
Biomarkers for C9orf7-ALS in Symptomatic and Pre-symptomatic Patients: State-of-the-art in the New Era of Clinical Trials
Abstract
The development of new possible treatments for C9orf72-related ALS and the possibility of early identification of subjects genetically at risk of developing the disease is creating a critical need for biomarkers to track neurodegeneration that could be used as outcome measures in clinical trials. Current candidate biomarkers in C9orf72-ALS include neuropsychology tests, imaging, electrophysiology as well as different circulating biomarkers. Neuropsychology tests show early executive and verbal function involvement both in symptomatic and asymptomatic mutation carriers. At brain MRI, C9orf72-ALS patients present diffuse white and grey matter degeneration, which are already identified up to 20 years before symptom onset and that seem to be slowly progressive over time, while regions of altered connectivity at fMRI and of hypometabolism at [18F]FDG PET have been described as well. At the same time, spinal cord MRI has also shown progressive decrease of FA in the cortico-spinal tract over time. On the side of wet biomarkers, neurofilament proteins are increased both in the CSF and serum just before symptom onset and tend to slowly increase over time, while poly(GP) protein can be detected in the CSF and probably used as target engagement marker in clinical trials.
ABBREVIATIONS:
ATS | Andersen-Tawil syndrome |
AAV | adeno-associated virus |
ALS | Amyotrophic Lateral Sclerosis |
ASO | Antisense oligonucleotide |
AD | axial diffusivity |
BAC | bacterial artificial chromosome |
bvFTD | behavioral fronto-temporal dementia |
CSF | cerebro-spinal fluid |
CNS | central nervous system |
CST | cortico-spinal tract |
DPR | dipeptide-repeat protein |
DTI | diffusion tensor imaging |
EMG | electromyography |
FA | fractional anisotropy |
FDA | Food and Drug Administration |
fMRI | functional magnetic imaging |
FTD | fronto-temporal dementia |
GA | glycine-alanine |
GP | glycine-proline |
MD | mean diffusivity |
mi-RNA | micro RNA |
MRI | magnetic resonance imaging |
MUNE | motor unit number estimation |
MUNIX | motor unit number index |
PET | positron emission tomography |
pNfL | phosphorylated Neurofilament Light Chain |
pNfH | phosphorylated Neurofilament Heavy Chain |
RCT | randomized clinical trial |
RD | radial diffusivity |
SC | spinal cord |
TDP43 | TAR-DNA binding protein 43 |
TMS | transcranial magnetic stimulation |
VBM | voxel-based morphometry |
INTRODUCTION
Amyotrophic Lateral Sclerosis (ALS) and Fronto-Temporal Dementia (FTD) are progressive neurodegenerative diseases displaying a remarkable clinical overlap and sharing common genetic bases and pathogenic mechanisms [1, 2]. The discovery, in 2011, that intronic GGGGCC hexanucleotide expansions in the C9orf72 gene localized on chromosome 9 justify almost half of familial cases of ALS and FTD, as well as up to 20%of apparently sporadic forms of the two diseases, further established the tight bond existing between these two clinical entities as part of a single spectrum [3, 4].
Over the last 10 years, knowledge about C9orf72 mutations and the clinical phenotype they can determine has been steadily increasing. Nowadays, it is well known that the hexanucleotide expansion is localized in the first intron of the C9orf72 gene and that a threshold higher than 30 repeats is usually pathogenic, with symptomatic patients having up to several hundred or thousand repeats [5]. The disease has an autosomal dominant transmission with elevated penetrance (up to 90%, increasing with age) [6]. Nevertheless, the relationship between repeats number and age of symptom onset is not clearly established yet [7], and no confirmed prognostic indicators currently exist to predict the projected clinical manifestation of GGGGCC repeat expansions as FTD or ALS at the asymptomatic stage. Subjects carrying the mutation may remain asymptomatic for a long time, knowing that the mean age of onset of the symptoms is between 50 and 70 years [8].
Pathogenic mechanisms underlying C9orf72-related diseases are on the other side not fully understood yet. The three most considered hypotheses are: loss of function caused by reduced C9Orf72 protein levels in the brain and the spinal cord (SC), toxicity of mutant RNA that aggregates into nuclear foci, and accumulation of dipeptide-repeat (DPR) proteins generated by non-ATG dependent translation of the expanded repeat [1]. At the pathological level, the mutation is mostly associated with the presence of widespread TAR-DNA binding protein 43 (TDP-43) and p62-positive inclusions, as well as with intranuclear RNA foci and cytoplasmic DPR inclusions [9]. At the pathological level, the mutation is mostly associated with FTLD-TDP type A or type B subtypes, or with a combination of both [10].
No effective treatment is currently available for C9orf72-related ALS-FTD. Nevertheless, some therapeutical strategies, including gene targeting approaches, are under study and new, more innovant, clinical trials for C9orf72-related ALS-FTD are on their way [11]. In this context, the identification of reliable biomarkers and outcome measures to be used in pre-clinical as well as in clinical stages of research will be needed to improve the responsiveness of clinical trials. Asymptomatic carriers of disease-causing GGGGCC hexanucleotide expansions represent an optimal study population to characterize early pathological changes, anatomical patterns of propagation and mechanisms of disease spread [12, 13]. More importantly, the presymptomatic phase of the disease provides an invaluable opportunity to widen the therapeutic window and introduce neuroprotective therapy at the time when central nervous system (CNS) integrity is still relatively preserved. Moreover, recent research has extensively described the presence of pre-clinical alterations in subjects carrying the expansion up to 20–25 years before the estimated onset of the symptoms [14].
The aim of this review is to describe actual and emerging biomarkers for C9orf72-related diseases starting from pre-clinical models and going through possible outcome measures for up-coming clinical trials.
Innovative Therapies For C9orf72-Als: From Pre-Clinical Development To Translation
In recent years, advances in the understanding of the molecular basis of C9orf72-related clinical phenotypes and the development of disease models have led to the identification of innovant disease modifying drugs [11, 15, 16]. Although currently available C9orf72 animal models lack some of the key features of the disease, such as reduced survival and neuromuscular decline [17, 18], they can recapitulate the molecular signature of the disease and were used for therapeutic pre-clinical development [19, 20]. Importantly, the accumulation of repeat-containing transcripts, RNA foci and/or DPRs, were exploited as therapeutic read-outs for the testing of two gene targeting approaches which recently entered clinical trials.
Antisense oligonucleotides (ASOs) are synthetic single-stranded deoxyribonucleotides which are complementary to an RNA target [21]. The goal of treatment with ASOs is the downregulation of a molecular target, usually leading to reduction of gene translation.
In C9orf72-related diseases, ASOs can be designed to bind toxic RNAs responsible of toxic gain-of-function, one of the central mechanism in the pathogenesis of the disease [22]. In fact, in mouse models, administration of ASOs targeting the C9orf72 hexanucleotide expansion selectively reduced the repeat-containing RNA levels, decreased both soluble and insoluble DPR proteins and significantly attenuated behavioral deficits in transgenic mice [19, 23]. Importantly, some of these ASOs are designed to specifically target expansions containing C9orf72 transcripts, reducing the risk of decreasing protein levels. One phase 1 clinical trial (NCT03626012) is ongoing with the aim of testing the safety and efficacy of an ASO (BIIB078, IONIS-C9Rx, Biogen) that selectively targets expansion-containing C9orf72 transcripts. Currently, no interim results on efficacy of this treatment have been published [11]. Other ASOs with a stabilized chemistry, called stereopure, selectively reduced the repeat-containing variants of C9orf72 in cellular and mouse models of the disease [24]. These ASOs (WVE-004, Wave Life Science) are now administered as investigational treatment in a phase 1b/2a clinical trial (FOCUS-C9, NCT04931862). The effects on molecular targets of the two translated therapies in mouse models are summarized in Table 1. Interestingly, the measurement of DPRs provided a valuable tool for therapeutic assessment not only in mouse tissues, but also in patients [25]. The analysis of a specific DPR, poly glycine-proline (GP), was used as pharmacodynamic marker in the cerebrospinal fluid of C9orf72-patients, to follow the ASO-therapy [25]. This work highlights the importance of the identification of “translatable” biomarkers in preclinical models and the need of synergizing the efforts of basic research and clinical research towards the development of precision medicine approaches.
Table 1
Effects on the C9Orf72-molecular signature of translated ASO-based therapies in mouse models
| Study | Model | ASOs | Reduction of repeat-containing transcripts after treatment | Reduction of sense RNA foci after treatment | Reduction of DPRs after treatment |
| Jiang et al. 2016 [19] | BAC mouse model harbouring 500 repeats | ASO 1 ASO 2 ASO 3 | ASO 1: 60%in cortex and 80%in spinal cord | 40–45%40–45%50% | ASO 1 and 2: PolyGP ∼100%in cortex and spinal cord PolyGA 80%in cortex and 60%in spinal cord |
| ASO 2: 60%–70%in cortex and spinal cord | |||||
| ASO 4 (at 4 weeks of treatment) | ASO 3 and 4: 60%in cortex and spinal cord | 50% In the hippocampal dentate gyrus | ASO 3 and 4: PolyGP 80%in cortex | ||
| Liu et al., 2021 [24] | BAC mouse model harbouring 1000 repeats (O’Rourke et al., 2015) [18] | ASO 631 (at 2 weeks of treatment) | 33%in cortex and 59%in spinal cord | 63%in motor neurons of the spinal cord | PolyGP 68.7%in spinal cord and 64.9%in cortex |
ASO: Antisense oligonucleotide, BAC: bacterial artificial chromosome, DPRs: dipeptide-repeat proteins, GP: proline –alanine, GA: glycine –arginine.
Another promising approach for genetic degenerative diseases and namely for C9orf72 ALS-FTD is viral vector-mediated gene therapy. In the last years, gene therapy has shown a great potential to modulate gene expression in different monogenic diseases including ALS [26] [27]. The use of viral vectors as, for example, adeno-associated viral (AAV) vectors with neural tropism, enables the correction of defective genes in affected tissues, replacing a dysfunctional gene or reducing the expression of toxic proteins. AAV-mediated expression of silencing sequences is being tested to treat C9orf72-ALS-FTD. Two studies sponsored by UniQure showed the feasibility of miRNA-based and AAV-delivered gene therapy in experimental models. In particular, an AAV serotype 5 (AAV5) expressing artificial miRNA, was injected in an ALS mouse model and showed significant reduction in repeat-containing C9orf72 transcripts and RNA foci after treatment [28]. However, no clinical trial in patients has started yet.
With the advent of these novel therapeutic entities, the identification of effective biomarkers is necessary for the design of proper clinical trials. In the next section, recent efforts in biomarker search for C9orf72 patients will be summarized. Importantly, clinical trials for gene targeting strategies in other diseases demonstrated the importance of precocious therapeutic interventions (Zolgensma) [29]. For this reason, one of the main focuses of this review will be on the search for diagnostic biomarkers in pre-symptomatic carriers. The early identification and follow-up of carriers amenable to a gene targeting approach, will likely ensure the efficacy of the upcoming therapies.
BIOMARKERS AND OUTCOME MEASURES
The term “biomarker” has been defined by the U.S. Food and Drug Administration (FDA) as a “characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathologic processes, or biological responses to a therapeutic intervention”. Four main categories of biomarkers are identified: diagnostic, prognostic, predictive and pharmacodynamic [30].
A diagnostic biomarker is a parameter that is related to the presence or absence of a pathophysiological state. Specific and sensitive diagnostic biomarkers may enable an early diagnosis of ALS-FTD, including pre-symptomatic carriers of pathogenic ALS-FTD mutations, and may be useful to differentiate ALS not just from healthy individuals but also from ALS-mimic diseases.
Prognostic biomarkers are baseline characteristics that inform about the natural history of the disease in the absence of therapy. The stratification of ALS-FTD patients into homogeneous prognostic groups has direct implications on clinical trials by improving statistical power, reducing trial duration and costs, and revealing a therapeutic effect in specific subgroups of patients. A predictive biomarker is a baseline characteristic that categorizes patients according to their likelihood of response to therapy.
Finally, pharmacodynamic biomarkers are measures that change in response to a therapeutic intervention [31].
In ALS-FTD, several categories of possible biomarkers have been studied and proposed, with the aim of better characterizing disease onset and progression and in the perspective of improving reliability of clinical trials [30, 32].
In the next paragraphs we will describe in detail different types of biomarkers and possible outcome measures which are of primary relevance both in pre-clinical and clinical research on C9orf72-related diseases.
Different biomarkers identified in C9orf72-related ALS are resumed in Fig. 1.
Fig. 1
Biomarkers in C9orf72-related ALS.
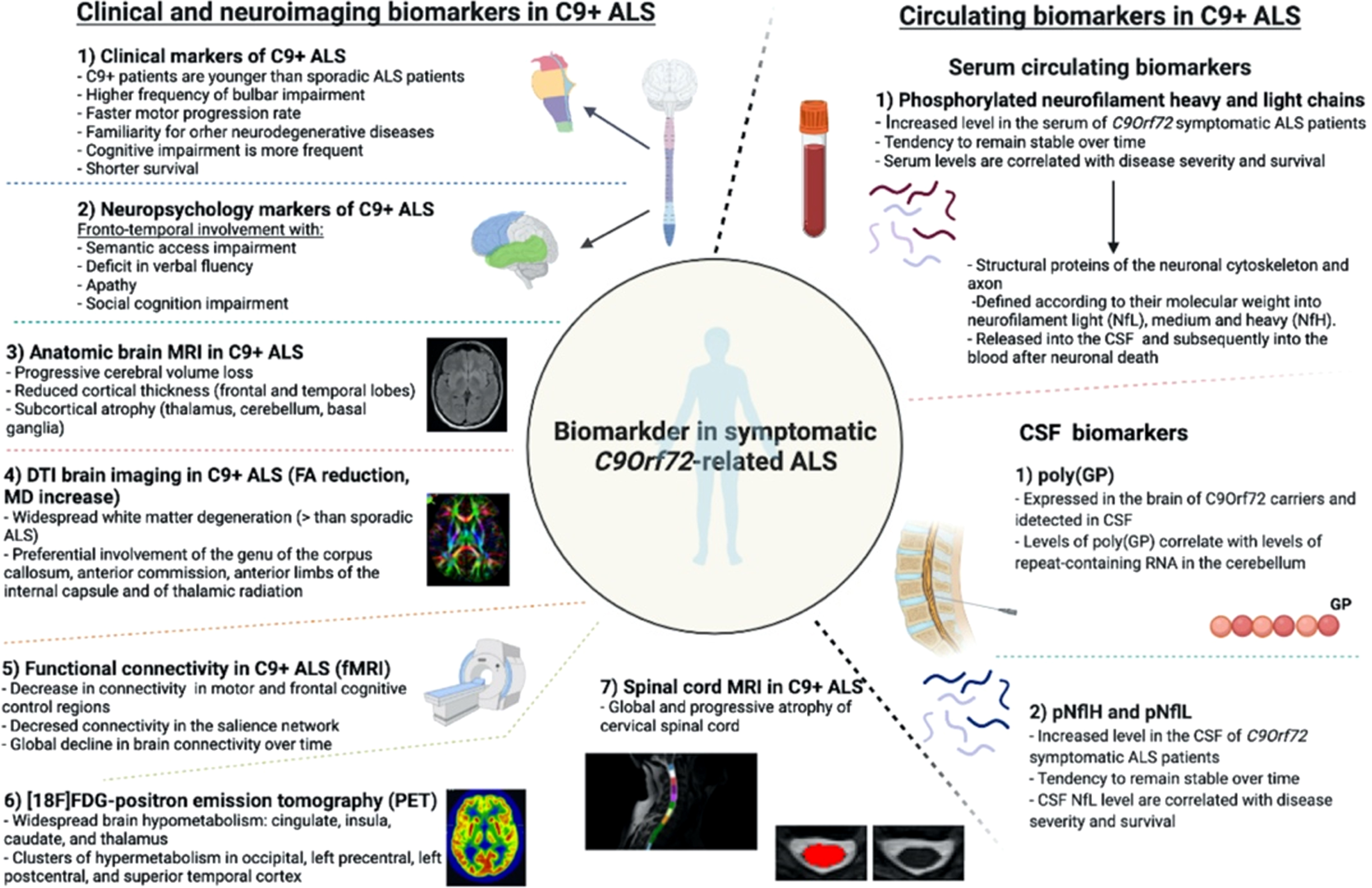
Clinical spectrum of C9orf72-related ALS-FTD and perspectives for clinical biomarkers in symptomatic and asymptomatic carriers
C9orf72 mutations are known to determine both ALS and FTD, inducing a large heterogeneity of phenotypes even in the same family.
The behavioral variant of FTD (bvFTD), characterized by behavior and social cognition alterations associated with semantic access and speech fluidity impairment, is the most frequent C9orf72-related form of FTD [33], while the aphasic presentation of FTD is rarer but anyway possible in association with this mutation.
In C9orf72 carriers presenting with ALS, the disease has no distinguishing features compared to sporadic patients. Nevertheless, C9orf72-mutated patients are in average younger than sporadic ones at onset of the symptoms and they present a higher frequency of bulbar onset and co-occurring frontal cognitive deterioration (around 50%of the patients) [34]. A significant faster motor progression rate is usually observed as well, which, together with the presence of cognitive impairment, finally leads to a shorter survival [34].
Extra-pyramidal involvement and psychiatric symptoms can sometimes also be considered as part of the clinical spectrum of C9orf72-related diseases. It is in fact now known that the mutation may produce phenotypes clinically mimicking idiopathic Parkinson disease, progressive supranuclear palsy, cortico-basal syndrome and Huntington disease [35]. Its implication in psychiatric disorders such as schizophrenia as well as in manifestations considered part of the autistic spectrum has been described in patients as well as in first-degree relatives [36, 37]. Interestingly, patients with schizophrenia associated with C9orf72 mutations often present later onset of clinically manifested psychosis than sporadic ones.
Studies investigating clinical biomarkers in pre-symptomatic carriers of C9orf72 suggest that the onset of motor symptoms is a late event in the history of the disease. Indeed, subjects that will later develop ALS have early cognitive alterations [38, 39].
An example of specific C9orf72-related impairment is the presence, in pre-symptomatic carriers, of deficits in cognitive inhibition (i.e., the ability to resist to interference from irrelevant stimuli) which seems to become more evident in individuals who are closer to symptom onset [40]. Some other studies have shown the presence of deficits in semantic access [41] as well as early involvement of praxis, which has been observed already in subjects younger than 40 years of age, supporting the hypothesis that frontal and prefrontal structures are early involved in degeneration [42]. Deficits in verbal fluency are also well recognized. Interestingly, they have been described to be stable over time, suggesting a possible neurodevelopmental origin for at least a part of the C9orf72-related syndrome [43].
Apathy is a frequent symptom of C9orf72-related bvFTD and is correlated with the extension of executive dysfunction. In pre-symptomatic C9orf72 mutation carriers, it occurs early in the disease history, worsens over time and predicts a subsequent subclinical deterioration of cognitive performance [44].
Impairment of social skills is also considered one of the main clinical manifestations of bvFTD. Late pre-symptomatic C9orf72 mutation carriers present significant alterations in social cognition associated with involvement of the insula and atrophy of the left frontal lobe, of basal ganglia and of the right amygdale. Such manifestations seem to be more evident in subjects that are closer to symptom onset, who also present a decrease in emotion processing abilities.
Neuroimaging biomarkers in symptomatic and asymptomatic C9orf72 carriers
Neuroimaging studies have gained much relevance in recent years thank to their ability to detect early degenerative changes and to track their evolution over time. To be considered an effective biomarker, an imaging measure should present measurable changes over a short observation period, good reliability, and effect size, and possibly be correlated to a clinical measure of disease progression.
Structural magnetic resonance imaging (MRI)
aims at describing volumetric modifications of cortical and sub-cortical structures (for example through voxel base morphometry studies, VBM, as well as through cortical thickness measurements). Similarly, at the SC level, white and grey matter atrophy can be quantified on T1 and T2-weigthed sequences.
All recent data on C9orf72 ALS-FTD symptomatic patients agree that widespread cortical and subcortical abnormalities are detectable all along the disease course. These abnormalities consist in cerebral volume loss with increased ventricular volume and reduced cortical thickness [45–47]. Cortical thinning involves mainly the superior and medial frontal cortex and temporal poles bilaterally. Compared to non-C9orf72 ALS patients, mutation carriers have more diffuse atrophy of extra-motor cortical regions, particularly of parieto-occipital areas including the cuneus and precuneus, while the primary motor cortex seems to be relatively spared. Such cortical changes are probably related to the presence of cognitive impairment and seem to be more severe in patients with clinical features of bvFTD [14, 44, 47].
Several studies have described the presence of marked and early subcortical atrophy involving the thalamus and especially the pulvinar [48]. Namely, pulvinar atrophy seems to be a typical feature of C9orf72-related diseases and could be considered as a relevant diagnostic biomarker [49, 50]. The thalamus plays a central role in several cortico-thalamic circuits, where the pulvinar has fundamental connections with the posterior parietal, prefrontal, and cingulate cortical areas [49, 51] and with executive functions more generally.
Another structure which is early involved in C9orf72-ALS is the cerebellum. Cerebellar volume loss is focal and involves regions connected to the dorsolateral prefrontal cortex and parietal association areas playing a role in executive function such as the posterior lobe and the vermis [45, 52]. Cerebellar and thalamic atrophy seem an interesting biomarkers since high levels of DPR proteins and RNA foci were observed in these structures and correlated with cognitive impairment [53].
Volumetric MRI has also pointed out the presence of basal ganglia nuclei atrophy as well as of the corpus callosum [45, 54].
Only a few MRI studies have followed up symptomatic C9orf72-ALS patients longitudinally, thus suggesting that cortical atrophy may evolve over time [46, 55]. The possibility that, at least in part, cortical and subcortical atrophy could be of neurodevelopmental origin, thus justifying its early presence in pre-symptomatic subjects and slow evolution over time, is under discussion as well [56, 57].
A few studies have analyzed the presence of brain MRI modifications in asymptomatic C9orf72 carriers, showing that they can be detected up to 20 years before the estimated onset of the symptoms [14]. Namely, one study described the presence of MRI alterations already in subjects younger than 40 years of age [42]. Morphometric analyses have focused over the years both on cortical and subcortical structures, showing the presence of atrophy of the cerebellum up to 10 years and of the thalamus up to 20 years before the estimated onset of the symptoms [14, 58, 59].
Diffusion tension imaging (DTI)
is the main approach used to study degeneration of white matter bundles. DTI is a mathematical model based on diffusion MRI that allows to compute four main parameters of degeneration (AD = axial diffusivity, MD = mean diffusivity, RD = radial diffusivity, FA = fractional anisotropy) [45].
DTI imaging studies showed that C9orf72-ALS patients have more widespread white matter degeneration than sporadic ALS and controls. Most commonly, white matter bundles degeneration is observed in frontal tracts and is described by reduced FA and increased MD. Involvement of the genu of the corpus callosum, of the anterior commission, of the anterior limbs of the internal capsule and of thalamic radiation seems to be specific to C9orf72-ALS and progressive over time [12, 45–47, 60]. Interestingly, FA decrease in the corpus callosum is correlated with functional scores, upper-motor neuron involvement clinical signs and disease duration [61].
DTI imaging has been used to confirm in vivo histopathological staging in ALS, showing that TDP-43 accumulation and degeneration start mainly along the cortico-spinal tract (CST) and evolves later into frontal and hippocampal areas [62, 63]. This model supports a corticofugal hypothesis of degeneration which apparently starts at brain level and progresses to the spinal cord and muscle only in more advanced phases of the disease [64]. Even if this corticofugal model is not fully confirmed and alternative hypotheses proposing a muscular origin of ALS exist, recent DTI data showed that degeneration follows the same stages in C9orf72-ALS patients, while it seems to present a different pattern in SOD1-mutated patients [65]. Furthermore, this study strongly supports the idea that DTI imaging constitute a reliable in vivo biomarker of disease status and progression in C9orf72-ALS.
Recent studies indicate that widespread cortical and subcortical abnormalities can be detected in asymptomatic C9orf72 carriers younger than 40 years of age [42, 66].
Functional connectivity
Functional connectivity is defined as the study of functional brain networks and is performed mostly by resting-state of task-based functional MRI (fMRI).
In C9orf72-ALS and FTD patients, connectivity alterations were noted using both task-based and resting state fMRI in the salience networks, frontal and temporal regions and thalamic networks [52], with regions of both increased and decreased connectivity being described by different groups. A recent study [67] demonstrated the presence of an overall decrease in connectivity with regions implicated in motor and frontal cognitive control being more impaired than others. Interestingly, no substantial reorganization of network hubs was shown in this study, indicating a relatively diffuse and possibly slow global decline in connectivity.
In pre-symptomatic carriers of the mutation, disruption of the salience network and of thalamic networks was detected, while other studies pointed out the presence of early alteration in visual related regions [68, 69]. Globally, alterations in brain networks are considered to be an early event in the history of the disease, further supporting the hypothesis of a neurodevelopmental origin of at least a part of the C9orf72-related syndrome. Alternatively, network disruption could be also interpreted as a progressive adaptation of brain networks over time [68].
[18F]FDG-positron emission tomography (PET)
assesses brain glucose metabolism and is retained as a useful diagnostic marker in the earliest stages of ALS and FTD [70], mainly showing hypometabolism in the frontal lobes already in early stages of the disease.
In C9orf72-ALS, brain hypometabolism is more widespread and involves different clusters in the cingulate, insula, caudate, and thalamus, with clusters of hypermetabolism in the occipital, left precentral, left postcentral, and superior temporal cortex [71].
In pre-symptomatic carriers of the mutation, hypometabolism is observed in frontotemporal and insular cortices, the basal ganglia, the thalamus as well as in the anterior cingulate cortex, supporting the hypothesis of an early silence network disruption [72]. At the same time, clusters of hypermetabolism are present in the precentral and superior frontal gyrus, suggesting the presence of a compensatory neuronal activity in response to ongoing neurodegeneration [72].
Spinal cord MRI
Spinal cord MRI has been used to describe the presence of white and grey matter atrophy in sporadic ALS carriers as well as to track longitudinal degeneration of cortico-spinal tracts (CST) at DTI [73–75]. Further studies showed that SC MRI can be useful in the classification of ALS patients from controls, suggesting that it could be an effective diagnostic biomarker [76].
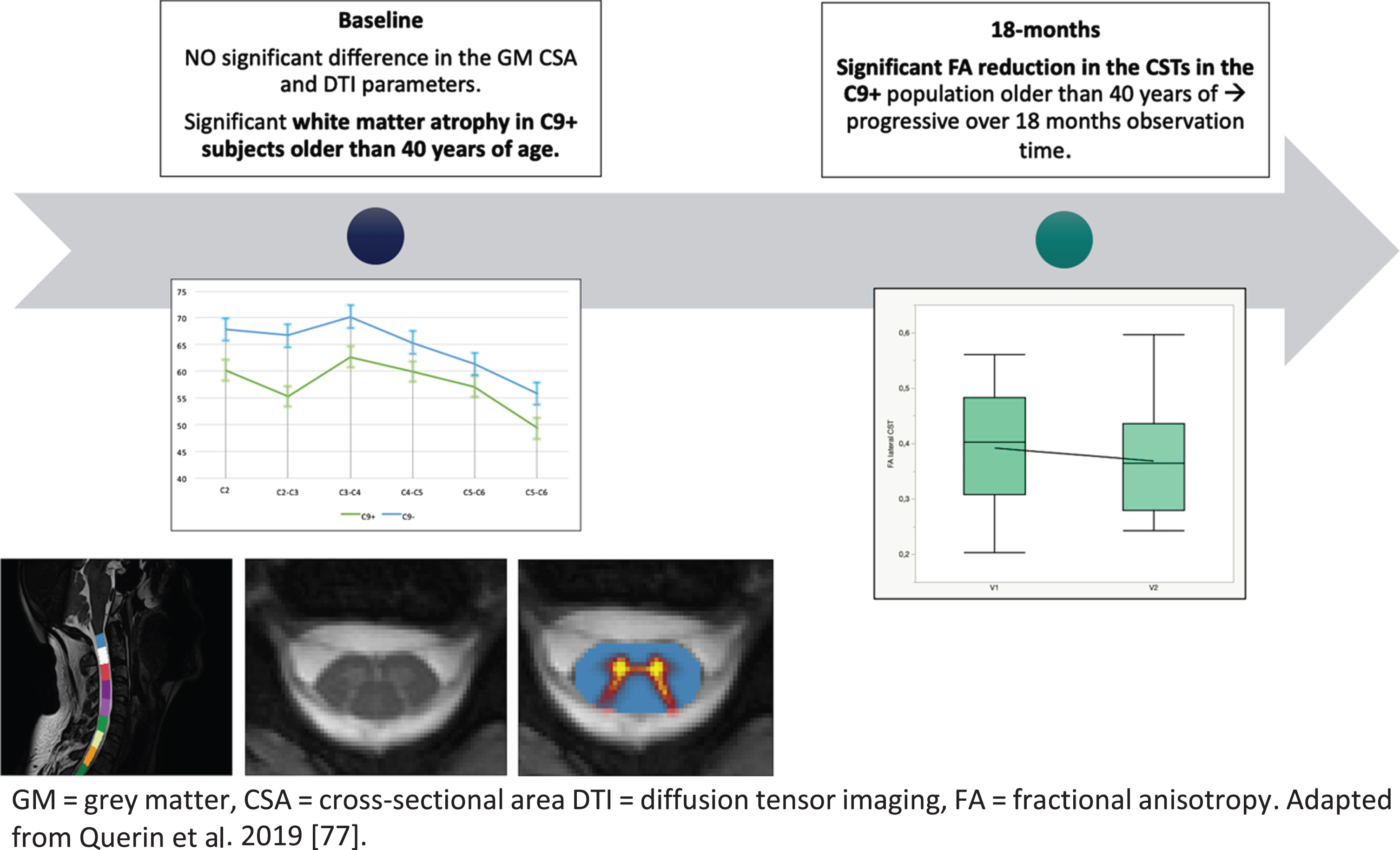
In C9orf72-ALS symptomatic patients, a recent study indicated the presence of global cervical SC atrophy which was relatively stable over a 6 months observation period [77].
In pre-symptomatic carriers of the disease, white matter atrophy was described in subjects older than 40 years of age, while no grey matter atrophy was detected. In the same cohort, progressive degeneration of the CSTs was described by the identification of a progressive decrease of FA [78]. SC MRI alterations in pre-symptomatic carriers of the mutation are graphically represented in Fig. 2.
Fig. 2
Longitudinal evolution of cervical SC alterations in pre-symptomatic C9 o rf72-mutation carriers.
Neurophysiology techniques are used in ALS to detect both upper (through transcranial magnetic simulation) and lower motor neuron degeneration (trough EMG) [79]. Transcranial magnetic stimulation (TMS) has been applied to C9orf72-ALS patients showing increased cortical excitability early in the course of the disease and loss of excitability in late stages. Nevertheless, pre-symptomatic carriers did not show any abnormalities [80].
Further techniques quantitatively assessing lower motor neuron loss, such as for example Motor Unit Number Estimation (MUNE and MUNIX), are being applied to ALS and techniques used as outcome measure in clinical trials [81, 82]. To date, no study has specifically used EMG and MUNIX to investigate LMN degeneration in symptomatic and pre-symptomatic C9orf72-ALS patients. Further explorations in this direction are needed to fully define the pathology.
Fluid-based and molecular biomarkers
Fluid-based biomarkers have been extensively studied in ALS, ranging from cerebrospinal fluid (CSF)-related molecules to markers of inflammation and altered metabolism, to micro-RNAs [32, 83].
One of the most promising fluid biomarkers are phosphorylated neurofilament heavy and light chains which can be measured both in the serum and in the CSF. Neurofilaments are neuron-specific structural proteins of the cytoskeleton and axon which are defined according to their molecular weight into neurofilament light (NfL), medium and heavy (NfH). Their release into the CSF and subsequently into the blood follows neuronal death, while their accumulation in motor neurons has been linked to neuronal dysfunction [84–86]. High levels of the neuronal subunits phosphorylated neurofilament heavy (pNfH) and neurofilament light chain (NfL) are found in a wide range of neurological diseases including ALS compared to healthy and neurological controls, suggesting that they could be an effective diagnostic marker of the disease [87].
In C9orf72-ALS, pNfH and pNfL are significantly higher both in the serum and CSF compared to controls as well as to sporadic ALS [88, 89], and tends to slowly increase over time, with higher levels predicting poorer survival and being related to a more severe phenotype [88, 90]. Interestingly, their serum level is less increased in patients with slowly progressive phenotypes and with psychiatric forms of the disease, suggesting that they could be used as a reliable predictor of aggressiveness and rapidity of disease progression [89].
Additionally, relevant studies have demonstrated that NfL levels change in pre-symptomatic carriers of the mutation 2–5 years before the fully symptomatic disease. They also suggest that NfL levels are a valuable predictor of clinical proximity in pre-symptomatic subjects, with a more rapid increase around 12 months before symptom onset [86, 88]. All these characteristics make neurofilaments levels a possible reliable biomarker which could be useful for patients’ stratification, for the identification of at-risk subjects closer to phenoconversion and for the monitoring of the response to treatment both in symptomatic and pre-symptomatic clinical trials.
Dipeptide repeat proteins
As mentioned above, a characteristic neuropathological feature of C9orf72 ALS and FTD is the presence of neuronal inclusions formed of DPR proteins synthesized from expanded C9orf72 repeats. One of these proteins, poly(GP), is abundantly expressed in the brain of C9orf72 carriers and is detected in CSF [91]. Given that levels of poly(GP) correlate with levels of repeat-containing RNA in the cerebellum of C9orf72 carriers, poly(GP) was investigated as a marker of target engagement for repeat RNA-based therapies [25].
CONCLUSION AND FUTURE DIRECTIONS
The landscape of biomarkers in ALS and FTD has dramatically changed in recent years with a radical transition from descriptive academic studies to precision clinical tools. In C9orf72-ALS and FTD development of “wet biomarkers” (neurofilaments and DPRs) and “dry biomarkers” (neuroimaging) can now be transferred to the clinical context. These unique tools will be central in this very exciting period of emerging promising treatment in C9orf72-ALS. However, we have to keep in mind that clinical efficacy, and not change in a biomarker such as neurofilament levels, will always be the criteria for the approval of a new treatment. In this context, we have to learn from the experience of randomized clinical trials (RCTs) in other neurodegenerative diseases, which showed some limits of biomarkers. In Alzheimer disease, anti-amyloid therapies dramatically reduced the amyloid burden on PET but did not demonstrate a parallel clinical efficacy [92]. In the same way, DPRs seem a useful target engagement marker [25], but their role in the neurodegenerative cascade remains controversial and we know nothing about their correlation with clinical severity. It can be assumed that in the future, evaluation of treatment will rely on a combination of wet and dry biomarkers, including the emerging digital health tools. In the context of rare diseases where RCTs cannot include large populations, biomarkers will also be essential in post-approval drug studies to provide real-world data about effectiveness, safety and definition of responder groups. Great progresses are expectable in a near future thanks to the technological and conceptual advances, collaborative multicentre research efforts as well as the strong involvement of pharmaceutical industry.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
M.G.B. and G.Q. are supported by the Association Française contre les Myopathies (AFM-Téléthon), the Association Institut de Myologie (AIM), Sorbonne Université, the Institut National de la Santé et de la Recherche Medicale (INSERM). PF Pradat received research study funding by the EU Joint Programme –Neurodegenerative Disease Research (JPND), Association Française pour la Recherche sur la SLA (ARSLA), Association Française contre les Myopathies (AFM-Téléthon), Institut pour la Recherche sur la Moelle Épinière et l’Encephale (IRME), Target ALS Foundation, Institut du Cerveau (ICM) and Institut des Neurosciences Translationnelles de Paris (IHU-A-ICM). GQ and MGB are members of the European Reference Network for rare neuromuscular diseases (EURO-NMD). M.G.B. is currently employed by Spark Therapeutics, Philadelphia (PA 19104, USA).
REFERENCES
[1] | Cruts M , Gijselinck I , Van Langenhove T , van der Zee J , Van Broeckhoven C . Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. (2013) ;36: (8):450–9s. doi:10.1016/j.tins.2013.04.010. |
[2] | Swinnen B , Robberecht W . The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. (2014) ;10: (11):661–70. doi:10.1038/nrneurol.2014.184. |
[3] | DeJesus-Hernandez M , Mackenzie IR , Boeve BF , Boxer AL , Baker M , Rutherford NJ , et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) ;72: (2):245–56. doi:10.1016/j.neuron.2011.09.011. |
[4] | Majounie E , Renton AE , Mok K , Dopper EGP , Waite A , Rollinson S , et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. (2012) ;11: (4):323–30. doi:10.1016/S1474-4422(12)70043-1. |
[5] | Balendra R , Isaacs AM . C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. (2018) ;14: (9):544–58. doi:10.1038/s41582-018-0047-2. |
[6] | Murphy NA , Arthur KC , Tienari PJ , Houlden H , Chiò A , Traynor BJ . Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. (2017) ;7: (1):2116. doi:10.1038/s41598-017-02364-1. |
[7] | Fournier C , Barbier M , Camuzat A , Anquetil V , Lattante S , Clot F , et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients andpresymptomatic carriers. Neurobiol Aging. (2018) ;74: :234.e1–234.e8. doi:10.1016/j.neurobiolaging.2018.09.010. |
[8] | Benussi L , Rossi G , Glionna M , Tonoli E , Piccoli E , Fostinelli S , et al. C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: a genotype-phenotype correlation study. J Alzheimers Dis. (2014) ;38: (4):799–808. doi:10.3233/JAD-131028. |
[9] | Mackenzie IRA , Frick P , Neumann M . The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. (2014) ;127: (3):347–57. doi:10.1007/s00401-013-1232-4. |
[10] | Mackenzie IRA , Neumann M , Baborie A , Sampathu DM , Du Plessis D , Jaros E , et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. (2011) ;122: (1):111–3. doi:10.1007/s00401-011-0845-8. |
[11] | Cappella M , Pradat P-F , Querin G , Biferi MG . Beyond the Traditional Clinical Trials for Amyotrophic Lateral Sclerosis and The Future Impact of Gene Therapy. J Neuromuscul Dis. (2021) ;8: (1):25–38. doi:10.3233/JND-200531. |
[12] | Schuster C , Elamin M , Hardiman O , Bede P . Presympto-matic and longitudinal neuroimaging in neurodegeneration–from snapshots to motion picture: a systematic review. J Neurol Neurosurg Psychiatry. (2015) ;86: (10):1089–96. doi:10.1136/jnnp-2014-309888. |
[13] | Floeter MK , Traynor BJ , Farren J , Braun LE , Tierney M , Wiggs EA , et al. Disease progression in C9orf72 mutation carriers. Neurology. (2017) ;89: (3):234–41. doi:10.1212/WNL.0000000000004115. |
[14] | Rohrer JD , Nicholas JM , Cash DM , van Swieten J , Dopper E , Jiskoot L , et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. The Lancet Neurology. (2015) ;14: (3):253–62. doi:10.1016/S1474-4422(14)70324-2. |
[15] | Cappella M , Ciotti C , Cohen-Tannoudji M , Biferi MG . Gene Therapy for ALS-A Perspective. Int J Mol Sci. (2019) ;20: (18):E4388. doi:10.3390/ijms20184388. |
[16] | Miccio A , Antoniou P , Ciura S , Kabashi E . Novel genome-editing-based approaches to treat motor neuron diseases: Promises and challenges. Mol Ther. 2021;S1525-0016(21)00190-8. doi:10.1016/j.ymthe.2021.04.003. |
[17] | Mordes DA , Morrison BM , Ament XH , Cantrell C , Mok J , Eggan P , et al. Absence of Survival and Motor Deficits in 500 Repeat C9ORF72 BAC Mice. Neuron. (2020) ;108: (4):775–83.e4. doi:10.1016/j.neuron.2020.08.009. |
[18] | Peters OM , Cabrera GT , Tran H , Gendron TF , McKeon JE , Metterville J , et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. (2015) ;88: (5):902–9. doi:10.1016/j.neuron.2015.11.018. |
[19] | O’Rourke JG , Bogdanik L , Muhammad AKMG , Gendron TF , Kim KJ , Austin A , et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. (2015) ;88: (5):892–901. doi:10.1016/j.neuron.2015.10.027. |
[20] | Liu Y , Pattamatta A , Zu T , Reid T , Bardhi O , Borchelt DR , et al. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. (2016) ;90: (3):521–34. doi:10.1016/j.neuron.2016.04.005. |
[21] | Di Fusco D , Dinallo V , Marafini I , Figliuzzi MM , Romano B , Monteleone G . Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Frontiers in Pharmacology. (2019) ;10: :305. doi:10.3389/fphar.2019.00305. |
[22] | Donnelly CJ , Zhang P-W , Pham JT , Haeusler AR , Heusler AR , Mistry NA , et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. (2013) ;80: (2):415–28. doi:10.1016/j.neuron.2013.10.015. |
[23] | Jiang J , Zhu Q , Gendron TF , Saberi S , McAlonis-Downes M , Seelman A , et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. (2016) ;90: (3):535–50. doi:10.1016/j.neuron.2016.04.006. |
[24] | Liu Y , Dodart J-C , Tran H , Berkovitch S , Braun M , Byrne M , et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. (2021) ;12: (1):847. doi:10.1038/s41467-021-21112-8. |
[25] | Gendron TF , Chew J , Stankowski JN , Hayes LR , Zhang Y-J , Prudencio M , et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. (2017) ;9: (383):eaai7866. doi:10.1126/scitranslmed.aai7866. |
[26] | Ly CV , Miller TM . Emerging antisense oligonucleotide and viral therapies for amyotrophic lateral sclerosis. Curr Opin Neurol. (2018) ;31: (5):648–54. doi:10.1097/WCO.0000000000000594. |
[27] | Martier R , Liefhebber JM , García-Osta A , Miniarikova J , Cuadrado-Tejedor M , Espelosin M , et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol Ther Nucleic Acids. (2019) ;16: :26–37. doi:10.1016/j.omtn.2019.02.001. |
[28] | Amado DA , Davidson BL . Gene therapy for ALS: A review. Molecular Therapy. 2021 S1525-0016(21)00195-7. doi:10.1016/j.ymthe.2021.04.008. |
[29] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. New England Journal of Medicine. (2017) ;377: (18):1713–22. doi:10.1056/NEJMoa1706198. |
[30] | Taga A , Maragakis NJ . Current and emerging ALS biomarkers: utility and potential in clinical trials. Expert Rev Neurother. (2018) ;18: (11):871–86. doi:10.1080/14737175.2018.1530987. |
[31] | Mendez EF , Sattler R . Biomarker development for C9orf72 repeat expansion in ALS. Brain Res. (2015) ;1607: :26–35. doi:10.1016/j.brainres.2014.09.041. |
[32] | Blasco H , Vourc’h P , Pradat PF , Gordon PH , Andres CR , Corcia P . Further development of biomarkers in amyotrophic lateral sclerosis. Expert Review of Molecular Diagnostics. (2016) ;16: (8):853–68. doi:10.1080/14737159.2016.1199277. |
[33] | van der Ende EL , Jackson JL , White A , Seelaar H , van Blitterswijk M , Van Swieten JC . Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J Neurol Neurosurg Psychiatry. (2021) ;92: (5):502–9. doi:10.1136/jnnp-2020-325377. |
[34] | Byrne S , Elamin M , Bede P , Shatunov A , Walsh C , Corr B , et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. (2012) ;11: (3):232–40. doi:10.1016/S1474-4422(12)70014-5. |
[35] | Rohrer JD , Isaacs AM , Mizielinska S , Mead S , Lashley T , Wray S , et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. The Lancet Neurology. (2015) ;14: (3):291–301. doi:10.1016/S1474-4422(14)70233-9. |
[36] | O’Brien M , Burke T , Heverin M , Vajda A , McLaughlin R , Gibbons J , et al. Clustering of Neuropsychiatric Disease in First-Degree and Second-Degree Relatives of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. (2017) ;74: (12):1425–30. doi:10.1001/jamaneurol.2017.2699. |
[37] | Devenney EM , Ahmed RM , Halliday G , Piguet O , Kiernan MC , Hodges JR , Psychiatric disorders in C9orf72 kindreds: Study of 1,414 family members. Neurology. (2018) ;91: (16):e1498–507. doi:10.1212/WNL.0000000000006344. |
[38] | Mahoney CJ , Beck J , Rohrer JD , Lashley T , Mok K , Shakespeare T , et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. (2012) ;135: (Pt 3):736–50. doi:10.1093/brain/awr361. |
[39] | Poos JM , Russell LL , Peakman G , Bocchetta M , Greaves CV , Jiskoot LC , et al. Impairment of episodic memory in genetic frontotemporal dementia: A GENFI study. Alzheimers Dement (Amst). (2021) ;13: (1):e12185. doi:10.1002/dad2.12185. |
[40] | Montembeault M , Sayah S , Rinaldi D , Le Toullec B , Bertrand A , Funkiewiez A , et al. Cognitive inhibition impairments in presymptomatic C9orf72 carriers. J Neurol Neurosurg Psychiatry. (2020) ;91: (4):366–72. doi:10.1136/jnnp-2019-322242. |
[41] | Moore K , Convery R , Bocchetta M , Neason M , Cash D , Greaves C , et al. A modified Camel and Cactus Test detects presymptomatic semantic impairment in genetic frontotemporal dementia within the GENFI cohort. Applied neuropsychology Adult 2020;1-8. doi:10.1080/23279095.2020.1716357. |
[42] | Bertrand A , Wen J , Rinaldi D , Houot M , Sayah S , Camuzat A , et al. Early Cognitive, Structural, and Microstructural Changes in Presymptomatic C9orf72 Carriers Younger Than 40 Years. JAMA Neurol. (2018) ;75: (2):236–45. doi:10.1001/jamaneurol.2017.4266. |
[43] | Lulé DE , Müller H-P , Finsel J , Weydt P , Knehr A , Winroth I , et al. Deficits in verbal fluency in presymptomatic C9orf72 mutation gene carriers-a developmental disorder. J Neurol Neurosurg Psychiatry. (2020) ;91: (11):1195–200. doi:10.1002/alz.12252. |
[44] | Malpetti M , Jones PS , Tsvetanov KA , Rittman T , van Swieten JC , Borroni B , et al. Apathy in presymptomatic genetic frontotemporal dementia predicts cognitive decline and is driven by structural brain changes. Alzheimers Dement. (2021) ;17: (6):969–83. doi:10.1002/alz.12252. |
[45] | Bede P , Bokde ALW , Byrne S , Elamin M , McLaughlin RL , Kenna K , et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology. (2013) ;81: (4):361–9. doi:10.1212/WNL.0b013e31829c5eee. |
[46] | Agosta F , Ferraro PM , Riva N , Spinelli EG , Domi T , Carrera P , et al. Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiology of Aging. (2017) ;57: :206–19. doi:10.1016/j.neurobiolaging.2017.05.024. |
[47] | Whitwell JL , Weigand SD , Boeve BF , Senjem ML , Gunter JL , DeJesus-Hernandez M , et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. (2012) ;135: :794–806. doi:10.1093/brain/aws001. |
[48] | Schönecker S , Neuhofer C , Otto M , Ludolph A , Kassubek J , Landwehrmeyer B , et al. Atrophy in the Thalamus But Not Cerebellum Is Specific for C9orf72 FTD and ALS Patients - An Atlas-Based Volumetric MRI Study. Front Aging Neurosci. (2018) ;10: :45. doi:10.3389/fnagi.2018.00045. |
[49] | Bocchetta M , Iglesias JE , Neason M , Cash DM , Warren JD , Rohrer JD . Thalamic nuclei in frontotemporal dementia: Mediodorsal nucleus involvement is universal but pulvinar atrophy is unique to C9orf72. Hum Brain Mapp. (2020) ;41: (4):1006–16. doi:10.1002/hbm.24856. |
[50] | Bocchetta M , Todd EG , Peakman G , Cash DM , Convery RS , Russell LL , et al. Differential early subcortical involvement in genetic FTD within the GENFI cohort. Neuroimage Clin. (2021) ;30: :102646. doi:10.1016/j.nicl.2021.102646. |
[51] | Lee SE , Khazenzon AM , Trujillo AJ , Guo CC , Yokoyama JS , Sha SJ , et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. (2014) ;137: :3047–60. doi:10.1093/brain/awu248. |
[52] | Bede P , Chipika RH , Christidi F , Hengeveld JC , Karavasilis E , Argyropoulos GD , et al. Genotype-associated cerebellar profiles in ALS: focal cerebellar pathology and cerebro-cerebellar connectivity alterations. J Neurol Neurosurg Psychiatry. (2021) ;92: (11):1197–205. doi:10.1136/jnnp-2021-326854. |
[53] | Schludi MH , May S , Grässer FA , Rentzsch K , Kremmer E , Küpper C , et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. (2015) ;130: (4):537–55. doi:10.1007/s00401-015-1450-z. |
[54] | Floeter MK , Gendron TF . Biomarkers for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Associated With Hexanucleotide Expansion Mutations in C9orf72. Front Neurol. (2018) ;9: :1063. doi:10.3389/fneur.2018.01063. |
[55] | Floeter MK , Bageac D , Danielian LE , Braun LE , Traynor BJ , Kwan JY . Longitudinal imaging in C9orf72 mutation carriers: Relationship to phenotype. Neuroimage Clin. (2016) ;12: :1035–43. doi:10.1016/j.nicl.2016.10.014. |
[56] | Mahoney CJ , Downey LE , Ridgway GR , Beck J , Clegg S , Blair M , et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. (2012) ;4: (5):41. doi:10.1186/alzrt144. |
[57] | Bede P , Siah WF , McKenna MC , Li Hi Shing S . Consideration of C9orf72-associated ALS-FTD as a neurodevel-opmental disorder: insights from neuroimaging. J Neurol Neurosurg Psychiatry. (2020) ;91: (11):1138. doi:10.1186/alzrt144. |
[58] | Walhout R , Schmidt R , Westeneng H-J , Verstraete E , Seelen M , van Rheenen W , et al. Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology. (2015) ;85: (20):1780–8. doi:10.1212/WNL.0000000000002135. |
[59] | Papma JM , Jiskoot LC , Panman JL , Dopper EG , den Heijer T , Donker Kaat L , et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology. (2017) ;89: (12):1256–64. doi:10.1212/WNL.0000000000004393. |
[60] | van der Burgh HK , Westeneng H-J , Walhout R , van Veenhuijzen K , Tan HHG , Meier JM , et al. Multimodal longitudinal study of structural brain involvement in amyotrophic lateral sclerosis. Neurology. (2020) ;94: (24):e2592–604. doi:10.1212/WNL.0000000000009498. |
[61] | Müller H-P , Lulé D , Roselli F , Behler A , Ludolph AC , Kassubek J . Segmental involvement of the corpus callosum in C9orf72-associated ALS: a tract of interest-based DTI study. Ther Adv Chronic Dis. 2021;12:20406223211002970. doi:10.1177/20406223211002969. |
[62] | Brettschneider J , Del Tredici K , Toledo JB , Robinson JL , Irwin DJ , Grossman M , et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. (2013) ;74: (1):20–38. doi:10.1002/ana.23937. |
[63] | Kassubek J , Müller H-P , Del Tredici K , Lulé D , Gorges M , Braak H , et al. Imaging the pathoanatomy of amyotrophic lateral sclerosis in vivo: targeting a propagation-based biological marker. J Neurol Neurosurg Psychiatry. (2018) ;89: (4):374–81. doi:10.1136/jnnp-2017-316365. |
[64] | Braak H , Brettschneider J , Ludolph AC , Lee VM , Trojanowski JQ , Del Tredici K . Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol. (2013) ;9: (12):708–14. doi:10.1038/nrneurol.2013.221. |
[65] | Müller H-P , Del Tredici K , Lulé D , Müller K , Weishaupt JH , Ludolph AC , et al. In vivo histopathological staging in C9orf72-associated ALS: A tract of interest DTI study. Neuroimage Clin. (2020) ;27: :102298. doi:10.1016/j.nicl.2020.102298. |
[66] | Wen J , Zhang H , Alexander DC , Durrleman S , Routier A , Rinaldi D , et al. Neurite density is reduced in the presymptomatic phase of C9orf72 disease. J Neurol Neurosurg Psychiatry. (2019) ;90: (4):387–94. doi:10.1136/jnnp-2018-318994. |
[67] | Smallwood Shoukry RF , Clark MG , Floeter MK . Resting State Functional Connectivity Is Decreased Globally Across the C9orf72 Mutation Spectrum. Front Neurol. (2020) ;11: :598474. doi:10.3389/fneur.2020.598474. |
[68] | Lee SE , Sias AC , Mandelli ML , Brown JA , Brown AB , Khazenzon AM , et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. (2017) ;14: :286–97. doi:10.1016/j.nicl.2016.12.006. |
[69] | Waugh RE , Danielian LE , Shoukry RFS , Floeter MK . Longitudinal changes in network homogeneity in presymptomatic C9orf72 mutation carriers. Neurobiology of Aging. (2021) ;99: :1–10. doi:10.1016/j.neurobiolaging.2020.11.014. |
[70] | Chew S , Atassi N . Positron Emission Tomography Molecular Imaging Biomarkers for Amyotrophic Lateral Sclerosis. Front Neurol. (2019) ;10: :135. doi:10.3389/fneur.2019.00135. |
[71] | Cistaro A , Pagani M , Montuschi A , Calvo A , Moglia C , Canosa A , et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging. (2014) ;41: (5):844–52. doi:10.1007/s00259-013-2667-5. |
[72] | Popuri K , Beg MF , Lee H , Balachandar R , Wang L , Sossi V , et al. FDG-PET in presymptomatic C9orf72 mutation carriers. Neuroimage Clin. (2021) ;31: :102687. doi:10.1016/j.nicl.2021.102687. |
[73] | De Vocht J , Blommaert J , Devrome M , Radwan A , Van Weehaeghe D , De Schaepdryver M , et al. Use of Multimodal Imaging and Clinical Biomarkers in Presymptomatic Carriers of C9orf72 Repeat Expansion. JAMA Neurol. (2020) ;77: (8):1–10. doi:10.1001/jamaneurol.2020.1087. |
[74] | Agosta F , Rocca MA , Valsasina P , Sala S , Caputo D , Perini M , et al. A longitudinal diffusion tensor MRI study of the cervical cord and brain in amyotrophic lateral sclerosis patients. Journal of Neurology, Neurosurgery & Psychiatry. (2009) ;80: (1):53–5. doi:10.1136/jnnp.2008.154252. |
[75] | El Mendili MM , Querin G , Bede P , Pradat P-F . Spinal Cord Imaging in Amyotrophic Lateral Sclerosis: Historical Concepts-Novel Techniques. Front Neurol. (2019) ;10: :350. doi:10.3389/fneur.2019.00350. |
[76] | Querin G , El Mendili M-M , Bede P , Delphine S , Lenglet T , Marchand-Pauvert V , et al. Multimodal spinal cord MRI offers accurate diagnostic classification in ALS. J Neurol Neurosurg Psychiatry. (2018) ;89: (11):1220–1. doi:10.1136/jnnp-2017-317214. |
[77] | van der Burgh HK , Westeneng H-J , Meier JM , van Es MA , Veldink JH , Hendrikse J , et al. Cross-sectional and longitudinal assessment of the upper cervical spinal cord in motor neuron disease. Neuroimage Clin. (2019) ;24: :101984. doi:10.1016/j.nicl.2019.101984. |
[78] | Querin G , Bede P , El Mendili MM , Li M , Pélégrini-Issac M , Rinaldi D , et al. Presymptomatic spinal cord pathology in c9orf72 mutation carriers: A longitudinal neuroimaging study. Ann Neurol. (2019) ;86: (2):158–67. doi:10.1002/ana.25520. |
[79] | Triggs WJ , Menkes D , Onorato J , Yan RS , Young MS , Newell K , et al. Transcranial magnetic stimulation identifies upper motor neuron involvement in motor neuron disease. Neurology. (1999) ;53: (3):605–11. doi:10.1212/wnl.53.3.605. |
[80] | Geevasinga N , Menon P , Özdinler PH , Kiernan MC , Vucic S . Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nature Reviews Neurology. (2016) ;12: (11):651–61. doi:10.1038/nrneurol.2016.140. |
[81] | Neuwirth C , Nandedkar S , Stålberg E , Barkhaus PE , Carvalho M de , Furtula J , et al. Motor Unit Number Index (MUNIX): reference values of five different muscles in healthy subjects from a multi-centre study. Clin Neurophysiol. (2011) ;122: (9):1895–8. doi:10.1016/j.clinph.2011.05.014. |
[82] | Neuwirth C , Barkhaus PE , Burkhardt C , Castro J , Czell D , de Carvalho M , et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the Motor Unit Number Index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatry. (2015) ;86: (11):1172–9. doi:10.1136/jnnp-2015-310509. |
[83] | Lanznaster D , de Assis DR , Corcia P , Pradat P-F , Blasco H . Metabolomics Biomarkers: A Strategy Toward Therapeutics Improvement in ALS. Front Neurol. (2018) ;9: :1126. doi:10.3389/fneur.2018.01126. |
[84] | Gaiani A , Martinelli I , Bello L , Querin G , Puthenparampil M , Ruggero S , et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: Neurofilament light chain levels in definite subtypes of disease. JAMA Neurology. (2017) ;74: (5). doi:10.1001/jamaneurol.2016.5398. |
[85] | Benatar M , Wuu J , Andersen PM , Lombardi V , Malaspina A . Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. (2018) ;84: (1):130–9. doi:10.1002/ana.25276. |
[86] | Van Damme P , Poesen K . Diagnostic and prognostic performance of neurofilaments in ALS. Front Neurol. 2018;doi:10.3389/fneur.2018.01167. |
[87] | Gendron TF , C9ORF72 Neurofilament Study Group, Daughrity LM , Heckman MG , Diehl NN , Wuu J , et al. Phosphorylated neurofilament heavy chain: A biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann Neurol. (2017) ;82: (1):139–46 |
[88] | Saracino D , Dorgham K , Camuzat A , Rinaldi D , Rametti-Lacroux A , Houot M , et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J Neurol Neurosurg Psychiatry. 2021;jnnp-2021-326914. |
[89] | Barschke P , Oeckl P , Steinacker P , Shweiki MRA , Weishaupt JH , Landwehrmeyer GB , et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J Neurol Neurosurg Psychiatry. (2020) ;91: (5):503–11. doi:10.1136/jnnp-2019-322476. |
[90] | Benatar M , Wuu J , Lombardi V , Jeromin A , Bowser R , Andersen PM , et al. Neurofilaments in Pre-Symptomatic ALS and the Impact of Genotype. Amyotroph Lateral Scler Frontotemporal Degener. (2019) ;20: (7-8):538–48. doi:10.1080/21678421.2019.1646769. |
[91] | Lehmer C , Oeckl P , Weishaupt JH , Volk AE , Diehl-Schmid J , Schroeter ML , et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med. (2017) ;9: (7):859–68. doi:10.15252/emmm.201607486. |
[92] | Avgerinos KI , Ferrucci L , Kapogiannis D . Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res Rev. (2021) ;68: :101339. doi:10.1016/j.arr.2021.101339. |


