
Concomitant Neuronal Tau Deposition and FKBP52 Decrease Is an Early Feature of Different Human and Experimental Tauopathies
Abstract
Background:
Pathological tau proteins constitute neurofibrillary tangles that accumulate in tauopathies including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), and familial frontotemporal lobar degeneration (FTLD-Tau). We previously showed that the FKBP52 immunophilin interacts functionally with tau and strongly decreases in AD brain neurons in correlation with tau deposition. We also reported that FKBP52 co-localizes with autophagy-lysosomal markers and an early pathological tau isoform in AD neurons, suggesting its involvement in autophagic tau clearance.
Objective:
Our objective was to evaluate if differences in neuronal FKBP52 expression levels and subcellular localization might be detected in AD, PSP, familial FTLD-Tau, and in the hTau-P301 S mouse model compared to controls.
Methods:
Cell by cell immunohistofluorescence analyses and quantification of FKBP52 were performed on postmortem brain samples of some human tauopathies and on hTau-P301 S mice spinal cords.
Results:
We describe a similar FKBP52 decrease and its localization with early pathological tau forms in the neuronal autophagy-lysosomal pathway in various tauopathies and hTau-P301 S mice. We find that FKBP52 decreases early during the pathologic process as it occurs in rare neurons with tau deposits in the marginally affected frontal cortex region of AD Braak IV brains and in the spinal cord of symptomless 1-month-old hTau-P301 S mice.
Conclusion:
As FKBP52 plays a significant role in cellular signaling and conceivably in tau clearance, our data support the idea that the prevention of FKBP52 decrease or the restoration of its normal expression at early pathologic stages might represent a new potential therapeutic approach in tauopathies including AD, familial FTLD-Tau, and PSP.
INTRODUCTION
Tau is a microtubule associated protein abundantly present in neurons of the central nervous system. It plays a key role in microtubule network stability and is essential for normal axonal transport [1, 2]. Abnormal tau conformations, induced by different post-translational modifications such as phosphorylation and truncation, can lead to tau self-aggregation with the formation of filamentous structures called neurofibrillary tangles (NFTs) and can hinder its activity on microtubule assembly with significant consequences on neuronal health. Pathological hyperphosphorylation and aggregation of the tau protein are the common feature of a wide range of neurodegenerative diseases known as tauopathies including Alzheimer’s disease (AD) and frontotemporal lobar degeneration with tau pathology (FTLD-Tau). Familial cases of FTLD-Tau, formerly termed FTDP-17 (frontotemporal dementia with Parkinsonism-17), are due to mutations in the tau gene (MAPT) on chromosome 17 [3–5]. Sporadic FTLD-Tau comports several pathological subtypes such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD), and others [6, 7]. While these tauopathies are all characterized by abnormal tau deposition in the brain, the affected regions, the type of the affected cells and the structural conformation of the tau aggregates differ [8]. In addition to the presence of NFTs, the pathology of PSP and CBD is also characterized by distinct tau inclusions in astrocytes (tufted astrocytes) and oligodendrocytes (coiled bodies) respectively. In PiD brains, intraneuronal tau aggregates assemble into typical spherical Pick bodies [9]. Recent studies have demonstrated the formation and propagation of different tau inclusions following the injection of homogenates from brains of patients suffering from different tauopathies (i.e., AD, PSP, CBD, PiD), into mouse brains, strongly suggesting the existence of distinct pathological tau strains [10]. There is some evidence suggesting that tau hyperphosphorylation is an early event in the progression of tau pathology [11, 12]. AD progresses from the trans-entorhinal and entorhinal cortex to spread to the hippocampus and eventually to the isocortex, gradually implicating previously healthy neurons in the pathological process. These newly diseased neurons are believed to go through several sequences of pathologic tau deposition and NFT building. The various anomalous tau forms are thought to follow a temporal deposition pattern from early forms such as Alz50, Tau-pS422, and truncated Tau-D421, recognized by specific antibodies, to more “mature” forms [13–17]. Proteolytic cleavage of tau by caspases has been demonstrated to initiate tau aggregates formation in vivo in a tau transgenic mouse model suggesting that truncated tau recruits normal tau inducing its misfolding and aggregation. These findings support the idea that tau truncation is an early event in tau pathology [18]. In eukaryotic cells, proteins are cleared by two major pathways: the ubiquitin-proteasome system (UPS), which degrades the majority of soluble proteins, and the autophagy-lysosomal pathway (ALP). Lysosomes are acidic cellular organelles (pH 4.8) responsible for cellular homeostasis and involved in intra and extracellular substrate degradation via the autophagic or the endocytic pathways, respectively [19, 20]. Truncated tau and tau aggregates are preferentially degraded by the ALP while full-length tau is degraded by the UPS [21, 22]. Several studies have underlined the importance of defective UPS and ALP function in pathological tau proteins accumulation in several tauopathies [23–25].
FKBP52 (FK506 Binding Proteins of 52 kDa) is a peptidyl-prolyl cis/trans isomerase implicated in the folding and function of its target proteins [26–28]. Besides its enzymatic activity, FKBP52 also displays a chaperone activity [29]. While FKBP52 has mainly been described as a chaperone of steroid receptors, it is also involved in many other biological processes. FKBP52 is largely distributed and particularly abundant in the nervous system [30]. We have previously shown that FKBP52 prevents in vitro microtubule formation through its interaction with tubulin and tau and that FKBP52 is able to interact with the “PHF6” sequences of tau by NMR experiments [31–33]. Moreover, we have shown that FKBP52 levels are strongly decreased in the frontal cortex of AD and familial FTLD-Tau brains and that this decrease is highly correlated with the accumulation and aggregation of pathological tau [34]. Both mechanisms and timeline of this FKBP52 decrease in AD and other tauopathic brains are still unknown. We proposed that FKBP52 might play a role in tau degradation [31, 35], and we showed that FKBP52 can promote in vitro oligomerization of different mutated or truncated tau species [36, 37]. Recently, we also showed in AD neurons that FKBP52 immunoreactivity colocalizes with ALP markers along with the toxic and highly fibrillogenic truncated tau (Tau-D421) that appears early in the process of tangle formation and suggested that FKBP52 could be involved in autophagic tau clearance [35]. Indeed, we have shown using dorsal root ganglion neurons from mice transgenic for the human Tau-P301S mutation, that FKBP52 deficiency impacts on the function of the ALP under tau-induced proteotoxic stress conditions and triggers insoluble tau accumulation [38]. On the other hand, it has been reported that overexpression of FKBP52 is associated with a modest increase of total caspase 12 and could modify tau phosphorylation and neurofibrillary tangle formation [39]. These observations have led us to think that FKBP52 might be implicated in the aggregation of tau in the endolysosomal environment leading to tau degradation through the ALP and to evoke the possibility that FKBP52 could be itself eliminated through this degradative activity.
In this study, we performed cell by cell immunohistofluorescence (IHF) analysis on postmortem brain samples of AD, PSP, and familial FTLD-Tau patients in order to evaluate if differences in FKBP52 expression levels and subcellular localization might be observed. We compared our observations on human brains with the results from studies on a tauopathy model, the transgenic mice homozygous for the human Tau-P301S mutation [40], at various stages of disease evolution. We detect an equivalent decrease of neuronal FKBP52 in the frontal cortex of different tauopathies studied and we show that the residual FKBP52 colocalizes with the early pathological phosphorylated Tau-pS422 in the neuronal autophagy endo-lysosomal system. We also find in hTau-P301S young mice that FKBP52 decrease happens concomitantly with the apparition and accumulation of pathological tau isoforms and starts quite early during the pathologic process analogously to our findings in the scarcely impacted frontal cortex from Braak IV AD patients. The obtained results from the different tauopathies are compared and discussed.
MATERIALS AND METHODS
Antibodies and chemicals
The concentrations and the provenance of the antibodies used in this study are listed in Table 1. We used antibodies against tau and different phosphorylation sites of tau such as AT180 (Thr231 and Ser235), AT270 (Thr 181), and HT7 (anti-human and bovine tau) (ThermoFisher). The caspase-cleaved Tau-D421 antibody (Tau-C3) was from SantaCruz Biotechnology. The phosphorylated Tau-pS422 antibody (2H9) was from 4BioDx. The anti-Tau PHF1 antibody was a king gift from Pr. P. Davies (Albert Einstein, College of Medicine, NY, USA). The FKBP52 antibodies used were from Abcam (EPR6618) or Enzo (EC1). For colocalization experiments, we used antibodies directed against the endo-lysosomal system: Cathepsin D (lysosomal marker, Santa Cruz Biotechnologies), LC3 (autophagosome marker, Novus), and Rab7 (late endosomes marker, Abcam). Donkey anti-mouse, rabbit, and goat IgG (H + L) secondary antibodies labeled with Alexa Fluor 488, 555, 546, and 647 by Life Technologies were purchased from Molecular Probes.
Table 1
List of antibodies
| Antibody | Antigen | Dilution | Dilution | Host | Source |
| IHF | WB | animal | |||
| EPR6618 | Human FKBP52 | 1/50. | x | R | Abcam |
| EC1 | human FKBP52 (130–260) | x | 1/1000 | M | Enzo |
| C-20 | Human C-terminus Cathepsin D | 0.5μg/ml | x | G | Santa Cruz |
| ab118493 | Human LC3 (97-106) | 15μg/ml | x | G | Abcam |
| ab74906 | Human Rab7 | 1/200 | x | R | Abcam |
| 56416 | Human P62/sqstm1 | 1μg/ml | x | M | Abcam |
| HT7 | Human tau | x | 1/1000 | M | ThermoFisher |
| AF3494 | Human tau | 1/150 | x | G | R&D Systems |
| 2H9 | pSer422 | 2μg/ml | 1/1000 | M | 4BioDx |
| AT270 | pThr181 | x | 1/1000 | M | ThermoFisher |
| AT180 | pThr231/Ser235 | 10μg/ml | 1/100 | M | ThermoFisher |
| TauC3 | Caspase-Cleaved tau at Asp421 | 10μg/ml | x | M | Abcam |
| PHF1 | pSer396/Ser404 | x | 1/500 | M | Dr. P. Davies |
| 6C5 | Human GAPDH | x | 1/1000 | M | ThermoFisher |
| ab53025 | Human NSE | 1/100 | 1/200 | R | Abcam |
IHF, immunohistofluorescence; WB, western blot; R, rabbit; M, mouse; G, goat.
Human brain samples
Brain samples from individuals without neurological disorders and from AD, PSP, and familial FTLD-Tau patients of comparable age and postmortem delay were obtained from the Brain Bank GIE NeuroCEB (Table 2). The samples were collected on behalf of a Brain Donation Program funded by a consortium of Patients Associations: ARSEP (association for research on multiple sclerosis), CSC (cerebellar ataxias), France Alzheimer, and France Parkinson. The consents were signed by the patients themselves or their next of kin in their name, in accordance with French Bioethical Laws. The Brain Bank GIE NeuroCEB (Bioresource Research Impact Factor number = BRIF BB-0033-00011) has been authorized to provide samples for research by the Ministry of Higher Education and Research (agreement AC-2013-1887). Prior to histological analysis, a brain hemisphere, randomly left or right, was fixed in buffered 4% formaldehyde. Samples from multiple regions were embedded in paraffin and cut at 5μm thickness. For diagnosis and staging, sections of affected brain regions were stained with hematoxylin-eosin, and immunostained with anti-Aβ (Dako, 6F/3D clone, 1 : 200), anti-AT8 (ThermoFisher, AT8, 1 : 500), anti-α-synuclein (Zymed, LB 509 clone, 1 : 250) monoclonal antibodies, and with an anti-ubiquitin antibody (polyclonal Dako, 1 : 500). AD cases were diagnosed according to the National Institute of Aging and Reagan Institute Criteria (1997). Control cases without known neurological disorders were systematically analyzed and used as negative control for pathological markers. For biochemical analysis, post-mortem tissues were snap-frozen in liquid nitrogen and stored at –80°C.
Table 2
List of cases studied (studied area is the middle frontal gyrus)
| Diagnostic | Sex | Age | Postmortem |
| delay (h) | |||
| Control | Male | 78 | 23 |
| Control | Male | 61 | 20 |
| Control | Male | 70 | 30 |
| Control | Male | 66 | 19 |
| Control | Female | 91 | 23 |
| AD VI | Female | 79 | 24 |
| AD VI | Male | 69 | 30 |
| AD VI | Male | 79 | 33 |
| AD VI | Male | 82 | 18 |
| AD VI | Male | 83 | 36 |
| AD VI | Male | 83 | 21 |
| AD VI | Female | 87 | 30 |
| AD VI | Female | 50 | 15 |
| AD VI | Female | 74 | 27 |
| AD VI | Female | 89 | 32 |
| AD VI | Female | 64 | 21 |
| AD VI | Male | 62 | 38 |
| AD VI | Male | 76 | 40 |
| AD VI | Female | 80 | 51 |
| AD VI | Male | 85 | 32 |
| AD VI | Male | 83 | 21 |
| AD VI | Female | 87 | 30 |
| AD IV | Male | 82 | 28 |
| AD IV | Female | 88 | 24 |
| AD IV | Male | 87 | 39 |
| AD IV | Male | 83 | 24 |
| AD IV | Male | 79 | 23 |
| FTLD-Tau | Female | 65 | 31 |
| FTLD-Tau | Male | 67 | 30 |
| FTLD-Tau | Male | 43 | 6 |
| FTLD-Tau | Female | 54 | Unknown |
| PSP | Female | 78 | 29 |
| PSP | Female | 70 | 24 |
| PSP | Female | 70 | 24 |
| PSP | Male | 77 | 32 |
| PiD | Male | 77 | Unknown |
| PiD | Female | 61 | 30 |
| CBD | Female | 59 | 30 |
| CBD | Female | 72 | 15 |
AD, Alzheimer’s disease; PSP, progressive supranuclear palsy; FTLD-Tau, frontotemporal lobar degeneration with tau pathology; PiD, Pick’s disease; CDB, cortico-basal degeneration.
Animal model
Brains and spinal cords were collected from transgenic mice homozygous for the human Tau-P301S mutation [40] at different ages. This mouse line, expressing the 383 amino acid isoform of human MAPT, displays a tauopathy phenotype characterized by severe paraparesis and a significant reduction in the number of motor neurons in the spinal cord at 5 months of age. Approval of the Inserm regional committee for animal experimentation, as established by the Scientific Research Ministry on January 1992, was previously obtained. Obligations required by the n° 86/609 European directive for use of laboratory animals were also observed. Mouse brains and spinal cords from animals aged from 1 to 5 months were fixed in buffered formalin for 24 hours maximum, paraffin-embedded and sectioned prior to immunolabeling experiments.
Immunohistofluorescence
Formalin fixed paraffin-embedded brain samples were sectioned at 8μm thickness. Sections were mounted on Superfrost+slides. After paraffin removal with three successive baths in xylene substitute for 5 minutes and three baths in absolute alcohol, the sections were rehydrated in 70% alcohol, rinsed in demineralized water and subjected to standard microwave antigen retrieval in pH6 Citrate buffer. Sections were then incubated with SEA BLOCK Blocking Buffer (ThermoFisher, Rockford, Il, USA) for 30 min prior to primary antibody incubation overnight at 4°C in a humid chamber. After PBS washing, the sections were then incubated with appropriate donkey anti-mouse, goat, or rabbit secondary antibodies labeled with AlexaFluor 488, AlexaFluor 555 or 546, or AlexaFluor 647 at a concentration of 1.6μg/ml for 90 min at room temperature. Pre-immune mouse, rabbit, and goat immunoglobulins were used in place of the primary antibodies as negative controls. For double and triple labeling experiments, overnight primary antibody incubations were repeated serially with successive appropriate secondary antibody incubations as we found that specific signal intensity was greater than using simultaneous application of primary and then secondary antibodies mixtures.
After antibody incubation and washing, lipofuscin autofluorescence on human brain sections was quenched with Millipore Autofluorescence Eliminator Reagent (Millipore, Cal USA) according to the manufacturer’s instructions. A DAPI (4’,6-diamidino-2-phenylindole) enriched mounting medium (Abcam) was used and provided a nuclear counterstain. Slides were examined with a Leica TCS SP8 confocal microscope with Z-stack imaging.
Western blotting
For each case studied (Table 2), a portion of medial frontal cortex (F2) was homogenized at 4°C using a Dounce homogenizer (glass-Teflon) in 5 volume (w/v) of buffer consisting of: Tris 10 mM, saccharose 0.32 M, and DTT 1 mM at pH 7.4 with “Complete” protease inhibitor cocktails (Roche). Homogenates were centrifuged 5 min at 1000 g and the protein estimation of the upper fraction was performed using a BCA protein assay kit (Pierce). Samples were analyzed on a 10% SDS Page gel and transferred onto iBlot™ Gel Transfer membranes (Invitrogen). Following transfer, membranes were incubated overnight with appropriate antibodies [FKBP52, mouse monoclonal EC1 (1 : 1000) (Enzo Life Sciences) and a Rabbit polyclonal HRP conjugated GAPDH (1 : 1000) (Abcam) rather than Actin for the loading control as recommended [41]. Images were recorded with the GeneGnome5 (Syngene). Quantification was performed using Genetools analysis software (Syngene).
Image analysis
The fluorescence profile was evaluated on a single plane of each confocal image using FIJI freeware image analysis program. The intensity of fluorescence in the green and red channels was measured along a line segment. To assess FKBP52 immunoreactivity in neurons with or without AT180, Tau-D421, Tau-pS422 positive NFTs, or P62 labeling, confocal images were analyzed with the FIJI image analysis program.
Statistical analysis
Statistical analysis was performed using one-way variance analysis (Student’s t test) comparing the mean of percentages (±SEM) of at least four independent experiments. Level of significance: N. S. not significant; p values of < 0.05 were considered statistically significant. The software GraphPad Prism 8 was used to determine the statistical analysis.
RESULTS
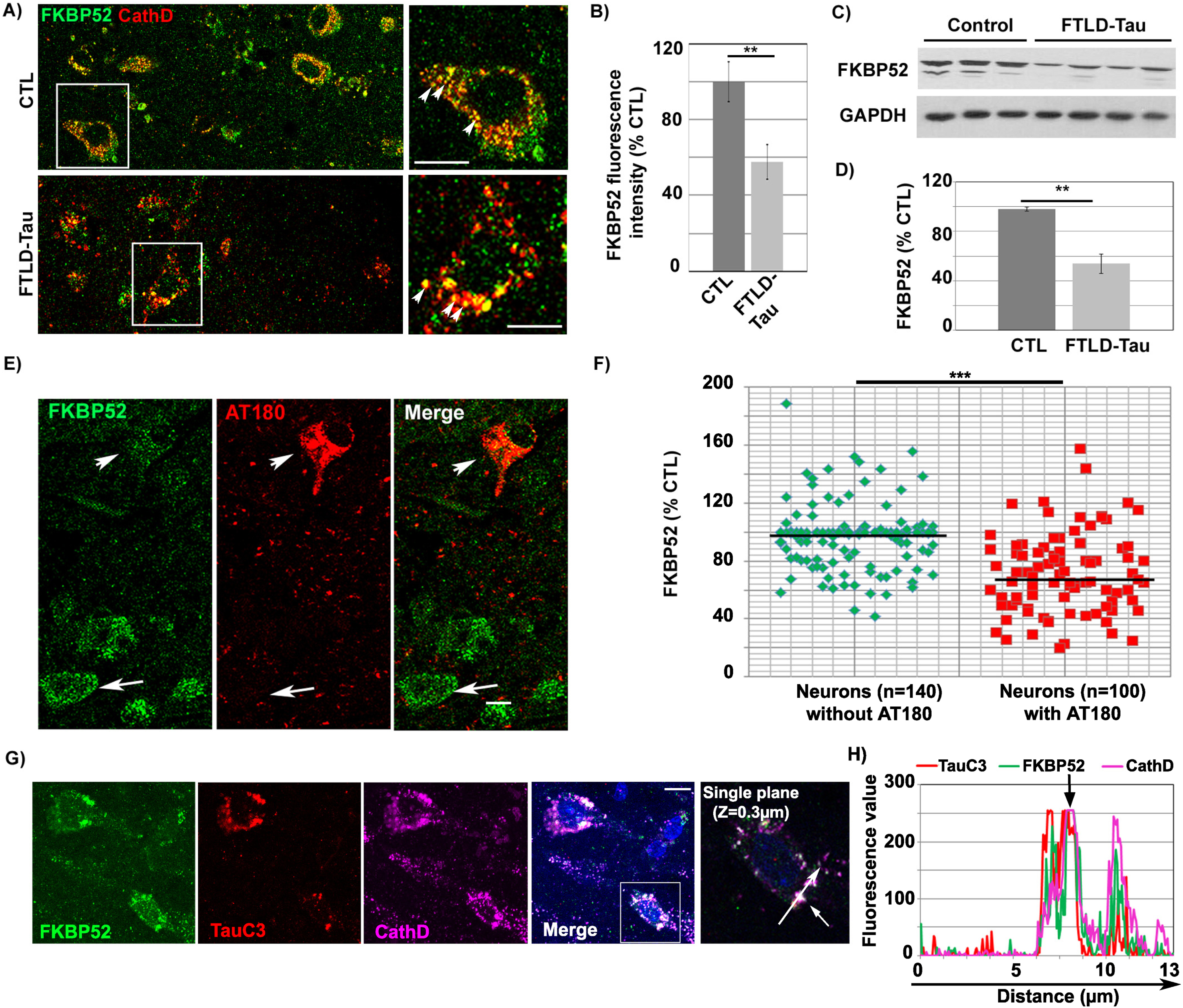
FKBP52 partially colocalizes with truncated Tau-D421 in the autophagy endo-lysosomal pathway (ALP) of familial FTLD-Tau brain neurons and its decrease is related to NFT deposition
IHF analysis performed in familial FTLD-Tau brains showed the colocalization of the FKBP52 signal with cathepsin D, an endosomal/lysosomal marker, as previously observed in AD and normal brain neurons [35]. It also showed a decrease of FKBP52 expression in isocortical neurons with pathological AT180 tau deposits (Fig. 1A) when compared to adjacent neurons without detectable deposits, analogously to what previously described in AD Braak VI. Residual FKBP52 in familial FTLD-Tau brain neurons was frequently localized into abnormally large endosomal/lysosomal vesicles (Fig. 1A (see magnifications) and Supplementary Figure 1). The fluorescent levels of FKBP52 immunoreactivity were examined in 108 neurons per sample in 4 familial FTLD-Tau cases (total: 432 neurons), and in 114 neurons per sample in 4 normal controls (total: 456 neurons). The mean value of the FKBP52 signal per cell was 100% ±10.5% [SEM] for the normal controls, 57.8% ±9.1% for the FTLD-Tau cases; the difference was highly significant (**p < 0.01; Fig. 1B). The signal per cell was, as a mean, inferior by 42% in familial FTLD-Tau neurons confirming the decrease of FKBP52 expression levels previously observed by western blotting on total frontal cortex homogenates (46.1% ±7.8% [SEM]; Fig. 1C, D) [34]. While FKBP52 was decreased in AT180 immuno-positive neurons in familial FTLD-Tau samples (Fig. 1E, arrowhead), neurons without apparent tau deposition exhibited FKBP52-positive endosomes/lysosomes similar to those seen in healthy control brains (arrow) [35]. In familial FTLD-Tau brains, the fluorescent level of FKBP52 immunoreactivity in neurons with AT180-positive NFTs was 69.5% (±4.1% [SEM]) of the value found in neurons without NFTs (96.4% ±1.6%). A mean of 35 neurons without NFTs and 25 NFTs-bearing neurons per case was analyzed from 4 familial FTLD-Tau cases, and the difference was highly significant (t = 6.13, df = n1 + n2-2 = 6; ***p < 0.001). The levels of FKBP52 labelling from each of the different neurons studied are presented in a scatter diagram (Fig. 1F). Triple labeling experiments demonstrated that FKBP52, cathepsin D, and caspase-cleaved Tau-D421 (TauC3 antibody) immunoreactivities colocalized in FTLD-Tau neurons as previously observed in AD neurons (Fig. 1G, H, see arrow in fluorescence profile, Z = 0.3 mm) [35]. This colocalization was also observed into large endo-lysosomal vesicles (Supplementary Figure 1). FKBP52 and Tau-D421 also colocalized in Rab7-positive late endosomes and LC3-positive autophagic vesicles (Supplementary Figure 2). Our findings demonstrate that residual FKBP52 is detected along with an early pathological and caspase-cleaved tau form in the ALP of FTLD-Tau neurons and suggest that the pathologic decrease of FKBP52 is associated with NFT apparition in affected neurons.
Fig. 1
FKBP52 partially colocalizes with tau in the lysosomes and is decreased in NFTs-bearing neurons of familial FTLD-Tau patients. A) Double labeling Immunofluorescence: The FKBP52 signal (green) colocalizes with the Cathepsin D signal (red) of endosomal lysosomal vesicles in neurons of FTLD-Tau patients. Normal lysosomes in healthy neurons (size range: 0.2–0.5μm) and enlarged lysosomes in AD neurons (size range: 1.5–3μm) are indicated by arrowheads (see magnification, Scale bar 10μm). B) Fluorescent levels of neuronal FKBP52 in familial FTLD-Tau neurons quantified by image analysis are significantly decreased compared with neurons in the frontal cortex of age matched controls. Statistical analysis was performed using Student’s t-test, n = 4; **p < 0.01;±SEM. C) Total frontal cortex homogenates of familial FTLD-Tau patients analyzed by western blot and immunoblotted with EC1 (FKBP52) and 6C5 (GAPDH) antibodies. D) FKBP52 quantification in total brain homogenates (FKBP52/GAPDH) in controls and familial FTLD-Tau patients. Student’s t-test, n = 4; **p < 0.01;±SEM. E) Double labeling of a frontal cortex section from a familial FTLD-Tau patient showing a visibly decreased FKBP52 expression (green, arrow and arrowhead) in neurons with AT180 deposits (red, arrowhead) but not in adjacent neurons without visible AT180 labeling (arrows). F) Scatter diagrams of image analysis results of frontal cortex samples from four familial FTLD-Tau patients showing a significant decrease of FKBP52 in neurons exhibiting AT180 deposits compared to neurons without apparent AT180 deposition. Student’s t-test, n = 4; ***p < 0.001;±SEM. G) Triple labeling of a single Z-stack plane (0.3μm) showing the colocalization of the FKBP52 signal (green) with the caspase-cleaved Tau-D421 (TauC3, red) and the lysosomal Cathepsin D (magenta) signals in familial FTLD-Tau brain neurons. H) Fluorescence profiles of FKBP52, TauC3, and Cathepsin D antibodies showing the colocalization of the three signals on a single plane. Scale bar 10μm.
Neuronal FKBP52 decrease and its colocalization with truncated Tau-D421 in the ALP are also detected in another human tauopathy such as PSP
In order to check if FKBP52 expression was also modified in the brains of patients with other tauopathies, we studied frontal cortex tissue samples from 5 PSP patients. Western blot analysis of FKBP52 in PSP brain homogenates showed a non-statistically significant decrease (48.3% ±17.5%) (t = 2.07, df = 6; p = 0.0837, Fig. 2A) very likely resulting from a strong heterogeneity of FKBP52 expression between PSP samples (n = 5; 7.6 to 82%). This difference might be explained by the pathological heterogeneity of PSP and the variable severity of tau pathology in the frontal cortex between patients [42, 43]. In contrast, cell by cell IHF analysis of AT180 NFTs-bearing neurons in the frontal cortex of PSP patients showed a significant decrease of FKBP52 (Fig. 2B, arrowhead) compared to neurons without apparent tau deposition (Fig. 2B, arrow). The fluorescent levels of FKBP52 immunoreactivity in PSP frontal cortex neurons with AT180 deposits were 66.6% (±4.43% [SEM]) of the value found in neurons devoid of deposits (104.4% ±1.96% [SEM]). A mean of 47 neurons without NFTs and 20 NFTs-bearing neurons per case was analyzed from five PSP cases resulting in a very significant difference (t = 7.8, df = 8; **p=0.0015). The levels of FKBP52 labelling from each of the different neurons studied are presented in a scatter diagram (Fig. 2C). We observed by IHF analysis a comparable interrelation between the decrease of FKBP52 and the presence of AT180-Tau deposits in frontal cortex neurons from CBD (n = 2) and PiD (n = 2) brains but the number of samples for both tauopathies was too low to determinate the significance of these observations (Supplementary Figure 3A). As previously observed in AD and familial FTLD-Tau neurons, Tau-D421 immunoreactivity also colocalizes with FKBP52 and CathD in PSP (Fig. 2D, E, see arrow in the fluorescence profile, Z = 0.3 mm), CBD and PiD neurons (Supplementary Figure 3B). Altogether, these findings suggest that neuronal FKBP52 decrease and its colocalization with Tau-D421 in the ALP is not specific to a single tauopathy as it is significantly detected in the brains of AD, familial FTLD-Tau, and PSP patients.
Fig. 2
Decreased expression of FKBP52 in the frontal cortex of PSP patients: FKBP52 decrease in neurons with pathologic tau deposits is not limited to a specific tauopathy. A) Representative western blot analysis of PSP frontal cortex homogenates showing a non-significant decrease of FKBP52 (lower half), probably due to the strong heterogeneity of the neuronal pathology in the frontal cortex of PSP patients [42, 43]. B) Double labeling experiment on the frontal cortex section of a PSP patient. FKBP52 (green) expression in neurons with AT180 deposits (red, arrowheads) is visibly decreased (merge) in comparison to neurons without visible AT180 labeling (arrows). C) Scatter diagrams of cell by cell image analysis of neurons with and without AT180 deposits showing a significantly lower FKBP52 expression in cells exhibiting AT180 NFTs. Statistical analysis was performed using Student’s t-test, n = 5; **p < 0.01;±SEM. D) Triple labeling experiment on a frontal cortex section from a PSP patient showing FKBP52 (green), TauC3 (red) and Cathepsin D (magenta) colocalization (merge). E) The single Z-stack plane (0.3μm) fluorescence profiles of a neuron show the superposition of the three signals (arrows on image and fluorescence profiles). Scale bar 10μm.
![Decreased expression of FKBP52 in the frontal cortex of PSP patients: FKBP52 decrease in neurons with pathologic tau deposits is not limited to a specific tauopathy. A) Representative western blot analysis of PSP frontal cortex homogenates showing a non-significant decrease of FKBP52 (lower half), probably due to the strong heterogeneity of the neuronal pathology in the frontal cortex of PSP patients [42, 43]. B) Double labeling experiment on the frontal cortex section of a PSP patient. FKBP52 (green) expression in neurons with AT180 deposits (red, arrowheads) is visibly decreased (merge) in comparison to neurons without visible AT180 labeling (arrows). C) Scatter diagrams of cell by cell image analysis of neurons with and without AT180 deposits showing a significantly lower FKBP52 expression in cells exhibiting AT180 NFTs. Statistical analysis was performed using Student’s t-test, n = 5; **p < 0.01;±SEM. D) Triple labeling experiment on a frontal cortex section from a PSP patient showing FKBP52 (green), TauC3 (red) and Cathepsin D (magenta) colocalization (merge). E) The single Z-stack plane (0.3μm) fluorescence profiles of a neuron show the superposition of the three signals (arrows on image and fluorescence profiles). Scale bar 10μm.](https://ip.ios.semcs.net:443/media/jad/2023/94-1/jad-94-1-jad230127/jad-94-jad230127-g002.jpg)
The abnormal decrease of FKBP52 and its colocalization with pathologic tau forms in the ALP of affected neurons in AD, FTLD-Tau, and PSP frontal cortex is not restricted to cells with caspase-cleaved Tau-D421 deposits
Caspase activation is often associated with apoptotic signals [44]. We detected the caspase cleaved Tau-D421 in association with FKBP52 in the ALP of affected neurons. This might suggest that the colocalization we observed is restricted to a population of apoptotic neurons. We thus decided to extend our study and focus our attention on another pathologic tau isoform. Tau cleavage by Caspases at serine 421 is inhibited by phosphorylation at serine 422 (Tau-pS422), which prevents tau autophagic clearance, triggering Tau-pS422 accumulation and possibly contributing to the escape of neurons from acute apoptotic death [45, 46]. It has been reported that Tau-pS422 is not preferentially degraded by autophagy unlike Tau-D421 [21]. We detected by IHF analysis a focal Tau-pS422 localization in the ALP of AD, familial FTLD-Tau, and PSP affected neurons (Fig. 3). Also, we observed a colocalization of FKBP52 with both Tau-pS422 and Cathepsin D suggesting that the association of FKBP52 with tau in the ALP in these tauopathies is not restricted to potentially apoptotic neurons bearing caspase-cleaved Tau-D421 (Fig. 3, see arrowheads in fluorescence profiles, Z = 0.3 mm). Tau-pS422 accumulation is considered an earlier tau pathologic event than formation of Tau-D421 aggregates [45, 47]. We thus measured by IHF analysis the fluorescent levels of FKBP52 immunoreactivity in neurons with Tau-pS422 deposits in AD Braak VI, familial FTLD-Tau, and PSP cases (Fig. 4). Tau-pS422 positive neurons in AD Braak VI, familial FTLD-Tau, and PSP cases exhibited a significant decrease of FKBP52 signal (Fig. 4A, C, and E; arrowheads) compared with neurons without apparent Tau-pS422 deposition (arrows). The fluorescent levels of FKBP52 immunoreactivity in neurons with Tau-pS422 deposits were 72% (±5.2%), 66% (±4.3%), and 73% (±7%) of the value found in neurons without aggregates (90% ±2.6%, 98% ±1.5%, and 98% ±2.9%) respectively in AD Braak VI, familial FTLD-Tau, and PSP brains (Fig. 4B, D, and F). A mean of 48 neurons without Tau-pS422 aggregates and 43 neurons bearing aggregates were analyzed from frontal cortex samples of 13 AD Braak VI patients showing a statistically significant decrease of FKBP52 signals in affected neurons (t = 2.54, df = 24; *p = 0.0177; Fig. 4B). Also, the analysis of a mean of 46 neurons without Tau-pS422 aggregates and of 40 neurons with Tau-pS422 deposits per patient from 4 familial FTLD-Tau frontal cortex samples evidenced a highly significant difference in FKBP52 signals between neurons with and without pathological deposits (t = 6.97, df = 6; ***p = 0.0004; Fig. 4D). Analogously, the analysis of a mean of 28 neurons without Tau-pS422 aggregates and 15 aggregate-bearing neurons per sample from 5 PSP patients, showed a statistically significant decrease of FKBP52 in neurons with Tau-pS422 deposits (t = 2.9, df = 8; *p = 0.0191; Fig. 4F). Overall, these results show that FKBP52 decrease is detected both in Tau-D421 and Tau-pS422 positive neurons from the frontal cortex of AD, familial FTLD-Tau, and PSP patients and suggest that the pathological decrease of FKBP52 we describe is not restricted to Tau-D421 formation and accumulation in pathologic neurons from these different tauopathies.
Fig. 3
Triple labeling experiments showing the lysosomal colocalization of FKBP52 and the early pathologic Tau-pS422 isoform in frontal cortex neurons of patients with different tauopathies. Triple labeling experiments of familial FTLD-Tau (A), AD Braak VI (B), and PSP (C) frontal cortex sections respectively: FKBP52 (green) and Tau-pS422 (red) colocalize (merge) with the endolysosomal marker Cathepsin D in the three tauopathies. Scale bar 10μm. At the lower right of each set of micrographs: superposition of the three respective single plane fluorescence profiles of a single neuron (lower left, scale bar 3μm).
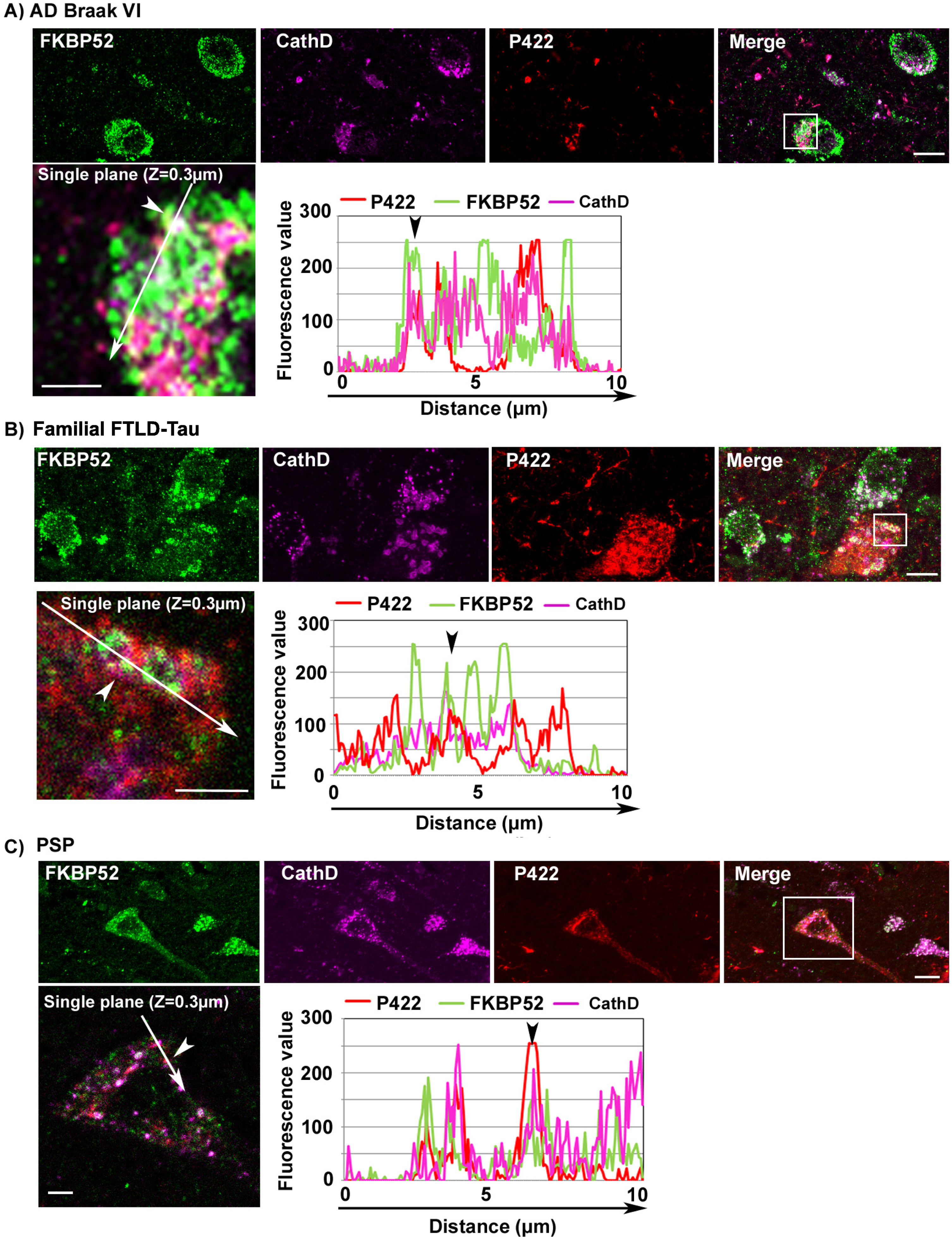
Fig. 4
Image analysis results showing the decrease of FKBP52 expression in neurons with Tau-pS422 deposits in the frontal cortex of patients with different tauopathies. A, C, E) FKBP52 expression (green) is visibly decreased in Tau-pS422 (P422, red) positive neurons of AD Braak VI, familial FTLD-Tau and PSP patients respectively (arrowheads) compared with adjacent neurons devoid of evident Tau-pS422 deposits (arrows). B, D, F) Scatter diagrams of image analysis results showing a significant decrease of FKBP52 expression in Tau-pS422 positive neurons of AD Braak VI, familial FTLD-Tau, and PSP patients respectively. Statistical analysis was performed using Student’s t-test, *p < 0.05, ***p < 0.001,±SEM.
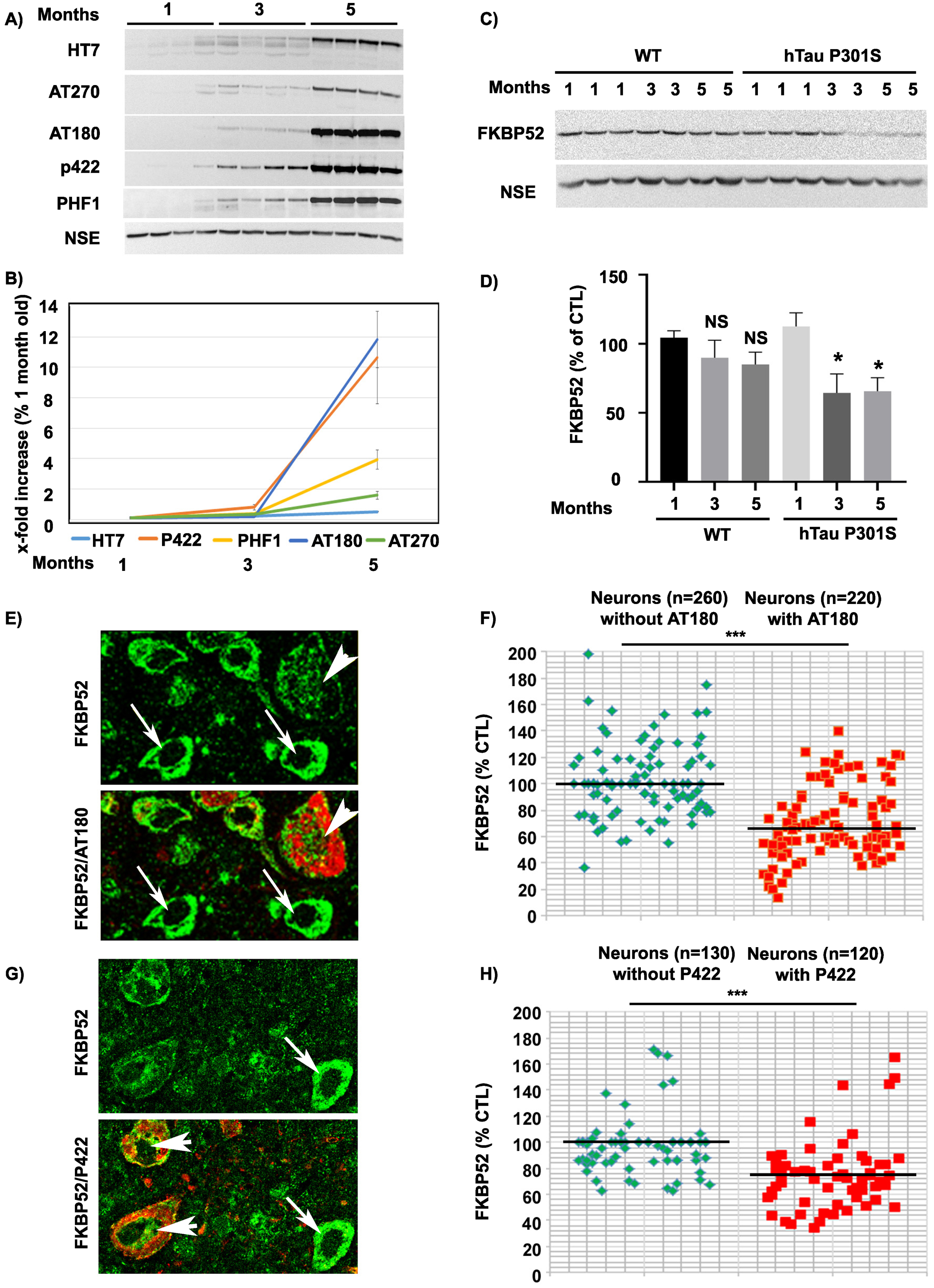
FKBP52 expression decreases in affected spinal neurons of hTau-P301S mice in parallel with the deposition of pathological tau and independently of Tau-D421 formation and accumulation
Next, we checked if the abnormal expression and subcellular localization of FKBP52 observed in familial FTLD-Tau brain neurons could be also detected in a transgenic mouse model of FTLD-Tau (hTau-P301S mice) [40]. At about 5 months of age, the homozygous hTau-P301S mice display a tauopathy phenotype characterized by severe paraparesis and the presence of abundant intraneuronal tau deposits in the brain and in the spinal cord [40]. Western blot analysis of spinal cords in 1- to 5-month-old hTau-P301S mice showed a progressive accumulation of human tau proteins, specifically recognized by the HT7 antibody, along with the detection of different pathological phosphorylated tau isoforms, recognized by several antibodies such as AT180 and Tau-pS422, already significantly present at 3 months of age (Fig. 5A, B). Others had previously reported a very low or absent caspase activity and a very low expression of Tau-D421 in hTau-P301S mice, which made it particularly suitable to exclude the possible influence of neuronal apoptosis on our results. Although several caspase-cleaved Tau-D421 positive neurons were detected in familial FTLD-Tau brain samples, only rare Tau-D421 positive neurons were present in the spinal cords and brains of hTau-P301S mice as previously described [40]. Despite the absence of this pathological tau form, western blot analysis of total spinal cord homogenates from mice of different ages showed a relevant decrease of FKBP52 already at 3 months (64.5% ±13.7% [SEM]) compared with 1-month-old mice (112% ±9.8% [SEM]; Fig. 5D) with little variation of its expression until 5 months (65% ±9.7% [SEM]). These results confirm our previous observations showing that FKBP52 decrease is independent of Tau-D421 accumulation but nevertheless happens in concomitance with the apparition of pathological tau deposits (Fig. 5A, C). IHF analysis of the spinal cord of 5-month-old mice supports our observations about the FKBP52 decrease both in AT180 and Tau-pS422-positive spinal neurons (Fig. 5E–H). The fluorescent levels of FKBP52 immunoreactivity in neurons with tau aggregates are 70.9% (±1.1%) and 70.3% (±3.5%) of the value found in neurons without aggregates (102.7% ±1.7%) and (100.1% ±1.8%) respectively in AT180 and Tau-pS422 positive neurons (Fig. 5F, H; [SEM]). A mean of 52 neurons without tau AT180 aggregates and of 44 aggregate-bearing neurons per sample were analyzed from 5 spinal cords of 5-month-old mice, and the difference was statistically significant (t = 12.47, df = 8; ***p = 0.0001; Fig. 5F). Analogously, a mean of 26 neurons devoid of Tau-pS422 aggregates and of 24 neurons with aggregates per sample were analyzed from 5 spinal cords of 5-month-old mice, and the difference was also statistically significant (t = 6.73, df = 8; ***p = 0.0001; Fig. 5H). Altogether, in the hTau-P301S mouse model of tauopathy, we observed an early pathological decrease of FKBP52 in the spinal cord neurons that occurred in parallel to tau accumulation. These results are analogous to the FKBP52 decrease in neurons with pathologic tau deposits we detected in the brains of patients with a pure tauopathy, such as familial FTLD-Tau, thus suggesting the possibility of an early decrease of FKBP52 at the preclinical stage in familial FTLD-Tau patients as well as in other human tauopathies.
Fig. 5
Expression and age variations of FKBP52 and pathologic tau isoforms in spinal cord neurons of hTau-P301S mice. A) Representative western blot analysis of the spinal cords of 1-, 3-, and 5-month-old hTau-P301S mice showing the progressive accumulation of human tau and of different pathologic tau isoforms. NSE was used as a loading control (n = 5). B) Graph illustrating the increase of human tau and some pathologic tau isoforms in spinal cords from hTau-P301S mice, beginning at 3 months and sharply increasing to peak at 5 months of age. C) Representative western blot analysis illustrating the decrease of FKBP52 neuronal expression beginning at 3 months of age in spinal cords from hTau-P301S mice, compared with control animals. NSE was used as a loading control. D) FKBP52 quantification in total spinal cord homogenates (FKBP52/NSE) in control and hTau-P301S mice. Statistical analysis was performed using Student’s t-test, n = 5; *p < 0.05;±SEM. E) Section of the spinal cord of a 5-month-old hTau-P301S mouse double labeled with FKBP52 (green) and AT180 (red) showing the decrease of FKBP52 expression in a neuron with AT180 deposits (arrowheads) compared with adjacent neurons devoid of AT180 (arrows). F) Scatter diagrams of image analysis results illustrating the significant decrease of FKBP52 in spinal cord neurons from 5-month-old hTau-P301S mice exhibiting AT180 deposits Statistical analysis was performed using Student’s t-test, n = 5; ***p < 0.001;±SEM. G) Spinal cord section of a 5-month-old hTau-P301S mouse double labeled with FKBP52 (green) and Tau-pS422 (red) showing the decrease of FKBP52 expression in two neurons exhibiting Tau-pS422 deposits (red, arrowheads) compared with an adjacent neuron devoid of Tau-pS422 deposits (arrows). H) Scatter diagrams of image analysis results illustrating the significant decrease of FKBP52 in spinal cord neurons from 5-month-old hTau-P301S mice exhibiting Tau-pS422 deposits. Statistical analysis was performed using Student’s t-test, n = 5; ***p < 0.001;±SEM.
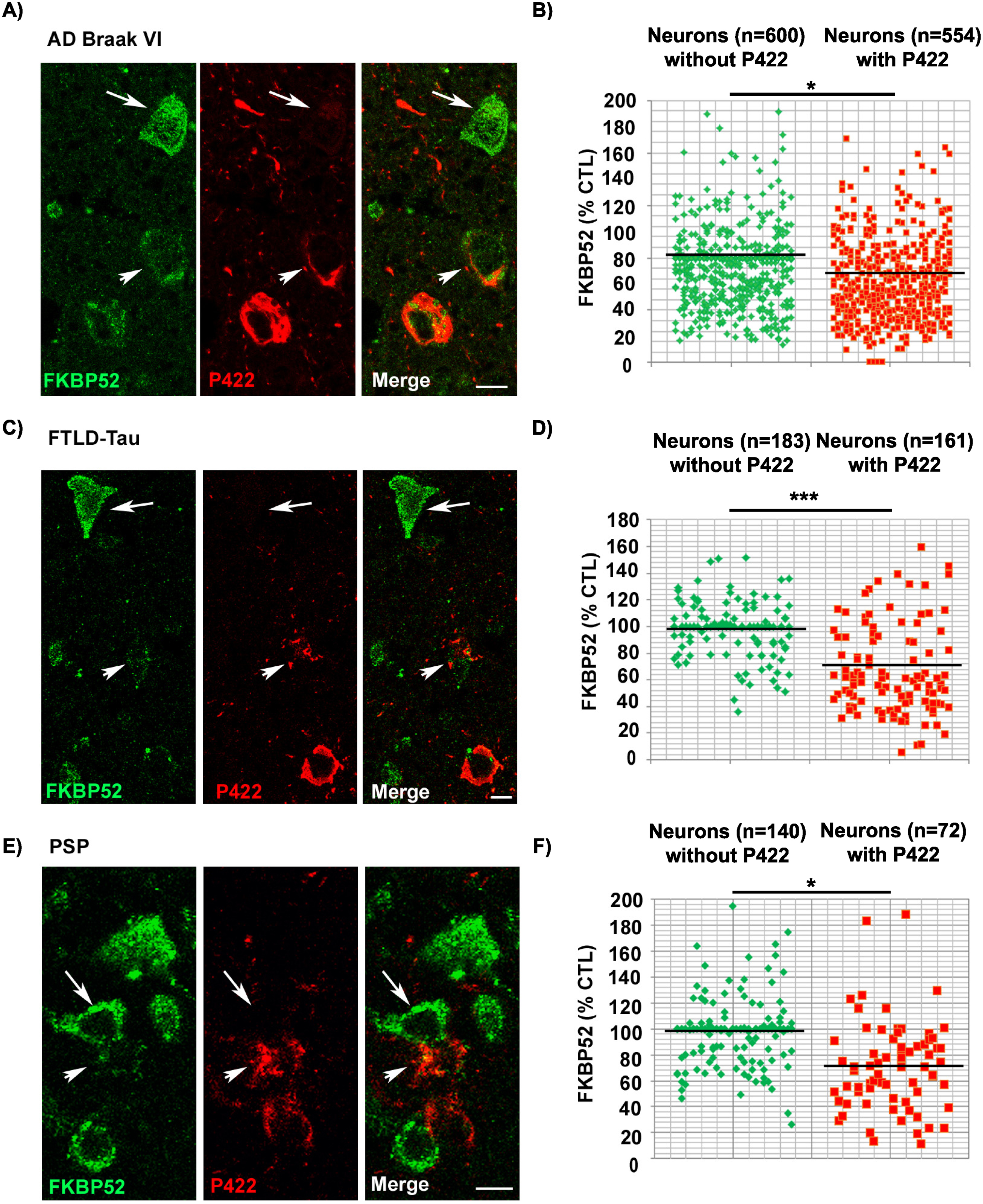
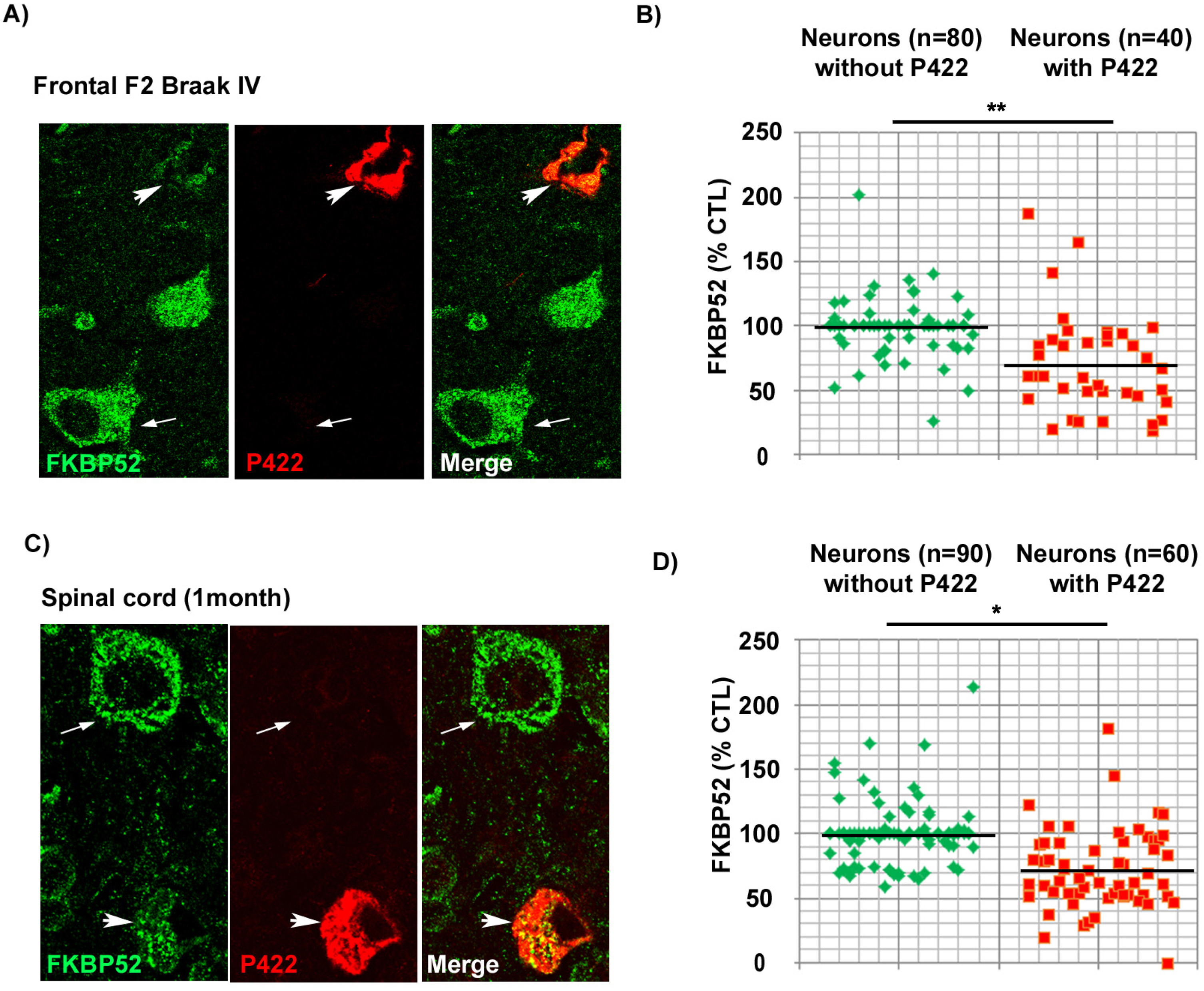
FKBP52 is decreased in pathologic neurons both from marginally affected AD Braak IV brain regions and from the rachis of symptomless 1-month-old hTau-P301S mice
The previously described results prompted us to investigate the levels of intraneuronal FKBP52 at a very early stage of disease progression. We thus examined the scarcely affected frontal cortex of AD patients with Braak IV stage (n = 5) and the spinal cords of symptomless 1-month-old hTau-P301S mice. Even if these samples comported few Tau-pS422 positive neurons, we were nevertheless able to analyze by IHF the variations of FKBP52 fluorescent signals in these rare cells (Fig. 6). Both Tau-pS422 positive neurons in AD Braak IV frontal cortex samples and in the spinal cord of symptomless 1-month-old hTau-P301S mice exhibited a significant decrease of FKBP52 (Fig. 6A, C) compared to neurons without Tau-pS422 deposition. The fluorescent level of FKBP52 immunoreactivity in neurons with Tau-pS422 aggregates was 72.6% (±6.8%) and 72.5% (±5.9%) of the value found in neurons without Tau-pS422 aggregates (100.1% ±3.1% and 99.9% ±5.5%) respectively in AD Braak IV and in the spinal cords of 1-month-old hTau-P301S mice (Fig. 6B, D). A mean of 16 neurons without apparent tau aggregates and of 8 Tau-pS422 aggregate-bearing neurons per patient was analyzed from 5 AD Braak IV frontal cortex samples, and the difference was highly significant (t = 3.65, df = 8; **p = 0.0065; Fig. 6B). The difference in FKBP52 expression was also significant when a mean of 18 neurons without apparent tau aggregates and of 12 neurons with Tau-pS422 aggregates per sample was analyzed from the spinal cords of three 1-month-old hTau-P301S mice (t = 3.37, df = 4; *p = 0.0279; Fig. 6D). The expression of FKBP52 measured by western blot analysis in the spinal cord of 1-month-old hTau-P301S mice is similar from that observed in wild-type mice of the same age (Fig. 5C, D), in contrast with our findings obtained by IHF and cell by cell image analysis showing a significant decrease of FKBP52 in the rare Tau-pS422 bearing neurons (Fig. 6C, D). This difference is very likely due to the rarity of affected neurons at such an early stage of disease: indeed our results suggest that FKBP52 decrease has already started in these tissues but is limited to a low number of affected neurons while its expression is unchanged in the unaffected cells. Altogether, these experiments show that the neuronal decrease of FKBP52 happens as early as Tau-pS422 deposition is detectable in marginally affected regions of AD brains and in the spinal cord of symptomless 1-month-old hTau-P301S mice.
Fig. 6
Decrease of neuronal FKBP52 at early asymptomatic stages of neurodegenerative tauopathy in human and mouse. A) Decrease of the FKBP52 signal (green) in the rare neurons exhibiting Tau-pS422 deposits (red, arrowheads), compared with adjacent neurons devoid of evident Tau-pS422 deposits (arrows), in the frontal cortex of AD Braak IV individuals after double labeling Immunofluorescence. B) Scatter diagrams of image analysis results of FKBP52 expression in individual neurons from the frontal cortex of AD Braak IV patients after double labeling immunofluorescence illustrating the significant decrease of FKBP52 expression in adjacent neurons with and without Tau-pS422 deposits. Statistical analysis was performed using Student’s t-test, n = 5; **p < 0.01;±SEM. C) Decrease of the FKBP52 signal (green) in the rare neurons exhibiting Tau-pS422 deposits (red, arrowheads), compared with adjacent neurons devoid of evident Tau-pS422 deposits (arrows), in the rachis of asymptomatic 1-month-old hTau-P301S mice after triple labeling immunofluorescence. D) Scatter diagrams of image analysis results of FKBP52 expression in individual neurons from asymptomatic 1-month-old hTau-P301S mice showing the significant decrease of FKBP52 expression in adjacent neurons with and without Tau-pS422 deposits. Statistical analysis was performed using Student’s t-test, n = 3; *p < 0.05;±SEM.
DISCUSSION
In this study, we extend previous findings showing that neuronal FKBP52 decrease and localization with truncated-Tau-D421 in the ALP are not limited to AD but are also detected in familial FTLD-Tau and PSP brains and are strongly linked with tau pathogenesis, suggesting a common mechanism for the abnormal FKBP52 depletion in different tauopathies. We also find by IHF analysis of pathologic human brains that FKBP52 decrease is not restricted to Tau-D421-bearing neurons, which might be apoptotic, but is also detected in Tau-pS422 immunoreactive cells. Tau cleavage by caspases at D421 is known to be inhibited by serine 422 phosphorylation (Tau-pS422), suggesting that the decrease of FKBP52 we observe is not necessarily related to Tau-D421 accumulation but might happen at an earlier pathological stage. Analysis of spinal cord neurons from transgenic hTau-P301S mice also shows that FKBP52 decrease happens concomitantly with pathological tau accumulation and confirms that this phenomenon is not restricted to caspase-cleaved Tau-D421 accumulation. Finally, using cell by cell IHF analysis, we find a significant FKBP52 depletion in the rare neurons with Tau-pS422 deposits present in scarcely affected frontal brain regions of AD Braak IV patients and in spinal cords of symptomless young hTau-P301S mice. These affected neurons are detected at an early stage of the pathologic process and in mostly spared areas that will nevertheless be strongly impacted later on with disease progression. This suggests that the FKBP52 decrease in Tau-pS422 positive neurons in these hardly affected regions is an early pathologic event.
Tau-D421 and Tau-pS422 lysosomal localization in different tauopathies
We report the association of FKBP52 with two early pathological tau forms, the caspase-cleaved Tau-D421 [18, 44, 48–50] and the Tau-pS422 phosphorylated at serine 422 [15, 47, 51–53] in the ALP of AD, PSP, and familial FTLD-Tau neurons. However, some studies propose a protective effect for both Tau-pS422 and Tau-D421 [45, 54]. This discordance might be explained by the possibility that tau forms, not deleterious in physiologic quantities, might abnormally accumulate in affected neurons through inefficient degradation, becoming pathogenic. It has been reported that Tau-D421 is preferentially degraded through autophagy [21], supporting our observations of its lysosomal localization, while tau phosphorylation at Serine 422 inhibits Tau-D421 formation preventing tau autophagic degradation [45, 55]. Nevertheless, we localize Tau-pS422 in the ALP which could suggest that the inhibition of caspase-induced tau cleavage by S422 phosphorylation might be removed locally in the endo-lysosomal environment allowing cathepsin cleavage of tau [56] in order to facilitate the autophagic degradation of its fragments. Indeed, while Tau-D421 immuno-labeling often presents a vesicular/lysosomal aspect (Figs. 1 and 2) [35], Tau-pS422 immunoreactivity exhibits mingled NFTs, pre-NFTs and vesicular/lysosomal patterns in each tauopathy we studied (Figs. 3 and 4). This observation agrees with other studies showing the infrequent presence of Tau-D421 positive neurofilamentous tau aggregates in AD, familial FTLD-Tau [57–59], and in PSP brain neurons [60]. A study even reports the absence of Tau-D421 signals in PSP brains [61], nevertheless we detect in PSP brain neurons the same vesicular/lysosomal aspect of Tau-D421 immunoreactivity we observed in AD and familial FTLD-Tau brains suggesting that this kind of tau caspase and/or lysosomal processing might occur in PSP neurons and contribute to tau pathogenesis. We do not detect a significant expression of Tau-D421 in the hTau-P301S mouse model as previously reported [40] whereas we detect Tau-pS422 in rare mouse spinal cord neurons at 1 month of age. This difference in Tau-D421 deposition between the hTau-P301S mouse model and the familial FTLD-Tau patients, both tauopathies exclusively caused by the human tau gene mutation, supports the idea that caspase-cleavage of tau at Asp421 is not an essential step towards neurodegeneration at least in pathologies linked to genetic dominant tau mutations.
FKBP52 expression is decreased in neurons with different tau deposits in different tauopathies
The presence of residual FKBP52 associated with pathological tau in the lysosomal environment of AD, familial FTLD-Tau, PSP, and in the hTau-P301S mice neurons suggests a common mechanism involved in FKBP52 decrease, potentially impacting on the pathologic process. The constant presence of FKBP52 in lysosomes and its constant association with early pathologic forms of tau in lysosomes of affected neurons suggests that its decrease might be mainly linked to lysosomal processing. Also, we have previously demonstrated the presence of extracellular release of FKBP52 in SH-SY5Y neurons under tau-induced proteotoxic stress [35]. It is therefore possible that different mechanisms might contribute to FKBP52 decrease in neurodegenerative diseases, albeit with a different impact. However, we also detect by IHC a little FKBP52 immunoreactivity embedded in big NFTs, thus a possible sequestration of this immunophilin in NFTs cannot be excluded, such a sequestration mechanism has been described for Pin1, another peptidyl-prolyl cis/trans isomerase deregulated in AD neurons [62–64]. The significant FKBP52 depletion in neurons bearing different pathologic tau deposits (Tau-pS422 and Tau-AT180) in the frontal cortex of AD Braak VI, familial FTLD-Tau, and PSP patients, without directly causing neurodegeneration, might nevertheless represent an aggravating consequence. Indeed, we have previously shown in dorsal root ganglion (DRG) neurons from hTau-P301S mice that FKBP52 deficiency impacts on ALP functions triggering insoluble tau accumulation under protracted tau-induced proteotoxic stress [38]. IHF analysis of brain neurons from patients with different tauopathies shows a range of FKBP52 decrease of approximately 27–34% in Tau-pS422-positive cells and 30–34% in Tau-AT180-positive cells. We also find a comparable FKBP52 decrease in the spinal cord neurons of tau transgenic mice with pathological tau deposits regardless of the absence of Tau-D421 deposition. Thus, the association of FKBP52 decrease and tau deposition is not specifically related toTau-D421 accumulation. All these observations support the hypothesis that FKBP52 decrease, as a consequence of tau dysfunction and accumulation, might contribute to tau pathogenesis in familial and possibly in sporadic tauopathies.
We detect a FKBP52 decrease in Tau-pS422-positive neurons in scarcely affected brain regions from AD Braak IV patients and in the spinal cords neurons of 1-month-old, symptomless hTau-P301S transgenic mice (Fig. 5). As Tau-pS422 is considered as an early tauopathy marker [47], these findings seem to suggest that FKBP52 decrease also happens very early and in regions that are scarcely affected at such an initial stage of disease progression. The percentage values of FKBP52 depletion measured by IHF were lower than those observed by western blot analysis showing a decrease of 54% and 70% in the frontal cortex of familial FTLD-Tau (Fig. 1) and in AD Braak VI [34] patients respectively. The different results between the techniques might be partly explained by the effects of formalin fixation on the preservation of epitopes revealed by IHF. This discordance might also be due to the fact that double labeling experiments for image analysis detect the deposits of one specific pathologic tau form and fail to detect other type of tau aggregates in adjacent neurons. This would obviously impact on the overall calculated FKBP52 decrease, without negating the significant reduction of FKBP52 in affected neurons. Despite these differences, we constantly observe a decrease of FKBP52 levels in pathological neurons. A minor to moderate decrease of FKBP52 might be in itself sufficient to induce some detrimental effects on tau function and accumulation over time as previously observed in DRG neurons from hTau-P301S mice [26]. We also observe a strong heterogeneity of FKBP52 expression by western blot in frontal cortex samples from PSP patients in contrast to cell by cell IHF analysis, this difference might be explained by the pathological heterogeneity of PSP and the variations in the severity of frontal cortex neuronal involvement among patients [42, 43].
It has been recently shown that FKBP52 overexpression accelerates hippocampal neuronal loss and memory defects in a tau transgenic mouse model and the study in question evokes the possibility that the decrease of FKBP52 expression in AD brains is due to the death of neurons expressing high FKBP52 levels during tau pathogenesis [39]. However, we show by image analysis after multiple labeling of AD Braak VI frontal cortex samples that FKBP52 decrease in neurons with tau deposits is not associated with the decrease of P62, another neuronal protein, whose levels are comparable to those found in non-affected adjacent neurons, excluding an effect of protein leakage from damaged or dying cells (Supplementary Figure 4). Nevertheless, an early rapid phase of neuronal FKBP52 increase preceding a successive slower decline cannot be totally excluded, especially as the distribution of FKBP52 expression values in the different scatter diagrams appears to be heterogeneous (see figures). Notably, we do not detect an increase of FKBP52 expression levels in spinal cords of hTau P301S mice different ages by western blot analysis (Fig. 5C, D).
Concluding, we show a consistent decrease of neuronal FKBP52 in the frontal cortex of different tauopathies such as PSP, familial FTLD-Tau, and AD. Additionally, we show by image analysis that FKBP52 decrease happens concomitantly with the apparition of some pathological tau isoforms including the early Tau-pS422 in spinal cord neurons from hTau-P301S mice. We also find that the FKBP52 decrease starts early during the pathologic process occurring in rare Tau-pS422 positive neurons in marginally affected frontal lobes of AD Braak IV brains and in the spinal cords of symptomless 1-month-old hTau-P301S mice. These observations lead us to think that the FKBP52 decrease in the brains of patients with some neurodegenerative diseases might be a new potential early marker of pathologic progression. Several imaging probes for the non-invasive detection of pathologic tau aggregates are currently used in positron emission tomography (PET) [65, 66]. However, actual tau probes do not well discriminate between early and late disease stages [67] and have difficulty in detecting tau deposition in non-AD tauopathies possibly owing to post-translational events and different NFTs maturation that might remove ligand binding sites and influence PET imaging results [65]. Besides, the new generation probe Flortaucipir shows off-target binding especially in hippocampus and basal ganglia, which might hinder its use in conditions such as early stage AD and corticobasal degeneration [68]. Thus, the detection of the prodromal decrease of a predominantly neuronal protein such as FKBP52 in the brain might helpfully contribute to the early diagnosis of some neuropathological diseases. Also, as FKBP52 plays an important role in cellular signaling and possibly in tau clearance [38], our data suggest that preventing FKBP52 decrease or restoring its normal expression in tauopathies including PSP, familial FTLD-Tau and AD might represent a new potential therapeutic approach.
ACKNOWLEDGMENTS
The authors would like to thank the national Brain Bank GIE NeuroCEB for providing us with the human brain samples of AD, PSP, and familial FTLD-Tau patients. We would like to particularly thank the late Pr. C. Duyckaerts (Laboratoire de Neuropathologie Escourolle, Hôpital de La Salpêtrière, AP-HP, Paris) for sharing with us his deep knowledge and expertise in neuropathology. This enlightened and esteemed neuropathologist shall be missed by all. We wish to thank Dr M.Goedert and Dr. MG. Spillantini (MRC Laboratory of Molecular Biology, Cambridge, UK) for the gift of the hTau-P301S mouse model. We also thank Dr. P. Davies (Albert Einstein, College of Medicine, NY, USA) for the gift of the PHF-1 antibody.
FUNDING
This project has been supported by Inserm and “Institut Professeur Baulieu”, le Kremlin-Bicêtre, France.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230127.
REFERENCES
[1] | Avila J , Lucas JJ , Perez M , Hernandez F ((2004) ) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84: , 361–384. |
[2] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[3] | Spillantini MG , Murrell JR , Goedert M , Farlow MR , Klug A , Ghetti B ((1998) ) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95: , 7737–7741. |
[4] | Hutton M , Lendon CL , Rizzu P , Baker M , Froelich S , Houlden H , Pickering-Brown S , Chakraverty S , Isaacs A , Grover A , Hackett J , Adamson J , Lincoln S , Dickson D , Davies P , Petersen RC , Stevens M , de Graaff E , Wauters E , van Baren J , Hillebrand M , Joosse M , Kwon JM , Nowotny P , Che LK , Norton J , Morris JC , Reed LA , Trojanowski J , Basun H , Lannfelt L , Neystat M , Fahn S , Dark F , Tannenberg T , Dodd PR , Hayward N , Kwok JB , Schofield PR , Andreadis A , Snowden J , Craufurd D , Neary D , Owen F , Oostra BA , Hardy J , Goate A , van Swieten J , Mann D , Lynch T , Heutink P ((1998) ) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: , 702–705. |
[5] | Forrest SL , Kril JJ , Stevens CH , Kwok JB , Hallupp M , Kim WS , Huang Y , McGinley CV , Werka H , Kiernan MC , Gotz J , Spillantini MG , Hodges JR , Ittner LM , Halliday GM ((2018) ) Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 141: , 521–534. |
[6] | Dickson DW , Kouri N , Murray ME , Josephs KA ((2011) ) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 45: , 384–389. |
[7] | Mackenzie IR , Neumann M , Bigio EH , Cairns NJ , Alafuzoff I , Kril J , Kovacs GG , Ghetti B , Halliday G , Holm IE , Ince PG , Kamphorst W , Revesz T , Rozemuller AJ , Kumar-Singh S , Akiyama H , Baborie A , Spina S , Dickson DW , Trojanowski JQ , Mann DM ((2010) ) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol 119: , 1–4. |
[8] | Lee VM , Goedert M , Trojanowski JQ ((2001) ) Neurodegenerative tauopathies. Annu Rev Neurosci 24: , 1121–1159. |
[9] | Buee L , Bussiere T , Buee-Scherrer V , Delacourte A , Hof PR ((2000) ) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33: , 95–130. |
[10] | Clavaguera F , Akatsu H , Fraser G , Crowther RA , Frank S , Hench J , Probst A , Winkler DT , Reichwald J , Staufenbiel M , Ghetti B , Goedert M , Tolnay M ((2013) ) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110: , 9535–9540. |
[11] | Luna-Munoz J , Chavez-Macias L , Garcia-Sierra F , Mena R ((2007) ) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis 12: , 365–375. |
[12] | Wang JZ , Xia YY , Grundke-Iqbal I , Iqbal K ((2013) ) Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33 Suppl 1: , S123–139. |
[13] | Cotman CW , Poon WW , Rissman RA , Blurton-Jones M ((2005) ) The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 64: , 104–112. |
[14] | Ferreira A , Bigio EH ((2011) ) Calpain-mediated tau cleavage: A mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17: , 676–685. |
[15] | Vana L , Kanaan NM , Ugwu IC , Wuu J , Mufson EJ , Binder LI ((2011) ) Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am J Pathol 179: , 2533–2550. |
[16] | Gamblin TC , Chen F , Zambrano A , Abraha A , Lagalwar S , Guillozet AL , Lu M , Fu Y , Garcia-Sierra F , LaPointe N , Miller R , Berry RW , Binder LI , Cryns VL ((2003) ) Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 100: , 10032–10037. |
[17] | Guillozet-Bongaarts AL , Garcia-Sierra F , Reynolds MR , Horowitz PM , Fu Y , Wang T , Cahill ME , Bigio EH , Berry RW , Binder LI ((2005) ) Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging 26: , 1015–1022. |
[18] | de Calignon A , Fox LM , Pitstick R , Carlson GA , Bacskai BJ , Spires-Jones TL , Hyman BT ((2010) ) Caspase activation precedes and leads to tangles. Nature 464: , 1201–1204. |
[19] | De Duve C , Pressman BC , Gianetto R , Wattiaux R , Appelmans F ((1955) ) Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J 60: , 604–617. |
[20] | Saftig P , Klumperman J ((2009) ) Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat Rev Mol Cell Biol 10: , 623–635. |
[21] | Dolan PJ , Johnson GV ((2010) ) A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem 285: , 21978–21987. |
[22] | Williams A , Jahreiss L , Sarkar S , Saiki S , Menzies FM , Ravikumar B , Rubinsztein DC ((2006) ) Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr Top Dev Biol 76: , 89–101. |
[23] | Ihara Y , Morishima-Kawashima M , Nixon R ((2012) ) The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006361. |
[24] | Myeku N , Clelland CL , Emrani S , Kukushkin NV , Yu WH , Goldberg AL , Duff KE ((2016) ) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 22: , 46–53. |
[25] | Piras A , Collin L , Gruninger F , Graff C , Ronnback A ((2016) ) Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4: , 22. |
[26] | Fischer G , Tradler T , Zarnt T ((1998) ) The mode of action of peptidyl prolyl cis/trans isomerases in vivo: Binding vs. catalysis. FEBS Lett 426: , 17–20. |
[27] | Daneri-Becerra C , Zgajnar NR , Lotufo CM , Ramos Hryb AB , Piwien-Pilipuk G , Galigniana MD ((2019) ) Regulation of FKBP51 and FKBP52 functions by post-translational modifications. Biochem Soc Trans 47: , 1815–1831. |
[28] | Chambraud B , Byrne C , Meduri G , Baulieu EE , Giustiniani J ((2022) ) FKBP52 in neuronal signaling and neurodegenerative diseases: A microtubule story. Int J Mol Sci 23: , 1738. |
[29] | Bose S , Weikl T , Bugl H , Buchner J ((1996) ) Chaperone function of Hsp90-associated proteins. Science 274: , 1715–1717. |
[30] | Steiner JP , Dawson TM , Fotuhi M , Glatt CE , Snowman AM , Cohen N , Snyder SH ((1992) ) High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature 358: , 584–587. |
[31] | Chambraud B , Sardin E , Giustiniani J , Dounane O , Schumacher M , Goedert M , Baulieu EE ((2010) ) A role for FKBP52 in tau protein function. Proc Natl Acad Sci U S A 107: , 2658–2663. |
[32] | Chambraud B , Belabes H , Fontaine-Lenoir V , Fellous A , Baulieu EE ((2007) ) The immunophilin FKBP52 specifically binds to tubulin and prevents microtubule formation. FASEB J 21: , 2787–2797. |
[33] | Kamah A , Cantrelle FX , Huvent I , Giustiniani J , Guillemeau K , Byrne C , Jacquot Y , Landrieu I , Baulieu EE , Smet C , Chambraud B , Lippens G ((2016) ) Isomerization and oligomerization of truncated and mutated tau forms by FKBP52 are independent processes. J Mol Biol 428: , 1080–1090. |
[34] | Giustiniani J , Sineus M , Sardin E , Dounane O , Panchal M , Sazdovitch V , Duyckaerts C , Chambraud B , Baulieu EE ((2012) ) Decrease of the immunophilin FKBP52 accumulation in human brains of Alzheimer’s disease and FTDP-17. J Alzheimers Dis 29: , 471–483. |
[35] | Meduri G , Guillemeau K , Dounane O , Sazdovitch V , Duyckaerts C , Chambraud B , Baulieu EE , Giustiniani J ((2016) ) Caspase-cleaved Tau-D(421) is colocalized with the immunophilin FKBP52 in the autophagy-endolysosomal system of Alzheimer’s disease neurons. Neurobiol Aging 46: , 124–137. |
[36] | Giustiniani J , Chambraud B , Sardin E , Dounane O , Guillemeau K , Nakatani H , Paquet D , Kamah A , Landrieu I , Lippens G , Baulieu EE , Tawk M ((2014) ) Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proc Natl Acad Sci U S A 111: , 4584–4589. |
[37] | Giustiniani J , Guillemeau K , Dounane O , Sardin E , Huvent I , Schmitt A , Hamdane M , Buee L , Landrieu I , Lippens G , Baulieu EE , Chambraud B ((2015) ) The FK506-binding protein FKBP52 in vitro induces aggregation of truncated tau forms with prion-like behavior. FASEB J 29: , 3171–3181. |
[38] | Chambraud B , Daguinot C , Guillemeau K , Genet M , Dounane O , Meduri G , Pous C , Baulieu EE , Giustiniani J ((2021) ) Decrease of neuronal FKBP4/FKBP52 modulates perinuclear lysosomal positioning and MAPT/tau behavior during MAPT/tau-induced proteotoxic stress. Autophagy 17: , 3491–3510. |
[39] | Criado-Marrero M , Gebru NT , Gould LA , Blazier DM , Vidal-Aguiar Y , Smith TM , Abdelmaboud SS , Shelton LB , Wang X , Dahrendorff J , Beaulieu-Abdelahad D , Dickey CA , Blair LJ ((2021) ) FKBP52 overexpression accelerates hippocampal-dependent memory impairments in a tau transgenic mouse model. NPJ Aging Mech Dis 7: , 9. |
[40] | Allen B , Ingram E , Takao M , Smith MJ , Jakes R , Virdee K , Yoshida H , Holzer M , Craxton M , Emson PC , Atzori C , Migheli A , Crowther RA , Ghetti B , Spillantini MG , Goedert M ((2002) ) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22: , 9340–9351. |
[41] | Gutala RV , Reddy PH ((2004) ) The use of real-time PCR analysis in a gene expression study of Alzheimer’s disease post-mortem brains. J Neurosci Methods 132: , 101–107. |
[42] | Williams DR , Lees AJ ((2009) ) Progressive supranuclear palsy: Clinicopathological concepts and diagnostic challenges. Lancet Neurol 8: , 270–279. |
[43] | Dickson DW , Ahmed Z , Algom AA , Tsuboi Y , Josephs KA ((2010) ) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23: , 394–400. |
[44] | Zhang Q , Zhang X , Sun A ((2009) ) Truncated tau at D421 is associated with neurodegeneration and tangle formation in the brain of Alzheimer transgenic models. Acta Neuropathol 117: , 687–697. |
[45] | Guillozet-Bongaarts AL , Cahill ME , Cryns VL , Reynolds MR , Berry RW , Binder LI ((2006) ) Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: In vitro evidence and implications for tangle formation in vivo. J Neurochem 97: , 1005–1014. |
[46] | Li HL , Wang HH , Liu SJ , Deng YQ , Zhang YJ , Tian Q , Wang XC , Chen XQ , Yang Y , Zhang JY , Wang Q , Xu H , Liao FF , Wang JZ ((2007) ) Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc Natl Acad Sci U S A 104: , 3591–3596. |
[47] | Voss K , Koren J , 3rd, Dickey CA ((2011) ) The earliest tau dysfunction in Alzheimer’s disease? Tau phosphorylated at s422 as a toxic seed. Am J Pathol 179: , 2148–2151. |
[48] | Rissman RA , Poon WW , Blurton-Jones M , Oddo S , Torp R , Vitek MP , LaFerla FM , Rohn TT , Cotman CW ((2004) ) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114: , 121–130. |
[49] | Perez MJ , Vergara-Pulgar K , Jara C , Cabezas-Opazo F , Quintanilla RA ((2018) ) Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol Neurobiol 55: , 1004–1018. |
[50] | Loon A , Zamudio F , Sanneh A , Brown B , Smeltzer S , Brownlow ML , Quadri Z , Peters M , Weeber E , Nash K , Lee DC , Gordon MN , Morgan D , Selenica MB ((2022) ) Accumulation of C-terminal cleaved tau is distinctly associated with cognitive deficits, synaptic plasticity impairment, and neurodegeneration in aged mice. Geroscience 44: , 173–194. |
[51] | Troquier L , Caillierez R , Burnouf S , Fernandez-Gomez FJ , Grosjean ME , Zommer N , Sergeant N , Schraen-Maschke S , Blum D , Buee L ((2012) ) Targeting phospho-Ser422 by active tau Immunotherapy in the THYTau22 mouse model: A suitable therapeutic approach. Curr Alzheimer Res 9: , 397–405. |
[52] | Collin L , Bohrmann B , Gopfert U , Oroszlan-Szovik K , Ozmen L , Gruninger F ((2014) ) Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 137: , 2834–2846. |
[53] | Tiernan CT , Combs B , Cox K , Morfini G , Brady ST , Counts SE , Kanaan NM ((2016) ) Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp Neurol 283: , 318–329. |
[54] | Chi H , Sun L , Shiu RH , Han R , Hsieh CP , Wei TM , Lo CC , Chang HY , Sang TK ((2020) ) Cleavage of human tau at Asp421 inhibits hyperphosphorylated tau induced pathology in a Drosophila model. Sci Rep 10: , 13482. |
[55] | Sandhu P , Naeem MM , Lu C , Kumarathasan P , Gomes J , Basak A ((2017) ) Ser(422) phosphorylation blocks human tau cleavage by caspase-3: Biochemical implications to Alzheimer’s Disease. Bioorg Med Chem Lett 27: , 642–652. |
[56] | Wang Y , Martinez-Vicente M , Kruger U , Kaushik S , Wong E , Mandelkow EM , Cuervo AM , Mandelkow E ((2009) ) Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum Mol Genet 18: , 4153–4170. |
[57] | Zhou Y , Shi J , Chu D , Hu W , Guan Z , Gong CX , Iqbal K , Liu F ((2018) ) Relevance of phosphorylation and truncation of tau to the etiopathogenesis of Alzheimer’s disease. Front Aging Neurosci 10: , 27. |
[58] | Tolkovsky AM , Spillantini MG ((2021) ) Tau aggregation and its relation to selected forms of neuronal cell death. Essays Biochem 65: , 847–857. |
[59] | Asadzadeh J , Ruchti E , Jiao W , Limoni G , MacLachlan C , Small SA , Knott G , Santa-Maria I , McCabe BD ((2022) ) Retromer deficiency in tauopathy models enhances the truncation and toxicity of tau. Nat Commun 13: , 5049. |
[60] | Zhao Y , Tseng IC , Heyser CJ , Rockenstein E , Mante M , Adame A , Zheng Q , Huang T , Wang X , Arslan PE , Chakrabarty P , Wu C , Bu G , Mobley WC , Zhang YW , St George-Hyslop P , Masliah E , Fraser P , Xu H ((2015) ) Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 87: , 963–975. |
[61] | Martinez-Maldonado A , Ontiveros-Torres MA , Harrington CR , Montiel-Sosa JF , Prandiz RG , Bocanegra-Lopez P , Sorsby-Vargas AM , Bravo-Munoz M , Floran-Garduno B , Villanueva-Fierro I , Perry G , Garces-Ramirez L , de la Cruz F , Martinez-Robles S , Pacheco-Herrero M , Luna-Munoz J ((2021) ) Molecular processing of tau protein in progressive supranuclear palsy: Neuronal and glial degeneration. J Alzheimers Dis 79: , 1517–1531. |
[62] | Lu PJ , Wulf G , Zhou XZ , Davies P , Lu KP ((1999) ) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399: , 784–788. |
[63] | Liou YC , Sun A , Ryo A , Zhou XZ , Yu ZX , Huang HK , Uchida T , Bronson R , Bing G , Li X , Hunter T , Lu KP ((2003) ) Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424: , 556–561. |
[64] | Lee TH , Pastorino L , Lu KP ((2011) ) Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med 13: , e21. |
[65] | Moloney CM , Lowe VJ , Murray ME ((2021) ) Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimers Dement 17: , 1554–1574. |
[66] | McCluskey SP , Plisson C , Rabiner EA , Howes O ((2020) ) Advances in CNS PET: The state-of-the-art for new imaging targets for pathophysiology and drug development. Eur J Nucl Med Mol Imaging 47: , 451–489. |
[67] | Pascoal TA , Therriault J , Benedet AL , Savard M , Lussier FZ , Chamoun M , Tissot C , Qureshi MNI , Kang MS , Mathotaarachchi S , Stevenson J , Hopewell R , Massarweh G , Soucy JP , Gauthier S , Rosa-Neto P ((2020) ) 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 143: , 2818–2830. |
[68] | Biel D , Brendel M , Rubinski A , Buerger K , Janowitz D , Dichgans M , Franzmeier N , Alzheimer’s Disease Neuroimaging I ((2021) ) Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res Ther 13: , 137. |


