
Muscle Cells Fix Breaches by Orchestrating a Membrane Repair Ballet
Abstract
Skeletal muscle undergoes many micro-membrane lesions at physiological state. Based on their sizes and magnitude these lesions are repaired via different complexes on a specific spatio-temporal manner. One of the major repair complex is a dysferlin-dependent mechanism. Accordingly, mutations in the DYSF gene encoding dysferlin results in the development of several muscle pathologies called dysferlinopathies, where abnormalities of the membrane repair process have been characterized in patients and animal models. Recent efforts have been deployed to decipher the function of dysferlin, they shed light on its direct implication in sarcolemma resealing after injuries. These discoveries served as a strong ground to design therapeutic approaches for dysferlin-deficient patients. This review detailed the different partners and function of dysferlin and positions the sarcolemma repair in normal and pathological conditions.
INTRODUCTION
Dysferlin is a transmembrane protein belonging to the ferlin family. It is coded by the gene DYSF located on chromosome 2p13.2. The dysferlin protein is comprised of multiple domains responsible for its various functionalities. Among these are multiple C2 domains, which are known to be calcium-sensitive and involved mainly in phospholipid interactions [1–3]. Mutations in DYSF [4, 5] cause a class of muscular disorders termed “dysferlinopathies”, including two principal muscular dystrophies: Limb Girdle Muscular Dystrophy type 2B (LGMD2B) and Miyoshi Myopathy (MM). Both disorders are inherited in an autosomal recessive way; LGMD2B affects mainly proximal muscle while MM manifests predominantly in distal muscle. Despite variation in age of onset, patients with either disorder are mostly healthy and athletic at young ages without any clinical sign of pathology. Disease onset usually becomes evident only in the second decade, for example with difficulties in ambulation or climbing stairs. The disease progress slowly but frequently results in wheelchair dependency.
Dysferlinopathies are clinically and genetically heterogeneous, with a broad spectrum of severity from asymptomatic to marked functional disability [6–8]. Patients with LGMD2B and MM are notable phenotypically for exhibiting massive increases in blood creatine kinase, up to 100-fold compared to normal values, associated with severe muscle inflammation [9] that can be misdiagnosed as polymyositis [10–12]. Because of such misdiagnoses and the recent introduction of the international patient registry within only the last few years (https://www.jain-foundation.org/patient-registration), the worldwide prevalence and incidence of dysferlinopathies are hard to estimate with much accuracy. However, some reviews suggest that dysferlinopathies may be the second most frequent type of LGMD (25% of the LGMD cases) after LGMD2A, with a prevalence of up to 1/50,000 [13].
In this review we describe the function of dysferlin and its network; while dysferlin functions are not exclusively limited to membrane repair, we decided to focus the review on this role in light of the recent discoveries in the field.
DYSFERLIN AND ANNEXINS DIRECT THE MEMBRANE REPAIR PROCESS
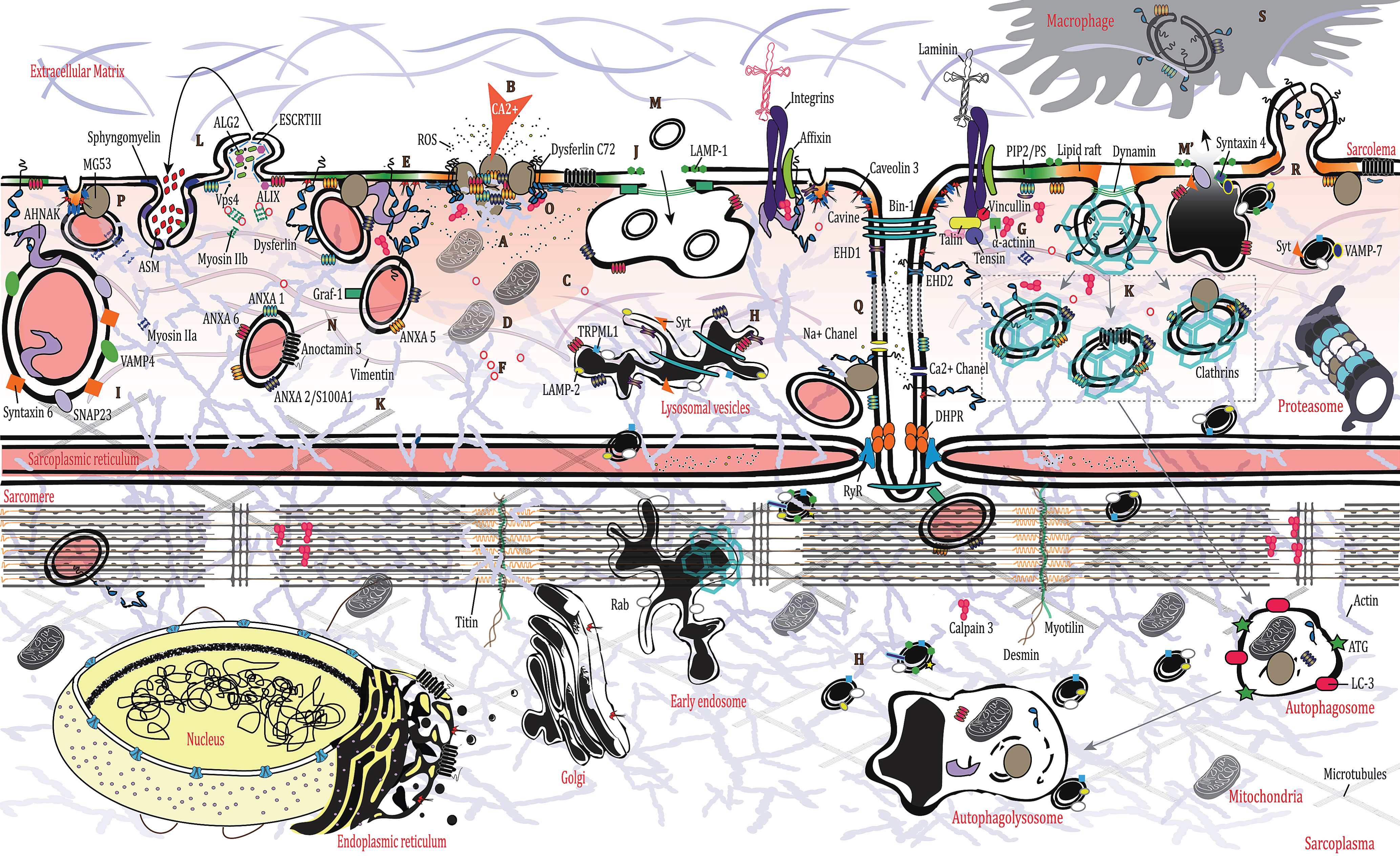
Due to its inherent function, skeletal muscle is a mechanically stressed organ and is thus exposed to frequent and sudden physiological tears at the membrane [14]. Membrane rupture is followed by a drastic increase in intracellular calcium levels, which in turn triggers plasma membrane repair (PMR) [15]. PMR is a multistep process that entails varied strategies aiming to repair the injury in a spatiotemporal manner. The cell creates an “emergency” patch to contain the damage (A on Fig), prior to other mechanisms (involving cytoskeletal and membrane remodeling) taking over the repair process. Calcium increase at the injury site lasts for several seconds (B on Fig), but it takes much longer to complete PMR (∼120 sec for myoblasts and up to several minutes for muscle fibers) [16, 17]. More specifically, a calcium gradient is formed after the injury that triggers the PMR events (C on Fig). This sets off a chain reaction involving recruitment and/or recycling of Ca2+-sensor proteins [synaptotagmins, ferlins, calpains, annexins, apoptosis-linked gene (ALG)-2], and additional proteins (ESCRT-III complex, MG53), membrane lipids, as well as cytoskeletal and organellar reorganization [18–25], which is absolutely essential for effective membrane repair [26]. Ca2+-triggered exocytosis likewise diminishes membrane tension, another key determinant of cell membrane resealing and effective repair [27, 28]. Despite some variation in the succession of events taking place during PMR, Ca2 + and ATP are indispensable [29] (D on Fig).
Cellular injuries also disturb the sarcomere, disorganize membrane lipid organization and composition, and influence the cytoskeletal dynamic [14] Phosphatidylserine (PS) becomes aggregated around wound sites over time while actin binds PS [30, 31] (E on Fig). Transitory actin cytoskeleton rearrangement is in turn essential for repair [14, 32]. Indeed, cytoskeletal actin facilitates the localized transport of internal vesicles to the wounded membrane, assisted by kinesin, while myosin motors promote exocytosis [33]. Also, consequent to the injury, proteins known to bind actin, like affixin, EDH2, and ANXA1, are increased at the lesion site together with F-actin in muscle cells [34–37] (A on Fig). Small GTPases (Rho, Rac, Cdc42) are also involved in distinct subcellular patterns by recruiting the cytoskeleton surrounding the lesion, as has been described in Drosophila or Xenopus oocytes [38–41] (F on Fig). PM depolarization is induced by the massive entry of calcium and the cleavage of two of actin’s partners, talin and vimentin, by calpains [42, 43] (G on Fig). Moreover, the disassembly of the microtubule mesh around the lesion, triggered by the calcium rise, may also influence the lysosomal transport and lipid traffic to the PM (H on Fig) [44, 45].
Intracellular vesicles
In the wake of cellular wounding, cells must undergo an emergency response (plugging) involving the cross-linkage of membranous compartments or vesicles, such as lysosomes, enlargeosomes, or other components to the plasma membrane such as membrane-deforming proteins (I on Fig) [46–48]. An elevation of Ca2 + in a 20 nm perimeter is required to trigger vesicle fusion. Following membrane rupture, there is an accumulation of small vesicles underneath the plasmalemma that function as the main reservoirs of Ca2 + required for membrane fusion. Among them, lysosomes represent one of the first actively recruited reservoirs, as confirmed by the presence of luminal epitopes of LAMP-1 (a lysosome-specific marker) on the cell surface after injury [29, 47] (J on Fig). This is also suggested by the observation that lysosomes form a pre-existing punctate distribution beneath the membrane [49, 50]. Indeed, lysosomes have the ability to fuse with the plasma membrane through a two-step approach, they are first recruited underneath the membrane independently from calcium and then they fuse with the plasma membrane in response to an increase in calcium via synaptotagmin VII which is present at the surface of the lysosome [47] (H on Figure). Therefore, lysosomes and lysosome-related organelles have been shown to participate in the membrane repair process [51, 52]. Calcium-dependent lysosomal exocytosis has been shown in injured muscle fibers, decreasing membrane tension and facilitating PMR [27] (M’ on Figure).
It is also widely acknowledged that the specific manner in which PMR takes place is dependent on the size of the lesion [29, 52]. Large injuries involve an increase of caveolar endocytosis [53] (K on Fig). Small lesions (<100 nm) meanwhile will trigger the ESCRT III complex, which promotes vesicle budding or membrane shedding via a complex involving apoptosis-linked gene 2 (ALG 2), ALG-2-interacting protein X (ALIX), ESCRT III and Vps4 [22, 25, 54] (L on Fig). Exocytosis releases acid sphingomyelinases (ASM), which will be turned into ceramide in the outer leaflet of the plasma membrane, leading to endosome formation [55, 56] (L on Fig). These domains change the binding affinities of annexins and caveolins for the plasmalemma, affect cytoskeleton dynamics, and bring about changes in plasmalemma architecture that play an important role in the progression of membrane repair [57, 58].
Intracellular vesicles undergo a rapid recycling in the region of the tears by a process of endocytosis/exocytosis turnover (M/M’ on Fig). Indeed, exocytosis of lysosomes, relying on synaptotagmin VII and calcium [47], is rapidly followed by massive endocytosis of small endosome vesicles that merge to form a larger endosome before joining the endocytic pathway [59, 60] (M on Fig). However, in dysferlinopathies, absence of dysferlin prevents the tethering of lysosomes to the membrane, thereby delaying their exocytosis [61]. This is especially problematic since lysosome exocytosis is a fundamental process occurring in all kinds of PMR independently of the type of injury involved (mechanical, toxin-induced perforation or other causes) [62].
The endocytic vesicles look highly similar to caveolae when observed via electronic microscopy as they are similar in appearance and size [63]. This is plausible since caveolar internalization could also function as a reservoir. Certainly, caveolae are highly expressed in tissue under mechanical stress or in a disease context [53, 63–65]. They could also merge to form a larger vesicle prior to their endocytosis or flattening, suggesting that an enlarged reservoir is required for membrane integrity [15, 53, 59, 66]. Markers of lysosomes or endosomes are also expressed on the surface of caveolae when fused after internalization [53]. Stretch-induced membrane rupture can also disrupt T-tubules by pulling them out of position [67] (Q on Fig). MG53, ANXA1, and dysferlin have been shown to localize at the T-tubule during stretch-induced injury, suggesting a role for the T-tubule as another membrane reservoir [68]. Moreover, EDH2 translocates to the lesion site via a T-tubule-dependent trafficking mechanism [36].
Finally, one other PMR mechanism of note is membrane blebbing, a temporary detachment of sub-membranous cytoskeleton from the plasma membrane that occurs when the PMR process needs more time to be fully completed or when the initial attempts at rescue have failed [69]. These blebs, which involve ANXA1 as a mediator, can be definitively excluded from the membrane if necessary [69].
Dysferlin
Dysferlin is a major mediator of PMR. Among other interactions, it has been reported to bind the cell membrane lipids phosphatidylserine (PS) and phosphatidylinositol bisphosphate (PIP2), and to co-localize with the SNARE protein syntaxin 4 and snap23 [70, 71]. Dysferlin-positive vesicles can fuse together via microtubule transport and accumulate underneath the membrane [72]. Additionally, it has been shown that dysferlin must be cleaved at its C-terminus by calpains to release an active fragment [composed of the last 2 C2 domains (out of 7) and the transmembrane domain] called minidysferlin-C72. Only the truncated dysferlin stays at the lesion site where it encircles the lesion [73, 74] (O on Fig). From there, as has been demonstrated, m- or μ-calpain can facilitate repair of damaged plasma membrane [75]. Dysferlin is also expressed at the PM via its interaction with caveolin 3, the main component of the caveolae [76, 77]. Dysferlin and MG53 are present at the surface of membrane vesicles, recruited by an actin-cytoskeleton–dependent mechanism during membrane damage [32, 78] (N on Fig).
MG53
Another protein found in caveolae and involved in membrane repair is MG53. This component binds directly to the PS at the plasma membrane or via Cavin-1/PTRF, probably in caveolae [79, 80]. MG53 and Cavin-1 may help in stabilizing the membrane by recruiting dysferlin and caveolin 3 [80, 81]. This mechanism also incorporates annexins (ANXA) 1 and 6 [82]. However, the direct involvement of this pathway in a physiological context is not clearly defined; it may involve prevention of cell death and therefore the maintenance of an environment favorable to membrane resealing with annexins as key factors [69, 82] (P in Fig). On the other hand, mutations in caveolin 3 and PTRF cause serious abnormalities in muscle where vesicles are often found underneath the PM [83, 84]. Finally, it has been suggested that MG53 could also function outside of the cell since the protein can participate to the PMR effort after intravenous injection of recombinant MG53 in a mouse model [85, 86].
Annexins
Annexins are a family of ubiquitously expressed soluble proteins binding negatively charged membrane lipids (mostly PS) in a Ca2+-dependent manner [87]. Among them, ANXA5 stands out since it can promote the formation of annexin-2D arrays below the membrane surface [88, 89] (A on Fig). Together with other components, ANXA (6, 1, 2) has been shown to promote several membrane-related processes like endocytosis or exocytosis, vesicle trafficking, and general membrane dynamics (including fusion, organization, and aggregation) [90]. ANXA2 seems to promote enlargeosomes’ movement toward the PM where they can fuse via SNAP23, VAMP4, and syntaxin6 or via a connecting ANXA2/S100A10-AHNAK complex [91–93] (I on Fig). Another protein interacting with ANXA2, S100A11, could play a role in the mesh formation by facilitating the actin polymerization at the wounding site [94, 95]. Enlargeosomes have also been involved in PMR with an undefined role; their main marker, AHNAK, is re-localized to the PM after injury [96]. ANXAs are therefore one of the major players in the membrane repair process [91]. ANXA 6, 1, 5, and 2 in particular are recruited at the outset to prevent extension of the wound by promoting emergency fusion [88, 91, 97] (A on Fig).
Additional proteins (MCOLN1/TRMPL1 and ANO5) have also been linked to PMR, but their roles are as yet not fully understood (A on Fig). It is believed that MCOLN1 may be involved in lysosomal exocytosis by promoting lysosomes’ trafficking to the membrane via a mechanism dependent on calcium [98]. The fact that ALG-2 and MCOLN1 interact in a calcium-dependent manner raises the question of possible involvement of MCOLN1 in membrane shedding [99]. In the same manner, ANO5 has been linked to PMR since it has been shown that recruitment of ANO5 vesicles to the membrane is triggered following the increase of calcium influx [100].
Altogether these results suggest that dysferlin and ANXAs, among other actors (caveolin 3, MG53, calpains, and others), are essential for the PMR process and more generally for muscle homeostasis, since damage to these components has been directly associated with pathologic processes or the exacerbation of numerous diseases [4, 5, 101–104]. They are highly active during the events following membrane rupture via vesicle recruitment and endocytosis/exocytosis events (K and R on Fig). Finally, it has been proposed recently that damaged membrane components could also be sequestered and removed from the membrane instead of being resealed, and we believe that in this context, dysferlin and ANXAs would likely be among the participants involved in this process [55]. It should also be noted that the vesicles involved in PMR are eliminated by macrophages (S on Fig) that have been recruited to the wound site via the increase of phosphatidylserine at the membrane triggered by dysferlin [105].
Fig.1
Membrane repair in muscle cell, Schematic representation of the different events from an emergency patch to a functional recovery in muscle cell after mechanic lesion. See text for description.
CONCLUDING REMARKS
PMR is an essential and conserved protective process found in multiple species and a variety of tissues. Several mechanisms have been proposed over the years but in light of recent progress in the field, it seems that the recycling of membrane vesicles in and out of the cells is crucial. Another consensus finding is that PMR is highly dependent on calcium and ATP and that the succession of events taking place has to be relatively fast (from an emergency response within seconds to a complete membrane repair within minutes). Studies suggest that the size and type of the lesion may trigger different mechanisms of repair, but one other possibility is that multiple mechanisms involved in the correction of the membrane damage can occur simultaneously within the same cell [106]. This hypothesis has served as the basis of our review, in which we seek to precisely depict the different players in the highly regulated PMR process in which we believe dysferlin plays a central role.
Furthermore, although a substantial body of knowledge about the PMR mechanisms has been attained in recent years, we are only beginning to fully comprehend the complexity of the events taking place, especially regarding the coordination of all the molecular participants. The knowledge accumulated in the past few years has been valuable to better understand the pathophysiology of numerous diseases involving defects in membrane repair. Nevertheless, numerous gaps still persist regarding our comprehension of the subtlety within each aspect of the pathology and in particular in dysferlinopathies where, although patients share a commonly mutated gene, the pathophysiology of the disease still varies significantly between patients independently of a given mutation for reasons not fully elucidated. Moreover, each domain of dysferlin has been shown to carry potential disease-causing mutations, resulting in a variety of natural histories in patients and demonstrating the importance of each domain in dysferlin’s functions. Based on the finding that the c-terminal portion of the protein is essential for membrane repair [73, 74, 107], mutations in this region could have dramatic consequences for muscle homeostasis. Therefore, new experiments (based on live imaging and models using mutated proteins) are still needed to fill in the gaps and help conceive more appropriate therapeutics in disorders resulting from malfunctioning PMR mechanisms, such as muscular dystrophy and cardiomyopathy.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We acknowledge J. Wes Ulm for proofreading.
This work was supported by the Association Francaise contre les Myopathies (AFM), Institut National de la Sante et de la Recherche Medicale (INSERM), Centre National de la Recherche Scientifique (CNRS), PPMD. A.D and F.B. have received PostDocs fellowship grants from the AFM and the UCLA Center for Duchenne Muscular Dystrophy-NIH NIAMS.
U54 AR052646 Wellstone Center of Excellence Training Fellowship respectively.
REFERENCES
[1] | Bazzi MD , Nelsestuen GL . Association of protein kinase C with phospholipid vesicles. Biochemistry. (1987) ;26: (1):115–22. |
[2] | Bazzi MD , Nelsestuen GL . Protein kinase C interaction with calcium: A phospholipid-dependent process. Biochemistry. (1990) ;29: (33):7624–30. |
[3] | Brose N , et al. Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science. (1992) ;256: (5059):1021–5. |
[4] | Bashir R , et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. (1998) ;20: (1):37–42. |
[5] | Liu J , et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. (1998) ;20: (1):31–6. |
[6] | Illarioshkin SN , et al. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology. (2000) ;55: (12):1931–33. |
[7] | Nguyen K , et al. Phenotypic study in 40 patients with dysferlin gene mutations: High frequency of atypical phenotypes. Archives of Neurology. (2007) ;64: (8):1176–82. |
[8] | Ueyama H , et al. Clinical heterogeneity in dysferlinopathy. Internal medicine (Tokyo, Japan). (2002) ;41: (7):532–36. |
[9] | Fanin M , Angelini C . Muscle pathology in dysferlin deficiency. Neuropathology and Applied Neurobiology. (2002) ;28: (6):461–70. |
[10] | Nguyen K , et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Human Mutation. (2005) ;26: (2):165. |
[11] | Selva-O’Callaghan A , et al. Muscle inflammation, autoimmune Addison’s disease and sarcoidosis in a patient with dysferlin deficiency. Neuromuscular Disorders: NMD. (2006) ;16: (3):208–09. |
[12] | Vinit J , et al. Dysferlin deficiency treated like refractory polymyositis. Clinical Rheumatology. (2010) ;29: (1):103–6. |
[13] | Mahmood OA , Jiang XM . Limb-girdle muscular dystrophies: Where next after six decades from the first proposal (Review). Mol Med Re. (2014) ;9: (5):1515–32. |
[14] | McNeil PL , Steinhardt RA . Plasma membrane disruption: Repair, prevention, adaptation. Annu Rev Cell Dev Biol. (2003) ;19: :697–731. |
[15] | Steinhardt RA , Bi G , Alderton JM . Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. (1994) ;263: (5145):390–3. |
[16] | Cooper ST , McNeil PL . Membrane Repair: Mechanisms and Pathophysiology. Physiological Reviews. (2015) ;95: (4):1205–40. |
[17] | Defour A , et al. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum Mol Genet. (2017) ;26: (11):1979–91. |
[18] | Cai C , et al. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. (2009) ;284: (23):15894–902. |
[19] | Draeger A , Monastyrskaya K , Babiychuk EB . Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem Pharmacol. (2011) ;81: (6):703–12. |
[20] | Eddleman CS , et al. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci U S A. (1997) ;94: (9):4745–50. |
[21] | Godell CM , et al. Calpain activity promotes the sealing of severed giant axons. Proc Natl Acad Sci U S A. (1997) ;94: (9):4751–6. |
[22] | Jimenez AJ , et al. ESCRT machinery is required for plasma membrane repair. Science. (2014) ;343: (6174):1247136. |
[23] | Lauritzen SP , Boye TL , Nylandsted J . Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin Cell Dev Biol. (2015) ;45: :32–8. |
[24] | Mellgren RL , Huang X . Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J Biol Chem. (2007) ;282: (49):35868–77. |
[25] | Scheffer LL , et al. Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. (2014) ;5: :5646. |
[26] | Mellgren RL . A plasma membrane wound proteome: Reversible externalization of intracellular proteins following reparable mechanical damage. J Biol Chem. (2010) ;285: (47):36597–607. |
[27] | Togo T , Krasieva TB , Steinhardt RA . A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell. (2000) ;11: (12):4339–46. |
[28] | Steinhardt RA . The mechanisms of cell membrane repair: A tutorial guide to key experiments. Ann N Y Acad Sci. (2005) ;1066: :152–65. |
[29] | Andrews NW , Corrotte M , Castro-Gomes T . Above the fray: Surface remodeling by secreted lysosomal enzymes leads to endocytosis-mediated plasma membrane repair. Semin Cell Dev Biol. (2015) ;45: :10–7. |
[30] | Kay JG , et al. Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell. (2012) ;23: (11):2198–212. |
[31] | Vaughan EM , et al. Lipid domain-dependent regulation of single-cell wound repair. Mol Biol Cell. (2014) ;25: (12):1867–76. |
[32] | McDade JR , Archambeau A , Michele DE . Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. (2014) ;28: (8):3660–70. |
[33] | Bi GQ , et al. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J Cell Biol. (1997) ;138: (5):999–1008. |
[34] | Alvarez-Martinez MT , et al. Characterization of the interaction between annexin I and profilin. Eur J Biochem. (1996) ;238: (3):777–84. |
[35] | Alvarez-Martinez MT , et al. Effects of profilin-annexin I association on some properties of both profilin and annexin I: modification of the inhibitory activity of profilin on actin polymerization and inhibition of the self-association of annexin I and its interactions with liposomes. Biochim Biophys Acta. (1997) ;1339: (2):331–40. |
[36] | Marg A , et al. Sarcolemmal repair is a slow process and includes EHD2. Traffic. (2012) ;13: (9):1286–94. |
[37] | Matsuda C , et al. Dysferlin interacts with affixin (beta-parvin) at the sarcolemma. J Neuropathol Exp Neurol. (2005) ;64: (4):334–40. |
[38] | Abreu-Blanco MT , Verboon JM , Parkhurst SM . Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J Cell Biol. (2011) ;193: (3):455–64. |
[39] | Abreu-Blanco MT , Verboon JM , Parkhurst SM . Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr Biol. (2014) ;24: (2):144–55. |
[40] | Benink HA , Bement WM . Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. (2005) ;168: (3):429–39. |
[41] | Mandato CA , Bement WM . Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J Cell Biol. (2001) ;154: (4):785–97. |
[42] | Mellgren RL , et al. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. (2007) ;282: (4):2567–75. |
[43] | Verstraeten SV , Mackenzie GG , Oteiza PI . The plasma membrane plays a central role in cells response to mechanical stress. Biochim Biophys Acta. (2010) ;1798: (9):1739–49. |
[44] | O’Brien ET , Salmon ED , Erickson HP . How calcium causes microtubule depolymerization. Cell Motil Cytoskeleton. (1997) ;36: (2):125–35. |
[45] | Togo T . Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J Cell Sci. (2006) ;119: (Pt 13):2780–6. |
[46] | McNeil PL , et al. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. (2000) ;113: (Pt 11):1891–902. |
[47] | Reddy A , Caler EV , Andrews NW . Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. (2001) ;106: (2):157–69. |
[48] | Suetsugu S , Kurisu S , Takenawa T . Dynamic shaping of cellular membranes by phospholipids and membrane-deforming proteins. Physiol Rev. (2014) ;94: (4):1219–48. |
[49] | Miyake K , McNeil PL . Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. (1995) ;131: (6 Pt 2):1737–45. |
[50] | Jaiswal JK , Andrews NW , Simon SM . Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. (2002) ;159: (4):625–35. |
[51] | McNeil PL . Repairing a torn cell surface: Make way, lysosomes to the rescue. J Cell Sci. (2002) ;115: (Pt 5):873–9. |
[52] | McNeil PL , Terasaki M . Coping with the inevitable: How cells repair a torn surface membrane. Nat Cell Biol. (2001) ;3: (5):E124–9. |
[53] | Corrotte M , et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife. (2013) ;2: :e00926. |
[54] | Jaiswal JK , et al. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat Commun. (2014) ;5: :3795. |
[55] | Andrews NW , Almeida PE , Corrotte M . Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. (2014) ;24: (12):734–42. |
[56] | Tam C , et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. (2010) ;189: (6):1027–38. |
[57] | Draeger A , Babiychuk EB . Ceramide in plasma membrane repair. Handb Exp Pharmacol. (2013) ;(216):341–53. |
[58] | Nichols B . Caveosomes and endocytosis of lipid rafts. J Cell Sci. (2003) ;116: (Pt 23):4707–14. |
[59] | Idone V , et al. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. (2008) ;180: (5):905–14. |
[60] | Lariccia V , et al. Massive calcium-activated endocytosis without involvement of classical endocytic proteins. J Gen Physiol. (2011) ;137: (1):111–32. |
[61] | Defour A , et al. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. (2014) ;5: :e1306. |
[62] | Draeger A , et al. Dealing with damage: Plasma membrane repair mechanisms. Biochimie. (2014) ;107: (Pt A):66–72. |
[63] | Parton RG , Simons K . The multiple faces of caveolae. Nat Rev Mol Cell Biol. (2007) ;8: (3):185–94. |
[64] | Bonilla E , Fischbeck K , Schotland DL . Freeze-fracture studies of muscle caveolae in human muscular dystrophy. Am J Pathol. (1981) ;104: (2):167–73. |
[65] | Repetto S , et al. Increased number of caveolae and caveolin-3 overexpression in Duchenne muscular dystrophy. Biochem Biophys Res Commun. (1999) ;261: (3):547–50. |
[66] | Sinha B , et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. (2011) ;144: (3):402–13. |
[67] | Proske U , Morgan DL . Muscle damage from eccentric exercise: Mechanism, mechanical signs, adaptation and clinical applications. J Physiol. (2001) ;537: (Pt 2):333–45. |
[68] | Waddell LB , et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J Neuropathol Exp Neurol. (2011) ;70: (4):302–13. |
[69] | Babiychuk EB , et al. Blebbing confers resistance against cell lysis. Cell Death Differ. (2011) ;18: (1):80–9. |
[70] | Codding SJ , et al. Dysferlin Binds SNAREs (Soluble N-Ethylmaleimide-sensitive Factor (NSF) Attachment Protein Receptors) and Stimulates Membrane Fusion in a Calcium-sensitive Manner. J Biol Chem. (2016) ;291: (28):14575–84. |
[71] | Therrien C , et al. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry. (2009) ;48: (11):2377–84. |
[72] | McDade JR , Michele DE . Membrane damage-induced vesicle-vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum Mol Genet. (2014) ;23: (7):1677–86. |
[73] | Lek A , et al. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2013) ;33: (12):5085–94. |
[74] | Redpath G , et al. Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair. Molecular Biology of the Cell. (2014) . |
[75] | Mellgren RL , et al. Calcium-dependent plasma membrane repair requires m- or mu-calpain, but not calpain-3, the proteasome, or caspases. Biochim Biophys Acta. (2009) ;1793: (12):1886–93. |
[76] | Hernandez-Deviez DJ , et al. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J Biol Chem. (2008) ;283: (10):6476–88. |
[77] | Matsuda C , et al. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet. (2001) ;10: (17):1761–6. |
[78] | Lin P , et al. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. (2012) ;26: (5):1875–83. |
[79] | Cai C , et al. MG53 regulates membrane budding and exocytosis in muscle cells. J Biol Chem. (2009) ;284: (5):3314–22. |
[80] | Zhu H , et al. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J Biol Chem. (2011) ;286: (15):12820–4. |
[81] | Cai C , et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. (2009) ;11: (1):56–64. |
[82] | Potez S , et al. Tailored protection against plasmalemmal injury by annexins with different Ca2+sensitivities. J Biol Chem. (2011) ;286: (20):17982–91. |
[83] | Rajab A , et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. (2010) ;6: (3):e1000874. |
[84] | Gazzerro E , et al. Caveolinopathies: From the biology of caveolin-3 to human diseases. Eur J Hum Genet. (2010) ;18: (2):137–45. |
[85] | Li H , et al. Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J Biol Chem. (2015) ;290: (40):24592–603. |
[86] | Liu J , et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J Mol Cell Cardiol. (2015) ;80: :10–19. |
[87] | Lizarbe MA , et al. Annexin-phospholipid interactions. Functional implications. Int J Mol Sci. (2013) ;14: (2):2652–83. |
[88] | Bouter A , et al. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat Commun. (2011) ;2: :270. |
[89] | Voges D , et al. Three-dimensional structure of membrane-bound annexin V. A correlative electron microscopy-X-ray crystallography study. J Mol Biol. (1994) ;238: (2):199–213. |
[90] | Gerke V , Creutz CE , Moss SE . Annexins: Linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. (2005) ;6: (6):449–61. |
[91] | Boye TL , Nylandsted J . Annexins in plasma membrane repair. Biol Chem. (2016) . |
[92] | Cocucci E , et al. The regulated exocytosis of enlargeosomes is mediated by a SNARE machinery that includes VAMP4. J Cell Sci. (2008) ;121: (Pt 18):2983–91. |
[93] | Rezvanpour A , Santamaria-Kisiel L , Shaw GS . The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J Biol Chem. (2011) ;286: (46):40174–83. |
[94] | Jaiswal JK , et al. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nature Communications. (2014) ;5: :3795. |
[95] | Demonbreun AR , et al. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol. (2016) ;213: (6):705–18. |
[96] | Borgonovo B , et al. Regulated exocytosis: A novel, widely expressed system. Nat Cell Biol. (2002) ;4: (12):955–62. |
[97] | Roostalu U , Strähle U . In vivo imaging of molecular interactions at damaged sarcolemma. Developmental Cell. (2012) ;22: (3):515–29. |
[98] | Cheng X , et al. The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat Med. (2014) ;20: (10):1187–92. |
[99] | Vergarajauregui S , Martina JA , Puertollano R . Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1. J Biol Chem. (2009) ;284: (52):36357–66. |
[100] | Tian Y , W J , Cebotaru L , Wang H , Guggino WB . Anoctamin5 is Related to Plasma Membrane Repair. JSM Regenerative Medicine & Bioengineering. (2015) ;3: (1):1015. |
[101] | Fatimathas L , Moss SE . Annexins as disease modifiers. HistolHistopathol. (2010) ;25: (4):527–32. |
[102] | Minetti C , et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. (1998) ;18: (4):365–8. |
[103] | Richard I , et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. (1995) ;81: (1):27–40. |
[104] | Saccone V , et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat. (2008) ;29: (2):240–7. |
[105] | Middel V , et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nature Communications. (2016) ;7: :12875. |
[106] | Atanassoff AP , et al. Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PLoS One. (2014) ;9: (2):e89743. |
[107] | Krahn M , et al. A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci Transl Med. (2010) ;2: (50):50ra69. |


