
Abnormal Excitation-Contraction Coupling and Calcium Homeostasis in Myopathies and Cardiomyopathies
Abstract
Muscle contraction requires specialized membrane structures with precise geometry and relies on the concerted interplay of electrical stimulation and Ca2+ release, known as excitation-contraction coupling (ECC). The membrane structure hosting ECC is called triad in skeletal muscle and dyad in cardiac muscle, and structural or functional defects of triads and dyads have been observed in a variety of myopathies and cardiomyopathies. Based on their function, the proteins localized at the triad/dyad can be classified into three molecular pathways: the Ca2+ release complex (CRC), store-operated Ca2+ entry (SOCE), and membrane remodeling. All three are mechanistically linked, and consequently, aberrations in any of these pathways cause similar disease entities. This review provides an overview of the clinical and genetic spectrum of triad and dyad defects with a main focus of attention on the underlying pathomechanisms.
INTRODUCTION
Muscle contraction relies on the conversion of an electrical stimulus into mechanical work. In skeletal muscle, the sequence of events begins with the excitation of a motor neuron and the subsequent release of the neurotransmitter acetylcholine (ACh) into the neuromuscular junction. Binding of ACh to ACh receptors in the motor endplate triggers Na+ influx, generating a local depolarization of the membrane [1]. In cardiac muscle, membrane depolarization is not initiated by nervous activity, but by sinoatrial node (SAN) cells in the right atrium, spontaneously generating 60–100 action potentials per minute [2]. The action potential then spreads along the skeletal and cardiac muscle cells and deep into membrane invaginations known as T (transverse)-tubules [3]. Together with the terminal cisternae of the sarcoplasmic reticulum (SR), T-tubules form a specialized complex known as triad in skeletal muscle and dyad in cardiac muscle, hosting the excitation-contraction machinery (ECC, Fig. 1) [4]. In detail, ECC is driven by the multiprotein Ca2+ release complex (CRC), composed of the voltage-gated L-type Ca2+ channel dihydropyridine receptor (DHPR) on the T-tubule, and ryanodine receptor (RyR), triadin, calsequestrin, and junctin, all residing in the SR. In skeletal muscle, membrane depolarization at the T-tubules causes the conformational change of the α1 subunit of DHPR, encoded by CACNA1S, enabling its physical interaction with the Ca2+ channel RyR1 across the membrane gap between the T-Tubule and the SR [5]. This is different to cardiac muscle, where membrane depolarization triggers influx of extracellular Ca2+ through the cardiac α1 subunit of DHPR (CACNA1C), and the increased local Ca2+ levels then activate RyR2 via a mechanism known as Ca2+-induced Ca2+ release (CICR) [6]. Skeletal muscle and cardiac ryanodine receptors are homotetrameric Ca2+ channels, and the amount of released Ca2+ is regulated by a quaternary complex with junctin, calsequestrin and triadin [7]. Ca2+ release finally triggers shortening of the contractile unit, the sarcomere, composed of myosin and actin filaments and various structural and regulatory elements. The myosin head thereby binds to actin and performs a power stroke under ATP hydrolysis, resulting in a relative movement of the filaments and the generation of force [8]. Muscle relaxation occurs when Ca2+ is removed from the contractile unit through the combined action of Ca2+ pumps and Na+/Ca2+ exchangers [9]. Repeated contractions require the maintenance of high Ca2+ gradients and the strict regulation of luminal and cytosolic Ca2+ balance. Store-operated Ca2+ entry (SOCE) is a major mechanism regulating Ca2+ homeostasis, and essentially depends on the interaction of the reticular Ca2+ sensor STIM1 and the plasma membrane Ca2+ channel ORAI1. Upon Ca2+ depletion, STIM1 undergoes a conformational change and activates ORAI1 to induce extracellular Ca2+ influx, and Ca2+ store refill is then ensured by the reticular SERCA Ca2+ pumps [10–14].
Fig.1
Schematic representation of the skeletal muscle triad and the cardiac muscle dyad and illustration of the fundamental key pathways. SOCE (store-operated Ca2+ entry) controls Ca2+ homeostasis through the combined activity of STIM1 and ORAI1, membrane remodeling depends on myotubularin (MTM1), amphiphysin 2 (BIN1), dynamin 2 (DNM2), caveolin-3 (CAV3), and dysferlin (DYSF), and the CRC (Ca2+ release complex) involves DHPR and the complex composed of ryanodine receptor (RyR), calsequestrin (CASQ), and triadin (TRDN), while junctophilin ties the sarcoplasmic reticulum with the T-tubule. Proteins marked in gray have not yet been associated with skeletal muscle or heart disorders.
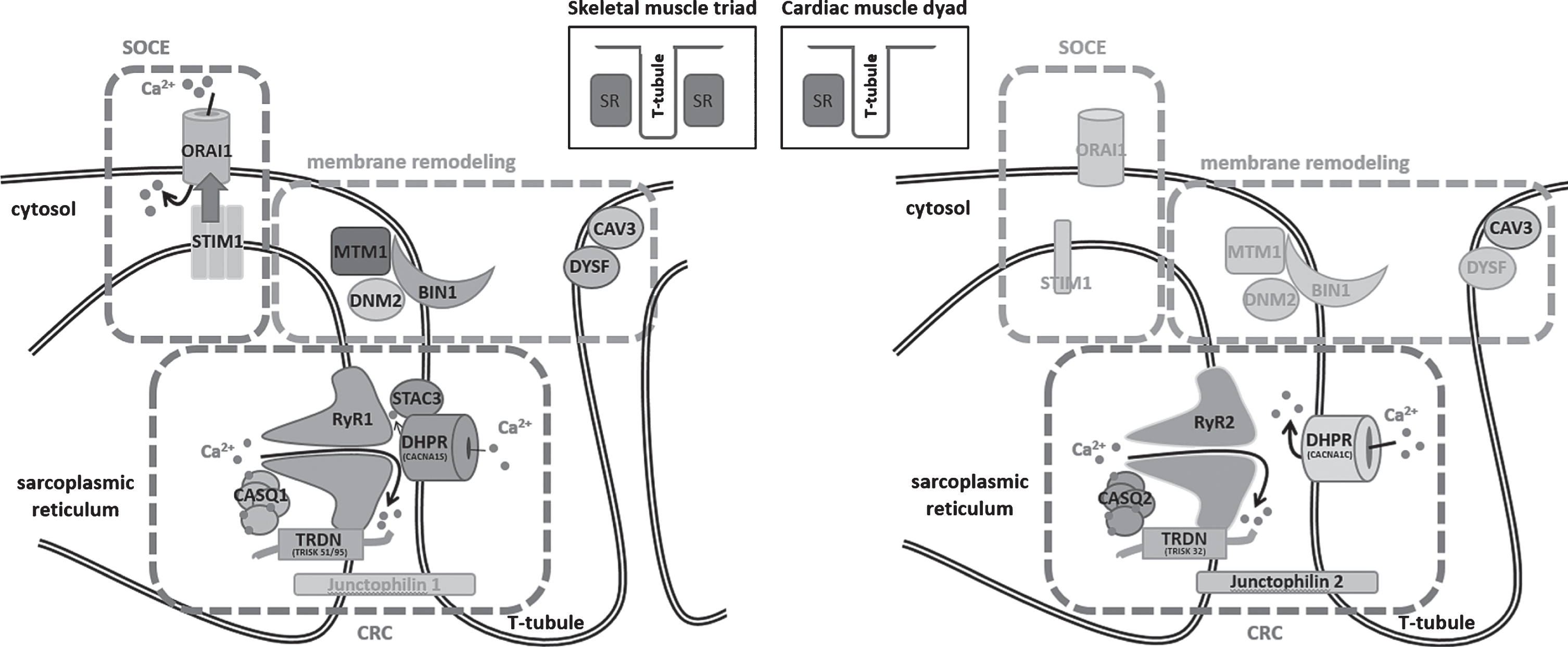
Disturbances of Ca2+ homeostasis can severely impact on muscle physiology, and structural or functional triad or dyad defects give rise to various myopathies and cardiac disorders (summarized in Table 1). This review aims to provide a gene-by-gene overview of the mutations leading to defects of the Ca2+ release complex (CRC), store-operated Ca2+ entry (SOCE), or membrane remodeling. The mutations thereby often affect paralogue genes with tissue-specific expression in skeletal muscle and heart, emphasizing the parallelism of myopathies and cardiomyopathies. We mainly focus on myopathies, and we draw an analogy between the pathomechanisms underlying skeletal muscle and heart dysfunction.
Table 1
Overview on the skeletal and cardiac muscle disorders, causative genes, and pathomechanisms associated with structural or functional triad and dyad defects
| Pathway | Skeletal muscle gene | Protein | Function | Disease (segregation) | Mutation impact | Cardiac muscle gene | Disease (segregation) | Mutation impact |
| CRC | CACNA1S | DHPR α1 subunit | Pore-forming subunit of voltage-gated Ca2+ channel | Malignant hyperthermia (MH, AD) | GOF | CACNA1C | Brugada syndrome (BrS, AD) | LOF |
| Hypokalemic periodic paralysis (HOKPP, AD) | LOF | Timothy syndrome (TS, AD) | GOF | |||||
| Congenital myopathy (AD, AR) | LOF | |||||||
| RYR1 | Ryanodine receptor | SR Ca2+ channel | Malignant hyperthermia (MH, AD) | GOF | RYR2 | Catecholaminergic polymorphic ventricular tachycardia (CPVT, AD) | GOF | |
| King-Denborough syndrome (AD) | GOF (?) | |||||||
| Central core disease (CCD, AD, AR) | LOF | |||||||
| Multi-minicore disease (MmD, AR) | LOF | Right ventricular dysplasia 2 (ARVD2, AD) | GOF | |||||
| Centronuclear myopathy (CNM, AR) | LOF | |||||||
| Congenital fiber type disproportion (CFTD, AR) | LOF | |||||||
| Samaritan myopathy (AR) | LOF | |||||||
| STAC3 | SH3 and cysteine-rich domains 3 | ECC regulator | Native American myopathy (NAM, AR) | LOF | – | – | – | |
| JPH1 | Junctophilin | Connects T-tubules with SR | – | – | JPH2 | Hypertrophic cardiomyopathy (CMH17, AD) | LOF | |
| TRDN | Triadin | RyR effector | – | – | TRDN | Catecholaminergic polymorphic ventricular tachycardia (CPVT, AR) | LOF | |
| CASQ1 | Calsequestrin | SR Ca2+ buffer | Vacuolar myopathy (AD) | GOF | CASQ2 | Catecholaminergic polymorphic ventricular tachycardia (CPVT, AR) | LOF | |
| Tubular aggregate myopathy (TAM, AD) | LOF | |||||||
| SOCE | STIM1 | Stromal interaction molecule 1 | SR Ca2+ sensor | Tubular aggregate myopathy (TAM, AD) | GOF | STIM1 | – | – |
| Stormorken syndrome (AD) | GOF | |||||||
| ORAI1 | ORAI1 | Plasma membrane Ca2+ channel | Tubular aggregate myopathy (TAM, AD) | GOF | ORAI1 | – | – | |
| Membrane remodeling | MTM1 | Myotubularin | Phosphoinositide phosphatase | Myotubular myopathy (MTM, XL) | LOF | MTM1 | – | – |
| BIN1 | Amphiphysin 2 | Membrane curvature | Centronuclear myopathy (CNM, AD, AR) | LOF | DNM2 | – | – | |
| DNM2 | Dynamin 2 | Membrane fission | Centronuclear myopathy (CNM, AD) | GOF | BIN1 | – | – | |
| CAV3 | Caveolin-3 | Caveolae formation | Rippling muscle disease 2 (RMD2, LGMD1C, AD) | LOF | CAV3 | Long QT syndrome 9 (LQT9, AD) | LOF | |
| Familial hypertrophic cardiomyopathy (AD) | LOF | |||||||
| Tateyama distal myopathy (AD) | LOF | |||||||
| DYSF | Dysferlin | Membrane repair | Miyoshi muscular dystrophy (MMD, AR) | LOF | DYSF | – | – | |
| Limb-girdle muscular dystrophy 2B (LGMDR2, AR) | LOF | |||||||
| Distal myopathy with anterior tibial onset (AR) | LOF |
AD = autosomal dominant; AR = autosomal recessive; XL = X-linked; GOF = gain-of-function; LOF = loss-of-function.
CA2+ RELEASE COMPLEX (CRC) DISORDERS
Mutations in CACNA1S and CACNA1C
The dihydropyridine receptor (DHPR) is composed of the five equimolar α1, α2, β, δ, and γ subunits [15]. The voltage sensor and pore-forming subunit α1, encoded by CACNA1S in skeletal muscle and by CACNA1C in heart, contains four transmembrane domains (I–IV), and the cytoplasmic loop between domains II and III mediates the activation of the ryanodine receptor in skeletal muscle [16, 17]. CACNA1S mutations have been associated with different disorders such as malignant hyperthermia (MH, OMIM #601887) [18], hypokalemic periodic paralysis (HOKPP, OMIM #170400) [19], and recently also with a new congenital myopathy entity [20], while CACNA1C mutations have been reported in Brugada syndrome (BrS, OMIM #611875) [21] and in Timothy syndrome (TS, OMIM #601005) [22].
Malignant hyperthermia (MH) is triggered by volatile anesthetics, and major symptoms are uncontrolled contractures, muscle rigidity, hyperthermia, cardiac arrhythmia, hyperkalemia, and hypermetabolism [23, 24]. MH is potentially fatal if not rapidly treated with the muscle relaxant dantrolene. CACNA1S mutations account for less than 10 % of the MH cases, and to date only three different heterozygous missense mutations have been reported [18, 25, 26]. It has been shown that these mutations increase the sensitivity of RyR1 to activation, leading to elevated resting Ca2+ levels in the cytosol [27, 28]. Muscle biopsies from MH patients were largely unremarkable and showed only minor alterations of the mitochondrial shape and also manifested mitochondrial breakdown, both potentially resulting from aberrant Ca2+ homeostasis in the muscle fibers [29]. Hypokalemic periodic paralysis is characterized by episodic and recurrent muscle weakness leading to paraparesis or tetraparesis lasting several hours to several days. The paralytic attacks are accompanied by hypokalemia, and treatment with the potassium channel opener acetazolamide has been shown to be beneficial [30]. A subset of HOKPP patients develop adult-onset myopathy, and the causative CACNA1S missense mutations affect a highly conserved arginine residue in the α1 voltage sensor domain [31]. Functional studies in the mouse model demonstrated that the loss of the positive charge impacts on the gating properties of the channel by causing an anomalous voltage-dependent inward current [32]. CACNA1S mutations were also associated with an unusual congenital myopathy with both dominant and recessive disease inheritance [20]. All patients had a comparable clinical presentation of generalized muscle weakness at birth, and the muscle biopsies exhibited cores, central nuclei, dilated triads, as well as a particular alveolar aspect of the intermyofibrillar network. DHPR stimulation resulted in significantly reduced reticular Ca2+ release in patient myotubes, suggesting an uncoupling of DHPR and RyR1 [20].
CACNA1C mutations have been found in patients with Brugada syndrome and Timothy syndrome. Brugada syndrome is a rare arrhythmia condition that can cause fast polymorphic ventricular tachycardia or ventricular fibrillation preceding sudden death [21]. The ECG pattern typically shows a persistent ST segment elevation with a right bundle branch block [21]. Heterozygous CACNA1C missense mutations only account for a subset of Brugada syndrome cases, and it has been shown that these mutations lead to decreased Ca2+ channel activity [21]. Timothy syndrome is a multi-systemic disorder combining a congenital heart disease - including patent ductus arteriosus, patent foramen ovale, ventricular septal defects, and tetralogy of Fallot - with immune deficiency, syndactyly, and cognitive abnormalities [22]. ECG typically reveals prolongation of the QT interval, leading to life-threatening arrhythmias. In contrast to Brugada syndrome, the Timothy syndrome mutations involve a gain-of-function and lead to sustained depolarizing L-type Ca2+ currents resulting from reduced channel inactivation [22].
Mutations in RYR1 and RYR2
The sarcoplasmic reticulum in skeletal and cardiac muscle is composed of structurally and functionally distinct domains, the junctional and the longitudinal SR. The junctional SR is primarily involved in Ca2+ release and hosts the ryanodine receptor (RyR). Each polypeptide of this homotetrameric protein is composed of more than 5000 amino acids forming six transmembrane domains. The C-terminal end of the RyR protein constitutes the Ca2+ channel, and the N-terminal cytoplasmic protrusion spans the narrow gap separating the junctional SR from the T-tubule [33–37]. Heterozygous missense mutations in RYR1 which account for the majority of molecularly diagnosed MH cases, render the channel hypersensitive to triggering agents, and lead to excessive Ca2+ release from the SR [38, 39]. Mutations in RYR1 have also been found to cause King-Denborough syndrome (OMIM #145600) [40], central core disease (CCD, OMIM #117000) [41, 42], multi-minicore disease (MmD, OMIM #255320) [43], centronuclear myopathy (CNM), congenital fiber type disproportion (CFTD), and the Samaritan myopathy [44], underlining the phenotypic variability arising from mutations in RYR1. RYR2 mutations have been described in the allelic diseases catecholaminergic polymorphic ventricular tachycardia (CPVT, OMIM #604772) [45] and arrhythmogenic right ventricular dysplasia 2 (ARVD2, OMIM # 600996) [46].
King-Denborough syndrome is a congenital myopathy associated with skeletal abnormalities, dysmorphic features, and susceptibility to malignant hyperthermia [47]. Muscle biopsies show a variety of myopathic features including atrophy, internalized nuclei, or cores [48]. It is not understood how the identified RYR1 mutations cause King-Denborough syndrome, but they reside in the same regions as MH mutations, indicating that both disorders might share a common pathomechanism [48].
Central core disease has been named after the central areas with reduced oxidative activity and a variable degree of sarcomeric disorganization running along the longitudinal muscle fiber axis. Clinically, CCD can be stable over long periods and typically presents with childhood-onset hypotonia, proximal muscle weakness, mild facial weakness, joint hypermobility, and scoliosis [49]. CCD is mainly caused by heterozygous missense mutations affecting amino acids in the C-terminal pore-forming domain of RyR1, and more severe cases with a fetal akinesia syndrome involving arthrogryposis and skeletal deformities have been described with recessive disease inheritance [50]. A subset of RYR1 mutations were found to generate a leaky channel leading to Ca2+ store depletion, others alter the interaction with DHPR and thereby uncouple excitation from contraction. In both cases, the amount of released Ca2+ upon depolarization is reduced [51–53].
Multi-minicore disease results from recessive RYR1 mutations dispersed over the entire gene, and is usually associated with an earlier disease onset and more severe clinical features with additional extraocular muscle involvement compared to CCD [43]. Biopsies from MmD patients display multiple areas of reduced mitochondrial activity and sarcomeric organization, and these minicores only span a limited zone on the longitudinal axis of the muscle fiber. Most MmD cases arise from missense mutations, but nonsense and splice site mutations have also been described. Patients with a clinicopathologic diagnosis of centronuclear myopathy (CNM) were found to carry recessive RYR1 missense or truncating mutations as well [54]. The affected individuals had an antenatal disease onset and were severely hypotonic at birth. Similarly to MmD, the patients had delayed motor milestones and showed facial weakness with extraocular muscle involvement. The muscle biopsies displayed cores as well as centralized nuclei, demonstrating a phenotypic overlap of RYR1-related MmD and CNM. Another muscle disorder associated with recessive missense and truncating RYR1 mutations is congenital fiber type disproportion (CFTD) [55]. The clinical presentation of the CFTD patients is comparable to RYR1-related MmD and CNM, and the muscle biopsies showed significantly smaller type I fibers as the main histological hallmark, whereas cores and centralized nuclei were not observed. The Samaritan myopathy is a unique form of multi-minicore disease, as it is characterized by an “inverse” course of disease: affected individuals present with a severe muscle phenotype at birth, but hypotonia and muscle weakness gradually improve and adults are only mildly affected [56]. This disease is caused by a single homozygous missense mutation in RYR1 and has only been described in the Samaritans, considering themselves as descendants of one of the twelve tribes forming the ancient kingdom of Israel in the 8th century BCE. In both multi-minicore disease and Samaritan myopathy, the RyR1 protein level was found to be strongly reduced in muscle extracts from affected individuals [44, 57], and siRNA-mediated downregulation of RYR1 in myotubes led to reduced CACNA1S expression and a disruption of the spatial organization of the EC coupling machinery, while IP3R was upregulated [58]. These findings illustrate that RyR1 deficiency alters the expression pattern of genes involved in Ca2+ homeostasis, and indicate that the upregulation of the alternative Ca2+ regulating IP3R system might be a compensatory mechanism.
Catecholaminergic polymorphic ventricular tachycardia is an arrhythmogenic disorder caused by heterozygous RYR2 mutations in more than 50% of the cases [45]. CPVT patients have a structurally normal heart and a normal resting ECG. Physical activity or stress can induce an increase of monomorphic and then polymorphic premature ventricular contractions (PVCs), followed by polymorphic ventricular tachycardia and ventricular fibrillation, possibly leading to syncope or sudden death [59]. It was shown that the RYR2 mutations alter the sensitivity of the channel, which tends to spontaneous Ca2+ release under conditions of reticular Ca2+ overload (SOICR, store-overload-induced Ca2+ release) [60, 61]. The mutations thereby do not appear to impact on ECC, as arrhythmias are not observed in unstimulated conditions. Dantrolene was shown to rescue arrhythmogenic RyR2 defects in cellular CPVT models, indicating a common pathomechanism for CPVT and malignant hyperthermia [62, 63]. Another heart disease caused by dominant RYR2 mutations is arrhythmogenic right ventricular dysplasia 2 (ARVD2), characterized by fibrous fatty replacements of the myocardium and in particular in the subepicardial layer of the right ventricle [46]. Physical effort can induce polymorphic ventricular tachycardia, representing a risk of sudden death. The ARVD2 mutations were shown to alter the RyR2 response to caffeine, resulting in an abnormal Ca2+ release [64]. The missense mutations causing either CPVT or ARVD2 are conjointly clustered in distinct N-terminal, central, and C-terminal regions of RyR2.
Mutations in STAC3
STAC3 is a recently discovered component of the excitation-contraction machinery, and mutations in STAC3 have been associated with the debilitating Native American myopathy (NAM, OMIM #255995) [65]. This autosomal recessive disorder was first reported in the Lumbee Indian tribe in North Carolina and is characterized by neonatal muscle weakness and arthrogryposis, dysmorphic facial features, and susceptibility to malignant hyperthermia with high mortality rate in childhood and adolescence [66]. Muscle biopsies from affected individuals revealed a non-specific myopathic pattern [65]. STAC3 possesses a functional interaction site for both the skeletal muscle and cardiac α1 subunit of DHPR, and promotes the localization of DHPR at the triad membrane [67, 68], demonstrating the importance of STAC3 in the regulation of excitation-contraction coupling. Indeed, the NAM STAC3 missense mutation was shown to reduce ECC [67], and the same observation was also made in myotubes from Stac3 knockout mice and morpholino-induced knockdown of stack3 in zebrafish embryos [65, 67].
Mutations in JPH2
The junctophilins are components of the junctional membrane complex (JMC), which physically and functionally connects the T-tubule with the SR to maintain the triad and dyad architecture [69]. The cardiac paralogue junctophilin 2 is encoded by JPH2, and mutations in this gene have been associated with hypertrophic cardiomyopathy (CMH17, OMIM #613873), one of the most common causes for sudden cardiac death in young athletes [70]. The JPH2 mutations were found to cause junctophilin 2 mislocalization, impaired Ca2+ handling, and cardiomyocyte hypertrophy in the cellular model. A murine model manifested pacing-induced spontaneous Ca2+ sparks followed by atrial fibrillations, indicating that junctophilin 2 also has a regulatory role on RyR2 [71].
Mutations in the ryanodine receptor effectors TRDN, CASQ1, and CASQ2
Triadin physically links the ryanodine receptor with calsequestrin [7], and recessive TRDN mutations cause catecholaminergic polymorphic ventricular tachycardia (CPVT, OMIM #615441) [72], a major cause of sudden death. All CPVT mutations were found in exons present in both the skeletal muscle and the cardiac isoform, and resulted in triadin protein loss. Accordingly, a subset of TRDN patients manifested an additional skeletal muscle disorder [73], and ablation of triadin in murine knockout models resulted in cardiac and skeletal muscle phenotypes [74, 75]. The hearts of the mice displayed lower expression levels of RYR2, CASQ2, and ASPH (junctin), abnormal contact sites between the T-tubules and the SR, as well as reticular Ca2+ overload resulting in spontaneous Ca2+ release (SOICR) and cardiac arrhythmias [74]. This emphasizes the importance of triadin in the structural and functional integrity of the cardiac Ca2+ release units. Physiological studies on skeletal muscle revealed an important reduction in muscle strength, and ultrastructural investigations uncovered abnormally oriented T-tubules, strongly suggesting that ECC is impaired [75].
Calsequestrin is the major Ca2+ storage protein in the sarcoplasmic reticulum. It binds Ca2+ with moderate affinity (Kd ∼ 103 M–1) and high capacity (40–50 ions/protein), and forms higher-order polymers with increasing Ca2+-binding capacities [76]. Calsequestrin also actively participates in the Ca2+ release process by providing a large Ca2+ pool in close proximity to the RyR release channel [77]. Heterozygous CASQ1 missense mutations were found in vacuolar myopathy (OMIM #616231) [78] and in tubular aggregate myopathy (TAM) [79, 80], both involving mild muscle weakness, but differing at the histopathological level. Vacuolar myopathy biopsies show vacuoles containing diverse SR proteins and enlargement of the terminal SR cisternae [78], while TAM muscle fibers typically accumulate densely packed membrane tubules [81]. Both disorders arise from adverse mutational effects: the TAM-related CASQ1 mutations were shown to significantly impair calsequestrin polymerization, resulting in reduced Ca2+ storage abilities of the SR, while the vacuolar myopathy mutation promoted the formation of high molecular mass polymers [78–80]. Noteworthy, Casq1 null mice exhibit a susceptibility to heat- and anesthetics-induced sudden death resembling malignant hyperthermia [82]. Recessive mutations in the cardiac paralogue CASQ2 have been associated with catecholamine-induced polymorphic ventricular tachycardia (CPVT, OMIM #114251) [83]. Whilst dominant RYR2 mutations are found in about half of the CPVT patients, recessive mutations in CASQ2 and TRDN are more seldom and account for less than 5% of the cases. Patients with CASQ2 mutations exhibit a childhood-onset relative resting bradychardia and a mild prolongation of the QTc segment, with Holter monitoring showing polymorphic ventricular tachycardia during exercise [83, 84]. In line with the functional impact of CASQ1 mutations, it was shown that the CASQ2 missense mutations alter the polymerization dynamics of calsequestrin, resulting in reduced Ca2+ binding capacities [85]. It has been proposed that the reduced Ca2+ buffering leads to a higher level of unbound Ca2+ in the SR with an increased risk for SOICR [86]. Indeed, mouse models harboring human CASQ2 mutations displayed stress-induced spontaneous Ca2+ release leading to ventricular tachycardia [87].
STORE-OPERATED CA2+ ENTRY (SOCE) DISORDERS
Upon stimulation of the ryanodine receptor, Ca2+ is released to the cytoplasm to trigger muscle contraction and to regulate various signaling pathways. The Ca2+ stores are refilled through store-operated Ca2+ entry (SOCE), an essential mechanism for prolonged tetanic stimulation that limits the effects of fatigue. The key players of SOCE are the reticular Ca2+ sensor STIM1 and the plasma membrane Ca2+ channel ORAI1. STIM1 contains an intraluminal region with Ca2+-sensing EF-hands, and a cytosolic part able to interact with ORAI1 [13, 88]. Ca2+ store depletion initiates STIM1 unfolding, oligomerization, and accumulation of the oligomers at the SR/plasma membrane junctions, where they bind and activate ORAI1 and thereby induce Ca2+ entry [11, 12, 89–92]. Ca2+ reuptake to the SR is ensured by the ATP-dependent SERCA pumps.
Mutations in STIM1
Heterozygous missense mutations in STIM1 were found in patients with tubular aggregate myopathy (TAM, OMIM #160565), a rare muscle disorder involving weakness, cramps, and myalgia, which is characterized by the presence of densely packed membrane tubules on biopsies [93]. TAM clinically and genetically overlaps with Stormorken syndrome (STRMK, OMIM #185070), a multi-systemic disorder associating TAM with miosis, thrombocytopenia, ichthyosis, asplenia, dyslexia, and short stature [94, 95]. Although TAM and STRMK form a clinical continuum, the Ca2+-binding EF-hand domains appear to be a mutation hotspot in TAM patients [93, 96–100], while the full clinical picture of Stormorken sydrome essentially arises from a substitution of a single amino acid in the cytosolic domain of STIM1 [101–103]. Nevertheless, TAM and STRMK mutations were both shown to induce constitutive STIM1 oligomerization and ORAI1 activation without SR Ca2+ depletion in the cellular model, and accordingly, TAM and STRMK myoblasts featured increased basal Ca2+ levels in the cytosol [93, 100–103]. The multi-systemic phenotype of Stormorken syndrome might result from a different mutational impact on Ca2+-dependent inactivation (CDI) of the ORAI1 Ca2+ channel. Electrophysiological studies on transfected cells demonstrated that the STRMK mutation especially suppresses CDI, suggesting a prolonged Ca2+ influx compared to TAM [103]. SOCE is not restricted to skeletal or cardiac muscle, but instead regulates Ca2+ homeostasis in various tissues, explaining the broad phenotypical presentation of Stormorken syndrome. A recently generated mouse models harboring the STRMK mutation recapitulated the main clinical features of the human disorder [104]. The mice were smaller than the control littermates, and displayed muscle weakness, thrombocytopenia, spleen dysplasia, skin irritations, and abnormal Ca2+ homeostasis in the myotubes.
Mutations in ORAI1
ORAI1 is a highly Ca2+-specific plasma membrane channel composed of hexamers forming three concentric rings around the central pore, and each ORAI1 subunit contains 4 alpha-helical transmembrane domains [105, 106]. Channel opening is mediated through the interaction of STIM1 with the cytoplasmic N- and C-termini of the ORAI1 subunits [107]. Similarly to STIM1, heterozygous missense mutations in ORAI1 cause TAM with additional signs of Stormorken syndrome (OMIM #615883). All mutations affect highly conserved amino acids in the transmembrane domains [103, 108–110]. Exogenous expression of mutant ORAI1 resulted in elevated basal Ca2+ levels and significant Ca2+ influx in the absence of Ca2+ store depletion [108–110], and similar results were also obtained in patient myotubes [109, 110]. Mutations in the first transmembrane domain were especially shown to cause a long-lasting Ca2+ influx, correlating with a more severe disease phenotype [108].
MEMBRANE REMODELING DISORDERS
Remodeling of membranes is essential for normal muscle function [111]. The skeletal muscle triad and the cardiac dyad are specialized membrane structures with precise geometry, and the mechanical stress exerted by steady contractions require efficient membrane repair mechanisms. Consequently, defects in proteins acting in membrane trafficking and remodeling result in muscle disorders. The triad and dyad formation and maintenance are coordinated by the concerted action of various proteins including myotubularin, amphiphysin 2, dynamin 2, caveolin, and dysferlin, and mutations in any of the respective coding genes lead to structural T-tubule defects in animal models and humans [112].
Mutations in MTM1
Myotubularin, encoded by MTM1, is a 3-phosphatase dephosphorylating the phosphoinositides PtdIns3P and PtdIns(3,5)P2 [113], which confer a local signature to the membrane and play a key role in the recruitment of proteins, in endocytosis, and in membrane trafficking [114, 115]. MTM1 mutations cause X-linked myotubular (or centronuclear) myopathy (XLMTM, OMIM #310400), a severe and generalized neonatal muscle disorder predominantly affecting boys and involving atrophic muscle fibers, type I fiber predominance, centralized nuclei, and sometimes basophilic rings known as necklace fibers on muscle biopsies [116, 117]. Women can also be affected, especially in case of skewed X-inactivation, but the clinical presentation is often milder [118]. More than 280 different causative XLMTM mutations have been reported, and most are nonsense mutations, splice-site mutations, or deletions, and result in a strong reduction of the myotubularin protein level and the dysregulation of the phosphoinositide metabolism [119, 120]. Mice, dogs, or zebrafish devoid of myotubularin exhibited a disorganization of the triad structure, reduced levels of RyR1 and DHPR, and defects in excitation-contraction coupling [121–123].
Mutations in BIN1
Amphiphysin 2, encoded by BIN1, is able to sense and induce membrane curvature in endocytosis and recycling. It interacts with dynamin 2, and connects the actin and microtubule cytoskeleton to the nuclear envelope LINC (linker of nucleoskeleton and cytoskeleton) complex [124–126]. The skeletal muscle-specific isoform of amphiphysin 2 contains a phosphoinositide-binding domain and plays a role in T-tubule biogenesis [127]. BIN1 mutations cause either dominant (ADCNM) [128] or recessive (ARCNM, OMIM #255200) [129] centronuclear myopathy, and both disorders presumably involve a different pathomechanism. ARCNM occurs as a severe childhood-onset disorder predominantly affecting the proximal muscles, and the biopsies display atrophy, type I fiber predominance, and nuclear centralization and clustering [129, 130]. ADCNM is clinically milder, has been described with adulthood-onset muscle weakness, and the biopsies of affected individuals exhibit nuclear centralization and clustering as well as ultrastructural triad abnormalities [128]. Comparative functional studies have shown that both dominant and recessive BIN1 mutations prevent membrane tubulation and interfere with protein-protein interactions, and that only the defects caused by the recessive mutations can be rescued by wild type amphiphysin 2 [128]. This is in accordance with the observation that heterozygous carriers of recessive BIN1 mutations are healthy.
Mutations in DNM2
Dynamin 2 is a mechanochemical enzyme involved in membrane fission and acting as a key player for vesicle formation and membrane trafficking at the plasma membrane and the trans-Golgi network [131, 132]. It binds PtdIns(4,5)P2, interacts with amphiphysin 2, and is able to self-assemble into rings [132–135]. Heterozygous missense mutations restricted to hotspots in the MID (middle), PH (phospholipid binding), and GED (GTPase effector) domains have been found in patients with autosomal dominant centronuclear myopathy (ADCNM, OMIM #160150) [136, 137]. The clinical presentation thereby correlates with the position of the mutation, and ranges from severe neonatal hypotonia and respiratory insufficiency to adult-onset distal muscle weakness and mild eye movement defects [138]. Histological findings on muscle biopsies include myofiber atrophy, central nuclei, type I fiber predominance, as well as prominent fiber size variability and radial arrangements of sarcoplasmic strands [130, 138]. Biochemical studies indicated that the DNM2 mutations increase dynamin 2 oligomer stability and GTPase activity [139, 140], and in vivo investigations suggested that dynamin 2 is negatively regulated by myotubularin and amphiphysin 2 [141, 142]. Knock-in mice harbouring the most common human mutation R465W manifested a mild myopathy with a structural disorganization of the muscle fibers, abnormal contractile properties, and elevated basal Ca2+ concentrations in the cytosol [143, 144]. AAV-mediated expression of the same mutation resulted in abnormal T-tubule structures and a swollen sarcoplasmic reticulum [145], and expression of the severe S619L mutation in zebrafish revealed defective excitation-contraction coupling [146].
Mutations in CAV3
Caveolin-3 is the principal protein component of caveolae in muscle. Caveolae are cholesterol and sphingolipid-rich invaginations of the plasma membrane playing an important role in signal transduction and vesicular transport [147]. Caveolin-3 belongs to the dystrophin-glycoprotein complex conferring stability to the sarcolemma [148], and also localizes to the developing T-tubules during muscle differentiation [149]. Mutations in CAV3 give rise to various skeletal and cardiac muscle disorders including rippling muscle disease 2 (RMD2, also known as limb-girdle muscular dystrophy 1C, OMIM #606072) [150, 151], Tateyama distal myopathy (OMIM #614321) [152], long QT syndrome 9 (LQT9, OMIM #611818) [153], and familial hypertrophic cardiomyopathy (CMH, OMIM #192600) [154]. RMD2 is a childhood-onset muscle disorder characterized by mild to moderate proximal muscle weakness, cramps, myalgia, visible ripples moving over the muscle following mechanical stimulation, and elevated CK levels, and muscle biopsies from affected individuals display dystrophic changes and almost complete loss of caveolin-3 expression [150, 155]. Tateyama distal myopathy mainly affects the small muscles of the hands and feet, and the very same CAV3 mutation can cause RMD2 or Tateyama myopathy, suggesting that both disorders are part of a clinical continuum [151, 152]. Long QT syndrome 9 is characterized by a prolonged QT interval and polymorphic ventricular arrhythmias potentially causing syncope, seizure, or sudden death, and the causative CAV3 mutations were shown to induce an increased late sodium current [153]. Hereditary ventricular hypertrophy is an idiopathic cardiomyopathy that manifests with diastolic dysfunction of the cardiac ventricles, and can entail arrhythmia, congestive heart failure, and sudden death. As for the skeletal muscle disorders, the CMH-related CAV3 mutation resulted in a strongly reduced cell surface expression of caveolin-3 [154]. Mice lacking caveolin-3 showed a RMD2-like mild myopathy, and investigations of the skeletal muscle revealed disorganized T-tubule [156], highlighting the importance of caveolin-3 in T-tubule biogenesis.
Mutations in DYSF
Dysferlin plays a crucial role in the process of membrane repair, and co-localizes with caveolin-3 at the site of the injury [157, 158]. It is also found in the cytoplasm, and localizes with DHPR in the developing T-tubule system [159]. Mutations in DYSF have been associated with Miyoshi muscular dystrophy (MMD, OMIM #254130) [160], limb-girdle muscular dystrophy 2B (LGMDR2, formerly LGMD2D, OMIM #253601) [160], and distal myopathy with anterior tibial onset (OMIM #606768) [160]. Miyoshi muscular dystrophy is an early-adult-onset disorder involving distal muscle weakness of the upper and lower limbs with sparing of the intrinsic hand muscles [161]. Serum creatine kinase levels are increased, and biopsies show a dystrophic pattern with necrosis. LGMDR2 patients generally present with a less distal muscle involvement, and distal myopathy with anterior tibial onset has been described in a single consanguineous family with a rapidly progressive course [160]. However, Miyoshi, LGMDR2, and distal myopathy biopsies are similar, and all three disorders can arise from identical DYSF mutations resulting in strong reduction of dysferlin [160]. They are therefore often conjointly referred to as dysferlinopathies. Dysferlin-deficient mice developed a progressive muscular dystrophy closely resembling the human disease, and histological and ultrastructural analyses of muscle samples revealed T-tubule anomalies similar to what has been seen in Cav3 null mice [157].
LINK BETWEEN CRC, SOCE AND MEMBRANE REMODELING
CRC, SOCE and membrane remodeling are mechanistically interconnected, as CRC and SOCE concur and require the formation and maintenance of specialized membrane structures. This explains why defects in these key pathways cause similar disease entities. It was proposed that amphiphysin 2 is essential for the microtubule-dependent transport of DHPR to the T-tubules in heart [162], and accordingly, Bin1 knockdown in murine cardiomyocytes resulted in decreased surface DHPR, leading to impaired Ca2+ transients [163]. Mtm1-deficient mice displayed a reduction in protein levels and an abnormal localization of RyR1 and the α1 subunit of DHPR, most probably contributing to the altered triad structure and the defective Ca2+ transients observed in the animals [121, 164]. DHPR also interacts with dysferlin and caveolin-3, and it has been suggested that the formation of this oligomeric complex plays a role in the fusion of caveolin-3-containing vesicles with T-tubules [159].
Phosphoinositides are critical for triad and dyad structure, excitation-contraction coupling, as well as SOCE. The membrane repair capacities of dysferlin largely depend on its Ca2+-dependent binding of PtdIns4P and PtdIns(4,5)P2 [165], and the latter is also crucial for the recruitment of amphiphysin 2 and dynamin 2 to the membrane [133, 166]. PtdIns(3,5)P2 modulates skeletal muscle and heart contractibility through activation of RyR1 and RyR2 [167–169], and increased levels of PtdIns(3,5)P2 were shown to induce chronic Ca2+ leakage from the sarcoplasmic reticulum, and promoted the degradation of DHPR [167, 168, 170]. Other studies highlighted the importance of phosphoinositides in SOCE: STIM1 interacts with PtdIns(4,5)P2 through its polybasic C-terminus, and inhibition of phosphatidylinositol 4-kinase prevents the formation of STIM1 oligomers at the plasma membrane [171]. The inhibition of phosphatidylinositol 4-kinases but not isolated PtdIns(4,5)P2 depletion also significantly reduced store-operated Ca2+ entry, indicating that PtdIns4P is a determinant for SOCE activation [172]. STIM1 was also found to bind the α1 subunit of DHPR in cardiac cells and to suppress the depolarization-induced channel opening [173]. In turn, STIM1 is negatively regulated by low polymeric forms of calsequestrin, which interfere with STIM1-ORAI1 interactions [174], demonstrating a reciprocal regulation of CRC and SOCE and the importance of the membrane structures these pathways rely on.
CONCLUSIONS
Cardiac and skeletal muscle contraction rely on similar membrane structures and the coordinated action of identical or evolutionary related proteins. Although a variety of cardiac and skeletal muscle disorders arise from structural or functional dyad or triad defects, only mutations affecting the Ca2+ release complex were found in malignant hyperthermia and in both cardiopathies and congenital myopathies. Mutations in CACNA1C, RYR2, and CASQ2 cause life-threatening arrhythmias primarily resulting from spontaneous SR Ca2+ release following exercise, and mutations in the skeletal muscle paralogues CACNA1S, RYR1, and CASQ1 cause muscle weakness resulting from reduced Ca2+ release upon ECC activation. With CAV3 as the only exception, mutations in genes implicated in membrane remodeling or SOCE were found in patients with congenital myopathies, but weren’t associated with hyperthermia or heart diseases. Noteworthy, mutations in the SOCE genes STIM1 and ORAI1, as well as in the membrane remodeling genes BIN1 and DNM2 mostly involve amino acid substitutions, and do not result in total protein loss [93, 109, 129, 137]. Knockout animal models for these genes exist, and some of them displayed cardiac defects. Cardiomyocyte-restricted knockout of Stim1 in mice resulted in a progressive decline of left ventricular function and the development of a dilated cardiomyopathy [175], and inactivation of the zebrafish orthologue of ORAI1 entailed reduced ventricular systolic function, bradycardia, and heart failure [176]. A critical role of STIM1 and ORAI1 has also been suggested in the pathogenesis of cardiac hypertrophy in humans [177]. Mice with heart-specific Bin1 deletion exhibited decreased T-tubule folding at the dyad and were susceptible to ventricular arrhythmias resulting from prolonged action potential duration [178]. In zebrafish hearts, bin1 knockdown significantly reduced Ca2+ transients and led to severe ventricular contractile dysfunction [163], and in humans, BIN1 expression was shown to be significantly decreased in failing cardiomyocytes [163]. Lastly, mice with dysferlin deficiency developed a mild cardiomyopathy that was exacerbated by stress exercise [179]. These examples demonstrate that SOCE and membrane remodeling are critical for normal skeletal and cardiac muscle function, and that mutations in the genes encoding key proteins of these pathways might account for molecularly yet uncharacterized heart diseases [121, 133, 162–174].
ACKNOWLEDGMENTS
We thank Valérie Biancalana for her comments and suggestions. This work was supported by Inserm and Association Française contre les Myopathies (AFM 21267). The authors have no conflict of interest to report.
REFERENCES
[1] | Edman KA , Schild HO . Interaction of acetylcholine, calcium and depolarization in the contraction of smooth muscle. Nature. (1961) ;190: :350–2. |
[2] | Shih HT . Anatomy of the action potential in the heart. Tex Heart Inst J. (1994) ;21: :30–41. |
[3] | Adrian RH , Costantin LL , Peachey LD . Radial spread of contraction in frog muscle fibres. J Physiol. (1969) ;204: :231–57. |
[4] | Franzini-Armstrong C , Jorgensen AO . Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol. (1994) ;56: :509–34. |
[5] | Rios E , Brum G . Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. (1987) ;325: :717–20. |
[6] | Nabauer M , Callewaert G , Cleemann L , Morad M . Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. (1989) ;244: :800–3. |
[7] | Zhang L , Kelley J , Schmeisser G , Kobayashi YM , Jones LR . Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. (1997) ;272: :23389–97. |
[8] | Ebashi S . Regulatory mechanism of muscle contraction with special reference to the Ca-troponin-tropomyosin system. Essays Biochem. (1974) ;10: :1–36. |
[9] | Parekh AB , Penner R . Store depletion and calcium influx. Physiol Rev. (1997) ;77: :901–30. |
[10] | Liou J , Fivaz M , Inoue T , Meyer T . Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci U S A. (2007) ;104: :9301–6. |
[11] | Luik RM , Wu MM , Buchanan J , Lewis RS . The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. (2006) ;174: :815–25. |
[12] | Park CY , Hoover PJ , Mullins FM , Bachhawat P , Covington ED , Raunser S , et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. (2009) ;136: :876–90. |
[13] | Stathopulos PB , Zheng L , Li GY , Plevin MJ , Ikura M . Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. (2008) ;135: :110–22. |
[14] | Wu MM , Buchanan J , Luik RM , Lewis RS . Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. (2006) ;174: :803–13. |
[15] | Catterall WA . Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. (2011) ;3: :a003947. |
[16] | Lu X , Xu L , Meissner G . Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J Biol Chem. (1994) ;269: :6511–6. |
[17] | Wu J , Yan Z , Li Z , Yan C , Lu S , Dong M , et al. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. (2015) ;350: :aad2395. |
[18] | Monnier N , Procaccio V , Stieglitz P , Lunardi J . Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. (1997) ;60: :1316–25. |
[19] | Ptacek LJ , Tawil R , Griggs RC , Engel AG , Layzer RB , Kwiecinski H , et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. (1994) ;77: :863–8. |
[20] | Schartner V , Romero NB , Donkervoort S , Treves S , Munot P , Pierson TM , et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. (2017) ;133: :517–33. |
[21] | Brugada P , Brugada J . Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. (1992) ;20: :1391–6. |
[22] | Splawski I , Timothy KW , Sharpe LM , Decher N , Kumar P , Bloise R , et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. (2004) ;119: :19–31. |
[23] | Denborough MA , Forster JF , Lovell RR , Maplestone PA , Villiers JD . Anaesthetic deaths in a family. Br J Anaesth. (1962) ;34: :395–6. |
[24] | MacLennan DH , Phillips MS . Malignant hyperthermia. Science. (1992) ;256: :789–94. |
[25] | Pirone A , Schredelseker J , Tuluc P , Gravino E , Fortunato G , Flucher BE , et al. Identification and functional characterization of malignant hyperthermia mutation TS in the outer pore of the Cavalpha1S-subunit. Am J Physiol Cell Physiol. (2010) ;299: :C1345–54. |
[26] | Toppin PJ , Chandy TT , Ghanekar A , Kraeva N , Beattie WS , Riazi S . A report of fulminant malignant hyperthermia in a patient with a novel mutation of the CACNA1S gene. Can J Anaesth. (2010) ;57: :689–93. |
[27] | Eltit JM , Bannister RA , Moua O , Altamirano F , Hopkins PM , Pessah IN , et al. Malignant hyperthermia susceptibility arising from altered resting coupling between the skeletal muscle L-type Ca2+ channel and the type 1 ryanodine receptor. Proc Natl Acad Sci U S A. (2012) ;109: :7923–8. |
[28] | Weiss RG , O’Connell KM , Flucher BE , Allen PD , Grabner M , Dirksen RT . Functional analysis of the RH malignant hyperthermia mutation in the DHPR reveals an unexpected influence of the III-IV loop on skeletal muscle EC coupling. Am J Physiol Cell Physiol. (2004) ;287: :C1094–102. |
[29] | Lavorato M , Gupta PK , Hopkins PM , Franzini-Armstrong C . Skeletal muscle microalterations in patients carrying malignant hyperthermia-related mutations of the e-c coupling machinery. Eur J Transl Myol. (2016) ;26: :6105. |
[30] | Tricarico D , Camerino DC . Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies. Front Pharmacol. (2011) ;2: :8. |
[31] | Matthews E , Labrum R , Sweeney MG , Sud R , Haworth A , Chinnery PF , et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. (2009) ;72: :1544–7. |
[32] | Wu F , Mi W , Hernandez-Ochoa EO , Burns DK , Fu Y , Gray HF , et al. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest. (2012) ;122: :4580–91. |
[33] | Franzini-Armstrong C , Protasi F . Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol Rev. (1997) ;77: :699–729. |
[34] | Wagenknecht T , Grassucci R , Frank J , Saito A , Inui M , Fleischer S . Three-dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature. (1989) ;338: :167–70. |
[35] | Efremov RG , Leitner A , Aebersold R , Raunser S . Architecture and conformational switch mechanism of the ryanodine receptor. Nature. (2015) ;517: :39–43. |
[36] | Yan Z , Bai X , Yan C , Wu J , Li Z , Xie T , et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. (2015) ;517: :50–5. |
[37] | Zalk R , Clarke OB , des Georges A , Grassucci RA , Reiken S , Mancia F , et al. Structure of a mammalian ryanodine receptor. Nature. (2015) ;517: :44–9. |
[38] | Manning BM , Quane KA , Ording H , Urwyler A , Tegazzin V , Lehane M , et al. Identification of novel mutations in the ryanodine-receptor gene (RYR1) in malignant hyperthermia: Genotype-phenotype correlation. Am J Hum Genet. (1998) ;62: :599–609. |
[39] | Gillard EF , Otsu K , Fujii J , Khanna VK , de Leon S , Derdemezi J , et al. A substitution of cysteine for arginine 614 in the ryanodine receptor is potentially causative of human malignant hyperthermia. Genomics. (1991) ;11: :751–5. |
[40] | D’Arcy CE , Bjorksten A , Yiu EM , Bankier A , Gillies R , McLean CA , et al. King-denborough syndrome caused by a novel mutation in the ryanodine receptor gene. Neurology. (2008) ;71: :776–7. |
[41] | Quane KA , Healy JM , Keating KE , Manning BM , Couch FJ , Palmucci LM , et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. (1993) ;5: :51–5. |
[42] | Zhang Y , Chen HS , Khanna VK , De Leon S , Phillips MS , Schappert K , et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet. (1993) ;5: :46–50. |
[43] | Monnier N , Ferreiro A , Marty I , Labarre-Vila A , Mezin P , Lunardi J . A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet. (2003) ;12: :1171–8. |
[44] | Bohm J , Leshinsky-Silver E , Vassilopoulos S , Le Gras S , Lerman-Sagie T , Ginzberg M , et al. Samaritan myopathy, an ultimately benign congenital myopathy, is caused by a RYR1 mutation. Acta Neuropathol. (2012) ;124: :575–81. |
[45] | Priori SG , Napolitano C , Tiso N , Memmi M , Vignati G , Bloise R , et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. (2001) ;103: :196–200. |
[46] | Tiso N , Stephan DA , Nava A , Bagattin A , Devaney JM , Stanchi F , et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. (2001) ;10: :189–94. |
[47] | King JO , Denborough MA . Anesthetic-induced malignant hyperpyrexia in children. J Pediatr. (1973) ;83: :37–40. |
[48] | Dowling JJ , Lillis S , Amburgey K , Zhou H , Al-Sarraj S , Buk SJ , et al. King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. (2011) ;21: :420–7. |
[49] | Jungbluth H , Sewry CA , Muntoni F . Core myopathies. Semin Pediatr Neurol. (2011) ;18: :239–49. |
[50] | Romero NB , Monnier N , Viollet L , Cortey A , Chevallay M , Leroy JP , et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain. (2003) ;126: :2341–9. |
[51] | Avila G , Dirksen RT . Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J Gen Physiol. (2001) ;118: :277–90. |
[52] | Dirksen RT , Avila G . Altered ryanodine receptor function in central core disease: Leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc Med. (2002) ;12: :189–97. |
[53] | Kraeva N , Zvaritch E , Rossi AE , Goonasekera SA , Zaid H , Frodis W , et al. Novel excitation-contraction uncoupled RYR1 mutations in patients with central core disease. Neuromuscul Disord. (2013) ;23: :120–32. |
[54] | Wilmshurst JM , Lillis S , Zhou H , Pillay K , Henderson H , Kress W , et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. (2010) ;68: :717–26. |
[55] | Clarke NF , Waddell LB , Cooper ST , Perry M , Smith RL , Kornberg AJ , et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat. (2010) ;31: :E1544–50. |
[56] | Lev D , Sadeh M , Watemberg N , Dabby R , Vinkler C , Ginzberg M , et al. A benign congenital myopathy in an inbred Samaritan family. Eur J Paediatr Neurol. (2006) ;10: :182–5. |
[57] | Ducreux S , Zorzato F , Ferreiro A , Jungbluth H , Muntoni F , Monnier N , et al. Functional properties of ryanodine receptors carrying three amino acid substitutions identified in patients affected by multi-minicore disease and central core disease, expressed in immortalized lymphocytes. Biochem J. (2006) ;395: :259–66. |
[58] | Zhou H , Rokach O , Feng L , Munteanu I , Mamchaoui K , Wilmshurst JM , et al. RyR1 deficiency in congenital myopathies disrupts excitation-contraction coupling. Hum Mutat. (2013) ;34: :986–96. |
[59] | Sumitomo N , Harada K , Nagashima M , Yasuda T , Nakamura Y , Aragaki Y , et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. (2003) ;89: :66–70. |
[60] | Jiang D , Wang R , Xiao B , Kong H , Hunt DJ , Choi P , et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. (2005) ;97: :1173–81. |
[61] | Jiang D , Xiao B , Zhang L , Chen SR . Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. (2002) ;91: :218–25. |
[62] | Jung CB , Moretti A , Mederos y Schnitzler M , Iop L , Storch U , Bellin M , et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. (2012) ;4: :180–91. |
[63] | Kobayashi S , Yano M , Suetomi T , Ono M , Tateishi H , Mochizuki M , et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol. (2009) ;53: :1993–2005. |
[64] | Thomas NL , George CH , Lai FA . Role of ryanodine receptor mutations in cardiac pathology: More questions than answers? Biochem Soc Trans. (2006) ;34: :913–8. |
[65] | Horstick EJ , Linsley JW , Dowling JJ , Hauser MA , McDonald KK , Ashley-Koch A , et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun. (2013) ;4: :1952. |
[66] | Stamm DS , Aylsworth AS , Stajich JM , Kahler SG , Thorne LB , Speer MC , et al. Native American myopathy: Congenital myopathy with cleft palate, skeletal anomalies, and susceptibility to malignant hyperthermia. Am J Med Genet A. (2008) ;146A: :1832–41. |
[67] | Polster A , Nelson BR , Olson EN , Beam KG . Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc Natl Acad Sci U S A. (2016) ;113: :10986–91. |
[68] | Wong King Yuen SM , Campiglio M , Tung CC , Flucher BE , Van Petegem F . Structural insights into binding of STAC proteins to voltage-gated calcium channels. Proc Natl Acad Sci U S A. (2017) ;114: :E9520–E8. |
[69] | Takeshima H , Komazaki S , Nishi M , Iino M , Kangawa K . Junctophilins: A novel family of junctional membrane complex proteins. Mol Cell. (2000) ;6: :11–22. |
[70] | Landstrom AP , Weisleder N , Batalden KB , Bos JM , Tester DJ , Ommen SR , et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. (2007) ;42: :1026–35. |
[71] | Beavers DL , Wang W , Ather S , Voigt N , Garbino A , Dixit SS , et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. (2013) ;62: :2010–9. |
[72] | Roux-Buisson N , Cacheux M , Fourest-Lieuvin A , Fauconnier J , Brocard J , Denjoy I , et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. (2012) ;21: :2759–67. |
[73] | Engel AG , Redhage KR , Tester DJ , Ackerman MJ , Selcen D . Congenital myopathy associated with the triadin knockout syndrome. Neurology. (2017) ;88: :1153–6. |
[74] | Chopra N , Yang T , Asghari P , Moore ED , Huke S , Akin B , et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. (2009) ;106: :7636–41. |
[75] | Oddoux S , Brocard J , Schweitzer A , Szentesi P , Giannesini B , Brocard J , et al. Triadin deletion induces impaired skeletal muscle function. J Biol Chem. (2009) ;284: :34918–29. |
[76] | Damiani E , Salvatori S , Margreth A . Characterization of calsequestrin of avian skeletal muscle. J Muscle Res Cell Motil. (1990) ;11: :48–55. |
[77] | Lee KW , Maeng JS , Choi JY , Lee YR , Hwang CY , Park SS , et al. Role of Junctin protein interactions in cellular dynamics of calsequestrin polymer upon calcium perturbation. J Biol Chem. (2012) ;287: :1679–87. |
[78] | Rossi D , Vezzani B , Galli L , Paolini C , Toniolo L , Pierantozzi E , et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum Mutat. (2014) ;35: :1163–70. |
[79] | Barone V , Del Re V , Gamberucci A , Polverino V , Galli L , Rossi D , et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum Mutat. (2017) ;38: :1761–73. |
[80] | Bohm J , Lornage X , Chevessier F , Birck C , Zanotti S , Cudia P , et al. CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol. (2018) ;135: :149–51. |
[81] | Chevessier F , Bauche-Godard S , Leroy JP , Koenig J , Paturneau-Jouas M , Eymard B , et al. The origin of tubular aggregates in human myopathies. J Pathol. (2005) ;207: :313–23. |
[82] | Dainese M , Quarta M , Lyfenko AD , Paolini C , Canato M , Reggiani C , et al. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB J. (2009) ;23: :1710–20. |
[83] | Lahat H , Pras E , Olender T , Avidan N , Ben-Asher E , Man O , et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. (2001) ;69: :1378–84. |
[84] | di Barletta MR , Viatchenko-Karpinski S , Nori A , Memmi M , Terentyev D , Turcato F , et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. (2006) ;114: :1012–9. |
[85] | Kim E , Youn B , Kemper L , Campbell C , Milting H , Varsanyi M , et al. Characterization of human cardiac calsequestrin and its deleterious mutants. J Mol Biol. (2007) ;373: :1047–57. |
[86] | Priori SG , Chen SR . Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. (2011) ;108: :871–83. |
[87] | Song L , Alcalai R , Arad M , Wolf CM , Toka O , Conner DA , et al. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. (2007) ;117: :1814–23. |
[88] | Stathopulos PB , Li GY , Plevin MJ , Ames JB , Ikura M . Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem. (2006) ;281: :35855–62. |
[89] | Hogan PG , Lewis RS , Rao A . Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. (2010) ;28: :491–533. |
[90] | Li Z , Lu J , Xu P , Xie X , Chen L , Xu T . Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. (2007) ;282: :29448–56. |
[91] | Luik RM , Wang B , Prakriya M , Wu MM , Lewis RS . Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. (2008) ;454: :538–42. |
[92] | Xu P , Lu J , Li Z , Yu X , Chen L , Xu T . Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. (2006) ;350: :969–76. |
[93] | Bohm J , Chevessier F , Maues De Paula A , Koch C , Attarian S , Feger C , et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet. (2013) ;92: :271–8. |
[94] | Stormorken H , Sjaastad O , Langslet A , Sulg I , Egge K , Diderichsen J . A new syndrome: Thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin Genet. (1985) ;28: :367–74. |
[95] | Bohm J , Laporte J . Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium. (2018) ;76: :1–9. |
[96] | Bohm J , Chevessier F , Koch C , Peche GA , Mora M , Morandi L , et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet. (2014) ;51: :824–33. |
[97] | Harris E , Burki U , Marini-Bettolo C , Neri M , Scotton C , Hudson J , et al. Complex phenotypes associated with STIM1 mutations in both coiled coil and EF-hand domains. Neuromuscul Disord. (2017) ;27: :861–72. |
[98] | Hedberg C , Niceta M , Fattori F , Lindvall B , Ciolfi A , D’Amico A , et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J Neurol. (2014) ;261: :870–6. |
[99] | Noury JB , Bohm J , Peche GA , Guyant-Marechal L , Bedat-Millet AL , Chiche L , et al. Tubular aggregate myopathy with features of Stormorken disease due to a new STIM1 mutation. Neuromuscul Disord. (2017) ;27: :78–82. |
[100] | Walter MC , Rossius M , Zitzelsberger M , Vorgerd M , Muller-Felber W , Ertl-Wagner B , et al. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul Disord. (2015) ;25: :577–84. |
[101] | Misceo D , Holmgren A , Louch WE , Holme PA , Mizobuchi M , Morales RJ , et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum Mutat. (2014) ;35: :556–64. |
[102] | Morin G , Bruechle NO , Singh AR , Knopp C , Jedraszak G , Elbracht M , et al. Gain-of-function mutation in STIM1 (P.R304W) is associated with stormorken syndrome. Hum Mutat. (2014) ;35: :1221–32. |
[103] | Nesin V , Wiley G , Kousi M , Ong EC , Lehmann T , Nicholl DJ , et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci U S A. (2014) ;111: :4197–202. |
[104] | Silva-Rojas R , Treves S , Jacobs H , Kessler P , Messaddeq N , Laporte J , et al. STIM1 over-activation generates a multi-systemic phenotype affecting skeletal muscle, spleen, eye, skin, bones, and the immune system in mice. Hum Mol Genet. 2018. |
[105] | Cai X , Zhou Y , Nwokonko RM , Loktionova NA , Wang X , Xin P , et al. The orai1 store-operated calcium channel functions as a hexamer. J Biol Chem. (2016) ;291: :25764–75. |
[106] | Hou X , Pedi L , Diver MM , Long SB . Crystal structure of the calcium release-activated calcium channel Orai. Science. (2012) ;338: :1308–13. |
[107] | Zheng H , Zhou MH , Hu C , Kuo E , Peng X , Hu J , et al. Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J Biol Chem. (2013) ;288: :11263–72. |
[108] | Bohm J , Bulla M , Urquhart JE , Malfatti E , Williams SG , O’Sullivan J , et al. ORAI1 mutations with distinct channel gating defects in tubular aggregate myopathy. Hum Mutat. (2017) ;38: :426–38. |
[109] | Endo Y , Noguchi S , Hara Y , Hayashi YK , Motomura K , Miyatake S , et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels. Hum Mol Genet. (2015) ;24: :637–48. |
[110] | Garibaldi M , Fattori F , Riva B , Labasse C , Brochier G , Ottaviani P , et al. A novel gain-of-function mutation in ORAI1 causes late-onset tubular aggregate myopathy and congenital miosis. Clin Genet. (2017) ;91: :780–6. |
[111] | Towler MC , Kaufman SJ , Brodsky FM . Membrane traffic in skeletal muscle. Traffic. (2004) ;5: :129–39. |
[112] | Al-Qusairi L , Laporte J . T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle. (2011) ;1: :26. |
[113] | Blondeau F , Laporte J , Bodin S , Superti-Furga G , Payrastre B , Mandel JL . Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. (2000) ;9: :2223–9. |
[114] | Di Paolo G , De Camilli P . Phosphoinositides in cell regulation and membrane dynamics. Nature. (2006) ;443: :651–7. |
[115] | Vicinanza M , D’Angelo G , Di Campli A , De Matteis MA . Phosphoinositides as regulators of membrane trafficking in health and disease. Cell Mol Life Sci. (2008) ;65: :2833–41. |
[116] | Laporte J , Hu LJ , Kretz C , Mandel JL , Kioschis P , Coy JF , et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. (1996) ;13: :175–82. |
[117] | Bevilacqua JA , Bitoun M , Biancalana V , Oldfors A , Stoltenburg G , Claeys KG , et al. “Necklace” fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol. (2009) ;117: :283–91. |
[118] | Biancalana V , Scheidecker S , Miguet M , Laquerriere A , Romero NB , Stojkovic T , et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. (2017) ;134: :889–904. |
[119] | Ketel K , Krauss M , Nicot AS , Puchkov D , Wieffer M , Muller R , et al. A phosphoinositide conversion mechanism for exit from endosomes. Nature. (2016) ;529: :408–12. |
[120] | Laporte J , Kress W , Mandel JL . Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol. (2001) ;50: :42–6. |
[121] | Al-Qusairi L , Weiss N , Toussaint A , Berbey C , Messaddeq N , Kretz C , et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A. (2009) ;106: :18763–8. |
[122] | Dowling JJ , Vreede AP , Low SE , Gibbs EM , Kuwada JY , Bonnemann CG , et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. (2009) ;5: :e1000372. |
[123] | Beggs AH , Bohm J , Snead E , Kozlowski M , Maurer M , Minor K , et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A. (2010) ;107: :14697–702. |
[124] | Peter BJ , Kent HM , Mills IG , Vallis Y , Butler PJ , Evans PR , et al. BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science. (2004) ;303: :495–9. |
[125] | Grabs D , Slepnev VI , Songyang Z , David C , Lynch M , Cantley LC , et al. The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J Biol Chem. (1997) ;272: :13419–25. |
[126] | D’Alessandro M , Hnia K , Gache V , Koch C , Gavriilidis C , Rodriguez D , et al. Amphiphysin 2 orchestrates nucleus positioning and shape by linking the nuclear envelope to the actin and microtubule cytoskeleton. Dev Cell. (2015) ;35: :186–98. |
[127] | Lee E , Marcucci M , Daniell L , Pypaert M , Weisz OA , Ochoa GC , et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. (2002) ;297: :1193–6. |
[128] | Bohm J , Biancalana V , Malfatti E , Dondaine N , Koch C , Vasli N , et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. (2014) ;137: :3160–70. |
[129] | Nicot AS , Toussaint A , Tosch V , Kretz C , Wallgren-Pettersson C , Iwarsson E , et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. (2007) ;39: :1134–9. |
[130] | Romero NB . Centronuclear myopathies: A widening concept. Neuromuscul Disord. (2010) ;20: :223–8. |
[131] | Jones SM , Howell KE , Henley JR , Cao H , McNiven MA . Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. (1998) ;279: :573–7. |
[132] | Warnock DE , Baba T , Schmid SL . Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol Biol Cell. (1997) ;8: :2553–62. |
[133] | Kojima C , Hashimoto A , Yabuta I , Hirose M , Hashimoto S , Kanaho Y , et al. Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J. (2004) ;23: :4413–22. |
[134] | Lin HC , Barylko B , Achiriloaie M , Albanesi JP . Phosphatidylinositol (4,5)-bisphosphate-dependent activation of dynamins I and II lacking the proline/arginine-rich domains. J Biol Chem. (1997) ;272: :25999–6004. |
[135] | Faelber K , Posor Y , Gao S , Held M , Roske Y , Schulze D , et al. Crystal structure of nucleotide-free dynamin. Nature. (2011) ;477: :556–60. |
[136] | Bitoun M , Bevilacqua JA , Prudhon B , Maugenre S , Taratuto AL , Monges S , et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. (2007) ;62: :666–70. |
[137] | Bitoun M , Maugenre S , Jeannet PY , Lacene E , Ferrer X , Laforet P , et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. (2005) ;37: :1207–9. |
[138] | Bohm J , Biancalana V , Dechene ET , Bitoun M , Pierson CR , Schaefer E , et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat. (2012) ;33: :949–59. |
[139] | Kenniston JA , Lemmon MA . Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. (2010) ;29: :3054–67. |
[140] | Wang L , Barylko B , Byers C , Ross JA , Jameson DM , Albanesi JP . Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J Biol Chem. (2010) ;285: :22753–7. |
[141] | Cowling BS , Chevremont T , Prokic I , Kretz C , Ferry A , Coirault C , et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest. (2014) ;124: :1350–63. |
[142] | Cowling BS , Prokic I , Tasfaout H , Rabai A , Humbert F , Rinaldi B , et al. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J Clin Invest. (2017) ;127: :4477–87. |
[143] | Durieux AC , Vignaud A , Prudhon B , Viou MT , Beuvin M , Vassilopoulos S , et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet. (2010) ;19: :4820–36. |
[144] | Fraysse B , Guicheney P , Bitoun M . Calcium homeostasis alterations in a mouse model of the Dynamin 2-related centronuclear myopathy. Biol Open. (2016) ;5: :1691–6. |
[145] | Cowling BS , Toussaint A , Amoasii L , Koebel P , Ferry A , Davignon L , et al. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am J Pathol. (2011) ;178: :2224–35. |
[146] | Gibbs EM , Davidson AE , Telfer WR , Feldman EL , Dowling JJ . The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis Model Mech. (2014) ;7: :157–61. |
[147] | Smart EJ , Graf GA , McNiven MA , Sessa WC , Engelman JA , Scherer PE , et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. (1999) ;19: :7289–304. |
[148] | Song KS , Li S , Okamoto T , Quilliam LA , Sargiacomo M , Lisanti MP . Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem. (1996) ;271: :9690–7. |
[149] | Parton RG , Way M , Zorzi N , Stang E . Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. (1997) ;136: :137–54. |
[150] | Minetti C , Sotgia F , Bruno C , Scartezzini P , Broda P , Bado M , et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. (1998) ;18: :365–8. |
[151] | Betz RC , Schoser BG , Kasper D , Ricker K , Ramirez A , Stein V , et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. (2001) ;28: :218–9. |
[152] | Tateyama M , Aoki M , Nishino I , Hayashi YK , Sekiguchi S , Shiga Y , et al. Mutation in the caveolin-3 gene causes a peculiar form of distal myopathy. Neurology. (2002) ;58: :323–5. |
[153] | Vatta M , Ackerman MJ , Ye B , Makielski JC , Ughanze EE , Taylor EW , et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. (2006) ;114: :2104–12. |
[154] | Hayashi T , Arimura T , Ueda K , Shibata H , Hohda S , Takahashi M , et al. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. (2004) ;313: :178–84. |
[155] | Figarella-Branger D , Pouget J , Bernard R , Krahn M , Fernandez C , Levy N , et al. Limb-girdle muscular dystrophy in a 71-year-old woman with an R27Q mutation in the CAV3 gene. Neurology. (2003) ;61: :562–4. |
[156] | Galbiati F , Engelman JA , Volonte D , Zhang XL , Minetti C , Li M , et al. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. (2001) ;276: :21425–33. |
[157] | Bansal D , Miyake K , Vogel SS , Groh S , Chen CC , Williamson R , et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. (2003) ;423: :168–72. |
[158] | Cai C , Weisleder N , Ko JK , Komazaki S , Sunada Y , Nishi M , et al. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. (2009) ;284: :15894–902. |
[159] | Ampong BN , Imamura M , Matsumiya T , Yoshida M , Takeda S . Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. (2005) ;24: :134–44. |
[160] | Liu J , Aoki M , Illa I , Wu C , Fardeau M , Angelini C , et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. (1998) ;20: :31–6. |
[161] | Miyoshi K , Kawai H , Iwasa M , Kusaka K , Nishino H . Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy. Seventeen cases in eight families including an autopsied case. Brain. (1986) ;109: (Pt 1):31–54. |
[162] | Hong TT , Smyth JW , Gao D , Chu KY , Vogan JM , Fong TS , et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. (2010) ;8: :e1000312. |
[163] | Hong TT , Smyth JW , Chu KY , Vogan JM , Fong TS , Jensen BC , et al. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. (2012) ;9: :812–20. |
[164] | Toussaint A , Cowling BS , Hnia K , Mohr M , Oldfors A , Schwab Y , et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. (2011) ;121: :253–66. |
[165] | Fuson K , Rice A , Mahling R , Snow A , Nayak K , Shanbhogue P , et al. Alternate splicing of dysferlin C2A confers Ca(2)(+)-dependent and Ca(2)(+)-independent binding for membrane repair. Structure. (2014) ;22: :104–15. |
[166] | Dong J , Misselwitz R , Welfle H , Westermann P . Expression and purification of dynamin II domains and initial studies on structure and function. Protein Expr Purif. (2000) ;20: :314–23. |
[167] | Shen J , Yu WM , Brotto M , Scherman JA , Guo C , Stoddard C , et al. Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca(2+) homeostasis. Nat Cell Biol. (2009) ;11: :769–76. |
[168] | Touchberry CD , Bales IK , Stone JK , Rohrberg TJ , Parelkar NK , Nguyen T , et al. Phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) potentiates cardiac contractility via activation of the ryanodine receptor. J Biol Chem. (2010) ;285: :40312–21. |
[169] | Rodriguez EG , Lefebvre R , Bodnar D , Legrand C , Szentesi P , Vincze J , et al. Phosphoinositide substrates of myotubularin affect voltage-activated Ca(2)(+) release in skeletal muscle. Pflugers Arch. (2014) ;466: :973–85. |
[170] | Tsuruta F , Green EM , Rousset M , Dolmetsch RE . PIKfyve regulates CaV1.2 degradation and prevents excitotoxic cell death. J Cell Biol. (2009) ;187: :279–94. |
[171] | Walsh CM , Chvanov M , Haynes LP , Petersen OH , Tepikin AV , Burgoyne RD . Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem J. (2009) ;425: :159–68. |
[172] | Korzeniowski MK , Popovic MA , Szentpetery Z , Varnai P , Stojilkovic SS , Balla T . Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem. (2009) ;284: :21027–35. |
[173] | Park CY , Shcheglovitov A , Dolmetsch R . The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. (2010) ;330: :101–5. |
[174] | Wang L , Zhang L , Li S , Zheng Y , Yan X , Chen M , et al. Retrograde regulation of STIM1-Orai1 interaction and store-operated Ca2+ entry by calsequestrin. Sci Rep. (2015) ;5: :11349. |
[175] | Collins HE , He L , Zou L , Qu J , Zhou L , Litovsky SH , et al. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am J Physiol Heart Circ Physiol. (2014) ;306: :H1231–9. |
[176] | Volkers M , Dolatabadi N , Gude N , Most P , Sussman MA , Hassel D . Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish. J Cell Sci. (2012) ;125: :287–94. |
[177] | Voelkers M , Salz M , Herzog N , Frank D , Dolatabadi N , Frey N , et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J Mol Cell Cardiol. (2010) ;48: :1329–34. |
[178] | Hong T , Yang H , Zhang SS , Cho HC , Kalashnikova M , Sun B , et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. (2014) ;20: :624–32. |
[179] | Han R , Bansal D , Miyake K , Muniz VP , Weiss RM , McNeil PL , et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Invest. (2007) ;117: :1805–13. |


