
Presynaptic Congenital Myasthenic Syndromes: Understanding Clinical Phenotypes through In vivo Models
Abstract
Presynaptic congenital myasthenic syndromes (CMS) are a group of genetic disorders affecting the presynaptic side of the neuromuscular junctions (NMJ). They can result from a dysfunction in acetylcholine (ACh) synthesis or recycling, in its packaging into synaptic vesicles, or its subsequent release into the synaptic cleft. Other proteins involved in presynaptic endplate development and maintenance can also be impaired.
Presynaptic CMS usually presents during the prenatal or neonatal period, with a severe phenotype including congenital arthrogryposis, developmental delay, and apnoeic crisis. However, milder phenotypes with proximal muscle weakness and good response to treatment have been described. Finally, many presynaptic genes are expressed in the brain, justifying the presence of additional central nervous system symptoms.
Several animal models have been developed to study CMS, providing the opportunity to identify disease mechanisms and test treatment options. In this review, we describe presynaptic CMS phenotypes with a focus on in vivo models, to better understand CMS pathophysiology and define new causative genes.
INTRODUCTION
Congenital myasthenic syndromes (CMS) are a group of genetically inherited diseases, resulting in impaired function of the neuromuscular junction (NMJ). CMS can be classified into presynaptic, synaptic, and postsynaptic defects depending on the localization of the affected protein. A fourth CMS group comprises disorders related to glycosylation [1].
The prevalence of CMS is highly variable worldwide, but it has been estimated at 1.8 to 14.8 per million under those aged 18 years [2–4].
Clinical onset is usually within the first two years of life with non-progressive fluctuating muscle weakness and fatigability involving ocular, bulbar, and limb muscles. Less frequently CMS presents at birth with respiratory discomfort, feeding difficulties, poor cry, joint contractures, and other typical symptoms and signs [5]. However, it is not uncommon that some symptoms appear later in life, resulting in an “adult-onset” CMS. This occurs in the case of a few postsynaptic (e.g., slow channel syndrome, DOK7, and RAPSN) and glycosylation-defect related forms (e.g., GFPT1) [6]. Some patients suffer from subtle muscular manifestations in infancy, but do not exhibit a clear distal or proximal muscle weakness until early or late adulthood [7].
CMS diagnosis is based on personal and family history, clinical examination, and neurophysiological studies. A decrement of the compound muscle action potential (CMAP) amplitude is typically detected in response to repetitive nerve stimulation (RNS), and single fibre electromyography (SFEMG) can reveal abnormal jitter and blocking. However, these findings are not always observed due to the fluctuating nature of CMS symptoms, so gene sequencing remains crucial in confirming the diagnosis [1].
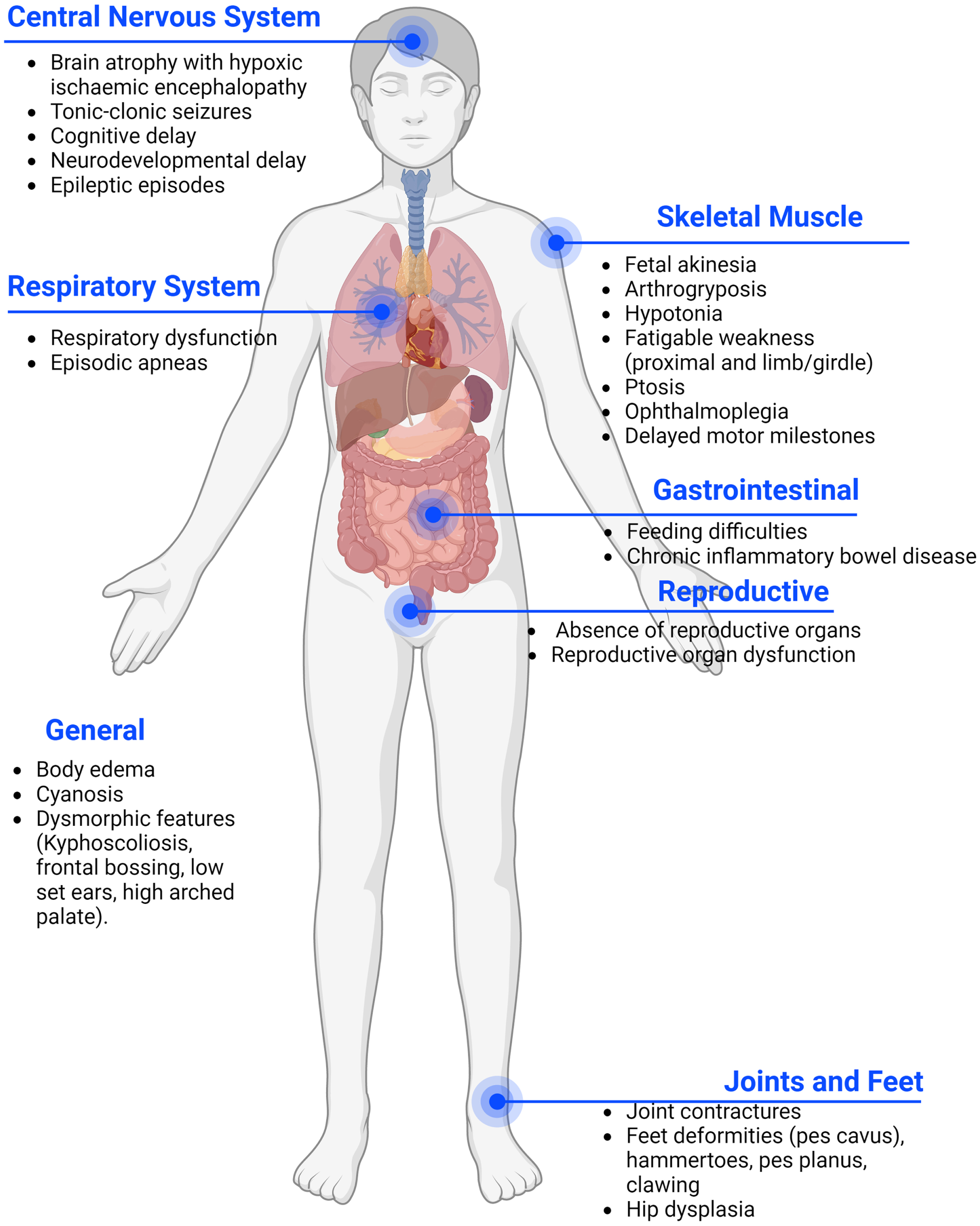
Presynaptic forms are deemed at around 5% -10% of all CMS and usually present earlier than postsynaptic forms, at birth or during the prenatal period. CHAT defects are the most frequent among presynaptic CMS, amounting to 4-5% of all CMS. Each of the other presynaptic CMS represents less than 1% of the total [5]. In pre-synaptic forms the phenotype is more severe including congenital arthrogryposis, joint contractures, developmental delay, and apnoeic crisis that can result in death [5], The abnormal cognitive development may also be impacted by hypoxic brain damage, as suggested by the radiological presence of hypoxic ischaemic encephalopathy in several CMS patients affected by repeated apneas [8]. Moreover, many presynaptic genes are also expressed in the brain, leading to additional central nervous system (CNS) symptoms which are uncommon in postsynaptic CMS (Fig. 1) [9–11]. However, milder cases of presynaptic CMS have been described, presenting with mild proximal weakness and good response to treatment [11].
Fig. 1
Common phenotypes of pre-synaptic forms of CMS. In pre-synaptic forms of CMS the phenotype is frequently more severe due to the presence of affected genes in the peripheral and central nervous system. Patients often present with a multi-systemic condition.
Presynaptic CMS can result from a dysfunction in acetylcholine (ACh) synthesis or recycling, in its packaging into synaptic vesicles, or its subsequent release into the synaptic cleft [12]. Recently, new proteins have been discovered that are involved in presynaptic endplate development and maintenance, although their function is not yet fully explained [13–15].
Increasingly animal models have been used to study the NMJ, aided by the fact that all mammals and many other research organisms use ACh for neuromuscular transmission. Presynaptic CMS caused by a mutated pathway directly associated with ACh can only be modelled in species using ACh as a neurotransmitter. In contrast, other CMS can be evaluated in a wider range of species, resulting in many discoveries with respect to NMJ formation and function.
In Zebrafish, which use ACh for synaptic transmission, morpholino oligonucleotides (MOs) have been used to knock down genes of interest at the NMJ. More recently CRISPR/Cas9 technology has been employed. Mouse models of CMS have also been extensively used to determine pathomechanisms and investigate potential drug treatments. These have been both tissue specific and whole-body knock outs. Caenorhabditis elegans (C. elegans) is another animal using ACh as the principal excitatory transmitter at the NMJ with a well-conserved synaptic machinery and a simple genetic system [16]. Drosophila melanogaster (D. melanogaster) is an additional good model for studying synaptic transmission. Indeed, many NMJ proteins are conserved between Drosophila and vertebrates, as well as synaptic molecular development. However, flies use glutamate as an excitatory neurotransmitter, highlighting the necessity of identifying the right animal model to use [17].
In this review, we provide an update on presynaptic CMS phenotypes with a focus on in vivo models which are important to better understand NMJ disease mechanisms and for defining new causative genes.
We conducted a PubMed literature search, considering papers of any design, except for reviews, published in English from January 2000 to March 2022. Each presynaptic CMS gene was checked as single search term and in association with “congenital myasthenic syndromes”, while the relative encoded protein was searched in combination with “models”. We selected studies focused on the evaluation of altered protein effects, mutant animal phenotypes, and CMS clinical case reports, assessing their reference lists to identify other relevant articles. Additional information for the characterization of genes and proteins was obtained from https://www.genecards.org/ and https://www.ncbi.nlm.nih.gov/omim/.
We provide details about the function and tissue expression of each gene. Then we proceed to delineate the clinical picture of the related CMS, highlighting how the use of animal models has allowed a better understanding of the pathophysiological mechanisms of the disease.
ACETYLCHOLINE SYNTHESIS AND RECYCLING
SLC5A7
Solute Carrier Family 5 Member 7 gene (SLC5A7), also called Choline Transporter (CHT), is located on chromosome 2q12.3, and encodes a high-affinity choline transporter (CHT). CHT is involved in the resynthesis of ACh, taking up the choline from the extracellular space into presynaptic terminals (GC02P107969, GeneCards). (Fig. 2).
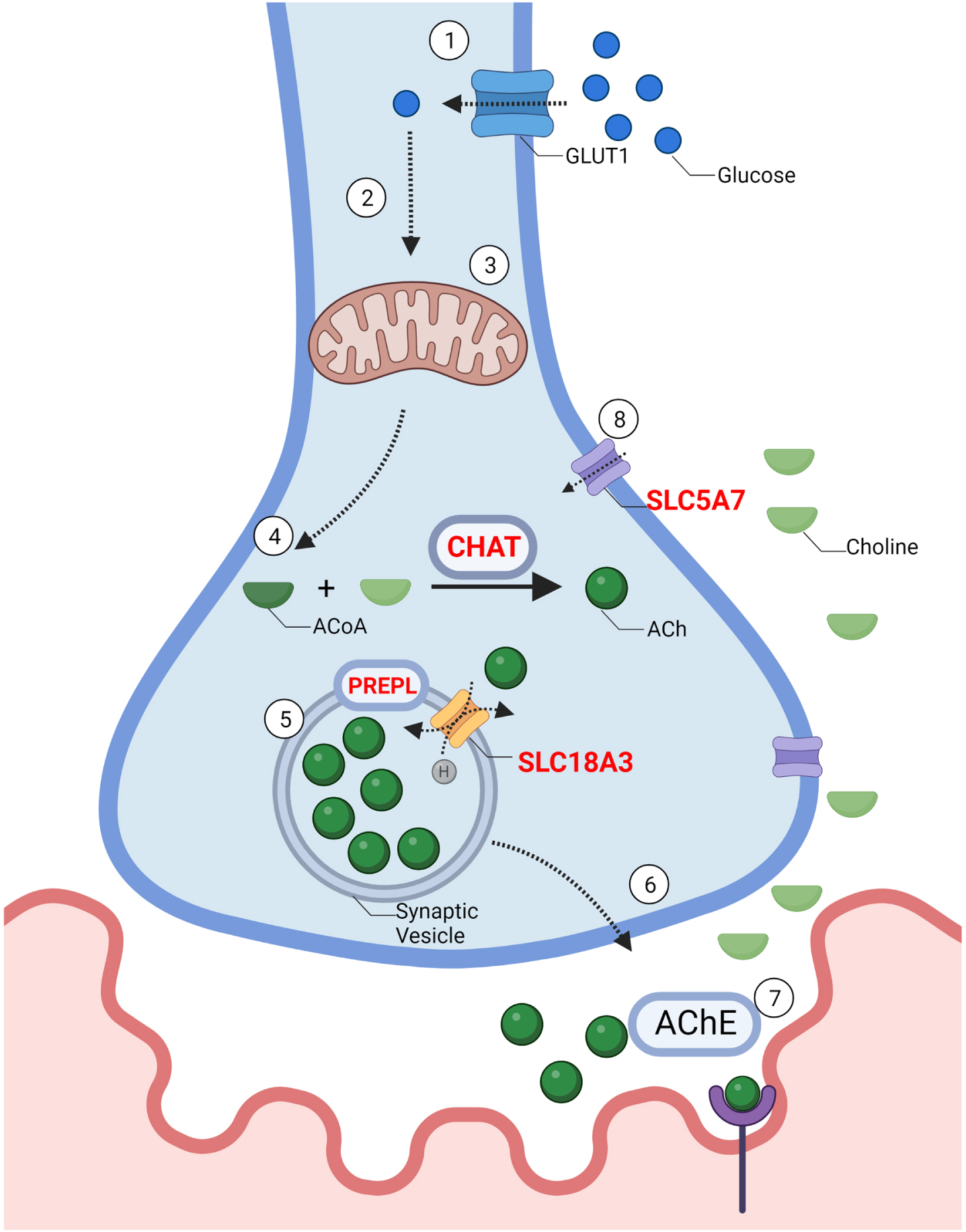
Fig. 2
–The synthesis and recycling of acetylcholine: 1. Synthesis of ACh requires the influx of glucose through a glucose transporter (GLUT) into the cell. 2. Intracellular glucose is then metabolized by glycolysis to produce pyruvate 3. Pyruvate then enters the mitochondria where it is further metabolized to Acetyl-CoA. 4. Choline acetyltransferase (CHAT) combines Acetyl-CoA with choline to produce acetylcholine (ACh). 5. ACh is packaged into vesicles through Vesicular Acetylcholine Transporter (SLC18A3), a 12-membrane protein located on the synaptic vesicle. Prolyl Endopeptidase (PREPL), plays a role in the packaging of ACh and other pre-synaptic machinery such as SNAREs into the synaptic vesicle, although the exact mechanism remains unknown. 6. Vesicles travel towards the pre-synaptic membrane and undergo exocytosis. 7. ACh is released from the pre-synaptic membrane through vesicle exocytosis. ACh diffuses across the synaptic cleft and binds to acetylcholine receptors (AChR). Acetylcholine esterase (AChE) breaks down ACh. 8. SLC5A7 is a Na+ and Cl- dependent high-affinity transporter that mediates the uptake of choline at the motor neuron terminal. Genes implicated in CMS are in red.
SLC5A7 CMS is typically inherited in an autosomal recessive manner presenting at birth, however, prenatal signs like antenatal hydramnios, reduced foetal movements, and arthrogryposis have been described [8, 9, 18–20]. Respiratory dysfunction with life-threatening episodic apnoeas is a constant feature, in variable association with hypotonia, fatigable weakness, oculo-bulbar symptoms and gastrointestinal alterations [8, 9, 18–21]. RNS reveals a decremental response, and brain MRI may show brain atrophy with hypoxic ischaemic encephalopathy [8, 9, 18, 20, 21]. Cognitive defects and neurodevelopmental delay are not unusual features in SLC5A7 CMS, underlying the link between cortical cholinergic neurotransmission and CHT [9, 20, 21] (Table 1).
Table 1
Summary of clinical and instrumental features of published patients affected by pre-synaptic CMS
| Gene Chromosome Inheritance | Frequency | Onset | Phenotype | Brain MRI | RNS/SFEMG | Muscle biopsy | Genotype | Drugs | Ref. |
| SLC5A7 2q12.3 AD | 14 pts | con | Hypotonia, apneas, swallowing difficulties, developmental delay, ptosis, ophtalmoplegia, muscle atrophy (rare: hydramnios, arthrogriposis, facial dysmorphism, seizures) | brain atrophy (rare: intraventricular and putamen hemorrage) | decremental response to RNS | Myopathic changes, atrophic fibres, targetoid areas | c.282T>A/c.282T>A (p.Ser94Arg/Ser94Arg) c.335T>A/c.335T>A (p.Val112Glu/Val112Glu) c.629 C>T/c.629 C>T (p.Pro210Leu/Pro210Leu) c.194 G>A/c.313 C>T ((p.Gly65Glu/Pro105Ser) | some response to pyridostigmine and ephedrine; no response to 3,4-DAP and salbutamol | [8, 9, 18–21] |
| CHAT 10q11.23 AR | 52 pts | con or early infancy | reduced fetal movements, hypotonia, apneas, swallowing and feeding difficulties, developmental delay, fluctuated ptosis, ophthalmoparesis, muscle fatigability | (rare: mild delay in myelination of the centrum semiovale, and corpus callosum atrophy) | decremental response to RNS after prolonged high frequency; increased jitter and blocking at SFEMG | unspecific myopathic changes, type 1 fibre predominance | c.1007T>C/c.1007T>C (p.Ile336Thr/Ile336Thr) c.629T>C/ 631 C>G (p.Leu210Pro/Pro211Ala) c.678_684delTGGCACC/2078T>G (p.*/Ile693Ser) c.1078 G.A/1078 G.A (p.Gly360Arg/Gly360Arg) | variable response to pyridostigmine and 3,4-DAP | [11, 30–41] |
| SLC18A3 10q11.23 AR | 4 pts | con | hypotonia, cyanosis, feeding difficulties, global developmental disability, ptosis, ophthalmoplegia, fatigable weakness (rare: arthrogryposis, severe necrotizing enterocolitis) | brain atrophy, demyelination (rare: small hemorrhages in the frontal and parietal lobes) | decremental response to RNS | n.p. | c.557 G>C/557 G>C (p.Gly186Ala/Gly186Ala)+10q11.22-q11.23del c.1192 G>C/1192 G>C (p.Asp398His/Asp398His) c.1078 G.A/1078 G.A (p.Gly360Arg/Gly360Arg) | slight response to pyridostigmine, and 3,4- DAP, distigmine and ephedrine | [10, 47–51] |
| SNAP25 20p12.12 AD | 2 pt | con | fetal hypomotility, cyanosis, fluctuating ptosis, dysarthria, ataxia, fatigable weakness, arthrogryposis and fatal respiratory failure (1 pt) | normal (1 pt), n.p. (1 pt) | decremental response to RNS (1 pt), n.p. (1 pt) | normal (1 pt), mild fibres size variation (1 pt) | c.200T>A (p.Ile67Asn) c.529 C>T (p.Gln177X) | no response to pyridostigmine; slight response to 3,4-DAP | [74] |
| VAMP1/ SYB1 12p13.21 AR | 9 pts | con | hypotonia, feeding difficulties, delayed motor development, ophthalmoparesis, facial and bulbar muscle weakness, generalized muscle weakness, joint contractures | n.p | decremental response to RNS | myopathic features and borderline low complex IV activity, type 2 fibres atrophy | c.200T>A/200T>A (p.Ile67Asn/Ile67Asn) c.146 G>C/146 G>C (p.Arg49Pro/Arg49Pro) c.51_64delAGGTGGGG GTCCCC/ c.51_64delAGGTGGGG GTCCCC (p.Gly18TrpfsTer5*/Gly18TrpfsTer5*) c.340delA/340delA (p.Ile114SerfrsTer72 for isoform A, p.Ser114ValfsTer32 for isoform D) | slight response to pyridostigmine and salbutamol | [67–70] |
| SYT2 1q32.1 AD | 10 pts | childhood | ptosis, ophthalmoparesis, limb weakness, muscle fatigue, gait difficulties, foot deformities, reduced or absent deep tendon reflexes | n.p | decremental response to RNS | n.p | c.920A>C (p.Asp307Ala) c.923 C>T (p.Pro308Leu) | no response to pyridostigmine; slight response to 3,4-DAP | [81–87] |
| UNC13A 19 AR | 1 pt | con | premature birth, hypotonia, ventilator dependent, facial dysmorphism, ptosis | thin corpus callosum | decrement response to RNS | marked type 2 fibres atrophy | c.304 C>T/304 C>T (p.Gln102*/Gln102*) | no response to pyridostigmine; modest response to 3,4-DAP | [94] |
| MYO9A 15q23 AR | 3 pts | con | fetal hypomotility, swallowing difficulties, apneas, delayed cognitive functions, ptosis, ophthalmoplegia, muscle weakness | normal | decremental response to RNS; abnormal jitter at SFEMG | n.p. | p.Arg1517His/Arg2283His p.Asp1698Gly/Asp1698Gly | good response to pyridostigmine and 3,4-DAP | [15] |
| PREPL 2p21 AR | 11 pts | con | hypotonia, feeding difficulties, respiratory distress, cognitive impairment, ptosis, bulbar signs, muscle weakness, short stature (rare: absence of uterus and ovaries, microcephaly) | normal | normal response to RNS (rare: decremental response) | irregular intracellular vacuoles and perimysium fibrosis | c.616 + 1 G>T/616 + 1 G>T (p.Glu206Glyfs*22/Glu206Glyfs*22) c.1528 C>T/2094 G>T (p.Arg510*/Lys698Asn) c.1529 + 1 G>A/c.1784delinsAA (p.?/Thr595Lysfs * 19) c.1282_1285delTTTG/c.1282_1285delTTTG (p.Phe428Argfs*18/Phe428Argfs*18) c.342delA/342delA (p.Val115Leufs*39/Val115Leufs*39) | slight response to pyridostigmine | [103–109] |
| LAMA5 20q13.33 AR | 1 pt | con | hypotonia, respiratory failure, scoliosis, chronic inflammatory bowel disease, ptosis, ophthalmoplegia, muscles weakness, dysmorphic features, myopia, tics | mild cerebral atrophy | decremental response to RNS | type I fibres predominance; EM: increased postsynaptic folding, moderate reduction of SVs | p.Arg2659Trp/Arg2659Trp | response to pyridostigmine and 3,4-DAP | [14, 112] |
| RPH3A 12q24.13 AR/AD? | 1 pt | childhood | limb weakness, fatigability, hand tremors, hands incoordination, postural imbalance, nasal speech, learning disabilities | normal | Incremental response to high frequency RNS; abnormal jitter at SFEMG | type I fibres predominance; EM: reduction of SVs, degenerative lamellar bodies | c.806 G>A/ c.1390 G>T (p.Arg269Gln/p.Val464Leu) | response to albuterol sulfate | [119] |
| SLC25A1 22q11.21 AR | 19 pts | childhood | ptosis, ophthalmoplegia, some dysmorphisms, dysarthria, chewing difficulties, generalized weakness, mild intellectual disability | normal | decremental response to RNS; abnormal jitter and blockings at SFEMG | non-specific myopathic features; enlarged mitochondria and increased in number on EM | c.205 G>T/c.205 G>T (p.Asp69Tyr/p.Asp69Tyr) c.740 G>A/c.740 G>A (p.Arg247Gln/p.Arg247Gln) c.628 C>T/c.145 G>A (p.Arg210X/p.Val49Met) | partial response to pyridostigmine and 3,4-DAP | [122–126] |
MRI: magnetic resonance imaging; RNS: repetitive nerve stimulation, SFEMG: single fiber electromyography; AD: autosomal dominant, AR: autosomal recessive; 3,4-DAP: 3,4-diaminopyridine; SVs: synaptic vescicles: con: congenital: pts: patients; n.p.: not performed; ?: inheritance pattern not clear.
SLC5A7 CMS can be referred to as a multisystem disorder, encompassing the central, peripheral, and possibly the autonomic nervous system. This multisystemic involvement was apparent in animal models prior to the first case reports, confirming a causal link between clinical features and the CHT mutation.
The presence of CHT in both the central [22, 23] and peripheral nervous system was apparent when Ferguson., et al. created Cht-knockout (KO) mice (Cht-/-) which became paralyzed and cyanotic a few minutes after birth and died of hypoxia probably due to failure of diaphragm neurotransmission [24]. When these mice were later transfected with the Cht gene under control of the motor neuron promoter Hb9, they were able to move and breathe for about 24 hours [25]. Additionally, intracellular recordings from muscle fibres revealed only initial evoked and spontaneous responses, supporting the hypothesis that Cht-/- mice are not able to synthetize ACh after depleting initial stores. The attempt to rescue the phenotype, by limiting the degradation of ACh with AChEI physostigmine and neostigmine, failed in these animals [24]. In humans, the response to AChEI is variable, resulting in some improvements in most cases, sometimes in addition with ephedrine or salbutamol [8, 9, 20, 21]. However, in one patient, presenting with a delayed motor development and MRI brain atrophy, pyridostigmine was not efficient after 3-days neonatally [21]. This was also the case in an older 14-year-old patient with ocular and bulbar involvement [8]. Finally, three patients died despite treatment with AchEI [19, 20].
Heterozygous Cht+/- mice survive and exhibit lower, but functionally acceptable CHT levels compared with WT [24]. They present normal performance on multiple sensorimotor, anxiety and cognitive tasks, except under physical or pharmacological stress [26]. Similar results have been obtained by Paolone G. et al., who tested cognitive performance in mice to examine cortical cholinergic neurotransmission. They postulated the presence of mechanisms that could compensate for reduced cholinergic activity in Cht+/– mice during moderate cognitive challenges, but not in conditions requiring a sudden ACh increase. Some behavioural and mood disorders in humans were correlated with CHT gene mutations, especially the p.Ile89Val variant [27]. Indeed, a new mouse model expressing the Cht –p.Ile89Val variant has been generated, resulting in a reduction of choline clearance in the striatum and cortex, decreased ACh release after prolonged neuronal depolarization, and altered attentional performance [28]. These attentional and behavioural deficits reflect cognitive alterations found in humans. Indeed, a few patients have presented with global developmental delay, resulting in speech, learning and social interaction impairment with no awareness of surroundings [20, 21].
Cht+/– mice also exhibited diminished parasympathetic heart effects with basal resting tachycardia and hypertension, because of depleted cardiac ACh levels [29]. There is no evidence of cardiac involvement in patients harbouring mutations in CHT, but autonomic dysfunction has been noted with core temperatures as low as 32 °C [20].
In contrast to mice, homozygous null alleles of C. elegans CHT (cho-1) are viable but only release around 40% ACh compared to WT animals. However, this is enough to permit thrashing under normal conditions, but with marked motor alterations when stressed [16] (Table 2).
Table 2
Published literature for animal models available for pre-synaptic CMS genes
| Gene | Animal | Technology Used | Model | Phenotype | Strength | Weakness | References |
| SLC5A7 | Mouse | Homologous recombination deletion (exon 2 to intron 4) | CHT(-/-) | Paralysis at birth, only spontaneous quantal release of ACh. Crossing with HB9-Cre increased survivability to 24hrs | ACh quantal release deficiency | Not viable; paralysis at birth | [16, 22–29] |
| CHT(-/+) | Improved survivability, cardiac involvement | Improved survival in heterozygotes, | Cardiac involvement noted in model, not observed, or studied in CMS patients | ||||
| CRISPR/Cas9 | CHTI89V | Developmental and behavioural defects, reduced ACh release and choline clearance | Diminished ACh release in cortex and striatum; neurological deficit observed | Impact on muscle morphology unknown | |||
| C. elegans | Cas9-sgRNA | Cho-1 | Viable, reduced ACh release, worsening phenotype when stressed | Reduced ACh release, stressing the model worsens phenotype | Impact on muscle morphology unknown | ||
| CHAT | Mouse | Injection of J1 Embryonic Stem Cell in C57BL/6 blastocyst with loss of Exon 11, 12 and 13 | ChAT(-/-) | Not viable at birth, paralysis, absent spontaneous and induced neurotransmission | Severe phenotype, absent neurotransmission | Not viable; paralysis at birth | [42–46] |
| ChAT(-/+) | Developmental impairment and behavioural defects, reduced lifespan, reduced muscle strength, motor function decreased | Viable, severe phenotype | Further work required to examine NMJs structure and muscle morphology | ||||
| D. renio | Point mutation (A to C) to the splice acceptor site of ChAT intron 2 | Bajan | Tail bend, behavioural defects, no evoked touch response, decreased spontaneous synaptic responses | Viable, CMS-like phenotype | NMJ structure and muscle morphology not investigated | ||
| Missense mutation S102R | Chata | Viable, CMS-like phenotype | NMJ structure and muscle morphology not investigated | ||||
| Canine | None (Spontaneous Mutation) | Exon 6 | Reduced run distance, stride length and recovery after exercise, decremental response at 3Hz | Exercise-induced muscle fatigue observed with decremental response | Spontaneous mutation, reproducibility challenging | ||
| SLC18A3 | Mouse | Homologous recombination Neo/tk knockdown of VAChT | VAChT(-/-) | Reduced spontaneous and induced ACh release, behavioural defects, loss of muscle strength | Knockdown of SLC18A3; Impaired ACh release, and loss of muscle strength; CMS-like phenotype | No major weakness detected; due to CMS-like phenotype | [52–58, 60–65] |
| VAChT(-/+) | Slight impairment in spontaneous ACh release, reduced memory output (maze test) and olfactory function | Impaired ACh release, and loss of muscle strength; CMS-like phenotype | Phenotype is mild, not consistent with human CMS patients | ||||
| Cre/LoxP creation of VAChT(six3 - Crefl/l) created and crossed with above line | VAChT(-/-) | Not viable at birth, paralysis, respiratory insufficiencies, cardiac development delay between parasympathetic and sympathetic tone | Severe phenotype observed | Not viable; paralysis at birth, cardiac dysfunction not documented in CMS patients | |||
| Motor neuron specific Cre/LoxP ablation of VAChT | VAChT(-/-) | Smaller motor neurons, decreased strength, hypoactive, impaired NMJ function and formation, atrophic muscle, kyphosis | Motor neuron specific, Severe phenotype, with impaired NMJ function, improved lifespan from other VAChT models | Motor neuron specific | |||
| D. melanogaster | Deletion of Cha, VAChT present in 1st exon of Cha | Cha(-/-) | Lethal phenotype at larval stage | Severe phenotype, NMJ development not studied | Not viable | ||
| C. elegans | Mutational screen revealed 19 independent unc-17 mutants | VAChTG347R | Reduced growth, poor coordination; other mutations reveal lethality that ablated gene function | Phenotype in some unc-17 mutants | Lethality in some mutations, viable mutations NMJ structure and function not studied | ||
| SYB1/ VAMP1 | Mouse | Nonsense mutation truncating SYB1 and VAMP1 | VAMP1(lew/lew) | Developmental and behavioural defects, muscle wasting, reduced survival (<3 weeks), decreased ACh release and synaptic transmission | Severe phenotype, with impaired NMJ function, and delayed motor development | Reduced lifespan, joint contractures not noted in model | [71–73] |
| SNAP25 | Mouse | Cre/LoxP knockout at Exon 4 | SNAP25(-/-) | Embryonic lethal, development delay, impaired NMJ formation, with reduced ACh release | Severe phenotype, with impaired NMJ function | Not viable | [76–80] |
| SNAP25(-/+) | Viable and fertile, minimal behavioural defects observed | Viable | No significant behavioural defects observed. Severe phenotype reported in SNAP25 CMS patients | ||||
| Missense mutation I67T | Blind-Drunk Mouse | Ataxic gait, behavioural impairment consistent with a schizophrenia-like symptoms | Neurological phenotype linked to schizophrenia | CMS phenotype not observed | |||
| B-6-Snap25tm3mc2 LoxP sites flanking Exon 5a/5b | SNAP25(-/-) | Fragmented axonal projections, axonal deterioration over time, behavioural defects | Impaired axonal projections, with deterioration suggesting neurological phenotype | More studies need to be determined to link to a CMS-phenotype | |||
| Rat | rAAV2/5 Over-expression in dorsal hippocampus | Male Wistar Rats (WT) | Cognitive defects, behavioural impairments | Neurological phenotype observed | Overexpression model with AAV, not observed in patients | ||
| SYT2 | Mouse | Homologous recombination inserting LacZ/Neo marker at Exon 2 | SYT2(-/-) | Developmental defects, impaired NMJ form and function, ataxic, seizures, non-viable | Severe phenotype with impairment of NMJ form and function | Non-viable, severe neurological phenotype observed with seizures, not consistent to CMS patients | [88–92] |
| D. melanogaster | Mutation of aspartate residues in the C2B domain | SYT2null | Decrease in evoked transmitter release | Decreased neurotransmitter release | Further studies are required to investigate CMS-like phenotype | ||
| L308P mutation in C2B domain | SYT2(-/-) | Developmental and behavioural defects, homozygous insertion of L308P resulted in lethal at 3 weeks | Severe neurological phenotype | Reduced lifespan | |||
| SYT2(-/+) | Developmental and behavioural defects, increased fatigability, reduced evoked transmitter release | Moderate neurological phenotype, CMS-like | CMS like however, NMJ structure not investigated | ||||
| D. renio | Morpholino injection of splice and translation blocking sequences | MO SYT2 | Loss of synchronous transmission at NMJs, asynchronous transmission not impacted | Impaired synchronous transmission | Mosaic expression of knockdown | ||
| UNC13A | Mouse | Homologous recombination in embryonic stem cells | MUNC13-1/2/3 (-/-) | Paralysis at birth, only spontaneous quantal release of Ach, small NMJs | Severe phenotype observed, NMJ form and function impaired | Not viable | [93, 95, 96] |
| C. elegans | Single copy transgenes introduced to EC6699 strain using mos1-mediated single copy insertion | MUNC13(-/-) | Developmental defects, paralysis due to impaired synaptic transmission | Severe phenotype observed, NMJ form and function impaired | Not viable | ||
| e1091, an amber mutant obtained by ethyl methansulfonate mutagenesis | MUNC13 mutant | Developmental and behavioural defects, incoordination, impaired neurotransmission | Viable model with impaired neurotransmission | Model requires more studies to confirm CMS-like phenotype | |||
| MYO9A | D. renio | Morpholino injection of splice and translation blocking sequences | MO myo9aa and myo9ab | Developmental delay, bent tail, behavioural defects, impaired NMJ structure and function | CMS phenotype, impaired NMJ structure and function | Mosaic expression system with morpholino | [15, 97–101] |
| Mouse | Mouse Exon 2 flanked with Cre/LoxP | MYO9A(-/-) | Retarded growth, behavioural defects, severe hydrocephalus, trembling gait, impaired kidney function | Neurological phenotype observed, muscle function impairment | NMJ form and function not studied | ||
| MYO9A(-/+) | Behavioural defects, impaired hippocampal synapse formation, memory and learning impairment | Neurological phenotype observed | NMJ form and function not studied | ||||
| PREPL | Mouse | Cre/LoxP targeting Exon 10 of PREPL | PREPL(-/-) | Reduced body weight, behavioural defects, impaired mitochondrial function, hypotonia | Viable, Muscle morphology dysfunction, with behavioural changes | Mitochondrial deficits not confirmed in patients | [110, 111] |
| LAMA5 | D. renio | Morpholino injection of splice and translation blocking sequences | MO LAMA5 | Muscle and nerve structural abnormalities, developmental delay, behavioural and developmental defects | CMS phenotype, impaired NMJ structure and function | Mosaic expression system with morpholino | [113–118] |
| Mouse | CRISPR/Cas9 creation of R291 L mutant | LAMApm | Die at birth, severe developmental defects, impaired kidney, and lung function | Severe phenotype with developmental defects | Not viable, NMJs not studied | ||
| Inducible Cre/LoxP (SP-Clama5fl/-) with doxycycline | SP-CLama5(fl/-) | Lethal shortly after birth, hypoxic, impaired epithelial differentiation and lung development | Severe phenotype, with developmental delay, respiratory issues consistent with patients | Early lethality, more studies required to examine NMJs of this model | |||
| Chimeric transgene Mr51 and Mr5G2H | LAMA(-/-) | Embryonic lethal, impaired lung and limb development; Mr5G2 H mice were viable but have reduced body size, and kidney function. | Mr5G2 H are viable and have impaired development | Not viable; Mr5G2 H viable but CMS phenotype not studied, lacks severe CMS phenotype seen in LAMA5-CMS patients | |||
| RPH3A | Mouse | Neomycin cassette flanking between exons of Rph3a, electroporated into ES cells | Rph3atm1Sud/J | Fertile, Enhanced axonal regeneration; rescues behavioural deficits after spinal cord injury. | Neuronal phenotype observed, mice are fertile | NMJs specifically not investigated, more information is needed to confirm CMS phenotype | [142–146] |
| Mouse | αsyn-PFF injected into C57Bl6 with AAV9-Rph3A overexpression | hSyn-GFP-Rph3a-WPRE-AAV9 | αsyn-PFF reduced Rph3A interaction with GluN2a-contiaing NMDRs, inducing neurological behavioural dysfunction, rescued with AAV9-Rph3a. | Neuronal phenotype observed, impacting Rph3A | NMJ phenotype not observed, not linked to CMS phenotype | ||
| Rat | RNAi knockdown in Middle cerebral artery occulusion (MCAO) model | LV-Rph3A-RNAi | Exacerbates cerebral infarction and neuronal death leading to neurological behavioural dysfunction | Neurological phenotype observed | MCAO model investigated, effect on NMJ unknown | ||
| C. elegans | Null mutation - 1500bp deletion including the promoter and 3 exons | Rbf-1 | Appeared lethargic, no impact on NMJ morphology, defecation, pharyngeal pumping and mating | Lethargic model, appears weak. | Did not alter NMJ morphology, | ||
| SLC25A1 | D. renio | Morpholino injection of splice and translation blocking sequences | MO SLC25A1 | Motor impairment, altered tail morphology, decreased swim and touch-evoked response, stunted motor neuron branching, with incomplete synapses | Neuromuscular phenotype observed, viable | Mosaic expression system with morpholino | [123, 126, 127] |
| Mouse | SLC25A1 overexpression | TRE-SLC25A1 nTg | Autistic-like phenotype (aberrant behaviours and repetitive jumping), altered synaptic structure and white matter content | Neurological phenotype observed, limited to brain involvement | SLC25A1 overexpression not observed in patients | ||
| SLC25A1 liver-restricted knockout (SLC25A1tm1a (EUCOMM) Wtsi +tg (Alb - cre) 21Mgn | SLC25A1-/- | Viable, normal liver histology, body weight and liver fat content, protected from steatosis, improved blood glucose clearance | Model restricted to liver assessment | NMJ phenotype not observed, not linked to CMS phenotype | |||
| D. melanogaster | Mutation to seaEP(reduced expression) | I(3)EP3364 | Increased chromosome aberrations, no functional characteristics noted | Viable model, more studies need to examine impact on NMJ neurotransmission | No phenotype observed |
CHAT
The Choline-O-Acetyltransferase (CHAT) gene encodes the homonym enzyme which catalyses ACh synthesis from acetyl CoA and choline at cholinergic synapses. It is expressed both in the central and peripheral nervous system. (GC10P049609, GeneCards) (Fig. 2).
CHAT CMS is the most common presynaptic form, accounting for 4–5% of all CMS cases. It is inherited in an autosomal recessive manner, and also known as CMS with episodic apnoea (CMS-EA) [30, 31]. Sudden episodes of respiratory crises and bulbar weakness may be provoked by infection, fever, or other stressful conditions in birth or early infancy, and in several cases this has resulted in death [32–34]. Occasionally EA is accompanied by loss of consciousness and tonic–clonic seizures [35]. Other reported symptoms comprise ptosis, fatigable weakness, proximal muscle weakness, aggravated by exposure to cold, delayed motor milestones and intellectual disability [11, 36–41]. RNS usually detects no decrement at low-frequency stimulation, but it appears after prolonged high-frequency stimulation [11]. AChEI are effective in moderating respiratory crises and improving other symptoms in some cases [11] (Table 1).
Mice with targeted deletion of the Chat gene at the NMJ appear normal in a heterozygous state, while homozygous pups die at birth, displaying flaccid paralysis. Both spontaneous and nerve-evoked postsynaptic potentials are absent in mutants, and several morphology and developmental abnormalities have been noted at the NMJ (i.e., marked excess of nerve terminal branching, altered synapse distribution with widened endplate, thinner muscles, and precocious appearance of larger AChR-rich sites at the postsynaptic membrane) [42, 43].
Similar results came from two mutant zebrafish lines, bajan and chatatk64. Bajan zebrafish have a single point mutation (A to C) at the splice acceptor site of Chat intron 2, while chatatk64 carry a missense mutation changing a highly conserved serine to an arginine (p.S102 R). Both exhibited no coiling movements and reduced touch response in a homozygous state [44, 45]. Spontaneous synaptic currents as well as evoked synaptic responses were markedly decreased [44] (Table 2).
Finally, an example of a naturally occurring CHAT mutant animal is the Old Danish Pointing Dog, carrying a valine to methionine substitution at position 29 of the CHAT exon 6. Affected dogs can walk and run for only 5–30 min before manifesting fore- and hind leg fatigability. Moreover, electrophysiology shows a decrement at 3 Hz induced by a long stimulation train [46].
ACETYLCHOLINE PACKAGING INTO SYNAPTIC VESICLES
SLC18A3
Solute Carrier Family 18 Member A3 (SLC18A3) mapped to chromosome 10q11.2, encodes for vesicular acetylcholine transporter (VAChT) that mediates ACh internalization into vesicles in the presynaptic terminal (600336, OMIM). Mutations of this gene leads to an autosomal recessive inherited CMS form (Fig. 2).
Clinical onset of SLC18A3 CMS occurs in the neonatal period, and is often associated with apnoeic episodes, hypotonia, variable degrees of joint contractures, and feeding difficulties [10, 47, 48]. However, four patients also displayed prenatal alterations [49, 50]. Ptosis, ophthalmoplegia, mild facial weakness and generalized fatigable weakness are other common symptoms. Hakoken A.H. et al. described two cases of severe and lethal CMS characterized by foetal akinesia, arthrogryposis, generalized body edema and dysmorphic features [51].
Cognitive function, especially learning capacity, attention and working memory, may also be affected [10, 47]. A decremental response to RNS is usually detected. Pyridostigmine improves clinical manifestations, and 3,4-Diaminopyridine (3,4-DAP) and ephedrine may be added in some cases [10, 48, 50]. Intriguingly, a mild reduction of left ventricular systolic function was rescued after treatment with AChEI in one patient [10] (Table 1).
One of the first animal models of the VAChT mutation was generated in Drosophila melanogaster (common fruit fly). Homozygous mutants were lethal, and those that survived till the larval stage showed reduced motility. Moreover, heterozygous flies exhibited electrophysiological defects in the giant fibre pathway after high-frequency stimulation [52]. More recently, Drosophila models have been tested with an overexpression of VAChT, demonstrating its negative impact on longevity, locomotor ability in an age-dependent way, learning and memory [53]. Surprisingly, reduced VAChT function produced a similar effect on lifespan and locomotive behavior [54]. There are currently no specific reports on VAChT mutations and life expectancy of patients, but clinical and electrophysiological features of affected Drosophila resemble those recognized in patients.
The effects of a reduced expression of VAChT in the central and peripheral nervous systems were later confirmed by Prado V.F. et al. in a Slc18a3 knockdown (KD) mouse model. They detected a reduction in VAChT levels in several central nervous system regions both in heterozygous KD mice and more markedly in homozygous ones. Moreover, miniature end-plate potentials (MEPPs) recorded at NMJs were smaller and less frequent in homozygous mutants.
Homozygous KD mice were significantly limited in physical activity, while both mutant genotypes showed difficulty in learning or remembering features for discriminating a novel object or recognizing a littermate [55]. The reduced cholinergic tone due to a slow refilling of synaptic vesicles in homozygous KD mice was displayed through an alteration in basic cognitive capacity, especially functions requiring a prolonged cholinergic involvement [56].
In addition, homozygous KD mice displayed impaired cardiac function related to autonomic imbalance between parasympathetic and sympathetic tone that was rescued by pyridostigmine [57, 58].
Further proof that VAChT is rate-limiting for neurotransmission is derived from Slc18a3 KO mice who died of respiratory insufficiency within 2 to 5 minutes following birth. However, small-amplitude and low-frequency MEPPs were detected in embryonic NMJs, probably due to a spontaneous ACh entry into presynaptic vesicles. Indeed, a lack of VAChT leads to the increase of CHAT and CHT expression, and subsequently of cytoplasmic ACh levels. Although, they demonstrated alterations in neuromuscular development like an absence of ACh, proving that passive ACh uptake by vesicles cannot compensate for VAChT deficiency [59]. Similar feedback mechanisms were detected in C. elegans by Zhu H. et al [60].
Conversely, heterozygous KO mice presented no locomotor activity dysfunction, no alteration in MEPPs or in NMJ morphology [59].
To avoid the interference from impaired peripheral cholinergic function, Martin-Silva C. et al. generated novel mutant lines with intact VAChT expression in the somatomotor cholinergic neurons, but significant reductions in the other cholinergic neurons. This technique allowed them to alter only brain cholinergic activity, resulting in the hyperactive behaviour of mice [61]. Deleting Slc18a3 from their forebrain impaired spatial memory, cognitive flexibility, and sustained attention [62, 63]; and knocking it out of the striatum caused different degrees of short-term memory deficit [64]. These studies provide evidence that ACh is fundamental to control locomotor function and to modulate hippocampal memory.
Finally, Cre-loxP technology has allowed for the deletion of Slc18a3 in select groups of motoneurons (mnVAChT-KD). Mutant mice have atrophy in muscle fibres innervated by mnVAChT-KD and appear hypoactive, leaner and with kyphosis and less muscle strength [65] (Table 2).
Findings from mice models trace once again the human phenotype from a motor, cardiac and cognitive point of view. Notably, a compromised cardiac pump function was found both in homozygous KD mice and in an affected patient, benefiting from AchEI [10]; as well as compromised hippocampal and other superior functions, that were identified as significant deficits in attention span, working memory, and immediate and delayed visual memory in a formal psychometric assessment of the same patient [10].
ACETYLCHOLINE VESICLES EXOCYTOSIS
VAMP1/SYB1
The vesicle-associated membrane protein (VAMP), also called synaptobrevin (SYB) is part of the soluble N-ethylmaleimide sensitive factor attachment receptor (SNARE) complex, in association with syntaxin1 and synaptosomal-associated protein 25 kilodalton (kDa). The SNARE complex is involved in the fusion of synaptic vesicles with the presynaptic plasma membrane for the subsequent ACh release into the synaptic cleft [66] (Fig. 3).
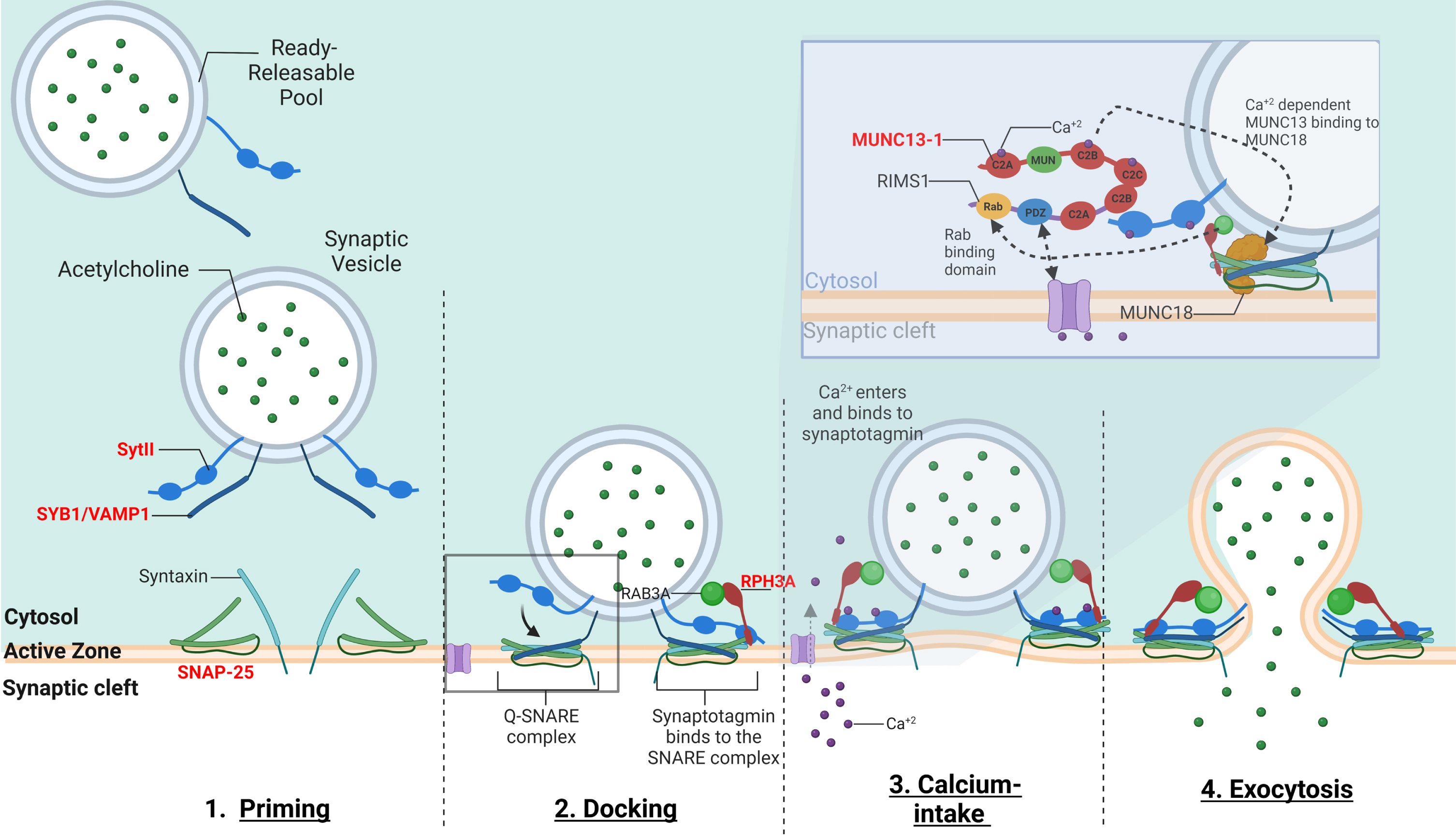
Fig. 3
–The mechanism of calcium-dependent synaptic vesicle exocytosis at the NMJ: The ready releasable pool (RRPs) of synaptic vesicles containing ACh. When activated, the synaptic vesicle moves towards the pre-synaptic membrane terminal where calcium influx releases the synaptic vesicle bound to actin and other cytoskeletal elements (not shown in image). The “Active Zone” is the area at the pre-synaptic terminal membrane where exocytosis occurs. 1. The pool of synaptic vesicles is termed the “readily releasable pool” (RRP) and is necessary to respond to an electrical impulse. Synaptotagmin II (SytII) and Synaptobrevin/VAMP1 (SYB1/VAMP1) proteins attach to the outer synaptic vesicle membrane forming the soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) complex. The cell membrane contains SNAP-25 and syntaxin components essential to the fusion of the vesicle and the cell membrane. 2. Docking –Here as the synaptic vesicle moves closer to the cell membrane, the SNARE complex begins to form 3. When an action potential depolarizes the pre-synaptic membrane, this causes the excitation of voltage-gated calcium channels and calcium entry. Calcium binds to both C2-domains (C2a and C2b) of SytII which induces a simultaneous binding of the vesicle to the cell membrane while completing the formation of the SNARE complex. Calcium induced binding of the SNARE complex requires additional proteins for vesicle exocytosis. MUNC18-1 inhibits the formation of the SNARE complex, locking syntaxin in a closed conformation, upon calcium induction MUNC18-1 interacts with MUNC13-1 opening the syntaxin conformation enabling the formation of the SNARE complex. This change in syntaxin conformation enables the binding of SYB1/VAMP1 initiating the fusion of the synaptic vesicle and cell membrane. In addition, MUNC13-1 contains C1, C2A, C2B, and C2 C domains which sequester calcium. MUNC13-1 also contains a central MUN domain. Importantly, Rab3-interacting molecule (RIMS1), which serves as an active zone scaffolding protein has been shown to interact with proteins ELKs and RIM-BPs. RIMS1 also interacts with MUNC13-1 by preventing homodimerization and recruiting MUNC13 s towards the active zone. RIMS1 also serves as a key regulator as it acts upon MUNC13 and 18, which are essential in determining the number of release sites and in the fusion of competent vesicles. RIMS1 can interact directly with the voltage-gated calcium channels at the presynaptic membrane, but altering the density of calcium channels present at the active zone, hereby effecting the size of the RRP. 4. The vesicle and cell membrane fuse together, the SNARE complex “zippers” the vesicle tightly to the cell membrane releasing ACh and other proteins into the synaptic cleft. Genes implicated in CMS are in red.
SYB1/VAMP1 CMS is an autosomal recessive disorder, occurring at birth. To date, nine patients have been described with a typical phenotype characterized by generalized hypotonia and muscle atrophy, bulbar dysfunction, feeding difficulties, delayed motor milestones, joint contractures or laxity, kyphoscoliosis and respiratory distress, perhaps caused by pulmonary infections [67–69]. Elongated myopathic face with ophthalmoparesis, frontal bossing, low set ears and high arched palate have also been reported [69, 70]. RNS revealed a decremental response in one case, with synaptic facilitation indicating a presynaptic defect [67] and an incremental response in others [68, 70]. Therapy with pyridostigmine led to a variable degree of improvement. In one of the most recently reported patients, salbutamol was administered in addition with pyridostigmine with some benefit [69] (Table 1).
Several VAMPs have been targeted in mice and different phenotypes elicited depending on the mutated isoform. Two highly homologous isoforms, VAMP1/SYB1 and VAMP2/SYB2, have been detected in neurons and in nerve terminals of the NMJ [71].
A nonsense mutation in Vamp1/Syb1 truncated nearly half the protein, resulting in a lethal-wasting mutant phenotype (lew) in mice. Vamp1/Syb1lew/lew mice displayed altered movements of paws and legs, becoming progressively immobile before death within three weeks [72]. While motor endplates developed with no delay in Vamp1/Syb1lew/lew mice, spontaneous and evoked synaptic activities were reduced [71]. The minimal registered activity was probably due to the presence of SYB2, which can partially compensate for the absence of SYB1, but its presence in the spinal cord is not enough to guarantee Vamp1/Syb1lew/lew mice survival. However, SYB2 plays a critical role in the sensitivity to calcium-triggered vesicle release [71]. Indeed, the complete lack of SYB2 drastically reduced evoked synaptic activity in hippocampal neurons of Syb2-/- mouse embryos, while spontaneous miniature excitatory currents had normal amplitude and rise time. Mice died immediately after birth, exhibiting body shape dysmorphism [66].
To examine the link between Syb1 and Syb2, Liu Y. et al. compared Syb2-/- to Syb1lew/lewSyb2-/- mice. Both presented with increased intramuscular nerve branching. Moreover, in the latter, evoked synaptic transmission was completely abolished and spontaneous ACh release was insensitive to calcium [73] (Table 2).
Human clinical features are relatively milder than the mouse model. Salpietro V. et al. suggested the possibility of a species-specific compensation of vesicle fusion and release at the nerve terminal to explain this difference [68]. Electrophysiological results in mouse models can explain the synaptic facilitation detected in some patients, but there is no evidence of increased intramuscular nerve branching. Since the molecular pathways involving VAMP1 are still not clear, we need research to deeply understand its role at the NMJ.
SNAP25
The Synaptosomal-associated protein 25 kDa (SNAP25) gene is located on chromosome 20p12.2 and encodes for a protein that is part of the SNARE complex (SNAP25). SNAP25 and syntaxin1 mediate the fusion of synaptic vesicles to presynaptic plasma membrane where they are docked. (600322, OMIM). (Fig. 3) An obligate alternative splicing of two exon 5 sequences in SNAP25 can result in two isoforms of SNAP25 (SNAP25A and SNAP25B), which are identical except for 9 residues. In the mouse brain, SNAP25B replaces SNAP25A by the second postnatal week, as the adult-specific isoform [74].
The first report of SNAP25 CMS dates to 2014, when a de novo dominant mutation of the isoform SNAP25B (c.200T>A; p.Ile67Asn) was identified in a patient who presented with foetal hypomotility, cyanosis, and joint contractures at birth. During childhood, she exhibited developmental delay with epileptic absence episodes, confirmed by EEG, and developed eyelid ptosis, fatigable weakness, dysarthria, and ataxic gait. The response to RNS was decremental. Therapy with pyridostigmine was ineffective, while 3,4-DAP along with levetiracetam for seizures, resulted in some improvements [74] (Table 1).
While not in our original literature search time frame (from January 2000 to March 2022), a recent publication has reported another de novo SNAP25B mutation (c.529 C>T; p.Gln177X). The patient died in the sixth day of life due to respiratory failure. Prenatal ultrasound revealed multiple anomalies including severe polyhydramnios and multiple findings consistent with arthrogryposis such as micrognathia, right clubfoot, left rocker-bottom foot, diffuse skin thickening, and hypomobility [75] (Table 1).
Like other forms of presynaptic CMS, the phenotype observed in the mouse models was more pronounced than in the patient. Heterozygous Snap25 null mutant mice were phenotypically indistinguishable from WT, while homozygous mice were embryologically lethal with smaller body size and no spontaneous movements. Evoked ACh release was abolished at NMJs, proving that the SNARE complex is crucial for Ca2+-triggered synaptic transmission and explaining the lethality as linked to failure of diaphragm contraction [76].
As well as the peripheral nervous system, SNAP25 is highly expressed by neurons in the central nervous system. Which may explain the presence of central neurologic manifestations such as seizures and ataxic gait in patients.
The Snap25 mouse model obtained by creating a missense mutation in a highly conserved domain of the SNAP25b isoform has been termed the “blind-drunk mouse”. These mice display ataxic gait with behavioural impairment resembling that of schizophrenia. They also have a lower frequency of evoked cortical excitatory postsynaptic potentials [77].
A brain-specific Snap25 KO mouse also demonstrated behaviour signs related to schizophrenia. The reduction of SNAP25 expression in the cortex caused a remarkable elevation of extracellular glutamate levels, resulting in abnormal hyperactivity and stereotypical behavior [78]. Hoerder-Suabedissen et al. produced a transgenic mouse with cell-specific loss of Snap25 from cortical projection neurons (cKO). cKO mice were viable at birth with normal neuronal development, but axonal projections became fragmented and deteriorated three weeks later [79].
In 2010, McKee A.G. et al. over-expressed Snap25 in the adult rat dorsal hippocampus by infusion of a recombinant adeno-associated virus vector. They noticed a defect in synaptic function resulting in cognitive deficits, such as delayed acquisition of spatial and fear memory consolidation. Hence, both reduced and excessive Snap25 activity has been implicated in memory and cognitive function impairment [80] (Table 2).
The only reported patient did not suffer from cognitive and behavioural alterations. However, we should consider these features as part of SNAP25 CMS clinical pictures in order to reduce misdiagnoses of this type of CMS.
SYT2
The Synaptotagmin 2 (SYT2) gene encodes for a synaptic vesicle membrane protein (SYT2) that works as a calcium sensor in vesicular trafficking and exocytosis (GC01M202559, GeneCards) (Fig. 3).
SYT2 CMS can be inherited in either an autosomal dominant or a recessive manner. The former pattern was described in patients mainly with a childhood onset [81–83], although not exclusively [84]. The latter, was associated with early onset and a more severe phenotype, characterized by reduced foetal movements, hypotonia at birth, joint laxity, and delayed motor milestone [85–87]. Typical manifestations include muscle weakness and fatigability, variable muscle atrophy and reduced or absent deep tendon reflexes, partially reversible after exercise [81]. Ocular involvement has been reported in a few families [82]. Other common features are foot deformities, such as pes cavus and hammer toe, and lower limbs sensitivity alterations [81, 82]. Sometimes electromyography of distal muscles detects a slight reinnervation, that is consistent with a motor neuropathy [81, 86]. Electrophysiological studies revealed low-amplitude CMAPs and a certain degree of post exercise amplitude facilitation [81, 83]. Therapy with 3,4-DAP ameliorated both clinical and electrophysiological manifestations, as did pyridostigmine and salbutamol in some patients [82, 84–87] (Table 1).
The role of SYT2 was first examined in Drosophila, in which a decrease of evoked transmitter release and its calcium-affinity was obtained through mutating two aspartate residues in the C2B calcium-binding pocket of SYT2 [88]. More recently, Shields. et al. reproduced the human substitution of a leucine with a proline in position 308 (sytL - P), in the same domain of Drosophila SYT2, via pan-neuronal expression of syt transgenes in the sytnull background. SYT2 is substituted by other isoforms in flies, but sytL - P results in lethality in the absence of WT syt. Heterozygous sytL - P were viable and showed marked fatigability, less motor capacity and reduced evoked transmitter release, similar to humans [89].
Analogous results were obtained from mice with a point mutation in Syt2, resulting in a decreased neurotransmitter calcium-dependent release in the Calyx of Held. Mice were viable but infertile, presenting smaller and severely ataxic in a homozygous state [90]. These results were further confirmed in Syt2 homozygous KO mice, which displayed severe motor dysfunction two weeks after birth and died after three weeks [91].
Finally, experiments on syt2 MO KD zebrafish showed a reduction in synchronous release that was responsible for around 90% of the neuromuscular transmission [92] (Table 2).
UNC13A
UNC13A encodes the protein MUNC13-1 which is a protein of the presynaptic active zone in cholinergic neuromuscular synapses and glutamatergic brain synapses that is involved in synaptic vesicle priming [93]. It binds the SNARE protein syntaxin 1B, shifting in closed conformation the position of Munc18-1, that normally stabilizes syntaxin 1B. In this way, syntaxin 1B is active and contributes to form the SNARE complex for vesicles exocytosis [94] (Fig. 3).
UNC13A CMS has been reported in only one patient with a homozygous nonsense mutation. The patient was born prematurely, presenting with hypotonia, feeding difficulties and respiratory distress with cyanotic episodes. She developed some dysmorphisms (marked frontotemporal narrowing, high-arched palate, small jaw, thoracic kyphoscoliosis, clinodactyly and joint contractures), delay in motor function acquisition, eyelid ptosis and reduced oculomotion. Clinical features comprised microcephaly and cortical hyperexcitability. EMG revealed low-amplitude CMAPs at rest, decremental response to low-frequency RNS and an incremental response to high-frequency. Therapy with pyridostigmine and 3,4-DAP slightly improved her symptoms, but the child died around 4 years old [94] (Table 1).
This type of CMS looks like a syndromic disorder, comprising a marked involvement of the central nervous system supported by the fact that MUNC13-1 has been abundantly shown in rat brain. Two other minor isoforms of the protein are MUNC13-2 and MUNC13-3 that are expressed in the rostral brain region and cerebellum, respectively. MUNC13-1 mice died postnatally because of a deficiency in synaptic vesicle exocytosis in the central nervous system. While Munc13-3 mice presented parallel fibre–Purkinje cell synapses of normal morphology but altered function, with a negative impact on motor learning [93]. Varoqueaux et al. created mice with a double deletion of Munc13-1/2 and triple deletion of Munc13-1/2/3 and, once more, the absence of MUNC13-1 resulted in lethality for both. Experiments on embryos, presenting paralyzed, showed morphological alterations in NMJs such as abnormal nerve terminal branching, larger endplates and improper alignment of muscle fibres. Evoked synaptic transmission was reduced while spontaneous release persisted [95].
UNC13A was also evaluated in C. elegans as the ortholog unc-13, which shares similar C1 and C2B domains that are essential for neurotransmission regulation. Removal of either the C1 or C2B domain enhanced calcium-triggered synaptic transmission, while deletion of both domains stopped synaptic transmission. Indeed, unc-13 null mutants were paralyzed, but the rescue of a single copy of unc-13 or of the protein lacking only C2B domain reverted to normal locomotion [96].
Motor impairments, altered learning, and reduced synaptic transmission from mouse and C. elegans models reflect UNC13A CMS, resulting in lethality in a homozygous state.
OTHER PRE-SYNAPTIC MECHANISMS
MYO9A
The Myosin IXA (MYO9A) gene encodes for an unconventional myosin that is an actin-based motor molecule that inhibits RhoA by stimulating its GTPase activity (MYO9A) (GeneCards, GC15M071822) (Fig. 4A).
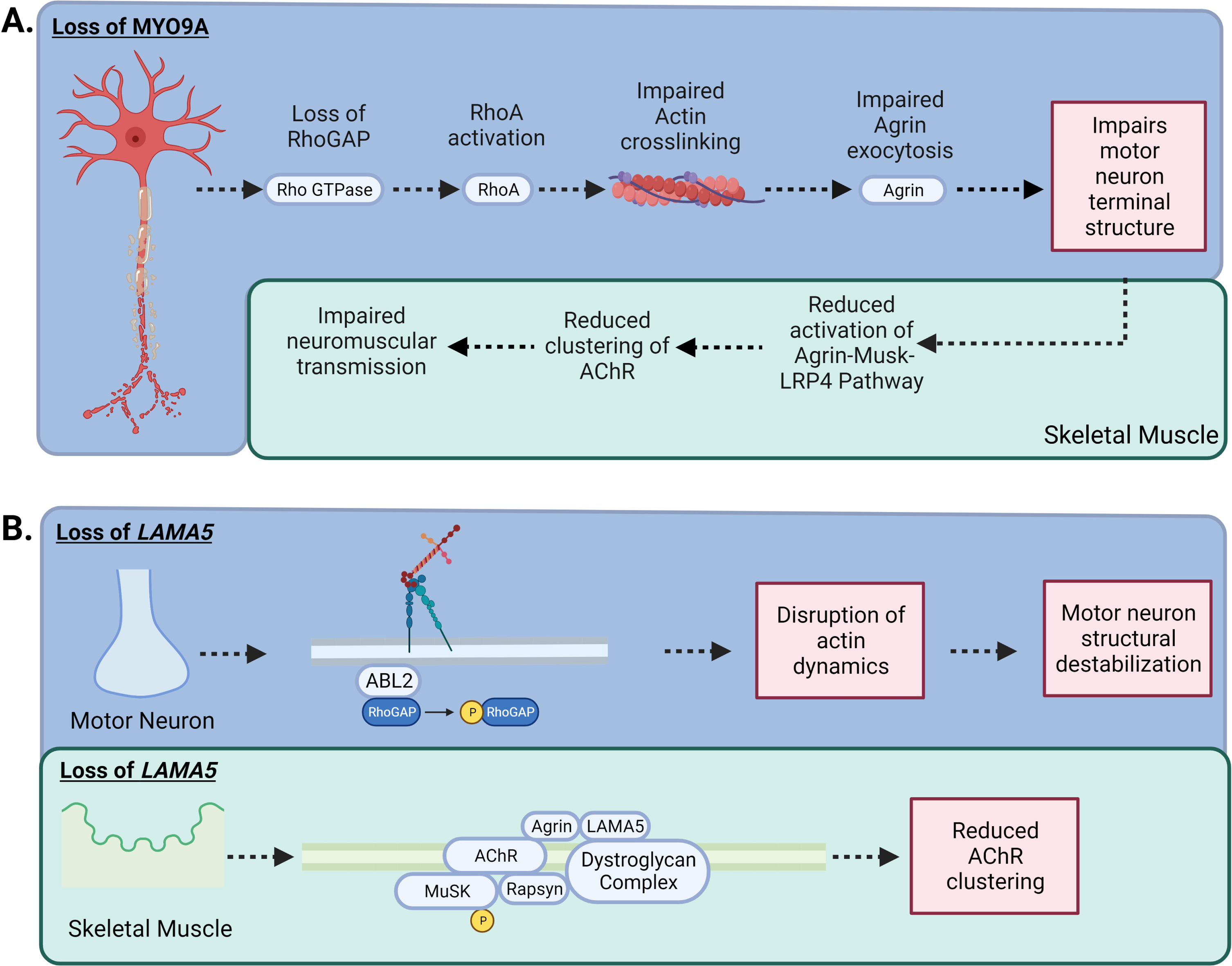
Fig. 4
–MYO9A and LAMA5 in CMS: A) Loss of MYO9A has been shown to impair actin cross-linking, causing an alteration in the growth of axons and terminal structure. In addition, RhoGAP activity is impeded, increasing the activation of the RhoA pathway, impairing the nerve cytoskeleton and intracellular transport resulting in compromised secretion of agrin. A reduction in agrin secretion results in less activation of the Agrin-MuSK-LRP4 AChR clustering pathway. B) Laminins are the most abundant non-collagenous protein in the basal lamina. There are five extracellular matrix heterotrimeric glycoproteins composed of α, β, and γ subunits. Laminins are involved in cell signalling, development and maintenance of the NMJ, and regeneration of the nerve terminal. While Laminin α5 (LAMA5) function has not been fully elucidated, CMS patients with mutations in LAMA5 have a reduction in the number of synaptic vesicles present for release due to the small size in nerve terminals and a reduction in the number of active zones. In addition, specific mutation to the coiled-coil domain LAMA5 may play a role in preventing interaction with other laminins causing dysfunctional agrin secretion.
To date, three patients of German and Turkish origin have been described in a single publication [15]. They variably presented with ptosis and ophthalmoplegia, bulbar dysfunction, generalized muscle weakness, hypotonia, episodic apnoea and respiratory failure at birth. During follow-up, developmental delay and some cognitive abnormalities were evident. SFEMG detected abnormal jitter and RNS revealed a decremental response in one patient. The best treatment approach remains ambiguous as the German patient responded to pyridostigmine and 3,4-DAP, but the Turkish siblings suffered from a respiratory crisis after oral intake of 3.4-DAP and fluoxetine [15] (Table 1).
MYO9A CMS presents a complex clinical picture, probably due to the ubiquitous expression of this protein. Rho activity is involved in morphology and differentiation of ciliated epithelial cells and renal proximal tubules, reflecting different aspects of MYO9A mutated animal models.
Myo9a deficient mice developed severe hydrocephalus with enlargement of lateral and third ventricles and stenosis of the Sylvian aqueduct. As a result, they displayed hopping and a trembling gait in adulthood [97]. They also developed bilateral renal disease, characterized by dilated renal calyces and parenchyma fibrosis, leading to polyuria and proteinuria [98]. Folci et al. demonstrated that MYO9A plays a role in hippocampal synapses and cognitive behaviour. Myo9a heterozygous KO mice presented with structural and molecular changes in hippocampal synapses, affecting learning and memory tasks as in reported patients [99].
Recently, the function of MYO9A at the mammalian NMJ was examined and while its exact location has still not been determined, several pieces of evidence suggest a presynaptic role [15]. A zebrafish model, in which the MYO9A orthologues, myo9aa and myo9ab, were knocked down using morpholinos developed curly tails, cardiac oedema, and abnormal swimming in response to tactile stimulation. The axons were aberrant, branching to the adjacent myotome beyond their target [15].
Based on the results from Myo9a-depleted NSC-34 cells (mouse motor neuron-derived cells), the same group demonstrated an impaired trafficking and release of proteins in the absence of Myo9a. One of these proteins, agrin, is crucial for the proper development and function of the postsynaptic endplate. It was hypothesized that some of the aberrations in the myo9a morphant fish were due to its reduction. This theory was confirmed in deficient myo9aa/ab zebrafish, obtained through a CRISPR/Cas9 approach where injection of morphants and crispants with agrin improved their locomotion and NMJ morphology [100, 101].
Animal models’ motor and behaviour alterations partly match the human phenotype. Patients’ hypotonia and muscle weakness assume the same pathophysiological mechanism of zebrafish abnormal swimming. In addition, patients present with cognitive impairment, similar to mice who display learning and memory defects. However, there is no evidence of hydrocephalus in reported patients, whose brain MRIs are normal, and there is no evidence of renal dysfunction.
PREPL
Prolyl Endopeptidase-Like (PREPL) is a ubiquitous serine protease, highly expressed in the brain, kidney and muscle. Its function remains partly elusive, but it is probably involved in the control of the trans-Golgi network morphology and sorting, as well as in the regulation of synaptic vesicle exocytosis (GC02M044281, GeneCards; 609557, OMIM) (Fig. 2).
Deletions of PREPL in combination with other genes at the 2p21 chromosomal region are associated with different variants of the hypotonia-cystinuria syndrome (HCS), characterized by cystinuria, neonatal hypotonia, eyelid ptosis, occasional bulbar symptoms, and growth hormone deficiency [102]. Some of these features suggest a neuromuscular transmission defect, but the first case diagnosed with a myasthenic syndrome related to PREPL deficiency was reported by Régal L. et al. The patient presented with marked hypotonia and feeding difficulties at birth, later associated with ptosis and daily fluctuating proximal muscle weakness. Electrophysiology studies revealed low amplitude MEPPs. Additionally, growth hormone blood levels were deficient. Genetic analysis detected compound heterozygosity for a paternal nonsense mutation in PREPL (p.Met270Ter) and a maternal deletion involving PREPL and SLC3A1 (one of the genes causing HCS). The patient improved with pyridostigmine treatment and did not worsen after weaning off the medication [103]. Other patients have been recently reported with similar features and various combinations of facial dysmorphism, developmental delay, hypergonadotropic hypogonadism, and childhood obesity [104–108]. One patient with a splicing mutation in the PREPL gene was characterized by the absence of a uterus and ovaries, microcephaly and short neck [109] (Table 1).
PREPL is a unique protein, different from the others in the family, because it plays a role in protein-protein interactions rather than in peptide proteolysis [110]. It shows homology with prolyl endopeptidase (PREP), whose knock-down in mice produced an impairment in spatial learning and memory, due to its expression in hippocampus and entorhinal cortex [111]. Deletion of Prepl in mice led to hypotonia in five-day old pups and impaired growth, as reported in patients. Therefore, it could be also hypothesized an underlying reduced growth hormone signalling, remarking patients’ phenotype [110] (Table 2).
LAMA5
Laminin alpha-5 (LAMA5) is part of the laminins family, a group of extracellular matrix glycoproteins that participate in several biological processes. LAMA5 is widely expressed in vertebrate tissues and mediates cellular attachment and migration during embryonic development (GC20M062307, GeneCards) (Fig. 4b).
Currently, the only described LAMA5 CMS was caused by a homozygous missense mutation, resulting in a deficient number of releasable SVs. Indeed, the mutant LAMA5 reduces cell adhesion and neurite outgrowth, and influences agrin attachment to the extracellular matrix. Furthermore, the reported mutation impairs the interaction between LAMA5 and the synaptic vesicle glycoprotein 2A (SV2A), which in turn binds to synaptotagmin, regulating the Ca2 + dependent SVs exocytosis.
LAMA5 CMS patient presented with a multi-faceted phenotype. Clinical features comprised congenital hypotonia and respiratory failure, scoliosis, chronic inflammatory bowel disease, neck and limb muscles weakness. Dysmorphic features, myopia, bilateral ptosis, facial muscles weakness and tics were also observed. RNS showed a marked decremental response and facilitation occurred after maximal muscle activation. The patient responded well to pyridostigmine and 3,4 DAP [14, 112] (Table 1).
A more complex phenotype occurred in mouse models, which also demonstrated the role of LAMA5 in pulmonary [113, 114] and renal development [115]. For instance, Lama5 KO mice died at the embryonic stage. Embryos presented with a defective anterior neural tube closure, syndactyly, aberrant genesis of kidneys and an absence of visceral pleura basement membrane with reduced lobar lung septation [113]. To avoid early lethality, LAMA5 deficiency was restricted to lung epithelial cells but mice still died perinatally of respiratory failure due to marked abnormalities in lung development [114].
However, a specific neuromuscular phenotype was obtained by creating a muscle specific KO Lama5 mouse, showing a delayed pre- and postsynaptic differentiation in NMJs [116].
In addition, a missense mutation of the Lama5 gene was generated in mouse models via CRISPR-Cas9 genome editing, further elucidating the importance of the role of laminin in tissue development and homeostasis. Mutant mice showed defective foetal growth and developmental abnormalities. Similar alterations occurred in the reported patient, such as elongated face, elevated hard palate, and eye defects, reflecting the role of LAMA5 in embryonic development [117].
Finally, comparable results were obtained from zebrafish, in which LAMA5 deficiency led to an altered formation of pectoral fins and a lack of fin folds, highlighting its role in basement membrane integrity [118] (Table 2).
RPH3A
Rabphilin 3a (RPH3A) encodes for the protein RPH3A, which is involved in docking and fusion steps for neurotransmitter release. Specifically, at the presynaptic level, RPH3A is recruited to the synaptic vesicle membrane binding 14-3-3 protein, Ca2+, phosphatidylinositol 4,5-bisphosphate (PIP2) and SNAP25 to modulate vesicle trafficking and calcium-triggered exocytosis (GC12P112570, GeneCards) [119].
An 11-year-old girl with a heterozygous mutation in RPH3A was described by Maselli R.A. et al. During early childhood, she presented with limb weakness and fatigability, later followed by hand tremors, hands incoordination, postural imbalance, and nasal speech. She also had learning disabilities with no abnormalities at brain MRI and EEG. High frequency RNS revealed and increment of 30% of CMAP areas and SFEMG detected an increased jitter. A favourable response to treatment with albuterol sulfate was shown. The authors demonstrated a synaptic vessicle density reduction, an increase in nonvesicular synaptic membranes, and the presence of degenerative lamellar bodies in the patient NMJ [119] (Table 1).
The exact function of RPH3A in the NMJ has not been completely elucidated. A null mutation in rbf-1, the RPH3A homolog in C. elegans, did not alter NMJ general morphology, nor defecation, pharyngeal pumping, and mating assays. Rbf-1 mutants appeared lethargic in absence of exogenous stimulation. Indeed, RPH3A alterations seemed synergic with SNARE complex’s ones, but rbf-1 mutants did not enhance the behavioural defects of synaptotagmin mutants. Thus, the role of RPH3A in the Ca2+-sensitive SVs release is still controversial [120] (Table 2).
PRE- AND POST-SYNAPTIC MECHANISMS
Mitochondrial genes
It has long been thought that mitochondria play a critical role at the NMJ, partly due to their clustering at the synapse. Solute carrier Family 25 Member 1 (SLC25A1) encodes the mitochondrial citrate carrier across the inner mitochondrial membrane. (GC22M019407, GeneCards) Hence, SLC25A1 mutations result in an impaired citrate/isocitrate export from mitochondria, and subsequently increased excretion of 2-hydroxyglutaric acid. This condition was reported in an 18-month-old female manifesting poor sucking, hypotonia, and prolonged apnoeas at birth, followed by psychomotor retardation, epilepsy, sensorineural deafness, and radiological evidence of corpus callosum agenesia and optic nerves hypoplasia [121]. Other patients with a variable associations of speech and motor developmental delay, learning difficulties, fatigable weakness, ptosis, and diplopia were later described, recognizing a link between SLC25A1 and altered NMJ transmission [122–126]. Indeed, SFEMG examination showed increased jitter or jitter with blocking and RNS detected a decremental response with some clinical improvement after therapy with 3,4-DAP or AChEI. Interestingly, reflexes were reduced at rest with post-exercise potentiation in one patient, suggesting a presynaptic alteration [123, 124].
This hypothesis was corroborated by the SLC25A1 zebrafish orthologues knocking down via antisense MOs injection. The animal model presented a motor impairment similarly to humans, displaying altered tail morphology, swimming, and touch-evoked escape responses. Furthermore, histological NMJ evaluation revealed short motor axon terminals with incomplete synapse formation [123]. Indeed, mitochondria are crucial for the presynaptic nerve terminal function since it requires a high energy demand [124] (Table 2) [127–129]. To date the SLC25A1 mutation is the only mitochondrial mutation described to cause CMS. However, given the importance of mitochondria to neurotransmission it is likely future mutations will be described necessitating the creation of a fifth ’mitochondrial CMS’ group.
CONCLUSIONS
Presynaptic CMS are very rare neuromuscular disorders, only comprising 5-10% all CMS types. Most proteins involved in presynaptic function are highly expressed in the central nervous system and other tissues besides the motor endplate, complicating the clinical picture. Some types of CMS classified in other groups, such as postsynaptic or glycosylation defects CMS, result from the alteration of proteins which may partly influence the presynaptic site. A low or no presence of motor neuron axon branches was demonstrated at the presynaptic zone, as well as a reduced quanta number available for release, resulting in a decreased endplate quantal content in two cases of glycosylation CMS [130, 131]. Frequently, CMS phenotypical and genotypical heterogeneity makes diagnosis challenging and delays treatment.
Clinicians can exploit animal models to deeply consider the pathophysiology of CMS, from which the broad spectrum of their clinical features derives. Recent technologies have allowed us to create ever more valid models, limiting off-target effects and providing a reliable substrate to test new drugs.
Many different animals have been used to study the organization and function of the NMJ, including C. elegans, D. melanogaster, zebrafish, and mice. Since the mouse NMJ is large and easily accessible, mouse models are currently the most functional option to investigate histologically and electro-physiologically. Moreover, they allow testing the clinical disease severity by objective measurement of muscle strength and fatigue, repeating the evaluation during the disease course and after treatment [132].
However, the localization of many presynaptic proteins even in the central nervous system has made presynaptic animal models hard to produce. The central clinical manifestations may cover or alter the NMJ features. Additionally, since most presynaptic proteins are essential for animal survival, specific techniques of NMJ gene modulation are needed. In general, presynaptic gene-KO mice are lethal at the embryonic stage in a homozygous state, preventing the possibility of studying the genetic pathomechanisms. Although, Cre-loxP technology enables gene deletion in select groups of cells such as motoneurons. There are however differences between human and mouse NMJs: anatomically human NMJs are smaller, have a higher folding index and active zone density, lower quantal content, and lower safety factor; finally, protein expression is quite different, influencing NMJ function. Proteomic profile analysis shows no significant variation between the two species. However, the expression profile highlights distinct expression levels for 97% of at least 36 distinct nervous system-related molecular pathways impacting on NMJs. They include synaptic signalling pathways, particularly, those related to agrin [133, 134].
C. elegans is another useful model to examine neurotransmission mechanisms, in particular at the presynapse, due to the conservation of many pathways between the nematode and mammals. C. elegans is a simple and transparent model to analyse gene and protein expression patterns using fluorescence [135].
Zebrafish is an advantageous model as many pathways of muscle development and differentiation are conserved between zebrafish and mammals. Zebrafish muscle has similarities to human, sharing some components of the sarcomere and contraction machinery [136]. NMJs are cholinergic with pentameric nicotinic ACh receptors at the postsynaptic membrane like humans. In addition, zebrafish present many synaptic molecules with a similar function in mammals. Finally, the transparent nature of this animal is useful for the characterization of genes and proteins’ activity in vivo.
Morpholino oligonucleotides (MOs) are an efficient method of gene-specific antisense knockdown, frequently used in zebrafish. However, the knock-down effectiveness is not always clear and off-target effects may complicate interpretation of findings. MOs could inhibit an irrelevant gene instead of, or in addition to, the intended gene [138]. Moreover, they are a short acting and must be injected at the embryos stage. Recently, crispr-technology has been employed for both short-term observations (“crispants”) as well as generating stable zf lines with specifically engineered mutations [139].
However, in some situations animal models may be inappropriate to provide evidence of the entire human genotypic heterogeneity. For example, most animal models have been created via a knocking out mechanism mimicking a null gene mutation. However, the majority of patients present with missense mutations which preserves the function of the encoded protein at least in part. Thereby, the distinct phenotypes between humans and animal models depend on both species properties and different types of mutations with their impact on the relative protein structure and function. While many presynaptic CMS causative genes have recently been discovered, we lack reproducible and reliable animal models. However, they remain a valid tool to determine drugs’ mechanism of action and response [140].
Finally, the NMJ has also been studied across a wider range of mammalian species such as cat, dog, pig, and sheep, showing that the latter is most similar to humans [141]. Hence, the opportunity to use large animal models for a more accurate NMJ diseases study should be considered.
In conclusion, animal models still present several limitations in reproducing human genotypes and phenotypes and many molecular details about NMJs need to be elucidated. However, a bilateral exchange between clinical and preclinical studies offers the only promising way to increase our recognition of CMS.
ACKNOWLEDGMENTS
All figures were created with BioRender.com.
FUNDING
HL receives support from the Canadian Institutes of Health Research (Foundation Grant FDN-167281, Team Grant ERT-174211), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant for NMD4 C NG2-170044), the Canada Foundation for Innovation (CFI-JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279). AP, HL and CR are members of the European Reference Network for rare, neuromuscular diseases ERN EURO-NMD –Project ID No 739543. SH received support from the Bob Korneluk CHEO Trainee Research Grant and Ontario Graduate Scholarship (OGS).
REFERENCES
[1] | Engel AG , Shen XM , Selcen D , Sine SM . Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. (2015) ;14: (4):420–434. doi:10.1016/S1474-4422(14)70201-7 |
[2] | Mihaylova V , Scola RH , Gervini B , et al. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J Neurol Neurosurg Psychiatry. (2010) ;81: (9):973–977. doi:10.1136/jnnp.2009.177816 |
[3] | Natera-de Benito D , Töpf A , Vilchez JJ , et al. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscular Disorders. (2017) ;27: (12):1087–1098. doi:10.1016/j.nmd.2017.08.003 |
[4] | Parr JR , Andrew MJ , Finnis M , Beeson D , Vincent A , Jayawant S . How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child. (2014) ;99: (6):539–542. doi:10.1136/archdischild-2013-304788 |
[5] | Abicht A , Müller JS , Lochmüller H . Congenital Myasthenic Syndromes Overview. 2003 May 9 [updated 2021 Dec 23]. In: Adam MP, Ardinger HH, Pagon RA,Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. Accessed March 14, 2022. |
[6] | Finsterer J . Congenital myasthenic syndromes. Orphanet J Rare Dis. (2019) ;14: (1). doi:10.1186/s13023-019-1025-5 |
[7] | Iyadurai SJP . Congenital Myasthenic Syndromes. Neurol Clin. (2020) ;38: (3):541–552. doi:10.1016/j.ncl.2020.03.004 |
[8] | McMacken G , Whittaker RG , Evangelista T , Abicht A , Dusl M , Lochmüller H . Congenital myasthenic syndrome with episodic apnoea: clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J Neurol. (2018) ;265: (1):194–203. doi:10.1007/s00415-017-8689-3 |
[9] | Bauché S , O’Regan S , Azuma Y , et al. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. Am J Hum Genet. (2016) ;99: (3):753–761. doi:10.1016/j.ajhg.2016.06.033 |
[10] | O’Grady GL , Verschuuren C , Yuen M , et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology. (2016) ;87: (14):1442–1448. doi:10.1212/WNL.0000000000003179 |
[11] | Maselli RA , Chen D , Mo D , Bowe C , Fenton G , Wollmann RL . Choline acetyl transferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve. (2003) ;27: (2):180–187. doi:10.1002/mus.10300 |
[12] | Ramdas S , Beeson D . Congenital myasthenic syndromes: where do we go from here? Neuromuscular Disorders. (2021) ;31: (10):943–954. doi:10.1016/j.nmd.2021.07.400 |
[13] | Régal L , Shen XM , Selcen D , et al. PREPL Deficiency with orwithout Cystinuria Causes a Novel Myasthenic Syndrome. Neurology. (2014) Apr;82: (14):1254–60. Doi:10.1212/WNL.0000000000000295 |
[14] | Maselli RA , Arredondo J , Vázquez J , et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am J Med Genet A. (2017) ;173: (8):2240–2245. doi:10.1002/ajmg.a.38291 |
[15] | O’Connor E , Töpf A , Müller JS , et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain. (2016) ;139: (8):2143–2153. doi:10.1093/brain/aww130 |
[16] | Matthies DS , Fleming PA , Wilkes DM , Blakely RD . The Caenorhabditis elegans choline transporter CHO-1 sustains acetylcholine synthesis and motor function in an activity-dependent manner. J Neurosci. (2006) ;26: (23):6200–6212. doi:10.1523/JNEUROSCI.5036-05.2006 |
[17] | Keshishian H , Broadie K , Chiba A , Bate M . 19%. 19545-75 Copyrighl 0 1996 by Annual Reviews Inc. All Righfs Reserved. www.annualreviews.org |
[18] | Pardal-Fernández JM , Carrascosa-Romero MC , Álvarez S , Medina-Monzón MC , Caamaño MB , de Cabo C . A new severemutation in the SLC5A7 gene related to congenital myasthenicsyndrome type 20. Neuromuscular Disorders. (2018) ;28: (10):881–884. doi:10.1016/j.nmd.2018.06.020 |
[19] | Banerjee M , Arutyunov D , Brandwein D , et al. The novel p.Ser263Phe mutation in the human high-affinity choline transporter 1 (CHT1/SLC5A7) causes a lethal form of fetal akinesia syndrome. Hum Mutat. (2019) ;40: (10):1676–1683. doi:10.1002/humu.23828 |
[20] | Rodríguez Cruz PM , Hughes I , Manzur A , et al. Presynaptic congenital myasthenic syndrome due to three novel mutations in SLC5A7 encoding the sodium-dependant high-affinity cholinetransporter. Neuromuscular Disorders. (2021) ;31: (1):21–28. doi:10.1016/j.nmd.2020.10.006 |
[21] | Wang H , Salter CG , Refai O , et al. Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization. Brain. (2017) ;140: (11):2838–2850. doi:10.1093/brain/awx249 |
[22] | Allen DD , Smith QR . Characterization of the Blood-Brain Barrier Choline Transporter Using the in Situ Rat Brain Perfusion Technique. J Neurochem. (2001) Feb;76: (4):1032–41. Doi: 10.1046/j.1471-4159.2001.00093.x. Erratum in: J Neurochem 2001 Apr;77(2):704.; 2001. |
[23] | Murakami H , Sawada N , Koyabu N , Ohtani H , Sawada Y . Characteristics of Choline Transport across the Blood-Brain Barrier in Mice: Correlation with in Vitro Data. Pharm Res. (2000) Dec;17: (12):1526–30. doi:10.1023/a:1007613326759; 2000 |
[24] | Ferguson SM , Bazalakova M , Savchenko V , Carlos Tapia J , Wright J , Blakely RD . Lethal Impairment of Cholinergic Neurotransmission in Hemicholinium-3-Sensitive Choline Transporter Knockout Mice.; 2004. www.pnas.orgcgidoi10.1073pnas.0401667101 |
[25] | Lund D , Ruggiero AM , Ferguson SM , et al. Motor neuron-specific overexpression of the presynaptic choline transporter: Impact on motor endurance and evoked muscle activity. Neuroscience. (2010) ;171: (4):1041–1053. doi:10.1016/j.neuroscience.2010.09.057 |
[26] | Bazalakova MH , Wright J , Schneble EJ , et al. Deficits in acetylcholine homeostasis, receptors and behaviors in choline transporter heterozygous mice. Genes Brain Behav. (2007) ;6: (5):411–424. doi:10.1111/j.1601-183X.2006.00269.x |
[27] | Paolone G , Mallory CS , Koshy Cherian A , Miller TR , Blakely RD , Sarter M . Monitoring cholinergic activity during attentional performance in mice heterozygous for the choline transporter: A model of cholinergic capacity limits. Neuropharmacology. (2013) ;75: :274–285. doi:10.1016/j.neuropharm.2013.07.032 |
[28] | Donovan E , Avila C , Klausner S , et al. Disrupted choline clearance and sustained acetylcholine release in vivo by a common choline transporter coding variant associated with poor attentional control in humans. The Journal of Neuroscience. Published online March 1, 2022:JN-RM-1334-21. doi: 10.1523/JNEUROSCI.1334-21.2022 |
[29] | English BA , Appalsamy M , Diedrich A , et al. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am J Physiol Heart Circ Physiol. (2010) ;299: :799–810. doi:10.1152/ajpheart.00170.2010.-Healthy |
[30] | Ohno K , Tsujino A , Brengman JM , et al. Choline Acetyltransferase Mutations Cause Myasthenic Syndrome Associated with Episodic Apnea in Humans. www.pnas.org |
[31] | Shen XM , Crawford TO , Brengman J , et al. Functional consequences and structural interpretation of mutations of human choline acetyltransferase. Hum Mutat. (2011) ;32: (11):1259–1267. doi:10.1002/humu.21560 |
[32] | Byring RF , Pihko H , Tsujino A , et al. Congenital Myasthenic Syndrome Associated with Episodic Apnea and Sudden Infant Death. www.elsevier.com/locate/nmd |
[33] | Liu Z , Zhang L , Shen D , et al. Compound heterozygous CHAT gene mutations of a large deletion and a missense variant in a Chinese patient with severe congenital myasthenic syndrome with episodic apnea. Front Pharmacol. (2019) ;10: (MAR). doi:10.3389/fphar.2019.00259 |
[34] | Zhang Y , Cheng X , Luo C , et al. Congenital Myasthenic Syndrome Caused by a Novel Hemizygous CHAT Mutation. Front Pediatr. (2020) ;8: . doi:10.3389/fped.2020.00185 |
[35] | Barisic N , Müller JS , Paucic-Kirincic E , et al. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. European Journal of Paediatric Neurology. (2005) ;9: (1):7–12. doi:10.1016/j.ejpn.2004.10.008 |
[36] | Schmidt C , Abicht A , Krampfl K , et al. Congenital myasthenic syndrome due to a novel missense mutation in the gene encoding choline acetyltransferase. doi: 10.1016/S0 |
[37] | Kraner S , Laufenberg I , Straßburg HM , Sieb JP , Steinlein OK . Congenital Myasthenic Syndrome With Episodic Apnea in Patients Homozygous for a CHAT Missense Mutation. http://archneur.jamanetwork.com/ |
[38] | Schara U , Christen HJ , Durmus H , et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. European Journal of Paediatric Neurology. (2010) ;14: (4):326–333. doi:10.1016/j.ejpn.2009.09.009 |
[39] | Stankiewicz P , Kulkarni S , Dharmadhikari A V , et al. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are Likely Mediated by Complex Low-Copy Repeats. Hum Mutat. (2012) ;33: (1):165–179. doi:10.1002/humu.21614 |
[40] | Arredondo J , Lara M , Gospe SM , et al. Choline Acetyltransferase Mutations Causing Congenital Myasthenic Syndrome: Molecular Findings and Genotype-Phenotype Correlations. Hum Mutat. (2015) ;36: (9):881–893. doi:10.1002/humu.22823 |
[41] | Dhasakeerthi T , Aravindhan A , Woodall A , Mills W , Veerapandiyan A . Congenital Myasthenic Syndrome due to a Novel Mutation in CHAT Gene. J Clin Neuromuscul Dis. (2021) ;23: (1):54–55. doi:10.1097/CND.0000000000000336 |
[42] | Misgeld T , Burgess RW , Lewis RM , Cunningham JM , Lichtman JW , Sanes JR . Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. (2002) ;36: (4):635–648. doi:10.1016/s0896-6273(02)01020-6 |
[43] | Brandon EP , Lin W , D’amour KA , et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci.. (2003) ;23: (3):539–549. doi:10.1523/JNEUROSCI.23-02-00539.2003 |
[44] | Wang M , Wen H , Brehm P . Function of neuromuscular synapses in the zebrafish choline- acetyltransferase mutant bajan. J Neurophysiol. (2008) ;100: (4):1995–2004. doi:10.1152/jn.90517.2008 |
[45] | Joshi S , Virdi S , Etard C , Geisler R , Strähle U . Mutation of a serine near the catalytic site of the choline acetyltransferase a gene almost completely abolishes motility of the zebrafish embryo. PLoS One. (2018) ;13: (11). doi:10.1371/journal.pone.0207747 |
[46] | Proschowsky HF , Flagstad A , Cirera S , Joergensen CB , Fredholm M . Identification of a mutation in the CHAT gene of Old Danish Pointing Dogs affected with congenital myasthenic syndrome. In: Journal of Heredity. 1314 Vol 98.; 2007:539-543. doi: 10.1093/jhered/esm026 |
[47] | Aran A , Segel R , Kaneshige K , et al. Vesicular acetylcholine transporter defect underlies devastating congenital myasthenia syndrome. Neurology. (2017) ;88: (11):1021–1028. doi:10.1212/WNL.0000000000003720 |
[48] | Lamond A , Buckley D , O’Dea J , Turner L . Variants of SLC18A3 leading to congenital myasthenic syndrome in two children with varying presentations. BMJ Case Rep. (2021) ;14: (1). doi:10.1136/bcr-2020-237799 |
[49] | Schwartz M , Sternberg D , Whalen S , et al. How chromosomal deletions can unmask recessive mutations? Deletions in 10q11. 2 associated with CHAT or SLC18A3 mutations lead to congenital myasthenic syndrome. Am J Med Genet A. (2018) ;176: (1):151–155. doi:10.1002/ajmg.a.38515 |
[50] | Della Marina A , Arlt A , Schara-Schmidt U , et al. Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3. Cells. (2021) ;10: (12):3481. doi:10.3390/cells10123481 |
[51] | Hakonen AH , Polvi A , Saloranta C , et al. SLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy. Am J Med Genet A. (2019) ;179: (7):1362–1365. doi:10.1002/ajmg.a.61186 |
[52] | Kitamoto T , Xie X , Wu CF , Salvaterra PM . Isolation and Characterization of Mutants for the Vesicular Acetylcholine Transporter Gene in Drosophila Melanogaster. Vol 42.; 2000. |
[53] | Showell SS , Martinez Y , Gondolfo S , Boppana S , Lawal HO . Overexpression of the vesicular acetylcholine transporter disrupts cognitive performance and causes age-dependent locomotion decline in Drosophila. Molecular and Cellular Neuroscience. (2020) ;105: . doi:10.1016/j.mcn.2020.103483 |
[54] | White D , de Sousa Abreu RP , Blake A , et al. Deficits in the vesicular acetylcholine transporter alter lifespan and behavior in adult Drosophila melanogaster. Neurochem Int. (2020) ;137: . doi:10.1016/j.neuint.2020.104744 |
[55] | Prado VF , Martins-Silva C , de Castro BM , et al. Mice Deficient for the Vesicular Acetylcholine Transporter Are Myasthenic and Have Deficits in Object and Social Recognition. Neuron. (2006) ;51: (5):601–612. doi:10.1016/j.neuron.2006.08.005 |
[56] | Schmid S , Azzopardi E , De Jaeger X , Prado MAM , Prado VF . VAChT knock-down mice show normal prepulse inhibition but disrupted long-term habituation. Genes Brain Behav. (2011) ;10: (4):457–464. doi:10.1111/j.1601-183X.2011.00686.x |
[57] | Lara A , Damasceno DD , Pires R , et al. Dysautonomia Due to Reduced Cholinergic Neurotransmission Causes Cardiac Remodeling and Heart Failure. Mol Cell Biol. (2010) ;30: (7):1746–1756. doi:10.1128/mcb.00996-09 |
[58] | Durand MT , Becari C , S Tezini V GC , et al. Autonomic cardiocirculatory control in mice with reduced expression of the vesicular acetylcholine transporter. Am J Physiol Heart Circ Physiol. (2015) ;309: :655–662. doi:10.1152/ajpheart.00114.2015.-In |
[59] | de Castro BM , de Jaeger X , Martins-Silva C , et al. The Vesicular Acetylcholine Transporter Is Required for Neuromuscular Development and Function. Mol Cell Biol. (2009) ;29: (19):5238–5250. doi:10.1128/mcb.00245-09 |
[60] | Zhu H , Duerr JS , Varoqui H , McManus JR , Rand JB , Erickson JD . Analysis of Point Mutants in the Caenorhabditis elegans Vesicular Acetylcholine Transporter Reveals Domains Involved in Substrate Translocation. Journal of Biological Chemistry. (2001) ;276: (45):41580–41587. doi:10.1074/jbc.M103550200 |
[61] | Martins-Silva C , de Jaeger X , Guzman MS , et al. Novel strains of mice deficient for the vesicular acetylcholine transporter: Insights on transcriptional regulation and control of locomotor behavior. PLoS One. (2011) ;6: (3). doi:10.1371/journal.pone.0017611 |
[62] | Martyn AC , De Jaeger X , Magalhães AC , et al. Elimination of thevesicular acetylcholine transporter in the forebrain causeshyperactivity and deficits in spatial memory and long-termpotentiation. Proc Natl Acad Sci U S A. (2012) ;109: (43):17651–17656. doi:10.1073/pnas.1215381109 |
[63] | Kolisnyk B , Al-Onaizi MA , Hirata PHF , et al. Forebrain deletion of the vesicular acetylcholine transporter results in deficits in executive function, metabolic, and RNA splicing abnormalities in the prefrontal cortex. Journal of Neuroscience. (2013) ;33: (37):14908–14920. doi:10.1523/JNEUROSCI.1933-13.2013 |
[64] | Palmer D , Creighton S , Prado VF , Prado MAM , Choleris E , Winters BD . Mice deficient for striatal Vesicular Acetylcholine Transporter (VAChT) display impaired short-term but normal long-term object recognition memory. Behavioural Brain Research. (2016) ;311: :267–278. doi:10.1016/j.bbr.2016.05.050 |
[65] | Joviano-Santos J V , Kljakic O , Magalhães-Gomes MPS et al. Motoneuron-specific loss of VAChT mimics neuromuscular defects seenin congenital myasthenic syndrome. FEBS Journal. (2021) ;288: (18):5331–5349. doi:10.1111/febs.15825 |
[66] | Schoch S , Deák F , Königstorfer A , et al. SNARE functionanalyzed in synaptobrevin/VAMP knockout mice. Science (1979). (2001) ;294: (5544):1117–1122. doi:10.1126/science.1064335 |
[67] | Shen XM , Scola RH , Lorenzoni PJ , et al. Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann Clin Transl Neurol. (2017) ;4: (2):130–138. doi:10.1002/acn3.387 |
[68] | Salpietro V , Lin W , Delle Vedove A , et al. Homozygous Mutations in VAMP1 Cause a Presynaptic Congenital Myasthenic Syndrome. doi:10.1002/ana |
[69] | Polavarapu K , Vengalil S , Preethish-Kumar V , et al. Recessive VAMP1 mutations associated with severe congenital myasthenic syndromes –A recognizable clinical phenotype. European Journal of Paediatric Neurology. (2021) ;31: :54–60. doi:10.1016/j.ejpn.2021.02.005 |
[70] | Al-Muhaizea MA , AlQuait L , AlRasheed A , et al. Pyrostigmine therapy in a patient with VAMP1-related congenital myasthenic syndrome. Neuromuscular Disorders. (2020) ;30: (7):611–615. doi:10.1016/j.nmd.2020.04.007 |
[71] | Liu Y , Sugiura Y , Lin W . The role of Synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. Journal of Physiology. (2011) ;589: (7):1603–1618. doi:10.1113/jphysiol.2010.201939 |
[72] | Nystuen AM , Schwendinger JK , Sachs AJ , Yang AW , Haider NB . A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. (2007) ;8: (1):1–10. doi:10.1007/s10048-006-0068-7 |
[73] | Liu Y , Sugiura Y , Südhof TC , Lin W . Ablation of all synaptobrevin vSNAREs blocks evoked but not spontaneous neurotransmitter release at neuromuscular synapses. Journal of Neuroscience. (2019) ;39: (31):6049–6066. doi:10.1523/JNEUROSCI.0403-19.2019 |
[74] | Shen XM , Selcen D , Brengman J , Engel AG . Mutant SNAP25B Causes Myasthenia, Cortical Hyperexcitability, Ataxia, and Intellectual Disability.; 2014. |
[75] | Reynolds HM , Wen T , Farrell A , et al. Rapid genome sequencing identifies a novel de novo SNAP25 variant for neonatal congenital myasthenic syndrome. Cold Spring Harb Mol Case Stud. (2022) ;8: (7):a006242. doi:10.1101/mcs.a006242 |
[76] | Washbourne P , Thompson PM , Carta M , et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. (2002) ;5: (1):19–26. doi:10.1038/nn783 |
[77] | Jeans AF , Oliver PL , Johnson R , et al. A Dominant Mutation in Snap25 Causes Impaired Vesicle Trafficking, Sensorimotor Gating, and Ataxia in the Blind-Drunk Mouse.; 2007. www.pnas.orgcgidoi10.1073pnas.0610222104 |
[78] | Yang H , Zhang M , Shi J , et al. Brain-Specific SNAP-25 Deletion Leads to Elevated Extracellular Glutamate Level and Schizophrenia-Like Behavior in Mice. Neural Plast. (2017) ;2017: . doi:10.1155/2017/4526417 |
[79] | Hoerder-Suabedissen A , Korrell K V , Hayashi S , et al. Cell-specific loss of SNAP25 from cortical projection neurons allows normal development but causes subsequent neurodegeneration. Cerebral Cortex. (2019) ;29: (5):2148–2159. doi:10.1093/cercor/bhy127 |
[80] | McKee AG , Loscher JS , O’Sullivan NC , et al. AAV-mediated chronic over-expression of SNAP-25 in adult rat dorsal hippocampus impairs memory-associated synaptic plasticity. J Neurochem. (2010) ;112: (4):991–1004. doi:10.1111/j.1471-4159.2009.06516.x |
[81] | Herrmann DN , Horvath R , Sowden JE , et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet. (2014) ;95: (3):332–339. doi:10.1016/j.ajhg.2014.08.007 |
[82] | Whittaker RG , Herrmann DN , Bansagi B , et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology. (2015) ;85: (22):1964–1971. doi:10.1212/WNL.0000000000002185 |
[83] | Fionda L , Turon-Sans J , Fuentes Prior P , et al. A new de novo SYT2 mutation presenting as distal weakness. Neuropathy or neuromuscular junction dysfunction? Journal of the Peripheral Nervous System. (2021) ;26: (1):113–117. doi:10.1111/jns.12425 |
[84] | Maselli RA , Wei DT , Hodgson TS , et al. Dominant and recessive congenital myasthenic syndromes caused by SYT2 mutations. Muscle Nerve. (2021) ;64: (2):219–224. doi:10.1002/mus.27332 |
[85] | Donkervoort S , Mohassel P , Laugwitz L , et al. Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am J Med Genet A. (2020) ;182: (10):2272–2283. doi:10.1002/ajmg.a.61765 |
[86] | Maselli RA , van der Linden H , Ferns M . Recessive congenital myasthenic syndrome caused by a homozygous mutation in SYT2 altering a highly conserved C-terminal amino acid sequence. Am J Med Genet A. (2020) ;182: (7):1744–1749. doi:10.1002/ajmg.a.61579 |
[87] | Bauché S , Sureau A , Sternberg D , et al. New recessive mutationsin SYT2 causing severe presynaptic congenital myasthenic syndromes. Neurol Genet. (2020) ;6: (6). doi:10.1212/NXG.0000000000000534 |
[88] | Mackler JM , Drummond JA , Loewen CA , Robinson IM , Reist NE . The C2B Ca2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. (2002) ;418: (6895):340–344. doi:10.1038/nature00846 |
[89] | Shields MC , Bowers MR , Fulcer MM , et al. Drosophila studies support a role for a presynaptic synaptotagmin mutation in a human congenital myasthenic syndrome. PLoS One. (2017) ;12: (9). doi:10.1371/journal.pone.0184817 |
[90] | Pang ZP , Sun J , Rizo J , Maximov A , Südhof TC . Genetic analysis of synaptotagmin 2 in spontaneous and Ca 2+-triggered neurotransmitter release. EMBO Journal. (2006) ;25: (10):2039–2050. doi:10.1038/sj.emboj.7601103 |
[91] | Pang ZP , Melicoff E , Padgett D , et al. Synaptotagmin-2 is essential for survival and contributes to Ca 2+triggering of neurotransmitter release in central and neuromuscular synapses. Journal of Neuroscience. (2006) ;26: (52):13493–13504. doi:10.1523/JNEUROSCI.3519-06.2006 |
[92] | Wen H , Linhoff MW , McGinley MJ , et al. Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A. (2010) ;107: (31):13906–13911. doi:10.1073/pnas.1008598107 |
[93] | Augustin I , Korte S , Rickmann M , et al. The Cerebellum-Specific Munc13 Isoform Munc13-3 Regulates Cerebellar Synaptic Transmission and Motor Learning in Mice.; 2001. |
[94] | Engel AG , Selcen D , Shen XM , Milone M , Harper CM . Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet. (2016) ;2: (5). doi:10.1212/NXG.0000000000000105 |
[95] | Varoqueaux F , Sons MS , Plomp JJ , Brose N . Aberrant Morphology and Residual Transmitter Release at the Munc13-Deficient Mouse Neuromuscular Synapse. Mol Cell Biol. (2005) ;25: (14):5973–5984. doi:10.1128/mcb.25.14.5973-5984.2005 |
[96] | Michelassi F , Liu H , Hu Z , Dittman JS . A C1-C2 Module in Munc13 Inhibits Calcium-Dependent Neurotransmitter Release. doi: 10.1016/j.neuron |
[97] | Abouhamed M , Grobe K , Leefa Chong San I v , et al. Myosin IXa Regulates Epithelial Differentiation and Its Deficiency Results in Hydrocephalus. Mol Biol Cell. (2009) ;20: :5074–5085. doi:10.1091/mbc.E09 |
[98] | Thelen S , Abouhamed M , Ciarimboli G , Edemir B , Bähler M . Rho GAP myosin IXa is a regulator of kidney tubule function. Am J Physiol Renal Physiol. (2015) ;309: :501–513. doi:10.1152/ajprenal.00220.2014.-Mammalian |
[99] | Folci A , Murru L , Vezzoli E , et al. Myosin IXa binds AMPAR and regulates synaptic structure, LTP, and cognitive function. Front Mol Neurosci. (2016) ;9: (JAN). doi:10.3389/fnmol.2016.00001 |
[100] | O’Connor E , Phan V , Cordts I , et al. MYO9A deficiency in motor neurons is associated with reduced neuromuscular agrin secretion. Hum Mol Genet. (2018) ;27: (8):1434–1446. doi:10.1093/hmg/ddy054 |
[101] | O’connor E , Cairns G , Spendiff S , et al. Modulation of agrin and RhoA pathways ameliorates movement defects and synapse morphology in MYO9A-depleted Zebrafish. Cells. (2019) ;8: (8). doi:10.3390/cells8080848 |
[102] | Boonen K , Régal L , Jaeken J , Creemers JWM . PREPL, a Prolyl Endopeptidase-Like Enzyme by Name Only?-Lessons from Patients. Vol 10.; 2011. |
[103] | Régal L , Shen XM , Selcen D , et al. Supplemental Data at Neurology.Org PREPL Deficiency with or without Cystinuria Causes a Novel Myasthenic Syndrome.; 2014. |
[104] | Régal L , Mårtensson E , Maystadt I , et al. PREPL deficiency:Delineation of the phenotype and development of a functional bloodassay. Genetics in Medicine. (2018) ;20: (1):109–118. doi:10.1038/gim.2017.74 |
[105] | Zhang P , Wu B , Lu Y , et al. First maternal uniparental disomy for chromosome 2 with PREPL novel frameshift mutation of congenital myasthenic syndrome 22 in an infant. Mol Genet Genomic Med. (2020) ;8: (3). doi:10.1002/mgg3.1144 |
[106] | Silva S , Miyake N , Tapia C , Matsumoto N . The second point mutation in PREPL: A case report and literature review. J Hum Genet. (2018) ;63: (5):677–681. doi:10.1038/s10038-018-0426-y |
[107] | Laugwitz L , Redler S , Buchert R , et al. Isolated PREPL deficiency associated with congenital myasthenic syndrome-22. Klin Padiatr. (2018) Sep;230: (5):281–283. 20. doi:10.1055/a-0605-3659 |
[108] | Shchagina O , Bessonova L , Bychkov I , Beskorovainaya T , Poliakov A . A family case of congenital myasthenic syndrome-22 induced by different combinations of molecular causes in siblings. Genes (Basel). (2020) ;11: (7):1–6. doi:10.3390/genes11070821 |
[109] | Yang Q , Hua R , Qian J , et al. PREPL Deficiency: A Homozygous Splice Site PREPL Mutation in a Patient With Congenital Myasthenic Syndrome and Absence of Ovaries and Hypoplasia of Uterus. Front Genet. (2020) ;11: . doi:10.3389/fgene.2020.00198 |
[110] | Lone AM , Leidl M , McFedries AK , Horner JW , Creemers J , Saghatelian A . Deletion of prepl causes growth impairment and hypotonia in mice. PLoS One. (2014) ;9: (2). doi:10.1371/journal.pone.0089160 |
[111] | D’Agostino G , Kim JD , Liu ZW , et al. Prolyl endopeptidase-deficient mice have reduced synaptic spine density in the CA1 region of the hippocampus, impaired LTP, and spatial learning and memory. Cerebral Cortex. (2013) ;23: (8):2007–2014. doi:10.1093/cercor/bhs199 |
[112] | Maselli RA , Arredondo J , Vázquez J , et al. A presynapticcongenital myasthenic syndrome attributed to a homozygous sequencevariant in LAMA5. Ann N Y Acad Sci. (2018) ;1413: (1):119–125. doi:10.1111/nyas.13585 |
[113] | Nguyen NM , Miner JH , Pierce RA , Senior RM . Laminin α5 is required for lobar septation and visceral pleural basement membrane formation in the developing mouse lung. Dev Biol. (2002) ;246: (2):231–244. doi:10.1006/dbio.2002.0658 |
[114] | Nguyen NM , Kelley DG , Schlueter JA , Meyer MJ , Senior RM , Miner JH . Epithelial laminin α5 is necessary for distal epithelial cell maturation, VEGF production, and alveolization in the developing murine lung. Dev Biol. (2005) ;282: (1):111–125. doi:10.1016/j.ydbio.2005.02.031 |
[115] | Kikkawa Y , Miner JH . Molecular dissection of laminin α5 in vivo reveals separable domain-specific roles in embryonic development and kidney function. Dev Biol. (2006) ;296: (1):265–277. doi:10.1016/j.ydbio.2006.04.463 |
[116] | Nishimune H , Valdez G , Jarad G , et al. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. Journal of Cell Biology. (2008) ;182: (6):1201–1215. doi:10.1083/jcb.200805095 |
[117] | Jones LK , Lam R , McKee KK , et al. A mutation affecting laminin alpha 5 polymerisation gives rise to a syndromic developmental disorder. Development (Cambridge). (2020) ;147: (21). doi:10.1242/dev.189183 |
[118] | Webb AE , Sanderford J , Frank D , Talbot WS , Driever W , Kimelman D . Laminin α5 is essential for the formation of the zebrafish fins. Dev Biol. (2007) ;311: (2):369–382. doi:10.1016/j.ydbio.2007.08.034 |
[119] | Maselli RA , Vázquez J , Schrumpf L , et al. Presynaptic congenitalmyasthenic syndrome with altered synaptic vesicle homeostasis linkedto compound heterozygous sequence variants in RPH3A. Mol GenetGenomic Med. (2018) ;6: (3):434–440. doi:10.1002/mgg3.370 |
[120] | Staunton J , Ganetzky B , Nonet ML . Rabphilin Potentiates Soluble N-Ethylmaleimide Sensitive Factor Attachment Protein Receptor Function Independently of Rab3.; 2001. |
[121] | Edvardson S , Porcelli V , Jalas C , et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J Med Genet. (2013) ;50: (4):240–245. doi:10.1136/jmedgenet-2012-101485 |
[122] | Nota B , Struys EA , Pop A , et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet. (2013) ;92: (4):627–631. doi:10.1016/j.ajhg.2013.03.009 |
[123] | Chaouch A , Porcelli V , Cox D , et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. (2014) ;1: (1):75–90. doi:10.3233/JND-140021 |
[124] | Balaraju S , Töpf A , McMacken G , et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurrent SLC25A1 variant. European Journal of Human Genetics. (2020) ;28: (3):373–377. doi:10.1038/s41431-019-0506-2 |
[125] | Al-Futaisi A , Ahmad F , Al-Kasbi G , Al-Thihli K , Koul R , Al-Maawali A . Missense mutations in SLC25A1 are associated with congenital myasthenic syndrome type 23. Clin Genet. (2020) ;97: (4):666–667. doi:10.1111/cge.13678 |
[126] | Li W , Zhang M , Zhang L , et al. A case report of an intermediate phenotype between congenital myasthenic syndrome and D-2- And L-2-hydroxyglutaric aciduria due to novel SLC25A1 variants. BMC Neurol. (2020) ;20: (1). doi:10.1186/s12883-020-01854-6 |
[127] | Rigby MJ , Orefice NS , Lawton AJ , et al. Increased expression of SLC25A1/CIC causes an autistic-like phenotype with altered neuron morphology. Brain. (2022) ;145: (2):500–516. doi:10.1093/brain/awab295 |
[128] | Tan M , Mosaoa R , Graham GT , et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. (2020) ;27: (7):2143–2157. doi:10.1038/s41418-020-0491-6 |
[129] | Morciano P , Carrisi C , Capobianco L , et al. A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum Mol Genet. (2009) ;18: (21):4180–4188. doi:10.1093/hmg/ddp370 |
[130] | Nicolau S , Liewluck T , Shen XM , Selcen D , Engel AG , Milone M . A homozygous mutation in GMPPB leads to centronuclear myopathy with combined pre- and postsynaptic defects of neuromuscular transmission. Neuromuscular Disorders. (2019) ;29: (8):614–617. doi:10.1016/j.nmd.2019.07.001 |
[131] | Senderek J , Müller JS , Dusl M , et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. (2011) ;88: (2):162–172. doi:10.1016/j.ajhg.2011.01.008 |
[132] | Webster RG . Animal models of the neuromuscular junction, vitally informative for understanding function and the molecular mechanisms of congenital myasthenic syndromes. Int J Mol Sci. (2018) ;19: (5). doi:10.3390/ijms19051326 |
[133] | Jones RA , Harrison C , Eaton SL , et al. Cellular and Molecular Anatomy of the Human Neuromuscular Junction. Cell Rep. (2017) ;21: (9):2348–2356. doi:10.1016/j.celrep.2017.11.008 |
[134] | Slater CR . Reliability of Neuromuscular Transmission and How It Is Maintained. |
[135] | Wang ZW . Origin of quantal size variation and high-frequency miniature postsynaptic currents at the Caenorhabditis elegans neuromuscular junction. J Neurosci Res. (2010) ;88: (16):3425–3432. doi:10.1002/jnr.22468 |
[136] | Maves L . Recent advances using zebrafish animal models for muscle disease drug discovery. Expert Opin Drug Discov. (2014) ;9: (9):1033–1045. doi:10.1517/17460441.2014.927435 |
[137] | Bayés À , Collins MO , Reig-Viader R , et al. Evolution ofcomplexity in the zebrafish synapse proteome. Nat Commun. (2017) ;8: . doi:10.1038/ncomms14613 |
[138] | Eisen JS , Smith JC . Controlling morpholino experiments: Don’t stop making antisense. Development. (2008) ;135: (10):1735–1743. doi:10.1242/dev.001115 |
[139] | Albadri S , del Bene F , Revenu C . Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods. (2017) ;122: :121–77-85. doi:10.1016/j.ymeth.2017.03.005 |
[140] | Fralish Z , Lotz EM , Chavez T , Khodabukus A , Bursac N . Neuromuscular Development and Disease: Learning From in vitro and invivo Models. Front Cell Dev Biol. (2021) ;9: . doi:10.3389/fcell.2021.764732 |
[141] | Boehm I , Alhindi A , Leite AS , et al. Comparative anatomy of the mammalian neuromuscular junction. J Anat. (2020) ;237: (5):827–836. doi:10.1111/joa.13260 |
[142] | Sekine Y , Kannan R , Wang X , Strittmatter SM . Rabphilin3A reduces integrin-dependent growth cone signaling to restrict axon regeneration after trauma. Exp Neurol. (2022) ;353: :114070. |
[143] | Schlüter OM , Schnell E , Verhage M , et al. Rabphilin Knock-Out Mice Reveal That Rabphilin Is Not Required for Rab3 Function in Regulating Neurotransmitter Release. The Journal of Neuroscience. (1999) ;19: (14):5834–5846. |
[144] | Ferrari E , Scheggia D , Zianni E , et al. Rabphilin-3A as a novel target to reverse α-synuclein-induced synaptic loss in Parkinson’s disease. Pharmacol Res. (2022) ;183: . doi:10.1016/j.phrs.2022.106375 |
[145] | Zhu X , Li H , You W , et al. Role of Rph3A in brain injury induced by experimental cerebral ischemia-reperfusion model in rats. CNS Neurosci Ther. (2022) ;28: (7):1124–1138. doi:10.1111/cns.13850 |
[146] | Feng WJ , Liang T , Yu JW , et al. RAB-27 and its effector RBF-1 regulate the tethering and docking steps of DCV exocytosis in C. elegans. Sci China Life Sci. (2012) ;55: (3):228–235. doi:10.1007/s11427-012-4296-9 |


