
Study Partner Type and Adverse Event Reporting in Mild-to-Moderate Alzheimer’s Disease Clinical Trials
Abstract
Background:
In randomized clinical trials (RCTs), monitoring adverse events (AEs) and serious AEs (SAEs) is critical. All Alzheimer’s disease (AD) RCTs require participants to enroll with a study partner.
Objective:
We examined AE reporting rates in mild-to-moderate AD trials and their associations with study partner type.
Methods:
We estimated AE reporting rates using placebo data from seven independent RCTs conducted by the Alzheimer’s Disease Cooperative Study. We assessed the heterogeneity of reporting rates as a function of visits using generalized estimating equations. In the primary analysis, we tested the hypotheses that the rates of reporting differed by study partner type and time they spent with the participant weekly using Poisson regression with robust variance estimation. In all regression models, log-transformed total patient years was included.
Results:
The estimated reporting rates were 2.83 (95% CI: 2.66, 3.02), 1.18 (95% CI: 1.09, 1.28), 0.23 (95% CI: 0.19, 0.27), and 0.28 (95% CI: 0.24, 0.33) events per participant year for grade 1–3 AEs and SAEs, respectively. We estimated that greater number of visits per year was associated with increased reporting for grade 1–2 AEs and SAEs. We did not find evidence to suggest that AE reporting differed by study partner type or by time the study partner spent with the participant.
Conclusions:
Study partner type and time the study partner spent with the participant did not appear to impact AE reporting. Estimated reporting rates may be useful to evaluate safety in future studies, particularly those with no control arm and similar visit frequencies.
INTRODUCTION
In randomized clinical trials (RCTs), consistent monitoring of adverse events (AEs) and serious AEs (SAEs) is critical to maximizing patient safety, assessing risk-benefit, and maintaining trial integrity [1]. AEs are defined as any unfavorable changes in patient health. AEs are often recorded with Common Terminology Criteria for AEs of grade 1–5 corresponding to events that are mild, moderate, severe, potentially life threatening, and leading to death, respectively [2]. SAEs are AEs that result in hospitalization or prolonged hospital stay, are deemed life-threatening, or result in death. AE monitoring is critical to understanding and quantifying toxicity and safety of an investigated treatment, particularly in placebo-controlled trials [3]. Comparison of AE rates between treatment and control arms represents a rigorous assessment of treatment safety and is critical to assessing the risk-benefit profile of a new experimental therapy.
Alzheimer’s disease (AD) is an active area of intervention research. AD clinical trials traditionally enroll patients with dementia who experience a loss of cognitive and/or functional ability that interferes with activities of daily life. Because of this, AD clinical trials require participants to enroll with a study partner who is often the primary caregiver [4]. Study partners may support the informed consent process or may provide surrogate consent on the participant’s behalf in some cases. They also accompany participants to study visits and often oversee administration of study treatment and other medications. Study partners provide critical information on changes in patients’ health and serve as a source of study data, often including reporting AEs.
Most AD caregivers are not spouses [5]. In AD RCTs, however, the majority of enrolled participants have a spousal study partner [6]. Patients with non-spousal study partners may be less likely to meet trial eligibility criteria when compared to patients with spousal study partners [7]. Furthermore, attitudes toward participation may differ between caregiver types; for example, adult children may be less willing to participate in trials and may see their burden as caregivers as greater than spouses [8, 9]. Non-spousal caregivers are also less likely to co-reside with patient participants and are more likely to be working and caring for additional dependents. Collectively, these observations may suggest that the rate of reporting AEs in AD trials could vary by study partner type.
Within a trial, placebo AE rates are essential to understanding treatment safety. Assessing AE reporting rates among participants in the control arms of multiple trials may provide guidance and expectations for studies sharing similar features, such as the number of protocol-defined visits and study length. In this work we sought to quantify AE reporting rates in mild-to-moderate AD RCTs and associations with study partner type. We first estimated raw reporting rates for AEs of different severity and SAEs, then quantified the relationship between rate of reporting and the number of protocol-defined annualized visits across trials. In the primary and secondary analyses, we tested the hypotheses that AE reporting rates differ by study partner type and the amount of time a study partner spends with the participant.
MATERIALS AND METHODS
Data source/Included trials
We conducted a retrospective analysis of placebo data from seven mild-to-moderate AD RCTs conducted by the Alzheimer’s Disease Cooperative Study (ADCS). The included trials tested the omega-3 fatty acid docosahexaenoic acid (DHA) (ClinicalTrials.gov NCT00440050) [10], vitamin B supplementation (VITB) (ClinicalTrials.gov NCT00056225) [11], Chinese herb Huperzine (HU) (ClinicalTrials.gov NCT00083590) [12], nonsteroidal anti-inflammatory drugs (NSAIDs) rofecoxib and naproxen (ClinicalTrials.gov NCT00004845) [13], resveratrol (RES) (ClinicalTrials.gov NCT01504854) [14], simvastatin (SIM) (ClinicalTrials.gov NCT00053599) [15], and valproate (VALPO) (ClinicalTrials.gov NCT00071721) [16]. The HU trial used a 4-month protocol. The remaining trials had protocols ranging from 12 to 26 months. DHA, VITB, and VALPO had 5 protocol-specified visits per year. For SIM, NSAIDs, RES, and HU, the number of annualized visits were 4, 6, 9, and 15, respectively. Each trial was performed under independent approval by an Institutional Review Board. Participants and their study partners granted informed consent to participate and for the data collected to be used in subsequent investigations. The current analyses were performed on deidentified data and therefore do not meet the standard definition of human subjects research.
Participants
Participants in each trial were required to meet National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association’s criteria for probable AD [17], with the exception of the VALPO trial that allowed participants with probable or possible AD. The range of minimum ages for participants was 50 to 55 years. Only the VALPO trial implemented an upper age limit (90 years). Participants in the trials were permitted to have screening Mini-Mental State Examination (MMSE) scores as low as 10 and as high as 26. Standard exclusion criteria were applied in each trial. Individuals were excluded if they had recently taken drugs with significant central anticholinergic effects, sedatives, anti-Parkinsonian medications, or any investigational treatment for AD. Individuals also could not have another neurologic or psychiatric diagnosis that contributed to cognitive impairment. Some exclusion criteria of the trials were specific to their study designs. For example, participants in DHA could not be taking DHA or omega-3 fatty acid supplements. Participants in VITB could not have levels of vitamin B12 or folate below normal or renal insufficiency. In the SIM study, participants could not be taking lipid-lowering drugs. At screening, study partner type and the average number of hours per week spent with the participant were recorded. Study partners were categorized as spouse, adult child, or other.
Statistical analysis
Data from the placebo arms of the trials were combined resulting in a sample of N = 934 participants. Baseline characteristics of participants including age (years), education (years), sex as a biological variable (male versus female), and race and ethnicity were collected in all trials. In the NSAIDs trial, race and ethnicity were recorded as one variable with 6 categories: American Indian or Alaskan Native, Asian or Pacific Islander, Hispanic, non-Hispanic Black, non-Hispanic White, and Other or Unknown. For consistency, we applied this categorization for race and ethnicity for the rest of the trials by first assigning ethnicity to the participants (Hispanic or non-Hispanic), then assigning race to non-Hispanic participants. As there was sparsity in some racial and ethnic groups, we collapsed American Indian or Alaskan Native, Asian or Pacific Islander, and Other or Unknown into Other race and ethnicity category. For the primary analysis, we removed one individual in the VITB trial who had missing race and ethnicity. The analysis was therefore performed on 933 trial participants. Information on study partners of the participants was collected at screening and when there was a change in study partner after screening. Two participants changed their study partners from spousal and other at screening to adult child at baseline. Thirty-two participants had at least one change of study partner after their baseline visit. For the purpose of this analysis, we used the study partner type at baseline and did not consider study partner change thereafter. The number of hours per week the study partner spent with the participant was also collected for most of the trials. Since this variable was unavailable for the SIM trial, the secondary analysis with time spent with participant as a predictor was conducted on 732 participants from the rest of the trials. Descriptive statistics for the patients and study partners were reported as mean (standard deviation) for continuous variables and count (percent) for categorical variables and were stratified by trial.
AE reporting in the trials followed Food and Drug Administration guidance [2] with Common Terminology Criteria for AEs of grade 1–5 corresponding to events that were mild, moderate, severe, potentially life threatening, and leading to death, respectively. SAEs were defined as events that result in hospitalization or prolonged hospital stay, were deemed life-threatening, or resulted in death. Per protocol, AEs were recorded after the beginning of study enrollment. We defined the AE reporting period as enrollment until 30 days after the last visit date for those who completed the study, or until 30 days after the treatment discontinuation date for those who prematurely discontinued treatment or study. We excluded AEs or SAEs that first occurred more than 30 days post treatment or study discontinuation. Besides official records of hospitalization or death, participants were asked at each visit or follow-up whether they had experienced any changes in their health since the previous visit. We identified when a subject completed or discontinued from the study to account for the total amount of time a participant was enrolled. For a discontinued participant, we adapted the last available exam date as the date of study discontinuation. The difference between the baseline exam date and the study discontinuation date, adding 30 days post study discontinuation, yielded the total amount of days the participant was in the study (or AE reporting period) that we considered. We then computed the total participant-years of a trial by summing the total days the trial participants spent in that trial and dividing by 365.25 to convert to years.
We calculated reporting rates per participant per follow-up year for AEs of different grades and SAEs at the trial level and across trials. An unadjusted AE reporting rate per participant per year respective to an AE grade in a trial was calculated by dividing the number of events reported during the recording period for all patients by the total participant-years in the trial. Poisson regression with robust variance estimation [18] for inference was used to quantify the uncertainty of the reporting rates.
From our observations on the estimated AE reporting rates in the exploratory analysis, we further described whether the rates were higher for trials with greater numbers of protocol-defined annualized visits for different AE severity and SAE. We assessed the heterogeneity of reporting rates across the trials as a function of protocol-defined visit frequency using generalized estimation equations (GEE) with a log-link function and Poisson working mean-variance relationship with independence working correlation structure [19]. The models allowed us to assess the marginal effect of visit frequency on the incident rate ratio (IRR) for reporting AEs and SAEs given other covariates. Log-transformed total patient follow-up years was included as an offset in the models. We adjusted for study partner type and a priori specified potential confounders including participant age, years of education, sex, and race and ethnicity. Study partner type, sex, and race and ethnicity were adjusted for as categorical variables. Age and years of education were adjusted for as a continuous variable.
In the primary analysis, we aimed to quantify the association between study partner type and AE reporting rate. We used Poisson regression to assess the IRR for reporting AEs and SAEs comparing participants with non-spousal study partners to those with spousal study partners. The outcome was AE count corresponding to AE grade or SAE. Log-transformed total patient follow-up years was included as an offset. The predictor of interest was study partner type (adult child, spouse, and other). The models adjusted for a priori specified covariates including participant age, years of education, sex, and race and ethnicity. Study partner type, sex, and race and ethnicity were adjusted for as categorical variables. Age and years of education were adjusted for as a continuous variable. We also performed sub-analyses, in which we adjusted for years of education as a categorical variable (<12 years, 12 years, 12–16 years, and >16 years) for interpretability and to assess deviations from linearity in the association between years of education categories and AE reporting. We presented results from models adjusting for years of education as a continuous variable, followed by the estimates for years of education categories.
To further assess whether AE reporting rates differed by study partner type and by AE severity, we fit a Poisson regression model adjusting for study partner type, AE grade, their interaction, and previously stated potential confounding factors. The outcome in this model was the individual counts of AE grade 1–5. We performed a multivariate Wald test to simultaneously test whether the interaction was significant to address the hypothesis. In a secondary analysis to address the hypothesis that the hours per week the study partner spent with participant was associated with reporting rates for AE of different grades and SAEs, we repeated the primary analysis with time the study partner spent with the participant as the predictor of interest. We did not adjust for study partner type in these models to avoid multicollinearity between study partner type and time spent with the participant. A robust variance estimator was used in Poisson regression models to obtain valid inference [20, 21]. All analyses were performed using R version 4.1.2 for Mac OS.
RESULTS
Table 1 displays summary statistics of participant and study partner characteristics in the placebo arms of each trial. Across the studies, mean participant age ranged from 73.0 to 78.6 years. The majority of participants had 12 to 16 years of education. The majority of participants in each trial were female. Non-Hispanic White participants accounted for 82.7% to 90.9% of study participants. Mean MMSE at screening was lowest in the VALPO trial (16.80, sd 2.82), compared to a range of 19.12 to 21.25 across all remaining trials. A total of 607 (65.0%) participants enrolled with spousal, 256 (27.4%) enrolled with adult child, and 71 (7.6%) enrolled with other study partners. The hours per week a study partner spent with the participants across the seven trials was 104.09 hours on average and was the highest among participants enrolled with spousal study partners (see Supplementary Table 1).
Table 1
Characteristics of participants (N = 934), study partners, and the trials
| Trial | |||||||
| HU | RES | NSAIDs | VALPO | DHA | SIM | VITB | |
| (N = 73) | (N = 55) | (N = 111) | (N = 160) | (N = 164) | (N = 202) | (N = 169) | |
| Participants | |||||||
| Age (y) (mean (SD)) | 78.59 (8.34) | 73.00 (8.22) | 73.82 (8.02) | 76.99 (7.47) | 76.01 (7.82) | 75.15 (8.96) | 77.74 (7.91) |
| Education (y) (mean (SD)) | 13.18 (2.90) | 14.58 (2.90) | 14.32 (3.34) | 13.57 (3.54) | 14.29 (2.68) | 14.22 (3.29) | 13.87 (3.23) |
| Education (n (%)) | |||||||
| <12 y | 8 (11.0) | 2 (3.6) | 14 (12.6) | 28 (17.5) | 12 (7.3) | 23 (11.4) | 26 (15.4) |
| 12 y | 29 (39.7) | 14 (25.5) | 31 (27.9) | 48 (30.0) | 36 (22.0) | 55 (27.2) | 41 (24.3) |
| 12-16 y | 30 (41.1) | 24 (43.6) | 40 (36.0) | 59 (36.9) | 95 (57.9) | 79 (39.1) | 75 (44.4) |
| >16 y | 6 (8.2) | 15 (27.3) | 26 (23.4) | 25 (15.6) | 21 (12.8) | 45 (22.3) | 27 (16.0) |
| Sex (n (%)) | |||||||
| Female | 47 (64.4) | 28 (50.9) | 62 (55.9) | 101 (63.1) | 98 (59.8) | 121 (59.9) | 91 (53.8) |
| Male | 26 (35.6) | 27 (49.1) | 49 (44.1) | 59 (36.9) | 66 (40.2) | 81 (40.1) | 78 (46.2) |
| Race and ethnicity (n (%)) | |||||||
| Non-Hispanic White | 65 (89.0) | 50 (90.9) | 100 (90.1) | 144 (90.0) | 146 (89.0) | 174 (86.1) | 139 (82.2) |
| Non-Hispanic Black | 3 (4.1) | 3 (5.5) | 4 (3.6) | 8 (5.0) | 11 (6.7) | 9 (4.5) | 15 (8.9) |
| Hispanic | 3 (4.1) | 1 (1.8) | 7 (6.3) | 4 (2.5) | 5 (3.0) | 18 (8.9) | 9 (5.3) |
| Other | 2 (2.7) | 1 (1.8) | 0 (0.0) | 4 (2.5) | 2 (1.2) | 1 (0.5) | 5 (3.0) |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) |
| MMSE score (mean (SD)) | 19.12 (4.00) | 21.25 (3.65) | 20.81 (3.59) | 16.80 (2.82) | 20.31 (3.65) | 20.56 (4.17) | 20.91 (3.69) |
| Study partner | |||||||
| Relationship with participant (n (%)) | |||||||
| Spouse | 45 (61.6) | 43 (78.2) | 75 (67.6) | 106 (66.2) | 106 (64.6) | 127 (62.9) | 105 (62.1) |
| Adult Child | 23 (31.5) | 8 (14.5) | 27 (24.3) | 41 (25.6) | 45 (27.4) | 62 (30.7) | 50 (29.6) |
| Other | 5 (6.8) | 4 (7.3) | 9 (8.1) | 13 (8.1) | 13 (7.9) | 13 (6.4) | 14 (8.3) |
| Time spent with participant (hours per week) (mean (SD)) | 110.56 (63.38) | 115.82 (51.22) | 79.05 (32.03) | 156.90 (29.10) | 115.62 (61.37) | n/a | 101.92 (61.82) |
| Study characteristics | |||||||
| Visits per year (n) | 15* | 9 | 6 | 6 | 5 | 5 | 5 |
*Since the HU trial had a 4-month protocol, we standardized its number of study visits to a full year for comparison among trials.
Figure 1 provides estimated reporting rates of AEs (per participant per year) by severity. There were no AEs of grade 4 or 5 reported in any of the analyzed trials. For grade 1 AEs, the estimated reporting rate across all trials was 2.83 events per participant per year (95% CI: 2.66, 3.02). There were fewer grade 2 and 3 AEs than grade 1 reported in each trial. The overall grade 2 and 3 reporting rates were 1.18 (95% CI: 1.09, 1.28) and 0.23 (95% CI: 0.19, 0.27) events per participant per year, respectively. The overall estimated reporting rate for SAEs was 0.28 (95% CI: 0.24, 0.33) events per participant per year. We also observed that AE reporting rates increased as the number of annualized visits increased for AE grade 1–3 but not SAEs. After adjusting for study partner type, age, year of education, sex, and race and ethnicity, we estimated that every additional protocol-specified visit per year was associated with higher reporting rates for AE grade 1 (IRR = 1.12, 95% CI: 1.07, 1.17, p < 0.001) and grade 2 (IRR = 1.08, 95% CI: 1.05, 1.12, p < 0.001). At the same time, we estimated that SAE reporting rate was lower by 6% for every additional visit per year (IRR = 0.94, 95% CI: 0.88, 1.00, p = 0.040). We did not find evidence of an association between grade 3 AE reporting rates and the number of visits per year (IRR = 1.03, 95% CI: 0.96, 1.11, p = 0.364).
Fig. 1
Estimated reporting rates per participant per year for AE by severity and SAE. After adjusting for study partner type, age, year of education, sex, and race and ethnicity, we estimated that each additional visit per year was associated with higher reporting rate for AE grade 1–3 (grade 1 IRR = 1.12, 95% CI: 1.07, 1.17, p < 0.001; grade 2 IRR = 1.08, 95% CI: 1.05, 1.12, p < 0.001; grade 3 IRR = 1.03, 95% CI: 0.96, 1.11, p = 0.364), and lower reporting rate for SAE (IRR = 0.94, 95% CI: 0.88, 1.00, p = 0.040) (see Supplementary Table 2 for estimates corresponding to all covariates).
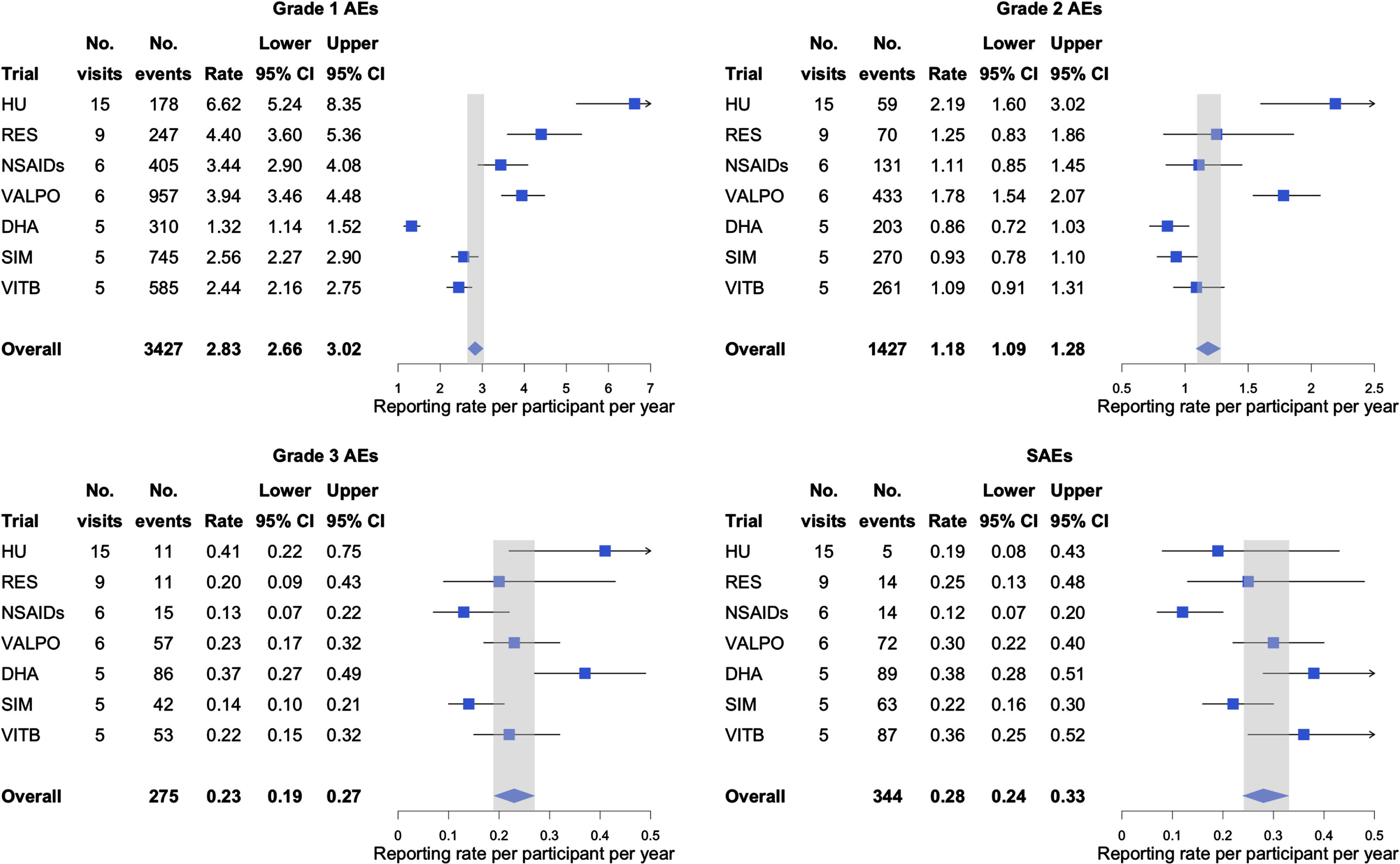
Table 2 presents the estimated IRR of reporting AEs by study partner type and severity. We found few statistically significant differences in AE reporting between partner types, though we did observe from the data that for AEs of grade 2 and 3, and SAEs, participants with non-spousal study partners had higher reporting rates than participants with spousal study partners. For instance, we estimated that participants with non-spousal study partners had a 26% higher grade 3 AE reporting rate compared to those with a spouse. In addition, we estimated that a 10-year increase in participant age was associated with a 39% higher SAE reporting rate (IRR = 1.39, 95% CI: 1.16, 1.67, p < 0.001). There was little evidence to suggest associations between age and reporting rates for grade 1–3 AEs. We did not observe significant associations between participant race and ethnicity and AE reporting rates, with the exception of grade 2 AEs, where Hispanic participants were estimated to have a 42% lower reporting rate compared to non-Hispanic White participants (IRR = 0.58, 95% CI: 0.41, 0.83, p = 0.003). We estimated that compared to the HU trial, trials with fewer visits per year had 35–80% lower grade 1 AE reporting rate (p-values≤0.006), with similar observations for AE grades 2 and 3. There was insufficient evidence to conclude an association between study partner type and reporting rate that differed by AE grade (
Table 2
Estimated IRR of reported AEs by study partner type and grade/severity
| Grade 1 AE | Grade 2 AE | Grade 3 AE | SAE | |||||
| Est. (95% CI) | p | Est. (95% CI) | p | Est. (95% CI) | p | Est. (95% CI) | p | |
| Study partner | ||||||||
| Spouse | 1 | 1 | 1 | 1 | ||||
| Adult Child | 0.98 (0.83, 1.17) | 0.848 | 1.16 (0.93, 1.43) | 0.185 | 1.26 (0.85, 1.86) | 0.248 | 1.27 (0.87, 1.85) | 0.214 |
| Other | 1.16 (0.94, 1.43) | 0.172 | 1.13 (0.89, 1.44) | 0.312 | 1.26 (0.73, 2.18) | 0.407 | 1.25 (0.82, 1.92) | 0.300 |
| Age (per 10 y) | 0.98 (0.92, 1.06) | 0.655 | 0.98 (0.88, 1.08) | 0.636 | 1.17 (0.95, 1.45) | 0.144 | 1.39 (1.16, 1.67) | <0.001 |
| Education (per year) | 1.01 (0.98, 1.03) | 0.598 | 0.99 (0.97, 1.02) | 0.561 | 0.95 (0.90, 1.00) | 0.051 | 1.00 (0.95, 1.05) | 0.898 |
| <12 y | 1 | 1 | 1 | 1 | ||||
| 12 y | 1.13 (0.91, 1.40) | 0.259 | 1.25 (0.97, 1.61) | 0.088 | 0.90 (0.56, 1.44) | 0.667 | 1.06 (0.61, 1.85) | 0.840 |
| 12–16 y | 1.03 (0.84, 1.27) | 0.758 | 1.05 (0.81, 1.35) | 0.728 | 0.72 (0.46, 1.13) | 0.153 | 1.06 (0.61, 1.83) | 0.832 |
| >16 y | 1.14 (0.89, 1.45) | 0.291 | 1.16 (0.87, 1.54) | 0.319 | 0.82 (0.49, 1.37) | 0.444 | 1.02 (0.54, 1.93) | 0.941 |
| Sex | ||||||||
| Female | 1 | 1 | 1 | 1 | ||||
| Male | 1.02 (0.90, 1.16) | 0.706 | 1.13 (0.95, 1.34) | 0.163 | 1.09 (0.77, 1.55) | 0.632 | 1.02 (0.74, 1.41) | 0.918 |
| Race and ethnicity | ||||||||
| Non-Hispanic White | 1 | 1 | 1 | 1 | ||||
| Non-Hispanic Black | 0.98 (0.73, 1.31) | 0.882 | 1.08 (0.80, 1.46) | 0.605 | 0.96 (0.58, 1.59) | 0.872 | 1.17 (0.70, 1.97) | 0.540 |
| Hispanic | 0.89 (0.68, 1.16) | 0.391 | 0.58 (0.41, 0.83) | 0.003 | 0.53 (0.25, 1.14) | 0.105 | 0.55 (0.26, 1.16) | 0.116 |
| Other | 1.25 (0.84, 1.84) | 0.268 | 0.68 (0.35, 1.35) | 0.274 | 0.96 (0.43, 2.15) | 0.921 | 0.91 (0.25, 3.28) | 0.889 |
| Trial | ||||||||
| HU | 1 | 1 | 1 | 1 | ||||
| RES | 0.65 (0.47, 0.89) | 0.006 | 0.56 (0.34, 0.94) | 0.027 | 0.57 (0.21, 1.55) | 0.270 | 1.68 (0.59, 4.75) | 0.331 |
| NSAIDs | 0.52 (0.39, 0.69) | <0.001 | 0.50 (0.33, 0.76) | 0.001 | 0.36 (0.16, 0.82) | 0.016 | 0.77 (0.29, 2.07) | 0.610 |
| VALPO | 0.59 (0.45, 0.77) | <0.001 | 0.81 (0.57, 1.15) | 0.240 | 0.60 (0.30, 1.20) | 0.151 | 1.73 (0.72, 4.18) | 0.221 |
| DHA | 0.20 (0.15, 0.26) | <0.001 | 0.39 (0.27, 0.56) | <0.001 | 0.97 (0.49, 1.94) | 0.935 | 2.23 (0.92, 5.37) | 0.075 |
| SIM | 0.39 (0.30, 0.51) | <0.001 | 0.43 (0.30, 0.62) | <0.001 | 0.40 (0.19, 0.83) | 0.014 | 1.35 (0.56, 3.28) | 0.506 |
| VITB | 0.36 (0.28, 0.47) | <0.001 | 0.49 (0.34, 0.70) | <0.001 | 0.56 (0.27, 1.15) | 0.113 | 2.04 (0.82, 5.04) | 0.123 |
Across six trials with available data and after accounting for age, years of education, participant sex, and trial, we did not find a statistically significant association between the time study partners spent with participants and the rate of reporting AEs (Table 3). We did observe that for grade 2–3 AEs and SAEs, the reporting rates were slightly lower when the study partner spent more days per week with the participant (grade 2 AE: IRR = 0.96, 95% CI: 0.93, 1.01; grade 3 AE: IRR = 0.98, 95% CI: 0.90, 1.06; SAE: IRR = 0.95, 95% CI: 0.88, 1.02). We also estimated that, given time spent with the participant and other potential confounders, every 10-year increase in age was associated with a 48% higher SAE reporting rate (IRR = 1.48, 95% CI: 1.19, 1.84, p < 0.001).
Table 3
Estimated IRR of reported AEs by grade/severity with time spent with participants as the predictor of interest*
| Grade 1 AE | Grade 2 AE | Grade 3 AE | SAE | |||||
| Est. (95% CI) | p | Est. (95% CI) | p | Est. (95% CI) | p | Est. (95% CI) | p | |
| Time spent with participant (per day per week) | 1.00 (0.97, 1.04) | 0.844 | 0.96 (0.93, 1.01) | 0.087 | 0.98 (0.90, 1.06) | 0.559 | 0.95 (0.88, 1.02) | 0.150 |
*Models adjusted for age, years of education, sex, and trials (see Supplementary Table 3 for estimates corresponding to all covariates).
DISCUSSION
It is crucial to monitor AEs and SAEs for patients’ safety in RCTs. SAEs have severe impacts on patients that ultimately could result in death. AEs of grade 1–4 are not life-threatening but are as important to be reported to help examine the risk of the investigated treatment. In this analysis of data from participants in placebo arms of mild-to-moderate AD clinical trials, we found that the reporting rates of AEs were higher for lower severity AEs. We also found that trials with greater numbers of visits per year had higher reporting rates of grade 1–2 AEs, and lower rates of reporting SAEs. The data did not suggest that AE reporting rates differed by study partner type and by AE severity, though we did observe that participants with non-spousal study partners had more AEs reported than participants with spousal study partners at most AE grades. We also observed that AE and SAE reporting rates were observed to be lower when the study partner spent more days per week with the participant, though this association was not statistically significant.
The need for safety assessments frequently determines visit frequency in trial protocols. The trials included in this study were multi-site, randomized, double-blind, and placebo-controlled and included mild-to-moderate AD patients. In this sample, we observed that the reporting rate of grade 1–3 AEs was higher for trials with more follow-up visits. This reporting pattern might occur due to the frequency of the participant, or their study partner, being asked if the participant had experienced any unfavorable health changes since their previous visit. For instance, HU had 15 annualized visits, and the estimated reporting rates for grade 1–3 AEs were the highest compared to other trials, all of which had 9 or fewer annualized visits. We did not observe this reporting pattern when it came to SAEs, suggesting that less severe events may be missed with infrequent protocol visits while more significant health matters are less prone to reporting oversights or biases. The observed patterns reinforce necessary caution that must be taken when comparing AE rates across studies, particularly those that incorporate differential visit frequencies for trials of similar agents that might be compared for safety outcomes.
In contrast to observations for lower grade AEs, we in fact estimated a lower reporting rate for SAEs with each additional visit per year after adjusting for other potential confounders. This may suggest that in addition to maximizing quantification of lower grade AEs, additional study visits may be key to participant safety, especially for trials of risky interventions or trials enrolling participant samples that may be at increased risk for serious health challenges. This may particularly include older participants, who in these analyses were also more likely to be reported as experiencing SAEs.
Since participants in this study were from placebo arms, the reported AEs can be assumed to be not related to the treatment being tested. The raw estimated AE reporting rates we provide may therefore give valuable insights to investigators designing future trials. For example, in single-arm and active-controlled studies, these estimates may provide valuable comparisons to better evaluate the safety profile of the investigated treatment, albeit with the many important caveats that accompany the use of historical controls [22].
Study partners play an important role in reporting AEs in AD trials. We did not find statistically significant evidence to conclude that reporting rates were different by study partner type. We did, however, observe that participants with spousal partners had lower reporting rates compared to those with non-spousal partners for AEs of grades 2–3 and SAEs. Our data do not address the causes of this discrepancy between the dyad types, but several possibilities exist. Spousal study partners, who tend to spend more time with participants, may be less likely to deem low-grade events as worth being reported. On the other hand, adult child and other study partners, who spend less time with participants on average, may be more inclined to notice subtle differences between interactions or may have a lower threshold for reporting changes as abnormal and untoward. A parallel can be drawn to the appreciation of disease progression, where individuals distant from a loved one with dementia are inclined to notice decline at intermittent visits, which may differ from the perception of family members who live with the person with dementia and provide care on daily basis [23, 24].
Limitations
We acknowledge that there are multiple limitations to our study. The trials we analyzed were federally funded and conducted primarily by academic sites. As such, the generalizability of our findings to larger trials supported by industry and conducted at private sites is uncertain. Next, the trial samples were composed mainly of non-Hispanic White and highly educated individuals making it similarly challenging to generalize to trials enrolling more diverse populations. We were unable to account for seasonality in our analyses and AEs may increase in certain seasons, particularly in specific regions of the country [25]. Finally, we lacked demographic information for study partners beyond their relationship to the participant, such as their age, race and ethnicity, sex, and education. These factors, along with study partners’ occupation, employment or retirement status, number of dependents, involvement with the participant’s health services, past RCT experience, and caregiver burden could have confounded the associations between study partner type and AE reporting. Future trials should consider collecting this information to permit such analyses.
Conclusions
We found that, adjusting for other confounders, the reporting rate of lower grade AEs was associated with the number of visits per year in mild-to-moderate AD trials. We did not find evidence to suggest that AE reporting rates differed by study partner type and by the amount of time the study partner spent with the participant on a weekly basis. In addition to these inferential findings, the estimated reporting rates presented here may be useful when evaluating safety profiles in future studies having similar visit frequency.
AUTHOR CONTRIBUTIONS
Thuy V. Lu (Formal analysis; Methodology; Visualization; Writing – original draft; Writing – review & editing); Joshua D. Grill (Conceptualization; Formal analysis; Methodology; Visualization; Writing – review & editing); Daniel Gillen (Conceptualization; Formal analysis; Methodology; Visualization; Writing – review & editing).
ACKNOWLEDGMENTS
We thank the participants, study partners, and researchers of the trials and the ADCS Legacy data project (supported by National Institute on Aging grant number U19 AG010483) that made our analysis possible. We thank Mr. Adam Birnbaum for his support with merging race and ethnicity data across the trials in this study.
FUNDING
This work was supported by the National Institute on Aging (grant numbers P30 AG066519, RF1 AG059407).
CONFLICT OF INTEREST
JDG discloses research support from the National Institute of Health, Alzheimer’s Association, Bright Focus Foundation, Eli Lilly, Biogen, and Eisai. He has provided consulting to Site Rx and served on the Editorial Board of Alzheimer’s & Dementia journal. JDG is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review. DLG and TVL declare that they have no conflicting interests.
DATA AVAILABILITY
The datasets analyzed were made available through the University of California, San Diego ADCS Legacy database. The datasets supporting the conclusions of this article are included within the article and its additional files.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-231283.
REFERENCES
[1] | Yao B , Zhu L , Jiang Q , Xia HA ((2013) ) Safety monitoring in clinical trials. Pharmaceutics, 5: , 94–106. |
[2] | Food and Drug Administration ((2007) ) Guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. Food and Drug Administration: Silver Spring, MD, USA. |
[3] | Kennedy F , Shearsmith L , Ayres M , Lindner OC , Marston L , Pass A , Danson S , Velikova G ((2021) ) Online monitoring of patient self-reported adverse events in early phase clinical trials: Views from patients, clinicians, and trial staff. Clin Trials 18: , 168–179. |
[4] | Grill JD , Karlawish J ((2010) ) Addressing the challenges to successful recruitment and retention in Alzheimer’s disease clinical trials. Alzheimers Res Ther 2: , 34. |
[5] | ((2023) ) 2023 Alzheimer’s disease facts and figures. Alzheimers Dement 19: , 1598–1695. |
[6] | Grill JD , Raman R , Ernstrom K , Aisen P , Karlawish J ((2013) ) Effect of study partner on the conduct of Alzheimer disease clinical trials. Neurology 80: , 282–288. |
[7] | Grill JD , Monsell S , Karlawish J ((2012) ) Are patients whose study partners are spouses more likely to be eligible for Alzheimer’s disease clinical trials. Dement Geriatr Cogn Disord 33: , 334–340. |
[8] | Cary MS , Rubright JD , Grill JD , Karlawish J ((2015) ) Why are spousal caregivers more prevalent than nonspousal caregivers as study partners in AD dementia clinical trials? Alzheimer Dis Assoc Disord 29: , 70–74. |
[9] | Conde-Sala JL , Garre-Olmo J , Turró-Garriga O , Vilalta-Franch J , López-Pousa S ((2010) ) Differential features of burden between spouse and adult-child caregivers of patients with Alzheimer’s disease: An exploratory comparative design. Int J Nurs Stud 47: , 1262–1273. |
[10] | Quinn JF , Raman R , Thomas RG , Yurko-Mauro K , Nelson EB , Van Dyck C , Galvin JE , Emond J , Jack CR , Weiner M , Shinto L , Aisen PS ((2010) ) Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 304: , 1903–1911. |
[11] | Aisen PS , Schneider LS , Sano M , Diaz-Arrastia R , van Dyck CH , Weiner MF , Bottiglieri T , Jin S , Stokes KT , Thomas RG , Thal LJ , Alzheimer Disease Cooperative Study ((2008) ) High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 300: , 1774–1783. |
[12] | Rafii MS , Walsh S , Little JT , Behan K , Reynolds B , Ward C , Jin S , Thomas R , Aisen PS , Alzheimer’s Disease Cooperative Study ((2011) ) A phase II trial of huperzine A in mild to moderate Alzheimer disease. Neurology 76: , 1389–1394. |
[13] | Aisen PS , Schafer KA , Grundman M , Pfeiffer E , Sano M , Davis KL , Farlow MR , Jin S , Thomas RG , Thal LJ , Alzheimer’s Disease Cooperative Study ((2003) ) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 289: , 2819–2826. |
[14] | Turner RS , Thomas RG , Craft S , Dyck CH van , Mintzer J , Reynolds BA , Brewer JB , Rissman RA , Raman R , Aisen PS , Alzheimer’s Disease Cooperative Study ((2015) ) A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 85: , 1383–1391. |
[15] | Sano M , Bell KL , Galasko D , Galvin JE , Thomas RG , Dyck CH van , Aisen PS ((2011) ) A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77: , 556–563. |
[16] | Tariot PN , Schneider LS , Cummings J , Thomas RG , Raman R , Jakimovich LJ , Loy R , Bartocci B , Fleisher A , Ismail MS , Porsteinsson A , Weiner M , Jack CR , Thal L , Aisen PS , Alzheimer’s Disease Cooperative Study Group ((2011) ) Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry 68: , 853–861. |
[17] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–939. |
[18] | White H ((1980) ) A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica 48: , 817–838. |
[19] | Liang K-Y , Zeger SL ((1986) ) Longitudinal data analysis using generalized linear models. Biometrika 73: , 13–22. |
[20] | Zou G ((2004) ) A modified poisson regression approach to prospective studies with binary data. Am J Epidemiol 159: , 702–706. |
[21] | Yelland LN , Salter AB , Ryan P ((2011) ) Performance of the modified Poisson regression approach for estimating relative risks from clustered prospective data. Am J Epidemiol 174: , 984–992. |
[22] | Pocock SJ ((1976) ) The combination of randomized and historical controls in clinical trials. J Chronic Dis 29: , 175–188. |
[23] | Roberto KA , Blieszner R , McCann BR , McPherson MC ((2011) ) Family triad perceptions of mild cognitive impairment. J Gerontol B Psychol Sci Soc Sci 66B: , 756–768. |
[24] | Greenop KR , Xiao J , Almeida OP , Flicker L , Beer C , Foster JK , van Bockxmeer FM , Lautenschlager NT ((2011) ) Awareness of cognitive deficits in older adults with cognitive-impairment-no-dementia (CIND): Comparison with informant report. Alzheimer Dis Assoc Disord 25: , 24. |
[25] | Marrero O , Hung EY , Hauben M ((2016) ) Seasonal and geographic variation in adverse event reporting. Drugs Real World Outcomes 3: , 297–306. |


