
Gut Microbiota as a Modifier of Huntington’s Disease Pathogenesis
Abstract
Huntingtin (HTT) protein is expressed in most cell lineages, and the toxicity of mutant HTT in multiple organs may contribute to the neurological and psychiatric symptoms observed in Huntington’s disease (HD). The proteostasis and neurotoxicity of mutant HTT are influenced by the intracellular milieu and responses to environmental signals. Recent research has highlighted a prominent role of gut microbiota in brain and immune system development, aging, and the progression of neurological disorders. Several studies suggest that mutant HTT might disrupt the homeostasis of gut microbiota (known as dysbiosis) and impact the pathogenesis of HD. Dysbiosis has been observed in HD patients, and in animal models of the disease it coincides with mutant HTT aggregation, abnormal behaviors, and reduced lifespan. This review article aims to highlight the potential toxicity of mutant HTT in organs and pathways within the microbiota-gut-immune-central nervous system (CNS) axis. Understanding the functions of Wild-Type (WT) HTT and the toxicity of mutant HTT in these organs and the associated networks may elucidate novel pathogenic pathways, identify biomarkers and peripheral therapeutic targets for HD.
INTRODUCTION
The expansion of a CAG repeat (>35) in the exon 1 of huntingtin (HTT) gene is the root cause of Huntington’s disease (HD), a neurodegenerative disorder exemplified by motor and cognitive deficits, psychiatric symptoms, and peripheral complications. The expanded CAG repeat produces an abnormal polyglutamine (polyQ) tract, which increases the amyloidogenic properties of the HTT exon1 and forms oligomeric assemblies disrupting numerous cellular pathways in the nervous system [1, 2]. However, the aberrant functions of mutant HTT (mHTT) in the peripheral organs like the innate immune system and gastrointestinal tract are also gaining attention as modifiers of HD pathology [3].
HTT is ubiquitously expressed, and it is predicted that the toxicity of mHTT in peripheral organs may contribute to neurodegeneration in the nervous system of HD patients [3]. One notable example is the elevated activation of the innate immune system. Activated microglia, infiltration of proinflammatory TH17.1 cells from the periphery into the CNS, and hyperactive monocytes and macrophages in the circulation are observed in pre-manifest HD patients several years before any noticeable neurological symptoms emerge [4–7]. Recent studies suggest that innate immune pathways promote the loss of corticostriatal synapses in the brains of postmortem HD patients and in animal models, and activation of the intraneuronal immune modulators contribute to neurodegeneration in the striatum [8, 9]. mHTT expressed in microglia and peripheral immune cells may promote an inflammatory intracellular milieu through changes in the activity of genes implicated in cell proliferation. Mechanistically, mHTT directly binds to and activates the inflammatory IKK/NF-κB pathway, which is present in most cell types and is a central node in the innate immune development and response to microorganisms [10–13].
The gastrointestinal tract (gut) hosts thousands of microbial species, collectively known as the microbiota, and over three million genes and regulatory DNA sequence, collectively identified as microbiome. Compared to the human genome, the gut microbiome contains approximately 100-fold more genes [14, 15]. The gut microbiota also produces and regulates the production of neuromodulators and neurotransmitters, which influence the physiology of the nervous system [16]. A sizable portion of gut microbiota contains inflammatory molecules known as inflammogens, which trigger defense pathways in the gut epithelium and innate immune cells to eliminate excessive growth of pathobionts and maintain microbial homeostasis [17]. The IKK/NF-κB pathway is one of the core pathways implicated in gut-microbiota communications and plays a role in the reorganization of gut epithelium in response to external signals from diet or exposure to pathogens [18, 19]. The aberrant interaction of mHTT with the IKK/NF-κB could potentially modify the microbiota-gut-immune networks in HD [11, 12].
The gut microbiota is remarkably adaptable, and its composition and physiology are influenced by factors such as diet, lifestyle, medications, and genetic makeup of the host. The cellular and humoral components of gut microbiota play a significant role in human physiology and metabolic homeostasis [20]. Recent studies have brought the role of gut microbiota in brain development and neurological disorders to the forefront of neuroscience. This has led to exploration of gut-based therapies for complex human diseases such as autism spectrum disorders (ASD), neurodegeneration and brain aging [21–23]. Several studies suggest that changes in the homeostasis of gut microbiota known as dysbiosis, may influence the pathogenesis of HD [24–30]. These studies herald new research directions to better understand the impact of gene-environment interactions in disease progression. Recent reviews of this literature have elaborated the importance of gut dysbiosis and microbial metabolites on the pathogenesis of HD and have speculated on how the gut-brain pathways could help to better understand the role of gene-environment interactions in disease onset and severity, as well as the development of gut-based therapeutics [31–34]. I refer the readers to these excellent reviews.
In this article, the focus is on exploring the potential mechanistic toxicity of mHTT within the microbiota-gut-immune-nervous systems axis (Fig. 1). Emphasis will be placed on the aberrant interaction between mHTT and the IKK/NF-κB, which is one the prominent pathways in the microbiota-gut-immune communications. Recent findings on the Drosophila melanogaster models of HD are discussed to highlight the efficacy of this model in mapping the molecular networks implicated in a pathogenic gut-brain pathways in HD, which also shed light on how individual bacterial species or their component, like microbial amyloids in the gut, may alter the proteostasis of mHTT and brain chemistry, thereby accelerating the neurological symptoms of HD. Furthermore, the utility of the HD Drosophila model for high-throughput screening of gut-based therapeutic molecules is discussed. Reference to studies conducted in other human neurodegenerative disorders will also be made to provide insights for the development of future therapeutics and dietary strategies aimed at promoting a healthy gut environment and mitigating the systemic toxicity of mHTT.
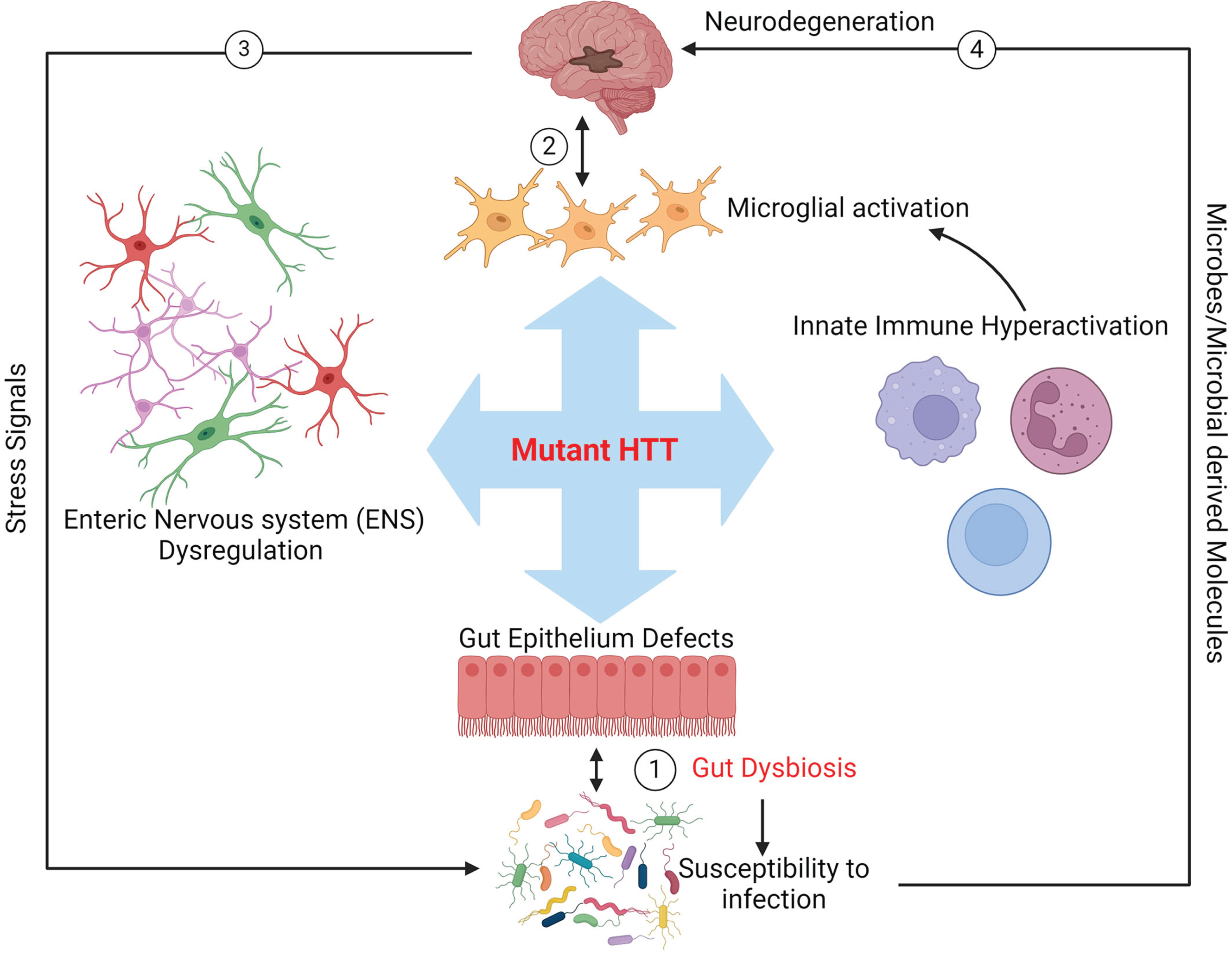
Fig.1
Schematic representation of potential mutant HTT-mediated gut dysbiosis in HD. The toxicity of mHTT may impact the biology of innate immunity, enteric nervous system (ENS), and gut epithelium, leading to aberrant blooming of pathobionts in the gut microbiota and enhancing colonization of pathogens (dysbiosis, arrow 1). Dysbiosis may in turn cause a feedback loop further exacerbating the toxic effects of mHTT in these organs and ultimately promoting neurodegeneration in the brain (arrow 2). Signals from stressed neurons in the CNS may also cause gut dysbiosis in HD (arrow 3). Furthermore, gut microbes and or microbial compounds produced in the gut could affect brain health directly (arrow 4).
GUT DYSBIOSIS IN HD
Gut dysbiosis has been associated with accelerated aging and the pathogenesis of several neurological and neurodegenerative disorders, including ASD, multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [21–23, 35–37]. In some cases, fecal transplants from ASD, AD, and PD patients into mouse models produce symptoms of disease further strengthening the impact of gut microbiota in neurodevelopment and neurodegeneration [22, 35, 38]. Two studies have demonstrated the presence of gut dysbiosis in HD patients (Table 1) [26, 27]. In an examination of fecal matters from 42 symptomatic and pre-manifest HD patients in Australia, Wasser et al., found a shift in bacterial community characterized by a lower number of species (alpha diversity) [26]. Male HD subjects displayed variations among the abundance of resident bacterial species (beta diversity) at the phylum and family levels and exemplified by lower abundance of Firmicutes, Lachnospiracea, and Akermansiacea, some of which are linked to gut barrier function, inflammation, and the production of essential metabolites like short chain fatty acids [16, 17]. Among the premanifest carriers, changes in the levels of Eubacteria halli correlated with cognitive performance (Table 1) [26].
Table 1
Clinical studies on gut dysbiosis in HD
| Ref | |
| 1. Gut dysbiosis in Huntington’s disease: associations among gut microbiota, cognitive performance and clinical outcomes (Wasser et al. 2020). | [26] |
| Major findings: | |
| •Reduced number of microbial species (alpha diversity) in the fecal microbiome of HD in comparison to healthy controls. | |
| •Altered levels among different microbial species (beta diversity). | |
| •Lowe levels of Firmicutes, Lachnospiraceae and Akkermansiaceae in males. | |
| •Variations in microbial enzymes and metabolic pathways. | |
| •High levels of Eubacterium halli correlates with diminished cognitive performance. | |
| 2. Altered gut microbiota related to inflammatory responses in patients with Huntington’s disease (Du et al. 2021) | [27] |
| Major findings: | |
| •Increased alpha and beta diversity in the microbiome of HD patients compared to healthy controls. | |
| •Associations between gut dysbiosis and systemic inflammatory biomarkers. | |
| •Abundance of Intestinimonas correlates with IL-4 concentration. | |
| •Abundance of Biolophila correlates with IL-6 concentration. | |
| •Abundance of Lactobacillus negatively correlated with Mini-Mental State Examination. |
Notably, a similar study examining the microbiome of 33 HD patients from China showed an increase in microbial diversity and altered levels of bacteria at the genus levels when compared with healthy controls in both genders [27]. The opposing findings on the alpha and beta diversities between the two studies highlight the complexity and adaptability of gut microbiota and the potential impact of geography, sampling protocols, diet and medications on the composition and physiology of gut microbiota [26, 27]. In the latter study, the abundance of gram-negative Bilophila and Intestinimonas species overlapped with elevated levels of inflammatory cytokines in the plasma of HD subjects (Table 1) [27]. Blooming of Bilophila species in the gut has consistently been linked to high fat diets, which also coincides with gut inflammation, elevated levels of inflammatory cytokines and defects in the gut barrier function (leaky gut) [39]. The correlation between the abundance of inflammatory gut bacteria and elevated cytokines in HD patients aligns with the systemic inflammation including activated microglia and proinflammatory TH17.1 immune cells in the brains of pre-manifest HD [4–7].
GUT DYSBIOSIS IN PRECLINICAL MODELS OF HD
Mouse models of HD expressing the neurotoxic mutant huntingtin exon1 (mHTTex1) also display gut dysbiosis (Table 2) [24, 25, 28]. For example, twelve-week-old R6/1 transgenic HD show an increase in species diversity and elevated levels of the phylum Bacteroidetes (gram-negative) but reduction in the phylum Firmicutes (gram- positives). Interestingly, in male R6/1 mice, gut dysbiosis coincided with lower body weight despite higher food intake, motor deficit and loose stool [24, 28]. Metabolomic studies prior to significant motor and cognitive decline also showed increased abundance of butanoate, a byproduct of butyrate metabolism produced by some members of gut bacteria [28]. The R6/2 HD mouse model, which expresses mHTTex1 with a polyQ repeat longer than that of R6/1, also has a higher ratio of Bacteroides/Firmicutes, which coincides with weight loss, shorter colon length and a leaky gut [25]. R6/1 HD mice also display changes in the gut mycobiome (fungi), which may integrate into bacterial dysbiosis (Table 2) [29]. Cross-communications between gut bacteria and fungi are important for the homeostasis of the gut microbiome. These new studies provide new directions to better understand the mechanism of gut of dysbiosis in HD and whether manipulation of the resident fungi is a therapeutic target.
Table 2
A list studies on gut dysbiosis in preclinical models of HD
| Ref | |
| 1. Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington’s disease (Kong et al. 2020) | [24] |
| Major findings | |
| •Gut dysbiosis in 12-week-old R6/1 HD mice. | |
| •Increased alpha diversity of gut microbiota in female HD mice. | |
| •‘Elevated Bacteroides and a potential decrease in Firmicutes in the fecal matters of HD mice. | |
| Gut dysbiosis coincides with weight loss and motor deficits. | |
| 2. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington’s disease (Stan et al. 2020) | [25] |
| Major findings | |
| •Gut dysbiosis in R6/2 mice. | |
| •increased relative abundance of Bacteroidetes and decreased of Firmicutes. | |
| •Increased Intestinal permeability independent of changes in the levels of tight junction proteins (occludin, zonula occludens). | |
| •Reduced body weight, body length and a decrease in colon lengths | |
| •Gene-environment-gut interactions in Huntington’s disease mice are associated with environmental modulation of the gut microbiome. | |
| 3. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease (Kong et al. 2021) | [28] |
| Major findings | |
| •Gut dysbiosis is detectable at 12 weeks of age in R6/1 HD mice prior to onset of motor symptoms. | |
| •The composition of Gut microbiome is unstable f in the pre-symptomatic HD mice. | |
| •A correlation between gut dysbiosis and changes in the plasma metabolomes of HD mice including butanoate. | |
| 4. Alterations in the Gut Fungal Community in a Mouse Model of Huntington’s Disease (Kong et al. 2022) | [29] |
| Major findings | |
| •The HD mice gut mycobiome beta diversity was significantly different from that of wild-type littermates at 12 weeks of age. | |
| •Increased alpha diversity in mycobiome of HD mice by 12 weeks of age. | |
| •Elevation of Malassezia restricta, Yarrowia lipolytica, and Aspergillus species negatively associated with the onset of symptoms in HD mice. | |
| •An inverse correlation between the elevation of fungal species and the probiotic bacterium Lactobacillus ruteri | |
| 5. Gut Bacteria Regulate the Pathogenesis of Huntington’s Disease in Drosophila Model (Chongtham et al. 2022) | [30] |
| Major findings | |
| •Gut dysbiosis in transgenic Drosophila expressing human mHTTex1 or full-length mHTT exemplified by the elevation of gram-negative and reduction of gram-positive bacteria. | |
| •Elimination of gut bacteria by antibiotics reduces the oligomerization of mHTTex1 and ameliorates motor defects in full-length mHTT transgenic flies. | |
| •Colonization with the pathobiont E. coli accelerates the oligomerization of mHTT-586, promotes motor defects and induces mortality. | |
| •The beta-carotenoid crocin, reduces the aggregation of mHTTex1, ameliorates motor defects and prolongs lifespan in transgenic flies with full-lengths mHTT. | |
| 6. Dietary fibre confers therapeutic effects in a preclinical model of Huntington’s disease (Gubert et al. 2023) | [109] |
| Major findings | |
| •Relative abundance of the phyla Actinobacteriota, Campylobacterota and Proteobacteria were decreased the relative abundance of the families. Bacteroidaceae, Oscillospiraceae and Ruminococcaceae were increased in HD mice when compared to wild-type mice on similar diet. | |
| •Fiber in the diet improved cognition and the depressive behavior in HD mice. | |
| 7. Faecal microbiota transplant ameliorates gut dysbiosis and cognitive deficits in Huntington’s disease mice (Gubert et al. 2022) | [113] |
| Major findings | |
| •Fecal microbial transplants (FMTs) from WT to HD mice improves cognition especially in females. | |
| •Males did not show any effects since FMTs did not stabilize likely due to variations in the immune status and elevated acetate. |
The origin of signals triggering gut dysbiosis in HD mice remains unknown and requires expressing the mHTT exclusively in the gut or the nervous system to identify the direction of the initial trigger. These studies would be a complex and expensive experiment to perform in mammalian models. Drosophila melanogaster with a simple gut microbiota is emerging as a powerful tool to investigate mechanistic questions for microbiota-gut-brain pathways and is a rapid and cost-effective model [40]. To better understand how mechanistically bacteria affect CNS pathology in HD, we have been utilizing Drosophila models, which emerged instrumental in understanding the role of full-length human mHTT or its N-terminal fragments on the host-microbiota communications pathways systemically or in an organ-exclusive manner. For example, transgenic Drosophila models expressing mHTTex1 exclusively in the nervous system display gut dysbiosis suggesting that disruptions in the nervous system may be a trigger of dysbiosis [30]. In this model, elimination or modification of gut microbiota lowers the aggregation and neurotoxicity of mHTTex1 further suggesting a bidirectional gut-brain loop (Table 2). Since mHTTex1 progressively accumulates in the neurons of HD patients, one possibility is that gut dysbiosis may be triggered by the nervous system. However, pre-manifest HD patients also show signs of gut dysbiosis suggesting that like the innate immune activation, dysbiosis may precede the development of neurological and psychiatric symptoms [4–7, 26, 27]. Drosophila models expressing full-length human mHTT systemically display gut dysbiosis exemplified by elevated growth of gram-negative bacteria and proportional reduction of gram-positive species prior to the development of motor defects [30]. Dysbiosis ensues in the absence of any detectable mHTT aggregates, which is predominantly formed by the abundance of mHTTex1. Thus, full-length mHTT and mHTTex1 may promote gut dysbiosis at different stages of the disease and potentially by different mechanisms, which remains to be investigated. It is worth mentioning that the assembly of gut microbiota begins with the vertical acquisition of bacterial species from the birth canal and reaches maturity in adulthood with the most diverse number of microorganisms [23]. Since mHTT is expressed from the embryonic stage and induces neurodevelopmental abnormalities [41], it is tempting to speculate that it may also affect microbial colonization in neonates and subsequent time-dependent inclusion of other members of gut microbiota. Notably, in the R6/1 HD mice major microbial shifts in the gut microbiome are seen only at ∼12 weeks of age, which coincides with the development of motor symptoms [24, 28]. However, it is important to note that neurotoxicity of mHTTex1 and fluctuations in the gut microbiota may commence long before the onset of motor defects. Conducting similar longitudinal studies in HD models expressing full-length mHTT or examination of fecal samples from HD patients, particularly juvenile HD cases, may offer insights into the mechanisms of gut dysbiosis in HD.
PATHOBIONTS AND PATHOGENS AS TRIGGERS OF NEUROLOGICAL SYMPTOMS IN HD
The notion that brain disorders may have a microbial origin is not new. Over two decades ago, Braak et al., proposed that PD may be induced by pathogenic microorganisms [42]. Since then, a substantial body of research has implicated bacteria and viruses in the etiology of PD [43]. For instance, in genetic mouse models of PD (Pink1–/–, or α-synuclein overexpression), gut infection with Citrobacter rodentium or E. coli accelerates motor impairment and the loss of dopaminergic neurons in the midbrain [44, 45]. E. coli also promotes the pathogenesis of ALS in a superoxide dismutase mutant mouse model [46]. These findings are consistent with similar dysbiotic bacteria present in the gut of some PD patients. Staining of intestinal biopsies of early diagnosed PD patients show elevated levels of Escherichia coli (E. coli). Furthermore, fecal samples of PD patients exhibit elevated levels of Enterobacteriaceae species, which are associated with inflammatory diseases [48, 49]. Collectively, these studies identify specific bacterial species in the gut as promoters of neurodegeneration in the CNS and periphery.
The role of infectious agents in the pathophysiology of HD remains unknown. Interestingly, a recent study reported on unmasking of two cases of HD by the COVID-19 infection [50]. Moreover, two different HD mouse models infected with Toxoplasma gondii, displayed aberrant activation of the inflammatory kynurenine pathway, reduced number of CD8 + T cells, elevated levels of soluble mHTT in brain, increased striatal neurodegeneration, and accelerated death [51, 52]. Colonization of transgenic Drosophila models of HD with E. coli accelerates the aggregation of an amyloidogenic mutant HTT 586 (N-terminal 586 amino acids with 120Qs) fragment, induces motor impairment and shortens lifespan. Elevated levels of Acetobacter, another gram-negative bacterium and a member of Drosophila gut microbiota, or E. coli also accelerate HD pathology and shorten lifespan in models expressing full-length mHTT [30]. In other studies, colonization of C. elegans with several gram-negative bacteria promotes proteotoxicity and the aggregation of expanded polyQ peptides, which is consistent with E. coli mediated aggregation of mHTT in Drosophila [30, 53]. These findings support a role for elevated gram-negative gut bacteria as modifiers of mHTT oligomerization and potentially HD pathogenesis in preclinical animal models. However, this does not negate the potential pathogenic contribution of gram-positive bacteria. The mechanism of how gram-negative bacterial may influence HD pathology remains to be investigated. Recent findings from our laboratory suggest that the brains of transgenic Drosophila with mHTT infected with E. coli display significant reductions in the expression of genes implicated in neurodevelopment, neurotransmission and synaptogenesis suggesting that gut infections may accelerate the mHTT-induced neurotoxicity [54]. Further investigation on “infection-driven HD pathology” and the mechanisms by which gram-negative bacteria and their metabolites change brain chemistry and the development neurological symptoms in HD will provide knowledge on the role of gut bacteria in the pathogenesis of HD.
TRANSLOCATION OF GUT BACTERIA IN THE CNS OF HD PATIENTS
One growing area of research is whether gut bacteria gain access to the CNS and promote neurodegeneration. Recent studies support gut-brain translocation of bacteria through the vagus nerve in preclinical rodents’ models of PD and AD. In these models, the presence of bacteria in the brain coincides with inflammation, microglial activation, and the accumulation aggregated proteins [55]. In AD subjects, the oral bacterial pathogen Porphyromonas gingivalis, implicated in periodontal disease, enters the CNS, and promotes AD pathology [56]. Notably, fungal antigens and bacterial DNA sequence have been detected in the post-mortem brain tissue of HD patients. Sequencing of the isolated DNA shows selective enrichment of gram-negative bacterial species like Pseudomonas, Acinetobacter, and Burkholderia in HD brains compared to other neurodegenerative disorders [57]. The notion that bacteria may gain access to the nervous system of HD patients remains a worthy area of investigation. However, it should be noted that bacteria and their components may also affect brain chemistry indirectly through neuroimmune pathways. For example, hyperactivation of Toll-like receptor 4 (TLR4) signaling in the gut has been implicated in neurodegeneration in PD and HD [58, 59].
THE ROLE OF MICROBIAL AMYLOIDS IN HD PATHOGENESIS
Cross-seeding of human amyloid proteins by microbial amyloids is another exciting area of research in neurodegenerative disorders. Functional bacterial amyloids Curli produced by members of Enterobacteriaceae like E. coli, Salmonella, and Shigella, seed the aggregation of a-synuclein protein and accelerate PD pathology in several animal models like C. elegans, mice, and rats [44, 60, 61]. Curli amyloids also activate the inflammatory pathways and are linked to inflammation in the gut, urinary tracts, and autoimmunity [62, 63]. It is noteworthy that inflammation and the induction of autoimmunity by curli amyloids involve the IKK/NF-κB pathway, which may be exacerbated by the binding of mHTT in immune cells [11, 12, 62]. Curli amyloids enhance the aggregation of mHTTex1 in mammalian cells, and in Drosophila models of HD it significantly reduces survival [30]. The potential aberrant interactions of mHTT with Curli and its downstream signaling pathways in the host may represent an example of how microbial products may contribute to the formation of neurotoxic assemblies and inflammation-mediated neurodegeneration in HD. Curli amyloids also induce disease in a mouse model of ALS suggesting that bacterial amyloids may play a significant role in the pathogenesis of multiple neurological disorders [46]. Curli is just one example of trans-kingdom modulation of proteins implicated in brain disorders as the human gut microbiota contains numerous amyloid-producing bacteria and thousands of prion-like coding sequences with the propensity to oligomerize [64]. Recent studies indicate yeast prions trigger α-synuclein- and tau-mediated pathologies in mouse models [65, 66]. Interestingly, yeast prions also seed mHTTex1 and promote the formation of toxic oligomers [67]. Thus, further investigation into the mechanism of how microbial amyloids may affect the proteostasis and neurotoxicity of mHTT may reveal valuable insights into the impact of gut-brain pathways on the pathogenesis of HD. Since bacterial amyloids enhance gut colonization and have the propensity to trigger gut inflammation, it will be interesting to examine whether HD patients harbor a higher abundance amyloid producing bacteria. Such bacterial strains may serve as useful gut biomarkers.
POTENTIAL TOXICITY OF mHTT IN GUT-IMMUNE-ENS NETWORKS
Gut epithelium, innate immune cells, and the enteric nervous system (ENS) collectively maintain the homeostasis of gut microbiota and defend against pathogenic infections [68]. At the molecular level, a list of germ line-encoded pattern recognition receptors (PRRs) such as Toll-like receptors (TLR), nucleotide-binding oligomerization (NOD)-like receptors (NLR) expressed by gut, immune, and ENS cells, monitor microbial balance by detecting pathogen-associated molecular patterns (PAMPs) including lipopolysaccharides (LPS), peptidoglycans and bacterial amyloids [69]. These MAMPs bind to PRRs and activate the IKK/NF-κB, leading to the expression of cytokines, antimicrobial peptides, and other regulatory molecules that prevent excessive growth of gut bacteria [69]. PRRs are also expressed in the CNS. For example, binding of microbial antigens to NOD-like receptor in the brains of mice regulates complex behaviors like appetite and body temperature [70]. Interestingly, in Drosophila brain, Toll receptors bind neurotrophin ligands and influence structural and behavioral mediated neuroplasticity, reinforcing the notion that microbial products may directly influence brain physiology [71]. I will discuss how mHTT may potentially disrupt these and additional pathways in the gut, immune and ENS responses to gut microbiota.
SPECULATIVE TOXICITY OF mHTT IN THE GUT EPITHELIUM
Little is known about the functions of mHTT in the gut epithelium, however, hypothetically, mHTT may alter the microbiota-gut communication pathways, namely the PRRs signaling pathways. The amplified signaling of PRR-MAMPs by the potential interaction of mHTT with the IKK/NF-κB pathway in the gut may intensify and prolong gut inflammation, which could contribute to dysbiosis. HTT plays a critical role in the development and morphogenesis of the brain and mammary tissue in mammals, and human embryos with mHTT exhibit neurodevelopmental abnormalities [41, 72, 73]. With this knowledge in mind, mHTT could hypothetically disrupt the biogenesis of gut epithelium. The gut epithelium is highly dynamic and continuously regenerates itself from a pool of intestinal stem cells (ISCs) located within the crypt of villi in the intestinal epithelium of mammals [74]. Signals from dietary cues, microbiota, and pathogens stimulate the proliferation and differentiation of ISCs, reshaping the gut architecture to adapt to various triggers [75]. mHTT is abundant in the crypt cells of Q140 and its derivative ZQ170 HD knock-in mice, however, whether it disrupts gut regeneration is unknown [54]. Notably, gastric mucosa biopsies from a small number of HD patients show reduced expression of gastrin, coinciding with decreased number of gastric cells, and a trend towards lower numbers of endocrine cells [76]. Since ISCs proliferation and differentiation into various lineages including endocrine cells is dependent on the activation of IKK/NF-κB, theoretically disruption of this pathway by the mHTT could alter the physiology of ISCs [11, 12, 18, 19].
ISCs must produce the proper ratios of different cell lineages such as enterocytes (ECs), enteroendocrine cells (EECs) and disruptions in their numbers or functions could produce various systemic complications [74]. ECs primarily play a role in diet digestion, nutrient sensing, and absorption [77]. HD patients often experience unintended weight loss, which is attributed to metabolic changes, mitochondrial dysfunction, and chorea [78]. Malabsorption of nutrients and the resulting weight loss have been reported in the R6/2 mouse model of HD [79]. ECs also serve as a fundamental component of gut barrier function, and disruption in their arrangement and physiology may contribute to a leaky gut [77]. Further research into the toxicity of mHTT in ECs is warranted since it may provide knowledge on the nutritional deficiencies and leaky gut in HD.
EECs in the gut epithelium exhibit neuronal properties and establish direct connection with neighboring vagal neurons through projections named neuropods, which transmit gut signals to the brain in mammals [80]. EECs also secrete various gut hormones, regulating food digestion, absorption, glucose homeostasis, satiety, and systemic metabolic adaptation across species [81]. Notable examples include ghrelin crucial for the regulation of satiety, as well as incretins responsible for insulin and glucagon production, both of which have been implicated in HD [82, 83]. EECs have also garnered attention in the gut-brain pathways for their potential role in propagating a-synuclein oligomers and their subsequent transport to the CNS through vagus nerve in PD models [84, 85]. Importantly, vagotomy has been associated with a lower risk of PD in humans [86]. Exploring the toxicity of mHTT in EECs along with the impact of gut microbiota on its misfolding and aggregation, and probable spread to CNS may expand our understanding of gut-triggered proteostasis of mHTT and the molecular mechanisms of how gut defects may contribute to HD pathogenesis.
LIKELY CONTRIBUTION GUT DYSBIOSIS TO ABERRANT ACTIVATION OF INNATE IMMUNITY IN HD
Innate immunity plays a crucial role in health and disease as it represents the main body’s initial defense against various pathogens and cellular damage. However, exaggerated innate immune activation has been implicated in most neurodegenerative disorders like tauopathies, AD, PD, and HD [6, 7, 22, 87]. Dysregulated innate immune activation in HD patients was reported ∼2 decades ago [6]. While acute activation of innate immune cells may offer protection in HD, persistent activation is considered pathogenic. Recent studies indicate that activation of innate immunity in the central nervous system of HD patients may lead to synaptopathies and neurodegeneration [8, 9]. It is noteworthy that ∼70% of innate immune cells are present in the gut [88]. Macrophages, monocytes, and TH17.1 cells, which directly and indirectly communicate with the gut microbiota, are activated in HD patients [4–7]. While further investigation is required, hyperactivation of innate immune cells in the gut of HD patients by microbial antigens like LPS could potentially be a significant contributor to gut dysbiosis. Deletion of wild-type huntingtin protein (WT HTT) in monocytes and phagocytes reduces lipopolysaccharide (LPS)-mediated expression and secretion of inflammatory cytokines, enhances phagocytosis, and decreases cellular resilience to stress [89]. Conversely, monocytes from individuals in the premanifest and manifest stages of HD exhibit an exaggerated response to LPS, resulting in elevated levels of inflammatory cytokines like TNF-α, IL-1β, and IL-6 [6, 12, 90, 91]. In a transgenic R6/1 HD mouse model, reducing the levels of TLR4, which primarily binds to LPS, delays the onset of HD pathology, and presents a potential gut-based therapeutic targets [59]. Understanding the delicate balance between the beneficial and detrimental aspects of innate immunity in the gut is vital for developing gut-based strategies that harness protective mechanisms while mitigating harmful inflammation, potentially slowing, or even preventing the progression of HD.
PROBABLE ENS DEFECTS IN HD
The ENS, a constellation of various neuronal and glial cell lineages in humans, is situated along the length of gut and regulates physiological systems such as blood flow, gut motility, intestinal secretion, inflammation, and microbial homeostasis [68]. Given the neurotoxicity of mHTT in the CNS, it is probable that it may also impair the development and physiology of ENS. mHTT aggregates in the ENS of Q140 and ZQ170 and R6/2 HD mice [54, 92]. However, the specific neuronal lineages that are susceptible to mHTT proteotoxicity in the ENS and the potential influence of gut microbiota on ENS degeneration remain to be investigated. mHTT may also disrupt the physiology of enteric glial cells, which play a central role in gut inflammation [93]. For instance, the activation of TLR4 in enteric glial cells has been shown to promote necrotizing colitis and is associated with reduced production of brain-derived neurotropic factor (BDNF), which is also reduced in HD [2, 94]. Although more research is needed, one may speculate that the cumulative interference of mHTT with microbial antigens interactions with PRRs signaling pathways in the gut epithelium, innate immune cells including microglia, and ENS may contribute to gut dysbiosis and the development of a pathogenic gut-brain axis in HD. Amelioration of HD pathology in TLR4 knockout mice is consistent with a pathogenic role of gut-immune pathways [59].
SPECULATIVE INVOLVEMENT OF GUT DYSBIOSIS IN MICROGLIA DYSREGULATION IN HD
The development and maturation of microglia, the innate immune cells of the central nervous system (CNS), are dependent on signals from the gut microbiota. Germ-free mice exhibit underdeveloped microglia, a condition that can be reversed by fecal transplants from pathogen-free counterparts [95]. Gut dysbiosis has been linked to microglial-mediated neuroinflammation and neuropathology in PD and AD [22, 96]. In HD activated microglia are observed years before the onset of neurological symptoms. This activation coincides with abnormal levels of proinflammatory cytokines like IL-1β, TNF-α, and IL-6, along with synaptic degeneration and cognitive decline [5, 6, 9, 97]. Additionally, post-mortem examinations of pre-manifest HD patients’ brains indicate microglial-dependent degeneration of corticostriatal synapses [8].
The mechanisms underlying microglial activation in HD are not fully understood, and whether gut dysbiosis plays a role remains to be investigated. However, since the development, maturation and inflammatory or protective functions of microglia are dependent on gut microbiota [95], one could speculate that gut dysbiosis in HD may be connected to aberrant elevation of microglia. Although indirect, studies have shown that IPSC-derived microglia from HD patients’ stem cells or microglia of HD mice are hyperactive, producing excessive proinflammatory cytokines when stimulated with microbial antigens like LPS [98, 99]. Gut microbiota, including bacteria in other body sites, are the predominant source of systemic LPS in mammals, and LPS has been identified as a major promoter of microglial and astrocytes activation in the nervous system [100]. Data on the presence of LPS in the CNS of HD patients is not available, however bacterial DNA has been detected in the brains of some HD patients [57]. Bacterial DNA are potent inducer of TLR3 and TLR9 expressed by microglia and other CNS cells, activating the cGAS-STING pathway, which is a key promoter of inflammasome activation [101]. Prolonged cGAS-STING has been implicated in neuroinflammation and the loss of dopaminergic neurons in preclinical models of PD [102]. cGAS is found in elevated levels in post-mortem brain tissue of human HD brains, and its inhibition in mouse models has shown promise in ameliorating neuroinflammation and HD pathology [103]. Notably, activation of IKK/ NF-κB pathway is a central node in the cGAS-STING pathway, which may further be exacerbated by the aberrant interaction of mHTT [101].
Microbial metabolites such as short-chain fatty acids (SCFA)—acetate, propionate, and butyrate, produced from fiber fermentation in the colon—also activate microglia and have been implicated in neuroinflammation and neurodegeneration in PD and AD [22, 104]. Surprisingly, a fiber-rich diet, metabolized by gut bacteria into SCFAs, promotes a healthy gut microbiota, ameliorates motor defects, reduces α-synuclein aggregation, and enhances the anti-inflammatory phenotype of microglia [105]. In these studies, the depletion of microglia in the CNS diminishes the protective effects of the fiber-rich diet, emphasizing the impact of gut dysbiosis on the inflammatory behavior of microglia and the progression of PD [105]. Therefore, SCFA may wield a double-edged sword, having both protective and detrimental effects. Since inflammatory microglia and TH17.1 cells in HD develop several years before the onset of neurological symptoms [4, 5, 7, 97], future studies on the link between gut microbiota and the activation of CNS immune cells may provide novel strategies to reduce their toxic phenotypes.
DIETARY AND ENVIRONMENTAL MODULATION OF GUT MICROBIOTA IN HD
Accumulating studies have identified diet as a modulator of gut microbial composition and physiology. For example, recent clinical studies indicate that diet-mediated alteration of gut microbiota influence the development immune system and the activation of the inflammatory pathways at a personal level [106]. Relevant to HD are the effects of fermented foods on increasing microbial diversity and reducing systemic inflammation [106]. Fiber-rich diets, which are predicted to increase the levels of SCFAs, are also potential candidates for influencing gut microbial diversity and reducing inflammation [105, 106]. Notably, before the gut microbiome was identified as pathogenic modality in neurodegenerative disorders, studies showed that treatment with butyrate has protective effects in mouse and Drosophila models of HD [107, 108]. However, whether these effects are mediated by changing gut microbiota and physiology remains unknown. A recent study showed that a fiber-rich diet enhances gut function, alters gut microbiota, and boosts cognition, and partially ameliorates the affective behavioral deficit in R6/1 HD mice (Table 2) [109]. Unexpectedly, in healthy humans a fiber-rich diet alone for a short period did not alter markers of inflammation or the diversity of gut microbiota [106]. Thus, the effects of SCFAs on the composition of gut microbiota, how they may integrate into the environmental regulation of HD pathology demand further investigation. Amelioration of gut dysbiosis in brain disorders by probiotics dubbed as “psychobiotics” has proven protective in depression and sleep disorders in pilot clinical studies [110]. However, a recent small clinical pilot trial with probiotics in HD patients did not ameliorate gut dysbiosis or any of the clinical parameters analyzed thus, requiring further investigations on the benefits of probiotics in HD [111]. Clinical investigations of fecal microbial transplants (FMTs) as remedies for various disorders including PD are gaining momentum [112]. Interestingly, FMT from WT to R6/1 HD mice improves cognition in female mice, highlighting the need for further investigations [113]. Replication of these studies may provide a relatively safe therapeutic strategy for HD. Prebiotics (compounds, which promote the growth of beneficial bacteria in the host) offer novel tools and candidate therapeutics to modulate gut microbiota and lower the levels of inflammatory biomarkers, as was reported for fermented food products in clinical trials [106]. Nutraceuticals are also attractive gut-modifying therapeutic candidates for HD. For example, crocin, a beta carotenoid promotes the growth of beneficial bacteria in the gut, reduces inflammation, increases the expression of neurotrophins like BDNF, prevents neurodegeneration in rodent brain ischemia models, and enhances memory in AD models [114–116]. Furthermore, crocin ameliorates LPS-induced neuroinflammation in PD models by reducing the level and activity of the inflammasome pathways, which are also elevated in HD [98, 117]. Notably, in Drosophila models of HD, crocin reduces the levels of gram- negative bacteria, prevents colonization of exogenous human pathobiont E. coli, reduces the buildup of toxic oligomers of mHTT, ameliorates motor defects, increases lifespan, and prevents mHTT mediated downregulation of gene products in the nervous system [30, 54]. These findings are exciting since crocin has proven protective in clinical trials and other preclinical models [113–118]. The speed and low cost of Drosophila allow for simultaneous testing of a large number of gut-based compounds or probiotics, which will expedite the discovery of therapeutics to treat the neurological and psychiatric symptoms of HD.
CONCLUDING REMARKS
The concept that mHTT may play a disruptive role in organs involved in gut microbiota homeostasis, gut health and development, nutrition, immunity, and gut-brain communications, adds to the functional repertoire of both WT and mHTT. Extending these studies could help to define how gene-environment interactions may impact the stability, turnover and toxicity of mHTT systemically. These studies also hold the promise of introducing novel gut-based therapeutic interventions for HD and other brain disorders with overlapping symptoms. One could imagine reducing the levels and systemic toxicity of mHTT from the gut with dietary compounds or specific small molecules. Simultaneously, implementing strategies to enhance the protective properties of WT HTT in the gut-brain organs may prove beneficial.
While we have knowledge of various biological properties of HTT in the nervous system, little is known of its functions in other organs. The presence of HTT in the gut and gut stem cells heralds various functions, which are yet to be discovered. Intriguingly, when WT human HTT is expressed systemically or selectively in the gut of Drosophila, it alters the composition and physiology of the gut microbiota. One noteworthy phenotype of WT HTT is its ability to lower the abundance of gram-negative bacteria like Acetobacter, a member of Enterobacteriaceae, while simultaneously elevating the levels of gram-positive Lactobacillus species [30]. An interesting possibility is that HTT in the gut epithelium influences pathways that determine the abundance of specific bacterial species, a function which is lost in mHTT. Given the known functions of HTT in neurodevelopment, it would be valuable to investigate whether its presence in gut stem cells affects development and the heterogeneity of various professional epithelial cells, including EECs, which exhibit neuronal properties and communicate with the nervous system [41, 80].
mHTT accelerates and prolongs the response of innate immune cells from HD patients to microbial antigens by augmenting the activity of the IKK/NF-κB pathway, which plays a crucial role in microbiota-immune system homeostasis. IKKs are key players in the communication between microbiota in the gut epithelium and innate immune cells. Understanding the outcomes of IKK/mHTT interactions in these organs, within the context of gut homeostasis, could provide valuable insights for drug development. It is noteworthy that IKKs or their intermediary kinases phosphorylate HTT at its N-terminus, impacting turnover and the proteotoxicity of mHTT [119–121]. Targeting these interactions may provide opportunities to influence the levels of WT and mHTT in the nervous system through interventions in the gut. In this context, it is noteworthy that genetic modulation of IKKβ in the brain regulates neurotoxicity and HD pathology in R6/1 HD mouse model [122].
It is likely that WT and mHTT serve overlapping functions in the CNS and ENS. mHTT misfolds and oligomerizes in the ENS, potentially enhancing its neurotoxic properties [54, 92]. In the context of gut microbiota, it would be worthy to investigate whether dysbiosis and gut pathogens influence the oligomerization of mHTT in the ENS and whether interventions could delay this process. Moreover, identifying the specific neuronal lineages in the ENS affected by mHTT may offer valuable insights into gut pathology and disruptions of microbial homeostasis in HD. Ultimately, these studies will provide novel and specific targets to develop precise and effective gut-based therapies.
ACKNOWLEDGMENTS
The author is grateful to Dr. Marian Difiglia for editing suggestions, and Reet Chobey for the artwork.
FUNDING
Research on gut-brain pathways in HD in the author’s laboratory is supported by an award from the Hereditary Disease Foundation.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
REFERENCES
[1] | Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: :971–83. |
[2] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) ;1: :15005. |
[3] | Chuang CL , Demontis F . Systemic manifestation and contribution of peripheral tissues to Huntington’s disease pathogenesis. Ageing Res Rev. (2021) ;69: :101358. |
[4] | Tai YF , Pavese N , Gerhard A , Tabrizi SJ , Barker RA , Brooks DJ , et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. (2007) ;130: (Pt 7):1759–66. |
[5] | Politis M , Lahiri N , Niccolini F , Su P , Wu K , Giannetti P , et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol Dis. (2015) ;83: :115–21. |
[6] | Björkqvist M , Wild EJ , Thiele J , Silvestroni A , Andre R , Lahiri N , et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. (2008) ;205: :1869–77. |
[7] | von Essen MR , Hellem MN , Vinther-Jensen T , Ammitzboll C , Hansen RH , Hjermind LE , et al. Early intrathecal T helper 17.1 cell activity in Huntington disease. Ann Neurol. (2020) ;87: :246–55. |
[8] | Wilton DK , Mastro K , Heller MD , Gergits FW , Willing CR , Fahey JB , et al. Microglia and complement mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington’s disease. Nat Med. (2023) ;29: (11):2866–84. |
[9] | Lee H , Fenster RJ , Pineda SS , Gibbs WS , Mohammadi S , Davila-Velderrain J , et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron. (2020) ;107: (5):891-908.e8. |
[10] | Crotti A , Benner C , Kerman BE , Gosselin D , Lagier-Tourenne C , Zuccato C , et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. (2014) ;17: (4):513–21. |
[11] | Khoshnan A , Ko J , Watkin EE , Paige LA , Reinhart PH , Patterson PH . Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci. (2004) ;24: :7999–8008. |
[12] | Trager U , Andre R , Lahiri N , Magnusson-Lind A , Weiss A , Grueninger S , et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain. (2014) ;137: :819–33. |
[13] | Khoshnan A , Sabbaugh A , Calamini B , Marinero SA , Dunn DE , Yoo JH , et al. IKKbeta and mutant huntingtin interactions regulate the expression of IL-34: implications for microglial-mediated neurodegeneration in HD. Hum Mol Genet. (2017) ;26: :4267–77. |
[14] | Donaldson GP , Lee SM , Mazmanian SK . Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. (2016) ;14: (1):20–32. |
[15] | Leviatan S , Shoer S , Rothschild D , Gorodetski M , Segal E . An expanded reference map of the human gut microbiome reveals hundreds of previously unknown species. Nat Commun. (2022) ;13: :3863. |
[16] | Miri S , Yeo J , Abubaker S , Hammami R . Neuromicrobiology, an emerging neurometabolic facet of the gut microbiome? Front Microbiol. (2023) ;14: :1098412. |
[17] | Belkaid Y , Hand TW . Role of the microbiota in immunity and inflammation. Cell. (2014) ;157: (1):121–41. |
[18] | Zaph C , Troy AE , Taylor BC , Berman-Booty LD , Guild KJ , Du Y , et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. (2007) ;446: (7135):552–6. |
[19] | Brischetto C , Krieger K , Klotz C , Krahn I , Kunz S , Kolesnichenko M , et al. NF-κB determines Paneth versus goblet cell fate decision in the small intestine. Development. (2021) ;148: (21):dev199683. |
[20] | Spencer SP , Fragiadakis GK , Sonnenburg JL . Pursuing human-relevant gut microbiota-immune interactions. Immunity. (2019) ;51: (2):225–39. |
[21] | Hsiao EY , McBride SW , Hsien S , Sharon G , Hyde ER , McCue T , et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) ;155: :1451–63. |
[22] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. (2016) ;167: :1469–80. |
[23] | Ratsika A , Cruz Pereira JS , Lynch CMK , Clarke G , Cryan JF . Microbiota-immune-brain interactions: A lifespan perspective. Curr Opin Neurobiol. (2023) ;78: :102652. |
[24] | Kong G , Cao KL , Judd LM , Li S , Renoir T , Hannan AJ . Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington’s disease. Neurobiol Dis. (2020) ;135: :104268. |
[25] | Stan TL , Soylu-Kucharz R , Burleigh S , Prykhodko O , Cao L , Franke N , et al. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington’s disease. Sci Rep. (2020) ;10: :18270. |
[26] | Wasser CI , Mercieca EC , Kong G , Hannan AJ , McKeown SJ , Glikmann-Johnston Y , et al. Gut dysbiosis in Huntington’s disease: associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. (2020) ;2: :fcaa110. |
[27] | Du G , Dong W , Yang Q , Yu X , Ma J , Gu W , et al. Altered gut microbiota related to inflammatory responses in patients with Huntington’s disease. Front Immunol. (2021) ;11: :603594. |
[28] | Kong G , Ellul S , Narayana VK , Kanojia K , Ha HTT , Li S , et al. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease. Neurobiol Dis. (2021) ;148: :105199. |
[29] | Kong G , Lê Cao KA , Hannan AJ . Alterations in the gut fungal community in a mouse model of Huntington’s disease. Microbiol Spectr. (2022) ;10: (2):e0219221. |
[30] | Chongtham A , Yoo JH , Chin TM , Akingbesote ND , Huda A , Marsh JL , et al. Gut bacteria regulate the pathogenesis of Huntington’s disease in Drosophila model. Front Neurosci. (2022) ;16: :902205. |
[31] | Martínez-Lazcano JC , González-Guevara E , Boll C , Cárdenas G . Gut dysbiosis and homocysteine: a couple for boosting neurotoxicity in Huntington disease. Rev Neurosci. (2022) ;33: (7):819–27. |
[32] | Love CJ , Masson BA , Gubert C , Hannan AJ . The microbiota-gut-brain axis in Huntington’s disease. Int Rev Neurobiol. (2022) ;167: :141–184. |
[33] | Sharma G , Biswas SS , Mishra J , Navik U , Kandimalla R , Reddy PH , Bhatti GK , Bhatti JS . Gut microbiota dysbiosis and Huntington’s disease: Exploring the gut-brain axis and novel microbiota-based interventions. Life Sci. (2023) ;328: :121882. |
[34] | Ekwudo MN , Gubert C , Hannan AJ . The microbiota-gut-brain axis in Huntington’s disease: pathogenic mechanisms and therapeutic targets. FEBS J. (2024) ; doi: 10.1111/febs.17102. |
[35] | Grabrucker S , Marizzoni M , Silajdžić E , Lopizzo N , Mombelli E , Nicolas S , et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain. (2023) ;18: :awad303. |
[36] | Zeng Q , Shen J , Chen K , Zhou J , Liao Q , Lu K , et al. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci Rep. (2020) ;10: :12998. |
[37] | López-Otín C , Blasco MA , Partridge L , Serrano M , Kroemer G . Hallmarks of aging: An expanding universe. Cell. (2023) ;19;186: (2):243–78. |
[38] | Sharon G , Cruz NJ , Kang DW , Gandal MJ , Wang B , Kim YM , et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. (2019) ;177: (6):1600-1618.e17. |
[39] | Natividad JM , Lamas B , Pham HP , Michel ML , Rainteau D , Bridonneau C , et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat Commun. (2018) ;9: (1):2802. |
[40] | Ludington WB , Ja WW . Drosophila as a model for the gut microbiome. PLoS Pathog. (2020) ;16: (4):e1008398. |
[41] | Barnat M , Capizzi M , Aparicio E , Boluda S , Wennagel D , Kacher R , et al. Huntington’s disease alters human neurodevelopment. Science. (2020) ;369: (6505):787–93. |
[42] | Braak H , de Vos RA , Bohl J , Del Tredici K . Gastric alpha-synuclein immunoreactive inclusions In Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. (2006) ;396: (1):67–72. |
[43] | Cannon T , Gruenheid S . Microbes and Parkinson’s disease: from associations to mechanisms. Trends Microbiol. (2022) ;30: (8):749–60. |
[44] | Sampson TR , Challis C , Jain N , Moiseyenko A , Ladinsky MS , Shastri GG , et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife. (2020) ;9: :e53111. |
[45] | Matheoud D , Cannon T , Voisin A , Penttinen AM , Ramet L , Fahmy AM , Ducrot C , et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1-/- mice. Nature. (2019) ;571: (7766):565–9. |
[46] | Kurlawala Z , McMillan JD , Singhal RA , Morehouse J , Burke DA , Sears SM , et al. Mutant and curli-producing E.coli enhance the disease phenotype in a hSOD1-G93A mouse model of ALS. Sci Rep. (2023) ;13: (1):5945. |
[47] | Nakahara K , Nakane S , Ishii K , Ikeda T , Ando Y . Gut microbiota of Parkinson’s disease in an appendectomy cohort: a preliminary study. Sci Rep. (2023) ;13: (1):2210. |
[48] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One. (2011) ;6: (12):e28032. |
[49] | Baldelli V , Scaldaferri F , Putignani L , Del Chierico F . The role of enterobacteriaceae in gut microbiota dysbiosis in inflammatory bowel diseases. Microorganisms. (2021) ;9: (4):697. |
[50] | Palermo G , Di Fonzo A , Francesconi A , Unti E , Ceravolo R . Two cases of Huntington’s disease unmasked by the COVID-19 pandemic. Neurol Sci. (2023) ;44: :811–3. |
[51] | Donley DW , Olson AR , Raisbeck MF , Fox JH , Gigley JP . Huntington’s disease mice infected with toxoplasma gondii demonstrate early kynurenine pathway activation, altered CD8+T-cell responses, and premature mortality. PLoS One. (2016) ;11: (9):e0162404. |
[52] | Donley DW , Jenkins T , Deiter C , Campbell R , Realing M , Chopra V , et al. Latent Toxoplasma gondii infection increases soluble mutant huntingtin and promotes neurodegeneration in the YAC128 mouse model of Huntington’s disease. bioRxiv. (2019) ; doi: https://doi.org/10.1101/550624 [Preprint]. Posted February 15, 2019. |
[53] | Walker AC , Bhargava R , Vaziriyan-Sani AS , Pourciau C , Donahue ET , Dove AS , et al. Colonization of the Caenorhabditis elegans gut with human enteric bacterial pathogens leads to proteostasis disruption that is rescued by butyrate. PLoS Pathog. (2021) ;17: (5):e1009510. |
[54] | Khoshnan A . Hereditary Disease Foundation meeting. (2022) . Boston, USA. |
[55] | Thapa M , Kumari A , Chin CY , Choby JE , Jin F , Bogati B , et al. Translocation of gut commensal bacteria to the brain. bioRxiv. (2023) ; doi: https://doi.org/10.1101/2023.08.30.555630 [Preprint]. Posted September 01, 2023. |
[56] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) ;5: (1):eaau3333. |
[57] | Alonso R , Pisa D , Carrasco L . Brain cmicrobiota in Huntington’s disease patients. Front Microbiol. (2019) ;10: :2622. |
[58] | Perez-Pardo P , Dodiya HB , Engen PA , Forsyth CB , Huschens AM , Shaikh M , et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. (2019) ;68: (5):829–43. |
[59] | Martínez-Gopar PE , Pérez-Rodríguez MJ , Angeles-López QD , Tristán-López L , González-Espinosa C , Pérez-Severiano F . Toll-like receptor 4 plays a significant role in the biochemical and neurological alterations observed in two distinct mice models of Huntington’s disease. Mol Neurobiol. (2023) ;60: (5):2678–90. |
[60] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep. (2016) ;6: :34477. |
[61] | Wang C , Lau CY , Ma F , Zheng C . Genome-wide screen identifies curli amyloid fibril as a bacterial component promoting host neurodegeneration. Proc Natl Acad Sci U S A. (2021) ;118: (34):e2106504118. |
[62] | Tursi SA , Lee EY , Medeiros NJ , Lee MH , Nicastro LK , Buttaro B , et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. (2017) ;13: (4):e1006315. |
[63] | Luna-Pineda VM , Moreno-Fierros L , Cázares-Domínguez V , Ilhuicatzi-Alvarado D , Ochoa SA , Cruz-Córdova A , et al. Curli of uropathogenic Escherichia coli enhance urinary tract colonization as a fitness factor. Front Microbiol. (2019) ;10: :2063. |
[64] | Balistreri A , Goetzler E , Chapman M . Functional amyloids are the rule rather than the exception in cellular biology. Microorganisms. (2020) ;8: (12):1951. |
[65] | Meng L , Liu C , Li Y , Chen G , Xiong M , Yu T , et al. The yeast prion protein Sup35 initiates α-synuclein pathology in mouse models of Parkinson’s disease. Sci Adv. (2023) ;9: (44):eadj1092. |
[66] | Meng L , Liu C , Liu M , Chen J , Liu C , Zhang Z , Chen G , Zhang Z . The yeast protein Ure2p triggers Tau pathology in a mouse model of tauopathy. Cell Rep. (2023) ;42: (11):113342. |
[67] | Gropp MHM , Klaips CL , Hartl FU . Formation of toxic oligomers of polyQ-expanded Huntingtin by prion-mediated cross-seeding. Mol Cell. (2022) ;82: (22):4290-4306.e11. |
[68] | Macpherson AJ , Pachnis V , Prinz M . Boundaries and integration between microbiota, the nervous system, and immunity. Immunity. (2023) ;56: (8):1712–26. |
[69] | Keogh CE , Rude KM , Gareau MG . Role of pattern recognition receptors and the microbiota in neurological disorders. J Physiol. (2021) ;599: (5):1379–89. |
[70] | Gabanyi I , Lepousez G , Wheeler R , Vieites-Prado A , Nissant A , Chevalier G , et al. Bacterial sensing via neuronal Nod2 regulates appetite and body temperature. Science. (2022) ;376: (6590):eabj3986. |
[71] | Li G , Hidalgo A . The toll route to structural brain plasticity. Front Physiol. (2021) ;12: :679766. |
[72] | Capizzi M , Carpentier R , Denarier E , Adrait A , Kassem R , Mapelli M , et al. Developmental defects in Huntington’s disease show that axonal growth and microtubule reorganization require NUMA1. Neuron. (2022) ;110: (1):36–50. |
[73] | Elias S , Thion MS , Yu H , Sousa CM , Lasgi C , Morin X , Humbert S . Huntingtin regulates mammary stem cell division and differentiation. Stem Cell Reports. (2014) ;2: (4):491–506. |
[74] | Stojanović O , Miguel-Aliaga I , Trajkovski M . Intestinal plasticity and metabolism as regulators of organismal energy homeostasis. Nat Metab. (2022) ;4: (11):1444–58. |
[75] | Deng H , Gerencser AA , Jasper H . Signal integration by Ca(2+) regulates intestinal stem-cell activity. Nature. (2015) ;528: (7581):212–7. |
[76] | McCourt AC , O’Donovan KL , Ekblad E , Sand E , Craufurd D , Rosser A , et al. Characterization of gastric mucosa biopsies reveals alterations in Huntington’s disease. PLoS Curr. (2015) ;7: :ecurrents.hd.858b4cc7f235df068387e9c20c436a79. |
[77] | Duca FA , Waise TMZ , Peppler WT , Lam TKT . The metabolic impact of small intestinal nutrient sensing. Nat Commun. (2021) ;12: (1):903. |
[78] | Ogilvie AC , Nopoulos PC , Schultz JL . Quantifying the onset of unintended weight loss in Huntington’s disease: a retrospective analysis of Enroll-HD. J Huntingtons Dis. (2021) ;10: (4):485–92. |
[79] | van der Burg JM , Winqvist A , Aziz NA , Maat-Schieman ML , Roos RA , et al. Gastrointestinal dysfunction contributes to weight loss in Huntington’s disease mice. Neurobiol Dis. (2011) ;44: (1):1–8. |
[80] | Kaelberer MM , Buchanan KL , Klein ME , Barth BB , Montoya MM , Shen X , et al. A gut-brain neural circuit for nutrient sensory transduction. Science. (2018) ;361: (6408):eaat5236. |
[81] | Gribble FM , Reimann F . Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat Rev Endocrinol. (2019) ;15: :226–37. |
[82] | Sjögren M , Duarte AI , McCourt AC , Shcherbina L , Wierup N , Björkqvist M . Ghrelin rescues skeletal muscle catabolic profile in the R6/2 mouse model of Huntington’s disease. Sci Rep. (2017) ;7: (1):13896. |
[83] | Montojo MT , Aganzo M , González N . Huntington’s disease and diabetes: chronological sequence of its association. J Huntingtons Dis. (2017) ;6: (3):179–88. |
[84] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. (2019) ;103: (4):627-641.e7. |
[85] | Challis C , Hori A , Sampson TR , Yoo BB , Challis RC , Hamilton AM , et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. (2020) ;23: (3):327–36. |
[86] | Svensson E , Horváth-Puhó E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. (2015) ;78: (4):522–9. |
[87] | Sanford SAI , Miller LVC , Vaysburd M , Keeling S , Tuck BJ , Clark J , et al. The type-I interferon response potentiates seeded tau aggregation and exacerbates tau pathology. Alzheimers Dement. (2024) ;20: (2):1013–25. |
[88] | Bostick JW , Schonhoff AM , Mazmanian SK . Gut microbiome-mediated regulation of neuroinflammation. Curr Opin Immunol. (2022) ;76: :102177. |
[89] | O’Regan GC , Farag SH , Ostroff GR , Tabrizi SJ , Andre R . Wild-type huntingtin regulates human macrophage function. Sci Rep. (2020) ;10: (1):17269. |
[90] | Träger U , Andre R , Magnusson-Lind A , Miller JR , Connolly C , Weiss A , et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol Dis. (2015) ;73: :388–98. |
[91] | Miller JR , Lo KK , Andre R , Hensman Moss DJ , Träger U , Stone TC , et al. RNA-Seq of Huntington’s disease patient myeloid cells reveals innate transcriptional dysregulation associated with proinflammatory pathway activation. Hum Mol Genet. (2016) ;25: (14):2893–904. |
[92] | Moffitt H , McPhail GD , Woodman B , Hobbs C , Bates GP . Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington’s disease. PLoS One. (2009) ;4: (11):e8025. |
[93] | Margolis KG , Gershon MD . Enteric neuronal regulation of intestinal inflammation. Trends Neurosci. (2016) ;39: (9):614–24. |
[94] | Kovler ML , Gonzalez Salazar AJ , Fulton WB , Lu P , Yamaguchi Y , Zhou Q , et al. Toll-like receptor 4-mediated enteric glia loss is critical for the development of necrotizing enterocolitis. Sci Transl Med. (2021) ;13: (612):eabg3459. 9. |
[95] | Erny D , Hrabě de Angelis AL , Jaitin D , Wieghofer P , Staszewski O , et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) ;18: (7):965–77. |
[96] | Subhramanyam CS , Wang C , Hu Q , Dheen ST . Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. (2019) ;94: :112–20. |
[97] | Politis M , Pavese N , Tai YF , Kiferle L , Mason SL , Brooks DJ , et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. (2011) ;32: (2):258–70. |
[98] | O’Regan GC , Farag SH , Casey CS , Wood-Kaczmar A , Pocock JM , Tabrizi SJ , et al. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J Neuroinflammation. (2021) ;18: (1):94. |
[99] | Connolly C , Magnusson-Lind A , Lu G , Wagner PK , Southwell AL , Hayden MR , et al. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience. (2016) ;325: :74–88. |
[100] | Kalyan M , Tousif AH , Sonali S , Vichitra C , Sunanda T , Praveenraj SS , et al. Role of endogenous lipopolysaccharides in neurological disorders. Cells. (2022) ;11: (24):4038. |
[101] | Patel DJ , Yu Y , Xie W . cGAMP-activated cGAS-STING signaling: its bacterial origins and evolutionary adaptation by metazoans. Nat Struct Mol Biol. (2023) ;30: (3):245–60. |
[102] | Szego EM , Malz L , Bernhardt N , Rösen-Wolff A , Falkenburger BH , Luksch H . Constitutively active STING causes neuroinflammation and degeneration of dopaminergic neurons in mice. Elife. (2022) ;11: :e81943. |
[103] | Jauhari A , Baranov SV , Suofu Y , Kim J , Singh T , Yablonska S , et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Invest. (2020) ;130: (6):3124–36. |
[104] | Colombo AV , Sadler RK , Llovera G , Singh V , Roth S , Heindl S , et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. Elife. (2021) ;10: :e59826. |
[105] | Abdel-Haq R , Schlachetzki JCM , Boktor JC , Cantu-Jungles TM , Thron T , Zhang M , et al. A prebiotic diet modulates microglial states and motor deficits in α-synuclein overexpressing mice. Elife. (2022) ;11: :e81453. |
[106] | Wastyk HC , Fragiadakis GK , Perelman D , Dahan D , Merrill BD , Yu FB , et al. Gut-microbiota-targeted diets modulate human immune status. Cell. (2021) ;184: (16):4137–53. |
[107] | Ferrante RJ , Kubilus JK , Lee J , Ryu H , Beesen A , Zucker B , et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. (2003) ;23: (28):9418–27. |
[108] | Steffan JS , Bodai L , Pallos J , Poelman M , McCampbell A , Apostol BL , et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. (2001) ;413: (6857):739–43. |
[109] | Gubert C , Kong G , Costello C , Adams CD , Masson BA , Qin W , et al. Dietary fibre confers therapeutic effects in a preclinical model of Huntington’s disease. Brain Behav Immun. (2024) ;116: :404–418. |
[110] | Berding K , Bastiaanssen TFS , Moloney GM , Boscaini S , Strain CR , Anesi A , et al. Feed your microbes to deal with stress: a psychobiotic diet impacts microbial stability and perceived stress in a healthy adult population. Mol Psychiatry. (2023) ;28: (2):601–10. |
[111] | Wasser CI , Mercieca EC , Kong G , Hannan AJ , Allford B , McKeown SJ , et al. A randomized controlled trial of probiotics targeting gut dysbiosis in Huntington’s disease. J Huntingtons Dis. (2023) ;12: (1):43–55. |
[112] | Cheng Y , Tan G , Zhu Q , Wang C , Ruan G , Ying S , et al. Efficacy of fecal microbiota transplantation in patients with Parkinson’s disease: clinical trial results from a randomized, placebo-controlled design. Gut Microbes. (2023) ;15: (2):2284247. |
[113] | Gubert C , Choo JM , Love CJ , Kodikara S , Masson BA , Liew JJM , et al. Faecal microbiota transplant ameliorates gut dysbiosis and cognitive deficits in Huntington’s disease mice. Brain Commun. (2022) ;4: (4):fcac205. |
[114] | Zhang Y , Geng J , Hong Y , Jiao L , Li S , Sun R , et al. Orally administered crocin protects against cerebral ischemia/reperfusion injury through the metabolic transformation of crocetin by gut microbiota. Front Pharmacol. (2019) ;10: :440. |
[115] | Moghadasi M , Akbari F , Najafi P . Interaction of aerobic exercise and crocin improves memory, learning and hippocampal tau and neurotrophins gene expression in rats treated with trimethyltin as a model of Alzheimer’s disease. Mol Biol Rep. (2024) ;51: (1):111. |
[116] | Salem M , Shaheen M , Tabbara A , Borjac J . Saffron extract and crocin exert anti-inflammatory and anti-oxidative effects in a repetitive mild traumatic brain injury mouse model. Sci Rep. (2022) ;12: (1):5004. |
[117] | Alizadehmoghaddam S , Pourabdolhossein F , Najafzadehvarzi H , Sarbishegi M , Saleki K , Nouri HR . Crocin attenuates the lipopolysaccharide-induced neuroinflammation via expression of AIM2 and NLRP1 inflammasome in an experimental model of Parkinson’s disease. Heliyon. (2024) ;10: (3):e25523. |
[118] | Kouchaki E , Rafiei H , Ghaderi A , Azadchehr MJ , Safa F , Omidian K , et al. Effects of crocin on inflammatory biomarkers and mental health status in patients with multiple sclerosis: A randomized, double-blinded clinical trial. Mult Scler Relat Disord. (2024) ;83: :105454. |
[119] | Thompson LM , Aiken CT , Kaltenbach LS , Agrawal N , Illes K , Khoshnan A , et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. (2009) ;187: (7):1083–99. |
[120] | Bustamante MB , Ansaloni A , Pedersen JF , Azzollini L , Cariulo C , Wang ZM , et al. Detection of huntingtin exon 1 phosphorylation by Phos-Tag SDS-PAGE: Predominant phosphorylation on threonine 3 and regulation by IKKβ. Biochem Biophys Res Commun. (2015) ;463: (4):1317–22. |
[121] | Cariulo C , Azzollini L , Verani M , Martufi P , Boggio R , Chiki A , et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc Natl Acad Sci U S A. (2017) ;114: (50):E10809–E10818. |
[122] | Ochaba J , Fote G , Kachemov M , Thein S , Yeung SY , Lau AL , et al. IKKβ slows Huntington’s disease progression in R6/1 mice. Proc Natl Acad Sci U S A. (2019) ;116: (22):10952–61. |

