelman J. eP01.05.05, S167
Kohlschmidt N. eP01.01.01, S133
Kokosali E. eP04.04.06, S287
Kok R. eP03.05.01, S248
Kok W. eP01.04.03, S158
Kommerell I. PS01.06, S91
Kononets O. eP04.01.05, S269
Kools J. SS02.03, S61
Koopman L. eP03.05.01, S248
Korideck H. eP03.04.01, S239
Kostera-Pruszczyk A. eP01.04.05, S160, PS06.01, S112
Kovalchuk M. eP02.01.06, S181, eP02.01.06, S181
Krag T.O. eP03.05.11, S258
Krasinski D. PS06.02, S113
Kroon R. PS01.06, S91
Krueger A. eP03.04.05, S242
Kruijshaar M. eP03.05.03, S251, eP03.05.04, S252
Kruja J. eP04.02.04, S272
Kuepper H. eP02.01.01, S177
Kuijpers T. PS05.03, S109, PS05.04, S110
Kulanthaivelu K. PS07.05, S120, SS20.02, S81
Kumar P. PS01.05, S90
Kumar U. eP01.05.07, S168
Kumar V. eP02.06.07, S214
Kummer K. eP01.05.01, S163, eP01.05.03, S165
Kummer L. PS05.03, S109, PS05.04, S110
Kummer S. eP01.05.01, S163
Kuntz N.L. eP04.04.01, S282, eP04.05.01, S291, PS01.01, S87, PS01.02, S87, PS03.06, S100
Kuqo A. eP04.02.04, S272
Kurihara Y. eP01.04.10, S163
Kurjane N. eP02.02.07, S188
Kvalsund M. PS04.02, S103
Kwon S. eP02.03.06, S195
Kwon Y. eP03.04.10, S247
Kyelem J. eP04.02.03, S271
Kågström V. eP03.04.07, S244
Labarthe F. eP03.05.04, S252
Lafont R. PS06.03, S114
Laforet P. eP03.01.06, S219, eP03.05.08, S256, WS02.02, S44, WS05.02, S49
Laing N. OS05.03, S33
Lakshman N. eP04.04.01, S282
LaMarca N. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Lambert E. eP03.04.04, S242
Lammens M. PS07.03, S119
Lanens D. PS08.02, S122
Langeslay R. eP01.06.05, S173
Lang S. PS07.01, S117
Larkin M. eP01.02.02, S142, eP01.02.04, S144
Larkin M. eP01.02.08, S146
La Serna J. eP04.05.07, S295
Latil M. PS06.03, S114
Latsoudis H. eP04.04.06, S287
Laurent J. eP03.06.03, S261
Lavrnic D. eP01.05.09, S170
Lawlor M.B. PS03.06, S100
Lawrence J. PS03.02, S97
Lazarte C. eP04.05.07, S295
LeBlanc P. PS07.03, S119
Lee D. PS03.03, S98
Lee H. eP01.06.07, S174
Lee J. eP01.06.09, S176, eP02.03.06, S195, eP03.04.10, S247
Lee N. eP01.01.10, S140
Lee S. eP01.06.07, S174, eP03.01.01, S216, eP04.05.05, S294
Lee Y. eP02.03.06, S195, PS08.01, S122
Lehman K. eP02.05.01, S201, PS03.01, S97, PS03.04, S99
Leinonen M. PS01.02, S87
Leite M. eP03.04.03, S241, SS22.03, S85
Lemiere J. eP04.01.04, S268
Lemmen D. PS04.06, S106
Lennox A. eP04.05.02, S291
Leonardis L. OS10.02, S39
Leone D. eP04.05.03, S293
Leu J. eP03.04.02, S240
Levine T. PS07.04, S120
Lewis S. eP02.05.01, S201, PS03.01, S97, PS03.04, S99
Lievens I. eP02.02.01, S185
Li F. eP03.04.05, S242
Lika A. eP03.05.03, S251
Lim J. eP01.05.02, S164
Ling L. eP03.04.02, S240
Lin S. WS01.03, S43, WS12.03, S55
Liouta E. eP01.02.09, S147, eP03.02.02, S224
Lipowska M. eP01.04.05, S160
Liu B. eP03.04.05, S242
Liu W. PS05.06, S111
Li X. PS03.04, S99
Li Z. eP03.02.04, S225
Ljubicic V. eP02.06.09, S215
Lleixà C. PS04.05, S105
Llewellyn S. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Lloyd T. PS07.04, S120
Lochmuller H. eP02.06.09, S215, OS06.01, S34, PS02.04, S94, SS06.03, S64, SS21.03, S84, TC07.03, S17
Lohmann K. eP01.01.05, S136
Lopes Abath Neto O. PS03.06, S100
Lopez-de Muniain A. eP01.06.06, S174
Lopez-Jimenez F. eP03.06.06, S263
Lopez-Lobato M. eP01.03.07, S151
Lorenz H. eP01.05.01, S163
Lowes L.P. PS03.01, S97, PS03.04, S99
Lozano Ros A. eP01.05.08, S169, eP04.05.09, S296
Lucia A. eP03.05.11, S258
Luckin J. eP04.06.06, S302
Ludolph A. eP04.03.10, S281
Lundin F. eP03.04.07, S244
Lunt P. eP04.05.10, S298
Lurz E. PS03.06, S100
Luyckx K. eP04.01.03, S268
Lynch O. eP04.05.02, S291
López-Martínez A. eP01.01.08, S139
López García J. eP02.05.10, S208
López Muñoz S. eP01.05.08, S169
Löscher W. eP03.04.04, S242
Maas M. eP03.06.07, S264
Machado P. PS07.04, S120
Madden M. eP01.03.05, S150
Madrigal I. eP04.05.04, S293
Maertens A. eP01.02.06, S145
Maertens de Noordhout A. eP02.02.01, S185
Maes F. eP01.06.01, S170
Maggi L. eP01.04.09, S162
Maggi R. eP01.06.10, S176
Magri F. eP02.04.08, S199, eP04.04.03, S284
Mahajan A. PS06.06, S116
Mah J. PS01.01, S87, PS01.02, S87
Maier A. PS02.04, S94
Major Z. eP04.04.08, S288
Makepeace C. eP01.03.03, S149
Malandrini A. eP03.01.11, S222
Maldonado Slootjes S. eP01.04.08, S162
Malfatti E. eP03.06.04, S262, eP04.04.08, S288, eP04.05.07, S295
Malhotra J. eP02.05.01, S201, eP02.05.08, S206
Mamada N. eP01.04.10, S163
Manca L. eP01.03.06, S151
Manganelli F. PS08.03, S123
Manini A. eP01.02.01, S142
Mankodi A. PS03.5, S99
Manousakis G. PS01.01, S87
Manta A. eP02.06.09, S215
Mantegazza R. eP03.04.03, S241, eP03.04.04, S242, eP03.04.08, S245, PS05.01, S106, PS05.02, S108, SS20.03, S82
Manuel M. eP04.03.10, S281
Manzur A. eP02.05.03, S202, eP04.01.02, S267, PS01.04, S89
Marais T. eP04.03.03, S277
Marchand-Pauvert V. PS02.2, S92
Marco C. eP01.03.07, S151, eP03.03.03, S232
Marcus H. PS04.02, S103
Margeta M. PS03.06, S100
Marini-Bettolo C. eP01.03.05, S150, eP02.06.06, S213, eP03.01.03, S217, eP04.05.02, S291, eP04.05.10, S298, PS02.03, S93
Marini M. OS02.02, S26
Marques A. eP02.06.05, S211
Marrak S. eP02.04.09, S200
Marsden D. eP03.05.07, S255
Marteau T. PS07.01, S117
Martens W. PS01.3, S89
Martic V. eP02.04.01, S195
Martin C. eP03.03.01, S230, PS06.01, S112
Martinez-Salcedo E. eP01.03.07, S151
Martins da Silva A. PS04.04, S104
Martins F. eP02.06.03, S210
Martinuzzi A. PS02.06, S96
Marti P. eP01.01.06, S137
Martín-Aguilar L. PS04.05, S105
Martín A.M. eP03.05.11, S258
Martínez-Esteban P. eP01.03.10, S154, eP03.06.04, S262, eP04.05.07, S295
Martínez-Vicente L. eP04.02.06, S274, PS08.04, S125
Martí Y. PS06.06, S116
Marzouk B. eP03.03.10, S238
Mason J. eP03.01.03, S217
Mason S. eP02.05.01, S201, eP02.05.08, S206, PS03.01, S97
Masrori P. eP04.03.01, S275, PS02.01, S92
Massade L. SS18.02, S79
Mateus T. eP02.06.03, S210, eP02.06.05, S211
Mathews K. eP03.06.03, S261
Mathioudakis L. eP04.04.06, S287
Matthews E. eP04.05.10, S298
Mattos-Castillo J. eP01.03.10, S154
Matà S. eP03.01.11, S222
Matías-Guiu J. eP04.02.06, S274, PS08.04, S125
Maulet T. eP03.01.06, S219
Mavroudakis N. eP04.06.07, S303
Maxwell S. PS05.06, S111
Mayhew A. eP01.03.05, S150, eP01.06.02, S171, eP03.01.03, S217, eP03.01.05, S218, PS01.3, S89
Mazanec R. eP03.05.05, S253
Mazurová S. eP03.05.05, S253
Mazzone E. PS06.01, S112
McCallum M. eP03.01.03, S217
McCauley J. PS01.04, S89
McDermott C. eP04.03.10, S281
McDermott M. PS01.3, S89, PS07.04, S120
McDonald C. eP02.05.01, S201, eP02.05.08, S206, PS01.01, S87, PS01.02, S87, PS01.3, S89
McElhanon K PS07.02, S118
McLaughlin H. eP03.05.07, S255
McMacken G. SS06.03, S64
McMillan H. PS01.02, S87
Meadows A. PS03.04, S99
Medard L. eP01.04.01, S156
Medina-Inojosa J. eP03.06.06, S263
Medina J. eP03.03.03, S232
Meek B. eP01.04.03, S158
Mehrabyan A. eP02.04.03, S196
Mehta L. eP04.03.10, S281
Meisel A. eP03.04.08, S245, PS05.02, S108, PS05.05, S110, SS20.03, S82
Mellion M. eP04.05.11, S299
Melnik E. eP03.02.05, S226
Mendell J. eP01.06.02, S171, PS03.04, S99
Mendell J.R. eP02.05.01, S201, eP02.05.08, S206, PS03.01, S97
Menendez M. PS02.05, S95
Meng H.M. PS03.06, S100
Menier M. eP03.03.05, S234
Mensova L. eP03.05.05, S253
Merckx C. eP04.06.05, S302
Mercuri E. eP03.03.01, S230, eP03.03.05, S234, eP04.05.03, S293, eP04.06.04, S301, PS06.01, S112
Mergenthaler P. PS05.05, S110
Merico E. OS09.01, S37
Merkies I. PS04.05, S105
Mesa S. eP03.03.04, S233
Meyer S. eP01.05.01, S163
Meylemans A. PS07.03, S119
Meznaric M. eP03.05.06, S254
Miladi N. eP02.04.09, S200
Milisenda J. eP01.05.06, S168
Millan J. eP03.02.07, S228
Miller N. eP03.05.07, S255
Miller W. eP04.04.01, S282,eP04.05.01, S291, PS03.06, S100
Millogo A. eP04.02.03, S271
Milone M. eP03.06.06, S263
Mion M. eP03.01.09, S221
Mitchell S. eP02.02.02, S185
Mittelbronn M. eP01.05.03, S165
Mittendorfer B. PS09.04, S129
Moat D. eP03.01.03, S217, eP04.05.02, S291
Moens T. eP04.03.01, S275
Moggio M. eP01.02.01, S142
Mohammad S.A. PS03.06, S100
Mohassel P. SS08.03, S67
Moldovan O. eP02.06.08, S215
Molnar M. OS02.04, S28
Molnar V. OS02.04, S28
Molnár M. eP04.06.02, S300
Monduy M. PS01.02, S87
Monforte M. eP04.05.03, S293, PS02.03, S93
Mongini T. eP03.05.04, S252, OS02.03, S27
Montagnese F. eP02.06.04, S211
Montague-Johnson C. SS12.03, S73
Montanaro D. PS02.06, S96
Montes J. PS06.02, S113
Moore U. eP01.06.02, S171
Morales-Gonzales S. PS07.01, S117
Moran J. eP01.02.07, S145
Moreno-Lozano P. eP01.05.06, S168
Moreno A. eP01.03.07, S151
Moreno T. eP01.01.03, S134, eP02.06.08, S215
Mori-Yoshimura M. eP01.06.02, S171
Moris G. PS02.05, S95
Morkuniene A. eP01.06.03, S172
Morozova D. eP02.03.04, S193
Morris C. PS03.02, S97
Morris K. eP01.03.11, S156
Morrow J. eP03.03.01, S230, PS02.03, S93
Moschou M. eP01.02.09, S147, eP03.02.02, S224
Moss C. eP01.03.05, S150
Mouly V. eP04.03.03, S277
Moura J. PS04.04, S104
Moya O. eP03.03.03, S232
Mozaffar T. eP03.04.01, S239, PS07.04, S120
Mrabet S. eP01.01.10, S141, eP02.02.03, S186, eP04.03.02, S276, eP04.03.07, S279
Muelas N. eP01.01.06, S137
Mueller S.R. PS03.06, S100
Muley S. eP02.02.09, S189
Munell F. eP01.03.07, S151
Muni-Lofra R. eP01.03.05, S150, eP03.01.03, S217, eP04.05.10, S298
Munot P. PS01.04, S89
Muntoni F. eP01.03.05, S150, eP01.03.09, S153, eP02.05.03, S202, eP03.03.01, S230, eP03.03.05, S234, eP03.03.06, S234, eP04.01.02, S267, eP04.06.04, S301, PS01.04, S89
Muppidi S. eP03.04.01, S239
Murai H. SS20.03, S82
Murdock B. SS09.01, S67
Murphy L. eP01.03.05, S150, eP03.06.03, S261, eP01.01.02, S134
Murtagh M. PS03.06, S100
Musatova E. eP02.01.10, S184
Musso G. eP03.01.09, S221
Musumeci O. eP01.04.09, S162
Muñoz Blanco J. eP01.05.08, S169, eP03.03.11, S238, eP04.05.09, S296
Méndez-Dávalos C. eP01.03.10, S154
Múnera L. eP03.03.04, S233
Müller-Felber W. eP04.04.01, S282, eP04.05.01, S291, PS03.06, S100
Müller J. eP04.06.08, S304
Müller-York A. PS03.04, S99
Nadaj-Pakleza A. eP03.05.08, S256
Nair N. eP01.01.02, S134
Najim J. eP02.06.01, S208
Nalini A. eP02.01.07, S182, eP02.06.07, S214, PS07.05, S120, SS20.02, S81
Napoli L. eP01.02.01, S142
Napoli M. eP04.06.01, S299
Napon C. eP04.02.03, S271
Narayanappa G. eP02.06.07, S214, PS01.05, S90
Narayanaswami P. eP03.04.01, S239, eP03.04.05, S242, OS07.03, S35, TC03.01, S10, WS11.03, S54
Nascimento A. eP01.03.02, S148, eP01.03.07, S151, eP03.03.03, S232, eP04.05.04, S293, PS06.01, S112, eP01.06.06, S174
Nascimiento-Osorio A. PS01.02, S87
Nashi S. eP02.01.07, S182, eP02.06.07, S214, PS01.05, S90, PS07.05, S120, SS20.02, S81, eP01.01.10, S141, eP02.02.03, S186, eP04.03.02, S276, eP04.03.07, S279
Natera D. eP03.03.03, S232, eP04.05.04, S293
Natera De Benito D. eP01.06.06, S174
Nava S. eP04.04.03, S284
Nazari Hashemi P. PS06.03, S114
Neff L. eP02.05.05, S204, eP02.05.06, S204, eP02.05.07, S205
Nelson L. PS06.04, S114
Neuhaus S. PS03.04, S99
Neurath M. PS02.04, S94
Neves Cardoso M. PS04.04, S104
Nevmerzhitskaya K. eP02.03.04, S193
Nevo Y. PS01.02, S87
Nicita F. eP03.01.04, S218
Nicolaisen E. eP01.02.07, S145
Niewold I. eP01.05.02, S164
Nigro V. eP02.05.04, S203, eP04.06.01, S299
Niizawa G. PS01.01, S87
Nikolic L. eP02.06.02, S209
Niks E. eP04.01.04, S268, eP04.04.03, S284, PS01.02, S87
Nishimura A. eP01.01.01, S133
Nivelle E. PS05.02, S108
Nobie-Orazio E. PS04.03, S103
Nogales-Gadea G. eP01.01.08, S139
Nogara L. PS09.03, S128
Nolte-Buchholtz S . OS04.03, S31
Nomikos G. eP02.03.03, S193
Norwood F. eP04.05.10, S298
Notas K. eP03.02.02, S224
Notermans N. PS04.1, S101
Noureldein M. SS14.01, S74
Nowak R. eP03.04.01, S239
Nungo C. eP01.03.07, S151
Obermaier C. PS08.03, S123
Odom G. PS03.03, S98
Ogawa R. eP01.04.10, S163
Oh J. eP04.04.09, S289
Olive M. PS02.03, S93, SS03.01, S61
Oliver D. OS04.02, S30
Olivier P. eP03.02.06, S226
Olivé M. HO02.04, S5
Ollendorf D. eP03.04.06, S243
Olsson T. eP03.04.07, S244
Olthoff A. PS01.06, S91
Onofri A. OS03.03, S29
Onore M. eP04.06.01, S299
Oommen A. eP02.06.07, S214
Ormandzhiev S. eP04.03.05, S278
Orrell R. eP02.06.06, S213, eP04.05.10, S298
Ortez C. eP01.06.06, S174, eP03.03.03, S232, eP04.05.04, S293
Ortiz Corredor F. eP03.02.03, S224, eP03.06.05, S263, eP04.03.06, S279
Ortiz F. eP01.06.04, S173, eP03.03.02, S231, eP04.02.07, S275, eP04.03.09, S281
Oskoui M. PS06.01, S112
Osman N. eP02.03.03, S193
Ospitalieri S. eP04.03.01, S275
Ouaja R. eP03.02.11, S230, PS04.03, S103
Ouedraogo M. eP04.02.03, S271
Oulhissane Omar A. eP02.01.02, S178
O’Connor E. SS06.03, S64
O’Donnell E. eP04.04.02, S284
O’Neil D. SS06.03, S64
O’Neil J. eP02.03.03, S193
P. Lemos J. eP04.03.03, S277
Paci S. eP03.04.08, S245, PS05.02, S108
Paganoni S. eP04.03.10, S281
Palace J. eP03.04.08, S245, PS05.02, S108, SS22.02, S85
Palacios Mendoza M. eP04.05.09, S296
Palasti A. OS02.04, S28
Palfreeman L. PS06.04, S114
Palibrk A. eP01.05.09, S170, eP02.06.02, S209
Palmeri J. PS03.5, S99
Palvadeau R. eP01.01.05, S136
Pandey B. eP04.04.04, S285, eP04.04.05, S286
Pane M. eP04.05.03, S293
Paniga C. eP01.06.10, S176
Paoli X. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Paquay S. eP04.01.01, S266
Paradas C. eP01.06.02, S171
Paradis A. eP01.03.03, S149, PS06.02, S113
Pareja A. eP01.03.07, S151
Pareja I. eP03.03.04, S233
Parenti G. eP03.05.04, S252
Pareyson D. PS08.03, S123, SS15.01, S74, TC06.01, S15
Parini R. eP01.04.09, S162, eP03.05.04, S252
Park H. eP03.01.01, S216, eP04.05.05, S294
Park J. eP04.04.09, S289
Park K. eP01.06.07, S174
Parman Y. PS08.03, S123
Parmová O. eP03.05.05, S253
Pascual-Goñi E. PS04.05, S105
Pascual I. eP01.06.06, S174
Pascual S. eP01.03.07, S151
Pasnoor M. eP01.04.07, S162, WS11.01, S53
Patel J. eP02.03.03, S193
Pathak D. eP01.01.09, S139
Pathak P. PS03.5, S99
Patiño V. eP03.02.07, S228
Paul P. eP04.03.08, S280
Pauly M. eP01.01.05, S136
Pawlitzki M. PS05.05, S110
Pechmann A. PS06.02, S113
Peduto C. eP04.06.01, S299
Peeters R. eP01.06.01, S170
Pegoraro E. eP01.06.02, S171, eP03.01.09, S221
Pelagatti S. OS02.02, S26
Pelegrín-Durá L. eP03.06.01, S259
Perecz B. eP03.01.10, S222
Pereira C. eP02.06.03, S210
Pereira I. eP01.01.03, S134
Perez Siles G. eP01.03.11, S156
Peric S. eP01.05.09, S170, eP02.06.02, S209, SS15.03, S76
Perlman S. PS01.02, S87
Perry L. PS01.04, S89
Peréz-Ruixo J. eP03.04.02, S240
Peschke G. eP02.01.09, S183
Pesovic J. eP02.06.02, S209
Pestronk A.C. eP01.06.02, S171, HO02.02, S4
Petermann O. eP04.06.03, S300
Petersson M. eP03.04.07, S244
Petroska D. eP01.06.03, S172
Petrovic S. eP01.04.06, S161
Pezet S. eP04.03.03, S277
Phillips G. eP03.04.06, S243, eP03.04.08, S245, PS05.02, S108
Phonekeo K. PS07.04, S120
Picariello T. eP02.06.01, S208
Picillo E. eP04.06.01, S299
Piehl F. eP03.04.07, S244
Piepers S. PS04.1, S101
Pikó H. eP04.06.02, S300
Pilcevic D. eP02.04.01, S195
Pilling K. eP04.06.06, S302
Piluso G. eP04.06.01, S299
Pinelli M. eP04.06.01, S299
Pinto M. eP02.02.08, S189
Pinós T. eP03.05.11, S258
Piperno A. eP01.04.09, S162
Pitarch I. eP01.01.06, S137, eP01.03.07, S151
Planté-Bordeneuve V. SS17.01, S77
Plebani M. eP03.01.09, S221
Poch M. eP01.06.06, S174
Poesen K. eP04.03.01, S275, PS02.01, S92
Poggetti F. eP02.04.08, S199
Polavarapu K. eP02.06.07, S214, SS20.02, S81
Poleur M. eP01.02.06, S145, eP02.02.01, S185
Porras L. eP02.04.04, S197
Portela Sanchez S. eP03.03.11, S238, eP01.05.08, S169, Portela Sánchez S., eP04.05.09, S296
Porter B. eP02.04.07, S198
Potter R.A. eP02.05.01, S201, PS03.01, S97
Potters W. eP01.05.05, S167
Poulidou V. eP01.02.09, S147, eP03.02.02, S224
Povedano M. eP01.03.07, S151, eP03.03.03, S232
Poza J. SS03.01, S61
Pozsgai E. PS03.04, S99, PS03.01, S97
Pradat P. PS02.2, S92
Prada V. eP01.02.07, S145
Prasad S. eP04.04.01, S282, eP04.05.01, S291, PS03.06, S100
Preethish-Kumar V. SS20.02, S81
Preusse C. PS07.01, S117, PS07.06, S121, eP03.06.02, S260
Previtali S. PS08.03, S123, WS01.01, S42, eP04.01.03, S268
Proud C. eP01.03.09, S153, eP02.05.01, S201, eP02.05.08, S206, eP03.03.05, S234eP03.03.06, S234
Prufer A. PS06.04, S114
Pujol S. PS04.03, S103
Putzeys G. eP01.06.01, S170
Pyatkov A. eP02.01.06, S181
Pál E. eP03.01.10, S222
Péllegrini-Issac M. PS02.2, S92
Pérez Pérez H. eP03.03.09, S237
Qi C. eP03.04.06, S243, eP03.04.08, S245, PS05.02, S108
Qiu Q. eP02.06.01, S208
Querin G. PS02.2, S92, SS11.01, S70
Querol Gutiérrez L. TC02.03, S9, PS04.05, S105
Quijano-Roy S. eP02.01.02, S178, eP02.03.01, S191, eP03.03.05, S234, eP03.03.06, S234
Quinlivan R. eP04.01.02, S267, OS03.02, S28, PS01.04, S89
Quinonez M. SS01.02, S58
Quiroga M. eP01.06.04, S173
Raaphorst J. eP01.04.03, S158, eP01.05.02, S164, eP01.05.05, S167, eP03.06.07, S264, PS05.03, S109, PS05.04, S110
Raasing L. eP03.02.09, S229
Race V eP04.03.01, S275
Rager L. PS07.06, S121
Rahman O. eP03.04.04, S242
Rajabally Y. PS04.03, S103
Raju D. eP01.03.09, S153, eP03.03.06, S234
Rakhade S. eP01.05.04, S166
Rakocevic-Stojanovic V. eP01.05.09, S170, eP02.06.02, S209
Ramana V. eP04.05.11, S299
Ramchandren S. eP03.04.02, S240
Ramdharry G. PS02.03, S93
Ramón-Azcón J. PS09.01, S127
Rangel Miller V. eP03.05.07, S255
Ranjan S. SS20.02, S81
Rankovic M. eP01.05.09, S170
Rao V. PS01.02, S87
Rattay T. eP01.01.05, S136
Rautenberg M. eP01.01.05, S136
Ravaglia S. eP01.04.09, S162, eP01.03.03, S149, PS06.02, S113
Real-Martinez A. eP03.05.11, S258
Reash N.F. PS03.01, S97, PS03.04, S99
Rebelo A. PS08.01, S122
Rebelo S. eP02.06.03, S210, eP02.06.05, S211
Reddel S. eP01.03.11, S156
Redican S. PS03.02, S97
Reed R. PS03.06, S100
Regensburger A. PS02.04, S94
Reid C. eP02.05.01, S201, eP02.05.08, S206
Reilly M. eP01.02.03, S143, PS02.03, S93, PS08.01, S122, PS08.03, S123
Reimann J. TC08.01, S18
Reinhard J. WS01.03, S43
Reka E. eP04.02.04, S272
Remiche G. eP03.05.10, S257, eP04.02.05, S273, eP04.06.07, S303, WS05.01, S48
Rendu J. eP04.05.07, S295
Restrepo D. eP02.05.10, S208
Reyes D. PS02.03, S93
Reyngoudt H. eP02.01.08, S182, eP02.01.10, S184
Riahi S. eP04.03.07, S279
Ribeiro-Jr A. eP04.06.10, S305
Ricci G. OS09.01, S37, eP01.03.06, S151, eP02.05.09, S207, OS02.02, S26
Richardson J. eP02.05.08, S206
Richardson M. eP03.01.03, S217
Rico S. eP04.04.01, S282, eP04.05.01, S291, PS03.06, S100
Ridout D. eP02.05.03, S202
Rietveld A. PS01.06, S91
Rinaldi D. PS02.2, S92
Ripolone M. eP02.04.08, S199, eP04.04.03, S284
Rispens T. PS05.03, S109, PS05.04, S110
Rizopoulos D. eP03.05.03, S251, eP03.05.04, S252
Rizzo F. eP01.02.01, S142
Robb S. PS01.04, S89
Roberts M. eP04.05.10, S298
Robertson L. eP03.01.02, S217
Robledo S. eP04.04.10, S289
Roca S. eP03.03.03, S232
Roccella S. eP03.01.11, S222
Roddate M. eP02.02.07, S188
Rodino-Klapac L.R. eP02.05.01, S201, eP02.05.08, S206, PS03.01, S97, PS03.04, S99
Rodney S. eP04.01.02, S267
Rodrigues E. eP03.04.01, S239
Rodriguez-Sanchez C. eP01.03.07, S151
Rodriguez Cruz P. SS21.02, S83
Rodriguez H. eP01.06.05, S173
Rodriguez Rodriguez M. eP03.03.07, S236
Rodríguez Hernández L. eP01.04.02, S157
Roepcke S. eP03.02.04, S225
Rogers M. eP02.06.06, S213
Rohwer A. eP01.03.05, S150
Rojas-Garcia R. PS02.03, S93, PS08.03, S123
Rojas S. eP02.05.10, S208
Romano F. eP04.06.01, S299
Roos A. eP01.01.01, S133, eP02.06.09, S215, eP03.04.07, S244, eP03.06.02, S260, PS02.04, S94, PS07.01, S117
Roosen J. PS05.04, S110
Ros-Arlanzón P. eP03.06.01, S259
Rose M. eP03.01.01, S216
Rosenbohm A. eP04.04.04, S285
Rossello J. eP02.03.03, S193
Rossor A. eP01.02.07, S145, PS02.03, S93, SS08.02, S66, TC06.04, S16
Rots D. eP02.02.07, S188
Rouyer A. eP03.05.08, S256
Rroji A. eP04.02.04, S272
Ruano Hernández A. eP04.02.02, S271
Ruck T. PS05.05, S110
Rudnik Schoeneborn S. TC04.04, S13
Ruegg M. WS01.03, S43, WS12.03, S55
Rufibach L. eP01.06.02, S171
Ruiter A. PS05.03, S109, PS05.04, S110
Ruiz A. eP01.02.03, S143
Ruiz E. eP01.06.04, S173, eP03.03.02, S231, eP03.06.05, S263, eP04.02.07, S275, eP04.03.06, S279, eP04.03.09, S281
Ruiz Ospina E. eP03.02.03, S224
Russell J. eP01.03.01, S147, PS08.06, S126
Russo R. eP02.06.01, S208, eP04.04.02, S284
Ryan M. PS01.02, S87, PS06.02, S113
Ryu H. eP02.03.06, S195
Saade D.E. PS03.06, S100
Saade D. eP04.04.01, S282
Saade N.D. eP04.05.01, S291
Saad Y. eP04.03.07, S279
Saak A. eP03.01.08, S220
Saavedra Ruiz M. eP03.06.04, S262
Sabatelli M. PS02.03, S93
Sabbatini D. eP02.05.02, S202
Sabo B. PS03.04, S99
Sabu-Kurian A. PS03.5, S99
Sacchetto R. PS09.02, S128, PS09.03, S128
Sacchini M. eP01.04.09, S162
Sacconi S. OS02.01, S25, PS05.01, S106, WS04.02, S47
Saccà F. eP03.04.08, S245, PS05.02, S108
Saegert C. eP04.05.02, S291
Sahenk Z. PS03.01, S97
Saianda A. eP01.01.03, S134, eP02.06.08, S215
Saito K. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234, PS06.01, S112
Sakbaeva G. eP04.01.06, S270
Salachas F. PS02.2, S92
Salazar Lengua C. eP03.02.03, S224
Salcedo M. eP01.06.04, S173, eP03.03.02, S231, eP03.06.05, S263, eP04.03.06, S279
Saldys N. eP04.06.10, S305
Salimian M. PS08.06, S126
Salort-Campana E. eP01.06.02, S171
Samsuddin S. eP01.03.05, S150
Samtani M. eP03.04.02, S240
Samões R. PS04.04, S104
Sanchez D. eP01.06.04, S173, eP03.06.05, S263, eP04.03.06, S279, eP04.03.09, S281
Sanchez Riera C. eP01.06.10, S176
Sanchez Soblechero A. eP03.03.11, S238
Sanders D. eP03.04.05, S242, WS11.03, S54, PS09.02, S128, PS09.03, S128
Sansone F. eP03.01.11, S222
Sansone V. PS06.02, S113
Santalla eP03.05.11, S258
Santamaría Montero P. eP03.03.07, S236
Santirso D. PS02.05, S95
Santorelli F. eP01.01.05, S136, eP03.01.11, S222
Santos E. PS04.04, S104
Sapego E. eP02.03.04, S193
Saracino D. PS02.2, S92
Sariego A. eP01.06.06, S174
Saris C. PS01.06, S91
Sarkozy A. eP02.05.03, S202, eP04.01.02, S267, OS11.02, S40, PS01.04, S89, WS01.02, S42
Sarycheva A. eP02.01.06, S181
Savarese M. SS03.01, S61, SS03.03, S62 Savic-Pavicevic D. eP02.06.02, S209
Savino W. eP04.03.03, S277
Sbrocchi A. PS01.02, S87
Scalco R. eP03.03.01, S230, PS06.01, S112, PS06.04, S114, PS06.05, S115
Scano M. PS09.03, S128
Scapozza L. eP02.05.05, S204, eP02.05.06, S204, eP02.05.07, S205, eP04.06.03, S300
Scarpa M. eP01.04.09, S162
Schaefer J. eP03.01.08, S220
Schalke B. PS05.05, S110
Schara-Schmidt U. eP01.01.01, S133, eP04.05.02, S291, TC07.04, S18
Schara U. PS05.05, S110
Scheffers L. eP03.05.01, S248
Schenone A. PS08.03, S123
Scherer S. eP01.02.03, S143, PS08.01, S122, PS08.03, S123
Schiava M. PS01.3, S89, PS02.03, S93
Schirinzi E. OS02.02, S26
Schlaefke L. eP02.06.01, S208
Schlereth M. PS02.04, S94
Schlotter-Weigel B. PS08.03, S123
Schmidt J. eP01.05.01, S163, eP01.05.03, S165, eP04.06.05, S302
Schmidt J. PS01.06, S91
Schmitt V. eP02.06.04, S211
Schneider-Gold C. eP03.04.04, S242, eP04.04.04, S285
Schneider J. PS03.02, S97
Scholle L. eP01.05.01, S163
Schoser B. eP02.06.04, S211, PS03.06, S100, TC04.02, S13, WS07.01, S50
Schreurs M. eP01.04.03, S158
Schroeter M. PS05.05, S110
Schuermans N. PS07.03, S119
Schuessler S. PS02.04, S94
Schulz J. eP02.01.09, S183
Schänzer A. eP01.01.01, S133, PS07.06, S121
Schönecker A. eP01.01.01, S133
Schöne U. eP02.01.09, S183
Schüle R. eP01.01.05, S136
Schülke-Gerstenfeld M. PS07.01, S117
Sciacco M. eP02.04.08, S199
Scorza F. eP01.04.04, S160
Scoto M. eP01.03.05, S150, PS01.04, S89
Scott D. PS06.06, S116
Sebastiani G. eP04.05.04, S293
Sechi A. eP01.04.09, S162
Sedehizadeh S. eP04.04.04, S285
Seeman P. PS08.03, S123
Seferian A. eP04.04.01, S282, eP04.06.04, S301
Seferian M.A. eP04.05.01, S291
Seghaier I. eP04.03.02, S276, eP04.03.07, S279
Segovia S. eP01.03.02, S148, eP01.03.05, S150, eP01.03.07, S151
Sehmisch S. eP01.05.01, S163
Seif A. PS01.06, S91
Selby K. PS01.02, S87
Selcen D. SS05.02, S63, TC07.01, S16
Sereda M. SS18.01, S79
Servais L. eP01.02.06, S145, eP01.03.09, S153, eP01.04.01, S156, eP03.03.01, S230, eP03.03.05, S234, eP03.03.06, S234, eP04.04.01, S282, eP04.05.01, S291, eP04.06.04, S301, OS01.03, S25, OS05.02, S32, PS06.01, S112, PS06.04, S114, SS12.02, S72, SS12.03, S73, SS12.1, S71
Sevilla T. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146, PS08.03, S123
Shaibani A. PS07.04, S120
Shamim U. eP01.01.07, S138, eP01.01.09, S139
Sharma M. eP01.01.07, S138, eP01.01.09, S139, eP01.05.07, S168
Sheih P. PS01.02, S87
Shell R. PS03.01, S97
Shelly S. eP03.06.06, S263
Shieh P.B. eP02.05.08, S206, eP04.04.01, S282, eP04.05.01, S291, PS03.02, S97, PS03.06, S100
Shim Y. eP02.03.06, S195
Shin J. eP01.06.09, S176, eP04.03.04, S278
Shoskes J. eP04.05.11, S299
Shy M. eP01.02.03, S143, eP01.02.07, S145, PS08.01, S122, PS08.03, S123
Siciliano G. eP01.03.06, S151, eP01.04.09, S162, eP02.05.09, S207, eP03.01.11, S222, eP03.03.08, S236, OS02.02, S26, OS09.01, S37, WS03.02, S45
Sickmann A. eP01.01.01, S133, PS07.01, S117
Siddiqui S. SS20.02, S81
Sifi Y. eP01.06.08, S175
Simmons K. eP03.05.07, S255
Singh T. eP02.05.08, S206
Sipos A. eP03.01.10, S222
Sivera R. eP01.02.02, S142, eP01.02.04, S144
Sivera R. eP01.02.08, S146
Skolarus L. PS04.02, S103
Skoula I. eP04.04.06, S287
Sleurs C. eP04.01.04, S268
Smeets N. eP04.01.01, S266
Smeriglio P. eP04.03.03, S277, SS11.01, S70
Smith E. PS01.01, S87, PS01.02, S87
Smithuis F. eP01.04.03, S158, eP01.05.05, S167
Smyth B. eP03.04.09, S246
Snezhko E. eP02.01.08, S182
Sobieszczuk E. eP01.04.05, S160
Soblechero-Martín P. eP01.01.08, S139
Sobrido M. eP01.01.05, S136
Sodhi J. eP02.06.06, S213, eP03.01.03, S217
Sola Vendrell E. eP01.05.08, S169
Somers V. eP03.06.06, S263
Song G. eP02.03.03, S193
Sorarù G. eP03.01.09, S221
Soto D. eP01.06.04, S173, eP03.03.02, S231, eP03.06.05, S263, eP04.03.06, S279
Souissi A. eP01.01.10, S141, eP02.02.03, S186, PS04.04, S104
Sousa L. SS17.03, S78
Souslov V. eP04.01.06, S270
Souza B. eP04.06.10, S305
Souza L. eP04.06.10, S305
Spendiff S. eP02.06.09, S215, SS06.03, S64
Spigarelli M. eP02.03.01, S191
Spiliopoulos K. eP02.01.03, S179
Spilioti M. eP01.02.09, S147
Spinazzi M. eP03.05.08, S256
Spinty S. PS01.02, S87, eP04.04.02, S284
Sproule D. eP01.06.05, S173
Spuler S. eP01.06.02, S171
Stadelmann C. eP01.05.01, S163
Stalens C. eP02.04.05, S197
Stalman E. PS05.03, S109, PS05.04, S110
Stascheit F. PS05.05, S110
Steck A. WS09.02, S51
Steel D. PS01.04, S89
Steenhuis M. PS05.03, S109, PS05.04, S110
Stenzel W. eP03.06.02, S260, PS07.01, S117, PS07.06, S121
Stepanov A. eP04.01.06, S270
Sternberg D. eP04.05.07, S295
Stettner G. eP02.03.05, S194
Stevanin G. eP01.01.05, S136
Stevenson H. eP04.06.04, S301
Stevic Z. eP01.05.09, S170
Stimpson G. eP02.05.03, S202
Stock F. eP01.01.05, S136
Stojanov A. eP01.04.06, S161
Stojanov J. eP01.04.06, S161
Stojkovic T. eP01.06.02, S171, PS08.03, S123
Stork S. eP01.02.07, S145
Straub V. eP01.06.02, S171, eP03.01.02, S217, eP03.01.03, S217, eP03.01.05, S218, eP03.06.03, S261, PL01.01, S20, PS01.02, S87, PS02.03, S93
Stromer D. PS02.04, S94
Suarez F. eP01.06.04, S173
Suarez V. PS02.05, S95
Suh B. eP03.01.01, S216
Sumner C. SS10.01, S68
Sundgreen C. PS07.04, S120
Sundström P. eP03.04.07, S244
Sung J. eP01.06.09, S176, eP03.04.10, S247, eP03.05.09, S257, eP04.03.04, S278, eP04.04.09, S289
Sun H. eP03.04.02, S240
Sun R. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Supiot F. eP04.02.01, S270
Suponeva N. eP03.02.05, S226
Suri V. eP01.01.07, S138, eP01.01.09, S139, eP01.05.07, S168
Suslov V. eP02.01.08, S182, eP02.01.10, S184
Svaren J. eP01.02.07, S145
Svetlove A. eP03.01.07, S220
Swoboda K. PS06.02, S113
Syed S. eP03.04.09, S246, eP03.04.09, S246
Symoens S. eP04.05.08, S296, PS07.03, S119
Synofzik M. eP01.01.05, S136
Sánchez D. eP03.02.03, S224
Sánchez Mateos P. eP01.05.08, S169
Sánchez Soblechero A. eP01.05.08, S169, eP04.05.09, S296
Sénéchal T. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
’t Hoen P. SS06.01, S64
Tahon V. eP01.03.08, S152
Tajuddin A. eP01.04.07, S162
Tamaoui L. eP01.02.05, S144, eP02.01.04, S180, eP03.03.10, S238
Tang K. eP04.04.02, S284
Tan L. PS02.04, S94
Tard C. eP03.05.08, S256, eP04.04.04, S285
Tardieu M. eP03.05.04, S252
Tartaglione I. eP02.05.04, S203
Tasca G. PS02.03, S93, SS02.02, S60
Tatillo C. eP03.05.10, S257
Taurisano R. eP01.04.09, S162
Tausan D. eP02.04.01, S195
Tavosanis M. OS02.02, S26
Tejedera-Villafranca A. PS09.01, S127
Teunissen L. PS04.1, S101
Thal D. eP04.03.01, S275
Thangarajh M. PS01.01, S87
Thiele S. eP03.06.03, S261
Thieme A. PS05.05, S110
Thokala P. SS12.02, S72
Thomas A. PS01.05, S90, PS07.05, S120
Thomas F. eP01.02.02, S142, eP01.02.04, S144, eP01.02.08, S146
Thürkauf M. WS12.03, S55
Tiar F. eP01.03.03, S149
Tilleux S. eP04.02.05, S273
Timmerman V. eP01.01.05, S136, PS08.02, S122
Timmons J. eP04.03.10, S281
Tirolien S. eP02.03.01, S191
Tizzano E. eP01.03.09, S153, eP03.03.05, S234, eP03.03.06, S234
Tobías E. eP01.05.06, S168
Todorova A. eP04.03.05, S278
Todorov T. eP04.03.05, S278
Tonacci A. eP03.01.11, S222
Torella A. eP02.05.04, S203, eP04.06.01, S299
Toro W. eP02.03.02, S192
Torrente Y. eP01.06.10, S176
Torri F. eP01.03.06, S151, eP02.05.09, S207, OS09.01, S37
Toscano A. eP01.04.09, S162
Tosi M. eP03.01.04, S218
Tournev I. eP04.03.05, S278
Toussaint M. eP01.01.04, S135
Toussaint M. PS09.06, S131
Trifunov S. eP03.03.03, S232
Trochet D. SS04.02, S63
Trollmann R. PS02.04, S94
Tsai L. eP01.01.10, S140
Tsarenko A. PS09.06, S131
Tsargush V. eP02.01.10, S184
Tsiverdis I. eP04.04.06, S287
Tsuji H. eP01.04.10, S163
Tulinius M. PS01.02, S87
Tupler R. eP03.01.11, S222
Turner C. eP02.04.07, S198, eP02.06.06, S213, eP03.01.02, S217, eP04.01.02, S267
Turner T. eP03.04.09, S246
Turon-Sans J. PS02.03, S93
T’joen C. SS20.03, S82
Udd B. SS03.01, S61, SS03.03, S62
Udovicic I. eP02.04.01, S195
Udvari S. eP04.06.02, S300
Uecker M. PS01.06, S91
Ulrichts P. SS20.03, S82
Umakhanova Z. eP02.01.10, S184
Unnikrishnan G. eP02.06.07, S214, PS07.05, S120
Upadhyay S. eP04.06.04, S301
Upadhyayula S. PS01.01, S87
Urban P. PS05.05, S110
Urtizberea A. eP04.05.07, S295
Urtizberea J. eP03.06.04, S262, eP04.04.08, S288
Utkus A. eP01.06.03, S172
Utsugisawa K. eP03.04.03, S241, PS05.01, S106, SS20.03, S82
Uysal H. eP01.01.05, S136
Vainzof M. eP04.06.10, S305
Valenzuela B. eP03.04.02, S240
Vallejo D. eP03.02.07, S228, eP04.04.10, S289
Valls-Roca L. eP01.05.06, S168
Vanakker O. PS07.03, S119
van Benthem J. eP04.04.03, S284
Van Boxstael E. eP01.04.08, S162
Van Damme P. eP04.03.01, S275, PS02.01, S92, SS09.02, S68, TC05.01, S14
van Dam P. PS05.03, S109, PS05.04, S110
van den Anker J. PS01.02, S87
Van den Bergh P. PS04.06, S106, TC02.02, S9, WS10.02, S52
van den Berg L. eP03.05.01, S248, eP04.03.10, S281, PS04.06, S106
Vandenborne K. eP04.04.03, S284
Vanden Brande L. eP04.01.01, S266
van den Hout H. eP03.05.02, S250, eP03.05.04, S252
van den Hout J. eP03.05.01, S248
van der Beek N. eP03.05.02, S250, eP03.05.03, S251, eP03.05.04, S252, OS01.01, S24, WS05.03, S49
Vanderkelen M. eP03.04.03, S241
van der Kooi A. eP01.04.03, S158, eP01.05.02, S164, eP01.05.05, S167, eP03.06.07, S264, PS05.03, S109, PS05.04, S110
van der Maarel S. WS08.03, S51
van der Meulen M.G.F. PS04.1, S101
van der Molen R. eP01.04.03, S158
Vandernoot I. eP03.05.10, S257, eP04.06.07, S303
van der Ploeg A. eP03.05.01, S248, eP03.05.02, S250, eP03.05.03, S251, eP03.05.04, S252, WS02.03, S44
van der Pol L. PS05.03, S109
van der Pol W. PS04.06, S106, PS05.04, S110
van der Star G. PS04.1, S101
van der Ven P. eP01.01.01, S133
van der Weele L. eP01.05.02, S164
van de Scheur J. PS04.06, S106
van de Velde N. eP04.04.03, S284
Van de Vondel L. eP01.01.05, S136
van de Wijdeven G. PS04.1, S101
van Doormaal P. PS04.1, S101
van Doorn P. PS04.1, S101, PS05.03, S109, PS05.04, S110
van Eijk R. eP04.03.10, S281, PS04.1, S101
Vanello N. OS02.02, S26
van Engelen B. SS02.03, S61, SS06.01, S64
van Ham M. PS05.03, S109
van Ham S. PS05.04, S110
Van Hoorick B. eP03.04.04, S242
van Kampen A. eP01.05.02, S164
van Kooten H. eP03.05.02, S250
Van Lander A. eP04.01.01, S266
van Leeuwen E. eP01.04.03, S158, eP01.05.05, S167
van Lent J. PS08.02, S122
van Lochem E. eP01.04.03, S158
van Onna M. eP01.04.03, S158
Van Parys V. eP04.02.05, S273
van Schaik B. eP01.05.02, S164
van Schaik I. eP01.05.05, S167, eP03.06.07, S264
Van Wermeskerken T. PS08.02, S122
Varga D. eP03.01.10, S222
Vasconcelos F. eP04.06.10, S305
Vaugier I. eP02.03.01, S191
Vavla M. PS02.06, S96
Vazquez J. eP01.03.07, S151
Veillet S. PS06.03, S114
Velardo D. eP01.02.01, S142, eP04.04.03, S284
Velez J. eP03.02.07, S228
Veltkamp M. eP03.02.09, S229
Veltsista D. eP02.01.03, S179, eP02.02.04, S187
Vengalil S. eP02.01.07, S182, eP02.06.07, S214, PS01.05, S90, PS07.05, S120, SS20.02, S81
Venitz J. eP03.04.05, S242
Venkatesan R. eP04.04.02, S284
Venugopalan Y.V. eP01.01.07, S138, eP01.01.09, S139
Verdugo R. RG.02, S56
Vergadi E. eP04.04.06, S287
Verhamme C. eP01.05.05, S167, eP03.06.07, S264
Verhoeven K. PS07.03, S119
Vermeulen A. eP03.04.02, S240
Verrecchia E. eP01.04.09, S162
Verschuuren J. eP03.04.04, S242, PS05.03, S109, PS05.04, S110
Versluys L. PS07.03, S119
Verstraete E. PS04.1, S101
Vesperinas A. PS02.03, S93
Vicari S. eP03.01.04, S218, eP04.04.04, S285
Viegas D. eP02.06.03, S210, eP02.06.05, S211
Vila-Bedmar S. eP01.06.06, S174
Vilchez-Padilla J. PS01.02, S87
Vilchez J. eP01.01.06, S137
Vilimiene R. eP01.06.03, S172
Villapando N. eP01.02.07, S145
Villarreal-Salazar M. eP03.05.11, S258
Villart M. eP02.03.01, S191
Vincent A. PS05.06, S111, SS22.03, S85
Viscidi E. eP01.03.03, S149
Vishnu V. eP01.05.07, S168
Vissing J. eP02.04.02, S196, eP02.04.06, S198, eP03.04.11, S248, eP03.05.11, S258, eP03.06.03, S261, PS05.01, S106, WS02.01, S43, WS07.03, S50
Vittore D. PS08.05, S125
Vivier Y. eP02.02.05, S187
Vlachopoulos G. eP02.01.03, S179
Vlodavets D. PS06.04, S114
Voermans N. OS11.02, S40, SS02.03, S61
Vogels O. eP03.02.09, S229
Vohra R. PS03.03, S98
Voháňka S. eP03.05.05, S253
Vola B. eP01.06.10, S176
von der Hagen M. OS04.03, S31
von Moers A. eP03.06.02, S260
Vorgia P. eP04.04.06, S287
Vrancken A. PS04.1, S101
Vucic S. eP01.03.11, S156
Vuillerot C. PS06.01, S112
Vukojevic Z. eP03.02.01, S223
Vural A. eP01.01.05, S136
Vu T. eP03.04.03, S241, PS05.01, S106, SS20.03, S82
Vélez Santamaria V. eP02.04.04, S197
Wadhwani N. PS03.06, S100
Wagner A. PS02.04, S94
Wagner K. eP03.03.01, S230
Waldner M. PS02.04, S94
Walker H. eP02.04.07, S198, eP02.06.06, S213, eP04.05.10, S298, eP01.04.03, S158, eP01.05.02, S164, eP01.05.05, S167, eP03.06.07, S264
Walter M. eP01.06.02, S171, eP03.06.03, S261
Wandel C. eP02.05.01, S201, eP02.05.08, S206
Wang F. PS03.02, S97
Wang N. eP01.03.03, S149
Wang S. eP02.05.01, S201, PS09.04, S129
Wang Y. PS06.04, S114
Ward L. PS01.02, S87
Waters P. eP03.04.10, S247
Watt S. eP04.05.10, S298
Wayand M. eP01.01.05, S136
Webster R. PS01.02, S87, PS05.06, S111
Weeden T. eP02.06.01, S208, eP04.04.02, S284
Weidenmüller M. PS01.06, S91
Weidlich D. eP02.03.02, S192
Weinberg J. SS03.01, S61
Weis J. SS07.02, S66
Weisleder N, PS07.02, S118
Whitney E. eP04.03.10, S281
Wicklund M. PS01.01, S87, PS07.04, S120
Wiendl H. PS05.05, S110
Wiersma M. PS04.1, S101
Wieske L. eP01.04.03, S158, PS05.03, S109, PS05.04, S110
Wijntjes J. HO01.02, S3
Willcocks R. eP04.04.03, S284
Willems M. eP01.04.03, S158
Willen J. eP04.01.03, S268
Williams D. eP02.06.06, S213
Willison H. WS10.03, S52
Willis T. eP04.05.10, S298
Winter N. PS08.05, S125
Witoonpanich R. RG.04, S57
Witting N. eP02.04.02, S196, eP02.04.06, S198, eP03.04.11, S248
Woelfle J. PS02.04, S94
Wolfe G. WS11.03, S54
Woltering F. PS05.01, S106
Wong J. eP04.01.02, S267
Wong K. eP03.01.03, S217
Won S. eP01.02.07, S145
Woodhall M. eP03.04.10, S247
Woods L. eP03.06.03, S261
Wozniak L. PS03.06, S100
Wu F. SS01.02, S58
Wu X. eP01.02.07, S145
Xhelili M. eP04.02.04, S272
Xu I. eP01.02.03, S143
Yang C. eP01.01.10, S140
Yang M. PS01.02, S87
Yel L. eP03.02.04, S225
Yeung W. PS06.01, S112
Yong J. eP03.05.07, S255
Yoo I. eP02.01.05, S180
Youn B. eP01.03.03, S149
Young P. OS10.03, S40
Yu L. eP02.05.08, S206
Zaganas I. eP04.04.06, S287
Zaidman C. eP02.05.01, S201, eP02.05.08, S206
Zammit P. SS02.01, S59
Zaninotto M. eP03.01.09, S221
Zanni G. eP01.01.05, S136
Zanobio M. eP04.06.01, S299
Zanoteli E. PS06.04, S114
Zanotti S. eP02.04.08, S199, eP02.06.01, S208, eP04.04.02, S284, eP01.05.01, S163, eP01.05.03, S165
Zekeridou A. WS12.01, S54
Zeng R. PS01.06, S91, eP04.06.01, S299
Zhang B. eP03.03.01, S230
Zhang R. PS08.03, S123
Zhang W. eP04.06.04, S301
Zhao L. SS09.01, S67
Zhu C. PS06.02, S113
Zhu Y. eP03.04.02, S240
Ziemssen T. eP03.01.08, S220
Zierz S. eP01.05.01, S163
Zlatar J. eP02.06.02, S209
Zogby I. eP04.06.10, S305
Zozulya-Weidenfeller A. eP04.04.04, S285, eP04.04.05, S286
Zschüntzsch J. eP01.05.01, S163, eP03.01.07, S220, eP04.06.05, S302
Zuccarino R. eP01.02.07, S145
Zuchner S. PS08.01, S122
Zukova V. eP02.02.07, S188
Zulaica M. eP01.06.06, S174
Züchner S. eP01.01.05, S136, eP01.02.03, S143, eP02.01.09, S183, PS08.03, S123
Ørstavik K. eP03.06.03, S261


