
The Inflammation-Induced Dysregulation of Reelin Homeostasis Hypothesis of Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) accounts for most dementia cases, but we lack a complete understanding of the mechanisms responsible for the core pathology associated with the disease (e.g., amyloid plaque and neurofibrillary tangles). Inflammation has been identified as a key contributor of AD pathology, with recent evidence pointing towards Reelin dysregulation as being associated with inflammation. Here we describe Reelin signaling and outline existing research involving Reelin signaling in AD and inflammation. Research is described pertaining to the inflammatory and immunological functions of Reelin before we propose a mechanism through which inflammation renders Reelin susceptible to dysregulation resulting in the induction and exacerbation of AD pathology. Based on this hypothesis, it is predicted that disorders of both inflammation (including peripheral inflammation and neuroinflammation) and Reelin dysregulation (including disorders associated with upregulated Reelin expression and disorders of Reelin downregulation) have elevated risk of developing AD. We conclude with a description of AD risk in various disorders involving Reelin dysregulation and inflammation.
INTRODUCTION
Dementia is a symptom of neurodegenerative diseases associated with significant quality of life impairment for patients and families [1–3]. Alzheimer’s disease (AD) typically results in progressive memory decline preceding dysfunction to numerous cognitive domains including spatial navigation, executive functioning (e.g., attention, decision making, and planning) and language [1]. Although treatments for AD provide temporary symptom relief at various stages of the disease and evidence consistent with slower disease progression has been shown with currently approved treatments, these treatments do not cease disease progression in terms of clinical presentation or neuropathology [4–6]. This is partly related to our incomplete understanding of the mechanisms responsible for the development of AD. Reelin, an extracellular matrix protein, could be a valuable therapeutic target for AD as a variety of studies show dysregulation of Reelin cleavage and Reelin signaling occurs in AD [7–9]. Promoting Reelin homeostasis could be a valuable therapeutic avenue for preventing disease onset and/or reversing AD pathology, although additional work is needed to support Reelin-based therapeutics in the context of AD.
Key pathological features of AD include senile plaques composed of amyloid-β (Aβ) peptides and neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein [10, 11]. Aβ is a by-product of cleavage of amyloid-β precursor protein (AβPP) which is a protein necessary for healthy neuronal development and function [12]. Tau is a microtubule-protein that confers microtubule stability and phosphorylation of tau allows microtubule dissociation, which is critical for healthy neuronal vesicular transport [13]. Hyperphosphorylation of tau destabilizes microtubules and leads to the formation of NFTs [14]. Reelin is a stress-sensitive and inflammation-sensitive protein that inhibits cleavage of AβPP into Aβ, reduces the toxicity of Aβ and inhibits the phosphorylation of tau [15–21]. Within AD and experimental models of AD pathology, research indicates inflammatory processes play a key role in the development of the disease [22]. A role of Reelin in inflammatory conditions has recently been identified, suggesting enhancement of Reelin cleavage may serve an important anti-inflammatory function that, over time, promotes core pathologies of AD due to a reduction of effective Reelin signaling [23, 24]. We propose a mechanism whereby peripheral inflammation and/or neuroinflammation compromise Reelin homeostasis and leads to the development of AD pathology, as outlined in Fig. 1. In this review, connections between Reelin, inflammation and AD are described and the basis for the loss of Reelin homeostasis hypothesis of AD is outlined.
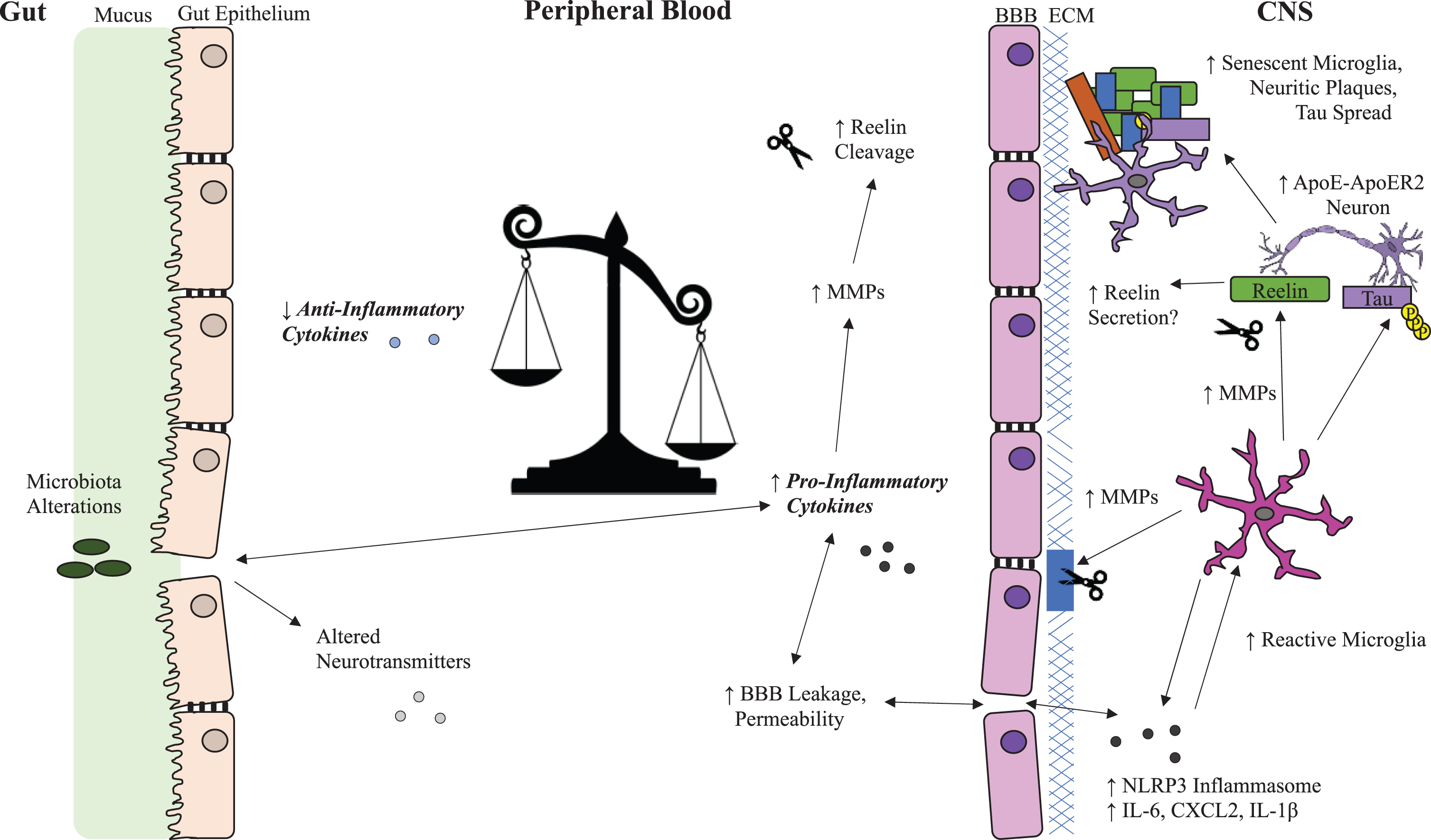
Fig. 1
Possible Reelin and Inflammation Systems in Alzheimer’s Disease. Overview of Reelin dysregulation in Alzheimer’s disease (AD) related to the potential impact of peripheral inflammatory cytokines on blood brain barrier integrity, microglial reactivity, and selectively vulnerable neurons in the brain. Inflammation promotes Reelin cleavage in both the periphery and the CNS through upregulation of MMPs, which break down the BBB and increase infiltration of inflammatory cytokines into the CNS. This creates a feedback loop that enhances microglia reactivity and Reelin cleavage and potentially disrupts Reelin secretion from selectively vulnerable neurons leading to aggregation of Reelin fragments in amyloid plaques. ApoE, apolipoprotein E; ApoER2, ApoE receptor 2; BBB, blood-brain barrier; CXCL2, chemokine (C-X-C motif) ligand 2; ECM, extracellular matrix; IL-1β, interleukin 1-beta; IL-6, interleukin 6; MMP, matrix metalloproteases; NLRP3, NLR family pyrin domain containing 3; p-tau, phosphorylated tau; VLDLR, very low-density lipoprotein receptor.
Alzheimer’s disease and inflammation
Inflammation is a tightly regulated protective process involving activation of immune and non-immune cells in response to infection, toxic compounds, damaged cells, and irradiation with the purpose of restoring homeostasis [25]. When dysregulated, inflammation can persist beyond the days or weeks required to resolve an inflammatory stimulus. Inflammation is presently considered a key promoter of AD pathology as reviewed elsewhere [26]. Inflammation is observed in the early stages of AD and exacerbates AD pathology experimentally, suggesting inflammation is a key pathological feature of AD rather than a by-product of the disease [27, 28]. Phosphatidylinositol 3-kinase (PI3K) and glycogen synthase kinase-3β (GSK3β) play important roles in both promoting and resolving inflammation [29, 30]. GSK3β also plays a central role in AD as a regulator of tau phosphorylation [28, 31–33]. GSK3β activity is regulated by Reelin signaling and is further described in the Reelin overview section. One of the most well-established genetic risk factors for late onset AD is the ɛ4 variant of apolipoprotein E, a protein that regulates cholesterol, lipoproteins, and Aβ metabolism [34–36]. The ɛ4 variant has reduced capacity for Aβ clearance and promotes inflammation through dysregulation of nuclear factor-kB (NF-κB) signaling [35, 37–40].
Microglia, the resident immune cells of the central nervous system (CNS), are considered the major drivers of neuroinflammation and are responsible for a variety of important regulatory functions in both healthy and disease states [41]. Microglia produce reactive oxygen species and secrete pro-inflammatory cytokines and matrix metalloproteinases (MMP) that contribute to blood-brain barrier (BBB) breakdown [41–46]. Functions of MMPs include cleaving extracellular matrix proteins which increases permeability of the BBB and allows infiltration of peripheral immune cells and inflammatory cytokines into the CNS, which have been reported in AD [47–52]. Microglial proliferation, morphology, and survival are regulated by cytokine signaling and are susceptible to inflammation [53, 54]. Hyperinflammation promotes microglial priming, increasing susceptibility to evoke an exaggerated inflammatory response to secondary inflammatory stimuli and also makes microglia prone to cellular senescence, an arrest of cell cycle that impairs organelle function and halts proliferation [22, 55–57]. Microglia senescence prevents microglia from effectively serving their numerous functions required for healthy neural activity [58]. The impact of microglial senescence and microglial dysfunction (involving impaired phagocytosis, spreading of amyloid beta and tau aggregates, and maladaptive synaptic element pruning) on AD progression was recently reviewed by Lau et al. [55]. Tau pathology causes neurons to externalize phosphatidylserine which triggers microglial phagocytosis, promotes microglial senescence, and causes a positive feedback loop of seeding tau aggregates and spreading of tau-based pathology [59].
Experimental evidence shows inflammation is a key regulator of AD pathology. In AβPP/PS1 transgenic mice (which express mutant human AβPP with the Swedish mutation (K6790N, M671L) and human Presenilin 1 with the M146V mutation) and 5xFAD mice (which express mutant human AβPP with the Swedish, Florida (I716V) and London (V717I) familial AD mutations and human PS1 harboring two familial AD mutations, M146L and L286V), interferon (IFN)-γ and IL-17 secreting T-cells infiltrate the brain parenchyma near Aβ deposits [49, 60, 61]. Immune challenge in AβPP/PS1 mice elevates infiltration of Th1 and Th17 cells, increases microglial reactivity and enhances Aβ accumulation [49]. Additionally, 10-month-old AβPP/PS1 mice show elevated cortical T-cell infiltration 8-weeks post infection not seen in 4-month-old AβPP/PS1 mice after infection [49]. This age-dependent process is likely explained by a combination of increased BBB permeability, heightened inflammation, and inflammatory priming of microglia [22, 62, 63]. In addition, adoptive transfer of IFN-γ secreting Th1 cells to AβPP/PS1 mice increases microglial reactivity, Aβ deposition, and induces cognitive decline [60]. Elsewhere, gestational exposure of CD-1 mice to lipopolysaccharide (LPS) increased the number of errors during the radial 6-arm water maze and decreased object recognition memory at 12, 18 and 22 months of age, while also increasing levels of p-tau and Aβ42 in the hippocampus at these ages [64].
The viral mimetic polyinosinic-polycytidylic acid (Poly(I:C)) has also been applied experimentally to elevate inflammatory cytokines. Prenatal exposure to Poly(I:C) predisposes wild-type mice to develop increased hippocampal AβPP and proteolytic fragments of amyloid, increased phosphorylated tau and impairment of working memory in adulthood [27]. A subsequent immune challenge in adult mice exposed prenatally to Poly(I:C) exacerbated neuropathological features of AD including AβPP proteolytic fragment deposition, tau aggregation, reactive astrogliosis, and microglial reactivity suggesting inflammatory challenges, particularly in early life, predispose animals to develop AD-like pathology upon future inflammatory events [27]. Peripheral exposure of 4–6 month old C57/BL/6J mice to Poly (I:C) for 7 days also impaired contextual fear conditioning and increased hippocampal Aβ levels 72 h after the final Poly(I:C) exposure, suggesting Poly(I:C) exposure does not just increase AD-like pathology when delivered during the gestational period, as adult-mice also developed greater AD-like pathology and induced cognitive impairment after Poly(I:C) exposure [65]. Splenectomy exacerbates AD-like pathology in AβPP/PS1 mice (which express APP695 with the Swedish mutation and human Presenilin1 lacking exon 9) further implicating the peripheral immune system to AD [66, 67]. In contrast to the various studies showing inflammation promotes or exacerbates AD pathology, Th-2 biased Aβ-specific T-cell infusions reduced plaque-associated microglial reactivity, vascular amyloidosis, and cognitive impairment without infiltrating into the CNS, suggesting modulation of peripheral inflammation can be a valuable therapeutic target for AD [68].
There exists a complex interaction between peripheral and central inflammation in the context of neurodegenerative diseases including AD. Chronic peripheral inflammation, indicated by elevated C-reactive protein (CRP) levels, is associated with age-related cognitive decline and all-cause dementia [69]. The gut microbiome modifies peripheral inflammation and mice transplanted with gut microbiota from human patients with AD show activation of the NLRP3 inflammasome, heightened hippocampal microglial reactivity and cognitive deficits [70]. Cognitively impaired adult humans with brain amyloidosis have higher levels of cytokines (IL-6, CXCL2, NLRP3, IL-1β) compared to controls and cognitively impaired individuals with no amyloidosis, and these alterations were associated with higher abundance of proinflammatory gut microbiota, specifically Escherichia and Shigella taxa, and lower levels of Eubacterium rectale, suggesting diet is a modifiable risk factor for AD [71]. Supporting a role of diet in modifying peripheral inflammation and reducing AD risk, adherence to the Mediterranean diet, which has antioxidant and anti-inflammatory properties, is associated with reduced risk of developing AD [72–76]. Future treatments might involve regulation of peripheral inflammation and/or BBB permeability given the various lines of evidence showing peripheral inflammation contributes to AD pathology.
Reelin overview
Reelin is a pleiotropic extracellular matrix protein associated with regulating neuronal migration in embryonic development and in adulthood, promotes synaptic plasticity, cell adhesion and cell proliferation [77–79]. Reelin dysregulation is seen in various neuropsychiatric conditions including schizophrenia, bipolar disorder, major depression, and AD [80–83]. Reelin is a large multi-domain protein consisting of a C-terminal of predominantly basic amino acids, eight Reelin repeat domains of 350–390 amino acids, and a F-spondin-like N-terminal domain [84]. Outside the CNS, Reelin is expressed in the liver, kidney, blood (erythrocytes, platelets), bone marrow, lymphatic tissues, enteric nervous system, and the yolk sac during development, serving a plethora of functions including regulating cell growth, cell cycle, cell motility, cellular adhesivity, hemostasis and platelet spreading [85]. Although the full-length Reelin molecule is large, at approximately 410 kDa, evidence of Reelin in caveolar vesicles of brain endothelial cells suggests Reelin could be actively trafficked into the brain from the periphery [86]. Reelin was first recognized for its important roles in embryonic neurodevelopment, and therefore, most of our understanding of Reelin relates to the central nervous system wherein Reelin has been considered a homeostatic regulator of synaptic plasticity [87]. In the brain parenchyma Reelin promotes cortical synaptogenesis, dendritic spine development and maturation, memory formation, hippocampal plasticity, neurogenesis, neuronal migration, and maturation and integration of dentate granule neurons, as reviewed elsewhere [88].
Reelin signaling is transduced through binding of Reelin to the apolipoprotein E receptor ApoER2, very-low density lipoprotein receptors (VLDLR), and interactions with α3/β1 integrin [89–91]. Reelin receptor binding leads to phosphorylation of the adaptor protein Dab1 which activates Src family tyrosine kinases that regulate the composition of synaptic NMDA receptors through modification of NR2B subunit surface mobility [92–95]. Reelin-induced modification of synaptic NMDA receptors increases calcium influx following subsequent glutamatergic transmission and, ultimately, modifies neural transmission required for long-term potentiation (LTP) and synaptic strength maintenance, which are perturbed in many neuropsychiatric disorders including AD [91, 93, 96–98]. Loss or reduction of Reelin-mediated NMDA receptor subunit trafficking to the synapse may contribute to disrupted distribution of synaptic and extrasynaptic NMDA receptors, which contributes to excitotoxicity [99]. In support of a key role of Reelin in limiting excitotoxicity, an in vitro study showed Reelin reduced glutamate-induced excitotoxicity in cultured neurons, although these results should also be evaluated in vivo to better understand if loss of Reelin signaling contributes to excitotoxicity in disorders of depleted Reelin signaling and excitotoxicity, such as AD [100]. Memantine, an available treatment for AD, reduces excitotoxicity by restoring the balance of synaptic/extrasynaptic glutamatergic signaling through antagonizing extrasynaptic NMDA receptors and sparing synaptic NMDA receptors [101–103]. In addition, mice with reduced Reelin expression are more susceptible to stress-induced alterations to neuronal nitric oxide synthase associated with excitotoxicity, suggesting reduced Reelin signaling increases risk of developing excitotoxicity [104].
Reelin signaling also leads to phosphorylation of PI3K which inactivates GSK3β through phosphorylation of GSK3β at serine 9 [105]. As mentioned, GSK3β signaling is a key source of tau phosphorylation that is dysregulated in AD and is modified by inflammation [28, 31–33, 106]. Reelin signaling also regulates mechanistic target of rapamycin (mTOR), which regulates metabolism, immune response, and autophagy, as well as extracellular signal-regulated protein kinases 1 and 2 (ERK 1 and 2), which control cell proliferation and cell death [107–110]. Both mTOR and ERK1/2 are dysregulated in AD and expression of mTOR has been correlated to both total and phosphorylated tau in AD [107, 110–113]. Figure 2 outlines Reelin signaling pathways in the context of AD. An extensive review of Reelin signaling pathways beyond the scope of AD is described elsewhere [114].
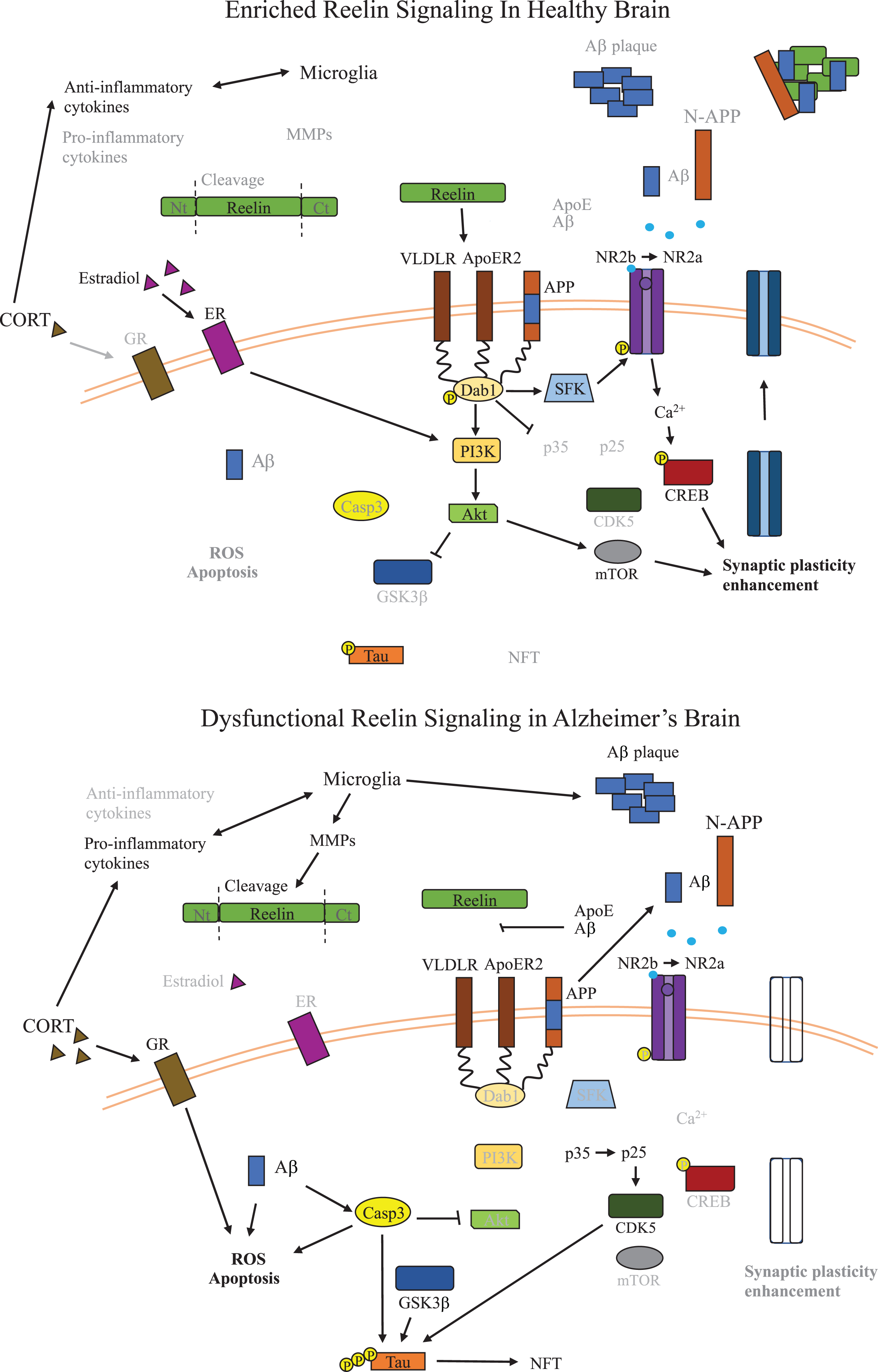
Fig. 2
Dysfunctional Reelin Signaling in Alzheimer’s Disease Brain. Overview of the signaling pathways relevant to Reelin dysfunction and inflammation in AD relative to healthy brains. Reelin signaling leads to inactivation of GSK3β and loss of Reelin signaling in AD promotes formation of NFTs by dysregulating phosphorylation of tau. Reelin also promotes synaptic plasticity, which is critical for learning and cognitive function, and in AD, a reduction of Reelin signaling leads to disruption of synaptic plasticity. The potential role of microglia secreted MMPs on Reelin proteolytic processing are also shown, resulting in reduced Reelin signaling. The impact of alterations to glucocorticoids and estrogen on kinases along the Reelin signaling pathway are also shown in the context of AD, and both reduce activity of kinases downstream of Reelin leading to less inhibition of GSK3β. Aβ, amyloid-β; ApoE, apolipoprotein E; ApoER2, ApoE receptor 2; CDK5, cyclin-dependent kinase 5; CORT, corticosterone; CREB, cAMP-response element binding protein; CXCL2, chemokine (C-X-C motif) ligand 2; Dab1, disabled-1; ECM, extracellular matrix; GSK3β, glycogen synthase kinase 3-beta; IL-1β, interleukin 1-beta; IL-6, interleukin 6; GR, glucocorticoid receptor; MMP, matrix metalloproteases; mTOR, metabolic target of rapamycin; NFT, neurofibrillary tangles; NLRP3, NLR family pyrin domain containing 3; NR2b/a, N-methyl D-aspartate receptor subtype 2B/2A; N-APP, N-terminal amyloid β precursor protein; PI3K, phosphoinositide 3-kinase; p-tau, phosphorylated tau; ROS, reactive oxygen species; SERT, serotonin transporter; VLDLR, very low density lipoprotein receptor.
Reelin in cognition
Although many disorders involving Reelin dysregulation, including but not limited to depression, schizophrenia, AD, epilepsy, and multiple sclerosis, are widely recognized to present cognitive alterations, whether Reelin dysregulation is responsible for these alterations is not yet clear [7, 80, 115–117]. For example, although AD and major depression involve reduced plasticity, lower spine density and disruption of associative learning and memory, it is unclear whether Reelin dysregulation is a major driving force for such dysfunction in these contexts. In addition, the impact of Reelin therapeutics have not been evaluated in humans and likewise, direct roles of Reelin treatment in normalizing cognition in humans have yet to be shown. Therefore, rather than speculate on the roles of Reelin in known neuropsychiatric disorders in which Reelin is dysregulated and cognition is affected, we focus here on the impact of Reelin signaling on cognition in rodent models.
Two relevant animal models for studying the roles of Reelin in cognition are the heterozygous reeler mouse (HRM) and models of chronic stress. HRM have a mutation leading to deletion of roughly 126 kb in one Reelin allele resulting in approximately 50% reduced Reelin expression [118]. Although there exists the Reeler mouse line, which are homozygous Reelin mutants devoid of Reelin expression, these mice have major brain malformations [119, 120]. While the Reeler mouse has been pivotal to identifying the roles of Reelin in brain development, it is difficult to conclude whether any behavioral deficits observed are a result of insufficient Reelin signaling and not resulting from the major brain malformations. Therefore, the HRM model, which has normal neuronal migration and brain development, is a more applicable model for assessing the role of Reelin signaling in cognition across the lifespan. Deficits observed in the HRM model include reduced dendritic spine density, altered synaptic plasticity and behaviorally deficits have been observed in associative learning, memory, and pre-pulse inhibition [121–124]. Reversal learning, the process of learning to inhibit previously rewarded actions, is also impaired in HRM [123, 124]. Impairment of HRM in reversal learning, particularly with increased perseverative responses, implicates Reelin in inhibitory processing and impulsivity, consistent with the roles of Reelin in regulating synaptic plasticity [96, 125, 126].
Reelin supplementation through intraventricular injection can also influence behavior and normalize various deficits associated with Reelin depletion in rodent models such as the HRM, including enhancement of hippocampal synaptic function and normalization of LTP, fear conditioning, associative learning, and spatial memory [98, 127, 128]. Supplementation of wild-type mice with Reelin through bilateral intraventricular injection has also been shown to improve context-dependent learning and memory and to enhance LTP [98]. In a mouse model of Angelman syndrome (a condition characterized by synaptic dysfunction and reduced Reelin expression) caused by a UBE3A null mutation, bilateral intraventricular injection of Reelin rescued dendritic spine density, synaptic function, learning and memory [125]. Reelin levels are also reduced in the dentate gyrus sub-granular zone in rodent models of chronic stress which induce behavioral changes including despair-like behavior and spatial memory impairment [129–131]. Either intravenous or intraventricular injection of recombinant Reelin resolves stress-induced depression-like behavior in rodents, stress-induced spatial memory deficits and Reelin cell counts in the dentate gyrus [129–131]. Additionally, a single peripheral Reelin injection reverses alterations to LTP induced by chronic stress [132]. Reversal of behavioral and neurobiological alterations with an intravenous injection of Reelin suggests that either Reelin crosses the BBB and/or peripheral Reelin indirectly normalizes Reelin expression in the brain to restore neural functions of Reelin following chronic stress. That Reelin supplementation resolves behavioral and neurobiological impairments of low Reelin signaling suggests Reelin therapeutics could be valuable for treatment of AD and other disorders where Reelin signaling is disrupted.
Reelin in AD
As mentioned above, Reelin signaling regulates a variety of pathways important for AD. In support of a key role of Reelin signaling in the context of AD, recently a gain of function mutation to the Reelin gene, Reelin-COLBOS, was identified as providing resistance in familial AD, and when inserted into the human tau transgenic mouse model (which express human tau with a P301L substitution), the Reelin-COLBOS mutation reduced tau hyperphosphorylation and intensity of neurofibrillary tangles [133]. In addition, a large single nucleus RNA sequencing study identified the loss of Reelin inhibitory neurons in the prefrontal cortex as a key factor for predicting level of cognitive impairment in AD, as individuals with maintained cognitive functioning had higher levels of Reelin expressing inhibitory neurons than those with lower scores in cognitive testing [134]. Reelin has been shown to be reduced in the entorhinal cortex at all stages of AD and was undetectable in severe AD entorhinal cortex samples [135]. Another study showed fewer Reelin positive cells in post-mortem frontal cortex, hippocampus, and entorhinal cortex in AD, as well as lower levels of full length Reelin [7]. Co-staining for Reelin, tau and Aβ in AD brain samples showed dense aggregations of Reelin positive cells within and surrounding amyloid plaques [7]. Reelin also binds to AβPP and reduces β cleavage, which may relate to accumulation of Reelin in amyloid plaques [7, 15]. Dab1 phosphorylation reduces β-cleavage of AβPP to Aβ in vitro, wherein Reelin only decreased Aβ when Dab1 was present; this suggests that reduced Reelin signaling impacts AβPP proteolysis and contributes to Aβ plaque aggregation[136].
Understanding Reelin cleavage and fragmentation is important for interpreting the various findings of Reelin expression in AD as a variety of studies show altered proteolytic processing of Reelin occurs in AD [8, 9, 83]. The full-length Reelin molecule is composed of 8 epidermal growth factor repeats that can be cleaved at Reelin repeat 3, or the N-terminal cleavage site, and C-terminal cleavage between repeats 6 and 7 [88, 137]. The N-terminal cleavage at repeat 3 leaves an approximately 180 kDa and 250 kDa Reelin fragment whereas the C-terminal cleavage between repeats 6 and 7 leaves an approximately 310 kDa fragment and 100 kDa Reelin fragment [9, 137]. The Reelin central fragment composed of repeats 3–6 is required for effectively inducing downstream signaling and N-terminal cleavage reduces the duration and range of Reelin-mediated Dab1 phosphorylation [138–141]. A variety of molecules are responsible for Reelin cleavage, many of which also serve anti-inflammatory functions [142]. Increasing the activity of these enzymes may serve to restore inflammatory homeostasis, but in chronic inflammatory conditions such as AD, increased enzymatic activity could promote development of AD through Reelin cleavage. Identified Reelin proteases include tissue plasminogen activator, meprin α and β, a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-2, ADAMTS-3, ADAMTS-4, and ADAMTS-5 [47, 139, 140, 143–147]. Several of these molecules also regulate cleavage of AβPP, and their dysregulation contributes to production of the pathogenic Aβ fragment [148–150]. Various modulators of Reelin cleavage have also been identified including MMP-9 and tissue inhibitors of metalloproteinases [47]. MMP-9, for example, is upregulated in the brain and plasma of AD patients, and elevation of MMP-9 is associated with faster decline of both cognitive function and hippocampal volume in AD, again suggesting Reelin cleavage is enhanced in AD [151–153].
Multiple studies support Reelin cleavage being dysregulated in AD. Increased levels of the inactive 180 kDa N-terminal Reelin fragment were found in the cerebrospinal fluid (CSF) of individuals diagnosed with frontotemporal dementia and AD, where the expression of the 180 kDa Reelin fragment correlated to changes in MMSE score [8]. Another study also reported increased levels of the 180 kDa Reelin fragment in the CSF in AD, which was also seen in frontotemporal dementia, progressive supranuclear palsy, and Parkinson’s dementia [154]. Elevation of Reelin mRNA and the 180 kDa Reelin fragment were also observed in frontal cortex samples of patients with AD, again suggesting proteolytic processing of Reelin is dysregulated in AD [83]. In contrast, a recent study evaluating Reelin in the CSF of individuals with AD showed a reduction of full-length Reelin, elevated levels of the 310 kDa fragment and the 100 kDa fragment, lower levels of the 250 kDa fragment, but no change to the 180 kDa fragment [9]. Although these results also show dysregulated Reelin cleavage in AD, they conflict with the previously mentioned studies observing elevated N-terminal cleavage products in AD, instead showing evidence of elevated C-terminal cleavage products. Reports of Reelin aggregates within and surrounding amyloid plaques and various studies showing dysregulation of Reelin cleavage, particularly increased expression of the N-terminal Reelin fragments that have reduced signaling capacity, indicate a reduction of Reelin bioavailability and signaling capacity occurs in AD [7–9, 83]. Altered levels of Reelin fragments strongly suggests Reelin cleavage is dysregulated in the context of AD, which has been reviewed elsewhere [137, 154]. Table 1 provides an overview of human studies linking AD to Reelindysregulation.
Table 1
Overview of human studies of Alzheimer’s disease evaluating relationships between Reelin and AD pathology
| Species | Sample(s) | Methods of Reelin quantification | Reelin alterations | Key findings summary | Reference |
| Human | CSF – FTD, AD & HC | WB | Upregulated 180 kDa fragments | Elevated 180 kDa Reelin fragments in AD & FTD; suggestive of altered proteolytic processing of Reelin | [8] |
| Human | EC – AD (severe) vs. HC | IHC | No change | No change to # of EC CR expressing Reelin in severe AD | [135] |
| Human | FC, cerebellum, CSF & Plasma of AD, MCI & HC | WB & Lectin Binding Assay | Increased 180 kDa Reelin fragments in FC and CSF but not in cerebellum or plasma. Altered glycosylation patterns in CSF, but not plasma | Elevated 180 kDa Reelin in AD relative to controls and MCI; altered glycosylation patterns in CSF. Correlation between tau burden and 180 kDa Reelin levels | [83] |
| Human | Human AD brain, HPC, FC and EC at various Braak stages and age-matched HC | cDNA; WB & IHC | Reelin decreased early in AD and in late stages; Reelin transcription downregulated in AD in all regions; Decreased Reelin expression in EC of late-stage AD relative to early-AD but no age-related decreases in HC | Reelin depletion occurs early in AD and in various structures, decreased transcription of Reelin and upregulated processing of Reelin | [7] |
| Human | FC and HPC from AD and HC | WB | Increased Reelin mRNA expression in late AD Braak stages; no change in earlier Braak stages | FC and HPC have increased Reelin mRNA expression in late but not early Braak stages; both full length 420 kDa and 180 kDa Reelin expression increased in late Braak stages; reduced Dab1 phosphorylation in AD | [154] |
CSF, cerebrospinal fluid; FTD, frontotemporal dementia; HC, healthy controls; WB, western blotting; EC, entorhinal cortex; IHC, immunohistochemistry; CR, Cajal Retzius cells; FC, frontal cortex; MCI, mild cognitive impairment; HPC, hippocampus
Evidence derived from experimental rodent models of AD also suggests Reelin signaling regulates the two hallmark pathological features of AD, amyloid and tau pathology. As mentioned above, N-terminal cleavage by ADAMTS-3, which is secreted by excitatory neurons, dysregulates Reelin-mediated Dab1 signaling [143, 144]. Relatedly, although Reelin is predominantly expressed in inhibitory neurons, Reelin is uniquely expressed in excitatory neurons in the entorhinal cortex, precisely where AD pathology is most severe, and atrophy first presents [20, 135]. The mechanisms regulating ADAMTS-3 secretion in the context of AD are not yet clear, but experimentally reducing ADAMTS-3 reduced Aβ levels in the cerebral cortex of 3-month-old APPNL - F mice (which express APP with the Swedish and Iberian (I716F) mutations) which otherwise display Aβ accumulation, increased synaptic loss, microgliosis, and astrogliosis around 6 months of age [150, 155]. Alike human samples, Reelin immunostaining has been shown in amyloid plaques in the hippocampus and cortex of wild-type mice and transgenic mice expressing mutant human AβPP (with the Swedish, Dutch (E693Q) and London mutations) and mutant human presenilin-1 (with mutation M146L) [156, 157]. Conditional Reelin knockout causing loss of Reelin beyond 2 months of age, when crossed with Tg2576 mice (which express APP with the Swedish mutation), was associated with deficits in the Morris water maze and in contextual and cued fear conditioning at 7 months of age that were not seen in Tg2576 mice or in mice with conditional Reelin knockout not crossed with Tg2576 mice [16].
Loss of Reelin signaling has also been shown to exacerbate tau hyperphosphorylation in rodent models of AD [15, 18, 19, 105, 158]. For example, relative to double human AβPP transgenic mice (which express the human APP695 gene containing the Swedish and Arctic (E693G) mutations) not crossed with HRM, double human AβPP transgenic crossed with HRM had elevated levels of amyloid plaque distribution, accumulation of hyperphosphorylated tau and elevated microglia reactivity [18]. In contrast to studies showing the impact of Reelin depletion, Reelin overexpression in transgenic mice expressing mutant human AβPP (with Swedish and Indiana (V717F) mutations) delayed Aβ plaque formation and normalized deficits in recognition memory [17]. Similarly, in the VLW transgenic mouse model of tauopathy (which overexpress a 4-repeat isoform of human tau bearing three frontotemporal dementia linked with parkinsonism-17 mutations (G272V, P301L and R406W)), overexpressing Reelin normalized tau phosphorylation, resolved deficits in latent inhibition, and enhanced LTP [19]. Evidence of Reelin overexpression and supplementation reversing AD pathologies experimentally again suggests that Reelin-based therapeutics could have value in treatment of human AD, although this requires empirical validation. Table 2 shows key in vivo animal studies demonstrating relationships between Reelin and AD-like pathology.
Table 2
Overview of key animal studies evaluating relationships between Reelin and AD-like pathology in vivo
| Species | Samples | Methods of Reelin quantification | Reelin alterations | Key findings summary | Reference |
| Mice | Tg mice lacking Reelin & Tg Mice lacking VLDLR and APoER2 | WB | Not applicable | Phosphorylated tau is upregulated in Reelin KOs and VLDLR/ApoER2 KOs | [91] |
| Mice | Tg AD mice expressing human mutant AβPP and presenilin-1 | IHC | Not applicable | Reelin colocalization in neuritic plaques in cortex and hippocampus | [156] |
| Mice | HRM, 2xTg AD mice, 2xTg AD mice crossed with HRM | IHC | Reelin plaque in HPC of HRM by 3 months; Aβ plaque in HPC of double mutants by 9 months; Cortical thinning in double mutants & worsened Aβ plaques by 15 months relative to Tg AD mice; Increased inflammation in crossbred mice not seen in AD Tg or HRM; Elevated p-tau in aged double mutants | Combining HRM with Tg AD mice exacerbates various aspects of AD-like pathology in an age-dependent manner | [18] |
| Mice | 3x Tg AD mice & age-matched WT | cDNA; WB & IHC | Decreased Reelin expression and increased Reelin processing in Tg mice; genotype and age-dependent depletion of Reelin positive HPC neurons; No age- or genotype-related change to Reelin mRNA levels | Reelin depletion occurs in Tg AD mice; decreased transcription of Reelin and upregulated processing of Reelin | [7] |
| Mice | 2x Transgenic AD mice & Tg Reelin-overexpression mice; and Crosses | IHC; WB | Not applicable | Reelin overexpression reduced amyloid plaque burden in aged AD mice in HPC and EC | [17] |
| Mice | Tg mice overexpressing Reelin crossed with 4x Tg AD mice; 4x Tg AD mice with normal Reelin expression | IHC; WB; Electrophysiology | Not applicable | Reelin-dependent reduction of phosphorylated-Tau levels; Reelin overexpression normalized LTP in 4x Tg mice | [19] |
| Monkey | Aged (27–38 years) and young adult (8–10 years) rhesus monkeys | IHC | Decreased # of Reelin expressing cells in Layer II of EC in aged monkeys with cognitive decline | Decreased Reelin expressing Layer II EC neurons in cognitively impaired aged monkeys relative to cognitively intact aged monkeys | [211] |
| Mice | AβPP KO mice | Golgi Staining | Not applicable | Intraventricular Reelin did not increase dendritic spine density in AβPP KO mice, suggesting the interaction between Reelin and AβPP may be necessary for upregulating spine density with Reelin | [210] |
Tg, transgenic; WB, western blotting; KO, knock-out; IHC, immunohistochemistry; HRM, heterozygous Reeler mice; HPC hippocampus; WT, wild-type; EC, entorhinal cortex; LTP, long-term potentiation.
Reelin and inflammation
Although the roles of Reelin in inflammation are not yet completely clear, several reviews describe roles of Reelin in inflammation [159, 160]. A potential role of Reelin in immunity was identified in the HRM model as altered splenic T lymphocyte proliferation and down-regulation of numerous immunity-related genes including IRF8, which is required for development and maturation of myeloid cells, and NFIL3, which controls regulatory T-cell function and stability, were observed [161–164]. Reeler-, Dab1- and Apoer- mutants were also shown to lack microglial immunoglobulin G Fc receptors in the cerebellum, suggesting healthy microglia might require Reelin signaling [165]. In the periphery, hepatic stellate cells, which respond to and release various inflammatory molecules, are thought to the be major source of peripheral Reelin [166]. A variety of studies show dysregulation of Reelin signaling in inflammatory conditions. Evidence from inflammatory disorders of elevated peripheral Reelin expression suggest elevated Reelin levels increase NF-κB and enhance leukocyte-endothelial cell adhesion through upregulation of various cell adhesion molecules (ex. E-selectin, ICAM-1, and VCAM-1) [23, 24]. How Reelin influences NF-κB signaling in AD remains unclear, but evidence from 5xFAD Tg mice shows depleted Reelin in the hippocampus and elevated levels of NF-κB that could be reversed with bergenin, which also attenuated Aβ aggregation, prevented spatial memory deficits and suppressed pro-inflammatory cytokines [167].
In the dextran sulphate sodium (DSS) model of ulcerative colitis, colonic Reelin expression was upregulated in wild-type mice due to decreased methylation of the Reelin promoter following DSS exposure, and Reeler mice exposed to DSS presented with elevated disease index scores and higher mortality rates [168]. Elevated peripheral Reelin levels have also been shown in COVID-19 and experimental autoimmune encephalitis (EAE) [23, 24]. Reelin upregulation correlated with elevation of pro-inflammatory cytokines in humans with mild and severe COVID-19 symptoms, whereas asymptomatic COVID-19 infection was associated with normal Reelin expression [23]. Experimentally, treatment with CR50, an antibody that reduces peripheral Reelin levels without affecting Reelin in the CNS, in transgenic mice expressing human angiotensin-converting enzyme 2, protected against COVID infection [23]. Similarly, depleting peripheral Reelin levels in EAE with the CR50 antibody normalized monocyte infiltration into the CNS through reducing cell adhesion molecule expression and prevented development of EAE symptoms [24]. In contrast to these disorders, where inflammation is linked to elevated Reelin signaling, systemic inflammation through exposure to LPS or maternal administration of Poly(I:C) has been shown to reduce CNS Reelin levels and induce learning and memory impairment, suggesting peripheral inflammation can, over time, lead to a depletion of Reelin in the CNS [21, 169, 170]. As described above, chronic stress also depletes Reelin in the brain, and we found atrophied spleen white pulp induced by chronic stress could be reversed by a peripheral Reelin injection at the acute dosage associated with antidepressant-like effects [131, 171]. In this model, promoting Reelin homeostasis through a single peripheral injection of Reelin recovered both stress-induced atrophy of lymphatic germinal zones as well as hippocampal Reelin, suggesting returning Reelin to homeostatic levels promotes healthy inflammatory responses in disorders of low Reelin expression [131, 171]. As Reelin is a pleiotropic molecule, evidence suggests the anti-inflammatory mechanisms responsible for promoting Reelin homeostasis in models of low Reelin expression through Reelin supplementation are very different from the anti-inflammatory mechanisms of Reelin depletion in inflammatory disorders of elevated Reelin levels. Evidence suggests restoring Reelin homeostasis, either through supplementing Reelin in disorders of low Reelin levels or depleting Reelin in disorders of high Reelin levels, both normalize inflammatory changes.
MICROGLIA AND REELIN DYSREGULATION HYPOTHESIS OF AD
As described above, there is an intricate relationship between immune dysregulation, inflammation and Reelin homeostasis in the context of AD. As mentioned above, microglia are involved in a variety of functions in both healthy and disease states and are implicated in a variety of pathological features associated with AD [41, 172]. Microglia are key regulators of inflammation and BBB integrity that produce various MMPs, particularly at sites of inflammation, although understudied in the context of Reelin dysfunction [45, 46]. MMPs increase BBB permeability through regulating cleavage of extracellular matrix proteins, including Reelin, which increases infiltration of peripheral immune cells and inflammatory molecules into the CNS [47]. Treatments such as resveratrol, an antioxidant commonly associated with red wine, and microRNA miR126a-5p reduce production of microglial MMP-9 and protect against inflammation and inflammation-induced BBB permeability [173, 174]. As the tight regulation of extracellular matrix proteins is disrupted and the permeability of the BBB is increased via increased MMP activity, peripheral inflammatory signaling molecules can elevate neuroinflammation by promoting inflammatory secretions from microglia. Additionally, as mentioned above, various molecules sensitive to inflammation cleave both Reelin and AβPP. This suggests that over time, initially protective anti-inflammatory mechanisms become destructive in the context of AD [148, 149]. Additionally, CXCL12/CXCR4 signaling regulates microglia reactivity, and plasma CXCL-12 is depleted in both early AD and the Tg2576 mouse model of AD [175, 176]. In cultured SH-SY5Y neurons, exposure to CXCL-12 dose-dependently increased Reelin secretion and depleted intracellular Reelin, which was blocked by a CXCR4 antagonist [177]. Disruption of CXCL12/CXCR4 signaling in AD could also alter Reelin secretion and intracellular Reelin expression in the absence of enhancement of pro-inflammatory microglia secretions.
The Reelin receptor VLDLR is expressed on microglia in brain tissue derived from individuals diagnosed with AD and in healthy controls, and VLDLR is upregulated in microglia at sites of plaques in AD [178]. Although direct interactions between Reelin and microglia have not been reported, to the authors’ knowledge, dysregulation of Reelin signaling could impact microglia motility and/or kinase activity through VLDLR. VLDLR is integral for regulating the radial distribution and proliferation of oligodendrocyte precursor cells and may have similar functions for microglia [77]. Microglia may selectively migrate to vulnerable neurons presenting increased Reelin secretion, ApoER2 upregulation, and containing initial tau aggregates. Microglial phagocytosis of selectively vulnerable neurons and their surrounding environments likely contributes to enriched Reelin deposits around neuritic plaques, as microglia engage in partial exocytosis of digested contents when exhausted and/or senescent [55].
In AD, inflammation and ongoing oxidative stress likely build into positive feedback loops to cause lipid peroxidation, a process by which reactive oxygen species react with lipids [55]. Enriched lipid peroxidation permanently cross-links ApoE into ApoER2 in the medial temporal lobe of individuals diagnosed with AD or mild cognitive impairment [179]. Although this requires empirical confirmation, if Reelin is unable to bind to cross-linked ApoE-ApoER2, lipid peroxidation of selectively vulnerable neurons could lead to compensation of reduced Reelin signaling through increasing secretion of Reelin and ApoER2 upregulation. These mechanisms account for enriched ApoER2 levels, inhomogeneous Reelin distribution via enriched Reelin deposits found in dystrophic neurites, most likely around vulnerable neurons involved in developing tau pathology, and Reelin homeostasis conferring protection against tau pathology and cognitive decline associated with AD. Increased Reelin production and cleavage in selectively vulnerable neurons may initially protect against inflammation but also inadvertently sustain progression of AD.
This hypothesis predicts disorders of both inflammation (both peripheral inflammation and neuroinflammation) and Reelin dysregulation (including disorders associated with upregulated Reelin expression and disorders of Reelin downregulation) would be associated with elevated risk of developing AD. Some research indicates inflammatory diseases altogether are not associated with increased AD risk, suggesting an additional factor is involved in increased risk of AD in specific inflammatory diseases [180]. Disruption of Reelin homeostasis could be an important secondary factor as a variety of disorders of both Reelin dysregulation and inflammation are associated with elevated AD risk including depression, epilepsy, autism, and multiple sclerosis [81, 117, 181–191]. Menopause, a midlife period of neuroendocrine alterations manifesting in reproductive senescence, is also associated with both significant AD risk and inflammatory alterations [192–195]. Reelin expression is coregulated with estrogen in ovarian follicles, with highest levels of Reelin co-occurring with the highest expression of estradiol, suggesting loss of estrus cycles could alter Reelin production, impacting the elevated risk for development of AD [196]. Although in healthy aging plasma Reelin levels are not affected by sex or age on the group level, longitudinal data and association with future disease risk should be evaluated to address whether individuals with transient dysregulation of Reelin during menopause are at greater risk of developing AD [24].
Temporal lobe epilepsy, which is associated with decreased RELN RNA, has also been linked to both heightened inflammation and elevated AD risk [116, 185, 186, 197–199]. Pathogenic variants of the Reelin gene are seen in roughly 1/5th of families with autosomal dominant lateral temporal epilepsy, and depletion of Reelin has been shown experimentally in rodents following long-term kindling, which induces seizures and an epilepsy-like state through repeated electrical stimulation of epileptogenic brain regions [185, 186, 200]. Autism spectrum disorder (ASD) is marked by both inflammation and RELN methylation, is frequently associated with RELN polymorphisms and is a condition associated with elevated risk of developing AD [181, 182, 201]. Relapsing remitting multiple sclerosis (MS) is another inflammatory disorder associated with downregulated RELN expression and a recent epidemiological study showed a 2-fold increased risk of AD in MS [117, 185, 202].
As mentioned above, COVID-19 has been linked to Reelin dysregulation and COVID-19 infection is associated with worsened AD pathology and elevated risk of developing AD, although further epidemiological evidence is still required to validate this association [23, 203–206]. The role of Reelin dysregulation in COVID-19 and AD risk also requires further evaluation. Reelin dysregulation was also described above in the context of experimental colitis, and some experimental and epidemiological evidence shows colitis is linked to inflammation and AD risk, although connections between colitis and elevated AD risk are not consistent across epidemiological studies and may be modifiable with treatment [207–209]. Although Reelin dysregulation in these disorders is not clearly linked to altered risk of AD, the numerous conditions linked to both inflammation and Reelin dysregulation associated with increased risk for developing AD warrants investigation into the potential causal role(s) of Reelin dysregulation in the development of AD, particularly in the context of these disorders and other disorders where both inflammation and Reelin dysregulation are observed. Given the numerous lines of evidence showing dysregulation of Reelin cleavage in AD and the direct connection between Reelin signaling in the hallmark pathological features of AD, evaluation of Reelin therapeutics in the context of these disorders is warranted to evaluate whether treatments that restore Reelin homeostasis influence risk of developingAD.
CONCLUSIONS
As we learn of the causal factors responsible for AD pathology, multiple lines of evidence suggest dysregulation of Reelin homeostasis and inflammation are key to development of the pathological features of AD. Recent evidence suggests inflammation, which has widely been recognized for contributing to AD, disrupts Reelin homeostasis. Improving our understanding of how Reelin homeostasis is affected by inflammation, and whether Reelin therapeutics that restore Reelin homeostasis can regulate inflammation and restore microglia function, could lead to the development of novel treatments for AD. Further research is warranted into the mechanisms responsible for altered Reelin signaling in AD and the evaluation of Reelin-based therapeutics should continue in the context of preclinical models of AD.
AUTHOR CONTRIBUTIONS
Brady S. Reive (Conceptualization; Writing – original draft; Writing – review & editing); Victor Lau (Visualization; Writing – review & editing); Carla Sánchez-Lafuente (Visualization; Writing – review & editing); Alex Henri-Bhargava (Writing – review & editing); Lisa E. Kalynchuk (Funding acquisition; Supervision; Writing – review & editing); Marie-Eve Tremblay (Conceptualization; Funding acquisition; Supervision; Writing – review & editing); Hector J. Caruncho (Conceptualization; Funding acquisition; Supervision; Writing – review & editing).
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
We would like to thank the financial support from NSERC, CIHR, CGS-M and Dr. Tremblay and Dr. Caruncho’s Canada Research Chairs which helps to make this research possible.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Thies W , Bleiler L ((2012) ) 2012 Alzheimer’s disease facts and figures, Alzheimer’s Association. Alzheimers Dement 8: , 131–168. |
[2] | Lyketsos CG , Sheppard J-ME , Steinberg M , Tschanz JA , Norton MC , Steffens DC , Breitner JCS ((2001) ) Neuropsychiatric disturbance in Alzheimer’s disease clusters into three groups: The Cache County study. Int J Geriatr Psychiatry 16: , 1043–1053. |
[3] | Livingston G , Sommerlad A , Orgeta V , Costafreda SG , Huntley J , Ames D , Ballard C , Banerjee S , Burns A , Cohen-Mansfield J , Cooper C , Fox N , Gitlin LN , Howard R , Kales HC , Larson EB , Ritchie K , Rockwood K , Sampson EL , Samus Q , Schneider LS , Selbaek G , Teri L , Mukadam N ((2017) ) Dementia prevention, intervention, and care. Lancet 390: , 2673–2734. |
[4] | McDade E , Cummings JL , Dhadda S , Swanson CJ , Reyderman L , Kanekiyo A , Irizarry M , Kramer LD , Bateman RJ ((2022) ) Lecanemab in patients with early Alzheimer’s disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res Ther 14: , 191. |
[5] | van Dyck CH , Swanson CJ , Aisen P , Bateman RJ , Chen C , Gee M , Kanekiyo M , Li D , Reyderman L , Cohen S , Froelich L , Katayama S , Sabbagh M , Vellas B , Watson D , Dhadda S , Irizarry M , Kramer LD , Iwatsubo T ((2023) ) Lecanemab in early Alzheimer’s disease. N Engl J Med 388: , 9–21. |
[6] | Kishi T , Matsunaga S , Oya K , Nomura I , Ikuta T , Iwata N ((2017) ) Memantine for Alzheimer’s disease: An updated systematic review and meta-analysis. J Alzheimers Dis 60: , 401–425. |
[7] | Herring A , Donath A , Steiner KM , Widera MP , Hamzehian S , Kanakis D , Kolble K , ElAli A , Hermann DM , Paulus W , Keyvani K ((2012) ) Reelin depletion is an early phenomenon of Alzheimer’s pathology. J Alzheimers Dis 30: , 963–979. |
[8] | Sáez-Valero J , Costell M , Sjögren M , Andreasen N , Blennow K , Luque JM ((2003) ) Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J Neurosci Res 72: , 132–136. |
[9] | Lopez-Font I , Lennol MP , Iborra-Lazaro G , Zetterberg H , Blennow K , Saez-Valero J ((2022) ) Altered balance of Reelin proteolytic fragments in the cerebrospinal fluid of Alzheimer’s disease patients. Int J Mol Sci 23: , 7522. |
[10] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Comm 120: , 885–890. |
[11] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[12] | Zheng H , Koo EH ((2011) ) Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegen 6: , 27. |
[13] | Guo T , Noble W , Hanger DP ((2017) ) Roles of tau protein in health and disease. Acta Neuropathol 133: , 665–704. |
[14] | Muralidar S , Ambi SV , Sekaran S , Thirumalai D , Palaniappan B ((2020) ) Role of tau protein in Alzheimer’s disease: The prime pathological player. Int J Bio Mol 163: , 1599–1617. |
[15] | Hoe H-S , Lee KJ , Carney RSE , Lee J , Markova A , Lee J-Y , Howell BW , Hyman BT , Pak DTS , Bu G , Rebeck GW ((2009) ) Interaction of Reelin with amyloid protein promotes neurite outgrowth. J Neurosci 29: , 7459–7473. |
[16] | Lane-Donovan C , Philips GT , Wasser CR , Durakoglugil MS , Masiulis I , Upadhaya A , Pohlkamp T , Coskun C , Kotti T , Steller L , Hammer RE , Frotscher M , Bock HH , Herz J ((2015) ) Reelin protects against amyloid β toxicity in vivo. Sci Signal 8: , ra67. |
[17] | Pujadas L , Rossi D , Andrés R , Teixeira CM , Serra-Vidal B , Parcerisas A , Maldonado R , Giralt E , Carulla N , Soriano E ((2014) ) Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat Comm 5: , 3443. |
[18] | Kocherhans S , Madhusudan A , Doehner J , Breu KS , Nitsch RM , Fritschy J-M , Knuesel I ((2010) ) Reduced Reelin expression accelerates amyloid-β plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J Neurosci 30: , 9228–9240. |
[19] | Rossi D , Gruart A , Contreras-Murillo G , Muhaisen A , Avila J , Delgado-Garcia JM , Pujadas L , Soriano E ((2019) ) Reelin reverts biochemical, physiological and cognitive alterations in mouse models of tauopathy. Prog Neurobiol 186: , 101743. |
[20] | Chin J , Massaro CM , Palop JJ , Thwin MT , Yu G-Q , Bien-Ly N , Bender A , Mucke L ((2007) ) Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J Neurosci 27: , 2727–2733. |
[21] | Depino AM ((2015) ) Early prenatal exposure to LPS results in anxiety- and depression-related behaviors in adulthood. Neuroscience 299: , 56–65. |
[22] | Heneka MT , Carson MJ , Khoury JE , Landreth GE , Brosseron F, Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[23] | Calvier L , Drelich A , Hsu J , Tseng C-T , Mina Y , Nath A , Kounnas MZ , Herz J ((2023) ) Circulating Reelin promotes inflammation and modulates disease activity in acute and long COVID-19 cases. Front Immunol 14: , 1185748. |
[24] | Calvier L , Alexander A , Marckx AT , Kounnas MZ , Durakoglugil M , Herz J ((2024) ) Safety of anti-Reelin therapeutic approaches for chronic inflammatory diseases. Cells 13: , 583. |
[25] | Oronsky B , Caroen S , Reid T ((2022) ) What exactly is inflammation (and what is it not?). Int J Mol Sci 23: , 14905. |
[26] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WST , Hampel H , Hull M , Landreth G , Lue L-F , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2014) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[27] | Krstic D , Madhusudan A , Doehner J , Vogel P , Notter T , Imhof C , Manalastas A , Hilfiker M , Pfister S , Schwerdel C , Riether C , Meyer U , Knuesel I ((2012) ) Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation 9: , 151. |
[28] | Hoozemans JJM , Veerhuis R , Rozemuller JM , Eikelenboom P ((2006) ) Neuroinflammation and regeneration in the early stages of Alzheimer’s disease pathology. Int J Dev Neurosci 24: , 157–165. |
[29] | Hoffmeister L , Diekmann M , Brand K , Huber R ((2020) ) GSK3: A kinase balancing promotion and resolution of inflammation. Cells 9: , 820. |
[30] | He X , Li Y , Deng B , Lin A , Zhang G , Ma M , Wang Y , Yang Y , Kang X ((2022) ) The PI3K/AKT signaling pathway in inflammation, cell death and glial scar formation after traumatic spinal cord injury: Mechanisms and therapeutic opportunities. Cell Prolif 55: , e13275. |
[31] | Razani E , Pourbagheri-Sigaroodi A , Safaroghli-Azar A , Zoghi A , Shanaki-Bavarsad M , Bashash D ((2021) ) The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 26: , 871–887. |
[32] | Lauretti E , Dincer O , Pratico D ((2020) ) Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res 1867: , e118664. |
[33] | Hooper C , Killick R , Lovestone S ((2007) ) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104: , 1433–1439. |
[34] | Mann KM , Thorngate FE , Katoh-Fukui Y , Hamanaka H , Williams DL , Fujita S , Lamb BT ((2004) ) Independent effects of APOE on cholesterol metabolism and brain Aβ levels in an Alzheimer disease mouse model. Hum Mol Genet 13: , 1959–1968. |
[35] | Tiraboschi P , Hansen LA , Masliah E , Alford M , Thal LJ , Corey-Bloom J ((2004) ) Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62: , 1977–1983. |
[36] | Ophir G , Amariglio N , Jacob-Hirsch J , Elkon R , Rechavi G , Michaelson DM ((2005) ) Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol Dis 20: , 709–718. |
[37] | Liu C-C , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[38] | Avramopoulos D ((2009) ) Genetics of Alzheimer’s disease: Recent advances. Genome Med 1: , 34. |
[39] | Lane-Donovan C , Herz J ((2017) ) The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J Lipid Res 58: , 1036–1043. |
[40] | Marottoli FM , Katsumata Y , Koster KP , Thomas R , Fardo DW , Tai LM ((2017) ) Peripheral inflammation, Apolipoprotein E4, and amyloid-β Interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro 9: , 1759091417719201. |
[41] | Wolf SA , Boddeke HWGM , Kettenmann H ((2017) ) Microglia in physiology and disease. Ann Rev Physiol 79: , 619–643. |
[42] | Dwir D , Giangreco B , Xin L , Tenenbaum L , Cabungcal J-H , Steullet P , Goupil A , Cleusix M , Jenni R , Chtarto A , Baumann PS , Klauser P , Conus P , Tirouvanziam R , Cuenod M , Do KQ ((2019) ) MMP9/RAGE pathway overactivation mediates redox dysregulation and neuroinflammation, leading to inhibitory/excitatory imbalance: A reverse translation study in schizophrenia patients. Mol Psychiatry 25: , 2889–2904. |
[43] | Giovannoni F , Quintana FJ ((2020) ) The role of astrocytes in CNS inflammation. Trends Immunol 41: , 805–819. |
[44] | Rempe RG , Hartz AMS , Bauer B ((2016) ) Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab 36: , 1481–1507. |
[45] | Ruan Z , Zhang D , Huang R , Sun W , Hou L , Zhao J , Wang Q ((2022) ) Microglial activation damages dopaminergic neurons through MMP-2/MMP-9-Mediated increase of blood-brain barrier permeability in a Parkinson’s disease mouse model. Int J Mol Sci 23: , 2793. |
[46] | Konnecke H , Bechmann I ((2013) ) The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clin Dev Immunol 2013: , 914104. |
[47] | Krstic D , Rodriguez M , Knuesel I ((2012) ) Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5 and their modulators. PloS One 7: , e47793. |
[48] | Blau CW , Cowley TR , O’Sullivan J , Grehan B , Browne TC , Kelly L , Birch A , Murphy N , Kelly AM , Kerskens CM , Lynch MA ((2012) ) The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol Aging 33: , 1005.e23–1005.e35. |
[49] | McManus RM , Higgins SC , Mills KHG , Lynch MA ((2014) ) Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging 35: , 109–121. |
[50] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood-brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders. Nat Rev Neurol 14: , 133–150. |
[51] | Welcome MO , Mastorakis NE ((2020) ) Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol Res 157: , 104769. |
[52] | Mietelska-Porowska A , Wojda U ((2017) ) T Lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer’s disease: Potential pools of new biomarkers. J Immunol Res 2017: , 4626540. |
[53] | Milner R , Campbell IL ((2003) ) The extracellular matrix and cytokines regulate microglial integrin expression and expression. J Immunol 170: , 3850–3858. |
[54] | Kreisel T , Frank MG , Licht T , Reshef R , Ben-Menachem-Zidon O , Baratta MV , Maier SF , Yirmiya R ((2013) ) Dynamic microglial alterations underlie stress-induced depressive-like behaviour and suppressed neurogenesis. Mol Psychiatry 19: , 699–709. |
[55] | Lau V , Ramer L , Tremblay ME ((2023) ) An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Comm 14: , 1670. |
[56] | Lin Y-F , Wang L-Y , Chen C-S , Li C-C , Hsiao Y-H ((2021) ) Cellular senescence as a driver of cognitive decline triggered by chronic unpredictable stress. Neurobiol Stress 15: , 100341. |
[57] | Park M-J , Park H-S , You M-J , Yoo J , Kim SH , Kwon M-S ((2018) ) Dexamethasone induces a specific form of ramified dysfunctional microglia. Mol Neurobiol 56: , 1421–1436. |
[58] | Tay TL , Savage JC , Hui CW , Bisht K , Tremblay ME ((2017) ) Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J Physiol 595: , 1929–1945. |
[59] | Brelstaff JH , Mason M , Katsinelos T , McEwan WA , Ghetti B , Tolkovsky AM , Spillantini MG ((2021) ) Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv 7: , eabg4980. |
[60] | Browne TC , McQuillan K , McManus RM , O’Reilly J-A. Mills KHG , Lynch MA ((2013) ) IFN-γ production by amyloid-β-specific Th1 cells promotes microglia activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol 190: , 2241–2251. |
[61] | Ferretti MT , Merlini M , Späni C , Gericke C , Schweizer N , Enzmann G , Engelhardt B , Kulic L , Suter T , Nitsch RM ((2016) ) T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav Imm 54: , 211–225. |
[62] | Minogue AM , Jones RS , Kelly RJ , McDonald CL , Connor TJ , Lynch MA ((2014) ) Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol Aging 35: , 1442–1452. |
[63] | Franceschi C , Campisi J ((2014) ) Chronic Inflammation (Inflammaging) and its potential contribution to age-associated diseases. J Gerontol 69: , 4–9. |
[64] | Wang F , Zhang Z-Z , Cao L , Yang Q-G , Lu Q-F , Chen G-H ((2020) ) Lipopolysaccharide exposure during late embryogenesis triggers and drives Alzheimer-like behavioural and neuropathological changes in CD-1 mice. Brain Behav 10: , e01546. |
[65] | Weintraub MK , Kranjac D , Eimerbrink MJ , Pearson SJ , Vinson BT , Patel J , Summers WM , Parnell TB , Boehm GW , Chumley MJ ((2014) ) Peripheral administration of poly I:C leads to increased hippocampal amyloid-beta and cognitive deficits in a non-transgenic mouse. Behav Brain Res 266: , 183–187. |
[66] | Yu Z-Y , Chen D-W , Tan C-R , Zeng G-H , He CY , Wang J , Bu X-L , Wang YJ ((2022) ) Physiological clearance of Aβ by spleen and splenectomy aggravates Alzheimer-type pathogenesis. Aging Cell 21: , e13533. |
[67] | Drechsler S , Zipperle J , Rademann P , Jafarmadar M , Klotz A , Bahrami S , Osuchowski MF ((2018) ) Splenectomy modulates early immune-inflammatory responses to trauma-hemorrhage and protects mice against secondary sepsis. Sci Rep 8: , 14890. |
[68] | Cao C , Arendash GW , Dickson A , Mamcarz MB , Lin X , Ethell DW ((2009) ) Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiol Dis 34: , 63–70. |
[69] | Dev SI , Moore RC , Soontornniyomkij B , Achim CL , Jeste DV , Eyler LT ((2016) ) Peripheral inflammation related to lower fMRI activation during a working memory task and resting functional connectivity among older adults: A preliminary study. Int J Geriatr Psychiatry 32: , 341–349. |
[70] | Shen H , Guan Q , Zhang X , Yuan C , Tan Z , Zhai L , Hao Y , Gu Y , Han C ((2020) ) New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry 100: , 109884. |
[71] | Cattaneo A , Cattane N , Galluzzi S , Provasi S , Lopizzo N , Festari C , Ferrari C , Guerra UP , Paghera B , Muscio C , Bianchetti A , Volta GD , Turla M , Cotelli MS , Gennuso M , Prelle A , Zanetti O , Lussignoli G , Mirabile D , Bellandi D , Gentile S , Belotti G , Villani D , Harach T , Bolmont T , Padovani A , Boccardi M , Frisoni GB ((2017) ) Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 49: , 60–68. |
[72] | Shannon OM , Ranson JM , Gregory S , Macpherson H , Milte C , Lentjes M , Mulligan A , McEvoy C , Griffiths A , Matu J , Hill TR , Adamson A , Siervo M , Minihane AM , Muniz-Tererra G , Ritchie C , Mathers JC , Llewellyn DJ , Stevenson E ((2023) ) Mediterranean diet adherence is associated with lower dementia risk, independent of genetic predisposition: Findings from the UK Biobank prospective cohort study. BMC Med 21: , 81. |
[73] | Chrysohoou C , Panagiotakos DB , Pitsavos C , Das UN , Stefanadis C ((2004) ) Adherence to the Mediterranean diet attenuates inflammation and coagulation process in health adults: The Attica study. J Am Coll Cardiol 44: , 152–158. |
[74] | Scarmeas N , Stern Y , Tang M-X , Mayeux R , Luchsinger JA ((2011) ) Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol 59: , 912–921. |
[75] | Tsigalou C , Konstantinidis T , Paraschaki A , Stavropoulou E , Voidarou C , Bezirtzoglou E ((2020) ) Mediterranean diet as a tool to combat inflammation and chronic diseases: An overview. Biomedicines 8: , 201. |
[76] | Ballarini T , van Lent DM , Brunner J , Schroder A , Wolfsgrubber S , Altenstein S , Brosseron F , Buerger K , Dechent P , Dobisch L , Duzel E , Ertl-Wagner B , Fliessbach K , Freiesleben SD , Frommann I , Glanz W , Hauser D , Haynes JD , Heneka MT , Janowitz D , Killmann I , Laske C , Maier F , Metzger CD , Munk MH , Perneczky R , Peters O , Priller J , Ramirez A , Rauchmann B-S , Roy N , Scheffler K , Schneider A , Spottke A , Spruth EJ , Teipel SJ , Vukovich R , Wiltfang J , Jessen F , Wagner M ((2021) ) Mediterranean diet, Alzheimer disease biomarkers, and brain atrophy in old age. Neurology 96: , e2920–e2932. |
[77] | Ogino H , Nakajima T , Hirota Y , Toriuchi H , Aoyama M , Nakajima K , Hattori M ((2020) ) The secreted glycoprotein Reelin suppresses the proliferation and regulates the distribution of oligodendrocyte progenitor cells in the embryonic neocortex. J Neurosci 40: , 7625–7636. |
[78] | Leemhuis J , Bouché E , Frotscher M , Henle F , Hein L , Herz J , Meyer DK , Pichler M , Roth G , Schwan C , Bock HH ((2010) ) Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. J Neurosci 30: , 14759–14772. |
[79] | Utsunomiya-Tate N , Kubo K-I , Tate S-I , Kainosho M , Katayama E , Nakajima K , Mikoshiba K ((2000) ) Reelin molecules assemble together to form a large protein complex, which is inhibited by the function-blocking CR-50 antibdoy. Proc Natl Acad Sci U S A 97: , 9729–9734. |
[80] | Fatemi SH , Earle JA , McMenomy T ((2000) ) Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry 5: , 654–663. |
[81] | Ishii K , Kubo KI , Nakajima K ((2016) ) Reelin and neuropsychiatric disorders. Front Cell Neurosci 10: , 229. |
[82] | Hornig T , Haas C , Sturm L , Fiebich B , van Elst LT ((2015) ) Increased blood-reelin-levels in first episode schizophrenia. PloS One 10: , e0142247. |
[83] | Botella-López A , Burgaya F , Gavín R , García-Ayllón MS , Gomez-Tortosa E , Pena-Casanova J , Urena JM , Del Rio JA , Blesa R , Soriano E , Saez-Valero J ((2006) ) Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5573–5578. |
[84] | D’Arcangelo G , Miao GG , Chen S-C , Scares HD , Morgan JI , Curran T ((1995) ) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374: , 719–723. |
[85] | Khialeeva E , Carpenter EM ((2016) ) Nonneuronal roles for the reelin signaling pathway. Dev Dyn 246: , 217–226. |
[86] | Perez-Costas E , Fenton EY , Caruncho HJ ((2015) ) Reelin expression in brain endothelial cells: An electron microscopy study. BMC Neurosci 16: , 16. |
[87] | Wasser CR , Herz J ((2017) ) Reelin: Neurodevelopmental architect and homeostatic regulator of excitatory synapses. J Biol Chem 292: , 1330–1338. |
[88] | Jossin Y ((2020) ) Reelin functions, mechanisms of action and signaling pathways during brain development and maturation. Biomolecules 10: , 964. |
[89] | D’Arcangelo G , Homayouni R , Keshvara L , Rice DS , Sheldon M , Curran T ((1999) ) Reelin is a ligand for lipoprotein receptors. Neuron 24: , 471–479. |
[90] | Dulabon L , Olson EC , Taglienti MG , Eisenhuth S , McGrath B , Walsh CA , Kreidberg JA , Anton ES ((2000) ) Reelin binds α3β1 integrin and inhibits neuronal migration. Neuron 27: , 33–44. |
[91] | Hiesberger T , Trommsdorff M , Howell BW , Goffinet A , Mumby MC , Cooper JA , Herz J ((1999) ) Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of Disabled-1 and modulates tau phosphorylation. Neuron 24: , 481–489. |
[92] | Bock HH , Herz J ((2003) ) Reelin activates SRC family tyrosine kinases in neurons. Curr Biol 13: , 18–26. |
[93] | Chen Y , Beffert U , Ertunc M , Tang T-S , Kavalali ET , Bezprozvanny I , Herz J ((2005) ) Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci 25: , 8209–8216. |
[94] | Qui S , Zhao LF , Korwek KM , Weeber EJ ((2006) ) Differential Reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci 26: , 12943–12955. |
[95] | Groc L , Choquet D , Stephenson A , Verrier D , Manzoni OJ , Chavis P ((2007) ) NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J Neurosci 27: , 10165–10175. |
[96] | Weeber EJ , Beffert U , Jones C , Christian JM , Forster E , Sweatt JD , Herz J ((2002) ) Reelin and apoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277: , 39944–39952. |
[97] | Chai X , Förster E , Zhao S , Bock HH , Frotscher M ((2009) ) Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine. J Neurosci 29: , 288–299. |
[98] | Rogers JT , Rusiana I , Trotter J , Zhao L , Donaldson E , Pak DTS , Babus LW , Peters M , Banko JL , Chavis P , Rebeck GW , Hoe H-S , Weeber EJ ((2011) ) Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn Mem 18: , 558–564. |
[99] | Sattler R , Xiong Z , Lu W-Y , MacDonald JF , Tymianski M ((2000) ) Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci 20: , 22–33. |
[100] | Dal Pozzo V , Crowell B , Briski N , Crockett DP , D’Arcangelo G ((2020) ) Reduced Reelin expression in the hippocampus after traumatic brain injury. Biomolecules 10: , 975. |
[101] | Dominguez E , Chin T-Y , Chen C-P , Wu T-Y ((2011) ) Management of moderate to severe Alzheimer’s disease: Focus on memantine. Taiwan J Obstet Gynecol 50: , 415–423. |
[102] | Kutzing MK , Luo V , Firestein BL ((2012) ) Protection from glutamate-induced excitotoxicity by memantine. Ann Biomed Eng 40: , 1170–1181. |
[103] | Xia P , Chen H-S , Zhang D , Lipton SA ((2010) ) Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor current in hippocampal autapses. J Neurosci 30: , 11246–11250. |
[104] | Romay-Tallon R , Rivera-Baltanas T , Kalynchuk LE , Caruncho HJ ((2015) ) Differential effects of corticosterone on the colocalization of reelin and neuronal nitric oxide synthase in the adult hippocampus in wild type and heterozygous reeler mice. Brain Res 1594: , 274–283. |
[105] | Beffert U , Morfini G , Bock HH , Reyna H , Brady ST , Herz J ((2002) ) Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3β. J Biol Chem 277: , 49958–49964. |
[106] | Toral-Rios D , Pichardo-Rojas PS , Alonso-Vanegas M , Campos-Pena V ((2020) ) GSK3β and tau protein in Alzheimer’s disease and epilepsy. Front Cell Neurosci 14: , 19. |
[107] | Jossin Y , Goffinet AM ((2007) ) Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and mTor to regulate dendritic growth. Mol Cell Biol 27: , 7113–7124. |
[108] | Mebratu Y , Tesfaigzi Y ((2009) ) How ERK1/2 activation controls cell proliferation and cell death is localization the answer? Cell Cycle 8: , 1168–1175. |
[109] | Panwar V , Singh A , Bhatt M , Tonk RK , Azizov S , Raza AS , Sengupta S , Kumar D , Garg M ((2023) ) Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther 8: , 375. |
[110] | Kherzi MR , Yousefi K , Esmaeili A , Ghasemnejad-Berenji M ((2022) ) The role of ERK1/2 pathway in the pathophysiology of Alzheimer’s disease: An overview and update on new developments. Cell Mol Neurobiol 43: , 177–191. |
[111] | Tramutola A , Triplett JC , Domenico F , Niedowicz DM , Murphy MP , Coccia R , Perluigi M , Butterfield DA ((2015) ) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133: , 739–749. |
[112] | Li X , Alafuzoff I , Soininen H , Winblad B , Pei JJ ((2005) ) Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J 272: , 4211–4220. |
[113] | Caccamo A , Magrì A , Medina DX , Wisely EV , Lopez-Aranda MF , Silva AJ , Oddo S ((2013) ) mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 12: , 370–380. |
[114] | Bock HH , May P ((2016) ) Canonical and non-canonical Reelin signaling. Front Cell Neurosci 10: , 166. |
[115] | Machado RA , Benjumea-Cuartas V , Berruecos JFZ , Agudelo-Flores PM , Salazar-Pelaez LM ((2019) ) Reelin, tau phosphorylation and psychiatric complications in patients with hippocampal sclerosis and structural abnormalities in temporal lobe epilepsy. Epilepsy Behav 96: , 192–199. |
[116] | Kobow K , Jeske I , Hildebrandt M , Hauke J , hahnen E , Buslei R , Buchfelder M , Weigel D , Stefan H , Kasper B , Pauli E , Blumcke I ((2009) ) Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol 68: , 356–364. |
[117] | Talebian S , Gharsesouran J , Ghafouri-Fard S , Esfahani BS , Arsang-Jang S , Omrani MD , Taheri M , Rezazadeh M ((2019) ) Assessment of expression of RELN signaling pathway in multiple sclerosis patients. Immunobiology 224: , 402–407. |
[118] | Buret L , van den Buuse M ((2014) ) Corticosterone treatment during adolescence induces down-regulation of reelin and NMDA receptor subunit GLUN2C expression only in male mice: Implications for schizophrenia. Int J Neuropsychopharmacol 17: , 1221–1232. |
[119] | Guy J , Staiger JF ((2017) ) The functioning of a cortex without layers. Front Neuroanat 11: , 54. |
[120] | D’Arcangelo G ((2005) ) The Reeler mouse: Anatomy of a mutant. Int Rev Neurobiol 71: , 383–417. |
[121] | Salinger WL , Ladrow P , Wheeler C ((2003) ) Behavioral phenotype of the Reeler mutant mouse: Effects of Reln gene dosage and social isolation. Behav Neurosci 117: , 1257–1275. |
[122] | Larson J , Hoffman JS , Guidotti A , Costa E ((2003) ) Olfactory discrimination learning deficit in heterozygous reeler mice. Brain Res 971: , 40–46. |
[123] | Brigman JL , Padukiewicz KE , Sutherland ML , Rothblat LA ((2006) ) Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav Neurosci 120: , 984–988. |
[124] | Krueger DD , Howell JL , Hebert BF , Olausson P , Taylor JR , Nairn AC ((2006) ) Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology 189: , 95–104. |
[125] | Hethorn WR , Ciarlone SL , Filonova I , Rogers JT , Aguirre D , Ramirez RA , Grieco JC , Peters MM , Gulick D , Anderson AE , Banko JL , Lussier AL , Weeber EJ ((2015) ) Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur J Neurosci 41: , 1372–1380. |
[126] | Dalley JW , Cardinal RN , Robbins TW ((2004) ) Prefrontal executive and cognitive functions in rodents: Neural and neurochemical substrates. Neurosci Biobehav Rev 28: , 771–784. |
[127] | Rogers JT , Zhao L , Trotter JH , Rusiana I , Peters MM , Li Q , Donaldson E , Banko JL , Keenoy KE , Rebeck GW , Hoe H-S , D’Arcangelo G , Weeber EJ ((2013) ) Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J Psychopharmacol 27: , 386–395. |
[128] | Morrill NK , Qingyou L , Joly-Amado E , Weeber EJ , Nash KR ((2023) ) A novel Reelin construct, R36, recovered behavioural deficits in the heterozygous reeler mouse. Euro J Neurosci 57: , 1657–1670. |
[129] | Allen J , Romay-Tallon R , Michell MA , Brymer KJ , Johnston J , Sanchez-Lafuente CL , Pinna G , Kalynchuk LE , Caruncho HJ ((2022) ) Reelin has antidepressant-like effects after repeated or singular peripheral injections. Neuropharmacology 211: , 109043. |
[130] | Brymer KJ , Johnston J , Botterill JJ , Romay-Tallon R , Mitchell MA , Allen J , Pinna G , Caruncho HJ , Kalynchuk LE ((2020) ) Fast-acting antidepressant-like effects of Reelin evaluated in the repeated-corticosterone chronic stress paradigm. Neuropsychopharmacology 45: , 1707–1716. |
[131] | Reive BS , Johnston J , Sanchez-Lafuente CL , Scheil K , Kurz K , Kalynchuk LE , Caruncho HJ ((2024) ) Intravenous Reelin rescues despair-like behaviour, Reelin cells in the dentate sub-granular zone, and spleen atrophy in the cyclic corticosterone model of recurring depressive episodes. Front Pharmacol 15: , 1368620. |
[132] | Johnston JN , Allen J , Shkolnikov I , Sanchez-Lafuente CL , Reive BS , Scheil K , Liang S , Christie BR , Kalynchuk LE , Caruncho HJ ((2023) ) Reelin rescues behavioural, electrophysiological, and molecular metrics of a chronic stress phenotype in a similar manner to ketamine. eNeuro 10: , eneuro.0106-23.2023. |
[133] | Lopera F , Marino C , Chandrahas AS , O’Hare M , Villalba-Moreno ND , Aguillon D , Baena A , Sanchez JS , Vila-Castelar C , Gomez LR , Chmielewska N , Oliveira GM , Littau JL , Hartmann K , Park K , Krasemann S , Glatzel M , Schoemaker D , Gonzalez-Buendia L , Delgado-Tirado S , Arevalo-Alquichire S , Saez-Torres KL , Amarnani D , Kim LA , Mazzarino RC , Gordon H , Bocanegra Y , Villegas A , Gai X , Bootwalla M , Ji J , Shen L , Kosik KS , Su Y , Chen Y , Schults A , Sperling RA , Johnson K , Reiman EM , Sepulveda-Falla , Arboleda-Velasquez JF , Quiroz YT ((2023) ) Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat Med 29: , 1243–1252. |
[134] | Mathys H , Peng Z , Boix CA , Victor MB , Leary N , Babu S , Abdelhady G , Jiang X , Ng AP , Ghafari K , Kunisky AK , Mantero J , Galani K , Lohia VN , Fortier GE , Lotfi Y , Ivey J , Brown HP , Patel PR , Chakraborty N , Beaudway JI , Imhoff EJ , Keeler CF , McChesney MM , Patel HH , Patel SP , Thai MT , Bennet DA , Kellis M , Tsai L-H ((2023) ) Single-cell atlas reveals correlates of high cognitive function, dementia and resilience to Alzheimer’s disease pathology. Cell 186: , 4365–4385. |
[135] | Riedel A , Miettinen R , Stieler J , Mikkonen M , Alafuzoff I , Soininen H , Arendt T ((2003) ) Reelin-immunoreactive Cajal-Retzius cells: The entorhinal cortex in normal aging and Alzheimer’s disease. Acta Neuropathol 106: , 291–302. |
[136] | Hoe H-S , Tran TS , Matsuoka Y , Howell BW , Rebeck GW ((2006) ) DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem 281: , 35176–35185. |
[137] | Lussier AL , Weeber EJ , Rebeck GW ((2016) ) Reelin proteolysis affects signaling related to normal synapse function and neurodegeneration. Front Cell Neurosci 10: , 75. |
[138] | Jossin Y , Ignatova N , Hiesberger T , Herz J , Lambert de Rouvroit C , Goffinet AM ((2004) ) The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci 24: , 514–521. |
[139] | Koie M , Okumura K , Hisanaga A , Kamei T , Sasaki K , Deng M , Baba A , Kohno T , Hattori M ((2014) ) Cleavage with Reelin repeat 3 regulates the duration and range of the signaling activity of Reelin protein. J Biol Chem 289: , 12922–12930. |
[140] | Kohno S , Kohno T , Nakano Y , Suzuki K , Ishii M , Tagai H , Baba A , Hattori M ((2009) ) Mechanism and significance of specific proteolytic cleavage of Reelin. Biochem Biophys Res Commun 380: , 93–97. |
[141] | Okugawa E , Ogino H , Shigenobu T , Yamakage Y , Tsuiji H , Oishi H , Kohno T , Hattori M ((2020) ) Physiological significance of proteolytic processing of Reelin revealed by cleavage-resistant Reelin knock-in mice. Sci Rep 10: , 4471. |
[142] | Herzog C , Haun RS , Kaushal GP ((2020) ) Role of meprin metalloproteinases in cytokine processing and inflammation. Cytokine 114: , 18–25. |
[143] | Ogino H , Hisanaga A , Kohno T , Kondo T , Okumura K , Kamei T , Sato T , Asahara H , Tsuiji H , Fukata M , Hattori M ((2017) ) Secreted metalloproteinase ADAMTS-3 inactivates Reelin. J Neurosci 37: , 3181–3191. |
[144] | Yamakage Y , Kato M , Hongo A , Ogino H , Ishii K , Ishizuka T , Kamei T , Tsuiji H , Miyamoto T , Oishi H , Kohno T , Hattori M ((2019) ) A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates Reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol Cell Neurosci 100: , 103401. |
[145] | Trotter JH , Lussier AL , Psilos KE , Mahoney HL , Sponaugle AE , Hoe H-S , Rebeck GW , Weeber EJ ((2014) ) Extracellular proteolysis of Reelin by tissue plasminogen activator following synaptic potentiation. Neuroscience 274: , 299–307. |
[146] | Sato Y , Kobayshi D , Kohno T , Kidani Y , Prox J , Becker-Pauly C , Hattori M ((2015) ) Determination of cleavage site of Reelin between its sixth and seventh repeat and contribution of meprin metalloproteases to the cleavage. J Biochem 159: , 305–312. |
[147] | Hisanaga A , Morishita S , Suzuki K , Sasaki K , Koie M , Kohno T , Hattori M ((2012) ) A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett 586: , 3349–3353. |
[148] | Schonherr C , Bien J , Isbert S , Wichert R , Prox J , Altmeppen H , Kumar S , Walter J , Lichtenthaler SF , Weggen S , Glatzel M , Becker-Pauly C , Pietrzik CU ((2016) ) Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site. Mol Neurodegener 11: , 19. |
[149] | Matsuzaki M , Yokoyama M , Yoshizawa Y , Kaneko N , Naito H , Kobayashi H , Korenaga A , Sekiya S , Ikemura L , Opoku G , Hirohata S , Iwamoto S , Tanaka K , Tomita T ((2023) ) ADAMTS4 is involved in the production of the Alzheimer disease amyloid biomarker APP669-711. Mol Psychiatry 28: , 1802–1812. |
[150] | Yamakage Y , Tsuiji H , Kohno T , Ogino H , Saito T , Saido TC , Hattori M ((2019) ) Reducing ADAMTS-3 inhibits amyloid β deposition in App Knock-in mouse. Biol Pharm Bull 42: , 354–356. |
[151] | Lorenzl S , Albers DS , Relkin N , Nguyen T , Hilgenberg SL , Chirichigno J , Cudkowicz ME , Beal MF ((2003) ) Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem Int 43: , 191–196. |
[152] | Hernandes-Alejandro M , Montano S , Harrington CR , Wischik CM , Salas-Casas A , Cortes-Reynosa P , Perez-Salazar E , Cazares-Apatiga J , Apatiga-Perez R , Ontiveros-Torres MA , Perry G , Pacheco-Herrero M , Luna-Munoz J ((2020) ) Analysis of the relationship between metalloprotease-9 and tau protein in Alzheimer’s disease. J Alzheimers Dis 76: , 553–569. |
[153] | Abe K , Hattori S , Yoshimi A , Asami T , Katsuse O , Suda A , Hishimoto A ((2020) ) Influence of plasma matrix metalloproteinase levels on longitudinal changes in Alzheimer’s disease (AD) biomarkers and cognitive function in patients with mild cognitive impairment due to AD registered in the Alzheimer’s Disease Neuroimaging Initiative database. J Neurol Sci 416: , 116989. |
[154] | Cuchillo-Ibanez I , Balmaceda V , Mata-Balaguer T , Lopez-Font I , Sáez-Valero J ((2016) ) Reelin in Alzheimer’s Disease, increased levels but impaired signaling: When more is less. J Alzheimers Dis 52: , 403–416. |
[155] | Saito T , Matsuba Y , Mihira N , Takano J , Nilsson P , Itohara S , Iwata N , Saido TC ((2014) ) Single APP knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17: , 661–663. |
[156] | Wirths O , Multhaup G , Czech C , Blanchard V , Tremp G , Pradier L , Beyreuther K , Bayer TA ((2001) ) Reelin in plaques of β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 316: , 145–148. |
[157] | Knuesel I , Nyffeler M , Mormede C , Muhia M , Meyer U , Pietropaolo S , Yee BK , Pryce CR , LaFerla FM , Marighetto A , Feldon J ((2009) ) Age-related accumulation of Reelin in amyloid-deposits. Neurobiol Aging 30: , 697–716. |
[158] | Leroy K , Yilmaz Z , Brion J-P. ((2007) ) Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 33: , 43–55. |
[159] | Alexander A , Herz J , Calvier L ((2023) ) Reelin through the years: From brain development to inflammation. Cell Rep 42: , 112669. |
[160] | Micera A , Balzamino BO , Biamonte F , Esposito G , Marino R , Fanelli F , Keller F ((2016) ) Current progress of Reelin in development, inflammation and tissue remodeling: From nervous to visual systems. Curr Mol Med 16: , 620–630. |
[161] | Greenjohnson JM , Zalcman S , Vriend CY , Nance DM , Greenberg AH ((1995) ) Suppressed T cell function and macrophage function in the “Reeler” (rl/rl) mutant, a murine strain with elevated cerebellar norepinephrine concentration. Brain Behav Immun 9: , 47–60. |
[162] | Garcia-Miranda P , Vazquez-Carretero MD , Gutierrez G , Peral MJ , Ilundain AA ((2012) ) Lack of reelin modifies the genes expression in the small intestine of mice. J Physiol Biochem 68: , 205–218. |
[163] | Salem S , Salem D , Gros P ((2020) ) Role of IRF8 in immune cells functions, protection against infections and susceptibility to inflammatory diseases. Hum Genet 139: , 707–721. |
[164] | Kim HS , Sohn H , Jang SW , Lee GR ((2019) ) The transcription factor NFIL3 controls regulatory t-cell function and stability. Exp Mol Med 51: , 1015. |
[165] | Rahimi-Balaei M , Jiao X , Dalvand A , Shabanipour S , Chung SH , Amiri A , Kong J , Marzban H ((2020) ) Mutations in the Reelin pathway are associated with abnormal expression of microglial IgG FC receptors in the cerebellar cortex. Mol Biol Rep 47: , 5323–5331. |
[166] | Kobold D , Grundmann A , Piscaglia F , Eisenbach C , Neubauer K , Steffgen J , Ramadori G , Knittel T ((2002) ) Expression of reelin in hepatic stellate cells and during hepatic tissue repair: A novel marker for the differentiation of HSC from other liver myofibroblasts. J Hepatol 36: , 607–613. |
[167] | Shal B , Khan A , Khan AU , Ullah R , Ali G , Islam SU , Haq IU , Ali H , Seo E-K , Khan S ((2021) ) Alleviation of memory deficit by Bergenin via the regulation of Reelin and Nrf-2/NF-kB pathways in transgenic mouse model. Int J Mol Sci 22: , 6603. |
[168] | Carvajal AE , Vázquez-Carretero MD , García-Miranda P , Peral MJ , Calonge ML , Ilundain AA ((2017) ) Reelin expression is up regulated in mice colon in response to acute colitis and provides resistance against colitis. Biochim Biophys Acta Mol Basis Dis 1863: , 462–473. |
[169] | Meyer U , Nyffeler M , Yee BK , Knuesel I , Feldon J ((2008) ) Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun 22: , 469–486. |
[170] | Ratnayake U , Quinn TA , Castillo-Melendez M , Dickinson H , Walker DW ((2012) ) Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behav Immun 26: , 1288–1299. |
[171] | Reive BS , Johnston JN , Sánchez-Lafuente CL , Zhang L , Chang A , Zhang J , Allen J , Romay-Tallon R , Kalynchuk LE , Caruncho HJ ((2023) ) Intravenous Reelin treatment rescues atrophy of spleen white pulp and correlates to rescue of forced swim test immobility and neurochemical alterations induced by chronic stress. Chronic Stress (Thousand Oaks) 7: , 24705470231164920. |
[172] | Yeh FL , Wang Y , Tom I , Gonzalez LC , Sheng M ((2016) ) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates update of amyloid-beta by microglia. Neuron 91: , 328–340. |
[173] | Yu Z , Fang X , Liu W , Sun R , Zhou J , Pu Y , Zhao M , Sun D , Xiang Z , Liu P , Ding Y , Cao L , He C ((2022) ) Microglia regulate blood-brain barrier integrity MiR-126a-5p/MMP9 axis during inflammatory demyelination. Adv Sci 9: , e2105442. |
[174] | Zhang H , Zhao W ((2022) ) Resveratrol alleviates ischemic brain injury by inhibiting the activation of pro-inflammatory microglia via the CD147/MMP-9 pathway. J Stroke Cerebrovasc Dis 31: , 106307. |
[175] | Parachikova A , Cotman CW ((2007) ) Reduced CXCL12/CXCR4 results in impaired learning and is downregulated in a mouse model of Alzheimer disease. Neurobiol Dis 28: , 143–153. |
[176] | Laske C , Stellos K , Eschweiler G , Leyhe T , Gawaz M ((2008) ) Decreased CXCL12 (SDF-1) plasma levels in Alzheimer’s disease: A contribution to a deficient hematopoietic brain support? J Alzheimers Dis 15: , 83–95. |
[177] | Cassidy AW , Carolan D , Pickering M , Regan C ((2010) ) Cytokine mediated regulation of Reelin expression and secretion in cultured SH-SY5Y neurons. Ir J Med Sci 179: , S296. |
[178] | Christie RH , Chung H , Rebeck GW , Strickland D , Hyman BT ((1996) ) Expression of the very low-density lipoprotein receptor (VLDL-r), an Apolipoprotein-E receptor, in the central nervous system and in Alzheimer’s disease. J Neuropath Exp Neur 55: , 491–498. |
[179] | Ramsden CE , Keyes GS , Calzada E , Horowitz MS , Zamora D , Jahanipour J , Sedlock A , Indig FE , Moaddel R , Kapogiannis D , Maric D ((2022) ) Lipid peroxidation induced ApoE receptor-ligand disruption as a unifying hypothesis underlying sporadic Alzheimer’s disease in humans. J Alzheimers Dis 87: , 1251–1290. |
[180] | Booth MJ , Kobayashi LC , Janevic MR , Clauw D , Piette JD ((2021) ) No increased risk of Alzheimer’s disease among people with immune-mediated inflammatory diseases: Findings from a longitudinal cohort study of U.S. older adults. BMC Rheumatol 5: , 48. |
[181] | Serajee FJ , Zhong H , Mahbubul Huq AH ((2006) ) Association of Reelin gene polymorphisms with autism. Genomics 87: , 75–83. |
[182] | Vivanti G , Tao S , Lyall K , Robins DL , Shea LL ((2021) ) The prevalence and incidence of early-onset dementia among adults with autism spectrum disorder. Autism Res 14: , 2189–219. |
[183] | Luczynski P , Laule C , Hsiung G-Y , Moore GR , Tremlett H ((2019) ) Coexistence of multiple sclerosis and Alzheimer’s disease: A review. Mult Scler Relat Dis 27: , 232–238. |
[184] | Cho EB , Jung SY , Jung J-H , Yeo Y , Kim HJ , Han K , Shin DW , Min J-H ((2023) ) The risk of dementia in multiple sclerosis and neuromyelitis optica spectrum disorder. Front Neurosci 17: , 1214652. |
[185] | Dazzo E , Fanciulli M , Serioli E , Minervini G , Pulitano P , Binelli S , Bonaventura CD , Luisi C , Pasini E , Striano S , Striano P , Coppola G , Chiavegato A , Radovic S , Spadotto A , Uzzau S , Neve AL , Giallonardo AT , Mecarelli O , Tosatto SCE , Ottman R , Michelucci R , Nobile C ((2015) ) Heterozygous Reelin mutations cause Autosomal-Dominant Lateral Temporal Epilepsy. Am J Hum Genet 96: , 992–1000. |
[186] | Dazzo E , Nobile C ((2021) ) Epilepsy-causing Reelin mutations result in impaired secretion and intracellular degradation of mutant proteins. Hum Mol Genet 31: , 665–673. |
[187] | Li B-Y , Chen S-D. ((2015) ) Potential similarities in temporal lobe epilepsy and Alzheimer’s disease: From clinic to pathology. Am J Alzheimers Dis Other Demen 30: , 723–728. |
[188] | Song H , Sieurin J , Wirdefeldt K , Pedersen NL , Almqvist C , Larsson H , Valdimarsdottir UA , Fang F ((2020) ) Association of stress-related disorders with subsequent neurodegenerative diseases. JAMA Neurol 77: , 700–709. |
[189] | Saiz-Vazquez O , Gracia-Garcia P , Ubillos-Landa S , Puente-Martinez A , Casado-Yusta S , Olaya B , Santabarbara J ((2021) ) Depression as a risk factor for Alzheimer’s disease: A systematic review of longitudinal meta-analyses. J Clin Med 10: , 1809. |
[190] | Byers AL , Yaffe K ((2012) ) Depression and risk of developing dementia. Nat Rev Neurol 7: , 323–331. |
[191] | Richmond-Rakerd LS , D’Souza S , Milne BJ , Caspi A , Moffitt TE ((2022) ) Longitudinal associations of mental disorders with dementia 30-year analysis of 1.7 million New Zealand citizens. JAMA Psychiatry 79: , 333–340. |
[192] | Mosconi L , Rahman A , Diaz I , Wu X , Scheyer O , Hristov HW , Vallabhajosula S , Isaacson RS , de Leon MJ , Brinton RD ((2018) ) Increased Alzheimer’s risk during the menopause transition: A 3-year longitudinal brain imaging study. PloS One 13: , e0207885. |
[193] | Wium-Anderson MK , Jorgensen TSH , Halvorsen AH , Hartsteen BH , Jorgensen MB , Osler M ((2022) ) Association of hormone therapy with depression during menopause in a cohort of Danish women. JAMA Netw Open 5: , e2239491. |
[194] | Coughlan GT , Betthauser TJ , Boyle R , Koscik RL , Klinger HM , Chibnik LB , Jonaitis EM , Yau W-YW , Wenzel A , Christian BT , Gleason CE , Saelzler UG , Properzi MJ , Schultz AP , Hanseeuw BJ , Manson JE , Rentz DM , Johnson KA , Sperling R , Johnson SC , Buckley RF ((2023) ) Association of age at menopause and hormone therapy use with tau and β-amyloid positron emission tomography. JAMA Neurol 80: , 462–473. |
[195] | Malutan AM , Dan M , Nicolae C , Carmen M ((2014) ) Proinflammatory and anti-inflammatory cytokine changes related to menopause. Prz Menopauzalny 13: , 162–168. |
[196] | Meseke M , Prois F , Schmahl C , Seebo K , Kruse K , Brandt N , Fester L , Zhou L , Bender R , Rune GM ((2018) ) Reelin and aromatase cooperate in ovarian follicle development. Sci Rep 8: , 8722. |
[197] | Gaitatzis A , Carroll K , Majeed A , Sander JW ((2004) ) The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 45: , 1613–1622. |
[198] | Dai Y-J , Wu D-W , Feng B , Chen B , Tang Y-S , Jin M-M , Zhao H-W , Dai H-B , Wang Y , Chen Z ((2019) ) Prolonged febrile seizures induce inheritable memory deficits in rats through DNA methylation. CNS Neurosci Ther 25: , 601–611. |
[199] | Mukhtar I ((2020) ) Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 82: , 65–79. |
[200] | Fournier NM , Andersen DR , Botterill JJ , Sterner EY , Lussier AL , Caruncho HJ , Kalynchuk LE ((2010) ) The effect of amygdala kindling on hippocampal neurogenesis coincides with decreased reelin and DISC1 expression in the adult dentate gyrus. Hippocampus 20: , 659–671. |
[201] | Depino AM ((2013) ) Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci 53: , 69–76. |
[202] | Haase S , Linker RA ((2021) ) Inflammation in multiple sclerosis. Ther Adv Neurol Disor 14: , 17562864211007687. |
[203] | Rudnicka-Drożak E , Drożak P , Mizerski G , Zaborowski T , Ślusarska B , Nowicki G , Drożak M ((2023) ) Links between COVID-19 and Alzheimer’s disease-what do we already know? Int J Environ Res Public Health 20: , 2146. |
[204] | Naughton SX , Raval U , Pasinetti GM ((2020) ) Potential novel role of COVID-19 in Alzheimer’s disease and preventative mitigation strategies. J Alzheimers Dis 76: , 21–25. |
[205] | Golzari-Sorkheh M , Weaver DF , Reed MA ((2023) ) COVID-19 as a risk factor for Alzheimer’s disease. J Alzheimers Dis 91: , 1–23. |
[206] | Nawaz AD , Haider MZ , Akhtar S ((2024) ) COVID-19 and Alzheimer’s disease: Impact of lockdown and other restrictive measures during the COVID-19 pandemic. Biomol Biomed 24: , 219–229. |
[207] | Kaneko R , Matsui A , Watanabe M , Harada Y , Kanamori M , Awata N , Kawazoe M , Takao T , Kobayashi Y , Kikutake C , Suyama M , Saito T , Saido TC , Ito M ((2023) ) Increased neutrophils in inflammatory bowel disease accelerate the accumulation of amyloid plaques in the mouse model of Alzheimer’s disease. Inflamm Regen 43: , 20. |
[208] | Xing Y , Li P , Jia Y , Zhang K , Liu M , Jiang J ((2022) ) Association of inflammatory bowel disease and related medication exposure with risk of Alzheimer’s disease: An updated meta-analysis. Front Aging Neurosci 14: , 1082575. |
[209] | Sohrabi M , Pecoraro HL , Combs CK ((2021) ) Gut inflammation induced by dextran sulfate sodium exacerbates amyloid-β plaque deposition in the AppNL-G-F mouse model of Alzheimer’s disease. J Alzheimers Dis 79: , 1235–1255. |
[210] | Lee H-J , Park J-H , Trotter JH , Maher JN , Keenoy KE , Jang YM , Lee Y , Kim J-I , Weeber EJ , Hoe H-S ((2023) ) Reelin and APP cooperatively modulate dendritic spine formation in vitro and in vivo. Exp Biol 32: , 42–55. |
[211] | Long JM , Perez EJ , Roberts JA , Roberts MT , Rapp PR ((2020) ) Reelin in the years: Decline in the number of reelin immunoreactive neurons in layer II of the entorhinal cortex in aged monkeys with memory impairment. Neurobiol Aging 87: , 132–137. |

