
DEVOTE Study Exploring Higher Dose of Nusinersen in Spinal Muscular Atrophy: Study Design and Part A Results
Abstract
Background:
Pharmacokinetic/pharmacodynamic modeling indicates that the higher dose of nusinersen may be associated with a clinically meaningful increase in efficacy above that seen with the 12-mg approved dose.
Objective:
Here we describe both the design of DEVOTE (NCT04089566), a 3-part clinical study evaluating safety, tolerability, and efficacy of higher dose of nusinersen, and results from the initial Part A.
Methods:
DEVOTE Part A evaluates safety and tolerability of a higher nusinersen dose; Part B assesses efficacy in a randomized, double-blind design; and Part C assesses safety and tolerability of participants transitioning from the 12-mg dose to higher doses.
Results:
In the completed Part A of DEVOTE, all 6 enrolled participants aged 6.1–12.6 years have completed the study. Four participants experienced treatment-emergent adverse events (TEAEs), the majority of which were mild. Common TEAEs of headache, pain, chills, vomiting, and paresthesia were considered related to the lumbar puncture procedure. There were no safety concerns regarding clinical or laboratory parameters. Nusinersen levels in the cerebrospinal fluid were within the range of modeled predictions for higher dose of nusinersen. While Part A was not designed for assessing efficacy, most participants showed stabilization or improvement in motor function. Parts B and C of DEVOTE are ongoing.
Conclusions:
The findings from Part A of the DEVOTE study support further development of higher dose of nusinersen.
INTRODUCTION
Spinal muscular atrophy (SMA), an autosomal recessive neurodegenerative disease that affects the lower motor neurons of the spinal cord and brain stem, is characterized by progressive and often severe muscle weakness and atrophy [1]. For most individuals with SMA, onset occurs in infancy and early childhood, although in milder forms, the disease can begin in adulthood [1]. The disease is caused by homozygous deletions or mutations in the survival motor neuron 1 (SMN1) gene that prevent the production of full-length functional SMN protein [2, 3]. A paralogous gene, SMN2, undergoes aberrant splicing most of the time, resulting in the exclusion of exon 7 and the production of a truncated, dysfunctional protein [1, 3]. However, approximately 10% of the SMN2 transcripts include exon 7, resulting in the production of the full-length SMN protein, and a higher number of SMN2 copies is associated with a less severe phenotype [1, 2]. SMA is categorized into 4 subtypes based on the age at onset and the maximum motor function achieved without treatment [1].
Nusinersen, approved for the treatment of SMA in the United States, Europe, and other countries, is an antisense oligonucleotide administered intrathecally that alters the splicing of the SMN2 pre-messenger RNA (mRNA), promoting the expression of full-length SMN2 mRNA and functional SMN protein [4–7]. The recommended nusinersen dosing regimen includes four 12-mg loading doses (3 doses separated by 2 weeks, followed by a fourth dose 1 month later), followed by maintenance doses every 4 months [4, 5]. Results from clinical trials and real-world studies show clinically meaningful efficacy on motor function and survival endpoints across a broad spectrum of SMA populations, from pre-symptomatic and infantile-onset symptomatic infants to children and adults with later-onset SMA [8–14]. Moreover, long-term outcomes from various studies show durability of response and continued improvements in motor function with no reports of treatment-related serious adverse events (AEs) [8, 9, 11, 15–18]. Thus, nusinersen has a well-established, long-term, favorable benefit–risk profile [11–14].
The favorable safety profile of nusinersen provides an opportunity to explore whether higher dose can offer greater improvements in clinical outcomes, which may include both motor and bulbar function. Previous results indicated a dose-response relationship between the levels of nusinersen in the cerebrospinal fluid (CSF) and the decrease in the levels of plasma phosphorylated neurofilament heavy chain, an axonal degeneration biomarker for SMA [19]. Furthermore, pharmacokinetic/pharmacodynamic (PK/PD) analysis of nusinersen CSF levels and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scores indicated that higher dose of nusinersen may be associated with a clinically meaningful increase in efficacy above that seen with the 12-mg approved dose [19].
Here we present the design of the ongoing 3-part DEVOTE study (NCT04089566) evaluating the clinical efficacy and safety of higher dose of nusinersen, as well as the final results from Part A.
MATERIALS AND METHODS
DEVOTE is an ongoing, global, 3-part, Phase 2/3 study designed to evaluate the clinical efficacy, safety, and tolerability of higher-dose regimens of nusinersen intrathecally administered to participants with SMA. The study adheres to the ethical principles outlined in the Declaration of Helsinki and signed consent from the parent or guardian in the case of young children, or the participant if indicated per participant’s age, is mandatory. (See Supplementary Materials4 for a list of investigators, site study personnel, and study sites.)
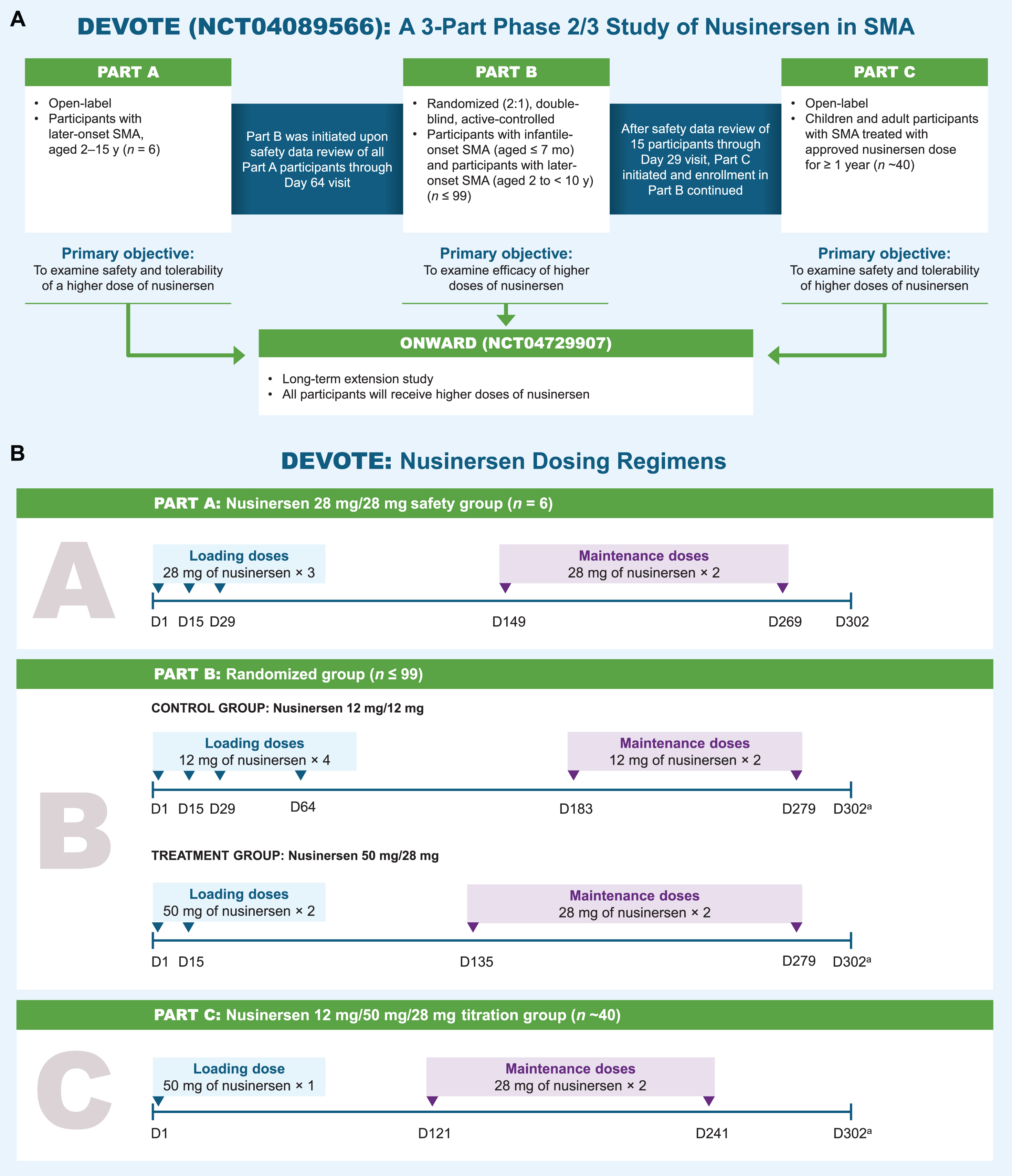
DEVOTE was designed so safety data from Part A were assessed before Part B was initiated. Similarly, initial safety data from Part B were assessed before Part C was initiated. Participants who complete DEVOTE may consider enrolling in the long-term extension study ONWARD (NCT04729907), where all participants receive higher dose of nusinersen. All higher-dose regimens in Parts A, B, and C are expected to achieve similar nusinersen concentrations in the CSF (Ctrough).
Part A
Study design
In Part A, participants aged 2–15 years with SMA symptom onset at≥6 months and an estimated life expectancy > 2 years were eligible for inclusion. Participants were naive to treatment with any SMN2-splicing modifier, gene therapy, or cell transplantation. Excluded participants were those with severe scoliosis, severe contractures, medical necessity for ventilation > 6 hours per 24-hour period, or medical necessity for most nutrition given by gastric feeding tube (i.e., the majority of feeds were given by this route, as assessed by the investigator). Eligible participants received 3 loading doses of intrathecally administered nusinersen 28 mg (Days 1, 15, and 29) followed by 2 maintenance doses of 28 mg (Days 149 and 269). The end-of-study follow-up visit for all participants in Part A was Day 302.
After enrolling the first participant, 72 hours of safety data were reviewed by the investigator and the study sponsor after the first loading dose prior to enrolling any additional participants, with only 1 participant receiving the first dose on a given day. After all 6 participants completed the Day 64 visit, safety data were reviewed by an independent data monitoring committee (IDMC) and enrollment in Part B was initiated. (See Supplementary Materials for a list of the IDMC members.) The first participant received the first dose in study on March 26, 2020, and the last participant visit in Part A took place on June 28, 2021.
Outcome measures
The primary objective of Part A was to assess the safety and tolerability of nusinersen based on the incidence of treatment-emergent AEs (TEAEs), including serious AEs, and clinical and laboratory safety parameters. Clinical safety assessments included electrocardiograms (ECGs), vital signs, physical examination, and neurological examination. Laboratory safety assessments included coagulation parameters, hematology, blood chemistry (which also included evaluation of liver and renal function), CSF (cell count, total protein, and glucose), and urinalysis.
Secondary outcomes included efficacy assessments of motor function and motor milestones, including the change from baseline in the Hammersmith Functional Motor Scale –Expanded (HFMSE) [20] and Revised Upper Limb Module (RULM) scores [21], and total number of new World Health Organization (WHO) motor milestones [22]. Exploratory outcomes included change from baseline in 10-Meter Walk/Run Test (10MWR) and 6-minute walk test (6MWT) in ambulatory participants only, and PK analyses (CSF and plasma levels of nusinersen). PK simulations were conducted for virtual subjects with weights consistent for the age range of the DEVOTE Part A population using a previously published model [19, 23].
Signs and symptoms of dysphagia were assessed by changes on the Parent Assessment of Swallowing Ability (PASA) scale. The PASA questionnaire consists of 33 items across 4 domains that cover General Feeding, Liquid Swallowing, Solid Swallowing, and Parental Assessment of Swallowing Concerns. The first 3 domains are assessed through 5 response (score) options consisting of never (4); rarely (3); sometimes (2); often (1); and always (0), although 2 items were assessed with a simple “yes” or “no” answer. The fourth domain assessing swallowing concerns has 4 response (score) options consisting of strongly disagree (3); disagree (2); agree (1); strongly agree (0). In answering each item, the caregiver is directed to consider the child’s swallowing function in the previous 7 days. The PASA questionnaire was developed by the study sponsor for use in SMA clinical trials and has not yet been validated.
Although Part A assessments included efficacy assessments, it was not designed to draw any conclusions on efficacy due to a limited number of participants and relatively broad inclusion/exclusion criteria.
Statistical analysis
Safety, PK, and efficacy data were summarized using descriptive statistics. The baseline for all HFMSE and RULM assessments was defined for each item as the last non-missing assessment prior to the first dose of treatment. In the event that any items were missing for HFMSE, if a score for the item was available from the previous visit, then this item’s score was utilized. In the event that any items were missing for RULM, if a score for the item was available from the previous visit, then this item’s score was utilized. For items on the RULM with left/right scores, the best of the left and right score was used for calculation of total score. If score for only 1 side was available, the score for this side was used.
Part B
Part B is a randomized, double-blind, active-controlled treatment period examining the clinical efficacy of nusinersen administered intrathecally at higher dose in treatment-naive children with infantile- or later-onset SMA relative to the approved nusinersen 12-mg dose regimen. The study population in Part B comprises participants with infantile- or later-onset SMA similar to prior ENDEAR and CHERISH studies [9, 10]. Eligible participants include children with infantile-onset SMA aged > 1 week and up to 7 months at screening, with symptom onset between 1 week and 6 months and 2 SMN2 copies; and children with later-onset SMA from 2 to < 10 years of age with symptom onset at > 6 months who could sit independently but have never had the ability to walk independently (most likely to develop Type II or Type III SMA). Using interactive response technology, participants are randomized in a 2:1 ratio to either higher dose of nusinersen or the approved nusinersen 12-mg dose regimen. The higher-dose regimen in Part B includes two 50-mg loading doses and two 28-mg maintenance doses. This dosing regimen is predicted to achieve similar nusinersen CSF Ctrough as in Part A, with the 2 higher loading doses of 50 mg in Part B relative to Part A predicted to achieve target concentrations faster. To maintain blinding, sham procedures are administered to ensure the approved dose and higher dose of nusinersen treatment groups have the same dosing visit schedule. Part B aims to enroll up to 99 participants.
The primary outcome measure for Part B is the change from baseline to Day 183 in CHOP INTEND total score in participants with infantile-onset SMA, accounting for mortality using the joint-rank test. Further details will be reported in future publication with data.
Part C
Part C is an open-label safety and tolerability evaluation of participants transitioning from the approved nusinersen dose schedule to a higher dose schedule, who have been receiving nusinersen for≥1 year. Participants receive a single nusinersen 50-mg loading dose followed by two 28-mg maintenance doses 4 months apart. The primary outcome in Part C is to assess safety and tolerability. Further details will be reported in future publication with data.
RESULTS
Final results from Part A
The overall DEVOTE study design and nusinersen dosing regimens used in the study are shown in Fig. 1.
Fig. 1
Study design. aParticipants who meet the protocol criteria for contraception use will complete additional visit (Day 399 in Part B, Day 361 in Part C). D, day. mo, months. SMA, spinal muscular atrophy. y, years.
Study population
Part A (now completed) enrolled 6 participants. Baseline characteristics are shown in Table 1. The majority (5 out of 6 [83.3%]) of participants were male. Four participants (66.7%) were white (1 Hispanic or Latino; 3 non-Hispanic or Latino), while the remaining 2 participants (33.3%) were Asian. Age at screening ranged from 6.1–12.6 years, with a median age of 9.4 years. Age at SMA symptom onset ranged from 8.0–36.0 months (median age of 19.5 months). Age at SMA diagnosis ranged from 9.0–139.0 months (median age of 50.5 months), and time from SMA diagnosis to first study dose ranged from 0.3–7.4 years (median of 5.5 years). Three participants had 3 SMN2 copies, and 3 had 4 SMN2 copies.
Table 1
Baseline characteristics
| Characteristic | Participants |
| N = 6 | |
| Male, n (%) | 5 (83) |
| Age at first screening, years, range | 6.1–12.6 |
| Age at SMA symptom onset, months, range | 8–36 |
| Age at SMA diagnosis, months, range | 9–139 |
| SMN2 gene copies, n (%) | |
| 3 | 3 (50) |
| 4 | 3 (50) |
| Motor milestones ever achieved, n (%) | |
| Sitting without support | 6 (100) |
| Standing without support | 5 (83) |
| Walking with support | 5 (83) |
| Walking without supporta | 5 (83) |
| Motor milestones at screening, n (%) | |
| Sitting without support | 6 (100) |
| Standing without support | 3 (50) |
| Walking with support | 3 (50) |
| Walking without supportb | 3 (50) |
| Wheelchair use at screening, n (%) | 4 (67) |
| Baseline HFMSE score,c range | 6–63 |
| Baseline RULM score,d range | 7–37 |
aBased on achieving walking 15 feet without support. bBased on walking 15 feet without support and achieving WHO milestone of walking (5 steps). The same three participants achieved these motor milestones at screening. cMaximum score possible on the scale: 66. dMaximum score possible on the scale: 37. HFMSE, Hammersmith Functional Motor Scale –Expanded. RULM, Revised Upper Limb Module. SMA, spinal muscular atrophy. SMN2, survival motor neuron 2.
All participants previously achieved and maintained sitting without support. Five participants were previously ambulatory (likely Type III SMA), with 2 losing ambulation prior to screening at 11 and 6 years of age. Three ambulatory participants had high HFMSE and RULM total scores at baseline, reflecting a higher level of motor function, with 2 HFMSE scores near the maximum (scores 62, 63, and 52 out of a maximum of 66), and RULM scores near or equal to the maximum (scores 35, 37, 35 out of a maximum of 37). One nonambulatory participant, the only participant with right and left elbow contractures, had baseline HFMSE and RULM score of 6 points and 7 points, respectively. All participants received all 5 planned study doses of nusinersen and enrolled in the long-term extension study of higher dose of nusinersen, ONWARD.
Safety and tolerability
The 28-mg dose regimen of nusinersen was generally safe and well tolerated. The majority of TEAEs were mild in severity, and none were considered by the investigators to be related to nusinersen. Four of the 6 participants (Participants 1, 2, 3, and 5) together experienced 23 TEAEs, of which 14 were considered related to the lumbar puncture procedure (Table 2). Participant 3 reported 1 event of increased CSF pressure prior to study treatment at Day 1. Participant 4 and Participant 6 did not report any TEAEs. Among a total of 30 lumbar puncture procedures performed in DEVOTE Part A, 7 (23%) were associated with at least 1 AE considered related to the procedure. The most common TEAEs were headache and procedural pain (3 occurrences each), and upper respiratory tract infection, vomiting, chills, paresthesia, and foot deformity (2 occurrences each). One participant (Participant 2, nonambulatory at baseline) had 2 serious TEAEs, a fall and a fracture to the femur, which were not related to study treatment and were resolved.
Table 2
Summary of adverse events by participant
| Participants | AE by MedDRA PT | Related to LPa | Severity | Days in Studyb |
| Participant 1 | Headache | Yes | Mild | 3–5 |
| Vomiting | Yes | Mild | 3–5 | |
| Tonsillitis | No | Mild | 57–72 | |
| Gingivitis | No | Mild | 176–181 | |
| Participant 2 | Procedural painc | Yes | Mild | 2 and 30–32c |
| Headachec | Yes | Mild | 30–31 and 156–157c | |
| Falld | No | Severe | 287 | |
| Femur fractured | No | Severe | 287–289 | |
| Participant 3 | Increased CSF pressuree | No | Mild | 1 |
| Procedural pain | Yes | Mild | 14–18 | |
| Procedural headache | Yes | Moderate | 15–18 | |
| Participant 4 | None reported | – | – | – |
| Participant 5 | Chillsc | Yes | Mild | 3 and 15–16c |
| Paresthesiac | Yes | Mild | 3 and 15–16c | |
| Vomiting | Yes | Mild | 15–16 | |
| Decreased appetite | Yes | Mild | 15–18 | |
| Foot deformityc | No | Moderate; Severec | 34- and 216f | |
| Upper respiratory tract infectionc | No | Mild | 54–63 and 152–160c | |
| Urticaria | No | Mild | 233–266 | |
| Participant 6 | None reported | – | – | – |
aAssessed by the Investigator as related to the LP procedure. bIndicates start and end date of AE. When same AE occurred twice, 2 ranges are provided. cTwo separate events of AE were reported in the same participant. dReported as serious AEs. Not related to study treatment and were resolved. ePrior to study treatment. fThe AE of foot deformity, reported initially on Day 34, did not resolve and was upgraded to severe on Day 216; not resolved prior to the end of study visit. All AEs were classified using MedDRA Version 24.0. AE, adverse event. CSF, cerebrospinal fluid; LP, lumbar puncture. MedDRA PT, Medical Dictionary for Regulatory Activities; Preferred Term.
No TEAEs led to treatment discontinuation or resulted in death. Over the course of the study, there were no clinically meaningful changes in clinical safety measures such as ECG, vital signs, or neurological examination. There were no clinically relevant changes to laboratory safety measures including coagulation parameters, hematology, blood chemistry, or urinalysis, and no clinically relevant CSF abnormalities (as assessed through glucose and protein levels, as well as erythrocyte and leukocyte counts).
Pharmacokinetics
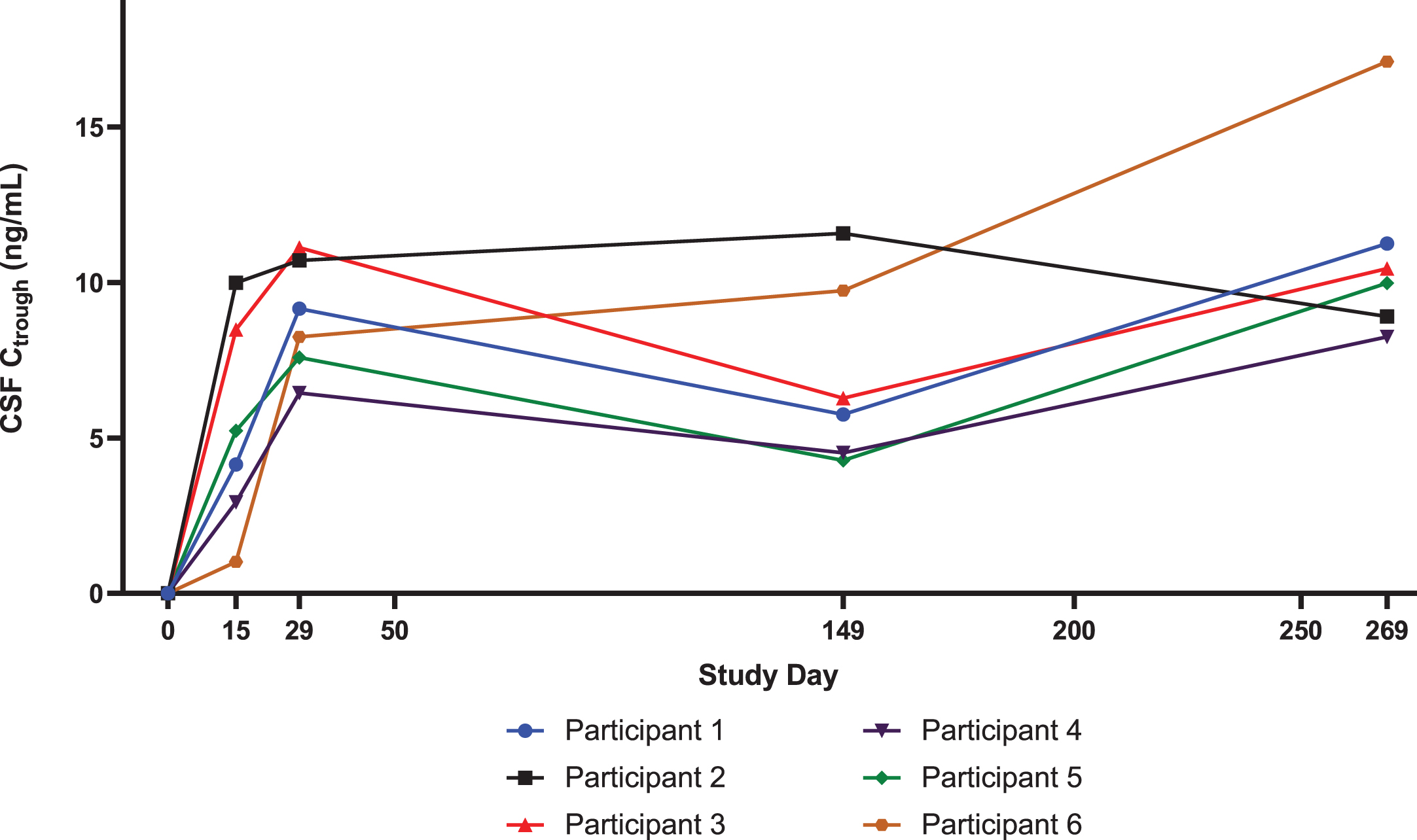
For all participants, nusinersen pre-dose CSF concentrations increased steadily through the loading dose period (i.e., up to Day 29) as expected (Fig. 2). On Day 269 (last measurement in Part A), the observed median pre-dose CSF concentration of 10.2 ng/mL was similar to the median value of 13.1 ng/mL predicted for this higher dose of nusinersen regimen using a previously published population PK model [19, 23]. The observed concentration range at Day 269 (8.3–17.1 ng/mL) was within the 95% predicted range of the population Ctrough simulated at steady state. On Day 29 (after 2 loading doses of 28 mg), the observed median pre-dose CSF concentration of 8.7 ng/mL in DEVOTE Part A was also higher than the observed value of 5.4 ng/mL in ENDEAR [19] (after two loading doses of 12 mg).
Fig. 2
Nusinersen levels in the CSF in DEVOTE Part A are greater than those with the 12-mg dose, as predicted by the population PK model. Individual level DEVOTE Part A data of nusinersen concentrations in the CSF (Ctrough) show steady increase through the loading dose period and reaching median concentration of 8.7 ng/mL on Day 29. On Day 269, the last measurement in Part A, the observed median pre-dose CSF Ctrough was 10.2 ng/mL. The predicted CSF Ctrough for the 12-mg dose regimen are 6.1 ng/mL on Day 29 and 5.6 ng/mL on Day 269. CSF levels of nusinersen increased during loading. CSF, cerebrospinal fluid.
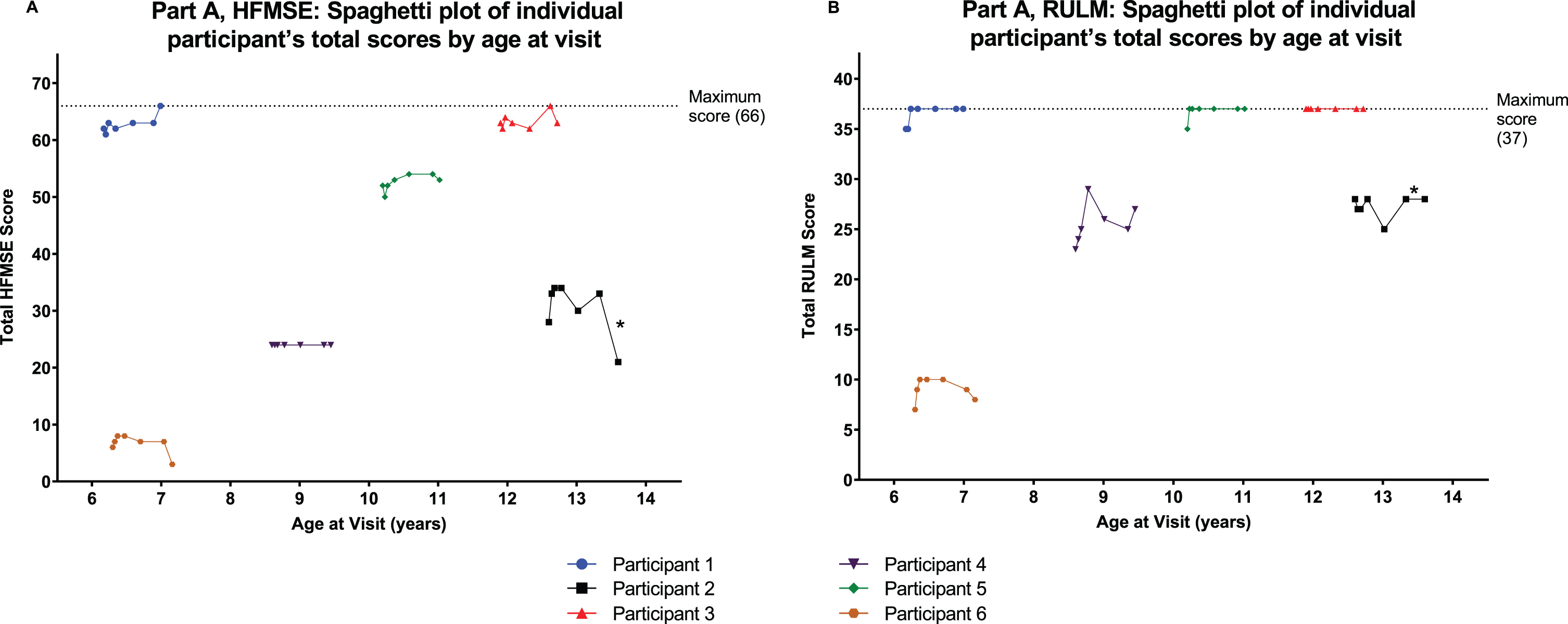
Motor function and motor milestones
Most participants in Part A showed improvement or stabilization in motor function after treatment with nusinersen 28 mg, as assessed by change in HFMSE and RULM scores from baseline to Day 302 (Fig. 3A and 3B; Supplementary Table 1). Participant 2 had a 7-point decrease in HMFSE from baseline to end of study, likely due to a fall and a fractured femur occurring on Day 287, prior to the end of study visit.
Fig. 3
Individual level data for HMFSE and RULM from Day 1 to Day 302 in Part A with x-axis showing age of participants. A: HFMSE total score. B: RULM total score. Most participants in Part A showed improvement or stabilization in motor function after treatment with nusinersen 28 mg. *Participant 2 reported a fall and fractured femur as treatment-emergent adverse events on Day 287, prior to the end of study visit; this participant had a 7-point decrease in HMFSE score from baseline to end of study, likely due to these adverse events. HFMSE, Hammersmith Functional Motor Scale –Expanded. RULM, Revised Upper Limb Module.
Three participants who were able to walk independently at baseline (Participants 1, 3, and 5) were assessed with the 10MWR test. Two participants (Participants 1 and 3) were stable in 10MWR time from baseline to the end of study (Participant 1, –0.34 seconds; Participant 3, +0.68 seconds). Participant 5, who experienced moderate and severe AEs of foot deformity (not resolved at last visit), had a 22-second increase from baseline to the end of study.
All but 1 participant maintained WHO motor milestones from baseline through the end of study. One participant (Participant 2) lost the ability to crawl on hands and knees (reported during the Day 269 study visit), with other milestones unaffected. This loss of milestone by Participant 2 may reflect limitations due to the bilateral Achilles and right hamstring tenotomies performed on Day 214.
Dysphagia
Changes in the PASA scale suggested maintenance of functioning with occasional signs of possible improvement. On Section 1 to Section 3 of the PASA scale, which covered general feeding, drinking liquids, and eating solid foods, most participants had maximum scores at baseline and Day 302 for most items. For the remaining items, improvement or stabilization was observed for those participants with less than maximum scores. In Section 4, which covered parental swallowing concerns, baseline scores varied across items, with most participants exhibiting stable or improved scores from baseline to Day 302 (Supplementary Figure 1).
Parts B and C
Safety data for the first 15 participants in Part B were assessed by an IDMC when they had completed Day 29 visit to determine if further enrollment into Part B could continue, and Part C be initiated. As of November 21, 2022, no safety concerns were identified, and Part B and Part C are ongoing.
DISCUSSION
The previously described PK/PD model indicates that the higher dose of nusinersen may be associated with a clinically meaningful increase in efficacy above that seen with the 12-mg approved dose of nusinersen [19]. The DEVOTE study has been designed to assess safety, tolerability, and efficacy of a higher dose of nusinersen. The nusinersen 28-mg dose regimen studied in Part A was generally safe and well tolerated in the 6 participants studied. The majority of the TEAEs reported in Part A were mild and reported as related to the lumbar puncture procedure by the investigator, and included headache, back pain, vomiting, and chills. Headache, back pain, and vomiting were also commonly reported in clinical trials of later-onset SMA, and the systematic safety review concluded that rates and types of AEs observed with nusinersen treatment were consistent with the lumbar puncture procedure or SMA itself [24]. Out of 30 lumbar puncture procedures in DEVOTE, 7 (23%) were associated with at least 1 AE considered related to lumbar puncture. While this is based on a small sample size (only 6 participants in Part A), making estimation of true percentage rate with this dosing regimen less certain, the frequency is consistent with that previously reported in later-onset participants treated with nusinersen 12 mg (21% within 168 hours) [24]. Of note, the current assessment in DEVOTE Part A relies on investigator’s assessment of relationship to lumbar puncture procedures while prior trials analyzed relationship to lumbar puncture based on specific AEs (by preferred terms) and certain time intervals since lumbar puncture [24].
The nusinersen exposures measured in Part A were similar to those predicted by the previously published population PK model for higher dose of nusinersen [19, 23] and were greater than those measured for the 12-mg dose. On Day 269 (last measurement in Part A), the observed median pre-dose CSF concentration of 10.2 ng/mL was similar to the median value of 13.1 ng/mL predicted for this higher dose of nusinersen regimen using population PK model. The previously reported values for steady-state concentrations of higher dose of nusinersen (12.4 ng/mL) are predicted to be achieved after 6 maintenance doses, or approximately 24 months after treatment initiation [19], a timepoint beyond the DEVOTE study. Importantly, the observed nusinersen CSF exposures in DEVOTE Part A are above those noted with the approved 12-mg dose regimen. Specifically, the population PK model leveraging data from all trials indicated that the approved 12-mg dose at Day 269 led to a median value of 5.6 ng/mL. The values observed at Day 29 with DEVOTE Part A (median of 8.7 ng/ml) were also higher than those observed with 12 mg at the same timepoint in ENDEAR (median of 5.4 ng/mL) and predicted by the model (median of 6.1 ng/mL) [19, 23]. Thus, the observed nusinersen CSF levels are consistent with the model and support the validity of testing the higher-dose regimen in Parts B and C of the DEVOTE study.
Part A was not designed to assess efficacy of the 28-mg dose given the small sample size (n = 6) and broad inclusion/exclusion criteria. This included allowing participants with scores near maximum of HFMSE/RULM scales, making an improvement difficult to observe in these scales. Furthermore, none of the 6 participants in Part A would have met the inclusion criteria for CHERISH, a Phase 3 trial in participants with later-onset SMA (e.g., can sit independently, never walked independently, HFMSE score 10–54) [9]. All 6 Part A participants had a wide range of baseline HFMSE and RULM scores. High baseline scores on the HFMSE and RULM scales were observed in 2 and 3 participants, respectively, which likely limited the opportunity to observe additional motor function improvements in these participants. In addition, 1 participant had low motor function at baseline, which may also impact observed changes in these scales over the short period of time in this trial. Over the course of the study, most participants receiving a higher 28-mg dose of nusinersen showed improvement or stabilization of motor function on HFMSE and RULM measures. Evaluation on the 10MWR test was limited owing to only 3 participants being ambulatory. The 6MWT data were incomplete without pre-dose data, making interpretation difficult. Five participants had no change in their WHO motor milestones. One participant (Participant 2) lost the ability to crawl on hands and knees, likely related to tenotomy procedures. Thus, given the limited number of participants in Part A, and their heterogeneous clinical presentation of SMA as shown by the range in baseline motor function (with some scores at the very high or low end of the spectrum), conclusions on efficacy cannot be drawn from Part A. Indeed, it is the objective of the ongoing blinded Part B to assess efficacy of the higher dose. A higher loading dose may also generate a more rapid initial clinical response, a clinically relevant topic which Part B can potentially address.
The final results from Part A of the DEVOTE study (n = 6) show that the 28-mg dose regimen of nusinersen studied was generally safe and well tolerated. The concentrations of nusinersen in the CSF are higher than those reported for 12-mg and consistent with predicted values for higher dose of nusinersen [19]. Overall, the data from Part A support continued development of higher dose of nusinersen. The DEVOTE study in its entirety will provide valuable information on the efficacy, pharmacokinetics, and safety of higher dose of nusinersen in a broad population of participants with SMA.
CONFLICT OF INTEREST
RSF: is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review; consultant to AveXis, Biogen, Capricor, Cytokinetics, Genentech, Novartis, Roche, and Scholar Rock on SMA related topics and with no financial interests in these companies; received SMA clinical trial funding from Biogen/Ionis, AveXis/Novartis, Genentech/Roche, Cytokinetics, and Scholar Rock; research funding from Biogen, Cure SMA, Genentech, SMA Foundation, Muscular Dystrophy Association, National Institutes of Health; data safety monitoring boards for the AveXis AVXS-101 START study, Roche MOONFISH study, and Ionis Angelman HALOS study; received royalty payments from Children’s Hospital of Philadelphia for licensing fees obtained for use of the CHOP INTEND motor function scale.
JWD: is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review; consultant for Avidity Biosciences; Biogen; BioMarin; Cytokinetics; Epirium Bio; Ionis Pharmaceuticals; Kate Therapeutics; Novartis Gene Therapies; PepGen; Roche/Genentech Pharmaceuticals; Sarepta Therapeutics; Scholar Rock; Shift Therapeutics; Vertex Pharmaceuticals, with no financial interest in these companies; research grant support from AMO Pharma; Avidity Biosciences; Biogen; Cure SMA; Cytokinetics; Genentech; Ionis Pharmaceuticals; Muscular Dystrophy Association; Novartis Gene Therapies; Roche Pharmaceuticals; Sanofi-Genzyme; Sarepta Therapeutics; Scholar Rock; SMA Foundation. He receives royalties and licensing fees from Athena Diagnostics for patents related to genetic testing of myotonic dystrophy type 2 (US patent 7442782) and spinocerebellar ataxia type 5 (US patent 7527931) assigned to Regents of the University of Minnesota and licensed to Athena Diagnostics.
SIPP: has participated in advisory boards for SMA studies for AveXis, Biogen, Ionis, Novartis, and Roche; has served as a Principal Investigator for Biogen/Ionis and Roche clinical trials.
MMR: advisory boards for nonprofit organizations: FSHD Global Research Foundation, Muscular Dystrophy Foundation, and Save Our Sons Duchenne Foundation; honoraria from Biogen and BioMarin; research funding from FSHD Global Research Foundation and Save Our Sons Duchenne Foundation; grants/advisor fees from Biogen and Genzyme.
EM: advisory boards for SMA studies for AveXis, Biogen, Ionis, Novartis, and Roche; Principal Investigator for ongoing Biogen/Ionis and Roche clinical trials; funding from Famiglie SMA Italy, Italian Telethon, and SMA Europe.
DCD: advisor/consultant for AveXis, Biogen, Cytokinetics, Ionis, METAFORA, Roche, Sanofi, Sarepta, Scholar Rock, SMA Foundation, and Ultragenyx, with no financial interests in these companies; grants from Cure SMA, Department of Defense, Glut1 Deficiency Foundation, Hope for Children Research Foundation, National Institutes of Health, and SMA Foundation; research funding from Department of Defense, Glut1 Deficiency Foundation, Hope for Children Research Foundation, iSMAC initiative (Biogen), National Institutes of Health, Sanofi, and SMA Foundation; clinical trial funding from Biogen, Ionis, Mallinckrodt, PTC, Santhera, Sarepta, Scholar Rock, and Ultragenyx.
JM: advisory boards for Biogen, Roche, and Scholar Rock; consultant for Biogen and Scholar Rock; research support from Eunice Kennedy Shriver National Institute for Child Health and Human Development (K01HD084690) and Muscular Dystrophy Association (575870 and 629259).
JG-G: advisory boards for AveXis/Novartis, Biogen, BioMarin, PTC, Roche, and Sarepta; Principal Investigator for ongoing clinical PTC and Sarepta trials; research funding from CAPES, CNPq, and FAPEMIG (Brazil).
MM, GG, CM, RF, and ZB: employees of and hold stock/stock options in Biogen.
ACKNOWLEDGMENTS
The authors thank the children and their parents/guardians and family members for their participation in this study, without whom this effort would not succeed. We also thank the many people who are contributing to this study, including the study site principal investigators, clinical monitors, study coordinators, physical therapists, and laboratory technicians. Vanessa Ducas from Excel Scientific Solutions provided writing assistance in the development of the first and subsequent drafts based on input from authors, and Cara Farrell from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. The authors had full editorial control of the paper and provided their final approval of all content.
FUNDING
Biogen provided funding for medical writing support in the development of this report. This study was sponsored by Biogen (Cambridge, MA, USA).
DATA AVAILABILITY STATEMENT
Request for the data supporting this manuscript should be submitted to https://vivli.org/.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-221667.
REFERENCES
[1] | Mercuri E , Sumner CJ , Muntoni F , Darras BT , Finkel RS . Spinal muscular atrophy. Nat Rev Dis Primers. (2022) ;8: (1):52. DOI: 10.1038/s41572-022-00380-8. |
[2] | Talbot K , Tizzano EF . The clinical landscape for SMA in a new therapeutic era. Gene Ther. (2017) ;24: (9):529–33. DOI: 10.1038/gt.2017.52. |
[3] | Darras BT , Monani UR , De Vivo DC . Genetic disorders affecting the motor neuron: Spinal muscular atrophy. In: Swaiman KF, Ashwal S, Ferriero DM, Schor NF, Finkel RS, Gropman AL, et al., editors. Swaiman’s Pediatric Neurology: Principles snd Practice 6th ed. Edinburgh: Elsevier; 2017. pp. 1057-64. |
[4] | SPINRAZA (nusinersen) injection, for intrathecal use. Prescribing Information. Cambridge, MA: Biogen; 2016 [updated 2023 February; cited 2023 April 19]. Available from: www.spinraza.com/content/dam/commercial/specialty/spinraza/caregiver/en_us/pdf/spinraza-prescribing-information.pdf. |
[5] | SPINRAZA (nusinersen). Summary of Product Characteristics. The Netherlands: Biogen; 2017 [updated 2022 February 02; cited 2023 April 19]. Available from: https://www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_en.pdf |
[6] | Passini MA , Bu J , Richards AM , Kinnecom C , Sardi SP , Stanek LM , et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. (2011) ;3: (72):72ra18. DOI: 10.1126/scitranslmed.3001777. |
[7] | Hua Y , Sahashi K , Hung G , Rigo F , Passini MA , Bennett CF , et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. (2010) ;24: (15):1634–44. DOI: 10.101/gad.1941310. |
[8] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. ENDEAR Study GrouNusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1723–32. DOI: 10.056/NEJMoa1702752. |
[9] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. CHERISH Study GrouNusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) ;378: (7):625–35. DOI: 10.1056/NEJMoa1710504. |
[10] | Darras BT , Chiriboga CA , Iannaccone ST , Swoboda KJ , Montes J , Mignon L , et al. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology. (2019) ;92: (21):e2492–e506. DOI: 10.1212/WNL.0000000000007527. |
[11] | De Vivo DC , Bertini E , Swoboda KJ , Hwu WL , Crawford TO , Finkel RS , et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. (2019) ;29: (11):842–56. DOI: 10.1016/j.nmd.2019.09.007. |
[12] | Hagenacker T , Wurster CD , Gunther R , Schreiber-Katz O , Osmanovic A , Petri S , et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. (2020) ;19: (4):317–25. DOI: 10.1016/S1474-4422(20)30037-5. |
[13] | Szabo L , Gergely A , Jakus R , Fogarasi A , Grosz Z , Molnar MJ , et al. Efficacy of nusinersen in type 1, 2 and 3 spinal muscular atrophy: Real world data from Hungarian patients. Eur J Paediatr Neurol. (2020) ;27: :37–42. DOI: 10.1016/j.ejpn.2020.05.002. |
[14] | Walter MC , Wenninger S , Thiele S , Stauber J , Hiebeler M , Greckl E , et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3 - A Prospective Observational Study. J Neuromuscul Dis. (2019) ;6: (4):453–65. DOI: 10.3233/JND-190416. |
[15] | Darras BT , Chiriboga CA , Farrar MA , Mercuri E , Kirschner J , Kuntz NL , et al. Longer-term treatment with nusinersen: Results in later-onset spinal muscular atrophy from the SHINE study. Cure SMA - 2020 Annual Conference (Virtual Presentation). June 11-14, 2020. |
[16] | Kirschner J , Crawford TO , Ryan MM , Finkel RS , Swoboda KJ , De VivoDC, et al. Nusinersen Effect in presymptomatic SMA infants: 4.9 year interim of the NURTURE study. SMA Europe - 3rd International Scientific Congress on Spinal Muscular Atrophy. October 21-23, 2022; Barcelona, Spain. |
[17] | Acsadi G , Crawford TO , Müller-Felber W , Shieh PB , Richardson R , Natarajan N , et al. Safety and efficacy of nusinersen in spinal muscular atrophy: The EMBRACE study. Muscle Nerve. (2021) ;63: (5):668–77. DOI: 10.1002/mus.27187. |
[18] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: Final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc Health. (2021) ;5: (7):491–500. DOI: 10.1016/S2352-4642(21)00100-0. |
[19] | Finkel RS , Ryan MM , Pascual Pascual SI , Day JW , Mercuri E , De Vivo DC , et al. Scientific rationale for a higher dose of nusinersen. Ann Clin Transl Neurol. (2022) ;9: (6):819–29. DOI: 10.1002/acn3.51562. |
[20] | O’Hagen JM , Glanzman AM , McDermott MP , Ryan PA , Flickinger J , Quigley J , et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord. (2007) ;17: (9-10):693–7. DOI: 10.1016/j.nmd.2007.05.009. |
[21] | Mazzone ES , Mayhew A , Montes J , Ramsey D , Fanelli L , Young SD , et al. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve. (2017) ;55: (6):869–74. DOI: 10.1002/mus.25430. |
[22] | WHO Multicentre Growth Reference Study Group. WHO Motor Development Study: Windows of achievement for six gross motor development milestones. Acta Paediatr Suppl. (2006) ;450: :86–95. DOI: 10.1111/j.1651-2227.2006.tb02379.x. |
[23] | MacCannell D , Berger Z , East L , Mercuri E , Kirschner J , Muntoni F , et al. Population pharmacokinetics-based recommendations for a single delayed or missed dose of nusinersen. Neuromuscul Disord. (2021) ;31: (4):310–8. DOI: 10.1016/j.nmd.2021.02.014. |
[24] | Darras BT , Farrar MA , Mercuri E , Finkel RS , Foster R , Hughes SG , et al. An integrated safety analysis of infants and children with symptomatic spinal muscular atrophy (SMA) treated with nusinersen in seven clinical trials. CNS Drugs. (2019) ;33: (9):919–32. DOI: 10.1007/s40263-019-00656-w. |


