
Simvastatin Treatment Does Not Ameliorate Muscle Pathophysiology in a Mouse Model for Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy is an X-linked, recessive muscular dystrophy in which the absence of the dystrophin protein leads to fibrosis, inflammation and oxidative stress, resulting in loss of muscle tissue. Drug repurposing, i.e. using drugs already approved for other disorders, is attractive as it decreases development time. Recent studies suggested that simvastatin, a cholesterol lowering drug used for cardiovascular diseases, has beneficial effects on several parameters in mdx mice. To validate properly the effectiveness of simvastatin, two independent labs tested the effects of 12-week simvastatin treatment in either young (starting at 4 weeks of age) or adult (starting at 12 weeks of age) mdx mice. In neither study were benefits of simvastatin treatment observed on muscle function, histology or expression of genes involved in fibrosis, regeneration, oxidative stress and autophagy. Unexpectedly, although the treatment protocol was similar, simvastatin plasma levels were found to be much lower than observed in a previous study. In conclusion, in two laboratories, simvastatin did not ameliorate disease pathology in mdx mice, which could either be due to the ineffectiveness of simvastatin itself or due to the low simvastatin plasma levels following oral administration via the food.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a progressive muscle wasting disorder affecting around 1 in 5000 new-born boys [1, 2]. Patients are usually diagnosed around 3–4 years of age, lose ambulation around 10 years of age and die before 30 years of age due to respiratory and/or cardiac failure [3]. Out-of-frame mutations or premature stop codons in the DMD gene cause the absence of a functional dystrophin protein, which is crucial for muscle fibre stability [4, 5]. The absence of dystrophin triggers pathological pathways leading to fibrosis and chronic inflammation, which in turn results in reduced regeneration and disrupted calcium homeostasis [6].
Although corticosteroids have increased the life expectancy of Duchenne boys, no effective treatment is currently available [7, 8]. Only four medicines aiming to correct the absence of the dystrophin protein have been authorised by regulatory authorities, but these are only applicable to a subset of patients and have moderate effects [9, 10]. This underlines the medical need for Duchenne patients.
In the absence of primary therapies aiming at re-storing the missing protein, therapies addressing other parts of the disease pathology are of interest. These compounds could be beneficial on their own or, in the future, support dystrophin-restoring therapies. Repurposing of drugs authorised for other disorders is especially attractive, since these drugs can be developed quicker, as there is already knowledge on their safety and pharmacokinetics [11]. However, proper investigation of these drugs in non-clinical studies is essential before moving into clinical trials [12].
Statins are 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors which have the ability to lower LDL cholesterol synthesis. They are therefore used for the treatment of cardiovascular disorders, also in children [13]. They also have been reported to have inhibitory effects on inflammation, oxidative stress and fibrosis and to enhance autophagy [14–16], making them a potential repurposing compound for the treatment of DMD.
Previous studies by Whitehead et al. have shown that simvastatin treatment results in improved muscle function and decreased muscle damage in the mdx mouse model [17] and acts on several parts of the disease pathology, i.e. fibrosis, inflammation and oxidative stress [18]. However, it is good practise to validate promising findings in independent laboratories [19–21]. This manuscript describes the findings of two studies (conducted in two independent laboratories) to evaluate the potential effect of simvastatin in mdx mice, using a set of complementary experiments. Surprisingly, despite using similar dosing and administration routes to those used in the previous studies, we were unable to replicate the previous findings. In addition, the serum levels of simvastatin found in our experiments were very low compared to those reported by Whitehead et al., which may contribute to the lack of reproducibility.
MATERIALS AND METHODS
Animals and simvastatin treatment
Experiments performed in the Wells laboratory were approved by the Royal Veterinary College Animal Welfare and Ethical Review Body and by the UK Home Office (PPL 70/7777). Experiments in the Aartsma-Rus laboratory were approved by the Animal Experiment Committee (Dierexperimentencommissie) of the Leiden University Medical Center (permit #13211) and executed following EU-guidelines. The Wells laboratory used mdx male mice (C57Bl/10ScSn background) from a long-standing internal colony and C57Bl/10ScSn male wild type controls from Envigo. Mice were held in a minimum disease unit in open-top cages with environmental enrichment, at 20.5°C with 12-hour dark-light cycles. Water was provided ad libitum. A power calculation based on previously observed variation determined a sample size of a minimum of 10 mice per group for a 20% effect size, allowing for a 20% loss of some samples due to unexpected problems (e.g. incomplete muscle physiology recordings, sample losses or any animal deaths). Personnel performing any of the tests listed below were blinded to group identity and any decisions to remove abnormal data were made prior to unblinding. There were no deaths during dosing and no sample losses. However, one recording for the TA physiology in the mdx control group was lost due to the mouse dying during anaesthesia and one set of recordings of TA physiology in the mdx control group was removed as grossly abnormal (a loss of force at the higher frequencies). A failure of the force transducer for the in vitro system led to incorrect recordings for two mice each in the simvastatin and control mdx groups and these data were removed.
In the Aartsma-Rus laboratory, C57Bl/10ScSn-DMDmdx/J (mdx) and C57Bl/10ScSn-DMD/J (wild type) males were used for all experiments. Mice were bred by the animal facility of the Leiden University Medical Center and were housed in individually ventilated cages at 20.5°C with 12-hour dark-light cycles. Water was provided ad libitum. All efforts were made to minimize suffering. Group sizes were based on previously observed variation in fibrosis levels in the diaphragm of mdx mice. To demonstrate a 20% effect size, a group size of 6 mice was required. Where possible personnel were blinded to group identity. During the experiment one mouse in the simvastatin-treated group and one wild type died, due to problems unrelated to treatment. All other animals are included in the analyses.
The simvastatin used at the Royal Veterinary College was purchased from Sigma-Aldrich, Switzerland (#PHR1438), and was mixed with breeding rodent diet containing 5.5 kcal% fat (#3336, Provimi Kliba SA, Switzerland) at a concentration of 80 mg/kg. At LUMC simvastatin powder (#S0509, TCI America, United States) was mixed with standard rodent diet containing 10 kcal% fat (Research Diets, Inc, United States) at a concentration of 80 mg/kg.
For the oral gavage experiment mice were treated with 8 mg/kg simvastatin activated by hydrolysis of the lactone group as previously described [17].
Study design
A schematic overview of the study design at the RVC and LUMC respectively, is shown in Fig. S1.
Male mdx mice were randomly divided into 2 groups at the age of 12 weeks (RVC) or 4 weeks (LUMC), whereby mice of the same litter were assigned to different treatment groups. One group was fed the diet containing 80 mg/kg simvastatin and the other group control diet (identical but without simvastatin). A wild type control group fed with control diet was taken along as a reference group. After 12 weeks of treatment (age 24 or 16 weeks) mice were sacrificed and muscles were isolated. For an overview of the outcome measures determined at the RVC and LUMC respectively, see Table S1.
For determination of plasma levels separate 14-week old male mice were gavaged in the afternoon with 8 mg/kg of simvastatin, activated as previously described [17], and blood samples were taken 30, 60 and 135 min later by cardiac puncture under very deep isoflurane anaesthesia. Blood was placed immediately in Lithium Heparin BD Microtainer Tubes, inverted several times and placed on ice. Mice were killed by cervical dislocation. Bloods were spun for 5 min at 2000g before plasma was removed and stored at –80°C before being sent to Keystone Bioanalytical, Inc. (North Wales, PA 19454, USA).
Functional tests
At the RVC, at 4 and 8 weeks of treatment mice were subjected in the morning to a four paw hang test as described previously [22] and TREAT-NMD standard operating procedure DMD_M.2.1.005 [23]. The mouse was placed on a grid, which was then turned upside down above a cage filled with bedding. The test ended when a hanging time of 600 s was achieved or, upon earlier fall, after three sessions. The holding impulse associated with the hang test equals the hang time multiplied by the body weight (given as Newtons sec; conversion factor –9.806 x 10-3 Newtons/gm).
Respiratory function analysis
At the LUMC, after 3, 6, 9 and 12 weeks of treatment respiratory rate and tidal amplitude were assessed with non-invasive whole-body plethysmography in unrestrained animals (RM-80; Columbus Instruments, OH, USA). After 30 s acclimatization, the respiration signal was recorded for 120 s. The signal was digitized using a Minidigi digitizer and AXOSCOPE 10 software (Axon Instruments/Molecular Devices, CA, USA) and analysed with the event detection feature of the Clampfit 10 program (Axon Instruments/Molecular Devices).
Muscle physiology
At the RVC, treated mdx mice, control mdx mice and control wild type mice were tested at 24 weeks old with the analysis of tibialis anterior (TA) and diaphragm physiology performed as previously described [24–26]. Briefly, in mice anaesthetised with a mixture of fentanyl/fluanisone (Hypnorm, Vetapharma, Leeds, UK) and midazolam (Hypnovel, Roche, Welwyn Garden City, UK) delivered intraperitoneally, the distal tendon of the TA was released and tied to a 4.0 braided surgical silk loop. The sciatic nerve was exposed and the connection to the spinal cord was cut as well as all the branches except for the common peroneal nerve. The anaesthetised mouse was placed on a thermopad (Harvard Apparatus, Edenbridge, UK) to maintain body temperature at 37°C. The foot was secured to a platform and the knee immobilized using a large sewing needle underneath the patella tendon. Bipolar platinum electrodes were placed on the sciatic nerve to stimulate the muscle.
Once surgically prepared, the loop on the TA tendon was attached to a 305B dual-mode servomotor transducer (Aurora Scientific, Aurora, Ontario, Canada) via a custom-made steel s-hook. Optimal muscle length (Lo) was determined by increasing muscle length and stimulation voltage until the maximal twitch force was achieved. To measure the force–frequency relationship, TA muscles were stimulated at different frequencies, delivered 1 min apart (1, 10, 30, 40, 50, 80, 100, 120, 150 and 180 Hz) using a 701C stimulator (Aurora, Scientific). Maximal isometric force (Po) was determined from the plateau of the force–frequency curve. Muscle fibre cross-sectional area (CSA in cm2) was determined by using the following formula: muscle weight (g)/[dia fibre length (Lf; cm) x 0.6 x 1.06 (g/cm3)]. The 0.6 is the correction for the pennation angle [27]. Specific isometric force (N/cm2) was calculated by dividing the absolute force (N) at each stimulation frequency by TA muscle cross-sectional area. Once Po had been obtained, the muscle was subject to eccentric (lengthening) contractions. The TA muscle was stimulated at 120 Hz for 500 ms before lengthening the muscle by 10% of the Lo at a velocity of 0.5 Lo s–1 for a further 200 ms, once the stimulation had ended the Lo returned at a rate of –0.5 Lo s–1. Between each contraction a 2 min rest period was permitted to avoid muscle fatigue. A total of 10 eccentric contractions were performed on each mouse. After each eccentric contraction, the maximum isometric force was measured and expressed as a percentage of the initial maximum isometric force achieved at the start of the protocol, prior to the first eccentric contraction.
After completing the TA muscle physiology, the mouse was killed by cervical dislocation. TA muscles were removed and immediately weighed prior to snap-freezing in isopentane pre-chilled in liquid nitrogen. The whole diaphragm including the attached ribs was removed and placed in a wax based dissection dish containing cold Ringer’s buffer. The diaphragm was pinned, and a 3–4 mm strip was cut from the central tendon to the ribs along the orientation of the muscle fibres. A custom made 4 mm wide stainless steel wire coat hanger was tied to the ribs using 4-0 surgical braided silk thread and a custom made elongated steel s-hook ( 4 cm) was attached to the central tendon. The muscle was suspended between two platinum electrodes in a water jacketed organ bath containing Ringer’s solution, which was continuously perfused with 95% O2/5% CO2 and maintained at 28°C. The central tendon was anchored to the platform base (via the elongated S-hook), whilst the costal margin was attached to the lever arm of a 300C dual-mode servomoter transducer through the stainless steel coat hanger in the 800A in vitro muscle apparatus (both Aurora Scientific). Electrical stimulation was generated through the two platinum electrodes, using square-wave pulses of 0.02 ms. Data acquisition and control of the servomotor were conducted using a Lab-View-based DMC program (Dynamic muscle control and Data Acquisition, Aurora Scientific).
The diaphragm strip was left to equilibrate for 10 min prior to a warm-up protocol consisting of 3 submaximal contractions at 50 Hz, 500 ms duration, with each contraction delivered one minute apart. After defining the optimal voltage (16 V), muscle length (Lo) was determined by increasing the muscle length until a maximal twitch force was achieved. The force-frequency relationship was evaluated by stimulating the muscle at the same set of frequencies as for the TA. Maximal isometric force (Po) was determined from the plateau of the force-frequency curve. Resistance to eccentric contraction-induced muscle damage was assessed by stimulating the diaphragm in the same protocol as the TA but at 100 Hz.
Diaphragm strips were removed from the water bath and the muscle carefully dissected away from the rib and central tendon. The muscle was blotted to remove excess liquid and immediately weighed prior to snap-freezing in isopentane pre-chilled in liquid nitrogen.
Muscle fibre cross-sectional area (CSA in cm2) was determined as for the TA but without a correction for pennation angle as the diaphragm is not a pennate muscle. Specific isometric force (N/cm2) was calculated by dividing the absolute force (N) at each stimulation frequency by diaphragm muscle fibre cross-sectional area.
At the LUMC, directly after killing the mice using CO2 asphyxiation, the contraction force of left phrenic nerve–hemidiaphragms with a strip of thorax left attached was recorded in Ringer’s medium at room temperature (20–22°C), continuously bubbled with 95% O2 and 5% CO2. The muscles were pinned down with multiple small pins through the thorax strip into the silicone rubber-lined bottom of a 10 mL Ringer’s medium containing Petri dish. The central tendon was attached via a hook and thread to a force transducer (type K30, Harvard Apparatus–Hugo Sachs Elektronik, Germany). The signal was amplified by a TAM-A bridge amplifier (Harvard Apparatus) and digitized by a Digidata 1440 digitizer (Axon Instruments/Molecular Devices) connected to a personal computer running Axoscope 10 data-acquisition software (Axon Instruments/Molecular Devices). The phrenic nerve was stimulated via a bipolar electrode with rectangular voltage pulse of 100μs duration from a computer-controlled programmable stimulator via an ISO-flex stimulus isolator/amplifier (AMPI). First, the minimal stimulus voltage amplitude at which the preparation started to contract, using 1 s/40 Hz tetanic stimulations, was determined. This was mostly around 0.5–1 V. The stimulus amplitude was then increased in small voltage steps to determine the amplitude at which the tetanic contraction no longer increased (typically 2 V). The voltage was then set to 10 V to assure supramaximal stimulation of all axons in the nerve during the entire experiment. To determine the optimal length of the muscle for producing maximal tetanic contraction, the muscle was stretched in small increments, using a Vernier control, each time followed by a 1 s/40 Hz tetanic stimulation. When the tetanic force no longer increased upon a length increment, no further lengthening was performed, and this optimal length was kept during the following experiment. Subsequently, every 5 min a 40 Hz nerve stimulus train of 7 s duration was applied and the tetanic contraction was recorded when it remained stable for at least 15 min. Thereafter, a stimulus protocol was applied of 7 s stimulations at 1, 2, 5, 10, 20, 40, 60, 80 and 100 Hz, with a 30 s pause between each stimulation. Peak values and area-under-the-curves of the responses were determined using Clampfit 10 software (Axon Instruments/Molecular Devices).
Simvastatin measurement in plasma
For the RVC, simvastatin and simvastatin acid levels were measured in mouse lithium heparin plasma using electrospray ionisation mass liquid chromatography with tandem mass spectrometry (ESI LC-MS/MS). LC-MS/MS for the RVC samples was performed by Keystone Bioanalytical, Inc. (North Wales, Pennsylvania, USA) and analysed by Solid Biosciences Inc. (Cambridge, Massachusetts, USA).
At LUMC, blood was taken via the vena cava directly after sacrifice and collected in K2EDTA BD Microtainer™ tubes (Thermo Fisher Scientific Inc., the Netherlands), and centrifuged at 13000 rpm for 5 min at 4°C to generate plasma. Citric acid (Citric acid ACS reagent, Sigma Aldrich, the Netherlands) was added at a concentration of 10 mg/mL and stored at –80°C until further processing. Simvastatin and internal standards were isolated by liquid-liquid extraction using methyl t-butyl ether as a solvent and dried supernatant was reconstituted in MeOH/water/acetic acid. An autosampler (SIL-30AC, Shimadzu, Japan), ZORBAX StableBond Phenyl columns (Agilent) HPLC pump (LC-20AD, Shimadzu) and tandem MS (API 5500 QTRAP, AB Sciex LLC) were used for mass spectrometry analysis.
Hydroxyproline assay
Hydroxyproline content was assayed essentially as described by Reddy and Enwemeka [28]. Diaphragm muscles (from treated and untreated mdx mice, and wild type controls) were pulverised under liquid nitrogen. Frozen tissue powders (10–20 mg) were mixed with 20 volumes (v/w) of distilled water and a 5μL aliquot was collected for protein assay (DC, BioRad). Remaining tissue suspension was mixed with an equal volume of 10 N HCl and transferred to gas-tight glass vials, then heated to 130°C for at least 3 hours to allow complete acid hydrolysis. 50μL aliquots of hydrolysed solution were transferred (in duplicate) to clear, flat-bottomed 96-well plates (Corning) and placed in a well-ventilated 60°C oven to dry (30–40 min). A standard dilution series of pure hydroxyproline (dissolved in distilled water) was included on every plate. Hydroxyproline was oxidised by addition of 100μL chloramine-T buffer (prepared as described below) followed by incubation at room temperature for 20 min with gentle shaking. Colour was developed by addition of 100μL of p-dimethylaminobenzaldehyde (DMAB, Ehrlich’s reagent, see below) and incubation at 60°C for 20 min. Absorbance was read at 550 nm using a plate reader (Tecan). Chloramine-T buffer (prepared fresh): 1.27 g chloramine-T dissolved in 20 mL of 50% n-propanol, then added to 80 mL of acetate-citrate buffer (120 g sodium acetate trihydrate, 12 mL acetic acid, 34 g sodium hydroxide, 46 g citric acid, 1 L final volume, pH 6.5).
Ehrlich’s reagent (prepared fresh): 15 g DMAB in 100 mL of 2 : 1 (v/v) n-propanol:perchloric acid. All reagent volumes can be adjusted accordingly depending on number of plates to be assayed.
RNA isolation and quantitative PCR
At RVC, RNA was isolated from frozen muscle powders (as prepared for hydroxyproline assay, above) using TRIzol reagent (Thermofisher) according to manufacturer’s instructions, with inclusion of an additional chloroform extraction (1 : 1) following phase separation. RNA yield and purity were confirmed by nanodrop (ND1000). cDNA was synthesised from 800 ng RNA using the Nanoscript2 RT kit (Primerdesign, United Kingdom). SYBR green qPCR was performed using PrecisionPLUS mastermix (Primerdesign, United Kingdom) in a LightCycler CFX384 (BioRad, United Kingdom), using 8 ng cDNA per well (assuming 1 : 1 conversion). Following 3 min hot-start, PCR used 40 cycles of 95°C (15 s), 60°C (20 s) and 72°C (20 s). All qPCR runs included a melt curve following the final cycle. Cq values were determined via regression and normalized to the geometric mean of CDC40 and Fbxo38 (taken from the geNormPLUS set, Primerdesign, United Kingdom).
At LUMC, muscle sections were collected in 1.4 mm Zirconium Beads prefilled tubes (OPS Diagnostics, Lebanon, USA) and disrupted in TRIsure isolation reagent (GCBioetech, the Netherlands) using a MagNA Lyser (Roche Diagnostics, the Netherlands). Total RNA was isolated and cleaned up by applying a NucleoSpin RNA II kit (Macherey-Nagel, Germany) according to the manufacturer’s instruction. cDNA was synthesised from 0.5μg of total RNA using random N6 primers (Thermo fisher scientific) and Bioscript enzyme (GCBiotech, the Netherlands) according to the manufacturer’s instructions. qPCR was performed using SensiMix reagents (GCBiotech) and the LightCycler 480 (Roche), using a program consisting of 45 cycles of 95°C (10 s), 58°C (30 s) and 72°C (20 s). The LinReg qPCR method was used to analyse gene expression levels and values were normalized utilizing the housekeeping gene Hmbs after confirming equal expression between samples from wild type and mdx, and simvastatin-treated and untreated mdx mice.
Gene expression levels were determined for Fbxo32 (Atrogin-1), Cd68, Col1a1, Col3a1, Lgals3, Map1lc3b, Myh1, Myh2, Myh3, Myh4, Myh7, Myh8, Nox2, Nox4. Primer sequences are provided in Table S2.
Sirius Red staining
At LUMC, directly after sacrifice diaphragm muscles were, embedded in Optimal Cutting Temperature (OCT) compound (TissueTek; Sakura Finetek, CA,USA), snap frozen in liquid nitrogen cooled isopentane. Sections (8μm thickness) were cut using with a cryostat (Leica CM3050 S Research Cryostat; the Netherlands). Interleaving sections were collected for RNA and protein analysis. Levels of collagen were quantified by Sirius Red staining. Sections were fixed in 4% paraformaldehyde for 10 min, fixed in 100% ethanol for 5 min and air dried for 30 min. Sections were washed in deionised water and stained for 45 min using Sirius Red solution (Direct Red 80; Sigma-Aldrich). Thereafter, they were washed with 0.5% acetic acid water for 5 min and rinsed in deionized water. Sections underwent dehydration steps using ethanol (80% –90% –100%) and after incubation in xylene for two times 5 min, were mounted in Pertex mounting medium (VWR International B.V). The stained muscle sections were imaged using a BZ-X700 fluorescent microscope (Keyence) at ten times magnification and processed and stitched using BZ-X Analyzer (Keyence). Adobe Photoshop CC 2018 (Adobe Systems Corporation, San Jose, CA, United States) was used for background correction and ImageJ software (NIH) for analysis. Sirius Red positive areas were normalised to the total area. Two examiners independently assessed the images and the average of their results was used for analysis.
Protein analysis
At LUMC, TA muscles were homogenised in buffer containing 100 mM Tris-HCl (pH 6.8)- 20% (w/v) sodium dodecyl sulfate (SDS) in 1.4 mm Zirconium Beads prefilled Tubes (OPS Diagnostics LLC, NJ, United States), using a MagNaLyzer (Roche). Protein concentrations were determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, MA, United States) according to manufacturer’s instructions. Samples containing 30μg of total protein were prepared in buffer containing 75 mM Tris-HCl (pH 6.8), 15% (w/v) SDS, 20% (v/v) glycerol, 5% (v/v) β-Mercaptoethanol, and 0.001% (w/v) Bromophenol blue and heated for 5 min at 95°C. A wild type control containing 5–50% of protein was included. Samples were loaded on 1.0 mm thick Criterion XT Tris acetate (poly-acrylamide) gels with a linear resolving gel gradient of 3–8% (the Netherlands). Gels were run at 75 V ( 0.07 A) for 1 hour and at 150 V ( 0.12 A) for 2 hours on ice. Proteins were blotted onto ready-to-use Trans-Blot Turbo transfer pack using the Trans-Blot Turbo system (BioRad) at 2.5 A ( 25 V) for 10 min. Membranes were blocked for 1 hour in buffer containing 5% Non-fat dried milk powder (Elk; Campina, the Netherlands). Membranes were washed once in TBST buffer containing 10 mM Tris-HCl (pH 8.0), 0.15 M NaCl and 0.005% (v/v) Tween20, incubated overnight at 4°C in primary antibody, washed 3 times in TBST buffer, incubated for 1 hour at 4°C in secondary antibody, and washed twice in TBST and once in TBS. As primary antibodies mouse anti-gp91[phox] (1 : 4000; #611415, BD Biosciences, the Netherlands) and rabbit anti-Gapdh [EPR6256] (1 : 50 000; #ab128915, Abcam, the Netherlands), and as secondary antibodies IRDye 800CW goat-anti-mouse IgG (1 : 5000; #P/N 926-32210; LI-COR Biotechnology, Germany) and IRDye 680TL goat-anti-rabbit IgG (1 : 10 000; #P/N 926-68021, LI-COR) were used. Proteins were visualized using the Odyssey system and software (LI-COR).
Statistical analysis
For statistical analysis RStudio [29] and Prism 7 (GraphPad Software Inc., CA, USA) were used. Values are presented as means±standard deviation (SD). Statistical significance was set at p < 0.05.
Holding impulse, endpoint bodyweight and gene expression were analysed using a nonparametric Kruskal-Wallis test and Dunn’s post-hoc correction for multiple comparison. Protein expression was compared with an unpaired, two-tailed t-test with Welch’s correction. In order to model the data most accurately for bodyweight, eccentric contraction and tetanic contraction force a log-transformed mixed-effects model analysis, using the lme4 package [30] was performed using fixed effects for treatment and fixed and quadratic effects for contraction number, stimulation frequency or weight (including their interactions). Since eccentric contraction curves showed variability in curvature between mice, a random intercept and a random contraction number-effect per mouse was included. Treatments were compared with an ANOVA test using a likelihood ratio test. The baseline and maximum force of the diaphragm and TA were compared using a one-way ANOVA between treatments groups with Tukey’s multiple comparison test to correct for multiple testing.
RESULTS
The dose and route of administration chosen for these studies (food containing 80 mg/kg simvastatin) were similar to the previously published study [17]. This led to an intake of 7–10 mg/kg/day, whereby no differences in total food intake were seen between the different groups. A daily dose of around 10 mg/kg would correspond to a dose of 1.2 mg/kg in children [31] and is thereby within the dose recommended for the treatment of conditions for which simvastatin has been approved [13, 32].
Since the diaphragm is the most severely affected muscle in mdx mice, these were used for most analyses [33].
The effect of simvastatin after onset of muscle damage
First the effect of simvastatin after the onset of muscle damage was investigated by starting simvastatin treatment at 12 weeks of age (RVC). Mdx mice were heavier than wild type, but no effect of 12 weeks of simvastatin treatment was seen on bodyweight (data not shown).
Functional performance
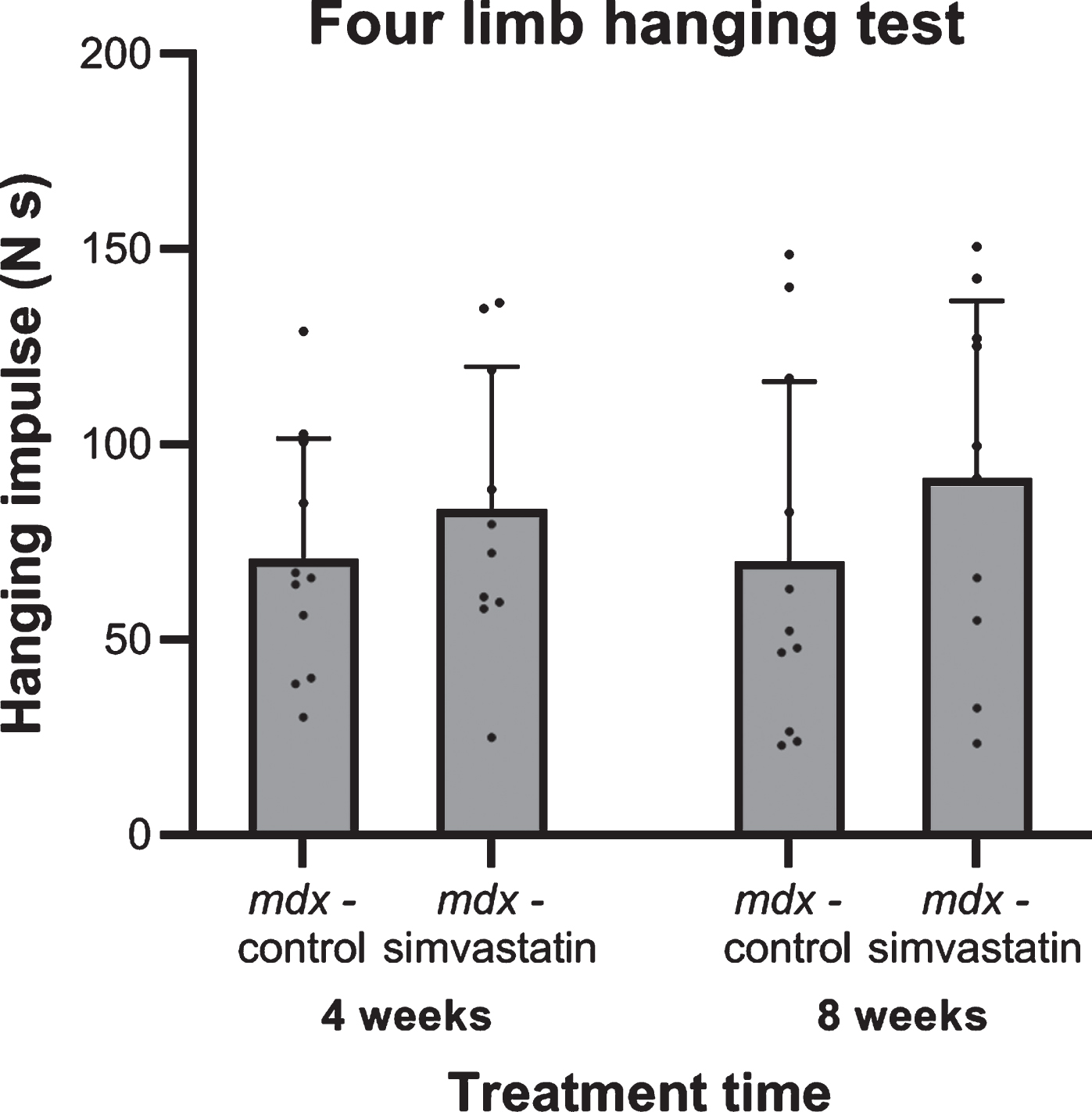
After 4 and 8 weeks of treatment (16 and 20 weeks of age respectively) the endurance of the mice was tested using a four limb hanging test. Although holding impulses were shorter in mdx mice compared to their wild type counterpart, as has been seen in previous studies [22, 34], no differences in holding impulse were seen between the control and simvastatin-treated mdx mice, and there was no change over time within the groups (Fig. 1).
Fig. 1
Four limb hanging test after 4 and 8 weeks of treatment for mice started simvastatin treatment at 12 weeks of age (n = 10–11). No significant differences were seen between the mdx controls and simvastatin treated mdx mice at either time.
Muscle physiology
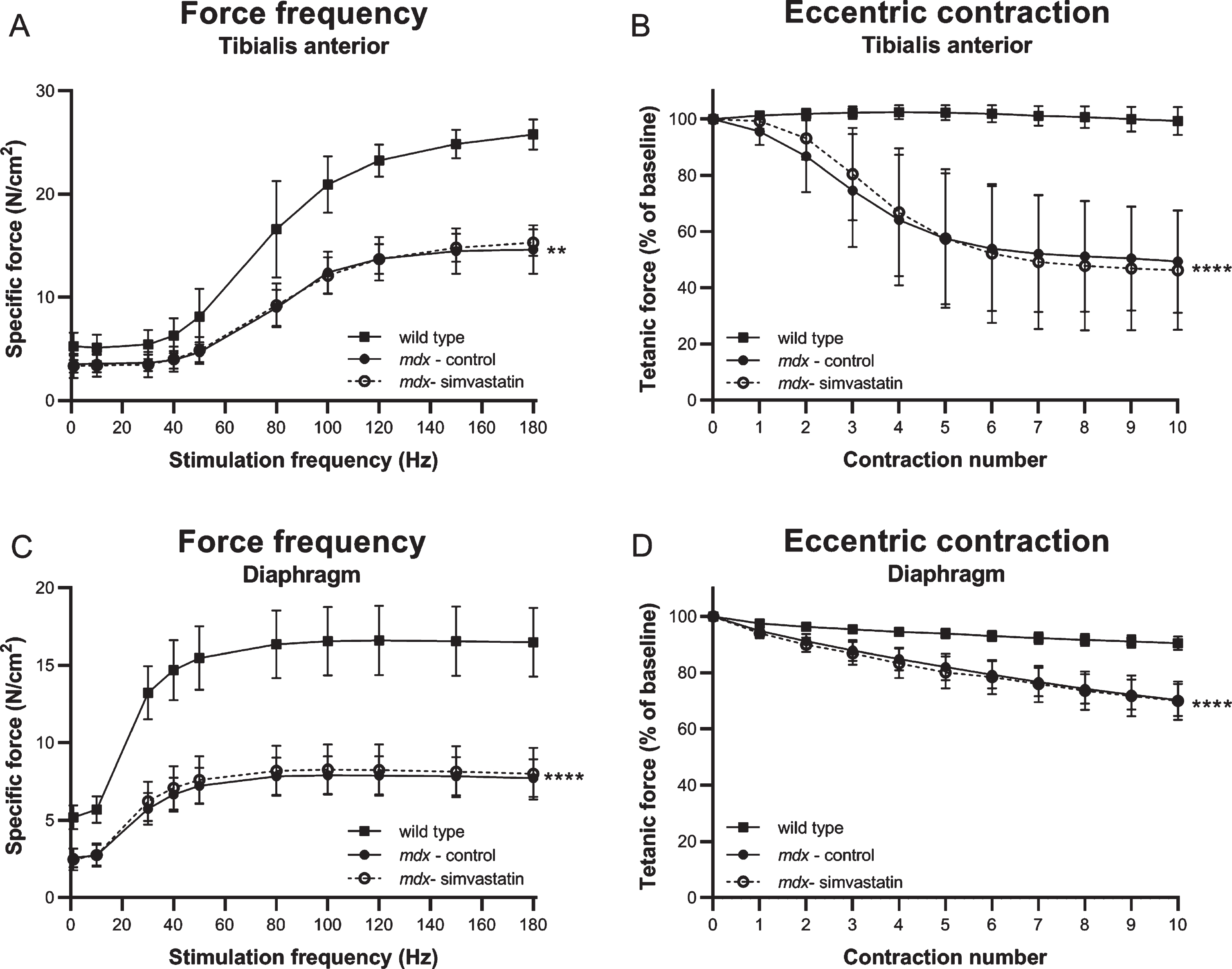
At the endpoint, force frequency relationship (a measure of muscle strength), and response to eccentric contractions (a measure of membrane integrity), were determined in the TA by stimulation of the sciatic nerve in situ in anaesthetised mice, and in diaphragm strips ex vivo by direct muscle stimulation.
Mdx TA muscles produced significantly less force and exhibited the expected significant drop in tetanic strength following eccentric contraction. The force deficit and sensitivity to eccentric damage was not prevented by simvastatin treatment (Fig. 2A–B). In the diaphragm, muscle function was similarly impaired in mdx mice and again no amelioration was observed after simvastatin treatment (Fig. 2C–D).
Fig. 2
Muscle strength and membrane integrity after direct muscle stimulation. Force frequency and response to eccentric contractions in 24–week old mice after 12 weeks of simvastatin treatment. (A–B) Tibialis anterior (in situ). (C–D) Diaphragm, (in vitro) (n = 8–12). **p < 0.01, ****p < 0.0001 compared to wild type mice.
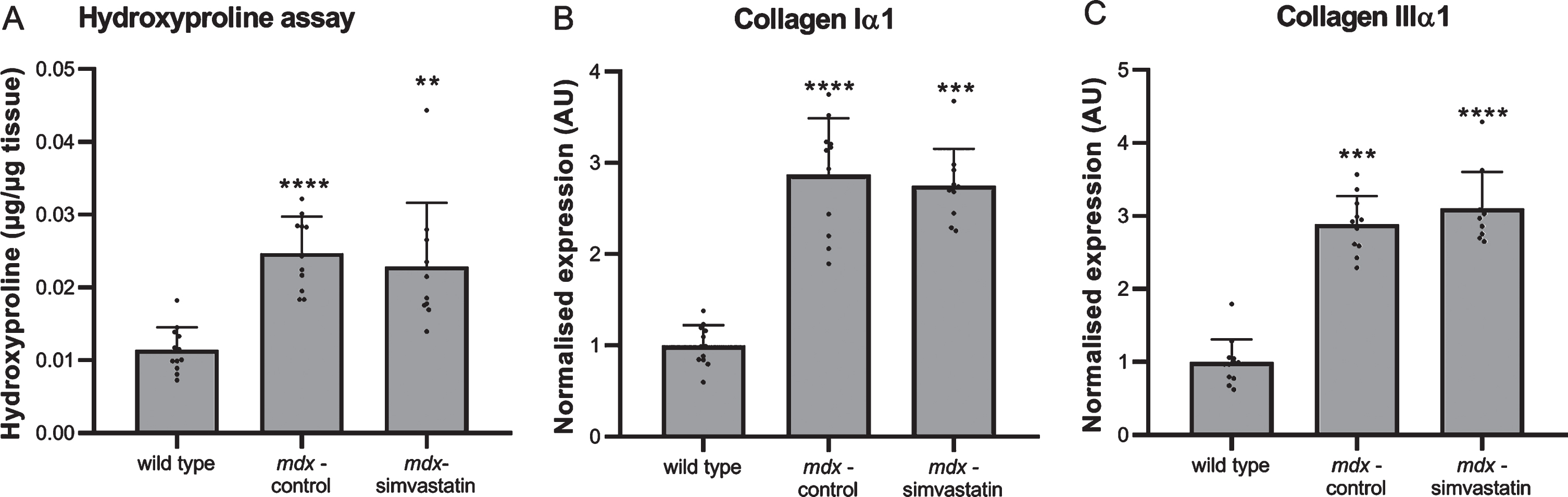
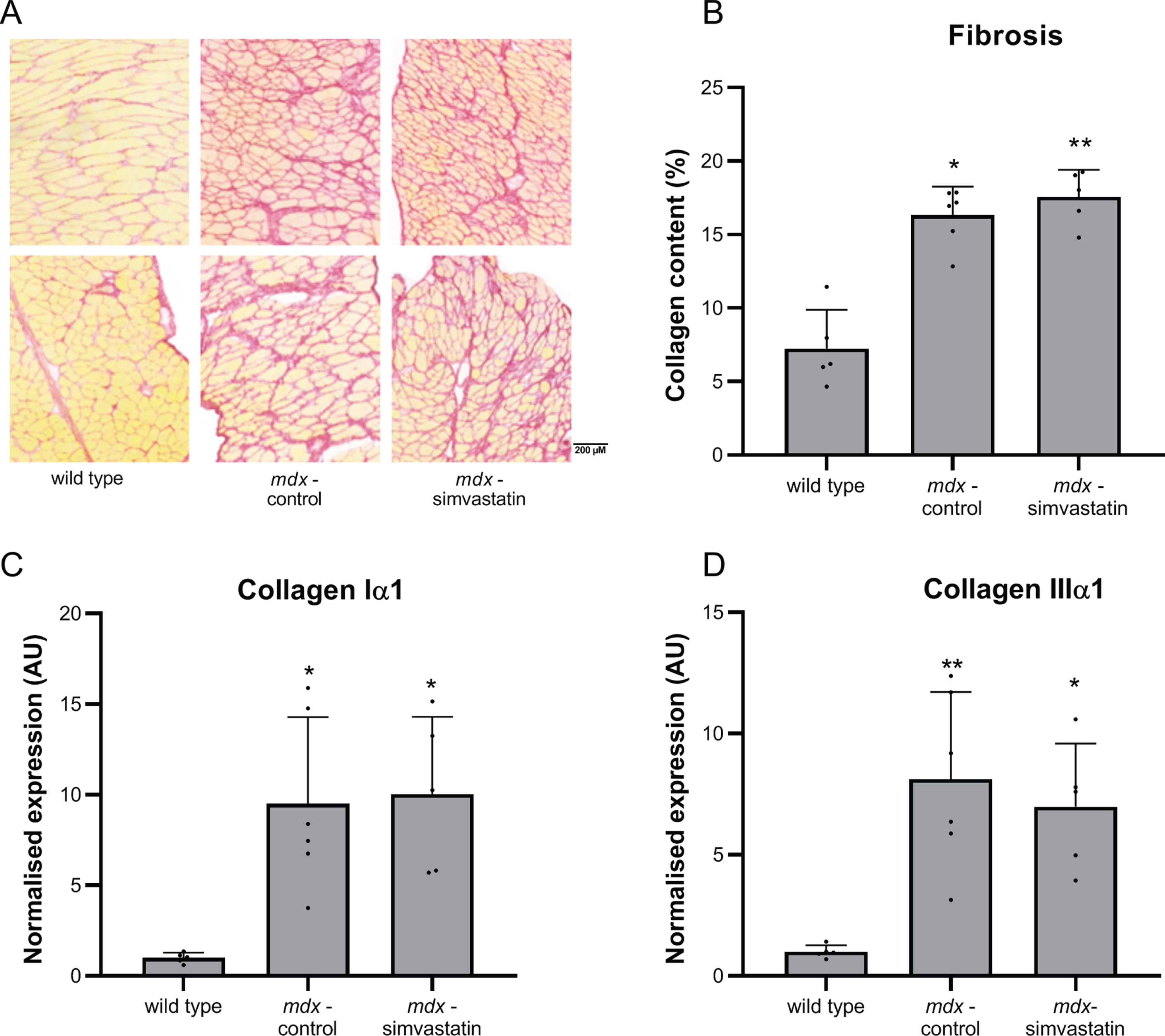
Fibrosis
Fibrosis is a hallmark of DMD, and diaphragms of mdx mice typically show severe fibrotic accumulation. We measured fibrosis in the diaphragm by determining the collagen content using a hydroxyproline assay, and by expression of collagen type I and III using qPCR (Fig. 3). As expected, extensive fibrosis is present in mdx mouse diaphragm, which show an increased collagen content (Fig. 3A) and expression of fibrotic markers when compared with wild type diaphragms (Fig. 3B–C). Simvastatin treatment did not affect the level of fibrosis.
Fig. 3
Fibrosis. Diaphragm of 24–week old mice after 12 weeks of simvastatin treatment. (A) hydroxyproline content. (B–C) expression of collagen type I (B) and III (C) (n = 10–12). **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to wild type mice.
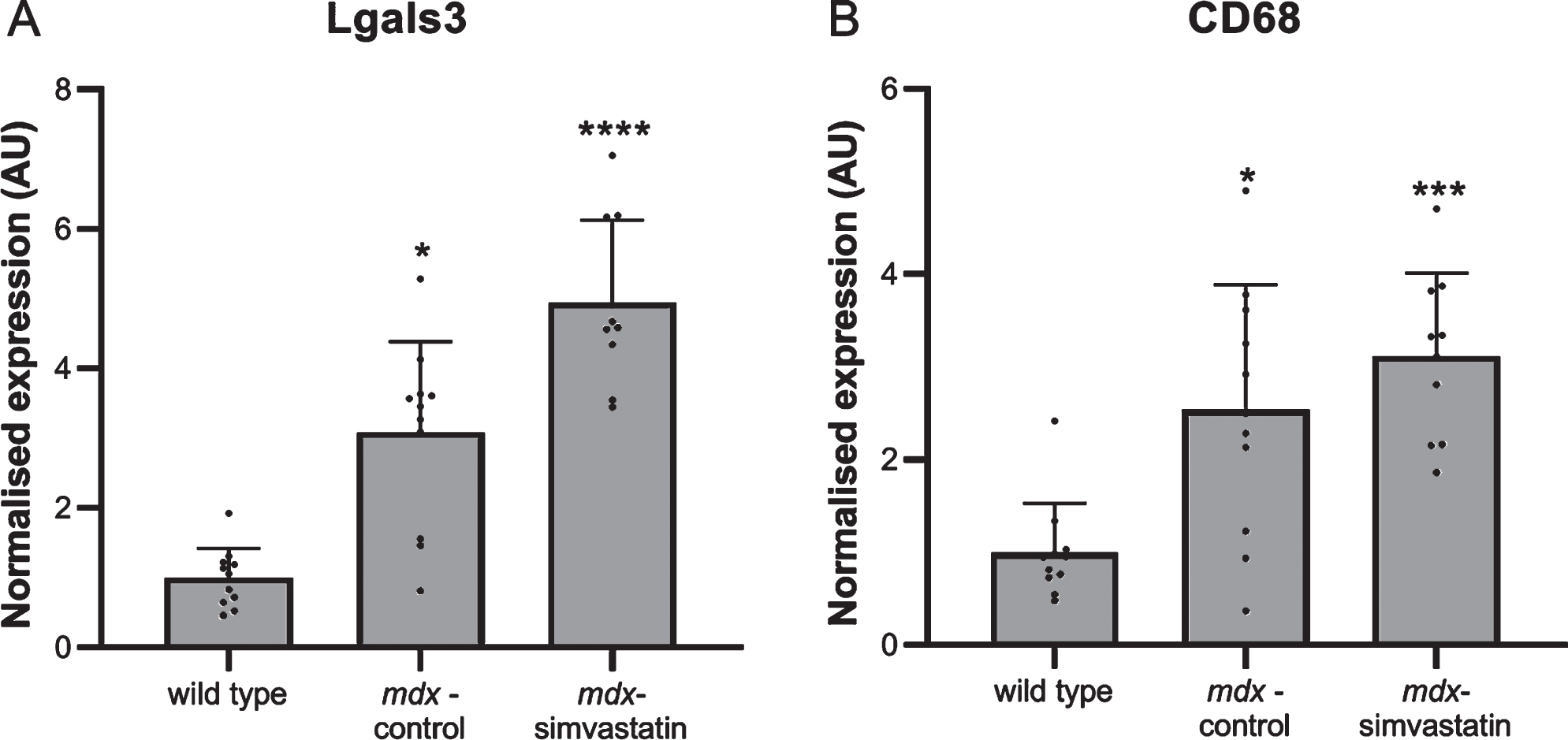
Inflammation
Inflammation in the muscles is often seen in DMD. Again a large increase in the expression of the inflammatory markers lectin, galactoside binding soluble 3 (Lgals3) and cluster of differentiation 68 (Cd68) was seen in the diaphragm of the mdx mice. This was not prevented by simvastatin treatment (Fig. 4).
Fig. 4
Inflammation. Expression of (A) Lgals3 and (B) CD68 in the diaphragm of 24-week old mice after 12 weeks of simvastatin treatment (n = 10–12). *p < 0.05, ***p < 0.001, ****p < 0.0001 compared to wild type mice.
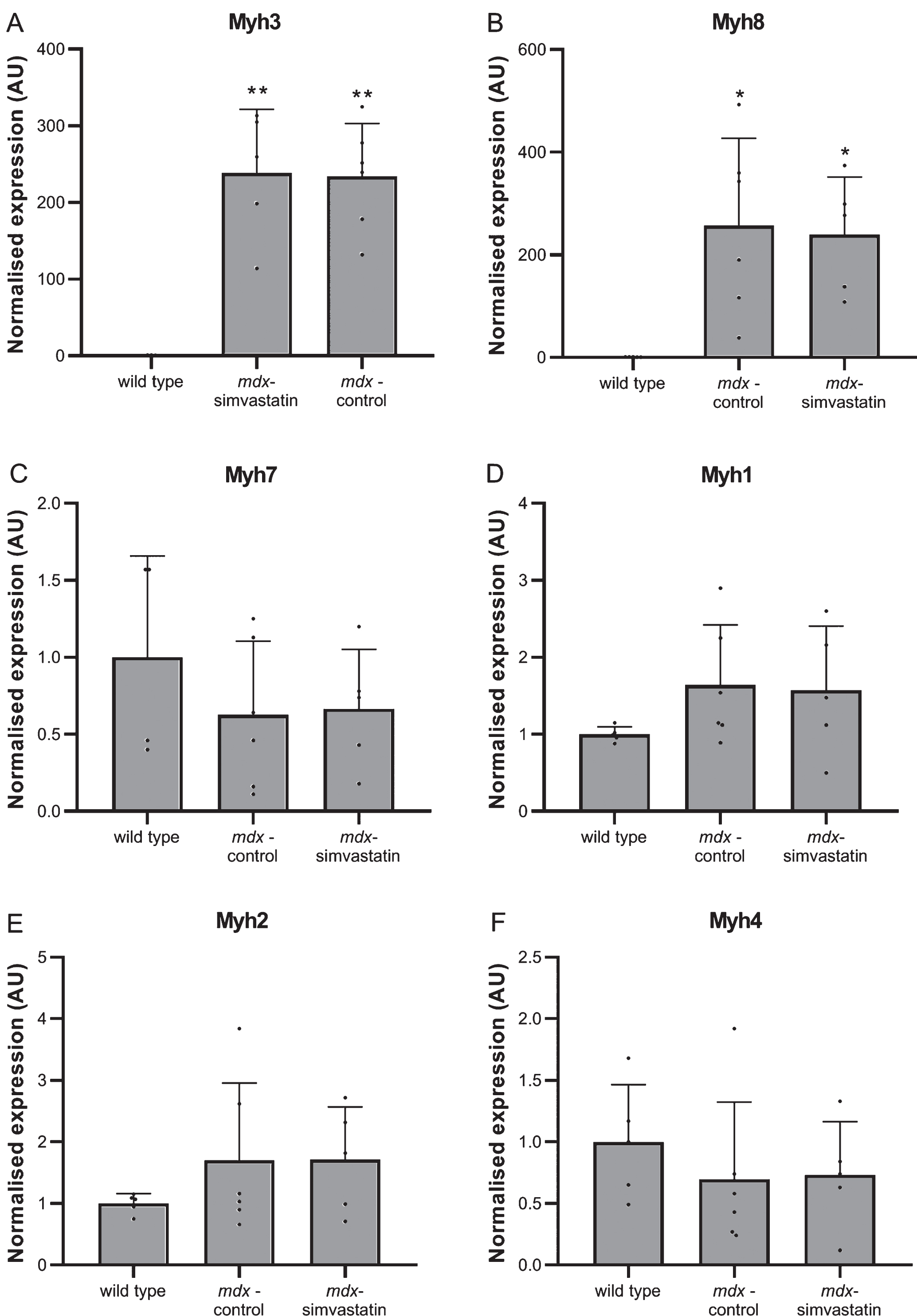
Degeneration and regeneration
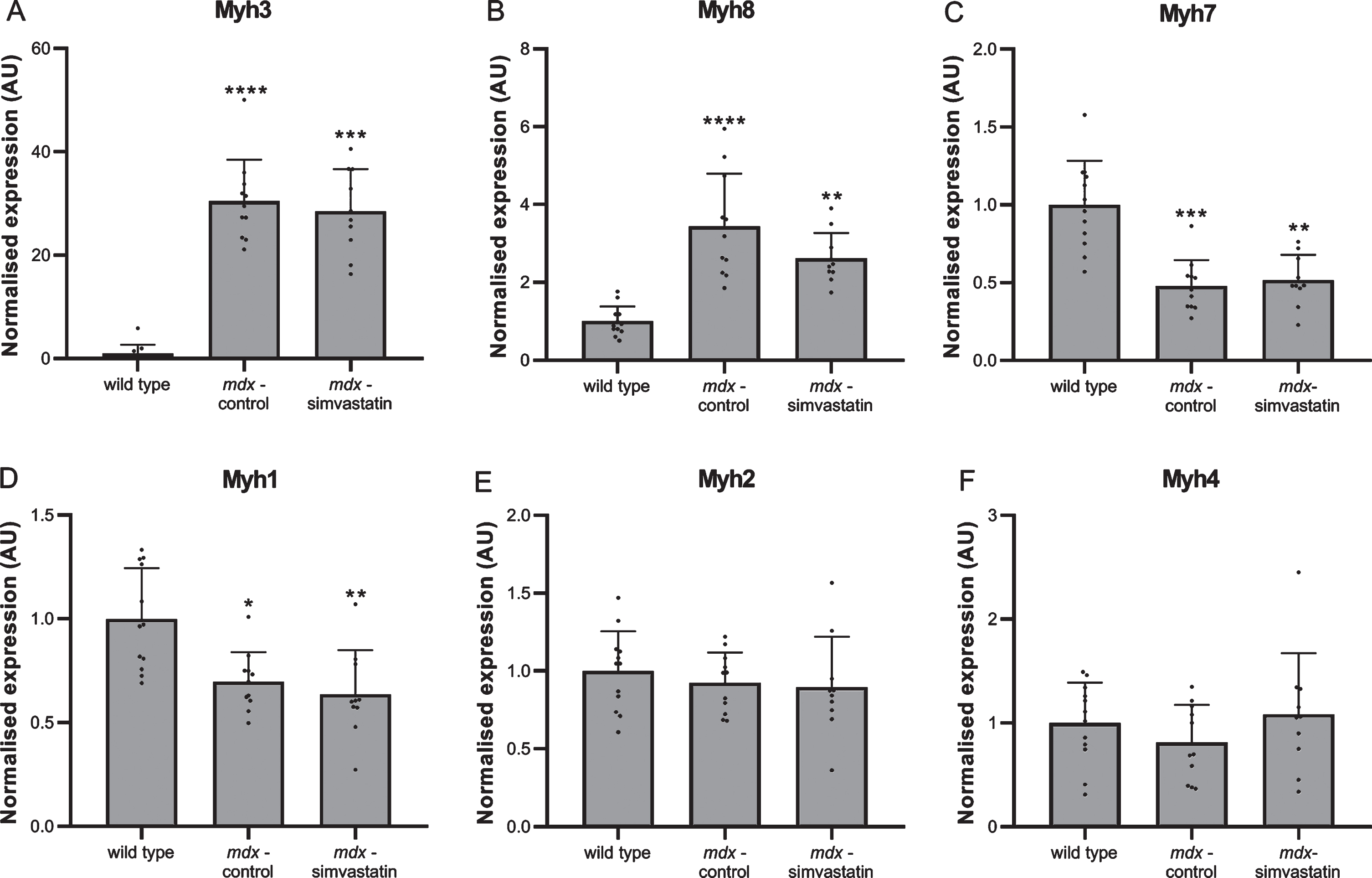
Muscle fibre degeneration and regeneration was assessed using myosin heavy chain (MHC) expression. Levels of several different isoforms were measured in the diaphragm [35, 36]. The expression of both embryonic (Myh3) and neonatal (Myh8) MHC was very low in wild type mice, indicating negligible levels of regeneration are present. In mdx mice continuous cycles of degeneration and regeneration are ongoing, leading to high levels of both forms of MHC (Fig. 5A–B). In adult muscle, different isoforms are expressed depending on the type of muscle. Type Iβ (Myh7) is present in slow-twitch fibres and type IIx (Myh1), IIa (Myh2) and IIb (Myh4) in fast-twitch fibres. Both the slow-twitch MHCIβ and fast-twitch MHCIIx were decreased in mdx diaphragms relative to wild type (Fig. 5C–D), while no differences were seen in MHCIIa and MHCIIb (Fig. 5E–F). Simvastatin-treated diaphragms were essentially indistinguishable from untreated mdx.
Fig. 5
De- and regeneration. Expression of different isoforms of myosin heavy chain in the diaphragm of 24–week old mice after 12 weeks of simvastatin treatment. (A) embryonic (Myh3). (B) neonatal (Myh8). (C) adult slow-twitch type Iβ (Myh7). (D–F) adult fast-twitch type IIx (Myh1) (D), IIa (Myh2) (E) and IIb (Myh4) (F) (n = 10–12). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to wild type mice.
Autophagy and oxidative stress
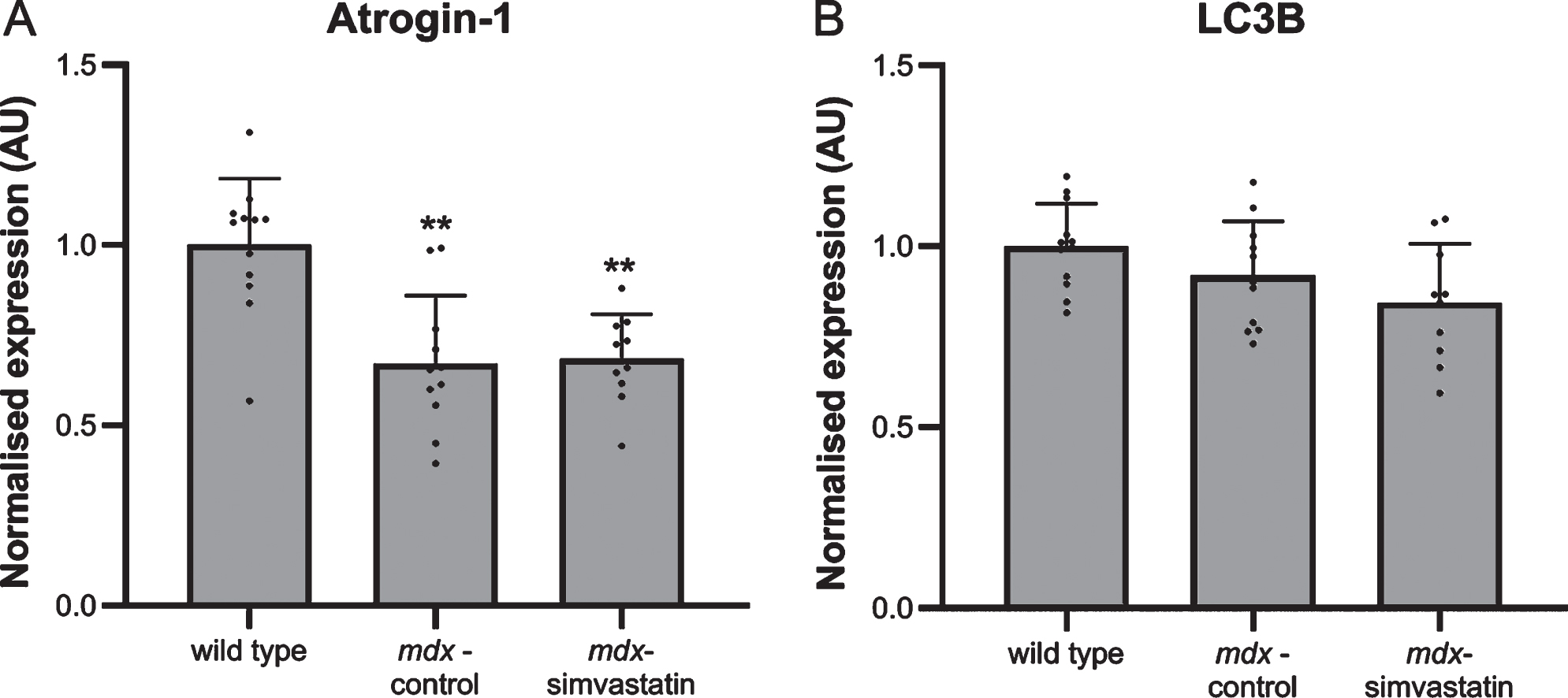
Autophagy plays a role in the DMD pathology. Autophagy is impaired in mdx muscles, leading to muscle atrophy [37]. Induction of atrogin–1 (FBXO32) expression is one of the pathways via which statins may induce muscle atrophy [38]. In the quadriceps of mdx mice, however, such an effect has not been seen [17]. Indeed, atrogin–1 levels in diaphragm were shown to be lower in mdx mice compared to wild type mice and were not influenced by simvastatin treatment (Fig. 6A).
Increases in autophagy have been shown to be associated with induction of microtubule-associated protein 1 light chain 3β (LC3B) [16, 39]. Indeed, Whitehead et al. reported an increase in LC3B protein levels after simvastatin treatment of mdx mice (though the authors did not examine healthy controls) [17]. In the present study, no differences in LC3B gene expression were seen both between mdx and wild type mice and between simvastatin-treated and non-treated mdx mice (Fig. 6B).
Fig. 6
Autophagy. Expression of (A) atrogin–1 and (B) LC3B in the diaphragm of 24–week old mice after 12 weeks of simvastatin treatment (n = 10–12). **p < 0.01 compared to wild type mice.
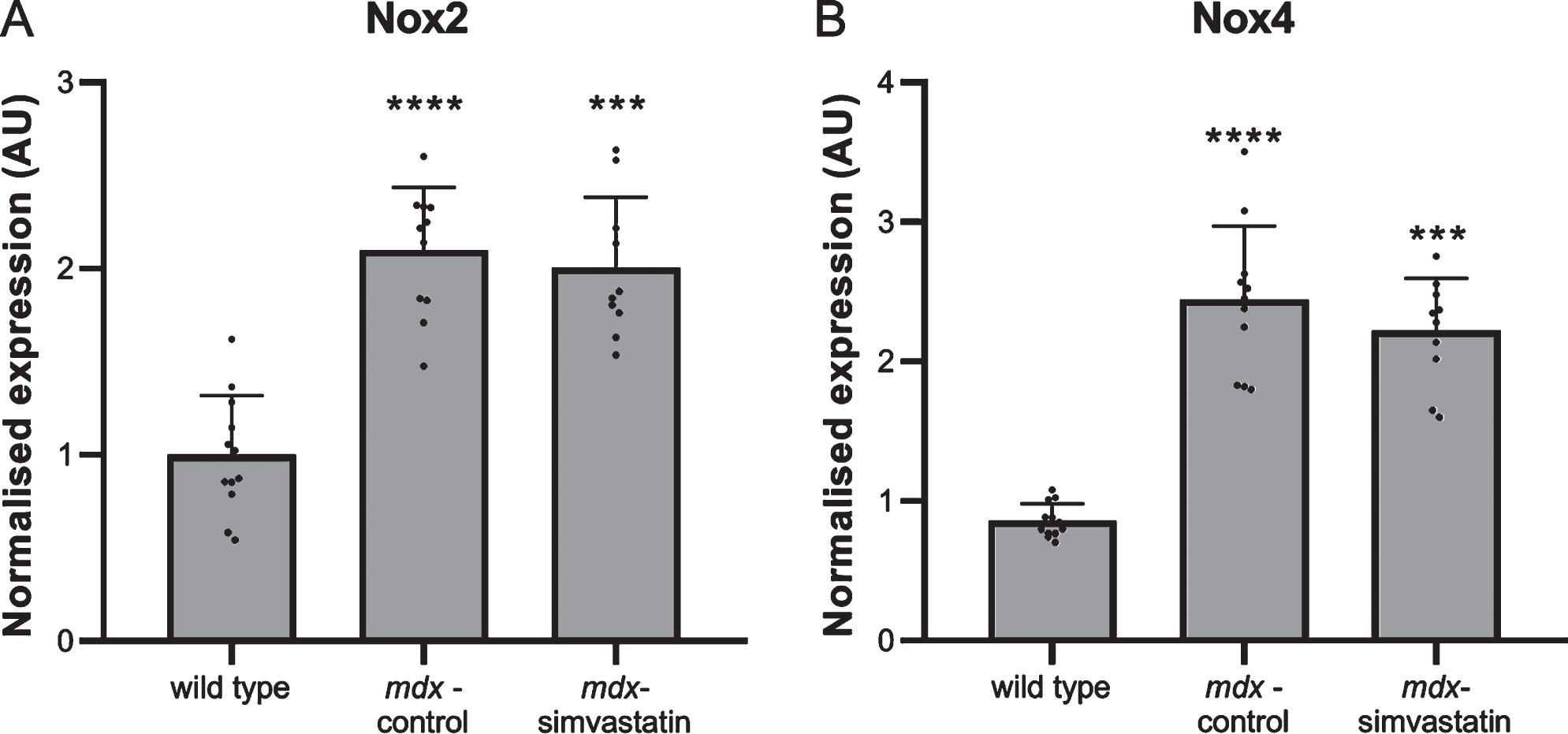
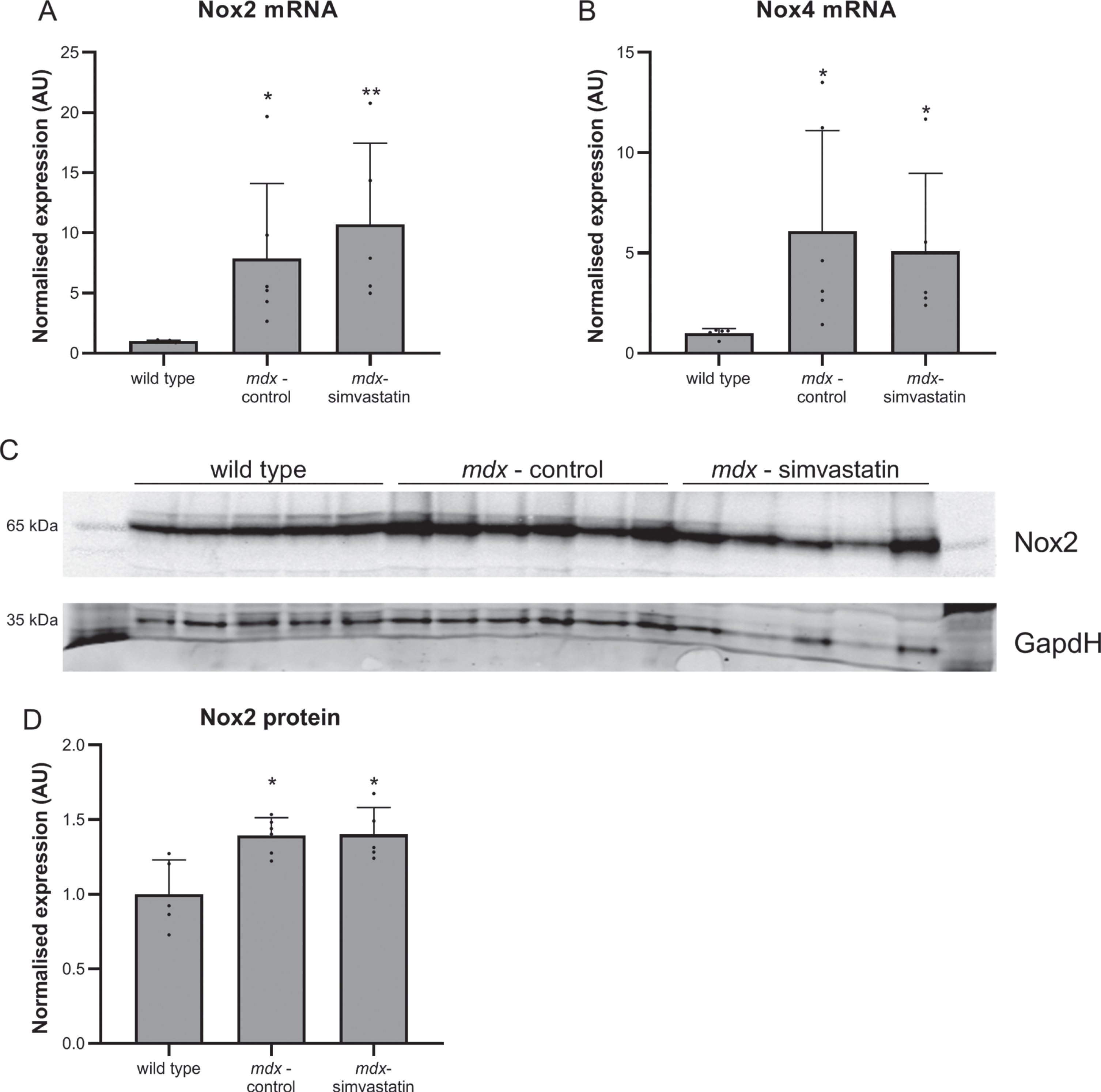
Another mechanism leading to impaired auto-phagy is oxidative stress [40, 41]. NADPH oxidases (Nox) are involved in the production of reactive oxygen species (ROS). Nox2 and Nox4 are two homologs found in skeletal muscle [42]. Statins are reported to decrease Nox2 and Nox4, thereby decreasing ROS and rescuing autophagy [43–45]. Both Nox2 and Nox4 expression were increased in diaphragms of mdx mice, but these increases were unaffected by simvastatin treatment (Fig. 7).
Fig. 7
Oxidative stress. Expression of (A) Nox2 and (B) Nox4 in the diaphragm of 24–week old mice after 12 weeks of simvastatin treatment (n = 10–12). ***p < 0.001, ****p < 0.0001 compared to wild type mice.
The effect of simvastatin during development of muscle damage
In mdx mice the first signs of muscle damage (necrosis, apoptosis and inflammation) are seen around three weeks of age. Thereafter, cycles of degeneration and regeneration are ongoing until around 12 weeks of age, after which the cycling rate falls. In contrast to DMD patients, mdx muscle recovers relatively well [46]. To investigate the effect of simvastatin treatment starting at the onset of muscle damage, four-week old mdx mice were treated with simvastatin for 12 weeks (LUMC). Although mdx mice gained more weight than wild type mice, simvastatin treatment did not affect weight gain (data not shown).
Respiratory function and muscle physiology
During treatment, the respiratory function (rate and amplitude) of the mice was followed. This was not affected by simvastatin treatment (data not shown).
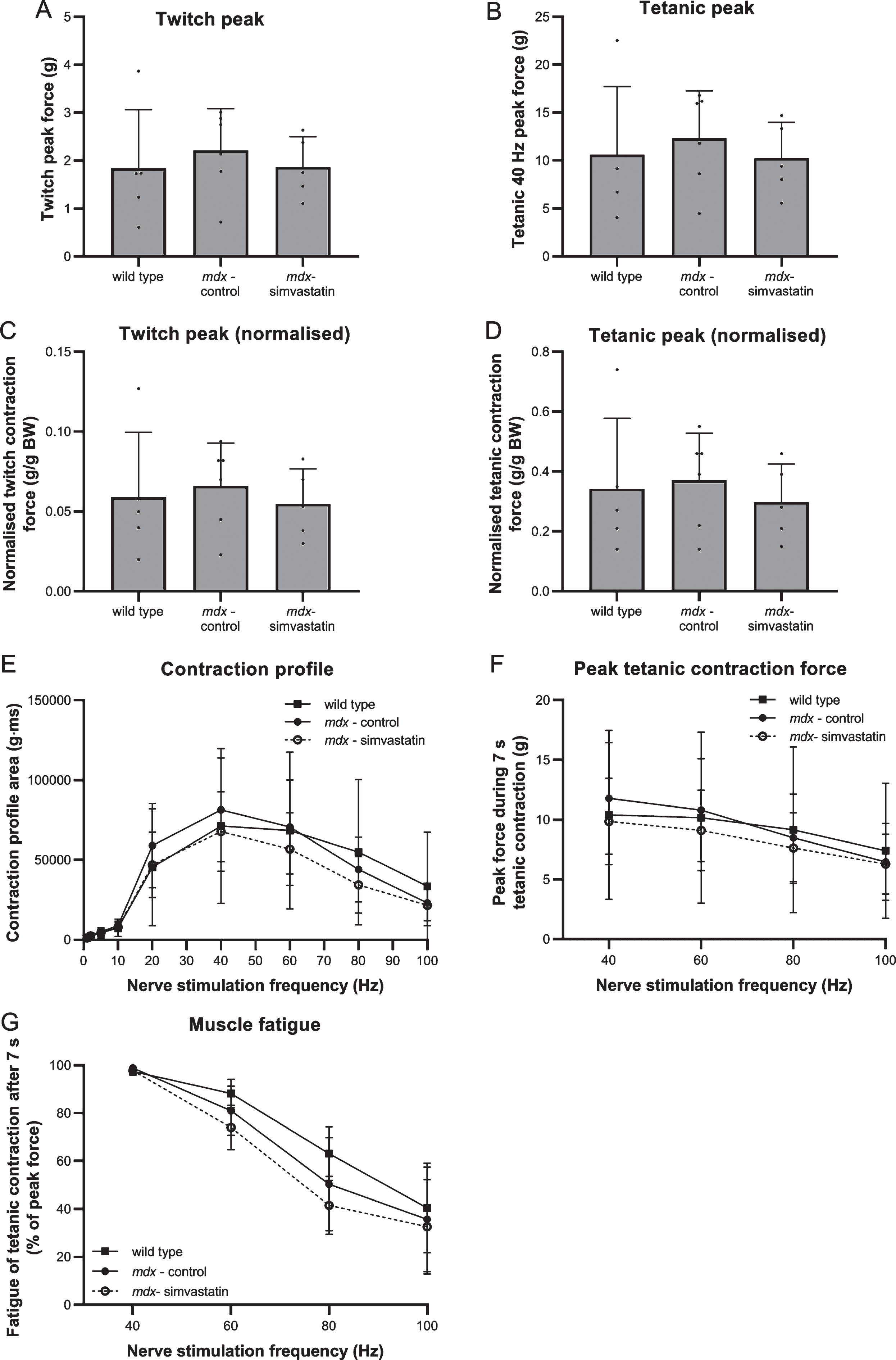
After 12 weeks of treatment starting at four weeks of age, nerve-stimulated contraction force of the diaphragm was measured ex vivo. No clear benefits of simvastatin were observed; however, no reduction in peak twitch contraction force nor in peak tetanic contraction force during 7 s at 40 Hz was seen in mdx muscles (simvastatin-treated and untreated) as compared to wild type (Fig. 8A–B), even after normalisation for bodyweight (Fig. 8C–D). Similarly, no clear differences were observed in area-under-the-curve analysis of the repetitive twitch contractions or tetanic contractions evoked by multiple frequencies of repetitive nerve stimulation (Fig. 8E). No differences between the groups were seen in peak force during tetanic contractions at 40, 60, 80 and 100 Hz nerve stimulation (Fig. 8F). However, a tendency towards a more pronounced muscle fatigue in the mdx (simvastatin-treated as well as untreated) muscles, as compared to wild type, was observed at the higher frequencies (60, 80 and 100 Hz) (Fig. 8G).
Fig. 8
Nerve-stimulated muscle strength. Diaphragm of 16–week old mice after 12 weeks of simvastatin treatment. (A) Twitch contraction force. (B) Peak of the tetanic contraction force evoked by 7 s stimulation at 40 Hz. (C–D) Twitch (C) and tetanic (D) contraction force normalised to bodyweight. (E) Stimulation frequency-contraction relationship. Area-under-the-curve of 7 s stimulation at the indicated frequency (F) Peak tetanic contraction force during 7s of 40, 60, 80 and 100 Hz nerve stimulation. (G) Fatigue after 7 s of tetanic contraction at 40, 60, 80 and 100 Hz nerve stimulation, expressed as percentage of the peak force (n = 5–6). There are no significant differences between any of the groups.
Muscle histology
Fibrosis in 16-week old mice was examined by Sirius Red staining of sections of the diaphragm muscle. As with 24-week old mice (described above), fibrotic content is increased in mdx mice compared to wild type mice. Similarly, simvastatin treatment did not prevent the development of fibrosis. Similar levels of fibrosis were detected in both treated and untreated mdx mice (Fig. 9A–B).
Fig. 9
Fibrosis. Diaphragm of 16–week old mice after 12 weeks of simvastatin treatment. (A) Representative images of Sirius Red stained diaphragms (10x magnification) (B) quantification of collagen content by Sirius Red staining. (C–D) expression of (C) collagen I and (D) collagen III (n = 5–6). *p < 0.05, **p < 0.01 compared to wild type mice.
Pathological pathway analysis
In agreement with the changes seen in 24-week old mice, there was clear deregulation of genes related to the disease pathology when comparing mdx and wild type mice. Increased expression of the fibrotic markers collagen type I and III was seen in mdx mice (Fig. 9C-D), as were markers of inflammation (Fig. 10) and MHC-related markers of de- and regeneration (Fig. 11). None of these markers were affected by simvastatin treatment.
Fig. 10
Inflammation. Expression of (A) Lgals3 and (B) CD68 in the diaphragm of 16-week old mice after 12 weeks of simvastatin treatment (n = 5–6). *p < 0.05 compared to wild type mice.
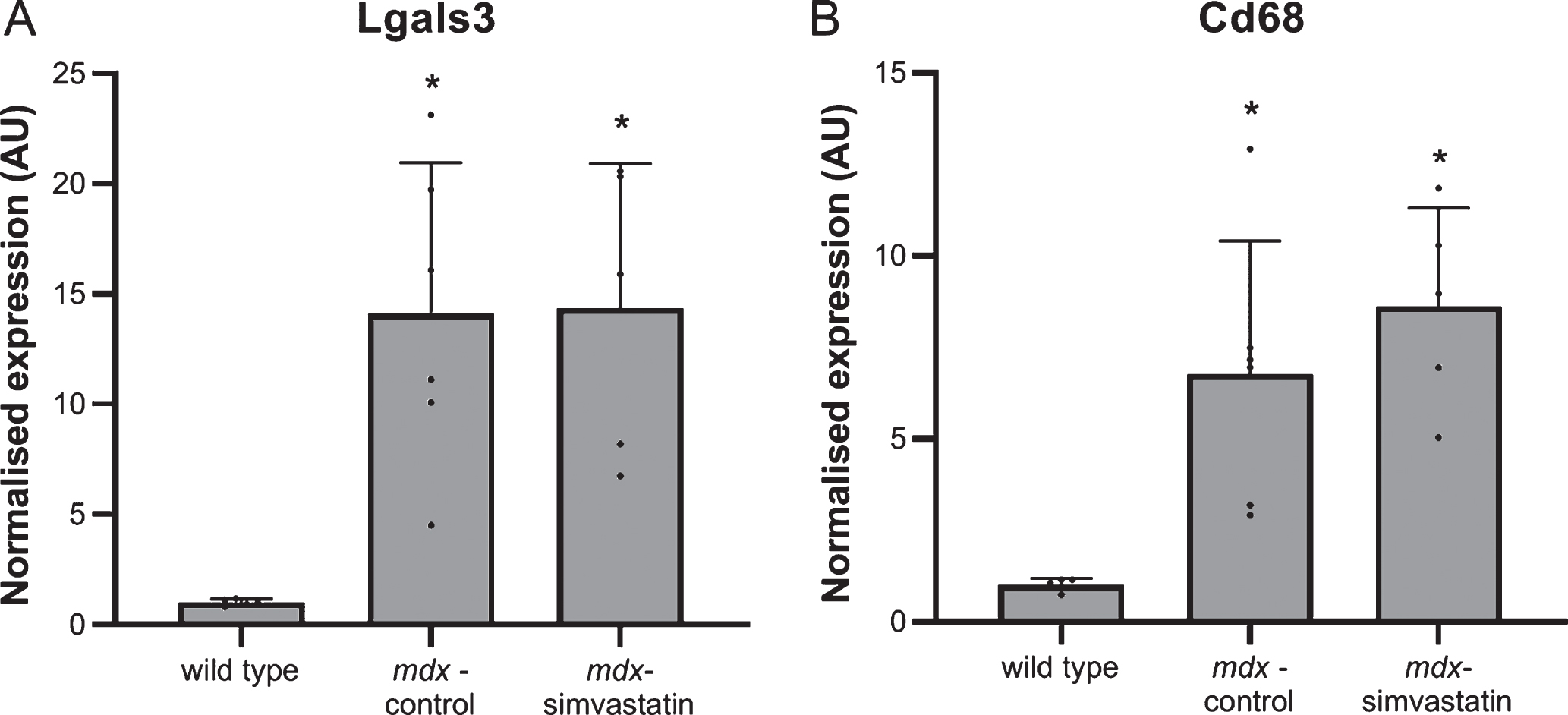
Fig. 11
De- and regeneration. Expression of different isoforms of myosin heavy chain in the diaphragm of 16-week old mice after 12 weeks of simvastatin treatment. (A) embryonic (Myh3). (B) neonatal (Myh8). (C) adult slow-twitch type Iβ (Myh7). (D–F) adult fast-twitch type IIx (Myh1) (D), IIa (Myh2) (E) and IIb (Myh4) (F) (n = 5–6). *p < 0.05, **p < 0.01 compared to wild type mice.
In 24–week old mice a downregulation of atrogin–1 compared to age-matched wild type mice (indicating impaired autophagy) was seen. In younger mice a similar trend towards decreased expression in mdx samples was observed (though this did not achieve statistical significance), and simvastatin treatment was comparable with untreated mdx mice (Fig. 12A). As with 24-week old mice, LC3B expression was comparable between all groups (Fig. 12B). As with older mice (Fig. 7), expression of Nox2 and Nox4 was markedly elevated in mdx diaphragms when compared with wild type (Fig. 13A–B), and western blotting for Nox2 showed this increase was also reflected at the protein level (Fig. 13C–D). Simvastatin treatment did not inhibit this upregulation.
Fig. 12
Autophagy. Expression of (A) atrogin–1 and (B) LC3B in the diaphragm of 16–week old mice after 12 weeks of simvastatin treatment (n = 5–6). There are no significant differences between any of the groups.
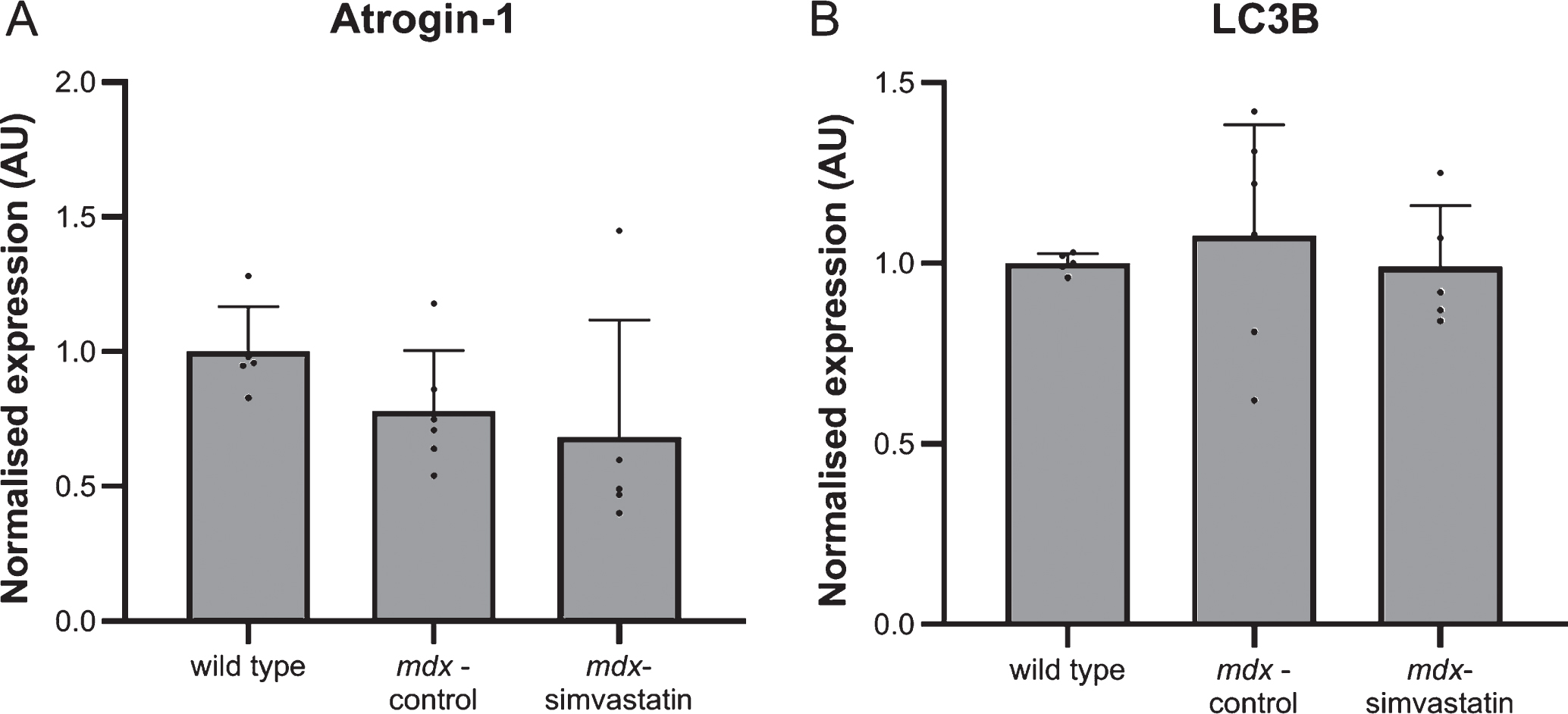
Fig. 13
Oxidative stress. 16–week old mice after 12 weeks of simvastatin treatment. (A–B) Gene expression of Nox2 (A) and Nox4 (B) in the diaphragm. (C–D) Expression of Nox2 protein in the tibialis anterior. Each lane represents an individual mouse (n = 5–6). *p < 0.05, **p < 0.01 compared to wild type mice.
Simvastatin plasma levels
Since, in contrast to previously reported results [17], no effects of simvastatin treatment were ob-served, it was decided to investigate simvastatin plasma levels to determine the effectiveness of simvastatin administration routes.
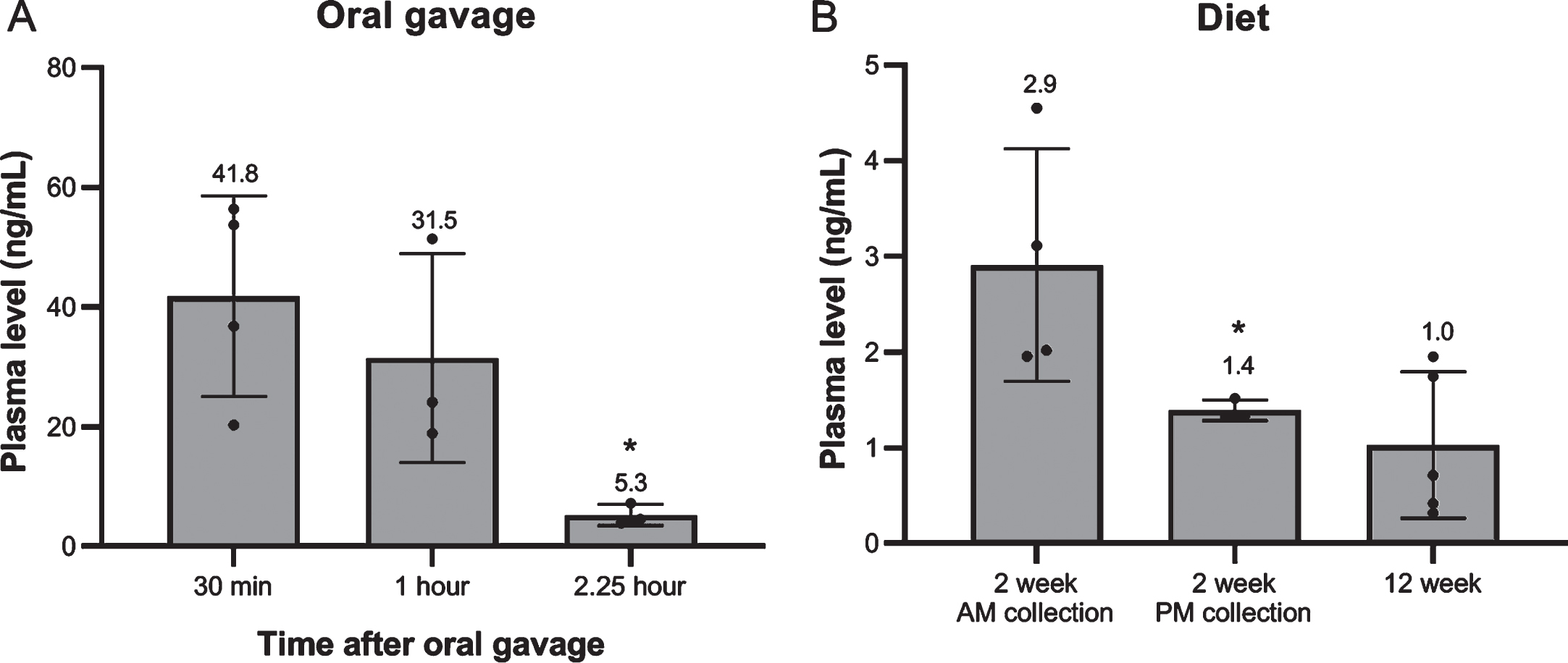
Administration via oral gavage led to high plasma levels directly after administration, but these declined rapidly. Two hours and 15 min after oral gavage only 12.6% of levels measured after 30 min remained (Fig. 14A). No later time points were included.
When simvastatin was administered via the food, only low levels of simvastatin were observed (Fig. 14B). In the first part of the experiment plasma levels were measured two weeks after initiation of the experiment at two different times points of the day. Levels were somewhat higher in blood collected in the morning during the last part of the dark phase than in the afternoon during the last part of the light phase (2.9 vs 1.4 ng/mL), probably since mice are nocturnal animals and mostly feed during the dark phase. Levels did not accumulate during treatment. After 12 weeks of treatment (in the second part of the experiment) plasma levels were still around 1 ng/mL (plasma collected during the day) (Fig. 14B).
Fig. 14
Simvastatin plasma levels. (A) At 30 min, 1 hour and 2.25 hour after single oral gavage (8 mg/kg) (n = 3–4). (B) After 2 week (AM/PM plasma collection) and 12 week treatment with 80 mg/kg simvastatin in the diet. (n = 5–8). *p < 0.05
DISCUSSION
Since promising results were seen with simvastatin treatment in the mdx mouse model at different stages of the muscle pathology [17], our study tested simvastatin treatment in mdx mice at different ages and thereby pathological stages. Clear signs of muscle damage were observed in mdx mice compared to wild type mice, but in both studies, starting during and after the onset of muscle damage, no preservation of muscle function or amelioration of pathology was seen due to simvastatin treatment. It is known that up to 30% of humans taking simvastatin experience side effects in skeletal muscle [47]. In our study in mice no obvious deleterious effects on muscle were seen, but given these are dystrophic animals, mild effects may be masked by more profound disease-related muscle pathology.
Although previous studies showed a modestly decreased peak contraction force of mdx diaphragms during nerve stimulation [48], no clear differences in comparison to wild type mice could be detected in the LUMC study. Variation between individual mice within the groups was substantial, precluding detection of minor changes (making it difficult to judge any possible effect of simvastatin). In contrast, using direct muscle stimulation of muscle strips a large decrease in contraction force and resistance was observed in mdx diaphragm when compared to their wild type counterpart in the RVC study. Similarly, nerve stimulation showed a large decrease in mdx TA contraction force and resistance (vs wild type mice). This is in line with previous results [24, 25, 49]. However, even with this more sensitive method, no effect of simvastatin treatment was observed.
Marked alterations in expression of genes involved in fibrosis, regeneration, autophagy and oxidative stress were seen in both 12- and 24-week old mdx mice, relative to age-matched wild type mice. Although these changes were largely consistent between both studies, they were more statistically robust at 24 weeks. This could both be due to the age of the mice and to the larger group size allowing detection of smaller differences. One of the proposed mechanisms by which simvastatin could act on DMD pathology is counteracting the impaired autophagy caused by increased oxidative stress [50]. Markers for autophagy and oxidative stress were, however, not altered by simvastatin treatment. In addition, no amelioration of muscle pathology was observed histologically.
Recently a new study, using the same treatment protocol as the original study [17], showed a positive effects of simvastatin on cardiomyopathy [51]. However, signs of cardiomyopathy only become clear in ten-month-old mdx mice and therefore we were unable to study these aspects here [52].
The lack of simvastatin efficacy revealed by our studies could be explained by low plasma levels of drug. Therefore, we investigated the pharmacokinetics of simvastatin. Simvastatin is known to have a short in vivo half-life [53]. Indeed, in our hands, after a single dose given via oral gavage plasma levels dropped rapidly (from 40 ng/mL after 30 min to 5 ng/mL after 2.25 hours). Administration via the food led to levels of only 1 to 3 ng/mL. Crucially, this is much lower than levels observed in the study reported by Whitehead and colleagues, where administration at the same dose in the food resulted in plasma levels of 400 nM (170 ng/mL), despite total food intake of the mice being comparable between the studies [17]. It is possible that such high plasma levels are necessary for the beneficial effects reported, but these levels (obtained after administration via the food) seem surprisingly high, given we and others were unable to reach such levels even with oral gavage. Other studies have reported a considerable range of plasma levels following oral gavage. Panajatovic and colleagues used this approach to dose three strains of mice with 5 mg/kg daily and found plasma samples collected one hour after treatment were between 7.05 and 14.7 nM, equivalent to 3 to 6.25 ng/mL [54]. In contrast, Famey reported plasma levels of 54.7 ng/mL (equivalent to 129 nM) in mice treated 1 hour earlier by oral gavage with 2 mg/kg simvastatin [55]. While these values are closer to those reported by Whitehead et al., our data suggests it is very unlikely that similar plasma levels can be achieved via the food, even when adjusted to an intake of 8–10 mg/kg. Other routes of administration might lead to higher plasma levels (and could thereby potentially ameliorate muscle pathology and function). Oral administration is, however, most relevant from a clinical perspective [13].
In conclusion, our results show that, in contrast to previous studies, no beneficial effects of simvastatin treatment were seen in either young or adult mdx mice.
ACKNOWLEDGMENTS
This study was supported by grants from Duchenne UK. We would like to thank Jelle Goeman (Department of Medical Statistics, Leiden University Medical Center, Leiden, the Netherlands) for assistance with statistical analysis.
CONFLICT OF INTEREST
IV, OC, CTdW, JJPs, SN, KW and JH have no conflict of interest to report. DB is research director of Duchenne UK. AAR discloses being employed by LUMC which has patents on exon skipping technology, some of which has been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents AAR is entitled to a share of royalties. AAR reports that she is on the scientific advisory board of Duchenne UK, but excused herself from commenting on this application when it was submitted for evaluation. AAR further discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, CRISPR Therapeutics, Summit PLC, Alpha Anomeric, BioMarin Pharmaceuticals Inc., Eisai, Astra Zeneca, Santhera, Audentes, Global Guidepoint and GLG consultancy, Grunenthal, Wave and BioClinica, having been a member of the Duchenne Network Steering Committee (BioMarin) and being a member of the scientific advisory boards of ProQR, hybridize therapeutics, silence therapeutics, Sarepta therapeutics and Philae Pharmaceuticals. Remuneration for these activities is paid to LUMC. LUMC also received speaker honoraria from PTC Therapeutics and BioMarin Pharmaceuticals and funding for contract research from Italfarmaco and Alpha Anomeric. Project funding is received from Sarepta Therapeutics. DJW has been an ad hoc consultant for a large number of companies and is on the Scientific Advisory Board for Akashi Therapeutics. Studies in the Wells laboratory have been funded by Proximagen and Shire Pharmaceuticals.
SUPPLEMENTARY MATERIALS
[1] Fig. S1 Study set-up used at the RVC and LUMC, respectively
Table S1 Outcome measures used at the RVC and LUMC, respectively
Table S2 Primer sequences used for qPCR
Supplementary material can be found here: http://dx.doi.org/10.3233/JND-200524.
REFERENCES
[1] | Mendell JR , Lloyd-Puryear M . Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. (2013) ;48: (1):21–6. |
[2] | Moat SJ , Bradley DM , Salmon R , Clarke A , Hartley L . Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. (2013) ;21: (10):1049–53. |
[3] | Emery AEH , Muntoni F , Quinlivan RCM . Duchenne muscular dystrophy. 4th ed: Oxford University Press; 2015May (2015) . |
[4] | Hoffman EP , Brown RH Jr , Kunkel LM . Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. (1987) ;51: (6):919–28. |
[5] | Koenig M , Monaco AP , Kunkel LM . The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. (1988) ;53: (2):219–28. |
[6] | Blake DJ , Weir A , Newey SE , Davies KE . Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. (2002) ;82: (2):291–329. |
[7] | McDonald CM , Gordish-Dressman H , Henricson EK , Duong T , Joyce NC , Jhawar S , et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul Disord. (2018) ;28: (11):897–909. |
[8] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet (London, England). (2018) ;391: (10119):451–61. |
[9] | Verhaart IEC , Aartsma-Rus A . Therapeutic developments for Duchenne muscular dystrophy. Nature reviews Neurology. (2019) ;15: (7):373–86. |
[10] | Aartsma-Rus A , Corey DR . The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. (2020) ;30: (2):67–70. |
[11] | Parvathaneni V , Kulkarni NS , Muth A , Gupta V . Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov Today. (2019) ;24: (10):2076–85. |
[12] | Heslop E , Csimma C , Straub V , McCall J , Nagaraju K , Wagner KR , et al. The TREAT-NMD advisory committee for therapeutics (TACT): An innovative de-risking model to foster orphan drug development. Orphanet J Rare Dis. (2015) ;10: (1):49. |
[13] | Vuorio A , Kuoppala J , Kovanen PT , Humphries SE , Tonstad S , Wiegman A , et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. (2019) ;2019: (11). |
[14] | Takemoto M , Liao JK . Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arteriosclerosis, thrombosis, and vascular biology. (2001) ;21: (11):1712–9. |
[15] | Oesterle A , Laufs U , Liao JK . Pleiotropic Effects of Statins on the Cardiovascular System. Circulation research. (2017) ;120: (1):229–43. |
[16] | Gu W , Cui R , Ding T , Li X , Peng J , Xu W , et al. Simvastatin alleviates airway inflammation and remodelling through up-regulation of autophagy in mouse models of asthma. Respirology (Carlton, Vic). (2017) ;22: (3):533–41. |
[17] | Whitehead NP , Kim MJ , Bible KL , Adams ME , Froehner SC . A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc Natl Acad Sci U S A. (2015) ;112: (41):12864–9. |
[18] | Whitehead NP , Kim MJ , Bible KL , Adams ME , Froehner SC . Simvastatin offers new prospects for the treatment of Duchenne muscular dystrophy. Rare Dis. (2016) ;4: (1):e1156286. |
[19] | Baker M . 1,500 scientists lift the lid on reproducibility. Nature. (2016) ;533: (7604):452–4. |
[20] | editors N . In praise of replication studies and null results. Nature. (2020) ;578: (7796):489–90. |
[21] | Ioannidis JP . Contradicted and initially stronger effects in highly cited clinical research. JAMA. (2005) ;294: (2):218–28. |
[22] | Aartsma-Rus A , van Putten M . Assessing functional performance in the mdx mouse model. J Vis Exp. 2014(85). |
[23] | Carlson G . DMD M.2.1.005: The use of four limb hanging tests to monitor muscle strength and condition over time: TREAT-NMD Neuromuscular Network; 2011 [updated May 27th 2019]. Available from: https://treat-nmd.org/wp-content/uploads/2016/08/MDX-DMD_M.2.1.005.pdf. |
[24] | Godfrey C , Muses S , McClorey G , Wells KE , Coursindel T , Terry RL , et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet. (2015) ;24: (15):4225–37. |
[25] | Sharp PS , Bye-a-Jee H , Wells DJ . Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol Ther. (2011) ;19: (1):165–71. |
[26] | Terry RL , Kaneb HM , Wells DJ . Poloxamer [corrected] 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS One. (2014) ;9: (3):e91221. |
[27] | Burkholder TJ , Fingado B , Baron S , Lieber RL . Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J Morphol. (1994) ;221: (2):177–90. |
[28] | Reddy GK , Enwemeka CS . A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem. (1996) ;29: (3):225–9. |
[29] | RStudio Team. RStudio: Integrated Development for R. In: RStudio Inc., editor. Boston, MA: RStudio Inc.; 2019. |
[30] | Bates D , Machler M , Bolker BM , Walker SC . Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software. (2015) ;67: (1):1–48. |
[31] | Reagan-Shaw S , Nihal M , Ahmad N . Dose translation from animal to human studies revisited. FASEB J. (2008) ;22: (3):659–61. |
[32] | O’Gorman CS , Higgins MF , O’Neill MB . Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: Evaluation of cholesterol changes and side effects. Pediatr Cardiol. (2009) ;30: (4):482–9. |
[33] | Stedman HH , Sweeney HL , Shrager JB , Maguire HC , Panettieri RA , Petrof B , et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. (1991) ;352: (6335):536–9. |
[34] | van Putten M , Hulsker M , Nadarajah VD , van Heiningen SH , van Huizen E , van Iterson M , et al. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. (2012) ;7: (2):e31937. |
[35] | Pette D , Staron RS . Myosin isoforms, muscle fiber types, and transitions. Microscopy research and technique. (2000) ;50: (6):500–9. |
[36] | Schiaffino S , Reggiani C . Fiber types in mammalian skeletal muscles. Physiol Rev. (2011) ;91: (4):1447–531. |
[37] | De Palma C , Morisi F , Cheli S , Pambianco S , Cappello V , Vezzoli M , et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. (2012) ;3: :e418. |
[38] | Hanai J , Cao P , Tanksale P , Imamura S , Koshimizu E , Zhao J , et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. (2007) ;117: (12):3940–51. |
[39] | Wei YM , Li X , Xu M , Abais JM , Chen Y , Riebling CR , et al. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cellular physiology and biochemistry: International journal of experimental cellular physiology, biochemistry, and pharmacology. (2013) ;31: (6):925–37. |
[40] | Kim JH , Kwak HB , Thompson LV , Lawler JM . Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. Journal of muscle research and cell motility. (2013) ;34: (1):1–13. |
[41] | Pal R , Palmieri M , Loehr JA , Li S , Abo-Zahrah R , Monroe TO , et al. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun. (2014) ;5: :4425. |
[42] | Bedard K , Krause KH . The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. (2007) ;87: (1):245–313. |
[43] | Takeno A , Kanazawa I , Tanaka K , Notsu M , Yokomoto-Umakoshi M , Sugimoto T . Simvastatin rescues homo-cysteine-induced apoptosis of osteocytic MLO-Y4 cells by decreasing the expressions of NADPH oxidase 1 and 2. Endocrine journal. (2016) ;63: (4):389–95. |
[44] | Yan J , Qiao L , Wu J , Fan H , Sun J , Zhang Y . Simvastatin Protects Dopaminergic Neurons Against MPP+-Induced Oxidative Stress and Regulates the Endogenous Anti-Oxidant System Through ERK. Cellular physiology and biochemistry: International journal of experimental cellular physiology, biochemistry, and pharmacology. (2018) ;51: (4):1957–68. |
[45] | Huang W , Shang WL , Li DH , Wu WW , Hou SX . Simvastatin protects osteoblast against H2O2-induced oxidative damage via inhibiting the upregulation of Nox4. Molecular and cellular biochemistry. (2012) ;360: (1-2):71–7. |
[46] | Turk R , Sterrenburg E , de Meijer EJ , van Ommen GJ , den Dunnen JT , t Hoen PA . Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC genomics. (2005) ;6: :98. |
[47] | Bouitbir J , Sanvee GM , Panajatovic MV , Singh F , Krahenbuhl S . Mechanisms of statin-associated skeletal muscle-associated symptoms. Pharmacol Res. (2020) ;154: :104201. |
[48] | van der Pijl EM , van Putten M , Niks EH , Verschuuren JJ , Aartsma-Rus A , Plomp JJ . Characterization of neuromuscular synapse function abnormalities in multiple Duchenne muscular dystrophy mouse models. Eur J Neurosci. (2016) ;43: (12):1623–35. |
[49] | Moorwood C , Liu M , Tian Z , Barton ER . Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J Vis Exp. (2013) (71):e50036. |
[50] | Whitehead NP . Enhanced autophagy as a potential mechanism for the improved physiological function by simvastatin in muscular dystrophy. Autophagy. (2016) ;12: (4):705–6. |
[51] | Kim MJ , Bible KL , Regnier M , Adams ME , Froehner SC , Whitehead NP . Simvastatin provides long-term improvement of left ventricular function and prevents cardiac fibrosis in muscular dystrophy. Physiol Rep. (2019) ;7: (6):e14018. |
[52] | Verhaart IE , van Duijn RJ , den Adel B , Roest AA , Verschuuren JJ , Aartsma-Rus A , et al. Assessment of cardiac function in three mouse dystrophinopathies by magnetic resonance imaging. Neuromuscul Disord. (2012) ;22: (5):418–26. |
[53] | Plosker GL , McTavish D . Simvastatin. A reappraisal of its pharmacology and therapeutic efficacy in hypercholesterolaemia. Drugs. (1995) ;50: (2):334–63. |
[54] | Panajatovic MV , Singh F , Roos NJ , Duthaler U , Handschin C , Krahenbuhl S , et al. PGC-1alpha plays a pivotal role in simvastatin-induced exercise impairment in mice. Acta Physiol (Oxf). (2020) ;228: (4):e13402. |
[55] | Fahmy UA . Quantification of simvastatin in mice plasma by near-infrared and chemometric analysis of spectral data. Drug Des Devel Ther. (2016) ;10: :2507–13. |


