
Interaction Between Arteriosclerosis and Amyloid-β on Cognitive Function
Abstract
Background:
Dementia is a multifactorial disease, with Alzheimer’s disease (AD) and vascular pathology often co-occurring in many individuals with dementia. Yet, the interplay between AD and vascular pathology in cognitive decline is largely undetermined.
Objective:
The aim of the present study was to examine the joint effect of arteriosclerosis and AD pathology on cognition in the general population without dementia.
Methods:
We determined the interaction between blood-based AD biomarkers and CT-defined arteriosclerosis on cognition in 2,229 dementia-free participants of the population-based Rotterdam Study (mean age: 68.9 years, 52% women) cross-sectionally.
Results:
Amyloid-β (Aβ)42 and arterial calcification were associated with cognitive performance. After further adjustment for confounders in a model that combined all biomarkers, only arterial calcification remained independently associated with cognition. There was a significant interaction between arterial calcification and Aβ42 and between arterial calcification and the ratio of Aβ42/40. Yet, estimates attenuated, and interactions were no longer statistically significant after adjustment for cardio metabolic risk factors.
Conclusions:
Arteriosclerosis and AD display additive interaction-effects on cognition in the general population, that are due in part to cardio metabolic risk factors. These findings suggest that joint assessment of arteriosclerosis and AD pathology is important for understanding of disease etiology in individuals with cognitive impairment.
INTRODUCTION
Alzheimer’s disease (AD) and vascular pathology are the most common causes of dementia, and can been found jointly in about 40% of individuals with dementia at time of death [1, 2]. Arteriosclerosis is a generic term that encompasses the loss of arterial wall elasticity related to stiffening of vessels. While the prevalence of both arteriosclerosis and AD pathology steeply increases with age, individually neither amyloid deposition nor arteriosclerosis by itself are generally sufficient to cause the cognitive impairment [3–5], as many individuals with either of these pathologies never develop dementia. Against this backdrop, the co-occurrence of different pathologies is an important determinant of developing cognitive impairment and dementia [6–8], with potential implications for diagnosis as well as treatment indication.
In recent years, various plasma biomarkers have been developed into valid markers of central nervous system pathology. With the availability of these less expensive and less invasive plasma alternatives to cerebrospinal fluid and PET investigation, it becomes possible to study the interplay between different pathophysiological processes in vivo in adequately powered studies of unselected individuals. Plasma levels of amyloid-β (Aβ)40 and Aβ42 can identify AD pathology and risk of AD [9, 10], whereas brain magnetic resonance imaging (MRI) and computed tomography (CT) provide good markers for small-vessel disease and arteriosclerosis, respectively. Using non-contrast CT, we have recently shown in non-demented individuals that plasma Aβ40 is associated with arteriosclerosis [11], suggesting an interplay between these pathologies in the preclinical disease stage. This adds to earlier MRI studies showing that Aβ was associated with subclinical markers of vascular brain disease [12–14].
Aβ is considered a hallmark of AD, but it remains unclear what the exact cause and consequence of accumulation is [15], and while much evidence indicates that vascular pathology is a major risk factor for AD and dementia, it is unknown if vascular brain disease causes, amplifies or precedes development of AD [15]. Furthermore, the impact of a joint contribution of vascular and amyloid pathology on cognitive outcomes in vivo is largely undetermined [14].
The aim of the present study was to examine the joint effect of arteriosclerosis and AD pathology on cognition in the general population without dementia.
METHODS
Study population
This study is embedded in the Rotterdam Study, an ongoing population-based cohort study in the Netherlands, details of which have been described previously [16]. The original study population in 1990 consisted of 7,983 participants aged≥55 years from the Ommoord area, a suburb of Rotterdam. In 2000, the cohort was expanded with 3,011 persons who had reached age 55 years or had moved into the study area. Follow-up examinations take place at a dedicated research center every 4 years. Baseline of the current study was the 4th examination round of the original cohort, and the 2nd examination round of the expansion cohort. Between 2002 and 2005, 5,094 participants had blood samples taken during their visit to the study center. We excluded 518 participants with missing or invalid test results for plasma Aβ40 or plasma Aβ42. Of the remaining 4,576 participants, a subsample of 2,252 underwent on-contrast CT examination of the heart, aorta, and carotid arteries up till the level of the circle of Willis. Measures of arterial wall elasticity were not available in this examination round of the Rotterdam Study cohort. Of these participants, we excluded 23 with extreme values (exceeding±3.5 SD), the remaining sample consisted of 2,229 participants.
Ethics statement
The Rotterdam Study has been approved by the Medical Ethics Committee of the Erasmus MC (registration number MEC 02.1015) and by the Dutch Ministry of Health, Welfare and Sport (Population Screening Act WBO, license number 1071272-159521-PG). The Rotterdam Study Personal Registration Data collection is filed with the Erasmus MC Data Protection Officer under registration number EMC1712001. The Rotterdam Study has been entered into the Netherlands National Trial Register (NTR; https://www.trialregister.nl) and into the WHO International Clinical Trials Registry Platform (ICTRP; https://apps.who.int/trialsearch/) under shared catalogue number NL6645/NTR6831. All participants provided written informed consent to participate in the study and to have their information obtained from treating physicians.
Measurement of plasma biomarkers
EDTA plasma was sampled, aliquoted, and frozen at –80°C according to standard procedures. Measurements were carried out in two separate batches; the first batch included 2,000 samples, obtained from a random selection of 1,000 participants from the fourth visit of RS-I and 1,000 from the second visit of RS-II. The second batch included samples from all the remaining participants of these two study waves. All measurements were performed at Quanterix (Lexington, MA, USA) on a single molecule array (Simoa) HD-1 analyzer platform [17]. Detailed methods have been described previously [10].
Quantification of arteriosclerosis
A 16-slice (n = 663) or 64-slice (n = 1,589) multidetector CT scanner (Somatom Sensation 16 or 64; Siemens, Forchheim, Germany) was used to perform non-contrast CT scanning. We obtained images of the coronary arteries, aortic arch, extracranial internal carotid arteries, and intracranial internal carotid arteries. Detailed information regarding imaging parameters has been described previously [18–20]. Calcification in the coronary arteries, aortic arch and the extracranial internal carotid arteries was quantified using commercially available software (SyngoCalciumScoring; Siemens). Calcification was measured in the left main, left anterior descending, left circumflex, and right coronary arteries. The aortic arch was measured from the origin to the first centimeter of the common carotid arteries, the vertebral arteries, and the subclavian arteries beyond the origin of the vertebral arteries. The extracranial carotid arteries were measured at both sides, within 3 centimeters proximal and distal of the bifurcation [18]. For the intracranial internal carotid arteries we used a separate, semi-automated scoring method to quantify calcification [21–23]. Briefly, after manually delineating calcification in the trajectory of the intracranial internal carotid artery, the calcification was calculated by multiplying the number of pixels above the threshold for calcium (i.e., 130 Hounsfield units) [24] with the pixel size and slice increment [19, 20]. Scoring of calcification was done semi-automatically by 3 trained raters, the interrater reliability was very good (intraclass correlation coefficient, 0.99) [21]. Examples of calcification in examined vessel beds can be seen in Supplementary Figure 1. Next, we used these calcification s from the four arteries to determine a single measure of the systemic burden of calcification using a summary metric for arterial calcification, the “C-factor” [25]. The C-factor is calculated using a principal component analysis including calcification levels in the coronary artery, aortic arch, extracranial carotid artery, and intracranial carotid artery. A more detailed description of the calculation method can be found elsewhere [25].
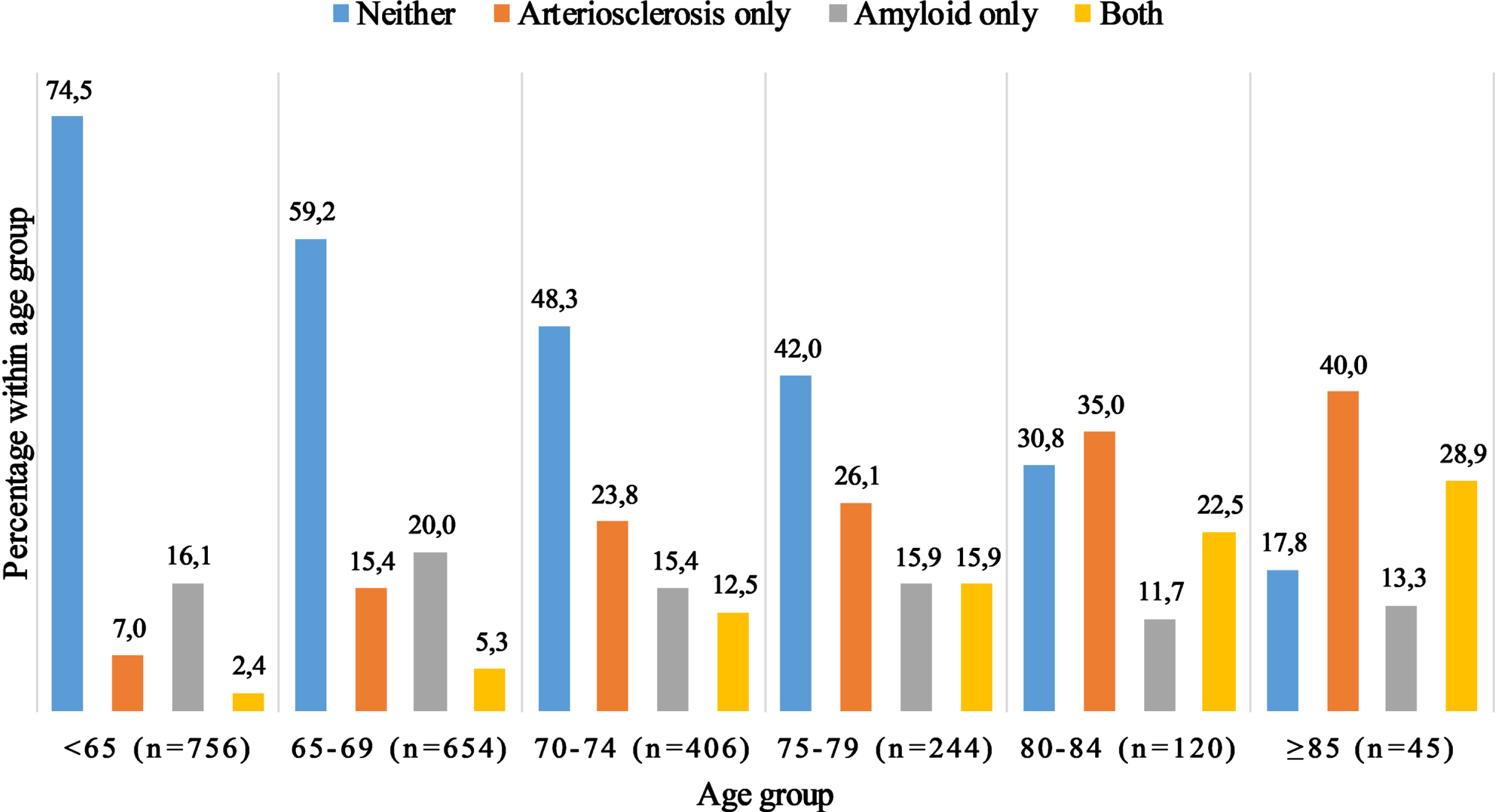
Fig. 1
Co-occurrence of arteriosclerosis and amyloid pathology. The figure depicts the number of participants with neither pathology, arteriosclerosis only, amyloid pathology only or co-occurrence of both pathologies, stratified by age groups. Presence of arteriosclerosis was defined as the highest quartile of the C-factor, whereas presence of amyloid was defined as the lowest quartile of the Aβ42/40 ratio.
Measures of cognition
Cognitive function was assessed with a neuropsychological test battery comprising the verbal fluency test, the letter-digit substitution task, a 15-word learning test (immediate and delayed recall), the Stroop test, and Purdue pegboard task [26]. For all participants, z-scores were calculated for each test separately by dividing the difference between the individual and mean test scores by the standard deviation. To obtain a measure of global cognitive function, we calculated a standardized compound score (g-factor) using principal component analysis [26]. We calculated scores for cognitive domains for memory (word learning test, immediate and delayed recall), executive function (Stroop interference task, verbal fluency test, and letter-digit substitution task [weighted half]), information processing (Stroop reading and color naming task and letter-digit substitution task [weighted half]), and motor function (Purdue pegboard test).
Covariables
For each participant, we obtained information on highest level of completed education (classified into lower, intermediate, and higher education), history of smoking (i.e., current, former, or never), and alcohol consumption. Hypertension was defined as a blood pressure >140/90mmHg or the use of blood-pressure lowering medication for this indication. Body mass index was computed from measurements of height and weight (kg/m2). Blood samples were obtained at baseline to determine serum total cholesterol and glucose. Apolipoprotein E4 (APOE ɛ4) genotype was determined by polymerase chain reaction on coded DNA samples in the original cohort, and by biallelic Taqman assays (TaqMan Gene Expression Assays; Thermo Fisher Scientific, Waltham, Massachusetts) (rs7412 and rs429358) for the expansion cohort. APOE genotype was classified as APOE ɛ4 carrier (≥1 ɛ4 allele) or noncarrier.
Statistical analysis
Because Aβ42 had a skewed distribution, we used natural log-transformed values. We subsequently standardized Aβ40, Aβ42, the Aβ42/40 ratio, and cognition measurements by dividing the difference between the individual value and the population mean by the population standard deviation. Missing data ranged from 0.1% for hypertension to 9.4% for creatinine level and were imputed 10 times with 20 iterations using chained equations (MICE R package). Distribution of covariables was similar in the imputed and non-imputed datasets.
We determined the association of arterial calcification, C-factor and per calcification tertiles (no calcification present and calcification present in tertiles), and plasma biomarkers with global cognition using linear regression models. We adjusted the association of the Aβ40 measurements for Aβ42, and Aβ42 for Aβ40 in all models. To adjust for confounding, we constructed two models. In the first model, we adjusted for age, age2, sex, batch number of the plasma analysis, scanner type used for CT, educational level and the time interval between scan acquisition and cognitive assessment. In the second model, we additionally adjusted for cholesterol, HDL, body mass index, smoking status, diabetes, lipid lowering medication, systolic blood pressure, diastolic blood pressure, blood pressure lowering medication, APOEɛ4 carriership, creatinine level and alcohol consumption. In a final model, we assessed independent effects of Aβ and calcification by mutual adjustment for arterial calcification and the plasma biomarkers.
Finally, we assessed the interaction effects between the C-factor and plasma biomarkers on global cognition and cognitive domains by addition of an interaction term to the linear regression models and stratifying on the biomarker level. Plots for the biomarker levels were stratified on low, mean, and high tertiles of the biomarker level. All statistical analyses were performed in R version 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Characteristics of the study population are shown in Table 1. The mean age was 68.9 years (SD 6.6), and 1,158 (52%) participants were women. Prevalence of calcification ranged from 73% in the extracranial carotid arteries to 93% in the aorta, both increasing steeply with age. The C-factor explained 66% of variation in calcification across the different arteries in this population. Severe calcification (highest quartile) was present in combination with a low plasma Aβ42/40-ratio (lowest quartile) in 183 (8.2%) of participants, and co-occurrence increased with age from 2.4% under the age of 65 to 28.9% after age 85 (Fig. 1). A correlation matrix of Aβ, calcification and all covariables is presented in Supplementary Figure 2. Levels of plasma Aβ40 and Aβ42 were highly correlated (r = 0.58). Aβ40 and the C-factor were positively correlated with age (Supplementary Fig. 2).
Table 1
Characteristics of the study population (N = 2,229), average over 10 imputed datasets
| Study population | |
| (N = 2,229) | |
| Age (y) | 68.9 (±6.6) |
| Female sex | 1,158 (52.0%) |
| Education | |
| Lower | 695 (31.2%) |
| Intermediate | 1,104 (49.5%) |
| Higher | 430 (19.3%) |
| Smoking | |
| Never | 640 (28.7%) |
| Former | 1,246 (55.9%) |
| Current | 343 (15.4%) |
| Alcohol (grams/day; median, IQR) | 9.3 (1.4–20.0) |
| Body mass index (kg/m2) | 27.6 (±3.9) |
| Systolic blood pressure (mmHg) | 146.5 (±20.1) |
| Diastolic blood pressure (mmHg) | 80.3 (±10.7) |
| Blood pressure lowering medication | 880 (39.5%) |
| Serum total cholesterol (mmol/L) | 5.7 (±1.0) |
| Serum high-density lipoprotein (mmol/L) | 1.4 (±0.4) |
| Lipid lowering medication | 979 (43.9%) |
| Diabetes | 230 (10.3%) |
| Creatinine level (μmol/L) | 82.1 (±18.8) |
| APOE ɛ4 carriers | 628 (28.2%) |
| Aβ40 (pg/mL; median, IQR) | 247.9 (222.9–279.8) |
| Aβ42 (pg/mL; median, IQR) | 10.1 (8.7–11.7) |
SD, standard deviation; IQR, interquartile range. Data are presented as frequency (%) for categorical and mean±SD for continuous variables unless indicated otherwise.
Amyloid-β, calcification, and cognition
Arterial calcification was associated with worse cognitive performance (β[95% CI] per standard deviation increase: –0.06 [–0.08; –0.04]), independent of cardiovascular risk factors and concurrent plasma levels of Aβ in multivariable analyses (–0.04 [–0.07; –0.01]) (Table 2). Plasma Aβ42 was associated with cognition in the age- and sex-adjusted model (β[95% CI]: 0.06 [0.01; 0.11]), but estimates attenuated and were no longer statistically significant in the further adjusted model (Table 2). Aβ40 and the ratio of Aβ42/40 were not significantly associated with cognitive performance (Table 2).
Table 2
Association of plasma biomarkers (Aβ40, Aβ42) and systemic arterial calcification (C-factor) on global cognition outcome (g-factor)
| Global cognition (g-factor) | |||
| Adjusted mean difference (95% Confidence Interval) | |||
| Model I | Model II | Model III | |
| Aβ40 | –0.04 (–0.09; 0.01) | –0.01 (–0.06; 0.04) | –0.01 (–0.05; 0.05) |
| Aβ42 | 0.06 (0.01; 0.11)* | 0.02 (–0.04; 0.07) | 0.01 (–0.04; 0.07) |
| Aβ42/40 ratio | 0.03 (–0.01; 0.07) | 0.001 (–0.05; 0.05) | 0.001 (–0.05; 0.04) |
| Arterial calcification (C-factor) | –0.06 (–0.08; –0.04)** | –0.04 (–0.07; –0.01)** | –0.04 (–0.07; –0.01)** |
*p < 0.05; **p < 0.01. Model I: adjusted for age, age2, sex, education, batch number (for plasma analysis only), and scanner used and time interval between scanning and cognitive assessment (for calcification analysis only). Aβ40 measurements were adjusted for Aβ42, and Aβ42 for Aβ40. Model II: model I + smoking habits, alcohol consumption, systolic and diastolic blood pressure, blood pressure lowering medication, diabetes mellitus, serum total cholesterol and HDL cholesterol, lipid-lowering medication, body mass index, creatinine level, APOE ɛ4 carrier status. Model III: model II + mutual adjustment for AD biomarkers and calcification.
When stratifying on age, associations of arterial calcification with cognition were most profound prior age 70, attenuating thereafter (Supplementary Table 1). Associations of plasma Aβ with cognition did not differ substantially by age, except for somewhat stronger yet non-significant risk estimates in the protective direction in the oldest old (Supplementary Table 1).
Interaction between Aβ and calcification on cognition
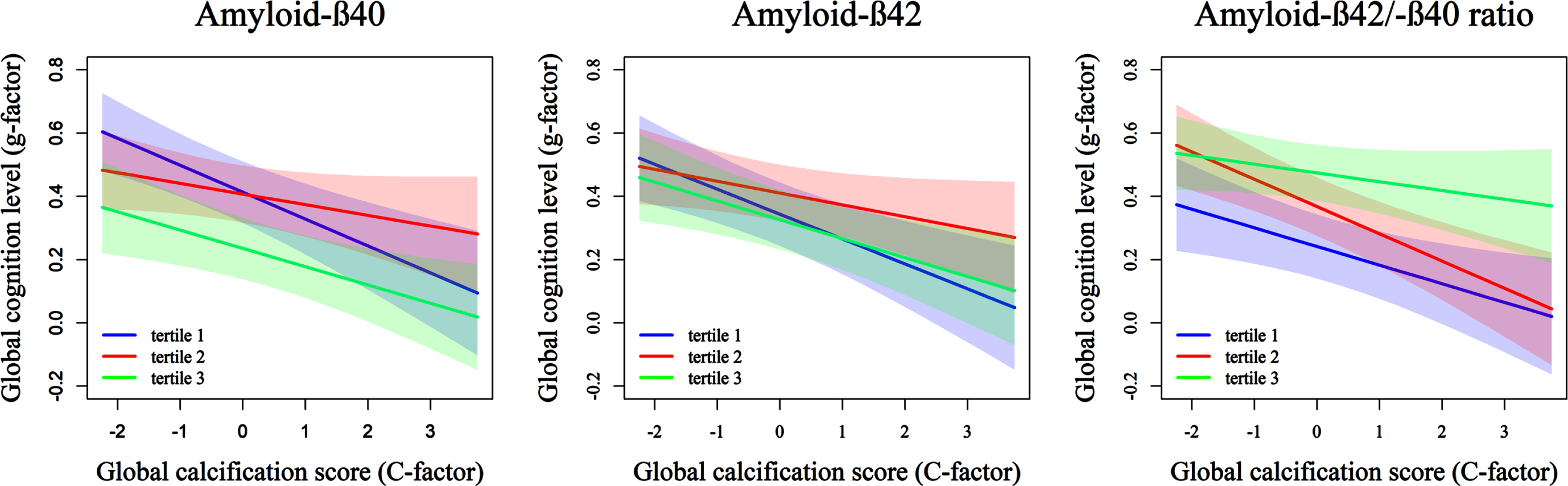
We observed a significant interaction between Aβ42 and calcification on global cognition in age- and sex-adjusted models (Table 3; pinteraction = 0.03), such that the associations of amyloid with cognition were more profound with higher levels of arterial calcification (Fig. 2). This interaction on global cognition was also present in the Aβ42/40 ratio (Table 3; pinteraction = 0.02). The associations attenuated, and interaction terms were no longer statistically significant after adjustment for cardio metabolic risk factors (Table 3). The interaction effects between Aβ42 and the C-factor were most profound for motor function, followed by the memory and information processing domains (Supplementary Table 2). In terms of location of arteriosclerosis, similar patterns were observed across sites, but with strongest evidence of interaction in the presence of coronary artery calcification, followed by extracranial and intracranial carotid artery calcification (Supplementary Table 3).
Table 3
Association and interaction of plasma biomarkers (Aβ40, Aβ42) and systemic arterial calcification (C-factor) on global cognition outcome (g-factor)
| Global cognition (g-factor) | ||
| Adjusted mean difference (95% Confidence Interval) | ||
| Model I | Model II | |
| Aβ40 | ||
| Aβ40 | –0.03 (–0.07; 0.02) | –0.01 (–0.06; 0.05) |
| Arterial calcification (C-factor) | –0.06 (–0.08; –0.04)** | –0.04 (–0.07; –0.01)** |
| Aβ40 * C-factor | 0.01 (–0.02; 0.03) | –0.004 (–0.03; 0.02) |
| Aβ42 | ||
| Aβ42 | 0.05 (–0.003; 0.10) | 0.01 (–0.04; 0.07) |
| Arterial calcification (C-factor) | –0.06 (–0.08; –0.04)** | –0.04 (–0.07; –0.01)** |
| Aβ42 * C-factor | 0.03 (0.003; 0.05)* | 0.01 (–0.01; 0.04) |
| Aβ42/40 ratio | ||
| Aβ42/40 ratio | 0.02 (–0.02; 0.06) | –0.01 (–0.05; 0.04) |
| Arterial calcification (C-factor) | –0.06 (–0.08; –0.04)** | –0.04 (–0.07; –0.01)** |
| Aβ42/40 ratio * C-factor | 0.03 (0.001; 0.05)* | 0.02 (–0.01; 0.04) |
*p < 0.05; **p < 0.01. Model I: adjusted for age, age2, sex, education, and plasma analysis batch. Mutual adjustment for AD biomarkers. Model II: model I + smoking habits, alcohol consumption, systolic and diastolic blood pressure, blood pressure lowering medication, diabetes mellitus, serum total cholesterol and HDL cholesterol, lipid-lowering medication, body mass index, creatinine level, APOE ɛ4 carrier status.
Fig. 2
Association of systemic calcification with global cognition, stratified by tertile of plasma levels of Aβ40, Aβ42, and the Aβ42/40 ratio. The figure depicts the association of systemic arterial calcification level (C-factor) and the cognition outcome (g-factor) per tertile of plasma Aβ40, Aβ42, and the Aβ42/40 ratio.
DISCUSSION
In this cross-sectional population-based study, we found that arterial calcification and plasma AD biomarkers display additive effects on cognition in the general population. Plasma Aβ was associated with worse cognition particularly in the presence of arterial calcification. These interactive patterns were explained in part by differences in cardiovascular risk profiles.
Our findings underline that in many individuals in the general population, AD and vascular disease contribute jointly to cognitive decline, through either separate or intertwined pathways. Previous study has shown a similar interaction of neurodegenerative markers on MRI [27]. Others have hinted at a vascular contribution to the pathogenesis of AD, reporting higher burden of Aβ on PET imaging in participants with hypertension and diabetes [28, 29]. Decline in cerebral blood flow later in life may be a mediating factor between vascular pathology and Aβ accumulation [30]. Evidence has shown that impairments in blood flow occur before the more common neurological biomarkers associated with AD [31, 32]. Driven by observations that arterial calcification contributes to AD risk [33], the “vascular hypothesis of AD” was coined, stating that impaired function of the neurovascular unit affects neuronal health and cognitive ability before the classical AD biomarkers are affected and can promote the accumulation of Aβ in the brain [34–36]. Similarly, vascular dysregulation and stiffening may affect clearance of Aβ from the brain [15, 32]. However, the role of arteriosclerosis in the etiology of AD remains uncertain [37], and associations of cardiovascular risk factors with cerebral amyloid deposition have not been consistent across cohort studies [38, 39]. The attenuation of interactions after adjustment for cardiovascular risk factors in the present study suggests a role at least in both AD and vascular cognitive impairment and decline that warrants further exploration in longitudinal studies capturing change in cognitive ability over time.
The co-occurrence of Aβ and arterial calcification steeply increased with age, in line with high prevalence of different pathologies in post-mortem studies of both cognitively healthy and demented individuals [40]. In the Religious Orders Study and the Memory and Aging Project, autopsy showed that >94% of participants had at least one neuropathology at death, with the most common neuropathology being AD (65.3%) although it very rarely occurred in isolation (9%), often AD was comorbid with at least one other neurodegenerative and vascular pathology (44%) or with at least one vascular pathology (40%) [2]. While the lack of established cut-offs for plasma Aβ to determine cerebral amyloid burden render the percentages in our study a poor men’s alternative to postmortem study, they do illustrate that findings from postmortem study translate into in vivo effects on cognition. The high co-occurrence of AD and vascular pathology may be reflected particularly by impairment on the memory and executive domains.
Our results suggest that measurement of both arterial calcification and Aβ in the diagnostic work-up could provide additional insight in the etiology of cognitive decline in the majority of older patients with combined pathologies, above and beyond measurement of a single disease marker. Simultaneous measurement could help to determine the most likely cause for cognitive decline in patients and personalize treatment. While we operationalized the vascular component as a joint measure of arteriosclerosis across four different vessel beds, we have previously shown that a more pragmatic approach across two vessels maybe just as reliable to map the arteriosclerotic burden [25]. Further studies are needed to determine whether similar interactions are observed for cerebral small-vessel disease, and to which extent these are related to vascular stiffening due to arterial calcification. This may also clarify the role of cardio metabolic risk factors in the interplay between amyloid and vascular pathology.
Strengths of this study include the use of the high sensitivity Simoa measurements in the setting of a large population-based sample. There are limitations also. First, although recent studies have shown good validity of plasma biomarkers of AD for detecting amyloid pathology in the brain and cerebral spinal fluid, their specificity remains somewhat lower and may be affected by comorbid pathology. In the absence of an absolute measure to determine presence of amyloid in the central nervous system, we relied on the lower quartiles of Aβ (and arteriosclerosis) to demonstrate to increasing prevalence of multipathology with advancing age. Second, CT-imaging was done on average 6 months prior to measurement of cognition and plasma markers, which may have introduced some measurement error. Third, the cross-sectional design hampers inference about the interplay of the different markers in the trajectory of cognitive decline. Fourth, the vast majority of participants (>96%) were of European ancestry, rendering results potentially less generalizable to other ancestries.
In conclusion, arteriosclerosis and Aβ display additive interaction-effects on cognitive performance in the general population, that are due in part to cardio metabolic risk factors. These findings suggest that joint assessment of vascular and AD pathology is crucial for understanding disease etiology in individuals with cognitive impairment.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. The Netherlands Consortium of Dementia Cohorts (NCDC) is funded in the context of DeltaplanDementie from ZonMw Memorabel and Alzheimer Nederland (project number 73305095005). This work is part of the Heart-Brain Connection crossroads (HBCx) consortium of the Dutch CardioVascular Alliance (DCVA). HBCx has received funding from the Dutch Heart Foundation under grant agreements 2018-28 and CVON 2012-06. Dr. Bos is supported by a grant (A2017424F) from the BrightFocus Foundation and a grant from the Alzheimer’s Association (AARG-21-846504).
CONFLICT OF INTEREST
M. Arfan Ikram is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
Anonymized data are available on reasonable request. Requests for access to the data reported in this paper can be directed to data manager Frank J.A. van Rooij ().
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230604.
REFERENCES
[1] | Attems J , Jellinger KA ((2014) ) The overlap between vascular disease and Alzheimer’s disease - lessons from pathology.. BMC Med 12: , 206. |
[2] | Boyle PA , Yu L , Wilson RS , Leurgans SE , Schneider JA , Bennett DA ((2018) ) Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83: , 74–83. |
[3] | Musiek ES , Holtzman DM ((2015) ) Three dimensions of the amyloid hypothesis: Time, space and “wingmen.” . Nat Neurosci 18: , 800–806. |
[4] | Mormino EC , Betensky RA , Hedden T , Schultz AP , Amariglio RE , Rentz DM , Johnson KA , Sperling RA ((2014) ) Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol 71: , 1379–1385. |
[5] | Rabin JS , Yang HS , Schultz AP , Hanseeuw BJ , Hedden T , Viswanathan A , Gatchel JR , Marshall GA , Kilpatrick E , Klein H , Rao V , Buckley RF , Yau WYW , Kirn DR , Rentz DM , Johnson KA , Sperling RA , Chhatwal JP ((2019) ) Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol 85: , 272–279. |
[6] | Albrecht D , Isenberg AL , Stradford J , Monreal T , Sagare A , Pachicano M , Sweeney M , Toga A , Zlokovic B , Chui H , Joe E , Schneider L , Conti P , Jann K , Pa J , Alzheimer Disease Neuroimaging Initiative ((2020) ) Associations between vascular function and tau PET are associated with global cognition and amyloid. J Neurosci 40: , 8573–8586. |
[7] | Forrest SL , Kovacs GG ((2023) ) Current concepts of mixed pathologiesin neurodegenerative diseases. Can J Neurol Sci 50: , 329–345. |
[8] | Mcaleese KE , Colloby SJ , Thomas AJ , Al-sarraj S , Francis PT , Ansorge O , Neal J , Attems J , Roncaroli F , Love S ((2021) ) Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimers Dement 17: , 1121–1133. |
[9] | van Oijen M , Hofman A , Soares HD , Koudstaal PJ , Breteler MM ((2006) ) Plasma Aβ1-40 and Aβ1-42 and the risk of dementia: A prospective case-cohort study. Lancet Neurol 5: , 655–660. |
[10] | de Wolf F , Ghanbari M , Licher S , McRae-McKee K , Gras L , Weverling GJ , Wermeling P , Sedaghat S , Kamran Ikram M , Waziry R , Koudstaal W , Klap J , Kostense S , Hofman A , Anderson R , Goudsmit J , Ikram MA ((2020) ) Plasma tau, neurofilament light chain and amyloid-β levels and risk of dementia; a population-based cohort study. Brain 143: , 1220–1232. |
[11] | Wolters FJ , Hilal S , Leening MJG , Kavousi M , Ghanbari M , Franco OH , Hofman A , Koudstaal PJ , Vernooij MW , Ikram MK , Bos D , Ikram MA ((2022) ) Plasma amyloid-β40 in relation to subclinical atherosclerosis and cardiovascular disease: A population-based study. Atherosclerosis 348: , 44–50. |
[12] | Hilal S , Akoudad S , van Duijn CM , Niessen WJ , Verbeek MM , Vanderstichele H , Stoops E , Ikram MA , Vernooij MW ((2017) ) Plasma amyloid-β levels, cerebral small vessel disease, and cognition: The Rotterdam Study. J Alzheimers Dis 60: , 977–987. |
[13] | Wong FCC , Saffari SE , Yatawara C , Ng KP , Kandiah N ((2021) ) Influence of white matter hyperintensities on baseline and longitudinal amyloid- in cognitively normal individuals. J Alzheimers Dis 84: ,91–101. |
[14] | Kim SE , Kim HJ , Jang H , Weiner MW , DeCarli C , Na DL , Seo SW ((2022) ) Interaction between Alzheimer’s disease and cerebral small vessel disease: A review focused on neuroimaging markers. Int J Mol Sci 23: , 10490. |
[15] | Eisenmenger LB , Peret A , Famakin BM , Spahic A , Roberts GS , Bockholt JH , Johnson KM , Paulsen JS ((2023) ) Vascular contributions to Alzheimer’s disease. Transl Res 254: , 41–53. |
[16] | Ikram MA , Brusselle G , Ghanbari M , Goedegebure A , Ikram MK , Kavousi M , Kieboom BCT , Klaver CCW , de Knegt RJ , Luik AI , Nijsten TEC , Peeters RP , van Rooij FJA , Stricker BH , Uitterlinden AG , Vernooij MW , Voortman T ((2020) ) Objectives, design and main findings until 2020 from the Rotterdam Study. Eur J Epidemiol 35: , 483–517. |
[17] | Rissin DM , Kan CW , Campbell TG , Howes SC , Fournier DR , Song L , Piech T , Patel PP , Chang L , Rivnak AJ , Ferrell EP , Randall JD , Provuncher GK , Walt DR , Duffy DC ((2010) ) Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol 28: , 595–599. |
[18] | Odink AE , Lugt Van Der A , Hofman A , Hunink MGM , Breteler MMB , Krestin GP , Witteman JCM ((2007) ) Association between calcification in the coronary arteries, aortic arch and carotid arteries: The Rotterdam study. Atherosclerosis 193: , 408–413. |
[19] | Odink AE , Van Der Lugt A , Hofman A , Hunink MGM , Breteler MMB , Krestin GP , Witteman JCM ((2010) ) Risk factors for coronary, aortic arch and carotid calcification; The Rotterdam Study. J Hum Hypertens 24: , 86–92. |
[20] | Bos D , Vernooij MW , Elias-Smale SE , Verhaaren BFJ , Vrooman HA , Hofman A , Niessen WJ , Witteman JCM , Van Der Lugt A , Ikram MA ((2012) ) Atherosclerotic calcification relates to cognitive function and to brain changes on magnetic resonance imaging. Alzheimers Dement 8: , S104–S111. |
[21] | Bos D , Ikram MA , Elias-Smale SE , Krestin GP , Hofman A , Witteman JCM , van der Lugt A , Vernooij MW ((2011) ) Calcification in major vessel beds relates to vascular brain disease. Arterioscler Thromb Vasc Biol 31: , 2331–2337. |
[22] | Bos D , Van Der Rijk MJM , Geeraedts TEA , Hofman A , Krestin GP , Witteman JCM , Van Der Lugt A , Ikram MA , Vernooij MW ((2012) ) Intracranial carotid artery atherosclerosis: Prevalence and risk factors in the general population. Stroke 43: , 1878–1884. |
[23] | De Weert TT , Cakir H , Rozie S , Cretier S , Meijering E , Dippel DWJ , Van Der Lugt A ((2009) ) Intracranial internal carotid artery calcifications: Association with vascular risk factors and ischemic cerebrovascular disease. Am J Neuroradiol 30: , 177–184. |
[24] | Agatston AS , Janowitz WR , Hildner FJ , Zusmer NR , Viamonte M , Detrano R ((1990) ) Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol 15: , 827–832. |
[25] | Kuiper LM , Ikram MK , Kavousi M , Vernooij MW , Ikram MA , Bos D ((2021) ) C-factor: A summary measure for systemic arterial calcifications. BMC Cardiovasc Disord 21: , 317. |
[26] | Hoogendam YY , Hofman A , Van Der Geest JN , Van Der Lugt A , Ikram MA ((2014) ) Patterns of cognitive function in aging: The Rotterdam Study. Eur J Epidemiol 29: , 133–140. |
[27] | Keuss SE , Coath W , Nicholas JM , Poole T , Barnes J , Cash DM , Lane CA , Parker TD , Keshavan A , Buchanan SM , Wagen AZ , Storey M , Harris M , Malone IB , Sudre CH , Lu K , James SN , Street R , Thomas DL , Dickson JC , Murray-Smith H , Wong A , Freiberger T , Crutch S , Richards M , Fox NC , Schott JM ((2022) ) Associations of β-amyloid and vascular burden with rates of neurodegeneration in cognitively normal members of the 1946 British Birth Cohort. Neurology 99: , E129–E141. |
[28] | Gottesman RF , Schneider ALC , Zhou Y , Coresh J , Green E , Gupta N , Knopman DS , Mintz A , Rahmim A , Sharrett AR , Wagenknecht LE , Wong DF , Mosley TH ((2017) ) Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317: , 1443–1450. |
[29] | van Arendonk J , Neitzel J , Steketee RME , van Assema DME , Vrooman HA , Segbers M , Ikram MA , Vernooij MW ((2023) ) Diabetes and hypertension are related to amyloid-beta burden in the population-based Rotterdam Study. Brain 146: , 337–348. |
[30] | Yan S , Qi Z , An Y , Zhang M , Qian T , Lu J ((2019) ) Detecting perfusiondeficit in Alzheimer’s disease and mild cognitive impairmentpatients by resting-state fMRI.. J Magn Reson Imaging 49: , 1099–1104. |
[31] | Barnes JN , Pearson AG , Corkery AT , Eisenmann NA , Miller KB ((2021) ) Exercise, arterial stiffness, and cerebral vascular function:Potential impact on brain health. J Int Neuropsychol Soc 27: , 761–775. |
[32] | Gupta A , Iadecola C ((2015) ) Impaired Aβ clearance: A potential link between atherosclerosis and Alzheimer’s disease. Front Aging Neurosci 7: , 115. |
[33] | Xie B , Shi X , Xing Y , Tang Y ((2020) ) Association between atherosclerosis and Alzheimer’s disease: A systematic review and meta-analysis.. Brain Behav 10: , e01601. |
[34] | Nelson AR , Sweeney MD , Sagare AP , Zlokovic B V. ((2016) ) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 1862: , 887–900. |
[35] | Kapasi A , Schneider JA ((2016) ) Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim Biophys Acta 1862: , 878–886. |
[36] | Beishon L , Panerai RB ((2021) ) The neurovascular unit in dementia: An opinion on current research and future directions. Front Aging Neurosci 13: , 721937. |
[37] | Gottesman RF , Mosley TH , Knopman DS , Hao Q , Wong D , Wagenknecht LE , Hughes TM , Qiao Y , Dearborn J , Wasserman BA ((2020) ) Association of intracranial atherosclerotic disease with brain β-amyloid deposition. JAMA Neurol 77: , 350. |
[38] | Lane CA , Barnes J , Nicholas JM , Sudre CH , Cash DM , Parker TD , Malone IB , Lu K , James SN , Keshavan A , Murray-Smith H , Wong A , Buchanan SM , Keuss SE , Gordon E , Coath W , Barnes A , Dickson J , Modat M , Thomas D , Crutch SJ , Hardy R , Richards M , Fox NC , Schott JM ((2019) ) Associations between blood pressure across adulthood and late-life brain structure and pathology in the neuroscience substudy of the 1946 British birth cohort (Insight 46): An epidemiological study. Lancet Neurol 18: , 942–952. |
[39] | Lane CA , Barnes J , Nicholas JM , Baker JW , Sudre CH , Cash DM , Parker TD , Malone IB , Lu K , James SN , Keshavan A , Buchanan S , Keuss S , Murray-Smith H , Wong A , Gordon E , Coath W , Modat M , Thomas D , Hardy R , Richards M , Fox NC , Schott JM ((2021) ) Investigating the relationship between BMI across adulthood and late life brain pathologies. Alzheimers Res Ther 13: , 91. |
[40] | Boyle PA , Yu L , Leurgans SE , Wilson RS , Brookmeyer R , Schneider JA , Bennett DA ((2019) ) Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol 85: ,114–124. |


