
Characteristics of Japanese Patients with Becker Muscular Dystrophy and Intermediate Muscular Dystrophy in a Japanese National Registry of Muscular Dystrophy (Remudy): Heterogeneity and Clinical Variation
Abstract
Background:
Obtaining an adequate number of patients to conduct a natural history study for rare diseases such as Becker muscular dystrophy (BMD) is difficult.
Objectives:
The present study used data from Remudy, a national registry for neuromuscular diseases in Japan, to conduct a phenotypic analysis of BMD.
Methods:
We analyzed Remudy data of participants with dystrophinopathy. All participants who were aged 17 and older and were ambulant at age 13 were included in this study. Participants were divided into two groups: those with BMD who were ambulant at age 17, and those with intermediate muscular dystrophy (IMD) who lost ambulation by age 17. Frequent mutations were analyzed by age at ambulation, cardiopulmonary function, and genotype. For clinical comparisons, participants who were administered steroids were excluded.
Results:
From July 2009 through September 2015, 192 participants had registered with Remudy. Mean participant age was 34.80±13.3 (range, 17–78) years, and 52.1% of participants were ambulant. Of the entire study population, 50.5% had cardiomyopathy and 35.9% had respiratory failure. Three participants required invasive ventilation and 30 required non-invasive ventilation. Nineteen of the 30 non-invasive ventilator users were part-time users. In total, 138 (71.9%) had BMD and 54 (28.1%) had IMD. The most frequent mutation was ex45_ex47del (36 participants). Among participants with frequent in-frame mutations, those with the ex45-49del mutation lost their ambulation earlier than those with the ex45_ex47del mutation. A total of 67 different exon deletions and duplications were identified in the study population.
Conclusion:
We clarified the clinical phenotypes of Japanese patients with BMD/IMD using data from Remudy. Our results suggest that not only IMD but also BMD are associated with risk of respiratory dysfunction.
ABBREVIATIONS
BMD | Becker muscular dystrophy |
DMD | Duchenne muscular dystrophy |
IMD | intermediate muscular dystrophy |
AONs | Antisense oligonucleotides |
Remudy | Registry of muscular dystrophy |
NCNP | National Center of Neurology and Psychiatry |
SD | standard deviation |
INTRODUCTION
Becker muscular dystrophy (BMD, OMIM 300376) is an X-linked recessive form of muscular dystrophy caused by mutations in the dystrophin gene (DMD) on chromosome Xp21.2 [1]. The DMD gene is the largest gene identified in humans and contains 79 exons. Mutations in this gene prevent the production of functional dystrophin protein [2]. Since the discovery of the DMD gene, efforts have been ongoing to develop effective strategies to treat Duchenne muscular dystrophy (DMD) and BMD.
DMD is associated with mutations that interrupt the open reading frame of the DMD gene, resulting in severe muscle weakness and, typically, death before the age of 30 years [3, 4]. In contrast, BMD is typically associated with mutations that maintain the open reading frame, producing an altered, but partially functional, dystrophin protein with no change in its C-terminal domain [3, 5]. Patients with BMD exhibit variable phenotypes, but are less severely affected than those with DMD, and have a much longer normal life expectancy [5]. Antisense oligonucleotides (AONs), a promising experimental line of molecular therapy for DMD, aim to restore gene expression by altering splicing at the pre-mRNA level [6]. These AONs hybridize to a specific target sequence and can cause the skipping of a targeted exon, resulting in restoration of the open reading frame, production of an internally-truncated dystrophin protein, and conversion of the DMD phenotype to BMD. Eteplirsen (Exondys 51®) is an antisense oligonucleotide designed to induce exon 51 skipping [7, 8]. Intravenous eteplirsen has received accelerated approval from the US FDA for the treatment of Duchenne muscular dystrophy (DMD) in patients with a confirmed mutation of the DMD gene amenable to exon 51 skipping [9]. Phase I clinical trials on exon 53 skipping have just been completed, and phase II trials are currently underway (NCT02081625) [10]. To assess the likely outcomes of exon-skipping therapies for patients with DMD, natural history studies as well as studies which examine phenotype-genotype correlations are important. In addition, investigating the heterogeneity of the BMD phenotype may further clarify the function and structure of the dystrophin protein [11]. Prediction of clinical outcomes is also important for both patients and physicians.
In 2009, we developed a national registry of Japanese patients with dystrophinopathy (Registry of Muscular Dystrophy; Remudy. http://www.remudy.jp/) in collaboration with TREAT-NMD [12]. Registry data can provide detailed information regarding the natural history and epidemiology of a disease, as well as information pertaining to clinical care. Several studies have used TREAT-NMD [13] and other patient registry data to investigate DMD, including our previous study [12]. However, no studies have described in detail the mild phenotypes of dystrophinopathy. Here, we describe the clinical and molecular genetic characteristics of Japanese patients with BMD and intermediate muscular dystrophy (IMD) using data from Remudy. To clarify the clinical characteristics of the population showing mild phenotypes (i.e., BMD) and those with less mild phenotypes (i.e., milder than DMD), we defined BMD and IMD separately.
METHODS
Institution, organization, registration method, data collection, and ethical approval
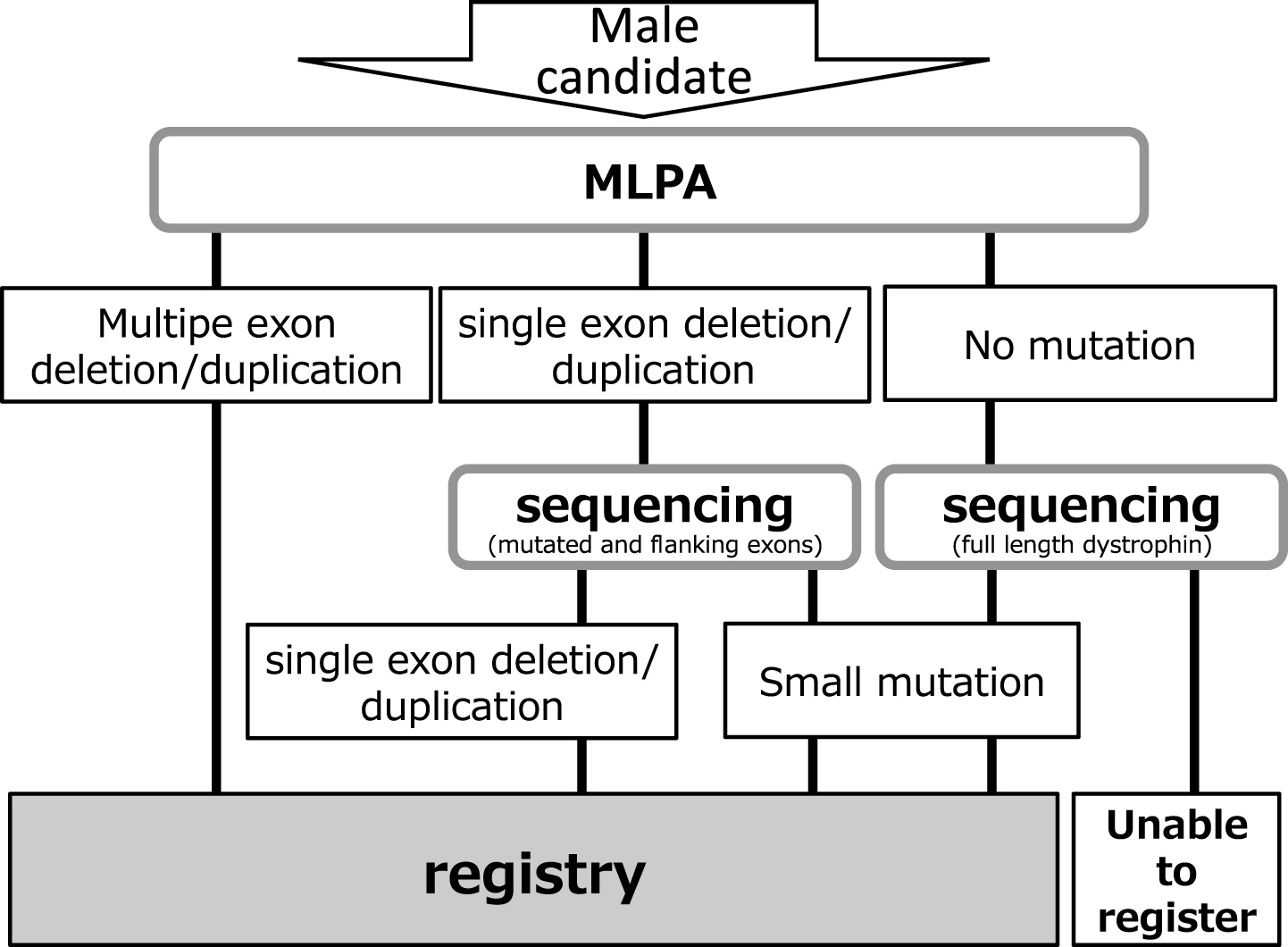
Remudy is supported by Intramural Research Grants (23-5/24-6) for Neurological and Psychiatric Disorders from the National Center of Neurology and Psychiatry (NCNP). Information relating to the development and management of the registry, registration method, and data collection has been published previously [12]. All data were collected using the registrant’s self report after obtaining physician confirmation. Inclusion criteria were men with dystrophinopathy, and that the diagnosis was confirmed genetically and/or pathologically by immunohistochemistry (IHC). Genetic diagnosis was confirmed by the original report. We used MLPA for multiple exon deletions and duplications, MLPA and sequencing of the bordering area for single exon deletions or duplications, and full-sequencing for small mutations (Fig. 1). Cardiac failure was defined as 1) EF <55% and/or 2) diagnosis by the attending physician. Respiratory dysfunction was defined as 1) % FVC <80 and/or 2) ventilator use. Since continuous positive airway pressure (CPAP) therapy of obstructive sleep apnea is distinguished from ventilator therapy according to the Japanese insurance system, no patients with CPAP were included in ventilator users. If patients had updated their annual registrant forms, we used the information on their original registration. Approval for this study was obtained from the Medical Ethics Committee of the NCNP. Study objectives, design, risks, and benefits of participation were explained to all patients, and their written informed consent was obtained prior to enrollment.
Fig.1
Patient registry system.
Patients
Among registered patients with dystrophinopathy at age 17 and older, data from patients with BMD and IMD were reviewed. All patients with IMD who were ambulant at age 13 were defined as registrants who had lost ambulation between ages 13 and 17, whereas those with BMD were ambulant at age 17 [14]. In addition, each mutation found in more than seven patients was further analyzed by age at lost ambulation, cardiopulmonary function, and genotype. For clinical comparisons, patients administered steroids were excluded.
Data analysis
Data were summarized using descriptive statistics, including mean, standard deviation (SD), median, range, frequency, and percentage. Spearman correlations were used to determine the association between each variable. The t-test was used for group comparisons of continuous data and Fisher’s exact test was used for binary data. Age at loss of ambulation was evaluated using the Kaplan–Meier method with log-rank test. All statistical analyses were performed using SPSS for Macintosh (Version 18; SPSS Inc., Chicago, IL).
RESULTS
General characteristics at study entry (Table 1, Fig. 2)
Fig.2
Participant distribution by (a) ambulation and (b) steroid usage.
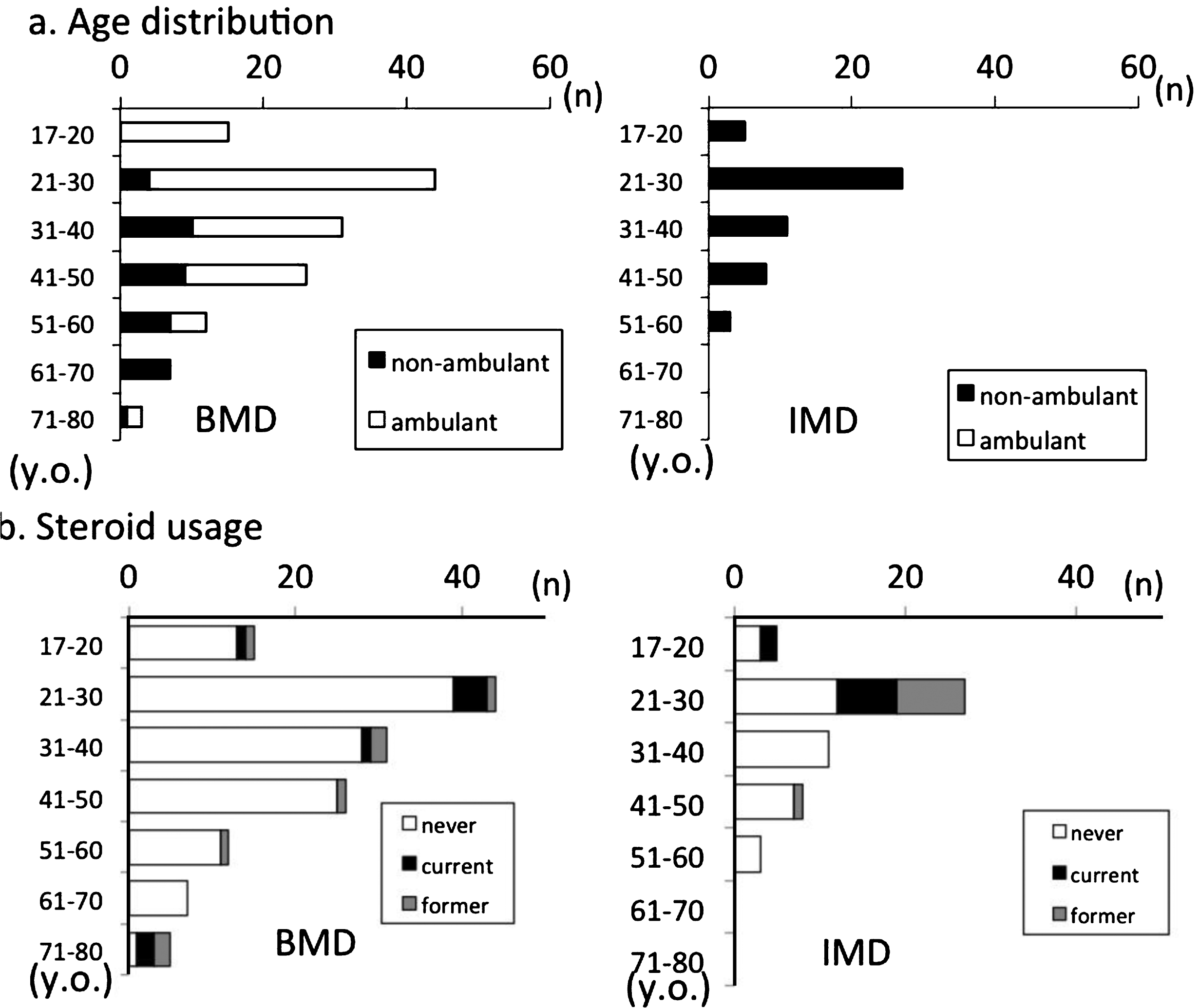
Table 1
Characteristics of BMD registrants
| Total | BMD | IMD | |||||||
| n | (%) | Age (mean±SD (median, range)) | n | (%) | Age (mean±SD (median, range)) | n | (%) | Age (mean±SD (median, range)) | |
| Patients | 192 | 100 | 34.80 +/–13.3 (32, 17–78) | 138 | 71.9 | 36.2 +/–14.0§ (33, 17–78) | 54 | 28.1 | 31.3 +/–10.5§(27, 16–60) |
| dead | 5 | 2.6 | 37.4 +/–11.8 (37.5, 17–60) | 0 | 0 | 5 | 100 | 37.4 +/–11.8 (37.5, 17–60) | |
| Walking capability | |||||||||
| ambulant | 100 | 52.1 | 32.1 +/–12.3 (30,17–78) | 100 | 72.5 | 32.1 +/–12.3 (30, 17–78) | 0 | 0 | |
| non-ambulant | 92 | 47.9 | 37.7 +/–13.7 (36, 17–72) | 38 | 27.5 | 46.8 +/–12.6 (45.5, 23–72) | 54 | 100 | 31.3 +/–10.5 (27, 17–60) |
| Wheelchair use | |||||||||
| never | 83 | 43.2 | 30.9 +/–11.4 (29, 17–78) | 38 | 27.5 | 30.9 +/–11.4 (29, 17–78) | 0 | 0 | |
| part-time | 36 | 18.8 | 39.1 +/–ç 14.1(36.5, 17–72) | 24 | 17.4 | 42.5 +/–14.4 (43, 17–72) | 12 | 22.2 | 32.3 +/–11.0 (30, 21–510) |
| full time | 66 | 34.4 | 35.6 +/–13.1(34, 17–65) | 28 | 20.3 | 44.6 +/–12.8 (44.5, 21–65) | 38 | 70.4 | 29.0 +/–8.8 (27, 17–53) |
| bedridden | 7 | 3.6 | 50.9 +/–13.7(49, 30–72) | 3 | 2.2 | 53.0 +/–21.3 (53, 30–72) | 4 | 7.4 | 49.3 +/–8.2 (48.5, 40–60) |
| Steroid use | |||||||||
| never | 160 | 83.3 | 35.7 +/–12.9 (35, 17–72) | 124 | 89.9 | 36.1 +/–13.4 (33.5, 17–72) | 36 | 66.7 | 34.3 +/–10.9 (35, 17–60) |
| yes | 32 | 16.7 | 30.3 +/–14.5 (25.5, 17–78) | 14 | 10.1§ | 36.9 +/–19.0 (31.5, 17–78) | 18 | 33.4§ | 29.2 +/–6.2 (23, 19–48) |
| former | 15 | 7.8 | 30.6 +/–10.4 (27, 20–53) | 6 | 4.3 | 36.0 +/–11.4 (34.5, 20–53) | 9 | 16.7 | 27.0 +/–8.4(25, 21–48) |
| current | 17 | 8.9 | 30.1 +/–17.7(23, 17–78) | 8 | 5.8 | 37.6 +/–24.0 (26.5, 17–78) | 9 | 16.7 | 23.3 +/–3.7 (23, 19–30) |
| Respiratory functions | |||||||||
| normal | 94 | 49.0 | 35.5 +/–13.4 (33, 17–72) | 83 | 60.1 | 36.2 +/–13.3 (33, 17–72) | 5 | 9.4 | 23.0 +/–6.8 (23, 17–34) |
| decreased | 79 | 41.1 | 34.8 +/–12.6(34, 17–78) | 35 | 25.4§ | 37.6 +/–14.5 (36.0, 19–78) | 44 | 83.0§ | 33.0 +/–10.7 (29.5, 18–60) |
| not assessed | 19 | 9.9 | 31.1 +/–13.3 (32, 17–78) | 20 | 14.5 | 32.1 +/–15.8 (23, 19–72) | 4 | 7.5 | 23.3 +/–2.9 (22.5, 21–27) |
| mechanical ventilation | 33 | 17.2 | 39.5 +/–11.3 (40, 22–64) | 7 | 5.1§ | 46.9 +/–11.7 (44, 30–64) | 26 | 48.1§ | 37.5 +/–10.6 (37, 22–60) |
| part-time | 19 | 57.6* | 36.4 +/–11.3 (38, 22–64) | 5 | 71.4* | 48.2 +/–10.3 (44, 40–60) | 14 | 53.8* | 32.1 +/–8.5 (29.5, 22–47) |
| full time | 13 | 39.4* | 43.7 +/–10.6 (48, 26–60) | 2 | 28.6* | 43.5 +/–19.1 (43.5, 30–57) | 11 | 42.3* | 43.8 +/–10.9 (48.0 26–60) |
| unknown | 1 | 3.0* | 17 | 0 | 0.0* | – | 1 | 1.9* | 17 |
| Cardiac functions | |||||||||
| normal | 95 | 49.5 | 33.0 +/–13.4 (30, 17–72) | 83 | 60.1 | 34.0 +/–13.7 (31, 17–72) | 12 | 22.2 | 26.0 +/–8.9 (22.5, 17–47) |
| decreased | 97 | 50.5 | 36.5 +/–13.0 (35, 19–78) | 55 | 39.9§ | 39.4 +/–14.0 (36, 20–78) | 42 | 77.8§ | 32.8 +/–10.6 (29.5–19) |
| Cardiac medication | |||||||||
| prescribed | 106 | 55.2 | 35.8 +/–12.7 (34.5, 18–78) | 60 | 43.5§ | 38.2 +/–13.7 (36, 19–78) | 46 | 85.2§ | 32.8 +/–10.6 (30, 18–60) |
| not prescribed | 86 | 44.8 | 33.5 +/–13.9 (30, 17–72) | 78 | 56.5 | 34.6 +/–14.1 (31.5, 17–72) | 8 | 14.8 | 22.4 +/–3.3 (21.5, 17–27) |
| Drug | |||||||||
| ACE-inhibitor | 70 | 66.0** | 37 | 71.7** | 33 | 71.8** | |||
| β-blocker | 66 | 62.3** | 34 | 56.7** | 32 | 69.6** | |||
| ARB | 17 | 16.0** | 13 | 21.7** | 4 | 8.7** | |||
| Diuretics | 10 | 9.4** | 4 | 6.7** | 6 | 13.0** | |||
| Other | 10 | 9.4** | 5 | 8.3** | 5 | 10.9** | |||
| Muscle biopsy | |||||||||
| performed | 120 | 62.5 | 36.9 +/–13.0 (35, 17–78) | 87 | 63 | 38.9 +/–13.4 (36, 17–78) | 33 | 61.1 | 31.8 +/–10.5 (27, 17–60) |
| not performed | 72 | 37.5 | 31.1 +/–13.0 (25.5, 17–72) | 51 | 37 | 31.4 +/–13.9 (25, 17–72) | 21 | 38.9 | 30.4 +/–10.8 (26, 18–53) |
| Dystrophin staining | |||||||||
| faint and patchy | 49 | 73.1 + | 39 | 83.0 + | 10 | 50.0 + | |||
| negative | 18 | 26.9 + | 8 | 17.0 +§ | 10 | 50.0 +§ | |||
| Family history | |||||||||
| yes | 80 | 41.7 | 37.2 +/–14.8 (35, 17–78) | 67 | 48.6§ | 37/5 +/–15.2 (35, 17–78) | 13 | 24.1§ | 46.0 +/–23.9 (38, 22–60) |
| no | 112 | 58.3 | 33.0 +/–11.9 (30, 17–65) | 71 | 51.4 | 34.9 +/–17.8 (33, 17–65) | 41 | 75.9 | 29.7 +/–9.3 (27, 17–51) |
| sibling | 43 | 22.4 | 34 | 50.74626866 | 8 | 61.53846154 | |||
| maternal relatives | 32 | 16.7 | 28 | 41.79104478 | 4 | 30.76923077 | |||
| mother | 6 | 3.1 | 5 | 7.5 | 1 | 7.692307692 | |||
| sibling’s children | 4 | 2.1 | 3 | 4.5 | 1 | 7.692307692 | |||
| grandchildren | 2 | 1 | 2 | 3.0 | 0 | 0 | |||
| unknown | 1 | 0.5 | 1 | 1.5 | 0 | 0 | |||
| Genotyping | |||||||||
| deletion | 137 | 71.4 | 36.2 +/–13.9 (35.0, 17–78) | 102 | 73.9 | 38.1 +/–14.5 (37, 18–78) | 35 | 64.8 | 30.5 +/–10.3 (27, 17–53) |
| duplication | 16 | 8.3 | 32.5 +/–12.3 (31.5 17–60) | 6 | 4.3 | 28.3 +/–9.9 (27.5, 17–43) | 10 | 18.5 | 35.0 +/–13.4 (33.5, 18–60) |
| small mutation | 39 | 20.3 | 30.9 +/–10.4 (17–63) | 30 | 21.7 | 31.1 +/–14.0 (33, 17–78) | 9 | 16.7 | 30.2 +/–9.0 (27, 21–47) |
| in-flame | 108 | 70.6 # | 96 | 89.7 | 12 | 26.1 | |||
| out-of-flame | 45 | 28.4 # | 11 | 10.3§ | 34 | 75.9§ | |||
| stop-codon | 16 | 13 | 3 | ||||||
*percentage among participants with ventilator use. **percentage among participants with cardiac medication. +percentage among participants with IHC. #percentage among participants with deletion/duplication mutations. §:significant difference between BMD and IMD.
A total of 1,298 Japanese patients with dystrophinopathy who were aged 17 and older had been registered in Remudy from July 2009 through September 2015. Among these were 1,106 patients with DMD, 138 with BMD, and 54 with IMD. Only patients with BMD and IMD were included for further analysis, excluding those with DMD (i.e., those who could not walk after age 13). Mean age±SD at the time of data collection was 34.8±13.3 (median, 32 years; range, 17–78 years).
The age distribution for ambulation is shown in Fig. 2a. The oldest ambulant patient was aged 78 years. Of the study population, 49.5% (n = 95) had normal cardiac function and 55.2% (n = 106) were prescribed at least one cardiac medication, and β-blockers were the most frequently prescribed medication for cardiac failure. Patients with cardiac medication had cardiac dysfunction significantly more frequently than those without (81.1% vs. 12.8%; p < 0.001). EF data were obtained from 92 participants. EF data were defective in 5 of 97 cardiac dysfunction participants and 65 of 95 normal participants.
Among registrants, 17 (8.9%) were treated or had been administered steroids. Among current steroid users, 11 were non-ambulant and 6 were ambulant. When former steroid users were included, 32 (16.7%) of BMD/IMD registrants had used steroids (Fig. 2b).
Muscle biopsies were obtained from 120 (62.5%) participants. The results of dystrophin IHC were available for 67 (55.8%) participants. Among these 67 participants, a faint and patchy pattern or dystrophin negative IHC staining pattern was observed in 49 (73.1%) and 18 (26.8%) participants, respectively. There were 80 (41.7%) patients with family members affected by dystrophinopathy, including 43 siblings, 6 mothers, and 32 other maternal kindred.
Comparison between BMD and IMD (Table 1)
Compared to participants with BMD, participants with IMD were younger (BMD vs. IMD, mean age, 36.2 years vs. 31.3 years; p = 0.009) and had more frequent steroid usage (BMD vs. IMD, 10.1% vs. 33.4%; p < 0.001), more frequent respiratory failure, (BMD vs. IMD, 21.7% vs. 72.2%; p < 0.001), more frequent ventilator usage (BMD vs. IMD, 5.1% vs. 48.1%; p < 0.001), more prevalent cardiac dysfunction (BMD vs. IMD, 39.9% vs. 77.8%; p < 0.001), and more treatment (BMD vs. IMD, 43.5% vs. 85.2%; p < 0.001). In addition, fewer participants with BMD had out-of-frame mutations (BMD vs. IMD, 28.4% vs. 75.9%; p < 0.001), had a positive family history (BMD vs. IMD, 24.1% vs. 48.6%; p = 0.002), and had dystrophin negative IHC (BMD vs. IMD 26.9% vs. 50.0%; p = 0.007) compared to participants with IMD.
Respiratory dysfunction (Table 2, Supplementary Table 1)
Table 2
Respiratory dysfunction
| Total | BMD | IMD | |||||||
| n | % | n | % | n | % | ||||
| FVC (%) | 60 | 75.9 | 48.5 +/–20.9 | 31 | 88.6 | 59.2 +/–14.8 | 29 | 65.9 | 36.4 +/–17.3 |
| (50.8, 5.9–80.4) | (59.9, 14.0–80.4) | (37.0. 5.9–63.0) | |||||||
| no FVC data | 19 | 24.1 | 4 | 11.3 | 15 | 34.1 | |||
| ambulation | |||||||||
| ambulant | 17 | 21.5 | 17 | 48.6 | 0 | 0.0 | |||
| non-ambulant | 62 | 78.5 | 18 | 51.4 | 44 | 100.0 | |||
| age at loss of ambulation (y.o.) | 63 | 79.7 | 18.9 +/–11.2 | 18 | 51.4 | 29.4 +/–5.6 | 44 | 100.0 | 14.0 +/–1.0 |
| (14, 13–65) | (20, 17–65) | (14, 13–16) | |||||||
| Steroid | |||||||||
| never | 58 | 73.4 | 28 | 80.0 | 30 | 68.2 | |||
| current | 11 | 13.9 | 4 | 11.4 | 7 | 15.9 | |||
| former | 10 | 12.7 | 3 | 8.6 | 7 | 15.9 | |||
| cardiac function | |||||||||
| dysfunction | 53 | 67.1 | 19 | 54.3 | 34 | 77.3 | |||
| normal | 26 | 32.9 | 16 | 45.7 | 10 | 22.7 | |||
| genotyping | |||||||||
| in-frame | 28 | 35.4 | 19 | 54.3 | 9 | 20.5 | |||
| out-of-frame | 35 | 44.3 | 6 | 17.1 | 29 | 65.9 | |||
| Small mutation | 16 | 20.3 | 10 | 28.6 | 6 | 13.6 | |||
| IHC | |||||||||
| faint and patchy | 11 | 13.9 | 5 | 14.3 | 5 | 11.4 | |||
| negative | 10 | 12.7 | 4 | 11.4 | 7 | 15.9 | |||
| no data | 58 | 73.4 | 26 | 74.3 | 32 | 72.7 | |||
Of the study population, 88 (45.8%) had normal respiratory function. However, respiratory function was not assessed in 25 (13.0%) participants. % FVC data were obtained from 60 participants, excluding 19 participants (18 were ventilator-dependent; five were dependent part-time, and 13 were dependent full-time). with respiratory dysfunction. Among 35 BMD participants with respiratory dysfunction, 18 were non-ambulant and 17 were ambulant. Nineteen patients had confirmed BMD (i.e., based on clinical pathology, in-frame mutations, and a faint and patchy dystrophin IHC pattern). Three of these patients were ambulant until age 17 and had respiratory insufficiency (Supplementary Table 1). Among 50 IMD participants, 44 had respiratory dysfunction; 33 (17.2%) were ventilator users. Of these, 19 were part-time and 13 were full-time users (one unknown). Three participants required invasive ventilation, and 30 participants required non-invasive ventilation. Among 33 ventilator users (both IMD and BMD patients), only one 44-year-old BMD participant was ambulant. The remaining 32 ventilator users lost ambulation at a median age of 14 years (range, 13–55 years), which was earlier than non-ambulant participants who were not ventilator users (median age, 17.5 years; range, 13–65 years; p < 0.001).
Dystrophin mutations
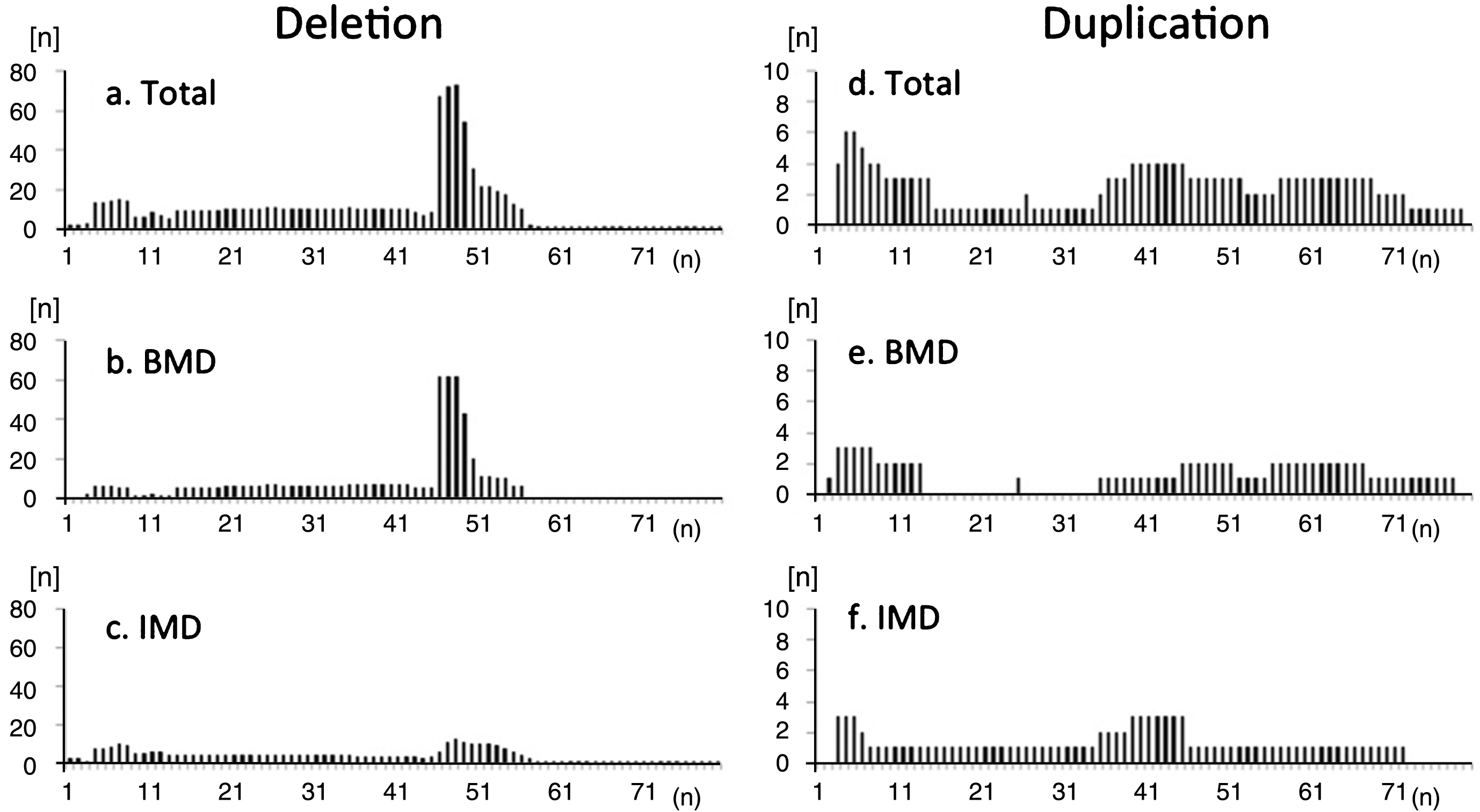
There were 137 participants with deletion mutations, 16 with duplication mutations, and 39 with small mutations (Supplementary Table 2). The distribution of exon deletions revealed common hot spot regions in exons 45–54, but no hot-spots for duplications (Fig. 3). Deletion mutations included the ex45_ex47del mutation in 26.3% (36/137), ex45_ex48del mutation in 12.4% (17/137), ex45_ex49del mutation in 6.6% (9/137), ex3_ex7del mutation in 5.1% (7/137), ex48del mutation in 2.9% (4/137), and ex45_ex55del mutation in 2.9% (4/137). In contrast, 58 deletion and duplication mutations (67 participants, 48.9%) were only observed in one or two participants. In total, we identified 67 different exon deletions and duplications comprising all mutations.
Fig.3
Frequency of deleted exons observed in registrants with (a-c) deletion and (d-f) duplication mutations in the total study population (a, d), participants with BMD (b, e), and participants with IMD (c, f). Distribution of exon deletions shows common hot-spot regions in exons 45–54. There were no “hot spots” for duplications. Among IMD participants, hotspot deletion was less frequent compared to BMD participants.
Genotype-phenotype correlations for deletion mutations (Table 3)
Table 3
Kaplan–Meier analysis for median age at loss of ambulation
| Median | |||
| Median age at loss of ambulation (years) | 95%CI | ||
| ex45_ex47del | 53 | 47.5 | 58.5 |
| ex45_ex48del | 60 | 52.5 | 67.5 |
| ex45_ex49del | 39.7 | . | . |
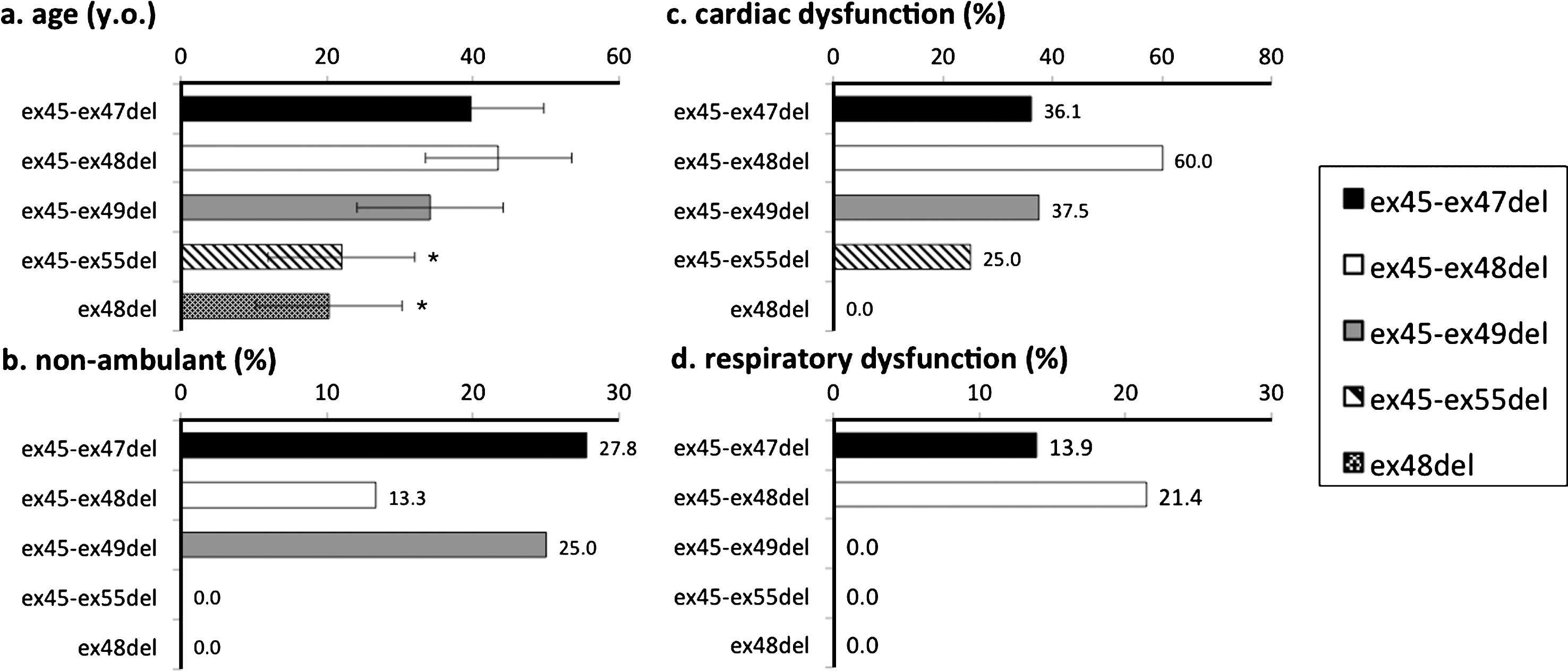
To compare clinical features of participants with common mutations, we analyzed data from those with ex45_ex47del, ex45_ex48del, ex45_ex49del, ex45_ex55del, and ex48del mutations, as these five mutations were the most frequently observed in our study population. Participants who had used steroids before were excluded, as steroid use itself may influence phenotype. The numbers of participants who had never used steroids were 36 for ex45_ex47del, 16 for ex45_ex48del, 8 for ex45_ex49del, 4 for ex45_ex55del, and 4 for ex48del mutations. The most frequent mutation, ex45-47del, was used as a reference for comparisons of other mutations. Among the five mutations, those with ex 45-ex55del and ex48del mutations were significantly younger than those with the ex45_ex47del (Fig. 4a) mutation. None of the non-ambulant participants had the ex45-55del or ex48del mutation. There were no significant differences between the other three mutation groups, with respect to the ratio of non-ambulant participants (Fig. 4b).
Fig.4
Genotype-phenotype correlation among common mutations of BMD and IMD. Cardiac dysfunction and respiratory dysfunction were the ratio of numbers of participants with cardiac/respiratory dysfunction carrying each mutation subtended to the entire population of each mutations. *significant difference relative to ex45-47del (p = 0.05). (a) Participant age; only those with ex48del were significantly younger. (b) Proportion of non-ambulant participants; no participants with ex45-55del and ex48del lost ambulation. (c) Proportion of participants with cardiac dysfunction; no significant difference among mutations. (d) Proportion of participants with respiratory dysfunction; a higher proportion of those with ex3-ex7del had respiratory dysfunction.
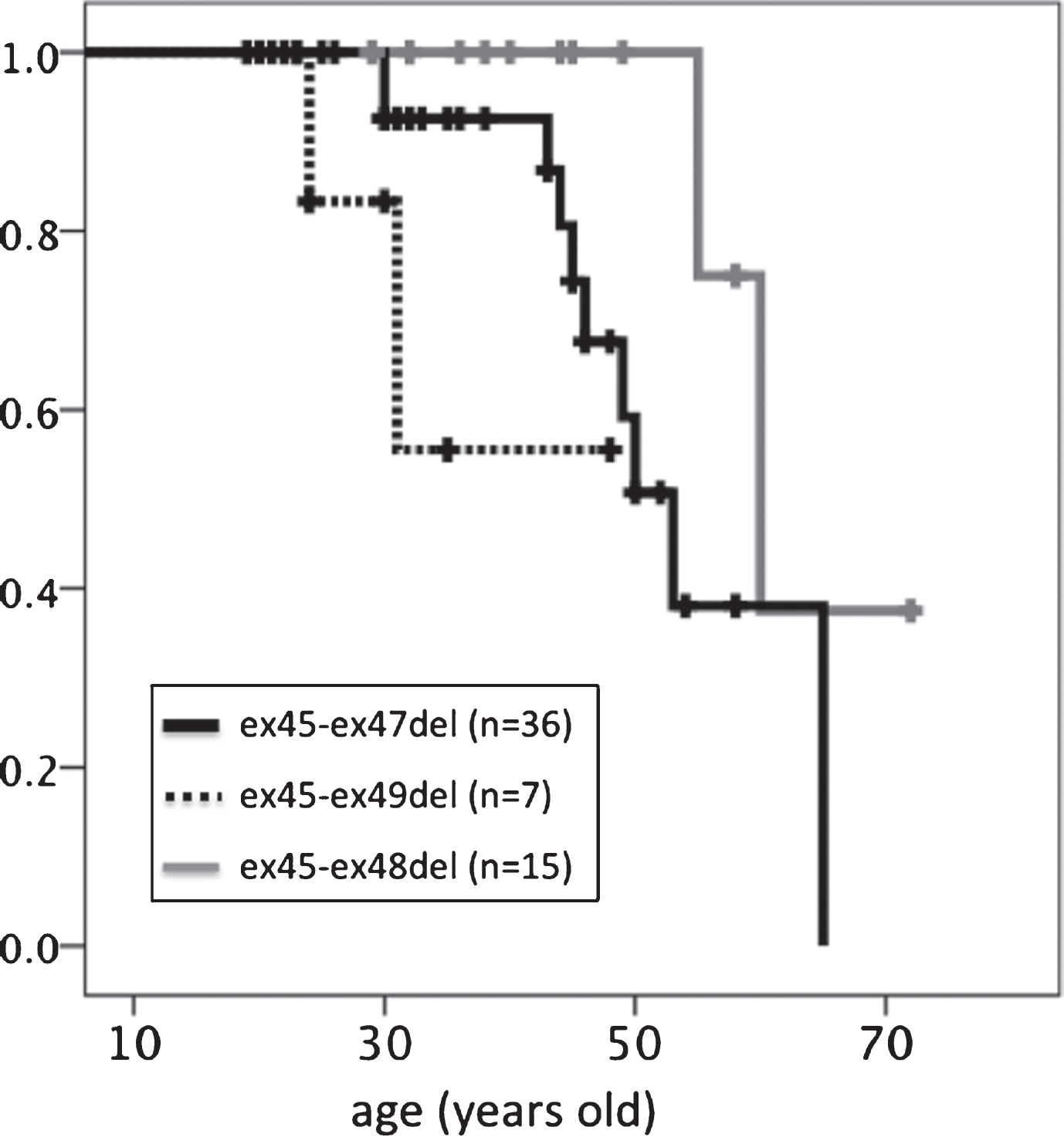
With regard to median age at loss of ambulation (Table 3, Fig. 5), participants with the ex45-ex49del mutation (39.7 years) lost ambulation earlier than those with the ex45-47del (53 years, p = 0.215) or ex45-48del (60 years, p = 0.018) mutation. Those with the ex45-ex48 mutation had the longest ambulation duration compared to those with the other two in-frame mutations.
Fig.5
Kaplan–Meier analysis of age at loss of ambulation. Significant differences were detected between ex45-ex47del and ex45-ex49del genotypes.
Percentages of participants who had cardiac dysfunction were 36.1% for ex45-ex47del, 60.0% for ex45-48del, 37.5% for ex45-49del, 25% for ex45-55del, and 0% for ex48del. No significant difference was found in cardiac dysfunction (Fig. 4c). Percentages of participants who had respiratory dysfunction were 13.9% for ex45-ex47del, 21.4% for ex45-48del, and 0% for ex45-49del, ex45-55del, and ex48del (Fig. 4d).
Only three participants carrying ex45-48del currently used steroids; however, no significant changes were found when these participants were included in the analysis.
DISCUSSION
Recent advances in exon skipping and other molecular therapies have brought focus to BMD, a potential model for DMD treated with exon skipping. The reasons underlying the heterogeneity of BMD phenotypes, however, remain unclear. No linear relation has been found between dystrophin levels (range, 3%–78%) and muscle strength or age at different disease milestones, in both the whole group and subgroups of exon 45–47 deleted patients. However, patients with less than 10% dystrophin levels have been found to follow a severe disease course. [15]. Genotype-phenotype correlations have been previously studied. For instance, deletions in the N-terminal region are associated with an earlier onset of symptoms compared to mutations in the distal region [16]. Patients with a deletion of exons 45–55 appear to have less severe muscular involvement, and only a few cases of dilated cardiomyopathy have been described in the literature [17, 18]. A previous comparison of the frequency of mutations, such as ex45-ex47del, ex45-ex48del, ex45-ex49del, and ex45-ex51del, found that the age of dilated cardiomyopathy onset was delayed in those with the ex45–ex48del or ex45–ex49del mutation compared to those with the ex45–ex47del mutation [11]. Earlier wheelchair dependency was also observed in patients with the ex45–ex47del or ex45–ex49del mutation compared to those with the ex45–ex48del mutation, which is consistent with our present findings. In their study, muscle dystrophin levels were moderately reduced, but without a clear correlation with the type of deletion. Therefore, they concluded that disease severity might be attributed to differences in structure of the mutated dystrophin protein and not dystrophin levels [9]. In the present study, we found that the ambulation duration of participants with the ex45-47del or ex45-48del mutation was significantly longer compared to those with the ex45-ex49del mutation (p < 0.05). As we did not assess parameters such as ejection fraction or vital capacity in all participants, we may not have had sufficient power to detect differences in genotype among those with cardiac or respiratory dysfunction.
We used clinical diagnostic criteria to classify participants with mild phenotypes of dystrophinopathy. At present, immunohistochemical diagnosis is not routinely performed in the first step of BMD diagnosis, as MLPA has been commercially available and covered by health insurance in Japan. In some cases, however, it is difficult to predict prognosis based on genetic diagnosis. Therefore, we suspect that neither genetic nor pathological diagnosis alone was sufficient to detect mild phenotypes of dystrophinopathy in the present study. On the other hand, given that steroid usage is one of the factors associated with prolonged ambulation status in DMD, clinical criteria alone may not be sufficient for defining phenotypes. In the present study, 26.7% of patients with BMD had negative dystrophin IHC, which seemed rather high. As our participants included those who carried out-of-frame and stop-codon mutations, the percentage of IHC-negative patients might have been higher compared to the past investigation, which included only BMD participants with in-frame mutations.
Although Remudy collects nationwide data, we could not amass a sufficient sample size to enable a statistical comparison of mutations. Thus, a broader study, e.g., one that uses an international registry, is warranted in the future.
This study is the first to find that some patients with BMD had been administered steroids. Interestingly, both children and adults were treated with steroids. However, no data relating to complications were collected in this study to determine whether steroids were administered for conditions other than dystrophinopathy. In this context, future studies on the significance of this finding, as well as the utility of steroids in treating patients with BMD, may yield interesting results. This study is also the first to report on respiratory dysfunction in patients with BMD. One may imagine that ventilator use occur only with IMD, however, an ambulant participant and a participant who kept ambulation until his 50’s were included. Those with typical BMD, ambulation over age 17, in-frame mutations, and faint and patchy IHC pattern also had respiratory insufficiency. Physicians should regularly monitor the respiratory function of patients with pulmonary function tests in order to detect early signs of respiratory dysfunction, perform respiratory training, and induce mechanical ventilation if necessary, as they do for patients with neuromuscular diseases who present with respiratory failure [19, 20].
CONCLUSIONS
We clarified the clinical phenotypes of Japanese patients with BMD/IMD using data from Remudy. Some limitations relating to the clarification of the natural history of the disease and genotype-phenotype correlations might be resolved through international collaborations. Physicians should monitor the respiratory function of BMD/IMD patients.
CONFLICT OF INTEREST
None to report.
ACKNOWLEDGMENTS
We thank all participants, families, and physicians involved in Remudy. This work was partly supported by Health and Labour Sciences Research Grants for Research on Rare and Intractable Diseases, and an Intramural Research Grant (26-7, 29-3) for Neurological and Psychiatric Disorders from the NCNP.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND170225.
REFERENCES
[1] | Davies KE , Smith TJ , Bundey S , Read AP , Flint T , Bell M , Speer A . Mild and severe muscular dystrophy associated with deletions in Xp21 of the human X chromosome. J Med Genet. (1988) ;25: :9–13. |
[2] | Muntoni F , Torelli S , Ferlini A . Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: :731–40. |
[3] | Emery AE . The muscular dystrophies. Lancet. (2002) ;359: :687–95. |
[4] | Passamano L , Taglia A , Palladino A , et al. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. (2012) ;31: :121–5. |
[5] | Monaco AP , Bertelson CJ , Liechti-Gallati S , Moser H , Kunkel LM . An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. (1988) ;2: :90–5. |
[6] | van Deutekom JC , van Ommen GJ . Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet. (2003) ;4: :774–83. |
[7] | Cirak S , Arechavala-Gomeza V , Guglieri M , et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. (2011) ;378: :595–605. |
[8] | Mendell JR , Goemans N , Lowes LP , et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) ;79: :257–71. |
[9] | Syed YY . Eteplirsen: First Global Approval. Drugs. (2016) ;76: :1699–704. |
[10] | https://clinicaltrials.gov/ct2/show/NCT02081625?term=japan+DMD&rank=2 |
[11] | Nicolas A , Raguénès-Nicol C , Ben Yaou R , et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Human Molecular Genetics. (2015) ;24: :1267–79. |
[12] | Nakamura H , Kimura E , Mori-Yoshimura M , et al. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy). Orphanet J Rare Dis. (2013) ;19: (8):60. |
[13] | Bladen CL , Salgado D , Monges S , et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: :395–402. |
[14] | Bushby K , Finkel F , Birnkrant BJ , et al. Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: :77–93. |
[15] | van den Bergen JC , Wokke BH , Janson AA , et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry. (2014) ;85: :747–53. |
[16] | Magri F , Govoni A , D’Angelo MG , et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. (2011) ;258: :1610–23. |
[17] | Anthony K , Cirak S , Torelli S , Tasca G . Dystrophin quantification and clinical correlations in Becker muscular dystrophy: implications for clinical trials. Brain. (2011) ;134: :3547–59. |
[18] | Taglia A , Petillo R , D’Ambrosio P . Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. (2015) ;34: (1):9–13. |
[19] | Bach JR . Noninvasive Respiratory Muscle Aids. In: Bach JR, ed. Management of Patients with Neuromuscular Disorders. Philadelphia: Hanley & Belfus, (2004) :211–69 |
[20] | Bach JR . Pulmonary defense mechanisms and cough peak flow. In: Bach JR, ed. Management of Patients with Neuromuscular Disorders. Philadelphia: Hanley & Belfus, (2004) :193–9. |

