
Modifiers of Somatic Repeat Instability in Mouse Models of Friedreich Ataxia and the Fragile X-Related Disorders: Implications for the Mechanism of Somatic Expansion in Huntington’s Disease
Abstract
Huntington’s disease (HD) is one of a large group of human disorders that are caused by expanded DNA repeats. These repeat expansion disorders can have repeat units of different size and sequence that can be located in any part of the gene and, while the pathological consequences of the expansion can differ widely, there is evidence to suggest that the underlying mutational mechanism may be similar. In the case of HD, the expanded repeat unit is a CAG trinucleotide located in exon 1 of the huntingtin (HTT) gene, resulting in an expanded polyglutamine tract in the huntingtin protein. Expansion results in neuronal cell death, particularly in the striatum. Emerging evidence suggests that somatic CAG expansion, specifically expansion occurring in the brain during the lifetime of an individual, contributes to an earlier disease onset and increased severity. In this review we will discuss mouse models of two non-CAG repeat expansion diseases, specifically the Fragile X-related disorders (FXDs) and Friedreich ataxia (FRDA). We will compare and contrast these models with mouse and patient-derived cell models of various other repeat expansion disorders and the relevance of these findings for somatic expansion in HD. We will also describe additional genetic factors and pathways that modify somatic expansion in the FXD mouse model for which no comparable data yet exists in HD mice or humans. These additional factors expand the potential druggable space for diseases like HD where somatic expansion is a significant contributor to disease impact.
INTRODUCTION
Huntington’s disease (HD) is a neurodegenerative disorder characterized most typically by abnormal involuntary movements or chorea, together with pro-gressive and debilitating motor, behavioral and cognitive impairments (reviewed in [1]). HD is inherited in an autosomal dominant manner and is invariably caused by the expansion of a CAG repeat located in exon 1 of the HTT gene [2]. Expansion results in the production of a mutant HTT protein with an expanded polyglutamine tract, aberrant HTT splicing isoforms [3], novel HTT antisense transcripts [4], elevated levels of CAG-containing microRNAs [5], and a number of abnormal proteins generated by repeat-associated non-AUG (RAN) translation [6], with expansion ultimately resulting in neuronal dysfunction and death. Pathology is seen in carriers of alleles with >35 CAG repeats, with alleles having 36–39 CAGs showing re-duced penetrance [2]. The length of the expanded repeat is a major determinant of both the likelihood of further expansion and the age at disease onset [7–11]. Progressive increases in repeat number on germline transmission account for the genetic anticipation seen in HD families [11, 12], as well as transitions from high normal alleles (27–35 CAGs) to disease alleles (i.e., de novo mutations) and transitions from alleles associated with incomplete penetrance to those causing completely penetrant disease [13–15].
The CAG repeat is also highly unstable in some somatic tissues, expanding progressively over time in a length-dependent and cell type/tissue-specific manner [16]. Expansions occur in postmitotic neurons [17, 18], with expansions in some brain regions like cortex and striatum being typically more extensive than expansions in blood [19]. A number of lines of evidence support the idea that somatic expansion is an important disease modifier. This includes the fact that larger somatic expansions in HD postmortem brain are associated with an earlier disease onset [20], and the observation that HD patients with higher levels of somatic expansion measured in blood have worse HD outcomes [21]. Furthermore, individuals with pure CAG repeat tracts have an earlier age at onset than individuals with CAA interruptions at the 3′ end of the repeat tract [21–23]. While such interruptions do not change the number of glutamines in the PolyQ tract, they result in a reduction in somatic expansion [23]. This suggests that the rate of further repeat expansion during an individual’s lifetime is an important contributor to HD onset. Finally, as will be discussed later in this review, compelling evidence from recent genome-wide association (GWA) and transcription-wide association (TWA) studies strongly implicates known genetic modifiers of somatic expansion as significant modifiers of the onset of HD motor symptoms, accounting for 30–50% of the variation in age at onset [21, 22, 24–27].
A large number of other degenerative diseases are also caused by repeat expansion, with the largest group being those that are also caused by expanded CAG/CTG repeats such as in Myotonic Dystrophy type 1 (DM1) and numerous Spinocerebellar ataxias (SCAs) (see Fig. 1). In addition, many other disorders are caused by the expansion of trinucleotide repeats other than CAG/CTG or repeats with a a variety of other unit sizes and sequences (reviewed in [28]). Furthermore, there is some evidence to suggest that tandem repeats may contribute to autism spectrum disorders [29], and in addition to those cases where the repeat is sufficient to cause disease, variations in the number of tandem repeats can also be a modifier of disease severity or age at disease onset, as in X-linked dystonia parkinsonism [30]. The disease-associated repeats share some common features including the ability to form non-canonical nucleic acid secondary structures as illustrated at the bottom of Fig. 1 (reviewed in [31]). Notably, despite the wide range of structures formed, these structures all have regions of single-strandedness that may make them prone to DNA damage. The repetitive nature of the repeat tract, coupled with the ability to form secondary structures, also increases the possibility of out-of-register reannealing during replication, transcription or repair that could potentially generate a substrate upon which the expansion process can act.
Fig. 1
The repeat expansion diseases.
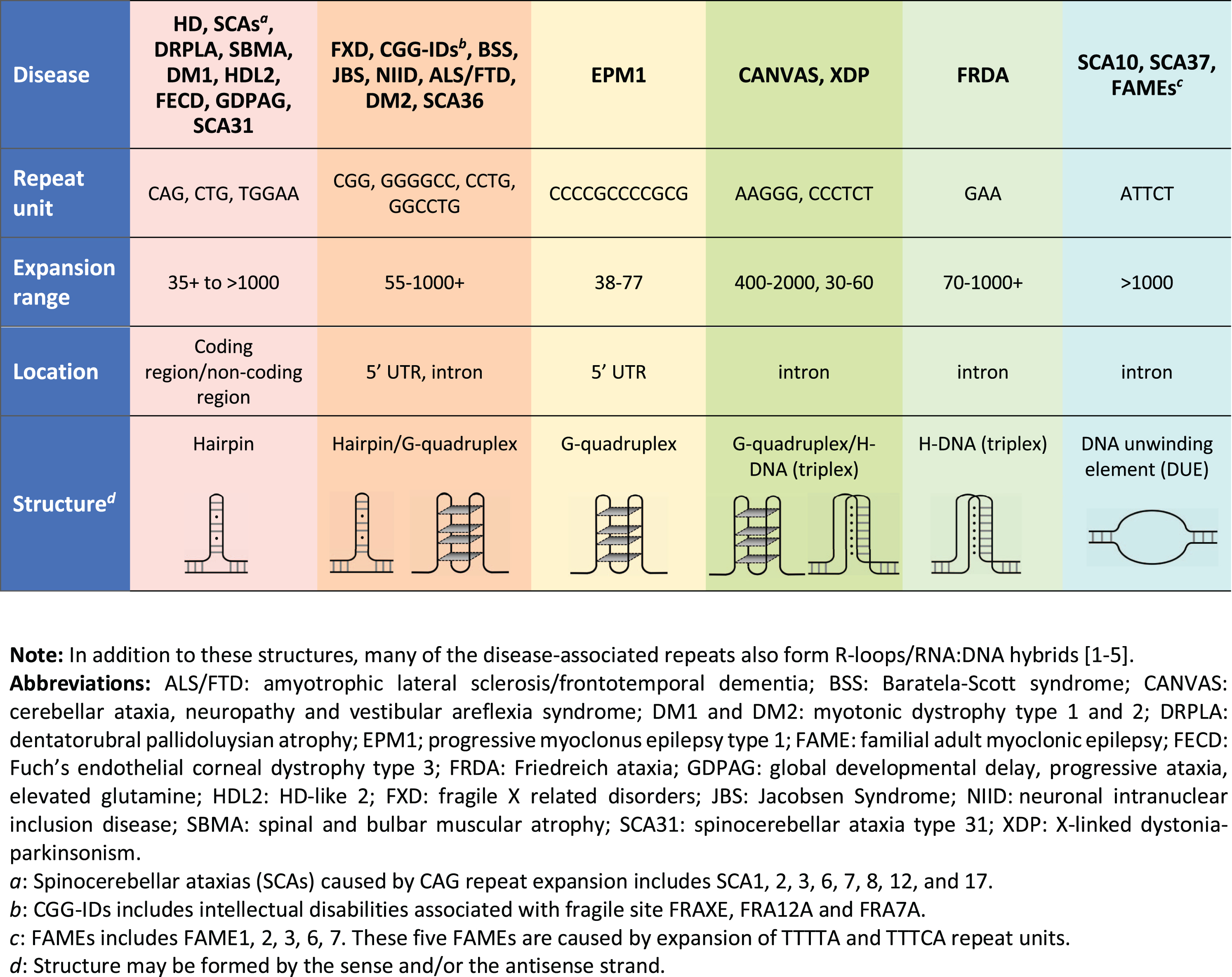
The expansion process in the non-CAG/CTG repeat expansion disorders has been most intensively studied in Friedreich ataxia (FRDA) and the Fragile X-related disorders (FXDs; aka the FMR1-associated disorders). FRDA is the most common hereditary ataxia and is typically caused by homozygosity for an expanded GAA-repeat tract in intron 1 of the frataxin (FXN) gene [32]. The FXDs are caused by the expansion of a CGG-repeat tract in the 5′ untranslated region of the X-linked FMR1 gene. This group of disorders includes the neurodegenerative disorder, Fragile X associated tremor/ataxia, a form of female infertility, Fragile X-associated primary ovarian insufficiency, and Fragile X syndrome, the most common inherited cause of intellectual disabilities and monogenic cause of autism worldwide [33]. Expansions in these diseases share a number of common features with expansions in HD. The FXDs and FRDA, like HD, show expansion both on germ-line transmission and somatically during the lifetime of the individual [34–42]. Although the magnitude of the changes in repeat number that are generally associated with HD is much smaller than those seen in the FXDs and FRDA, the starting repeat sizes are also much smaller (Fig. 1). Nonetheless, large expansions are seen in some tissues like the striatum (prior to pathological cell loss), liver and testis [20, 43]. Larger expansions are also seen in multiple tissues in juvenile HD cases where the inherited repeat size is significantly larger [43]. While the significance of somatic instability for FXD and FRDA disease severity is unclear at this time, emerging evidence suggests that the mechanism responsible for somatic expansions in these disorders is relevant for efforts to ameliorate somatic expansion in HD. This review will focus on what can be learned about the mechanism responsible for somatic CAG expansion in HD from mouse models of FRDA and the FXDs. For a broader perspective, the reader is referred to an excellent recent review that discusses findings from other model systems [44].
MOUSE MODELS OF FRDA AND THE FXDs
A number of FRDA mouse models with expan-ded GAA repeats have been generated including a knock-in (KI) mouse containing a (GAA)230 repeat expansion in the first intron of the endogenous Fxn gene [45] and two yeast artificial chromosome (YAC) transgenic mouse lines—YG8R and YG22R—containing different numbers of copies of a randomly integrated human FXN transgene (370kb of human genomic sequence) with 90–190 GAA repeats [46]. More recently, through natural breeding of the YG8R line, a new line, YG8sR, has been developed harboring a single copy of the FXN transgene and a single (GAA)120 repeat tract [47]. Of these mouse lines, the YAC-based lines have been most extensively studied [46, 47]. These mice also display age-dependent and tissue-specific expansion of the GAA repeat in brain, cerebellum, dorsal root ganglia and liver tissues [46–48], similar to that seen in FRDA patient autopsy tissues [40–42].
The most intensively studied FXD mouse model is one containing 130+ uninterrupted CGG repeats in the endogenous murine Fmr1 gene [49]. These mice show both germline and somatic instability with a strong expansion bias as seen in humans [35]. The dynamics of somatic expansion in these animals resembles that seen in human carriers of expansion-prone FMR1 alleles [50]. They are also similar to those seen in HD patients [16–20] and in mouse models of HD [51–55] and individuals with DM1 [56], although with some differences in the extent of expansion in different tissues. The expansion profiles in all cases are consistent with a high frequency of relatively small expansions (1–3 repeats) [54], although larger expansions are also occasionally seen [57, 58]. For expansion-prone cell types like the mucosal cells of the small intestine, a ∼170 repeat allele expands as often as once every 5–6 days in the majority of cells in the population [59].
Several lines of evidence demonstrate that origin-dependent chromosomal replication is not required for expansion at the FMR1 locus in either mice or humans. For example, expansion is seen in mouse oocytes [50], a cell-type that is non-dividing. Expansion in oocytes is consistent with the maternal age effect seen for expansion risk in humans [60]. Furthermore, expansion in somatic cells of the FXD mouse does not correlate with the tissue proliferation rate [35]. In mouse models of other repeat expansion disorders and in humans with such diseases expansion is seen in post-mitotic somatic cells such as neurons [17, 18, 38, 40, 61, 62].
Because the FMR1 gene is located on the X chromosome, the FXDs provide a particularly clear demonstration that expansion requires transcription or open chromatin. Specifically, expansion in female FXD mice, as in women with an expansion-prone FMR1 allele, only occurs when the expanded FMR1 allele is on the active X chromosome [63]. A depen-dence on transcription for expansion is consistent with correlations between transcriptional activity and expansion that are seen in transgenic mouse models of some of the CAG repeat expansion dis-orders [51, 64]. Thus, many characteristics of somatic expansion in the FRDA and FXD mice, including their small average size and high frequency, their transcription-dependence and replication-in-dependence, are similar to those of mouse models of HD and other repeat expansion disorders, as well as human patients with these diseases.
Many genetic factors that affect somatic expansion have been identified in the FXD and FRDA mouse models. These include factors that are required for, or play a role in, promoting expansion and factors that protect against expansion. In principle, if there is a single expansion mechanism, then the factors essential for expansion should act in all cell types where expansion is observed. For factors that play an auxiliary role in expansion or those that are protective, their effect in different cell types may reflect the relative levels of other protective or promoting factors, as well as the stoichiometry and functional redundancy of such factors.
THE ROLE OF MutS AND MutL COMPLEXES IN THE FRDA AND FXD MOUSE MODELS
Somatic expansions in both the FXD KI and FRDA YAC mice involve proteins that are critical for normal mismatch repair (MMR) (Table 1). This includes one or both of the MutS complexes MutSα and MutSβ, that are involved in mismatch recognition; as well as one or more of the three mammalian MutL complexes, MutLα, MutLβ and MutLγ, that are involved in lesion processing. Specifically, MSH2, a constituent of both MutS complexes, plays an important role in repeat expansion in FRDA [65] and is essential for expansion in the FXD mice [66]. Loss of MSH6, the MSH2-binding partner in the MutSα complex, leads to a sharp reduction in expansions in the cerebellum of FRDA mice [65] and a > 50% reduction in the extent of expansion in most FXD mouse tissues [67]. However, in the FXD mouse loss of MSH3, the MSH2-binding partner in the MutSβ complex, results in the loss of almost all expansions [68]. This suggests that MutSα may act by facilitating MutSβ dependent expansions in the FXD mice and by extension, perhaps in the FRDA mice as well.
Table 1
The role of MutS and MutL proteins in somatic instability in mouse models of the FXDs, FRDA and HD
| Protein | Effect | FXDs | FRDA | HD | |
| MutS | MSH2 | ↑ | [66] | [65] | [105, 106] |
| MSH3 | ↑ | [68] | [62, 108] | ||
| MSH6 | ↑ | [67] | [65] | ||
| – | [62] | ||||
| MutL | MLH1 | ↑ | [69] | [107] | |
| MLH3 | ↑ | [59] | [107] | ||
| PMS1 | ↑ | [70] | |||
| PMS2 | ↑ | [70] | |||
| ↓ | [65] |
↑: promotes expansion. ↓: prevents expansion; –: no effect.
MLH1, a protein present in all three MutL complexes, is also an important contributor to the ex-pansion process in the FRDA YAC mice [69]. However, PMS2, the MLH1-binding partner in the MutLα complex, protects against somatic expansion in these animals [65]. Given the importance of MLH1 in expansions, we can infer that one or more of the other MLH1-binding partners, PMS1 and/or MLH3, must play a role in expansions. In the case of the FXD mouse, all three MLH1-binding proteins, PMS1, PMS2 and MLH3, are required for expansion since the loss of any one of these proteins eliminates expansions either in vivo or in embryonic stem cells derived from these animals [59, 70]. Furthermore, a point mutation (D1185N) in the nuclease domain of MLH3 also eliminates all expansions in FXD mouse embryonic stem cells [71], suggesting that the nuclease activity of MutLγ is required.
Since MMR normally acts to prevent mismatches or insertion/deletions, it is generally thought that the role of these proteins in expansion reflects their ability to bind and process the secondary structures or loop-outs formed by the repeats that contain mismatched bases or regions of single-strandedness. However, given that MMR usually acts to prevent instability of similar tandem repeats or microsatellites, the processing of these structures presumably differs from canonical MMR. The specific requirement for MutLγ and its nuclease activity is interesting since it is much less abundant than MutLα [72], which typically plays a much larger role in MMR [73]. This suggests either that expansion involves an intermediate that is preferentially processed by MutLγ, or perhaps that MutLγ cleavage plays a unique and critical role in generating an intermediate that can be processed to generate an expansion. Notably, while MutLγ only plays a minor role in MMR, it plays a critical role in meiosis in resolving Holliday Junctions [74–76], which are cruciform-like structures that are also reminiscent of loop outs that could be formed by intrastrand structure formation by both strands of the repeats or perhaps simply by out-of-register reannealing. The role of MutLβ is also intriguing since it has no known nuclease motifs and, despite its abundance relative to MutLγ, its function is largely unknown [77].
OTHER GENETIC MODIFIERS OF SOMATIC EXPANSION RISK IN FXD MOUSE MODELS
In addition to MutS and MutL proteins, a variety of other genetic modifiers of somatic expansion risk have been identified in the FXD mouse (Table 2). Some of these factors promote expansion, whilst others are protective or neutral. The factors involved in the expansion process include DNA polymerase β (Polβ) [78]. Polβ is a DNA polymerase essential for base excision repair [79], as well as for gap-filling in other repair processes [80–82]. Its importance in the expansion process is evidenced by the fact that even heterozygosity for a hypomorphic allele resulted in a significant decrease in expansions in FXD mice [78]. Cockayne Syndrome B (CSB; aka ERCC6), a protein essential for transcription-coupled repair, contributes to, but is not required for, somatic expansion in older mice [83]. Since it is not essential for expansion, it is presumably acting outside of transcription-coupled repair to facilitate expansions, perhaps via its participation in other DNA processing pathways like base excision repair [84], chromatin remodeling [85–87] or R-loop induced double-strand break repair [88, 89].
Table 2
Role of other DNA repair genes in somatic repeat instability in FXD mouse model
| Protein | Repair Pathways | Effect | Ref |
| Polβ | base excision repair/other | ↑ | [78] |
| CSB | transcription coupled repair/other | ↑ | [83] |
| FAN1 | Fanconi anemia/other | ↓ | [58] |
| EXO1 | MMR/other | ↓ | [59] |
| Lig4 | non-homologous end-joining | ↓ | [57] |
| MRE11 | homologous recombination/other | – | [57] |
↑: promotes expansion. ↓: prevents expansion; –: no effect.
Factors protecting against expansion include EXO1 [59], a 5′- 3′ exonuclease that is involved in meiosis as well as MMR [90, 91]. Loss of EXO1 caused a significant increase in expansions in the germ line and in the small intestine, but not in the brain [59]. Moreover, a point mutation (D173A) in the active site of EXO1 also significantly increased the extent of expansion in small intestine and germ line, but not quite to the same extent as the EXO1 null mutation [59]. The D173A mutant protein is defective in MMR but retains the structural role of EXO1 in meiosis [92], suggesting that EXO1 protects against expansion both in a nuclease-dependent and a nuclease-independent manner. However, since the loss of EXO1 had no effect on somatic expansion in organs other than the small intestine [59], EXO1 may be more relevant for germline rather than somatic expansion in the FXD mouse.
The loss of FAN1, another nuclease that has both 5′-3′ exonuclease and 5′ flap endonuclease activities [93–97], also causes a significant increase in somatic expansion in multiple tissues of FXD mice, including the brain [58]. Although named for its role in the Fanconi anemia (FA) pathway, FAN1 interacts with MLH1 [98] and has been suggested to be able to substitute for EXO1 in MMR [99].
DNA ligase IV (LIG4) also protects against somatic expansion in liver [57]. LIG4 is required for non-homologous end-joining [100–102], a process that is particularly important for neuronal integrity [103] since it is the major form of double-strand break repair active outside of S-phase. It is also the major repair pathway able to repair double-strand breaks with 5′ overhangs without requiring signifi-cant end resectioning. The protective effect of LIG4 suggests that non-homologous end-joining and the expansion pathway compete for a common substrate and thus expansion likely involves an intermediate with a double-strand break. Other factors involved in other forms of double-strand break repair like MRE11, an exonuclease important for the end-resec-tioning required to generate the 3′ overhang necessary for homologous recombination and other related forms of double-strand break repair [104], does not affect repeat expansion in FXD mice [57]. In principle, the nucleases EXO1 and FAN1 might be able to compensate for the absence of MRE11. However, since these nucleases are both pro-tective, this seems unlikely. Thus, evidence sugge-sts that expansion in the FXD mouse involves a homologous recombination-independent and non-homologous end-joining-independent processing of a double-strand break intermediate, perhaps one generated by MutLγ cleavage.
The wide variety of different proteins that play a role in modulating repeat expansion presumably reflects the different ways that the same repeat DNA substrates can be processed, with competition between factors that promote expansion and those that protect against them. The relative abundance of these proteins/complexes in different cell types could contribute to the tendency of the repeat to expand more in some cell types and not others [35, 78]. For example, MSH6, which promotes expansion is highly expressed in brain, liver and testes, the some of the most expansion prone tissues, whilst MSH2, a protein essential for expansion, is difficult to detect at all in heart, a tissue that shows little or no expansion [35].
PARALLELS TO MOUSE AND PATIENT-DERIVED CELL MODELS OF OTHER REPEAT EXPANSION DISORDERS
MutS and MutL complexes have also been shown to play major roles in somatic expansion in HD mouse models [62, 105–109] as well as mouse models of DM1 [110, 111], with a good correlation being observed between the levels of MutSβ and the extent of repeat expansion [112]. A critical role for MutS proteins in expansion has also been reported in cells from FRDA, HD and DM1 patients and mammalian model systems [113–117]. MutLγ has also been implicated in expansion in FRDA fibroblasts, where a role for the MLH3 nuclease has also been proposed [118]. As in the FXD mouse, FAN1 also protects against expansion in HD KI mice [119] and in HD induced pluripotent stem cells [120].
Despite the similarities seen across different disease-associated repeat loci, some differences are seen. For example, loss of MSH6 results in a significant suppression of somatic expansions in both FXD and FRDA mice [65, 67], a phenomenon that is also seen in FRDA patient induced pluripotent stem cells [113]. In contrast, in a HD mouse model, knockout of MSH6 had no obvious effect on expansions in striatum [62], whilst loss of MSH6 increased the expansion frequency in a human cell line carrying a (CAG)800 construct [116]. Similarly, in the DM1 mouse, a protective effect of MSH6 was seen in some organs like liver, but not in others, including brain [111]. Another example of a difference between disease models is PMS2, which promotes repeat expansions in FXD mouse embryonic stem cells [70] and in multiple tissues of DM1 mice [121], yet it seems to protect against GAA expansions in multiple brain regions of FRDA mice [65].
How is it that the same gene can have apparently opposing effects at different repeat expansion loci? Given that all of these diseases share many unusual features, it is possible that the occasional differences do not represent fundamentally different expansion mechanisms. For example, the differential effect of MSH6 may reflect the fact that MutSα is able to promote MutSβ binding to mismatches [122] and to the hairpins formed by the FX repeats [67], an ability that may only be apparent when MutSβ is rate-limiting. Alternatively, since typical MMR lesions are bound by two or more MutS dimers [123] and MutSα and MutSβ cofractionate from human cell extracts [124], and can both bind to the same FX hairpin [67], MutSα may be able to contribute to the MutSβ-bound lesion when MutSβ is limiting. The fact that loss of MSH6 has a bigger effect in the liver than in the brain in the FXD mouse model, would be consistent with this idea since liver has less MSH3 and more MSH6 than brain [35]. Similarly, in testis which shows the largest effect of the loss of MSH6, MSH6 is more abundant than it is in either brain or liver [35]. The apparently paradoxical effect of PMS2 on repeat expansion may result from competition between MutLγ and MutLα for binding to the expansion substrate as illustrated in Fig. 2.
Fig. 2
MutL competition model for the differential effect of the loss of PMS2 seen in different cell types or disease models. A simple mathematical model was developed for the competition between MutL proteins for binding to the expansion substrates. This model used the following assumptions: 1) Only a small proportion of the total cellular MutL is actually available for binding to the repeat; 2) Any one of the three MutLs can be recruited to a MutS-bound substrate; 3) Three MutLs (a MutL trimer) are required to bind productively to a substrate [123]; 4) The available MutL is distributed across all the substrates in proportion to their levels/binding affinity. 5) Only those MutL trimers that contain at least one MutLγ complex results in an expansion (indicated by a check mark); 6) Trimers that lack MutLγ or lesions that are not bound by at least three MutL complexes do not produce an expansion (indicated by a cross). A) Diagrammatic representation of the model showing MutL binding when the expansion substrates are present at different levels in the presence or absence of PMS2, with tick marks indicating outcomes that lead to expansions and the crosses those that do not. The number of available MutL complexes was set at MutLα= 10; MutLβ= 5 and MutLγ= 2, a ratio similar to that reported in mammalian cells [72]. When expansion substrate levels are low and PMS2 is present, not all MutLγ is bound, since PMS2 competes effectively for binding to the expansion substrate. As a result many MutL trimers formed lack MutLγ and their substrates are repaired without expansion. In the absence of PMS2, more MutLγ is able to bind and MutLβ contributes to the formation of additional MutL trimers required for MutLγ-generated expansions. As a result, a net increase in expansions is seen relative to cells with PMS2. At intermediate levels of substrate more MutLγ is able to bind and when PMS2 is absent, the residual MutLβ is sufficient for trimer formation at all MutLγ-bound sites. This results in no net change in the expansion frequency relative to cells with PMS2. However, at high levels of substrate, MutLβ becomes rate-limiting when PMS2 is absent, resulting in a net decrease in expansions. B) Graphical representation of the expansion probabilities across the range of substrate levels in the presence or absence of PMS2 based on the average of 1000 independent tests of the chances of binding of MutLα, MutLβ and MutLγ for each of the substrate levels. The python script used to generate the data upon which the graph is based is provided in the Supplementary Material. As in panel A, the number of available MutL complexes used was MutLα= 10; MutLβ= 5 and MutLγ= 2. However, as shown in the Supplementary Material similar results in terms of the range of effects of the loss of PMS2 are seen with wide range of different proportions of MutLα, MutLβ and MutLγ and with a wide range of absolute levels of total MutL.
![MutL competition model for the differential effect of the loss of PMS2 seen in different cell types or disease models. A simple mathematical model was developed for the competition between MutL proteins for binding to the expansion substrates. This model used the following assumptions: 1) Only a small proportion of the total cellular MutL is actually available for binding to the repeat; 2) Any one of the three MutLs can be recruited to a MutS-bound substrate; 3) Three MutLs (a MutL trimer) are required to bind productively to a substrate [123]; 4) The available MutL is distributed across all the substrates in proportion to their levels/binding affinity. 5) Only those MutL trimers that contain at least one MutLγ complex results in an expansion (indicated by a check mark); 6) Trimers that lack MutLγ or lesions that are not bound by at least three MutL complexes do not produce an expansion (indicated by a cross). A) Diagrammatic representation of the model showing MutL binding when the expansion substrates are present at different levels in the presence or absence of PMS2, with tick marks indicating outcomes that lead to expansions and the crosses those that do not. The number of available MutL complexes was set at MutLα= 10; MutLβ= 5 and MutLγ= 2, a ratio similar to that reported in mammalian cells [72]. When expansion substrate levels are low and PMS2 is present, not all MutLγ is bound, since PMS2 competes effectively for binding to the expansion substrate. As a result many MutL trimers formed lack MutLγ and their substrates are repaired without expansion. In the absence of PMS2, more MutLγ is able to bind and MutLβ contributes to the formation of additional MutL trimers required for MutLγ-generated expansions. As a result, a net increase in expansions is seen relative to cells with PMS2. At intermediate levels of substrate more MutLγ is able to bind and when PMS2 is absent, the residual MutLβ is sufficient for trimer formation at all MutLγ-bound sites. This results in no net change in the expansion frequency relative to cells with PMS2. However, at high levels of substrate, MutLβ becomes rate-limiting when PMS2 is absent, resulting in a net decrease in expansions. B) Graphical representation of the expansion probabilities across the range of substrate levels in the presence or absence of PMS2 based on the average of 1000 independent tests of the chances of binding of MutLα, MutLβ and MutLγ for each of the substrate levels. The python script used to generate the data upon which the graph is based is provided in the Supplementary Material. As in panel A, the number of available MutL complexes used was MutLα= 10; MutLβ= 5 and MutLγ= 2. However, as shown in the Supplementary Material similar results in terms of the range of effects of the loss of PMS2 are seen with wide range of different proportions of MutLα, MutLβ and MutLγ and with a wide range of absolute levels of total MutL.](https://ip.ios.semcs.net:443/media/jhd/2021/10-1/jhd-10-1-jhd200423/jhd-10-jhd200423-g002.jpg)
The fact that the base excision repair polymerase, Polβ, plays an important role in promoting expansion in the FXD mouse is consistent with the fact that the loss of OGG1 and NEIL1, two DNA glycosylases involved in base excision repair, reduce expansion in HD mouse models [125, 126]. Since base excision repair is the major pathway by which oxidative damage to DNA is repaired in mammalian cells, this would also be consistent with the fact that oxidizing agents increase expansions in FXD mice [127] and in embryonic stem cells derived from HD mice [128]. The transcription-coupled repair protein CSB has also been reported to protect against expansion in HD mice [129]. However, this effect was only seen in an OGG1 background and thus its relationship to what is seen in the FXD mouse [83] is unclear. In contrast, the loss of XPA, a protein involved in the common steps of transcription-coupled repair and global-genome nucleotide excision repair, has been reported to reduce expansions in neuronal tissues of SCA1 mice, but not in gametes or liver [130]. This tissue-specific effect is reminiscent of the effect of CSB in FXD mice [83]. The fact that it is not essential for expansion suggests that, like CSB, it is acting independently of nucleotide excision repair, perhaps via its participation in other repair pathways [131]. Finally, recent work has shown that FAN1 has a clear protective role in HD patient cells [120] and HD KI mice [119], thus representing an additional parallel to the FXD mice [58].
Thus, in spite of some differences, a case can be made that many factors affecting somatic repeat instability in mouse models of FRDA and FXD are similar in key respects to the genetic factors shown to affect somatic expansions in mouse and cell models of other repeat expansion disorders.
THE RELEVANCE OF THESE MODELS TO SOMATIC EXPANSIONS IN HD PATIENTS AND AFFECTED INDIVIDUALS WITH OTHER REPEAT EXPANSION DISORDERS
As described in more detail elsewhere in this volume [132], recent GWA studies in HD patient cohorts have implicated loci containing some of the same DNA repair genes discussed above—FAN1, MSH3, MLH1, PMS1, and PMS2—s modifiers of somatic expansion, age at symptoms onset and disease progression [21, 22, 24–26]. Polymorphisms in MSH3 have also been shown to modify somatic expansion risk in DM1 patients [26, 133] and patients with CAG-repeat related spinocerebellar ataxias [25], while variants in FAN1 and PMS2 were also associated with variations in age at disease onset in patients with CAG-repeat related spinocerebellar ataxias [25]. Although CSB has not been implicated as a modifier of expansion risk in other diseases, a SNP in the CSB/ERCC6 gene has been shown to be associated with increased somatic instability in SCA3 [134].
In the case of FAN1, SNPs associated with earlier HD onset include missense variants within or near FAN1’s DNA-binding domain, consequently reducing its DNA-binding activity and capacity to rescue mitomycin C-induced cytotoxicity [135]. In addition, SNPs associated with later HD onset are associated with increased FAN1 expression in various brain regions [23, 135]. This is consistent with FAN1 activity protecting against expansions in humans, as in HD and FXD mice [58, 119, 120]. In contrast, again consistent with the results from model systems, SNPs associated with decreased MSH3 expression were associated with later disease onset, whilst SNPs associated with increased expression resulted in earlier ages of onset [22, 136]. TWAS has also shown a correlation between increased PMS2 expression and a later age at disease onset [23] which would be consistent with the protective effect of PMS2 in some mouse models. The situation is less clear for PMS1 where TWAS showed a variable effect, with an increase in cortex PMS1 expression being associated with a later HD onset [22], whilst data from other tissue sources suggest the opposite (P. Holmans, personal communication).
Thus, while loci associated with variations in expansion risk in patient GWA studies contain genes implicated in modifying expansion risk in mouse and patient-derived cell models, more work is needed to fully understand the contribution of these genes to expansion in disease-relevant organs like brain in humans. Additional factors that contribute to expansion risk in FXD mice may only become apparent with GWA studies on larger HD patient cohorts. However, given the already established similarities between expansion in the FXD mouse and HD models and patients, it is reasonable to think that many of these factors could well play a role in modulating expansion in HD as well as other repeat expansion disorders.
IMPLICATIONS FOR THE MECHANISM OF SOMATIC EXPANSION IN HD
Taken together, the available data suggests that most expansions occur via a transcription-dependent, replication-independent process, that involves components of multiple DNA repair pathways including MMR, base excision repair and some form of double-strand break repair. One way that all of these factors can be accommodated in a single model for repeat expansion is illustrated in Fig. 2.
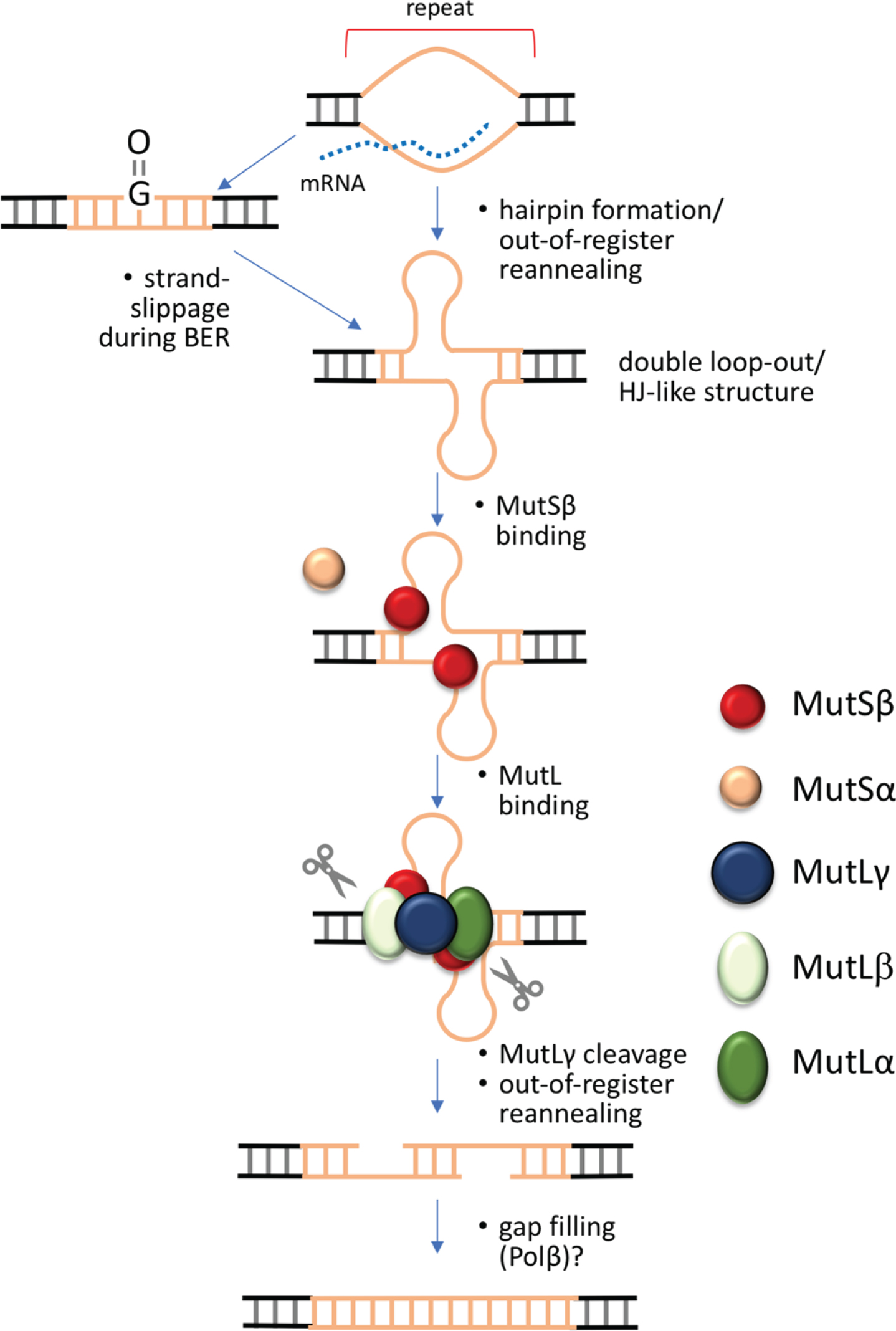
Oxidative damage has been proposed to initiate repair of the damaged base with strand loop-outs arising by strand-slippage during Long Patch base excision repair [125, 137]. The loop-outs could then bind the MutS proteins to ultimately generate expansions. However, whether oxidative stress can account for the extraordinarily high expansion frequency that is seen in some mouse cells remains unclear; and it is possible that oxidative damage is not the only trigger for expansion. This would be consistent with data showing that antioxidants have a relatively small effect on somatic expansion frequencies in HD mice [138, 139]. In theory, similar loop-outs could be generated any time the repeat region was unpaired, during transcription for example, when hairpins could form on the non-template strand. It is also possible that simple out-of-register reannealing of the template and non-template strand could occur after the transcription complex has moved on. This could be exacerbated by the formation of stable R-loops that are characteristic of many of the repeat expansion loci associated with diease [49, 140–143]. Loop-outs formed on both strands would resemble a Holliday Junction, a preferred MutLγ binding substrate [144]. These loop-outs would be bound by MutSβ, and perhaps in some cases by MutSα as well [67]. The MutS-bound loop-outs would then be cleaved by MutLγ. This reaction may be facilitated by MutLβ in some way. Since non-homologous end-joining protects against expansion in the FXD mouse [57], it suggests that expansion proceeds via a double-strand break that is then processed by a mechanism that is independent of non-homologous end-joining and homologous recombination. MutLγ is known to cleave the strand opposite loop-outs in vitro [145]. Loop-outs on both strands may result in off-set cleavages as illustrated in Fig. 3. This could result in staggered double-strand break with 5′ overhangs that could then anneal out-of-register resulting in small gaps. Simple gap filling, a reaction that can be carried out by Polβ [146, 147], followed by ligation would generate an expanded allele. CSB and XPA may affect expansion in a number of different ways, including via binding to R-loops [148] or the inhibition of non-homologous end-joining in the case of CSB [149], and the ability to bind to Holliday junctions [150] in the case of XPA. FAN1 and EXO1 may reduce the likelihood that this pathway is used by digesting the broken ends in such a way as to favor their processing to restore the original allele or to generate contractions. EXO1 also has a structural role in determining the orientation of MutLγ cleavage [92, 151] that may explain its nuclease-independent role in preventing expansions [59].
Fig. 3
Double-strand break model for the generation of repeat expansions. Expansion in this model is initiated when the repeat is transiently unpaired, as for example during transcription, replication or DNA damage repair. Out-of-register annealing of the two strands during this process could result in a double loop-out structure that resembles a Holliday Junction, the normal MutLγ meiotic substrate. This process may be exacerbated by the ability of the individual strands of some repeats to form stable intrastrand secondary structures like hairpins. Cleavage by MutLγ on either side of the double loop-out results in a double-strand break that can anneal out of register. Simple gap filling and ligation then results in expansions.
This particular expansion pathway may occur in parallel with other potential expansion pathways that have been described [44, 152–155]. However, since these pathways do not involve the MMR repair proteins implicated by GWAS in patient cohorts, their contribution to somatic expansion in the repeat expansion diseases is unclear.
CONCLUDING REMARKS
The genetic data from FRDA, FXD, and HD mice and human cell models, as well as GWA/TWA studies in HD patients, strongly implicate a subset of MMR components as major contributors to somatic repeat expansions. However, studies in the FXD mice suggest that there are a variety of additional DNA repair factors and pathways that could be targeted to reduce somatic expansion in HD, as well as other repeat expansion disorders in which somatic instability is a contributor to disease burden. This naturally increases the potentially druggable space for somatic expansion in HD patients and increases the chances that a suitable modifier can be identified that is amenable to safe modulation and efficient drug targeting. A common druggable target for all repeat expansion diseases may make the development of a more broadly useful drug a more economically feasible endeavor.
CONFLICT OF INTEREST
RMP has received sponsored research funding from Pfizer unrelated to the content of this manuscript.
Other authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors would like to thank Drs Peter Holmans and Tom Massey (Cardiff University) for helpful conversations. This work was made possible by funding from the Intramural Program of the National Institute of Diabetes, Kidney and Digestive Diseases to KU, and the Huntington’s Disease Society of America to RMP.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JHD-200423.
REFERENCES
[1] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) ;1: :15005. |
[2] | The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6):971–83. |
[3] | Sathasivam K , Neueder A , Gipson TA , Landles C , Benjamin AC , Bondulich MK , et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. (2013) ;110: (6):2366–70. |
[4] | Chung DW , Rudnicki DD , Yu L , Margolis RL . A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum Mol Genet. (2011) ;20: (17):3467–77. |
[5] | Banez-Coronel M , Porta S , Kagerbauer B , Mateu-Huertas E , Pantano L , Ferrer I , et al. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. (2012) ;8: (2):e1002481. |
[6] | Banez-Coronel M , Ayhan F , Tarabochia AD , Zu T , Perez BA , Tusi SK , et al. RAN translation in Huntington disease. Neuron. (2015) ;88: (4):667–77. |
[7] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theilmann J , Adam S , et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. (1993) ;4: (4):398–403. |
[8] | Duyao M , Ambrose C , Myers R , Novelletto A , Persichetti F , Frontali M , et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. (1993) ;4: (4):387–92. |
[9] | Telenius H , Kremer HP , Theilmann J , Andrew SE , Almqvist E , Anvret M , et al. Molecular analysis of juvenile Huntington disease: The major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet. (1993) ;2: (10):1535–40. |
[10] | Trottier Y , Biancalana V , Mandel JL . Instability of CAG repeats in Huntington’s disease: Relation to parental transmission and age of onset. J Med Genet. (1994) ;31: (5):377–82. |
[11] | Ranen NG , Stine OC , Abbott MH , Sherr M , Codori AM , Franz ML , et al. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am J Hum Genet. (1995) ;57: (3):593–602. |
[12] | Kremer B , Almqvist E , Theilmann J , Spence N , Telenius H , Goldberg YP , et al. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet. (1995) ;57: (2):343–50. |
[13] | Myers RH , MacDonald ME , Koroshetz WJ , Duyao MP , Ambrose CM , Taylor SA , et al. De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat Genet. (1993) ;5: (2):168–73. |
[14] | Hendricks AE , Latourelle JC , Lunetta KL , Cupples LA , Wheeler V , MacDonald ME , et al. Estimating the probability of de novo HD cases from transmissions of expanded penetrant CAG alleles in the Huntington disease gene from male carriers of high normal alleles (27-35 CAG). Am J Med Genet A. (2009) ;149A: (7):1375–81. |
[15] | Sequeiros J , Ramos EM , Cerqueira J , Costa MC , Sousa A , Pinto-Basto J , et al. Large normal and reduced penetrance alleles in Huntington disease: Instability in families and frequency at the laboratory, at the clinic and in the population. Clin Genet. (2010) ;78: (4):381–7. |
[16] | Kennedy L , Evans E , Chen CM , Craven L , Detloff PJ , Ennis M , et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) ;12: (24):3359–67. |
[17] | Shelbourne PF , Keller-McGandy C , Bi WL , Yoon SR , Dubeau L , Veitch NJ , et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. (2007) ;16: (10):1133–42. |
[18] | Gonitel R , Moffitt H , Sathasivam K , Woodman B , Detloff PJ , Faull RL , et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A. (2008) ;105: (9):3467–72. |
[19] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: (4):409–14. |
[20] | Swami M , Hendricks AE , Gillis T , Massood T , Mysore J , Myers RH , et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) ;18: (16):3039–47. |
[21] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. |
[22] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Electronic address: gusella@helix.mgh. harvard.edu; Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900 e14. |
[23] | Wright GEB , Caron NS , Ng B , Casal L , Casazza W , Xu X , et al. Gene expression profiles complement the analysis of genomic modifiers of the clinical onset of Huntington disease. Hum Mol Genet. (2020) ;29: (16):2788–802. |
[24] | Genetic Modifiers of Huntington’s Disease Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[25] | Bettencourt C , Hensman-Moss D , Flower M , Wiethoff S , Brice A , Goizet C , et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. (2016) ;79: (6):983–90. |
[26] | Flower M , Lomeikaite V , Ciosi M , Cumming S , Morales F , Lo K , et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. (2019) ;142: (7):1876–86. |
[27] | Gusella JF , MacDonald ME , Lee JM . Genetic modifiers of Huntington’s disease. Mov Disord. (2014) ;29: (11):1359–65. |
[28] | Paulson H . Repeat expansion diseases. Handb Clin Neurol. (2018) ;147: :105–23. |
[29] | Trost B , Engchuan W , Nguyen CM , Thiruvahindrapuram B , Dolzhenko E , Backstrom I , et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. (2020) ;586: (7827):80–6. |
[30] | Bragg DC , Mangkalaphiban K , Vaine CA , Kulkarni NJ , Shin D , Yadav R , et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci U S A. (2017) ;114: (51):E11020–E8. |
[31] | Mirkin SM . Expandable DNA repeats and human disease. Nature. (2007) ;447: (7147):932–40. |
[32] | Campuzano V , Montermini L , Molto MD , Pianese L , Cossee M , Cavalcanti F , et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. (1996) ;271: (5254):1423–7. |
[33] | Lozano R , Rosero CA , Hagerman RJ . Fragile X spectrum disorders. Intractable Rare Dis Res. (2014) ;3: (4):134–46. |
[34] | Nolin SL , Houck GE Jr , Gargano AD , Blumstein H , Dobkin CS , Brown WT . FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am J Hum Genet. (1999) ;65: (3):680–8. |
[35] | Lokanga RA , Entezam A , Kumari D , Yudkin D , Qin M , Smith CB , et al. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum Mutat. (2013) ;34: (1):157–66. |
[36] | Zhao X , Gazy I , Hayward B , Pintado E , Hwang YH , Tassone F , et al. Repeat instability in the Fragile X-related disorders: Lessons from a mouse model. Brain Sci. (2019) ;9: (3):52. |
[37] | De Michele G , Filla A , Criscuolo C , Scarano V , Cavalcanti F , Pianese L , et al. Determinants of onset age in Friedreich’s ataxia. J Neurol. (1998) ;245: (3):166–8. |
[38] | Montermini L , Kish SJ , Jiralerspong S , Lamarche JB , Pandolfo M . Somatic mosaicism for Friedreich’s ataxia GAA triplet repeat expansions in the central nervous system. Neurology. (1997) ;49: (2):606–10. |
[39] | Sharma R , Bhatti S , Gomez M , Clark RM , Murray C , Ashizawa T , et al. The GAA triplet-repeat sequence in Friedreich ataxia shows a high level of somatic instability in vivo, with a significant predilection for large contractions. Hum Mol Genet. (2002) ;11: (18):2175–87. |
[40] | De Biase I , Rasmussen A , Endres D , Al-Mahdawi S , Monticelli A , Cocozza S , et al. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. (2007) ;61: (1):55–60. |
[41] | De Biase I , Rasmussen A , Monticelli A , Al-Mahdawi S , Pook M , Cocozza S , et al. Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life. Genomics. (2007) ;90: (1):1–5. |
[42] | Long A , Napierala JS , Polak U , Hauser L , Koeppen AH , Lynch DR , et al. Somatic instability of the expanded GAA repeats in Friedreich’s ataxia. PLoS One. (2017) ;12: (12):e0189990. |
[43] | Mouro Pinto R , Arning L , Giordano JV , Razghandi P , Andrew MA , Gillis T , et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet. (2020) ;29: (15):2551–67. |
[44] | McGinty RJ , Mirkin SM . Cis- and trans-modifiers of repeat expansions: Blending model systems with human genetics. Trends Genet. (2018) ;34: (6):448–65. |
[45] | Miranda CJ , Santos MM , Ohshima K , Smith J , Li L , Bunting M , et al. Frataxin knockin mouse. FEBS Lett. (2002) ;512: (1-3):291–7. |
[46] | Al-Mahdawi S , Pinto RM , Ruddle P , Carroll C , Webster Z , Pook M . GAA repeat instability in Friedreich ataxia YAC transgenic mice. Genomics. (2004) ;84: (2):301–10. |
[47] | Anjomani Virmouni S , Ezzatizadeh V , Sandi C , Sandi M , Al-Mahdawi S , Chutake Y , et al. A novel GAA-repeat-expansion-based mouse model of Friedreich’s ataxia. Dis Model Mech. (2015) ;8: (3):225–35. |
[48] | Clark RM , De Biase I , Malykhina AP , Al-Mahdawi S , Pook M , Bidichandani SI . The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum Genet. (2007) ;120: (5):633–40. |
[49] | Entezam A , Biacsi R , Orrison B , Saha T , Hoffman GE , Grabczyk E , et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. (2007) ;395: (1-2):125–34. |
[50] | Zhao XN , Usdin K . Timing of expansion of Fragile X premutation alleles during intergenerational transmission in a mouse model of the Fragile X-related disorders. Front Genet. (2018) ;9: :314. |
[51] | Mangiarini L , Sathasivam K , Seller M , Cozens B , Harper A , Hetherington C , et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) ;87: (3):493–506. |
[52] | Mangiarini L , Sathasivam K , Mahal A , Mott R , Seller M , Bates GP . Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. (1997) ;15: (2):197–200. |
[53] | Wheeler VC , Auerbach W , White JK , Srinidhi J , Auerbach A , Ryan A , et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet. (1999) ;8: (1):115–22. |
[54] | Mollersen L , Rowe AD , Larsen E , Rognes T , Klungland A . Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. (2010) ;6: (12):e1001242. |
[55] | Lee JM , Pinto RM , Gillis T , St Claire JC , Wheeler VC . Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One. (2011) ;6: (8):e23647. |
[56] | Higham CF , Morales F , Cobbold CA , Haydon DT , Monckton DG . High levels of somatic DNA diversity at the myotonic dystrophy type 1 locus are driven by ultra-frequent expansion and contraction mutations. Hum Mol Genet. (2012) ;21: (11):2450–63. |
[57] | Gazy I , Hayward B , Potapova S , Zhao X , Usdin K . Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair (Amst). (2019) ;74: :63–9. |
[58] | Zhao XN , Usdin K . FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst). (2018) ;69: :1–5. |
[59] | Zhao X , Zhang Y , Wilkins K , Edelmann W , Usdin K . MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. (2018) ;14: (10):e1007719. |
[60] | Yrigollen CM , Durbin-Johnson B , Gane L , Nelson DL , Hagerman R , Hagerman PJ , et al. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with Fragile X syndrome. Genet Med. (2012) ;14: (8):729–36. |
[61] | Kovalenko M , Dragileva E , St Claire J , Gillis T , Guide JR , New J , et al. Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington’s disease knock-in mice. PLoS One. (2012) ;7: (9):e44273. |
[62] | Dragileva E , Hendricks A , Teed A , Gillis T , Lopez ET , Friedberg EC , et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. (2009) ;33: (1):37–47. |
[63] | Adihe Lokanga R , Zhao XN , Entezam A , Usdin K . X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: Implications for the mechanism of repeat expansion. Hum Mol Genet. (2014) ;23: (18):4985–94. |
[64] | Goula AV , Stys A , Chan JP , Trottier Y , Festenstein R , Merienne K . Transcription elongation and tissue-specific somatic CAG instability. PLoS Genet. (2012) ;8: (11):e1003051. |
[65] | Bourn RL , De Biase I , Pinto RM , Sandi C , Al-Mahdawi S , Pook MA , et al. Pms2 suppresses large expansions of the (GAA. TTC)n sequence in neuronal tissues. PLoS One. (2012) ;7: (10):e47085. |
[66] | Lokanga RA , Zhao XN , Usdin K . The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum Mutat. (2014) ;35: (1):129–36. |
[67] | Zhao XN , Lokanga R , Allette K , Gazy I , Wu D , Usdin K . A MutSbeta-dependent contribution of MutSalpha to repeat expansions in Fragile X premutation mice? PLoS Genet. (2016) ;12: (7):e1006190. |
[68] | Zhao XN , Kumari D , Gupta S , Wu D , Evanitsky M , Yang W , et al. Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum Mol Genet. (2015) ;24: (24):7087–96. |
[69] | Ezzatizadeh V , Sandi C , Sandi M , Anjomani-Virmouni S , Al-Mahdawi S , Pook MA . MutLalpha heterodimers modify the molecular phenotype of Friedreich ataxia. PLoS One. (2014) ;9: (6):e100523. |
[70] | Miller C , Kim G-Y , Zhao X , Usdin K . All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. (2020) ;16: (6):e1008902. |
[71] | Hayward BE , Steinbach PJ , Usdin K . A point mutation in the nuclease domain of MLH3 eliminates repeat expansions in a mouse stem cell model of the Fragile X-related disorders. Nucleic Acids Res. (2020) ;48: (14):7856–63. |
[72] | Cannavo E , Gerrits B , Marra G , Schlapbach R , Jiricny J . Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. (2007) ;282: (5):2976–86. |
[73] | Raschle M , Marra G , Nystrom-Lahti M , Schar P , Jiricny J . Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J Biol Chem. (1999) ;274: (45):32368–75. |
[74] | Nishant KT , Plys AJ , Alani E . A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics. (2008) ;179: (2):747–55. |
[75] | Rogacheva MV , Manhart CM , Chen C , Guarne A , Surtees J , Alani E . Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J Biol Chem. (2014) ;289: (9):5664–73. |
[76] | Zakharyevich K , Tang S , Ma Y , Hunter N . Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell. (2012) ;149: (2):334–47. |
[77] | Kadyrov FA , Dzantiev L , Constantin N , Modrich P . Endonucleolytic function of MutLalpha in human mismatch repair. Cell. (2006) ;126: (2):297–308. |
[78] | Lokanga RA , Senejani AG , Sweasy JB , Usdin K . Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet. (2015) ;11: (4):e1005181. |
[79] | Sobol RW , Horton JK , Kuhn R , Gu H , Singhal RK , Prasad R , et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. (1996) ;379: (6561):183–6. |
[80] | Ray S , Menezes MR , Senejani A , Sweasy JB . Cellular roles of DNA polymerase beta. Yale J Biol Med. (2013) ;86: (4):463–9. |
[81] | Crespan E , Pasi E , Imoto S , Hubscher U , Greenberg MM , Maga G . Human DNA polymerase beta, but not lambda, can bypass a 2-deoxyribonolactone lesion together with proliferating cell nuclear antigen. ACS Chem Biol. (2013) ;8: (2):336–44. |
[82] | London RE . The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst). (2015) ;30: :90–103. |
[83] | Zhao XN , Usdin K . Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum Mutat. (2014) ;35: (3):341–9. |
[84] | Menoni H , Wienholz F , Theil AF , Janssens RC , Lans H , Campalans A , et al. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. (2018) ;46: (15):7747–56. |
[85] | Beerens N , Hoeijmakers JH , Kanaar R , Vermeulen W , Wyman C . The CSB protein actively wraps DNA. J Biol Chem. (2005) ;280: (6):4722–9. |
[86] | Citterio E , Van Den Boom V , Schnitzler G , Kanaar R , Bonte E , Kingston RE , et al. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. (2000) ;20: (20):7643–53. |
[87] | Newman JC , Bailey AD , Weiner AM . Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. (2006) ;103: (25):9613–8. |
[88] | Sollier J , Stork CT , Garcia-Rubio ML , Paulsen RD , Aguilera A , Cimprich KA . Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell. (2014) ;56: (6):777–85. |
[89] | Tan J , Duan M , Yadav T , Phoon L , Wang X , Zhang JM , et al. An R-loop-initiated CSB-RAD52-POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. (2020) ;48: (3):1285–300. |
[90] | Wilson DM , 3rd , Carney JP , Coleman MA , Adamson AW , Christensen M , Lamerdin JE . Hex A new human Rad2 nuclease family member with homology to yeast exonuclease 1. Nucleic Acids Res. (1998) ;26: (16):3762–8. |
[91] | Schmutte C , Marinescu RC , Sadoff MM , Guerrette S , Overhauser J , Fishel R . Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. (1998) ;58: (20):4537–42. |
[92] | Zakharyevich K , Ma Y , Tang S , Hwang PY , Boiteux S , Hunter N . Temporally and biochemically distinct activities of Exo1 during meiosis: Double-strand break resection and resolution of double Holliday junctions. Mol Cell. (2010) ;40: (6):1001–15. |
[93] | Smogorzewska A , Desetty R , Saito TT , Schlabach M , Lach FP , Sowa ME , et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. (2010) ;39: (1):36–47. |
[94] | Castella M , Taniguchi T . The role of FAN1 nuclease in the Fanconi anemia pathway. Cell Cycle. (2010) ;9: (21):4259–60. |
[95] | Liu T , Ghosal G , Yuan J , Chen J , Huang J . FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. (2010) ;329: (5992):693–6. |
[96] | MacKay C , Declais AC , Lundin C , Agostinho A , Deans AJ , MacArtney TJ , et al. Identification of KIAA/ FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. (2010) ;142: (1):65–76. |
[97] | Yoshikiyo K , Kratz K , Hirota K , Nishihara K , Takata M , Kurumizaka H , et al. KIAA/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci U S A. (2010) ;107: (50):21553–7. |
[98] | Cannavo E , Marra G , Sabates-Bellver J , Menigatti M , Lipkin SM , Fischer F , et al. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. (2005) ;65: (23):10759–66. |
[99] | Desai A , Gerson S . Exo1 independent DNA mismatch repair involves multiple compensatory nucleases. DNA Repair (Amst). (2014) ;21: :55–64. |
[100] | Wei YF , Robins P , Carter K , Caldecott K , Pappin DJ , Yu GL , et al. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol Cell Biol. (1995) ;15: (6):3206–16. |
[101] | Lieber MR . The biochemistry and biological significance of nonhomologous DNA end joining: An essential repair process in multicellular eukaryotes. Genes Cells. (1999) ;4: (2):77–85. |
[102] | Pannunzio NR , Watanabe G , Lieber MR . Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. (2018) ;293: (27):10512–23. |
[103] | Rass U , Ahel I , West SC . Defective DNA repair and neurodegenerative disease. Cell. (2007) ;130: (6):991–1004. |
[104] | Haber JE . The many interfaces of Mre11. Cell. (1998) ;95: (5):583–6. |
[105] | Manley K , Shirley TL , Flaherty L , Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. (1999) ;23: (4):471–3. |
[106] | Wheeler VC , Lebel LA , Vrbanac V , Teed A , te Riele H , MacDonald ME . Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. (2003) ;12: (3):273–81. |
[107] | Pinto RM , Dragileva E , Kirby A , Lloret A , Lopez E , St Claire J , et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. (2013) ;9: (10):e1003930. |
[108] | Tome S , Manley K , Simard JP , Clark GW , Slean MM , Swami M , et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. (2013) ;9: (2):e1003280. |
[109] | Tomé S , Holt I , Edelmann W , Morris GE , Munnich A , Pearson CE , et al. MSH2 ATPase domain mutation affects CTG*CAG repeat instability in transgenic mice. PLoS Genet. (2009) ;5: (5):e1000482. |
[110] | Savouret C , Brisson E , Essers J , Kanaar R , Pastink A , te Riele H , et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. (2003) ;22: (9):2264–73. |
[111] | van den Broek WJ , Nelen MR , Wansink DG , Coerwinkel MM , te Riele H , Groenen PJ , et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. (2002) ;11: (2):191–8. |
[112] | Tomé S , Manley K , Simard JP , Clark GW , Slean MM , Swami M , et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. (2013) ;9: (2):e1003280. |
[113] | Du J , Campau E , Soragni E , Ku S , Puckett JW , Dervan PB , et al. Role of mismatch repair enzymes in GAA. TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem. (2012) ;287: (35):29861–72. |
[114] | Du J , Campau E , Soragni E , Jespersen C , Gottesfeld JM . Length-dependent CTG. CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet. (2013) ;22: (25):5276–87. |
[115] | Halabi A , Ditch S , Wang J , Grabczyk E . DNA mismatch repair complex MutSbeta promotes GAA. TTC repeat expansion in human cells. J Biol Chem. (2012) ;287: (35):29958–67. |
[116] | Nakatani R , Nakamori M , Fujimura H , Mochizuki H , Takahashi MP . Large expansion of CTG*CAG repeats is exacerbated by MutSbeta in human cells. Sci Rep. (2015) ;5: :11020. |
[117] | Gannon AM , Frizzell A , Healy E , Lahue RS . MutSbeta and histone deacetylase complexes promote expansions of trinucleotide repeats in human cells. Nucleic Acids Res. (2012) ;40: (20):10324–33. |
[118] | Halabi A , Fuselier KTB , Grabczyk E . GAA*TTC repeat expansion in human cells is mediated by mismatch repair complex MutLgamma and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res. (2018) ;46: (8):4022–32. |
[119] | Loupe JM , Pinto RM , Kim KH , Gillis T , Mysore JS , Andrew MA , et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet. 2020. |
[120] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: (4):650–61. |
[121] | Gomes-Pereira M , Fortune MT , Ingram L , McAbney JP , Monckton DG . Pms2 is a genetic enhancer of trinucleotide CAG. CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum Mol Genet. (2004) ;13: (16):1815–25. |
[122] | Tian L , Gu L , Li GM . Distinct nucleotide binding/hydrolysis properties and molar ratio of MutSalpha and MutSbeta determine their differential mismatch binding activities. J Biol Chem. (2009) ;284: (17):11557–62. |
[123] | Bradford KC , Wilkins H , Hao P , Li ZM , Wang B , Burke D , et al. Dynamic human MutSalpha-MutLalpha complexes compact mismatched DNA. Proc Natl Acad Sci U S A. (2020) ;117: (28):16302–12. |
[124] | Havugimana PC , Hart GT , Nepusz T , Yang H , Turinsky AL , Li Z , et al. A census of human soluble protein complexes. Cell. (2012) ;150: (5):1068–81. |
[125] | Kovtun IV , Liu Y , Bjoras M , Klungland A , Wilson SH , McMurray CT . OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. (2007) ;447: (7143):447–52. |
[126] | Mollersen L , Rowe AD , Illuzzi JL , Hildrestrand GA , Gerhold KJ , Tveteras L , et al. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum Mol Genet. (2012) ;21: (22):4939–47. |
[127] | Entezam A , Lokanga AR , Le W , Hoffman G , Usdin K . Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. (2010) ;31: (5):611–6. |
[128] | Jonson I , Ougland R , Klungland A , Larsen E . Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res. (2013) ;11: (3):1264–71. |
[129] | Kovtun IV , Johnson KO , McMurray CT . Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY). (2011) ;3: (5):509–14. |
[130] | Hubert L Jr , Lin Y , Dion V , Wilson JH . Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet. (2011) ;20: (24):4822–30. |
[131] | Borszekova Pulzova L , Ward TA , Chovanec M . XPA: DNA repair protein of significant clinical importance. Int J Mol Sci. (2020) ;21: (6):2182. |
[132] | Hong EP , MacDonald ME , Wheeler VC , Jones L , Holmans P , Orthe M , et al. Huntington’s disease pathogenesis: Two sequential components. J Huntingtons Dis. 2020;doi: 10.3233/JHD-200427. |
[133] | Morales F , Couto JM , Higham CF , Hogg G , Cuenca P , Braida C , et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet. (2012) ;21: (16):3558–67. |
[134] | Martins S , Pearson CE , Coutinho P , Provost S , Amorim A , Dube MP , et al. Modifiers of (CAG)(n) instability in Machado-Joseph disease (MJD/SCA3) transmissions: An association study with DNA replication, repair and recombination genes. Hum Genet. (2014) ;133: (10):1311–8. |
[135] | Kim KH , Hong EP , Shin JW , Chao MJ , Loupe J , Gillis T , et al. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am J Hum Genet. (2020) ;107: (1):96–110. |
[136] | Moss DJH , Pardinas AF , Langbehn D , Lo K , Leavitt BR , Roos R , et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. (2017) ;16: (9):701–11. |
[137] | Polyzos AA , McMurray CT . Close encounters: Moving along bumps, breaks, and bubbles on expanded trinucleotide tracts. DNA Repair (Amst). (2017) ;56: :144–55. |
[138] | Budworth H , Harris FR , Williams P , Lee DY , Holt A , Pahnke J , et al. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. (2015) ;11: (8):e1005267. |
[139] | Mollersen L , Moldestad O , Rowe AD , Bjolgerud A , Holm I , Tveteras L , et al. Effects of anthocyanins on CAG repeat instability and behaviour in Huntington’s disease R6/1 mice. PLoS Curr. 2016;8:ecurrents.hd.58d04209ab6d5de0844db7ef5628ff67. |
[140] | Loomis EW , Sanz LA , Chedin F , Hagerman PJ . Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. (2014) ;10: (4):e1004294. |
[141] | Kumari D , Usdin K . Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethy-lation of H3K27. Hum Mol Genet. (2016) ;25: (17):3689–98. |
[142] | Groh M , Lufino MM , Wade-Martins R , Gromak N . R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. (2014) ;10: (5):e1004318. |
[143] | Reddy K , Tam M , Bowater RP , Barber M , Tomlinson M , Nichol Edamura K , et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. (2011) ;39: (5):1749–62. |
[144] | Ranjha L , Anand R , Cejka P . The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem. (2014) ;289: (9):5674–86. |
[145] | Kadyrova LY , Gujar V , Burdett V , Modrich P , Kadyrov FA . Human MutLγ, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc Natl Acad Sci U S A. (2020) ;117: (7):3535–42. |
[146] | Nowak R , Kulik J , Siedlecki JA . The ability of DNA polymerase beta to synthesize DNA beyond the gap with displacement of the non-replicated strand. Acta Biochim Pol. (1987) ;34: (2):205–15. |
[147] | Singhal RK , Wilson SH . Short gap-filling synthesis by DNA polymerase beta is processive. J Biol Chem. (1993) ;268: (21):15906–11. |
[148] | Teng Y , Yadav T , Duan M , Tan J , Xiang Y , Gao B , et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat Commun. (2018) ;9: (1):4115. |
[149] | Batenburg NL , Thompson EL , Hendrickson EA , Zhu XD . Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. (2015) ;34: (10):1399–416. |
[150] | Camenisch U , Dip R , Schumacher SB , Schuler B , Naegeli H . Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat Struct Mol Biol. (2006) ;13: (3):278–84. |
[151] | Keelagher RE , Cotton VE , Goldman AS , Borts RH . Separable roles for Exonuclease I in meiotic DNA double-strand break repair. DNA Repair (Amst). (2011) ;10: (2):126–37. |
[152] | Khristich AN , Mirkin SM . On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem. (2020) ;295: (13):4134–70. |
[153] | Kononenko AV , Ebersole T , Vasquez KM , Mirkin SM . Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat Struct Mol Biol. (2018) ;25: (8):669–76. |
[154] | Neil AJ , Liang MU , Khristich AN , Shah KA , Mirkin SM . RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. (2018) ;46: (7):3487–97. |
[155] | Zhang Y , Shishkin AA , Nishida Y , Marcinkowski-Desmond D , Saini N , Volkov KV , et al. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell. (2012) ;48: (2):254–65. |

