
How Periodontitis or Periodontal Bacteria Can Influence Alzheimer’s Disease Features? A Systematic Review of Pre-Clinical Studies
Abstract
Background:
The negative effects of periodontitis on systemic diseases, including diabetes, cardiovascular diseases, and Alzheimer’s disease (AD), have been widely described.
Objective:
This systematic review aimed to gather the current understanding of the pathophysiological mechanisms linking periodontitis to AD.
Methods:
An electronic systematic search of the PubMed/MEDLINE, Scopus, and Embase databases was performed using the following PECO question: How can periodontitis or periodontal bacteria influence Alzheimer’s disease features?”. Only preclinical studies exploring the biological links between periodontitis and AD pathology were included. This study was registered at the International Prospective Register of Systematic Reviews (PROSPERO), and the Syrcle and Camarades protocols were used to assess the risk of bias.
Results:
After a systematic screening of titles and abstracts (n = 3,307), thirty-six titles were selected for abstract reading, of which 13 were excluded (k = 1), resulting in the inclusion of 23 articles. Oral or systemic exposure to periodontopathogens or their byproducts is responsible for both in situ brain manifestations and systemic effects. Significant elevated rates of cytokines and amyloid peptides (Aβ) and derivate products were found in both serum and brain. Additionally, in infected animals, hyperphosphorylation of tau protein, hippocampal microgliosis, and neuronal death were observed. Exposure to periodontal infection negatively impairs cognitive behavior, leading to memory decline.
Conclusions:
Systemic inflammation and brain metastatic infections induced by periodontal pathogens contribute to neuroinflammation, amyloidosis, and tau phosphorylation, leading to brain damage and subsequent cognitive impairment.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder that affects more than 35 million individuals worldwide and more than 10 million individuals in Europe [1]. AD is a multifactorial disease characterized by early synaptic and neuronal dysfunction, which induces amnesia, cognitive impairment, and subsequently dementia [2]. The AD brain physiopathology involves lesions characterized by the presence of extracellular insoluble senile plaques formed by amyloid-β (Aβ) peptide and the presence of intraneuronal neurofibrillary tangles (NFTs) formed by hyperphosphorylation of the tau protein (p-Tau) (tauopathy) [1, 3, 4]. Moreover, neuroinflammation is considered a key factor in both the onset [5] and progression of AD [6].
Periodontal disease (PD) is a chronic bacterial inflammatory disease [7, 8] that affects 20–50% of the overall population [9]. The disease is characterized by colonization by gram-negative bacteria, which induce local inflammation inside the periodontium, with progressive destruction of the supporting tissues around the teeth [10–12]. Second, owing to mastication and tooth brushing [13], periodontal bacteria or their byproducts can be released into the bloodstream, resulting in transient or prolonged bacteremia that influences systemic diseases or conditions, such as cardiovascular pathology or diabetes [14]. Furthermore, recent systematic reviews of clinical studies have shown a positive association between periodontitis and AD [15–17]. On the one hand, the presence of PD is associated with the presence of AD [15], while on the other, patients with dementia show significantly worse clinical periodontal variables than healthy individuals do [16]. Additionally, Nadim et al. [17] provided evidence that PD increased the risk of incident dementia and speculated that a 50% reduction in the current prevalence of PD in the population could save 850,000 patients from dementia worldwide while preventing and treating PD could contribute to controlling the global dementia epidemic.
Inflammation is a risk factor shared by these two diseases, influencing their onset and progression [18, 19]. Indeed, in AD, the brain accumulation of Aβ peptide activates glial cells, lymphocytes, and macrophages, which release inflammatory mediators such as cytokines, chemokines, neurotransmitters, and reactive oxygen species (ROS) [20, 21]. In addition, the tauopathy induces the synthesis of pro-inflammatory cytokines as tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and the production of C-reactive protein (CRP). Furthermore, PD induces local degradation of the extracellular matrix, infiltration of neutrophils, macrophages, and lymphocytes, and the production of proteases (matrix metalloproteases [MMPs] and serine proteases) inside the periodontium, leading to the genesis and progression of the disease [22, 23]. Systemically, PD induces, inside the bloodstream, the production of inflammatory molecules such as IL-1β, IL-6, IL-8, TNF-α, and CRP [24, 25], that in turn increase systemic inflammation and influence the progression of systemic diseases [26] such as AD [27–29].
However, the pathophysiological mechanisms that link AD and periodontitis remain poorly investigated [30, 31]. Therefore, the present systematic review aims to explore and gather the identified mechanisms linking the PD to AD pathology, throughout in vivo animal studies to answer the major question: “How periodontitis or periodontal bacteria can influence Alzheimer’s disease features?”
MATERIALS AND METHODS
Study design
The present study was designed as a systematic review focusing on the relationship between AD and PD. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA-SR) checklist and guidelines were followed. This study was initially registered as CRD42022257306 in the Prospective International Registry of Systematic Reviews (PROSPERO).
Focus question and search strategy
The focus question and PECO framework (P: population, E: exposure, C: control, O: outcome) were used in order to answer the following research question: “How periodontitis or periodontal bacteria can influence Alzheimer’s disease features?” This question was addressed according to the following criteria.
- Population: Animals with AD pathology
- Exposure: Exposed to periodontitis or periodontal bacteria
- Control: Not exposed to periodontitis or periodontal bacteria
- Outcome:
1. In the brain: neuroinflammation, expression of pro-inflammatory cytokines (TNF-α, IL-6, Il-1 β), microglial cell activity, accumulation of amyloid beta plaques, and intraneuronal neurofibrillary tangles.
2. In the blood: CRP and pro-inflammatory cytokines (TNF-α, IL-6, Il-1β).
3. Cognitive behavior.
The search strategy (Fig. 1) consisted of each database’s controlled vocabulary (MeSH terms) and free keywords. Supplementary Table 1 presents the search algorithm. An electronic search of the literature was performed using PubMed/MEDLINE, Scopus, and Embase databases. Selected publications were collected using the Mendeley Reference Manager software, and the Cochrane tool (COVIDENCE) was used for screening and data extraction.
Fig. 1
Search strategy. Diagram of article selection process.
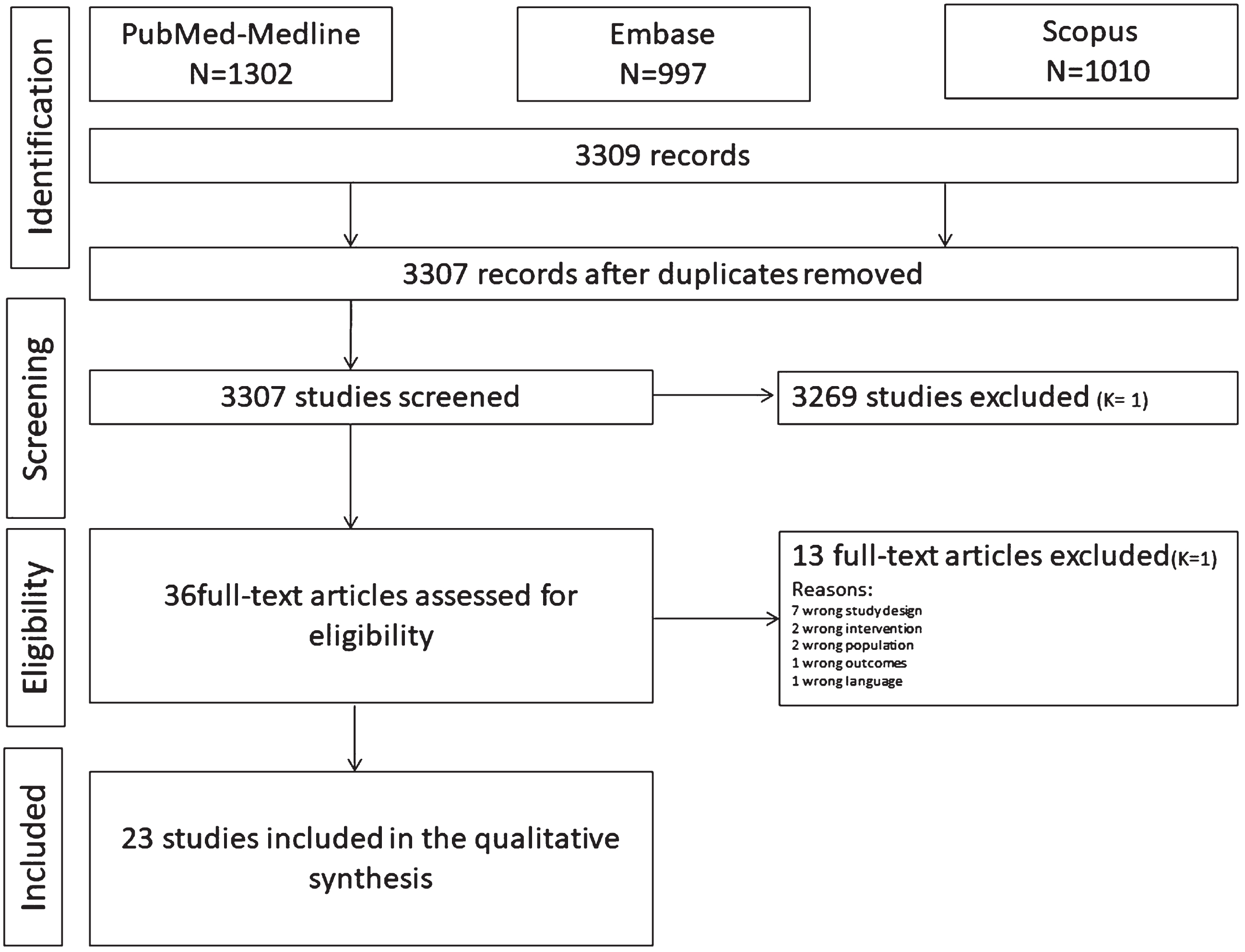
Table 1
Study characteristics
| Aim/Hypothesis | Study animal | Study design | AD model | Perio-model | ||||
| Intervention: C | Administration route | Dose: | Follow-up | |||||
| Gu 2020 [35] | Periodontitis aggravates AD by exacerbating bone loss and AD pathologies, with IL-6 and IL-17 as the common factors for bone loss and AD-like pathologies. | Female mice (n = 16) DBA/2 background | 2 groups: 1) test (n = 8): systemic exposure to Pg-LPS 2) control (n = 8): no systemic exposure to PgLPS | Open field test evaluate the general locomotor activity levels | Chronic systemic exposure to Pg-LPS | i.p. | 0.03 mg/mL, 1 mg/kg/day | 3 weeks |
| Zeng 2020 [52] | RAGE in cerebral endothelial cells contributes to the peripheral Aβ influx during chronic Pg infection. | Female mice (n = 12) C57BL/6JJmsSlcJapan SLC | 2 groups: 1) Test (n = 6): systemic Pg infection 2) Control (n = 6): no systemic Pg infection (saline) | AD assessment: Passive avoidance test (test memory decline): device included a bright compartment and a dark compartment | Chronic systemic infection to Pg | i.p. | 1×108 CFU/mouse, every 3 days | 3 weeks |
| Han 2019 [36] | Extracellular RNAs in periodontopathogenic (Aa) OMVs promote TNF-α production in human macrophages and cross the BBB. | Male mice C57BL, 6 weeks old | NA | NA | Injections of Aa OMV solution | cardiac injections | 100 ml of Aa OMV solution for 4 or 24 h | |
| Leira 2019 [15] | Determine the serum levels of Aβ1–40 and Aβ1–42 using the Pg-LPS model to induce periodontitis. | Male Sprague-Dawley rats of 7 weeks of age (n = 6) | 1 group: Experimental periodontitis (oral infection) group (n = 6) | Serum levels of Aβ1–40 and Aβ1–42 peptides were measured by ELISA technique | Oral infection/exposure to Pg-LPS | Gingival injections of a Pg-LPS saline solution | 10μL of a saline solution containing 1 mg/mL Pg-LPS, 3 days per weeks at 48-hours intervals during 2 weeks (6 injections) | 21 days |
| Wang 2021 [37] | To investigate neuroinflammation under different periodontal status. | 8-week-old C57BL/6 male mice (n = 80) | 80 mice randomly divided in 3 groups: 1) Control (n = 30) 2) Molar ligation group (Lig group n = 25) 3) LPS injection group (Lps group n = 25) | Neuroinflammation state assessed by glial cell proliferation and proinflammatory factor expression: 1) Microgliosis and astrogliosis were determined by IF (M) and IHC (A) 2) IL-1β were assessed by ELISA | Oral infection/exposure to E. coli-LPS | Molar ligation or periodontal injection of E. coli-LPS | 1μg/μL and 5 uL once every 2 days | 2, 4, 8, and 12 weeks |
| Diaz Zuniga 2020 [38] | The encapsulated Pg strains could trigger the appearance of brain AD-markers, neuroinflammation, and cognitive decline. The immune response in the brain differs depending on the infecting capsular Pg serotype (K1, K2, and K4 encapsulated strains). | Sprague-Dawley young male rats (4 weeks old, n = 24) | 4 experimental groups and 1 sham-infected group: 1) control (K1-isogenic non-encapsulated mutant (n = 6) 2) serotype K1 (n = 6) 3) serotype K2 (n = 6) 4) serotype K4 (n = 6) 5) sham-infected group (n = ?) | 1) Oasis Maze task to evaluate hippocampal-dependent spatial memory 2) Levels of Aβ1-42 peptide, IL-1beta, IL-4, IL-6, IL-10, TNF-alpha, IFN-gamma, in hippocampal homogenates, serum and CSF samples | Oral infection/exposure to multipe Pg strains | 2 palatal injections | 2 injections of 1010 CFU/ml at 1 week interval | 6 consecutive days after peridontitis inoculation (45 days) |
| Hayashi 2019 [54] | Brain exposure of Pg-LPS triggers the onset of AD. | 6 month-old and 13-month-old 5XFAD mice (n = 80) | n = 80 mice divided in 6 groups for experiment 1, 2, and 3:1) Experiment 1:6 months-old mice (n = 20) divided in 2 groups: a. Control (n = 10); b. Treated with Pg-LPS (n = 10) 2) Experiment 2:13 months-old mice (n = 20) divided in 2 groups: a. Control (n = 10); b. Treated with Pg-LPS 3) Experiment 3:6 months-old (n = 12) divided in 2 groups: a. Control (n = 6); b. Treated with Pg-LPS (n = 6) | AD model mice: Cognitive functions monitored by Y maze, nest building and Morris water maze tests | ICV injection of Pg-LPS | a. Experiment 1: Single ICV injection of Pg-LPS b. Experiment 2 and 3: continuous ICV injection of Pg-LPS in saline | a. Experiment 1: Single ICV injection of 2μg Pg-LPS/day at a rate of 0.2μL/min b. Experiment 2 and 3: continuous ICV injection of 2μg Pg-LPS/day at a rate of 0.11μL/h | NA |
| Hu 2020 [45] | To investigate the association between periodontitis and cognitive impairment. | 10-week-old male Sprague-Dawley rats (n = 32) | 4 groups: 1) control group 2) Pg-LPS group 3) Pg-LPS + TAK 242 group 4) TAK-242 group | AD assessment: Cognitive function (learning and memory) assessed using Morris water maze test | Oral infection/exposure to Pg-LPS | Topical application of Pg-LPS into the palatal gingival sulcus of the maxillary first molars | 0.5 mg/kg, twice a week | 10 weeks |
| Su 2021 [49] | To investigate the effect and mechanisms of Td on the accumulation of Aβ in the hippocampus | 8 week-old C57BL/6 background mice (n = ?) | 3 groups: 1) Td group (experimental group) 2) Pg group (positive control group) 3) negative control group | AD assessment: AD pathological marker: Intra- and extracellular Aβ1–40 and Aβ1–42 accumulation in the hippocampus | Oral infection/exposure to Pg and Td | Oral | NA, infection 3 times a week | 24 weeks |
| Poole 2015 [50] | To investigate that periodontalbacteria (Pg, Td, Tf, Fn) and/or their byproducts can access the brain and induce local CNS inflammation. | 8-week-old male ApoE-/- mice strain B6.129P2-Apoetm1Unc/J (n = 36) | 3 groups: 1) sham-infected or control (n = 12) 2) mono-infected (n = 12) 3) polymicrobial infected (n = 12) | AD assessment: Local CNS inflammatory/immune response: glial cells priming leading to neuronal lysis | Oral infection/exposure to Pg, Td, Tf, Fn either as mono- or polybacterial | Oral | NA | 24 weeks |
| Furutama 2020 [46] | To investigate the involvement of periodontal inflammation in neuroinflammation and BBB disruption. | Wild-type (WT) C57BL/6j mice (8- to 12-week-old females), (n = ?) | 2 groups: 1) ligature-induced PD mice group 2) control mice group | Cognitive impairment assessed through: 1) Inflammation in the hippocampus: increased IL-1beta, TNF-alpha, and IL-6 levels. 2) BBB disruption | Ligature induced periodontitis around the second maxillary molars | Oral | NA | NA |
| Wu 2017 [47] | To hypothese that Cat B plays a critical role in the initiation of neuroinflammation and neural dysfunction following chronic systemic exposure to LPS from PgLPS in mice. | Heterozygous CatB deficient mice on a C57BL/6 background Homozygous CatB-deficient mice (CatB-/-) mice (n = 30) | 5 groups (6 mice per group): 1) control group: heterozygous CatB-deficient mice 2) Wild-type (WT) adult/young mice (2 months old, heterozygous CatB-deficient) 3) WT middle-aged mice (12 months old, heterozygous CatB-deficient) 4) WT adult/young mice (2 months old, homozygous CatB deficient) 5) WT middle-aged mice (12 months old, homozygous CatB deficient) | Learning and memory function assessed using: passive avoidance test and expression of AβPP, CatB, TLR2, and IL-1beta in brain tissues | Chronic systemic exposure t Pg-LPS | i.p. | 1 mg/Kg daily | 5 weeks |
| Liu 2017 [39] | To evaluate the effect of Pg gingipains Arg-gingipain (Rgp) and Lys-gingipain (Kgp) on the cellular activation of brain-resident microglia. | CX3CR1+/GFP mice on a C57/BL6J background (8–10 weeks old) | NA | Intracerebral injection (somatosensory cortex): Pg (1μl, 1×106 CFU/ml) was injected | Intracerebral injection | Pg Intra-cerebral injection Pg | 1μl, 1×106 CFU/ml) | NA |
| Zhang 2020 [40] | To examine the association between oral health and cognition. | Male Sprague-Dawley rats, 12 weeks old (n = 24) | 4 groups (n = 6 per group): 1) Control 2) Ligature-induced periodontitis (Lig) 3) AD 4) Lig+AD | Aβ25-35-induced model of AD: Injection of 5μL of prepared Aβ25–35 per hemisphere into the bilateral hippocampus over 5 min AD assessment: Spatial memory and learning assessment through Morris Water Maze test | Ligature induced periodontitis around the second maxillary molars | Oral | NA | 18 days |
| Zhang 2018 [48] | To investigate the effects of Pg-LPS on cognitive function | 8-week-old male C57BL/6 mice (n = 60) | 5 groups: 1) Pg-LPS group 2) E. coli-LPS group 3) TAK (Toll-like receptor 4 inhibitor) group 4) Pg-LPS + TAK group 5) Control group | Cognitive function assessed through: Morris Water Maze test, Open Field test, and Passive Avoidance test | Systemic exposure to Pg-LPS | i.p. | 1 mg/Kg daily | 5 weeks |
| Singhrao 2017 [51] | To investiagte that Pg infection accelerates the appearance of age-related granules in ApoE–/–mice brains. | Male ApoE–/–mice (strain B6.129P2-Apoetm1Unc/J), 10 weeks of age (n = ?) | 2 groups: 1) infected group 2) sham-infected group | 1) Intra-cerebral localization of Pg detected by fluoresence in situ (FISH) 2) Age-related granules observed by periodic acid-Schiff (PAS) | Oral infection/exposure to Pg FDC 381 | Oral | 5×109 bacteria per mL | 24 weeks |
| Cueno 2018 [33] | To evaluate the effect of BA produced by periodontopathic bacteria on systemic inflammation and neuroinflammation in brain (hippocampus, pineal gland, hypothalamus, cerebrum, and cerebellum). | 10-week-old male Wistar rats (n = 12) | 2 groups: 1) BA-treated group 2) Control group | 1) AβPP and presinilin (PS1 and PS2) blood levels detection by western blotting 2) Brain levels of tau protein measured by ELISA | Oral infection/exposure to BA | Injection in the gingival mucosa | 10μL of 13C n-butyrate solution (1 M) | 6 and 12 h after BA injection |
| Kantarci 2020 [41] | To investigate that periodontitis impacts the inflammatory process in the brain and the microglia function. | 8-month-old male 5xFAD (n = 15) and B) non-transgenic male littermate (Wild Type; n = 15) mice | 2 groups: 1) AD-modeled mice (A); 5xFAD mice co-express human APP and presinilin 1 (PS1) 2) control group (B) 8 in each group mice were treated to induce experimental PD and 7 mice in each group were left untreated. | 1) Analysis of Aβ deposits and microglia by IHC 2) Analysis of Aβ brain levels by ELISA 3) Analysis of brain inflammatory cytokines and chemokines by multiplex immunoassay | Ligature induced periodontitis around the maxillary right and left second molars. | Oral | NA | NA |
| Hu 2021 [45] | To investigate the role of the STAT3 signaling pathway related to peridontitis on neuroinflammation and cognitive impairement. | 10-week-old male Sprague Dawley rats (n = 32) | 4 groups: 1) control 2) Chronic periodontitis (CP) 3) CP + pSTAT3 inhibitor cryptotanshinone (CTS) 4) CTS | 1) Locomotor activity and cognitive function of mice by open field test and Morris water maze test 2) Microglia and astrocytes activation in the mice cortex and hippocampus by IHC 3) Interleukins expression in the mice cortex via RT-PCR and ELISA 4) AβPP and its key secretases (including alpha-, beta-, and gamma- secretases) by RT-PCR. 5) Aβ protein levels and the ratio of Aβ1-40/1-42 in the plasma and cortex by ELISA 6) Aβ1-42 levels in the mice brains by IHC | Ligature induced periodontitis around the maxillary first molar | Oral | NA | 72 days |
| Intervention: C | Administration route | Dose: | Follow-up | |||||
| Ilievski 2018 [43] | To investigate the effect of the Pg exposition on neuroinflammation, neurodegeneration, microgliosis, astrogliosis and formation of intra- and extracellular amyloid plaque and NFTs. | 8-week-old C57BL/6 Wild Type (WT) mice (n = 20) | 2 groups (10 mice per group): 1) Mice oral administedred Pg/gingipain 2) control group | 1) Pg/gingipain detection in the hippocampus of mice via IF, confocal microscopy and qPCR 2) Neuroinflammation in the hippocampus observed with RT-PCR and IF microscopy 3) Neurodegeneration in the hippocampus via RT-PCR and Fluoro Jade C positivity 4) Aβ42 accumulation in the hippocampus and frontal cortex 5) Intracellular Aβ42 formation in astrocytes 6) Phospho-tau and NFT detection by IHC 7) Microgliosis and astrogliosis in the hippocampus | Oral infection/exposure to Pg/gingipain | Oral | 100μl of Pg in CMC containing 109 Pg | 22 weeks |
| Nie 2019 [53] | To investigate that Pg infection induces Aβ accumulation in monocytes/macrophages. | 12-month-old female mice on a C57BL/6J background (n = 20) | 2 groups (10 mice per group): 1) Pg infected group 2) control group | 1) Aβ producing molecules in the liver macrophages: q-PCR 2) AβPP, CatB, and Aβ production in the liver of macrophages: IF | Systemic exposure to Pg | i.p. | 1×108 CFU/mouse every 3 days | 3 weeks |
| Dominy 2019 [44] | To design and synthetize molecule inhibitors of Pg gingipains that contribute to modify AD. | Experiment I: Study the Aβ1-42 levels in the brains of orally infected mice a. Experiment I.1:40 female BALB/c mice, 43- to 44- week old (n = 8 per arm) b. Experiment I.2:100 female BALB/c mice, 8 week-old (n = 10 per arm) c. Experiment I.3:70 (n = 10 per arm) 8-week-old female BALB/c mice. | Experiment I: Study the Aβ1-42 levels in the brains of orally infected mice a. Experiment I.1:40 female BALB/c mice, 43- to 44- week old (n = 8 per arm). 2 groups: 1) mice infected with Pg + gingipains inhibitors (COR119) 2) Control group. b. Experiment I.2:100 female BALB/c mice, 8 week-old (n = 10 per arm). 3 groups: 1) mice infected with Pg + gingipains inhibitors (COR271 and COR286) or moxifloxacin 2) Mock-infected and Pg W83 (WT)-infected mice 3) control group c. Experiment I.3:70 (n = 10 per arm) 8-week-old female BALB/c mice. 2 groups: 1) mice infected with Pg + gingipains inhibitors (COR388 or COR271) 2) control group | AD features assessment: 1) Aβ and TNF-alpha mice brain levels quantification by ELISA 2) Pg detection in mice brain by qPCR 3) Quantification of hippocampal Gad67+ interneurons by IHC | Chronic Oral infection/exposure to Pg, by ligature placement around the upper maxillary left and right second molars + topical application of bacterial solution to the buccal surface of the maxillae | Oral | 100μl of the bacterial solution | For 6 weeks every other day |
| a. Experiment I.1:40 female BALB/c mice, 43- to 44- week old (n = 8 per arm) | Experiment II: Study neurodegeneration after stereotactic injection of gingipains (Kgp and RgpB combination) into mouse hippocampus (CA1 area). Fifteen 8-week-old male BALB/c mice. 2 groups: 1) mice infected with Pg gingipains + gingipains inhibitors (COR271 and COR286) 2) control group | Brain exposure (Hippocampus CA1 area) via stereotactic injections | Oral | Bilateral injections of 0.5μl of gingipains solution were made into coordinates from bregma: anteroposterior –2.0, lateral±1.5, and ventral –1.4 mm from dura at a rate of 0.1μl/min with a 5 min rest period | 7 days later | |||
| Tang 2020 [42] | To evaluate that Pg infection triggers neuroinflammation and induce tau hyperphosphorylation in brain. | b. Experiment I.2:100 female BALB/c mice, 8 week-old (n = 10 per arm) c. Experiment I.3:70 (n = 10 per arm) 8-week-old female BALB/c mice | 4 groups (10 rats per group): 1) experimental group 1 2) experimental group 2 3) control group 1 4) control group 2 | AD parameters assessed through: 1) Neuroinflammation by measuring the proinflammatory cytokines levels (IL-1B, TNF-alpha, and IL-6) in the hippocampus of rats 2) Tau hyperphosphorylation in the hippocampus of rats using western blotting | Systemic exposure to Pg | i.v. | 10∧8 CFU/ 200μL per rat 3 times a week | 24 h after the last injection. |
AD, Alzheimer disease; Aβ, amyloid-β peptide; AβPP, amyloid-β protein precursor; BA, butyric acid; BBB, blood-brain barrier; Cat, cathepsin; CFU, colony forming unit; CSF, cerebrospinal fluid; E. coli, Escherichia coli; ELISA, enzyme-linked immunosorbent assay; Fn, Fusobacterium nucleatum; ICV, intracerebroventricular; IF, immunofluorescence; IFN, interferon; IHC, immunohistochemistry; IL, interleukin; i.p., intraperitoneally; LPS, lipopolysaccharide; NFTs, neurofibrillary tangles; OMVs, outer membrane vesicles; PCR, polymerase chain reaction; Pg, Porphyromonas gingivalis; RAGE, receptor for advanced glycation endproducts; Td, Treponema denticola; Tf, Tannarella forsytensis; TLR, toll-like receptor; TNF, tumor necrosis factor.
Study selection and data extraction
Procedure for study selection
Two independent blinded reviewers (LS and YA) performed study selection using the online Cochrane platform (COVIDENCE). The reviewers (LS, YA,) first screened titles, then abstracts, and finally full texts of the 3307 articles identified by electronic and manual searches. Agreement between examiners was generated using Cohen’s kappa (κ) coefficient at each level of selection. Records were excluded only to enhance sensitivity if both reviewers excluded them based on their titles. Disagreements were resolved by discussion with a third reviewer (FL) resolved the issue.
Inclusion and exclusion criteria
• The inclusion criteria were the following:
- Animal studies (preclinical studies)
- Species: mice and rats
- In vivo studies
- Studies focusing on the relationship between periodontitis or periodontal bacteria and AD features.
- The studied animals must suffer from both AD pathology and periodontitis
- The experimental animals must display one or more of the following AD characteristics:
1. Cognitive impairment
2. Pro-inflammatory cytokines in the blood or brain (IL-1β, IL-6, and TNF-α): systemic inflammation and/or neuroinflammation
3. Neurodegeneration
4. Microgliosis and astrogliosis
5. Aβ peptides levels in blood or in the brain
6. NFTs and tau protein phosphorylation in the extracellular area of the brain
• Experimental animals must display periodontitis manifested through a ligature or by oral or systemic exposure to:
1. Periodontal bacteria or periodontal genetic materials (DNA, RNA)
2. Byproducts such as lipopolyssacharides (LPS) and endotoxins
• The exclusion criteria were the following:
1. Human study
2. In vitro study
3. Review articles
4. Studies in other species
5. In silico studies
6. Study not involving AD pathology and periodontitis
7. Any outcomes not related to both AD pathology and periodontitis
8. Animals with systemic disease (cardiovascular disease, diabetes)
9. Use of medical drugs (antibiotics)
Methods for data extraction
All selected studies were extracted from the three main databases and sorted according to each database using Mendeley software, a bibliographic tool. The data were then sent from Mendeley to the COVIDENCE platform for extraction. All information extracted from the articles (see the list below) was grouped, filled in, and checked by two authors to verify inconsistencies and ensure data accuracy using Microsoft Office Excel 2022.
In the case of missing data, the authors were contacted by e-mail to collect the required information.
Data to be extracted
• Authors and year of publication
• Aim/hypothesis
• Studied animals (specie, age, gender, background)
• Study design (sample size, treatment groups)
• Methods for AD features assessment and AD model induction
• Methods for periodontitis induction and characterization (intervention)
• Effects of periodontitis on both AD-related systemic inflammation and neuroinflammation
• Consequences of systemic and brain inflammation on animals’ cognitive behavior
• Conclusion of the authors
Statistical analysis
The level of agreement between the reviewers was calculated using the Cohen κ coefficient at each level of selection. The Kappa test was performed directly using the Covidence tool. The selection was performed according to inclusion and exclusion criteria.
Assessment of study quality
To assess the risk of bias and consistency of the studies, two reviewers rated each article independently based on its quality by 2 reviewers, using the criteria outlined in the Systematic Review Centre for Laboratory Animal Experimentation Office of Health Assessment (SYRCLE) bias tool. This tool contains 10 entries related to selection, performance, detection, attrition, reporting, and other biases (Supplementary Table 2).
Methodological quality assessment was also performed using the collaborative approach to meta-analysis and review of Animal Data from Experimental Studies (CAMARADES) checklist items. This checklist comprised 22 independent items (see Supplementary Table 3). To evaluate study quality, scores were calculated as 1 (V = fulfilling the criterion) or 0 (N = not fulfilling the criterion) for each item on the checklist. After providing scores for study quality, the scores of each study were summed (Supplementary Table 3).
RESULTS
Search and selection
The search strategy and characteristics of the included studies are shown in Fig. 1 and Table 1. The Embase, Scopus, and PubMed search strategies resulted in 3,307 articles. After discarding duplicates, two calibrate reviewers (YA and LS) screened the titles, and 3,271 articles were excluded by the first (LS) and second reviewers (YA), resulting in a Cohen κ coefficient of 1. Thirty-six titles were selected for the abstract reading, of which 13 were excluded (k = 1). After the full-text assessment, no additional articles were excluded. Ultimately, both reviewers agreed to include 23 articles in the systematic review. Studies were excluded, and the reasons for exclusion are shown in Fig. 1.
Assessment of study quality
Using the CAMARADES tool, 15 studies showed a low risk of bias, two exhibited a moderate risk of bias, and six displayed a high risk of bias.
Regarding the SYCRLE assessment, 15 studies were associated with a low risk of bias, 1 study presented a moderate risk of bias, and 7 studies demonstrated a high risk of bias.
For both tools, most studies (65.3%) had a low risk of bias.
Study characteristics
The relationship between AD pathology and periodontitis was explored by evaluating the effects of periodontal inflammation and periodontal bacteria on systemic inflammation (n = 13 studies) [32–34], brain tissue damage (n = 21 studies) [33–48], and the consequences on cognitive behavior (n = 9 studies) [34, 35, 38, 40, 45, 47–50].
Descriptions of the included articles are summarized in Table 1. The effects of periodontitis on systemic inflammation (SI), brain tissue damage, and their consequences on cognitive behavior are described in Table 2.
Periodontitis assessment
Periodontitis was established using experimental oral infection models (n = 14) [32–34, 37, 38, 40, 41, 43–45, 49–51] or by systemic injection of periodontal bacteria or their byproducts (n = 9) [35, 36, 42, 47, 48, 52, 53].
• Experimental oral infection models
Oral infection models were established either by gingival injection of periodontal bacteria (Porphyromonas gingivalis, Pg) and/or their byproducts (Pg-lipopolysaccharide (Pg-LPS), Escherichia coli-LPS (Ec-LPS), butyric acid (BA)) (n = 4) [32, 33, 37, 38] or molar ligation (ligature-induced periodontitis) (n = 5) [34, 40, 41, 44, 46], or both (n = 1) (37). Oral inoculation with periodontal bacteria (Pg, Treponema denticola (Td), Tannerella forsythia (Tf), and Fusobacterium nucleatum (Fn)) [43, 49–51] was also used in five studies (n = 5).
• Systemic injection of periodontal bacteria or their byproducts
The injections of periodontal bacteria (Pg, Td, Tf, Fn) and/or their byproducts (Pg-LPS), Aggregatibacter actinomycetemcomitans (Aa) outer membrane vesicles (Ec-LPS)) were done intraperitoneally (n = 5) [35, 47, 48, 52, 53], intracerebrally (n = 3) [39, 52, 54], or intracardially (n = 1) [36].
Alzheimer’s disease induction models and assessment
To establish an AD model, animals were either genetically modified (n = 19) [35] or exposed to Aβ peptides by brain injections (n = 4) [39].
AD pathology was assessed using cognitive function tests (n = 9) [34, 35, 38, 40, 45, 47, 48, 52, 54] by evaluating the neuroinflammatory state (n = 7) [34, 40–43, 46, 50] by measuring the concentration of Aβ peptides (or its precursors) in different samples, such as serum (n = 4) [32–34, 38], brain tissues (hippocampus, cerebral cortex, and periventricular area) (n = 9) [34, 41, 43, 44, 47–49], and cerebrospinal fluid (n = 1) [38]. Additionally, the brain levels of tau protein (tau hyperphosphorylation) (n = 4) [33, 38, 42, 43] and NFTs (n = 1) [43] were quantified to define AD pathology.
• Cognitive function
The cognitive function tests included the open field test (n = 3) [34, 35, 48], passive avoidance test (n = 3) [47, 48, 52], oasis maze (n = 1) [38], Morris water maze (n = 5) [34, 40, 45, 48, 54], Y maze (n = 1) [54], and the nest building test (n = 1) [54].
• Neuroinflammation
The neuroinflammatory state was assessed by quantification of pro-inflammatory molecules expression in the brain, such as TNF-α, IL-1β, IL-6, IL-17, IL-8, iNOS, CRP, MMP-9, free fatty acid, hydrogen peroxide, GM-CSF, IFN-gamma, MCP-1, IL-21, and Iba-1, (n = 16) [33–48] and by measuring the glial cell activity (n = 10) [34, 37, 39, 41–43, 45, 47, 48, 50].
• Blood quantification
The concentration of Aβ peptides and the ratio of Aβ1 - 40/1 -42 were investigated in the serum (n = 3) [32, 34, 45]. Furthermore, blood levels of amyloid-β protein precursor (AβPP), its key secretases (including α-, β-, and γ-secretases), and presenilin (PS1 and PS2) [33, 34, 43] (n = 3) were measured to characterize AD.
• Brain tissue
Aβ peptide concentration was investigated in the hippocampal neurons, cerebral cortex, and periventricular area (n = 8) by quantification of Aβ1–40 and Aβ1–42. Additionally, the ratio of Aβ1 - 40/1 -42 (n = 1) [34], as well as the expression of AβPP, Cathepsin B (CatB), and Toll-like receptor-2 (TLR2), were also quantified in the hippocampus and the cerebral cortex (n = 1) [47]. Moreover, the levels of hyperphosphorylation of the tau protein (n = 4) [33, 38, 42, 43], presence of NFTs (n = 1) [43], and hippocampal Gad67+ interneuron quantification (n = 1) [44] were used to characterize AD pathology in brain tissues.
Effect of periodontitis on AD physiopathology
The effects of periodontal inflammation or bacteria on the increase in systemic inflammation and brain tissue damage and the impairment of cognitive behavior were investigated in 13, 21, and six studies, respectively. Considering the heterogeneity of the models and data, a meta-analysis could not be performed. The study results are presented in Table 2.
• Effect on systemic inflammation
Table 2
Effect of periodontitis on systemic inflammation, brain inflammation, and cognitive behavior: results, proposed mechanisms, and conclusion
| Authors | Proposed mechanism | Conclusion | ||||
| Effect on systemic inflammation | Effect on brain | Effect on cognitive behavior | ||||
| in situ brain manifestation | systemic manifestation of brain damage | |||||
| Gu 2020 [35] | mRNA expression of both IL-17 and IL-6 | -In the cortex, PgLPS induces a PI3K-related IL-6 production | Memory decline associated with bone loss (45% to 47%) | PgLPS induces a PI3K-related IL-6 production in turn to promote IL-17 production in microglia | Chronic systemic exposure to PgLPS induces systemic inflammatory bone loss-related cognitive decline through upregulation of IL-6 and IL-17 | |
| -Microglia expression of IL-17 (2.8-fold increase) and IL-6 (1.9-fold increase) | In response to systemic exposure to PgLPS, microglia are: | |||||
| -Cortex mRNA expression of IL-17 (2.6-fold increase) and IL-6 (1.8-fold increase) | Bone loss (26%) associated with systemic IL-17 | 1. the main cell sources of IL-17 and IL-6 that amplifies neuroinflammation | ||||
| -Aβ accumulation in neurons correlated with IL-17 expression in microglia | 2. accumulates Aβ production by increasing production of AβPP and CatB in neurons | |||||
| -Increased expression of Aβ in neurons (2.5-fold increase) | ||||||
| Zeng 2020 [52] | N/A | -Increased RAGE expression in cerebral endothelial cells | Memory decline | CatB/NF-κB activation | Pg activates NF-κB pathway through binding TLR2/4 that triggers transcription of RAGE in brain | |
| -Increased of Aβ loads in brains, positively correlated with the RAGE expression in endothelial cells | Pg infection upregulated RAGE expression in cerebral endothelial cells that promotes the Aβ influx and amyloidogenesis | |||||
| -Induction of Aβ1-42 around cerebral endothelial cells | ||||||
| -CatB/NF-κB activation-dependent RAGE upregulation in cerebral endothelial cell | ||||||
| Han 2019 [36] | N/A | -Increased expression of TNF-α with neuroinflammation | N/A | Periodontal pathogens cross BBB and cause AD | ||
| -TNF-α was promoted by extracellular RNAs via the TLR-8 and NF-kB signaling pathways | exRNAs increasing brain expression of TNF-α | Extracellular RNAs in periodontopathogenic OMVs promote neuroinflammation through TNF-α production in macrophages and cross the BBB | ||||
| Leira 2019 [15] | N/A | N/A | Elevation of Aβ1–40 and Aβ1–42 serum levels | N/A | TLR-/NF-κB activation | Periodontitis induced PgLPS produced increased serum levels |
| Correlate with alveolar bone loss | of Aβ peptides | |||||
| TLR-4/CD14 activation by LPS leads to NF-κB mediated induction of cytokines in monocytes/neutrophils in blood and from microglia in brain | ||||||
| Wang 2021 [37] | Increased expressions of IL-1β and TNF-α | -Hippocampal microgliosis and astrogliosis | Increased expression of TNF-α and IL-1β | Inflammatory response in the hippocampus of mice can change depending on the periodontal inflammation status. | ||
| -Number of glial cells significantly correlated with the degree of periodontal inflammation | N/A | N/A | ||||
| Diaz Zuniga 2020 [38] | -Increased production of serum IL-1β, IL-6, INF-y | -IL-1β, IL-6, INF-γ production in the hippocampus memory deficits | Spatial memory decline and | The bacterial virulence factors constituted by capsular polysaccharides play a central role in activating innate immunity and inflammation in the AD-like pathology triggered by Pg. The infection by encapsulated serotypes of Pg (K1 and K2) leads to AD-like pathology compared to less virulent serotypes. | ||
| -Detection of 1×103 CFU/ml Pg serotypes in serum | -Changes in astrocytic morphology in the CA1, CA3, and DG regions of the hippocampus | N/A | Gingipains induced pro-inflammatory cytokine production, astrogliosis, Aβ42 secretion and tau hyperphosphorylation | |||
| -Increased Aβ1-42 levels in the hippocampus | ||||||
| -Increased tau hyperphosphorylation in the hippocampus and glial activation | ||||||
| -Detection of 1×105 CFU/ml Pg serotypes in hippocampus | ||||||
| -Detection of RgpA and Kgp gingipains genes in hippocampus | ||||||
| Hayashi 2019 [54] | N/A | Increase of Iba-1 and CD3 positive cells in periventricular area | N/A | N/A | N/A | Continuous brain exposure of PgLPS can deteriorate prognosis of AD patient |
| Hu 2020 [45] | -Increased IL-1β, IL-6, IL-8, IL-21, and LPS | -Increased IL-1β, IL-6, IL-8, IL-21 expression (related to LPS) in the cortex | Aβ ratio (Aβ1-40/Aβ1-42) was upregulated in the plasma | Learning and memory impairment | TLR4/NF-κB signaling pathway | Periodontitis was associated with learning and memory impairment, induced by neuroinflammation via activating the TLR4/NF-κB signaling pathway. Furthermore, abnormal AβPP processing could be involved in this progress |
| -Increased expression of TLR4 and CD14 mRNA in the peripheral blood mononuclear cells | -Microglia and astrocytes activation (related to LPS) in the cortex | |||||
| -Increased expression of TLR4 and CD14 mRNA in the cortex | ||||||
| -Aβ ratio (Aβ1-40/Aβ1-42) in cortex | ||||||
| -Promotion of β- and γ-secretase AβPP processing. | ||||||
| Su 2021 [49] | N/A | -Aβ1–40 and Aβ1–42 accumulation in the hippocampus | N/A | N/A | BACE1 and PS1 | Td increase expression of Aβ1-40 and Aβ1-42 accumulation in the hippocampus of mice, participating in the AD pathological process |
| -Hippocampus colonization by Pg and Td | ||||||
| -Increased expression of BACE1 and PS2 related to Pg and Td infection | ||||||
| Poole 2015 [50] | N/A | -Lower density of astrocytes in entorhinal cortex and hippocampus | N/A | N/A | Acute- phase proteins, cytokines and the complement cascade | Pg contribute to local inflammation in the CNS comprising complement activation and neuronal injury |
| -Activation of complement system (iC3b, C3b and C3d and C9) in microglia that lead to a chronic local inflammation in glial cells and neurons | ||||||
| -The pyramidal neurons of the hippocampus demonstrated opsonization with C3 activation fragments | ||||||
| - Pg DNA detection | ||||||
| Furutama 2020 [46] | Increased expression of IL-6 | -Increased expression of IL-1β in the hippocampus related to IL-6 serum level | N/A | N/A | IL-6 | Periodontal inflammation induced the expression of IL-6 that contribute: |
| -Increased BBB permeability in the hippocampus through the decrease of claudin 5 levels, related to IL-6 serum level | 1. to increase the IL-1β expression levels in the hippocampus 2. to decrease the claudin 5 levels, which resulted in an | |||||
| -IL-6 neutralizing antibody prevented decreases in claudin 5 level | increased BBB permeability | |||||
| Wu 2017 [47] | N/A | -Increased expression of CatB in both microglia and neurons in the hippocampus | N/A | Learning and memory deficits | CatB | Chronic systemic exposure to PgLPS induces AD-like phenotypes, including microglia-mediated neuroinflammation, intracellular Aβ accumulation in neurons and impairment of the learning and memory functions |
| -Increased expression of IL-1β in microglia in the hippocampus | ||||||
| -Increased expression of TLR2 and TLR4 in microglia in the hippocampus | ||||||
| -Intra-cellular Aβ accumulation in the hippocampus | ||||||
| Liu 2017 [39] | N/A | Arg-gingipain (Rgp) and Lys-gingipain (Kgp) cooperatively induced microglia cell migration and inflammation through: | N/A | Rgp and Kgp cooperatively contribute to: 1. Pg invasion into the brain 2. cell migration of microglia and induction of neuroinflammation | ||
| 1. the proteolytic activation of PAR2 in microglia | Activation of PAR2 | |||||
| with subsequent activation of phosphoinositide 3-kinase/Akt and mitogen-activated protein kinase/ERK kinase/ERK pat | N/A | TLR4/NF-κB signaling pathway | ||||
| 2. the activation of TLR2 that induces | ||||||
| the expression of proinflammatory mediators (IL-6, TNF-α, and iNOS) by microglial cells | ||||||
| Zhang 2020 [40] | N/A | -Neuronal damage and morphological abnormalities in the CA3 area of the hippocampus | N/A | Learning and memory decline | IL-1, IL-6, and TNF-α | Periodontitis is a risk factor for AD and may increase inflammatory cytokines in AD models |
| -In the hippocampus and cerebral cortex, increase of: | ||||||
| -Aβ1–40 in the hippocampus and cerebral cortex | ||||||
| -IL-1, IL-6, TNF-α, and CRP | ||||||
| Zhang 2018 [48] | N/A | Activation of microglia and astrocytes in the cortex and hippocampus | N/A | Spatial learning and memory | TLR4/NF-κB signaling pathway | Pg-LPS can lead to learning and memory impairment through the activation of the TLR4 signaling pathway |
| -Expression of inflammatory cytokines (TNF-α, IL-1, IL-6, and IL-8) and associated genes | ||||||
| - Activation of the TLR4/NF-κB signaling pathway, including the TLR4/CD14 receptor, IRAK1, NF-κB, and p-p65/p65 | ||||||
| Singhrao 2017 [51] | N/A | -Intracerebral dissemination of Pg in the frontotemporal lobe of the cerebral cortex | N/A | N/A | The diffuse staining in the hippocampus is attributed to physical tissue injury resulting from bacterial lysates and the high inflammatory burden | |
| -Dissemination of Pg gingipains in microvessels of the hippocampus | ApoE and inflammation related to Pg | Lack of functional/repair protein (ApoE) and inflammation related to Pg contribute to astrocytic processing of cellular debris containing serum protein components in the form of age-related granules | ||||
| -IgG to bacterial peptidoglycan in the capillaries of the cerebral parenchyma and pyramidal neurons | ||||||
| Cueno 2018 [33] | -Increased blood activity of MMP-9 | -Oxidative stress in different regions of the brain by increasing H2O2, glutathione reductase and free fatty acid brain levels | Increased blood levels of AβPP, PS1, and PS2 associated with elevated rate of TLR | N/A | Gingival PDL-BA can potentially cause systemic inflammation ascribable to prolonged systemic manifestations in the blood and localized detrimental effects within the brain organ. | |
| -Increased amounts of TLR2 | -Increased tau protein amounts in the hippocampus and cerebellum | TLR2 | ||||
| -Increased blood levels of AβPP, PS1 and PS2 that were associated with elevated rate of TLR | -ER stress in the brain that leads to neuronal cell damage or death. | |||||
| Kantarci 2020 [41] | High levels of MCP-1 | -Increased levels of insoluble Aβ42 in the pre-frontal cortex | N/A | Increased Iba1 and inflammatory cytokines | PD increases neuroinflammation and disrupts the neuroinflammatory response, suggesting that microglia is central to the association between PD and AD. | |
| -Increased Iba1 levels in cortical layers and in the hippocampus (CA1, hippocampal fissure, and DG) | N/A | N/A | ||||
| -Decreased percentage of PAM | ||||||
| -High brain levels of TNF-α and IL-10 | ||||||
| Hu 2021 [45] | -Expression of TLR/NF-κB, PPAR and ROS genes | -Activation of microglia and astrocytes in both cortex and hippocampus | -Increased expression of APLP2 and AβPP secretases (including α-, β-, and γ-secretases) | Spatial and learning memory impairment | STAT3 signaling pathway | Induced periodontitis increased the inflammation load both in the periphery and the CNS leading to cognitive impairment. STAT3 signaling pathway played an important role in promoting neuroinflammation and modulating AβPP processing |
| -Increased level of IL-1β, IL-6, IL-8 and IL-21 | -Increase in the expression of interleukins (IL-1β, IL-6, IL-8, IL-21) | -Increased plasma levels of Aβ and Aβ ratio (Aβ1-40/Aβ1-42) | ||||
| -Activation of the STAT3 signaling pathway | -Expression of TLR/NF-κB, PPAR, and ROS genes | |||||
| -Expression of APLP2 and AβPP secretases and Aβ and Aβ ratio (Aβ1-40/Aβ1-42) in the cortex | ||||||
| -Activation of the STAT3 signaling pathway in cortex | ||||||
| -Increased levels of Aβ1-42 in the hippocampus and cortex | ||||||
| Ilievski 2018 [43] | N/A | -Pg gingipains detection intracellularly in astrocytes, microglia, and neurons | Increased gene expression of AβPP and beta-site AβPP cleaving enzyme 1 (BACE1) | NA | Proinflammatory cytokines expression | Chronic oral infection of Pg can be an initiator of the development of neuropathology due to direct invasion of Pg/gingipain into neurons |
| Nie 2019 [53] | Increased expression of TLR2, IL-1β | -Increased inflammatory responses and Aβ-producing molecules in the liver | N/A | N/A | CatB/NF-κB signaling | Systemic Pg infection induces the production of Aβ in inflammatory macrophages via activating CatB/NF-κB signaling, and CatB plays critical roles in peripheral Aβ generation. |
| -Increased expression AβPP770 and CatB | ||||||
| Dominy 2019 [44] | N/A | N/A | Gingipains induced neurodegeneration | Pg and gingipains in the brain play a central role in the pathogenesis of AD. Oral administration of small-molecule gingipain inhibitors blocks gingipain- induced neurodegeneration, significantly reduces Pg load in the mouse brain, and significantly decreases the host Aβ1–42 response to Pg brain infection. | ||
| Tang 2020 [42] | Increased expression of IL-6, IL-1β, and TNF-α | -Tau hyperphosphorylation in the hippocampus | N/A | Tau hyperphosphorylation and attenuate the activity of PP2A | Pg infection could induce tau hyperphosphorylation through triggering systemic inflammation and neuroinflammation | |
| inhibition of the activity of PP2A | -Increase astrocytes activity | |||||
| -Increase of proinflammatory cytokines (IL-6, IL-1β, and TNF-α) in the hippocampus | ||||||
AD, Alzheimer’s disease; Aβ, amyloid-β; AβPP, amyloid-β protein precursor; BBB, blood-brain barrier; Cat, cathepsin; CFU, colony forming unit; CNS, central nervous system; CRP, C-reactive protein; DG, dentate gyrus; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; exRNAs, extracellular RNAs; IL, interleukin; IFN, interferon; LPS, lipopolysaccharide; MMP, matrix metalloproteinases; NF-κB, nuclear factor-κB; NFTs, neurofibrillary tangles; OMVs, outer membrane vesicles; PAM, plaque-associated microglia; PAR, protease-activated receptor; PD, periodontal disease; Pg, Porphyromonas gingivalis; PP2A, protein phosphatase 2A; PPAR, peroxisome proliferator-activated receptors; PS, presenilin; RAGE, receptor for advanced glycation endproducts; Td, Treponema denticola; TLR, toll-like receptor; TNF, tumor necrosis factor.
Gingival injection of Pg-LPS and BA contributed to the systemic elevation of Aβ1–40 levels, and MMP-9 was significantly associated with an increase in blood AβPP, PS1, and PS2 [33]. Additionally, periodontitis was associated with the increase of Aβ ratio (Aβ1 - 40/Aβ1 - 42) [34] and the plasma levels of Aβ and inflammatory cytokines (IL-1β, IL-6, IL-8, and IL-21) [34, 46]. Systemic periodontitis is induced in the bloodstream of infected animals and increases Aβ peptides [32, 34, 46] MMPs, and inflammatory cytokines (IL-1β, IL-6, IL-8 IL-21, and NF-kB) [32–34, 46, 55]. Furthermore, the Pg/Pg-LPS complex leads to its binding to toll-like receptors (TLR-4/CD14) on peripheral monocytes, macrophages, and neutrophils that induce the activation of the nuclear factor (NF)-κB pathway [32] and ultimately leading to the release of cytokines by circulating monocytes and neutrophils. After periodontitis induction, a significant increase in the serum levels of Aβ1–40 was observed [32], and an increase in Aβ1–42 was detected by ELISA [56]. Additionally, chronic systemic exposure to Pg-LPS induced mandibular [32] and tibia [35] bone loss that was positively correlated with circulating levels of Aβ1–40 [32] and memory decline [35], suggesting a relationship between periodontal infection and increased systemic levels of this peptide.
Hence, the authors concluded that periodontitis infection or periodontal pathogens could increase AD-specific and-nonspecific biomarkers of systemic inflammation.
• Effect on brain tissue inflammation/damage
The effects of periodontitis and periodontal pathogens on the brain tissue involve neuroinflammation and neuronal cell damage. Periodontal infections are also associated with hippocampal microgliosis and astrogliosis [35–37, 52]. Indeed, significantly increased expression of interleukins (IL-6,-17) [35, 48, 52], TNF-α [36, 46] and cytokine production [34, 41, 45, 47] have been found inside microglia. Additionally, inside the hippocampus, the cytokines production related to periodontitis was significantly associated with tau hyperphosphorylation [34] and Aβ levels [38, 41, 42, 45, 47]. Inside the neurons, the IL-6 and IL-17 expression was associated with Aβ peptide accumulation [35]. Furthermore, chronic local inflammation caused by periodontal pathogens contributes to neuronal damage [40] or neuronal death [50]. Moreover, the brain endothelial cells of periodontally infected animals harbored significant expression of receptors for advanced glycation end products (RAGE) associated with a significant increase in Aβ level [52]. Additionally, periodontal pathogen virulence factors (gingipains) in the cortex are associated with a significant increase in neuroinflammation [39, 51] and degenerative neuronal cells [43].
• Effect on cognitive behavior
Chronic systemic exposure to Pg [38, 52] or Pg-LPS [35, 42, 47, 48] is significantly associated with memory decline. Additionally, periodontal ligatures in AD animal models are significantly associated with deficits in cognitive function [34, 40].
DISCUSSION
The present systematic review of preclinical studies raises hypotheses regarding the pathophysiological mechanisms linking periodontitis-induced systemic inflammation and metastatic infection with AD progression.
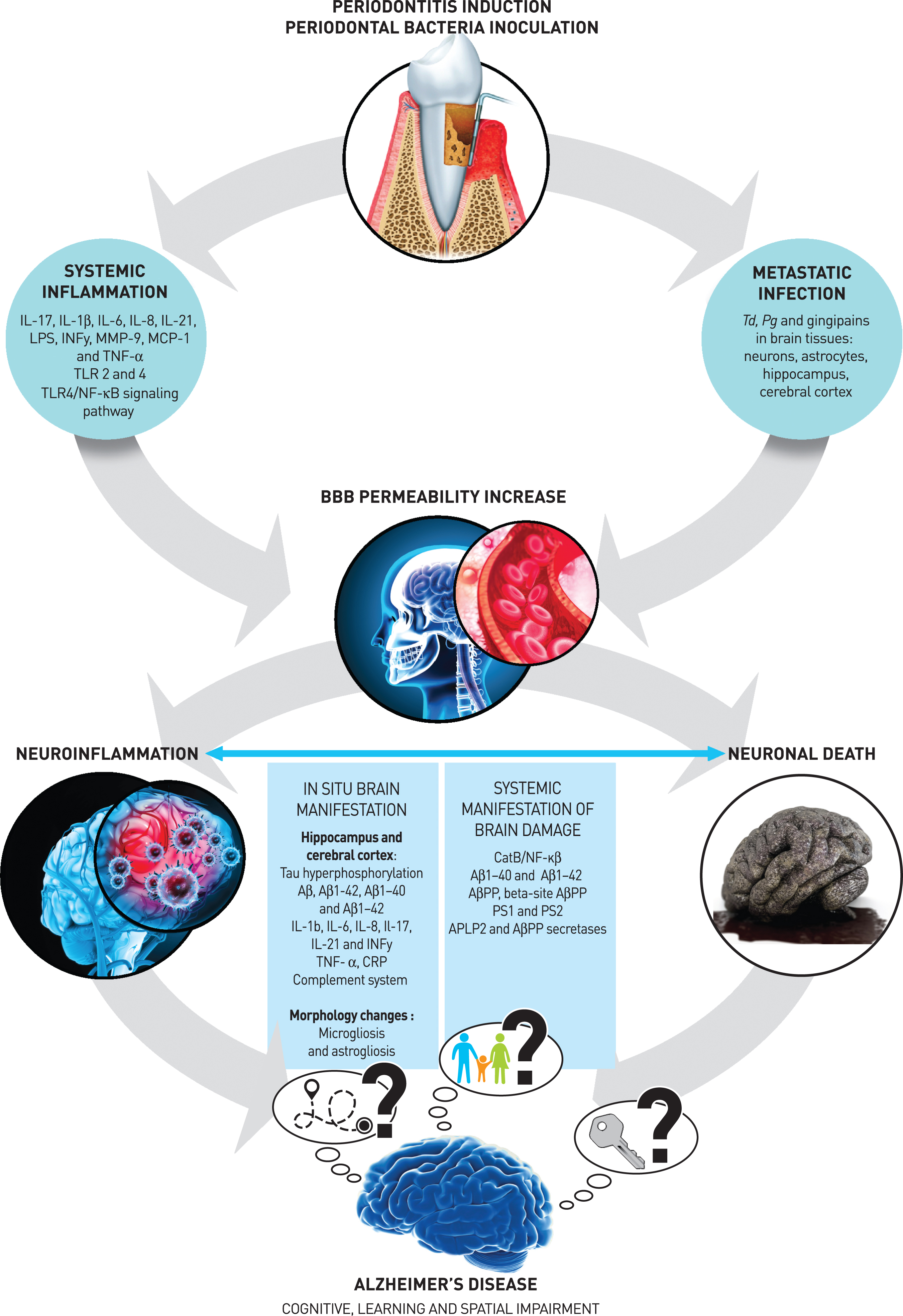
Indeed, it has been widely demonstrated that Gram-negative periodontal bacteria and their byproducts can invade the blood circulation and subsequently increase SI and/or colonize vulnerable sites called locus minoris resistentiae, where metastatic infection (MI) occurs [57–61]. Both mechanisms (SI and MI) are involved in the relationship between periodontitis and systemic diseases such as cardiovascular diseases [62, 63], diabetes [64, 65], and respiratory diseases [66]. Recent systematic reviews have suggested that periodontitis or periodontal bacteria lead to neuroinflammation with brain tissue damage and cognitive/behavioral impairment, consistent with AD [29, 67, 68]. Additionally, a potential bidirectional relationship between periodontitis and AD has been proposed, as the resulting changes in cognition and behavior may lead to a decrease in oral hygiene habits and, therefore, an increase in tooth loss and edentulism. Furthermore, the presence of periodontitis was associated with a six-fold increase in the rate of cognitive decline over a 6-month follow-up period [29, 67, 68]. If the level of clinical evidence remains limited, the potential pathophysiological mechanisms (Fig. 2) linking these two diseases will be investigated in a substantial number of the preclinical rodent studies included in the present systematic review.
Fig. 2
Hypothetical physiopathological mechanisms linking periodontitis and Alzheimer’s disease.
Effect of the periodontal pathogen on blood-brain barrier permeability
Several studies included in the present review have demonstrated in preclinical models that SI [33–35, 38, 41, 42, 45, 46, 48, 53] and/or brain MI [49–51] occur after periodontal induction or periodontal bacterial inoculation. Two studies demonstrated that both SI and MI induce the permeability of the blood-brain barrier (BBB), namely through a decrease in claudin 5 levels [46] and the destruction of collagen and fibronectin [39]. The resulting destruction of the components of the basal membrane of brain endothelial cells enhances vascular permeability [39] and therefore facilitates the penetration of other bacteria.
Effect of the periodontal pathogen on in situ brain manifestation
This review also highlights how periodontal pathogens may lead to in situ brain manifestations at the cellular and molecular levels. Periodontal bacteria (Pg and Td) [44, 49–51] and their virulence factors can access the central nervous system, particularly the hippocampus [49, 50] and cerebral cortex [50, 51] and lead to brain damage through neuroinflammation (1), amyloidogenesis (2), and phosphorylation of the tau protein (3), which are the characteristic pathological hallmarks of AD.
Neuroinflammation (1)
The molecular mechanisms of periodontitis-related neuroinflammation are linked to the activation of the NF-κB pathway, the involvement of CatB, the secretion of pro-inflammatory cytokines (as TNF-α, IL-1β, IL-6), and the activation of the complement system [50].
LPS [45], gingipains [39, 43, 44, 51], capsules, and fimbriae of Pg promote the entrance of bacteria into brain cells, disturb the phagocytic function of the host, and therefore contribute to the destruction of neuronal tissues. Furthermore, Pg and its byproducts bind to TLR-4/CD14 [32] and TLR 2/4 [48] on microglia, activating B, which in turn induces increased levels of intracerebral cytokines. Consequently, the secretion of pro-inflammatory and dysregulated cytokines occurs [69], contributing to the enhancement of neuroinflammation and cognitive impairment.
CatB is another molecule that plays a role in periodontitis-related neuroinflammation. Indeed, this molecule is involved in the NF-κB upregulation and further increases the expression of pro-inflammatory cytokines inside microglia and hippocampus [47] and that of RAGE in cerebral endothelial cells [52]. Additionally, Han et al. [36] suggested that the periopathogen could directly increase the brain expression of TNF (without the activation of NF-kB or CatB). Notably, extracellular RNAs located in the outer membrane vesicles of the periodontal pathogen are able to increase the brain expression of TNF-α.
Finally, neuroinflammation-related periodontitis can also be explained by complement system activation [50]. Indeed, Pg affects genes that regulate the complement system and impairs the onset of pro-inflammatory signaling cascades and the release of systemic pro-inflammatory mediators [32–34, 46, 55]. The resulting expression of cytokines in brain (IL-1β, IL-6, IL-8, Il-17, IL-21, TNF-α, and INF-γ) [34–36, 38, 39, 41–43, 45, 49] contributes to neuroinflammation [33–38, 41, 42, 45–47, 49, 50, 52, 54], neuronal death [37, 40, 43, 44], and increases AβPP as well as Aβ [32] in the brain tissues.
Amyloidogenesis (2)
Several studies have demonstrated that periodontitis authors [35, 47, 52, 53] that periodontitis infection can contribute to brain amyloidogenesis. Chronic systemic exposure to Pg-LPS induces Il-6 and Il-17 expression in the cortex and microglia, and the production of these cytokines was correlated to Aβ accumulation in neurons [35]. Furthermore, the exposition to periodontal pathogens increases TNF-α and IL-1β and induces a reactive-astrocyte expression with consecutive increased levels of the Aβ [70]. In addition to brain cytokine expression, it was demonstrated that CatB prolongs the NF-κB pathway, contributing to the Aβ influx [52, 53], the Aβ accumulation in neurons, and peripheral inflammatory monocytes/macrophages [47, 53]. Moreover, the induced production of antibodies in the brain can be used as a substrate for oral bacterial adhesion [71], favoring their colonization and enhancing in situ inflammation and amyloidogenesis.
Tau protein (3)
Finally, it was demonstrated that the presence of periodontal bacteria or virulence factors [38, 42, 43] induces hyperphosphorylation of the tau protein (p-Tau) [38, 42, 44] and the formation of NFTs. Consequently, microglial changes (microgliosis and astrogliosis) [34, 37–39, 42–45] and neuronal death [40, 43, 49, 50] lead to the progression of AD. The suggested molecular mechanism is that Pg infection induces tauopathies in the brain by inhibiting the activity of protein phosphatase 2A (PP2A), consequently leading to neuroinflammation [42] and AD progression.
Systemic manifestation of brain damage
The in situ brain damage [33–45, 47–54] was also investigated through systemic AD-specific biomarkers such as the tau, Aβ protein, and its derivate products (Aβ1–40, Aβ1–42, AβPP, beta-site AβPP, presenilins 1 and 2) [32–34, 43, 45, 52]. Furthermore, it has been suggested that the systemic manifestations of brain damage [32–34, 43, 45] are a consequence of the enhanced permeability of the BBB. Indeed, the increase in bacteria and their byproducts acts on TLR-4, creating a positive feedback loop that allows AD-specific biomarkers to exit the brain [32]. Therefore, neuronal degradation associated with AD [72] can be evaluated both extracellularly by amyloidosis (Aβ levels in the blood) and intracellularly by the phosphorylation of tau protein, which leads to the disruption of the microtubule network inside neurons, ultimately leading to neuronal death, neuroinflammation and neurotoxicity [44].
Effect of the periodontal pathogen on cognitive behavior
In addition to the above-mentioned biological and biomolecular findings, this systematic review also supports that chronic systemic exposure to Pg and Pg-LPS is associated with memory decline and loss of cognitive behavior [34, 35, 38, 40, 45, 47, 48, 52]. Cognitive impairment related to neuronal death was demonstrated using cognitive and functional tests, such as the Morris Water maze test, passive avoidance test, and passive avoidance test, which assesses learning and memory abilities [35, 41, 45, 47, 48, 52]. Indeed, AD symptomatology is mainly attributed to hippocampal destruction, as it is the area responsible for memory and learning capacity [41]. All studies in the present review investigating cognitive performance reported a negative effect of periodontitis or periodontal pathogens on animals’ cognitive behaviors, namely memory decline as well as loss of learning and spatial capabilities [34, 40, 45, 47, 48, 52, 54].
This systematic review had some limitations. A meta-analysis was not possible because of the heterogeneity of the studies, which had different aims and analyzed different mechanisms/markers and different outcomes. Therefore, the establishment of standardized and validated AD models in future studies would yield more consistent conclusions regarding the influence of periodontitis on AD progression. Moreover, the present data remain preclinical, and further clinical data are required to validate this hypothesis regarding the effects of periodontitis and periodontal treatment on AD progression. However, the preclinical study analyses in the present systematic review have the advantage of excluding confounding factors, such as diabetes and tobacco, which are often encountered in human clinical data.
Conclusion
Within the limits of preclinical models, the present systematic review contributes to the understanding of the potential mechanisms linking periodontal bacteria to AD. Systemic inflammation and brain metastatic infections induced by periodontal pathogens contribute to neuroinflammation, amyloidosis, and tau phosphorylation, leading to brain damage and subsequent cognitive impairment.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230478.
REFERENCES
[1] | Richards SS , Hendrie HC ((1999) ) Diagnosis, management, and treatment of Alzheimer disease: A guide for the internist. Arch Intern Med 159: , 789–798. |
[2] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[3] | Kumar A , Singh A , Ekavali ((2015) ) A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol Rep 67: , 195–203. |
[4] | McGeer EG , McGeer PL ((2010) ) Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimers Dis 19: , 355–361. |
[5] | Heneka MT , Kummer MP , Latz E ((2014) ) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: , 463–477. |
[6] | Wu Z , Nakanishi H ((2014) ) Connection between periodontitis and Alzheimer’s disease: Possible roles of microglia and leptomeningeal cells. J Pharmacol Sci 126: , 8–13. |
[7] | de Pablo P , Chapple IL , Buckley CD , Dietrich T ((2009) ) Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol 5: , 218–224. |
[8] | Allenspach-Petrzilka GE , Guggenheim B ((1983) ) Bacterial invasion of the periodontium; an important factor in the pathogenesis of periodontitis? J Clin Periodontol 10: , 609–617. |
[9] | Lee YT , Lee HC , Hu CJ , Huang LK , Chao SP , Lin CP , Su EC , Lee YC , Chen CC ((2017) ) Periodontitis as a modifiable risk factor for dementia: A nationwide population-based cohort study. J Am Geriatr Soc 65: , 301–305. |
[10] | Socransky SS , Haffajee AD , Cugini MA , Smith C , Kent RL Jr ((1998) ) Microbial complexes in subgingival plaque. J Clin Periodontol 25: , 134–144. |
[11] | Haffajee AD , Socransky SS ((1994) ) Microbial etiological agents of destructive periodontal diseases. Periodontol 2000 5: , 78–111. |
[12] | Tatakis DN , Kumar PS ((2005) ) Etiology and pathogenesis of periodontal diseases. Dent Clin North Am 49: , 491–516v. |
[13] | Silver JG , Martin AW , McBride BC ((1977) ) Experimental transient bacteraemias in human subjects with varying degrees of plaque accumulation and gingival inflammation. J Clin Periodontol 4: , 92–99. |
[14] | Fischer RG , Lira Junior R , Retamal-Valdes B , Figueiredo LC , Malheiros Z , Stewart B , Feres M ((2020) ) Periodontal disease and its impact on general health in Latin America. Section V: Treatment of periodontitis. Braz Oral Res 34: , e026. |
[15] | Leira Y , Domínguez C , Seoane J , Seoane-Romero J , Pías-Peleteiro JM , Takkouche B , Blanco J , Aldrey JM ((2017) ) Isperiodontal disease associated with Alzheimer’s disease? Asystematic review with meta-analysis. Neuroepidemiology 48: , 21–31. |
[16] | Maldonado A , Laugisch O , Bürgin W , Sculean A , Eick S ((2018) ) Clinical periodontal variables in patients with and without dementia-a systematic review and meta-analysis. Clin Oral Investig 22: , 2463–2474. |
[17] | Nadim R , Tang J , Dilmohamed A , Yuan S , Wu C , Bakre AT , Partridge M , Ni J , Copeland JR , Anstey KJ , Chen R ((2020) ) Influence of periodontal disease on risk of dementia: A systematic literature review and a meta-analysis. Eur J Epidemiol 35: , 821–833. |
[18] | Wang RP , Ho YS , Leung WK , Goto T , Chang RC ((2019) ) Systemic inflammation linking chronic periodontitis to cognitive decline. Brain Behav Immun 81: , 63–73. |
[19] | Liccardo D , Marzano F , Carraturo F , Guida M , Femminella GD , Bencivenga L , Agrimi J , Addonizio A , Melino I , Valletta A , Rengo C , Ferrara N , Rengo G , Cannavo A ((2020) ) Potential bidirectional relationship between periodontitis and Alzheimer’s disease. Front Physiol 11: , 683. |
[20] | Li Y , Tan MS , Jiang T , Tan L ((2014) ) Microglia in Alzheimer’s disease. Biomed Res Int 2014: , 437483. |
[21] | Hansen DV , Hanson JE , Sheng M ((2018) ) Microglia in Alzheimer’s disease. J Cell Biol 217: , 459–472. |
[22] | Kinane DF , Zhang P , Benakanakere M , Singleton J , Biesbrock A , Nonnenmacher C , He T ((2015) ) Experimental gingivitis, bacteremia and systemic biomarkers: A randomized clinical trial. J Periodontal Res 50: , 864–869. |
[23] | Zafiropoulos GG , Flores-de-Jacoby L , Plate VM , Eckle I , Kolb G ((1991) ) Polymorphonuclear neutrophil chemiluminescence in periodontal disease. J Clin Periodontol 18: , 634–639. |
[24] | Kempuraj D , Thangavel R , Natteru PA , Selvakumar GP , Saeed D , Zahoor H , Zaheer S , Iyer SS , Zaheer A ((2016) ) Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine 1: , 1003. |
[25] | Kempuraj D , Thangavel R , Selvakumar GP , Zaheer S , Ahmed ME , Raikwar SP , Zahoor H , Saeed D , Natteru PA , Iyer S , Zaheer A ((2017) ) Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation andneurodegeneration. Front Cell Neurosci 11: , 216. |
[26] | Dioguardi M , Caloro GA , Troiano G , Giannatempo G , Laino L , Petruzzi M , Lo Muzio L ((2016) ) Oral manifestations in chronic uremia patients. Ren Fail 38: , 1–6. |
[27] | Kamer AR , Craig RG , Dasanayake AP , Brys M , Glodzik-Sobanska L , de Leon MJ ((2008) ) Inflammation and Alzheimer’s disease: Possible role of periodontal diseases. Alzheimers Dement 4: , 242–250. |
[28] | Ho GJ , Drego R , Hakimian E , Masliah E ((2005) ) Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy 4: , 247–256. |
[29] | Borsa L , Dubois M , Sacco G , Lupi L ((2021) ) Analysis the link between periodontal diseases and Alzheimer’s disease: A systematic review. Int J Environ Res Public Health 18: , 9312. |
[30] | Costa MJF , de Araújo IDT , da Rocha Alves L , da Silva RL , Dos Santos Calderon P , Borges BCD , de Aquino Martins ARL , de Vasconcelos Gurgel BC , Lins R ((2021) ) Relationship of Porphyromonas gingivalis and Alzheimer’s disease: Asystematic review of pre-clinical studies. Clin Oral Investig 25: , 797–806. |
[31] | Parra-Torres V , Melgar-Rodríguez S , Muñoz-Manríquez C , Sanhueza B , Cafferata EA , Paula-Lima AC , Díaz-Zúñiga J ((2023) ) Periodontal bacteria in the brain-Implication for Alzheimer’sdisease: A systematic review. Oral Dis 29: , 21–28. |
[32] | Leira Y , Iglesias-Rey R , Gómez-Lado N , Aguiar P , Campos F , D’Aiuto F , Castillo J , Blanco J , Sobrino T ((2019) ) Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch Oral Biol 99: , 120–125. |
[33] | Cueno ME , Ochiai K ((2018) ) Gingival Periodontal Disease (PD) Level-butyric acid affects the systemic blood and brain organ: Insights into the systemic inflammation of periodontal disease. Front Immunol 9: , 1158. |
[34] | Hu Y , Zhang X , Zhang J , Xia X , Li H , Qiu C , Liao Y , Chen H , He Z , Song Z , Zhou W ((2021) ) Activated STAT3 signaling pathway by ligature-induced periodontitis could contribute to neuroinflammation and cognitive impairment in rats. J Neuroinflammation 18: , 80. |
[35] | Gu Y , Wu Z , Zeng F , Jiang M , Teeling JL , Ni J , Takahashi I ((2020) ) Systemic exposure to lipopolysaccharide from Porphyromonas gingivalis induces bone loss-correlated Alzheimer’s disease-like pathologies in middle-aged mice. J Alzheimers Dis 78: , 61–74. |
[36] | Han EC , Choi SY , Lee Y , Park JW , Hong SH , Lee HJ ((2019) ) Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J 33: , 13412–13422. |
[37] | Wang X , Tong Y , Zhang J , Khan N , Zhang K , Bai H , Zhang Q , Chen Y ((2021) ) Neuroinflammation changes with periodontal inflammation status during periodontitis in wild-type mice. Oral Dis 27: , 1001–1011. |
[38] | Díaz-Zúñiga J , More J , Melgar-Rodríguez S , Jiménez-Unión M , Villalobos-Orchard F , Muñoz-Manríquez C , Monasterio G , Valdés JL , Vernal R , Paula-Lima A ((2020) ) Alzheimer’s disease-like pathology triggered byPorphyromonas gingivalis in wild type rats is serotype dependent. Front Immunol 11: , 588036. |
[39] | Liu Y , Wu Z , Nakanishi Y , Ni J , Hayashi Y , Takayama F , Zhou Y , Kadowaki T , Nakanishi H ((2017) ) Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep 7: , 11759. |
[40] | Zhang S , Yang F , Wang Z , Qian X , Ji Y , Gong L , Ge S , Yan F ((2020) ) Poor oral health conditions and cognitive decline: Studies in humans and rats. PLoS One 15: , e0234659. |
[41] | Kantarci A , Tognoni CM , Yaghmoor W , Marghalani A , Stephens D , Ahn JY , Carreras I , Dedeoglu A ((2020) ) Microglial response to experimental periodontitis in a murine model of Alzheimer’s disease. Sci Rep 10: , 18561. |
[42] | Tang Z , Liang D , Cheng M , Su X , Liu R , Zhang Y , Wu H ((2021) ) Effects of Porphyromonas gingivalis and its underlying mechanisms on Alzheimer-like tau hyperphosphorylation in Sprague-Dawley rats. J Mol Neurosci 71: , 89–100. |
[43] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O’Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[44] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , Nguyen M , Haditsch U , Raha D , Griffin C , Holsinger LJ , Arastu-Kapur S , Kaba S , Lee A , Ryder MI , Potempa B , Mydel P , Hellvard A , Adamowicz K , Hasturk H , Walker GD , Reynolds EC , Faull RLM , Curtis MA , Dragunow M , Potempa J ((2019) ) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: , eaau3333. |
[45] | Hu Y , Li H , Zhang J , Zhang X , Xia X , Qiu C , Liao Y , Chen H , Song Z , Zhou W ((2020) ) Periodontitis induced by P. gingivalis-LPS is associated with neuroinflammation and learning and memory impairment in Sprague-Dawley rats. Front Neurosci 14: , 658. |
[46] | Furutama D , Matsuda S , Yamawaki Y , Hatano S , Okanobu A , Memida T , Oue H , Fujita T , Ouhara K , Kajiya M , Mizuno N , Kanematsu T , Tsuga K , Kurihara H ((2020) ) IL-6 induced by periodontal inflammation causes neuroinflammation and disrupts the blood-brain barrier. Brain Sci 10: , 679. |
[47] | Wu Z , Ni J , Liu Y , Teeling JL , Takayama F , Collcutt A , Ibbett P , Nakanishi H ((2017) ) Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65: , 350–361. |
[48] | Zhang J , Yu C , Zhang X , Chen H , Dong J , Lu W , Song Z , Zhou W ((2018) ) Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation 15: , 37. |
[49] | Su X , Tang Z , Lu Z , Liu Y , He W , Jiang J , Zhang Y , Wu H ((2021) ) Oral Treponema denticola infection induces Aβ(1-40) and Aβ(1-42) accumulation in the hippocampus of C57BL/6 mice. J Mol Neurosci 71: , 1506–1514. |
[50] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean S ((2015) ) Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimers Dis 43: , 67–80. |
[51] | Singhrao SK , Chukkapalli S , Poole S , Velsko I , Crean SJ , Kesavalu L ((2017) ) Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE(-)(/)(-) mice brains. J Oral Microbiol 9: , 1270602. |
[52] | Zeng F , Liu Y , Huang W , Qing H , Kadowaki T , Kashiwazaki H , Ni J , Wu Z ((2021) ) Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection. J Neurochem 158: , 724–736. |
[53] | Nie R , Wu Z , Ni J , Zeng F , Yu W , Zhang Y , Kadowaki T , Kashiwazaki H , Teeling JL , Zhou Y ((2019) ) Porphyromonasgingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimers Dis 72: , 479–494. |
[54] | Hayashi K , Hasegawa Y , Takemoto Y , Cao C , Takeya H , Komohara Y , Mukasa A , Kim-Mitsuyama S ((2019) ) Continuous intracerebroventricular injection of Porphyromonas gingivalis lipopolysaccharide induces systemic organ dysfunction in a mouse model of Alzheimer’s disease. Exp Gerontol 120: , 1–5. |
[55] | Keles Yucel ZP , Afacan B , Emingil G , Tervahartiala T , Kose T , Sorsa T ((2020) ) Local and systemic levels of aMMP-8 in gingivitis and stage 3 grade C periodontitis. J Periodontal Res 55: , 887–894. |
[56] | Sadrameli M , Bathini P , Alberi L ((2020) ) Linking mechanisms of periodontitis to Alzheimer’s disease. Curr Opin Neurol 33: , 230–238. |
[57] | Chapple IL , Genco R ((2013) ) Diabetes and periodontal diseases: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol 40 Suppl 14: , S106–112. |
[58] | Van Dyke TE , van Winkelhoff AJ ((2013) ) Infection and inflammatory mechanisms. J Periodontol 84: , S1–7. |
[59] | Van Dyke TE ((2009) ) Resolution of inflammation-unraveling mechanistic links between periodontitis and cardiovascular disease. J Dent 37: , S582–583. |
[60] | Szulc M , Kustrzycki W , Janczak D , Michalowska D , Baczynska D , Radwan-Oczko M ((2015) ) Presence of periodontopathic bacteria DNA in atheromatous plaques from coronary and carotid arteries. Biomed Res Int 2015: , 825397. |
[61] | Tonetti MS , Van Dyke TE ((2013) ) Periodontitis and atherosclerotic cardiovascular disease: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol 40: (Suppl 14), S24–29. |
[62] | Salhi L , Rompen E , Sakalihasan N , Laleman I , Teughels W , Michel JB , Lambert F ((2019) ) Can periodontitis influence the progression of abdominal aortic aneurysm? A systematic review. Angiology 70: , 479–491. |
[63] | Sanz M , Marco Del Castillo A , Jepsen S , Gonzalez-Juanatey JR , D’Aiuto F , Bouchard P , Chapple I , Dietrich T , Gotsman I , Graziani F , Herrera D , Loos B , Madianos P , Michel JB , Perel P , Pieske B , Shapira L , Shechter M , Tonetti M , Vlachopoulos C , Wimmer G ((2020) ) Periodontitis and cardiovascular diseases: Consensus report. J Clin Periodontol 47: , 268–288. |
[64] | Sanz M , Ceriello A , Buysschaert M , Chapple I , Demmer RT , Graziani F , Herrera D , Jepsen S , Lione L , Madianos P , Mathur M , Montanya E , Shapira L , Tonetti M , Vegh D ((2018) ) Scientificevidence on the links between periodontal diseases and diabetes:Consensus report and guidelines of the joint workshop on periodontaldiseases and diabetes by the International Diabetes Federation andthe European Federation of Periodontology. J Clin Periodontol 45: , 138–149. |
[65] | Luong A , Tawfik AN , Islamoglu H , Gobriel HS , Ali N , Ansari P , Shah R , Hung T , Patel T , Henson B , Thankam F , Lewis J , Mintline M , Boehm T , Tumur Z , Seleem D ((2021) ) Periodontitis and diabetes mellitus co-morbidity: A molecular dialogue. J Oral Biosci 63: , 360–369. |
[66] | Gomes-Filho IS , Cruz SSD , Trindade SC , Passos-Soares JS , Carvalho-Filho PC , Figueiredo A , Lyrio AO , Hintz AM , Pereira MG , Scannapieco F ((2020) ) Periodontitis and respiratory diseases: A systematic review with meta-analysis. Oral Dis 26: , 439–446. |
[67] | Elwishahy A , Antia K , Bhusari S , Ilechukwu NC , Horstick O , Winkler V ((2021) ) Porphyromonas gingivalis as a risk factor to Alzheimer’s disease: A systematic review. J Alzheimers Dis Rep 5: , 721–732. |
[68] | Kaliamoorthy S , Nagarajan M , Sethuraman V , Jayavel K , Lakshmanan V , Palla S ((2022) ) Association of Alzheimer’s disease and periodontitis - a systematic review and meta-analysis of evidence from observational studies. Med Pharm Rep 95: , 144–151. |
[69] | Reed-Geaghan EG , Savage JC , Hise AG , Landreth GE ((2009) ) CD14 and toll-like receptors 2 and 4 are required for fibrillar Abeta-stimulated microglial activation. J Neurosci 29: , 11982–11992. |
[70] | Frost GR , Li YM ((2017) ) The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol 7: , 170228. |
[71] | Ezzat K , Pernemalm M , Pålsson S , Roberts TC , Järver P , Dondalska A , Bestas B , Sobkowiak MJ , LevänenB , Sköld M , Thompson EA , Saher O , Kari OK , Lajunen T , Sverremark Ekström E , Nilsson C , Ishchenko Y , MalmT , Wood MJA , Power UF , Masich S , Lindén A , Sandberg JK , Lehtiö J , Spetz AL , El Andaloussi S ((2019) ) Theviral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun 10: , 2331. |
[72] | Bregaint S , Boyer E , Fong SB , Meuric V , Bonnaure-Mallet M , Jolivet-Gougeon A ((2022) ) Porphyromonas gingivalis outside the oral cavity. Odontology 110: , 1–19. |


