
Molecular Mechanisms of Skeletal Muscle Hypertrophy
Abstract
Skeletal muscle hypertrophy can be induced by hormones and growth factors acting directly as positive regulators of muscle growth or indirectly by neutralizing negative regulators, and by mechanical signals mediating the effect of resistance exercise. Muscle growth during hypertrophy is controlled at the translational level, through the stimulation of protein synthesis, and at the transcriptional level, through the activation of ribosomal RNAs and muscle-specific genes. mTORC1 has a central role in the regulation of both protein synthesis and ribosomal biogenesis. Several transcription factors and co-activators, including MEF2, SRF, PGC-1α4, and YAP promote the growth of the myofibers. Satellite cell proliferation and fusion is involved in some but not all muscle hypertrophy models.
INTRODUCTION
Skeletal muscle has a remarkable capacity to undergo hypertrophy, i. e. increase in size, in response to certain physical activities, such as those based on resistance exercise, or to hormones, such as androgens, responsible for the difference in muscle size between males and females. Muscle hypertrophy is an interesting object of study per se, as a model of growth in cell biology, but is also clinically relevant. The decreased muscle mass in old age is a risk factor for frailty, falls and fractures, and is also found in a wide range of chronic diseases. The recognition that muscle wasting is a widespread condition affecting millions of people has stimulated the study of the molecular mechanisms responsible for the maintenance of the muscle mass and the search for treatments able to induce muscle hypertrophy and increase in muscle force.
The hypertrophy process has been extensively analyzed in humans, using different training protocols based on resistance exercise, and in animal models, such as the overload hypertrophy induced by tenotomy or ablation of synergistic muscles. Muscle hypertrophy can be evaluated quantitatively at the macroscopic level using a variety of imaging techniques, including dual-energy x-ray absorptiometry (DXA), computed tomography (CT) scanning, magnetic resonance imaging (MRI), and ultrasound assessment, or at the microscopic level, by measuring the cross-sectional area (CSA) of the muscle fibers [see 1].
We have previously reviewed some aspects of muscle hypertrophy in the broader context of muscle adaptation and remodeling [2, 3]. The aim of this review is to focus on the molecular pathways underpinning the hypertrophic process. We will first consider the extracellular signals acting on muscle fibers and triggering the hypertrophic response. These include i) hormones and growth factors acting directly as positive regulators of muscle growth or indirectly by neutralizing negative regulators, and ii) mechanical signals acting at the level of the plasma membrane or through the muscle cytoskeleton. We will subsequently focus on two control hubs regulating muscle hypertrophy: the translational control responsible for protein synthesis, and the transcriptional control, which regulates the expression of two major sets of genes required for muscle hypertrophy, the genes of ribosomal RNAs and proteins and the muscle-specific genes coding for contractile, EC coupling and metabolic proteins.
PRO-HYPERTROPHIC HORMONES AND GROWTH FACTORS
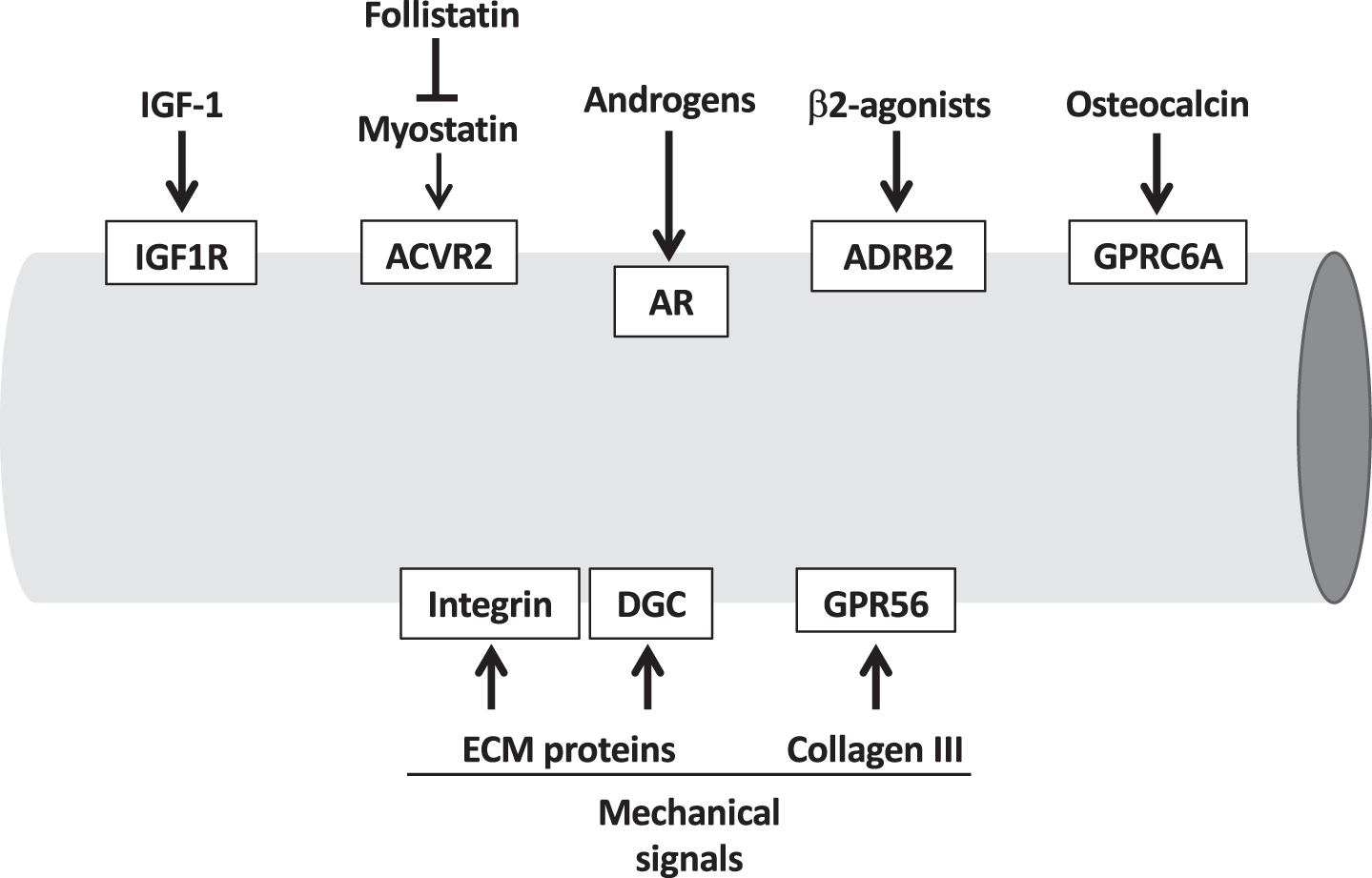
A selection of the most important hormones and growth factors which affect muscle mass and are able to induce skeletal muscle hypertrophy is shown in Fig. 1 and is briefly discussed below.
Fig. 1
Extracellular pro-hypertrophic signals, including hormo-nes and growth factors, and mechanical signals acting on the muscle cell membrane. The receptors mediating these signals are indicated in the white boxes. ACVR2, activin receptor type II; AR, androgen receptor; ADRB2, adrenergic receptor b2; DGC, dystrophin glycoprotein complex; GPRC6A, G Protein-Coupled ReceptorC6A; GPR56, G protein-coupled receptor 56; IGF1R, IGF-1 receptor.
IGF1
IGF1 is a potent growth factor affecting muscle growth during development and acting both systemically as a typical hormone produced by the liver under the control of growth hormone and locally as a paracrine/autocrine factor produced by skeletal muscle. IGF1 is able to induce muscle hypertrophy by binding a specific receptor (IGF1R) and activating the PI3K-Akt-mTOR signaling pathway, which is discussed below. The study of IGF1 is complicated by the existence of multiple isoforms with variable potency in inducing muscle hypertrophy, by the partial overlapping of activity with insulin (insulin can activate the IGF1 receptor and vice versa) and by the presence of binding factors that may act to enhance or attenuate IGF1 signaling. IGF1 released by muscle fibers has been implicated in different models of muscle hypertrophy; however, overload-induced hypertrophy is completely unaffected in mouse muscles lacking the IGF1 receptor [4].
Follistatin-Myostatin-BMP
Myostatin (GDF8) and activin A are members of the TGFβ superfamily which act as negative regulators of muscle mass by binding to the activin type II receptor (ActRII). Their effect is blocked by the endogenous inhibitor, follistatin, which acts as a pro-hypertrophic signal [5]. Myostatin inactivation or follistatin overexpression, induced in adult mouse muscles by injection of antibodies to myostatin or by injection of the follistatin variant Fst288, causes muscle hypertrophy with a slow-to-fast switch in fiber type composition [6, 7]. Muscle hypertrophy is also induced by a soluble form of ActRII in mice and monkeys, however interpretation of this model is complicated by the fact that soluble ActRII may sequester other ligands binding to this receptor, which explains the adverse effects seen in humans after this treatment [see 8]. Binding of myostatin to its receptor leads to Smad3 phosphorylation that interferes with the Akt-mTOR pathway whereas follistatin activates this pathway and promotes protein synthesis [9, 10, 11]. In contrast, the effect of phosphorylated Smad3 following nuclear translocation and binding to target genes has not yet been defined with respect to its role in muscle hypertrophy. Other members of the TGFβ superfamily, such as some bone morphogenetic proteins (BMPs), have opposite effects on muscle growth compared with myostatin [12]. In particular, overexpression of BMP7 was shown to induce mouse muscle hypertrophy [12, 13]. This effect is mediated by signaling through Smad1/5/8 and requires the activation of the Akt-mTOR pathway, as BMP-mediated hypertrophy of skeletal muscle was prevented by inhibition of mTOR signaling.
Androgens
Androgens, such as testosterone, are potent inducers of skeletal muscle hypertrophy by binding to the androgen receptor (AR), followed by nuclear translocation and target gene regulation. In addition, testosterone can be converted into dihydrotestosterone, which is the most powerful androgen due to its high affinity for the AR, through the activity of 5α-reductase type 1 (Srd5a1), which is expressed in skeletal muscle and is upregulated by physical exercise in rats [14]. In vivo studies indicate that androgen withdrawal in male mice decreases muscle myofibrillar protein synthesis through suppression of Akt/mTORC1 signaling, possibly mediated by downregulation of IGF-1 expression, and this is readily reversible by androgen administration [15]. Androgens can also act through a nongenomic signaling pathway mediated by the binding of androgens to surface receptors and leading to activation of Akt-mTOR, as suggested by the finding that the increase in myotube size and activation of Akt-mTOR in vitro are not inhibited by androgen receptor antagonists that block the genomic effects of androgens [16]. Androgens also induce satellite cell proliferation followed by fusion and myonuclear accretion, however, these processes are not required for androgen-induced muscle hypertrophy as muscle fiber size is increased in satellite cell-depleted mice treated with testosterone [17].
β2-agonists
Epinephrine interacts with the β2 adrenergic receptor (β2AR), a G protein-coupled receptor coded by the ADRB2 gene, which is the most abundant adrenergic receptor present in muscle fibers. Binding of β2 agonists to the receptor activates adenylate cyclase with generation of cyclic AMP and activation of protein kinase A (PKA). Chronic treatment with β2 agonists as clenbuterol leads to muscle hypertrophy through still poorly defined pathways, which appear to involve the IGF1-PI3K-Akt-mTOR cascade [18, 19]. The role of PKA-dependent phosphorylation of the transcription factor CREB (cAMP response element binding protein) and associated coactivators in mediating muscle hypertrophy is not known, although the pro-hypertrophic factor MEF2 (see below) could be involved, as a dominant-negative CREB in postnatal mouse muscles caused muscle wasting that was associated with reduced expression of MEF2 target genes [see 20]. Identification of the pathways mediating the β2AR signaling is complicated by the fact that, following agonist binding, β2ARs undergo rapid desensitization through receptor phosphorylation by G protein-coupled receptor kinases (GRKs) and subsequent recruitment of the adaptor protein, β-arrestin [21]. Both GRKs and β-arrestin initiate signaling cascades, which are G protein-dependent and receptor independent, that may affect the pro-hypertrophic pathway. For example, muscle-specific GRK2 knockout enhances clenbuterol-stimulated hypertrophy [22] whereas the hypertrophic response was abrogated in mice lacking β-arrestin 1, which is the predominant β-arrestin isoform in skeletal muscle [23].
Osteocalcin
The role of osteocalcin, a bone-derived hormone, as a pro-hypertrophic signal was revealed by the finding that osteocalcin null mice or mice with muscle-specific knockout of the osteocalcin receptor, GPRC6A, undergo muscle atrophy, and supported by the finding that treatment with exogenous osteocalcin for 4 weeks is sufficient to increase muscle mass of adult mice [24]. This effect may be due to the fact that osteocalcin promotes muscle protein synthesis, as indicated by studies in cultured myotubes, but the signaling pathway involved has not been determined. It was suggested that osteocalcin is a central component of a muscle-bone-muscle endocrine axis, whereby interleukin 6 (IL-6) released by skeletal muscle during exercise acts on osteoblasts to induce the release of bioactive osteocalcin that in turn acts on muscle cells [25]. However, recent studies using new osteocalcin knockout models have not confirmed an endocrine role of osteocalcin nor a pro-hypertrophic effect on skeletal muscle [26, 27].
MECHANOTRANSDUCTION AND MUSCLE HYPERTROPHY
Resistance exercise increases muscle mass in humans and animals, and the fact that only contractions against a load produce this effect suggests that mechanical signaling is involved. The search for specific mechanosensors responsible for muscle hypertrophy has mainly focused on the plasma membrane and on the sarcomeric cytoskeleton, however no clear signaling pathway leading from the sensors to the final translational and transcriptional targets has emerged so far [28].
Plasma membrane mechanosensors: dystrophin, integrin and GPR56.
Mechanical signals generated by muscle contraction or by passive stretch are transmitted through two multiprotein complexes spanning the plasma membrane and connecting extracellular matrix (ECM) with intracellular cytoskeleton: the dystrophin glycoprotein complex (DGC) and the integrin adhesion complex. These structures are especially abundant at sites of high longitudinal or lateral force transmission, the myotendinous junctions and costameres, and act as shock absorbers stabilizing the sarcolemma during contraction/stretch. Mutations of dystrophin or α7β1 integrin, the predominant integrin form present in adult skeletal muscle, cause contraction-induced muscle injury in mice. In addition to this structural role, both dystrophin and integrin act as scaffold for signaling proteins and are thus potentially involved in mediating the pro-hypertrophic effect of contractile activity against high load, as occurs in resistance exercise, or passive stretch.
Integrins are linked to actin through the adaptor protein talin and may activate different signaling pathway through focal adhesion kinase (FAK) and integrin linked kinase (ILK), two kinases potentially involved in mechanotransduction in striated muscle [29]. However, it is not clear whether FAK and ILK mediate muscle hypertrophy in response to resistance exercise or in overload models [30]. The integrin-associated protein, melusin, has been implicated as a load sensor, as the levels of melusin decrease in unloaded muscles in rats and humans and muscle atrophy induced by unloading can be prevented by melusin overexpression [31].
Other studies point to possible pathways connecting the dystrophin glycoprotein complex to growth-promoting signaling pathways and muscle hypertrophy. Muscle hypertrophy induced by functional overload is reduced in dystrophin-deficient mdx mice [32]. Neuronal nitric oxide synthase (nNOS), a DGC component bound to dystrophin through α-syntrophin, is rapidly activated after synergist ablation and overload hypertrophy is blunted in nNOS knockout mice [33]. The signaling cascade proposed involves peroxynitrite generated by nNOS activating the cation channel Trpv1, thus inducing an increase of intracellular Ca2 + concentration that subsequently triggers mTOR activation. Another study showed that γ1-syntrophin interacts with and regulates the activity of diacylglycerol kinase-zeta (DGKζ), which phosphorylates diacylglycerol to yield phosphatidic acid [34]. Interestingly, phosphatidic acid synthesized by DGKζ regulates the mechanical activation of mTOR signaling and muscle hypertrophy, and overexpression of DGKζ induces skeletal muscle hypertrophy [35] (Fig. 2). Another potential connection between dystrophin and muscle hypertrophy is based on the finding that the insulin receptors associate with DGC-rich clusters at costameres and this association, which is stabilized by plakoglobin, promotes insulin signaling through the PI3K-Akt pathway [36]. The finding that cancer cachexia is accompanied by reduced levels of dystrophin and is partly prevented in dystrophin transgenic mice suggests that DGC dysfunction plays a critical role in cancer-induced wasting [37].
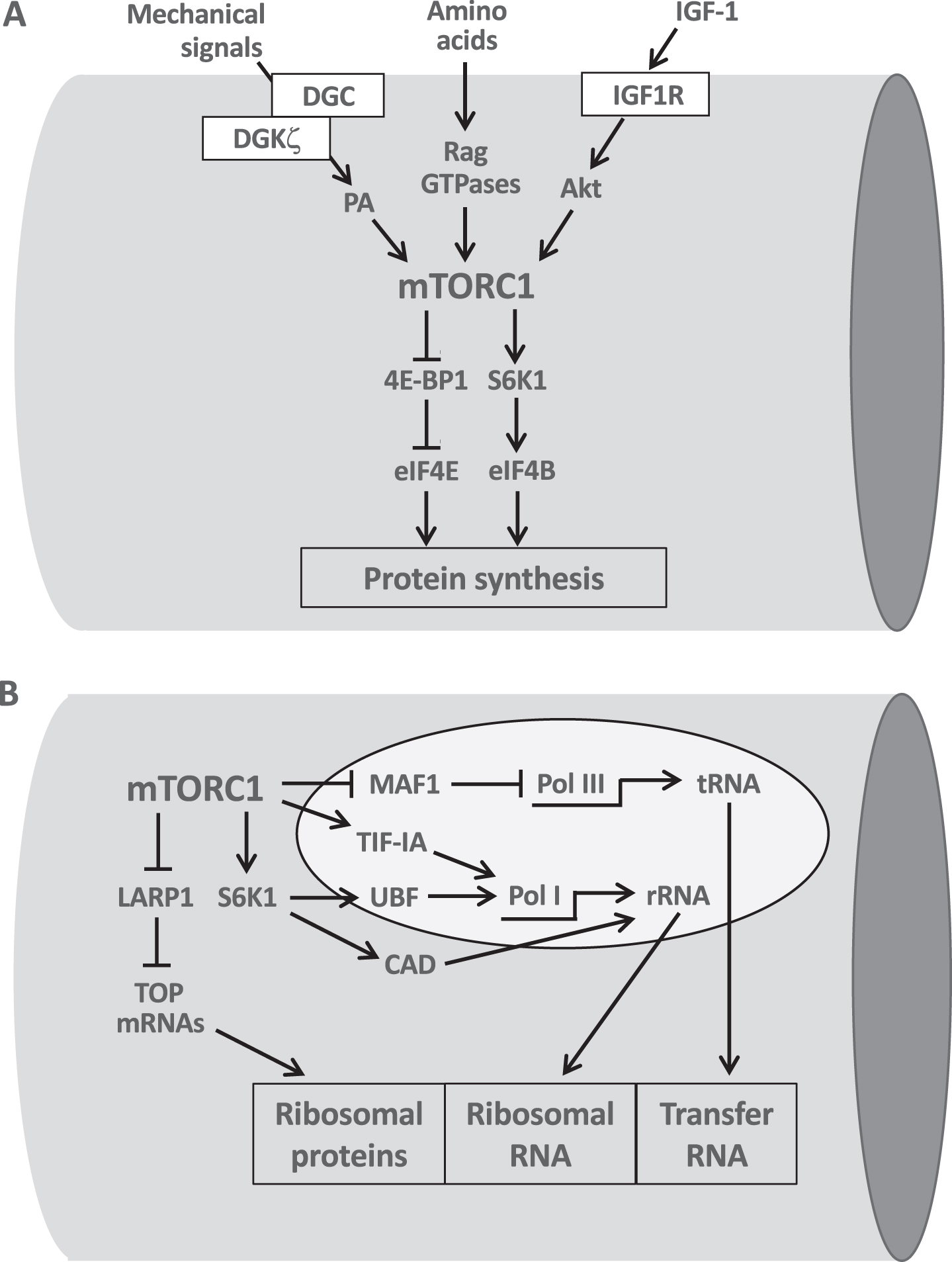
Fig. 2
The central role of mTORC1 in the translational and transcriptional control of protein synthesis. A. mTORC1 integrates the pro-hypertrophic input from growth factors, amino acids and mechanical signals. mTORC1 stimulates protein synthesis by acting at the translational level through phosphorylation of 4E-BP1 and S6K1 which in turn activate initiation factors eIF4E and eIF4B. B. mTORC1 controls ribosomal biogenesis at the transcriptional level by stimulating PolI-mediated synthesis of ribosomal RNA (rRNA) via TIF-1A and Pol III-mediated synthesis of transfer RNA (tRNA) via MAF1. S6K1 also activates PolI through UBF and stimulates pyrimidine biosynthesis required for rRNA synthesis by CAD phosphorylation. The translational activation of TOP mRNAs, controlled by mTORC1 via LARP1, leads to the formation of ribosomal proteins. See text for further details.
Another signaling pathway potentially involved in mechanotransduction at the plasma membrane is through GPR56, a member of the adhesion G protein-coupled receptor family, whose extracellular ligand is collagen type III (coded for by the COL3A1 gene). GPR56, whose expression is controlled by the transcriptional coactivator PGC-1α4 (see below), appears to drive muscle hypertrophy downstream of a Gα12/13–Rho pathway leading to mTOR activation and increase in protein synthesis [38]. The role of this pathway in mediating overload-induced hypertrophy is supported by the finding that the hypertrophic response is blunted in GPR56 knockout mice.
Sarcomeric mechanosensors
Mechanosensors embedded at different locations in the sarcomere have been implicated in the activation of signaling pathways leading to muscle hypertrophy [39]. These include proteins located in the Z-disk, such as muscle LIM protein (MLP, coded by CSRP3), which translocates to the nucleus in response to mechanical strain in cultured muscle cells [40, 41], at the I-band, like ankyrin-repeat domain 2 (ANKRD2) and other muscle ankyrin repeat proteins (MARPs), as well as four and a half LIM domain proteins (FHL1-4) implicated in cardiac hypertrophy [42], or at the M-band, where the kinase domain of titin may act as a sensor of mechanical signals leading to derepression of the transcription factor SRF [43]. Titin-based mechanosensing has also been recently described in a mouse model in which the denervated hemidiaphragm is passively stretched by the contralateral, innervated hemidiaphragm and undergoes hypertrophy: the degree of hypertrophy was found to vary in titin mutant models showing decreased and increased titin stiffness, high stiffness resulting in an exaggerated hypertrophy response [44].
TRANSLATIONAL CONTROL OF MUSCLE HYPERTROPHY: THE CENTRAL ROLE OF THE AKT-mTOR PATHWAY
It is known since the early studies of Goldberg [45] that protein synthesis is increased in experimental models of rat muscle hypertrophy and subsequent studies showed that the rate of muscle protein synthesis increases significantly following a single bout of resistance exercise in humans [46]. Several lines of evidence indicate that the rate of protein synthesis in skeletal muscle is controlled by a kinase cascade activated by growth factors such as IGF1 and comprising in sequence the phosphoinositide 3-kinase (PI3K), Akt (also called protein kinase B, PKB) and the mechanistic target of rapamycin (mTOR) [47]. That stimulation of this pathway leads to muscle hypertrophy was first suggested by the finding that transfection of adult mouse muscle with a RAS mutant selectively activating Akt leads to striking hypertrophy of transfected fibers [48]. Activation of this pathway is required for load-induced skeletal muscle hypertrophy, as shown by gain- and loss-of-function genetic approaches and by the finding that muscle hypertrophy is blocked by rapamycin, a selective inhibitor of mTOR [49, 50] (Fig. 2A). In particular, the raptor-binding, rapamycin sensitive mTOR complex 1 (mTORC1) can stimulate protein synthesis by phosphorylating the eukaryotic initiation factor 4E-binding proteins (4E-BPs) and the ribosomal S6 protein kinase 1 (S6K1) [see 51].
In addition, mTORC1 controls the translation of terminal oligopyrimidine (TOP) mRNAs, which code for ribosomal proteins and several initiation and elongation factors, by acting on LARP1, a key repressor of TOP mRNA translation [52]. The upstream activators of mTOR, in addition to growth factors such as IGF1 and insulin acting through the PI3K-Akt cascade, are different amino acids, acting via Rag GTPases, and mechanical signals, such as phosphatidic acid generated by DGKζ, as discussed in the previous section. However, the signaling pathways linking mechanical overload and mTOR activation during muscle hypertrophy induced by exercise are still unclear.
TRANSCRIPTIONAL CONTROL: RIBOSOMAL BIOGENESIS
The coordinated regulation of two major sets of genes is required for muscle hypertrophy: the genes coding for ribosomal RNAs, involved in ribosomal biogenesis, and the muscle-specific genes coding for contractile, EC coupling and metabolic proteins, whose expression is controlled by transcription factors such as MEF2 and SRF, or transcriptional co-regulators, such as PGC-1α4.
While mRNAs derived from protein-coding genes, including those coding for ribosomal proteins, are generated by RNA polymerase II (RNA Pol II), the synthesis of ribosomal RNAs requires RNA polymerase I (RNA Pol I), producing the 47 S ribosomal RNA (rRNA), which is subsequently processed to mature 5.8 S, 18 S and 28 S ribosomal components, and RNA polymerase III (RNA Pol III), which synthesizes 5 S rRNA and the transfer RNAs. pre-rRNA processing and ribosome assembly take place in the nucleolus. The ubiquitous transcription factor MYC controls ribosome biogenesis and translation through a variety of mechanisms [53]. In addition, as shown in Fig. 2B, mTORC1 activates ribosomal biogenesis by controlling the activity of MAF1, a key repressor of RNA Pol III [54, 55], and of transcription initiation factor 1A (TIF-IA), which regulates RNA Pol I transcription [56]. RNA PolI is also controlled by S6K1 through phosphorylation of the upstream binding factor (UBF) [57]. rRNA synthesis requires a continuous supply of nucleotides and it has been shown that S6K1 promotes nucleotide biosynthesis by phosphorylation of CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase), the enzyme that catalyzes the first three steps of de novo pyrimidine biosynthesis and thus increases the pool of nucleotides available for the RNA synthesis that accompanies cell growth [58, 59].
Muscle growth is accompanied by and dependent on ribosomal biogenesis [60, 61]. The rRNA pool, which accounts for more than 80% of total RNA, increases during muscle hypertrophy in response to the activity of different regulatory factors, including MYC and UBF, whose levels and activities are increased during resistance exercise [61]. Muscle hypertrophy caused by activation of an inducible Akt transgene is also accompanied by an increase in RNA levels and by increased CAD phosphorylation [62]. The role of the different mTORC1-dependent pathways controlling rRNA and tRNA transcription during muscle hypertrophy remains to be established. Interestingly, interindividual variability in the hypertrophic response to the same exercise training program appears to correlate with parallel changes in ribosomal biogenesis [63] (Fig. 3). Other studies in humans reveal a correlation between dose-dependent training responses, RNA accumulation and increases in muscle mass [64].
Fig. 3
Wide interindividual variability in the hypertrophic response. Percent change in type II myofiber cross-sectional area (CSA) from pre- to post-resistance exercise training in older adults (age 60–75 yr) subjected for 4 wk to the same resistance exercise protocol. Based on the hypertrophic response, 3 groups were identified: nonresponders, moderate responders and extreme responders. Variations in resistance training-induced myofiber hypertrophy were found to correlate with parallel changes in markers of ribosomal biogenesis (Modified from [63]).
![Wide interindividual variability in the hypertrophic response. Percent change in type II myofiber cross-sectional area (CSA) from pre- to post-resistance exercise training in older adults (age 60–75 yr) subjected for 4 wk to the same resistance exercise protocol. Based on the hypertrophic response, 3 groups were identified: nonresponders, moderate responders and extreme responders. Variations in resistance training-induced myofiber hypertrophy were found to correlate with parallel changes in markers of ribosomal biogenesis (Modified from [63]).](https://ip.ios.semcs.net:443/media/jnd/2021/8-2/jnd-8-2-jnd200568/jnd-8-jnd200568-g003.jpg)
TRANSCRIPTIONAL CONTROL: TRANSCRIPTION FACTORS AND COREGULATORS
Different transcription factors and coactivators/corepressors are involved in muscle hypertrophy. In this section, we will focus on the transcription factors MEF2 and SRF, their transcriptional coregulators, and the coactivators PGC-1α4 and YAP/TAZ.
MEF2 factors
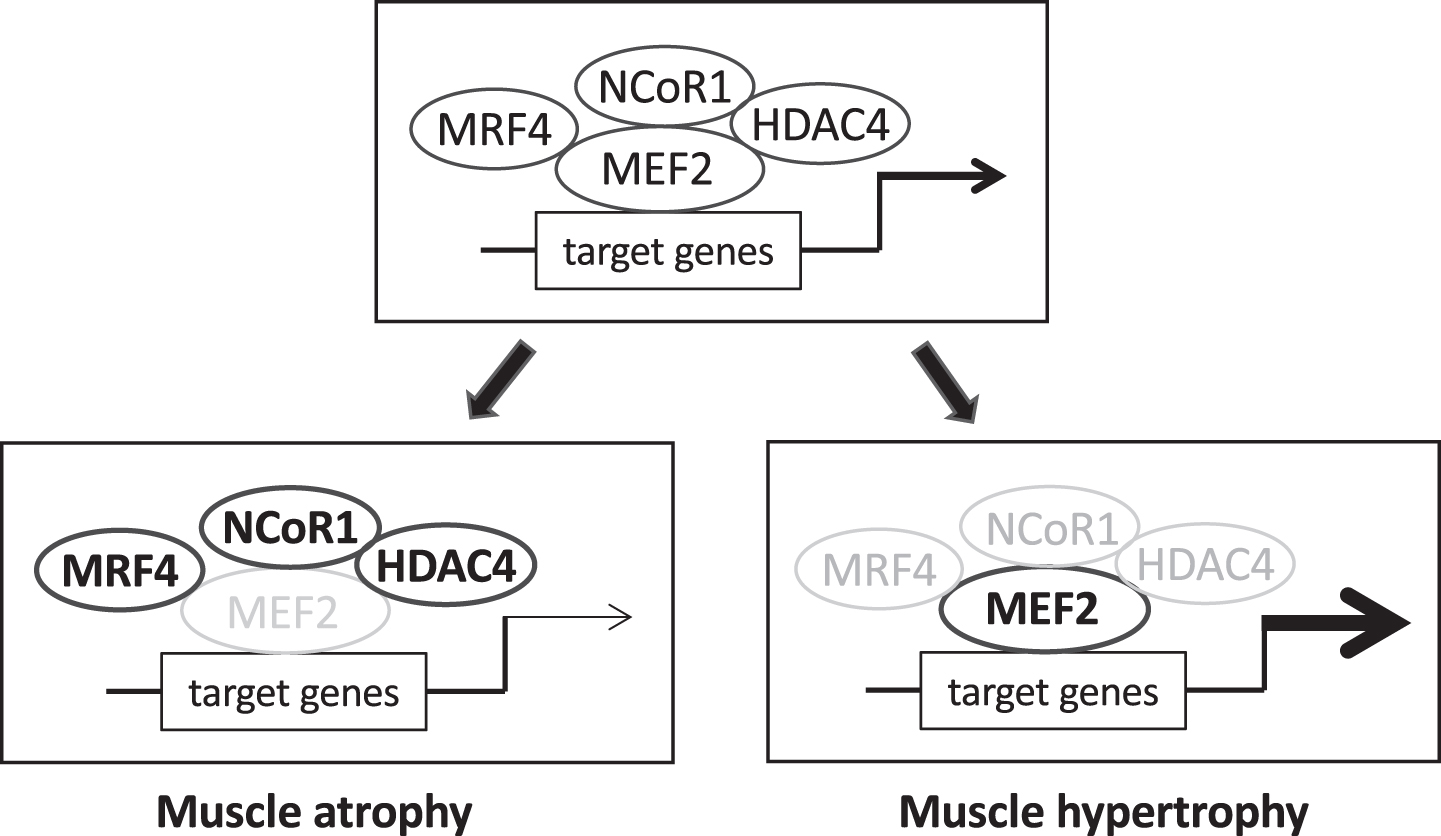
The transcription factors MEF2A, C and D are known to be major players in controlling muscle differentiation and growth during embryonic development by cooperating with myogenic regulatory factors of the MyoD family and regulating the expression of a large number of muscle-specific genes [65]. MEF2 factors are also essential during muscle regeneration and show redundant functions, as inactivation of all three MEF2A, C and D is required to block the regeneration process [66]. Although inducible MEF2 knockout models, in which MEF2 genes are exclusively inactivated at an adult stage specifically in skeletal muscle, have not yet been generated, recent studies suggest that MEF2 factors are also involved in the regulation of adult muscle mass in rats and mice. In vivo transfection experiments showed that muscle hypertrophy can be induced by a constitutively active MEF2 (caMEF2) mutant and also by shRNAs against the transcription factor MRF4, a member of the MyoD family [67]. MRF4 knockdown causes activation of MEF2 transcriptional activity and upregulation of muscle-specific genes known to be targets of MEF2. The existence of a MRF4-MEF2 axis, with MRF4 interacting with MEF2 and repressing MEF2 activity, was supported by the finding that a dominant negative MEF2 mutant prevents muscle hypertrophy induced by MRF4 knockdown. MRF4 appears to exert its repressive effect on MEF2 via a multiprotein repressive complex containing HDAC4 and the co-repressor NCoR1, as shown by the finding that MRF4 knockdown causes HDAC4 nuclear export [67], muscle gene expression is repressed by HDAC4 through direct binding and inhibition of MEF2 activity [68] and muscle atrophy upon denervation is partially prevented by double knockout of HDAC4 and HDAC5 [69]. Accordingly, activation of MEF2 and muscle hypertrophy are induced by muscle-specific knockout of NCoR1 [70] (Fig. 4).
Fig. 4
The transcriptional activity of myocyte enhancer factor-2 (MEF2) factors is controlled by different repressors. MEF2 factors promote muscle growth during development and in the adult by regulating the expression of muscle-specific genes. MEF2 transcriptional activity is controlled by different repressors, including muscle-specific repressors like myogenic regulatory factor 4 (MRF4, coded by MYF6) and ubiquitous repressor as nuclear receptor co-repressor 1 (NCoR1) and class II histone deacetylases (HDACs), like HDAC4. Under normal conditions (upper panel) muscle size is maintained in the adult by a balance between these inhibitory factors and different stimulatory influences, including MEF2 post-translational changes, not depicted in the scheme. Loss of repressor activity (lower right panel), such as muscle-specific knockout of NCoR1 or muscle-specific knockdown of MRF4, lead to upregulation of MEF2 transcriptional activity and muscle hypertrophy. Increased repressor function (lower left panel), e.g. denervation-induced up-regulation and nuclear translocation of MRF4 and HDAC4, reduce MEF2 transcriptional activity and contribute to muscle atrophy.
A role of MEF2 in maintaining muscle size and preventing muscle atrophy is supported by the finding that denervation atrophy is accompanied by markedly decreased MEF2 transcriptional activity and by upregulation and nuclear translocation of MRF4 [71] and HDAC4 [72]. MEF2 transcriptional activity is also decreased in human skeletal muscle following muscle unloading induced by prolonged bed rest [73]. Accordingly, a recent study has shown that MEF2 transcriptional activity and MEF2 target gene expression is decreased in a mouse model of spinal and bulbar muscular atrophy (SBMA) caused by polyglutamine (polyQ) tract expansion in the androgen receptor [74]. Under these conditions MEF2 function is apparently disrupted due to sequestration of MEF2 into the polyQ intranuclear aggregates and a similar effect is seen in other models of polyQ disease that exhibit skeletal muscle atrophy, such as a mouse model of Hungtinton disease. Interestingly, MEF2 transcriptional activity and muscle atrophy can be rescued by a caMEF2 mutant that shows less propensity for co-aggregation [74]. Another recent study reported that caMEF2 prevents muscle wasting in cachectic tumor-bearing mice [75]. This study identified the myocilin gene Myoc as a transcriptional target of MEF2 and showed that loss of myocilin mediates tumor-associated muscle wasting while caMEF2 prevents concomitant Myoc downregulation. The role of myocilin in the regulation of muscle size is supported by the muscle hypertrophy phenotype observed in transgenic mice overexpressing myocilin [76]. However, it is not clear how myocilin affects muscle size, one possibility being that myocilin binds to and stabilizes the DGC [76]. The DGC may be involved in the transmission of mechanical signals to pro-hypertrophic pathways such as Akt-mTOR, thus promoting protein synthesis and preventing protein degradation, as shown by a study on cancer cachexia in mice [37].
A simplified scheme of the regulation of MEF2 transcriptional activity by inhibitory factors such as HDAC4, NCoR1 and MRF4 is shown in Fig. 4. Interestingly, a similar scheme holds true also for cardiac muscle, with NCoR1 and class II HDACs repressing MEF2 activity and MEF2-dependent cardiac hypertrophy [see 77]. However, MRF4 is exclusively expressed in skeletal muscle, thus could be a specific therapeutic target to promote skeletal muscle hypertrophy. Future studies should clarify the role of alternative splicing of MEF2 factors [78], leading to MEF2 variants which have different effects on cardiac hypertrophy [79] and can also affect skeletal muscle hypertrophy [80]. It should be stressed that what is found in cardiac hypertrophy is not necessarily valid for skeletal muscle hypertrophy. For example, cardiac hypertrophy is controlled by the Ca2 +-calmodulin dependent phosphatase calcineurin and Ca2 +-calmodulin dependent kinase (CaMKII), acting via nuclear factor of activated T cell (NFAT) and MEF2 transcription factors, respectively [81, 82]. However, pharmacological inhibitors of calcineurin activity gave variable results in skeletal muscle according to the different experimental setting [83], and genetic loss of calcineurin did not block overload-induced skeletal muscle hypertrophy [84]. On the other hand, the role of CaMK-dependent pathways in skeletal muscle hypertrophy has not been determined by specific loss-of-function approaches.
Serum response factor (SRF)
The transcription factor SRF belongs, like MEF2, to the MADS (MCM1, Agamous, Deficiens, and SRF) box superfamily of transcription factors. SRF is essential for muscle growth, as muscle-specific deletion of mouse Srf causes severe skeletal muscle hypoplasia leading to perinatal lethality [85], and for muscle maintenance in adult stages, as loss of Srf induced in adult mice leads to progressive loss of muscle mass and a sarcopenia-like phenotype [86]. SRF is known to activate transcription of sarcomeric actins and actin-binding proteins by associating with myocardin-related transcription factors (MRTFs) and a scheme of the pathways regulating the MRTF-SRF axis is starting to emerge (Fig. 5). Muscles expressing a dominant negative form of MRTF-A display a phenotype similar to that of Srf knockout [81]. Double knockout of MRTF-A and B causes an even more severe phenotype with dysregulation of contractile protein expression [87].
Fig. 5
Signaling pathways regulating the activity of the transcription factor SRF (serum response factor). SRF is activated by high intensity resistance exercise via nuclear translocation of myocardin related transcription factor B (MRTF-B), which is induced by ERK-dependent phopshorylation on serine 66, and by actin polymerization induced by STARS and RhoA, thus relieving the G-actin inhibitory effect on MRTF. SRF activation also requires chromatin remodeling which is induced by histone 3 phosphorylation on serine 10 (H3S10). H3S10 phosphorylation is mediated by mitogen- and stress-activated kinases (MSK1/2), which are in turn activated by p38 MAPK. (Modified from [94]).
![Signaling pathways regulating the activity of the transcription factor SRF (serum response factor). SRF is activated by high intensity resistance exercise via nuclear translocation of myocardin related transcription factor B (MRTF-B), which is induced by ERK-dependent phopshorylation on serine 66, and by actin polymerization induced by STARS and RhoA, thus relieving the G-actin inhibitory effect on MRTF. SRF activation also requires chromatin remodeling which is induced by histone 3 phosphorylation on serine 10 (H3S10). H3S10 phosphorylation is mediated by mitogen- and stress-activated kinases (MSK1/2), which are in turn activated by p38 MAPK. (Modified from [94]).](https://ip.ios.semcs.net:443/media/jnd/2021/8-2/jnd-8-2-jnd200568/jnd-8-jnd200568-g005.jpg)
The MRTF-SRF axis is also regulated by STARS (striated muscle activator of Rho signaling), coded by ABRA (Actin-binding Rho-activating protein) gene. STARS, acting together with the small GTPase RhoA, binds G-actin promoting actin polymerization and thus relieving the G-actin inhibitory effect on MRTF, allowing the nuclear import of MRTFs and stimulation of SRF activity [88, 89, 90]. Interestingly, a study on cardiac muscle has shown that STARS is controlled by MEF2c in response to mechanical stress, suggesting that STARS couples MEF2 and SRF signaling [91]. In human skeletal muscle, resistance training-induced muscle hypertrophy is accompanied by upregulation of STARS, MRTFs and SRF [92]. In mouse skeletal muscle, hypertrophy induced by synergist elimination is blunted by Srf gene knockout induced specifically in the adult [93], as a result of factors released from myofibers and acting on associated satellite cells (see below).
A recent study has shown that a specific MRTF-B phosphorylation site on serine 66, detected by a phosphoproteomics screen and similar to an ERK-dependent MRTF-A phosphorylation in another cell system, is induced in human muscle by high intensity resistance exercise and is required for MRTF-B nuclear translocation and for transcription of SRF target genes [94] (Fig. 5). The same study revealed that exercise also induces chromatin remodeling at SRF target gene loci in myonuclei through histone 3 phosphorylation on serine 10 (H3S10), which was prevented by knockdown of the mitogen- and stress-activated kinases (MSK) 1/2, thus validating in skeletal muscle a p38 MAPK-dependent and MRTF-dependent SRF activation pathway previously described in other tissues. Interestingly, this pathway was found to control protein synthesis, thus providing a foundation for the muscle growth function of SRF.
PGC-1α4
PGC-1α4 is a splice variant of peroxisome pro-liferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α), coded by the PPARGC1A gene, a transcriptional coactivator with multiple roles in different tissues, including skeletal muscle [95]. PGC-1α factors act by binding to different transcriptional factors, including nuclear receptors, such as peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), and thyroid hormone receptors (TRs). Whereas the PGC-1α1 variant is induced by endurance training and affects mitochondrial biogenesis, PGC-1α4 was reported to be increased by resistance exercise and to induce muscle hypertrophy, possibly through increased IGF1 expression and reduced myostatin levels [96]. However, there are conflicting results about the induction of PGC-1α4 by resistance training in humans [97, 98] and in experimental models. PGC-1α4 was increased during the reloading/hypertrophic phase using the hindlimb suspension–reloading model [96], however, chronic overload induced by synergist ablation was unaffected by muscle-specific knockout of the PGC1α gene [99]. Interpretation of these results is difficult because appropriate mouse genetic models of PGC-1α isoform-specific gain- and loss-of-function are lacking [100]. As reported above, deletion of the PGC-1α4 target gene, GPR56, impairs muscle hypertrophy by reducing activation of mTORC1 signaling [38]. Muscle hypertrophy induced by overexpression of the mitochondrial calcium uniporter (MCU), leading to increased mitochondrial calcium uptake, was reported to be accompanied by a marked increase in PGC-1α4 expression, while viral-mediated MCU shRNA knockdown in adult muscle caused muscle atrophy [101]. However, a recent study using a genetic inducible model showed that ablation of MCU at fetal, postnatal, and adult stages had no significant impact on overall muscle growth [102]. An open issue with PGC-1α4 is that the transcription factor(s) mediating its pro-hypertrophic function have not been identified.
YAP/TAZ
The major components of the Hippo pathway, an evolutionary conserved pathway that controls tissue growth, are the kinases MST1/2 and LATS1/2, the transcriptional coactivators YAP and its analog TAZ and the transcription factors TEAD1-4. YAP is sequestered in the cytoplasm by LATS1/2-mediated phosphorylation, or by alternative pathways, while inactivation of the kinase allows the translocation of YAP into the nucleus and stimulation of TEAD transcriptional activity that promotes tissue growth. The identification of YAP/TAZ as crucial mediators of mechanotransduction in different cell types [103] has stimulated the study of these transcriptional coregulators in striated muscle. A role of YAP in promoting muscle hypertrophy was supported by the finding that YAP overexpression causes muscle hypertrophy and increased protein synthesis while YAP knockdown causes muscle atrophy by reducing protein synthesis [104, 105]. The physiological regulation of the pathways leading to YAP activation remains largely unknown. Interestingly, Rho GTPase activity, which is known to mediate mechanotransduction in muscle through different pathways, including GPR56, integrin and STARS (see above), is required to induce YAP nuclear localization and transcriptional activity in cultured cells [103]. YAP function may also be affected by the DGC as suggested by the finding that YAP directly binds dystroglycan, a component of the DGC, leading to inhibition of cardiomyocyte proliferation [106]. It was shown that YAP signaling is constitutively active in mdx skeletal muscle, which is unresponsive to loading, possibly due to aberrant increase in cytoskeletal and extracellular matrix stiffness in these muscles [107]. A recent study has shown that phosphatidic acid, generated by phospholipase D, suppresses YAP phosphorylation thereby inducing YAP transcriptional activity in cultured cells [108]. Phosphatidic acid has been implicated in the regulation of muscle size through mTOR signaling [35, see above]. However, the role of mTOR in mediating YAP activation remains to be established, as gain- and loss-of-function studies suggest that YAP positively regulate skeletal muscle size independently of mTOR signaling [104, 105].
SATELLITE CELL ACTIVATION AND MUSCLE HYPERTROPHY
Muscle growth during development and regeneration requires the obligatory participation of satellite cells (SCs), specialized stem cells located under the basal lamina of the muscle fibers, which undergo proliferation and fusion with the associated muscle fiber. On the other hand, the contribution of SCs during muscle hypertrophy in adult muscle appears to vary in different models of hypertrophy. SC proliferation and fusion is induced at early stages after synergist ablation [109, 110, 111], thus leading to an increase in the number of myonuclei [108], and can be induced by exercise in both human and experimental models [see 113, 114, 115]. In contrast, muscle hypertrophy induced by myostatin inactivation or Akt activation is not accompanied by SC activation [116, 117]. It is likely that the activation of SCs in certain muscle hypertrophy models, such as strenuous exercise in humans or overload hypertrophy in animals, is induced by muscle damage, thus is essentially similar to muscle regeneration or repair. A discussion of the factors and pathways that promote SC activation under these conditions is outside the scope of this review.
Different approaches were used to establish whether SC activation is required for mouse muscle hypertrophy. Hypertrophy induced by synergist ablation was unaffected by diphteria toxin-mediated elimination of SCs [118], however, a subsequent study using the same approach showed that hypertrophy does not occur in the absence of SCs [119], in agreement with previous studies based on muscle irradiation [120]. The notion that muscle hypertrophy requires SC fusion and myonuclear accretion is supported by the effect of SC-specific knockout (scKO) of myomaker, a muscle-specific membrane protein necessary for fusion of SCs. Both overload hypertrophy induced by synergist ablation and muscle hypertrophy induced by high-intensity interval training in the treadmill were abrogated by myomaker scKO induced by tamoxifen treatment in adult mouse muscles [121, 122].
An interesting though poorly explored area of investigation concerns the cross-talk between myofibers and SCs during muscle hypertrophy. As reported above, deletion of Srf induced in adult mice within myofibers but not in SCs was found to impair overload hypertrophy [93]. SC proliferation and fusion was also impaired in this model as SRF activation within myofibers leads to paracrine release of interleukin 6 (IL-6), affecting SC proliferation, and interleukin 4 (IL-4), which is required for SC fusion [93]. On the other hand, it remains to be established whether and how SC incorporation into hypertrophying myofibers affects the molecular mechanisms of protein synthesis leading to the increase in myofiber size. Ongoing single-nucleus RNA-seq (snRNA-seq) studies, which allow to identify distinct myonuclear populations in skeletal muscle fibers [123, 124, 125] have revealed the existence of specific myonuclear subsets during early postnatal development, when SCs are actively incorporated into muscle fibers [119]. It will be of interest to determine whether “immature” myonuclei, corresponding to newly fused SC, are also detected during muscle hypertrophy.
PERSPECTIVES AND OPEN ISSUES
In this short review we selected only major factors and pathways of muscle hypertrophy from the huge amount of information collected in a large number of human and animal studies. However, other factors and pathways potentially important for muscle hypertrophy should be explored and a number of open questions should be answered in future studies. Omics approaches, including the identification of genes whose gain or loss-of-function affects muscle mass [126], will further contribute to this research. Some open issues and new fields to be explored are briefly discussed below.
Non-coding RNAs
In addition to coding genes, muscle hypertrophy can also be modulated by non-coding RNAs, including microRNAs and long non-coding RNAs (lncRNAs), which have been mainly investigated in muscle atrophy. A number of lncRNAs were found to increase in skeletal muscle in different hypertrophy models, such as mechanical overload and deficiency of the myostatin gene [127], however in most cases their role has not been validated by gain- and loss-of-function experiments. A well characterized lncRNA involved in muscle hypertrophy is lnc-mg, which causes hypertrophy when overexpressed in vivo, whereas conditional knockout of lnc-mg in skeletal muscle results in muscle atrophy [128]. Lnc-mg acts as a competing endogenous RNAs (ceRNA) by acting as molecular sponge for a microRNA, miR-125b, which in turn controls protein abundance of Igf2. Another example is the lnc-RNA Chronos, which acts as an inhibitor of muscle growth as shown by the finding that in vivo knockdown causes muscle hypertrophy [129].
The callipyge phenotype in sheep, characterized by marked hypertrophy of hind limb and loin muscles, is also of interest in this respect. This phenotype is due to a point mutation in an imprinted locus close to the DLK1 (Delta-Like homolog 1), which is probably responsible for muscle hypertrophy because transgenic mice over-expressing Dlk1 have increased muscle mass and myofiber diameter [130]. Interestingly, the expression of DLK1 is controlled by a microRNA gene cluster (miR-379/miR-544) located in the same locus [131]. This microRNA gene cluster was also found to be markedly upregulated during the rapid muscle growth that occurs in mice around the second week of postnatal life [132].
Muscle pathology
Muscle hypertrophy is relevant for muscle pathology not only as a means to contrast muscle atrophy, but potentially also to reduce muscle pathology itself. For example, muscle hypertrophy induced by muscle-specific Akt activation was found to promote sarcolemma stability and prevent the force drop induced by eccentric contractions in dystrophin-deficient mouse skeletal muscle [133, 134]. However, this may be due to the upregulation of the utrophin-glycoprotein complex, as well as proteins associated with Z-disks and costameres, and proteins with anti-oxidant or chaperone function, rather than to muscle hypertrophy per se. On the other hand, loss of Akt-mTORC1 signaling in different pathologies can contribute to muscle wasting per se, but it can also impair other aspects important for restoring muscle mass, like fiber innervation [135, 136].
Muscle physiology
Sport science provides a variety of training protocols optimized to promote muscle growth, most of which have not been investigated at the molecular level. For example, there is evidence that blood flow restriction, when combined with exercise, even low-load exercise, leads to a significant increase in the degree of hypertrophy compared to exercise alone, however the mechanism of this effect is not clear. The study of the interindividual variability in the response to the same training protocol (responders vs non-responders, see Fig. 3) may provide useful information on the mechanism of muscle hypertrophy: in particular, omics techniques are likely to reveal novel clues through an unbiased approach. Age-dependent differences in the development of muscle hypertrophy are especially important in this respect given the importance in promoting muscle growth and prevent sarcopenia in the increasing aging population.
Muscle size and muscle force
Finally, a crucial question is whether the increase in muscle size is reflected in an increase in muscle force. Several studies indicate that the two parameters are not always correlated in different models of muscle hypertrophy. For example, a proportional increase in muscle size and force is not always seen following resistance exercise [see 137, 138], and extreme hypertrophy without corresponding increase in strength has been reported in body builders [139, 140, 141]. The same is true for animal models: for example, a constitutively actively Akt transgene causes a parallel increase in muscle size and force [117], whereas myostatin mutation induces muscle hypertrophy without a corresponding increase in force generation [142]. It will be important to investigate the molecular underpinnings of the different models of muscle hypertrophy to identify the pro-hypertrophic factors and pathways efficient in promoting muscle strength, because an increase in muscle size not accompanied by an increase in force is functionally meaningless.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was funded by ASI, MARS-PRE Project, n. DC-VUM-2017-006.
REFERENCES
[1] | Haun CT , Vann CG , Roberts BM , Vigotsky AD , Schoenfeld BJ , Roberts MD . A critical evaluation of the biological construct skeletal muscle hypertrophy: size matters but so does the measurement. Front Physiol. (2019) ;10: :247. |
[2] | Blaauw B , Schiaffino S , Reggiani C . Mechanisms modulating skeletal muscle phenotype. Compr Physiol. (2013) ;3: (4):1645–87. |
[3] | Schiaffino S , Dyar KA , Ciciliot S , Blaauw B , Sandri M . Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. (2013) ;280: (17):4294–314. |
[4] | Spangenburg EE , Le Roith D , Ward CW , Bodine SC . A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J Physiol. (2008) ;586: (1):283–91. |
[5] | Lee SJ , McPherron AC . Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. (2001) ;98: (16):9306–11. |
[6] | Girgenrath S , Song K , Whittemore LA . Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve. (2005) ;31: (1):34–40. |
[7] | Gangopadhyay SS . Systemic administration of follistatin288 increases muscle mass and reduces fat accumulation in mice. Sci Rep. (2013) ;3: :2441. |
[8] | Latres E , Mastaitis J , Fury W , Miloscio L , Trejos J , Pangilinan J , et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. (2017) ;8: :15153. |
[9] | Sartori R , Milan G , Patron M , Mammucari C , Blaauw B , Abraham R , et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. (2009) ;296: (6):C1248–57. |
[10] | Winbanks CE , Weeks KL , Thomson RE , Sepulveda PV , Beyer C , Qian H , et al. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. (2012) ;197: (7):997–1008. |
[11] | Han X , Møller LLV , De Groote E , Bojsen-Møller KN , Davey J , Henríquez-Olguin C , et al. Mechanisms involved in follistatin-induced hypertrophy and increased insulin action in skeletal muscle. J Cachexia Sarcopenia Muscle. (2019) ;10: (6):1241–57. |
[12] | Sartori R , Schirwis E , Blaauw B , Bortolanza S , Zhao J , Enzo E , et al. BMP signaling controls muscle mass. Nat Genet. (2013) ;45: (11):1309–18. |
[13] | Winbanks CE , Chen JL , Qian H , Liu Y , Bernardo BC , Beyer C , et al. The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J Cell Biol. (2013) ;203: (2):345–57. |
[14] | Aizawa K , Iemitsu M , Maeda S , Otsuki T , Sato K , Ushida T , et al. Acute exercise activates local bioactive androgen metabolism in skeletal muscle. Steroids. (2010) ;75: (3):219–23. |
[15] | White JP , Gao S , Puppa MJ , Sato S , Welle SL , Carson JA . Testosterone regulation of Akt/ mTORC1/FoxO3a signaling in skeletal muscle. Mol Cell Endocrinol. (2013) ;365: (2):174–86. |
[16] | Basualto-Alarcón C , Jorquera G , Altamirano F , Jaimovich E , Estrada M . Testosterone signals through mTOR and androgen receptor to induce muscle hypertrophy. Med Sci Sports Exerc. (2013) ;45: (9):1712–20. |
[17] | Englund DA , Peck BD , Murach KA , Neal AC , Caldwell HA , McCarthy JJ , et al. Resident muscle stem cells are not required for testosterone-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. (2019) ;317: (4):C719–C724. |
[18] | Goncalves DA , Silveira WA , Manfredi LH , Graca FA , Armani A , Bertaggia E , et al. Insulin/IGF1 Signalling Mediates the Effects of β 2 -Adrenergic Agonist on Muscle Proteostasis and Growth. J Cachexia Sarcopenia Muscle. (2019) ;10: (2):455–75. |
[19] | Chia LY , Evans BA , Mukaida S , Bengtsson T , Hutchinson DS , Sato M . Adrenoceptor regulation of the mechanistic target of rapamycin in muscle and adipose tissue. Br J Pharmacol. (2019) ;176: (14):2433–48. |
[20] | Berdeaux R , Hutchins C . Anabolic and Pro-metabolic Functions of CREB-CRTC in Skeletal Muscle: Advantages and Obstacles for Type 2 Diabetes and Cancer Cachexia. Front Endocrinol (Lausanne). (2019) ;10: :535. |
[21] | Smith JS , Rajagopal S . The beta-arrestins: multifunctional regulators of G protein-coupled receptors. J Biol Chem. (2016) ;291: (17):8969–77. |
[22] | Woodall BP , Woodall MC , Luongo TS , Grisanti LA , Tilley DG , Elrod JW , et al. Skeletal muscle-specific G protein-coupled receptor kinase 2 ablation alters isolated skeletal muscle mechanics and enhances clenbuterol-stimulated hypertrophy. J Biol Chem. (2016) ;291: (42):21913–24. |
[23] | Kim J , Grotegut CA , Wisler JW , Li T , Mao L , Chen M , et al. β-arrestin 1 regulates β2-adrenergic receptor-mediated skeletal muscle hypertrophy and contractility. Skelet Muscle. (2018) ;8: (1):39. |
[24] | Mera P , Laue K , Wei J , Berger JM , Karsenty G . Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol Metab. (2016) ;5: (10):1042–7. |
[25] | Chowdhury S , Schulz L , Palmisano B , Singh P , Berger JM , Yadav VK , et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J Clin Invest. (2020) ;130: (6):2888–902. |
[26] | Moriishi T , Ozasa R , Ishimoto T , Nakano T , Hasegawa T , Miyazaki T , et al. Osteocalcin is necessary for the alignment of apatite crystallites, but not glucose metabolism, testosterone synthesis, or muscle mass. PLoS Genet. (2020) ;16: (5):e1008586. |
[27] | Diegel CR , Hann S , Ayturk UM , Hu JCW , Lim KE , Droscha CJ , et al. An Osteocalcin-Deficient Mouse Strain Without Endocrine Abnormalities. PLoS Genet. (2020) ;16: (5):e1008361. |
[28] | Wackerhage H , Schoenfeld BJ , Hamilton DL , Lehti M , Hulmi JJ . Stimuli and sensors that initiate skeletal muscle hypertrophy following resistance exercise. J Appl Physiol. (2019) ;126: (1):30–43. |
[29] | Durieux AC , Desplanches D , Freyssenet D , Flück M . Mechanotransduction in striated muscle via focal adhesion kinase. Biochem Soc Trans. (2007) ;35: (Pt 5):1312–3. |
[30] | Boppart MD , Mahmassani ZS . Integrin signaling: linking mechanical stimulation to skeletal muscle hypertrophy. Am J Physiol Cell Physiol. (2019) ;317: (4):C629–C641. |
[31] | Vitadello M , Sorge M , Percivalle E , Germinario E , Danieli-Betto D , Turco E , et al. Loss of melusin is a novel, neuronal NO synthase/FoxO3-independent master switch of unloading-induced muscle atrophy. J Cachexia Sarcopenia Muscle. (2020) ;11: (3):802–19. |
[32] | Joanne P , Hourdé C , Ochala J , Caudéran Y , Medja F , Vignaud A , et al. Impaired adaptive response to mechanical overloading in dystrophic skeletal muscle. PLoS One. (2012) ;7: (4):e35346. |
[33] | Ito N , Ruegg UT , Kudo A , Miyagoe-Suzuki Y , Takeda S . Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med. (2013) ;19: (1):101–6. |
[34] | Abramovici H , Hogan AB , Obagi C , Topham MK , Gee SH . Diacylglycerol kinase-zeta localization in skeletal muscle is regulated by phosphorylation and interaction with syntrophins. Mol Biol Cell. (2003) ;14: (11):4499–511. |
[35] | You JS , Lincoln HC , Kim CR , Frey JW , Goodman CA , Zhong XP , et al. The role of diacylglycerol kinase ζ and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J Biol Chem. (2014) ;289: (3):1551–63. |
[36] | Eid Mutlak Y , Aweida D , Volodin A , Ayalon B , Dahan N , Parnis A , et al. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat Commun. (2020) ;11: (1):1381. |
[37] | Acharyya S , Butchbach ME , Sahenk Z , Wang H , Saji M , Carathers M , et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. (2005) ;8: (5):421–32. |
[38] | White JP , Wrann CD , Rao RR , Nair SK , Jedrychowski MP , You JS , et al. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc Natl Acad Sci U S A. (2014) ;111: (44):15756–61. |
[39] | Braun T , Gautel M . Transcriptional mechanisms regulating skeletal muscle differentiation, growth and home-ostasis. Nat Rev Mol Cell Biol. (2011) ;12: (6):349–61. |
[40] | Boateng SY , Senyo SE , Qi L , Goldspink PH , Russell B . Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol. (2009) ;47: (4):426–35. |
[41] | Vafiadaki E , Arvanitis DA , Sanoudou D . Muscle LIM Prote Master Regulator of Cardiac and Skeletal Muscle Functions. Gene. (2015) ;566: (1):1–7. |
[42] | Liang Y , Bradford WH , Zhang J , Sheikh F . Four and a half LIM domain protein signaling and cardiomyopathy. Biophys Rev. (2018) ;10: (4):1073–85. |
[43] | Lange S , Xiang F , Yakovenko A , Vihola A , Hackman P , Rostkova E , et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. (2005) ;308: (5728)1599–603. |
[44] | van der Pijl R , Strom J , Conijn S , Lindqvist J , Labeit S , Granzier H , et al. Titin-based mechanosensing modulates muscle hypertrophy. J Cachexia Sarcopenia Muscle. (2018) ;9: (5):947–61. |
[45] | Goldberg AL . Protein Synthesis During Work-Induced Growth of Skeletal Muscle. J Cell Biol. (1968) ;36: (3):653–8. |
[46] | Kumar V , Atherton P , Smith K , Rennie MJ . Human muscle protein synthesis and breakdown during and after exercise. J Appl Physiol. (2009) ;106: (6):2026–39. |
[47] | Schiaffino S , Mammucari C . Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle. (2011) ;1: (1):4. |
[48] | Murgia M , Serrano AL , Calabria E , Pallafacchina G , Lomo T , Schiaffino S . Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat Cell Biol. (2000) ;2: (3):142–7. |
[49] | Bodine SC , Stitt TN , Gonzalez M , Kline WO , Stover GL , Bauerlein R , et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. (2001) ;3: (11):1014–9. |
[50] | Pallafacchina G , Calabria E , Serrano AL , Kalhovde JM , Schiaffino S . A Protein Kinase B-dependent and Rapamycin-Sensitive Pathway Controls Skeletal Muscle Growth but Not Fiber Type Specification. Proc Natl Acad Sci U S A. (2002) ;99: (14):9213–8. |
[51] | Liu GY , Sabatini DM . mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. (2020) ;21: (4):183–203. |
[52] | Fonseca BD , Zakaria C , Jia JJ , Graber TE , Svitkin Y , Tahmasebi S , et al. La-related Protein 1 (LARP1) Represses Terminal Oligopyrimidine (TOP) mRNA Translation Downstream of mTOR Complex 1 (mTORC1). J Biol Chem. (2015) ;290: (26):15996–16020. |
[53] | van Riggelen J , Yetil A , Felsher DW . MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. (2010) ;10: (4):301–9. |
[54] | Michels AA , Robitaille AM , Buczynski-Ruchonnet D , Hodroj W , Reina JH , Hall MN , et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. (2010) ;30: (15):3749–57. |
[55] | Shor B , Wu J , Shakey Q , Toral-Barza L , Shi C , Follettie M , Yu K . Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III- dependent transcription in cancer cells. J Biol Chem. (2010) ;285: (20):15380–92. |
[56] | Mayer C , Zhao J , Yuan X , Grummt I . mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev.. (2004) ;18: (4):423–34. |
[57] | Hannan KM , Brandenburger Y , Jenkins A , Sharkey K , Cavanaugh A , Rothblum L , et al. mTOR- dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. (2003) ;23: (23):8862–77. |
[58] | Ben-Sahra I , Howell JJ , Asara JM , Manning BD . Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science. (2013) ;339: (6125):1323–8. |
[59] | Robitaille AM , Christen S , Shimobayashi M , Cornu M , Fava LL , Moes S , et al. Quantitative Phosphoproteomics Reveal mTORC1 Activates De Novo Pyrimidine Synthesis. Science. (2013) ;339: (6125):1320–3. |
[60] | von Walden F . Ribosome Biogenesis in Skeletal Muscle: Coordination of Transcription and Translation. J Appl Physiol (1985). (2019) ;127: (2):591–8. |
[61] | Figueiredo VC , McCarthy JJ . Regulation of Ribosome Biogenesis in Skeletal Muscle Hypertrophy. Physiology (Bethesda). (2019) ;34: (1):30–42. |
[62] | Marabita M , Baraldo M , Solagna F , Ceelen JJM , Sartori R , Nolte H , et al. S6K1 Is Required for Increasing Skeletal Muscle Force During Hypertrophy. Cell Rep. (2016) ;17: (2):501–13. |
[63] | Stec MJ , Kelly NA , Many GM , Windham ST , Tuggle SC , Bamman MM . Ribosome biogenesis may augment resistance training-induced myofiber hypertrophy and is required for myotube growth in vitro. Am J Physiol Endocrinol Metab. (2016) ;310: (8):E652–61. |
[64] | Hammarström D , Øfsteng S , Koll L , Hanestadhaugen M , Hollan I , Apró W , et al. Benefits of Higher Resistance-Training Volume Are Related to Ribosome Biogenesis. J Physiol. (2020) ;598: (3):543–65. |
[65] | Potthoff MJ , Olson EN . MEF a central regulator of diverse developmental programs. Development. (2007) ;134: (23):4131–40. |
[66] | Liu N , Nelson BR , Bezprozvannaya S , Shelton JM , Richardson JA , Bassel-Duby R , et al. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci U S A. (2014) ;111: (11):4109–14. |
[67] | Moretti I , Ciciliot S , Dyar KA , Abraham R , Murgia M , Agatea L , et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat Commun. (2016) ;7: :12397. |
[68] | Cohen TJ , Barrientos T , Hartman ZC , Garvey SM , Cox GA , Yao TP . The deacetylase HDAC4 controls myocyte enhancing factor-2-dependent structural gene expression in response to neural activity. FASEB J. (2009) ;23: (1):99–106. |
[69] | Moresi V , Williams AH , Meadows E , Flynn JM , Potthoff MJ , McAnally J , et al. Myogenin and Class II HDACs Control Neurogenic Muscle Atrophy by Inducing E3 Ubiquitin Ligases. Cell. (2010) ;143: (1):35–45. |
[70] | Yamamoto H , Williams EG , Mouchiroud L , Cantó C , Fan W , Downes M , et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. (2011) ;147: (4):827–39. |
[71] | Weis J , Kaussen M , Calvo S , Buonanno A . Denervation induces a rapid nuclear accumulation of MRF4 in mature myofibers. Dev Dyn. (2000) ;218: (3):438–51. |
[72] | Cohen TJ , Waddell DS , Barrientos T , Lu Z , Feng G , Cox GA , et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J Biol Chem. (2007) ;282: (46):33752–9. |
[73] | Rullman E , Fernandez-Gonzalo R , Mekjavić IB , Gustafsson T , Eiken O . MEF2 as upstream regulator of the transcriptome signature in human skeletal muscle during unloading. Am J Physiol Regul Integr Comp Physiol. (2018) ;315: (4):R799–R809. |
[74] | Nath SR , Lieberman ML , Yu Z , Marchioretti C , Jones ST , Danby ECE , et al. MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease. Acta Neuropathol. (2020) ;140: (1):63–80. |
[75] | Judge SM , Deyhle MR , Neyroud D , Nosacka RL , D’Lugos AC , Cameron ME , et al. MEF2c-dependent downregulation of myocilin mediates cancer-induced muscle wasting and associates with cachexia in patients with cancer. Cancer Res. (2020) ;80: (9):1861–74. |
[76] | Joe MK , Kee C , Tomarev SI . Myocilin interacts with syntrophins and is member of dystrophin-associated protein complex. J Biol Chem. (2012) ;287: (16):13216–27. |
[77] | Li C , Sun XN , Chen BY , Zeng MR , Du LJ , Liu T , et al. Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol Med. (2019) ;11: (11):e9127. |
[78] | Zhu B , Ramachandran B , Gulick T . Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J Biol Chem. (2005) ;280: (31):28749–60. |
[79] | Pereira AHM , Cardoso AC , Consonni SR , Oliveira RR , Saito A , Vaggione MLB , et al. MEF2C repressor variant deregulation leads to cell cycle re-entry and development of heart failure. EBioMedicine. (2020) ;51: :102571. |
[80] | Baruffaldi F , Montarras D , Basile V , De Feo L , Badodi S , Ganassi M , et al. Dynamic phosphorylation of the myocyte enhancer factor 2Ca1 splice variant promotes skeletal muscle regeneration and hypertrophy. Stem Cells. (2017) ;35: (3):725–38. |
[81] | Molkentin JD , Lu JR , Antos CL , Markham B , Richardson J , Robbins J , et al. A calcineurin- dependent transcriptional pathway for cardiac hypertrophy. Cell. (1998) ;93: (2):215–28. |
[82] | Passier R , Zeng H , Frey N , Naya FJ , Nicol RL , McKinsey TA , et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo . J Clin Invest. (2000) ;105: (10):1395–406. |
[83] | Serrano AL , Murgia M , Pallafacchina G , Calabria E , Coniglio P , Lømo T , Schiaffino S . Calcineurin controls nerve activity-dependent specification of slow skeletal muscle fibers but not muscle growth. Proc Natl Acad Sci U S A. (2001) ;98: (23):13108–13. |
[84] | Parsons SA , Millay DP , Wilkins BJ , Bueno OF , Tsika GL , Neilson JR , et al. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. (2004) ;279: (25):26192–200. |
[85] | Li S , Czubryt MP , McAnally J , Bassel-Duby R , Richardson JA , Wiebel FF , et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci U S A. (2005) ;102: (4):1082–7. |
[86] | Lahoute C , Sotiropoulos A , Favier M , Guillet-Deniau I , Charvet C , Ferry A , et al. Premature aging in skeletal muscle lacking serum response factor. PLoS One. (2008) ;3: (12):e3910. |
[87] | Cenik BK , Liu N , Chen B , Bezprozvannaya S , Olson EN , Bassel-Duby R . Myocardin-related transcription factors are required for skeletal muscle development. Development. (2016) ;143: (15):2853–61. |
[88] | Arai A , Spencer JA , Olson EN . STARS, a striated muscle activator of Rho signaling and serum response factor-dependent transcription. J Biol Chem. (2002) ;277: (27):24453–9. |
[89] | Kuwahara K , Barrientos T , Pipes GC , Li S , Olson EN . Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol. (2005) ;25: (8):3173–81. |
[90] | Olson EN , Nordheim A . Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. (2010) ;11: (5):353–65. |
[91] | Kuwahara K , Teg Pipes GC , McAnally J , Richardson JA , Hill JA , et al. Modulation of adverse cardiac remodeling by STARS, a mediator of MEF2 signaling and SRF activity. J Clin Invest. (2007) ;117: (5):1324–34. |
[92] | Lamon S , Wallace MA , Léger B , Russell AP . Regulation of STARS and its downstream targets suggest a novel pathway involved in human skeletal muscle hypertrophy and atrophy. J Physiol. (2009) ;587: (Pt 8):1795–803. |
[93] | Guerci A , Lahoute C , Hébrard S , Collard L , Graindorge D , Favier M , et al. Srf-dependent paracrine signals produced by myofibers control satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. (2012) ;15: (1):25–37. |
[94] | Solagna F , Nogara L , Dyar KA , Greulich F , Mir AA , Türk C , et al. Exercise-dependent increases in protein synthesis are accompanied by chromatin modifications and increased MRTF-SRF signalling. Acta Physiol (Oxf). 2020;e13496. |
[95] | Correia JC , Ferreira DM , Ruas JL . Intercellular: local and systemic actions of skeletal muscle PGC-1s. Trends Endocrinol Metab. (2015) ;26: (6):305–14. |
[96] | Ruas JL , White JP , Rao RR , Kleiner S , Brannan KT , Harrison BC , et al. A PGC-1alpha Isoform Induced by Resistance Training Regulates Skeletal Muscle Hypertrophy. Cell. (2012) ;151: (6):1319–31. |
[97] | Nader GA , von Walden F , Liu C , Lindvall J , Gutmann L , Pistilli EE , et al. Resistance exercise training modulates acute gene expression during human skeletal muscle hypertrophy. J Appl Physiol (1985). (2014) ;116: (6):693–702. |
[98] | Lundberg TR , Fernandez-Gonzalo R , Norrbom J , Fischer H , Tesch PA , Gustafsson T . Truncated splice variant PGC-1α4 is not associated with exercise-induced human muscle hypertrophy. Acta Physiol (Oxf). (2014) ;212: (2):142–51. |
[99] | Pérez-Schindler J , Summermatter S , Santos G , Zorzato F , Handschin C . The transcriptional coactivator PGC-1α is dispensable for chronic overload-induced skeletal muscle hypertrophy and metabolic remodeling. Proc Natl Acad Sci U S A. (2013) ;110: (50):20314–19. |
[100] | Martínez-Redondo V , Pettersson AT , Ruas JL . The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia. (2015) ;58: (9):1969–77. |
[101] | Mammucari C , Gherardi G , Zamparo I , Raffaello A , Boncompagni S , Chemello F , et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. (2015) ;10: (8):1269–79. |
[102] | Kwong JQ , Huo J , Bround MJ , Boyer JG , Schwanekamp JA , Ghazal N , et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. (2018) ;3: (22):e121689. |
[103] | Dupont S , Morsut L , Aragona M , Enzo E , Giulitti S , Cordenonsi M , et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) ;474: (7350):179–83. |
[104] | Goodman CA , Dietz JM , Jacobs BL , McNally RM , You JS , Hornberger TA . Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. (2015) ;589: (13):1491–7. |
[105] | Watt KI , Turner BJ , Hagg A , Zhang X , Davey JR , Qian H , et al. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat Commun. (2015) ;6: :6048. |
[106] | Morikawa Y , Heallen T , Leach J , Xiao Y , Martin JF . Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature. (2017) ;547: (7662):227–31. |
[107] | Iyer SR , Shah SB , Ward CW , Stains JP , Spangenburg EE , Folker ES , et al . Differential YAP nuclear signaling in healthy and dystrophic skeletal muscle. Am J Physiol Cell Physiol. (2019) ;317: (1):C48–C57. |
[108] | Han H , Qi R , Zhou JJ , Ta AP , Yang B , Nakaoka HJ , et al. Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction. Mol Cell. (2018) ;72: (2):328–40. |
[109] | Schiaffino S , Bormioli SP , Aloisi M . Cell proliferation in rat skeletal muscle during early stages of compensatory hypertrophy. Virchows Arch B Cell Pathol Incl Mol Pathol. (1972) ;11: (3):268–273. |
[110] | Schiaffino S , Bormioli SP , Aloisi M . The fate of newly formed satellite cells during compensatory muscle hypertrophy. Virchows Arch B Cell Pathol Incl Mol Pathol. (1976) ;21: (2):113–118. |
[111] | Snow MH . Satellite cell response in rat soleus muscle undergoing hypertrophy due to surgical ablation of synergists. Anat Rec. (1990) ;227: (4):437–46. |
[112] | Bruusgaard JC , Johansen IB , Egner IM , Rana ZA , Gundersen K . Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc Natl Acad Sci U S A. (2010) ;107: (34):15111–6. |
[113] | Snijders T , Nederveen JP , McKay BR , Joanisse S , Verdijk LB , van Loon LJ , et al. Satellite cells in human skeletal muscle plasticity. Front Physiol. (2015) ;6: :283. |
[114] | Murach KA , Fry CS , Kirby TJ , Jackson JR , Lee JD , White SH , et al. Starring or supporting role? satellite cells and skeletal muscle fiber size regulation. Physiology (Bethesda). (2018) ;33: (1):26–38. |
[115] | Bamman MM , Roberts BM , Adams GR . Molecular Regulation of Exercise-Induced Muscle Fiber Hypertrophy. Cold Spring Harb Perspect Med. (2018) ;8: (6):a029751. |
[116] | Amthor H , Otto A , Vulin A , Rochat A , Dumonceaux J , Garcia L , et al. Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc Natl Acad Sci U S A. (2009) ;106: (18):7479–84. |
[117] | Blaauw B , Canato M , Agatea L , Toniolo L , Mammucari C , Masiero E , et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. (2009) ;23: (11):3896–905. |
[118] | McCarthy JJ , Mula J , Miyazaki M , Erfani R , Garrison K , Farooqui AB , et al A. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. (2011) ;138: (17):3657–66. |
[119] | Egner IM , Bruusgaard JC , Gundersen K . Satellite cell depletion prevents fiber hypertrophy in skeletal muscle. Development. (2016) ;143: (16):2898–906. |
[120] | Rosenblatt JD , Yong D , Parry DJ . Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve. (1994) ;17: (6):608–13. |
[121] | Goh Q , Millay DP . Requirement of myomaker-mediated stem cell fusion for skeletal muscle hypertrophy. eLife. (2017) ;6: :e20007. |
[122] | Goh Q , Song T , Petrany MJ , Cramer AA , Sun C , Sadayappan S , et al. Myonuclear accretion is a determinant of exercise-induced remodeling in skeletal muscle. eLife. (2019) ;8: :e44876. |
[123] | Petrany MJ , Swoboda CO , Sun C , Chetal K , Chen X , Weirauch MT , et al. Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. bioRxiv. (2020) ; 04.14.041400. |
[124] | Dos Santos M , Backer S , Saintpierre B , Relaix F , Sotiropoulos A , Maire P . Single-nucleus RNA-seq and FISH reveal coordinated transcriptional activity in mammalian myofibers. bioRxiv. 2020;04.16.043620. |
[125] | Kim M , Franke V , Brandt B , Spuler S , Akalin A , Birchmeier C . Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. bioRxiv. 2020;04.14.041665. |
[126] | Verbrugge SAJ , Schönfelder M , Becker L , Yaghoob Nezhad F , Hrabe de Angelis M , Wackerhage H . Genes Whose Gain or Loss-Of-Function Increases Skeletal Muscle Mass in Mice: A Systematic Literature Review. Front Physiol. (2018) ;9: :553. |
[127] | Hitachi K , Nakatani M , Tsuchida K . Myostatin signaling regulates Akt activity via the regulation of miR-486 expression. Int J Biochem Cell Biol. (2014) ;47: :93–103. |
[128] | Zhu M , Liu J , Xiao J , Yang L , Cai M , Shen H , et al. Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat Commun. (2017) ;8: :14718. |
[129] | Neppl RL , Wu CL , Wals K . lncRNA Chronos is an aging-induced inhibitor of muscle hypertrophy. J. Cell Biol.. (2017) ;216: (11):3497–507. |
[130] | Yu H , Waddell JN , Kuang S , Tellam RL , Cockett NE , Bidwell CA . Identification of genes directly responding to DLK1 signaling in Callipyge sheep. BMC Genomics (2018) ;19: (1):283. |
[131] | Gao YQ , Chen X , Wang P , Lu L , Zhao W , Chen C , et al. Regulation of DLK1 by the maternally expressed miR-379/ miR-544 cluster may underlie callipyge polar overdominance inheritance. Proc Natl Acad Sci U S A. (2015) ;112: (44):13627–32. |
[132] | Pereira MG , Dyar KA , Nogara L , Solagna F , Marabita M , Baraldo M , et al. Comparative analysis of muscle hypertrophy models reveals divergent gene transcription profiles and points to translational regulation of muscle growth through Increased mTOR signaling. Front Physiol. (2017) ;8: :968. |
[133] | Blaauw B , Mammucari C , Toniolo L , Agatea L , Abraham R , Sandri M , Reggiani C , Schiaffino S . Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum Mol Genet. (2008) ;17: :3686–96. |
[134] | Peter AK , Ko CY , Kim MH , Hsu N , Ouchi N , Rhie S , et al. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum Mol Genet.. (2009) ;18: (2):318–27. |
[135] | Castets P , Rion N , Théodore M , Falcetta D , Lin S , Reischl M , et al. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat Commun. (2019) ;10: (1):3187. |
[136] | Baraldo M , Geremia A , Pirazzini M , Nogara L , Solagna F , Türk C , et al. Skeletal muscle mTORC1 regulates neuromuscular junction stability. J Cachexia Sarcopenia Muscle. (2020) ;11: (1):208–25. |
[137] | Loenneke JP , Buckner SL , Dankel SJ , Abe T . Exercise-Induced Changes in Muscle Size do not Contribute to Exercise-Induced Changes in Muscle Strength. Sports Med. (2019) ;49: (7):987–91. |
[138] | Reggiani C , Schiaffino S . Muscle hypertrophy and muscle strength: dependent or independent variables? Eur J Transl Myol. (2020) ;30: (3):9311. |
[139] | D’Antona G , Lanfranconi F , Pellegrino MA , Brocca L , Adami R , Rossi R , et al. Skeletal muscle hypertrophy and structure and function of skeletal muscle fibres in male body builders. J Physiol. (2006) ;570: (3):611–27. |
[140] | Meijer JP , Jaspers RT , Rittweger J , Seynnes OR , Kamandulis S , Brazaitis M , et al. Single muscle fibre contractile properties differ between body-builders, power athletes and control subjects. Exp Physiol. (2015) ;100: (11):1331–41. |
[141] | Monti E , Toniolo L , Marcucci L , Bondí M , Martellato I , Simunič B , et al. Are muscle fibres of body builders intrinsically weaker? A comparison with single fibres of aged-matched controls. Acta Physiol (Oxf). (2020) Sep 13: :e13557. |
[142] | Amthor H , Macharia R , Navarrete R , Schuelke M , Brown SC , Otto A , et al. (2007) Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. (2007) ;104: :1835–40. |


