
Rac1 Activity Is Modulated by Huntingtin and Dysregulated in Models of Huntington’s Disease
Abstract
Background:
Previous studies suggest that Huntingtin, the protein mutated in Huntington’s disease (HD), is required for actin based changes in cell morphology, and undergoes stimulus induced targeting to plasma membranes where it interacts with phospholipids involved in cell signaling. The small GTPase Rac1 is a downstream target of growth factor stimulation and PI 3-kinase activity and is critical for actin dependent membrane remodeling.
Objective:
To determine if Rac1 activity is impaired in HD or regulated by normal Huntingtin.
Methods:
Analyses were performed in differentiated control and HD human stem cells and HD Q140/Q140 knock-in mice. Biochemical methods included SDS-PAGE, western blot, immunoprecipitation, affinity chromatography, and ELISA based Rac activity assays.
Results:
Basal Rac1 activity increased following depletion of Huntingtin with Huntingtin specific siRNA in human primary fibroblasts and in human control neuron cultures. Human cells (fibroblasts, neural stem cells, and neurons) with the HD mutation failed to increase Rac1 activity in response to growth factors. Rac1 activity levels were elevated in striatum of 1.5-month-old HD Q140/Q140 mice and in primary embryonic cortical neurons from HD mice. Affinity chromatography analysis of striatal lysates showed that Huntingtin is in a complex with Rac1, p85α subunit of PI 3-kinase, and the actin bundling protein α-actinin and interacts preferentially with the GTP bound form of Rac1. The HD mutation reduced Huntingtin interaction with p85α.
Conclusions:
These findings suggest that Huntingtin regulates Rac1 activity as part of a coordinated response to growth factor signaling and this function is impaired early in HD.
INTRODUCTION
Huntingtin, the protein product of HTT, the gene mutated in Huntington’s disease (HD) [1], has an expanded polyglutamine tract. Huntingtin is required for embryonic survival [2–4] and its acute knockdown in adult neurons causes degeneration [5–8]. Moreover, the presence of mutant Huntingtin disrupts TrkA receptor signaling and neurite outgrowth [9]. Collectively these findings suggest that Huntingtin influences cellular responses to growth factor signaling.
Studies indirectly support Huntingtin’s involvement in signaling through receptor tyrosine kinases which drive actin dependent morphology changes. Huntingtin preferentially associates with particular phospholipids in vitro including phosphatidylinositol bis (3,4) phosphate and phosphatidylinositol tri (3,4,5) phosphate (PI(3,4)P2 and PI(3, 4, 5)P3) [10, 11]. Stimulation of receptor tyrosine kinases activate class IA PI 3-kinases causing a transient rise of PI(3, 4)P2 and PI(3, 4, 5)P3 [12, 13]. These phospholipids in turn accumulate at local sites on the plasma membrane and engage protein complexes that propagate signal cascades directing cell morphology changes driven by actin reorganization [14, 15]. Stimulation of receptor tyrosine kinases by PDGF and IGF1 caused Huntingtin to re-localize to the plasma membrane [10] suggesting a functional role for Huntingtin in PI 3-kinase pathways. In PC12 cells, Huntingtin was co-immunoprecipitated with TrkA receptors following nerve growth factor stimulation and with p85α regulatory subunit of PI 3-kinase and Grb2 [9, 16]. The interaction of Huntingtin with p85α was also identified in a yeast two-hybrid screen with Huntingtin fragments [17, 18].
The small GTPases Rac1, 2 and 3 are members of the Rho GTPase family and are important for actin-based changes that propel cell motility, protrusion formation and membrane ruffling [19]. Rac1 is known to be a downstream target of growth factor receptor /PI3kinase activity [14] and controls growth factor initiated morphology changes [14]. Transcripts for Rac1 are ubiquitously expressed whereas those for Rac2 are restricted to T-, B- and hemopoietic cell lineages [20, 21]. Rac3 transcripts are expressed most prominently in brain tissue but also in heart, pancreas and placenta [22]. Thus, total Rac activity levels may be due to contributions from more than one Rac protein especially in brain.
Results of investigations into Huntingtin binding partners using yeast two-hybrid or mass spectrometry (MS) after Huntingtin immunoprecipitation suggest that Huntingtin associates with proteins involved in actin cytoskeletal regulation and function; this includes actin itself [18, 23, 24]. In agreement with these findings, a purified N-terminal Huntingtin fragment directly interacted with actin in vitro via aa1–14 of Huntingtin [25]. In the nucleus, endogenous Huntingtin co-localized with actin and cofilin rods which form upon stress and Huntingtin is necessary for their formation; mutant Huntingtin has been shown to alter actin/cofilin rods [26]. Other evidence that mutant Huntingtin may impact actin related functions includes the finding that mutant Huntingtin exon1 toxicity can be modulated by inhibiting the kinase ROCK and its phosphorylation of the actin binding protein profilin [27]. However, there has been limited evidence linking endogenous Huntingtin localization or function with cytoplasmic actin in cells [18, 28].
Here we sought to determine in different wild-type (WT) and HD models the molecular basis for Huntingtin involvement in PI 3-kinase dependent functions. Our results show that mutant Huntingtin affects the activation of Rac1 and is in a complex with the active form of Rac1, the p85 regulatory subunit of PI-3 kinase, and α-actinin-2, a protein that crosslinks and bundles actin filaments [29, 30] and is enriched in striatum and present in spines [31].
MATERIALS AND METHODS
Animals
Brains from mice homozygous for a polyglutamine expansion and human exon1 knock-in within the endogenous mouse huntingtin genes (Hdh 140Q/140Q) [32] or WT mice with the same strain background of C57BL/6 were bred at the MGH animal facility. Genotypes were determined by PCR of tail DNA. Although named “Q140”, the average CAG repeat length of this colony is now 128 CAGs due to the instability at the locus. The animal protocol was approved by the MGH Subcommittee on Research Animal Care (SRAC)-OLAW Assurance # A3596–01. All procedures conform to the USD Animal Welfare Act, the ‘ILAR Guide for the Care and Use of Laboratory Animals’, PHS Policy on Humane Care and Use of Laboratory Animals, [33, 34]. Prior to rapid dissections on ice, mice were anesthetized with CO2 followed by cervical dislocation. Striatal and cortical tissues were rapidly frozen using liquid nitrogen then stored at –80°C until further use. Primary cortical neuron cultures were prepared and cultured exactly as previously described [35].
Cell culture
Primary human fibroblasts were obtained from Coriell Cell Repository and used within 10 passages. Cell line number and CAG repeat on each allele are as follows: GM08399, 17/19, WT; GM04837, 44/47, HD; GM04855 20/50, HD; GM04281, 17/69, HD; GM03440B, not determined, “unaffected” control. Cells were cultured using recommended medium. For PDGF experiments primary fibroblasts were made quiescent by incubating in serum-free medium for 48 hours prior to the addition of 100 ng/ml PDGF-BB (Biosource) for 15 min at 37C. The normal culture medium includes additional essential and non-essential amino acids. Cell serum starvation in this enriched medium showed no evidence of increased autophagy. The siRNAs targeting exon1 of HTT (named E1–4, 5’GACCCTGGAAAAGCTGATGAA’3) or GFP were previously described [36]. The sequence for GFP serves as a non-targeting sequence since the sequence does not exist in the human genome. Cells were plated in serum-containing medium onto coverslips plated at 2500 cells/coverslip. SiRNAs were added to cells plated at 2500 cells/coverslip at 20 nM (0.8μL Dharmafect reagent per 200 uL serum-free medium) on 12 mm glass coverslips coated with poly-L-lysine in serum free medium containing siRNA for 48 hrs then incubated for 15 min in serum-free media containing 100 ng/mL PDGF-BB (Gibco) or serum-free medium alone (control).
Human induced pluripotent stem cells (IPSCs) were acquired from NINDS Repository at the Coriell Institute for Medical Research: Cat# ND38554 (abbreviated as WT54) with 17 CAGs; Cat# ND36997 (abbreviated as WT97) with 32 CAGs; Cat# ND38551 (abbreviated as HD51) with 50 CAGs; Cat# ND38548 (abbreviated as HD48) with 41 CAGs. Genotypes were verified by PCR amplification of HTT locus in genomic DNA and by western blot. IPSCs were grown on mouse embryonic fibroblasts treated with mitomycin C in the following medium: Knockout DMEM/F12 (Life Technologies #12660–012) supplemented with 20% knockout serum replacement (Life Technologies #10828–028), 1 mM L-glutamine (Life Technologies #35050–061), 1x MEM non-essential amino acids (Life Technologies #11140–050), 100μM 2-mercaptoethanol and 50 units/mL penicillin/streptomycin (Life Technologies #15070–063). Adherent human neural stem cells (NSCs) were established as described [37–39] with modifications. Briefly, IPSC colonies were dislodged with 1 mg/mL collagenase type I (Life Technologies #17018–029) in DMEM (Life Technologies #11995–073) and re-plated onto 0.1% gelatin coated plates (Sigma #G1393) in NSC medium containing Advanced DMEM/F12 (Life Technologies #12634–028) with 2 mM L-glutamine, 50 units/mL penicillin/streptomycin, retinoic acid-free B27 supplement (Life Technologies #12587–010), N2 supplement; supplemented with 100 ng/mL FGF (Invitrogen #PHG0023), 100 ng/mL EGF (Invitrogen #PMG8041), 5μg/mL heparin (Sigma #H3149), and 1% knockout serum replacement. Bipolar cells streamed out of colonies during the next 5–14 days. When 60–80% confluent, cells were detached using Accutase (Sigma, #A6964) for 2–5 min and pelleted by centrifugation. Remaining cell clumps were disrupted with a p1000 micropipette tip and all cells were re-suspended in a single cell suspension and re-plated onto 0.1% gelatin coated plastic in NSC media containing 20 ng/mL FGF, 20 ng/mL EGF, and 5μg/mL heparin. Cells were passaged with Accutase as previously described 1:2–1:4 for the first four passages, then 1:2–1:10 during later passages. NSCs were routinely tested for expression of the radial glial marker Nestin by immunofluorescence and for normal and mutant Huntingtin expression using anti-huntingtin antibody Ab1 (against aa 1–17) by western blot.
Human neurons were differentiated from non-disease and HD iPSCs using dual TGFbeta and Smad inhibition and Activin A exactly as described [40]; the main neuron type produced is GABAergic with expression of DARPP32, characteristic of medium spiny neurons (MSNs) which are highly affected in HD. Using this procedure, we have about 80% cells positive for the neuronal marker Beta-III-tubulin with immunofluorescence and no detectable GFAP by western blot. To lower Huntingtin in human neurons, 1 or 3μM of a hydrophobically-modified siRNA (HTT10150, [41]) directed to position 10150 in HTT mRNA or a non-targeting control (NTC) was introduced into 38 day old neurons. Cultures were harvested at 43 days for analysis by western blot for huntingtin levels and for Rac activity.
Antibodies and cell stains
For western blot
Polyclonal anti-htt 1–17 Ab1(1μg/ml) [42], mAb 1C2 to polyglutamines (MAB1574, 1:1000; Millipore), mAb 3B5H10 to polyglutamines (P1874, 1:10,000; Sigma), p85 PI 3-kinase (1:500; #4292, Cell Signaling), AKT and phospho (Ser473) AKT (1:500; Cell Signaling); ERK and phospho ERK (1:1000; Cell Signaling); β-tubulin (1:4000; T8328, Sigma), beta3-tubulin (1:2000; Sigma), anti-GFAP (1:2000; Millipore), Rac1 (1:2000; Millipore), nestin (1:500; Millipore), DARPP32 (1:5000; Abcam), Islet1 (1:200; University of Iowa Developmental Studies Hybridoma Bank), GAPDH (1:6000; Millipore), α-actinin-2 (1:250; mAb clone EA-53, Sigma) or α-actinin-2 (1:2000; ab68167; Abcam; [31], α-actinin (2μg/ml; ab18061; Abcam; may cross react with isoforms 1–4).
For fluorescent labeling
Anti-Nestin (1:500; ABD69 rabbit polyclonal; Millipore), anti-DARPP32 (1:200; [EP720Y] ab40801 rabbit monoclonal; Abcam), and anti-BIII Tubulin Isotype III (1:500; T8660 mouse monoclonal; Sigma). Secondary antibodies include Cy3 goat anti-mouse secondary antibody (1:500; Jackson Immunoresearch); Cy3 goat anti-rabbit secondary antibody (1:500; Jackson Immunoresearch); Bodipy Green goat anti-mouse secondary antibody (1:500; Invitrogen). Stains include DAPI (1:500 Sigma) for nuclei.
Rac activity measurements
Rac activity was measured using ELISA-based kits: a colorimetric assay measuring active Rac1, 2, 3 and a luminescence assay specifically measuring active Rac1 (Cytoskeleton, Inc., Denver Colorado). These assays quantify the amount of Rac binding to the binding domain of its downstream effector, p21-activated kinase and are measured by absorbance at 490 nm or luminescence respectively on a Perkin Elmer Victor plate reader [43]. Fibroblasts and NSCs were grown to 50–70% confluence in 60 mm plates then fresh medium was introduced either with no growth factor but carrier, 100 ng/ml PDGF-BB, or 100 ng/ ml BDNF (Invitrogen) for 15 min at 37C. Cells were then lysed in 150μL in the kit lysis buffer on ice after three washes with ice cold 1X PBS containing 1 mM MgCl2 and 2 mM CaCl2. Mouse brain tissues were homogenized in provided lysis buffer, with the addition of three phosphatase inhibitors: 1 mM Na3VO4, 50 mM NaF and 20 mM Na4P2O7 and cleared by centrifugation for 1 min at 3,000 rpm. All lysates and supernatants were snap frozen in liquid nitrogen, and stored at –80C. Relative Rac1,2, 3 or Rac1 activity was measured per manufacturer instructions (Cytoskeleton, Inc., Denver, Colorado). Results are relative only to other samples run at the same time on the same ELISA plate. For each experimental set of lysates, samples were diluted to match that with the lowest protein concentration using lysis buffer, as per kit instructions. Thus, the exact amount of protein used varied from experiment to experiment. Equal volumes of lysates were loaded into 96 well plates pre-coated with the effector protein. Initial experiments were performed to determine that the concentrations of cell lysates resulted in signal that was above background and did saturate binding on the ELISA plate. Positive and negative controls were run for each experiment. All experimental results were repeated at least twice, except in Fig. 3b (using differentiated human neuronal cultures). Graphs show results of representative experiments. For relative Rac activity measurements from animals, WT and HD male mice at 1.5 and 4.5 months were sacrificed and processed randomly, but the investigator was not blinded to genotype; Rac activation assays on lysates from the same set of animals were performed in two independent experiments each with three technical replicates which were averaged for each animal.
Western blots and silver staining
Proteins were separated by SDS-PAGE using 3–8% Tris-acetate gels or 4–12% Tris-bis gels (Life Technologies); proteins were transferred to nitrocellulose using iBlot semi-dry transfer apparatus (Life Technologies) and developed using ECL or gels were stained using the Silver Stain Plus kit (BioRad). Statistical analysis was performed using Microsoft Excel or Prism (GraphPad Software). Pixel intensity quantification of 16-bit images was performed using ImageJ (NIH) and signal intensity for Huntingtin was standardized to signal intensity for β-tubulin or GAPDH. The bands were manually traced in ImageJ and the area and mean gray values of each band were determined. Total signal intensity was calculated by multiplying mean gray value by area to determine the total pixel intensity per band.
Immunoprecipitations
Primary human fibroblasts were serum-starved 24–48 h, treated with 100 ng/ml PDGF for 7 min then lysed in IP buffer at pH 7.4 (50 mM Tris, 150 mM NaCl, 1 mM MgCl2, 1 mM NaF, 1 mM Na3VO4, 1% NP 40, 10% glycerol) containing protease inhibitors (complete midi EDTA-free, Roche). The IP buffer was chosen to maximize interactions with membrane related signaling complexes [16]. 375μg of lysates, first pre-cleared with protein G-Sepharose 4-flastflow (GE Healthcare), were incubated overnight at 4C with 3μl anti-Htt monoclonal antibody MAB2166, clone 1HU-4C8 (Millipore). Lysates were incubated 2 h with protein-G-Sepharose then immune complexes were washed 3x with IP buffer and 1x in PBS. Immunoprecipitated proteins were eluted in 50μl sample buffer and analyzed by SDS-PAGE and western blot. For rapid IPs from mouse brain, the primary antibody MAB2166 was first bound to protein-G Sepharose for 3 h in IP buffer then pre-blocked with 2% BSA in PBS overnight at 4C. Brain tissues were homogenized in IP buffer, protein concentrations measured by Bradford Assay and 500μg of lysate per reaction were incubated with the whole antibody-protein-G-Sepharose complex for 30 min on ice with intermittent gentle shaking. Control reactions containing pre-blocked protein-G-Sepharose and lysate were included. Complexes were rapidly washed 3x in ice-cold IP buffer, washed 1x in PBS and eluted with 50μl sample buffer.
Statistical analysis
Confocal images file names or slides were encrypted and pixel intensity quantification and morphology analyses was performed by blinded observers. For animal studies, no statistical method was used to predetermine sample size number, however, a posthoc power analysis (G Power software) indicated n = 4 animals per group for the change between WT and HD in striatum at 1.5 months was Actual Power = 0.98. For comparisons of the same cell line treated or untreated, a paired t-test was used. For comparisons between two cell lines or two animal genotypes, unpaired t-tests were used. For comparisons among multiple samples ANOVA and Tukey’s HSD posthoc tests were performed in R or using GraphPad Prism.
Affinity chromatography
Control GST protein was expressed from pGEX-6P-1-FLAG-tag, a gift from Erich Wanker [45]. pGEX-2T-Rac1-wt was a gift from Gary Bokoch (Addgene plasmid # 12977). Plasmids were transformed into E.Coli strain BL21 by heat shock and plated onto LB agar, clones were isolated then grown in LB broth plus ampicillin at 37C with shaking until OD600 nm = 0.7, then cultures were induced with 0.5 mM IPTG and incubated another 4 h with shaking. Bacteria were collected by centrifugation at 4000x g for 45 min at 4°C. Pellets were re-suspended in 30 mL of buffer (10 mM Tris pH7, 50 mM NaCl, 50 mM KCl, 10% Glycerol) and centrifuged again at 10,000x g for 15 min at 4C. Pellets were preserved at –20°C until further use. Pellets were re-suspended in 10 mL of buffer (10 mM Tris pH 8.0, 50 mM NaCl, 50 mM KCl, Glycerol 10%, Lysozyme 1 mg.L–1, DTT 1 mM, 1 tablet of CompleteTM Mini EDTA-free). Suspensions were sonicated for 5 min (3 s “on”, 10 s “off”, amplitude of 40%). GST proteins were isolated using a gravity flow column purification with 400μL of Glutathione Sepharose 4B equilibrated in the same buffer, washed with 5 volumes 10 mM Tris pH 8.0, 50 mM NaCl, 50 mM KCl, Glycerol 10 % +Triton 0.5% followed by 5 volumes in the same buffer excluding Triton, then eluted with Tris 10 mM pH 8.0, 150 mM NaCl, 50 mM Glutathione. Fractions were analyzed on NuPAGE® Bis-Tris gels with MES buffer and evaluated for purity and integrity by SDS-PAGE and Coomassie stain. Most concentrated fractions were pooled and concentrated with Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-3 membrane to ∼100μL (13,000 rpm at 4°C during 15 min to concentrate 500μL to 100μL). The concentrated solution was brought to the original sample volume (∼500μL) with phosphate buffer 50 mM, pH7.4. This process of “washing out” was repeated 5 times (∼55 = 3,125 dilution fold) to realize buffer exchange.
Affinity chromatography was based on methods described by Christoforidis et al. [46]. For each condition, 750μg GST-Rac1 protein was incubated with 15μl Glutathione Sepharose 4B beads in buffer 1 as described with either 1 mM GTPγS (Sigma G8634) or 1 mM GDP (Sigma G7127) for 90 min at RT, then stabilized in buffer 2 with 1 mM GTPγS or GDP. Snap frozen dissected tissue from striatum of WT and HD Q140/Q140 mice at 1.5 months were lysed into buffer 2 with the addition of protease inhibitors, 1 mM NaF, and 1 mM Na3VO4, and without GTPγS or GDP. Lysates were centrifuged at 3000 g for 5 min to remove nuclei and supernatants (S1) were used for experiments. S1 sample concentrations were determined by Bradford, and 500μg of protein lysates were added to each GST-RAC1[GTPgammaS] or GST-RAC1[GDP] Glutathione sepharose complexes, incubated ON at 4C. Beads were spun down and flow through buffer (FT) was removed to avoid overloading the column. Beads were re-suspended in 3 volumes (45μl) buffer 2 and 10μM GTPγS or GDP, and loaded onto a micro-spin column (Pierce #89879). All spins were 2 min at 1000 g. Additional washes were completed (45μl, 30μl, 30μl) with buffer 2 and 10μM GTPγS or GDP. Additional washes 2×45μl and 2×30μl with buffer 2 containing 250 mM NaCl and 10μM GTPγS or GDP. One final wash was completed in 1 volume (15μl) (20 mM Hepes, 250 mM NaCl, 1 mm DTT, pH 7.5). A total of 5 serial dilutions (10μl each) were completed using elution buffer (20 mM Hepes, 250 mM NaCl, 20 mM EDTA, 50 mM Glutathione, 1 mM DTT, pH 7.5). GTPγS sample elution buffer additionally contained 5 mM GDP, while GDP sample elution buffer contained 1 mM GTPγS. Input (S1), flow through (FT) and five elution fractions (E1–5) were collected and analyzed by SDS-PAGE, silver stain and western blot analysis. WT brain was also incubated with GST alone with GTPγS or GDP buffers as controls.
RESULTS
Rac1 activity is impaired in HD fibroblasts and is positively regulated by Huntingtin lowering
Rac1 is involved in adhesion by promoting assembly of vinculin and α-actinin containing complexes [47] and requires PI(3,4,5)P3 to promote actin polymerization together with the WAVE regulatory complex [48–50]. Since Huntingtin interacts preferentially with lipid products of PI-3 kinase including PI(3, 4, 5)P3 [10], we asked if the presence of the HD mutation affected Rac activity. The ELISA assay with a peroxidase-based readout that we used did not discriminate between Rac1, Rac2 or Rac3 activity; however, northern blot studies show that Rac1 is ubiquitously expressed whereas Rac2 and 3 have restricted tissue distribution [20–22]. Thus, it is likely that the Rac activity measured in adult fibroblasts can be attributed to Rac1. Rac1, 2, 3 activity in a normal quiescent fibroblast line exposed to PDGF was significantly increased compared to no PDGF treatment as expected (Fig. 1a). However, in response to PDGF, Rac1, 2, 3 activity in three HD fibroblast cell lines failed to increase significantly (Fig. 1a).
Fig.1
Rac (1, 2, 3) activation is altered in human HD fibroblasts and by loss of Huntingtin. (a) Bar graph shows mean±SD of relative Rac (1, 2, 3) activity (OD490) in primary human control and HD fibroblasts. CAG repeat length for both alleles is indicated under the x-axis. PDGF treatment significantly increased relative Rac (1, 2, 3) activity in control fibroblasts but not in three HD cell lines (ANOVA and Tukey’s HSD posthoc tests, F = 5.03, *p < 0.01, n = 6 technical replicates except HD17/69 – PDGF which is n = 4). (b) Huntingtin lowering by siRNA (E1–4). Bar graph shows mean±SD for Huntingtin pixel intensity quantification standardized to tubulin in control fibroblasts as percent of mock by western blot (*p < 0.05, n = 3 biological replicates, unpaired t-test compared to GFP). Western blots are shown in Supplementary Figure 1e. (c) Relative Rac (1, 2, 3) activity (OD490) was measured in lysates from cells transfected with HTT siRNA compared to GFP siRNA. Bar graph shows mean±SD of relative Rac (1, 2, 3) activity with siRNA for GFP or Huntingtin. In the Huntingtin (E1–4) treated cells, there is a significant increase in the basal level of Rac (1, 2, 3) activation per microgram protein (*p < 0.005, n = 6 technical replicates, paired t-test). (d) Huntingtin interacts with PI 3 kinase p85α. Primary human fibroblasts were serum-starved for 48 h then stimulated with PDGF added directly to the medium for 7 minutes (100 ng/ml final concentration). The CAG repeat length for each allele of the HD gene is indicated in parenthesis at top. Anti-Huntingtin antibody MAB2166 was used to isolate Huntingtin from control and HD fibroblasts. Control lanes are B (buffer alone with protein G and primary antibody), and no ab control (protein G incubated with input lysates without primary antibody). Western blots probed with anti-Huntingtin Ab1 then re-probed with anti-p85 PI 3-kinase. β-tubulin was used as a loading control. Molecular mass markers are on the left. Black lines indicated where lanes were removed from the same gel. (e, f) Pixel intensity quantification of immunoprecipitation (IP) results for p85 PI 3-kinase standardized to signal for Huntingtin (e) and inputs standardized to β-tubulin (f) shows a significant increase in level of p85 PI 3-kinase in Huntingtin-immunoprecipitates with PDGF treatment for control fibroblasts but not HD fibroblasts and PI 3-kinase p85α levels were significantly reduced in HD cells compared to the normal control in the inputs. Bar graph shows mean ratio ± SD, *p < 0.05, n = 3 experiments, unpaired t-test.
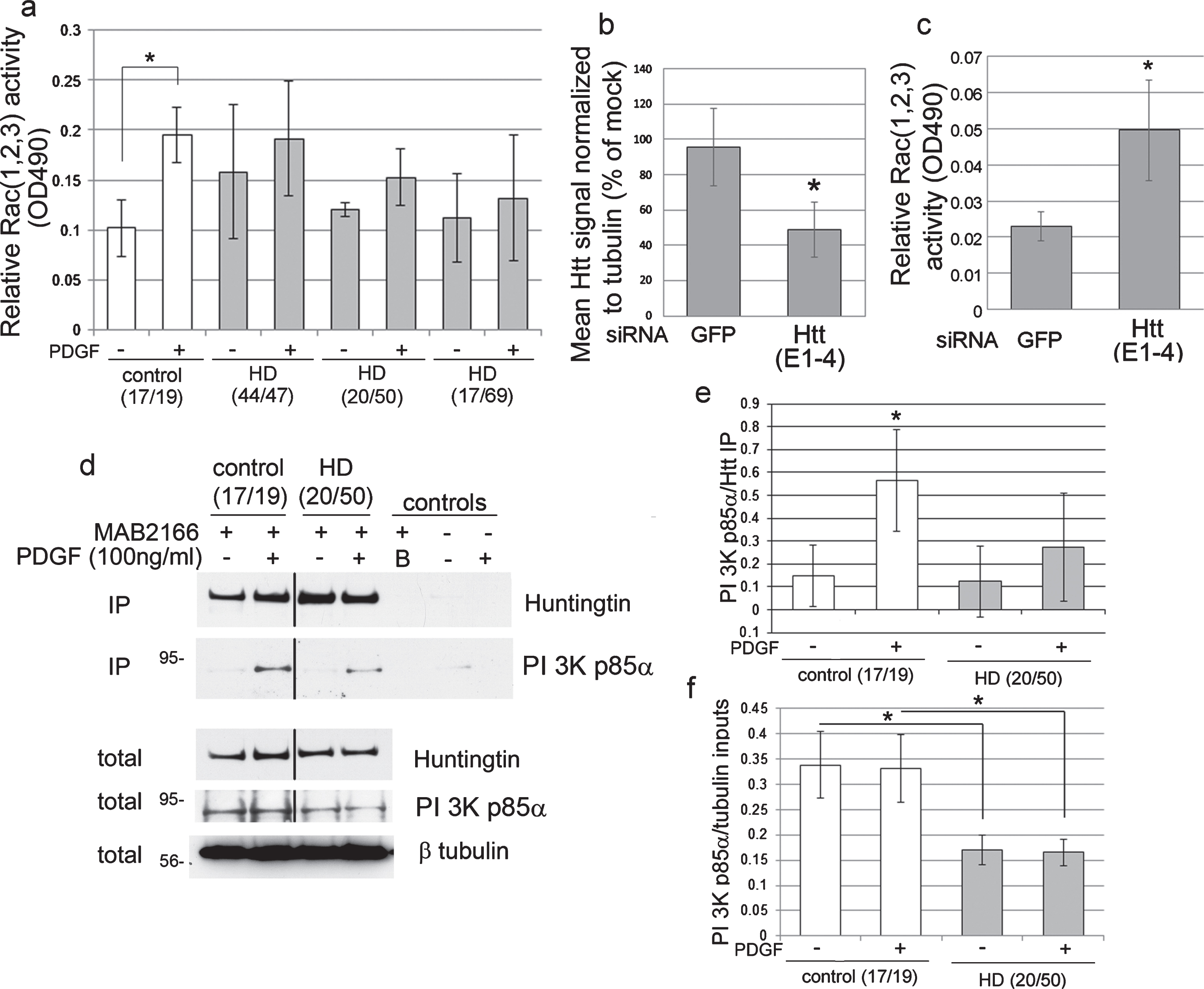
To determine if other downstream signaling targets of PDGF receptor were also affected, phosphorylation levels of AKT and ERK were examined. Western blot analysis showed normal levels of phosphoAKT and phosphoERKs in total lysates of control and HD fibroblasts in response to PDGF stimulation, except for the phosphoAKT response to PDGF in one HD line that had a very large CAG repeat in HTT (Supplementary Figure 1a-d). These data indicate that activation of PI 3-kinase occurs normally in human HD fibroblasts since AKT requires the PI 3-kinase products PI(3,4)P2 and PI(3,4,5)P3 for binding to the plasma membrane where it is phosphorylated by protein kinase D. To determine if the inability to activate Rac1, 2, 3 in HD fibroblasts was due to altered Rac1 protein levels, western blot analysis was performed. Levels of Rac1 were not significantly different between control and HD cell lines, however there was a trend for lower levels of Rac1 protein in two of the HD cell lines with the higher CAG repeats (20/50 and 17/69) (Supplementary Figure 1f).
To determine if Huntingtin is required for the activity of Rac1, 2, 3 we treated control human fibroblast cells with siRNA targeting HTT mRNA. At 48 hours post-transfection, western blot and pixel intensity quantification analysis showed that treatment with siRNA targeting HTT mRNA (E1-4) significantly lowered Huntingtin protein levels in the cultures by 50% compared to siRNA targeting GFP (Fig. 1b and Supplementary Figure 1e; p < 0.01, n = 3). Huntingtin lowering using siRNA E1-4 significantly increased the basal level of Rac1, 2, 3 activation compared to GFP siRNA (Fig. 1c; p < 0.005, n = 6, t-test). Western blot analysis showed levels of Rac1 were similar with siRNA E1-4 targeting HTT mRNA and siRNA GFP mRNA (Supplementary Figure 1 g). PDGF treatment of cells with reduced levels of Huntingtin rapidly induced about a 50% detachment of cells during the wash steps and precluded our ability to measure levels of active Rac following PDGF stimulation. These findings show that Huntingtin participates in the normal homeostasis of Rac activation.
Next, we looked for interactions between endogenous Huntingtin and components of the PI 3-kinase/Rac pathway using immunoprecipitation assay with anti-Huntingtin antibody MAB2166. From lysates of control human fibroblasts, negligible amounts of p85α co-immunoprecipitated with Huntingtin; however, upon stimulation with PDGF detectible levels of p85α co-precipitated with Huntingtin and were significantly higher than without PDGF (Fig. 1d, e; p < 0.05, n = 3 experiments), indicating a stimulus dependent interaction. In HD fibroblasts, the levels of p85α that co-immunoprecipitated with Huntingtin after PDGF stimulation were not statistically significant compared to non-stimulated HD cells (Fig. 1d, e). Pixel intensity quantification results of the input lysates showed that PI 3-kinase p85α levels were significantly reduced in HD cells compared to the normal control (Fig. 1d, f). These results show that endogenous Huntingtin associates with p85α, a major component of the receptor tyrosine kinase/PI3 kinase/Rac pathway. Polyglutamine expansion in endogenous Huntingtin reduced the association and stimulus dependent interaction with p85α in human fibroblasts.
Rac1 activity is altered in human HD NSCs and neuron cultures, and mouse HD striatum and is rescued by Huntingtin correction or lowering
Although HD affects peripheral cells, CNS cells are severely affected and the hallmark of HD is degeneration of cortical and striatal neurons. To determine if Rac activation was affected in more relevant cell types, we examined human neural stem cells (NSCs) derived from control or HD induced pluripotent stem cells (IPSCs). The presence of wild-type (WT) and mutant HTT (HD) alleles and protein were confirmed by PCR and western blot (Supplementary Figure 2a, b), as were levels of other markers of the neural lineage. Nestin-positive cells were assessed by immunofluorescence (Supplementary Figure 2c). The cells were all heterozygous and the polyglutamine lengths for the longest allele (including the CAA following the CAG tract) were 17Q for WT54, 33Q for WT97; 51Q for HD51, 42Q for HD48, 72Q for HD4, 21Q for HDc116. These experiments were performed using an ELISA specific for Rac1 and with a luminescent readout. Unlike fibroblasts, control NSCs did not respond to PDGF but instead responded to BDNF by significantly increasing their levels of active Rac1 (Fig. 2a). BDNF is a growth factor that also activates the PI 3-kinase pathway and can regulate morphology changes in neural cell types. HD NSCs had elevated basal levels of Rac1 activation that were equal to or higher than the BDNF-stimulated levels of the control NSCs (Fig. 2a). Treatment with BDNF failed to significantly increase Rac1 activation further in any of the HD lines tested suggesting that the HD cells were maximally stimulated in basal conditions. Crucially, an isogenic line (HDc116 NSCs) to HD4 in which the repeat was reduced to 21 CAGs via homologous recombination [51] had basal Rac1 activation levels that were significantly lower compared to HD4 (Fig. 2b).
Fig.2
Rac1 activity in human NSCs and neurons and effects of Huntingtin lowering. (a) Bar graph shows mean±SD of relative Rac1 activity (Luminescence RLUx106) for human control NSCs (WT54 and WT97) and HD NSCs (HD48, HD51, and HD4), without and with BDNF treatment. In WT cells Rac1 activity is increased with BDNF treatment as expected compared to unstimulated WT cells (ANOVA and Tukey’s HSD posthoc tests *p < 0.01; n = 3 technical replicates). In non-stimulated conditions (– BDNF), relative Rac1 activity is increased in HD NSCs compared to WT NSCs (+p < 0.01). (b) Graph shows mean±SE of relative Rac1 activity (Luminescence RLU×106) in NSCs from line HD116c (genetically corrected HD4 line) and HD4. Genetic correction significantly decreases relative Rac activity levels (*p < 0.01, n = 12 wells from 3 biological replicates, unpaired t-test). (c) Basal Rac1 activity in human HD (HD51) neurons is increased compared to WT (WT54) neurons. Graph shows mean±SD of relative Rac1 activity at DIV35 (*p < 0.05, unpaired t-test, n = 4 technical replicates). (d-g) Lowering Huntingtin increases Rac1 activity in WT neurons and reduces Rac1 activity to normal levels in HD neurons. WT97 and HD51 were treated with siRNA HTT10150 targeting HTT mRNA or with a non-targeting control (NTC) and harvested for Huntingtin detection by western blot with antibody directed to htt1–17 (Ab1) (d and e) and for Rac1 activity (f and g). Bar graphs show results for mean Huntingtin signal intensity based on pixel intensity quantification as percent of untreated control and relative Rac1 activity in siRNA HTT10150 (HTT) treated and non-targeting control (NTC) treated cells (*p < 0.05, ANOVA with Bonferroni’s Multiple Comparison test, n = 3 technical replicates). Black line in f and g shows Rac1 levels in the untreated cells measured on the same ELISA plate.
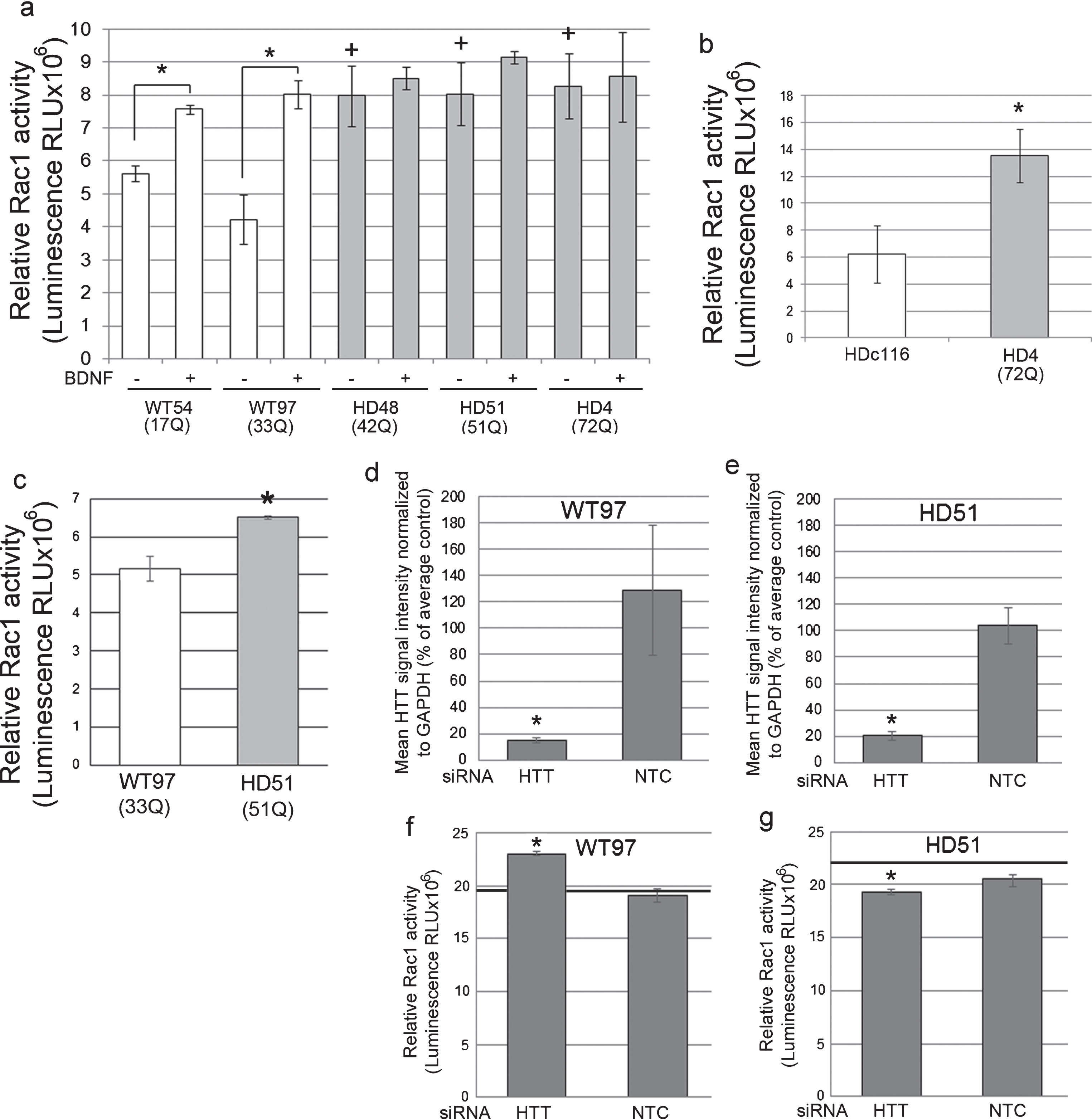
Rac1 activation was also measured in human control WT and HD neuron cultures that were differentiated from IPSC using the method described by Arber et al. [40]. Neuronal cultures were characterized for the presence of normal and mutant Huntingtin and for neuronal phenotypes using western blot and immunofluorescence (Supplementary Figure 2d, e). Like the HD NSCs, HD51 neuron cultures (51Q) had increased levels of basal active Rac1 compared to WT97 neurons (Fig. 2c) and compared to WT54 (Supplementary Figure 3a). Lowering Huntingtin in human WT97 neuron cultures with a chemically modified siRNA called HTT10150 targeting HTT mRNA significantly increased the levels of Rac1 activity compared to cultures treated with a non-targeting (NTC) control siRNA with the same chemical modification, indicating that Huntingtin in neuron cultures is normally required for the maintenance of Rac1 homeostasis (Fig. 2d, f; Supplementary Figure 3b). In HD51 neuron cultures, lowering Huntingtin (both normal and mutant alleles) attenuated the elevated levels of Rac1 activity to that of WT97 cells treated with the control siRNA (NTC treated group) (Fig. 2e, g; Supplementary Figure 3b). These results suggest that reducing total Huntingtin in the presence of the HD mutation can restore normal levels of Rac activity. Using independent neuronal cultures and siRNA HTT10150 to lower Huntingtin protein levels, the same results were found using the ELISA assay for Rac1, 2, 3 activity (Supplementary Figure 3c, d-g). The magnitude of change was slightly higher using the Rac1, 2, 3 assay possibly due to differences in the enzymatic readout between the assays or a contribution of Rac3 (see Discussion).
Finally, we examined levels of Rac1 activation in a mouse model of HD. Primary embryonic cortical neurons from knock-in Q140/Q140 HD mice had increased basal levels of Rac1, 2, 3 activity compared to WT neurons at 5, 7 and 11 days in vitro (Fig. 3a, published previously in [52]). Relative Rac1 activity was examined in the striatum and cortex of the WT and knock-in Q140/Q140 HD mice at 1.5 and 4.5 months. Compared to the striatum of WT mice, the striatum of Q140/Q140 HD mice had increased levels of Rac1 activation at 1.5 months (Fig. 3b). However, by 4.5 months, the level of Rac1 activity in HD striatum was significantly lower than in WT. Rac1 activity in striatum and cortex of WT mice increased significantly with age. Rac1 activity in the cortex was not different between WT and HD mice at the ages examined (Fig. 3b).
Fig.3
Rac1, 2, 3 and Rac1 activity in WT and HD mouse primary neurons, striatum and cortex. (a) Primary embryonic cortical neurons from knock-in Q140/Q140 HD mice have increased basal levels of Rac1, 2, 3 activity compared to WT neurons (*p < 0.01, unpaired t-test, n = 3–4). Reported before in a previous paper retracted for problems with other data. (b) Basal Rac1 activity in the striatum and cortex of WT and Q140/Q140 HD mice. Graphs show mean relative Rac1 activity±SD (ANOVA and Tukey’s HSD posthoc tests, F = 20.36 in “striatum” and 27.32 in “cortex”; *p < 0.001 **p < 0.05, n = 4 mice per genotype at 1.5 months and 4.5 months). In the striatum, relative Rac1 activity is increased in HD compared to WT at 1.5 months, but decreased at 4.5 months. In the cortex relative Rac1 activation is increased in WT and HD mice at 4.5 months compared to 1.5 months. (c) Western blot analysis of lysates from striatum of WT and HD mice at 1.5 and 4.5 months. Top blots probed with antibody to Huntingtin to verify genotype (10μg protein separated on 3–8% Tris-acetate SDS-PAGE). Middle and bottom blots probed with antibody to Rac1 and re-probed with antibody to GAPDH as a loading control (10μg protein separated on 4–12% Bis-tris SDS-PAGE). Space indicates separate gels. Pixel intensity quantification results at right show less Rac1 protein in HD mice at 1.5 months. Bars are mean±SD, *indicates p < 0.05, n = 4 mice, unpaired t-test.
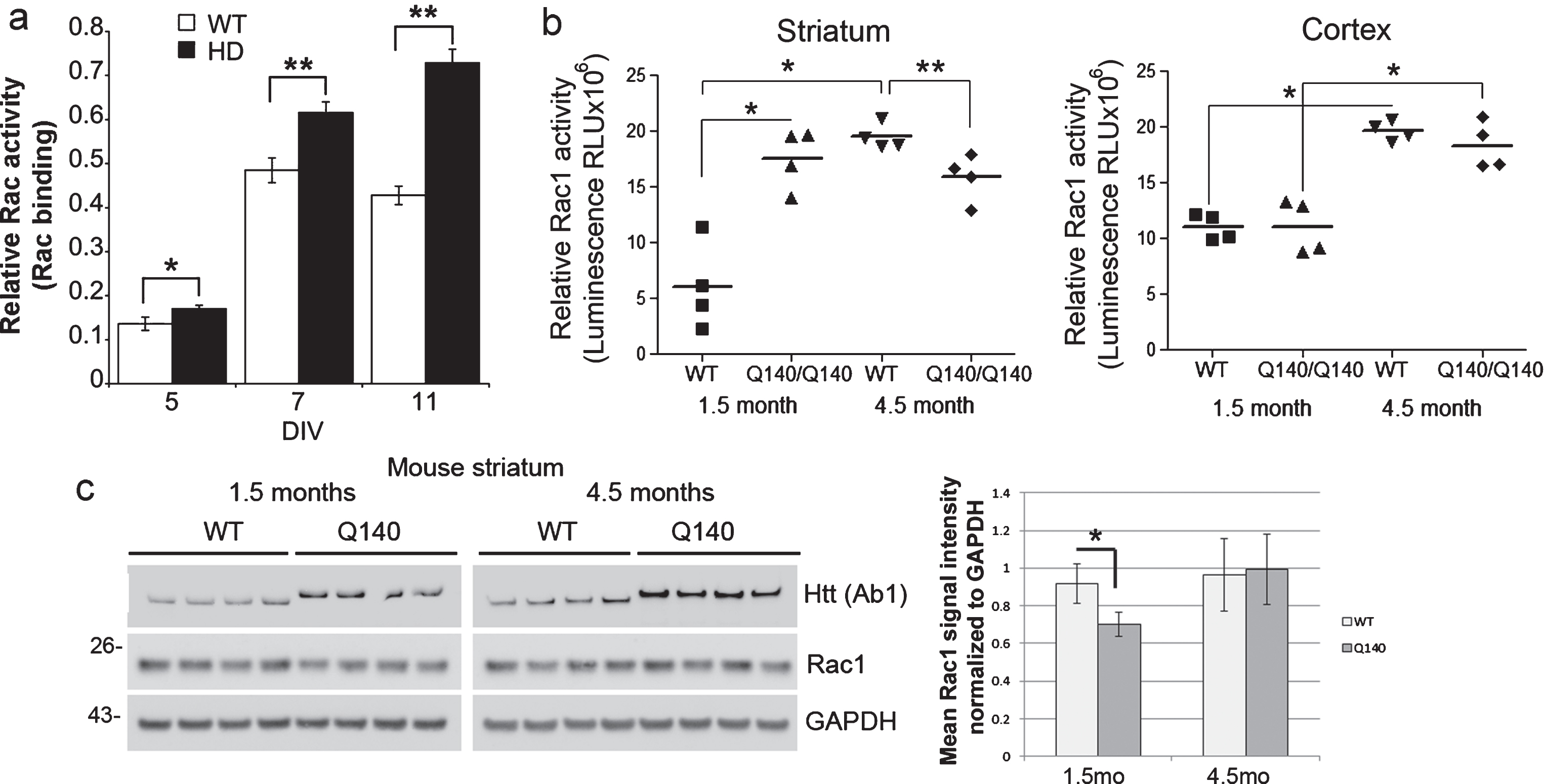
To ascertain if an increase in levels of Rac1 protein could explain the increases in Rac1 activation in NSCs and neurons, we detected Rac1 protein by western blot assay. No consistent change in levels of Rac1 protein was observed between WT and HD human NSCs or human neuron cultures (Supplementary Figure 2b, d). HD48 NSCs did have a higher level of Rac1 protein compared to the other NSCs, but both HD51 and HD4 had similar levels to WT NSCs. However, Rac1 protein levels were significantly lower at 1.5 months in striatum of Q140/Q140 HD mice compared to WT mice (Fig. 3c, middle row and bar graph). The levels of two surface receptors that can activate Rac1, p75NTR and the BDNF receptor TRKB, also were not different between WT and HD mice in 1.5-month cortex and striatum; however, a significant reduction in TRKB protein was apparent in striatum at 4.5 months in HD compared to WT (Supplementary Figure 4a, b). Finally, we did not observe any changes in levels of the GEFs for Rac1, Kalirin-7 or Kalirin-8, in striatum of Q140/Q140 HD mice (Supplementary Figure 4b) that might explain increased Rac1 activation.
Collectively, the results show that PI 3-kinase-dependent Rac1 activation is impaired by the presence of mutant Huntingtin in HD human NSCs and neuron cultures, primary embryonic cortical neurons of HD mice, and in the striatum of young HD mice. Furthermore, correction of the HD mutation by homologous recombination in human NSCs or by lowering overall levels of Huntingtin in human HD neuron cultures reduced basal Rac1 activity to normal levels. The elevated levels of Rac1 activation in HD cannot be explained by increased Rac1 protein or changes in upstream receptors tyrosine kinases, p75NTR or TRKB, or in levels of Kalirin-7 or Kalirin-8.
Huntingtin is in a complex with Rac1, p85α subunit of PI 3-kinase, and alpha-actinin in brain
To look for interactions between endogenous Huntingtin and components of the PI 3-kinase/Rac pathway in brain, we performed immunoprecipitations from WT and HD Q140/Q140 mice total brain lysates using anti-Huntingtin MAB2166 and a rapid immunoprecipitation protocol (see Methods). Consistent with previous findings in PC12 cells [9, 16] and in human fibroblasts (this study, Fig. 1d, e), the p85 regulatory subunit of PI 3-kinase (p85α) was detected in anti-Huntingtin immunoprecipitates (Fig. 4). In contrast to results found in human fibroblasts (this study, Fig. 1), no significant difference in the levels of pull down for either protein occurred between wild-type and Q140/Q140 HD mice (n = 4 experiments; unpaired t-test). These same Huntingtin immunoprecipitates from WT and HD mouse brains also contained Rac1 (Fig. 4). We were unable to find conditions allowing for immunoprecipitation of endogenous Huntingtin with Rac1 using a Rac1 antibody. To gain more information regarding the regulation of the interaction of Rac1 and Huntingtin, we performed affinity chromatography using bacterially expressed GST-Rac1 fusion protein loaded with GTPγS (the active state of Rac1) or loaded with GDP (the inactive state of Rac1) and lysates from dissected striatum from WT or Q140/Q140 HD mouse brains at 1.5 months (Fig. 5a-d). Western blots showed WT Huntingtin and mutant Huntingtin were eluted in the first fraction and preferentially pulled down with GST-Rac1[GTPγS] compared to GST-Rac1[GDP] (Fig. 5a, b, lanes E1 GTP and E1 GDP). Similarly, PI 3-kinase p85α pulled down with GST-Rac1[GTPγS] from both WT and HD lysates and co-eluted with Huntingtin (Fig. 5a, b). Silver stained gels of starting fraction (S1), flow-through (FT) fractions and eluates (E) of the same experiment are shown in Fig. 5c and d. Western blots from a control reaction using GST alone showed no detectible Huntingtin or PI 3-kinase pulled down (Fig. 5e, lanes E1 GTP and E1 GDP). A silver stained gel from the GST only column with WT mouse striatum S1 input also shows many fewer proteins eluting from the column (Fig. 5f). These results show that as in human fibroblasts (Fig. 1), endogenous Huntingtin in striatum associates with components of the receptor tyrosine kinase/PI3 kinase/Rac pathway (Rac1 and p85α) and that Huntingtin and PI 3-kinase p85α associate preferentially with the GTP bound state of Rac1.
Fig.4
Rac and p85α PI 3-kinase both co-immunoprecipitate with endogenous Huntingtin. (a) Western blot analysis of Huntingtin (Htt) immunoprecipitated from wild-type (WT) and Q140/Q140 (HD) adult mouse brain using anti-Huntingtin antibody MAB2166. Eluates were analyzed using two gel formats. Blot from a 3–8% SDS-PAGE gel was probed with polyclonal anti-Huntingtin Ab1 to confirm presence of full-length Huntingtin (∼350 kDa) (top). Blot from 4–12% SDS-PAGE gel was probed for p85 regulatory subunit of PI 3-kinase (middle blot, arrow) and Rac1 (bottom blot, arrow). The mouse IgG light chains (indicated) are recognized by the secondary antibody on the Rac1 probe. Vertical lines indicate where lanes were removed from same gel.
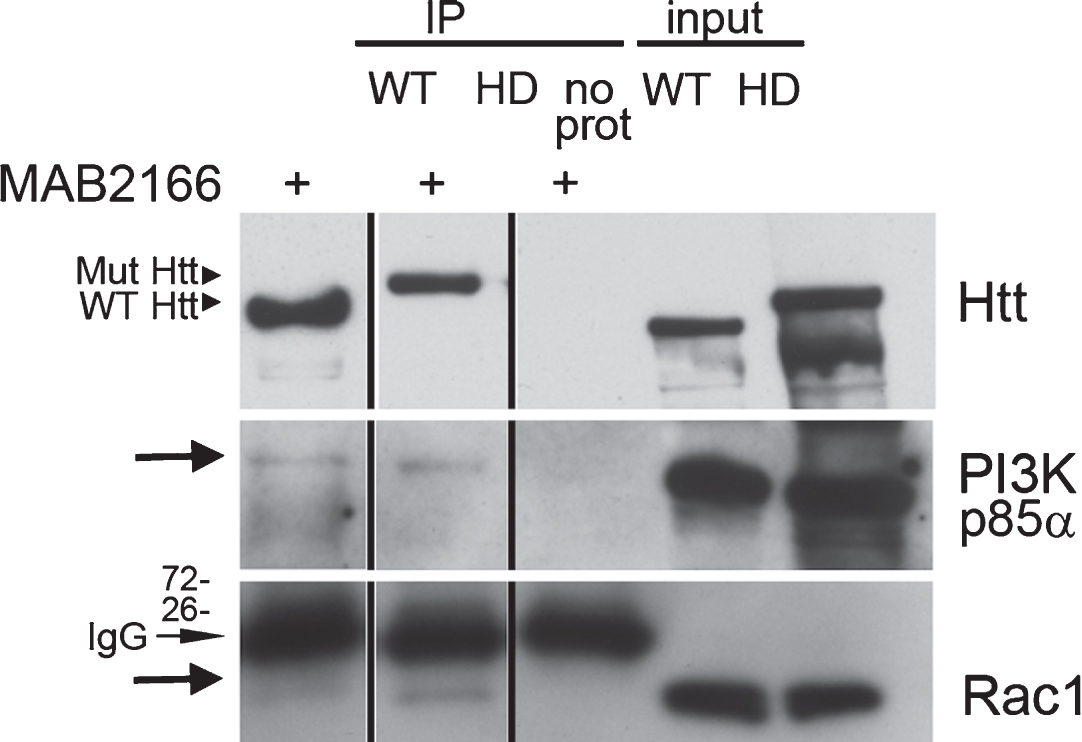
Fig.5
Affinity chromatography isolates a protein complex with GST-Rac1[GTPγS] including Huntingtin, p85α, and α-actinin-2. Affinity chromatography using GST-Rac1[GSTγS] or GST-Rac1[GDP] with lysates from wild-type (WT) or HD (Q140/Q140) mouse striatum at 1.5 months. Lanes (applies for a-f): P1, 2000 g pellet fraction; S1, 2000 g supernatant which was used as input for column; FT GTP and FT GDP, flow-through fractions; GTP E1–E5, elutions 1–5 from GST-Rac1[GSTγS] column. GDP E1–E5, elutions 1–5 from GST-Rac1[GDP] column; lanes at far right loaded with pure GST-Rac1 protein at indicated concentrations to identify fusion protein on the gels. (a, b) Western blots of fractions indicated at top of blots were probed for Huntingtin (Htt) with Ab1 targeting aa1–17 of Huntingtin, α-actinin-2, PI 3-kinase p85α regulatory subunit, and for Rac1 as indicated at right of blots. Relative molecular mass is indicated in kilodalton at left. Black lines indicate removal of marker lanes from samples that were run on the same gel exactly as in c and d. (c, d) Silver stained gels of the same samples shown in a and b. Molecular weight markers indicated on left or within gel lane at left. Marker 1 (EZ Run), Marker 2 (SeeBlue). Silver stains were overdeveloped to visualize high molecular weight proteins (Huntingtin). (e, f) Western blot and silver stained gel of affinity chromatography input, flow through and elutions using GST alone with lysates from wild-type (WT) mouse striatum at 1.5 months as a control. Low background binding was observed with α-actinin-2, but binding to GST-Rac1 was significantly higher. Vertical lines in a,b, and e indicate where lanes were removed from the same gel.
![Affinity chromatography isolates a protein complex with GST-Rac1[GTPγS] including Huntingtin, p85α, and α-actinin-2. Affinity chromatography using GST-Rac1[GSTγS] or GST-Rac1[GDP] with lysates from wild-type (WT) or HD (Q140/Q140) mouse striatum at 1.5 months. Lanes (applies for a-f): P1, 2000 g pellet fraction; S1, 2000 g supernatant which was used as input for column; FT GTP and FT GDP, flow-through fractions; GTP E1–E5, elutions 1–5 from GST-Rac1[GSTγS] column. GDP E1–E5, elutions 1–5 from GST-Rac1[GDP] column; lanes at far right loaded with pure GST-Rac1 protein at indicated concentrations to identify fusion protein on the gels. (a, b) Western blots of fractions indicated at top of blots were probed for Huntingtin (Htt) with Ab1 targeting aa1–17 of Huntingtin, α-actinin-2, PI 3-kinase p85α regulatory subunit, and for Rac1 as indicated at right of blots. Relative molecular mass is indicated in kilodalton at left. Black lines indicate removal of marker lanes from samples that were run on the same gel exactly as in c and d. (c, d) Silver stained gels of the same samples shown in a and b. Molecular weight markers indicated on left or within gel lane at left. Marker 1 (EZ Run), Marker 2 (SeeBlue). Silver stains were overdeveloped to visualize high molecular weight proteins (Huntingtin). (e, f) Western blot and silver stained gel of affinity chromatography input, flow through and elutions using GST alone with lysates from wild-type (WT) mouse striatum at 1.5 months as a control. Low background binding was observed with α-actinin-2, but binding to GST-Rac1 was significantly higher. Vertical lines in a,b, and e indicate where lanes were removed from the same gel.](https://ip.ios.semcs.net:443/media/jhd/2019/8-1/jhd-8-1-jhd180311/jhd-8-jhd180311-g005.jpg)
α-actinin-1 and Rac1 work together in the assembly of stress fibers during cell spreading and cell migration [53] and Rac1 promotes assembly of vinculin and α-actinin containing complexes [47]. Previous work by others suggested that Huntingtin might interact with α-actinin isoforms [18, 23, 24]. We found that α-actinin-2 which is enriched in striatum was pulled down with GST-Rac1 affinity chromatography using striatal lysates in the same fraction as Huntingtin (Fig. 5a, b, lanes E1 GTP and E1 GDP), suggesting that α-actinin-2, Rac1, PI 3-kinase and Huntingtin may co-exist in a complex in brain. A control experiment using lysates from WT striatum and GST alone did show some background binding of α-actinin-2, but the signal for GST-Rac1 was higher using the same mass protein input (Fig. 5e).
DISCUSSION
The preferential interactions of native Huntingtin with the lipid products of PI 3-kinase, PI(3,4)P2 and PI(3,4,5)P3, led us to examine the role of Huntingtin in the activity of Rac1 which is a downstream target of receptor tyrosine kinase/PI-3 kinase dependent signaling that regulates changes in morphology in response to growth factors. In this study, we demonstrated that endogenous Huntingtin interacts with endogenous Rac1, the p85α subunit of PI 3-kinase and may associate with α-actinin-2 in a protein complex. Acute reduction of Huntingtin increased basal Rac1 activity in human control fibroblasts and neuronal cultures, and the presence of mutant Huntingtin increased basal Rac1 activity and impaired growth factor dependent activation of Rac1. The increase in basal Rac1 activity occurred in HD cells of neuroectodermal origin (neural stem cells/glia and neurons) which could be a factor in HD pathogenesis in the CNS. Rac1 is ubiquitously expressed whereas Rac2 is restricted to T-, B- and hemopoietic cell lineages [20, 21]. Rac3 transcripts are expressed most prominently in brain tissue but also in heart, pancreas and placenta [22]. Here, Rac activation levels were measured using both an assay for Rac1, 2, 3 as well as one more specific to Rac1 in human neuron cultures and found to be changed in the same direction with both assays. These results show that Rac1 activity contributes in part to altered Rac activity in human neurons. However, since Rac3 is highly expressed in brain and a greater change was observed when measuring Rac1, 2, 3 activity, we cannot exclude contributions from Rac3 in NSCs and neurons and mouse brain. The Rac activity measured in fibroblasts using the Rac 1, 2, 3 method was likely arising from Rac1 since Rac2 and 3 have restricted expression patterns. A large scale proteomic study with GST-Huntingtin fragments and mouse brain lysates identified Rac1 as an interactor (Rac 2 and Rac 3 were not identified) [17] supporting the idea that Huntingtin regulates Rac1.
Our findings here together with work on the roles of Rac1 and α-actinin in other studies [53–55] suggest that Huntingtin regulates Rac activity as part of a coordinated response to growth factor signaling. A logical scenario is that upon growth factor stimulation, Huntingtin associates with p85α PI 3-kinase. PI(3,4,5)P3, which is created on the plasma membrane by the PI 3-kinase complex (regulatory plus catalytic domains) [56], causes dissociation of α-actinin from beta-integrin in adhesion plaques to allow restructuring of the plaques, as previously described [54]. Huntingtin then could act as a scaffold to support p85α PI 3-kinase, α-actinin and Rac1 at new focal regions on membranes for localized activation of Rac1 and the WAVE regulatory complex for Arp2/3 nucleation of actin polymerization [50, 57]. Using brain lysates, we found α-actinin-2 retained on a GST-Rac1 column together with Huntingtin and p85α. In neurons, the shape of spines is regulated by both α-actinin-2 [31] and Rac activity (see below). Our finding that reducing Huntingtin levels increased Rac1 activation, suggests that Huntingtin may also coordinate complexes to curtail Rac1 activation, perhaps through its interaction with p85α. p85α has been implicated in negative feedback regulation of Rac1 [58] and directly interacts with Huntingtin by yeast two-hybrid assay [17, 18]. Alternatively, Huntingtin may coordinate a guanine nucleotide exchange factor (GEF) or GTPase activating protein (GAP) activities to control Rac activation levels. Interactome data [18, 23, 24, 59] suggests that Huntingtin associates or interacts with numerous GAPs including ARHGAP 21, ARHGAP 23, ARHGAP 24, and ARHGAP 25, and SRGAP1–3. That Huntingtin in brain preferentially associates with Rac1 in its GTP bound state is most consistent with the idea that Huntingtin coordinates negative regulation through recruitment of a GAP or via PI 3-kinase p85α. Future experiments will be aimed at defining which molecules Huntingtin coordinates to achieve normal regulation of Rac1.
Basal levels of Rac1 activity were elevated compared to WT in human HD NSCs and neurons, HD mouse primary cortical neurons, and striatum of 1.5-month-old HD mice. In HD cells (human HD fibroblasts and human HD NSCs), growth factor stimulation failed to increase Rac activity but likely through different mechanisms since the resting level of Rac1, 2, 3 activity was normal in HD fibroblasts but elevated in HD NSCs compared to controls. The lack of Rac1, 2, 3 activation in response to stimulation in human HD fibroblasts is consistent with previous findings showing impaired ruffling response of peripheral HD monocytes to stimulation [60]. Our results suggest that the mechanism underlying these Rac1 signaling alterations are different between peripheral (fibroblasts and monocytes) and central nervous system cells (neural-derived cells). In fibroblasts, one possibility is that mutant Huntingtin has direct effects on PI 3-kinase activity. We found that HD fibroblasts had lower levels of PI 3-kinase p85α than control fibroblasts in total lysates and in co-precipitates with Huntingtin which could prevent full activation of Rac by the complex that contains Huntingtin. On the other hand, normal and mutant Huntingtin from mouse brain pulled down similar amounts of p85α, and yeast two hybrid studies have shown no change in interaction of p85 subunits with polyglutamine expansion in Huntingtin fragments [18]. Two HD fibroblasts lines also had trends for lower levels of Rac1 protein. Although basal levels of Rac1, 2, 3 activity were not changed compared to the control line, lower levels of total Rac1 protein may reduce the amount of Rac1 present at the correct location available for further activation with growth factor stimulation. Other factors may contribute to excessive Rac1 activity in HD neural cells such as inappropriate activation via GEFs, a reduced capacity to attenuate Rac activation via GAPs, or a failure to remove Rac1 from the membrane via the protein Rho GDi. The divergent response between peripheral cells and CNS cells also might be due to stimulation using different growth factors (PDGF in fibroblasts versus BDNF in NSCs).
We recently described an interaction with Huntingtin and Kalirin-7, a known GEF for Rac1 [61]. Our in vitro studies suggested that mutant Huntingtin can inhibit exchange of GTP onto GST-Rac1 facilitated by Kalirin-7. One would predict then that cells expressing mutant Huntingtin should have lower Rac1 activation, as observed in fibroblasts but not cells of neural ectoderm origin. It is possible that another GEF is upregulated in early developing HD neuroectodermal cells resulting in over-compensation. After the early increased activation of Rac1 in HD knock-in mice, we did observe a significant decrease in Rac activation. This is consistent with results by Puigdellívol et al. (2105) who found decreased Rac1 activation in cortex at 8 months in Q111/Q7 HD mice paralleling changes in number of synapses [62]. Gines and colleagues attributed the activation in Rac1 to decreased levels of Kalirin-7 at 8 months, which we did not observe in striatum of Q140/Q140 HD mice at 4.5 months. The same group also described an increased RhoA activation due to upregulation of p75NTR [63, 64]. We did not see differences in levels of p75NTR at 1.5 months in striatum when Rac1 activation levels were elevated in Q140/Q140 HD mice. We did see a small reduction in the BDNF receptor, TRKB, at 4.5 months when Rac1 activity was reduced. Thus, our results are consistent with the idea that decreased Rac1 activation levels at later ages may be due to an imbalance in signaling between p75NTR and TRKB. However, since Rac1 activation is elevated early in HD mice or in cells at developmental stages (NSCs or embryonic neurons), our results suggest an early imbalance in small GTPase activities. Altogether, findings point to ongoing disruption of the Rac pathway starting early with increased activity that could be either compensatory or a primary site of pathology followed by eventual degradation of Rac signalling capacity with age and disease progression.
Elevated basal Rac1 activity in HD neurons may have effects on neuronal development and/or survival in the disease. Tourette et al. showed that reducing Rac1 levels with siRNA in immortalized striatal mouse cells from HD mice (HdhQ111/Q111) reduced levels of active Caspase 3/7, demonstrating a neuroprotective effect [18] and suggesting increased Rac1 activation is not compensatory but pathogenic. In normal mouse brain, Rac activity and regulation by one of its GEFs, Kalirin7, are critical for neuronal development, dendritic arborization and spine morphogenesis in neurons [65–70]. Mouse neurons expressing constitutively active Rac in vivo displayed altered dendritic spine morphologies [71] reminiscent to those previously described in HD patient brains. McKinstry and colleagues showed an increased number of mature spines at 21 days in the dorsal striatum at the expense of intermediate spines in heterozygous KI HD mouse model, zQ175; however, by 5 weeks the total spine density was lower in the zQ175 dorsal striatum compared to WT with a reduction in mature spines, suggesting a dynamic process of dendrite morphology changes and a developmental component [72]. These findings are in line with ours in that increased Rac1 activation in striatum occurred early at 1.5 months then was lower at 4.5 months in homozygous Q140/Q140 HD brains. Our data suggest intrinsic excess Rac1 activation is a proximal event in HD and maybe the molecular mechanism underlying dendritic spine changes. We speculate that the reduction in Rac1 activation observed in older Q140/Q140 HD mice is either a compensatory response to the increased activation measured in young animals or an early indication of degeneration and loss of synaptic terminals. In human striatum and cortex from HD patient autopsy tissue, both increases and decreases in dendritic outgrowth and spine density of medium spiny neurons (MSNs) and cortical neurons have been reported using the Golgi-Cox method [73–75]. Although autopsy findings provide only a snapshot of ongoing disease process, even at death HD brains show a spectrum of cells undergoing regenerative and degenerative states.
WT and HD neuronal cultures responded in a profoundly different manner to lowering of total Huntingtin protein. In human control fibroblasts and neuron cultures, cells responded to lowered normal Huntingtin by increasing Rac1 activation. One interpretation of this response is that Huntingtin helps to negatively regulate of Rac1 activity. Another possibility is that Rac1 activation is an indirect effect from some other change produced in the cell by Huntingtin lowering. In contrast, lowering total Huntingtin levels (both normal and mutant Huntingtin) in HD neuronal cultures appeared to correct an aberrant increase in Rac1 activation. This finding shows that the homeostatic set point for WT and HD cells is very different. This is not surprising since transcriptional changes can be found even at the IPSC [51] and NSC stage [76] suggesting either a pathological problem or an adaptation to the presence of mutant Huntingtin early on in development. Our data support the notion that ongoing trials using Huntingtin lowering reagents may be beneficial to HD patients, but the same treatment might be quite toxic to a normal individual.
Conclusions
Our data support a role for Huntingtin in modulating Rac1 activity which occurs in response to growth factor signaling and is important for actin cytoskeletal reorganization at membranes. Changes in Rac1 activity occur early in the HD brain and could regulate plasticity of developing neurons and dendritic spines and contribute to the abnormalities in dendrite growth and spine density of medium spiny neurons described in the HD postmortem brain [74].
CONFLICT OF INTEREST
All purchases are compliant with Research Procurement standards that both individual and organizational conflicts of interest compliance with Uniform Guidance (UG) Procurement Standards issued by the Office of Management and Budget (OMB). No other authors have conflicts of interest to report.
ACKNOWLEDGMENTS
This work was supported by a contract from CHDI, Inc. to KKG and MD and the Dake Family Fund to MD and KKG. We are grateful for help with confocal microscopy from Yun Joon Kim and Luke Maynard.
KKG spouse owns less than 0.1% stock in the following companies: Bristol-Myers Squibb Company, Cisco Systems, Inc., GE Healthcare Life Sciences, Generex Biotechnology Corporation, GlaxoSmithKline, Metabolix Bioplastics, Nanogen, Inc., Nanometrics, inc., StemCells, Inc.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JHD-180311.
REFERENCES
[1] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes, The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6):971–83. |
[2] | Duyao MP , Auerbach AB , Ryan A , Persichetti F , Barnes GT , McNeil SM , et al., Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. (1995) ;269: (5222):407–10. |
[3] | Nasir J , Floresco SB , O’Kusky JR , Diewert VM , Richman JM , Zeisler J , et al., Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. (1995) ;81: (5):811–23. |
[4] | Zeitlin S , Liu JP , Chapman DL , Papaioannou VE , Efstratiadis A . Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. (1995) ;11: (2):155–63. |
[5] | Harjes P , Wanker EE . The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem Sci. (2003) ;28: (8):425–33. |
[6] | Saudou F , Humbert S . The biology of Huntingtin. Neuron. (2016) ;89: (5):910–26. |
[7] | O’Kusky JR , Nasir J , Cicchetti F , Parent A , Hayden MR . Neuronal degeneration in the basal ganglia and loss of pallido-subthalamic synapses in mice with targeted disruption of the Huntington’s disease gene. Brain Res. (1999) ;818: (2):468–79. |
[8] | Dragatsis I , Levine MS , Zeitlin S . Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. (2000) ;26: (3):300–6. |
[9] | Song C , Perides G , Liu YF . Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem. (2002) ;277: (8):6703–7. |
[10] | Kegel KB , Sapp E , Alexander J , Valencia A , Reeves P , Li X , et al., Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem. (2009) ;110: (5):1585–97. |
[11] | Kegel KB , Sapp E , Yoder J , Cuiffo B , Sobin L , Kim YJ , et al., Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem. (2005) ;280: (43):36464–73. |
[12] | Schlessinger J . Cell signaling by receptor tyrosine kinases. Cell. (2000) ;103: (2):211–25. |
[13] | Vanhaesebroeck B , Waterfield MD . Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. (1999) ;253: (1):239–54. |
[14] | Nobes CD , Hawkins P , Stephens L , Hall A . Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci. (1995) ;108: (Pt 1):225–33. |
[15] | Rodriguez-Viciana P , Warne PH , Khwaja A , Marte BM , Pappin D , Das P , et al., Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. (1997) ;89: (3):457–67. |
[16] | Liu YF , Deth RC , Devys D . SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes. J Biol Chem. (1997) ;272: (13):8121–4. |
[17] | Kaltenbach LS , Romero E , Becklin RR , Chettier R , Bell R , Phansalkar A , et al., Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. (2007) ;3: (5):e82. |
[18] | Tourette C , Li B , Bell R , O’Hare S , Kaltenbach LS , Mooney SD , et al., A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J Biol Chem. (2014) ;289: (10):6709–26. |
[19] | Burridge K , Wennerberg K . Rho and Rac take center stage. Cell. (2004) ;116: (2):167–79. |
[20] | Moll J , Sansig G , Fattori E , van der Putten H . The murine rac1 gene: cDNA cloning, tissue distribution and regulated expression of rac1 mRNA by disassembly of actin microfilaments. Oncogene. (1991) ;6: (5):863–6. |
[21] | Shirsat NV , Pignolo RJ , Kreider BL , Rovera G . A member of the ras gene superfamily is expressed specifically in T, B and myeloid hemopoietic cells. Oncogene. (1990) ;5: (5):769–72. |
[22] | Haataja L , Groffen J , Heisterkamp N . Characterization of RAC3, a novel member of the Rho family. J Biol Chem. (1997) ;272: (33):20384–8. |
[23] | Culver BP , Savas JN , Park SK , Choi JH , Zheng S , Zeitlin SO , et al., Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem. (2014) ;287: (26):21599–614. |
[24] | Shirasaki DI , Greiner ER , Al-Ramahi I , Gray M , Boontheung P , Geschwind DH , et al., Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. (2012) ;75: (1):41–57. |
[25] | Angeli S , Shao J , Diamond MI . F-actin binding regions on the androgen receptor and huntingtin increase aggregation and alter aggregate characteristics. PLoS One. (2010) ;5: (2):e9053. |
[26] | Munsie L , Caron N , Atwal RS , Marsden I , Wild EJ , Bamburg JR , et al., Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. (2011) ;20: (10):1937–51. |
[27] | Shao J , Welch WJ , Diprospero NA , Diamond MI . Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol Cell Biol. (2008) ;28: (17):5196–208. |
[28] | Miller JP , Yates BE , Al-Ramahi I , Berman AE , Sanhueza M , Kim E , et al., A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington’s disease. PLoS Genet. (2012) ;8: (11):e1003042. |
[29] | Djinovic-Carugo K , Young P , Gautel M , Saraste M . Structure of the alpha-actinin rod: Molecular basis for cross-linking of actin filaments. Cell. (1999) ;98: (4):537–46. |
[30] | Sjoblom B , Salmazo A , Djinovic-Carugo K . Alpha-actinin structure and regulation. Cell Mol Life Sci. (2008) ;65: (17):2688–701. |
[31] | Hodges JL , Vilchez SM , Asmussen H , Whitmore LA , Horwitz AR . alpha-Actinin-2 mediates spine morphology and assembly of the post-synaptic density in hippocampal neurons. PLoS One. (2014) ;9: (7):e101770. |
[32] | Menalled LB , Sison JD , Wu Y , Olivieri M , Li XJ , Li H , et al., Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J Neurosci. (2002) ;22: (18):8266–76. |
[33] | Valencia A , Reeves PB , Sapp E , Li X , Alexander J , Kegel KB , et al., Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J Neurosci Res. (2010) ;88: (1):179–90. |
[34] | Valencia A , Sapp E , Reeves PB , Alexander J , Masso N , Li X , et al., Reagents that block neuronal death from Huntington’s disease also curb oxidative stress. Neuroreport. (2012) ;23: (1):10–5. |
[35] | Kegel KB , Sapp E , Alexander J , Reeves P , Bleckmann D , Sobin L , et al., Huntingtin cleavage product A forms in neurons and is reduced by gamma-secretase inhibitors. Mol Neurodegener. (2010) ;5: : 58. |
[36] | DiFiglia M , Sena-Esteves M , Chase K , Sapp E , Pfister E , Sass M , et al., Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. (2007) ;104: (43):17204–9. |
[37] | Ritch JJ , Valencia A , Alexander J , Sapp E , Gatune L , Sangrey GR , et al., Multiple phenotypes in Huntington disease mouse neural stem cells. Mol Cell Neurosci. (2012) ;50: (1):70–81. |
[38] | Ebert AD , Shelley BC , Hurley AM , Onorati M , Castiglioni V , Patitucci TN , et al., EZ spheres: A stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. (2013) ;10: (3):417–27. |
[39] | Conti L , Pollard SM , Gorba T , Reitano E , Toselli M , Biella G , et al., Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. (2005) ;3: (9):e283. |
[40] | Arber C , Precious SV , Cambray S , Risner-Janiczek JR , Kelly C , Noakes Z , et al., Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. (2015) ;142: (7):1375–86. |
[41] | Alterman JF , Hall LM , Coles AH , Hassler MR , Didiot MC , Chase K , et al., Hydrophobically modified siRNAs silence huntingtin mrna in primary neurons and mouse brain. Mol Ther Nucleic Acids. (2015) ;4: : e266. |
[42] | DiFiglia M , Sapp E , Chase K , Schwarz C , Meloni A , Young C , et al., Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. (1995) ;14: (5):1075–81. |
[43] | Benard V , Bohl BP , Bokoch GM . Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. (1999) ;274: (19):13198–204. |
[44] | Hall DD , Dai S , Tseng PY , Malik Z , Nguyen M , Matt L , et al., Competition between alpha-actinin and Ca(2)(+)-calmodulin controls surface retention of the L-type Ca(2)(+) channel Ca(V)1.2. Neuron. (2013) ;78: (3):483–97. |
[45] | Scherzinger E , Lurz R , Turmaine M , Mangiarini L , Hollenbach B , Hasenbank R , et al., Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. (1997) ;90: (3):549–58. |
[46] | Christoforidis S , McBride HM , Burgoyne RD , Zerial M . The Rab5 effector EEA1 is a core component of endosome docking. Nature. (1999) ;397: (6720):621–5. |
[47] | Martinez JJ , Hultgren SJ . Requirement of Rho-family GTPases in the invasion of Type 1-piliated uropathogenic Escherichia coli. Cell Microbiol. (2002) ;4: (1):19–28. |
[48] | Miki H , Suetsugu S , Takenawa T . WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. (1998) ;17: (23):6932–41. |
[49] | Steffen A , Rottner K , Ehinger J , Innocenti M , Scita G , Wehland J , et al., Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. (2004) ;23: (4):749–59. |
[50] | Lebensohn AM , Kirschner MW . Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. (2009) ;36: (3):512–24. |
[51] | An MC , Zhang N , Scott G , Montoro D , Wittkop T , Mooney S , et al., Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. (2012) ;11: (2):253–63. |
[52] | Valencia A , Sapp E , Kimm JS , McClory H , Reeves PB , Alexander J , et al., Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington’s disease. Hum Mol Genet. (2013) ;22: (6):1112–31. |
[53] | Kovac B , Teo JL , Makela TP , Vallenius T . Assembly of non-contractile dorsal stress fibers requires alpha-actinin-1 and Rac1 in migrating and spreading cells. J Cell Sci. (2013) ;126: (Pt 1):263–73. |
[54] | Greenwood JA , Theibert AB , Prestwich GD , Murphy-Ullrich JE . Restructuring of focal adhesion plaques by PI 3-kinase. Regulation by PtdIns (3,4,5)-p(3) binding to alpha-actinin. J Cell Biol. (2000) ;150: (3):627–42. |
[55] | Guo F , Debidda M , Yang L , Williams DA , Zheng Y . Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem. (2006) ;281: (27):18652–9. |
[56] | Auger KR , Serunian LA , Soltoff SP , Libby P , Cantley LC . PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. (1989) ;57: (1):167–75. |
[57] | Takenawa T , Suetsugu S . The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. (2007) ;8: (1):37–48. |
[58] | Tolias KF , Cantley LC , Carpenter CL . Rho family GTPases bind to phosphoinositide kinases. J Biol Chem. (1995) ;270: (30):17656–9. |
[59] | Ratovitski T , Chighladze E , Arbez N , Boronina T , Herbrich S , Cole RN , et al., Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. (2012) ;11: (10):2006–21. |
[60] | Kwan W , Trager U , Davalos D , Chou A , Bouchard J , Andre R , et al., Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. (2012) ;122: (12):4737–47. |
[61] | McClory H , Wang X , Sapp E , Gatune LW , Iuliano M , Wu CY , et al., The COOH-terminal domain of huntingtin interacts with RhoGEF kalirin and modulates cell survival. Sci Re. (2018) ;8: (1):8000. |
[62] | Puigdellivol M , Cherubini M , Brito V , Giralt A , Suelves N , Ballesteros J , et al., A role for Kalirin-7 in corticostriatal synaptic dysfunction in Huntington’s disease. Hum Mol Genet. (2015) ;24: (25):7265–85. |
[63] | Brito V , Giralt A , Enriquez-Barreto L , Puigdellivol M , Suelves N , Zamora-Moratalla A , et al., Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J Clin Invest. (2014) ;124: (10):4411–28. |
[64] | Suelves N , Miguez A , Lopez-Benito S , Barriga GG , Giralt A , Alvarez-Periel E , et al., Early downregulation of p75(NTR) by genetic and pharmacological approaches delays the onset of motor deficits and striatal dysfunction in Huntington’s disease mice. Mol Neurobiol. (2018) ; doi: 10.1007/s12035-018-1126-5 . |
[65] | Corbetta S , Gualdoni S , Ciceri G , Monari M , Zuccaro E , Tybulewicz VL , et al., Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. (2009) ;23: (5):1347–57. |
[66] | Gualdoni S , Albertinazzi C , Corbetta S , Valtorta F , de Curtis I . Normal levels of Rac1 are important for dendritic but not axonal development in hippocampal neurons. Biol Cell. (2007) ;99: (8):455–64. |
[67] | Zhang H , Webb DJ , Asmussen H , Niu S , Horwitz AF . A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. (2005) ;25: (13):3379–88. |
[68] | Ma XM , Kiraly DD , Gaier ED , Wang Y , Kim EJ , Levine ES , et al., Kalirin-7 is required for synaptic structure and function. J Neurosci. (2008) ;28: (47):12368–82. |
[69] | Xie Z , Srivastava DP , Photowala H , Kai L , Cahill ME , Woolfrey KM , et al., Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. (2007) ;56: (4):640–56. |
[70] | Chakrabarti K , Lin R , Schiller NI , Wang Y , Koubi D , Fan YX , et al., Critical role for Kalirin in nerve growth factor signaling through TrkA. Mol Cell Biol. (2005) ;25: (12):5106–18. |
[71] | Luo L , Hensch TK , Ackerman L , Barbel S , Jan LY , Jan YN . Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. (1996) ;379: (6568):837–40. |
[72] | McKinstry SU , Karadeniz YB , Worthington AK , Hayrapetyan VY , Ozlu MI , Serafin-Molina K , et al., Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci. (2014) ;34: (28):9455–72. |
[73] | Ferrante RJ , Kowall NW , Richardson EP , Jr . Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. (1991) ;11: (12):3877–87. |
[74] | Graveland GA , Williams RS , DiFiglia M . Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. (1985) ;227: (4688):770–3. |
[75] | Sotrel A , Paskevich PA , Kiely DK , Bird ED , Williams RS , Myers RH . Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology. (1991) ;41: (7):1117–23. |
[76] | HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. (2012) ;11: (2):264–78. |

