
Beyond the Traditional Clinical Trials for Amyotrophic Lateral Sclerosis and The Future Impact of Gene Therapy
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating and incurable motor neuron (MN) disorder affecting both upper and lower MNs. Despite impressive advances in the understanding of the disease’s pathological mechanism, classical pharmacological clinical trials failed to provide an efficient cure for ALS over the past twenty years. Two different gene therapy approaches were recently approved for the monogenic disease Spinal muscular atrophy, characterized by degeneration of lower MNs. This milestone suggests that gene therapy-based therapeutic solutions could be effective for the treatment of ALS. This review summarizes the possible reasons for the failure of traditional clinical trials for ALS. It provides then a focus on the advent of gene therapy approaches for hereditary forms of ALS. Specifically, it describes clinical use of antisense oligonucleotides in three familial forms of ALS, caused by mutations in SOD1, C9orf72 and FUS genes, respectively.. Clinical and pre-clinical studies based on AAV-mediated gene therapy approaches for both familial and sporadic ALS cases are presented as well. Overall, this overview highlights the potential of gene therapy as a transforming technology that will have a huge impact on treatment perspective for ALS patients and on the design of future clinical trials.
INTRODUCTION – AMYOTROPHIC LATERAL SCLEROSIS
Amyotrophic Lateral Sclerosis (ALS) is a relentless and rare neurodegenerative disease with a mean survival of 3 to 5 years from symptom onset [1]. The disease is characterized by the degeneration of motor neurons in the brain, brainstem and anterior horn of the spinal cord which induces a progressive muscle paralysis leading to death after respiratory failure [2]. Although more than 80 randomized clinical trials (RCTs) have been conducted in the past 20 years investigating the effects of several categories of molecules, there is still a lack of effective treatments for ALS patients. This is likely due to several factors, including clinical and genetic complexity of the disease [3].
The objective of this review is to describe the emerging gene therapy approaches in light of the overall context of clinical trials in ALS. First, it will introduce clinical and genetic characteristics of ALS, then it will describe the possible reasons for the failure of traditional clinical trials (CTs). Finally, a clinical and pre-clinical overview of advances in the field of gene therapy will be presented. Therapeutic perspectives for ALS patients, based on the advent of these novel technologies, will be discussed.
CLINICAL PRESENTATION OF ALS
ALS is characterized by an extremely heterogeneous clinical presentation. Conventionally, three main phenotypes are identified according to the site of onset of the muscle weakness: spinal (65%), bulbar (30%), and respiratory ALS (5%) [4]. Some atypical phenotypes such as flail arm syndrome, progressive muscular atrophy or primary lateral sclerosis (PLS) have been described as well [5–7]. PLS patients have the longest survival, while classic and bulbar ALS patients have the worst prognosis [8, 9]. A clinical continuum between ALS and Frontotemporal dementia (FTD) is nowadays recognized [10], with up to 15% of ALS patients reaching the criteria for FTD [11]. Furthermore, almost 50% of ALS patients have some cognitive impairment, which is manifested as behavioral (ALS-bi) or executive and language dysfunction (ALSci) [12].
GENETIC OF ALS
ALS is considered a sporadic disease in 90% of the cases (sALS), while approximately 10% of patients have a positive family history with an autosomal dominant (AD) transmission (familial ALS, fALS) [13]. About 30 ALS-related genes are known so far [14, 15]. The first of them to be identified in 1993 was the superoxide dismutase 1 (SOD1) gene [16]. Nowadays, more then 180, mostly AD, SOD1 mutations have been characterized, accounting for 15–20% of fALS cases. From a clinical point of view, SOD1 mutations usually determine prevalent lower limbs involvement without cognitive alterations [17]. In contrast, several mutations are known in genes related both to ALS and FTD, such as TAR-DNA-binding protein (TDP-43), fused in sarcoma (FUS) and chromosome 9 open reading frame 72 (C9orf72) [18, 19]. Both TDP-43 and FUS mutations have AD transmission and induce misfolding of the corresponding protein and accumulation of cytosolic toxic aggregates [20]. A radical progress in the genetic of ALS-FTD was made in 2011 with the discovery of a pathologic hexanucleotide GGGGCC repeat expansion in the C9orf72 gene, which results to be the most frequent disease-causing genetic mutation in the Caucasian population (40% of fALS, 7% of sALS) [21–23]. The mutation has an AD transmission and high age-related penetrance (up to 90%). From a clinical point of view, C9orf72-ALS-FTD patients frequently present a younger age at symptom onset [24] and have a shorter survival. In addition to cognitive alterations, psychiatric and bulbar involvement are considered more frequent in C9orf72-positive patients. Furthermore, this mutation has been rarely associated also to other neurodegenerative diseases such as Parkinson disease, progressive supranuclear palsy and cortico-basal syndrome [25]. The incidence of neurodegenerative diseases, other than ALS-FTD, in C9orf72-positive families suggests that genetic cases of ALS could be more frequent than usually considered [26].
CLINICAL TRIALS IN ALS – FAILURE OF PHARMACOLOGICAL APPROACHES
Currently, the only drug approved by both Food and Drug Administration (FDA) and European Medicines Agency (EMA) to treat ALS is riluzole [27]. This molecule only modestly improves survival in ALS patients [27]. After its approval in 1995, almost 80 RCTs testing different therapeutic approaches have been performed. However, no drug has been demonstrated to have a statistically significant positive effect neither on motor function nor on disease progression and survival of ALS patients [3].
The main therapeutic strategy applied over the years in ALS CTs has been to target specific cellular pathways inducing neurodegeneration [28, 29]. One of the principal mechanisms that has been addressed to treat ALS is glutamate excitotoxicity, which induces cell death through excessive neuronal firing. Importantly, riluzole and several other drugs target this process [27, 30]. Another mechanism considered as one of the main pathological triggers in ALS is oxidative stress, which causes chronic damage of critical cellular macromolecules. Compounds targeting this pathway (as for example coenzyme Q10) were not proven effective, although some RCTs are still ongoing [3, 29]. Interestingly, edaravone, a free radical scavenger, was shown to slow down the functional decline in ALS patients with short disease duration [31]. Therefore, the drug has been approved by the FDA for the treatment of early-stage ALS patients, but its definitive efficacy is still under debate [32]. Several other molecules acting on mitochondrial dysfunction [33, 34], autophagy and protein quality control [35–38] were tested without definitive positive results. In parallel, treatment strategies focusing on neuroinflammation [39–42], apoptosis, as well as alterations of neuromuscular transmission were also studied [3, 29, 43, 44]. Although some of the tested compounds gave promising results in phase 2 studies, further investigations are needed to prove their efficacy in ALS.
To highlight the elements to take into consideration for the improvement of traditional CTs for ALS, some putative explanations for their failure are discussed below and summarized in Table 1.
Table 1
Summary of the main reasons for clinical trials failure in ALS and proposal of possible solutions
| Reasons of failure of ALS clinical trials | Proposed solutions to be developed: |
| Incomplete knowledge of pathogenic pathways and limited disease models of the disease | -Novel disease models encompassing the complexity of the human disease and its key pathology (i.e. cytoplasmic accumulation of TDP-43); |
| -Specific models to demonstrate target engagement of different treatment strategies; | |
| -Preclinical pharmacodynamics biomarkers for therapeutic candidates; | |
| -Multidrug approaches to affect different pathogenic cascades. | |
| Phenotypic and biological heterogeneity and rarity of the disease. | -Organization of multicentric clinical trials and collaboration with patients’ associations and caregivers to facilitate recruitment and reduce the drop-out rate; |
| -Effective stratification of the patients to improve statistical power according to disease phenotype and stage; | |
| -Extended genetic screening of the ALS population with the aim of improving stratification, treating pre-symptomatic subjects and developing targeted gene therapies. | |
| Limitations in clinical trials design, outcome measures and patient’s stratification. | -Development of sensitive outcome measures to be adapted to the specific clinical trial with standardized operating procedures; |
| -Implementation of predictive, prognostic and pharmacodynamic biomarkers including combined scores and multimodal composed outcome measures; | |
| -Development of innovative predictive models to demonstrate treatment efficacy and to take into account pharmacological interactions; | |
| -Implementation of alternative study designs such as adaptive designs, platform trials, enrichment design, multistage sequential design, futility design. |
Pre-clinical models and multiple pathological mechanisms
Considering that none of the recently conducted CTs was able to demonstrate the efficacy of one particular therapeutic approach in ALS patients, the reliability of some of the models used in these last years for pre-clinical research needs to be questioned. Furthermore, ALS is a multifactorial disease and the choice of focusing on one mechanism at a time should be reconsidered [45, 46].
Design and structure of CTs
A huge amount of work is currently conducted to address scientific, statistic and design limitations in ALS CTs. Efforts in this direction could lead to the organization of enriched studies and to a better selection of sensitive outcome measures. New recommendations [45] have been developed and considered in the most recent FDA guidance for implementation of CTs in ALS [46]. This, together with the development of CT platforms for the rapid test of several molecules, should increase the probability of therapeutic success in the future [46]. Nevertheless, the rarity of the disease still limits the possibility to include wide cohorts of patients over short time periods, thus reducing the capacity of performing adequate stratification of the subjects participating in RCTs and reducing statistical power [45].
Heterogeneity of ALS patients
ALS is a highly heterogeneous disease from a clinical and also from a genetic point of view [47]. This has implications for the efficient development of innovative therapeutics. For this reason, well defined inclusion or stratification criteria considering timing of the treatment and individual clinical features (e.g predictors of prognosis) might be considered with the aim of obtaining more homogeneous study cohorts [47, 48].
Concerning genetic heterogeneity, it has been clearly described how it can negatively influence the results of RCTs [45, 49]. In fact, some therapeutic approaches might be useful in patients carrying specific mutations but be less useful in others. This hypothesis is sustained also by the fact that in several recent CTs, small subpopulations of patients seem to have a better response to the treatment as compared to the global study cohort. For example, a post-hoc pharmacogenetic study grouping the results from three negative CTs with lithium in ALS suggests that the drug may be effective in patients carrying a genetic variation in UNC13A. This indicates that further stratification or selection of patients based on their genetic characteristics could pave the way to the development of highly targeted and effective treatments [45]. In this perspective, the design of gene therapy approaches seems to be of primary relevance for the development of efficient treatments for ALS.
GENE THERAPY IN ALS
Gene therapy is a promising therapeutic approach for ALS, in particular to tackle the genetic roots of the well characterized fALS forms. Gene therapy is classified in i) non-viral gene therapy, in which naked sequences of nucleic acids, chemically modified or not, can be directly delivered to the host cell to correct pathological mechanisms; and ii) viral gene therapy, where genetically-modified viruses are used as vectors to deliver therapeutic sequences. These molecular strategies and their potential for fALS treatment were recently reviewed by our group [50].
Gene therapy showed great potential to modulate gene expression in different monogenic diseases of the central nervous system (CNS) [51]. Non-viral mediated gene therapy approaches and viral-mediated treatments are currently developed for fALS. Importantly, gene therapy treatments were recently approved for spinal muscular atrophy (SMA), another motor neuron disorder (MND). In light of these advances, this section will present the outcomes of ongoing pre-clinical and clinical studies based on oligonucleotides and viral vector delivery for the treatment of fALS. To provide an updated overview, relevant information will be gained through data released in companies’ web sites, when peer-reviewed articles are not available yet.
Non-viral gene therapy: Antisense oligonucleotides for fALS
A major breakthrough for the treatment of monogenic disorders was the approval, in 2016, of nusinersen with the commercial name of Spinraza® (Biogen) as treatment for all subtypes of SMA. Spinraza® is a survival motor neuron-2 (SMN2)-directed antisense oligonucleotide (ASO) drug that induces the translation of a fully functional SMN protein [53]. This drug is not able to cross the blood brain barrier (BBB) and it is thus administrated through repeated intrathecal (IT) injections. Spinraza® was evaluated in two randomized, double-blind, sham-controlled studies of infantile and later-onset SMA (ENDEAR and CHERISH, respectively) and in ongoing open-label studies (NURTURE), which include pre-symptomatic infants. In the ENDEAR study, 70% of treated patients showed improvement in motor function and did not need permanent assisted ventilation. Interestingly, 51% of treated infants had also a motor-milestone response [54]. Significant improvement in motor function was also observed in later-onset SMA after Spinraza® treatment compared to the placebo group [55]. These positive results obtained using ASOs for the treatment of SMA open perspectives for the application of similar approaches in ALS patients [56].
ASOs were proven efficient in pre-clinical models of ALS (reviewed by [50]) and remarkably different tests using ASO are ongoing for SOD1-, C9orf72- and FUS- linked fALS.
ASOs for SOD1-fALS in clinical trials
Tofersen (BIIB067), previously called IONIS-SOD1Rx, is an ASO that specifically binds to SOD1 mRNA. It induces SOD1 mRNA degradation through RNase H and thus prevents toxic SOD1 protein production. Results of the phase 1/2 CT were recently published [57]. Tofersen was tested after multiple IT ascending doses in adult ALS patients carrying a documented SOD1 mutation. The primary outcomes of this trial were safety and pharmacokinetics. The second outcome was analysis of SOD1 concentration in the cerebrospinal fluid (CSF), at baseline and after treatment. Interestingly, SOD1 protein levels were decreased by 36% in the CSF of patients treated with the highest concentration of Tofersen. Neurofilaments were significantly reduced in CSF and plasma of the same group. A trend towards a slowing of clinical decline measured by the ALS Functional Rating Scale-Revised (ALSFRS-R, [58]) was also detected. Importantly, clinical decline was slower in patients with a SOD1 mutation associated to a fast progressing phenotype. This highlights the importance of considering the rate of disease progression in ALS CTs to effectively stratify patients. Despite these positive effects, adverse events (AE) related to the IT lumbar puncture, such as headache, post–lumbar puncture syndrome and back pain, were observed in most participants and CSF pleocytosis occurred in some of them [57]. To confirm the safety and to evaluate efficacy of Tofersen, a phase 3, randomized, double-blind, placebo-controlled clinical trial (NCT02623699) named VALOR was initiated in May 2019. The trial includes adult patients with confirmed diagnosis of ALS due to all type of SOD1 mutations and with all severity levels but having forced vital capacity > 50%. The study is still ongoing and no interim analysis was published before the writing of this review.
ASOs for C9orf72-fALS in clinical trial
Similar to SOD1, different groups developed ASOs-based approaches as treatments for C9orf72-linked ALS (reviewed by [50]). BIIB078 (Biogen), is an ASO that selectively targets expansion-containing C9orf72 transcripts. Proof-of-concepts studies demonstrated how single, intracerebroventricular (ICV), administration of a C9orf72-ASO produces sustained reductions in RNA foci and dipeptide-repeat proteins and ameliorate behavioural deficits in C9orf72 mice [59]. A phase 1 clinical trial (NCT03626012) assessing safety, tolerability, PK and the effect on clinical function of BIIB078 is ongoing. This is the first-in-human gene therapy study for this ALS form, which aims to enrol about 90 adult participants carrying C9orf72 mutation and having a diagnosis of probable or definite ALS. As in the previously described trial for SOD1-ALS patients, participants are divided in 6 cohorts, each of them received different doses of BIIB078 or placebo by repeated IT injections. Patients are monitored for approximately 8 months after the last dose. Currently, no interim results on efficacy of treatment have been published.
Patient-specific ASOs for FUS-fALS
Jacifusen is a recently developed ASO (Ionis Pharmaceutical and Columbia Medical centre) for patients with mutation in FUS gene. Mutations in this gene are causative of about 5% of fALS and less than 1% of sALS, with some juvenile onset forms [60]. Jacifusen is a patient-specific ASO targeting the FUS mutation p.P525 L, which produces a mutant and toxic protein that accumulate in MN inducing alteration in their functions [60, 61]. In May 2019, the FDA decided to approve the administration of this experimental ASO for a young woman affected by this specific mutation before the completion of toxicological study [61]. To date FDA approved the treatment as compassionate use for other three ALS patients with FUS mutations. This important achievement endorses the use of ASOs in personalized medicine approaches for rare diseases.
In conclusion, although efficacy data in patients are still needed, ASOs seem a promising tool to target the toxic gain of function mechanisms responsible of certain forms of fALS. This is based on preclinical data, comparison to effects in SMA as well as on the ability to reduce SOD1 levels in the CSF. However, the repeated direct injections into the CSF increases the possibility of complications associated with this delivery route, such as lumbar pain due to needle punction and related AE [57, 62].
6VIRAL GENE THERAPY FOR MNDS
The use of viral vectors and, in particular, of adeno-associated viral (AAV) vectors with neural tropism overcomes the need of repeated direct injections with ASOs, enabling persistent and global gene transfer after one single administration. Viral vectors can be used to replace faulty genes in affected tissues or to reduce the expression of toxic proteins. Several serotypes are investigated for efficient CNS delivery. In particular, AAV serotype 9 (AAV9) and rh.10 (AAVrh.10), are largely used as preferred vectors for CNS delivery, due to their neuronal tropism and increased ability to cross the BBB [63–65]. In 2019, a gene replacement approach in which the SMN cDNA is delivered by an AAV9 in order to rescue the normal production of functional SMN protein received approval by the FDA to treat all types of SMA [66]. The single intravenous (IV) administration of the AAV9-SMN (Zolgensma®, Novartis) in SMA type I patients, was reported as safe and well tolerated [66–68]. However, the impact of age, disease severity and progression rate on the treatment were demonstrated [66, 69], underscoring the importance of early intervention [69]. Currently, Novartis is also testing the possibility to treat older patients with SMA type 2 and type 3 by a single IT infusion. This phase 1/2 trial (NCT03381729) is based on pre-clinical work in animals [70], demonstrating that direct injection to the CSF mediates higher therapeutic effects with reduced viral load compared to IV delivery. However, pre-clinical pathology findings affecting the dorsal root ganglia, an area that is highly targeted with gene therapy, has led the FDA to put a partial hold on this CT to allow further review. Importantly, no indication of such pathology or sensory impairment has been found to date in patients that received IT injections of Zolgensma® [71].
In general, this ongoing effort in testing AAV-mediated gene therapy for SMA is pivotal for the development and refinement of similar approaches for ALS patients.
AAV-MEDIATED SILENCING APPROACHES FOR FALS (SOD1 AND C9ORF72): PRE-CLINICAL STUDIES AND THE FIRST-IN-HUMAN TRIAL
Over the past decade, different molecular strategies have been developed to reduce SOD1 in ALS, using AAV vectors [50].
AveXis (recently renamed Novartis Gene Therapies), the biotech company who developed Zolgensma®, is developing a potential therapy for SOD1-linked fALS. This approach is based on the use of a self-complementary AAV9 vector expressing a short hairpin (sh) RNA to silence SOD1. The research-grade vector was successfully tested through single IV injection in SOD1 mice at different ages [72]. However, like for SMA, efficient CNS targeting remains one of the major issues to be addressed for safe translation of this and similar approaches. Therefore, the AAV9-sh-SOD1 vector was further tested 1) via intra-cisterna magna in adult SOD1 mice [73] and 2) via subpial (SP) injections in mice, pigs and non-human primates [74]. The positive results obtained in these studies pointed to the AAV9-sh-SOD1 as a promising candidate for SOD1-linked ALS and its pre-clinical development is currently ongoing [75].
Other similar strategies are developed to silence SOD1 with artificial microRNA (miRNA), administrated via AAVrh.10 vectors and one single intra-CSF injection.
Based on the expression of artificial miRNA, the group of Robert H. Brown (University of Massachusetts) developed a one-time gene therapy designed to silence the activity of the mutated SOD1. A first preclinical study in monkeys showed that the research-grade vector blocks the expression of SOD1 by 93% in lower motor neurons [76]. Further studies on cynomolgus macaques (Macaca fascicularis), demonstrated the safety of a modified construct in pre-clinical tests. In this study, Borel et al., showed that IT infusion of a large volume of an AAVrh.10 vector (5 ml) was safe and well tolerated, up to 92 days after administration. The vector lowered SOD1 expression in the entire spinal cord motor neurons and was found to be well tolerated with no negative effects [77]. Importantly, a similar AAV vector was recently tested in a compassionate-use study on two SOD1-ALS patients who were respectively 22-years (patient 1) and 56-years-old (patient 2) [52]. The results of this study showed post-mortem decreased levels of SOD1 in spinal cord tissue of patient 1 compared to untreated patients with SOD1-linked ALS and healthy controls. In contrast to what observed with Tofersen, levels of SOD1 in CSF were transiently and only slightly lowered in patient 1 but were not affected in patient 2. Patient 1 developed meningoradiculitis after viral infusion and only transient improvement in the strength of his right leg was observed, with no change in vital capacity. In contrast, Patient 2, who received immunosuppressive drugs, had stable scores on a composite measure of ALS function and a stable vital capacity during a 12-month period [52]. The follow-up of this patient should give further indications on the effects of this novel therapeutic approach.
These results showed that intrathecal AAV-mediated microRNA delivery could be used as a potential treatment for SOD1-linked ALS, although additional studies and further improvements are required to determine whether the approach is safe and above all efficient.
Another approach relying on SOD1 silencing via microRNA and AAV vector is currently developed by Voyager Therapeutics. Their second-generation drug candidate, VY-SOD102 is currently tested through one-time infusion in the cervical spinal cord after laminectomy [78].
An innovative and efficient approach to silence SOD1 expression was also recently developed by our group. This is based on the combined use of AS sequences and AAV.rh10 vector for in vivo delivery. The approach was tested in adult SOD1 mice through a combined IV and ICV delivery [79]. The AAV.rh10-AS-SOD1 is currently further evaluated in pre-clinical studies for potential translation in the near future.
AAV-mediated expression of silencing sequences was also tested to treat C9orf72-ALS. Two studies sponsored by UniQure showed the feasibility of miRNA-based and AAV-delivered gene therapy in experimental models. In particular, an AAV serotype 5 (AAV5) expressing artificial miRNA, was IT injected in an ALS mouse model and showed significant reduction in repeat-containing C9orf72 transcripts and RNA foci after treatment [80].
The combination of the different molecular approaches described above and the use of different AAV serotypes with high tropism for CNS is promising for the development of effective treatments for ALS. However, one of the major issues faced for the development of these application is the delivery route of administration. It should take into account an efficient targeting of the CNS while limiting invasive surgical operations. Based on the property of some AAV serotypes and of the self-complementary genome to bypass the BBB, the preferred delivery route for AAV vector in CNS disorders was considered the IV delivery [63, 64]. Several pre-clinical studies indicated thereafter that a direct access to the CNS, i.e. via intra-CSF injections, outperforms the systemic administration for the treatment of MNDs using AAV vectors [70, 81–84]. For this reason, the majority of methods currently developed for the treatment of ALS are based on direct injections to the CNS (summarized in Fig. 1).
Fig. 1
Schematic representation of methods used to deliver gene therapy approaches directly into the CNS, in clinical and pre-clinical trials for ALS. Direct access to the central nervous system (CNS) is currently considered the best method for efficient delivery of gene therapy strategies for motor neuron disorders. To date, the preferred translational route of administration is the intrathecal (IT) delivery. This is a minimally invasive method and consents widespread distribution of gene therapies through the cerebrospinal fluid (CSF) and consequently to the CNS. Other two methods are currently investigated in pre-clinical studies for efficient access to the CSF: the intracerebroventricular (ICV) and intra-cisterna magna injections. Although these are easily applicable in experimental models (such as ALS mice), their translation to humans could be hard. Recently, the subpial injection was also reported as method allowing viral vector distribution to the spinal cord in animal models, including non-human primates. Further investigations are needed to define the best delivery route for efficient translation of gene therapy treatments to patients. The seek for non-invasive and efficient delivery methods, represent a key step for the future development of these innovative approaches.
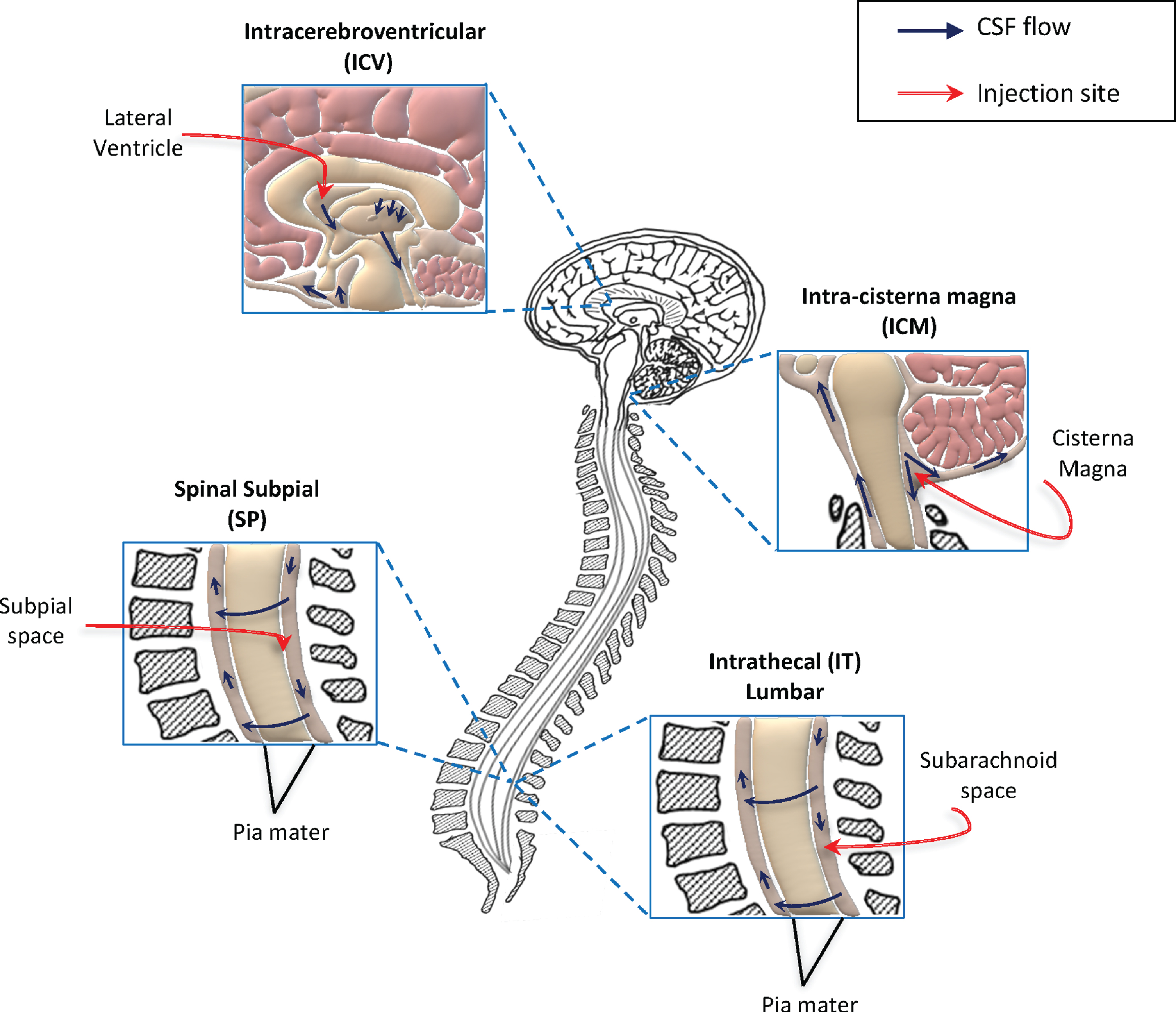
Multiple parameters (other than vector doses and site of administration) have to be considered for intra-CNS delivery of gene therapy vectors to patients. For example, the volume of injection, the formulation of the vector solution and the velocity of injection could be important factors to determine therapeutic outcomes. Indeed, differences between fast and slow IT injection of viral vector in SOD1-G93A mice were analysed and it was demonstrated that the slower one enhances transduction of spinal cord and therapeutic efficacy [85]. Alternative delivery routes of administration are also investigated, such as a combination of intralingual and intrapleural injections [86].
Overall, the increasing number of pre-clinical studies in non-human primates (NHP) and the generation of novel AAV vectors, more efficient in targeting CNS cells [87], will help assessing the best method for gene therapy delivery to ALS patients. Several other parameters will have to be considered to move forward the field, such as the long-term transgene expression, which produces sustained expression of silencing molecules. Indeed, AAV vectors persists over years in the patient [88–90] and it is still unclear whether the prolonged and unselective silencing of both wild-type and mutant alleles will have undesirable consequences in neurological patients. However, inducible transgene expression, i.e. using Tet-On/Off systems, in AAV vectors is under pre-clinical investigation and its development to clinic will likely limit potential detrimental effects at the long-term [91].
In conclusion, AAV-mediated gene therapy is on the way, together with ASOs, for the development of precision-medicine approaches for fALS. In particular, for the treatment of SOD1-ALS, both strategies were already tested in patients and the results have been recently published [52, 57]. These two studies highlight the advantages and disadvantages of each approach (summarized in Table 2). Even if further tests and refinements are still necessary, ASOs and AAV hold promise for the treatment of fALS.
Table 2
Summary of the advantages and possible disadvantages of gene therapy strategies translated to fALS patients
| Non-viral gene therapy (ASOs) | Viral gene therapy (artificial miRNA or shRNA) | ||
| Advantages | Disadvantages | Advantages | Disadvantages |
| -Efficient silencing of the target mRNA | -Rapidly degraded, thus repeated injections are needed, causing related AE | -High tropism for CNS -One single administration | -Long term expression of the transgene, the treatment cannot be stopped in case of AE |
| -Possibility to stop the treatment in case of AE | -Efficient silencing of target mRNA demonstrated in pre-clinical settings | -Possible immune response | |
ASOs = antisense oligonucleotides; shRNA = short hairpin RNA; miRNA = microRNA; AE = adverse events; CNS = central nervous system.
TESTS OF GENE THERAPY STRATEGIES TO TREAT BOTH FAMILIAL AND SPORADIC ALS
As mentioned in the introduction, the major part of ALS cases (90%) are sporadic and the precise pathological mechanisms inducing neurodegeneration are still not completely understood. The ideal therapeutic application should target common mechanisms to the different ALS forms. Many strategies are under development to target mechanisms common to both fALS and sALS [92, 93]. One of the preferred targets towards these applications, is TDP-43, an ubiquitously expressed DNA/RNA-binding protein involved in different aspects of RNA metabolism, such as transcription, splicing, stability and transport [94]. Pathological aspects linked to TDP-43 include mislocalization and aggregation of the protein in the cytoplasm (TDP-43 proteinopathy) and are present in more than 90% of ALS cases (fALS and sALS), but also in patients with FTD [95] and other neurodegenerative disorders, such as Alzheimer’s, Parkinson’s and Huntington’s disease [94]. Single-chain antibodies against TDP-43 were generated and delivered in the cortex of transgenic mice expressing ALS-linked TDP-43 mutations through the use of self-complementary AAV9 vectors. This approach mediated reduction of microgliosis, motor defects and of TDP-43 proteinopathy in mice [93]. This work represents an important step-forward for the development of viral-mediated, gene-targeting approaches for ALS and further studies are necessary to prove the clinical validity of this therapy.
Other strategies developed to target mechanisms common to both fALS and sALS are based on mechanisms of neuroprotection through the delivery of trophic factors. Some trophic factors have been demonstrated to promote cell survival and to be protective in both in vitro and in vivo models of neuronal degeneration. In the past few years, brain-derived neurotrophic factor (BDNF), glial cell line derived neurotrophic factor (GDNF), insulin-like growth factor 1 (IGF-1), vascular endothelial growth factor (VEGF), Neuregulin 1 and Neurturin were used to treat ALS under pre-clinical settings [92, 96–98]. Different studies showed that AAV mediated expression of these neurotrophic factors delayed disease onset and prolonged survival in SOD1 mice model [96–102].
Recently, a gene therapy approach was tested to correct cortical hyperexcitability. This might be caused by the reduction of inhibition activity of the pyramidal cells in layer 5 (L5) of the motor cortex. The bilateral injection in the motor cortex of an AAV5 vector, increasing interneuron activity, through a chemogenetic technology, rescued inhibition of L5 neural cells. This treatment delayed neurodegeneration and motor impairment, and increased survival in SOD1-ALS mice [103]. The alteration of the interneurons activity was described not only for SOD1 mouse models, but also in TDP-43 models, and interestingly, interneurons hypoactivity is also reduced in patients with C9orf72 mutation [104, 105]. These results open thus a novel perspective for the treatment of ALS, underlining the importance of the early treatment of the upper motor neuron, which might be also beneficial to lower motor neurons degeneration.
Overall, gene therapy might be effective in the treatment of both familial and sporadic ALS by acting on specific mutations and mechanisms of cellular damage. Furthermore, different approaches may be combined in the future to enhance the effects of gene targeting strategies.
In view of future CTs testing these innovative approaches, stratification of patients based on their genetic status and disease progression will be a relevant strategy to overcome genetic heterogeneity of ALS patients, thus improving the probability of identifying a positive effect of the treatment [106].
CONCLUSION
In conclusion, we highlight here the principal reasons of CTs failure, based on classical pharmacological approaches for ALS. We then present advances in the field of gene therapy and its potential for the development of future treatments for ALS patients.
These novel therapeutic entities will also benefit from improved clinical design relying on lessons learned from the previous trials, in which several aspects will have to be considered. Specifically, treatment of genetically homogeneous populations will increase the statistical power of the studies, together with the clear identification of target engagement, which will be easier to demonstrate for strictly targeted treatments [45, 57]. However, accurate stratification of the patients will still be needed even when considering cohorts of subjects carrying the same gene mutation. In fact, for the majority of ALS related genes, a multitude of mutations are identified as well as different phenotypes with variable disease progression [18, 57].
Along with the development of broader treatments, genetic screening of ALS patients will allow to identify gene mutations also in sALS subjects, widening the possibility of a targeted treatment for these patients. Furthermore, the experiences in SMA confirmed that the efficacy of gene-targeted treatment is highly dependent on an early administration [66, 69]. A future perspective would be to treat pre-symptomatic ALS gene carriers to potentially prevent or delay the onset of symptoms.
Another factor to take into account in the design of clinical trials for gene therapy approaches will be that, especially when using AAV vectors, once injected in the body the transgene will be continuously expressed in the treated patients. The risk of determining long term AE related to this procedure as well as of developing an immune response against the vector (or the transgene) need to be accurately analysed. Furthermore, gene therapy could influence the possibility of participating in others pharmacological CTs and this will have to be considered for patient’s inclusion.
Finally, one of the limits of gene therapy is to identify a safe route of administration to make sure that the treatment gets into the CNS and reaches as many affected cells as possible. IT injections are nowadays performed effectively and with small side effects in patients, although several different administration approaches, less invasive, are under study. This, together with the development of more efficient vectors, will likely facilitate the translation of gene therapy approaches for ALS patients.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
M.G.B. and G.Q. are supported by the Association Française contre les Myopathies (AFM), the Association Institut de Myologie (AIM), the Sorbonne Université, the Institut National de la Santé et de la Recherche Médicale (INSERM). M.C. is supported by the ANR grant N° ANR-19-CE18-0014-01 (Magenta 2020).
REFERENCES
[1] | Robberecht W , Philips T . The changing scene of amyotrophic lateral sclerosis. Nature Reviews Neuroscience. (2013) ;14: :248–64. https://doi.org/10.1038/nrn3430. |
[2] | Rowland LP , Shneider NA . Amyotrophic Lateral Sclerosis. New England Journal of Medicine. (2001) ;344: :1688–700. https://doi.org/10.1056/NEJM200105313442207. |
[3] | Petrov D , Mansfield C , Moussy A , Hermine O . ALS Clinical Trials Review: 20 Years of Failure. AreWe Any Closer to Registering a New Treatment? Frontiers in Aging Neuroscience. 2017;9. https://doi.org/10.3389/fnagi.2017.00068. |
[4] | Ravits J , Paul P , Jorg C . Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology. (2007) ;68: :1571. https://doi.org/10.1212/01.wnl.0000260965.20021.47. |
[5] | Gamez J , Cervera C , Codina A . Flail arm syndrome or VulpianBernhart’s form of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (1999) ;67: :258. |
[6] | Gordon PH , Cheng B , Katz IB , Pinto M , Hays AP , Mitsumoto H , et al. The natural history of primary lateral sclerosis. Neurology. (2006) ;66: :647. https://doi.org/10.1212/01.wnl.0000200962.94777.71. |
[7] | Rowland LP . Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle & Nerve. (2010) ;41: :161–5. https://doi.org/10.1002/mus.21565. |
[8] | Calvo A , Moglia C , Lunetta C , Marinou K , Ticozzi N , Ferrante GD , et al. Factors predicting survival in ALS: A multicenter Italian study. J Neurol. (2017) ;264: :54–63. https://doi.org/10.1007/s00415-016-8313-y. |
[9] | Swinnen B , Robberecht W . The phenotypic variability of amyotrophic lateral sclerosis. Nature Reviews Neurology. (2014) ;10: :661–70. https://doi.org/10.1038/nrneurol.2014.184. |
[10] | Goldstein LH , Abrahams S . Changes in cognition and behaviour in amyotrophic lateral sclerosis: Nature of impairment and implications for assessment. The Lancet Neurology. (2013) ;12: :368–80. https://doi.org/10.1016/S1474-4422(13)70026-7. |
[11] | Gordon PH , Delgadillo D , Piquard A , Bruneteau G , Pradat P-F , Salachas F , et al. The range and clinical impact of cognitive impairment in French patients with ALS: A cross-sectional study of neuropsychological test performance. Amyotrophic Lateral Sclerosis. (2011) ;12: :372–8. 737 https://doi.org/10.3109/17482968.2011.580847. |
[12] | Phukan J , Pender NP , Hardiman O . Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. (2007) ;6: :994–1003. https://doi.org/10.1016/S1474-4422(07)70265-X. |
[13] | Byrne S , Walsh C , Lynch C , Bede P , Elamin M , Kenna K , et al. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2011) ;82: :623–7. https://doi.org/10.1136/jnnp.2010.224501. |
[14] | Volk AE , Weishaupt JH , Andersen PM , Ludolph AC , Kubisch C . Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. (2018) ;30: :252–8. https://doi.org/10.1007/s11825-018-0185-3. |
[15] | Vijayakumar UG , Milla V , Cynthia Stafford MY , Bjourson AJ , Duddy W , Duguez SM-R . A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front Neurol. 2019;10. https://doi.org/10.3389/fneur.2019.00400. |
[16] | Rosen DR , Sapp P , O’Regan J , McKenna-Yasek D , Schlumpf KS , Haines JL , et al. Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am J Med Genet. (1994) ;51: :61–9. https://doi.org/10.1002/ajmg.1320510114. |
[17] | Corcia P , Couratier P , Blasco H , Andres CR , Beltran S , Meininger V , et al. Genetics of amyotrophic lateral sclerosis. Rev Neurol (Paris). (2017) ;173: :254–62. https://doi.org/10.1016/j.neurol.2017.03.030. |
[18] | Andersen PM , Al-Chalabi A . Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat Rev Neurol. (2011) ;7: :603–15. https://doi.org/10.1038/nrneurol.2011.150. |
[19] | Marangi G , Traynor BJ . Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res-earch. (2015) ;1607: :75–93. https://doi.org/10.1016/j.brainres.2014.10.009. |
[20] | Lagier-Tourenne C , Cleveland DW . Rethinking ALS: The FUS about TDP-43. Cell. (2009) ;136: :1001–4. https://doi.org/10.1016/j.cell.2009.03.006. |
[21] | DeJesus-Hernandez M , Mackenzie IR , Boeve BF , Boxer AL , Baker M , Rutherford NJ , et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. (2011) ;72: :245–56. https://doi.org/10.1016/j.neuron.2011.09.011. |
[22] | Majounie E , Renton AE , Mok K , Dopper EG , Waite A , Rollinson S , et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. (2012) ;11: :323–30. https://doi.org/10.1016/S1474-4422(12)70043-1. |
[23] | Renton AE , Majounie E , Waite A , Simón-Sánchez J , Rollinson S , Gibbs JR , et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. (2011) ;72: :257–68. https://doi.org/10.1016/j.neuron.2011.09.010. |
[24] | Byrne S , Elamin M , Bede P , Shatunov A , Walsh C , Corr B , et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. (2012) ;11: :232–40. https://doi.org/10.1016/S1474-4422(12)70014-5. |
[25] | Rohrer JD , Isaacs AM , Mizielinska S , Mead S , Lashley T , Wray S , et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. (2015) ;14: :291–301. https://doi.org/10.1016/S1474-4422(14)70233-9. |
[26] | Liu Y , Yu J-T , Zong Y , Zhou J , Tan L . C9ORF72 Mutations in Neurodegenerative Diseases. Mol Neurobiol. (2014) ;49: :386–98. https://doi.org/10.1007/s12035-013-8528-1. |
[27] | Bensimon G , Lacomblez L , Meininger V . A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. New England Journal of Medicine. (1994) ;330: :585–91. https://doi.org/10.1056/NEJM199403033300901. |
[28] | Wobst HJ , Mack KL , Brown DG , Brandon NJ , Shorter J . The clinical trial landscape in amyotrophic lateral sclerosis-Past, present, and future. Med Res Rev. 2020. https://doi.org/10.1002/med.21661. |
[29] | Chiò A , Mazzini L , Mora G . Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology. (2020) ;167: :107986. https://doi.org/10.1016/j.neuropharm.2020.107986. |
[30] | Blasco H , Mavel S , Corcia P , Gordon PH . The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr Med Chem. (2014) ;21: :3551–75. https://doi.org/10.2174/0929867321666140916120118. |
[31] | Takei K , Tsuda K , Takahashi F , Palumbo J . Post-hoc analysis of open-label extension period of study MCI186-19 in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2017) ;18: :64–70. https://doi.org/10.1080/21678421.2017.1365372. |
[32] | Rothstein JD . Edaravone: A new drug approved for ALS. Cell. (2017) ;171: :725. https://doi.org/10.1016/j.cell.2017.10.011. |
[33] | Cudkowicz ME , van den Berg LH , Shefner JM , Mitsumoto H , Mora JS , Ludolph A , et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomised, double-blind, phase 3 trial. Lancet Neurol. (2013) ;12: :1059–67. https://doi.org/10.1016/S1474-4422(13)70221-7. |
[34] | Lenglet T , Lacomblez L , Abitbol JL , Ludolph A , Mora JS , Robberecht W , et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. (2014) ;21: :529–36. https://doi.org/10.1111/ene.12344. |
[35] | Benatar M , Wuu J , Andersen PM , Atassi N , David W , Cudkowicz M , et al. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology. (2018) ;90: :e565–74. https://doi.org/10.1212/WNL.0000000000004960. |
[36] | Bella ED , Tramacere I , Antonini G , Borghero G , Capasso M , Caponnetto C , et al. Protein misfolding, amyotrophic lateral sclerosis and guanabenz: Protocol for a phase II RCT with futility design (ProMISe trial). BMJ Open. (2017) ;7: :e015434. https://doi.org/10.1136/bmjopen-2016-015434. |
[37] | Mandrioli J , D’Amico R , Zucchi E , Gessani A , Fini N , Fasano A , et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore). (2018) ;97: :e11119. https://doi.org/10.1097/MD.0000000000011119. |
[38] | Mandrioli J , Crippa V , Cereda C , Bonetto V , Zucchi E , Gessani A , et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open. (2019) ;9: :e028486. https://doi.org/10.1136/bmjopen-2018-028486. |
[39] | Cudkowicz ME , Shefner JM , Schoenfeld DA , Zhang H , Andreasson KI , Rothstein JD , et al. Trial of celecoxib in amyotrophic lateral sclerosis. Annals of Neurology. (2006) ;60: :22–31. https://doi.org/10.1002/ana.20903. |
[40] | Lauria G , Dalla Bella E , Antonini G , Borghero G , Capasso M , Caponnetto C , et al. Erythropoietin in amyotrophic lateral sclerosis: A multicentre, randomised, double blind, placebo controlled, phase III study. J Neurol Neurosurg Psychiatry. (2015) ;86: :879–86. https://doi.org/10.1136/jnnp-2014-308996. |
[41] | Miller RG , Block G , Katz JS , Barohn RJ , Gopalakrishnan V , Cudkowicz M , et al. Randomized phase 2 trial of NP001-a novel immune regulator: Safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. 2015;2. https://doi.org/10.1212/NXI.0000000000000100. |
[42] | Dupuis L , Dengler R , Heneka MT , Meyer T , Zierz S , Kassubek J , et al. A Randomized, Double Blind, Placebo-Controlled Trial of Pioglitazone in Combination with Riluzole in Amyotrophic Lateral Sclerosis. PLoS One. 2012;7. https://doi.org/10.1371/journal.pone.0037885. |
[43] | Patten SA , Aggad D , Martinez J , Tremblay E , Petrillo J , Armstrong GA , et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight. 2017;2. https://doi.org/10.1172/jci.insight.97152. |
[44] | Andrews JA , Cudkowicz ME , Hardiman O , Meng L , Bian A , Lee J , et al. VITALITY-ALS, a phase III trial of tirasemtiv, a selective fast skeletal muscle troponin activator, as a potential treatment for patients with amyotrophic lateral sclerosis: Study design and baseline characteristics. Amyotroph Lateral Scler Frontotemporal Degener. (2018) ;19: :259–66. https://doi.org/10.1080/21678421.2018.1426770. |
[45] | van den Berg LH , Sorenson E , Gronseth G , Macklin EA , Andrews J , Baloh RH , et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology. (2019) ;92: :e1610–23. https://doi.org/10.1212/WNL.0000000000007242. |
[46] | Andrews JA , Bruijn LI , Shefner JM . ALS drug development guidances and trial guidelines: Consensus and opportunities for alignment. Neurology. (2019) ;93: :66–71. https://doi.org/10.1212/WNL.0000000000007695. |
[47] | Goyal NA , Berry JD , Windebank A , Staff NP , Maragakis NJ , van den Berg LH , et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve. 2020. https://doi.org/10.1002/mus.26801. |
[48] | Kueffner R , Zach N , Bronfeld M , Norel R , Atassi N , Balagurusamy V , et al. Stratification of amyotrophic lateral sclerosis patients: A crowdsourcing approach. Sci Rep. (2019) ;9: :690. https://doi.org/10.1038/s41598-018-36873-4. |
[49] | van Eijk RPA , Jones AR , Sproviero W , Shatunov A , Shaw PJ , Leigh PN , et al. Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology. (2017) ;89: :1915–22. https://doi.org/10.1212/WNL.0000000000004606. |
[50] | Cappella M , Ciotti C , Cohen-Tannoudji M , Biferi MG . Gene Therapy for ALS— A Perspective. International Journal of Molecular Sciences. (2019) ;20: :4388. https://doi.org/10.3390/ijms20184388. |
[51] | Choong C-J , Baba K , Mochizuki H . Gene therapy for neurological disorders. Expert Opinion on Biological Therapy. (2016) ;16: :143–59. https://doi.org/10.1517/14712598.2016.1114096. |
[52] | Mueller C , Berry JD , McKenna-Yasek DM , Gernoux G , Owegi MA , Pothier LM , et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med. (2020) ;383: :151–8. https://doi.org/10.1056/NEJMoa2005056. |
[53] | Vita G , Vita GL , Musumeci O , Rodolico C , Messina S . Genetic neuromuscular disorders: Living the era of a therapeutic revolution. Part Diseases of motor neuron and skeletal muscle. Neurol Sci. (2019) ;40: :671–81. https://doi.org/10.1007/s10072-019-03764-z. |
[54] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. (2017) ;377: :1723–32. https://doi.org/10.1056/NEJMoa947752. |
[55] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. (2018) ;378: :625–35. https://doi.org/10.1056/NEJMoa1710504. |
[56] | Mejzini R , Flynn LL , Pitout IL , Fletcher S , Wilton SD , Akkari PA . ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019;13. https://doi.org/10.3389/fnins.2019.01310. |
[57] | Miller T , Cudkowicz M , Shaw PJ , Andersen PM , Atassi N , Bucelli RC , et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. (2020) ;383: :109–19. https://doi.org/10.1056/NEJMoa2003715. |
[58] | Cedarbaum JM , Stambler N , Malta E , Fuller C , Hilt D , Thurmond B , et al. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. (1999) ;169: :13–21. https://doi.org/10.1016/s0022-510x(99)00210-5. |
[59] | Jiang J , Zhu Q , Gendron TF , Saberi S , McAlonis-Downes M , Seelman A , et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. (2016) ;90: :535–50. https://doi.org/10.1016/j.neuron.2016.04.006. |
[60] | Conte A , Lattante S , Zollino M , Marangi G , Luigetti M , Del Grande A , et al. P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscular Disorders. (2012) ;22: :73–5. https://doi.org/10.1016/j.nmd.2011.08.003. |
[61] | ArnoldC. Tailored treatment for ALS poised to move ahead. Nature Medicine. (2019) . https://doi.org/10.1038/d41591-019-00013-w. |
[62] | Miller TM , Pestronk A , David W , Rothstein J , Simpson E , Appel SH , et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. The Lancet Neurology. (2013) ;12: :435–42. https://doi.org/10.1016/S1474-4422(13)70061-9. |
[63] | Duque S , Joussemet B , Riviere C , Marais T , Dubreil L , Douar A-M , et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. (2009) ;17: :1187–96. https://doi.org/10.1038/mt.2009.71. |
[64] | Foust KD , Nurre E , Montgomery CL , Hernandez A , Chan CM , Kaspar BK . Intravascular AAV9 preferentially targets neonatal-neurons and adult-astrocytes in CNS. Nat Biotechnol. (2009) ;27: :59–65. https://doi.org/10.1038/nbt.1515. |
[65] | Tanguy Y , Biferi MG , Besse A , Astord S , Cohen-Tannoudji M , Marais T , et al. Systemic AAVrh10 provides higher transgene expression than AAV9 in the brain and the spinal cord of neonatal mice. Front Mol Neurosci. (2015) ;8: :36. https://doi.org/10.3389/fnmol.2015.00036. |
[66] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. New England Journal of Medicine. (2017) ;377: :1713–22. https://doi.org/10.1056/NEJMoa1706198. |
[67] | Al-Zaidy S , Pickard AS , Kotha K , Alfano LN , Lowes L , Paul G , et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatric Pulmonology. (2019) ;54: :179–85. https://doi.org/10.1002/ppul.24203. |
[68] | Al-Zaidy SA , Kolb SJ , Lowes L , Alfano LN , Shell R , Church KR , et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA Comparative Study with a Prospective Natural History Cohort. Journal of Neuromuscular Disea-ses. (2019) ;6: :307–17. https://doi.org/10.3233/JND-190403. |
[69] | Lowes LP , Alfano LN , Arnold WD , Shell R , Prior TW , McColly M , et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatric Neurology. (2019) ;98: :39–45. https://doi.org/10.1016/j.pediatrneurol.2019.05.005. |
[70] | Meyer K , Ferraiuolo L , Schmelzer L , Braun L , McGovern V , Likhite S , et al. Improving Single Injection CSF Delivery of AAV9-mediated Gene Therapy for SMA: A Dose–response Study in Mice and Nonhuman Primates. Mol Ther. (2015) ;23: :477–87. https://doi.org/10.1038/mt.2014.210. |
[71] | AveXis presents AVXS-101 IT data demonstrating remarkable increases in HFMSE scores and a consistent clinically meaningful response in older patients with SMA Type 2. Novartis n.d. https://www.novartis.com/news/media-releases/avexis-presents-avxs-101-it-data-demonstrating-remarkable-increases-hfmse-scores-and-consistent-clinically-meaningful-response-older-patients-sma-type-2 (accessed July 15, 2020). |
[72] | Foust KD , Salazar DL , Likhite S , Ferraiuolo L , Ditsworth D , Ilieva H , et al. Therapeutic AAV9-mediated Suppression of Mutant SOD1 Slows Disease Progression and Extends Survival in Models of Inherited ALS. Mol Ther. (2013) ;21: :2148–59. https://doi.org/10.1038/mt.2013.211. |
[73] | Iannitti T , Scarrott JM , Likhite S , Coldicott IRP , Lewis KE , Heath PR , et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-free, and Biomarker-Supported Strategy for fALS. Molecular Therapy - Nucleic Acids. (2018) ;12: :75–88. https://doi.org/10.1016/j.omtn.2018.04.015. |
[74] | Bravo-Hernandez M , Tadokoro T , Navarro MR , Platoshyn O , Kobayashi Y , Marsala S , et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med. (2020) ;26: :118–30. https://doi.org/10.1038/s41591-019-0674-1. |
[75] | AveXis: A Gene Therapy Company. AvexisCom n.d. https://www.avexis.com (accessed July 15, 2020). |
[76] | Borel F , Gernoux G , Cardozo B , Metterville JP , Toro Cabreja GC , Song L , et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum Gene Ther. (2016) ;27: :19–31. https://doi.org/10.1089/hum.2015.122. |
[77] | Borel F , Gernoux G , Sun H , Stock R , Blackwood M , Brown RH , et al. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci Transl Med. 2018;10. https://doi.org/10.1126/scitranslmed.aau6414. |
[78] | Voyager Therapeutics Announces Preclinical Data for Huntington’s Disease and Amyotrophic Lateral Sclerosis Programs at the Congress of the European Society of Gene and Cell Therapy | Voyager Therapeutics Inc. n.d. https://ir.voyagertherapeutics.com/news-releases/news-release-details/voyager-therapeutics-announces-preclinical-data-huntingtons/ (accessed July 15, 2020). |
[79] | Biferi MG , Cohen-Tannoudji M , Cappelletto A , Giroux B , Roda M , Astord S , et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol Ther. (2017) ;25: :2038–52. https://doi.org/10.1016/j.ymthe.2017.05.017. |
[80] | Martier R , Liefhebber JM , García-Osta A , Miniarikova J , Cuadrado-Tejedor M , Espelosin M , et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Molecular Therapy - Nucleic Acids. (2019) ;16: :26–37. https://doi.org/10.1016/j.omtn.2019.02.001. |
[81] | Armbruster N , Lattanzi A , Jeavons M , Van Wittenberghe L , Gjata B , Marais T , et al. Efficacy and biodistribution analysis of intracerebroventricular administration of an optimized scAAV9-SMN1 vector in a mouse model of spinal muscular atrophy. Molecular Therapy - Methods & Clinical Development. (2016) ;3: :16060. https://doi.org/10.1038/mtm.2016.60. |
[82] | Bevan AK , Duque S , Foust KD , Morales PR , Braun L , Schmelzer L , et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. (2011) ;19: :1971–80. https://doi.org/10.1038/mt.2011.157. |
[83] | Hinderer C , Bell P , Katz N , Vite CH , Louboutin J-P , Bote E , et al. Evaluation of Intrathecal Routes of Administration for Adeno-Associated Viral Vectors in Large Animals. Hum Gene Ther. (2018) ;29: :15–24. https://doi.org/10.1089/hum.2017.026. |
[84] | Samaranch L , Salegio EA , San Sebastian W , Kells AP , Foust KD , Bringas JR , et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. (2012) ;23: :382–9. https://doi.org/10.1089/hum.2011.200. |
[85] | Li D , Liu C , Yang C , Wang D , Wu D , Qi Y , et al. Slow intrathecal injection of rAAVrh10 enhances its transduction of spinal cord and therapeutic efficacy in a mutant SOD1 model of ALS. Neuroscience. (2017) ;365: :192–205. https://doi.org/10.1016/j.neuroscience.2017.10.001. |
[86] | Keeler AM , Zieger M , Semple C , Pucci L , Veinbachs A , Brown RH , et al. Intralingual and Intrapleural AAV Gene Therapy Prolongs Survival in a SOD1 ALS Mouse Model. Mol Ther Methods Clin Dev. (2019) ;17: :246–57. https://doi.org/10.1016/j.omtm.2019.12.007. |
[87] | Hsu H-L , Brown A , Loveland AB , Lotun A , Xu M , Luo L , et al. Structural characterization of a novel human adeno-associated virus capsid with neurotropic properties. Nature Communications. (2020) ;11: :3279. https://doi.org/10.1038/s41467-020-17047-1. |
[88] | Buchlis G , Podsakoff GM , Radu A , Hawk SM , Flake AW , Mingozzi F , et al. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. (2012) ;119: :3038–41. https://doi.org/10.1182/blood-2011-09-382317. |
[89] | Nathwani AC , Reiss UM , Tuddenham EGD , Rosales C , Chowdary P , McIntosh J , et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. (2014) ;371: :1994–2004. https://doi.org/10.1056/NEJMoa1407309. |
[90] | Nathwani AC , Reiss U , Tuddenham E , Chowdary P , McIntosh J , Riddell A , et al. Adeno-Associated Mediated Gene Transfer for Hemophilia B:8 Year Follow up and Impact of Removing Empty Viral Particles on Safety and Efficacy of Gene Transfer. Blood. (2018) ;132: :491–491. https://doi.org/10.1182/blood-2018-99-118334. |
[91] | Guilbaud M , Devaux M , Couzinié C , Le Duff J , Toromanoff A , Vandamme C , et al. Five Years of Successful Inducible Transgene Expression Following Locoregional Adeno-Associated Virus Delivery in Nonhuman Primates with No Detectable Immunity. Hum Gene Ther. (2019) ;30: :802–13. https://doi.org/10.1089/hum.2018.234. |
[92] | Gouel F , Rolland A-S , Devedjian J-C , Burnouf T , Devos D . Past and Future of Neurotrophic Growth Factors Therapies in ALS: From Single Neurotrophic Growth Factor to Stem Cells and Human Platelet Lysates. Front Neurol. (2019) ;10: :835. https://doi.org/10.3389/fneur.2019.00835. |
[93] | Pozzi S , Thammisetty SS , Codron P , Rahimian R , Plourde KV , Soucy G , et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest. (2019) ;129: :1581–95. https://doi.org/10.1172/JCI123931. |
[94] | Buratti E . Chapter One - Functional Significance of TDP-43 Mutations in Disease. In: Friedmann T, Dunlap JC, Goodwin SF, editors. Advances in Genetics, vol. 91, Academic Press; 2015, pp. 1-53. https://doi.org/10.1016/bs.adgen.2015.07.001. |
[95] | Burrell JR , Halliday GM , Kril JJ , Ittner LM , Götz J , Kiernan MC , et al. The frontotemporal dementia-motor neuron disease continuum. The Lancet. (2016) ;388: :919–31. https://doi.org/10.1016/S0140-6736(16)00737-6. |
[96] | Gross SK , Shim BS , Bartus RT , Deaver D , McEachin Z , Bétourné A , et al. Focal and dose-dependent neuroprotection in ALS mice following AAV2-neurturin delivery. Experimental Neurology. (2020) ;323: :113091. https://doi.org/10.1016/j.expneurol.2019.113091. |
[97] | Lasiene J , Komine O , Fujimori-Tonou N , Powers B , Endo F , Watanabe S , et al. Neuregulin 1 confers neuroprotection in SOD1-linked amyotrophic lateral sclerosis mice via restoration of C-boutons of spinal motor neurons. Acta Neuropathol Commun. 2016;4. https://doi.org/10.1186/s40478-016-0286-7. |
[98] | Mòdol-Caballero G , Herrando-Grabulosa M , García-Lareu B , Solanes N , Verdés S , Osta R , et al. Gene therapy for overexpressing Neuregulin 1 type I in skeletal muscles promotes functional improvement in the SOD1G93A ALS mice. Neurobiol Dis. (2020) ;137: :104793. https://doi.org/10.1016/j.nbd.2020.104793. |
[99] | Azzouz M , Ralph GS , Storkebaum E , Walmsley LE , Mitrophanous KA , Kingsman SM , et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. (2004) ;429: :413. https://doi.org/10.1038/nature02544. |
[100] | Dodge JC , Treleaven CM , Fidler JA , Hester M , Haidet A , Handy C , et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol Ther. (2010) ;18: :2075–84. https://doi.org/10.1038/mt.2010.206. |
[101] | Kaspar BK , Lladó J , Sherkat N , Rothstein JD , Gage FH . Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science. (2003) ;301: :839–42. https://doi.org/10.1126/science.1086137. |
[102] | Lepore AC , Haenggeli C , Gasmi M , Bishop KM , Bartus RT , Maragakis NJ , et al. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. (2016) ;19: :256–65. https://doi.org/10.1016/j.brainres.2007.09.034. |
[103] | Khademullah CS , Aqrabawi AJ , Place KM , Dargaei Z , Liang X , Pressey JC , et al. Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain. (2020) ;143: :800–10. https://doi.org/10.1093/brain/awaa034. |
[104] | Geevasinga N , Menon P , Nicholson GA , Ng K , Howells J , Kril JJ , et al. Cortical Function in Asymptomatic Carriers and Patients With C9orf72 Amyotrophic Lateral Sclerosis. JAMA Neurol. (2015) ;72: :1268–74. https://doi.org/10.1001/jamaneurol.2015.1872. |
[105] | Zhang W , Zhang L , Liang B , Schroeder D , Zhang Z-W , Cox GA , et al. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat Neurosci. (2016) ;19: :557–9. https://doi.org/10.1038/nn.4257. |
[106] | Westeneng H-J , Debray TPA , Visser AE , Eijk RPA van , Rooney JPK , Calvo A , et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. The Lancet Neurology. (2018) ;17: :423–33. https://doi.org/10.1016/S1474-4422(18)30089-9. |


