
Effect of Lecanemab and Donanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective
Abstract
In clinical trials, lecanemab and donanemab showed statistically significant yet marginal slowdown of Alzheimer’s disease (AD)-associated cognitive decline. This could be due to their sub-optimal design and/or deployment; alternatively, their limited efficiency could be intrinsic. Distinguishing between the two is of great importance considering the acute need of efficient AD therapy and tremendous resources being invested in its pursuit. The present study analyzes the mode of operation of lecanemab and donanemab within the framework of recently proposed Amyloid Cascade Hypothesis 2.0 and concludes that the second possibility is correct. It suggests that substantial improvement of the efficiency of these drugs in symptomatic AD is unlikely and proposes the alternative therapeutic strategy.
Lecanemab, the recently approved drug for treatment of early stages of Alzheimer’s disease (AD), exhibited statistically significant, yet marginal reduction in the rate of AD-associated cognitive decline [1]. These findings were met with great enthusiasm and the hope that the drug can be improved to arrest the progression of or even to cure the disease [2, 3]. The question is whether the drug or its utilization in the trial have been suboptimal and outcomes could be significantly improved, or if its limited efficiency in early AD is intrinsic. The present study analyzes the mode of operation of lecanemab and donanemab within the framework of the recently proposed Amyloid Cascade Hypothesis 2.0 (ACH2.0) and concludes that the latter is correct. It suggests that, in their trials, both drugs acted preventively, not curatively, on only a small neuronal subpopulation, and that a substantial improvement of their efficiency in symptomatic AD is highly unlikely and proposes the alternative therapeutic strategy.
The initial Amyloid Cascade Hypothesis (ACH) [4] postulated that AD is caused by amyloid-β (Aβ) produced in the Aβ protein precursor (AβPP) proteolytic/secretory pathway and deposited extracellularly. Accordingly, two principal categories of ACH-based AD drugs are either those suppressing production and, consequently, secretion of AβPP-derived Aβ or agents sequestering or clearing extracellular Aβ; lecanemab belongs to the second group. In contrast, the recently proposed ACH2.0 posits that AD is a two-stage disease caused and driven by intraneuronal (rather than extracellular) Aβ, iAβ [5]. The first, asymptomatic, stage is a life-long accumulation of iAβ, which occurs via internalization of extracellular Aβ and through retention of Aβ produced by the gamma-cleavage of the C99 fragment of AβPP on intracellular, rather than on plasma, membranes [6–15] (reviewed in [5]). Upon reaching the T1 threshold in affected neurons in a narrow temporal window, iAβ triggers, presumably following the elicitation of PKR- and HRI-mediated integrated stress response, activation of the AβPP-independent iAβ generation pathway [5]. The bulk, if not the entire output of this pathway are retained intraneuronally and perpetuate the operation of the pathway [5]. iAβ levels rapidly increase and drive a devastating cascade that includes tau pathology; when they cross the T2 threshold, neurons commit apoptosis and AD symptoms start to manifest (Fig. 1A). When a sufficient fraction of neurons is lost, AD enters the end-stage (Fig. 1B). Thus, in the ACH2.0 paradigm, the primary therapeutic target is intraneuronal, rather than extracellular, Aβ.
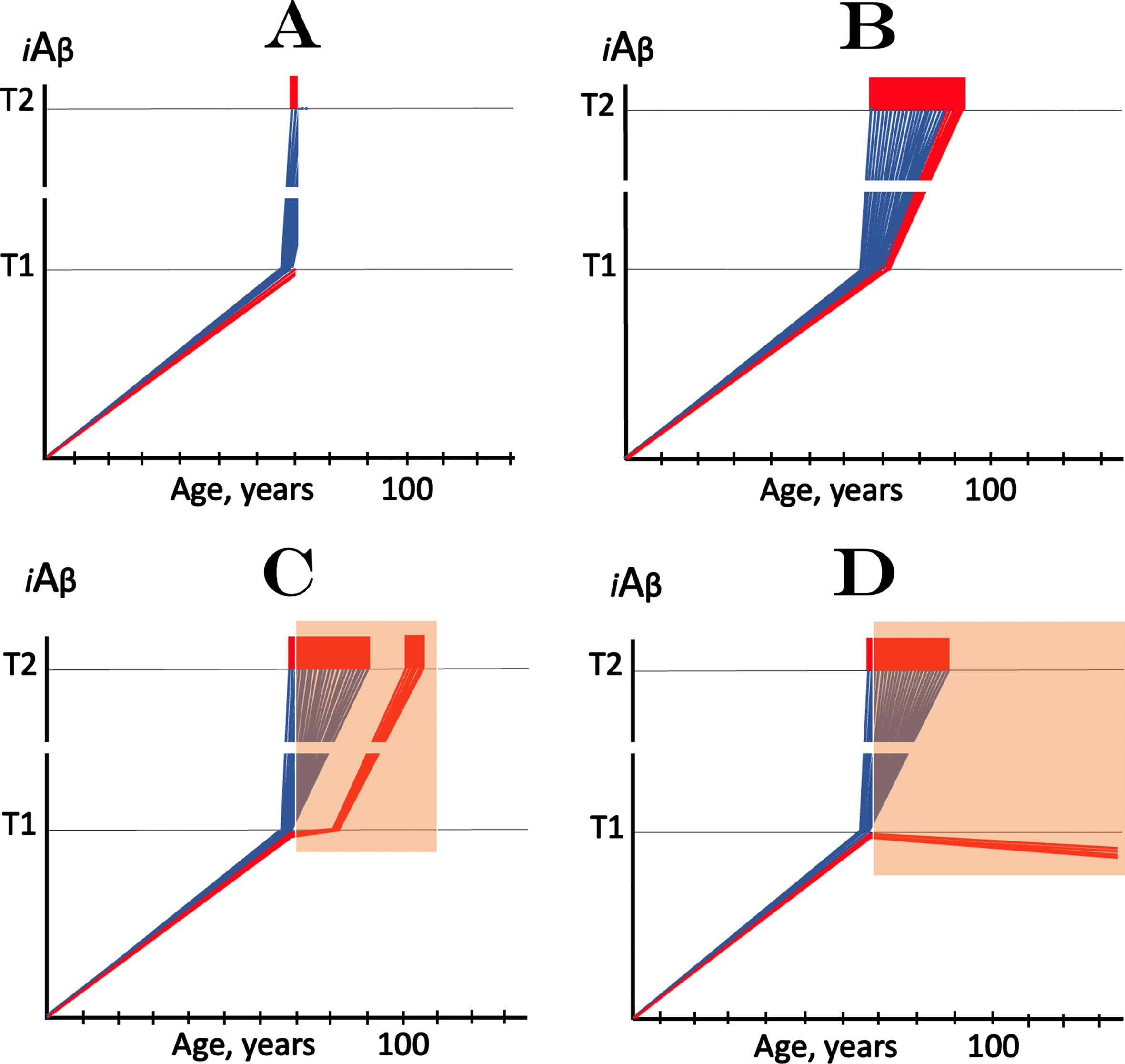
Fig. 1
Effect of lecanemab at early symptomatic AD. iAβ: Level of intraneuronal Aβ. T1 threshold: Levels of iAβ triggering, plausibly via activation of PKR and HRI kinases, elicitation of the ISR and initiation of AβPP-independent production of iAβ. T2 threshold: Levels of iAβ triggering neurons’ entrance into the apoptotic pathway. Blue and red lines: Individual affected neurons. Red lines: A fraction of affected neurons that did not reach the T1 threshold at the time of the commencement of lecanemab treatment. Red blocks: Apoptotic zone. Orange fields: The duration of lecanemab treatment. A: The “initial state” – iAβ dynamics in affected neurons at the commencement of lecanemab treatment. Note that a small neuronal fraction did not yet reach the T1 threshold (shown in red solely to distinguish it from the bulk of neurons that already crossed the T1 threshold; otherwise both fractions are identical). B: Result of the evolution of the initial state in the absence of a treatment. The “red” neuronal fraction reached the T1 threshold, iAβ levels in both neuronal fractions crossed the T2 threshold and AD entered the end-stage. C, D: Effect of lecanemab treatment in early AD. Note that neurons that crossed the T1 threshold by the commencement of the treatment remain unaffected by it and evolve as shown in B. C: The rate of accumulation of AβPP-derived iAβ is reduced but its levels continue to increase. Eventually, they reach the T1 threshold, cross the T2 threshold and cells commit apoptosis. The fate of the “red” neuronal population is the same as in B but occurs with a delay; these neurons are redeemed by the drug but only temporarily. D: The treatment arrests or reverses the accumulation of AβPP-derived iAβ. Levels of AβPP-derived iAβ do not reach the T1 threshold and the “red” neuronal fraction is redeemed permanently for the duration of the treatment.
Importantly, in the framework of the ACH2.0, ACH-based AD drugs (as defined above) could be effective only preventively by reducing the rate of AβPP-derived iAβ accumulation and delaying the T1 crossing or precluding it within the lifespan of an individual [5]. This is because the crossing of the T1 threshold enables the activation of the AβPP-independent iAβ production pathway, which is unaffected by ACH-based drugs [5]. Conceptually, drugs suppressing the production of AβPP-derived Aβ would inhibit both components of the influx of AβPP-derived iAβ, its retention and internalization: less Aβ is produced, less is retained; less Aβ is secreted, its extracellular pool is smaller and less is taken up. On the other hand, agents sequestering or clearing extracellular Aβ affect only one iAβ influx component: its cellular uptake; this is precisely what lecanemab does in two ways. First, it lowers internalization of extracellular Aβ simply by reducing its pool. More importantly, it acts in a specific and targeted manner. Lecanemab is, in essence, a monoclonal antibody (BAN2401), which specifically sequesters “protofibril” Aβ, i.e., soluble extracellular Aβ in oligomeric form [1]. Crucially, the latter is the intermediate in the cellular uptake of Aβ [6–11]. Consequently, sequestering oligomeric Aβ, lecanemab specifically suppresses internalization of extracellular Aβ, one of the two components of the steady-state influx of AβPP-derived iAβ in neuronal cells, and reduces the rate of its accumulation. After the T1 crossing, however, the utilization of ACH-based drugs, including lecanemab, would be futile because the activation of the AβPP-independent iAβ production pathway would render the contribution of AβPP-derived Aβ to the iAβ pool insignificant and the AβPP proteolytic pathway irrelevant for the progression of AD [5].
All participants of the lecanemab trial have exhibited early AD symptoms by the commencement of the treatment. By the time AD symptoms manifest, however, the bulk of the affected neurons have already crossed the T1 threshold and would be unresponsive to the drug [5]. Therefore, the only explanation of the observed effect of lecanemab [1] in the ACH2.0 perspective is that at the time of the commencement of the treatment a fraction of affected neurons had not yet reached the T1 threshold and was still responsive to the drug. It is this fraction of the neuronal population that was meaningfully targeted by and responded to the treatment with lecanemab, and positive results were marginal because of the marginal size of the neuronal fraction redeemed, although possibly only temporarily (see below), by the drug.
The presumed mode of the lecanemab’s action, described above, is illustrated in Fig. 1. Figure 1A shows the initial state of iAβ dynamics in the affected neuronal population at the time of the commencement of the treatment. The bulk of neurons have crossed the T1 threshold. Of those, a fraction have also crossed the T2 threshold and triggered the manifestation of AD symptoms; the majority has iAβ levels distributed between the T1 and T2 threshold. At this time, a minor subpopulation of affected neurons did not yet cross the T1 threshold (shown in red solely to graphically distinguish them from neurons that crossed the T1; otherwise both groups are identical). Figure 1B–D depict results of the evolution of the initial state in the absence or presence of the drug. No drug is administered in Fig. 1B. The “red” neuronal fraction crosses the T1 threshold, AβPP-independent production of iAβ occurs in all affected neurons, and iAβ levels ascend stochastically toward the T2 threshold. When a sufficient portion of neurons is lost, AD enters the end-stage shown in Fig. 1B.
Figure 1C and 1D show results of the evolution of the initial state in the presence of the drug (orange fields indicate the duration of the treatment). Neurons that crossed the T1 threshold (“blue” neuronal fraction) are not affected by the treatment and evolve toward the same outcome as shown in Fig. 1B. For the “red” neuronal fraction, two distinct outcomes are possible. In one, shown in Fig. 1C, the rate of accumulation of AβPP-derived iAβ is reduced but its levels continue to increase. Eventually, they would reach and cross the T1 threshold, and begin ascending toward the T2 threshold. In this scenario, the fate of the “red” neuronal population would be the same as in Fig. 1B but will occur with a delay; neurons would be redeemed by a drug but only temporarily. The outcome shown in Fig. 1D is the arrest or reversal (due to degradation and clearance of iAβ) of the accumulation of AβPP-derived iAβ in the “red” neuronal fraction. In this scenario (Fig. 1D), levels of AβPP-derived iAβ would not reach the T1 threshold and the “red” neuronal fraction would be redeemed permanently for the duration of the treatment. The published results of clinical trials of lecanemab [1] do not allow to distinguish between the two outcomes described above because its participants were not yet followed for a sufficient duration.
The therapeutic outcomes in both scenarios (Fig. 1C, D) would, nevertheless, be only marginal because the size of the targeted neuronal subpopulation (“red” neuronal fraction) would be only marginal since by the time AD symptoms manifest, the bulk of the affected neurons have already crossed the T1 threshold [5]. The only way to improve the therapeutic outcome of any treatment targeting AβPP-derived Aβ, including lecanemab, is by advancing the diagnosis and the commencement of a treatment, thus maximizing the “red” neuronal fraction. This approach, however, is limited, hence the intrinsic limitation of lecanemab or any other AβPP-derived Aβ-targeting drug in the treatment of symptomatic AD. On the other hand, the present interpretation of the results of the lecanemab’s trial asserts that drugs that cause the arrest or reversal of accumulation of AβPP-derived iAβ, or sufficient reduction of its levels, would be effective in preventing AD if their administration commences before symptomatic manifestation of the disease, more precisely before the crossing of the T1 threshold and activation of the AβPP-independent iAβ production. It also mandates clinical trials of lecanemab in prevention of AD using asymptomatic cohorts.
Conceptually similar, both quantitatively and qualitatively, results in clinical trials for treatment of very early symptomatic AD were recently obtained with donanemab, a humanized IgG1 antibody directed at an N-terminal pyroglutamate Aβ epitope that is present only in established plaques [20, 21]. By sequestering extracellular amyloid-beta deposits, donanemab shifts the equilibrium of extracellular Aβ processing toward formation of plaques and thus reduces the levels of extracellular soluble Aβ. This, in turn, suppresses the rate of extracellular Aβ internalization and inhibits the influx of iAβ. Therefore, the explanation of the observed effect of donanemab in early AD is identical to that of the effect of lecanemab, namely the suppression of the influx of iAβ and, consequently, the reduction or reversal of the rate of its accumulation. Likewise, in similarity with lecanemab, the observed effect of donanemab in clinical trials [21] was marginal because, in the trial participants, the drug impacted only the marginal neuronal subpopulations where the iAβ levels have not yet crossed the T1 threshold and the AβPP-independent iAβ production pathway was not yet activated.
A recent Nature commentary on the subject [2] posed a question: “Alzheimer’s drug slows mental decline in trial —but is it a breakthrough?” The ACH2.0 provides the unequivocally negative answer. However, it also suggests the strategy to actually achieve such a breakthrough. As expounded upon elsewhere [5], the only viable therapeutic option for symptomatic AD in the ACH2.0 framework is the reduction of the iAβ levels to those below the T1 threshold; this would cease the operation of the AD-driving AβPP-independent iAβ generation pathway and arrest the progression of the disease. Moreover, a sufficient depletion of iAβ levels would substantially reset them and force the resumption of its accumulation (only in the AβPP proteolytic pathway) from a low baseline. This opens an attractive possibility of a transient, once-in-a-lifetime-only treatment of symptomatic AD [5]. Indeed, sufficiently depleted AβPP-derived iAβ levels would not reach the T1 threshold within the lifespan of an AD patient. Such a therapy would be effective in symptomatic AD because it would potentially redeem all neurons that did not yet cross the T2 threshold and commit apoptosis. Even more attractively, the same strategy could be applied preventively prior to the manifestation of AD symptoms. Any agent capable of targeted degradation of iAβ and its sufficient depletion within a short duration would potentially be appropriate for enacting the above strategy. Two apparently suitable physiologically operating activities are actually built-into the two familiar actors in the AD play: BACE1 and BACE2. Both are capable of multiple cleavages within iAβ (reviewed in [5]), a capacity enhanced in BACE1 by the Icelandic AβPP mutation [16, 17] (explaining its protective action) and suppressed in BACE2 by the Flemish AβPP mutation [18] (thus causing familial AD). Activators of physiologically occurring iAβ-cleaving capabilities of BACE1 and/or BACE2 could potentially constitute potent AD drugs [5].
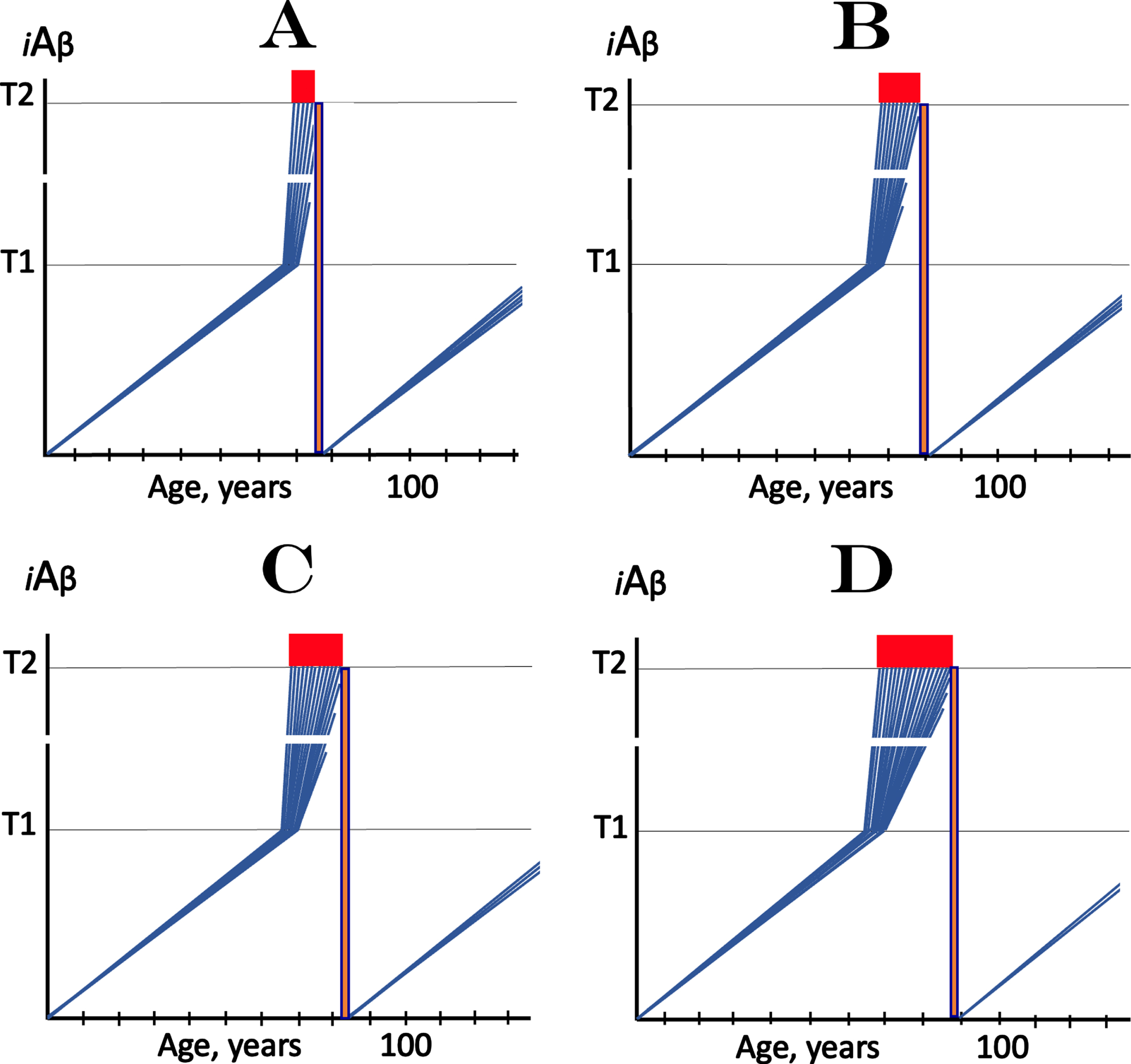
Application of the transient iAβ depletion therapy in sporadic AD is illustrated in Fig. 2. Figure 2A-D show progressive stages of the disease. In each, levels of iAβ have crossed the T1 threshold in all affected neurons. The transient iAβ depletion treatment resets its levels to a low baseline, switches off the now unsustainable AD-driving AβPP-independent iAβ production pathway, stops the progression of the disease and enables the still viable neurons to recover and reconnect. Since, with the progression of AD, less and less affected neurons with iAβ levels below the T2 threshold are left, progressively smaller neuronal fraction remains viable and can be redeemed. The levels of iAβ, now driven solely by the AβPP proteolysis and associated processes (internalization of secreted Aβ and retention of Aβ following the gamma-cleavage of C99 on intracellular membranes), are not expected to reach the T1 threshold and the disease is not expected to resume within the remaining lifetime of a patient. The best therapeutic outcome can be obtained if the transient iAβ depletion treatment is administered preventively, prior to the activation of the AβPP-independent iAβ production pathway and manifestation of AD symptoms (not shown in Fig. 2); in this case all neurons would be redeemed, potentially for the remaining lifetime of an individual.
Fig. 2
Effect of transient iAβ depletion therapy at various stages of symptomatic AD. iAβ: Level of intraneuronal Aβ. T1 threshold: Levels of iAβ triggering, plausibly via activation of PKR and HRI kinases, elicitation of the ISR and initiation of AβPP-independent production of iAβ. T2 threshold: Levels of iAβ triggering neurons’ entrance into the apoptotic pathway. Blue lines: Individual affected neurons. Red blocks: Apoptotic zone. Orange boxes: The duration of the transient iAβ depletion treatment; levels of iAβ are reset and the accumulation of AβPP-derived iAβ resumes from a low baseline. A: The transient iAβ depletion via its targeted degradation is implemented, via the enhancement of iAβ-cleaving activities of BACE1 and/or BACE2 or through employment of any suitable agent capable of iAβ depletion, at the early symptomatic stage of AD, with the bulk of the affected neurons still viable. Following the reset of iAβ levels, its build-up starts de-novo, supported only by the AβPP proteolytic pathway. It is anticipated that iAβ levels will not reach the T1 threshold and AD will not recur within the remaining lifetime of an SAD patient. Note that the reset occurs in neurons that already crossed the T1 but not yet the T2 thresholds. B- D: The transient iAβ depletion treatment is implemented at progressively advanced stages of AD. The results are analogous to those depicted in A. However, at these AD stages increasing number of affected neurons cross the T2 threshold and commit apoptosis. This leaves progressively smaller number of affected neurons that retained their viability and can be redeemed. Note that the best therapeutic outcome can be obtained if the transient iAβ depletion treatment is administered preventively, prior to the activation of the AβPP-independent iAβ production pathway and manifestation of AD symptoms (shown below); in this case all neurons would be redeemed, potentially for the remaining lifetime of an individual.
Both approaches, (a) targeting the influx of AβPP-derived iAβ and (b) depleting its levels via targeted degradation, can be effectively employed preventively. For the former, two conditions are crucial: 1) that the treatment commences prior to the T1 threshold crossing; any neurons that crossed would be unredeemable in this approach, and 2) that a drug employed causes the arrest or reversal of accumulation of AβPP-derived iAβ, or such reduction in the rate of the latter that would prevent the T1 crossing within individual’s lifetime; otherwise the relief would be only temporary. There is more leeway with the iAβ depletion via its targeted degradation since in this approach neurons are potentially redeemable until they cross the T2 threshold. It should be mentioned that the first approach reduces extracellular levels of Aβ and thus may interfere with its protective effect (e.g., antimicrobial function [19]). In contrast, the proposed transient iAβ depletion therapy would circumvent or minimize this potential problem due to its limited duration.
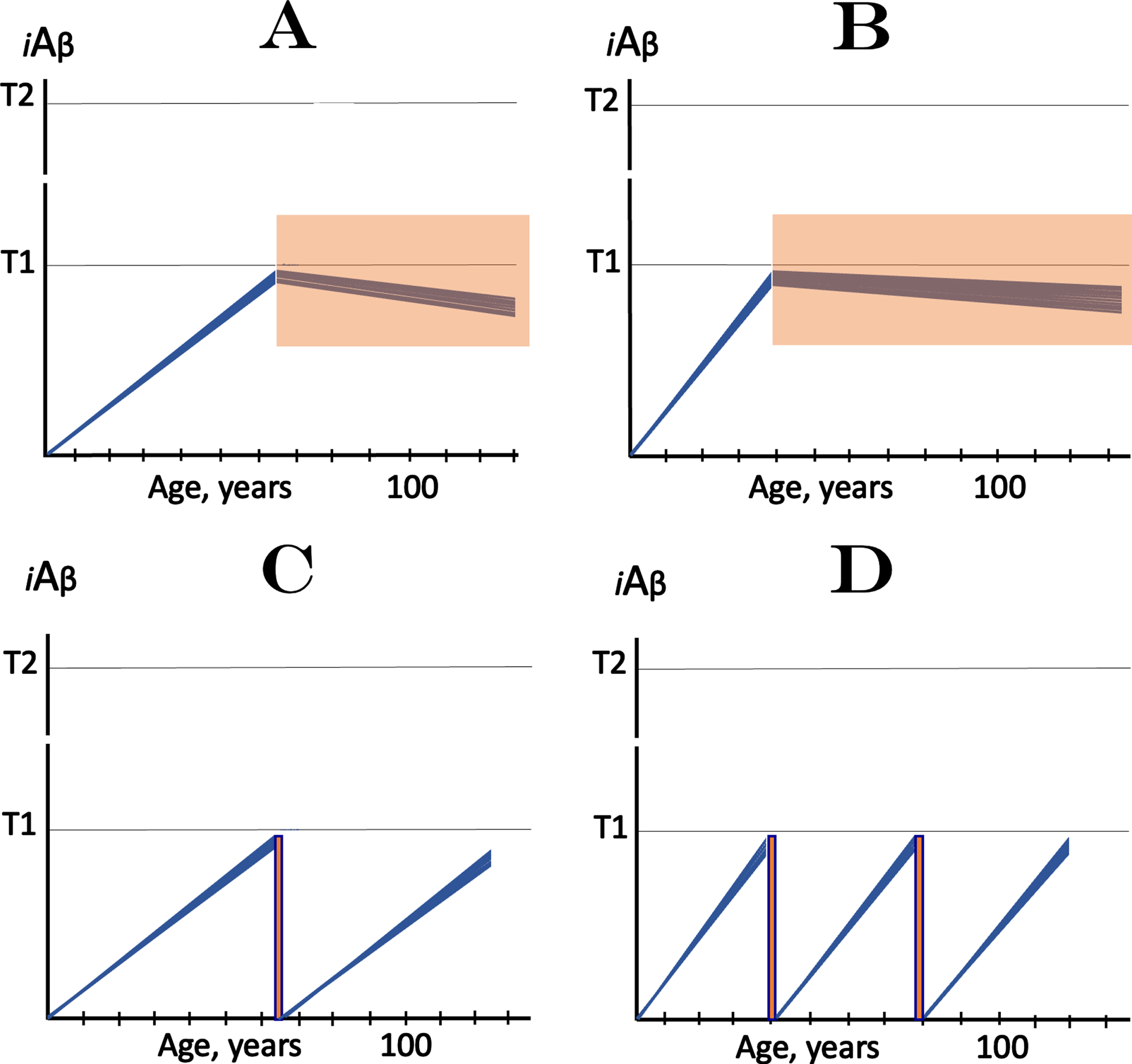
Preventive implementation of both strategies is illustrated in Fig. 3. Figures 3A and 3B depict the first approach for prevention of SAD and FAD respectively. The only difference is the timing of the commencement of treatment’s administration. In this approach, iAβ levels would not reach the T1 threshold and AD would not occur for the duration of the treatment (orange fields); the treatment constitutes, in effect, the AD “statin”. In Fig. 3C and 3D, the transient iAβ depletion therapy is deployed (orange boxes). A single transient treatment is potentially sufficient to prevent SAD within the remaining lifetime, and repeated treatments could be required for prevention of FAD.
Fig. 3
The prevention of AD: Two approaches. iAβ: Level of intraneuronal Aβ. T1 threshold: Levels of iAβ triggering, plausibly via activation of PKR and HRI kinases, elicitation of the ISR and initiation of AβPP-independent production of iAβ. T2 threshold: Levels of iAβ triggering neurons’ entrance into the apoptotic pathway. Blue lines: Individual affected neurons. Note that in all panels the commencement or the implementation of a treatment occurs prior to the crossing of the T1 threshold by AβPP-derived iAβ. A, B: Prevention of AD via suppression of the rate of AβPP-derived iAβ accumulation. It is assumed that a drug employed in this approach, which suppresses the influx of AβPP-derived iAβ is capable of arresting or reversing AβPP-derived iAβ accumulation or of reducing it (not shown) to the extent sufficient to prevent the T1 crossing within the lifetime of an individual. Under such a treatment, iAβ levels would not reach the T1 threshold, the AβPP-independent iAβ generation pathway would not be activated, and AD would not occur for the duration of the treatment (orange fields); in this scenario, the treatment continues for the remaining part of the lifespan of an individual and constitutes, in effect, the AD “statin”. A: Prevention of SAD; B: prevention of FAD (note the difference in the timing of the commencement of treatment). C, D: Prevention of AD by transient iAβ depletion via its targeted degradation (orange boxes; note drastic difference in duration of treatment in comparison with orange fields in A and B). The duration of the iAβ depletion treatment is defined by the desired extent of depletion and potentially could be as short as few days, a regiment possibly akin to that of an antibiotic treatment. It is assumed that, following the treatment, the iAβ pool would collapse, its levels would be reset to a low baseline, and the operation of the AβPP-independent iAβ production pathway would cease. The accumulation of AβPP-derived iAβ would resume at presumably constant rate; in prevention of SAD (C) its levels would not reach the T1 threshold, AβPP-independent production of iAβ would not be activated, and the disease would not occur within the remaining lifetime of an individual. For prevention of FAD, the treatment is implemented earlier (D); following the treatment, accumulation of AβPP-derived iAβ to near-T1 levels would require several decades but could occur within the lifetime of an individual and thus necessitate a repeat treatment. This approach could be implemented via the enhancement of iAβ-cleaving activities of BACE1 and/or BACE2 or through employment of any suitable agent causing selective degradation of iAβ.
In conclusion, lecanemab and donanemab are currently the only AD drugs that showed positive effect in clinical trials [1]. Even if they only reduce (rather than arrest or reverse) the rate of AβPP-derived iAβ accumulation, this could be sufficient, if initiated presymptomatically, to prevent the T1 crossing, and, consequently, the occurrence of AD within the individual’s lifetime. However, the mode of their administration (frequent infusions of large quantities of the antibody) substantially reduces the feasibility of their utilization as preventive agents. In the ACH2.0 perspective, any compound, possibly a small molecule, interfering with internalization of extracellular Aβ, either by preventing its oligomerization or through blocking its cellular receptors, would have effect similar to or exceeding that of lecanemab and donanemab, and could be feasible as an AD preventive agent. As an added benefit, such drug would not deplete extracellular Aβ thus preserving its protective potential. Likewise, the proposed transient iAβ depletion therapy [5] would minimize or circumvent the depletion of extracellular Aβ due to its limited duration. Moreover, whereas both strategies, the suppression of accumulation of AβPP-derived iAβ via the reduction of its influx and the depletion of iAβ through its targeted degradation can be employed preventively, only the latter is capable of meaningful treatment of AD at its symptomatic stages.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Bjorn R. Olsen (Harvard Medical School) for his support.
FUNDING
This work was supported by grants from the National Institutes of Health (R21 GM056179; R01 AR036819).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
REFERENCES
[1] | van Dyck CH , Swanson CJ , Aisen P , Bateman RJ , Chen C , Gee M , Kanekiyo M , Li D , Reyderman L , Cohen S , Froelich L , Katayama S , Sabbagh M , Vellas B , Watson D , Dhadda S , Irizarry M , Kramer LD , Iwatsubo T ((2023) ) Lecanemab in early Alzheimer’s disease. N Engl J Med 388: , 9–21. |
[2] | Prillaman M ((2022) ) Alzheimer’s drug slows mental decline in trial —but is it a breakthrough? Nature 610: , 15–16. |
[3] | Gallagher J ((2023) ) Alzheimer’s-slowing drug labelled historic. https://www.bbc.com/news/health-63060019. |
[4] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[5] | Volloch V , Rits-Volloch S ((2022) ) The Amyloid Cascade Hypothesis 2.0: On the possibility of once-in-a-lifetime-only treatment for prevention of Alzheimer’s disease and for Its potential cure at symptomatic stages. J Alzheimers Dis Rep 6: , 369–399. |
[6] | Chafekar S , Baas F , Scheper W ((2008) ) Oligomer-specific amyloid-beta toxicity in cell models is mediated by selective uptake. Biochem Biophys Acta 9: , 523–531. |
[7] | Wesen E , Jeffries G , Dzebo M , Esbjorner M ((2017) ) Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40). Sci Rep 7: , (2021) . |
[8] | Kumar-Singh S , Theuns J , Van Broeck B , Pirici D , Vennekens K , Corsmit E , Cruts M , Dermaut B , Wang R , Van Broeckhoven C ((2006) ) Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 27: , 686–695. |
[9] | Hu X , Crick SL , Bu G , Frieden C , Pappu RV , Lee JM ((2009) ) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A 106: , 20324–20329. |
[10] | Yajima R , Tokutake T , Koyama A , Kasuga K , Tezuka T , Nishizawa M , Ikeuchi T ((2015) ) ApoE-isoform-dependent cellular uptake of amyloid-β is mediated by lipoprotein receptor LR11/SorLA. Biochem Biophys Res Comm 456: , 482–488. |
[11] | Omtri RS , Davidson MW , Arumugam B , Poduslo JF , Kandimalla KK ((2012) ) Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: Ramifications for the Alzheimer’s drug discovery. Mol Pharmaceutics 9: , 1887. |
[12] | Cook DG , Forman MS , Sung JC , Leight S , Kolson DL , Iwatsubo T , Lee VM , Doms RW ((1997) ) Alzheimer’s A beta42 is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 3: , 1021–1023. |
[13] | Hartmann T , Bieger SC , Brühl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
[14] | Wild-Bode C , Yamazaki T , Capell A , Leimer U , Steiner H , Ihara Y , Haass C ((1997) ) Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J Biol Chem 272: , 16085–16088. |
[15] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: , 1437–1449. |
[16] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jonsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease andage-related cognitive decline. Nature 488: , 96–99. |
[17] | Harper AR , Nayee S , Topol EJ ((2015) ) Protective alleles and modifier variants in human health and disease. Nat Rev Genet 16: , 689–701. |
[18] | Farzan M , Schnitzler CE , Vasilieva N , Leung D , Choe H ((2000) ) BACE2, a β-secretase homolog, cleaves at the β site and within the amyloid-β region of the amyloid-β precursor protein. Proc Natl Acad Sci U S A 97: , 9712–9717. |
[19] | Gosztyla ML , Brothers HM , Robinson SR ((2018) ) Alzheimer’s amyloid-β is an antimicrobial peptide: A review of the evidence. J Alzheimers Dis 62: , 1495–1506. |
[20] | Mintun M , et al. ((2021) ) Donanemab in Early Alzheimer’s Disease. N Engl J Med 384: , 1691–1704 10.1056/NEJMoa2100708. |
[21] | Eli Lilly ((2023) ) Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease. Press release. https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-significantly-slowed-cognitive-and-functional. |


