
Evaluation of the Lipid-binding Properties of Recombinant Dystrophin Spectrin-like Repeat Domains R1-3
Abstract
Recombinant micro-dystrophin genes are designed to treat Duchenne muscular dystrophy (DMD) by retaining dystrophin domains believed to play key functional roles while fitting the packaging capacity of adeno-associated virus vectors. Domains R1-3 are important for muscle force generation and for association with the sarcolemma, but the nature of this interaction is not fully understood. We measured lipid-binding affinity of 3 peptides containing different spectrin-like repeat modules (R1-3; R1-2; and R1, 2, 22). Lipid-binding affinity was highest with R1-3, suggesting that the complete R1-R3 region could be beneficial and should be considered for inclusion in micro-dystrophin constructs.
ABBREVIATIONS
AAV | adeno-associated virus |
AGC | alpha-dystroglycan |
BDG | β-dystroglycan |
BT-PE | Biotinyl–1,2-dipalmitoyl phosphatidylethanolamine |
CR | cysteine-rich |
CT | carboxyl-terminus |
DAPC | dystrophin-associated protein complex |
Dbr | distrobrevin |
DMD | Duchenne muscular dystrophy |
H | hinge |
PBS | phosphate-buffered saline |
POPC | 1-palmitoyl,2-oleoyl phosphatidylcholine |
SG | sarcoglycan complex |
Syn | Syntrophin |
NT | amino-terminus |
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a rare, X-linked, fatal, degenerative neuromuscular disease caused by dystrophin gene (DMD) mutations that disrupt the production of functional dystrophin protein [1, 2]. DMD is the largest known human gene, with a > 11 kbp region that encodes dystrophin, a 427 kDa cytoskeletal protein required for sarcolemmal stability [2, 3]. Protein loss results in susceptibility to repeated cycles of necrosis and regeneration as well as diminished regenerative muscle capacity, resulting in fat and connective tissue replacement (ie, fibrosis) [4]. DMD is progressive: loss of ambulation occurs between ages 9 and 14 years, followed by respiratory complications, cardiac function decline, and, ultimately, premature death [5–7].
Fig. 1
Model of dystrophin–sarcolemma interaction. Binding to the sarcolemma is essential for dystrophin to protect muscle from contraction-induced injury, and in vivo evidence suggests that dystrophin contains four membrane-binding domains, including spectrin-like repeats (R)1-3 and R10-12; cysteine-rich (CR) domain; and carboxyl-terminus (CT) [14].
![Model of dystrophin–sarcolemma interaction. Binding to the sarcolemma is essential for dystrophin to protect muscle from contraction-induced injury, and in vivo evidence suggests that dystrophin contains four membrane-binding domains, including spectrin-like repeats (R)1-3 and R10-12; cysteine-rich (CR) domain; and carboxyl-terminus (CT) [14].](https://ip.ios.semcs.net:443/media/jnd/2021/8-4/jnd-8-4-jnd200622/jnd-8-jnd200622-g001.jpg)
Gene therapy with adeno-associated virus (AAV) vectors is a promising approach for treating monogenic diseases such as DMD and is being evaluated as a therapeutic strategy to restore production of a shortened but functional version of dystrophin (ie, micro-dystrophin). The gene’s large size (∼14 kb) relative to the packaging capacity of AAV-derived vectors (∼5 kb) along with other challenges makes replacement of the full-length gene infeasible with AAV vectors [8, 9]. Large deletions in the dystrophin gene that do not disrupt the open reading frame in patients with milder Becker muscular dystrophy result in partially normal muscle structure and function [10]. Several studies have demonstrated that not all domains seem to be essential and revealed that the functional performance of micro-dystrophin when including essential elements could be similar to that of full-length dystrophin in protecting muscle from damage and improving force production in preclinical dystrophic models [11–13]. Identification of the optimal shortened micro-dystrophin construct that most normalizes muscle function is a key translation question that could overcome the constraints of the AAV cloning capacity.
Dystrophin has four main functional domains: an N-terminus binding domain, which is needed for linkage to the internal actin cytoskeleton; a central rod domain composed of spectrin-like repeat units (R) and hinges (H); a cysteine-rich (CR) domain, which is needed for assembly of the dystrophin-associated protein complex (DAPC); and a carboxyl-terminus (CT) [3]. Interaction with the sarcolemma is central to how dystrophin protects the muscle, as it is this interaction that allows dystrophin to protect muscle from contraction-induced injury (Fig. 1) [3, 12, 14, 15]. Molecular, biochemical, and structural studies have shown that dystrophin membrane-binding domains include spectrin-like repeats R1-3, R10-12, CR domain, and CT [14].
In vivo, AAV-mediated delivery of shortened dystrophin genes demonstrates that the CT domain may not be essential due to redundant protein–protein interaction domains within the DAPC [16]. In addition, the R1-3 domain modulates radial force transmission and mechanical vulnerability, with a possible impact on shortened dystrophin functionality [17]. This evidence highlights a possible key role of the R1-3 module in dystrophin function, linked to its unique property of binding to the membrane, unlike other spectrin-like repeats [16–19]. Additional study is required to determine if this property is inherent to the whole module or if it depends on specific elements (ie, R1, R2 or R3).
This study evaluates the binding affinity of several spectrin-like repeats of dystrophin protein to lipid membranes in vitro to identify differences necessary to consider in designing a micro-dystrophin construct for AAV gene transfer therapy in patients with DMD. Our results shed light on differences in the lipid-binding properties of the R1-3 elements that could be critical for the design of AAV-based gene therapies for restoration of functional dystrophin.
MATERIALS AND METHODS
Reagents
The R1-3, R1-2, and R1,2,22 peptides were sourced from GenScript. Sequences for each peptide construct and apolipoprotein (Abcam [ab50239]) are shown in Supplementary Table 1.
Resuspension of lipids
Phosphatidylcholines, the most abundant phospholipid in the membrane and lipid vesicles, were chosen for these experiments [20]. 1-Palmitoyl,2-oleoyl phosphatidylcholine (POPC) lipids (Avanti Polar Lipids, 850457) were resuspended in chloroform at a concentration of 10 mg/ml. Biotinyl–1,2-dipalmitoyl phosphatidylethanolamine (BT-PE) lipids (Avanti Polar Lipids, 870282) were resuspended in chloroform at a concentration of 2 mg/ml. These resuspensions were mixed to a final concentration of POPC to BT-PE of 1:10. The 1:10 POPC:BT-PE mixture was dried under nitrogen gas until liquid was removed and a film was formed. The film was resuspended in phosphate-buffered saline (PBS), vortexed, and incubated at room temperature for 1 hour to ensure that the lipid was resuspended completely.
Preparation of lipid vesicles
The resuspended biotinylated lipid mixture was submerged in liquid nitrogen for 1 minute, then placed into a 60°C water bath for 3 minutes. This process was repeated for 10 freeze/thaw cycles, after which the lipids were passed through an extruder (NanoSizer extruder, T&T Scientific, TT-030-0009) with an 80 nm filter heated to 45 °C. The lipid solution was pushed through the extruder using syringes 11 times and was then stored at 4°C until use.
Incubation of peptides with vesicles
Mixtures of peptides and biotinylated lipid vesicles were prepared and left to incubate for at least 1 hour at room temperature. Peptide concentration was varied from 1:100 to 1:5, peptide:vesicle, and lipid concentration was held constant.
Lipid binding of peptides assessment
Once the incubation was complete, peptide/lipid mixtures were loaded into a streptavidin column (Fisher Scientific, PI20351) for at least 10 minutes. Columns were washed with 5 column volumes (cv) of PBS (pH 7.4). Protein was then eluted with an elution buffer (8 M guanidine HCl pH 1.5) into tubes containing neutralizing buffer (1 M Tris pH 9.0). The protein/lipid mixture was eluted using 5 cv of elution buffer. Wash and eluate were concentrated in Pierce protein concentrators (PES 10 K MWCO, Fisher Scientific, PI88528) using centrifugation at 4500XG for 15 minutes. The protein concentration of wash and eluate was determined by absorbance at 280 nm using NanoDrop (Thermo Scientific, NDNDLPRUSCA).
Data analysis
The protein concentration of each sample was run in duplicate to assess intra-assay variability. Concentrations of total protein were graphed against the concentration of bound protein/concentration of lipid using GraphPad Prism version 7.01 for Windows (GraphPad Software; www.graphpad.com) as described by Davidson and colleagues [20]. Using the one site-specific binding nonlinear fit program in GraphPad, an equilibrium dissociation constant (Kd) was calculated for each peptide.
RESULTS
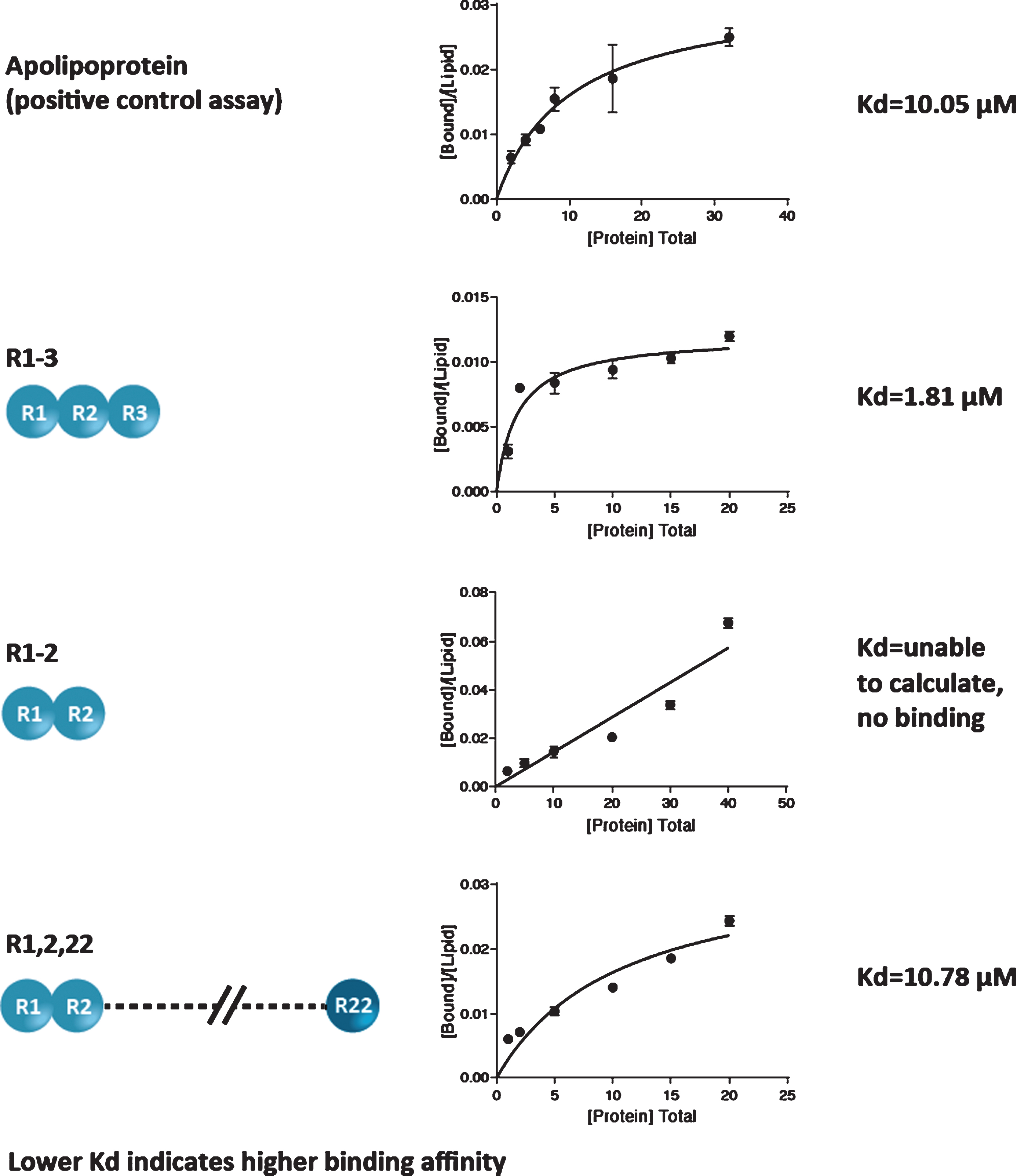
The lipid-binding affinity of 3 peptides containing different R modules (R1-3; R1-2; and R1,2,22) was measured by determination of Kd (lower Kd denotes a higher binding affinity).
Results demonstrated differences in lipid-binding affinities for the peptides containing different R modules. Apolipoprotein, the positive control assay, yielded a Kd value of 10.05±2.35μM; the R1-3 module yielded a Kd of 1.81±0.49μM; the R1,2,22 module yielded a Kd of 10.78±4.00μM; and the Kd for the R1-2 module could not be calculated because no protein binding was detected (Fig. 2). Based on the 95% confidence intervals, the differences in Kd between the R1-3 module and apolipoprotein and between the R1-3 and R1,2,22 modules were statistically significant (both p < 0.05). The difference in Kd between the R1,2,22 module and apolipoprotein was not statistically significant. Notably, among the 3 peptides, the R1-3 module yielded the lowest Kd value, indicating the highest lipid-binding affinity. These results show that R1-3 has the highest binding affinity for lipids, R1,2,22 has a moderate binding affinity, and R1-2 has a weak binding affinity.
Fig. 2
Lipid-binding affinity of 3 peptides containing different R modules.
DISCUSSION
Previous studies showed that the R1-3 region binds to the lipid component of the sarcolemma in vivo [14], and studies in mdx mice demonstrated that the inclusion of spectrin-like repeats R1-3 is important for protection against eccentric contraction-induced force drop [17].
This study demonstrates the high affinity of the module R1-3 in binding to the lipid membrane in vitro. Previous research has shown that peptides containing 3 spectrin-like repeats seem to bind more tightly than those with fewer repeats. Although both R1-3 and R1,2,22 peptides are able to bind lipids at clinical concentrations, R1-3 binds lipids more tightly, especially at concentrations below 10μM, which represents the majority of the clinical concentration range. However, this does not explain the superior binding of R1-3 compared with R1,2,22.
This study demonstrates the high affinity of the intact module R1-3 in binding directly to the lipid membrane, a property that seems to be disrupted when R3 is not present, suggesting a potential role for the R3 domain in the maintenance of muscular contractile force. We hypothesize that R1-3, and specifically R3, may contribute to maintaining structural integrity by its ability to interact with the lipid membrane, and this could explain the protection of muscle tissue from damage and elicitation of the optimal force generation observed in previous studies [17]. The lipid binding of this region seems to be disrupted when R3 is absent, suggesting that the complete R1-3 region may play an important role in dystrophin functionality. This may provide a functional benefit when included in the design of a micro-dystrophin construct for AAV gene therapy in patients with DMD. However, future studies are needed to elucidate the mechanism behind this functionality and to clarify the role of R3 in lipid binding. We plan to further investigate the importance of R3 in lipid binding using an in vivo–like system, such as a cell-based assay, in the future.
ACKNOWLEDGMENTS
This study was sponsored by Sarepta Therapeutics, Inc. Authors are employees of Sarepta Therapeutics and may have stock options. Medical writing and editorial support was provided by Lucia Quintana-Gallardo, PhD of Sarepta Therapeutics, Inc., and Purvi Kobawala Smith, MS, MPH and Clare Sonntag, MA of Health & Wellness Partners, LLC, funded by Sarepta Therapeutics.
AUTHOR CONTRIBUTIONS
GC-O and RAP created the study design. GC-O performed the study. GC-O, LRR-K and RAP wrote, edited and approved the final version of the manu-script.
FUNDING
This study was funded by Sarepta Therapeutics, Inc.
CONFLICTS OF INTEREST
GC-O, RAP, and LRR-K are employees of Sarepta Therapeutics, Inc., which funded the study.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200622.
REFERENCES
[1] | Hoffman EP , Brown RH Jr , Kunkel LM . Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. (1987) ;51: (6):919–28. |
[2] | Koenig M , Hoffman EP , Bertelson CJ , Monaco AP , Feener C , Kunkel LM . Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) ;50: (3):509–17. |
[3] | Gao QQ , McNally EM . The dystrophin complex: Structure, function, and implications for therapy. Compr Physiol. (2015) ;5: (3):1223–39. |
[4] | Mendell JR , Lloyd-Puryear M . Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. (2013) ;48: (1):21–6. |
[5] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S . CINRG investigators. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. (2016) ;87: (4):401–9. |
[6] | Birnkrant DJ , Bushby K , Bann CM , et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) ;17: (4):347–61. |
[7] | Birnkrant DJ , Bushby K , Bann CM , et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. (2018) ;17: (5):445–55. |
[8] | Mendell JR , Rodino-Klapac L , Sahenk Z , et al. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci Lett. (2012) ;527: (2):90–9. |
[9] | Duan D . Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther. (2018) ;26: (10):2337–56. |
[10] | England SB , Nicholson LV , Johnson MA , et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. (1990) ;343: (6254):180–2. |
[11] | Wells DJ , Wells KE , Asante EA , et al. Expression of human full-length and minidystrophin in transgenic mdx mice: Implications for gene therapy of Duchenne muscular dystrophy. Hum Mol Genet. (1995) ;4: (8):1245–50. |
[12] | Harper SQ , Hauser MA , DelloRusso C , et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat Med. (2002) ;8: (3):253–61. |
[13] | Rodino-Klapac LR , Janssen PML , Shontz KM ,et al.. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum Mol Genet. (2013) ;22: (24):4929–37. |
[14] | Zhao J , Kodippili K , Yue Y ,et al.. Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet. (2016) ;25: (17):3647–53. |
[15] | Crawford GE , Faulkner JA , Crosbie RH , Campbell KP , Froehner SC , Chamberlain JS . Assembly of the dystrophin-associated protein complex does not require the dystro-phin COOH-terminal domain. J Cell Biol. (2000) ;150: (6):1399–410. |
[16] | Ramos JN , Hollinger K , Bengtsson NE , Allen JM , Hauschka SD , Chamberlain JS . Development of novel micro-dystrophins with enhanced functionality. Mol Ther. (2019) ;27: (3):623–35. |
[17] | Nelson DM , Lindsay A , Judge LM ,et al.. Variable rescue of microtubule and physiological phenotypes in mdx muscle expressing different miniaturized dystrophins. Hum Mol Genet. (2018) ;27: (12):2090–100. Erratumin: Hum Mol Genet. 2018;27(15):2773. |
[18] | Legardinier S , Hubert JF , Le Bihan O ,et al.. Sub-domains of the dystrophin rod domain display contrasting lipid-binding and stability properties. Biochim Biophys Acta. (2008) ;1784: (4):672–82. |
[19] | Legardinier S , Raguénès-Nicol C , Tascon C ,et al.. Mapping of the lipid-binding and stability properties of the central rod domain of human dystrophin. J Mol Biol. (2009) ;389: (3):546–58. |
[20] | Davidson WS , Ghering AB , Beish L , Tubb MR , Hui DY , Pearson K . The biotin-capture lipid affinity assay: A rapid method for determining lipid binding parameters for apolipoproteins. J Lipid Res. (2006) ;47: (2):440–9. |


