
Refining the Language of Huntington’s Disease Progression with the Huntington’s Disease Integrated Staging System (HD-ISS)
In this issue of the Journal of Huntington’s Disease (JHD), the journal editors, together with a large group of co-authors, delve into the world of terminology and language specific to Huntington’s disease (HD) research [1]. This comprehensive effort aims to standardize the language used by contributors to the journal and provide clear guidance on the most appropriate technical terms for the journal’s readership and the wider HD research community. In areas such as protein and gene terminology, scientists in their respective fields have already achieved international consensus, leaving our primary task as adapting these established rules to the context of HD. However, within the clinical domain, some terminology remains idiosyncratic, leading to diverse interpretations of terms like “pre-symptomatic,” “prodromal,” and “pre-manifest. This variability underscores the critical need to establish a definitive rule set to ensure consistency and clarity in the discourse surrounding the progression of HD.
This editorial highlights the imprecision in the terminology related to HD progression, which impacts the ability to compare data across different studies—a significant limitation in research.
Neurodegenerative diseases follow a long, varying pathogenic process where compensatory mechanisms initially mask symptoms [2]. Over time, these compensatory mechanisms fail, and symptoms emerge, leading to progressive disability at rates that differ by disease. This process resembles an iceberg: the late, symptomatic stage is just the visible tip, while the larger, unseen portion represents the extensive, undetected period of underlying biological change.
In the realm of neurodegenerative diseases, HD stands out for its remarkable ability to be detected early, decades before symptoms appear. This is due to the availability of a highly accurate diagnostic test and the fully penetrant nature of the causal mutation when the length of the CAG repeat in the HTT gene is 40 or higher. In contrast, in Alzheimer’s disease and Parkinson’s disease there is no one marker that confirms the diagnosis in all the variable forms that these disorders present. Though amyloid/tau and alpha-synuclein levels can be helpful, the sensitivity of these measures and their correlation with disease presence and onset is low compared with the certainty that having a CAG repeat size of greater than 40 leads to the disease. This complexity of other neurodegenerative diseases highlights HD’s advantageous position in early disease detection within this group of disorders.
The ability to detect HD early, before any overt symptoms, became a reality only with the discovery of the causative gene mutation in 1993 [3]. which led to the development and dissemination of a standardized genetic test. Prior to this, diagnosing HD depended on the presence of specific clinical signs, primarily motor symptoms such as involuntary chorea-type movements. For a more precise definition of an HD case, scientists established criteria that included the presence of a movement disorder along with a relevant family history. Diagnostic tools like the Diagnostic Confidence Level (DCL) were incorporated into the Unified Huntington’s Disease Rating Scale (UHDRS) in its 1999 version [4]. However, these criteria, whilst aimed at high specificity, tended to identify HD cases relatively late in the symptomatic phase of the disease’s progression.
Despite accepting criteira for clinical diagnosis based on the presence of motor signs, clinicians quickly acknowledged that a variety of symptoms and signs often precede that formal clinical diagnosis of HD. Observational studies such as TRACK-HD [5], PREDICT-HD [6], and PHAROS [7] have provided substantial evidence for the existence of changes in brain structure and in symptomatology in the period before formal clinical diagnosis. During this period, not only motor, but also cognitive and behavioral changes can significantly impact the lives of people with HD (PwHD).
When reading various papers on HD research from different academic groups or institutions, one notices both subtle and more pronounced discrepancies in the usage of terminology. For instance, ‘premanifest’ (or pre-manifest) is sometimes used synonymously with ‘prodromal’ to describe the period before clinical motor diagnosis when subtle signs or symptoms are present. Other times, it refers to the entire period preceding clinical motor diagnosis, regardless of any signs or symptoms [8, 9]. The article [1] in this issue of the journal provides guidelines on how these terms should be utilized in its publications.
Interestingly, PubMed statistics show the term ‘Manifest HD’ has been in use since at least the 1960 s, likely as a shorthand for the clinical manifestations of HD. Around 1985, the term ‘disease onset’ began to appear in the literature, referring somewhat ambiguously to the point of clinical motor diagnosis (CMD). This usage is particularly misleading because, as outlined above, it is well established the disease doesn’t start at time of CMD but much earlier. It wasn’t until about 2007 that the term ‘premanifest’ came into use, specifically to describe the period prior to the clinical motor diagnosis, or when the DCL equals 4. The adoption of terminology related to HD progression seems to have evolved more through happenstance than through a structured approach. It wasn’t until the Movement Disorders task force published their viewpoint on diagnostic categories [10] that an attempt was made to formally define and standardize the meanings of terms like, pre-manifest, presymptomatic, and prodromal. However, the terms were not operationalized.
The absence of operational definitions further complicates the characterization of study populations, leading researchers to rely on idiosyncratic cut-offs, such as Total Motor Score (TMS) > 5 [5], or varying ranges of CAG-Age Product (CAP) [6] scores calculated using different formulas. This adds to the heterogeneity in how different studies define their populations. Recently, a standardized and updated formula for the CAP score has been published [11], which hopefully will bring more consistency to future research endeavors in this field.
The development and publication of the Huntington’s Disease Integrated Staging System (HD-ISS) [12] marks a significant milestone in the pursuit of standardizing and operationalizing terminology related to HD. Even more crucially, it contributes to the conceptual understanding of HD progression as a continuum that begins with the causal mutation in the HTT gene and continues through to the end of life. This system offers a more coherent and comprehensive framework to understand and study HD, particularly by enabling the clear definition of disease stages that occur before the emergence of clinical signs or symptoms. It enhances our ability to categorize and examine the disease’s progression in its earliest phases.
Fig. 1
Figure 1 provides a schematic representation of how the Huntington’s Disease Integrated Staging System (HD-ISS) aligns with the terminology traditionally used in HD research. In this diagram: •Clinical motor diagnosis, or a Diagnostic Confidence Level (DCL) of 4, corresponds to the latter part of HD-ISS stage 2. •The Shoulson and Fahn stages, which are applicable after clinical motor diagnosis and are based on specific Total Functional Capacity (TFC) scores, overlap with the end of HD-ISS stage 2 and encompass all of stage 3. The figure also illustrates various terms used to denote periods before clinical motor diagnosis. While their usage has not been strictly defined, we propose that HD-ISS stages 0 and 1 can be referred to as pre-symptomatic, whereas stages 2 and 3 of the HD-ISS can be considered symptomatic stages.
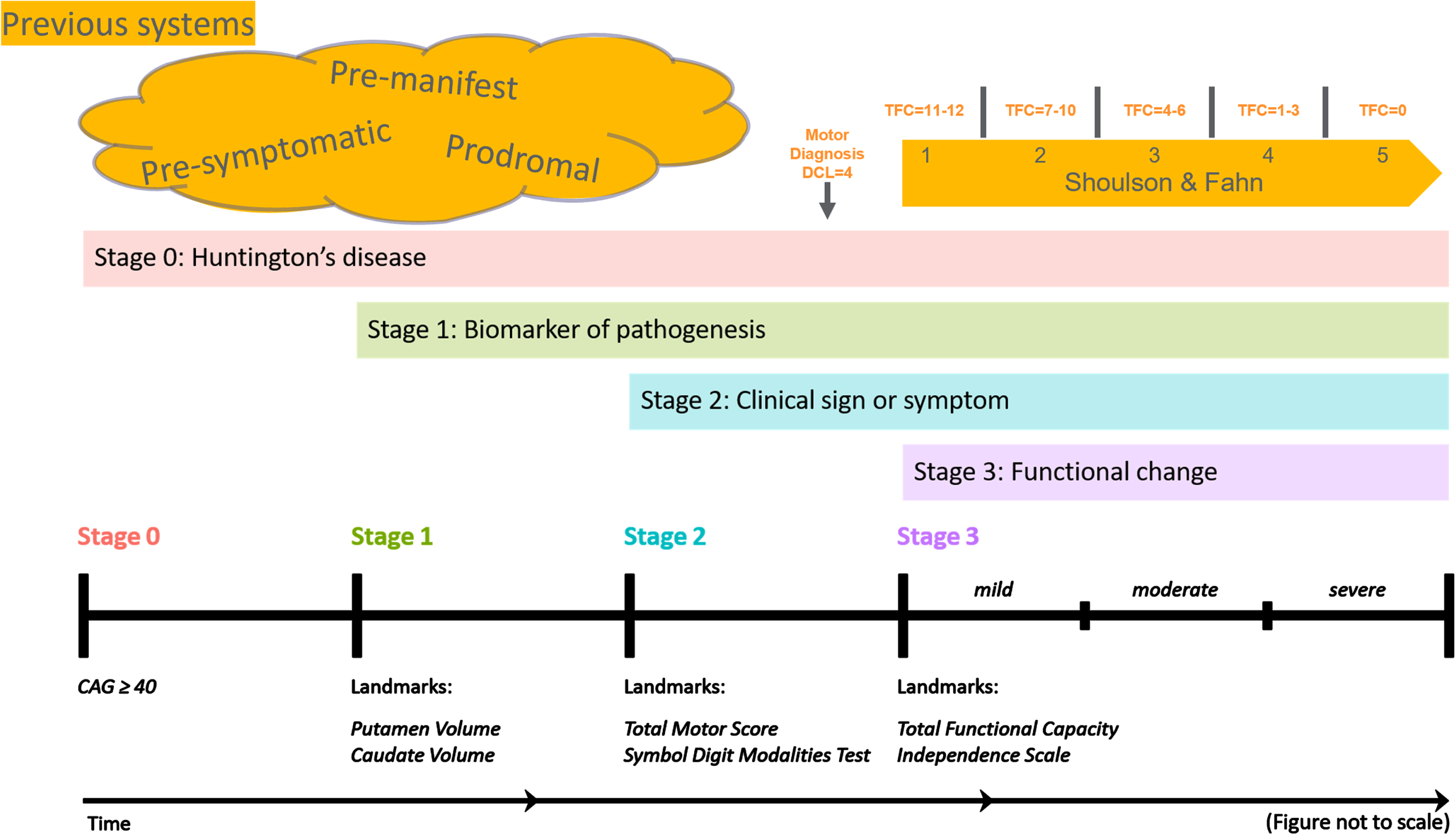
The HD-ISS was specifically designed for clinical research. When it has been systematically adopted, the resulting publications should employ a standardized terminology derived from the HD-ISS. This adoption will represent a natural evolution in the field. There will be a transition period while that adoption takes place and research utilizing this system is published. The guidelines provided by [1] are intended to bridge this gap, which could span five years or more, ensuring continuity and clarity in terminology during this transitional phase.
The basic features of the Huntington’s Disease Integrated Staging System (HD-ISS) can be summarized as follows:
1. HD is defined by a CAG expansion of 40 or more in exon 1 of the HTT gene. It’s important to note that CAG expansions between 36 and 39 cannot be formally classified using the HD-ISS without the presence of a specific biomarker or clinical syndrome, which is yet to be defined.
2. The progression of the disease is viewed as a continuum. It begins with the identification of the pathological gene expansion, placing individuals in stage 0. If there are signs of neurodegeneration, evidenced by atrophy of the caudate or putamen, individuals are assigned to stage 1. If disease signs are measurable by the TMS or Symbol Digit Modalities Test (SDMT), they are classified as stage 2. Finally, if there is an impact on daily functioning as assessed by the Total Functional Capacity (TFC) or the Independence Scale, individuals are assigned to stage 3.
The HD-ISS was developed through a formal consensus approach, grounded in a detailed, evidence-based analysis of data. This has garnered broad support from the community and established a strong scientific foundation. The system not only integrates a biological definition of each case but also describes progression in stages characterized by degrees of neurodegeneration or by the type and severity of clinical manifestations. Considering the progressive nature of the disease and our understanding of its progression (evaluated in predictive models of progression), each stage is prognostic for subsequent ones.
The adoption of the HD-ISS promises to resolve many inconsistencies in the clinical discourse surrounding HD. Simultaneously, this system equips the field with the tools to describe the continuum of HD progression more accurately, especially the very early stages. These initial stages, which have only recently become a focus of study, are particularly promising as they may reveal the most effective targets for disease-modifying therapies. Furthermore, for PwHD the recognition of the disease status in the pre-symptomatic phase is empowering and respectful of their right to autonomy in decisions related to their health.
ACKNOWLEDGMENTS
Thanks to Emily Gantman for assistance with editing and help with the figure.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Sarah Tabrizi is an Associate Editor and Anne Rosser is an Editorial Board Member of this journal but neither were involved in the peer-review process of the original article or of this editorial nor had access to any information regarding its peer-review.
REFERENCES
[1] | DiFiglia M , Leavitt BR , Macdonald D , Thompson LM , Huntington’s Disease Nomenclature Working Group. Towards standardizing nomenclature in Huntington’s disease research. J Huntingtons Dis. (2024) . 10.3233/JHD-240044. |
[2] | Gregory S , Long JD , Klöppel S , Razi A , Scheller E , Minkova L , et al. Operationalizing compensation over time in neurodegenerative disease. Brain. (2017) ;140: (4):1158–65. |
[3] | The Huntington’s Disease Collaborative Research Grou. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: (6):971–83. |
[4] | Unified Huntington’s Disease Rating Scale: Reliability and consistency. Mov Disord. (1996) ;11: (2):136–4. |
[5] | Tabrizi SJ , Langbehn DR , Leavitt BR , Roos RA , Durr A , Craufurd D , et al, TRACK-HD investigators. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: Cross-sectional analysis of baseline data. Lancet Neurol. (2009) ;8: (9):791–801. |
[6] | Biglan KM , Ross CA , Langbehn DR , Aylward EH , Stout JC , Queller S , et al. Motor abnormalities in premanifest persons with Huntington’s disease: The PREDICT-HD study. Mov Disord. (2009) ;24: (12):1763–72. |
[7] | Huntington Study Group PHAROS Investigators; Biglan KM , Shoulson I , Kieburtz K , Oakes D , Kayson E , et al. Clinical-Genetic Associations in the Prospective Huntington at Risk Observational Study (PHAROS): implications for clinical trials. JAMA Neurol. (2016) ;73: (1):102–10. |
[8] | Hersch SM , Rosas HD . Neuroprotection for Huntington’s disease: Ready, set, slow. Neurotherapeutics. (2008) ;5: (2):226–36. |
[9] | Roos RA . Huntington’s disease: A clinical review. Orphanet J Rare Dis. (2010) 5: :40. |
[10] | Ross CA , Reilmann R , Cardoso F , McCusker EA , Testa CM , Stout JC , et al. Movement Disorder Society Task Force Viewpoint: Huntington’s disease diagnostic categories. Mov Disord Clin Pract. (2019) ;6: (7):541–6. |
[11] | Warner JH , Long JD , Mills JA , Langbehn DR , Ware J , Mohan A , Sampaio C . Standardizing the CAP score in Huntington’s disease by predicting age-at-onset. J Huntingtons Dis. (2022) ;11: (2):153–71. |
[12] | Tabrizi SJ , Schobel S , Gantman EC , Mansbach A , Borowsky B , Konstantinova P , et al. Huntington’s Disease Regulatory Science Consortium (HD-RSC). A biological classification of Huntington’s disease: The Integrated Staging System. Lancet Neurol. (2022) ;21: (7):632–44. |

