
Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases in Inflammation and Fibrosis of Skeletal Muscles
Abstract
In skeletal muscles, levels and activity of Matrix MetalloProteinases (MMPs) and Tissue Inhibitors of MetalloProteinases (TIMPs) have been involved in myoblast migration, fusion and various physiological and pathological remodeling situations including neuromuscular diseases. This has opened perspectives for the use of MMPs’ overexpression to improve the efficiency of cell therapy in muscular dystrophies and resolve fibrosis. Alternatively, inhibition of individual MMPs in animal models of muscular dystrophies has provided evidence of beneficial, dual or adverse effects on muscle morphology or function. We review here the role played by MMPs/TIMPs in skeletal muscle inflammation and fibrosis, two major hurdles that limit the success of cell and gene therapy. We report and analyze the consequences of genetic or pharmacological modulation of MMP levels on the inflammation of skeletal muscles and their repair in light of experimental findings. We further discuss how the interplay between MMPs/TIMPs levels, cytokines/chemokines, growth factors and permanent low-grade inflammation favor cellular and molecular modifications resulting in fibrosis.
ABBREVIATIONS
ACE | Angiotensin Converting Enzyme |
ADAM | A Desintegrin And Metalloproteinase |
α-SMA | Alpha-Smooth Muscle Actin |
CA-MMP | Cysteine Array-MMP |
CCL2 | chemokine (C-C motif) ligand 2 (CCL2); |
MCP1 | Monocyte Chemotactic Protein 1 |
CCR2 | C-C chemokine Receptor type 2 |
CD | Cluster of Differentiation |
CTGF | Connective Tissue Growth Factor |
CXC | Cysteine X Cysteine |
CXCL | Cysteine X Cysteine Ligand |
CXCR | Cysteine X Cysteine Receptor |
DAMPs | Damage associated Molecular Patterns |
EGF | Epidermal Growth Factor |
EGFR | Epidermal Growth Factor Receptor |
cGMP | Cyclic Guanosine Monophosphate |
HE4 | Human Epididymis Protein 4 |
HGF | Hepatocyte Growth Factor also known as Scatter Factor |
IGF | Insulin Growth Factor |
IGFBP | Insulin Growth Factor Binding Protein |
IGFR | Insulin Growth Factor Receptor |
M-CSF | Macrophage Colony Stimulating Factor |
MMPs | Matrix MetalloProteinases |
MMPIs | Matrix MetalloProteinase Inhibitors |
MT-MMP | Membrane Type- MMP |
Musk | Muscle specific Kinase |
NO | Nitric Oxide |
NOS | Nitrogen Reactive Species |
NOX | NADPH Oxidase |
NRROS | Negative Regulator of ROS |
PAMPs | Pathogen Associated Molecular Patterns |
PDE5 | PhosphoDiesterase 5 |
PDGF | Platelet Derived Growth Factor |
PMN | Polymorphonuclear Leukocyte |
Prss (23) | Protease, Serine, (23) or (35) or Prss (35): |
RASI | Rheumatoid Arthritis Sign of Inflammation, |
RECK | Reversion-inducing-Cysteine-rich protein with Kazal motifs |
ROS | Reactive Oxygen Species |
SDF-1 | Stromal Derived Factor-1 or (CXCL12) |
SMAD | Sma and Mad Related Protein |
SOD | Super Oxide Dismuthase |
TACE | Tumour necrosis factor Alpha Converting Enzyme |
TGF-β: | Transforming Growth Factor beta |
TIMP | Tissue Inhibitor of MetalloProteinase |
TLR | Toll Like Receptor |
TNF-α: | Tumour Necrosis Factor alpha |
VEGF | Vascular Endothelial Growth Factor |
XMMP | Xenopus MMP |
PUMP-1 | Plant Uncoupling Mitochondrial Protein 1 |
INTRODUCTION
The balance between hydrolytic activity and its inhibition regulates tissue and extracellular matrix (ECM) homeostasis. Disruption of this balance occurs as an integral part of tissue response to remodeling stimuli but its long lasting perturbation deregulates many biological processes and leads to the development of diseases. High MMP levels characterize acute or chronic disease situations and correlate with disease severity suggesting they have detrimental effects. Hardly appreciated however, is the possibility that these proteins serve essential or beneficial functions. MMPs are major determinants in remodeling events such as placental development, embryo implantation- a highly invasive but tightly controlled process involving ECM degradation and cell migration-, angiogenesis, bone development and mammary involution. They participate in ECM degradation by direct cleavage of connective tissue collagen, activation of latent enzymes and processing or liberation of structural or signaling molecules from the ECM.
In normal skeletal muscles, the steady state situation is characterized by basal activity of hydrolytic enzymes titrated by their inhibitors. Modifications of functional demands, trauma or disease disrupt this balance and trigger an adaptive response that includes MMPs/TIMPs regulation [1–12]. In turn, these proteins alter cell-cell and/or cell-matrix interactions thereby affecting cell proliferation, migration and differentiation. Nevertheless, “guidance cues”, able to relocate and stop regenerating axons at original synaptic sites, are preserved during the denervation/reinnervation process [13] indicating that highly regulated and tightly controlled hydrolysis of ECM components characterize the accomplishment of a specific task.
The involvement of metallo-endopeptidase in myoblast fusion were published in the early eighties [14, 15] but their implication in pathophysiological processes [16–27] and therapeutic follow-up/perspectives [28–33] were published decades later. At present, we have indications of some of the functions accomplished by MMP-2, MMP-9, MMP-10 and MMP-14 but there is hardly a hint of the role played by other family members. However, considering the dysregulation of MMPs/TIMPs balance in disease situations, it seems important to identify the modification of expression pattern in identified muscle disease entities to sort out “IF” and “HOW” they affect clinical severity and determine potential targets for therapeutic interventions. By exploring the function(s) of each MMP and TIMP and identifying their degrading and regulatory actions one may depict the mechanism involved in their mode of action as described for MMP-9 and MT6-MMP [34, 35].
In a previous review, we have presented the involvement of MMPs/TIMPs in specific skeletal muscle diseases such as muscular dystrophies, inflammatory myopathies and neurogenic muscular diseases [36]. Here, we chose to focus on MMPs/TIMPs involvement in inflammation and fibrosis that occur in several muscle diseases namely inflammatory myopathies and muscular dystrophies. In this respect, the emblematic Duchenne Muscular Dystrophy or its animal models, characterized by the occurrence of both components, have been widely used to investigate the functional consequences of MMPs/TIMPs balance modulation and will, therefore, be referred to in a specific section to illustrate these points.
Matrix Metalloproteinases (MMPs)
The MMP family is composed of 23 members in human (24 in mouse) [37, 38] most of which are secreted and six are membrane bound (Table 1). They are inhibited by TIMPs and, collectively, degrade all ECM components. Their general characteristics and regulation have been reviewed in [36]. The regulation of MMPs activity and the modulation of MMPs/TIMPs activity/expression in inflammatory and wound conditions are schematically presented in Fig. 1.
MMP-2, MMP-10 and MMP-3 deficient mice are reported with no evident signs of malformation. Curiously, Mmp2-/- mice, originally characterized as overtly normal but with slower growth rate [39] revealed, upon closer examination, attenuated features of multicentric osteolysis and arthropathy [40] caused by MMP-2 mutations which lead to cardiac defects when the terminal hemopexin domain is deleted [41]. Such mutations may have phenotypic repercussions on skeletal muscles as well but has not been investigated in animal models. Mmp14 deficiency causes severe developmental defects and defective muscle maturation leading to premature death [42]; a phenotype aggravated by additional MMP-2 deficiency [43]. Mice deficient in MMP-3, known to degrade agrin, have high agrin levels and show increased size and number of AchR at neuromuscular junctions [44, 45]. Upon prolonged denervation, they maintain a normal topography with preserved agrin and Musk at denervated endplates [5].
MMPs have an essential role as regulators of microenvironmental changes. They participate to cell migration by hydrolyzing ECM components and releasing cryptic fragments with different biological activities. MMPs and their related ADAMs (a disintegrin and metalloproteases) and ADAM-TSs (ADAMs with thrombospondin repeats) families are involved in shedding growth factors or cell-surface-adhesion molecules such as syndecan-1. From simple hydrolytic enzymes, MMPs have evolved to regulators of signal transduction, of innate and adaptive immunity, and modifiers of cellular/molecular phenotype [46, 47].
In skeletal muscles, MMPs have been involved in cell migration and fusion. On one hand, MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, and MMP-14 enhance myoblast migration [48–54]. On the other hand, MMP-7, MMP-9, MMP-14 are involved in cell fusion [48, 49, 52, 53, 55–58]. Further, MMP-2, MMP-10, MMP-14 proved to be essential for successful muscle regeneration: MMP-2 and MMP-14 act on the maturation of muscle fibers by controlling angiogenesis [42, 43, 59] whereas MMP-10 is involved in CXCR4/SDF1 signaling axis that is essential for efficient skeletal muscle regeneration [60, 61]. Finally, MMP-2 and MMP-9 may potentially be involved in muscular dystrophies and muscle atrophy. Indeed, α and β dystroglycan are ascertained and direct targets for MMP-2 and MMP-9 as originally proposed for β-dystroglycan processing by MMPs [64]. Furthermore, intracellularly localized MMP-2 [65] is involved in muscle fibers atrophy in various physiological and pathological situations [66–70]. This activity probably relies on MMP-2 ability to hydrolyze sarcomeric proteins such as troponin I, myosin light chain-1, titin, and α-actinin [71–73].
MMPs Inhibitors: The physiological (TIMPs) and synthetic inhibitors (MMPIs)
The TIMP family is composed of four members (TIMP-1,-2,-3 and -4) with significant homology that inhibit MMPs with some specificity [74, 75]. Originally, TIMPs are thought to function exclusively as endogenous inhibitors of MMPs thereby modulating MMP-mediated ECM degradation. However accumulating evidence indicate they are multifunctional proteins involved in various biological activities that may or may not depend on their inhibitory function and range from cell growth and differentiation, to cell migration, invasion, angiogenesis, survival and apoptosis depending on cell and tissue context [76–82]. TIMP-1 promotes cell proliferation in a wide range of cell types and regulates apoptosis. TIMP-2 is involved in MMP-2 activation through association with MMP-14. It contributes to ECM protection from proteolysis and increases both fibroblast proliferation and collagen production. TIMP-3 has pro-apoptotic activity whereas TIMP-4, the most recently identified and least studied, is reported to modulate angiogenesis. TIMPs and MMPs are regulated in a similar or reciprocal manner whereas cytokines and growth factors regulate TIMPs in tissue-specific, constitutive, or inducible manner [78].
In skeletal muscles, different TIMPs have been implicated in myoblasts fusion and myofibers maturation. The involvement of TIMP-1 in cell fusion has been suggested by the concomitance between MMP-9 downregulation and TIMP-1 upregulation during cell fusion [83] whereas Timp2 can be involved in skeletal muscle maturation directly or via formation of the TIMP-2/MMP-14/MMP-2 complex leading to MMP-2 activation. Timp-2 Knockout mice have deficient motor function with abnormalities of neuromuscular junctions, increased sprouting of intramuscular nerves and decreased muscle mass [84, 85]. Timp-3 regulates myogenesis via miR-206-TIMP3-TACE-TNF-α−p38 signaling pathway and acts as an On/Off switch by regulating autocrine release of Tumor Necrosis Factor-α (TNF-α) [86].
Clinical trials have used synthetic MMP Inhibitors (MMPIs) to limit the progression of diseases. They have been unsuccessful [87, 88] but underscored the necessity for designing more selective inhibitors that discriminate between the different members of the MMP family [89, 90].
MMPs are involved in skeletal muscle inflammation
Several members of the MMP family have been involved in the inflammatory process occurring after injury or disease [91]. Inflammation is an essential step in the initiation and progression of tissue remodeling and entails the degradation and reorganization of the ECM scaffold to which MMPs are important contributors. Yet, despite its importance in host defense and tissue repair, if the inflammatory process becomes excessive or chronic, it associates with organ dysfunction and exacerbation of pathological features [92]. The pattern of MMPs/TIMPs regulation in non-pathological or pathological remodeling of skeletal muscles has been reviewed [36] and is summarized in (Table 2). Recently, experimental findings have characterized the involvement of MMP-10 (stromelysin-2) in muscle regeneration [61] and report transient MMP-13 upregulation during muscle injury [50].
MMP-9 is the most widely documented protease in the inflammatory process that characterizes the initial stages of muscle injury. MMP-9 increase correlates with the invasion of necrotic tissue by inflammatory cells, more particularly polymorphonuclear neutrophils (PMN) and activated satellite cells [7, 93]. Increased MMP-9 expression/activity quantitatively and qualitatively correlates with different stages of inflammation. An early phase of dramatic increase of MMP-9 protein corresponds to the initial flux of PMN into the necrotic tissue followed by a second phase of massive invasion by macrophages during which MMP-9 protein level is less intense but gelatinase activity more potent [7]. Such increase is predictable since white blood cells produce MMPs that facilitate their migration [94] and regulate their function [95, 96]. MMP-9 produced by these cells [97] is stored in granules [98–100] being, hence, immediately available for degranulation thereby facilitating transmigration through the vessel/capillary wall. These cells also regulate MMP-9 production in a time and phase specific manner [101] similar to sequential variations of MMP-9 at the early stages of muscle regeneration.
MMP upregulation correlates with inflammation in muscular dystrophies and inflammatory myopathies
In a number of muscle pathologies, MMP overexpression correlates particularly but not exclusively with inflammation (Table 2). In muscular dystrophies and inflammatory myopathies, MMP elevation is due, at least in part, to inflammation whereas in motor neuron and peripheral nervous system diseases with secondary muscle manifestations, the evidence points towards an association with tissue remodeling [102–105]. In muscles of Duchenne Muscular Dystrophy (DMD) patients, the presence of inflammatory cells [106–109] correlates with high MMP-9 [17, 20, 110] in blood vessels, mononuclear cells and regenerating fibers [111]. MMP-9 is also elevated in the serum of dystrophic mice [36] and DMD patients [33]. Intense MMP-1 signal is reported around individual or small groups of necrotic muscle fibers and areas containing a high density of macrophages [111]. TIMP-1 is elevated in the serum, plasma, and muscle biopsies of DMD patients [27, 33] and increased immunolabeling is observed in the endomysium (unpublished results). TIMP-1 and MMP-2 mRNAs localize to areas of degeneration/regeneration whereas TIMP-2 transcripts distribute more homogeneously in mesenchymal fibroblasts [27].
In inflammatory myopathies, there is no evidence of elevation of MMP or TIMP levels in the serum [22] but MMP-9 up-regulation is found in muscles of Polymyositis, Dermatomyosistis, and Inclusion Body Myositis patients [22, 25, 112]. Immunolabeling localizes MMP-9 to atrophic myofibers or is restricted to CD8+ cytotoxic T cells [18]. MMP-1 transcripts are also upregulated in these pathologies, and the protein localizes around the sarcolemma and in cells resembling fibroblasts. MMP-7 strongly labels myofibers invaded by inflammatory cells in polymyositis cases only [25] and MMP-2 has a similar distribution but weaker intensity.
Effects of MMP modulation in Duchenne muscular dystrophy
Overexpression or inhibition of individual MMPs may deteriorate or ameliorate skeletal muscle structure and function depending on the specific outcome one is investigating. Examples of deleterious effects are provided by dystrophic and Amyotrophic Lateral sclerosis (ALS) mouse models in which MMP-9 overexpression has been incriminated in the aggravation of dystrophy in mdx mice [110] and selective denervation of fast muscles in the SOD1 ALS mouse model [113]. In this context, MMP-9 inhibition in mdx mice was reported to reduce necrosis, fibrosis, increase the levels of β-dystroglycan and improve muscle force production [31, 32, 110, 114]. It is thought to act by favoring satellite cell proliferation, emergence of M2 pro-myogenic macrophages, increase of the expression of Notch ligands, receptors and components of canonical Wnt signaling while decreasing non-canonical Wnt signaling [30]. These characteristics represent circumstantial evidence that fit well with structural and functional improvement but the effect of MMP-9 inhibition on satellite cell proliferation and improvement of muscle regeneration remains controversial. High MMP-9 levels in myogenic cells favored cell migration, differentiation and engraftment upon grafting into dystrophic mice [57] and its inhibition delayed myoblast proliferation and differentiation [115] causing, in the long term, impairment of muscle regeneration, reduction of muscle force and the development of fibroadipogenic tissue [26]. L-arginine, Nitric Oxide (NO) donors, Doxycycline (Dox) and Minocycline (Min) or other natural substances have the same beneficial effect. These substances decrease MMP-9 levels, orchestrate inflammation towards the “repair mode”, favor preservation of structural integrity and reduce fibrosis in different mouse models [31, 116–118].
As far as we know today, the deficiency or inhibition of other MMPs have a detrimental effect on skeletal muscle regeneration/maturation. MMP-2 deficiency impairs the maturation of regenerating skeletal muscles by inhibiting angiogenesis [59] while MMP-10 deficiency is reported to worsen muscle dystrophy, delay muscle regeneration, impair the recruitment of endothelial cells and reduce the levels of ECM proteins by a mechanism associated with Vascular Endothelial Growth Factor (VEGF)/Akt signaling [61].
MMPs/TIMPs are involved in muscle fibrosis
Fibrosis is a multifactorial process that integrates multiple cellular and biochemical events between different cell types, growth factors, inflammatory/fibrogenic cytokines and proteolytic enzymes resulting in the alteration of the tissue microenvironment. The build-up of fibrosis reflects a disruption of the balance between synthesis and degradation of ECM components, accomplished by MMPs and proteases of the plasminogen activation system [36, 55, 119]. Myofibroblasts, the key cellular mediators of fibrosis, are increased in fibrotic tissues [111, 120] and contribute to ECM and protease production. Their existence is still debated because we lack reliable markers to discriminate between myoblasts and myofibroblasts [121, 122].
Myofibroblasts arise from independent sources [123–127] and help in the repair process. They induce re-emergence of embryonic ECM proteins that favor tissue reconstitution [128]. The activation of myofibroblasts, necessary for tissue repair, involves paracrine signals derived from lymphocytes and macrophages, autocrine factors, pathogen- (PAMPs) or damage- (DAMPs) associated molecular patterns [129] and mechanosensing [130]. Fibroblasts respond to signals from immune cells that produce cytokines, growth factors and proteases that modify the phenotype of neighboring cells. Similarly, the behavior of different cells types sharing the same environment is profoundly influenced by the disturbance and remodeling of ECM. Increased tissue stiffness and decreased elasticity generate a mechanical stress that exacerbates tissue injury and perpetuates the activation of fibroblasts [128, 131].
Persistence of inflammation favors the transformation of wound healing to fibrosis
The wound healing response involves transitory, highly orchestrated events consisting of interrelated dynamic phases with overlapping time course that lead to tissue replacement [132]. They necessitate high levels of extracellular proteolytic activity attributed to MMPs, serine proteases and cysteine proteases [133]. The fibrotic response is initiated by innate wound healing mechanisms involving inflammatory myeloid cells and adaptive immune activation that modulate synthesis and deposition of ECM by myofibroblasts [134]. The accumulation of fibrin and fibronectin, immediately after trauma, forms a provisional matrix that serves to fill in the lesion and allow inflammatory cells to migrate into the wound [135, 136]. The platelets present in the clot participate in the recruitment of inflammatory cells, by releasing growth factors such as Platelet Derived Growth Factor (PDGF), potent chemoattractant for inflammatory cells and TGF-β1 which stimulates ECM synthesis [134]. Innate immunity is activated in response to molecular signals such as cryptic fragments, liberated by the digestion of ECM components, endowed with chemoattractant properties due to structural relatedness to Cystein X Cystein (CXC) chemokines [137]. Endogenous myofiber proteins and mitochondrial-DAMPs, released into the circulation, generate a systemic inflammatory reaction and activate PMN through formyl peptide receptor-1 and toll-like receptor (TLR) 9 [138]. In turn, TLR activation initiates signaling cascades that converge at the NF-κB pathway activating the synthesis of inflammatory cytokines and β-defensins [139, 140]. TLR-9 activation by mitochondrial DAMPS induces the release of MMP-8, which, together with MMP-9, aid in PMN tissue penetration and recruitment [36, 138]. The second wave of inflammatory cells, the monocyte/macrophages, have a remarkable plasticity and ability to exacerbate, suppress or reverse fibrosis [141–143]. During the evolutionary phases of wound healing, macrophages with divergent functions arise progressively in response to exposure to fate determining mediators. Initial amplification of the inflammatory response is followed by a phase during which inflammation is resorbed and tissue repaired [144–146]. The neutrophil-specific protease Membrane-Type 6 Matrix Metalloproteinase (MT6-MMP/MMP-25) has a salient role in favoring monocyte chemotaxis and resolution of inflammation. It regulates the shift of macrophages from pro- to anti-inflammatory phenotype and contributes to efferocytosis by increasing phagocytic removal of neutrophils carrying “eat-me” signals [35]. Efferocytosis triggers specific downstream intracellular signal transduction pathways resulting in the production of anti-inflammatory, anti-protease and growth-promoting effects that favor the replacement of dead cells [147, 148]. Activated fibroblasts acquire a myofibroblast phenotype, synthesize and deposit ECM to replace the provisional matrix and contribute to the contraction and maturation of granulation tissue [149]. They also produce MMPs that disrupt the basement membrane allowing cell migration at the site of injury. Scar formation further involves progressive remodeling of the granulation tissue by proteolytic enzymes and their inhibitors, followed by a resolution phase leading to reconstitution of the damaged tissue and reduction of myofibroblast number by apoptosis [150]. Recurrent necrosis/regeneration, like in DMD, results in perpetuation of DAMPs release and chronic low-grade inflammation with persistence of neutrophils and monocytes at the injury site. These produce proteases, cytokines as well as reactive oxygen and nitrogen species (ROS/RNS) causing supplementary damage [151] and generating a prolonged inflammatory reaction, chemokine/cytokine production with consequent modifications of gene expression, cell functions and phenotype. In such conditions, wound healing is sustained with continuous degradation and deposition of ECM matrix resulting in fibrosis (Fig. 2).
Skeletal muscle fibrosis: The result of an interplay between cytokine/chemokines and growth factors which affect cell fate and levels of MMPs/TIMPs
Permanent induction of the wound healing response in muscles generates continuous exudation of plasma proteins, recruitment of inflammatory cells, activation of inflammatory mediators and increased expression of MMPs. These induce a cascade of molecular modifications that affect tissue reconstitution. During these modifications, the balance between hydrolysis of ECM components and their synthesis is deregulated in favour of hydrolysis in a first stage followed by a resolution phase during which inhibitors are upregulated. The end-result of these continuous cycles of necrosis-regeneration can, therefore, be associated with an increase of both MMPs and TIMPs, as occurs in DMD muscles.
Inflammatory cells contribute to increased fibrosis by producing cytokines/chemokines [109, 122, 146, 152] that regulate MMPs. These in turn, modulate the activities of cytokines [153, 154], their receptors and ligands, which are abundant in injured muscles [155–159]. Among them, SDF-1/CXCR, CCL2(MCP-1)/CCR2 and M-CSF/M-CSFR axis have a beneficial effect on muscle recovery [160, 161]. The first one improves cell mobilization and migration by increasing MMP-2 and MMP-9 expression [162] and interferes in muscle regeneration by a mechanism dependent on MMP-10 [60]. The second two [163, 164] participate in the recruitment of monocytes/macrophages that help to repair the injury by producing Insulin Growth Factor-1 (IGF-1). The mitogenic IGF/IGF receptor autocrine loop is partly regulated by a family of six IGF Binding Proteins (IGFBPs) proteolyzed by MMP-1, MMP-3 and MMP-9 [165–167]. In contrast, the reduction of IGF-I/insulin signaling promotes fibrosis by regulating the interaction between p-Akt and Smad3. This allows the dissociation of Smad-3 and its nuclear translocation and results in increased TGF-β1 signaling and fibrosis [168].
Growth factors also influence satellite cell activation, (trans)-differentiation and muscle regeneration. TGF-β induces phenotypic transformation of myoblasts into myofibroblasts, downregulates myogenic regulatory proteins [126], stimulates collagen synthesis and inhibits its degradation [169, 170] by reducing MMP activity and promoting TIMP expression [132]. TGF-β is activated by MMP-9 [171] and it is neutralized by decorin. The latter is cleaved by MMP-2, MMP-3 and MMP-9 that release TGF-β from the complex [172]. The action of TGF-β is synergized by Connective Tissue Growth Factor (CTGF), incriminated in multiple fibrotic diseases [173, 174] and upregulated in DMD muscles [175]. The modular domains composing the protein have independent functions and can be cleaved by MMP-1, -2, -3, -7, -9, -13, elastase, and plasmin [176]. CTGF overexpression in normal skeletal muscles induces dystrophic features [177] possibly through the c-terminal module IV shown to have an immuno-modulatory function [178]. Its inhibition reduces skeletal muscle impairment, reverses fibrosis and improves muscle strength without affecting TGF-β [179, 180] indicating that CTGF is, by itself, a critical modulator of fibrosis.
Activated metalloproteinases from both MMPs and A Desintegrin And Metalloproteinase (ADAMs) families signal through their receptors and downstream mitogen-activated kinases to activate the transcription of immediate-early genes, mediators of fibrosis [181]. Two ADAMs, notably ADAM-17 (Tumour Necrosis Factor-α Converting Enzyme TACE, or MT4-MMP) and ADAM-12 are key players in the pathogenesis of inflammatory and fibrous connective tissue diseases. TACE overexpression and activation in dermal fibroblasts, activates Epidermal Growth Factor Receptor (EGFR) by its ligands and stimulates type I collagen expression [182]. In a model of cardiac fibrosis induced by angiotensin II [183], TACE overexpression induces transcriptional regulation of MMP-2 and ADAM-12 that activate TGF-β signalling independently of its protease activity [184]. This leads one to question whether the blockade of fibrosis in dystrophic mdx mice by ACE inhibitors inhibits only CTGF expression as hypothesized [180] or whether it also affects TACE expression and activation. TACE release of TNF-α activates the myogenic program [185] and stimulates collagen synthesis in fibroblasts [186] exerting direct adverse effects on skeletal muscle function and regeneration. Its blockade reduces necrosis and contractile dysfunction in response to eccentric exercise [187, 188] and significantly reduces the levels of TGF-β1 and type I collagen mRNA in mdx mice [189].
TIMPs and other protease inhibitors interfere in the fibrotic process. Exposure of cardiac fibroblasts to any of the four TIMPs stimulates cell proliferation and induces a significant increase of α-SMA but only TIMP-2 increases both fibroblast proliferation and collagen production [81]. The occurrence of a similar process in skeletal muscles needs to be investigated. We also need to, precisely, define the effects of all four inhibitors on fibroblast and myoblast proliferation, differentiation and ECM production. Finally, a putative serine protease inhibitor HE4 (encoding human epididymis protein 4), that inhibits serine proteases Prss23 and Prss35 as well as collagenase, MMP-2, MMP-9 and trypsin, is upregulated in fibrosis-associated fibroblasts of mouse and human kidneys and in serum of patients with chronic kidney diseases [190]. Its inhibition accelerates collagen I degradation, inhibits fibrosis and restores higher levels of Prss23 and Prss35 indicating that HE4 serves as biomarker and therapeutic target for the treatment of renal fibrosis. To date, HE4 expression is still unexplored either in skeletal muscles and serum of dystrophic patients or in other fibrotic diseases.
CONCLUDING REMARKS
Although we have gained some insight about the expression and role of certain MMPs and TIMPs in skeletal muscles, we still have much to learn before being able to use them for therapeutic perspectives. Clearly, MMP-2 and its activator MMP-14 are linked to angiogenesis and vessel growth [59] and MMP-2 contributes to satellite cell activation by mediating HGF shedding from extracellular matrix in response to NO [191]. Furthermore, MMP-9 and MMP-10 are important for muscle regeneration and one can assume, although it remains to be experimentally proven, that deficiency of MMP-25, MMP-28 or MMP-24 would have a deleterious effect on muscle regeneration because MMP-25 and MMP-28 influence macrophage progression [35, 192] and MMP-24 regulates stem cell quiescence [193].
Another observation concerns the specific issue of inhibiting proteases in general and MMPs in particular as a complementary therapeutic approach to cell or gene therapy. Many reviews have highlighted the potential benefit of inhibiting diverse classes of proteases in muscular dystrophies placing “MMPs” among potential therapeutic targets [194–196]. This can be misleading in the absence of a comprehensive view of the role played by these proteins in skeletal muscles. Clearly, it is only short term inhibition of MMP-9 or its signaling cascade that was proven to improve skeletal muscle and tendon healing and ameliorate structure and function of dystrophic muscles [30, 32, 110]. What MMP-9 inhibition is, most likely, doing is limiting “excessive/prolonged” inflammation and attenuating its micro-environmental consequences possibly by cooling-off inflammation [197]. Alternatively, reduction of hydrolytic enzymes reduces inflammation and ROS production causing a reduction of pro-MMP-1, -8, -9 activation and oxidative cell injury [198, 199] expected to limit DAMPs release, reduce the activation/perpetuation of the immune response and decrease MMP-9 levels [198, 199]. Several substances that modulate p38-MAPK, NF-kB, NO-cGMP [116, 118, 200–203], inhibit TGF-β [204], reduce oxidative stress [205] or inflammation [206, 207] have had beneficial effects in muscular dystrophies and deserve to be tested, in preclinical settings, in combination with short-term, transitory MMP-9 inhibition. With the exception of MMP-9 that plays a dual role in skeletal muscle regeneration/dystrophy [110, 208, 209], the inhibition of other MMPs proved they are essential for efficient muscle regeneration -this review and [210]-, reduction of fibrosis [211] and amelioration of myoblast engraftment following implantation into dystrophic mice [212]. This confirms the necessity for questioning long-term inhibition of MMPs not only in cancer and inflammation [213] but also in muscular dystrophies and other muscle diseases.
ACKNOWLEDGMENTS
Hala Alameddine is supported by the AFM (Association Française contre les Myopathies). Jennifer Morgan is supported by the Great Ormond Street Hospital Children’s Charity, the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
REFERENCES
[1] | Barnes BR , Szelenyi ER , Warren GL , Urso ML . Alterations in mRNA and protein levels of metalloproteinases-2, -9, and -14 and tissue inhibitor of metalloproteinase-2 responses to traumatic skeletal muscle injury. Am J Physiol Cell Physiol. (2009) ;297: (6):C1501–8. |
[2] | Brown MD , Hudlicka O . Modulation of physiological angiogenesis in skeletal muscle by mechanical forces: Involvement of VEGF and metalloproteinases. Angiogenesis. (2003) ;6: (1):1–14. |
[3] | Carmeli E , Beiker R , Maor M , Kodesh E . Increased iNOS, MMP-2, and HSP-72 in skeletal muscle following high-intensity exercise training. J Basic Clin Physiol Pharmacol. (2010) ;21: (2):127–46. |
[4] | Carmeli E , Moas M , Lennon S , Powers SK . High intensity exercise increases expression of matrix metalloproteinases in fast skeletal muscle fibres. Exp Physiol. (2005) ;90: (4):613–9. |
[5] | Chao T , Frump D , Lin M , Caiozzo VJ , Mozaffar T , Steward O , et al. Matrix metalloproteinase 3 deletion preserves denervated motor endplates after traumatic nerve injury. Ann Neurol. (2013) ;73: (2):210–23. |
[6] | Kherif S , Dehaupas M , Lafuma C , Fardeau M , Alameddine HS . Matrix metalloproteinases MMP-2 and MMP-9 in denervated muscle and injured nerve. Neuropathol Appl Neurobiol. (1998) ;24: (4):309–19. |
[7] | Kherif S , Lafuma C , Dehaupas M , Lachkar S , Fournier JG , Verdiere-Sahuque M , et al. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: A study in experimentally injured and mdx muscles. Dev Biol. (1999) ;205: (1):158–70. |
[8] | Rullman E , Norrbom J , Stromberg A , Wagsater D , Rundqvist H , Haas T , et al. Endurance exercise activates matrix metalloproteinases in human skeletal muscle. J Appl Physiol. (2009) ;106: (3):804–12. |
[9] | Rullman E , Rundqvist H , Wagsater D , Fischer H , Eriksson P , Sundberg CJ , et al. A single bout of exercise activates matrix metalloproteinase in human skeletal muscle. J Appl Physiol. (2007) ;102: (6):2346–51. |
[10] | Thornton GM , Shao X , Chung M , Sciore P , Boorman RS , Hart DA , et al. Changes in mechanical loading lead to tendon specific alterations in MMP and TIMP expression: Influence of stress deprivation and intermittent cyclic hydrostatic compression on rat supraspinatus and achilles tendons. Br J Sports Med. (2010) ;44: (10):698–703. |
[11] | Urso ML , Pierce JR , Alemany JA , Harman EA , Nindl BC . Effects of exercise training on the matrix metalloprotease response to acute exercise. Eur J Appl Physiol. (2009) ;106: (5):655–63. |
[12] | Urso ML , Szelenyi ER , Warren GL , Barnes BR. Matrix metalloprotease-3 and tissue inhibitor of metalloprotease-1 mRNA and protein levels are altered in response to traumatic skeletal muscle injury. Eur J App Physiol. (2010) ;109: (5):963–72. |
[13] | Porter BE , Weis J , Sanes JR . A motoneuron-selective stop signal in the synaptic protein S-laminin. Neuron. (1995) ;14: (3):549–59. |
[14] | Couch CB , Strittmatter WJ . Rat myoblast fusion requires metalloendoprotease activity. Cell. (1983) ;32: (1):257–65. |
[15] | Couch CB , Strittmatter WJ . Specific blockers of myoblast fusion inhibit a soluble and not the membrane-associated metalloendoprotease in myoblasts. J Biol Chem. (1984) ;259: :5396–9. |
[16] | Anaya-Segura MA , Garcia-Martinez FA , Montes-Almanza LA , Diaz BG , Avila-Ramirez G , Alvarez-Maya I , et al. Non-invasive biomarkers for Duchenne Muscular Dystrophy and carrier detection. Molecules. (2015) ;20: (6):11154–72. |
[17] | Bani C , Lagrota-Candido J , Pinheiro DF , Leite PE , Salimena MC , Henriques-Pons A , et al. Pattern of metalloprotease activity and myofiber regeneration in skeletal muscles of mdx mice. Muscle Nerve. (2008) ;37: (5):583–92. |
[18] | Choi YC , Dalakas MC . Expression of matrix metalloproteinases in the muscle of patients with inflammatory myopathies. Neurology. (2000) ;54: (1):65–71. |
[19] | Devine RD , Bicer S , Reiser PJ , Velten M , Wold LE . Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am J Physiol Heart Circ Physiol. (2015) ;309: (4):H685–91. |
[20] | Fukushima K , Nakamura A , Ueda H , Yuasa K , Yoshida K , Takeda S , et al. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ). BMC Musculoskelet Disord. (2007) ;8: :54. |
[21] | Jenkins MH , Alrowaished SS , Goody MF , Crawford BD , Henry CA . Laminin and Matrix metalloproteinase 11 regulate Fibronectin levels in the zebrafish myotendinous junction. Skelet Muscle. (2016) ;6: (1):18. |
[22] | Kieseier BC , Schneider C , Clements JM , Gearing AJ , Gold R , Toyka KV , et al. Expression of specific matrix metalloproteinases in inflammatory myopathies. Brain. (2001) ;124: (Pt 2):341–51. |
[23] | Li H , Mittal A , Paul PK , Kumar M , Srivastava DS , Tyagi SC , et al. Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase 9 (MMP-9) production in skeletal muscle through the activation of nuclear factor-kappaB-inducing kinase and p38 mitogen-activated protein kinase: A potential role of MMP-9 in myopathy. J Biol Chem. (2009) ;284: (7):4439–50. |
[24] | Sardone F , Traina F , Bondi A , Merlini L , Santi S , Maraldi NM , et al. Tendon extracellular matrix alterations in Ullrich Congenital Muscular Dystrophy. Frontiers in Aging Neuroscience. (2016) ;8: (131: ). |
[25] | Schoser BGH , Blottner D , Stuerenburg HJ . Matrix metalloproteinases in inflammatory myopathies: Enhanced immunoreactivity near atrophic myofibers. Acta Neurol Scand. (2002) ;105: (4):309–13. |
[26] | Shiba N , Miyazaki D , Yoshizawa T , Fukushima K , Shiba Y , Inaba Y , et al. Differential roles of MMP-9 in early and late stages of dystrophic muscles in a mouse model of Duchenne muscular dystrophy. Biochim Biophys Acta. (2015) ;1852: (10 Pt A):2170–82. |
[27] | Von Moers A , Zwirner A , Reinhold A , Bruckmann O , van Landeghem F , Stoltenburg-Didinger G , et al. Increased mRNA expression of tissue inhibitors of metalloproteinase-1 and -2 in Duchenne muscular dystrophy. Acta Neuropathol. (2005) ;109: (3):285–93. |
[28] | Hurnaus S , Mueller-Felber W , Pongratz D , Schoser BG . Serum levels of matrix metalloproteinases-2 and -9 and their tissue inhibitors in inflammatory neuromuscular disorders. Eur Neurol. (2006) ;55: (4):204–8. |
[29] | Gargioli C , Coletta M , De Grandis F , Cannata SM , Cossu G . PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat Med. (2008) ;14: (9):973–8. |
[30] | Hindi SM , Shin J , Ogura Y , Li H , Kumar A . Matrix Metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One. (2013) ;8: (8):e72121. |
[31] | Hnia K , Gayraud J , Hugon G , Ramonatxo M , De La Porte S , Matecki S , et al. L-arginine decreases inflammation and modulates the nuclear factor-kappaB/matrix metalloproteinase cascade in mdx muscle fibers. Am J Pathol. (2008) ;172: (6):1509–19. |
[32] | Kumar A , Bhatnagar S , Kumar A . Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am J Pathol. (2010) ;177: (1):248–60. |
[33] | Nadarajah VD , van Putten M , Chaouch A , Garrood P , Straub V , Lochmuller H , et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul Disord. (2011) ;21: (8):569–78. |
[34] | Cauwe B , Martens E , Proost P , Opdenakker G . Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr Biol (Camb). (2009) ;1: (5-6):404–26. |
[35] | Starr AE , Bellac CL , Dufour A , Goebeler V , Overall CM . Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25): Chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J Biol Chem. (2012) ;287: (16):13382–95. |
[36] | Alameddine HS . Matrix metalloproteinases in skeletal muscles: Friends or foes? Neurobiol Dis. (2012) ;48: (3):508–18. |
[37] | Egeblad M , Werb Z . New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. (2002) ;2: (3):161–74. |
[38] | Nagase H , Woessner JF . Matrix metalloproteinases. J Biol Chem. (1999) ;274: (31):21491–4. |
[39] | Itoh T , Ikeda T , Gomi H , Nakao S , Suzuki T , Itohara S . Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. (1997) ;272: (36):22389–92. |
[40] | Mosig RA , Dowling O , DiFeo A , Ramirez MC , Parker IC , Abe E , et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. (2007) ;16: (9):1113–23. |
[41] | Tuysuz B , Mosig R , Altun G , Sancak S , Glucksman MJ , Martignetti JA . A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. (2009) ;17: (5):565–72. |
[42] | Holmbeck K , Bianco P , Caterina J , Yamada S , Kromer M , Kuznetsov SA , et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. (1999) ;99: (1):81–92. |
[43] | Oh J , Takahashi R , Adachi E , Kondo S , Kuratomi S , Noma A , et al. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene. (2004) ;23: (29):5041–8. |
[44] | VanSaun M , Herrera AA , Werle MJ . Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J Neurocytol. (2003) ;32: (9):1129–42. |
[45] | Werle MJ , VanSaun M . Activity dependent removal of agrin from synaptic basal lamina by matrix metalloproteinase 3. J Neurocytol. (2003) ;32: (5-8):905–13. |
[46] | Apte SS , Parks WC . Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Biology. (2015) ;44-46: :1–6. |
[47] | Adair-Kirk TL , Senior RM . Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. (2008) ;40: (6-7):1101–10. |
[48] | Caron NJ , Asselin I , Morel G , Tremblay JP . Increased myogenic potential and fusion of matrilysin-expressing myoblasts transplanted in mice. Cell Transplant. (1999) ;8: (5):465–76. |
[49] | El Fahime E , Torrente Y , Caron NJ , Bresolin MD , Tremblay JP . In vivo migration of transplanted myoblasts requires Matrix Metalloproteinase activity. Exp Cell Res. (2000) ;258: (2):279–87. |
[50] | Lei H , Leong D , Smith LR , Barton ER . Matrix-metalloproteinase 13 (MMP-13) is a new contributor to skeletal muscle regeneration and critical for myoblast migration. Am J Physiol Cell Physiol. (2013) ;305: (5):C529–C38. |
[51] | Nishimura T , Nakamura K , Kishioka Y , Kato-Mori Y , Wakamatsu J , Hattori A . Inhibition of matrix metalloproteinases suppresses the migration of skeletal muscle cells. J Muscle Res Cell Motil. (2008) ;29: (1):37–44. |
[52] | Torrente Y , El Fahime E , Caron NJ , Bresolin N , Tremblay JP . Intramuscular migration of myoblasts transplanted after muscle pretreatment with metalloproteinases. Cell Transplant. (2000) ;9: (4):539–49. |
[53] | Torrente Y , El Fahime E , Caron NJ , Del Bo R , Belicchi M , Pisati F , et al. Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. (2003) ;12: (1):91–100. |
[54] | Wang W , Pan H , Murray K , Jefferson BS , Li Y . Matrix metalloproteinase-1 promotes muscle cell migration and differentiation. Am J Pathol. (2009) ;174: (2):541–9. |
[55] | Chen X , Li Y . Role of matrix metalloproteinases in skeletal muscle: Migration, differentiation, regeneration and fibrosis. Cell Adh Migr. (2009) ;3: (4):337–41. |
[56] | Ito H , Hallauer PL , Hastings KE , Tremblay JP . Prior culture with concanavalin A increases intramuscular migration of transplanted myoblast. Muscle Nerve. (1998) ;21: (3):291–7. |
[57] | Morgan J , Rouche A , Bausero P , Houssaïni A , Gross J , Fiszman MY , et al. MMP-9 overexpression improves myogenic cell migration and engraftment. Muscle Nerve. (2010) ;42: (4):584–95. |
[58] | Ohtake Y , Tojo H , Seiki M . Multifunctional roles of MT1-MMP in myofiber formation and morphostatic maintenance of skeletal muscle. J Cell Sci. (2006) ;119: (Pt 18):3822–32. |
[59] | Miyazaki D , Nakamura A , Fukushima K , Yoshida K , Takeda S , Ikeda SI . Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum Mol Genet. (2011) ;9: (20):1787–99. |
[60] | Bobadilla M , Sainz N , Abizanda G , Orbe J , Rodriguez JA , Paramo JA , et al. The CXCR4/SDF1 axis improves muscle regeneration through MMP-10 activity. Stem Cells Dev. (2014) ;23: (12):1417–27. |
[61] | Bobadilla M , Sainz N , Rodriguez JA , Abizanda G , Orbe J , de Martino A , et al. MMP-10 is required for efficient muscle regeneration in mouse models of injury and muscular dystrophy. Stem Cells. (2014) ;32: (2):447–61. |
[62] | Bozzi M , Sciandra F , Brancaccio A . Role of gelatinases in pathological and physiological processes involving the dystrophin–glycoprotein complex. Matrix Biol. (2015) ;44-46: :130–7. |
[63] | Sbardella D , Sciandra F , Gioia M , Marini S , Gori A , Giardina B , et al. alpha-dystroglycan is a potential target of matrix metalloproteinase MMP-2. Matrix Biol. (2015) ;41: :2–7. |
[64] | Yamada H , Saito F , Fukuta-Ohi H , Zhong D , Hase A , Arai K , et al. Processing of beta-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum Mol Genet. (2001) ;10: (15):1563–9. |
[65] | Hadler-Olsen E , Solli AI , Hafstad A , Winberg JO , Uhlin-Hansen L . Intracellular MMP-2 activity in skeletal muscle is associated with type II fibers. J Cell Physiol. (2015) ;230: (1):160–9. |
[66] | Carmeli E , Moas M , Reznick AZ , Coleman R . Matrix metalloproteinases and skeletal muscle: A brief review. Muscle Nerve. (2004) ;29: (2):191–7. |
[67] | Carmeli E , Kodesh E , Nemcovsky C . Tetracycline therapy for muscle atrophy due to immobilization. J Musculoskelet Neuronal Interact. (2009) ;9: (2):81–8. |
[68] | Giannelli G , De Marzo A , Marinosci F , Antonaci S . Matrix metalloproteinase imbalance in muscle disuse atrophy. Histol Histopathol. (2005) ;20: (1):99–106. |
[69] | Joshi SK , Lee L , Lovett DH , Kang H , Kim HT , Delgado C , et al. Novel intracellular N-terminal truncated matrix metalloproteinase-2 isoform in skeletal muscle ischemia-reperfusion injury. J Orthop Res. (2016) ;34: (3):502–9. |
[70] | Liu X , Lee DJ , Skittone LK , Natsuhara K , Kim HT . Role of gelatinases in disuse-induced skeletal muscle atrophy. Muscle Nerve. (2010) ;41: (2):174–8. |
[71] | Wang W , Schulze CJ , Suarez-Pinzon WL , Dyck JR , Sawicki G , Schulz R . Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. (2002) ;106: (12):1543–9. |
[72] | Ali MA , Cho WJ , Hudson B , Kassiri Z , Granzier H , Schulz R . Titin is a target of matrix metalloproteinase-2: Implications in myocardial ischemia/reperfusion injury. Circulation. (2010) ;122: (20):2039–47. |
[73] | DeCoux A , Lindsey ML , Villarreal F , Garcia RA , Schulz R . Myocardial matrix metalloproteinase-2: Inside out and upside down. J Mol Cell Cardiol. (2014) ;77: :64–72. |
[74] | Brew K , Nagase H . The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim Biophys Acta- Mol Cell Res. (2010) ;1803: (1):55–71. |
[75] | Nagase H , Visse R , Murphy G . Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. (2006) ;69: (3):562–73. |
[76] | Arpino V , Brock M , Gill SE . The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biology. (2015) ;44-46: :247–54. |
[77] | Baker AH , Edwards DR , Murphy G . Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J Cell Sci. (2002) ;115: (Pt 19):3719–27. |
[78] | Clark IM , Swingler TE , Sampieri CL , Edwards DR . The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell B. (2008) ;40: (6-7):1362–78. |
[79] | Fernández CA , Moses MA . Modulation of angiogenesis by tissue inhibitor of metalloproteinase-4. Biochem Biophy Res Co. (2006) ;345: (1):523–9. |
[80] | Hornebeck W . Down-regulation of tissue inhibitor of matrix metalloprotease-1 (TIMP-1) in aged human skin contributes to matrix degradation and impaired cell growth and survival. Pathologie Biologie. (2003) ;51: (10):569–73. |
[81] | Lovelock JD , Baker AH , Gao F , Dong J-F , Bergeron AL , McPheat W , et al. Heterogeneous effects of tissue inhibitors of matrix metalloproteinases on cardiac fibroblasts. Am J Physiol - Heart C. (2005) ;288: (2):H461–H8. |
[82] | Visse R , Nagase H . Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ Res. (2003) ;92: (8):827–39. |
[83] | Lewis MP , Tippett HL , Sinanan AC , Morgan MJ , Hunt NP . Gelatinase-B (matrix metalloproteinase-9; MMP-9) secretion is involved in the migratory phase of human and murine muscle cell cultures. J Muscle Res Cell Motil. (2000) ;21: (3):223–33. |
[84] | Jaworski DM , Soloway P , Caterina J , Falls WA . Tissue inhibitor of metalloproteinase-2(TIMP-2)-deficient mice display motor deficits. J Neurobiol. (2006) ;66: (1):82–94. |
[85] | Lluri G , Langlois GD , Soloway PD , Jaworski DM . Tissue inhibitor of metalloproteinase-2 (TIMP-2) regulates myogenesis and beta1 integrin expression in vitro. Exp Cell Res. (2008) ;314: (1):11–24. |
[86] | Liu H , Chen SE , Jin B , Carson JA , Niu A , Durham W , et al. TIMP3: A physiological regulator of adult myogenesis. J Cell Sci. (2010) ;123: (Pt 17):2914–21. |
[87] | Coussens LM , Fingleton B , Matrisian LM . Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science. (2002) ;295: (5564):2387–92. |
[88] | Pavlaki M , Zucker S . Matrix metalloproteinase inhibitors (MMPIs): The beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. (2003) ;22: (2-3):177–203. |
[89] | Devel L , Beau F , Amoura M , Vera L , Cassar-Lajeunesse E , Garcia S , et al. Simple Pseudo-dipeptides with a P2’ Glutamate: A novel inhibitor family of matrix metalloproteases and other Metzincins. J Biol Chem. (2012) ;287: (32):26647–56. |
[90] | Johnson JL , Devel L , Czarny B , George SJ , Jackson CL , Rogakos V , et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. (2011) ;31: (3):528–35. |
[91] | Nissinen L , Kahari VM . Matrix metalloproteinases in inflammation. Biochim Biophys Acta. (2014) ;1840: (8):2571–80. |
[92] | Sbardella D , Fasciglione GF , Gioia M , Ciaccio C , Tundo GR , Marini S , et al. Human matrix metalloproteinases: An ubiquitarian class of enzymes involved in several pathological processes. Mol Aspects Med. (2012) ;33: (2):119–208. |
[93] | Dehne N , Kerkweg U , Flohe SB , Brune B , Fandrey J . Activation of hypoxia-inducible factor 1 in skeletal muscle cells after exposure to damaged muscle cell debris. Shock. (2011) ;35: (6):632–8. |
[94] | Goetzl EJ , Banda MJ , Leppert D . Matrix metalloproteinases in immunity. J Immunol. (1996) ;156: (1):1–4. |
[95] | Sopata I , Dancewicz AM . Presence of a gelatin-specific proteinase and its latent form in human leucocytes. Biochim Biophys Acta. (1974) ;370: (2):510–23. |
[96] | Mainardi CL , Hibbs MS , Hasty KA , Seyer JM . Purification of a type V collagen degrading metalloproteinase from rabbit alveolar macrophages. Coll Relat Res. (1984) ;4: (6):479–92. |
[97] | Opdenakker G , Van den Steen PE , Dubois B , Nelissen I , Van Coillie E , Masure S , et al. Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol. (2001) ;69: (6):851–9. |
[98] | Baram D , Vaday GG , Salamon P , Drucker I , Hershkoviz R , Mekori YA . Human mast cells release metalloproteinase-9 on contact with activated T cells: Juxtacrine regulation by TNF-alpha. J Immunol. (2001) ;167: (7):4008–16. |
[99] | Chakrabarti S , Zee JM , Patel KD . Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: Novel pathways for tertiary granule release. J Leukoc Biol. (2006) ;79: (1):214–22. |
[100] | Cowland JB , Borregaard N . The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol. (1999) ;66: (6):989–95. |
[101] | Kolaczkowska E , Arnold B , Opdenakker G . Gelatinase B/MMP-9 as an inflammatory marker enzyme in mouse zymosan peritonitis: Comparison of phase-specific and cell-specific production by mast cells, macrophages and neutrophils. Immunobiology. (2008) ;213: (2):109–24. |
[102] | Beuche W , Yushchenko M , Mader M , Maliszewska M , Felgenhauer K , Weber F . Matrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosis. Neuroreport. (2000) ;11: (16):3419–22. |
[103] | Lorenzl S , Albers DS , LeWitt PA , Chirichigno JW , Hilgenberg SL , Cudkowicz ME , et al. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci. (2003) ;207: (1-2):71–6. |
[104] | Niebroj-Dobosz I , Janik P , Sokołowska B , Kwiecinski H . Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur J Neurol. (2010) ;17: (2):226–31. |
[105] | Schoser BG , Blottner D . Matrix metalloproteinases MMP-2, MMP-7 and MMP-9 in denervated human muscle. Neuroreport. (1999) ;10: (13):2795–7. |
[106] | Gorospe JR , Tharp MD , Hinckley J , Kornegay JN , Hoffman EP . A role for mast cells in the progression of Duchenne muscular dystrophy? Correlations in dystrophin-deficient humans, dogs, and mice. J Neurol Sci. (1994) ;122: (1):44–56. |
[107] | Hodgetts S , Radley H , Davies M , Grounds MD . Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNF-alpha function with Etanercept in mdx mice. Neuromuscul Disord. (2006) ;16: (9-10):591–602. |
[108] | Cai B , Spencer MJ , Nakamura G , Tseng-Ong L , Tidball JG . Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am J Pathol. (2000) ;156: (5):1789–96. |
[109] | Spencer MJ , Montecino-Rodriguez E , Dorshkind K , Tidball JG . Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol. (2001) ;98: (2):235–43. |
[110] | Li H , Mittal A , Makonchuk DY , Bhatnagar S , Kumar A . Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. (2009) ;18: (14):2584–98. |
[111] | Zanotti S , Gibertini S , Di Blasi C , Cappelletti C , Bernasconi P , Mantegazza R , et al. Osteopontin is highly expressed in severely dystrophic muscle and seems to play a role in muscle regeneration and fibrosis. Histopathol. (2011) ;59: (6):1215–28. |
[112] | Rodolico C , Mazzeo A , Toscano A , Messina S , Aguennouz M , Gaeta M , et al. Specific matrix metalloproteinase expression in focal myositis: An immunopathological study. Acta Neurol Scand. (2005) ;112: (3):173–7. |
[113] | Kaplan A , Spiller Krista J , Towne C , Kanning Kevin C , Choe Ginn T , Geber A , et al. Neuronal Matrix Metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. (2014) ;81: (2):333–48. |
[114] | Tang Y , Reay DP , Salay MN , Mi MY , Clemens PR , Guttridge DC , et al. Inhibition of the IKK/NF-kappaB pathway by AAV gene transfer improves muscle regeneration in older mdx mice. Gene Ther. (2010) ;17: (12):1476–83. |
[115] | Zimowska M , Olszynski KH , Swierczynska M , Streminska W , Ciemerych MA . Decrease of MMP-9 activity improves soleus muscle regeneration. Tissue Eng Part A. (2012) ;18: (11-12):1183–92. |
[116] | Brunelli S , Sciorati C , D’Antona G , Innocenzi A , Covarello D , Galvez BG , et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci U S A. (2007) ;104: (1):264–9. |
[117] | Sciorati C , Buono R , Azzoni E , Casati S , Ciuffreda P , D’Angelo G , et al. Co-administration of ibuprofen and nitric oxide is an effective experimental therapy for muscular dystrophy, with immediate applicability to humans. Br J Pharmacol. (2010) ;160: (6):1550–60. |
[118] | Zordan P , Sciorati C , Campana L , Cottone L , Clementi E , Querini PR , et al. The nitric oxide-donor molsidomine modulates the innate inflammatory response in a mouse model of muscular dystrophy. Eur J Pharmacol. (2013) ;715: (1-3):296–303. |
[119] | Nagamine Y , Medcalf RL , Munoz-Canoves P . Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb Haemost. (2005) ;93: (4):661–75. |
[120] | Zwetsloot KA , Nedergaard A , Gilpin LT , Childs TE , Booth FW . Differences in transcriptional patterns of extracellular matrix, inflammatory, and myogenic regulatory genes in myofibroblasts, fibroblasts, and muscle precursor cells isolated from old male rat skeletal muscle using a novel cell isolation procedure. Biogerontology. (2012) ;13: (4):383–98. |
[121] | Lieber RL , Ward SR . Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am J Physiol Cell Physiol. (2013) ;305: (3):C241–52. |
[122] | Mann CJ , Perdiguero E , Kharraz Y , Aguilar S , Pessina P , Serrano AL , et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. (2011) ;1: (1):21. |
[123] | Dulauroy S , Di Carlo SE , Langa F , Eberl G , Peduto L . Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. (2012) ;18: :1262–70. |
[124] | Galligan CL , Fish EN . The role of circulating fibrocytes in inflammation and autoimmunity. J Leukoc Biol. (2013) ;93: (1):45–50. |
[125] | Hinz B , Phan SH , Thannickal VJ , Prunotto M , Desmouliere A , Varga J , et al. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am J Pathol. (2012) ;180: (4):1340–55. |
[126] | Li Y , Foster W , Deasy BM , Chan Y , Prisk V , Tang Y , et al. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: A key event in muscle fibrogenesis. Am J Pathol. (2004) ;164: (3):1007–19. |
[127] | Petrov VV , Fagard RH , Lijnen PJ . Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. (2002) ;39: (2):258–63. |
[128] | Klingberg F , Hinz B , White ES . The myofibroblast matrix: Implications for tissue repair and fibrosis. J Pathol. (2013) ;229: (2):298–309. |
[129] | Wynn TA . Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) ;214: (2):199–210. |
[130] | Hinz B , Mastrangelo D , Iselin CE , Chaponnier C , Gabbiani G . Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol. (2001) ;159: (3):1009–20. |
[131] | Hinz B , Celetta G , Tomasek JJ , Gabbiani G , Chaponnier C . Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. (2001) ;12: (9):2730–41. |
[132] | Micallef L , Vedrenne N , Billet F , Coulomb B , Darby IA , Desmouliere A . The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair. (2012) ;5: (Suppl 1):S5. |
[133] | Mignatti P , Rifkin DB . Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. (1993) ;73: (1):161–95. |
[134] | Wynn TA , Ramalingam TR . Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med. (2012) ;18: (7):1028–40. |
[135] | Fechner G , Bajanowski T , Brinkmann B . Immunohistochemical alterations after muscle trauma. Int J Legal Med. (1993) ;105: (4):203–7. |
[136] | Serrano AL , Munoz-Canoves P . Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res. (2010) ;316: (18):3050–8. |
[137] | O’Reilly PJ , Gaggar A , Blalock JE . Interfering with extracellular matrix degradation to blunt inflammation. Curr Opin Pharmacol. (2008) ;8: (3):242–8. |
[138] | Zhang Q , Raoof M , Chen Y , Sumi Y , Sursal T , Junger W , et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) ;464: (7285):104–7. |
[139] | Ono K , Han J . The p38 signal transduction pathway: Activation and function. Cell Signal. (2000) ;12: (1):1–13. |
[140] | Zhang Z , Reenstra W , Weiner DJ , Louboutin JP , Wilson JM . The p38 mitogen-activated protein kinase signaling pathway is coupled to Toll-like receptor 5 to mediate gene regulation in response to Pseudomonas aeruginosa infection in human airway epithelial cells. Infect Immun. (2007) ;75: (12):5985–92. |
[141] | Barron L , Wynn TA . Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. (2011) ;300: (5):G723–8. |
[142] | Bosurgi L , Manfredi AA , Rovere-Querini P . Macrophages in injured skeletal muscle: A perpetuum mobile causing and limiting fibrosis, prompting or restricting resolution and regeneration. Front Immunol. (2011) ;2: :62. |
[143] | Gordon S , Martinez FO . Alternative activation of macrophages: Mechanism and functions. Immunity. (2010) ;32: (5):593–604. |
[144] | Arnold L , Henry A , Poron F , Baba-Amer Y , van Rooijen N , Plonquet A , et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. (2007) ;204: (5):1057–69. |
[145] | Duffield JS , Lupher M , Thannickal VJ , Wynn TA . Host responses in tissue repair and fibrosis. Annu Rev Pathol. (2013) ;8: :241–76. |
[146] | Kharraz Y , Guerra J , Mann CJ , Serrano AL , Munoz-Canoves P . Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. (2013) ;2013: :491497. |
[147] | Ariel A , Serhan CN . New lives given by cell death: Macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front Immunol. (2012) ;3: :4. |
[148] | Korns D , Frasch SC , Fernandez-Boyanapalli R , Henson PM , Bratton DL . Modulation of macrophage efferocytosis in inflammation. Front Immunol. (2011) ;2: :57. |
[149] | Hinz B . The myofibroblast: Paradigm for a mechanically active cell. J Biomech. (2010) ;43: (1):146–55. |
[150] | Desmouliere A , Redard M , Darby I , Gabbiani G . Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. (1995) ;146: (1):56–66. |
[151] | Mantovani A , Sica A , Locati M . Macrophage polarization comes of age. Immunity. (2005) ;23: (4):344–6. |
[152] | Morrison J , Lu QL , Pastoret C , Partridge T , Bou-Gharios G . T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab Invest. (2000) ;80: (6):881–91. |
[153] | Ito A , Mukaiyama A , Itoh Y , Nagase H , Thogersen IB , Enghild JJ , et al. Degradation of interleukin 1beta by matrix metalloproteinases. J Biol Chem. (1996) ;271: (25):14657–60. |
[154] | Schonbeck U , Mach F , Libby P . Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. (1998) ;161: (7):3340–6. |
[155] | Chen YW , Nagaraju K , Bakay M , McIntyre O , Rawat R , Shi R , et al. Early onset of inflammation and later involvement of TGF beta in Duchenne muscular dystrophy. Neurology. (2005) ;65: (6):826–34. |
[156] | Chen YW , Zhao P , Borup R , Hoffman E . Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J Cell Biol. (2000) ;151: :1321–36. |
[157] | Demoule A , Divangahi M , Danialou G , Gvozdic D , Larkin G , Bao W , et al. Expression and Regulation of CC Class Chemokines in the Dystrophic (mdx) Diaphragm. Am J Respir Cell Mol Biol. (2005) ;33: (2):178–85. |
[158] | Hirata A , Masuda S , Tamura T , Kai K , Ojima K , Fukase A , et al. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: A role for osteopontin. Am J Pathol. (2003) ;163: :203–15. |
[159] | Porter JD , Khanna S , Kaminski HJ , Rao JS , Merriam A , Richmonds CR , et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. (2002) ;11: :263–72. |
[160] | Ge Y , Waldemer RJ , Nalluri R , Nuzzi PD , Chen J . RNAi screen reveals potentially novel roles of cytokines in myoblast differentiation. PLoS One. (2013) ;8: (7):e68068. |
[161] | De Paepe B , Creus KK , Martin JJ , De Bleecker JL . Upregulation of chemokines and their receptors in Duchenne muscular dystrophy: Potential for attenuation of myofiber necrosis. Muscle Nerve. (2012) ;46: (6):917–25. |
[162] | Brzoska E , Kowalewska M , Markowska-Zagrajek A , Kowalski K , Archacka K , Zimowska M , et al. Sdf-1 (CXCL12) improves skeletal muscle regeneration via the mobilisation of Cxcr4 and CD34 expressing cells. Biol Cell. (2012) ;104: (12):722–37. |
[163] | Lu H , Huang D , Saederup N , Charo IF , Ransohoff RM , Zhou L . Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. Faseb J. (2011) ;25: (1):358–69. |
[164] | Segawa M , Fukada S , Yamamoto Y , Yahagi H , Kanematsu M , Sato M , et al. Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp Cell Res. (2008) ;314: (17):3232–44. |
[165] | Rajah R , Nachajon RV , Collins MH , Hakonarson H , Grunstein MM , Cohen P . Elevated levels of the IGF-binding protein protease MMP-1 in asthmatic airway smooth muscle. Am J Respir Cell Mol Biol. (1999) ;20: (2):199–208. |
[166] | Fowlkes JL , Enghild JJ , Suzuki K , Nagase H . Matrix metalloproteinases degrade insulin-like growth factor-binding protein-3 in dermal fibroblast cultures. J Biol Chem. (1994) ;269: (41):25742–6. |
[167] | Manes S , Llorente M , Lacalle RA , Gomez-Mouton C , Kremer L , Mira E , et al. The matrix metalloproteinase-9 regulates the insulin-like growth factor-triggered autocrine response in DU-145 carcinoma cells. J Biol Chem. (1999) ;274: (11):6935–45. |
[168] | Dong Y , Lakhia R , Thomas SS , Dong Y , Wang XH , Silva KA , et al. Interactions between p-Akt and Smad3 in injured muscles initiate myogenesis or fibrogenesis. Am J Physiol Endocrinol Metab. (2013) ;305: (3):E367–75. |
[169] | Grande JP , Melder DC , Zinsmeister AR . Modulation of collagen gene expression by cytokines: Stimulatory effect of transforming growth factor-beta1, with divergent effects of epidermal growth factor and tumor necrosis factor-alpha on collagen type I and collagen type IV. J Lab Clin Med. (1997) ;130: (5):476–86. |
[170] | Ignotz RA , Massague J . Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. (1986) ;261: (9):4337–45. |
[171] | Yu Q , Stamenkovic I . Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. (2000) ;14: (2):163–76. |
[172] | Imai K , Hiramatsu A , Fukushima D , Pierschbacher MD , Okada Y . Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-beta 1 release. Biochem J. (1997) ;322: (Pt 3):809–14. |
[173] | Phanish MK , Winn SK , Dockrell ME . Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol. (2010) ;114: (3):e83–92. |
[174] | Leask A , Parapuram SK , Shi-Wen X , Abraham DJ . Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J Cell Commun Signal (2009) ;3: (2):89–94. |
[175] | Sun G , Haginoya K , Wu Y , Chiba Y , Nakanishi T , Onuma A , et al. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci. (2008) ;267: (1):48–56. |
[176] | Hashimoto G , Inoki I , Fujii Y , Aoki T , Ikeda E , Okada Y . Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. (2002) ;277: (39):36288–95. |
[177] | Morales MG , Cabello-Verrugio C , Santander C , Cabrera D , Goldschmeding R , Brandan E . CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J Pathol. (2011) ;225: (4):490–501. |
[178] | Rodrigues-Diez R , Rodrigues-Diez RR , Rayego-Mateos S , Suarez-Alvarez B , Lavoz C , Stark Aroeira L , et al. The C-terminal module IV of connective tissue growth factor is a novel immune modulator of the Th17 response. Lab Invest. (2013) ;93: (7):812–24. |
[179] | Morales MG , Gutierrez J , Cabello-Verrugio C , Cabrera D , Lipson KE , Goldschmeding R , et al. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet. (2013) ;22: (24):4938–495. |
[180] | Morales MG , Cabrera D , Cespedes C , Vio CP , Vazquez Y , Brandan E , et al. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. (2013) ;353: (1):173–87. |
[181] | Bhattacharyya S , Wu M , Fang F , Tourtellotte W , Feghali-Bostwick C , Varga J . Early growth response transcription factors: Key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. (2011) ;30: (4):235–42. |
[182] | Fukaya S , Matsui Y , Tomaru U , Kawakami A , Sogo S , Bohgaki T , et al. Overexpression of TNF-alpha-converting enzyme in fibroblasts augments dermal fibrosis after inflammation. Lab Invest. (2013) ;93: (1):72–80. |
[183] | Wang X , Oka T , Chow FL , Cooper SB , Odenbach J , Lopaschuk GD , et al. Tumor necrosis factor-alpha-converting enzyme is a key regulator of agonist-induced cardiac hypertrophy and fibrosis. Hypertension. (2009) ;54: (3):575–82. |
[184] | Atfi A , Dumont E , Colland F , Bonnier D , L’Helgoualc’h A , Prunier C , et al. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J Cell Biol. (2007) ;178: (2):201–8. |
[185] | Zhan M , Jin B , Chen SE , Reecy JM , Li YP . TACE release of TNF-alpha mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J Cell Sci. (2007) ;120: (Pt 4):692–701. |
[186] | Lurton J , Soto H , Narayanan A , Raghu G . Regulation of human lung fibroblast C1q-receptors by transforming growth factor-beta and tumor necrosis factor-alpha. Exp Lung Res. (1999) ;25: :151–64. |
[187] | Piers AT , Lavin T , Radley-Crabb HG , Bakker AJ , Grounds MD , Pinniger GJ . Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul Disord. (2011) ;21: (2):132–41. |
[188] | Radley HG , Davies MJ , Grounds MD . Reduced muscle necrosis and long-term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul Disord. (2008) ;18: (3):227–38. |
[189] | Gosselin LE , Martinez DA . Impact of TNF-alpha blockade on TGF-beta1 and type I collagen mRNA expression in dystrophic muscle. Muscle Nerve. (2004) ;30: (2):244–6. |
[190] | LeBleu VS , Teng Y , O’Connell JT , Charytan D , Muller GA , Muller CA , et al. Identification of human epididymis protein-4 as a fibroblast-derived mediator of fibrosis. Nat Med. (2013) ;19: (2):227–31. |
[191] | Yamada M , Sankoda Y , Tatsumi R , Mizunoya W , Ikeuchi Y , Sunagawa K , et al. Matrix metalloproteinase-2 mediates stretch-induced activation of skeletal muscle satellite cells in a nitric oxide-dependent manner. Int J Biochem Cell B. (2008) ;40: (10):2183–91. |
[192] | Ma Y , Halade GV , Zhang J , Ramirez TA , Levin D , Voorhees A , et al. Matrix Metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res. (2013) ;112: (4):675–88. |
[193] | Porlan E , Martí-Prado B , Morante-Redolat JM , Consiglio A , Delgado AC , Kypta R , et al. MT5-MMP regulates adult neural stem cell functional quiescence through the cleavage of N-cadherin. Nat Cell Biol. (2014) ;16: (7):629–38. |
[194] | Davis ME , Gumucio JP , Sugg KB , Bedi A , Mendias CL . MMP inhibition as a potential method to augment the healing of skeletal muscle and tendon extracellular matrix. J Appl Physiol. (2013) ;115: (6):884–91. |
[195] | Hollinger K , Selsby JT . The physiological response of protease inhibition in dystrophic muscle. Acta Physiol (Oxf). (2013) ;208: (3):234–44. |
[196] | Urso ML . Anti-inflammatory interventions and skeletal muscle injury: Benefit or detriment? J Appl Physiol (2013) ;115: :920–8. |
[197] | Noubade R , Wong K , Ota N , Rutz S , Eidenschenk C , Valdez PA , et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. (2014) ;509: (7499):235–9. |
[198] | Okamoto T , Akaike T , Sawa T , Miyamoto Y , van der Vliet A , Maeda H . Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem. (2001) ;276: (31):29596–602. |
[199] | Kim JH , Kwak HB , Thompson LV , Lawler JM . Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J Muscle Res Cell Motil. (2013) ;34: (1):1–13. |
[200] | Acharyya S , Villalta SA , Bakkar N , Bupha-Intr T , Janssen PM , Carathers M , et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. (2007) ;117: (4):889–901. |
[201] | Shin J , Tajrishi MM , Ogura Y , Kumar A . Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol. (2013) ;45: (10):2266–79. |
[202] | Hnia K , Hugon G , Rivier F , Masmoudi A , Mercier J , Mornet D . Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient mdx mice. Am J Pathol. (2007) ;170: (2):633–43. |
[203] | Percival JM , Whitehead NP , Adams ME , Adamo CM , Beavo JA , Froehner SC . Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol. (2012) ;228: (1):77–87. |
[204] | Taniguti AP , Matsumura CY , Rodrigues-Simioni L , Santo Neto H , Marques MJ . Suramin affects metalloproteinase-9 activity and increases beta-dystroglycan levels in the diaphragm of the dystrophin-deficient mdx mouse. Muscle Nerve. (2012) ;46: (5):810–3. |
[205] | Dorchies OM , Wagner S , Vuadens O , Waldhauser K , Buetler TM , Kucera P , et al. Green tea extract and its major polyphenol (-)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am J Physiol Cell Physiol. (2006) ;290: (2):C616–25. |
[206] | Girgenrath M , Beermann ML , Vishnudas VK , Homma S , Miller JB . Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann Neurol. (2009) ;65: (1):47–56. |
[207] | Pereira JA , Taniguti AP , Matsumura C , Marques MJ , Neto HS . Doxycycline ameliorates the dystrophic phenotype of skeletal and cardiac muscles in mdx mice. Muscle Nerve. (2012) ;46: (3):400–6. |
[208] | Dahiya S , Bhatnagar S , Hindi SM , Jiang C , Paul PK , Kuang S , et al. Elevated levels of active matrix metalloproteinase-9 cause hypertrophy in skeletal muscle of normal and dystrophin-deficient mdx mice. Hum Mol Genet. (2011) ;20: (22):4345–59. |
[209] | Mehan R , Greybeck B , Emmons K , Byrnes W , Allen D . Matrix metalloproteinase-9 deficiency results in decreased fiber cross-sectional area and alters fiber type distribution in mouse hindlimb skeletal muscle. Cells Tissues Organs. (2011) ;194: (6):510–20. |
[210] | Ogura Y , Tajrishi MM , Sato S , Hindi SM , Kumar A . Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front Cell Dev Biol. (2014) ;2: :11. |
[211] | Kaar JL , Li Y , Blair HC , Asche G , Koepsel RR , Huard J , et al. Matrix metalloproteinase-1 treatment of muscle fibrosis. Acta Biomater. (2008) ;4: (5):1411–20. |
[212] | Pan H , Vojnits K , Liu TT , Meng F , Yang L , Wang Y , et al. MMP1 gene expression enhances myoblast migration and engraftment following implanting into mdx/SCID mice. Cell Adh Migr. (2015) ;9: (4):283–92. |
[213] | Dufour A , Overall CM . Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci. (2013) ;34: (4):233–42. |
[214] | Sun G , Haginoya K , Chiba Y , Uematsu M , Hino-Fukuyo N , Tanaka S , et al. Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and their overexpression in muscle in human and mouse muscular dystrophy. J Neurol Sci. (2010) ;297: (1-2):19–28. |
[215] | Niebroj-Dobosz I , Madej-Pilarczyk A , Marchel M , Sokolowska B , Hausmanowa-Petrusewicz I . Matrix metalloproteinases in serum of Emery-Dreifuss muscular dystrophy patients. Acta Biochim Pol. (2009) ;56: (4):717–22. |
[216] | Morgan J , Alameddine HS . Stem cell based therapy for muscular dystrophies: Cell types and environmental factors influencing their efficacy. In: Hegde M, Ankala A, editors. Muscular Dystrophy. InTech; (2012) . DOI: 10.5772/30831. Available from: http://www.intechopen.com/books/muscular-dystrophy/stem-cell-based-therapy-for- |
[217] | Romi F , Helgeland G , Gilhus NE . Serum levels of matrix metalloproteinases: Implications in clinical neurology. Eur Neurol. (2012) ;67: (2):121–8. |
[218] | Sokolowska B , Jozwik A , Niebroj-Dobosz I , Janik P , Kwiecinski H . Evaluation of matrix metalloproteinases in serum of patients with amyotrophic lateral sclerosis with pattern recognition methods. J Physiol Pharmacol. (2009) ;60: (Suppl 5):117–20. |
[219] | Creange A , Sharshar T , Planchenault T , Christov C , Poron F , Raphael JC , et al. Matrix metalloproteinase-9 is increased and correlates with severity in Guillain-Barre syndrome. Neurology. (1999) ;53: (8):1683–91. |
[220] | Zanoteli E , van de Vlekkert D , Bonten EJ , Hu H , Mann L , Gomero EM , et al. Muscle degeneration in neuraminidase 1-deficient mice results from infiltration of the muscle fibers by expanded connective tissue. Biochim Biophys Acta. (2010) ;1802: (7-8):659–72. |
[221] | Soon CP , Crouch PJ , Turner BJ , McLean CA , Laughton KM , Atkin JD , et al. Serum matrix metalloproteinase-9 activity is dysregulated with disease progression in the mutant SOD1 transgenic mice. Neuromuscul Disord. (2010) ;20: (4):260–6. |
[222] | Helgeland G , Petzold A , Luckman SP , Gilhus NE , Plant GT , Romi FR . Matrix metalloproteinases in myasthenia gravis. Eur Neurol. (2011) ;65: (1):53–8. |
[223] | Luckman SP , Gilhus NE , Romi F . Matrix metalloproteinase-3 in myasthenia gravis compared to other neurological disorders and healthy controls. Autoimmune Dis. (2011) ;2011: :151258. |
[224] | Romi FR , Gilhus NE , Luckman SP . Serum matrix metalloproteinase-3 levels are elevated in myasthenia gravis. J Neuroimmunol. (2008) ;195: (1-2):96–9. |
Figures and Tables
Fig.1
Schematic representation of MMPs activation in inflammatory and wound conditions. Panel A: Overview of MMPs production, activation and their inhibition by TIMPs. Panel B: Modulation of MMPs/TIMPs production by reactive oxygen species, other proteases and by cytokines and growth factors released in inflammatory and wound conditions. These regulations are reported in the literature and may depend on cell types and tissue microenvironment. Pro-MMPs are the inactive MMPs forms; MT-MMPs membrane-type MMP; TIMPs, Tissue Inhibitor of Metalloproteinases. Thin curved arrow indicates MMP activation and ⊣ MMPs inhibition.
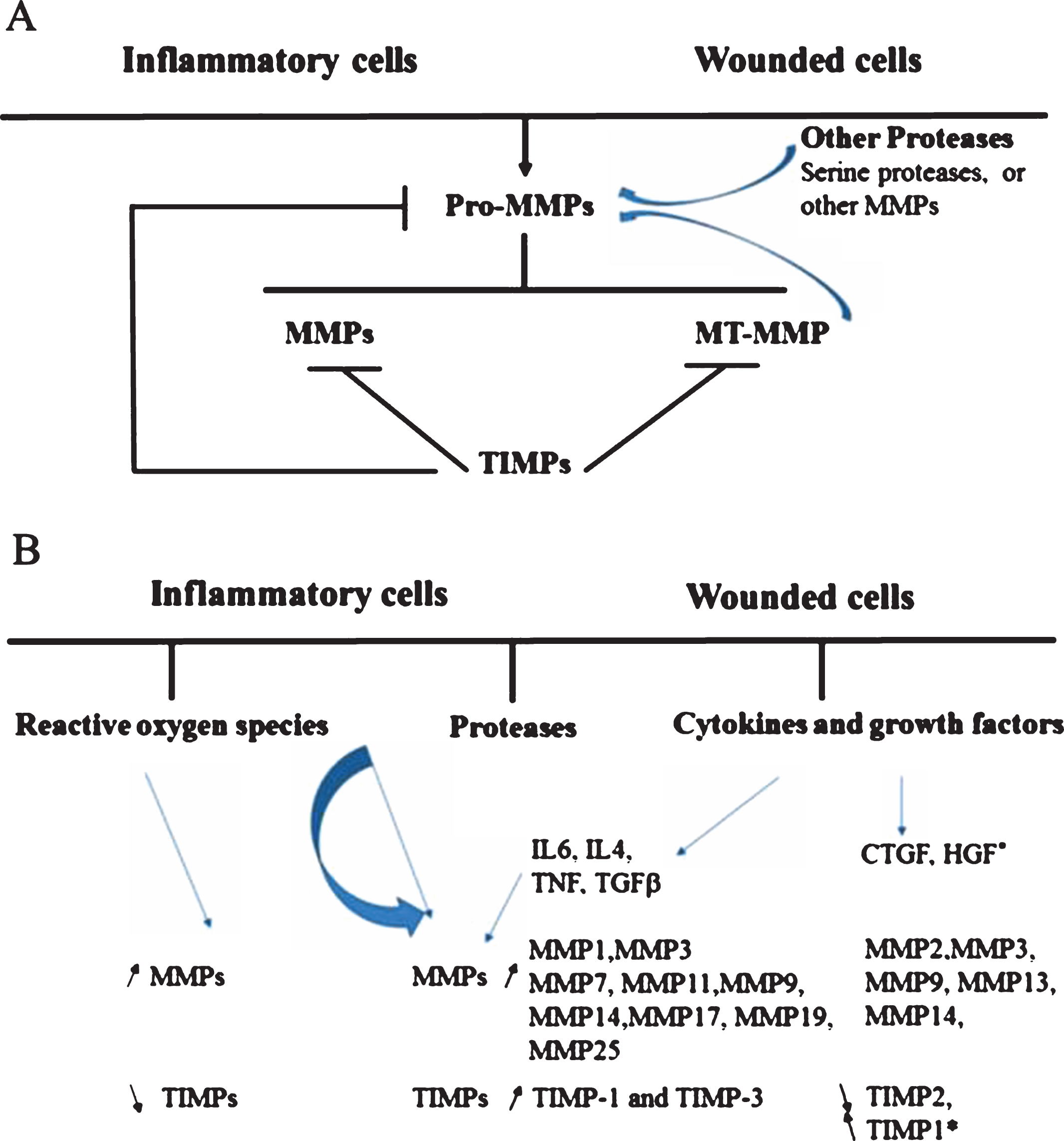
Fig.2
Schematic representation showing the involvement of inflammatory cells in the regulation of myofibroblast activation in wound healing and fibrosis. Tissue injury triggers a cascade of interconnected steps to restore tissue homeostasis. The initial activation of coagulation pathway is followed by an acute inflammation and activation of innate immune mediators including macrophages, neutrophils and dendritic cells. Cytokines liberated by injured and inflammatory cells subsequently regulate the activation of adaptive immune response. Inflammatory cells and immune mediators attempt to eliminate noxious stimuli and activate fibroblasts into myofibroblasts that orchestrate angiogenesis and regulation of ECM components. Failure to eliminate factors causing the injury perpetuates wound healing and inflammation ultimately resulting in fibrosis.
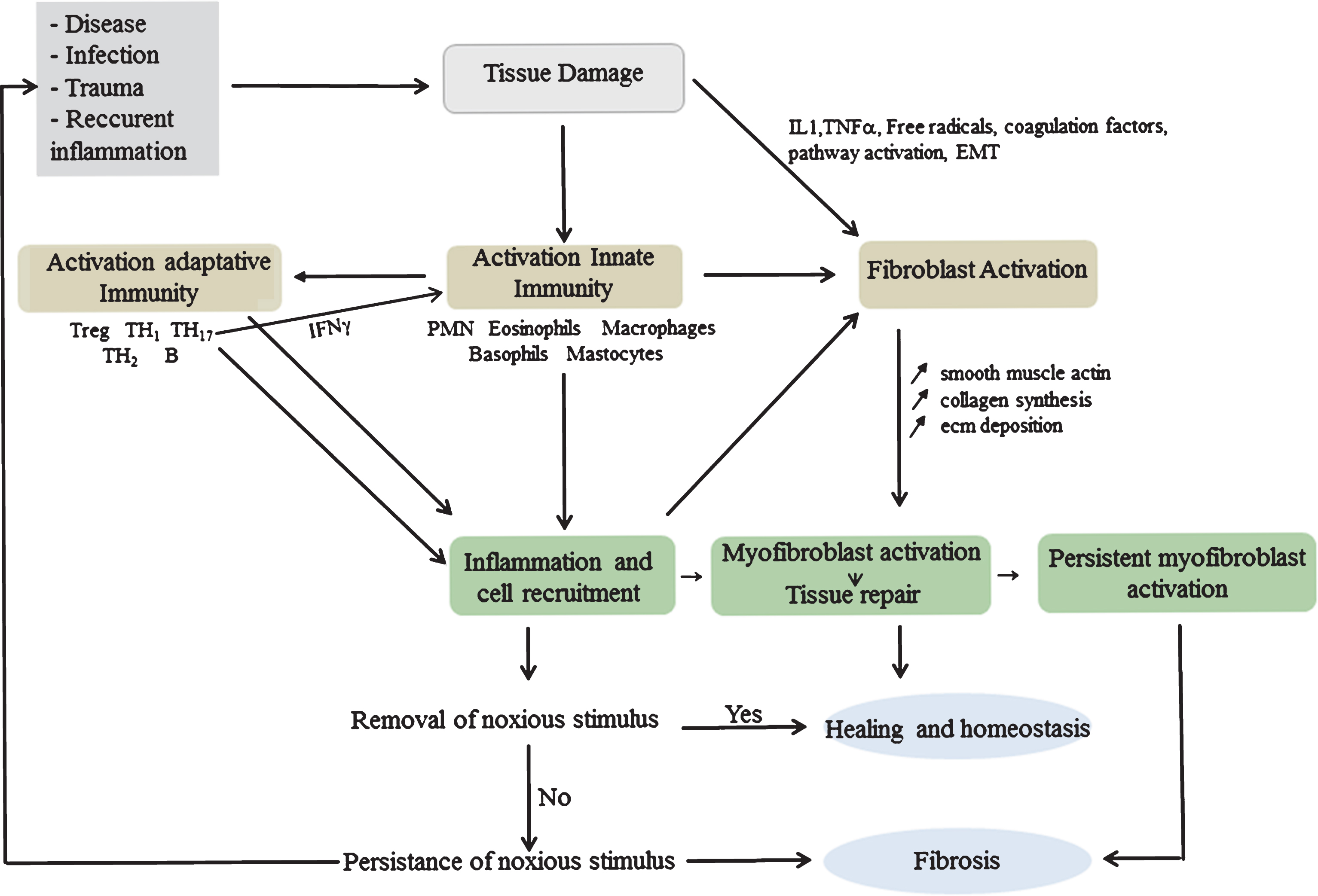
Table 1
Matrix Metalloproteinases: their alternative names, location within tissues and chromosomes and source of activation from a pro-form. MMP: Matrix Metalloproteinase; MT(n)-MMP: Membrane Type (n = 1,2,3,4,5,6)- MMP; RASI: Rheumatoid Arthritis Sign of Inflammation, XMMP: Xenopus MMP; CA-MMP: Cysteine Array-MMP; Pump-1: plant uncoupling mitochondrial protein 1
| Enzyme | MMP | Location | Activation | Chromosomal Location |
| Collagenases | ||||
| Interstitial collagenase; collagenase 1 | MMP-1 | Secreted | MMP-3 | 11q22-q23 |
| Neutrophil collagenase; collagenase 2 | MMP-8 | Secreted | MMP-3 | 11q21-q22 |
| Collagenase 3 | MMP-13 | Secreted | MMP-14/TIMP-2/MMP-3 | 11q22.3 |
| Collagenase 4 (Xenopus) | MMP-18 | Secreted | Unknown | NA |
| Gelatinases | ||||
| Gelatinase A | MMP-2 | Secreted | MMP-14/TIMP-2 | 16q13 |
| Gelatinase B | MMP-9 | Secreted | MMP-3/MMP-13 | 20q11.2-q13.1 |
| Matrilysin | ||||
| Matrilysin 1; Pump-1 | MMP-7 | Secreted | MMP-3 | 11q21-q22 |
| MMP-26 | Secreted | Unknown | 11p15 | |
| Membrane-type MMPs | ||||
| MT1-MMP | MMP-14 | Membrane | Furin | 14q11-q12 |
| MT2-MMP | MMP-15 | Membrane | Furin | 15q13-q21 |
| MT3-MMP | MMP-16 | Membrane | Furin | 8q21 |
| MT4-MMP, TACE | MMP-17 | Membrane | Furin | 12q24.3 |
| MT5-MMP | MMP-24 | Membrane | Furin | 20q11.2 |
| MT6-MMP | MMP-25 | Membrane | Furin | 16p13.3 |
| Metalloelastase | ||||
| Macrophage Elastase | MMP-12 | Secreted | Unknown | 11q22.2-q22.3 |
| Stromelysin | ||||
| Stromelysin 1 | MMP-3 | Secreted | MMP-14/TIMP-2 | 11q23 |
| Stromelysin 2 | MMP-10 | Secreted | Unknown | 11q22.3-q23 |
| Stromelysin 3 | MMP-11 | Secreted | Furin | 22q11.2 |
| Others | ||||
| RASI | MMP-19 | Secreted | Unknown | 12q14 |
| Enamelysin | MMP-20 | Secreted | Unknown | 11q22.3 |
| XMMP (Xenopus) | MMP-21 | Secreted | Unknown | ND |
| CA-MMP | MMP-23 | Secreted | Furin | 1p36.3 |
| CMMP (Gallus) | MMP-27 | Secreted | Unknown | 11q24 |
| Epilysin | MMP-28 | Secreted | Furin | 17q21.1 |
Table 2
MMPs and TIMPs expression in neuromuscular diseases and their animal models. ALS TgSOD1 (G93A): Transgenic mouse model of Amyotrophic Lateral Sclerosis carrying mutant Super Oxide Dismuthase gene, CSF: Cerebro Spinal Fluid; CXMD: canine x-linked muscular dystrophy; mdx: x-linked muscular dystrophy; ND: Not determined, Neu1-/Neu1- mice: Neuraminidase deficient mice
| Inflammatory Myopathies | MMP expressed | MMP localisation | TIMPs expressed | Circulating | References |
| – Inclusion body myositis | MMP-2, MMP-9  | MMP-9 in atrophic fibers &inflammatory cells | MMPs unchanged | [22, 25] | |
| MMP-1 and MMP-9 (mRNA)++ | TIMPs unchanged | ||||
| – Sporadic inclusion body myositis | MMP-2 and MMP-9 | MMP-2 in rimmed vacuoles | MMPs unchanged | [18] | |
| MMP-9 cytotoxic T cells | TIMPs unchanged | ||||
| – Polymyositis | MMP-2 and-9 MHC class1 + fibers | MMPs unchanged | [18] | ||
| MMP-7 in inflammatory cells | TIMPs unchanged | [22] | |||
| MMP-1 around sarcolemma &in fibroblasts | |||||
| MMP-1 and MMP-9 (mRNA)+++ | |||||
| – Dermatomyositis | MMP-2 | MMP-9 in perifascicular atrophic fibers | MMPs unchanged | [22, 25] | |
| MMP-1 around sarcolemma &in fibroblasts | TIMPs unchanged | ||||
| MMP-1 and MMP-9 (mRNA)+++ | |||||
| Muscular Dystrophies | |||||
| Human | |||||
| – Duchenne muscular dystrophy | MMP-1, MMP-2, MMP-9 | MMP-1 around individual or groups of muscle fibers | TIMP-1 &TIMP-2 | MMP-9 and TIMP-1 | [27, 33, 111, 214] |
| MMP-2 surface of few myofibers and around blood vessels | |||||
| MMP-9 in blood vessels, mononuclear cells and cytoplasm of regenerating fibers | |||||
| – Emery-Dreifuss muscular dystrophy | MMP-2, MMP-9, MMP-14 | MMP-2, MMP-9, MMP-14, TIMP-1 | [215] | ||
| Animal models | |||||
| – mdx | MMP-2, MMP-9, MMP-13 | MMP activity in ECM around myofibers and in the sarcoplasm MMP-9 mRNA in inflammatory and satellite cells | TIMP1 (mRNA) | MMP-9 | [7, 203, 216] |
| TIMP2,-3 (mRNA) | |||||
| MMP3,-8,-9,-10,-12,-14,-15 (mRNA) | [32, 61] | ||||
MMP11,  | |||||
| – CXMD | MMP-2, MMP-9, MMP-14 | [20] | |||
| Neurodegenerative Muscular disorders | |||||
| Human | |||||
| – Amyotrophic lateral sclerosis (affected patients) | MMP-2,-7,-9, MMP-14 | MMP + in normal and atrophic fibers | MMP-1, -2, -9, -14 | [105, 217, 218] | |
| – Spinal muscular atrophy | MMP-7, -9 | MMP + in normal and atrophic fibers | MMP-9 | [102] | |
| [103] | |||||
| – Chronic axonal neuropathies | MMP-9 | MMP + in normal and atrophic fibers | TIMP-1 (CSF) | [105] | |
| – Guillain Barre | MMP-9 | [219] | |||
| TIMP-1 | |||||
| Animal models | |||||
| – Neu1-/Neu1- mice | MMP-2,MMP-9 | ND | [220] | ||
| – ALS TgSOD1(G93A) low copy number and low progression | MMP-2, MMP-9 | MMP-2,-9 activity associate with disease onset | [221] | ||
| – ALS TgSOD1(G93A) high copy number and rapid progression | MMP-2, MMP-9 | MMP-2,-9 activity associate with disease onset | [221] | ||
| Autoimmune Myopathies | |||||
| – Myasthenia Gravis: Ocular and Generalized subgroup (17% seropositive and 10% seronegative) | MMP-2, MMP-3, MMP-9 | MMP-2,-3,-9 | [217, 222–224] |


