
A Case of Oculopharyngeal Muscular Dystrophy Caused by a Novel PABPN1 c.34G > T (p.Gly12Trp) Point Mutation without Polyalanine Expansion
Abstract
Immediately after the initial methionine codon, the PABPN1 gene encodes a stretch of 10 alanines, 1 glycine, and 2 alanines. Oculopharyngeal muscular dystrophy (OPMD) is caused by the expansion of the first 10 alanine stretches. The only exception is the missense mutation of glycine at the 12th residue into alanine, which elongates the stretch to 13 alanines by connecting the first and second stretch with the addition of one alanine in between, indicating that the expansion or elongation of the alanine stretch results in OPMD. We report a 77-year-old man with the novel missense mutation c.34G > T (p.Gly12Trp) in PABPN1 gene whose clinicopathological findings were compatible with OPMD. He presented with slowly progressive bilateral ptosis, dysphagia, and symmetrical proximal dominant muscle weakness. Magnetic resonance imaging revealed selective fat replacement of the tongue, bilateral adductor magnus, and soleus muscles. Immunohistochemistry studies of the muscle biopsy sample revealed PABPN1-posibive aggregates in the myonuclei which have been reported to be specific to OPMD. This is the first OPMD case caused by neither the expansion nor the elongation of alanine stretch. The present case suggests that OPMD may be caused not only by triplet repeats but also by point mutations.
INTRODUCTION
Oculopharyngeal muscular dystrophy (OPMD) is a late-onset muscle disease that is clinically characterized by slowly progressive ptosis, dysphagia, and proximal limb muscle weakness. The short expansion of a GCN trinucleotide repeat encoding a polyalanine tract within the N-terminus of the poly-A binding protein nuclear 1 (PABPN1) gene is a well-known common cause of OPMD [1]. However, four OPMD cases were previously reported to be caused by the PABPN1 c.35G > C point mutation, which changes a glycine codon to an alanine codon. This results in an increased in the number of contiguous polyalanine codons, mimicking the triplet repeat effect [2–4]. Here, we report a case of OPMD due to the novel point mutation c.34G > T (p. Gly12Trp) in the PABPN1 gene, which is not caused by alanine codon expansion or elongation.
CASE
A 77-year-old man presented with progressive bilateral ptosis, dysphagia, gait disturbance, and weight loss upon admission. He described experiencing slowly progressive bilateral ptosis which began when he was 60 years old; and, at the age of 73 years old, he underwent levator muscle resection to address this. However, ptosis gradually returned. One year later, he developed steadily progressive dysphagia, gait disturbance, proximal muscle weakness, and muscle atrophy in all extremities. He also reported a cumulative weight loss of 15 kg in the last 3 years. Although he had no clear family history, his father, who died of renal failure at 72 years old, was suspected of having ptosis. His son, who was 47 years old at the time of evaluation, had elevated serum creatine kinase levels ranging from 300 to 550 U/L (normal range 59–248 U/L); but claims to have no apparent neurological symptoms resembling ptosis, dysphagia, or muscle weakness.
Upon admission to our hospital, neurological examination revealed bilateral ptosis, slightly limited bilateral upward gaze, dysphagia, and muscle weakness. The ptosis severity level was determined by palpebral aperture, which measured at 4 mm in each eye. He had dysphagia without tongue atrophy and required texture-modified diet as well as thickened fluids. His muscle strength in Medical Research Council (MRC) grading system was MRC 3 for both iliopsoas muscles and MRC 4 for bilateral quadriceps and biceps femoris muscles. Muscle weakness was not observed in the neck, both upper extremities, tibialis anterior, and gastrocnemius muscles. However, the examination showed positive Gowers’ sign and gait disturbance with poor thigh elevation.
Laboratory tests revealed a normal serum creatine kinase level (153 U/L). Anti-acetylcholine receptor (AChR) and anti-muscle specific kinase (MuSK) antibodies were negative. Electrocardiography showed no abnormalities. Forced vital capacity was normal to 120.7% of the normal predicted value for patients of the same age. Needle electromyography showed myogenic changes with a low-amplitude motor unit potential in the right lower extremity. Axial T1-weighted magnetic resonance imaging revealed selective skeletal muscle fat replacement involving the tongue, bilateral adductor magnus, and soleus; along with bilateral adductor magnus muscle atrophy (Fig. 1A, E, F, arrow). In contrast, other truncal and limb muscles were preserved (Fig. 1B–F). Biopsy of the left rectus femoris muscle showed nonspecific myopathic change with fiber size variation on routine hematoxylin and eosin (H&E) staining, and with no inflammatory cells, necrotic fibers, or regenerating fibers (Fig. 2A). Modified Gömöri trichrome staining showed rimmed vacuoles in only one fiber (Fig. 2B, arrow). The p62 immunohistochemistry study showed cytoplasmic accumulation of p62 aggregates in only one fiber (Fig. 2C, arrow) and some p62-positive intramyonuclear inclusions (Fig. 2C, arrowheads). The immunohistochemistry study for anti-PABPN1 antibody revealed nuclear accumulation of insoluble PABPN1, which corresponds to the p62-positive intramyonuclear inclusions (Fig. 2D). DNA sequence analysis revealed a novel heterozygous point mutation, c.34G > T (p. Gly12Trp), in exon 1 of the PABPN1 gene (Fig. 3A).
Fig. 1
Axial T1-weighted muscle magnetic resonance imaging. (A) The tongue muscle tissue is replaced by fat (arrows), whereas other neck muscles are preserved. (B–D) The upper-arm, trunk, and hip region show no obvious muscle atrophy or fat replacement. (E) Bilateral adductor magnus muscle atrophy with fat replacement is observed at the thigh level (arrows). (F) The lower leg level shows bilateral soleus muscle fat replacement (arrows).
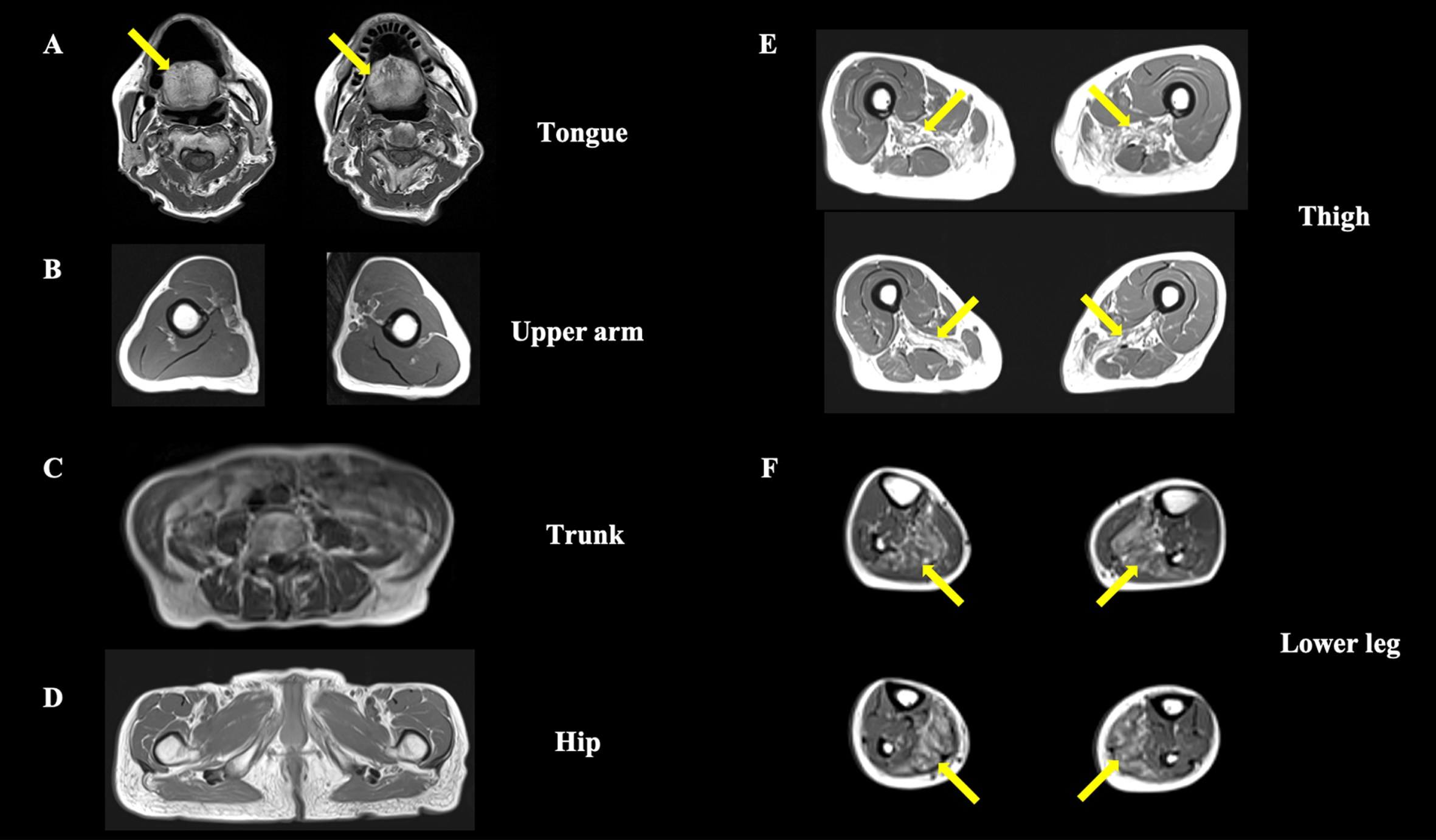
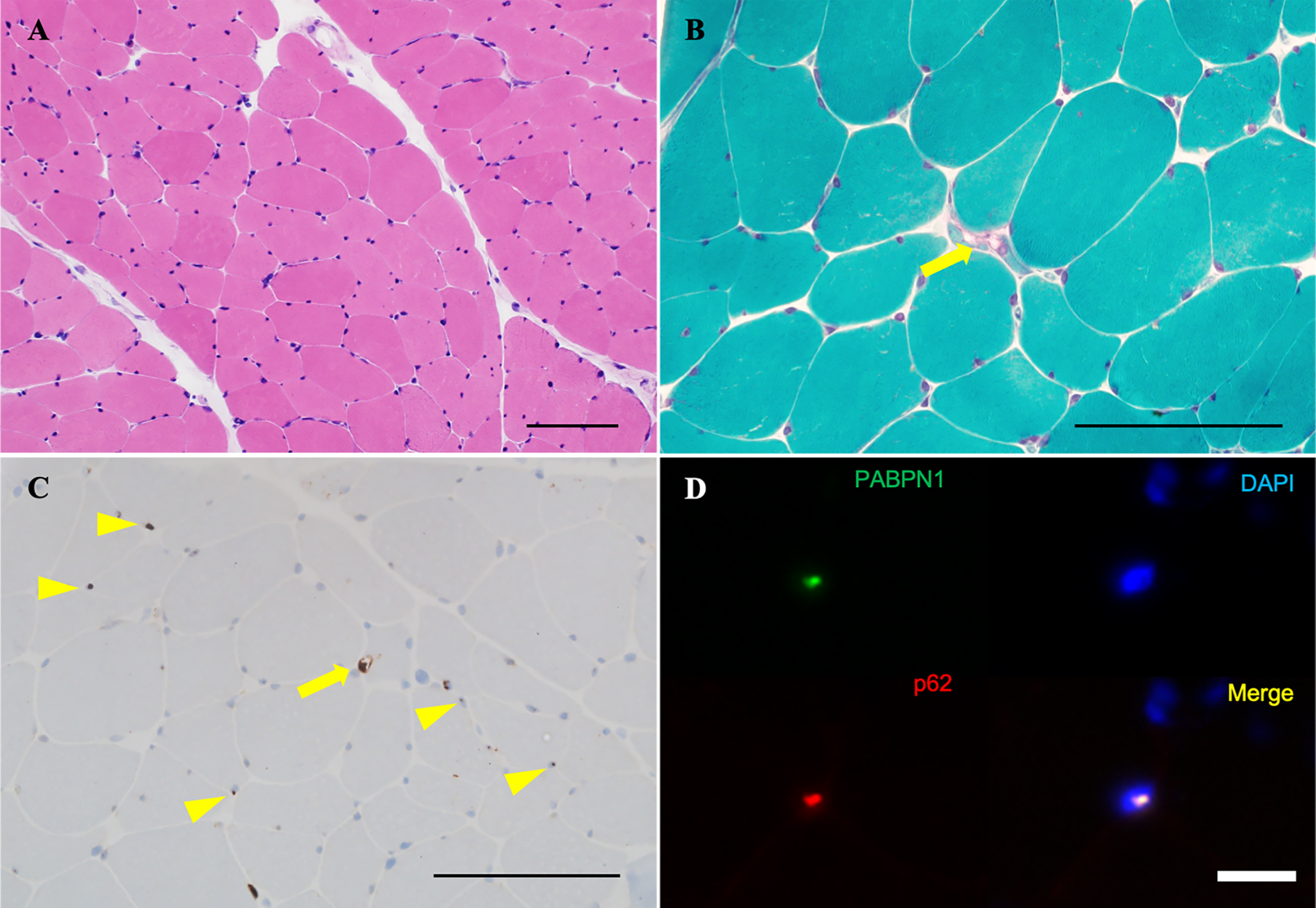
Fig. 2
Biopsy sample of the left rectus femoris. (A) Variable myofiber size and the absence of inflammatory cells, necrotic fibers, or regenerating fibers are observed on H&E. (B) On modified Gömöri trichrome staining, rimmed vacuoles are observed in only one fiber (arrow). (C) Intracytoplasmic accumulation of p62 aggregates are observed in only one fiber (arrow), along with some p62-positive intranuclear inclusions on immunohistochemistry (arrowheads). (D) Nuclear accumulation of insoluble PABPN1 is highlighted by anti-PABPN1 antibody in immunohistochemistry (arrows). Scale bars: 100μm.
DISCUSSION
This is the first report of a patient with OPMD that has been attributed to a novel point mutation, PABPN1 c.34G > T (p. Gly12Trp), which is not caused by alanine codon expansion or elongation. This illustrates a novel mutational mechanism that has not been previously documented.
Our patient presented with slowly progressive severe bilateral ptosis, dysphagia, and symmetrical proximal dominant muscle weakness without myocardial involvement, and the muscle imaging findings led to the suspicion of OPMD (Fig. 1A, E, F). A previous report described that fat replacement involving the tongue, adductor magnus, and soleus muscle has been observed in the magnetic resonance imaging studies of an OPMD patient. The similarity with the present report may allow us to consider the feature as characteristic of OPMD [5]. Our patient’s muscle biopsy showed rimmed vacuoles and p62-positive intranuclear inclusions (Fig. 2C), which is consistent with the known characteristics of OPMD [6]. Furthermore, immunohistochemistry for anti-PABPN1 revealed intranuclear accumulation of insoluble PABPN1 colocalized with p62 (Fig. 2D). The presence of intranuclear-insoluble PABPN1 accumulation has been reported as highly specific (96%) for OPMD [7].
The short expansion of a GCG or GCA trinucleotide repeat (from 10 repeat units to 11–17 repeat units) encoding a polyalanine tract at the N-terminus of the PABPN1 gene is a well-known common cause of OPMD [1]. Additionally, four OPMD cases without the gene expansion have been previously reported [2–4]. All of these cases showed a c.35G > C point mutation converting glycine to alanine at amino acid position 12 (p.Gly12Ala); thus, resulting in a sequence of 13 contiguous alanine codons (Fig. 3B). Therefore, this suggests that missense mutation may cause the disease by mimicking the effect of the common polyalanine expansion. In contrast, the c.34G > T point mutation found in our patient convers a glycine codon to tryptophan at amino acid position 12 (p.Gly12Trp), which does not make a long alanine stretch (Fig. 3A). Although it is unclear how p.Gly12Trp causes OPMD, one plausible explanation would be that tryptophan may have a similar effect as alanine, possibly due to its biophysical and chemical properties, and a stretch of 10 alanines, 1 tryptophan, and 2 alanines may behave similar to stretch of 13 alanines.
Fig. 3
DNA sequence analysis. (A) The upper panel reports the sequence trace of the patient, indicating the position of the c.34G > T base change (arrow). The lower panel reports the coding sequence of the first 18 codons in exon 1 of the PABPN1 gene of the patient. The c.34G > T mutation results in the substitution of glycine by tryptophan at codon 12, which does not cause expansion of polyalanine stretch. (B) The coding sequence of the 4 reported cases with the c.35G > C mutation. The c.35G > C mutation results in the substitution of glycine by alanine at codon 12, which generates a sequence of 13 contiguous alanines.
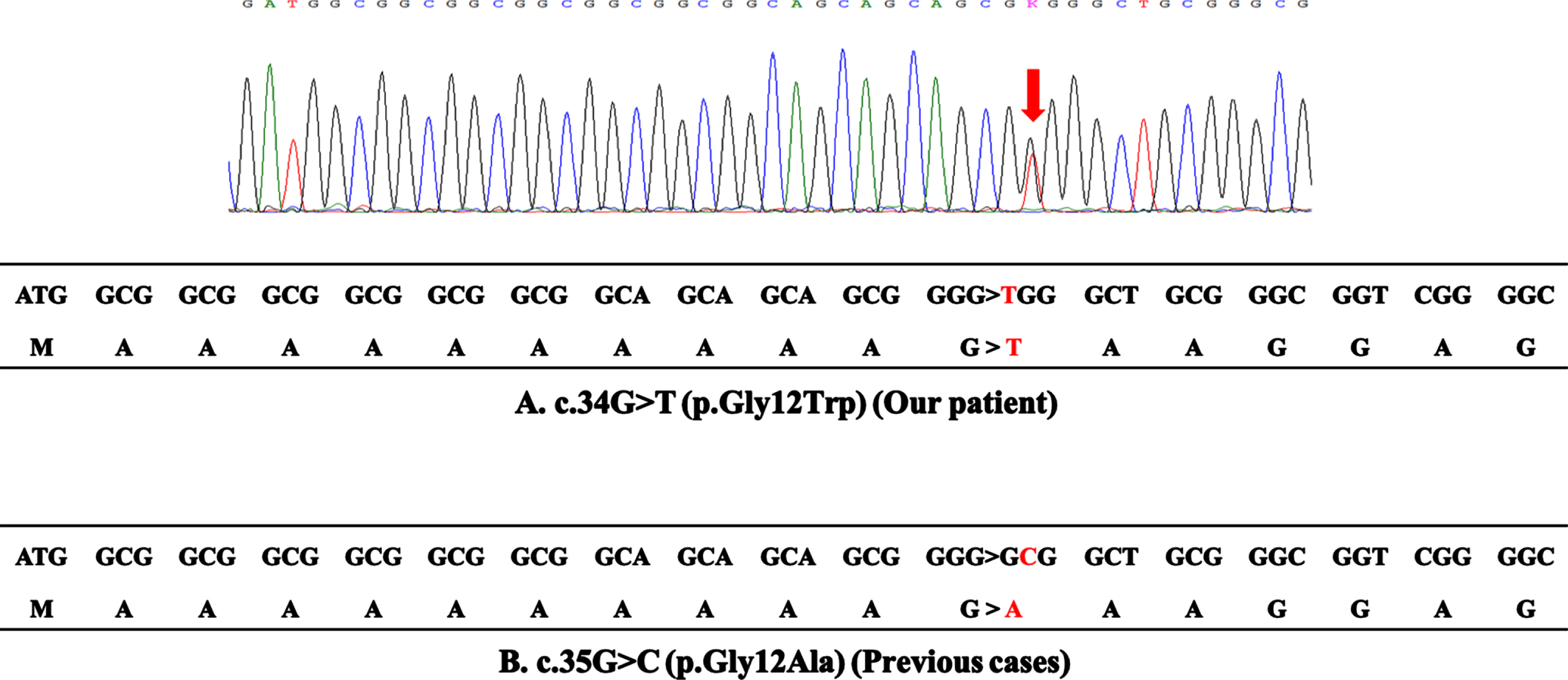
To support this notion, the age of onset of our patient (60 years old) is similar to those of the previously reported four cases with the c.35G > C point mutation. Characteristic symptoms of OPMD, e.g., slowly progressive ptosis, dysphagia, and proximal limb muscle weakness, were comparable to or presented with a milder phenotype than the reported cases with the c.35G > C point mutation (Table 1). The difference in clinical presentation may potentially be because of the difference in the properties of tryptophan compared to alanine.
Table 1
Comparison of the clinical summaries of our case with the c.34G > T mutation and previous cases with the c.35G > C mutation
| No | 1 | 2 | 3 | 4 | Present case |
| PABPN1gene mutation | c.35G > C | c.35G > C | c.35G > C | c.35G > C | c.34G > T |
| Sex | F | M | F | F | M |
| Initial symptom | Ptosis | Ptosis | Swallowing difficulties | Ptosis, dysphasia | Ptosis |
| Age at onset (years) | 61 | 65 | 60 | 62 | 60 |
| Age at diagnosis (years) | 65 | 67 | 75 | 78 | 77 |
| Clinical features | |||||
| ptosis | n.m. | Palpebral aperture was 6 mm at age 68 | Moderate bilateral ptosis at age 75 | Surgery at age 66 | Surgery at age 73 Palpebral aperture was 4 mm at age 77 |
| dysphagia | Ptosis followed by dysphagia | Admitted to choking at age 68 | Frequent choking episodes at age 75 | Repeated aspiration pneumonia at age 78 | Thickening agents in liquid and texture-modified diets at age 77 |
| Muscle weakness | Peripheral muscles were normal at age 65 | Normal at age 67 | Weakness in legs at early 70s | MRC 2–4 at age 78 | MRC 3-4 at age 77 |
| Serum creatine kinase (U/L) | 122 | n.m. | n.m. | n.m. | 153 |
| Reference (No) | Robinson et al., 2006 [2] | Robinson et al., 2011 [3] | Robinson et al., 2011 [3] | Nishii et al., 2021 [4] |
MRC, Medical Research Council; n.m., not mentioned.
CONCLUSION
This is the first report of OPMD due to the PABPN1 c.34G > T (p. Gly12Trp) mutation, which is not caused by alanine codon expansion or elongation. Our result indicates that OPMD-causing missense mutation at the 12th residue is not limited to only harboring a glycine to alanine substitution.
ACKNOWLEDGMENTS
We would like to thank Dr. Francia Victoria De Los Reyes, NCNP, for her critical reading.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Brais B , Bouchard JP , Xie YG , Rochefort DL , Chrétien N , Tomé FM , et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. (1998) ;18: (2):164–7. doi: 10.1038/ng0298-164. |
[2] | Robinson DO , Wills AJ , Hammans SR , Read SP , Sillibourne J Oculopharyngeal muscular dystrophy: A point mutation which mimics the effect of the PABPN1 gene triplet repeat expansion mutation. J Med Genet. (2006) ;43: :e23. doi: 10.1136/jmg.2005.037598. |
[3] | Robinson DO , Hilton-Jones D , Mansfield D , Hildebrand GD , Marks S , Mechan D , et al. Two cases of oculopharyngeal muscular dystrophy (OPMD) with the rare PABPN1 c.35G>C; Gly12Ala point mutation. Neuromuscul Disord. (2011) ;21: (11):809–11. doi: 10.1016/j.nmd.2011.06.003. |
[4] | Nishii Y-S , Noto Y , Yasuda R , Kitaoji T , Ashida S , Tanaka E , et al. A Japanese case of oculopharyngeal muscular dystrophy (OPMD) with PABPN1 c.35G>C; Gly12Ala point mutation. BMC Neurol. (2021) ;21: (1):265. doi: 10.1186/s12883-021-02300-x. |
[5] | Alonso-Jimenez A , Kroon RHMJM , Alejaldre-Monforte A , Nuñez-Peralta C , Horlings CGC , van Engelen BGM , et al. Muscle MRI in a large cohort of patients with oculopharyngeal muscular dystrophy. J Neurol Neurosurg Psychiatry. (2019) ;90: (5):576–85. doi: 10.1136/jnnp-2018-319578. |
[6] | Ogasawara M , Eura N , Iida A , Kumutpongpanich T , Minami N , Nonaka I , et al. Intranuclear inclusions in muscle biopsy can differentiate oculopharyngodistal myopathy and oculopharyngeal muscular dystrophy. Acta Neuropathol Commun. (2022) ;10: (1):176. doi: 10.1186/s40478-022-01482-w. |
[7] | Galimberti V , Tironi R , Lerario A , Scali M , Del Bo R , Rodolico C , et al. Value of insoluble PABPN1 accumulation in the diagnosis of oculopharyngeal muscular dystrophy. Eur J Neurol. (2020) ;27: (4):709–15. doi: 10.1111/ene.14131. |


