
Isolation and cultivation of primary keratinocytes from piglet skin for compartmentalized co-culture with dorsal root ganglion neurons
Abstract
Keratinocytes are the main cell population in the epidermis, where they coexist with a variety of other cell types. Their successful isolation and cultivation have afforded opportunities to study epidermal functions. Human keratinocytes have been studied most extensively, but their source is limited by the skin supply. In a previous work, we developed an in vitro co-culture model of porcine keratinocytes with porcine sensory neurites to investigate functional interaction. However, a detailed description of the isolation of porcine keratinocytes and their culture conditions has not been given in detail. Here, we present the isolation procedure and a characterization of keratinocytes derived from new-born piglets, using simple assays based on conventional and fluorescence microscopy. Media, coating substrates and plating densities were tested with respect to cell viability, proliferation and morphology. Growing keratinocytes in EpiLife keratinocyte growth medium (EKGM) on human collagen type I substrate was best to support proliferation. The minimum plated density was 500 viable cells/cm2 for primary and 1000 viable cells/cm2 for subcultured cells. Population doubling (PD) and generation time (tg) depended on the plating densities. Keratinocytes seeded at a density of 5000 viable cells/cm2 had a PD of 4.36±0.60 per passage and tg of 1.69±0.24 days. Our results show that the optimal isolation and culture conditions for keratinocytes from piglets differ from those for keratinocytes from adult pigs and humans. Thus, the information obtained from the characterization allowed the performance and optimization of a co-culture and contributes to further investigations in epidermal homeostasis and cutaneous sensation.
1Introduction
The skin is organized in three structural layers: epidermis, dermis and hypodermis. The epidermis, the outermost layer, is a stratified epithelium mainly composed of keratinocytes. It continually renews itself, maintaining cellular proliferation and differentiation in a constant balance, termed epidermal homeostasis. As a physical barrier it is of importance, e.g. for protection against pathogens and for the detection of environmental conditions, being linked via sensory endings to the nervous system to mediate tactile, temperature and noxious stimuli.
Keratinocyte isolation and culture procedures with human skin have been well documented [1–6]. However, ethical considerations and limited supply make human skin often inconvenient for in-vitro studies. Several investigations have revealed great similarity between skin from humans and pigs, e.g. in cutaneous nociception and pain signalling [7–11]. Therefore, we established a co-culture model of porcine keratinocytes and porcine DRG neurons [12]. Here, we present a detailed description for the keratinocyte isolation and culture of cutaneous keratinocytes from piglets.
Isolation and cultivation of keratinocytes from mouse, rat and human skin have revealed different requirements for an optimal cell culture and isolation procedure [2, 13–19]. In these studies most culture media are supplemented with EGF to promote cell proliferation and to avoid cell differentiation, hydrocortisone to induce colony formation, bovine pituitary extract or bovine serum albumin to provide several growth factors, and insulin to fulfil glucose metabolism requirements [2, 6, 20–22]. Serum supplementation is avoided in most of the cases in order to control final calcium concentrations, as well as the content of growth factors, growth inhibitors and other not absolutely defined serum component proteins potentially able to induce undesired differentiation processes [22, 23]. In addition to an optimized culture medium, keratinocyte proliferation can be further improved by the use of different coating substrates such as collagen, a constitutive protein in the basement membrane, which has been successfully used to enhance attachment and proliferation of the cells [4, 24]. Following the established isolation and culture protocols for skin from adult pig, we did not achieve satisfactory results [13, 18, 25–28]. Therefore, we combined and tested the different protocols in order to obtain high quality keratinocyte cultures from the skin of piglets.
Keratinocytes in the epidermis coexist not only with melanocytes, Merkel and Langerhans cells, but also with peripheral terminals of somatosensory neurons. Keratinocyte-neuronal interactions are involved in the modulation of functions such as sensation and inflammation [29–33]. Several studies have been performed using co-culture models with keratinocytes, neuronal somata and nerve endings in one single compartment or separated by membrane inserts, in consequence necessitating the use of only one culture medium for both types of cells [19, 28, 34–38]. This restriction prevents the use of different supplementation and growth factors for keratinocytes and neurons and hampers the performance of studies on segregated neurites as well. Moreover, a one-dish co-culture exposes both cell types to possible secreted substances. We established a co-culture of porcine keratinocytes and porcine sensory neurites [12, 39] by using a compartimentalized culture chamber [40, 41] which separates sensory somata on the one side and keratinocytes and outgrown neurites on the other side. To this end, optimal isolation procedure, plating density, and culture conditions had to be established, but were not detailed yet. With the aim of delivering the tools to adapt the co-culture settings to different necessities, we provided a comprehensive isolation protocol and culture procedure for keratinocytes from piglet. Characterization was based on morphology, adhesion properties, plating densities, generation time (tg), population doubling (PD), the differentiation process, and immunological criteria (cellular markers).
2Materials and methods
2.1Animals
Piglets (Sus scrofa domesticus) were sacrificed by sedation i. m. with Stresnil® (Janssen-Cilag GmbH) and Ketamine (Essex Pharma GmbH) followed by an intracardial lethal dose of Narcoren®. For isolation of keratinocytes, the age of the animals was 3 to 7 days, for isolation of neurons, 6 to 12 days. All experimental procedures were conducted in conformity with the German law for use and care of animals, in accordance with the European directive 86/609/EEC on the protection of animals used for experimental and other scientific purpose.
2.2Transport and culture medium
The transport medium (TM) for skin tissue consisted of EpiLife® medium (Gibco) supplemented with EpiLife Defined Growth Supplement (EDGS) (Gibco), 25 μg/mL gentamicin (antibiotic) and 2.5 μg/mL amphotericin B (fungicide).
For keratinocyte culture, three different media were used: (i) MCDB153 basal medium (Biochrom), (ii) complete EKGM composed of EpiLife® medium supplemented with EDGS (iii) KGM2 composed of Keratinocyte Growth medium 2 (Promocell). Final concentrations of supplements, growth factors and calcium chloride in the listed media are detailed in Table 1.
Table 1
Comparison of the different growth media used for porcine keratinocyte cultures
| Medium | Components and supplements | Concentrations |
| EKGM supplemented with EDGS | rec. human epidermal growth factor | 1 ng/mL |
| 60 μM calcium | ||
| rec. human insulin-like growth factor type-1 | 10 ng/mL | |
| hydrocortisone | 11 ng/mL | |
| purified bovine transferring | 5 μg/mL | |
| bovine serum albumin (BSA) | 30 μg/mL | |
| prostaglandin E2 | 18 ng/mL | |
| Keratinocyte Growth medium 2 (KGM2) | bovine pituitary extract | 0.4% (v/v) |
| 60 μM calcium | ||
| rec. human epidermal growth factor | 0.125 ng/mL | |
| human insulin | 5 μg/mL | |
| hydrocortisone | 0.33 μg/mL | |
| human transferring | 10 μg/mL | |
| epinephrine | 0.39 μg/mL | |
| MCDB153 basal medium 60 μM calcium | rec. human epidermal growth factor | 25 ng/mL |
| Hydrocortisone | 0.1 μg/mL | |
| rec. human insulin | 10 μg/mL | |
| bovine serum albumin | 2 mg/mL | |
| histidine | 0.23 mM | |
| isoleucine | 0.05 mM | |
| tryptophan | 0.015 mM | |
| threonine | 1.25 mM | |
| tyrosine | 0.031 mM | |
| alanine | 4.08 mM |
2.3Cultivation and cryopreservation of porcine keratinocytes
Keratinocytes were seeded in coated vessels and cultured at 37°C in 5% CO2 in a humidified atmosphere. The culture medium was replaced three days after isolation with the respective growth medium without antibiotics. Afterwards, medium change was performed every second day and keratinocyte growth was documented and imaged.
Subculture was performed at 60 to 70% confluence. Keratinocytes were trypsinized with 30 μL/cm2 pre-warmed 0.04% Trypsin/0.03% EDTA (Promocell) for 10 min at 37°C. Trypsinization was followed by smooth pipetting to separate cell clumps obtaining a cell suspension. Finally, cells were centrifuged at 150×g for 5 min, resuspended in corresponding growth medium and seeded at a density as indicated for each experiment.
For cryo-preservation, 3×106 viable freshly isolated keratinocytes or 1×106 viable passaged keratinocytes were pelleted and then resuspended in cold freezing medium composed of EKGM (70%), fetal calf serum (FCS, 20%) and DMSO (10%), frozen at –80°C at a cooling rate of –1°C/min overnight and stored in liquid nitrogen (–196°C).
For thawing, the medium was equilibrated to 37°C and 5% CO2 before the procedure in order to reach optimal physiological conditions (pH and temperature). Frozen keratinocytes were thawed by smoothly swirling the vial in a 37°C water bath until only small ice crystals remained in the vials. Afterwards, cells were quickly transferred to a tube and pre-warmed medium was added dropwise. Cells were centrifuged at 150×g for 5 min, resuspended in the corresponding pre-warmed growth medium and seeded at a density as indicated for each experiment. For cryopreservated cells, the first medium change was performed 3 days after cell seeding and thereafter every second day.
2.4Adhesion substrate for porcine keratinocyte cultures
Several substrates were tested for optimal adhesion and proliferation of porcine keratinocytes. Tissue culture flasks were coated with collagen I from rat (Roche), Coating matrix kit (Gibco), laminin (Boehringer Mannheim), p-L-ornithine (Sigma), collagen I bovine (Biochrom) or p-L-lysine (Sigma). Stock and working concentrations are detailed in Table 2. Coating was performed according to the respective manufacturer’s instructions for thin coatings. All coated surfaces were air dried and coated tissue culture vessels were used immediately or stored for not longer than one week at 4°C.
Table 2
Substrates tested for adhesion and proliferation of keratinocytes
| Substrate | Stock solution | Working concentration | Keratinocyte adhesion |
| and proliferation | |||
| Collagen I from rat-tail tendon (Roche) | 1 mg/ml | 5 μg/cm2 | + |
| Coating Matrix Kit (Gibco) | – | 0.7 μL collagen and 68 | +++ |
| μL dilution medium/cm2 | |||
| Bovine collagen I (Biochrom) | 5 mg/ml | 50 μg/mL; 5 μg/cm2 | + |
| Laminin (Boehringer Mannheim) | 25 mg/ml | 50 μg/mL; 5 μg/cm2 | + |
| p-L-ornithine (Sigma) | 0.1 mg/ml | 2 μg/cm2 | – |
| p-L-lysine (Sigma) | 0.1 mg/ml | 4 μg/cm2 | – |
| No substrate (plastic) | – | – | ++ |
+support adhesion but low proliferation capacities. ++support adhesion and proliferation only when using high plating densities. +++support adhesion and high proliferation capacities. -no adhesion.
2.5Immunocytochemistry and immunohistochemistry
For immunocytochemistry (ICC), 5×103 viable cells/cm2 were seeded on collagen-coated glass coverslips in 12-well plates. At 60 to 70% confluence cells were washed with PBS, fixed with 4% PFA for 15 min at RT and quenched for 5 min with 50 mM Tris-HCl (pH 8.0) and 100 mM NaCl to saturate remaining aldehyde groups, avoiding aldehyde-induced fluorescence. Afterwards, cells were permeabilized with 0.3% Triton X-100 in PBS for 15 min at RT and blocked with 5% goat serum (PAA) for 1 h at RT to avoid non-specific binding. Cells were then incubated with the respective first antibody diluted in 1% goat serum overnight at 4°C. Keratinocytes were characterized using mouse anti-cytokeratin 14 (1 : 400; Millipore-Chemicon) and mouse anti-cytokeratin 1/10 (1 : 400; Millipore-Chemicon) antibodies. After washing the cells 5 times with PBS, cells were incubated with the secondary antibody AlexaFluor® 488 (A488) goat anti-mouse IgG (H+L) highly cross-adsorbed (1 : 400; Invitrogen) for 1 h at RT in darkness. Nuclei were visualized by staining cells with 1 μg/ μL DAPI in PBS for 10 min. Finally, cells were washed 4 times with PBS and once with purified water (to remove the rest of the PBS), and coverslips were mounted with Roti®-Mount FluorCare (Roth) on glass slides.
For immunohistochemistry (IHC), fresh skin pieces were embedded in OCT (cryo-embedding media) in cryomolds and frozen in liquid nitrogen. Once the tissue matrix had solidified completely, the cryomold block was removed, and the samples were stored at –80°C until use. Prior to sectioning, the temperature of both the frozen tissue matrix and the cryostat were equilibrated to –20°C, then tissue was sectioned with a microtome (LEICA CM 3050S cryostat) to 20 μm thickness and placed onto superfrost slides. Samples were stored at –20°C until immunostaining. For immunostaining, tissue sections were fixed with 4% PFA for 15 min at RT and posterior procedure followed as described for ICC.
Cells and tissue sections were observed using an inverted epifluorescence microscope (Axiovert 200, Zeiss) equipped with a 75W Xenon lamp, the corresponding filters (excitation BP 500/20, FT 515, emission BP 535/30 for A488 fluorochrome and excitation G 365, FT 395, emission BP 445/50 for DAPI), a set of objectives (EC Plan-Neofluar 20x/0.5 Ph2, LD Plan-Neofluar 20x/0.40 Korr, LD Plan-Neofluar 40x/0.60 Korr and Plan-Neofluar 63x/1,25 Oil Ph3, Zeiss) and imaged using an AxioCam HR camera and Axiovision software (Zeiss). For differential interference contrast (DIC) images, the PlasDIC polarization-optical DIC for plastic materials from Zeiss was used. PlasDIC allows the use of DIC to observe samples maintained in plastic dishes. Controls were performed omitting the primary antibody.
As further negative controls, (1) primary keratinocyte cultures were incubated with an antibody against vimentin, a marker for fibroblasts, and (2) neurites were incubated with a cytokeratin antibody, a specific marker for keratinocytes.
2.6Growth rate, viability and population doubling (PD)
The growth rate, viability and population doubling was investigated with keratinocytes seeded at several densities in 8.8 cm2 collagen-coated dishes. Over a period of 10 days, every second day bright field images were captured before dye loading for documentation. The procedure consisted of incubating cells with 15 μg/mL Hoechst 33342 (Invitrogen) and 15 μg/mL propidium iodide (PI) (Roth) for 18 min at 37°C and 5% CO2. Afterwards, the medium was replaced with fresh medium and cells were imaged using an inverted epifluorescence microscope (described before). Thirteen images were captured per dish, using two different filters: filter A for PI staining (excitation BP 560/40, FT 585, emission BP 630/75) to visualize only dead cells and filter B for Hoechst staining to detect total cells (same filter used for DAPI staining) using a 10x objective (EC Plan-Neofluar 10x/0.3 Ph1; Zeiss). Cells were counted per image using the analyze particles plug-in from ImageJ (NIH). Briefly, two different images were opened, corresponding to the two different filters (A and B). For each image both brightness and contrast from the image settings were adjusted automatically with the option “Auto”. Using the threshold plug-in, cells were distinguished from the background. Then, using the analyze particles plug-in, the size of the smallest and largest particles was set up and numerical data were collected in an Excel table using the option “show summary”. Finally, values were plotted as viability and viable cells/cm2 against time. PD, also called generation number, was calculated as log2 (number of viable cells/number of plated cells). Generation time (tg) was calculated as t/PD, representing the time (hours) needed to double the cell population.
2.7Co-culture of keratinocytes and dorsal root ganglion neurites in a compartmented chamber
A compartmented Campenot chamber was used for co-culture [40, 41]. Briefly, a round teflon chamber of 21 mm inside diameter, divided into three compartments, was used (see Fig. 5a). The central compartment of 3 mm width was separated from the lateral compartments by 1 mm thick walls. The chambers were attached to the bottom of polystyrene petri dishes 35 mm in diameter. Before mounting the chamber, the dishes were coated with collagen (Coating Matrix Kit, Gibco). Then the bottom of the dishes was prepared with ca. 45 parallel scratches (ca. 300 μm interspace) to run perpendicularly to the dividing walls of the chamber. Adhesion and sealing to the dish was achieved by application of high viscosity silicone grease (Bayer). Underneath the two dividing walls, a thin layer of 1% methylcellulose in DMEM was applied before mounting the chamber. For a precise application of the silicone grease onto the chamber walls a glass syringe-based applicator was used, in which a fine-feed screw actuated the plunger.
Dorsal root ganglia were excised from the spinal cord and neurons were isolated as described previously [8]. Neurons were seeded into the central compartment at a density of approx. 5000 cells/cm2. Keratinocytes were seeded in the outer part of the lateral compartments at a density of either 5000 viable cells/cm2 for freshly isolated keratinocytes or 2000 viable cells/cm2 for passaged cells (P2-3). (Fig. 6). After allowing keratinocytes to attach to the bottom of the plate, 300 μL EKGM were added to the side compartment the day after keratinocyte seeding. Neurons were cultured in supplemented F12 medium containing NGF (50 ng/mL) [12], keratinocytes together with the outgrown neurites in supplemented EKGM medium containing NGF (50 ng/mL). Depending on the experiments, keratinocytes were seeded before or after neuron seeding. Half of the medium was replaced in each compartment every second day. Experiments were performed 5 to 7 days after seeding somata.
3Results
3.1Optimization of isolation and cultivation of porcine keratinocytes from piglets
3.1.1Isolation and morphology
For the isolation of epidermal keratinocytes from piglets, several previously described protocols for human keratinocytes [1–6], murine [2, 14–19] and adult porcine keratinocytes [13, 18, 25–28] were performed, however, with low cell yields, high percentages of senescent cells, slow growth rates, and hence the necessity of high plating densities. Adult porcine keratinocytes were successfully isolated using Dispase for tissue dissociation (3 h at 37°C) followed by further incubation with trypsin/EDTA for 30 min for cell disaggregation [18], whereas other protocols describe the successful isolation of porcine keratinocytes using a 0.25% trypsin solution for 2 h at 37°C for both tissue dissociation and cell disaggregation [13]. Therefore, taking the information from those published protocols, we combined different isolation procedures, in order to obtain the appropriate conditions for the isolation of porcine keratinocytes.
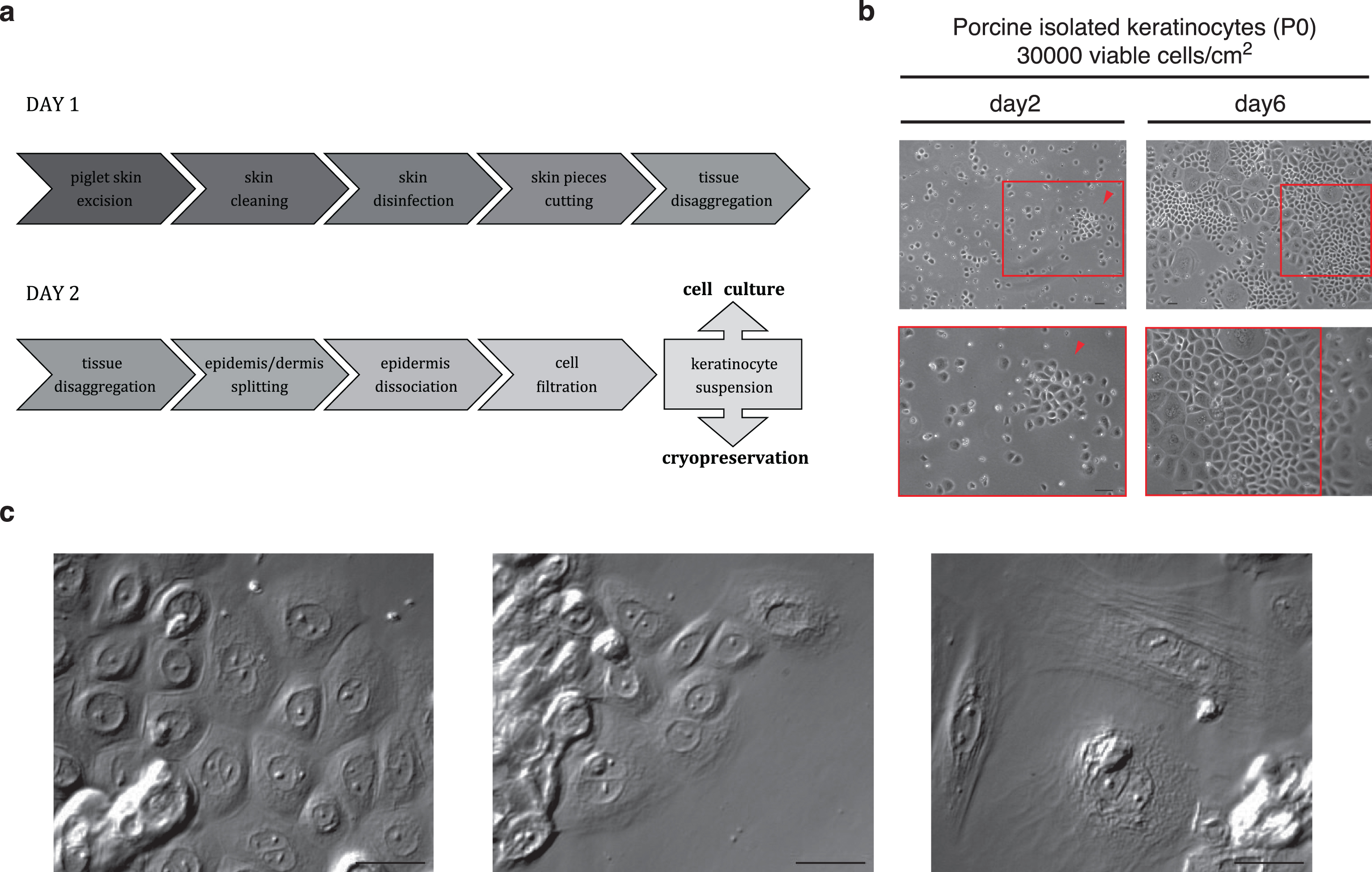
The optimized isolation procedure was performed as detailed at the end of this chapter and is schematized in Fig. 1a. Briefly, skin tissue was excised, cleaned, disinfected and then cut into small pieces (approx. 0.5 cm×0.5 cm). Epidermis was dissociated from dermis by incubation with Dispase II (17 h at 4°C) allowing subsequent mechanical separation. A single cell suspension of primary keratinocytes was finally obtained by incubation with Trypsin-EDTA (9 min at 37°C) and epidermal debris was removed by filtration through a cell strainer (70 μm). This provided a cell yield of 1.73×106±16% viable cells per cm2 of tissue with a viability of 88.01±2.04% (n = 6 animals). Cells were seeded in collagen-coated vessels (Coating Matrix Kit, Gibco) using 30 000 viable cells and 0.2 mL cell suspension per cm2 surface in EKGM. As keratinocytes in vivo are dependent of rich cell-cell and cell-ECM interactions, a minimal plating density is required for proper cell development in in vitro cultivation. Thus, to establish the isolation procedure, a high plating density was used for subsequent culturing. Lower seeding densities were also tested but showed slow growth or a low confluence, or both.
The first medium change was performed after 3 days to allow most of the cells to attach before removing the unattached cells and remaining debris. In the Fig. 1b, keratinocytes are shown at day 2 and day 6 after seeding. At day 1, only single cells or a few clumps were observed; proliferation mostly occurred from the second day on, showing the typical island association. At day 6, big islands with proliferating keratinocytes were observed (Fig. 1b, arrowhead). Cells reached 60–70% confluence after 1 week and were passaged until passage 10 using 1000 to 2000 viable cells/cm2. No morphological or immunological changes were seen either by optical microscopy or ICC. Nevertheless, the cell dissociation (trypsinization) became easier with the passages. When cells were kept in culture for more than 10 passages, they exhibited a different morphology, becoming longer and prickly (data not shown). Examples of cell morphology are shown with higher magnification in Fig. 1c. Proliferating keratinocytes show a polygonal shape containing little cytoplasm and a prominent nucleus, growing together in islands (Fig. 1c, left), clumped keratinocytes occurred as result of an incomplete enzymatic dissociation or inappropriate cell seeding. Senescent cells (estimated 20 to 30%) were observed in all cultures as oversized flattened multinucleate cells (Fig. 1c, right). They also showed vacuoles and an enlarged cytoplasm volume.
Fig.1
Primary porcine keratinocyte culture. As described in chapter results, in (a) the two days’ keratinocyte isolation procedure from newborn piglet skin is illustrated by a schematic representation. (b) Phase contrast images of primary porcine keratinocytes cultured in EKGM medium. Island formation was observed on the second day in culture (left, red arrowhead) and 60–70% confluency was reached at day 6 (right, red box). Scales indicate 50 μm. (c) DIC images show cultured porcine keratinocytes. The left panel shows cells displaying typical features of proliferating keratinocytes (e. g. island formation with high cell contact, small content of cytoplasm, prominent nucleus and cobblestone-like morphology), the central panel shows clumped keratinocytes as the result of an incomplete enzymatic dissociation or inappropriate cell seeding, and the right panel shows cells displaying typical features of senescent keratinocytes (e. g. oversized flattened form, enlarged cytoplasm volume and multinucleate phenotype). Scales indicate 30 μm. All keratinocytes were seeded onto collagen-coated surfaces.
3.1.2Medium-dependent proliferation and viability of keratinocytes
Different culture media have been used to improve cell culture yield and the viability of keratinocytes from humans, mice and adult pigs [2, 6, 20–22], but no medium has been described yet for keratinocytes isolated from neonatal pigs.
For the cultivation of keratinocytes from adult pigs Regauer and Compton used DMEM supplemented with 20% fetal bovine serum and 20 ng/ml hydrocortisone. They were able to culture freshly isolated cells on plastic surfaces without coating or feeder cells, but passaged cells required a 3T3 fibroblast feeder layer [18]. Bevan et al. used OptiMEM®1 medium (Gibco) supplemented with 0.5 mM calcium chloride, 2 mM L-glutamine, 1.25 μg/μl amphotericin and 1% fetal bovine serum. This method also necessitates the use 3T3 feeder cells [13].
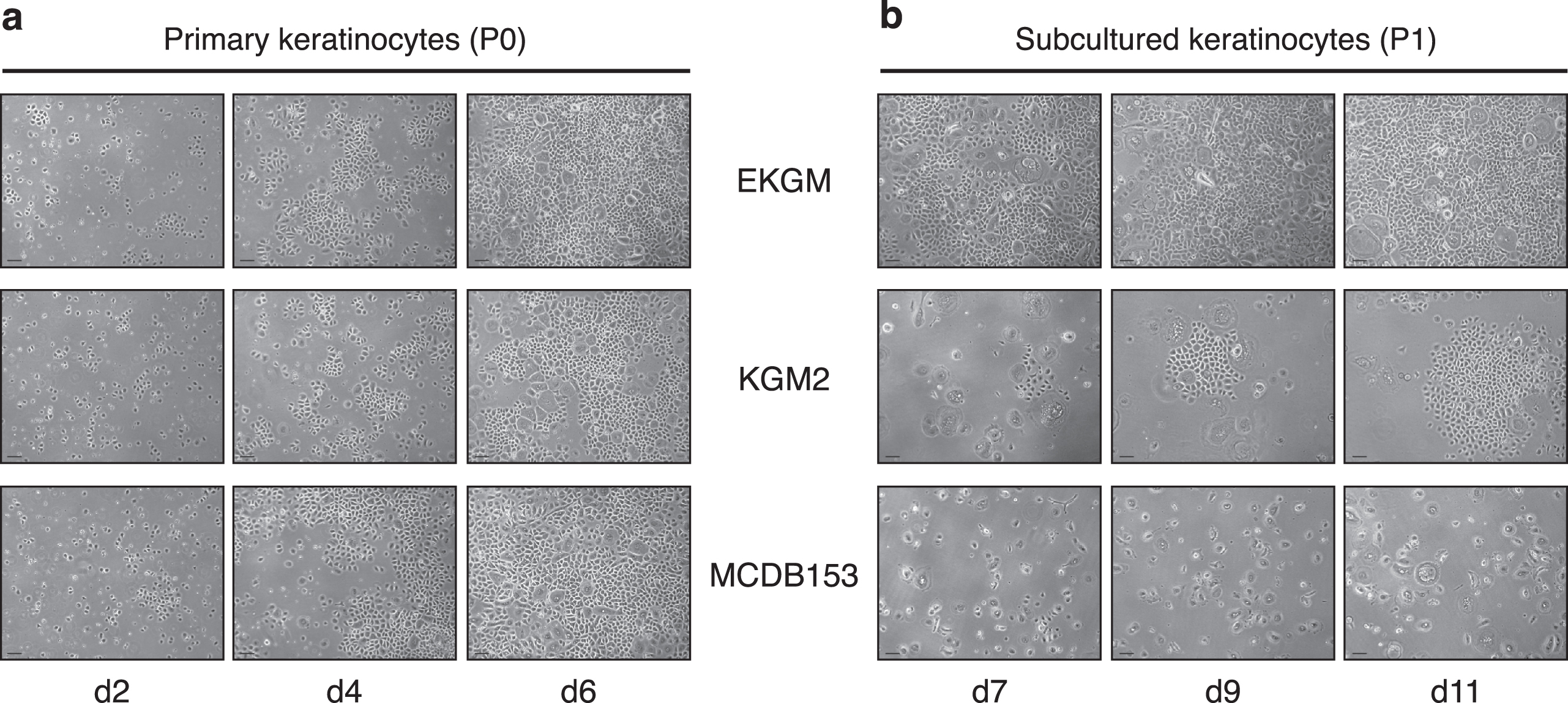
In order to determine the appropriate growth medium for keratinocytes from 3 to 7 days old piglets, cells were seeded in different culture media, that were developed for culturing human keratinocytes independently of a feeder layer in serum-free conditions: (i) MCDB153, one of the first well-established keratinocyte culture media [1], and two ready-to-use human keratinocyte media (ii) EKGM and (iii) KGM2 (Table 1). Differences in proliferation and viability were monitored and compared for primary keratinocytes (P0) and for secondary keratinocytes (P1). P0 were seeded at a density of 30 000 viable cells/cm2 (Fig. 2a), and 5000 viable cells/cm2 for P1 (Fig. 2b).
For P0, high proliferation was obtained, with EKGM reaching complete confluence at day 6. Cells cultured in MCDB153 medium also reached complete confluence at day 6. Nevertheless, more cells transformed to a senescent phenotype and died. The cultures of keratinocytes in KGM2 never reached total confluence; this was independently of the period the cells were cultured (Fig. 2a).
Subcultures (>P0) were performed when cells reached approx. 70% confluence and were kept in the corresponding medium. For P1, cell proliferation was observed only with EKGM and KGM2 media. Cells cultured in EKGM reached a nearly confluent culture at day 7, which became completely confluent at day 11. With KGM2 only clonal growth was observed, without confluence being reached at any time in culture. Fast cell adhesion was observed for keratinocytes cultured in both media. Keratinocytes cultured in KGM2 proliferated more slowly than those cultured in EKGM, as shown in Fig. 2b. The first islands were observed at day 7, whereas, using EKGM, the culture was almost confluent.
Fig.2
Comparison of different culture media on porcine keratinocyte growth and proliferation. Phase contrast images show in (a) primary keratinocytes at days 2, 4, and 6. All media used supported primary cell growth, resulting in high confluencies at day 6. In (b), phase contrast images show secondary keratinocytes (passage 1) at days 7, 9, and 11. EKGM effectively promoted secondary cell growth, resulting in high confluency already at day 7, KGM2 promoted growth in islands without leading to a complete confluence, whereas MCDB153 did not induce significant growth at all. Keratinocytes were seeded onto collagen-coated surfaces. Scales indicate 100 μm.
In sum, for primary and secondary keratinocyte cultures, isolated from piglets, the medium EKGM yielded the highest proliferation rate and the lowest percentage of senescent cells.
3.1.3Substrate dependent adhesion and proliferation of keratinocytes
Cultivating keratinocytes together with other cell types, e.g. sensory neurons, requires a substrate supporting adhesion of both types. For keratinocyte cultures 3T3 feeder layer and collagen are the preferred substrate [6, 27]; for sensory neurites, it is p-L-lysine or p-L-ornithin [8, 42]. We therefore tested different substrates to evaluate adhesion and the growth of keratinocytes (Table 2). The best cell adhesion and proliferation rates were achieved using human collagen I Coating Matrix Kit (Gibco). Laminin also allowed cell adhesion, as did rat collagen (Roche) and bovine collagen I (Biochrom). However, all three substrates resulted in a lower proliferation rate compared to human collagen I (data not shown). Keratinocytes did not attach when using p-L-lysine and p-L-ornithin. Without coating, on plastic or glass, cells attached poorly and only when the density was very high (>60000 viable cells/cm2). Porcine DRG neurons also attached well to human collagen I Coating Matrix Kit (Gibco). Due to the generated results, this matrix was used to perform the co-culture experiments.
3.1.4Plating density dependent clonal growth and proliferation of keratinocytes
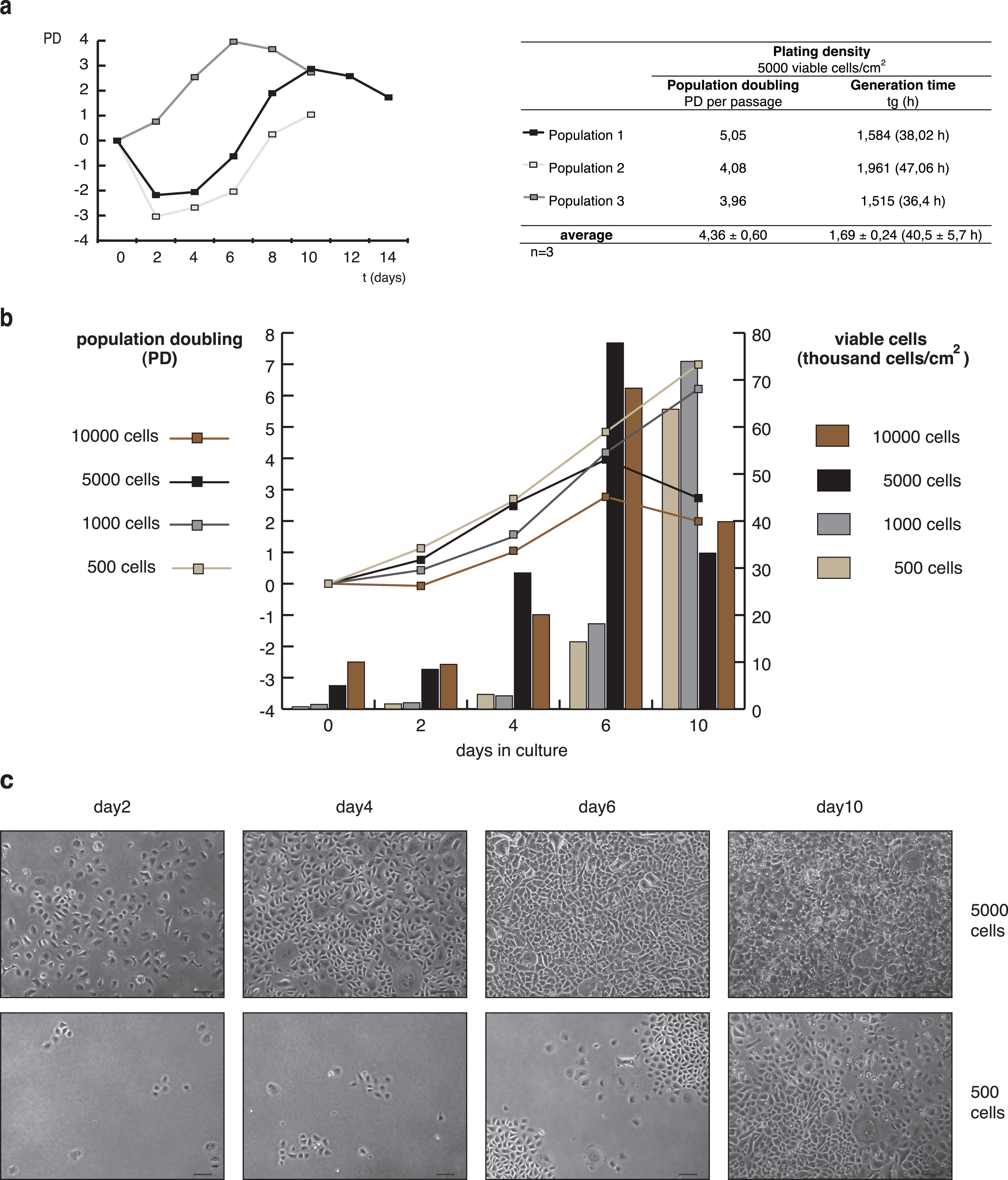
The proliferation of keratinocytes was tested with cells seeded on human collagen I coated 8.8 cm2 dishes in EKGM medium. In a first row of experiments, cells were seeded with a plating density of 5000 viable cells/cm2. In Fig. 3a, three growth curves from independent experiments of subcultured keratinocytes (P2) are shown as population doubling (PD) against time. PD and generation time (tg) values are detailed in the table aside. Similar growth curves were observed for populations 1 and 2, showing a partial loss of cell population the first two days after seeding, followed by a logarithmic increase in growth during one week. Population 3 showed a logarithmic increase in growth immediately after 1 day in culture, which extended for approx. 1 week, as also observed for populations 1 and 2. After keratinocyte proliferation had reached a maximum, the PD decreased, meaning that cell proliferation ceased. The value obtained for tg was 1.69±0.24 (40.5±5.7 h), indicating that every 40.5±5.7 h the population is doubled. These data, together with a PD of 4.36±0.60 determined that keratinocytes doubled their population 4.36±0.60 times per passage during the course of one week.
In a second row of experiments, different seeding densities were compared using subcultured keratinocytes (P2) (Fig. 3b,c). As shown in Fig. 2b, a culture of subcultured keratinocytes (P1) plated at a density of 5000 viable cells showed a high confluence at 7 d in culture, reaching complete confluence at day 11 (using EKGM). Cell-cell contact inhibits the proliferation of keratinocytes and starts the differentiation process [43]. Therefore, plating densities of 500, 1000, 5000 and 10000 viable cells/cm2 were used for a growth rate analysis. Two days after seeding, cell adhesion was achieved for all cultures. The quantity of viable cells and the PD rates are shown in Fig. 3b, while examples of island forming in Fig. 3c. Plating densities of 500 and 1000 viable cells/cm2 promoted sustained cell growth, until day 10 (Fig. 3b) and longer (data not shown). Cells seeded at higher densities only grew until day 6, followed by a decrease both in the PD and the quantity of viable cells, consistent with the inhibition by contact. Besides, no more surface area was available for further proliferation. At this point, cultures plated with 5000 viable cells/cm2 showed complete confluence, while the cultures plated with 500 viable cells/cm2 displayed well-defined islands even after day 10 (Fig. 3c). In cultures seeded at the highest densities, keratinocytes presented the typical morphology of differentiated and senescent cells (day 10). Prolonged culture of cells seeded at the lowest plating densities did not induce complete confluence (data not shown). Those cells died before they became confluent. All plated densities allowed clonal growth, visualized by trypan blue staining, commonly used to quantify colony formation (data not shown). Larger colonies were observed just for higher plating densities as a result of colony convergence. In summary, subcultured keratinocytes show proliferation dependent on plating density. The optimal seeding density fluctuates between 500 and 5000 viable cells/cm2.
Fig.3
Growth curve and propagation of porcine keratinocytes. (a) Left: Growth curves of 3 independent populations of secondary (P2) keratinocytes cultured in EKGM on collagen substrate, seeded at 5000 cells/cm2. The cells showed an increment in the population, reaching a plateau after 6 to 10 days, followed by a diminished proliferation capacity. Right: Tabular display of population doubling (PD) and generation time (tg) for each population. (b) Growth curves (graphs) and number of viable cells per cm2 (bars) of subcultured porcine keratinocytes (P2) seeded at different densities (500, 1000, 5000 and 10000 viable cells/cm2) and cultured in EKGM on collagen substrate. The results are presented as a mean (SD). (c) Phase contrast images of secondary keratinocyte cultures seeded at 5000 viable cells/cm2 (upper panels) and 500 viable cells/cm2 (lower panels) taken at different times (days 2, 4, 6, and 10) after seeding. Lower densities allowed clonal growth, whereas higher densities allowed more confluent cultures. Scales indicate 100 μm.
3.1.5Immunological characterization of keratinocytes
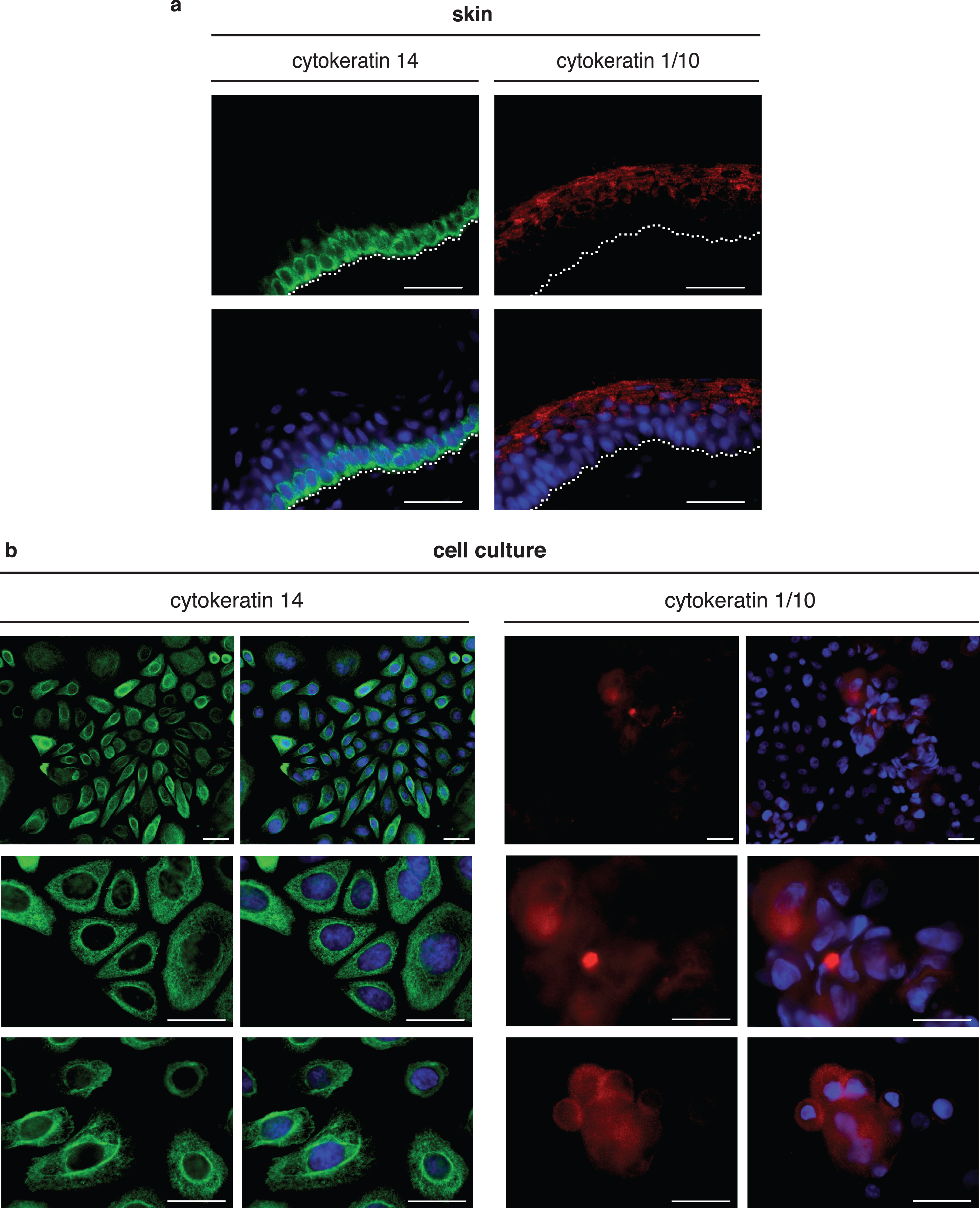
In keratinocyte cultures, we observed cells with different morphologies as shown in Fig. 1c. To determine the phenotype of these cells, we performed indirect immunostaining using antibodies against keratins. Keratinocytes of the basal layer can be immunostained with cytokeratin 14 antibody and keratinocytes from the suprabasal layer with cytokeratin 1/10 antibody [44]. Immunohistochemistry was performed with 20 μm porcine skin sections to validate the specificity of the antibodies (Fig. 4a) to be used in cultured keratinocytes (Fig. 4b). Cytokeratin 14 expression was observed in a single basal layer of the epidermis (Fig. 4a, left); the expression of cytokeratin 1/10 was observed in the upper outer layer of the epidermis (Fig. 4a, right). To stain cultured keratinocytes, cells from different passages were seeded on collagen-coated glas coverslips. After one week (preconfluent cells) and two weeks (confluent cells) in culture in EKGM medium, cells were fixed and immunocytochemistry was performed, using cytokeratin 14 and cytokeratin 1/10 antibodies. Preconfluent keratinocytes showed cytokeratin 14 staining (Fig. 4b, left panel), but not cytokeratin 1/10 staining (data not shown). In cultures with confluent keratinocytes, only a few cells were positive for cytokeratin 1/10. These cells were growing above other keratinocytes. The superposition of cells was detected by the nuclei being located at different levels as detected by DAPI staining (Fig. 4b, right panel, in blue). Senescent cells displayed positive staining for cytokeratin 14 but not for cytokeratin 1/10. Negative controls, either by omitting the first antibody or by using anti-vimentin antibody, did not show any staining of keratinocytes (data not shown).
Fig.4
Immunohistochemistry and immunocytochemistry of porcine skin and keratinocyte cell culture show differential expression of keratinocyte markers. (a) Immunohistochemical staining of skin cross-sectional slices (thickness 20 μm). The dashed white line indicates the basal membrane. Cytokeratin 14 immunoreactivity (green) was detected in the inner layers of the epidermis, indicating basal keratinocytes and cytokeratin 1/10 immunoreactivity (red) in the outer layers, indicating suprabasal keratinocytes. (b) Immunocytochemical staining of primary keratinocytes after 10 days in culture. Most cells exhibited cytokeratin 14 immunoreactivity (green). Only in overconfluent keratinocyte cultures (2 weeks in culture) did a few cells growing above other cells (multilayer-like structure) exhibit cytokeratin 1/10 immunoreactivity (red). Nuclei are visualized by DAPI staining (blue). Scales indicate 30 μm.
3.1.6Effect of calcium and serum on cell differentiation of keratinocytes
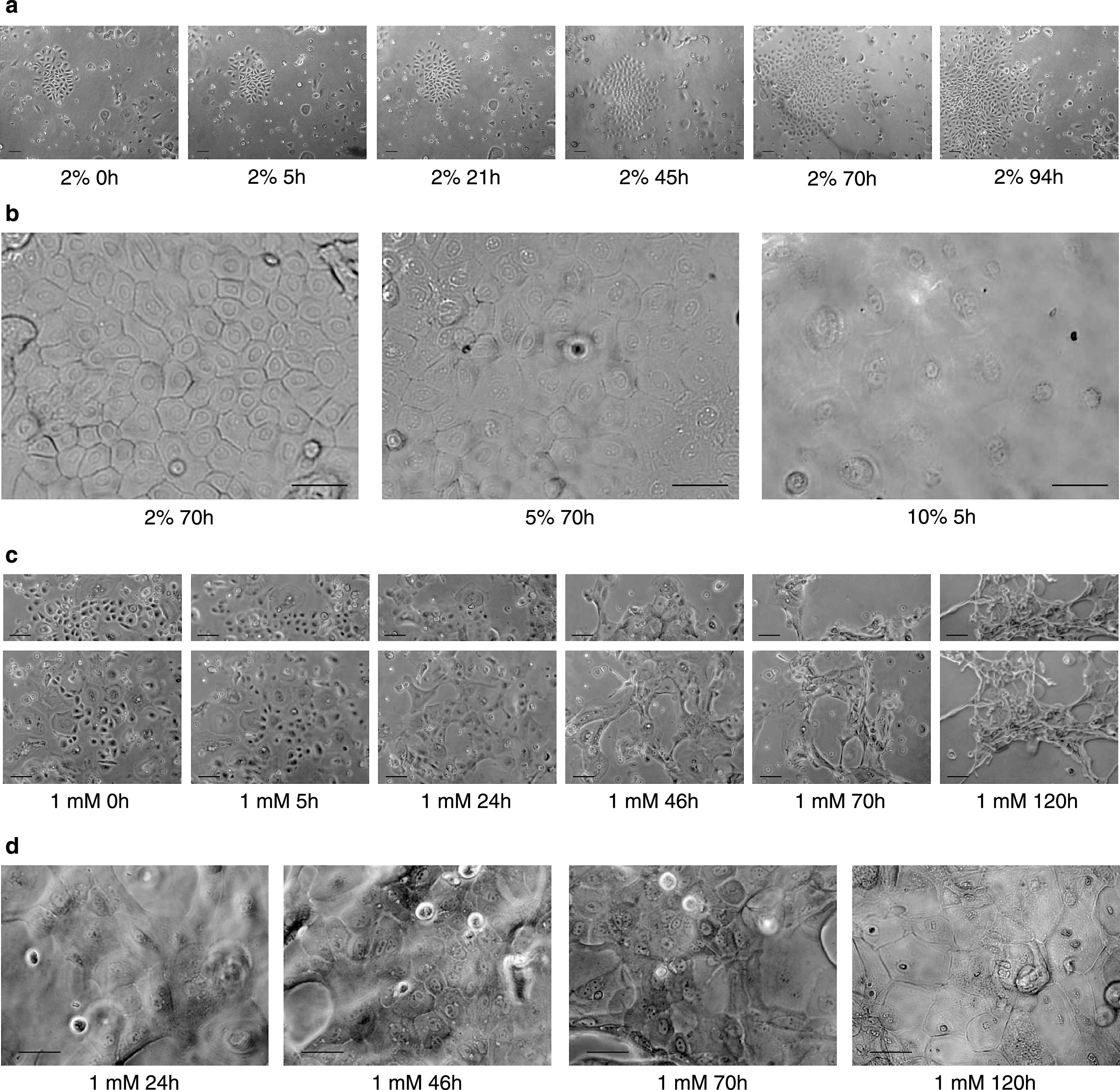
In keratinocytes, the differentiation process is essential for normal skin homeostasis [45]. In vitro, keratinocyte differentiation has been successfully induced by several factors, such as osmotic stress, growth factors, high calcium concentrations, or serum supplementation [16, 22, 23, 27, 46]. To investigate whether the keratinocytes isolated from piglets retain the ability to differentiate in vitro after the isolation procedure, normal keratinocyte growth medium (EKGM) was replaced with a differentiation medium. We tested 1%, 2%, 5% and 10% porcine serum supplemented EKGM (serum differentiation medium, SeDM) and calcium supplemented EKGM to a final concentration of 0.1 mM, 1 mM and 2 mM in EKGM (calcium differentiation medium, CaDM). Cells were cultivated in complete medium (EKGM) until 50–60% confluence. Afterwards, EKGM medium was replaced with the respective differentiation medium. Differentiation was evaluated for morphological and molecular changes by light microscopy, ICC and WB. The morphology of keratinocytes changed after switching to 2%,5%, and 10% SeDM (Fig. 5a-b) or 1 mM and 2 mM CaDM media (Fig. 5c-d). The islands lost their compact association (Fig. 5c). The cells enlarged and became flattened (Fig. 5b,d). Either longer incubation with 1 mM calcium CaDM or a higher percentage of serum (10% SeDM) induced in keratinocytes the formation of a cornified cell envelope, which is a marker for differentiated keratinocytes from the stratum corneum. Sometimes no effect by 2% SeDM on the growth of the keratinoctyes was observed, as shown in Fig. 5a. Keratinocytes did proliferate and the island became large, following the normal kinetic growth pattern previously observed in the growth curve studies. Calcium concentrations lower than 1 mM (0.1 mM) also induced morphological changes, as observed for 1% SeDM as well. However, a longer incubation time was necessary to observe comparative effects (data not shown). For cultures using 2 mM CaDM, morphological changes were observed already one hour after medium switch, followed by a generalized detachment of the cells (data not shown). The expression of keratin 14 was observed by ICC and WB assay in keratinocytes from all cultures, as shown previously with untreated medium (data not shown). In ICC, no staining was obtained with the cytokeratin 1/10 antibody for any culture condition.
Fig.5
Calcium-Serum-regulated differentiation of porcine keratinocytes. Cell cultures were exposed either to a growth medium containing serum (a-b) or increased calcium concentrations (c-d) to investigate cell differentiation. 5000 viable cells/cm2 were plated on collagen substrate and grown in EKGM until 50–60% confluence was attained. Then, EKGM medium was switched to SeDM (A-B) or CaDM (c-d) and either bright field or phase contrast images were captured in intervals for a period of 94 to 120 h. In (a) images show keratinocytes growing in a 2% SeDM. In (b) three images are shown with a higher magnification corresponding to three different serum supplementations. In (c) and (d) the images show the morphological changes appearing during culture in 1 mM CaDM. Scale indicates for a,c 100 μm and b,d 50 μm.
3.2Co-culture of porcine keratinocytes and porcine DRG neurons
3.2.1Tissue dissection
The venter of the sacrificed animal was shaved before removal of the tissues. The skin was disinfected with Braunol® solution before and after excision to avoid microbial contamination.
Skin tissue was excised from the lateral venter. Excised skin samples of approx. 10×5 cm were then washed with phosphate-buffered saline (PBS) containing 25 μg/mL gentamicin and 2.5 μg/mL amphotericin B, and were kept in TM medium on ice until isolation began after about 4 h. All the following steps were performed at RT under aseptic conditions in a laminar flow cabinet if no other instruction is stated. Tissue was transferred after each incubation using sterile forceps. First, tissue was washed with PBS, then placed in Braunol® for 20 min, followed by 5 min incubation with 0.05% sodium thiosulfate in PBS to reduce the excess of iodine contained in the Braunol®. Next, as much excess fatty and subconnective tissue as possible was removed. The cleaned tissue was cut into small pieces (∼ 0.5 cm2); these procedures facilitate the subsequent enzymatic reactions and further epidermal-dermal separation.
3.2.2Dissociation of keratinocytes
For tissue disaggregation, i.e. the separation of epidermis, containing a high percentage of keratinocytes, and dermis, mainly composed of connective tissue cells like fibroblasts, pieces of epidermis were incubated upside down, in 1.6 U/mL Dispase® II (Roche) for 17 h at 4°C. The epidermis could then easily be separated from the dermis, using a sterile forceps and was immediately submerged in PBS. When the epidermis could not be easily peeled off, those pieces were discarded to avoid damaging the cells. If most of the epidermis pieces did not separate easily from the dermis, a longer incubation with Dispase was performed until a gentle separation was possible. The epidermis was then incubated in pre-warmed 0.04% Trypsin/0.03% EDTA solution (Promocell) for 9 min at 37°C, followed by a smooth pipetting to dissociate the keratinocytes. Trypsin activity was inhibited with 1 : 1 Trypsin Neutralizing Solution (TNS, Promocell), because the inhibition with serum could induce cell differentiation. Keratinocytes were separated from epidermal debris by filtration through a cell strainer (70 μm), centrifuged for 5 min at 150×g and resuspended in the corresponding growth medium containing 25 μg/mL gentamicin and 2.5 μg/mL amphotericin B. Finally, a keratinocyte suspension was obtained, either for direct use in cell culture or kept in cryopreservation until needed.
3.2.3Co-culture of neurites of porcine DRG neurons and porcine keratinocytes
To study the interaction between keratinocytes and sensory neurites, a compartmented culture chamber was used [12]. Isolated DRG somata were seeded in the central compartment and cultured in NGF-supplemented (50 ng/mL) F12 medium. Keratinocytes were seeded in the outer perimeter of the lateral compartment and cultured in complete EKGM medium, additionally supplemented with NGF (50 ng/mL). Freshly isolated keratinocytes were seeded at a density of 5000 viable cells/cm2 and passaged cells (P2-3) at a density of 2000 viable cells/cm2.
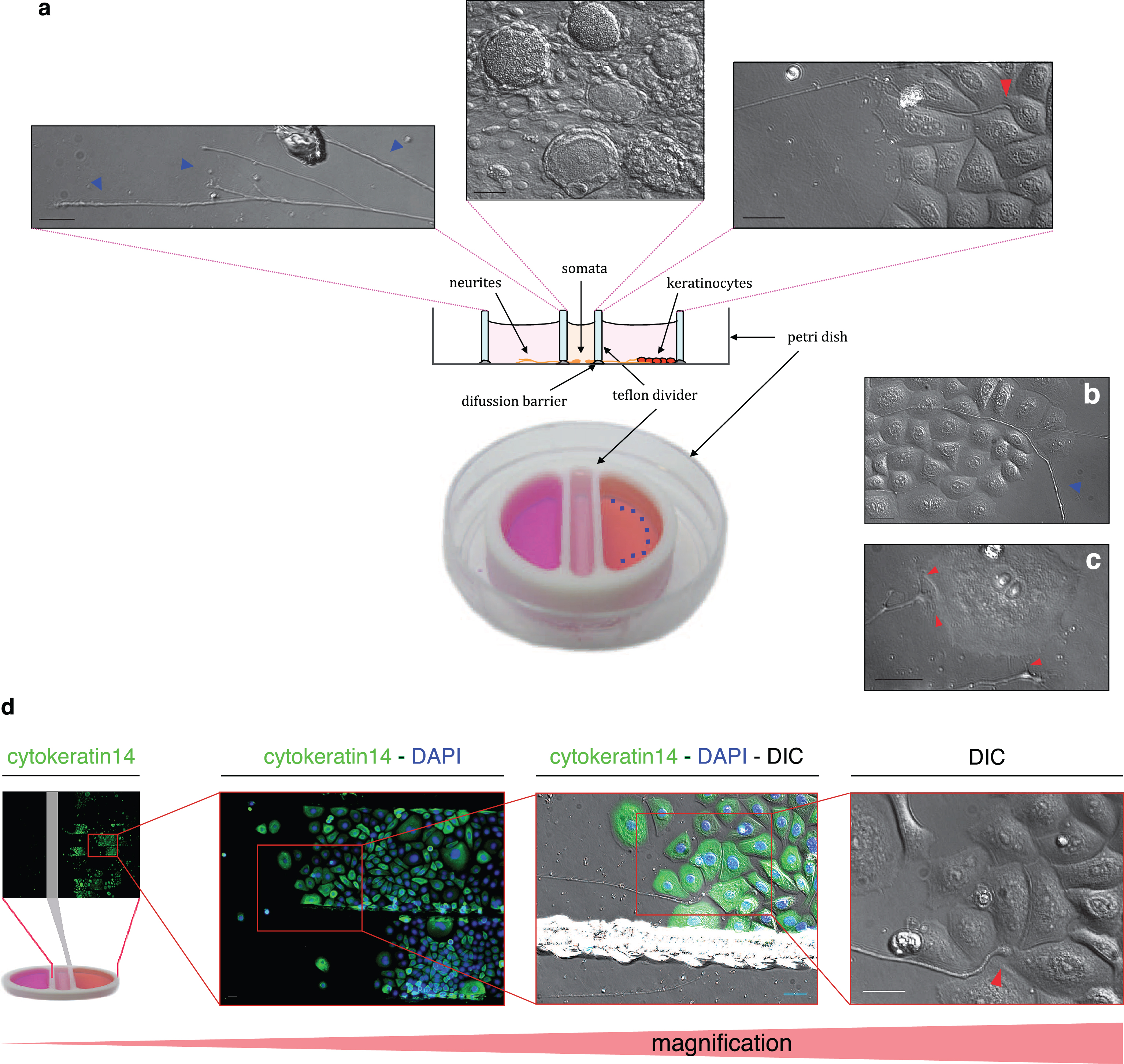
After about 3 to 4 days outgrowing neurites reach the side compartment, keratinocytes reached 60–70% confluence in about 6 days. Therefore, in most experiments, keratinocytes were seeded two days before somata plating. However, using different plating densities of keratinocytes, the time point for seeding can be shifted (see growth curve studies). DIC images show neurites (left, blue arrowhead), neuronal somata (middle), and neurites in contact with keratinocyte islands (right) in the lateral compartment of the compartmented co-culture (Fig. 6a). Neurites were observed in close vicinity with proliferating keratinocytes (Fig. 6b, red arrow), but also with senescent cells (Fig. 6c). Immunostaining for keratinocytes with cytokeratin 14 antibody (Fig. 6d, green) was performed in the co-culture of keratinocytes and neurites. Only keratinocytes were stained with CK14 (6d, green), but not neurites. Neurites can be observed by the corresponding DIC image. Using higher magnifications, it was possible to see neurites reaching confluent keratinocytes islands (Fig. 6d, right images).
Fig.6
Keratinocyte-neuron compartmentalized coculture. (a) Schematic diagram illustrating the compartmentalized coculture chamber. Keratinocytes were seeded in one side compartment in EKGM, whereas somata of DRG neurons were seeded in the central compartment in F12 medium. Three DIC images corresponding to the left, central and right compartments are shown above the schematic diagram. Neuronal somata are observed in fsthe central image, surrounded by satellite cells. Neurites cross the diffusion barrier to the side compartment, crossing the diffusion barrier to both side compartments. In the left side compartment without keratinocytes, only neurites were observed (blue arrowhead). In the right side compartment, keratinocytes and neurites grow in close vicinity. The red arrowhead shows the neurite ending. (b) A neurite growing above and between the keratinocytes (DIC image). (c) A neurite branch reaching a senescent keratinocyte. Red arrowheads show potential contact points between neurite endings and the membrane of the keratinocytes. (d) Immunostaining was performed for cytokeratin 14 in the coculture chamber. Nuclei were visualized in blue by DAPI staining. Enclosed sections show enlarged images (from left to right). Red arrowhead shows the neurite ending. Scale indicates 30 μm.
4Discussion
We established a procedure allowing isolation and cultivation of cutaneous keratinocytes from 3–7 days old piglets, for which previously published methods [1–6, 13–19, 25–28] appeared insufficient. We combined those protocols in order to obtain healthy proliferating cultures. Furthermore, we established a compartmentalized co-culture of primary porcine keratinocytes and neurites of porcine DRG neurons, allowing studies of epidermal interactions between keratinocytes and sensory neurites [12].
Porcine skin has proven to closely resemble human skin in many structural, cytological, physiological and immunohistological features. To isolate porcine keratinocytes from piglets several methods as described for humans [1–6], mice [2, 13–19], and adult pigs [13, 18, 25–28] were performed without satisfactory results. These methods resulted in low cell yields with rather high percentages of senescent cells. Cultivation of such isolated cells required high plating densities and still provided only minor growth rates.
In our study, senescent cells were observed in all cultures with a proportion of approximately 20–30%, even from freshly isolated keratinocytes. They are morphologically distinguishable by their flattened, oversized shapes, with an enlarged cytoplasm volume, and their tendency towards multinuclearity and vacuole formation [47]. In contrast, proliferating cells show a polygonal shape with little cytoplasm and a prominent nucleus and tend to form islands [16]. In our hands, the percentage of senescent cells was increased, when cells were subjected to stress conditions during the isolation procedure or cell culture. For the effective retrieval of healthy proliferating primary cultures from tissue sections, we found five factors to be critical: tissue disaggregation, cell dissociation, adhesion substrate, growth medium and seeding density.
Moreover, Trypsin incubation, cryopreservation, and thawing play an important role in avoiding premature senescence and differentiation in the cultures. The critical points in obtaining healthy cultures of proliferating keratinocytes from cryopreservated cells were the use of pre-warmed consumables like collagen-coated vessels, and culture medium equilibrated to at least room temperature, performing a fast procedure and providing an appropriate volume of medium. Thus, cells were quickly thawed in a water bath, diluted with culture medium, and centrifuged to remove DMSO from the freezing medium. Adhesion and proliferation is improved by seeding the cells in a volume of culture medium, that abbets quick cell sedimentation [4]. A small sample for counting the cells should therefore be taken before centrifugation, to define cell densities directly after centrifugation.
4.1Tissue disaggregation and cell dissociation
Skin thickness is an important issue for the enzymatic cell dissociation, influencing the required trypsin concentration and exposure times [4]. Thickness varies between age of the animal and body area (e. g. dorsal plane compared to ventral side) [48]. For our study, skin was taken only from the lateral venter of 3 to 7 d old animals. We also tested thinner skin from the belly using the same enzyme concentrations and incubation times, which, however, resulted in damaged cells, a lower cell yield, and poor cell attachment (data not shown). Thus, concentrations and incubation times of enzymes used were optimized for newborn porcine lateral ventral skin.
A first critical step for the isolation of primary porcine keratinocytes is the separation of epidermis from dermis. In established protocols, this step is performed after incubation with proteases like Trypsin or Dispase. Those enzymes exhibit profoundly different separating mechanisms. Dispase cleaves hemidesmosomes, allowing mechanical separation of dermis and epidermis as intact layers. Trypsin, in contrast, disrupts desmosomes, thus releasing single cells to the liquid supernatant for further harvesting [49], but is less effective for tissue disaggregation. Consequently, Dispase is well applicable for a prior isolation of epidermis from dermis, but not for single cell dissociation. For the isolation of keratinocytes from adult pigs, both enzymes have been previously used. Regauer and Compton used 0.25% Dispase for 3 h at 37°C for tissue disaggregation. The dermis was then mechanically removed using forceps, and cell dissociation was achieved by incubation in 0.1% Trypsin and 0.02% EDTA at 37°C for 30–45 min [18]. In another study the use of Dispase was avoided. They incubated skin pieces in 0.25% Trypsin at 37°C for 2 h and then mechanically separated dermis and epidermis using a scalpel. Subsequent cell dissociation was performed by fine mincing of epidermal pieces and shaking in Trypsin solution. Debris was removed by filtering through a fine mesh sieve [13].
For our approach, we found it most effective to initially isolate the epidermis by incubating small skin pieces (approx. 0.5 cm×0.5 cm) in 1.6 U/mL Dispase ® II (Roche) for 17 h at 4°C, which allowed a gently mechanical separation from the dermis. Performing this tissue disaggregation as described in the literature [13, 18] with either Trypsin/EDTA, higher Dispase concentrations or at higher temperatures (37°C) for only a few hours, resulted in an impaired cell culture.
For cell dissociation, the best results were obtained by incubating separated epidermis with pre-warmed 0.04% Trypsin/0.03% EDTA for 9 min at 37°C and subsequent smooth pipetting. Alternative protocols, like using Trypsin/EDTA (1 : 250, PAA) or incubation at room temperature, resulted in impaired cell yields (data not shown).
Sometimes, clumped cells were observed in cultures after seeding. In that case, either adherent keratinocytes were detached from the flasks, but not separated, or cell dissociation from the epidermis was not complete. This resulted in lower proliferation rates, which illustrates the importance of thorough cell dissociation.
The combination of the above-described steps resulted in a cell yield of 1.73×106 (±16%) viable cells per cm2 of tissue and a high cell viability of 88.01% (±2.04%). Both cell dissociation and cell yield were impaired when using larger pieces of tissue, incubating at room temperature instead of 37°C or using a 1 : 250 dilution of Trypsin-EDTA solution (PAA).
4.2Adhesion substrate
Several substrates were reported to support the adhesion of keratinocytes in cell cultures. The best cell adhesion and proliferation rate were achieved with collagen I [27]. In normal epidermis, keratinocytes express heteromeric transmembrane receptors, termed integrins. They consist of two different variable subunits, denoted α and β. Determined by their heterodimer composition and the cellular context, integrins can bind to different specific extracellular matrix ligands, like collagen, laminin, vitronectin and fibronectin [24, 50, 51]. Thus, in vivo expression and ECM binding of integrins allow the anchoring of the epidermis to the basement membrane. In vitro, this is mimicked by a coating matrix.
For our co-culture experiments, a coating matrix is compulsory for the adhesion of keratinocytes as well as neurites. Therefore, we tested for keratinocytes the commercially available collagen coatings of human, rat and bovine origin and the coatings laminin, poly-L-orinithine and poly-L-lysine mostly used for supporting adhesion of DRG neurons [8, 42].
We found human collagen I to effectively enable cell adhesion and the growth of keratinocytes and neurites of DRG neurons. Collagen of other species (bovine, rodent) and laminin also supported cell attachment, but only minor proliferation rates. In contrast, p-L-orinithine and p-L-lysine did not support keratinocyte adhesion at all. Without coating of the surface, keratinocytes attached poorly and only when the plating density was very high (>60,000 viable cells per cm2).
4.3Growth medium
We tested three different established media, known to support the growth of human keratinocytes. MCDB153 is one of the first published keratinocyte media [2]. EKGM and KGM2 are ready-to-use media developed by Gibco and Promocell, respectively, more recently.
Both, freshly isolated and passaged keratinocytes were tested as media requirements for primary and secondary cells might differ. Primary cells contain a heterogeneous mixture of cells in various differentiation states due to their origins from different epidermal layers. As only cells from the basal layer are expected to show considerable proliferative activity, secondary cultures are bound to be significantly enriched in basal keratinocytes. For primary cells, we found EKGM and MCDB153 to support growth until reaching confluency. The highest growth rate was observed with EKGM; with MCDB153 it was slightly lower, but still satisfying. In contrast, using KGM2 induced lower growth rates and failed to lead to confluent cultures. This might be explained by the observation that the proportion of senescent cells increased over culture time in all KGM2 cultures, while it stayed constant at approximately 20–30% in EKGM and MCDB153 cultures.
The medium showing the best cell cultures was EKGM, even when cell growth was observed with KGM2. EKGM allowed prolonged incubation times without increasing the population of senescent cells, which was always observed with KGM2 medium, independently of cell plating density. With MCDB153, primary cells exhibited even faster growth rates than cells cultured in KGM2, while subcultured cells did attach but not proliferate.
MCDB153 was the first chemically defined medium allowing keratinocyte growth independently of a feeder layer [1, 2]. For the experiment shown in Fig. 2, collagen-coated vessels were always used to compare cell growth using the same conditions, even for MCDB153. In some of those cultures, we observed very small islands of 2 to 3 cells for subcultured cells, but they did not propagate.
All three media are different in formulations and supplementations (see Table 1), influencing the proliferation of porcine keratinocytes. Suboptimal supplementation of nutrients, amino acids and vitamins can limit the growth of keratinocytes [2]. Minimal media composition with supplementation of epidermal growth factor (EGF) and hydrocortisone in an optimal ratio increases the culture lifetime, delaying cell senescence. Hydrocortisone increased lateral expansion of the islands and the rate of cell proliferation in human keratinocytes [6], whereas EGF not only increased the growth rate and colony-forming efficiency of keratinocytes, but also decreased the fraction of differentiated cells [21]. Bovine pituitary extract and insulin are other nutrients found in most keratinocyte media and can increase keratinocyte proliferation. BPE can stimulate keratinocyte proliferation in medium deficient in EGF (0.1 ng/ml) [22]. However, in combination with other nutrients they can also prevent growth. Thus, BPE can act synergistically to favour growth if EGF is low, but in the presence of high EGF (10 ng/ml), the growth decreases [22]. On the other hand, each nutrient alone is not sufficient to support clonal growth. The results obtained in this study support the observations made by the other groups. Thus, both supplements and concentrations contained in the media determined a fine-tuning in the proliferation rate.
4.4Seeding density
After keratinocyte isolation, differentiated keratinocytes of suprabasal layers showed poor adhesion properties and so, many of these cells were washed out after the first medium change, which decreased cell density and further enriched basal cells. Thus, the seeding density of primary keratinocytes (P0) was higher than of subcultured cells (P > 0). Besides, the loss of the ability to initiate DNA synthesis after the harsh isolation procedure [6] can also reduce the available proliferating cells in a primary culture, increasing the required plating density.
Our isolation procedure allows seeding of passaged keratinocytes (P2) over a very wide cell density range (500 to 10,000 cells/cm2), but still providing healthy proliferating cultures. In general, high seeding densities of 5,000 cm–2 and more provide confluent cultures and high cell numbers relatively soon (after approx. 6 days), while lower seeding densities of 1,000 cm–2 allow clonal growth over longer culture times (10 days and more).
In our coculture model, we determined that a seeding density of 5,000 cm–2 was applicable in the case of primary (P0) cells, while 2,000 cm–2 proved to be preferable in the case of passaged cells. Those seeding densities enabled us to seed the keratinocytes two days before the neurons, giving the neurites enough time to innervate the cell layer without being hampered by a too high keratinocyte density.
Higher plating densities resulted in lower numbers of PD events, due to a higher confluence of cells (Table 1, suppl. inf.). There were not only proliferating cells seeded, but also senescent cells and cells that were not viable any more, due to the aggressive trypsinization step (10 min and pipetting) or because they had exhausted their proliferative capacity. Besides, clumped keratinocytes were also present in some subcultures as a result of incomplete enzymatic dissociation or inappropriate cell seeding. Thus, some of those cells or groups of cells were either washed out or prevented the proper cell attachment lowering cell proliferation.
4.5Porcine keratinocytes retain their ability to differentiate
The epidermis is comprised of multiple layers of keratinocytes, expressing different cytokeratin proteins [52]. To determine the phenotype of porcine keratinocytes, we performed indirect immunostaining using antibodies against keratins.
In skin sections, cytokeratin 14 expression was observed in a single basal layer of the epidermis (Fig. 4a, green), representing undifferentiated proliferating basal cells. Expression of cytokeratin 1/10 was observed in the upper outer layer of the epidermis, representing the suprabasal layers with differentiated cells (Fig. 4a, red).
Under culture conditions, most of the keratinocytes expressed the basal marker cytokeratin 14 for proliferating cells. Only in overconfluent cultures did a few keratinocytes express cytokeratin 1 and 10, a marker for differentiated cells. In the skin, some differentiated keratinocytes move up to the surface. Those cells are replaced by new proliferative keratinocytes in the stratum basale.
In the porcine keratinocyte monocultures, differentiated cells are probably washed out after a change of medium, because there was no matrix or gel for the retention of the cells supporting a stratified cell culture (3D culture). We suggest that our porcine keratinocytes have retained the inherent ability to differentiate (Fig. 4b, red), as demonstrated by Sun and Green for human keratinocytes [21], but those differentiated cells could not stratify in the monolayer cultures. To confirm our hypothesis, we investigated the ability of porcine keratinocytes to induce a differentiation process. We defined morphological criteria to characterize differentiated keratinocytes, as described in the literature [21]. By adjusting the plane of focus of the microscope, we were able to classify enlarged cells with a flattened morphology and a cornified envelope as differentiated cells. To induce differentiation, the medium was supplemented either with serum (SeDM) [23] or calcium (CaDM) [16]. Keratinocytes growing as a monolayer in low calcium (60 μM) can be induced to terminally differentiate by adding calcium to the culture medium (>1 mM). In our work, with increased calcium concentrations, morphological changes were observed after 5 hours of incubation; cells enlarged, became flattened and did not stratify. Serum-induced keratinocyte differentiation (>5% serum, Fig. 5b) was also observed. It was also demonstrated that a low concentration of serum (0.5%) was sufficient to prevent clonal growth in the presence of high EGF concentrations (10 ng/ml) [22]. In porcine keratinocytes, low concentrations of serum (<2%) in the presence of 1 ng/ml EGF resulted sometimes in increased proliferation and large islands (Fig. 5a). Those results confirmed the hypothesis that serum prevents clonal growth only in the presence of high EGF concentrations. Besides, serum contains undefined growth and inhibitory factors, which could also potentially modulate keratinocyte proliferation. Although, we confirmed the differentiation of keratinocytes by morphological criteria, we did not succeed to observe the differentiation markers keratin 1 and 10 in differentiated cells by ICC, whereas they continued expressing keratin 14 (data not shown). This phenomenon has been discussed by Watt, who suggested that calcium allows the formation of desmosomes and other cell contacts necessary for stratification, but does not induce the synthesis of new proteins [53]. Therefore, other signaling pathways must be involved to turn the proliferating expression pattern into a differentiating one.
4.6Compartmented co-culture of keratinocytes and DRG neurons
The skin is innervated by sensory neurites that transduce the stimuli and conduct the signal to their cell bodies, located in the trigeminal and dorsal root ganglia (DRG) [54, 55]. Their endings are thin structures with a diameter of a few μm, which hampers in-vivo studies. Therefore, we established an in-vitro model to investigate interaction between sensory endings and keratinocytes.
In earlier studies, co-cultures were often maintained in a single reservoir [28, 34, 38] or with adapted inserts [19]. In these systems overgrowth was often observed between keratinocytes and neurons and the simultaneous use of different media for the individual cell types is hampered as they provide only one coherent medium room. However, knowing that keratinocytes and neurons require different media compositions and that serum and calcium are important differentiation factors for keratinocytes [2, 16, 22, 23, 27, 56], the establishment of a compartmented chamber provides a tool for co-culture studies.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the following grants: Ministry of Science, Research and Art – Baden Württemberg (“Innovative Projekte”; to M. H.); “ZAFH-ABIMAS”, Landesstiftung Baden-Württemberg and the European Fund for Regional Development (EFRE; to M. H. and Carsten Hopf, ABIMAS-Center, Mannheim).
References
[1] | Boyce ST , Ham RG . Cultivation, frozen storage, and clonal growth of normal human epidermal keratinocytes in serum-free media. Journal of Tissue Culture Methods (1985) ;9: :83–93. DOI: 10.1007/bf01797779 |
[2] | Boyce ST , Ham RG . Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. The Journal of Investigative Dermatology (1983) ;81: :33s–40s. |
[3] | Karasek MA . In vitro culture of human skin epithelial cells. The Journal of Investigative Dermatology (1966) ;47: :533–40. |
[4] | Karasek MA , Charlton ME . Growth of postembryonic skin epithelial cells on collagen gels. The Journal of Investigative Dermatology (1971) ;56: :205–10. |
[5] | Radtke C , Sinis N , Sauter M , Jahn S , et al. TRPV channel expression in human skin and possible role in thermally induced cell death. Journal of Burn Care & Research Official Publication of the American Burn Association (2011) ;32: :150–9. DOI: 10.1097/BCR.0b013e318203350c |
[6] | Rheinwald JG , Green H . Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell (1975) ;6: :331–43. |
[7] | Lynn B , Faulstroh K , Pierau FK . The classification and properties of nociceptive afferent units from the skin of the anaesthetized pig. The European Journal of Neuroscience (1995) ;7: :431–7. |
[8] | Obreja O , Klusch A , Ponelies N , Schmelz M , et al. A subpopulation of capsaicin-sensitive porcine dorsal root ganglion neurons is lacking hyperpolarization-activated cyclic nucleotide-gated channels. European Journal of Pain (London, England) (2008) ;12: :775–89. DOI: 10.1016/j.ejpain.2007.11.010 |
[9] | Obreja O , Ringkamp M , Namer B , Forsch E , et al. Patterns of activity-dependent conduction velocity changes differentiate classes of unmyelinated mechano-insensitive afferents including cold nociceptors, in pig and in human. Pain (2010) ;148: :59–69. DOI: 10.1016/j.pain.2009.10.006 |
[10] | Rukwied R , Dusch M , Schley M , Forsch E , et al. Nociceptor sensitization to mechanical and thermal stimuli in pig skin in vivo. European Journal of Pain (London, England) (2008) ;12: :242–50. DOI: 10.1016/j.ejpain.2007.05.007 |
[11] | Rukwied R , Schley M , Forsch E , Obreja O , et al. Nerve growth factor-evoked nociceptor sensitization in pig skin in vivo. Journal of Neuroscience Research (2010) ;88: :2066–72. DOI: 10.1002/jnr.22351 |
[12] | Klusch A , Ponce L , Gorzelanny C , Schäfer I , et al. Coculture model of sensory neurites and keratinocytes to investigate functional interaction: Chemical stimulation and atomic force microscope-transmitted mechanical stimulation combined with live-cell imaging. The Journal of Investigative Dermatology (2013) ;133: :1387–90. DOI: 10.1038/jid.2012.471 |
[13] | Bevan S , Woodward B , Ng RL , Green C , et al. Retroviral gene transfer into porcine keratinocytes following improved methods of cultivation. Burns Journal of the International Society for Burn Injuries (1997) ;23: :525–32. |
[14] | Chung M-K , Lee H , Mizuno A , Suzuki M , et al. TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. The Journal of Biological Chemistry (2004) ;279: :21569–75. DOI: 10.1074/jbc.M401872200 |
[15] | Hager B , Bickenbach JR , Fleckman P . Long-term culture of murine epidermal keratinocytes. The Journal of Investigative Dermatology (1999) ;112: :971–6. DOI: 10.1046/j.1523-1747.1999.00605.x |
[16] | Hennings H , Michael D , Cheng C , Steinert P , et al. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell (1980) ;19: :245–54. |
[17] | Papini S , Cecchetti D , Campani D , Fitzgerald W , et al. Isolation and clonal analysis of human epidermal keratinocyte stem cells in long-term culture. Stem cells (Dayton, Ohio) (2003) ;21: :481–94. DOI: 10.1634/stemcells.21-4-481 |
[18] | Regauer S , Compton C . Cultured porcine epithelial grafts: An improved method. The Journal of Investigative Dermatology (1990) ;94: :230–4. |
[19] | Ulmann L , Rodeau J-L , Danoux L , Contet-Audonneau J-L , et al. Trophic effects of keratinocytes on the axonal development of sensory neurons in a coculture model. The European Journal of Neuroscience (2007) ;26: :113–25. DOI: 10.1111/j.1460-9568.2007.05649.x |
[20] | Hewlett G . Strategies for optimising serum-free media. Cytotechnology (1991) ;5: :3–14. |
[21] | Sun TT , Green H . Differentiation of the epidermal keratinocyte in cell culture: Formation of the cornified envelope. Cell (1976) ;9: :511–21. |
[22] | Wille JJ , Pittelkow MR , Shipley GD , Scott RE . Integrated control of growth and differentiation of normal human prokeratinocytes cultured in serum-free medium: Clonal analyses, growth kinetics, and cell cycle studies. Journal of Cellular Physiology (1984) ;121: :31–44. DOI: 10.1002/jc1041210106 |
[23] | Alfandari J , Shnitman Magal S , Jackman A , Schlegel R , et al. HPV16 E6 oncoprotein inhibits apoptosis induced during serum-calcium differentiation of foreskin human keratinocytes. Virology (1999) ;257: :383–96. DOI: 10.1006/viro.1999.9675 |
[24] | Watt FM . Role of integrins in regulating epidermal adhesion, growth and differentiation. The EMBO Journal (2002) ;21: :3919–26. DOI: 10.1093/emboj/cdf399 |
[25] | Dame MK , Spahlinger DM , DaSilva M , Perone P , et al. Establishment and characteristics of Gottingen minipig skin in organ culture and monolayer cell culture: Relevance to drug safety testing. In vitro cellular & developmental biology. Animal (2008) ;44: :245–52. DOI: 10.1007/s11626-008-9091-3 |
[26] | Eisinger M . Regeneration of epidermis by cells grown in tissue culture. Journal of the American Academy of Dermatology (1985) ;12: :402–8. |
[27] | Hengge UR , Chan EF , Hampshire V , Foster RA , et al. The derivation and characterization of pig keratinocyte cell lines that retain the ability to differentiate. The Journal of Investigative Dermatology (1996) ;106: :287–93. |
[28] | Pereira U , Boulais N , Lebonvallet N , Lefeuvre L , et al. Development of an in vitro coculture of primary sensitive pig neurons and keratinocytes for the study of cutaneous neurogenic inflammation. Experimental Dermatology (2010) ;19: :931–5. DOI: 10.1111/j.1600-0625.2010.01119.x |
[29] | Boulais N , Misery L . The epidermis: A sensory tissue. European Journal of Dermatology EJD (2008) ;18: :119–27. DOI: 10.1684/ejd.2008.0348 |
[30] | Lumpkin EA , Caterina MJ . Mechanisms of sensory transduction in the skin. Nature (2007) ;445: :858–65. DOI: 10.1038/nature05662 |
[31] | Schmelz M . Neuronal sensitivity of the skin. European Journal of Dermatology EJD (2011) ;21: (Suppl 2):43–7. DOI: 10.1684/ejd.2011.1265 |
[32] | Ständer S , Schneider SW , Weishaupt C , Luger TA , et al. Putative neuronal mechanisms of sensitive skin. Experimental Dermatology (2009) ;18: :417–23. |
[33] | Zylka MJ , Rice FL , Anderson DJ . Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron (2005) ;45: :17–25. DOI: 10.1016/j.neuron.2004.12.015 |
[34] | Koizumi S , Fujishita K , Inoue K , Shigemoto-Mogami Y , et al. Ca2+ waves in keratinocytes are transmitted to sensory neurons: The involvement of extracellular ATP and P2Y2 receptor activation. The Biochemical Journal (2004) ;380: :329–38. DOI: 10.1042/BJ20031089 |
[35] | Mandadi S , Sokabe T , Shibasaki K , Katanosaka K , et al. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflügers Archiv European Journal of Physiology (2009) ;458: :1093–102. DOI: 10.1007/s00424-009-0703-x |
[36] | Pereira U , Garcia-Le Gal C , Le Gal G , Boulais N , et al. Effects of sangre de drago in an in vitro model of cutaneous neurogenic inflammation. Experimental Dermatology (2010) ;19: :796–9. DOI: 10.1111/j.1600-0625.2010.01090.x |
[37] | Tsutsumi M , Goto M , Denda S , Denda M . Morphological and functional differences in coculture system of keratinocytes and dorsal-root-ganglion-derived cells depending on time of seeding. Experimental Dermatology (2011) ;20: :464–7. DOI: 10.1111/j.1600-0625.2011.01276.x |
[38] | Ulmann L , Rodeau J-L , Danoux L , Contet-Audonneau J-L , et al. Dehydroepiandrosterone and neurotrophins favor axonal growth in a sensory neuron-keratinocyte coculture model. Neuroscience (2009) ;159: :514–25. DOI: 10.1016/j.neuroscience.2009.01.018 |
[39] | Roggenkamp D , Falkner S , Stäb F , Petersen M , et al. Atopic keratinocytes induce increased neurite outgrowth in a coculture model of porcine dorsal root ganglia neurons and human skin cells. The Journal of Investigative Dermatology (2012) ;132: :1892–900. DOI: 10.1038/jid.2012.44 |
[40] | Campenot RB . Local control of neurite development by nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America (1977) ;74: :4516–9. |
[41] | Campenot RB , Lund K , Mok S-A . Production of compartmented cultures of rat sympathetic neurons. Nature Protocols (2009) ;4: :1869–87. DOI: 10.1038/nprot.2009.210 |
[42] | Gold MS , Dastmalchi S , Levine JD . Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience (1996) ;71: :265–75. |
[43] | Charest JL , Jennings JM , King WP , Kowalczyk AP , et al. Cadherin-mediated cell-cell contact regulates keratinocyte differentiation. The Journal of Investigative Dermatology (2009) ;129: :564–72. DOI: 10.1038/jid.2008.265 |
[44] | Roop DR , Huitfeldt H , Kilkenny A , Yuspa SH . Regulated expression of differentiation-associated keratins in cultured epidermal cells detected by monospecific antibodies to unique peptides of mouse epidermal keratins. Differentiation; Research in Biological Diversity (1987) ;35: :143–50. |
[45] | Blanpain C , Fuchs E . Epidermal homeostasis: A balancing act of stem cells in the skin. Nature reviews. Molecular Cell Biology (2009) ;10: :207–17. DOI: 10.1038/nrm2636 |
[46] | Mammone T , Ingrassia M , Goyarts E . Osmotic stress induces terminal differentiation in cultured normal human epidermal keratinocytes. In vitro cellular & developmental biology. Animal (2008) ;44: :135–9. DOI: 10.1007/s11626-008-9087-z |
[47] | Gosselin K , Deruy E , Martien S , Vercamer C , et al. Senescent keratinocytes die by autophagic programmed cell death. The American Journal of Pathology (2009) ;174: :423–35. DOI: 10.2353/ajpath.2009.080332 |
[48] | Monteiro-Riviere NA , Bristol DG , Manning TO , Rogers RA , et al. Interspecies and interregional analysis of the comparative histologic thickness and laser Doppler blood flow measurements at five cutaneous sites in nine species. The Journal of Investigative Dermatology (1990) ;95: :582–6. |
[49] | Hybbinette S , Boström M , Lindberg K . Enzymatic dissociation of keratinocytes from human skin biopsies for in vitro cell propagation. Experimental Dermatology (1999) ;8: :30–8. |
[50] | Adams JC , Watt FM . Regulation of development and differentiation by the extracellular matrix. Development (Cambridge, England) (1993) ;117: :1183–98. |
[51] | Fortunel NO , Chadli L , Bourreau E , Cadio E , et al. Cellular adhesion on collagen: A simple method to select human basal keratinocytes which preserves their high growth capacity. European Journal of Dermatology EJD (2011) ;21: (Suppl 2):12–20. DOI: 10.1684/ejd.2011.1268 |
[52] | Watt FM . Terminal differentiation of epidermal keratinocytes. Current Opinion in Cell Biology (1989) ;1: :1107–15. |
[53] | Watt FM . Selective migration of terminally differentiating cells from the basal layer of cultured human epidermis. The Journal of Cell Biology (1984) ;98: :16–21. |
[54] | Arthur RP , Shelley WB . The innervation of human epidermis. The Journal of Investigative Dermatology (1959) ;32: :397–411. |
[55] | Hilliges M , Wang L , Johansson O . Ultrastructural evidence for nerve fibers within all vital layers of the human epidermis. The Journal of Investigative Dermatology (1995) ;104: :134–7. |
[56] | Hennings H , Holbrook KA , Yuspa SH . Factors influencing calcium-induced terminal differentiation in cultured mouse epidermal cells. Journal of Cellular Physiology (1983) ;116: :265–81. DOI: 10.1002/jc1041160303 |

