
AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort
Abstract
Background:
Spinal muscular atrophy type 1 (SMA1) is the leading genetic cause of infant mortality for which therapies, including AVXS-101 (onasemnogene abeparvovec, Zolgensma®) gene replacement therapy, are emerging.
Objective:
This study evaluated the effectiveness of AVXS-101 in infants with spinal muscular atrophy type 1 (SMA1) compared with a prospective natural history cohort and a cohort of healthy infants.
Methods:
Twelve SMA1 infants received the proposed therapeutic dose of AVXS-101 (NCT02122952). Where possible, the following outcomes were compared with a natural history cohort of SMA1 infants (n = 16) and healthy infants (n = 27) enrolled in the NeuroNEXT (NN101) study (NCT01736553): event-free survival, CHOP-INTEND scores, motor milestone achievements, compound muscle action potential (CMAP), and adverse events.
Results:
Baseline characteristics of SMA1 infants in the AVXS-101 and NN101 studies were similar in age and genetic profile. The proportion of AVXS-101–treated infants who survived by 24 months of follow-up was higher compared with the NN101 study (100% vs 38%, respectively). The average baseline CHOP-INTEND score for NN101 SMA1 infants was 20.3, worsening to 5.3 by age 24 months; the average baseline score in AVXS-101–treated infants was 28.2, improving to 56.5 by age 24 months. Infants receiving AVXS-101 achieved motor milestones, such as sitting unassisted and walking. Improvements in CMAP peak area were observed in AVXS-101–treated infants at 6 and 24 months (means of 1.1 and 3.2 mV/s, respectively).
Conclusions:
In this study, AVXS-101 increased the probability of survival, rapidly improved motor function, and enabled motor milestone achievement in SMA1 infants.
INTRODUCTION
Spinal muscular atrophy (SMA) is a devastating progressive neurodegenerative disease that results from biallelic survival motor neuron (SMN1) gene deletions and/or mutations [1]. Disease severity is modified by the number of SMN2 copies, a backup gene, which produces a small fraction of functional SMN protein; fewer copies of SMN2 correlate with more severe disease [2]. The majority of infants with SMA type 1 (SMA1) have 1 or 2 copies of SMN2 [2]. SMA1 symptoms typically begin in an infant before 6 months of age, and follow a rapidly progressive course that results in the inability to achieve motor milestones and a median age at time of death or requirement for permanent ventilation of 8–10 months [3–5]. The NeuroNEXT (National Network for Excellence in Neuroscience Clinical Trials) SMA Infant Biomarker Study (NN101) is a recent prospective natural history study in which early motor function decline (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders [CHOP-INTEND] scale, range 0–64 [6, 7], and electrophysiology measures) and death and tracheostomy (composite survival) were evaluated in infants with SMA1 treated with modern supportive care, with results demonstrating rapid early decline [4]. The NN101 study design follows the framework of a clinical trial, permitting comparison with prospective clinical trials, an important approach given the lethality of the disease and the ethical dilemma of including a control treatment arm.
Therapeutic approaches for SMA include increasing SMN protein levels. Nusinersen, an intrathecally delivered antisense oligonucleotide, is the first US FDA-approved therapy for SMA [8, 9]. The first-ever gene replacement therapy in clinical development for SMA (AVXS-101; onasemnogene abeparvovec, Zolgensma®) recently reported positive data in a single-arm, open-label trial in infants with SMA1 [10]. Treatment with AVXS-101 addresses the genetic root cause of SMA by intravenous delivery of human SMN via an adeno-associated virus 9 (AAV9) vector that can cross the blood–brain barrier, and has a rapid onset of effect because of its self-complementary feature. AVXS-101 is also designed to provide high and continuous expression of the SMN protein through a strong continuous promoter, which is essential for durable effects. In a phase 1 trial, the AVXS-101 study, all participants receiving a 1-time intravenous dose of AVXS-101 achieved permanent ventilation-free survival up to 24 months of age and further [10, 11]. Eleven (92%) of 12 infants receiving the proposed therapeutic dose achieved and maintained the ability to sit unassisted, and 2 were able to stand and walk independently [10]. AVXS-101 is currently being evaluated in an ongoing phase 3 trial in infants with SMA1 (NCT03306277).
The objective of this study was to compare survival and motor function data from infants receiving the proposed therapeutic dose of AVXS-101 in the full 24-month follow-up dataset to data reported in the NN101 infants with SMA1 natural history cohort and select data from a cohort of healthy infants, hypothesizing that AVXS-101–treated infants would have improved survival, motor function, and motor milestone achievement. This comparison was possible because infants with SMA1 in both studies were symptomatic before 6 months of age and all had similar genetic profiles (homozygous SMN1 exon 7 deletions and 2 SMN2 copies). Furthermore, infants in both studies received contemporary standard supportive care as increasingly adopted in the United States [12], and both studies were completed before the publication of the 2018 SMA management guidelines [13, 14].
METHODS
Study design
The objective of this analysis was to evaluate the efficacy of AVXS-101 in infants with SMA1 who received the proposed therapeutic dose of AVXS-101 and were enrolled in the first-in-human, open-label, single-arm phase 1/2a study of AVXS-101 (NCT02122952) compared with a natural history cohort of infants with SMA1 and a healthy infant cohort from a separate prospective natural history study (NCT01736553). This analysis compared survival, motor function, motor milestone achievements, and motor unit function as measured by compound muscle action potential (CMAP).
The details of the AVXS-101 study have been previously published, and the study was funded by AveXis, Inc., a Novartis Company [10]. Infants with SMA1 (n = 15) who had a genetically confirmed diagnosis of SMA1 (homozygous SMN1 exon 7 deletions and 2 copies of SMN2) were enrolled in a phase 1 study of AVXS-101. Results from study follow-up until all infant participants were at least 20 months of age have been published [10]. For the purposes of this comparison, the 12 infants who received the proposed therapeutic dose of AVXS-101 (1.1×1014 vg/kg based on droplet digital polymerase chain reaction [ddPCR™; Bio-Rad, Inc.], equivalent to 2.0 × 1014 vg/kg based on quantitative PCR [10]) between December 2014 and December 2015 and results from the complete 24 months of follow-up were included. Of the 12 infants, 11 received the proposed therapeutic dose at <6 months of age and 1 was dosed at 8 months of age [10]. At enrollment in the AVXS-101 study, infants requiring the use of non-invasive ventilation (NIV) ≥16 hours/day were excluded; this criterion was a defined endpoint of survival in natural history studies and applied to the clinical trial design [3]. AVXS-101 infant participants were experiencing symptoms at enrollment, with onset prior to 6 months of age.
Natural history cohort
Infants with SMA1 from a natural history study were identified from the NeuroNEXT prospective natural history study (NN101) and completed their first study visit between December 2012 and September 2012. Final results and detailed methods of the entire NN101 study were previously published [4, 8]. In the NN101 study, infants were ≤6 months of age at enrollment [8]. Infants were excluded if they were required to use NIV ≥12 hours/day [8]. To obtain a comparable group from the NN101 database, data analysis was restricted to infants with 2 confirmed copies of SMN2, excluding 4 NN101 infants of the 20 total because of the absence of confirmed genotype information, which was attributable to insufficient blood samples. Thus 16 infants with SMA1 were selected for this comparative study. Although symptomatic status was not specifically an exclusion criterion in the NN101 study, all infants with 2 SMN2 copies were experiencing symptoms at the first study visit. The use of a natural history cohort provides a frame of reference to evaluate the treatment benefits of AVXS-101 and places efficacy data in the context of the natural progression of the disease.
Healthy infant cohort
The healthy infant cohort was identified from the NN101 database as well. A cohort of 27 genetically confirmed healthy infants (siblings of children with SMA) were enrolled in the NN101 study and were matched for age, sex, birth weight, and height.
Ethical standards statement
The study protocols were approved by the institutional review boards at each participating institution, and study procedures were conducted according to the principles outlined in the Declaration of Helsinki. All parents or guardians provided written informed consent before any study procedures wereperformed.
Statistical analyses
Baseline descriptive statistics were generated using univariate analysis (means with standard deviations or frequencies with percentages) on all available assessments from infant participants’ first visits. Swallowing function and feeding ability were not systematically recorded in the NN101 study, which did not allow comparison; they were evaluated monthly in the AVXS-101 study. Safety analyses for adverse events were performed in all AVXS-101–treated infants. However, only adverse events related to study procedures were recorded in the NN101 study, thereby not allowing comparison. Statistical analyses were performed with SAS software, version 9.4 (Cary, North Carolina, USA). Because statistical analyses were post hoc and comparisons were made across independent studies, any P-values presented are nominal and for descriptive purposes.
Descriptive statistics of infants surviving event-free at 24 months was performed using proportions of patients in SMA1 cohorts. Graphical representations of time-to-event analyses were created using the Kaplan-Meier method, and the infants in the AVXS-101 study, NN101 infants with SMA1, and NN101 healthy infant cohorts were included. Of note, the NN101 study composite endpoint was death or tracheostomy [4, 8], while the composite endpoint for the AVXS-101 study was death or ≥16 hours of ventilation per day for >2 consecutive weeks [3, 10].
Motor function was compared graphically between the infants treated with AVXS-101 and the NN101 infants with SMA1 cohorts with the CHOP-INTEND scale [6]. For longitudinal assessments in the NN101 study, infants were evaluated at every 3 months of age (months 0, 3, 6, 12, 18, and 24); those in the AVXS-101 study were evaluated at monthly study visits. In the NN101 study, visit date and infant age were in alignment as follows: infants aged up to 1.5 months were categorized in the “0 month” age group, infants aged 1.5 to 4.5 months were categorized in the “3 months” age group, infants aged 4.5 to 6 (±2 weeks) months were categorized in the “6 months” age group, and for all other visits, infants were categorized according to age in months (±2 weeks). In the AVXS-101 study, it was necessary to pool patients into age groups to align with the NN101 groups. Therefore, infant age was rounded to the closest unit of 3 months as follows: infants aged up to 2 months were categorized in the “0 month” age group, infants aged 2 to 4.5 months were categorized in the “3 months” age group, infants aged 4.5 to 7.5 months were categorized in the “6 months” age group, etc. For those infants treated with AVXS-101 who reached a CHOP-INTEND score of ≥60 points, further gains in motor function were assessed using the Bayley Scales of Infant and Toddler Development, Third Edition (BSID-III), but are not reported in this study. Ten infants with SMA1 in the NN101 study and all 12 patients in the AVXS-101 study completed ≥2 CHOP-INTEND assessments. A change in CHOP-INTEND score for each study was determined by calculating the slope (changes over time) from the age at first to last visit. Slope was expressed as change per month (i.e., 30 days). The per-month slopes were compared with analysis of variance (ANOVA), yielding least-squares mean points per month in overall improvement (+) or worsening (–). Missing data were not imputed.
Regarding motor milestone comparison, because motor milestones were not expected to be achieved by infants with SMA1 in the NN101 study [3, 5], these milestones were not recorded. In the AVXS-101 study, sitting unassisted as a milestone was defined by BSID-III gross motor subtest scale (item 22 for ≥5 seconds and item 26 for ≥30 seconds) and World Health Organization criteria (≥10 seconds) [15, 16].
Ulnar CMAP measurements were obtained in both studies using the same electrophysiologic methods, so a comparison between all cohorts was completed. For this analysis, the infants were grouped with the same method described for motor function assessments. However, only the last CMAP reading for each infant was retained, resulting in at least 1 data point for each 3-month period. In the NN101 study, ulnar CMAP was measured at baseline and 3, 6, 12, 18, and 24 months of age [4, 8]. During the AVXS-101 study, ulnar CMAP was obtained at pre-dose baseline, and every 3 months thereafter; visits were timed based on study enrollment rather than age. For the purposes of this analysis, infant age was rounded to the closest unit of 3 months, with only the last reading retained for each age group. CMAP measurements at ages 0, 3, 6, 12, 18, and 24 months were used for analysis with NN101 infant participant ages.
RESULTS
Baseline characteristics
Key baseline characteristics are reported in Table 1. Infants in the AVXS-101–treated cohort, the NN101 infants with SMA1 cohort, and the NN101 healthy infant cohort were of similar age at dosing or first study visit (3.4 months, 4.0 months, and 3.4 months, respectively). Whereas all 12 infants began experiencing symptoms before 3 months of age in the AVXS-101 study, 14 infants with SMA1 in the NN101 cohort began experiencing symptoms before 3 months of age: 1 infant between 3 and 6 months of age, and 1 infant did not have data recorded(Table 2).
Table 1
Baseline demographics of infants in the AVXS-101 and NN101 cohorts
| Characteristic | AVXS-101 (n = 12) | NN101 - SMA1 (n = 16) | NN101 - healthy (n = 27) |
| Sex | |||
| Female, n (%) | 7 (58) | 8 (50) | 14 (52) |
| Age at first visit (mos) | |||
| Mean (SD) | 2.9 (2)a | 4.0 (2) | 3.4 (2) |
| Minimum–maximum | 0.4–7.3 | 0.4–6.0 | 0.6–6.1 |
| Age at dosing (mos) | |||
| Mean (SD) | 3.4 (2) | Not applicable | Not applicable |
| Minimum–maximum | 0.9–7.9 | Not applicable | Not applicable |
| Race, n (%) | |||
| White/not Hispanic or Latino | 11 (92) | 15 (94) | 21 (78) |
| Ethnicity, n (%) | |||
| White/non-Hispanic | 10 (83) | 11 (69) | 24 (89) |
| Age at SMA1 onset (mos) | |||
| Mean (SD) | 1.4 (1.0) | Not collectedb | Not applicable |
| Minimum–maximum | 0.0–3.0 | ||
| Did not require support of, n (%)c: | |||
| Nutrition | 7 (58) | 9 (56) | 26 (96) |
| Non-invasive ventilation | 10 (83) | 10 (63) | 25 (93) |
| CHOP-INTEND score | |||
| Mean (SD) | 28.2 (12.3) | 20.3 (7.3) | 51.1 (8.9) |
| Minimum–maximum | 12.0–50.0 | 10.0–33.0 | 32.0–62.0 |
| Ulnar CMAP peak area (mV/s) | |||
| Mean (SD) | 1.7 (2.1) | 1.1 (2.3) | 11.6 (4.8) |
| Minimum–maximum | 0.3–7.3 | 0.0–9.2 | 1.1–20.4 |
| Ulnar CMAP amplitude (mV) | |||
| Mean (SD) | 0.74 (1.07) | 0.48 (1.05) | 5.54 (1.96) |
| Minimum–maximum | 0.1–3.4 | 0.0–4.2 | 0.5–9.9 |
CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CMAP, compound muscle action potential; SD, standard deviation; SMA1, spinal muscular atrophy type 1. aIn the AVXS-101 study, this reflects age at enrollment. bExact age was not collected. However, age rounded to month was recorded and is reported in Table 2. cIn the NN101 study, use of nutritional support was not documented. However, difficulty swallowing was documented and an affirmation of this was used as a proxy for use of nutritional support.
Table 2
Age at symptom onset
| <1 month (n) | 1–2 months (n) | 3 months (n) | 4–5 months (n) | Not recorded (n) | |
| AVXS-101 (n = 12) | 2 | 8 | 2 | 0 | 0 |
| NN101 (n = 16) | 6 | 5 | 3 | 1 | 1 |
Survival
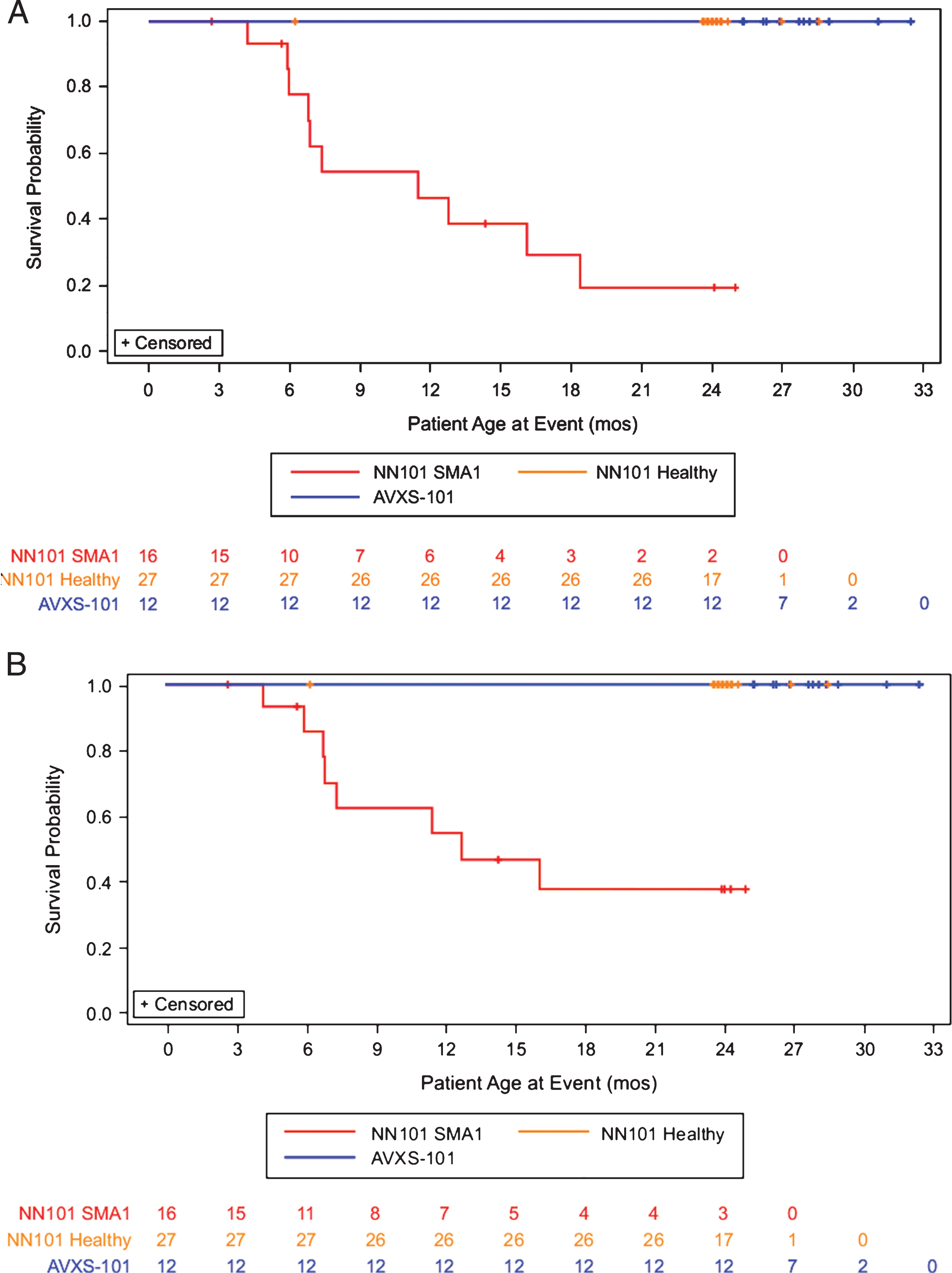
All 12 infants in the AVXS-101 study completed the 24-month follow-up period and reached a median age of 27.8 months at last follow-up. Whereas no infants in the AVXS-101 study reached the composite survival endpoint at the 24-month post-treatment visit, 10 infants (63%) in the NN101 study reached it by a mean age of 9.6 months. Of the 6 remaining infants, 5 were either removed from the study by a parent or guardian or were lost to follow-up (mean age, 10.6 months). The final participant completed the NN101 study per the protocol at 24.1 months of age without reaching the endpoint. No infants died in the AVXS-101 study, whereas 8 (50%) died in the NN101 study at a mean age of 8.9 months. Kaplan-Meier plots for survival and the composite endpoints are provided in Fig. 1.
Fig.1
Permanent ventilation-free survival probability analysis. Survival analysis of the (A) composite survival endpoint (death or permanent ventilation) or (B) survival alone for infants with SMA1 in the AVXS-101–treated (green line, n = 12), NN101 infants with SMA1 (blue line, n = 16), NN101 healthy infant (orange line, n = 27) cohorts. All infants in the AVXS-101 study completed this 24-month follow-up study without permanent ventilation. In the NN101 study, infants with SMA1 either reached the composite endpoint (n = 10), were removed from the study by a parent or guardian or were lost to follow-up (n = 5), or completed the study and reached 24 months of age without reaching the endpoint (n = 1). SMA1, spinal muscular atrophy type 1.
Motor function
At 0 months of age, the average CHOP-INTEND score was 19.0 in the NN101 infants with SMA1 cohort and 37.0 in the AVXS-101–treated cohort. By 12 months of age, the average CHOP-INTEND score of the NN101 infants with SMA1 and AVXS-101–treated cohorts was 10.1 and 50.0, respectively (Fig. 2A). By 24 months of age, CHOP-INTEND scores in the NN101 infants with SMA1 cohort decreased to 5.3, whereas the mean score increased to 56.5 points in the AVXS-101–treated cohort (Fig. 2B). The least-squares mean was –2.15 points/month (–12.9 points/6 months) for the NN101 infants with SMA1 cohort and +2.81 points/month (+16.9 points/6 months) for the AVXS-101–treated cohort. The difference between least-squares means was 4.96 (95% CI: –6.57 to –3.35) points/month (P < .0001).
Fig.2
Motor function analysis of the AVXS-101 and NN101 studies. (A) Maximum longitudinal CHOP-INTEND scores reached. Mean CHOP-INTEND scores by infant age are shown; shaded areas indicate the standard deviation for each mean at each study visit. (B) Change in longitudinal CHOP-INTEND score up to 24 months of age. CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders. SD, standard deviation. SMA1, spinal muscular atrophy type 1.
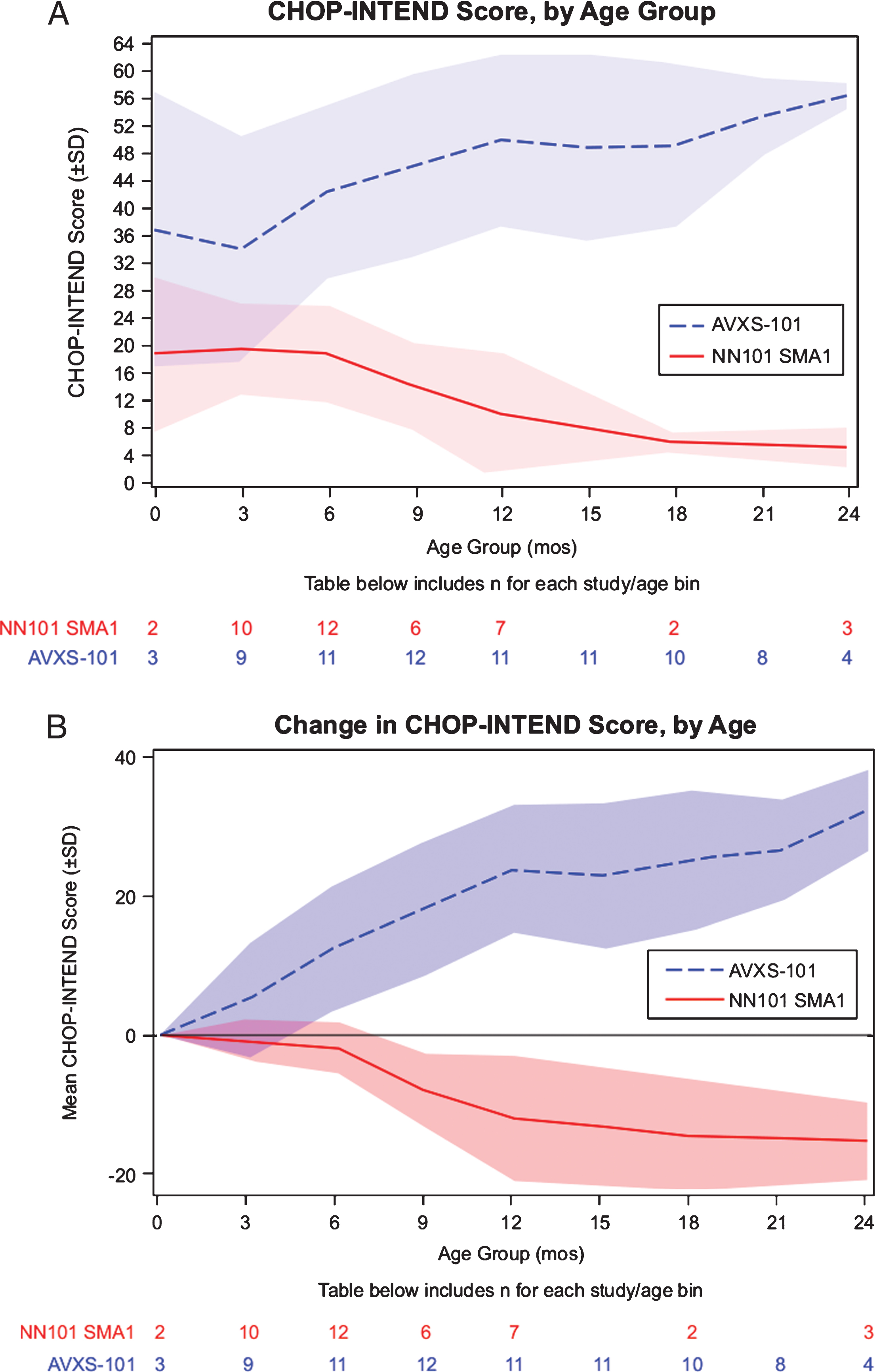
All 12 infants in the AVXS-101 study showed and maintained clinically significant improvements in motor function (≥4.0 points gained in CHOP-INTEND); 11 of 12 infants had CHOP-INTEND scores that surpassed and sustained >40.0 points. None of the NN101 infants with SMA1 cohort showed motor function improvements, with a maximum CHOP-INTEND of 33.0 points at first study visit at age 5.7 months.
Motor milestone achievements
Although motor milestones were not formally assessed in the NN101 study, the CHOP-INTEND scores in infants with SMA1 suggested that they did not achieve any motor milestones (Table 3). In contrast, 11 (92%) of the AVXS-101–treated infants achieved and maintained the milestone of sitting unassisted for ≥5 seconds, 10 (83%) achieved sitting unassisted for ≥10 seconds, and 9 (75%) could sit unassisted for ≥30 seconds by 24 months of follow-up [10]. In addition, 2 could stand and walk without assistance.
Table 3
Comparison of motor milestone achievements at end of study
| Motor milestone and motor function achievements, n (%) | AVXS-101 (n = 12) | NN101 (n = 16)a |
| Ever sit without support for ≥5 seconds (BSID-III) | 11 (92) | 0 |
| Ever sit without support for ≥10 seconds (WHO-MGRS) | 10 (83) | 0 |
| Ever sit without support for ≥30 seconds (BSID-III) | 9 (75) | 0 |
| Ever stand without support | 2 (17) | 0 |
| Ever walk alone | 2 (17) | 0 |
| CHOP-INTEND score >40 at any time | 11 (92) | 0 |
| ≥4.0-point improvement in CHOP-INTEND score | 12 (100) | 0 |
BSID-III, Bayley Scales of Infant and Toddler Development, Third Edition; CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; WHO-MGRS, World Health Organization-Multicentre Growth Study. aLack of motor milestone achievement was inferred from the participants’ CHOP-INTEND scores and not directly assessed.
Longitudinal CMAP assessments
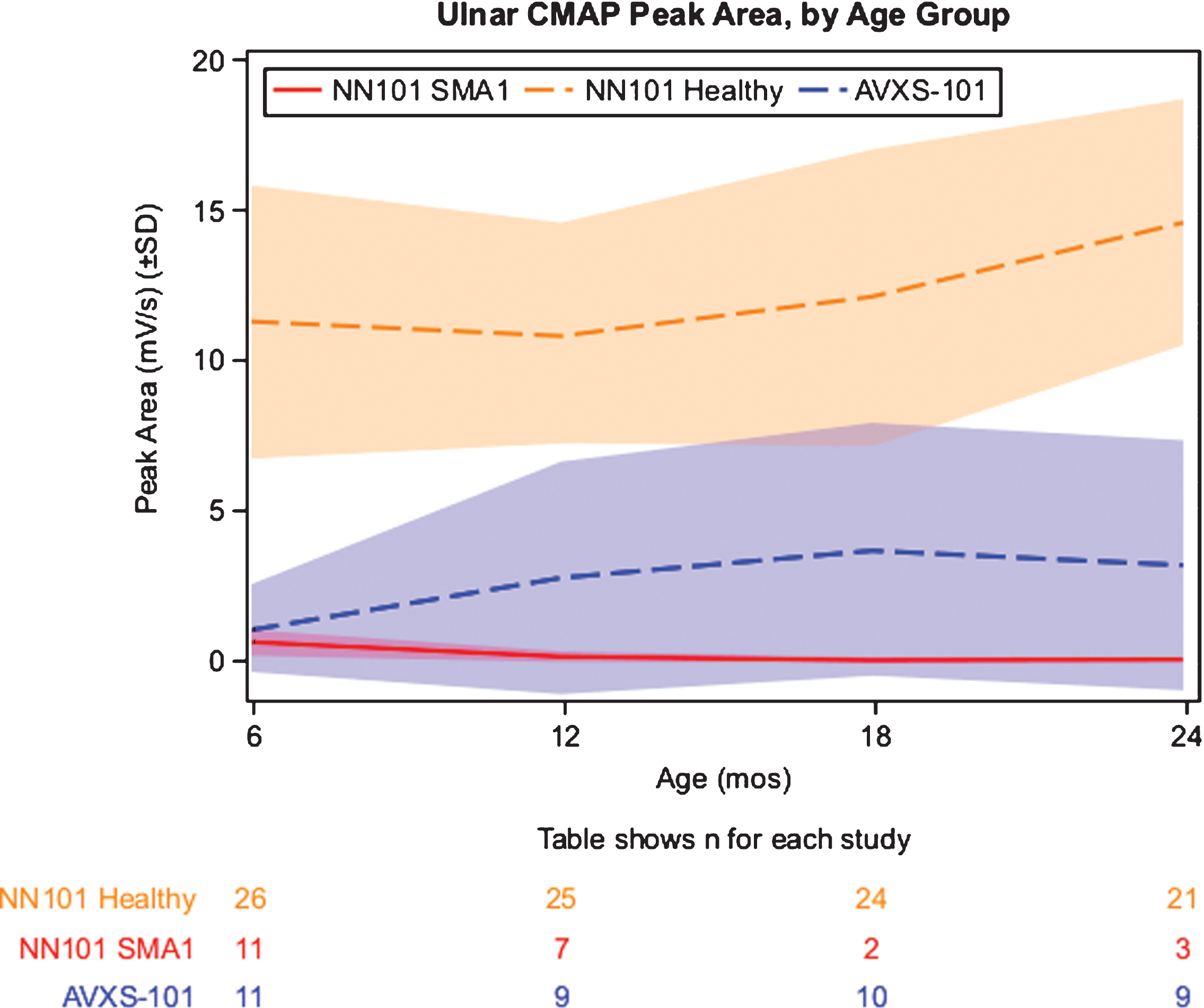
Ulnar CMAP peak area values decreased rapidly in the NN101 infants with SMA1 cohort, from a mean of 0.61 mV/s at 6 months of age to a mean of 0.12 mV/s at 12 months. In contrast, the AVXS-101–treated cohort demonstrated a rapid increase in CMAP peak area values, from a mean of 1.1 mV/s at 6 months of age to a mean of 2.8 mV/s at 12 months (Fig. 3). By 24 months of age, the NN101 infants with SMA1 cohort and the AVXS-101–treated cohorts had mean CMAP peak area values of 0.02 and 3.2 mV/s, respectively. Mean CMAP peak area values for the NN101 healthy infant cohort increased from 6 to 24 months of age (11.3 and 14.6 mV/s, respectively). CMAP peak amplitude values demonstrated similar trends (Supplemental Figure 1). Peroneal CMAP was not performed in the NN101 study; the AVXS-101–treated cohort demonstrated a substantial increase in peroneal CMAP peak area values, from a mean of 5.7 mV/s at 6 months of age to a mean of 11.5 mV/s at 12 months.
Fig.3
Longitudinal mean CMAP peak area. Mean CMAP peak areas (mV/s) are shown. Study visits were linked to infant age (in months, ±2 weeks for visits 6, 12, 18, and 24 months). The shaded areas describe the standard deviation for each mean at each study visit. CMAP, compound muscle action potential. SD, standard deviation. SMA1, spinal muscular atrophy type 1.
Adverse events after administration of AVXS-101
In the AVXS-101 study, a total of 275 adverse events, mostly related to the underlying disease, were detected, of which 53 were serious adverse events. Of the serious adverse events, 2 were treatment-related and consisted of asymptomatic elevation of serum aminotransferase levels (details of adverse events and prednisolone regimen were previously published [10]). No new treatment-related adverse event was observed since the last report (Table 4).
Table 4
Adverse events of AVXS-101–treated cohort at end of 24-month follow-up
| SMA1 AVXS-101 studya (n = 12) | ||
| Events (n) | Participants, n (%) | |
| Any adverse event | 275 | 12 (100) |
| Any serious adverse event | 53 | 10 (83) |
| Adverse event associated with treatment | 4 | 3 (25) |
| Upper respiratory tract infection | 28 | 10 (83) |
| Pyrexia | 12 | 7 (58) |
| Vomiting | 11 | 8 (67) |
| Pneumonia | 14 | 7 (58) |
| Constipation | 8 | 6 (50) |
| Nasal congestion | 9 | 6 (50) |
| Gastroesophageal reflux disease | 6 | 5 (42) |
| Cough | 11 | 5 (42) |
| Rhinovirus infection | 10 | 4 (33) |
| Enterovirus infection | 7 | 4 (33) |
| Rash | 6 | 5 (42) |
| Gastroenteritis viral | 5 | 5 (42) |
| Otitis media | 3 | 2 (17) |
| Respiratory failure | 5 | 3 (25) |
| Parainfluenza virus infection | 4 | 3 (25) |
| Atelectasis | 4 | 4 (33) |
| Transaminases increased | 3 | 3 (25) |
| Human rhinovirus test positive | 4 | 3 (25) |
| Rhinorrhea | 4 | 3 (25) |
| Bronchiolitis | 3 | 3 (25) |
| Diarrhea | 3 | 3 (25) |
| Ear infection | 2 | 2 (17) |
| Fall | 3 | 3 (25) |
| Pharyngitis streptococcal | 2 | 2 (17) |
| Pneumonia respiratory syncytial virus | 2 | 2 (17) |
| Respiratory syncytial virus bronchiolitis | 2 | 2 (17) |
| Viral upper respiratory tract infection | 3 | 3 (25) |
aIn the AVXS-101 study, 3 additional infants received a low dose of AVXS-101. Total adverse events for this cohort were reported previously [10].
DISCUSSION
This summary of the completed 24-month post-treatment follow-up from the AVXS-101 study demonstrated improved outcomes in AVXS-101–treated infants with SMA1. The results are a substantial departure from the comparable NN101 infants with SMA1 cohort, with differences in survival, motor function, and motor milestone achievement appearing to increase over time [11]. Along with a significant positive impact on event-free survival (100% vs 38% event-free survival), AVXS-101 treatment also had a rapid positive impact on the expected early motor function decline in infants with SMA1, as demonstrated by increased CHOP-INTEND scores (9.8 and 15.4 points at 1 and 3 months post-dose, respectively). The self-complementary attribute of the AVXS-101 transgene, which enables rapid protein production, likely contributed to the sharp early increases in the CHOP-INTEND scores [17]. At 14 months of age, mean CHOP-INTEND scores improved by 22.7 points in AVXS-101–treated infants, contrasting with the decline of 8.9 points in the NN101 infants with SMA1 cohort. The majority of AVXS-101–treated infants also achieved major motor milestones, such as sitting unassisted with improved feeding, swallowing, and ventilation compared with the natural history cohort previously described [10]. Although both cohorts of infants with SMA1 had similar baseline mean CMAP peak area values (AVXS-101, 1.7 mV/s and NN101, 1.1 mV/s), AVXS-101–treated infants had greater mean CMAP peak area values than the NN101 infants with SMA1 cohort (3.2 vs 0.02 mV/s, respectively), suggesting improved motor unit function. In contrast, the healthy infant cohort had a mean CMAP peak area of 11.3 mV/s and 14.6 mV/s at 6 and 24 months of age, respectively. It appears that AVXS-101 treatment halted further motor unit function decline, and presumably allowed an increase similar to that seen in the NN101 healthy infant cohort. Even with initial decreased motor unit function, 9 of the 12 participants treated with AVXS-101 ultimately achieved the milestone of sitting without support for ≥30 seconds.
The durability of AVXS-101 efficacy was also observed, with no waning of motor function, ulnar CMAP peak area, or motor milestone achievements. As of March 8, 2019, the age of participants in cohort 1 (n = 3) ranges from 61.5 to 64.6 months, with the longest follow-up at 58.7 months. The age of cohort 2 (proposed therapeutic dose, n = 10) ranges from 41.3 to 57.5 months, with the longest follow-up at 51.9 months. The mean age of patients in cohorts 1 and 2 is 62.4 and 47.2 months, respectively.
The durability of favorable safety results with AVXS-101 was also demonstrated. The additional 22 adverse events and 4 serious adverse events since the last report [10] were related to the disease, and were primarily respiratory issues; none were treatment-related. Previously described treatment-related adverse events were limited to transient asymptomatic elevated liver enzymes [10], which is consistent with other AAV gene replacement therapy trials [18–20].
Given that the infants in the AVXS-101 study were symptomatic at enrollment, some degree of irreversible motor neuron loss had already occurred that would potentially impact motor function. Infants with SMA1 who were treated with AVXS-101 did not develop as quickly as the healthy infant cohort in terms of motor milestone achievement and motor function. However, most eventually continued to gain motor function and developed new motor milestones [11]. The AVXS-101 study results and other interventional study reports suggest that intervention with disease-modifying treatment at the youngest possible age and early in the disease course, potentially before symptoms occur, might provide the best opportunity for optimal outcomes [21, 22].
Limitations of this analysis include dissimilar baseline disease severity, dissimilar composite survival endpoint definition, and potential bias in the approach to supportive care. At baseline, the NN101 infants with SMA1 cohort had more progressed motor symptoms according to CHOP-INTEND scores (20.3 vs 28.2, respectively), which may predict a more severe trajectory. However, the majority of the AVXS-101–treated cohort with low CHOP-INTEND scores (<20) also achieved major motor milestones. Of note, the age of symptom onset in the AVXS-101 and NN101 cohorts was relatively similar; specifically, all infants in the AVXS-101 cohort and the majority (14 of 16) of the NN101 cohort had an age of symptom onset ≤3 months. Definitions of composite endpoints are not identical; the AVXS-101 study had a composite endpoint that included NIV, which arguably is a criterion more easily attained, as ventilatory support is commonly used. In addition, families of infants in the NN101 natural history study may have favored palliative care instead of intense hospitalization; thus, application of supportive care may be different between the study cohorts and may contribute to the higher proportion of deaths in the NN101 study. Furthermore, in the NN101 study, 4 infants in the SMA1 cohort were excluded, as their SMN2 copy status was unknown because they had died or were lost to follow-up before they could be tested. Thus, the differences between the cohorts may be understated because of their exclusion. Nevertheless, comparison of the 2 study cohorts, although imperfect, demonstrates the profound positive impact on disease course of AVXS-101 treatment in infants with SMA1 in this study.
CONCLUSIONS
End-of-study analysis demonstrated that AVXS-101 treatment substantially improved permanent ventilation-free survival, and significantly improved motor function and motor milestone achievement in infants with SMA1 in the study compared with the outcomes observed in the NeuroNEXT NN101 natural history cohort.
FUNDING SOURCE
NN101 was funded by NINDS (U01NS079163), Cure SMA, Muscular Dystrophy Association, and SMA Foundation. AVXS-101 was sponsored by AveXis, Inc.
CONFLICTS OF INTEREST
Samiah A. Al-Zaidy and Jerry R. Mendell report consultancy fees from AveXis, Inc. Jerry R. Mendell also serves as a consultant for Sarepta Therapeutics and Exonics Therapeutics. Stephen J. Kolb received consultancy fees from AveXis, Inc., Biogen Idec, and F. Hoffmann-La Roche. Linda Lowes received consultancy fees from AveXis, Inc. Lindsay N. Alfano reports no disclosures. Richard Shell received consultancy fees from AveXis, Inc. Kathleen R. Church reports no disclosures. Douglas M. Sproule, Douglas E. Feltner, Francis Ogrinc, Melissa Menier, James L’Italien, and Brian K. Kaspar are employees or contractors of AveXis, Inc. Sukumar Nagendran and Courtney Wells were employees of AveXis, Inc., during the conduct of the study. W. David Arnold received consultancy fees from F. Hoffmann-La Roche AG. John T. Kissel has received consultancy fees from AveXis, Inc.
CONTRIBUTORS’ STATEMENT PAGE
Drs. Al-Zaidy and Mendell designed the AVXS-101 study. Drs. Al-Zaidy, Lowes, Alfano, Church, Shell, Kissel, Arnold, and Mendell conducted the AVXS-101 study.
Dr. Kolb designed the NN101 study. Drs. Lowes, Alfano, Shell, Arnold, and Kolb conducted the NN101 study.
Dr. Ogrinc and Ms. Menier performed the analy-sis with the direction of Drs. Al-Zaidy, Nagendran, Sproule, L’Italien, Feltner, Arnold, Kolb, and Mendell.
Drs. Al-Zaidy, Kolb, Lowes, Nagendran, Sproule, Feltner, Wells, L’Italien, Arnold, Kaspar, and Mend-ell interpreted the data.
All authors wrote the manuscript with writingassistance from Drs. Cardenas and Heitzer. All authors critically revised the manuscript and appro-ved the final manuscript as submitted and agree to be accountable for all aspects of the work.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-190403.
ACKNOWLEDGMENTS
The authors would like to acknowledge the commitment and contribution of the clinical trial coordinators involved in both studies, especially Kelly Lehman and Mark McColly of Nationwide Children’s Hospital, Amy Bartlett of The Ohio State Wexner Medical Center, and the coordinators in the NeuroNEXT Clinical Trial Network. The authors thank Jessica Cardenas, PhD, and Marjet D. Heitzer, PhD, of Syneos Health for writing and editorial assistance in preparing this manuscript.
REFERENCES
[1] | Kolb SJ , Kissel JT . Spinal muscular atrophy. Neurol Clin. (2015) ;33: :831–46. doi: 10.1016/j.ncl.2015.07.004 |
[2] | Feldkotter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. (2002) ;70: :358–68. doi: 10.1086/338627 |
[3] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: :810–7. doi: 10.1212/WNL.0000000000000741 |
[4] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) ;82: :883–91. doi: 10.1002/ana.25101 |
[5] | De Sanctis R , Coratti G , Pasternak A , Montes J , Pane M , Mazzone ES , et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord. (2016) ;26: :754–9. doi: 10.1016/j.nmd.2016.10.002 |
[6] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul Disord. (2010) ;20: :155–61. doi: 10.1016/j.nmd.2009.11.014 |
[7] | Glanzman AM , McDermott MP , Montes J , Martens WB , Flickinger J , Riley S , et al. Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr Phys Ther. (2011) ;23: :322–6. doi: 10.1097/PEP.0b013e3182351f04 |
[8] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol. (2016) ;3: :132–45. doi: 10.1002/acn3.283 |
[9] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: :1723–32. doi: 10.1056/NEJMoa1702752 |
[10] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) ;377: :1713–22. doi: 10.1056/NEJMoa1706198 |
[11] | Mendell J , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac L , Prior T , et al. AVXS-101 phase 1 gene replacement therapy clinical trial in SMA type 1: Continued event free survival and achievement of developmental milestones (S29.001). Neurology. (2018) ;90: (15 suppl), S29.001. |
[12] | Oskoui M , Levy G , Garland CJ , Gray JM , O’Hagen J , De Vivo DC , et al. The changing natural history of spinal muscular atrophy type 1. Neurology. (2007) ;69: :1931–6. doi: 10.1212/01.wnl.0000290830.40544.b9 |
[13] | Mercuri E , Finkel RS , Mutoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) ;28: (2):103–15. doi: 10.1016/j.nmd.2017.11.005 |
[14] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2018) ;28: (3):197–207. doi: 10.1016/j.nmd.2017.11.004 |
[15] | Bayley N . Bayley Scales of Infant and Toddler Development. 3rd ed (Bayley III). San Antonio, TX: Pearson Education; (2005) . |
[16] | Wijnhoven TM , de Onis M , Onyango AW , Wang T , Bjoerneboe GE , Bhandari N , et al. Assessment of gross motor development in the WHO Multicentre Growth Reference Study. Food Nutr Bull. (2004) ;25: (1 suppl):S37–45. doi: 10.1177/15648265040251S105 |
[17] | Wang Z , Ma HI , Li J , Sun L , Zhang J , Xiao X . Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. (2003) ;10: :2105–11. doi: 10.1038/sj.gt.3302133 |
[18] | Colella P , Ronzitti G , Mingozzi F . Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev. (2018) ;8: :87–104. doi: 10.1016/j.omtm.2017.11.007 |
[19] | George LA , Sullivan SK , Giermasz A , Rasko JEJ , Samelson-Jones BJ , Ducore J , et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. (2017) ;377: :2215–27. doi: 10.1056/NEJMoa1708538 |
[20] | Miesbach W , Meijer K , Coppens M , Kampmann P , Klamroth R , Schutgens R , et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. (2018) ;131: :1022–31. doi: 10.1182/blood-2017-09-804419 |
[21] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. (2018) ;5: :145–58. doi: 10.3233/JND-180304 |
[22] | Servais L , Farrar M , Finkel RS , Kirschner J , Muntoni F , Sun P , et al. Nusinersen demonstrates greater efficacy in infants with shorter disease duration: End of study results from the ENDEAR study in infants with spinal muscular atrophy (SMA). Neuromuscul Disord. (2017) ;27: (suppl 2):S211. doi: https://doi.org/10.1016/j.nmd.2017.06.421 |


