
An Antibody to Detect Alanine-Expanded PABPN1: A New Tool to Study Oculopharyngeal Muscular Dystrophy
Abstract
Background:
Oculopharyngeal muscular dystrophy (OPMD), a late onset disorder affecting specific skeletal muscles, is caused by a (GCG)n expansion mutation in the gene encoding the mRNA processing protein, polyadenylate binding protein nuclear 1 (PABPN1). The expansion in PABPN1 leads to an increase in a stretch of N-terminal alanine residues in the PABPN1 protein from the normal 10 to 12-18. Given this modest change, detection of mutant protein has not been possible without the use of tagged constructs.
Objective:
We sought to generate a polyclonal antibody that recognizes alanine-expanded but not wild type PABPN1 with the goal of making possible analysis of expression and localization of alanine-expanded PABPN1.
Methods:
We immunized rabbits with a GST-tagged alanine peptide and tested the resulting serum against alanine-expanded PABPN1 expressed in cell culture as well as in animal models of OPMD.
Results:
The resulting α-alanine antibody detected PABPN1 proteins that contained 14 or more alanine residues. Importantly, the α-alanine antibody could be used to detect alanine-expanded PABPN1 in muscles from either a mouse or Drosophila model of OPMD.
Conclusions:
This α-alanine antibody provides a new tool that will allow for more in-depth study of how alanine expansion affects aggregation, localization, and steady-state levels of alanine-expanded PABPN1 levels in vivo, providing new insight into the molecular mechanisms underlying OPMD.
INTRODUCTION
Oculopharyngeal muscular dystrophy (OPMD) is a late onset disorder that affects specific skeletal muscles including the levator palpebrae superioris, pharyngeal muscles, and proximal limb muscles [1]. Patients typically develop symptoms in the fifth decade that include ptosis, dysphagia, and loss of mobility [1]. OPMD decreases quality of life for affected individuals as they are prone to choking, aspiration pneumonia, and may be confined to a wheelchair in later stages of disease [1]. Currently, no pharmacologic treatments are available for OPMD.
The majority of individuals with OPMD harbor an autosomal dominant (GCG)n expansion mutation in the gene encoding the polyadenylate-binding proteinnuclear 1 (PABPN1) [2]. This mutation causes the PABPN1 protein, which normally contains ten N-terminal alanine residues immediately following the initial methionine, to expand to 12–18 alanine residues [2, 3]. The alanine expansions in PABPN1 that cause OPMD are rather modest and cause little change in the molecular weight or net charge of PABPN1 so distinguishing the mutant protein from the wild type one has not been possible [4]. Therefore, one of the biggest challenges for study of alanine-expanded PABPN1 is the inability to specifically detect the mutant protein. Previous in vitro studies have relied on tagged fusion proteins for detection of mutant PABPN1 [5–7]. In vivo studies in flies and mice that have focused on localization of PABPN1 have used antibodies to the wild type protein only and thus have not been able to assess localization of the alanine-expanded protein specifically [8, 9]. To provide more detailed analysis of PABPN1, new tools are required that can distinguish wild type from alanine-expanded PABPN1.
A number of diseases are caused by amino acid expansions of varying lengths [10, 11]. Such diseases are not limited to the modest increase found in OPMD but are often due to large expansions. For example, polyglutamine expansions in huntingtin protein cause Huntington’s disease [12]. As polyglutamine expansions in the huntingtin protein number in the tens to hundreds of additional residues, the expanded protein can be readily distinguished from wild type by SDS-PAGE and immunoblotting [12]. However, polyglutamine-specific antibodies have been developed as potential therapeutics against aggregated huntingtin [13, 14]. The anti-polyglutamine antibodies were produced by immunizing animals with tagged polyglutamine fusion proteins and could specifically detect expanded huntingtin [13]. Unlike huntingtin, the modest alanine expansion in PABPN1 thatcauses OPMD does not result in a major size or charge difference. Thus, creating a reagent to specifically detect alanine-expanded PABPN1 presents a challenge.
In this study, we sought to generate antibodies against alanine-expanded PABPN1. We applied a similar strategy to that previously employed to raise anti-glutamine antibodies [13, 14] and used a GST-tagged alanine peptide as the antigen. The resulting α-alanine antibody detects alanine-expanded PABPN1 overexpressed in cultured cells as well as in animal models of OPMD. This newly developed antibody to alanine-expanded PABPN1 provides a new tool for the study of OPMD.
MATERIALS AND METHODS
Plasmids, cloning, constructs
An oligonucleotide encoding a 20 Alanine peptide was cloned into pGEX4T-3 (Amersham Biosciences) to create the construct containing glutathione-S-transferase containing 20 carboxy-terminal alanine residues (GST-A20). Constructs were confirmed by dideoxynucleotide sequencing using the pGEX-F primer (Amersham Biosciences). The PABPN1 A10 (WT) and PABPN1 A13-17 constructs were cloned into the pCDH expression vector (System Biosciences, Mountain View, CA) containing a 6x-His tag (Table 1). Constructs encoding RUNX2 WT and RUNX2 A27 were cloned into the expression vector pTL1-HA, which contains an HA tag [15].
Recombinant protein expression and purification
GST-A20 was expressed from pGEX4T-3 in Escherichia coli BL21(DE3) as described [16]. Recombinant GST-A20 was purified from soluble cell extracts with glutathione-sepharose as described [16]. Expression and purification of recombinant GST-A20 was confirmed by SDS-PAGE followed by Coomassie staining. For expression of WT and alanine-expanded PABPN1 and RUNX2, HEK 293 cells were transfected using lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Transfected cells were grown for 48 hours at 37°C after which total protein was extracted using RIPA-2 cell lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) [17].
Animals
Two New Zealand white rabbits were used for antibody production. As a mouse model of OPMD, two-month-old male wild type, A10.1, and A17.1 transgenic mice (kindly provided by Dr. David Rubinsztein) were used for immunoblots and immunofluorescence studies [8]. Mice were genotyped using the following primers: F: 5′-GAGCGACATCATGGTATTCCC-3′; R: 5′-AGGACTGACACGTGCTACGA-3′ to detect the A10.1 or A17.1 Pabpn1 allele. Experiments involving animals were performed in accordance with approved guidelines and ethical approval from Emory University’s Institutional Animal Care and UseCommittee.
All fly stocks and crosses were maintained under standard conditions. UAS-PABPN1 and UAS-PABPN1-17ala flies were a gift from the Simonelig laboratory [9]. The Mhc-Gal4 driver was used to drive transgene expression in muscles.
SDS-PAGE and immunoblot
Sera, cell lysates, and homogenized tissue were prepared and analyzed by SDS-PAGE as described [18] using Any kDtrademark Mini-PROTEAN ® TGX gels (Bio-Rad Laboratories, Hercules, CA). Unless otherwise noted, 50 μg cell lysate containing recombinant proteins or 20 μg of mouse muscle extracts were loaded onto gels. For flies, 10 adult flies of each genotype were homogenized in sample buffer (50 mM Tris HCl, 100 mM DTT, 2% SDS, 0.1% Bromophenol Blue, 10% Glycerol) and equal amounts of total protein were loaded onto gels. Resolved proteins were transferred to nitrocellulose paper and blocked with 10% nonfat milk in Tris-buffered saline pH 7.4, with 0.1% Tween-20 (TBS-T). Blots were probed overnight at 4°C with primary antibody diluted 1:5000 in TBS-T with 5% nonfat milk unless otherwise noted. Primary antibodies used were rabbit polyclonal α-alanine 20 diluted (1:1000), rabbit polyclonal α-PABPN1 [19], rabbit polyclonal α-His tag (Cell Signaling Technology, Beverley, MA), mouse monoclonal α-HSP90 (Santa Cruz Biotechnology, Dallas, TX), mouse monoclonal α-HA(Covance, Princeton, NJ), mouse monoclonal α- αTubulin (Sigma Aldrich, St. Louis, MO), or mouse monoclonal α-lamin D (1:250, ADL84.12, Developmental Studies Hybridoma Bank, Iowa City, Iowa). Blots were washed in TBS-T and probed for 1–3 hours at room temperature with secondary antibodies diluted 1:5000 in TBS-T. Secondary antibodies used were horseradish peroxidase (HRP) conjugated donkey α-rabbit IgG or HRP conjugated goat α-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA).
Antibody production
Polyclonal antibodies were produced in rabbits as described [20]. Rabbits were given one primary immunization with 100 μg purified GST-A20 in phosphate buffered saline (PBS) with complete Freund’s adjuvant. Three booster immunizations containing 50 μg GST-Ala20 in PBS with incomplete Freund’s adjuvant were administered at 21, 42, and 63 days after initial immunization. Blood was collected via central ear artery, allowed to clot for one hour at 37°C, and serum was removed by centrifugation. Small volume (10 ml) blood collection was performed prior to immunization and at days 32 and 53 after immunization. Large volume (50 ml) blood collection and euthanasia was performed 74 days after immunization.
Immunostaining
Mouse tibialis anterior muscles were dissected and immediately flash frozen in TFM Tissue Freezing Medium (Triangle Biomedical Sciences, Durham, NC). Tissues were cryosectioned at 14 μm thickness every 140 μm on a Leica CM1850 cryostat and fixed with 4% paraformaldehyde for 5 minutes. All reactions were performed at room temperature unless otherwise noted. Antigen retrieval was performed in a pressure cooker using sodium citrate buffer as described [21]. After antigen retrieval, tissue sections were treated with mouse IgG blocking reagent mouse kit (Vector Laboratories, Burlingame, CA) and incubated overnight at 4°C in primary antibody diluted 1:200 in blocking buffer. In all cases, rabbit pre-immune serum was used as a negative control. Slides probed with α-WT PABPN1 primary antibody were washed and treated with FITC-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted 1:200 in PBS with 0.2% Triton X-100 (PBS-Tx). Tyramide signal amplification kits (Invitrogen, Carlsbad, CA) were used for slides treated with anti-Ala primary antibody according to the manufacturer’s protocol. Briefly, slides were incubated with biotin-conjugated donkey anti-rabbit IgG diluted 1:200 in PBS-Tx, treated with Tris-NaCl blocking buffer (TNB), and incubated with streptavidin-HRP diluted 1:100 in TNB followed by fluorescein tyramide reagent diluted 1:300 in manufacturer provided diluent. To visualize nuclei, all sections were labeled with 4′, 6-diamidino-2-phenylindole (DAPI) to stain DNA prior to visualization by fluorescence microscopy.
RESULTS
Generation of polyclonal antibody in rabbit
Developing an antibody to detect mutant PABPN1 is a major challenge as OPMD results from a very modest expansion from 10 to 12–18 alanine residues in the PABPN1 protein [3, 4]. Given this small increase in the number of alanine residues in PABPN1, we chose to raise an antibody against an alanine peptide rather than the alanine-expanded PABPN1 protein. To maximize the chances of detecting alanine expansions of various lengths and to avoid the production of antibodies to the alanine-GST tag interface, a 20-alanine residue peptide was used. As described in Materials and Methods, we generated an expression construct encoding the Ala 20 peptide attached to an N-terminal GST tag (GST-A20) and expressed it in E. coli. Bacterial lysates were separated by SDS-PAGE and the produced protein was detected by Coomassie staining (Fig. 1A). The GST-A20 was then purified using glutathione sepharose for use in immunization (Fig. 1B).
Two rabbits, designated rabbit 1 and rabbit 2, were used for antibody generation. To ensure that rabbits had no endogenous antibodies to alanine peptides, sera from initial bleeds of pre-immunized animals were tested by immunoblot against recombinant PABPN1 expanded to 17 alanine residues, as A17-PABPN1 is most commonly used for in vitro and in vivo studies of mutant PABPN1 [8, 9, 22–25]. Neither serum sample showed any reactivity with A17-PABPN1 indicating that neither animal had previously mounted an immune response to alanine peptides (not shown). As described in Materials and Methods, Rabbits 1 and 2 were given one primary and three booster immunizations of GST-A20 plus adjuvant. To determine whether animals mounted an immune response to the GST-A20 fusion protein, blood samples were taken subsequent to the first boost. Sera from the first bleed were tested by immunoblot for reaction with recombinant GST-A20. At this initial test, both animals showed robust immune response to GST-A20. After the final bleed, sera from both animals were tested against equal loads of lysates from HEK 293 cells transfected with empty vector, wild type (A10) PABPN1, or A17-PABPN1. One animal (rabbit 2) showed specific reactivity with A17-PABPN1 with no background signal in empty vector or A10-PABPN1 samples (Fig. 1C). Therefore, serum from rabbit 2 was used as the polyclonal α-alanine antibody for all subsequent analyses.
Anti-alanine antibody detects alanine repeats
Multiple approaches were used to test the specificity of the polyclonal α-alanine antibody. The majority of OPMD-associated expansions of PABPN1 range from 12–17 alanine repeats [2] though patients with A18-PABPN1 have been recently identified [2, 4]. To test immunoreactivity to alanine-expanded PABPN1, we transfected HEK 293 cells with plasmids encoding His-tagged PABPN1 constructs with increasing numbers of N-terminal alanine residues and performed immunoblots to detect wild type or A13-17 PABPN1 with the α-alanine antibody. Robust reaction was detected with A16- and A17-PABPN1 while milder reactivity was observed for A14- and A15- PABPN1 (Fig. 2A). No bands were detected in lanes containing lysates from cells expressing wild type (A10) or A13-PABPN1.
Multiple polyalanine proteins exist in mammalian cells, many of which can be subject to alanine expansions that cause disease [11]. To further analyze the specificity of the α-alanine antibody, we tested another polyalanine protein, RUNX2. The transcription factor RUNX2, which is required for normal skeletal development [26], contains a stretch of 17 alanine residues. Expansion mutations resulting in A27-RUNX2 are associated with the congenital disease cleidocranial dysplasia [27]. To assess whether the α-alanine antibody detects wild type or alanine-expanded RUNX2, HEK 293 cells were transfected with plasmids encoding HA-tagged wild type (A17) and expanded (A27) RUNX2. Immunoblotting with an α-HA antibody confirmed that both proteins are expressed in HEK 293 cells (Fig. 2B). When probed with the α-alanine antibody, both A17- and A27-RUNX2 proteins were detected (Fig. 2B), indicating that the α-alanine antibody can detect alanine stretches in multiple proteins.
Plasmid-mediated expression of PABPN1 in HEK 293 cells results in much higher PABPN1 levels than the level of endogenous PABPN1 in these cells (Fig. 2A). To assess the sensitivity of the α-alanine antibody, lysates from HEK 293 cells transfected with plasmid encoding A17-PABPN1 were serially diluted until PABPN1 levels, as detected by α-PABPN1 antibody that detects WT and A17-PABPN1 [19], were similar to expression of the endogenous protein in HEK 293 cells. At all dilutions tested, A17-PABPN1 could be detected with the α-alanine antibody (Fig. 3). This result indicates that the α-alanine antibody is sufficiently sensitive to detect endogenous levels of A17-PABPN1.
Use of α-alanine antibody in animal models of oculopharyngeal muscular dystrophy
To test the utility of the α-alanine antibody, we sought to detect alanine-expanded PABPN1 in animal models of OPMD [8, 9]. We first analyzed a transgenic mouse model of OPMD. These mice contain transgenic wild type (GCN) 10-PABPN1 (A10.1) or expanded (GCN) 17-PABPN1 (A17.1) alleles under the control of the human skeletal actin promoter [8], which allows for constitutive transgene expression in skeletal muscle tissue. As detected using an antibody to wild type PABPN1, transgenic A10.1 and A17.1 animals express A10-PABPN1 or A17-PABPN1 at levels 10-fold or 30-fold higher, respectively, than endogenous PABPN1 [8]. We analyzed whole muscle lysates from A10.1 and A17.1 mice by immunoblot using the α-alanine antibody. As shown in Fig. 4A, an immunoreactive band corresponding to the size of A17-PABPN1 was detected specifically in muscle tissue from A17.1 animals. Immunoblots were also probed with a polyclonal α-PABPN1 antibody to confirm the presence of PABPN1 in both A10.1 and A17.1 lysates and detection of HSP-90 was used as a loading control.
To determine whether the α-alanine antibody can recognize A17-PABPN1 in tissue sections, we immunostained muscle sections from A10.1 and A17.1 mice with either α-alanine or α-PABPN1 antibodies [19]. A17-PABPN1 was detected in sections from A17.1 mice as a fluorescent signal that localized near the DAPI-stained chromatin in the nuclei (Fig. 4B). We observed no signal in muscle sections from A10.1 mice probed with α-alanine antibody. However, PABPN1 was detected in A10.1 sections probed with α-PABPN1 antibody (Fig. 4B). These results indicate that the α-alanine antibody can be used to detect A17-PABPN1 in tissues from the most commonly used transgenic mouse model of OPMD.
To further demonstrate the utility of the α-alanine antibody in animal models, we tested it in a Drosophila model of OPMD. The OPMD fly model contains transgenic alleles encoding mammalian A10-PABPN1 or A17-PABPN1 under the control of a muscle-specific, inducible Mhc-Gal4 promoter [9]. As shown in Fig. 4C, these flies express comparable levels of transgenic PABPN1; however the α-alanine antibody specifically detected A17-PABPN1. Immunoblots were also probed with α-lamin D to ensure equal loading. This result demonstrates that the α-alanine antibody can be used to detect A17-PABPN1 in multiple animal models of OPMD.
DISCUSSION
One of the major deficits in the study of OPMD has been the lack of a reagent to specifically detect alanine-expanded PABPN1 protein. Generating such a reagent has been an ongoing challenge as the modest expansions associated with OPMD cause very little change in the size, charge, or biophysical properties of the PABPN1 protein. We generated a polyclonal antibody to an alanine peptide, which is a strategy similar to that used by other groups to generate antibodies to glutamine-expanded huntingtin protein [13, 14]. The α-alanine antibody detects polyalanine stretches in A14-A17 PABPN1 as well as another polyalanine protein, RUNX2. Most importantly, this valuable reagent can distinguish alanine-expanded PABPN1 from the wild type protein in muscle tissue from both fly and mouse models of OPMD.
This newly developed α-alanine antibody has the potential for multiple applications in multiple models. The most commonly studied OPMD models are cell culture, fly, and mouse models that overexpress 17 alanine-expanded PABPN1 [8, 9, 22–24]. Multiple studies have revealed that PABPN1 is present in nuclear aggregates in these models as well as OPMD patients [8, 22, 23, 28]. Other studies have demonstrated that wild type PABPN1 localizes to sub-nuclear structures called nuclear speckles and that overexpression of alanine-expanded PABPN1 affects PABPN1 localization to nuclear speckles [5, 6]. While in vitro studies using tagged PABPN1 constructs have shown that both wild type and alanine-expanded PABPN1 accumulate in nuclear aggregates, no studies have demonstrated the half-life or sub-nuclear localization of endogenous mutant protein specifically. Furthermore, how localization and levels of endogenous alanine-expanded PABPN1 are affected relative to wild type PABPN1 protein is unknown. The α-alanine antibody described here provides an excellent tool to study mutant PABPN1 stability and localization independent of wild type PABPN1 in addition to allowing for confirmation of alanine-expanded PABPN1 expression. Perhaps when genetic models that express nativelevels of mutant PABPN1 become available, the α-alanine antibody can be used to glean more information about the in vivo characteristics of mutant PABPN1.
The α-alanine antibody is a valuable new tool in analysis of A17-PABPN1 in cell culture and animal models of OPMD. However, it is limited in that it cannot be used as a diagnostic tool or as a means to study tissue samples from the majority of OPMD patients, who harbor a mutation encoding A13-expanded PABPN1 [4]. In the majority of OPMD cases, there is little correlation between the length of alanine expansion and disease severity [1] suggesting that the molecular pathology across all alanine expansions of PABPN1 is conserved. Thus, samples from rare patients expressing A14-A18 PABPN1 could be used for localization and stability studies. Additionally, as sequencing and diagnostic methods improve, patients with larger alanine expansions may be identified in the future. For example, patients with an expansion to 18 alanine residues in PABPN1 were recently described [3]. However, as a tool to specifically detect mutant PABPN1 in animal models or in limited patient tissues, the α-alanine antibody has the potential to provide greater understanding of the mechanisms that underlie OPMD pathology.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
Funding provided by National Institutes of Health grants (NS084870, AR061987) to GKP and AHC and by a Muscular Dystrophy Association Development grants MDA157523 to LHA and MDA255856 to AB.
REFERENCES
1 | Trollet C, Gidaro T, Klein P, Perie S, Butler-Browne G, Lacau St Guily J (1993) Oculopharyngeal Muscular Dystrophy Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH Seattle (WA) GeneReviews(R) |
2 | Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM (1998) Short GCG expansions in the PABP2 gene causeoculopharyngeal muscular dystrophy Nature Genetics 18: 2 164 167 |
3 | Jouan L, Rocheford D, Szuto A, Carney E, David K, Dion PA (2014) An 18 alanine repeat in a severe form of oculopharyngeal musculardystrophy The Canadian Journal of Neurological Sciences LeJournal Canadien Des Sciences Neurologiques 41: 4 508 511 |
4 | Banerjee A, Apponi LH, Pavlath GK, Corbett AH (2013) PABPN Molecularfunction and muscle disease The FEBS Journal 280: 17 4230 4250 |
5 | Bengoechea R, Tapia O, Casafont I, Berciano J, Lafarga M, Berciano MT (2012) Nuclear speckles are involved in nuclear aggregation of PABPN1 and in the pathophysiology of oculopharyngeal muscular dystrophy Neurobiology of Disease 46: 1 118 129 |
6 | Klein AF, Ebihara M, Alexander C, Dicaire MJ, Sasseville AM, Langelier Y (2008) PABPN1 polyalanine tract deletionandlong expansions modify its aggregation pattern andexpression Experimental Cell Research 314: 8 1652 1666 |
7 | Wang Q, Bag J (2008) Induction of expression and co-localization of heatshock polypeptides with the polyalanine expansion mutant of poly(A)-binding protein N1 after chemical stress Biochemical and Biophysical Research Communications 370: 1 11 15 |
8 | Davies JE, Wang L, Garcia-Oroz L, Cook LJ, Vacher C, O’Donovan DG (2005) Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice Nature Medicine 11: 6 672 677 |
9 | Chartier A, Benoit B, Simonelig M (2006) A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1 The EMBO Journal 25: 10 2253 2262 |
10 | Cummings CJ, Zoghbi HY (2000) Fourteen and counting: Unraveling trinucleotide repeat diseases Human Molecular Genetics 9: 6 909 916 |
11 | Albrecht A, Mundlos S (2005) The other trinucleotide repeat: Polyalanine expansion disorders Current Opinion in Genetics & Development 15: 3 285 293 |
12 | Ross CA, Wood JD, Schilling G, Peters MF, Nucifora FCJr, Cooper JK (1999) Polyglutamine pathogenesis. Philosophical transactionsof the Royal Society of London Series B Biological Sciences 354: 1386 1005 1011 |
13 | Ko J, Ou S, Patterson PH (2001) New anti-huntingtin monoclonalantibodies: Implications for huntingtin conformation and itsbinding proteins Brain Research Bulletin 56: 3-4 319 329 |
14 | Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL (2009) Monoclonal antibodies recognize distinctconformational epitopes formed by polyglutamine in a mutanthuntingtin fragment The Journal of Biological Chemistry 284: 32 21647 21658 |
15 | Albrecht AN, Kornak U, Boddrich A, Suring K, Robinson PN, StiegeAC (2004) A molecular pathogenesis for transcription factorassociated poly-alanine tract expansions Human Molecular Genetics 13: 20 2351 2359 |
16 | Apponi LH, Kelly SM, Harreman MT, Lehner AN, Corbett AH, Valentini SR (2007) An interaction between two RNA binding proteins,Nab2 and Pub1, links mRNA processing/export and mRNA stability Molecular and Cellular Biology 27: 18 6569 6579 |
17 | Sefton BM, Hunter T, Beemon K (1980) Temperature-sensitivetransformation by Rous sarcoma virus and temperature-sensitiveprotein kinase activity Journal of Virology 33: 1 220 229 |
18 | Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4 Nature 227: 5259 680 685 |
19 | Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, Pavlath GK (2010) Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis Human Molecular Genetics 19: 6 1058 1065 |
20 | Hendriksen CaH, J (2003) Production of polyclonal and monoclonalantibodies Hau JaVH Handbook of Laboratory AnimalScience: Essential Principles and Practices. I 391 411 Boca Raton, FL CRC Press |
21 | Shi SR, Chaiwun B, Young L, Cote RJ, Taylor CR (1993) Antigen retrievaltechnique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor informalin-fixed paraffin sections The journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society 41: 11 1599 15604 |
22 | Davies JE, Sarkar S, Rubinsztein DC (2006) Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy Human Molecular Genetics 15: 1 23 31 |
23 | Trollet C, Anvar SY, Venema A, Hargreaves IP, Foster K, Vignaud A (2010) Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscularatrophy restricted to fast glycolytic fibres Human Molecular Genetics 19: 11 2191 2207 |
24 | Wang Q, Bag J (2006) Ectopic expression of a polyalanine expansion mutant of poly(A)-binding protein N1 in muscle cells in culture inhibits myogenesis Biochemical and Biophysical Research Communications 340: 3 815 822 |
25 | Fan X, Dion P, Laganiere J, Brais B, Rouleau GA (2001) Oligomerizationof polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death Human Molecular Genetics 10: 21 2341 2351 |
26 | Komori T (2002) Runx2, a multifunctional transcription factor inskeletal development Journal of Cellular Biochemistry 87: 1 1 8 |
27 | Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia Cell 89: 5 773 779 |
28 | Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E (2000) Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A)RNA Human Molecular Genetics 9: 15 2321 2328 |
Figures and Tables
Fig.1
Production and purification of GST-A20 construct and generation of polyclonal antibody. A) Coomassie stained gel of bacterial lysates showing presence of the 26 kDa recombinant GST-A20 construct after inducing expression with IPTG for the indicated number of hours. B) Affinity purification of the GST-A20 construct by fractionation over glutathione sepharose resin and subsequent elution with reduced glutathione. The 26 kDa construct was detected in flow-through (FT) and the wash (W) and the majority was eluted in Fractions 4 and 5. C) Immunoblot of 50 μg of lysates from HEK 293 cells transfected with empty plasmid (Ctrl) or plasmid encoding wild type (A10) and alanine-expanded (A17) PABPN1 probed with sera from immunized rabbits. PABPN1 protein was detected as a 50 kDa band as indicated [19].
![Production and purification of GST-A20 construct and generation of polyclonal antibody. A) Coomassie stained gel of bacterial lysates showing presence of the 26 kDa recombinant GST-A20 construct after inducing expression with IPTG for the indicated number of hours. B) Affinity purification of the GST-A20 construct by fractionation over glutathione sepharose resin and subsequent elution with reduced glutathione. The 26 kDa construct was detected in flow-through (FT) and the wash (W) and the majority was eluted in Fractions 4 and 5. C) Immunoblot of 50 μg of lysates from HEK 293 cells transfected with empty plasmid (Ctrl) or plasmid encoding wild type (A10) and alanine-expanded (A17) PABPN1 probed with sera from immunized rabbits. PABPN1 protein was detected as a 50 kDa band as indicated [19].](https://ip.ios.semcs.net:443/media/jnd/2015/2-4/jnd-2-4-jnd150111/jnd-2-4-jnd150111-g001.jpg)
Fig.2
Specificity of the α-alanine antibody. A) Immunoblot of lysates from HEK 293 cells transfected with empty plasmid (Ctrl) or plasmid encoding His-tagged wild type (A10) PABPN1 or PABPN1 expanded to 13–17 alanines probed with the α-Alanine antibody. Blots were probed with α-His to confirm expression of recombinant protein and α-PABPN1 to detect PABPN1 protein. An α-Tubulin antibody was used to detect tubulin as a loading control. B) Immunoblot of lysates from untransfected HEK 293 cells (Ctrl) or HEK 293 cells transfected with pTL-1 plasmid encoding HA-tagged wild type (A17) RUNX2 or alanine-expanded (A27) RUNX2 probed with the α-Alanine antibody. Blots were probed with α-HA to confirm protein expression and α-HSP90 was used to detect HSP90 as a loading control.
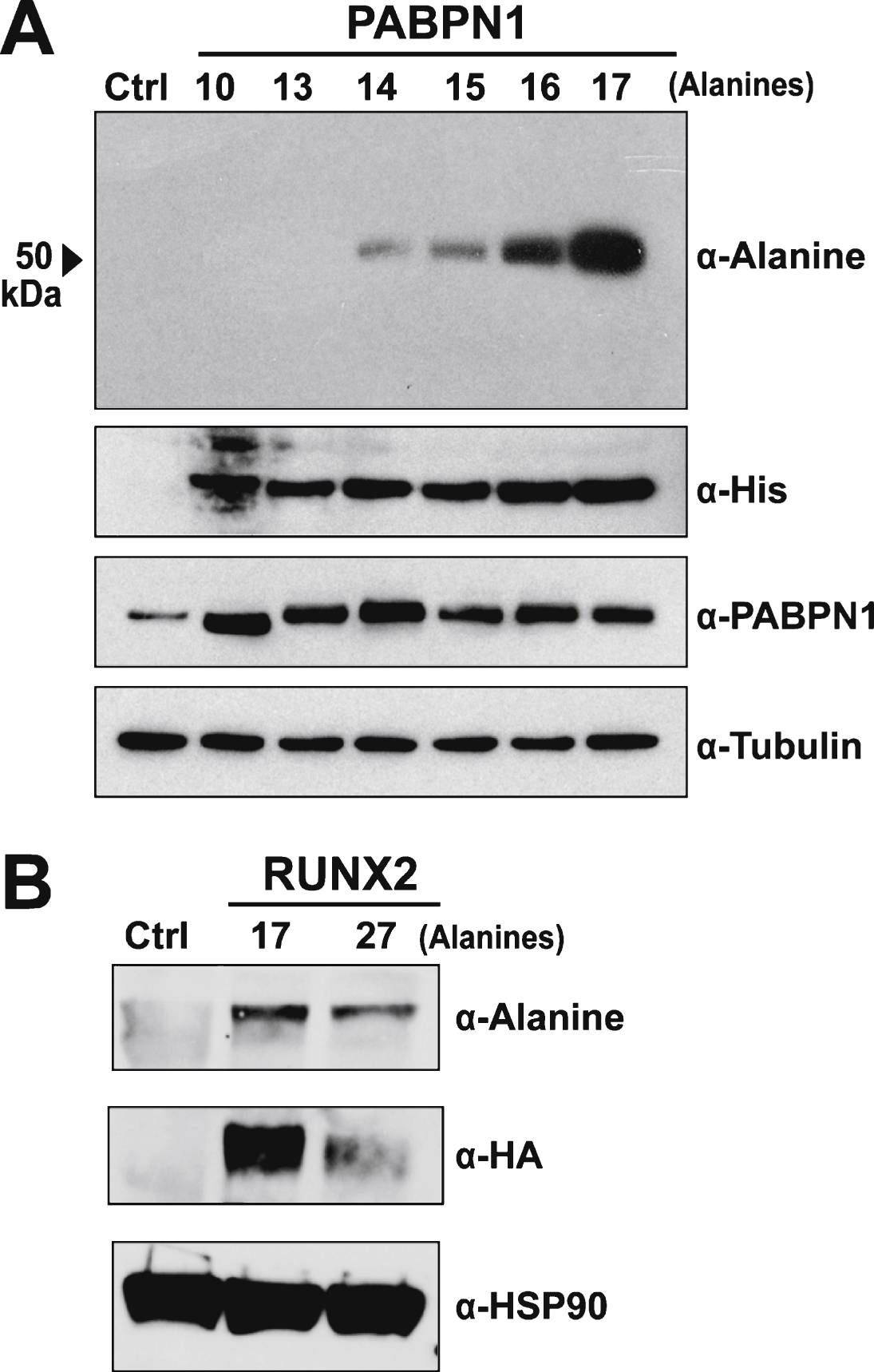
Fig.3
Sensitivity of the α-alanine antibody. Immunoblots of serial dilutions of lysates from HEK 293 cells transfected with plasmid encoding A17-PABPN1 probed with α-Alanine antibody. The total amount of lysate ( μg protein) loaded in each lane is indicated below the immunoblot panels. Blots were probed with α-PABPN1 to detect endogenous and recombinant PABPN1 protein and α-Tubulin was used to detect tubulin as a loading control.
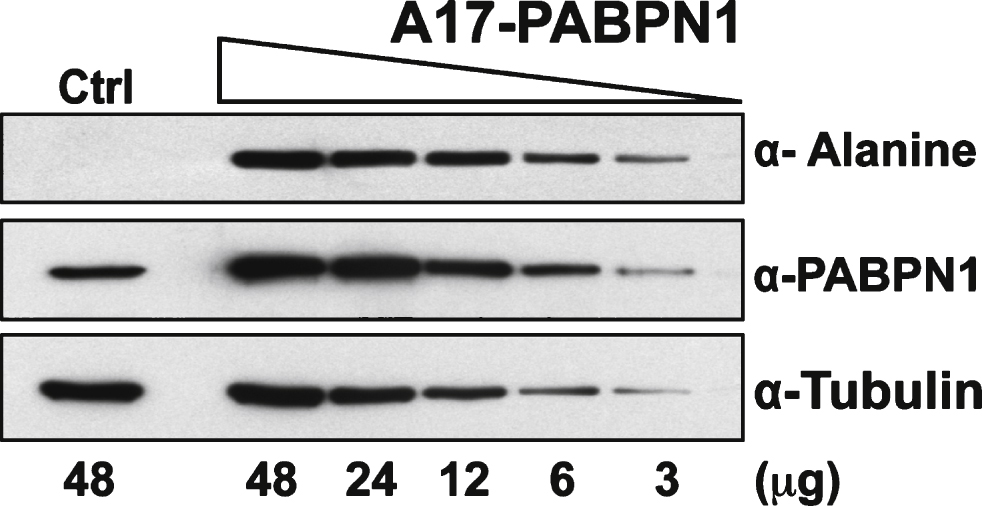
Fig.4
Use of the α-alanine antibody in a transgenic mouse model of OPMD. A) Immunoblots of whole muscle lysate from mice expressing transgenic wild type (A10.1) or alanine-expanded PABPN1 (A17.1) were probed with α-Alanine antibody. Samples were also probed with α-PABPN1 and α-HSP90 as controls. B) Immunostaining of tibialis anterior muscle sections from A10.1 and A17.1 animals stained with α-Alanine antibody. Immunostaining with α-PABPN1 was used to confirm the presence of PABPN1 protein in A10.1 animals and DAPI was used to stain nuclei. Merge image (bottom panels) is included to show co-localization of both A17 and wild type PABPN1 with nuclei. Bar = 5 μm. C) Immunoblots of whole fly lysates from flies expressing muscle-specific A10- or A17-PABPN1 probed with α-Alanine antibody. Samples were also probed with α-PABPN1 to detect PABPN1 and with α-Lamin D to detect lamin D as a loading control.
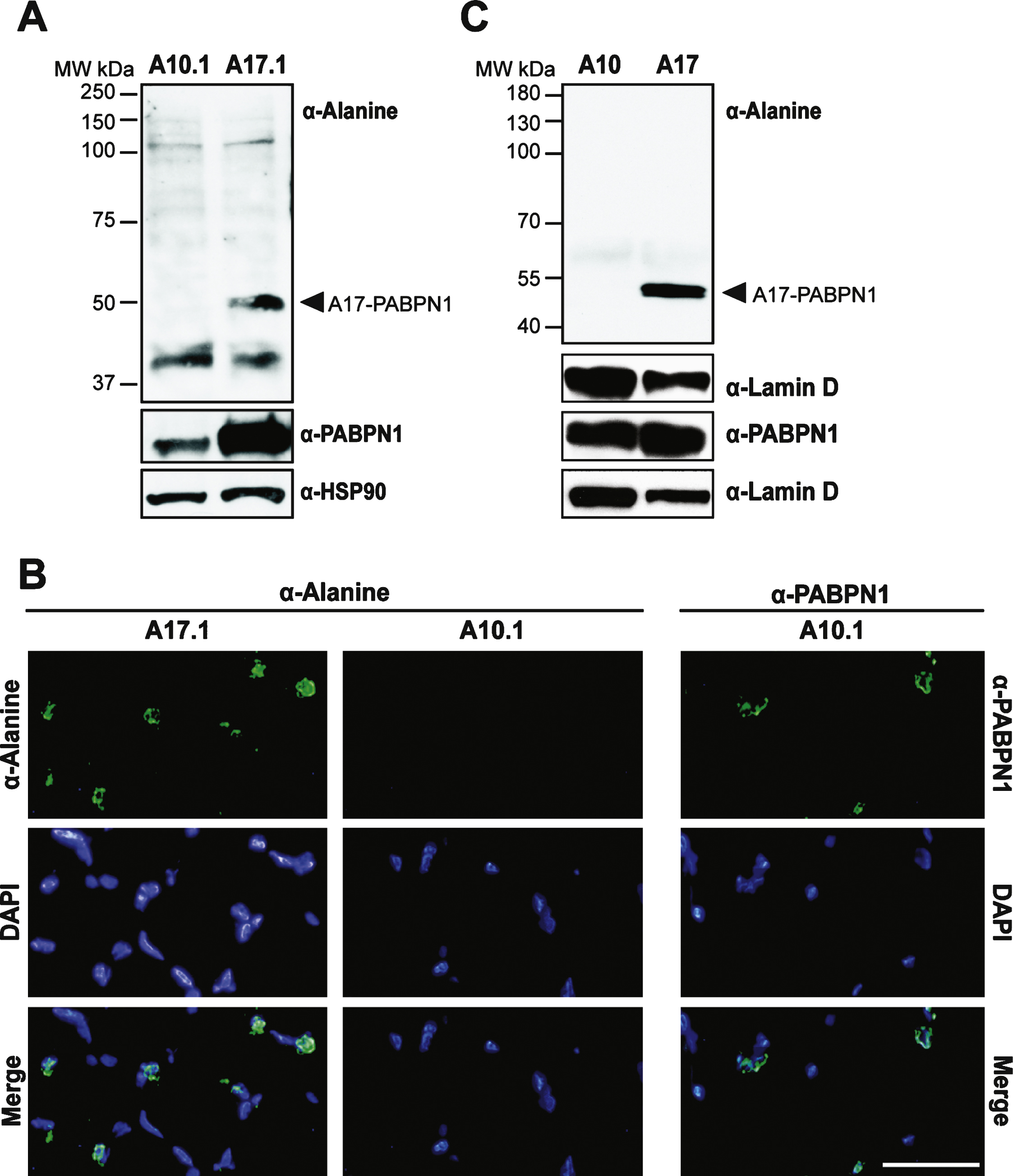
Table 1
Primers Used for Cloning WT and Alanine-Expanded PABPN1
| Primer | Sequence |
| PABPN1 | 5′CGGAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCAGCAGCAGCGGGGGCT |
| WT F | GCGGGCGGTCGGGGCTCCGGGCCGGGGCGGCGGCGCCATCTTGTG-3′ |
| PABPN1 | 5′-GAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCAGCAGCAGCAGCAGCAGC |
| A13 F | GGGGGCTGCGGGCGGTCGGGGCTCCGGGCCGGGGCGGCGGCGCCATC-3′ |
| PABPN1 | 5′-GAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCGGCAGCAGCAGCAGCAGC |
| A14 F | AGCGGGGGCTGCGGGCGGTCGGGGCTCCGGGCCGGGGCGGCGGCGC-3′ |
| PABPN1 | 5′GAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCGGCGGCAGCAGCAGCAGC |
| A15 F | AGCAGCGGGGGCTGCGGGCGGTCGGGGCTCCGGGCCGGGGCGGCGG-3′ |
| PABPN1 | 5′GAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCAGCAGCAGC |
| A16 F | AGCAGCAGCGGGGGCTGCGGGCGGTCGGGGCTCCGGGCCGGGGCGG-3′ |
| PABPN1 | 5′CGGAATTCGCCACCATGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCGGCG |
| A17 F | GCGGCAGCAGCAGCGGGGGCTGCGGGCGGTCGGGGCTCCGGGCCGGG-3′ |
| PABPN1 R | 5′-TAAGGGGAATACCATGATG-3′ |


