A Large Deletion Affecting TPM3, Causing Severe Nemaline Myopathy
Abstract
Background and Objectives:
Nemaline myopathy may be caused by pathogenic variants in the TPM3 gene and is then called NEM1. All previously identified disease-causing variants are point mutations including missense, nonsense and splice-site variants. The aim of the study was to identify the disease-causing gene in this patient and verify the NM diagnosis.
Methods:
Mutation analysis methods include our self-designed nemaline myopathy array, The Nemaline Myopathy Comparative Genomic Hybridisation Array (NM-CGH array), whole-genome array-CGH, dHPLC, Sanger sequencing and whole-exome sequencing. The diagnostic muscle biopsy was investigated further by routine histopathological methods.
Results:
We present here the first large (17–21 kb) aberration in the α-tropomyosinslow gene (TPM3), identified using the NM-CGH array. This homozygous deletion removes the exons 1a and 2b as well as the promoter of the TPM3 isoform encoding Tpm3.12st. The severe phenotype included paucity of movement, proximal and axial weakness and feeding difficulties requiring nasogastric tube feeding. The infant died at the age of 17.5 months. Muscle biopsy showed variation in fibre size and rods in a population of hypotrophic muscle fibres expressing slow myosin, often with internal nuclei, and abnormal immunolabelling revealing many hybrid fibres.
Conclusions:
This is the only copy number variation we have identified in any NM gene other than nebulin (NEB), suggesting that large deletions or duplications in these genes are very rare, yet possible, causes of NM.
INTRODUCTION
Nemaline myopathy (NM) is one of the most common congenital myopathies and currently there are ten genes known to cause NM: TPM3, NEB, ACTA1, TNNT1, TPM2, CFL2, KBTBD13, KLHL40, KLHL41 and LMOD3 [1]. The TPM3 gene (MIM:191030, geneID:7170) was the first of these to be identified [2]. Pathogenic variants in TPM3 are also known to cause cap myopathy and congenital fibre type disproportion (CFTD) [3, 4]. Recently, Marttila and co-workers summarized the findings in 35 clinically and histologically characterized families with 22 different TPM3 variants. The majority of the families (30/35) had missense mutations segregating in an autosomal dominant fashion, or arising de novo [5].
Recessive variants in TPM3 have previously been described in five families. A homozygous nonsense mutation in the exon 1a of TPM3 in a patient with a severe form of NM was published in 1999 [6]. A NM patient compound heterozygous for two different TPM3 variants was described in 2002; a splice-site mutation causing skipping of exon 9b, and a point mutation abolishing the stop codon in exon 9b, causing elongation of the protein by 57 amino acids [7]. The same point mutation abolishing the stop codon of exon 9b has also been found in homozygous form in a patient with CFTD [8]. A deletion removing the last nucleotide before the stop codon in exon 9b, causing a frameshift and elongation of the protein by 73 amino acids, was identified in two families of Turkish origin. The patients were homozygous for the deletion and had severe or intermediate forms of NM [9].
Tropomyosins are actin-binding proteins that are found in all eukaryotic cells. There are four different genes (TPM1, TPM2, TPM3 and TPM4) in humans encoding the different tropomyosins, creating more than 40 different isoforms by alternative splicing and alternative promoters. The different isoforms are expressed in different tissues and cell types, and their expression is highly regulated temporally and spatially during development. The different isoforms can substitute for each other to some extent but certain isoforms and exons are known to be essential [10]. In skeletal muscle, the tropomyosins stabilize actin and regulate actin-myosin interactions of the thin actin filament of the muscle sarcomere. TPM1 encodes α-tropomyosinfast, which is expressed in cardiac and fast skeletal muscles, whereas TPM3 encodes α-tropomyosinslow, which is expressed in slow, type 1 muscle fibres [11]. Non-muscle isoforms of TPM3 are known to be essential for embryonic development [10, 12]. The exons are numbered according to a recently published tropomyosin nomenclature [13].
CASE REPORT
The Proband
This female infant was born to first-cousin parents. Paucity of movement was noted from birth, and proximal and axial weakness was confirmed at 5 months. The best motor function achieved was rolling from side to side. Failure to thrive necessitated nasogastric tube feeding at ten months followed by gastrostomy insertion at 13 months of age. CK levels as well as ECG were normal. At the age of 11 months, the infant had ankle contractures which required surgical release and ankle foot orthosis. She died at the age of 17.5 months. A quadriceps muscle biopsy at 5 months (Fig. 1) showed myopathic features with marked abnormal variation in fibre size with many abnormally small fibres (approximately 10 μm or less in diameter) and a population of larger fibres of normal size for age (approximately 20–30 μm in diameter). Internal or central nuclei were present in several small fibres which expressed slow myosin, and some fibres showed aggregates of mitochondria. Fibres of normal size for age expressed fast myosin or were hybrid fibres co–expressing fast and slow myosin. Several small fibres expressed fetal myosin and co-expressed developmental myosin and cardiac actin, probably related to immaturity and underdevelopment. The Gömöri trichrome stain identified red-staining inclusions in several fibres, which electron microscopy confirmed as nemaline rods. Rods were prominent with the Gömöri trichrome stain in the small fibres expressing only slow myosin but the presence of any very small rods in other fibres could not be determined by light microscopy as they could not be distinguished from mitochondria. Ultrastructurally these small fibres with rods showed pronounced disruption/malformation of sarcomeres in contrast to adjacent fibres without rods (Fig. 1G).
Genetic studies
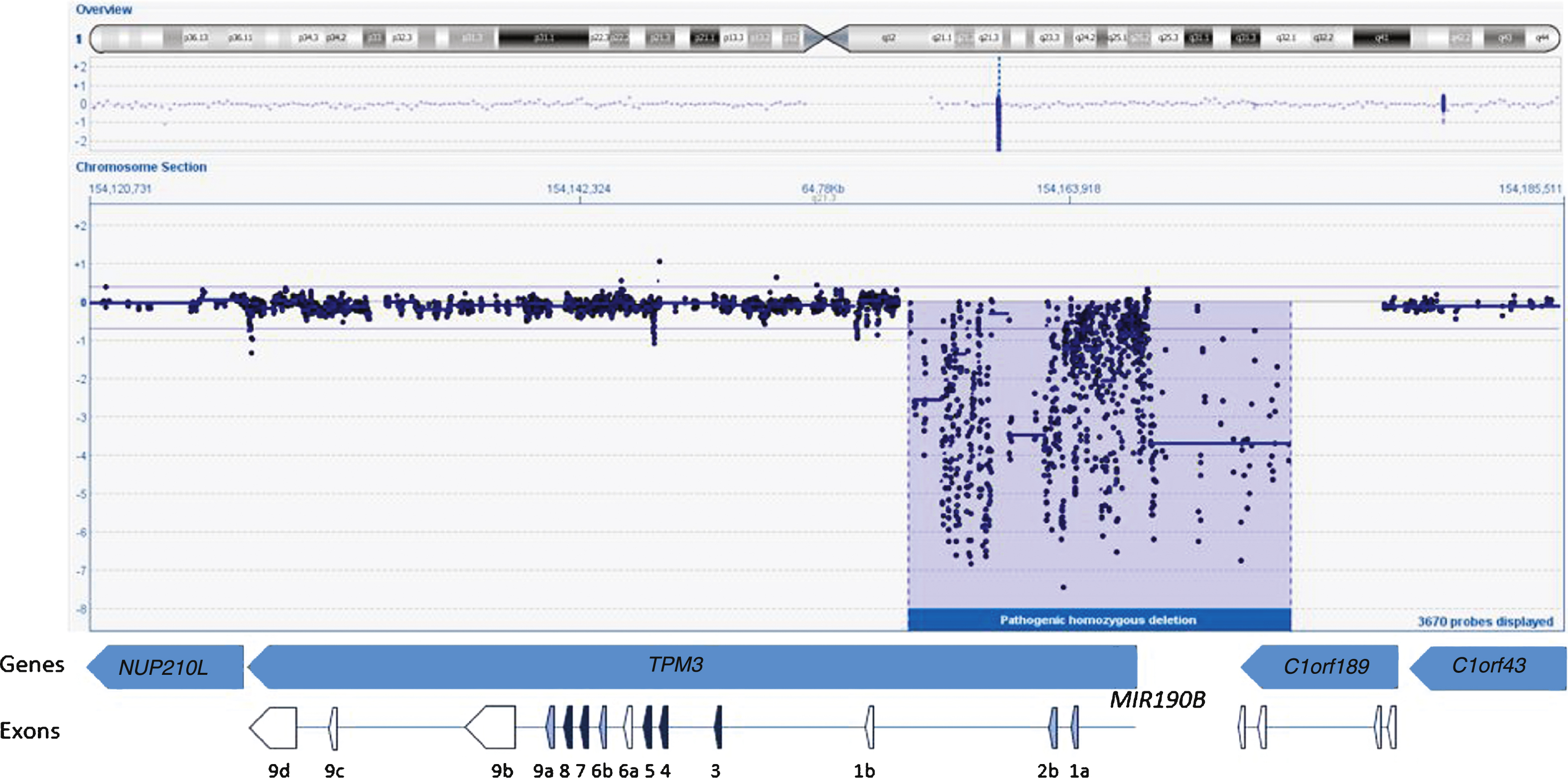
We started the analysis by screening the DNA sample of the patient for mutations in the first five known NM genes (NEB, ACTA1, TPM2, TPM3, and TNNT1) using dHPLC and Sanger sequencing and identified one heterozygous nonsense mutation in NEB exon 42 (c.5060 G>A; p.W1687X)(Reference Sequence NM_001271208.1). A second disease-causing variant, in NEB or any other NM gene, remained unidentified using these methods. Sequencing of the 5’end and the first exons of TPM3 failed repeatedly despite several attempts. Subsequently, we studied the sample using our targeted 8×60k NM-CGH microarray (Oxford Gene Technology Ltd, Oxford, UK), self-designed to detect large copy number variations (CNV) in the known NM genes [14]. We identified a large (17–21 kb) homozygous copy number variation in TPM3, deleting the promoter as well as the exons 1a and 2b of the gene (Fig. 2). According to HGVS nomenclature this rearrangement is a homozygous deletion of hg19 chr1:g.(154,156,325_154,156,028)_(154,173,059_154,177,712). The promoter and exon 1b of the non-muscle isoforms seem to be intact. The breakpoint resides in a 4.7 kb region that is lacking probes, because this region contains various Alu sequence repeats, hindering the designing of unique probes. The deletion also spans the micro-RNA miR-190b encoding MIR190B gene and the last two exons of the C1orf189 gene upstream of TPM3. A whole-genome 4×180k ISCA+SNP array (Oxford Gene Technology Ltd) showed that chromosomal region 1q21.3 including TPM3 resides inside a loss of heterozygosity (LOH) region, but 2q23.3 including NEB does not, explaining the homozygous variant in TPM3 and the heterozygous variant in NEB. Furthermore, the homozygous deletion was verified using whole-exome sequencing, which showed a complete lack of reads in the region corresponding to the deletion shown by the NM-CGH array. Exome capture and sequencing was done using the Agilent SureSelectXT All Exon 50Mb Target Enrichment Kit (protocol v1.2; Agilent Technologies, Santa Clara, CA, USA) on an Illumina HiSeq2000 platform (Illumina, San Diego, CA, USA) and analysis was completed using the Oxford Gene Technology exome sequencing pipeline. Whole-exome sequencing revealed no other pathogenic variants.
DISCUSSION
To the best of our knowledge, we report here the first large deletion characterized in TPM3. This novel 17–21 kb homozygous deletion in TPM3 deletes exons 1a and 2b as well as the promoter of the skeletal muscle isoform. Based on Sanger sequencing, TPM3 exon 1b and its promoter are present, indicating that the non-muscle isoforms should be expressed. This is most likely the case, as non-muscle isoforms of TPM3 are essential for normal embryonic development. Furthermore, it was not possible to generate knock-out mice lacking exon 1b, and this exon was shown to be required for embryo preimplantation and embryonic stem cell viability [12, 15]. Thus, we conclude that this deletion was the cause of the NM in the patient presented herein.
Previously, Tan and co-workers described a NM patient with a homozygous nonsense mutation in TPM3, changing codon 31, glutamine, to a stop codon in the exon 1a [6]. Their patient had a similar severe NM phenotype as the current patient, likewise showing extremely delayed and impaired motor development, where the best motor function achieved was being able to roll over. Contrary to the case of our patient, no feeding problems occurred. This patient died at the age of 21 months. There was type 1 fibre hypotrophy and mild predominance of type 2 fibres. Nemaline bodies were only present in type 1 fibres. The authors hypothesized that either no TPM3 peptide would be produced from the mutant allele, due to instability of the mutant mRNA, or, if a truncated peptide was produced, it could act as a poison protein, preventing proper formation of the tropomyosin polymer chain [6]. Because, in the current case, the exon 1a and the promoter of the isoform were deleted from both alleles, we assume that no Tpm3.12st (a.b.b.a.) was produced. This makes the two cases comparable, despite the differences in mutational mechanisms, and the null state for the striated muscle-specific isoform may explain the similarity and severity of the patients’ phenotypes.
Nemaline bodies affecting only type 1 fibres can occur in several forms of NM, but this phenomenon seems to be consistent in patients with TPM3 mutations. This is in agreement with the uniform expression of TPM3 in type 1, slow skeletal muscle fibres which, in TPM3-mutated patients, are often severely hypotrophic. As type 2 fast muscle fibres do not express TPM3 but TPM1, they remain relatively unaffected [5–7, 9].
Fibre type disproportion has been defined on ATPase stained sections and requires fibres to be defined as either type 1 or of type 2 and the type 1 fibres to be 25% smaller than type 2 [16, 17]. Although type 1 and type 2 fibres often equate to fibres with slow or fast myosin respectively, classification of fibre types is difficult if hybrid fibres expressing more than one isoform are present. The patient presented here had small fibres with slow myosin, but a population of large fibres were hybrid fibres. This can also be seen in other congenital myopathies. Therefore, the patient’s biopsy did not fulfill the formal criteria for fibre type disproportion, although a size disproportion of more than 25 % was present.
In the current patient, in addition to the homozygous deletion in TPM3, we also found a heterozygous nonsense variant in NEB exon 42. In accordance with the lack of clinical symptoms in other carriers of heterozygous NEB mutations, this mutation is unlikely to influence the clinical picture of the patient. Parental samples were not available to investigate this. Furthermore, the MIR190B gene upstream of TPM3 was also deleted. It has been shown that miR-190b regulates the expression of myotubularin-related protein 6 (MTMR6) in CD4+ T cells and macrophages [18], but the role, if any, of miR-190b in skeletal muscle is unknown. The deletion also disrupts the C1orf189 gene located upstream of TPM3, the function of which is unknown.
It has been suggested that the nemaline body accumulation in type 1 fibres in NEM1 might result from a disturbed ratio of functional tropomyosin in relation to other thin filament components. The severity of the disease might relate to the extent to which the underlying pathogenic variants disrupt protein expression and stability [5–7, 9].
To date, we have screened 266 samples from 196 NM and related myopathy families using the NM-CGH-array, and identified nine large aberrations in NEB in ten families. We have not detected large copy number variations in any of the other NM genes, except for the TPM3 deletion discussed here. However, Friedman and co-workers have described one patient with a large deletion of the ACTA1 gene causing recessive NM [19]. In summary, it is likely that large aberrations in known NM genes other than NEB are very rare causes of NM. Nevertheless, the possibility of such large aberrations should be taken into consideration.
ETHICS APPROVAL
The study has been approved by the Ethics Committee of the Children’s Hospital, University of Helsinki, Helsinki, Finland. The family had given informedconsent.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank Marilotta Turunen for excellent technical assistance and Nina Horelli-Kuitunen from United Medix Laboratories Ltd for help with the whole-genome array-CGH study. This study was supported by grants from the Sigrid Jusélius Foundation, the Association Francaise contre les Myopathies, the Finska Läkaresällskapet and the Medicinska understödsföreningen Liv och Hälsa, FP7 2012 Neuromics; the NSCT support to the Dubowitz Neuromuscular Centre is also acknowledged, as the MRC Neuromuscular Centre Grant Biobank and the Great Ormond Street Hospital Biomedical researchCentre.
REFERENCES
1 | Lehtokari VL, Kiiski K, Sandaradura SA, Laporte J, Repo P, Frey JA, Donner K, Marttila M, Saunders C, Barth PG, den Dunnen JT, Beggs AH, Clarke NF, North KN, Laing NG, Romero NB, Winder TL, Pelin K, Wallgren-Pettersson C (2014) Mutation update: The spectra of nebulin variants and associated myopathies Hum Mutat 35: 12 1418 1426 |
2 | Laing NG, Wilton SD, Akkari PA, Dorosz S, Boundy K, Kneebone C, Blumbergs P, White S, Watkins H, Love DR (1995) A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy Nat Genet 9: 1 75 79 |
3 | Clarke NF, Kolski H, Dye DE, Lim E, Smith RL, Patel R, Fahey MC, Bellance R, Romero NB, Johnson ES, Labarre-Vila A, Monnier N, Laing NG, North KN (2008) Mutations in TPM3 are a common cause of congenital fiber type disproportion Ann Neurol 63: 3 329 337 |
4 | De Paula AM, Franques J, Fernandez C, Monnier N, Lunardi J, Pellissier JF, Figarella-Branger D, Pouget J (2009) A TPM3 mutation causing cap myopathy Neuromuscul Disord 19: 10 685 688 |
5 | Marttila M, Lehtokari VL, Marston S, Nyman TA, Barnerias C, Beggs AH, Bertini E, Ceyhan-Birsoy O, Cintas P, Gerard M, Gilbert-Dussardier B, Hogue JS, Longman C, Eymard B, Frydman M, Kang PB, Klinge L, Kolski H, Lochmuller H, Magy L, Manel V, Mayer M, Mercuri E, North KN, Peudenier-Robert S, Pihko H, Probst FJ, Reisin R, Stewart W, Taratuto AL, de Visser M, Wilichowski E, Winer J, Nowak K, Laing NG, Winder TL, Monnier N, Clarke NF, Pelin K, Gronholm M, Wallgren-Pettersson C (2014) Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies Hum Mutat 35: 7 779 790 |
6 | Tan P, Briner J, Boltshauser E, Davis MR, Wilton SD, North K, Wallgren-Pettersson C, Laing NG (1999) Homozygosity for a nonsense mutation in the alpha-tropomyosin slow gene TPM3 in a patient with severe infantile nemaline myopathy Neuromuscul Disord 9: 8 573 579 |
7 | Wattanasirichaigoon D, Swoboda KJ, Takada F, Tong HQ, Lip V, Iannaccone ST, Wallgren-Pettersson C, Laing NG, Beggs AH (2002) Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy Neurology 59: 4 613 617 |
8 | Lawlor MW, Dechene ET, Roumm E, Geggel AS, Moghadaszadeh B, Beggs AH (2010) Mutations of tropomyosin 3 (TPM3) are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion Hum Mutat 31: 2 176 183 |
9 | Lehtokari VL, Pelin K, Donner K, Voit T, Rudnik-Schoneborn S, Stoetter M, Talim B, Topaloglu H, Laing NG, Wallgren-Pettersson C (2008) Identification of a founder mutation in TPM3 in nemaline myopathy patients of Turkish origin Eur J Hum Genet 16: 9 1055 1061 |
10 | Gunning PW, Schevzov G, Kee AJ, Hardeman EC (2005) Tropomyosin isoforms: Divining rods for actin cytoskeleton function Trends Cell Biol 15: 6 333 341 |
11 | Gunning P, Gordon M, Wade R, Gahlmann R, Lin CS, Hardeman E (1990) Differential control of tropomyosin mRNA levels during myogenesis suggests the existence of an isoform competition-autoregulatory compensation control mechanism Dev Biol 138: 2 443 453 |
12 | Hook J, Lemckert F, Qin H, Schevzov G, Gunning P (2004) Gamma tropomyosin gene products are required for embryonic development Mol Cell Biol 24: 6 2318 2323 |
13 | Geeves MA, Hitchcock-DeGregori SE, Gunning PW (2015) A systematic nomenclature for mammalian tropomyosin isoforms J Muscle Res Cell Motil 36: 2 147 153 |
14 | Kiiski K, Laari L, Lehtokari VL, Lunkka-Hytonen M, Angelini C, Petty R, Hackman P, Wallgren-Pettersson C, Pelin K (2013) Targeted array comparative genomic hybridization–a new diagnostic tool for the detection of large copy number variations in nemaline myopathy-causing genes Neuromuscul Disord 23: 1 56 65 |
15 | Hook J, Lemckert F, Schevzov G, Fath T, Gunning P (2011) Functional identity of the gamma tropomyosin gene: Implications for embryonic development, reproduction and cell viability Bioarchitecture 1: 1 49 59 |
16 | Brooke MH, Engel WK (1969) The histographic analysis of human muscle biopsies with regard to fiber types. 4. Children’s biopsies Neurology 19: 6 591 605 |
17 | Clarke NF, North KN (2003) Congenital fiber type disproportion–30 years on J Neuropathol Exp Neurol 62: 10 977 989 |
18 | Mohan M, Chandra LC, Torben W, Aye PP, Alvarez X, Lackner AA (2014) miR-190b is markedly upregulated in the intestine in response to simian immunodeficiency virus replication and partly regulates myotubularin-related protein-6 expression J Immunol 193: 3 1301 1313 |
19 | Friedman B, Simpson K, Tesi-Rocha C, Zhou D, Palmer CA, Suchy SF (2014) Novel large deletion in the ACTA1 gene in a child with autosomal recessive nemaline myopathy Neuromuscul Disord 24: 4 331 334 |
Figures and Tables
Fig.1
Histological stainings and immunolabelling of frozen sections and an electron micrograph of the muscle biopsy from the Proband. A) Haematoxylin and eosin staining showing a wide variation in fibre size with internal nuclei in several small fibres. Excess endomysial connective tissue or necrosis were not detected. B) Gömöri trichrome staining showing a population of small fibres containing red stained rods. C) Cytochrome C oxidase staining showing a population of darkly stained small type 1 fibres and some small weakly stained fibres. D) Immunolabelling with a monoclonal antibody to fetal myosin showing several very small positive fibres. E-F) Immunolabelling with monoclonal antibodies to fast (E) and slow (F) myosin heavy chains showing a population of small fibres that only express slow myosin (*), larger fibres that express fast (f) and several hybrid fibres with varying intensity that express both fast and slow myosin (+). G) Electron micrograph showing a fibre with severely disrupted myofibrils, an internal nucleus, small rods (arrow) and clusters of mitochondria (m), and less severely affected fibres either side (*) without rods.
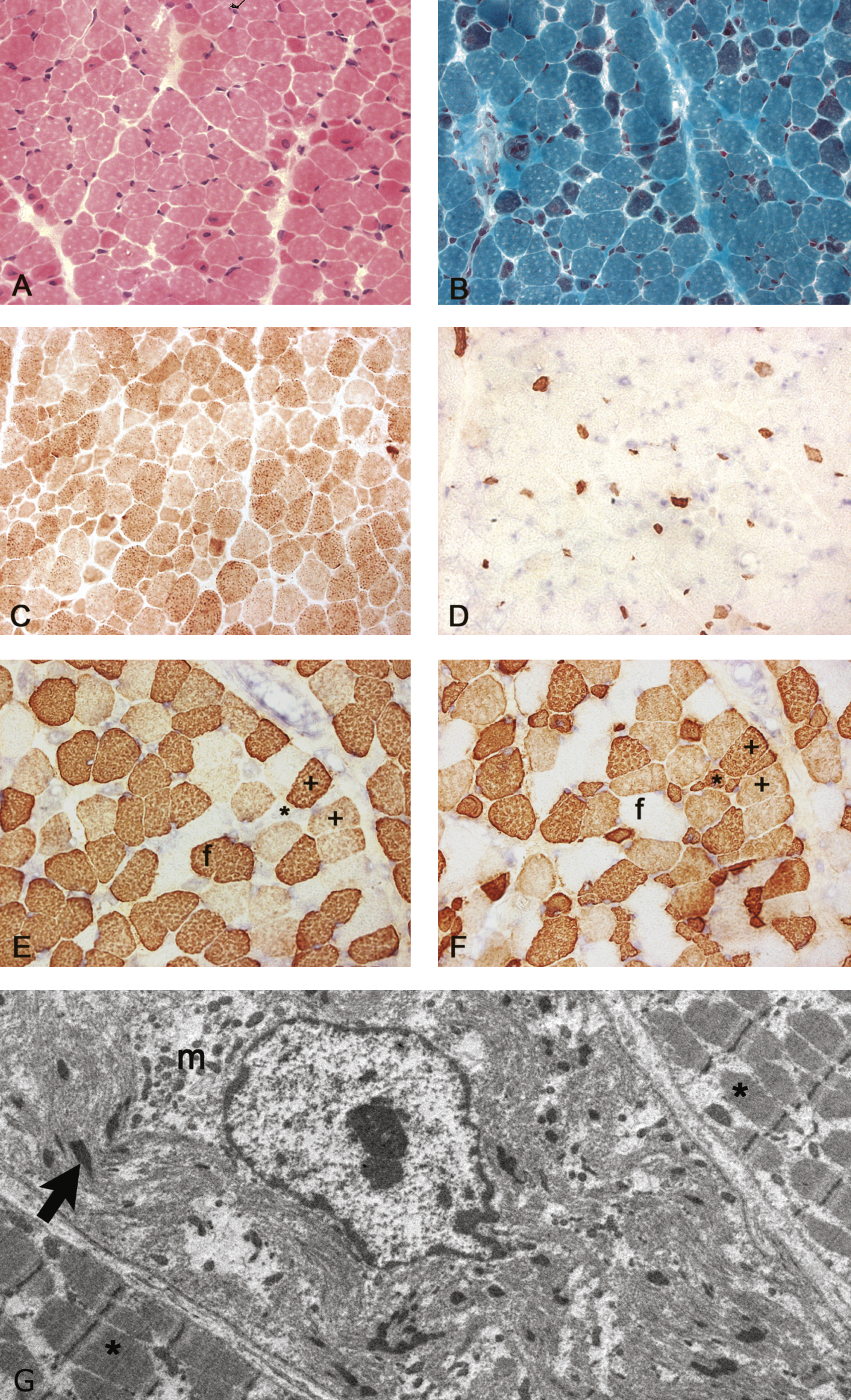
Fig.2
NM-CGH profile of the TMP3 gene. The DNA sample of the proband shows a 17–21 kb homozygous deletion in chromosome region 1q21.3 removing TPM3 exons 1a and 2b as well as the promoter of the isoform. The deletion covers also the first two exons of MIR190B as well as the last two exons of C1orf189. The genes, exons and their orientation are indicated in the schematic overview in the lower part of the figure. The exons expressed in all TPM3 isoforms are marked with dark blue, exons not expressed in striated muscle are marked with white and alternatively spliced exons present in the striated muscle isoform are marked with pale blue colour. TPM3 geneID:7170, concerned protein isoform Tpm3.12st (a.b.b.a.).