
Hippocampal Reduction of α-Synuclein via RNA Interference Improves Neuropathology in Alzheimer’s Disease Mice
Abstract
Background:
Alzheimer’s disease (AD) cases are often characterized by the pathological accumulation of α-synuclein (α-syn) in addition to amyloid-β (Aβ) and tau hallmarks. The role of α-syn has been extensively studied in synucleinopathy disorders, but less so in AD. Recent studies have shown that α-syn may also play a role in AD and its downregulation may be protective against the toxic effects of Aβ accumulation.
Objective:
We hypothesized that selectively knocking down α-syn via RNA interference improves the neuropathological and biochemical findings in AD mice.
Methods:
Here we used amyloid precursor protein transgenic (APP-Tg) mice to model AD and explore pathologic and behavioral phenotypes with knockdown of α-syn using RNA interference. We selectively reduced α-syn levels by stereotaxic bilateral injection of either LV-shRNA α-syn or LV-shRNA-luc (control) into the hippocampus of AD mice.
Results:
We found that downregulation of α-syn results in significant reduction in the number of Aβ plaques. In addition, mice treated with LV-shRNA α-syn had amelioration of abnormal microglial activation (Iba1) and astrocytosis (GFAP) phenotypes in AD mice.
Conclusion:
Our data suggests a novel link between Aβ and α-syn pathology as well as a new therapeutic angle for targeting AD.
INTRODUCTION
Neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Lewy body disease (LBD) are characterized by the progressive accumulation of protein aggregates that initially trigger synaptic damage and network dysfunction and eventually lead to loss of selected neuronal populations [1]. AD is characterized by the accumulation of amyloid-β (Aβ) and phosphorylation of the microtubule associate protein, tau [2]. In addition to Aβ and tau, recent studies have shown α-synuclein (α-syn) may also contribute to AD pathogenesis [3–6].
Pathological comorbidity is common in neurodegenerative diseases. For example, the co-occurrence of Aβ and α-syn at autopsy is found in over 50% of patients diagnosed with AD [7, 8]. Co-morbid patients show an increased rate of cognitive decline and shorter life span compared to classic AD, suggesting that accumulation of these two proteins together is synergistic in contributing to neuronal degeneration and pathology. In addition, studies demonstrate that cortical Aβ plaque load correlates with α-syn load and overall Lewy body quantity in the cortex and is associated with cognitive decline [9]. Similarly, patients with autosomal dominant APP mutations show α-syn accumulations in Lewy bodies without having mutations in any known PD or associated genes [10, 11]. Peripherally, α-syn and Aβ have been found together in blood cells of normal patients [12, 13] and in exosomes of Lewy body disease patients [14], suggesting that α-syn and Aβ co-aggregate outside of the CNS. These data suggest that α-syn can have a significant effect on the aggregation and accumulation of Aβ. α-syn has been extensively studied in the context of PD and dementia with Lewy bodies (DLB) where it accumulates as discrete neuropathological structures (Lewy bodies and Lewy neurites) that are directly linked to neuronal degeneration [15]. In AD and APP transgenic models, α-syn interacts with both Aβ and tau and accumulates in selected regions of the limbic system [16]. Aβ promotes α-syn aggregation [17] and toxicity and levels of soluble α-syn are elevated in cerebrospinal fluid (CSF) in patients with AD [18–21], whereas in PD and DLB patients, CSF α-syn is not altered compared to control patients [21]. However, aggregated forms of α-syn are indeed elevated in patients suffering from synucleinopathies, as shown by RT-QuIC assays which are currently some of the most promising methods for amplification of aggregates from in vivo samples and thereby diagnosis of disease [22–25]. Moreover, studies demonstrate that α-syn administration enhances synaptic degeneration in APP-Tg mice [26].
Our laboratory recently reported that enhanced expression of α-syn in AD mice led to increased degeneration of cholinergic neurons in the nucleus basalis and hippocampal glutamatergic neurons [27]. We have expanded these studies and showed that genetic ablation of α-syn (by crossing with an α-syn knockout mouse) prevents loss of cholinergic neurons and ameliorates the functional memory deficits in an APP-Tg mouse [27]. Thus, reducing α-syn expression may be a disease-modifying therapeutic strategy for AD, though this hypothesis needs further elucidation. Here we show, for the first time, that specifically targeting α-syn in vivo in an AD model can be a successful strategy in ameliorating neuropathological lesions and rescuing learning and memory deficits.
While many studies are focused only on targeting Aβ, tau or inflammation, we are targeting the concept of co-morbidity in AD. To this end we utilize shRNA molecules to downregulate α-syn in vivo. shRNAs can bind specifically to target RNAs and deliver them for degradation and this approach has shown promising results clinically for spinal muscular atrophy and amyotrophic lateral sclerosis [28, 29]. Here we followed the concepts of such studies and applied this gene therapy principle to AD in a non-conventional way, i.e., by targeting α-syn in order to investigate the neuropathological link between these two neurodegenerative diseases.
MATERIALS AND METHODS
Ethical statement
All experimental studies involving animals were approved by the Institutional Animal Care and Use Committee of University of California San Diego and performed in accordance with relevant guidelines and regulations established by NIH Guide for the Care and Use of Laboratory Animals under protocol #S02221.
Mouse line APP-Tg
In this study we used the PSAPP mouse line as an AD transgenic model (APP-Tg). APP-Tg mice are double transgenic, expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9), both directed to CNS neurons [30]. Both mutations are associated with early-onset AD [31]. These mice begin to accumulate Aβ at a young age and develop large numbers of fibrillar Aβ deposits in the cerebral cortex and hippocampus at about six months of age [32–34]. Plaque pathology is not only accelerated, but enhanced, with Aβ deposits eventually occupying a large area of the neocortex and hippocampus by 16 months. In parallel, there is a substantial increase in plaque-associated astrocytes and microglia, suggesting an overall increase in neuroinflammation between the ages of six and 16 months of age. In summary, these mice display early Aβ deposition, behavioral deficits, and degeneration of the limbic and cholinergic systems [35].
Stereotaxic delivery to the hippocampus
Mice were sterotaxically injected with 3μl of the lentiviral preparations (2.5×107 TU) into the temporal cortex and hippocampus (using a 5μl Hamilton syringe). Briefly, as previously described, mice were placed under anesthesia on a Koft stereotaxic apparatus and coordinates (hippocampus: AP –2.0 mm, lateral 1.5 mm, depth 1.3 mm and cortex: AP –0.5 mm, lateral 1.5 mm, depth 1.0 mm) were determined as per the Franklin and Paxinos Atlas [36]. The lentiviral vectors were delivered using a Hamilton syringe connected to a hydraulic system to inject the solution at a rate of 1μl every 2 min. To allow diffusion of the solution into the brain tissue, the needle was left for an additional 5 min after the completion of the injection. As an additional control for LV injection, age-matched littermates were injected with LV-shLuc.
Construction and packaging of lentivirus vector
The shα-syn (5’GAC UUU CAA AGG CCA AGG A) corresponding to nucleotides 168–186 of α-syn was chosen because we have previously shown this targets both mouse and human α-syn [27]. As a control we used the control shRNA lentivector (LV-shLuc) contains an shRNA directed against firefly luciferase. Lentiviruses were prepared by transient transfection in 293T cells as previously described [37].
Tissue preparation
Following National Institutes of Health guidelines for the humane treatment of animals, mice were anesthetized with isoflurane and flush-perfused transcardially with 0.9% saline. Brains were removed and divided in sagittal sections. The right hemibrain was postfixed in phosphate-buffered 4% PFA, pH 7.4, at 4°C for 48 h for neuropathological analysis and the left hemibrain was snap-frozen and stored at –70°C for subsequent protein analysis.
Immunohistochemical analysis
Analysis was performed using free floating, 40μm-thick, vibratome-cut, blind-coded sections, as described previously [38]. Briefly, sections were incubated overnight at 4°C with antibodies against total α-syn (1:500, affinity-purified mouse monoclonal syn211, Millipore [18]), phosphorylated α-syn (1:1000, rabbit monoclonal, abcam ab51253), Aβ (82E1, mouse monoclonal; BioLegend), ChAT (1:100, rabbit monoclonal, Invitrogen), NeuN (1:500, affinity-purified monoclonal; Millipore), GFAP (1:500, affinity purified monoclonal; Millipore), Iba1 (1:500, mouse monoclonal, ThermoFisher) followed by biotin-tagged anti-rabbit or anti-mouse IgG1 (1:100; Vector Laboratories) secondary antibodies, avidin D-HRP (1:200, ABC Elite; Vector Laboratories), and visualized with diaminobenzidine (DAB). For detection of the injected lentivirus construct in tissue, two different strategies were used: 1) we used an anti-copGFP antibody (AB501, Evrogen), followed by goat anti-rabbit-AF488 secondary antibody (Invitrogen cat11008) and 2) we did nickel enhancement of anti-copGFP DAB stained sections. We then controlled for α-syn reduction in these tissues by staining for phosphorylated α-syn (1:500, phospho S129, affinity-purified rabbit monoclonal, EP1536Y). Sections were scanned with a NanoZoomer S60 Digital slide scanner C13210-01 and images were analyzed using ImageJ [39] for quantification of optical densities and cell counts, by correcting for background staining.
Immunoblot analysis
The levels of α-syn in hippocampus and cortex from mouse brains were analyzed using lysates that were extracted and fractioned into membrane and cytosolic fractions by ultracentrifugation. Briefly, previously frozen hemibrains were sub-dissected to obtain the cortex and the hippocampus for further analysis. Tissues were homogenized in RIPA (lysis buffer, Thermofisher) buffer with the addition of protease inhibitor cocktails (HaltTM and PMSF –Thermofisher) followed by ultracentrifugation at 30,000 rpm after each step. Protein (20μg/lane) was loaded onto 4 –12% SDS/PAGE gels and blotted onto PVDF membranes and incubated with total α-syn antibody (1:500, affinity-purified mouse monoclonal syn211, Millipore16), followed by HRP-tagged secondary antibodies (1:5000; Santa Cruz Biotechnology). Bands were visualized by enhanced chemiluminescence (PerkinElmer) and analyzed with a quantitative Chemidoc MP imaging apparatus (Bio-Rad). β-actin (1:3000) was the loading control.
Statistical analysis
All analyses were performed using GraphPad Prism (version 9.0) software. Differences among means were assessed by one-way ANOVA with Dunnett’s post hoc test when comparing treated versus non-treated and transgenic versus WT mice.
RESULTS
Delivery of shα-syn via intracranial injection of a lentiviral vector reduces the accumulation of α-syn in mouse brains
In our previous work we demonstrated that treatment of neuronal cells with shRNA constructs is an effective strategy to lower α-syn levels in vitro. In this study, we went a step further and confirmed whether we could selectively reduce α-syn levels in neuronal tissue in vivo. We administered bilateral stereotaxic injections of either the lentiviral vector expressing shα-syn or the control vector (shCtrl) expressing an shRNA against firefly luciferase into the hippocampus of mice, as previously described [36]. The lentivirus construct was built such that it expressed copGFP, to facilitate later detection in the tissues tested. This is shown in Fig. 1B, where both the LV-shαsyn- and LV-shScr-injected mice had copGFP-positive signal in and around the hippocampus. This is also visible by DAB and Ni+ staining, in Fig. 1B, with more representative images in Supplementary Figure 1. Furthermore, we quantified the effect of our construct to lower alpha-syn levels in Fig. 1C using Western blot. In this way we were able to ascertain that α-syn levels are significantly reduced in the hippocampus, but the difference was not significant in the cortex and striatum regions. To understand whether RNA interference of α-syn is an effective strategy for reducing α-syn-associated pathology in an AD model. We hypothesized that a highly effective shRNA would be sufficient in reducing α-syn pathology.
Fig. 1
Validation of in vivo knockdown of α-syn using a lentiviral vector to deliver shα-syn intracranially. A) Line PSAPP (APP-Tg) was intracranially injected with LV-shαSyn or LV-shLuc at 6 months of age. At 9 months of age, brains were harvested. B) Representative high magnification images of brain slices from LV-shαSyn- or PBS-injected mice, immunostained for copGFP (upper panel) and double immunostained for copGFP-Ni/Psyn (bottom panels). C) Hemibrains from APP-Tg were processed using punches collected from each sub-region of the hippocampus and homogenized for quantification of total α-syn by Western blotting, using Syn211antibody. Quantification of band intensities, normalized for β-actin control. One-way Anova with post-hoc Dunnett’s comparisons test revealed for hippocampus regions p = 0.0253. D) Brain slices from APP-Tg mice injected with LV-sh-scr, LV-shα-syn, and PBS were stained for phosphorylated α-syn (S129) – DAB staining. Rows reflect zoomed in panels of each hippocampal region (CA1, CA3, and DG). *p < 0.05. Scale bars, 200μm in low-power images and 40μm in high-power images.
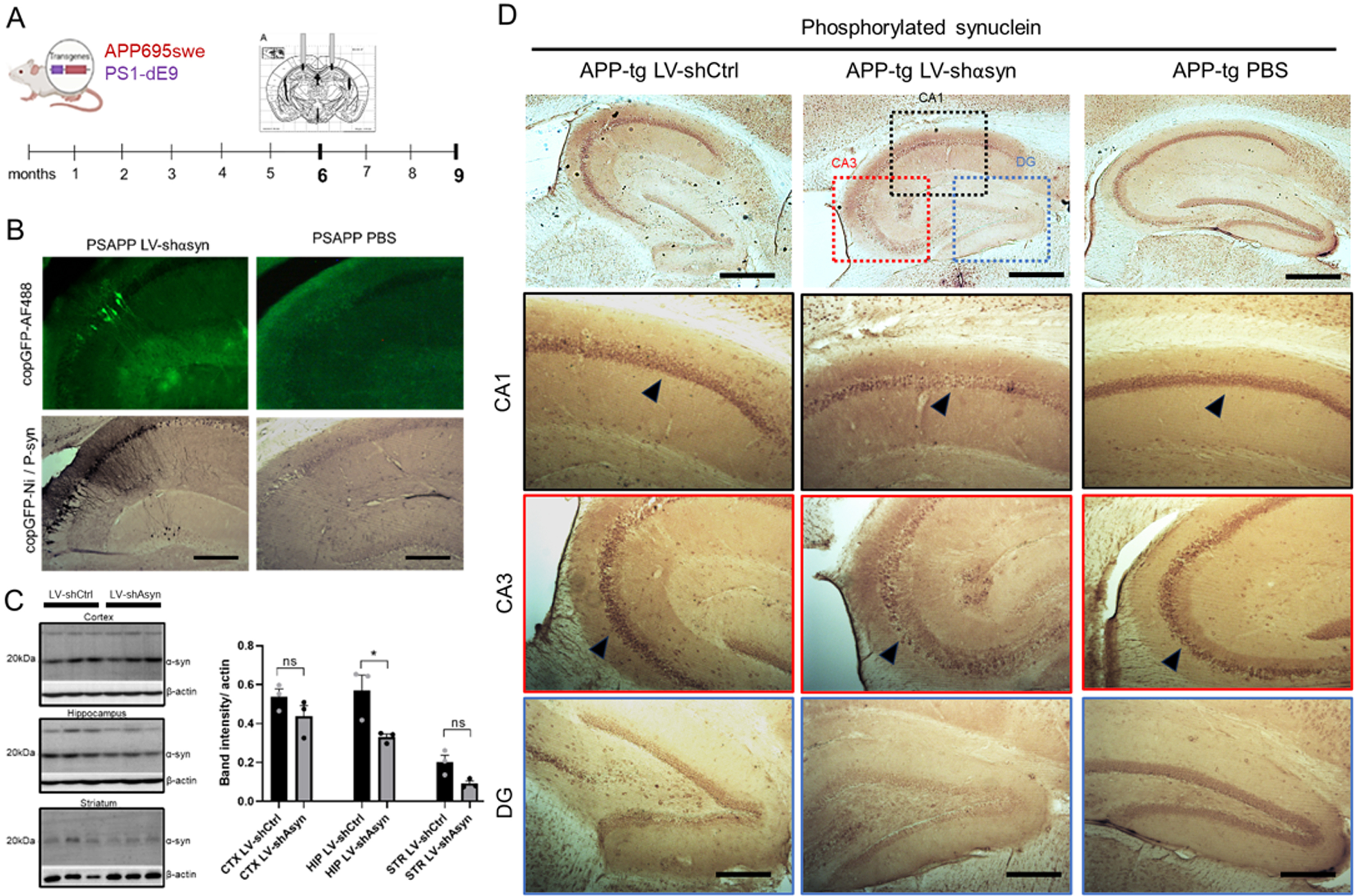
Bilateral stereotaxic intracranial injections in both hippocampi were performed at 6 months of age, a timepoint where APP-Tg mice start to develop pathological plaques (Fig. 1A). Mice were sacrificed after 3 months and immunohistochemistry for α-syn was performed using two antibodies: Syn211 (Invitrogen), which cross-reacts with both total human and mouse α-syn; and PSyn, which cross-reacts with human and mouse α-syn phosphorylated at the S129 residue, a post-translational modification that is associated with pathological protein aggregation [40]. Quantification of Psyn immunostaining indicated a significant reduction in phosphorylated α-syn in the hippocampus in mice injected with LV-shα-syn compared to the control LV-shLuc (Fig. 1D). No significant reduction in immunostaining was observed in the cortex or striatum, attesting to the efficacy of injections and the LV delivery method.
Delivery of an α-syn shRNA to APP-Tg mice reduces the pathological accumulation of Aβ
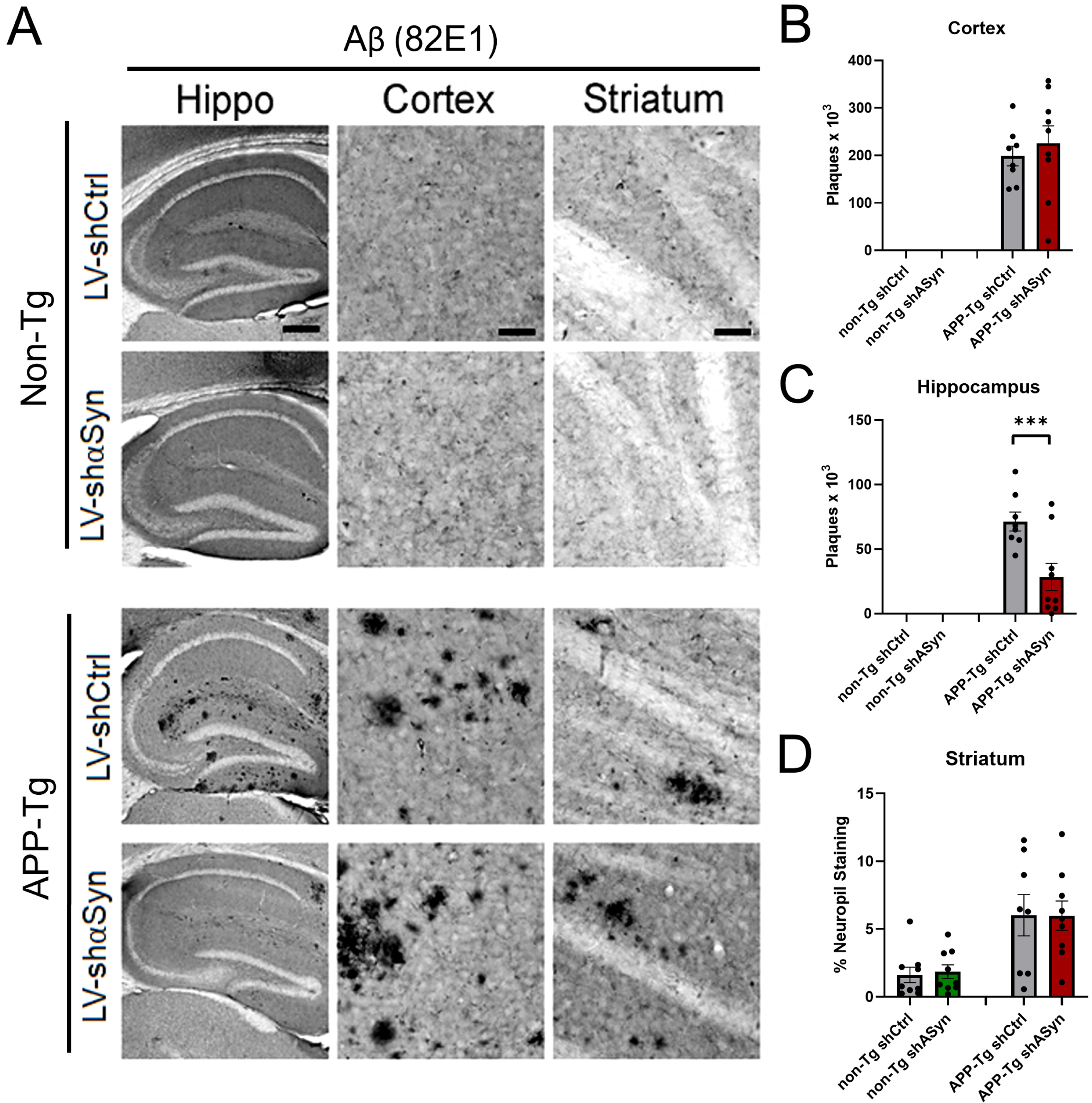
Having successfully established a method for the knockdown of α-syn levels in vivo, we then tested our hypothesis that, by reducing α-syn, we would be able to diminish pathological accumulations of Aβ in the brains of APP-Tg mice. As stated above, mice received bilaterally injections in the hippocampus with either a LV-shα-syn or LV-shLuc at 6 months of age. Three months later, mice were sacrificed and the number of Aβ plaques was counted in the hippocampus, cortex, and striatum. Immunohistochemistry using the anti-human monoclonal antibody 82E1 against the N-terminus of human Aβ showed, as expected, the formation of Aβ plaques in APP-Tg mice (Fig. 2). Importantly, a significant reduction in the number of plaques was observed in mice that were injected with the LV-shα-syn. This reduction occurred only within the hippocampus with no changes observed in the cortex or striatum of these mice (Fig. 2B–D). These results suggest that α-syn and Aβ co-exist and co-accumulate in specific brain regions under pathological conditions.
Fig. 2
Knockdown of α-syn via LV-shα-syn intracranial injection reveals significant reduction of Aβ plaques in APP-Tg mice. A) Immunohistochemistry using 82E1 antibody that detects human Aβ. B–D) Quantification of Aβ plaques in hippocampus, cortex, and neuropil staining in striatum using 82E1 antibody. ***p < 0.001 One-way ANOVA with post-hoc Dunnett’s multiple comparisons test. Scale bars, 100μm in low-power images and 40μm in high-power images.
Immunohistochemical analysis of pan-neuronal (NeuN) and cholinergic (ChAT) systems upon reduction of α-syn
Next, we analyzed the effects of reducing α-syn levels on the degeneration of specific neuronal populations in the hippocampus and cortex. At 9 months of age, we did not find significant neuronal loss in APP-Tg mice as compared to non-Tg, based on immunohistochemistry against pan-neuronal populations using NeuN antibody (Fig. 3). We observed that LV-shα-syn treatment showed a lower NeuN staining in CA3 in APP-Tg mice, which might suggest an effect of the method itself, since injections targeted this area (Fig. 3B, C).
Fig. 3
Reduction of α-syn by lentivirus delivery to the hippocampus of APP-Tg mice results in increased cholinergic neuronal populations while not affecting pan neuronal cell populations. Vibratome sections were immunostained with an antibody against NeuN and ChAT. A) Representative low- and high-magnification photomicrographs of the frontal cortex, CA3, and CA1 regions of the hippocampus from non-Tg and APP-Tg, mice immunoreacted with anti-NeuN. B, C) Quantification of adjusted optical density in the hippocampus (CA3) and cell counts per 103 in the cortex revealed lower number of NeuN-positive cells in LV-shα-syn-treated mice. D) Representative photomicrographs of ChAT immunoreactivity in the basal forebrain and striatum at high magnification. E, F) APP-Tg Mice treated with LV-shα-syn showed a higher number of ChAT-positive cells in the basal forebrain as compared to LV-shCtrl-treated mice. Statistical analysis was conducted using one-way ANOVA post hoc Dunnett’s test *p < 0.05, **p < 0.005. For analysis, 10 mice (5M, 5F) 6 months of age were used. Scale bars, 100μm in low-power images and 40μm in high-power images.
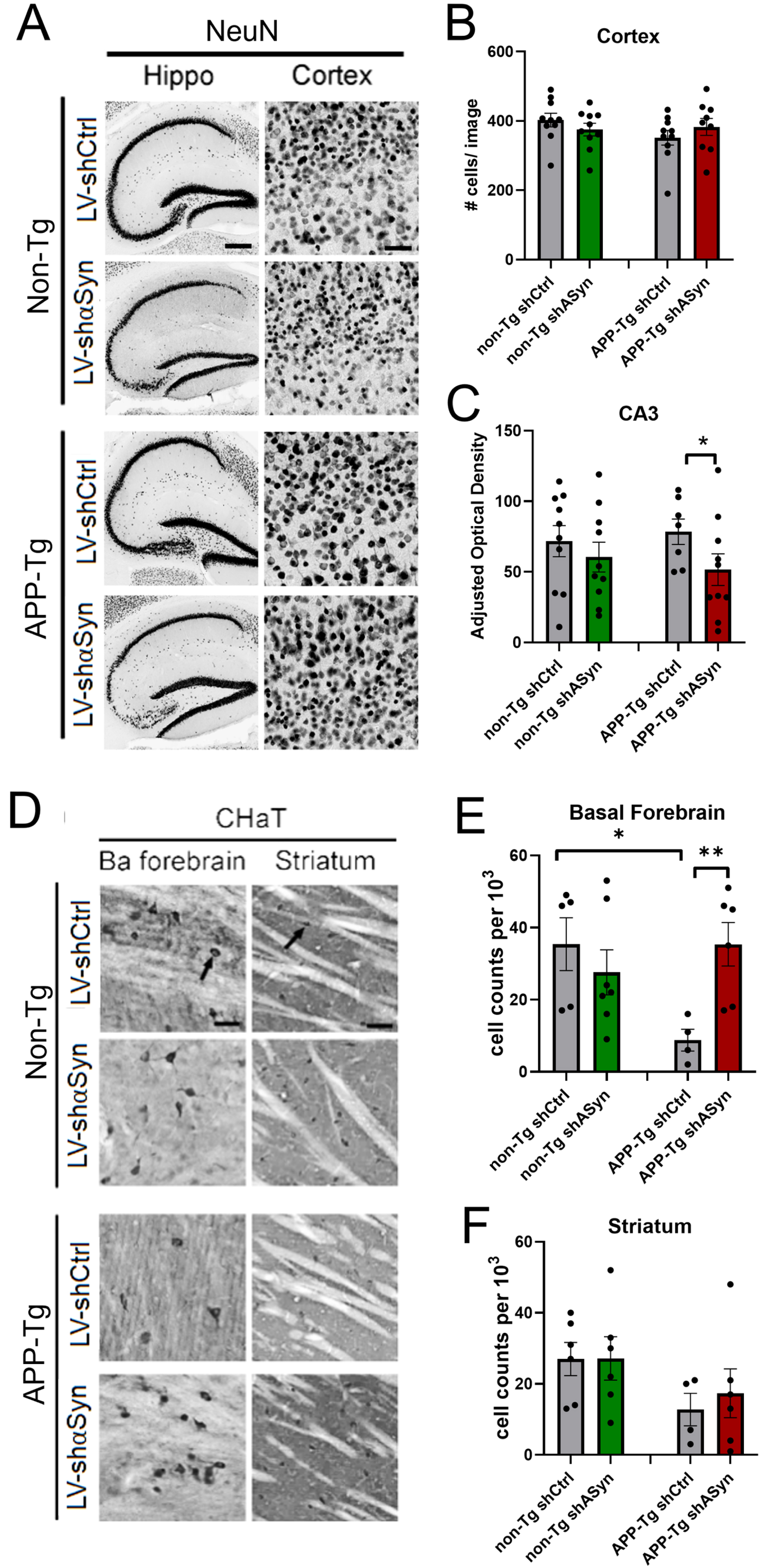
Previous studies have reported that α-syn can interact and aggravate Aβ pathology leading to cholinergic-specific neurodegeneration. For this reason, we next investigated whether reducing α-syn might protect selective neuronal populations from the neurotoxic effects of Aβ. Consistent with previous studies, analysis of the cholinergic system with an antibody against ChAT showed that, compared with non-Tg mice, APP-Tg mice displayed a reduction in the number of cholinergic neurons in the basal forebrain (Fig. 3D–F). Importantly, APP-Tg mice that were treated with LV-shα-syn at 6 months of age showed similar number of ChAT-positive neurons as the non-Tg group, supporting the efficacy of this treatment in preventing the early loss of cholinergic neurons in AD. Finally, given that α-syn has been reported to accelerate neurodegeneration of the dopaminergic system [41], we also performed immunohistochemistry using an antibody against tyrosine hydroxylase, however we did not find differences between APP-Tg and non-tg mice at 9 months, nor between treated and untreated groups following delivery of the LV-shα-syn to the hippocampus (data not shown).
Downregulation of α-syn prevents glial-mediated neuroinflammatory phenotypes in AD
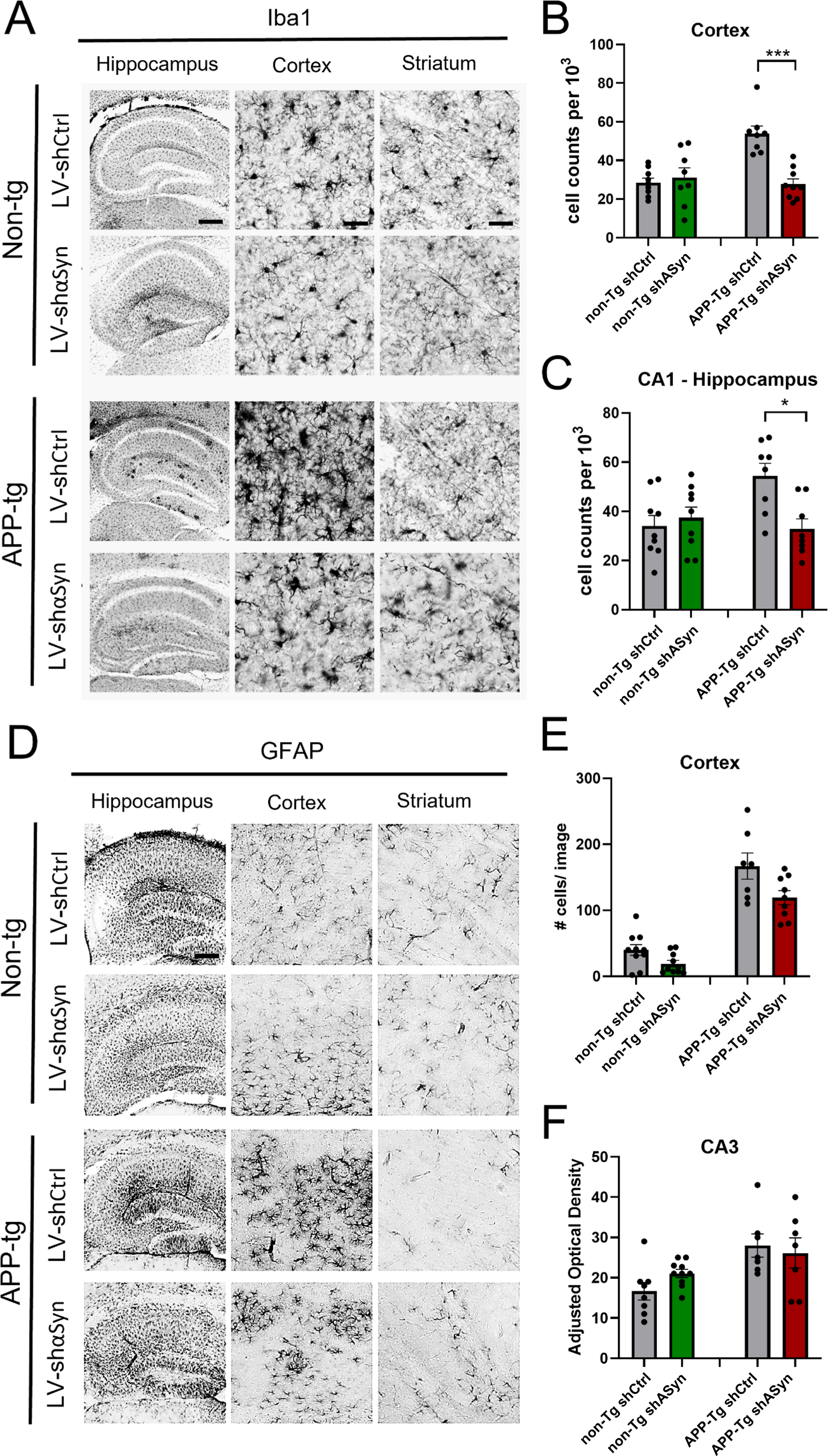
The role of glial cells in the pathogenesis of several neurodegenerative diseases has been extensively studied [42–45]. Astrocytes in particular have been demonstrated to associate closely with Aβ plaques, and reactive astrocytes are increased both in human AD patient brains and AD mouse models [46, 47]. We hypothesized that by alleviating Aβ burden via reduction of α-syn, we could also provide a means to reduce neuroinflammation in the brains of AD mice. Our results show that both Iba1 (Fig. 4A) and GFAP (Fig. 4D) are increased in APP-Tg mouse brains (cortex), when compared to non-Tg mice. Remarkably, when APP-Tg mice receive the treatment with LV-shα-syn, this difference is significantly abrogated for both Iba1 and GFAP, demonstrating the potential for the downregulation of α-syn in preventing inflammatory responses in mouse models of AD.
Fig. 4
Immunohistochemical analysis of changes in microglial (Iba1) and astrocytic activation (GFAP) in non-Tg and APP-Tg mice treated with LV-shα-syn as compared to LV-shCtrl. A, D) Representative low- and high-magnification photomicrographs of the frontal cortex, CA3, and CA1 regions of the hippocampus –Iba1 and GFAP immunostains respectively. B, C) Quantification of anti-Iba1 immunostains for cell counts/103 mm in the hippocampus (CA3), cortex and striatum, showing a statistically significant lower number of Iba1-positive cells in the cortex of APP-Tg mice treated with LV-shα-syn as compared with LV-shCtrl. E, F) Quantification of GFAP-positive signal using adjusted optical density in the CA3 and cell counts per 103 mm in the cortex and striatum. Statistical analysis was conducted using one-way ANOVA post hoc Dunnett’s test *p < 0.05, ***p < 0.001. Scale bars, 100μm in low-power images and 40μm in high-power images.
Thus, the aggregate of these experiments shows that reduction of α-syn by lentivirus vector delivery to the hippocampus has the effect of reducing the accumulation of Aβ, reducing cholinergic neuron loss and reducing cortical inflammation in a mouse model of AD.
DISCUSSION
This work describes the use of shRNAs to reduce levels of α-syn in vivo in different mouse models of neurodegenerative diseases. Our main goal was to explore the involvement of α-syn in the selective vulnerability triggered by Aβ in AD mouse models, given the extensive evidence in literature on comorbidities caused by both proteins in these mice [48–50]. To that end, we manipulated the expression of α-syn in AD mice and asked the question whether the neuropathological of AD would be ameliorated.
Pathological interactions between Aβ and α-syn
Our work shows that in vivo (intracranial) delivery of shα-syn using a lentiviral vector successfully clears Aβ plaques in APP-Tg mice. These findings support the notion that α-syn and Aβ are inter-related in their neuropathological accumulation. This is supported by the fact that treatment with shα-syn also led to a significantly lower loss of cholinergic neurons in the basal forebrain in our study, a neuropathological finding that has been shown to be the main correlating factor to cognitive and memory deficits in AD [51, 52].
In line with this, an increasing body of evidence suggests that α-syn increases the aggregation and toxicity of Aβ [4]. In vitro and computer simulations have shown α-syn may interact with Aβ through the repeat lysine domains in the α-syn ‘KTKEGV’ domain [17, 53]. Additionally, inter-aggregations between α-syn and Aβ may occur through hydrophobic domains in the NAC domain of α-syn and the core domain of Aβ42 [17, 53, 54]. These inter-protein interactions have been shown to promote cross-aggregation and toxicity in neuronal cells [54], and this may explain why Lewy bodies are present in many AD patients and why lowering the expression levels of one protein could prevent the aggregation of the other. This has been tested before, as mice transgenic for α-syn and APP developed more severe neuronal degeneration and learning deficits than mice singly transgenic for either gene alone [18]. Double α-syn/APP transgenic mice have also been shown to accumulate α-syn to a greater extent than singly α-syn transgenic mice [27]. Interestingly, increased α-syn expression has also been shown to promote AβPP expression as well as β- and γ-secretase processing of AβPP to increase Aβ production [55]. Therefore, higher levels of α-syn may increase Aβ which may be promoting at least some of the aggregation of Aβ into plaques that we observe under normal levels of α-syn expression.
Inflammatory responses to α-syn
We observed less Aβ plaques in mice treated with LV-shα-syn. Importantly, these results correlate with an amelioration of glial-mediated neuroinflammation, as shown by the rescue of immunostaining against Iba1 (activated microglia) and astrocytes (GFAP) to wild-type (non-transgenic) levels. Interestingly, we observed this reduction in the cortex, a region outside of the injection region (Fig. 4). Previous studies have shown that in AD, resident microglia undergo proinflammatory activation, resulting in an increased capacity to convert resting astrocytes to reactive astrocytes [56, 57]. Therefore, microglia are a major therapeutic target for AD and blocking microglia-astrocyte activation has provided promising results in limiting neurodegeneration in AD [56]. Recent studies supporting this idea have shown that α-syn accumulation in microglia induces selective neuronal degeneration by promoting phagocytic exhaustion, an excessively toxic environment and the selective recruitment of peripheral immune cells [58]. This inflammatory state creates a molecular feed-forward vicious cycle between microglia and IFNγ-secreting immune cells infiltrating the brain parenchyma [57, 58]. Our results are promising insofar as we postulate that targeting α-syn could represent a proxy to an anti-neuroinflammatory treatment by blocking this detrimental microglia-astrocyte activation in AD.
It is still unclear whether Aβ plaques originate from neuroinflammation, or mostly contribute to it [57, 59]. Other cells in the brain may also play a role in spreading pathology. Astrocytes are recruited to the vicinity of plaques, engulf them and eventually are lysed, contributing to the dispersal of Aβ42, leading to the deposition of GFAP-rich plaques in the cortex, as we observed in our study [59, 60]. Microglia, congregate within plaques to try and clear these accumulations [59, 60].
Study limitations
Our study is limited by the lack of behavior analysis and the shRNA delivery method. While Aβ plaques and neurofibrillary tangles define an AD diagnosis, cognitive impairment impacts the everyday lives of AD patients. Removal of the α-syn gene improved behavioral performance of APP-Tg mice [27]. However, the observed behavioral results from these mice may arise from developmental compensations physiologically irrelevant to the human disease. To further understand the consequences of hippocampal α-syn-mediated plaque reduction on cognitive impairment, future studies should focus on measuring both motor ability and memory.
Selective reduction of α-syn was mediated through lentiviral delivery to ensure stable expression of shα-syn in mouse brain. Lentiviral gene delivery involves the integration of the virus into the host genome, but aberrant chromosome positioning may result in gene suppression [61, 62]. The modest reduction in α-syn protein observed in LV-shα-syn treated mice may be due to inappropriate gene integration which led to stunted shRNA expression (Fig. 1). The low viral viral titer delivered in our study (2.5×107 TU) could contribute to the variating results. Additionally, lentiviral transduction localizes to the site of injection [63], which may add variability when injecting a large brain region such as the hippocampus. However, delivery of high viral titers resulted in reduced gene expression and cell density [64]. A modest reduction in Iba1+ cells in the cortex of LV-shα-syn injected mice suggests that localized changes in the hippocampus may impact the state of other cells in neighboring regions (Fig. 4). Measuring transcript production, whether it be shRNA transcript or even α-syn, may indicate the success of gene transduction and further support our conclusions. While lentiviral gene delivery allows for robust expression, the limits of the technique influence overall expression, and may not be a suitable vehicle for shRNA-mediated therapeutics for AD. Alternative methods to shRNA delivery, such as protein-mediated gene delivery may increase the success of delivery without the potential side effects [27].
Although the reduction of α-syn levels was achieved in this study as shown by immunocytochemistry and immunoblotting, it was relatively modest. However, perhaps the most interesting finding of this study is that, despite this hippocampal reduction being subtle, the reduction in the number of Aβ plaques in this region is noticeable. It should be mentioned that we were not able to confirm this reduction via immunoblotting using different Aβ antibodies, due to the overall unchanged levels of Aβ in tissue (Supplementary Figure 2). Further studies should be performed to confirm that the presence of insoluble Aβ (plaques) is lowered when α-syn levels are reduced.
Our study highlights the potential pathogenic relationship between Aβ and α-syn rather than suggest a therapeutic method of targeting Aβ plaques. These data build upon studies by demonstrating that local reduction of endogenous α-syn through shRNA sufficiently altered overall hippocampal AD pathogenesis in a mouse line overexpressing several AD-associated. To further investigate the role of co-morbid expression in AD pathogenesis, future studies should focus on α-syn interactions with other proteins involved in AD such as tau. Interactions between α-syn and the C-terminus of tau resulted in accelerated toxic aggregates and increased spread in vitro and in vivo [65–69]. Understanding how the modulation of either α-syn or tau alters pathological spread or behavioral symptoms would open additional therapeutic paths to treating aggregate protein neurodegenerative diseases.
In this work we present a strategy to therapeutically target α-syn in AD, a protein which is traditionally not associated with this disease. Neurodegeneration is increasingly regarded as a multifactorial phenomenon, where several proteins co-accumulate in the brain and lead to functional deficits. Here we show that α-syn can be detrimental in the initiation and/or progression of AD and that its reduction leads to improvement in neuropathological phenotypes.
ACKNOWLEDGMENTS
The authors thank Kimberley Mullen, James Barlow, Jazmin Florio, Michael Mante, Estefani Guzman, Sahar Salehi, Bao Quach, and Carlos Arias from the Rissman lab for technical assistance with this project. The authors would also like to thank the UCSD Microscopy Core for helping with imaging immunohistochemistry slides.
FUNDING
This work was supported by NIH/NIA grants AG018440 and AG072053 to RAR. The Microscopy core was funded by NINDS NS047101.
CONFLICT OF INTEREST
Robert Rissman is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
DATA AVAILABILITY
All the data supporting the findings of this study, namely the full quantification of immunohistochemical slides, are available on request from the corresponding author.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230232.
REFERENCES
[1] | Fu H , Hardy J , Duff KE ((2018) ) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21: , 1350–1358. |
[2] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[3] | Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM ((2010) ) Synergistic interactions between Aβ,tau, and α-synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 30: , 7281–7289. |
[4] | Mandal PK , Pettegrew JW , Masliah E , Hamilton RL , Mandal R ((2006) ) Interaction between Aβ peptide and α synuclein: Molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res 31: , 1153–1162. |
[5] | Jellinger KA ((2011) ) Interaction between alpha-synuclein and other proteins in neurodegenerative disorders. ScientificWorldJournal 11: , 1893–1907. |
[6] | Twohig D , Nielsen HM ((2019) ) α-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener 14: , 23. |
[7] | Hamilton RL ((2000) ) Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol 10: , 378–384. |
[8] | Rabinovici GD , Carrillo MC , Forman M , DeSanti S , Miller DS , Kozauer N , Petersen RC , Randolph C , Knopman DS , Smith EE ((2017) ) Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimers Dement (N Y) 3: , 83–91. |
[9] | Lashley T , Holton JL , Gray E , Kirkham K , O’Sullivan SS , Hilbig A , Wood NW , Lees AJ , Revesz T ((2008) ) Cortical α-synuclein load is associated with amyloid-β plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol 115: , 417–425. |
[10] | Leverenz JB , Fishel MA , Peskind ER , Montine TJ , Nochlin D , Steinbart E , Raskind MA , Schellenberg GD , Bird TD , Tsuang D ((2006) ) Lewy body pathology in familial Alzheimer disease: Evidence for disease-and mutation-specific pathologic phenotype. Arch Neurol 63: , 370–376. |
[11] | Rosenberg CK , Pericak-Vance MA , Saunders AM , Gilbert JR , Gaskell PC , Hulette CM ((2000) ) Lewy body and Alzheimer pathology in a family with the amyloid-β precursor protein APP717 gene mutation. Acta Neuropathol 100: , 145–152. |
[12] | Daniele S , Frosini D , Pietrobono D , Petrozzi L , Lo Gerfo A , Baldacci F , Fusi J , Giacomelli C , Siciliano G , Trincavelli ML ((2018) ) α-Synuclein heterocomplexes with β-amyloid are increased in red blood cells of Parkinson’s disease patients and correlate with disease severity. Front Mol Neurosci 11: , 53. |
[13] | Daniele S , Pietrobono D , Fusi J , Lo Gerfo A , Cerri E , Chico L , Iofrida C , Petrozzi L , Baldacci F , Giacomelli C ((2018) ) α-Synuclein aggregated with tau and β-amyloid in human platelets from healthy subjects: Correlation with physical exercise. Front Aging Neurosci 10: , 17. |
[14] | Ngolab J , Trinh I , Rockenstein E , Mante M , Florio J , Trejo M , Masliah D , Adame A , Masliah E , Rissman RA ((2017) ) Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun 5: , 46. |
[15] | Mahul-Mellier A-L , Burtscher J , Maharjan N , Weerens L , Croisier M , Kuttler F , Leleu M , Knott GW , Lashuel HA ((2020) ) The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 117: , 4971–4982. |
[16] | Ferman TJ , Aoki N , Crook JE , Murray ME , Graff-Radford NR , van Gerpen JA , Uitti RJ , Wszolek ZK , Graff-Radford J , Pedraza O ((2018) ) The limbic and neocortical contribution of α-synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimers Dement 14: , 330–339. |
[17] | Tsigelny IF , Crews L , Desplats P , Shaked GM , Sharikov Y , Mizuno H , Spencer B , Rockenstein E , Trejo M , Platoshyn O ((2008) ) Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PloS One 3: , e3135. |
[18] | Masliah E , Rockenstein E , Veinbergs I , Sagara Y , Mallory M , Hashimoto M , Mucke L ((2001) ) β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 12245–12250. |
[19] | Bate C , Gentleman S , Williams A ((2010) ) α-synuclein induced synapse damage is enhanced by amyloid-β1-42. Mol Neurodegener 5: , 55. |
[20] | Korff A , Liu C , Ginghina C , Shi M , Zhang J , Alzheimer’s Disease Neuroimaging Initiative ((2013) ) α-Synuclein in cerebrospinal fluid of Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis 36: , 679–688. |
[21] | Slaets S , Vanmechelen E , Le Bastard N , Decraemer H , Vandijck M , Martin JJ , De Deyn PP , Engelborghs S ((2014) ) Increased CSF α-synuclein levels in Alzheimer’s disease: Correlation with tau levels. Alzheimers Dement 10: , S290–S298. |
[22] | Concha-Marambio L , Farris CM , Holguin B , Ma Y , Seibyl J , Russo MJ , Kang UJ , Hutten SJ , Merchant K , Shahnawaz M ((2021) ) Seed amplification assay to diagnose early parkinson’s and predict dopaminergic deficit progression. Mov Disord 36: , 2444. |
[23] | Kang UJ , Boehme AK , Fairfoul G , Shahnawaz M , Ma TC , Hutten SJ , Green A , Soto C ((2019) ) Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov Disord 34: , 536–544. |
[24] | Russo MJ , Orru CD , Concha-Marambio L , Giaisi S , Groveman BR , Farris CM , Holguin B , Hughson AG , LaFontant D-E , Caspell-Garcia C ((2021) ) High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun 9: , 179. |
[25] | Bhumkar A , Magnan C , Lau D , Jun ESW , Dzamko N , Gambin Y , Sierecki E ((2021) ) Single-molecule counting coupled to rapid amplification enables detection of α-synuclein aggregates in cerebrospinal fluid of Parkinson’s disease patients. Angew Chem Int Ed Engl 133: , 11981–11990. |
[26] | Rockenstein E , Nuber S , Overk CR , Ubhi K , Mante M , Patrick C , Adame A , Trejo-Morales M , Gerez J , Picotti P ((2014) ) Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137: , 1496–1513. |
[27] | Spencer B , Desplats PA , Overk CR , Valera-Martin E , Rissman RA , Wu C , Mante M , Adame A , Florio J , Rockenstein E ((2016) ) Reducing endogenous α-synuclein mitigates the degeneration of selective neuronal populations in an Alzheimer’s disease transgenic mouse model. J Neurosci 36: , 7971–7984. |
[28] | Mathis S , Le Masson G ((2018) ) RNA-targeted therapies and amyotrophic lateral sclerosis. Biomedicines 6: , 9. |
[29] | Trülzsch B , Davies K , Wood M ((2004) ) Survival of motor neuron gene downregulation by RNAi: Towards a cell culture model of spinal muscular atrophy. Mol Brain Res 120: , 145–150. |
[30] | JankowskyJL, SluntHH, RatovitskiT, JenkinsNA, CopelandNG, BorcheltDR ((2001) ) Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 17: , 157–165. |
[31] | Holcomb L , Gordon MN , McGowan E , Yu X , Benkovic S , Jantzen P , Wright K , Saad I , Mueller R , Morgan D ((1998) ) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: , 97–100. |
[32] | Campbell SN , Zhang C , Roe AD , Lee N , Lao KU , Monte L , Donohue MC , Rissman RA ((2015) ) Impact of CRFR1 ablation on amyloid-β production and accumulation in a mouse model of Alzheimer’s disease. J Alzheimers Dis 45: , 1175–1184. |
[33] | Zhang C , Kuo C-C , Moghadam SH , Monte L , Campbell SN , Rice KC , Sawchenko PE , Masliah E , Rissman RA ((2016) ) Corticotropin-releasing factor receptor-1 antagonism mitigates beta amyloid pathology and cognitive and synaptic deficits in a mouse model of Alzheimer’s disease. Alzheimers Dement 12: , 527–537. |
[34] | Zhang C , Kuo C-C , Moghadam SH , Monte L , Rice KC , Rissman RA ((2015) ) Corticotropin-releasing factor receptor-1 antagonism reduces oxidative damage in an alzheimer’s disease transgenic mouse model. J Alzheimers Dis 45: , 639–650. |
[35] | Havas D , Hutter-Paier B , Ubhi K , Rockenstein E , Crailsheim K , Masliah E , Windisch M ((2011) ) A longitudinal study of behavioral deficits in an AβPP transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 25: , 231–243. |
[36] | Sapru MK , Yates JW , Hogan S , Jiang L , Halter J , Bohn MC ((2006) ) Silencing of human α-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp Neurol 198: , 382–390. |
[37] | Tiscornia G , Singer O , Verma IM ((2006) ) Production and purification of lentiviral vectors. Nat Protoc 1: , 241–245. |
[38] | Zwilling D , Huang S-Y , Sathyasaikumar KV , Notarangelo FM , Guidetti P , Wu H-Q , Lee J , Truong J , Andrews-Zwilling Y , Hsieh EW ((2011) ) Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 145: , 863–874. |
[39] | Rasband W (2011) US National Institutes of Health. http://imagej.nih.gov/ij/ |
[40] | Oueslati A ((2016) ) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: What have we learned in the last decade? J Parkinsons Dis 6: , 39–51. |
[41] | Michel PP , Hirsch EC , Hunot S ((2016) ) Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90: , 675–691. |
[42] | Mrak RE , Griffin WST ((2005) ) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26: , 349–354. |
[43] | Brück D , Wenning GK , Stefanova N , Fellner L ((2016) ) Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis 85: , 262–274. |
[44] | Gleichman AJ , Carmichael ST ((2020) ) Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol Dis 142: , 104957. |
[45] | Bennett ML , Viaene AN ((2021) ) What are activated and reactive glia and what is their role in neurodegeneration? Neurobiol Dis 148: , 105172. |
[46] | Funato H , Yoshimura M , Yamazaki T , Saido TC , Ito Y , Yokofujita J , Okeda R , Ihara Y ((1998) ) Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol 152: , 983. |
[47] | Nagele RG , D’Andrea MR , Lee H , Venkataraman V , Wang H-Y ((2003) ) Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971: , 197–209. |
[48] | Visanji NP , Lang AE , Kovacs GG ((2019) ) Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl Neurodegener 8: , 1–13. |
[49] | Matej R , Tesar A , Rusina R ((2019) ) Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin Biochem 73: , 26–31. |
[50] | Spina S , La Joie R , Petersen C , Nolan AL , Cuevas D , Cosme C , Hepker M , Hwang J-H , Miller ZA , Huang EJ ((2021) ) Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 144: , 2186–2198. |
[51] | Francis PT , Palmer AM , Snape M , Wilcock GK ((1999) ) The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg Psychiatry 66: , 137–147. |
[52] | Hampel H , Mesulam M-M , Cuello AC , Farlow MR , Giacobini E , Grossberg GT , Khachaturian AS , Vergallo A , Cavedo E , Snyder PJ ((2018) ) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141: , 1917–1933. |
[53] | Jose JC , Chatterjee P , Sengupta N ((2014) ) Cross dimerization of amyloid-β and αSynuclein proteins in aqueous environment: A molecular dynamics simulations study. PloS One 9: , e106883. |
[54] | Kazmierczak A , Strosznajder JB , Adamczyk A ((2008) ) α-Synuclein enhances secretion and toxicity of amyloid beta peptides in PC12 cells. Neurochem Int 53: , 263–269. |
[55] | Roberts HL , Schneider BL , Brown DR ((2017) ) α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PloS One 12: , e0171925. |
[56] | Park J-S , Kam T-I , Lee S , Park H , Oh Y , Kwon S-H , Song J-J , Kim D , Kim H , Jhaldiyal A ((2021) ) Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol Commun 9: , 78. |
[57] | Halliday GM , Stevens CH ((2011) ) Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26: , 6–17. |
[58] | Bido S , Muggeo S , Massimino L , Marzi MJ , Giannelli SG , Melacini E , Nannoni M , Gambarè D , Bellini E , Ordazzo G ((2021) ) Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat Commun 12: , 6237. |
[59] | Nagele RG , Wegiel J , Venkataraman V , Imaki H , Wang K-C , Wegiel J ((2004) ) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25: , 663–674. |
[60] | Serrano-Pozo A , Mielke ML , Gómez-Isla T , Betensky RA , Growdon JH , Frosch MP , Hyman BT ((2011) ) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol 179: , 1373–1384. |
[61] | Persons DA , Hargrove PW , Allay ER , Hanawa H , Nienhuis AW ((2003) ) The degree of phenotypic correction of murine β-thalassemia intermedia following lentiviral-mediated transfer of a human γ-globin gene is influenced by chromosomal position effects and vector copy number. Blood 101: , 2175–2183. |
[62] | Ellis J ((2005) ) Silencing and variegation of gammaretrovirus and lentivirus vectors. Human Gene Ther 16: , 1241–1246. |
[63] | Deroose CM , Reumers V , Gijsbers R , Bormans G , Debyser Z , Mortelmans L , Baekelandt V ((2006) ) Noninvasive monitoring of long-term lentiviral vector-mediated gene expression in rodent brain with bioluminescence imaging. Mol Ther 14: , 423–431. |
[64] | Lowenstein PR , Castro MG ((2003) ) Inflammation and adaptive immune responses to adenoviral vectors injected into the brain: Peculiarities, mechanisms, and consequences. Gene Ther 10: , 946–954. |
[65] | Bassil F , Meymand ES , Brown HJ , Xu H , Cox TO , Pattabhiraman S , Maghames CM , Wu Q , Zhang B , Trojanowski JQ , Lee VM-Y ((2020) ) α-Synuclein modulates tau spreading in mouse brains. J Exp Med 218: , e20192193. |
[66] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM-Y ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[67] | Pan L , Li C , Meng L , Tian Y , He M , Yuan X , Zhang G , Zhang Z , Xiong J , Chen G , Zhang Z ((2022) ) Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain 145: , 3454–3471. |
[68] | Nübling G , Bader B , Levin J , Hildebrandt J , Kretzschmar H , Giese A ((2012) ) Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol Neurodegener 7: , 35. |
[69] | Dasari AKR , Kayed R , Wi S , Lim KH ((2019) ) Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry 58: , 2814–2821. |


