
Neuroinflammation: A Common Pathway in Alzheimer’s Disease and Epilepsy
Abstract
Background:
Neuroinflammation is an innate immunological response of the central nervous system that may be induced by a brain insult and chronic neurodegenerative conditions. Recent research has shown that neuroinflammation may contribute to the initiation of Alzheimer’s disease (AD) pathogenesis and associated epileptogenesis.
Objective:
This systematic review aimed to investigate the available literature on the shared molecular mechanisms of neuroinflammation in AD and epilepsy.
Methods:
The search included in this systematic review was obtained from 5 established databases. A total of 2,760 articles were screened according to inclusion criteria. Articles related to the modulation of the inflammatory biomarkers commonly associated with the progression of AD and epilepsy in all populations were included in this review.
Results:
Only 7 articles met these criteria and were chosen for further analysis. Selected studies include both in vitro and in vivo research conducted on rodents. Several neuroinflammatory biomarkers were reported to be involved in the cross-talk between AD and epilepsy.
Conclusion:
Neuroinflammation was directly associated with the advancement of AD and epilepsy in populations compared to those with either AD or epilepsy. However, more studies focusing on common inflammatory biomarkers are required to develop standardized monitoring guidelines to prevent the manifestation of epilepsy and delay the progression of AD in patients.
INTRODUCTION
Amyloid-β (Aβ) plaques, the key hallmark of Alzheimer’s disease (AD), were first reported in epileptic patients long before the acknowledgement of AD by Alois Alzheimer [1]. The relative risk of unprovoked seizure surges significantly up to 10-fold greater occurrence in late-onset AD in comparison to the general population of the same age [2, 3], and an 80% of seizure recurrence risk after a first seizure episode was reported in an elderly AD population [3]. The linkage between AD and epilepsy pathologies has since then remained obscure [2]. To date, pharmaceutical treatments available for AD and epilepsy are mainly targeting symptomatic control for the disease/disorder instead of curative treatment [4].
The most common form of dementia is AD, which is a neurodegenerative disease characterized by memory loss and progressive neurocognitive impairment [5]. The widely recognized hypothesis on the predominant cause of AD is due to the aberrance in the processing and polymerization of regularly soluble proteins. As a result, a misfolded protein with modified conformation is produced which could lead to abnormal or loss of neuronal functions [6, 7]. The key pathologies for AD are the presence of amyloid-β (Aβ) and hyperphosphorylated tau [4, 5, 8]. In brief, amyloid pathogenesis is initiated with altered cleavage of an integral protein on the plasma membrane, amyloid-β protein precursor (AβPP), by β-secretases (BACE1) and γ-secretases to produce insoluble Aβ fibrils [9, 10]. Consecutively, Aβ oligomerizes, diffuses into the synaptic cleft and disrupts synaptic signaling. The polymerization of Aβ oligomers into insoluble amyloid fibrils leads to aggregation into plaques, leading to kinase activation, hyperphosphorylation of microtubule-associated tau protein, and the formation of intracellular neurofibrillary tangles (NFTs). Accordingly, the clinical characteristics of AD include the presence of extracellular aggregates of Aβ and NFTs constituted of hyperphosphorylated tau protein in the cortical and limbic regions of the brain [5]. On the other hand, epilepsy is one of the most prevailing neurologic conditions with an occurrence of one in every 2000 population [11]. Epilepsy is a condition defined as at least two unprovoked seizures developing more than 24 hours apart or one unprovoked seizure with a 60% probability of developing further seizures similar to the general recurrence risk after two unprovoked seizures over the next 10 years [12]. The manifestation of seizure is due to the disproportionate, hypersynchronous firing of neurons, which disrupts the excitatory and inhibitory imbalance in the cerebral cortex of the brain.
Recent studies have shown that non-convulsive epileptiform activity appears to be an under-reported comorbidity of AD [13]. Besides that, shared pathological similarities such as hippocampal atrophy, neuronal death, gliosis, neuroinflammation, and neuropsychiatric and cognitive comorbidities were also observed in AD and epilepsy patients [14]. Following this, several experimental studies have demonstrated a lowered threshold for seizures in AD models, suggesting neuronal hyperexcitability may contribute to the behavior deficit in AD [2]. Although the exact mechanisms of associating seizures with the onset and trajectory of AD pathogenesis remain unclear, numerous biomarkers and mechanisms have been proposed. Aβ and tau protein, GABAergic and glutamatergic alterations, the role of the noradrenergic nucleus Locus Coeruleus, and neuroinflammation are possible pathological mechanisms to underlie the increased risk of seizures in AD [15–17]. Among the proposed mechanisms, increasing evidence has found neuroinflammation was presented in both AD [2, 18] and epilepsy [16, 17, 19], potentially linking the pathogenesis between these two conditions.
Neuroinflammation is triggered when there is a brain insult, such as traumatic brain injury, infections, or status epilepticus (SE) [20]. Neuroinflammation is described by the initiation of the central nervous system (CNS) immune response, mediated by astrocytes, microglia, endothelial cells of the blood-brain barrier (BBB), neurons, and peripheral immune cells extravasation in the CNS parenchyma [20]. For decades, AD has been tightly linked to neuroinflammation [21–23]. Aβ interacts directly with receptors on microglia and astrocytes including receptors for advanced glycation end products (RAGEs), Toll-like receptors (TLR), chemokine-like receptor 1, and the α6β1 integrin [24, 25], inducing an acute inflammatory response, and triggering the complement and inflammatory enzymes [2]. Consequently, the microglia and astrocytes are activated, releasing pro-inflammatory chemokines and cytokines [26]. Extracellular and intracellular Aβ and tangles produce intense toxicity that causes synaptic damage [2], increasing oxidative stress [27], which in turn causes the infiltration of microglial surrounding the plaques regions [24, 28]. Microglia can also activate T-mediated neurotoxic immune response as antigen-presenting cells and exacerbate neurodegeneration [28]. Persistent secretion of pro-inflammatory cytokines from reactive microglial may, in return, further promote amyloid deposition [29]. Thus, the unavailing inflammatory escalation response towards Aβ proteins predating the onset of cognitive declination in AD has been hypothesized [26].
In some instances, proinflammatory conditions such as autoimmune disorder, fever as well as the underlying neuroinflammation in AD, is a risk factor for seizures [2]. According to prior evidence from in vitro and in vivo research, a bidirectional association between epilepsy and aberrant inflammation was presented [30]. Seizures that were triggered by neuroinflammation may consecutively promote or exacerbate neuronal hyperexcitability and facilitate further seizure generation. Cytokines such as Interleukin 6 (IL-6), IL-1β, and tumor necrosis factor-α (TNF-α) along with other cytokines were implicated in the development of seizures due to the disproportion in inhibitory-excitatory balance [16, 17]. Overexpression of TNF- α and IL-6 in transgenic mice was shown to have lowered seizure threshold and exhibited spontaneous seizures [31, 32] whereas intrahippocampal administration of IL-1β into the rat prolonged seizurogenic-induced seizures [33]. Likewise, depending on the phenotype of microglia activated, microglia have also been hypothesized to be pro-epileptogenic particularly if inhibitory synapses were affected in the complement-dependent synaptic engulfment [34].
The aforementioned findings have supported the role of neuroinflammation in promoting seizures and cognitive decline seen in AD. Both AD and epilepsy precipitate aberrant neuroinflammation; in which neuroinflammation is seen as a risk determinant for seizures and may exacerbate AD. However, a concise depiction of the responsible inflammatory biomarkers and the subsequent pathology that correlates with epilepsy and AD have not been summarized previously. Thus, this systematic review aimed to critically evaluate the currently available literature investigating the association of neuroinflammation in both AD and epilepsy, which could potentially serve as a guide in exploring alternative therapeutic strategies.
MATERIALS AND METHODS
Search strategy and studies identification
A literature search was conducted using databases namely PubMed, Ovid Medline, Scopus, and EMBASE. The search was limited to publications from the last 5 years, January 2018 to August 2022 for the retrieval of recent, relevant, and updated studies. The search terms “Neuroinflammation”, “Alzheimer’s Disease”, “Epilepsy”, “Seizure”, and “Cognitive Decline” were used to extract relevant articles from the databases. A title, abstract, and keyword search were performed using the search terms. The Boolean operator “AND” and “OR” was used to link the terms together on all databases. Articles were first screened through their titles and abstracts before proceeding with the full-text screening of relevant articles. Additionally, electronic searches were restricted to the availability of abstracts and studies published in the English language.
Study selection
The study selection for this systematic review is based on Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) protocol. Studies eligible for inclusion include peer-reviewed original research articles in in vitro and in vivo research performed in all populations with AD or epilepsy/epileptiform discharge and the availability of the full-text article in English. All the included studies evaluated the association of neuroinflammation with the conditions’ progressions directly and indirectly. Regardless of the methods of assessments utilized by the studies, the main outcome measures were the modulated inflammatory biomarkers associated with the progression of AD and epilepsy. Non-original research articles such as conference abstracts, case reports, reviews, book chapters and editorials, as well as articles that did not investigate neuroinflammation in relation to AD and seizure within a study were excluded from this systematic review. The whole methodological process was independently performed by two researchers, who reached a consensus before the selection of the final number of articles for critical evaluation.
Data extraction, quality assessment, and data analysis
Key study data were extracted from each study. These include first author, study model, specimens used, measure outcome, and biomolecules studied. Systematic Review Centre for Laboratory Animal Experimentation Risk of Bias (SYRCLE RoB) tool was used to evaluate the quality and risk of bias of the animal interventional studies that were included in this review [35]. “Yes” indicates a low risk of bias and ‘No’ indicates a high risk of bias in this quality item whereas ‘Unclear” indicates insufficient data was provided for evaluating the degree of bias. The literature search, selection and quality analysis of the selected articles were performed by two independent researchers. Meta-analysis was not conducted in this review due to variability in statistical analysis utilized, methodologic diversity (e.g., study design, including procedures, measured outcome), and heterogeneity (e.g., type of specimens) of the selected studies.
RESULTS
Study characteristics
The initial systematic search and reference tracking yielded 2,760 articles (Fig. 1). Of these initial 2,760 articles, 288 duplicates were removed and 2,472 articles were included after screening of the abstracts. Most of these 2,440 articles were either conference abstracts, or review papers, or do not fulfil the study inclusion criteria. Hence, a total of 32 full-text articles were screened and 25 articles that do not measure the association of either AD, epilepsy, and seizure together with neuroinflammation were excluded. A total of 7 articles were finally used for the analyses and summary. The characteristics, findings, and quality of articles based on SYRCLE’s risk of bias checklist of each study (Supplementary Table 1) were summarized and tabulated in Tables 1 and 2, based on their experiment design. Although most of the included study has a high risk of selection bias, the overall quality remained unbiased. Out of the included studies, there were only one research of the included articles was conducted in in vitro whereas in vivo research was performed in the other six studies. The predominant model used in the included literature was AD model (71%, n = 5), performed in transgenic mice.
Fig. 1
A systematic PRISMA flowchart for articles identification
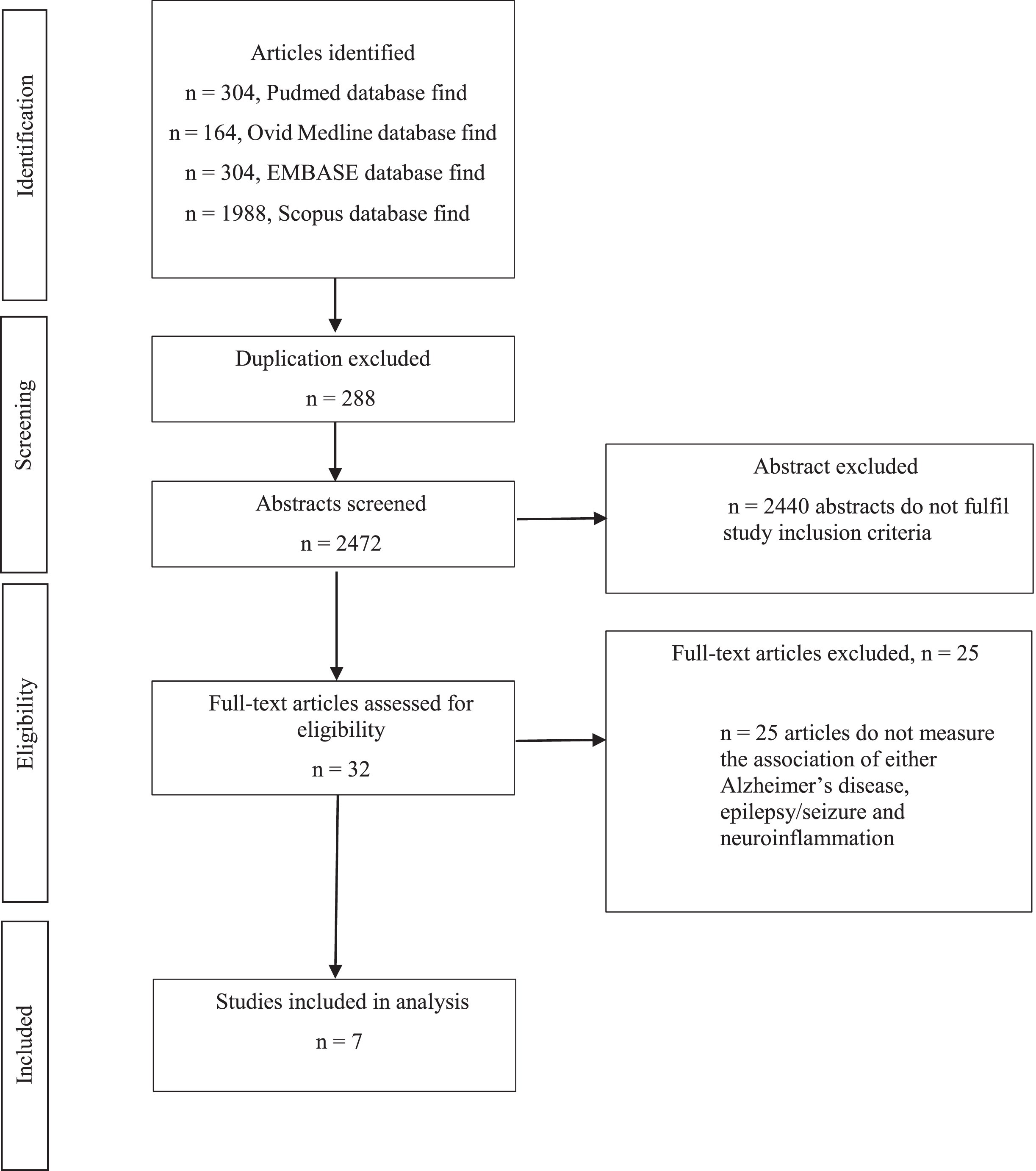
Table 1
Ex vivo research studies
| First author (year) | Specimens | Inflammatory biomolecules/markers | Pathological outcomes | SYRCLE’s risk of bias assessmenta |
| Aldabbagh (2022) [36] | APP NL-F/NL-F knock-in male mice (12–16 months old) hippocampal (CA1 and DG) tissue | Increased GFAP in human and mice | Seizurogenic: Bicucilline | Unbiased |
| Increased baseline spontaneous synaptic excitation, Decreased phasic spontaneous inhibitory events, and Increased background tonic inhibition. | ||||
| Increased GABA and glutamate level, Increased amplitude and frequency of sEPSPs |
AD, Alzheimer’s disease; CA, cornu ammonis; DG, dentate gyrus; GFAP, glial fibrillary acidic protein; sEPSPs, spontaneous excitatory postsynaptic potentials; a Detailed items presented in the Supplementary Material.
Table 2
In vivo research studies
| First author (Year) | Animal type and disease model | Brain region | Progression | SYRCLE’s risk of bias assessmenta | |||
| Neuropathological | Electrophysiological | Neurodegeneration/neurogenesis | Behavior | ||||
| Canet (2022) [37] | MTLE model (C57BL/6J male mice (8–10 weeks old) | Hippocampus | Epileptogenesis | Seizurogenic: Kainic acid | No information available | Decreased well-being, short and long-term memory in AD and post-SE mice | Unbiased |
| Hallmarks of AD: | |||||||
| Increased AT100, PHF-1, CP13, Cdk5 activity, and overexpression of APP, C99, BACE1 | |||||||
| Decreased ADAM10 level | |||||||
| Inflammation: Increased GFAP and Iba1 level | |||||||
| Spontaneous seizures | |||||||
| Hallmarks of AD: | |||||||
| Increased AT100, PHF- 1, CP13, GSK3β activity, and overexpression of APP and C99 | |||||||
| BACE1 and PS1 were unchanged | |||||||
| Inflammation: Increased GFAP and Iba1 level | |||||||
| SE | |||||||
| Hallmarks of AD: | |||||||
| Increased transiently in PHF1, CP13, APP, C99, ADAM10, PS1, and long term overexpression in AT100, BACE1 and GSK-3β activity | |||||||
| Inflammation: Increased GFAP, Iba1, IL-1β, TNF-α, CCL-12 transiently and corticosterone plasma levels | |||||||
| J20 model (J20 (hAPPSw/Ind) male mice (9 months old)) | Hippocampus | Hallmarks of AD: | |||||
| Increased PHF1, CP13, BACE1, GSK-3β, and CDK5 activity | |||||||
| AT100 level unchanged | |||||||
| Inflammation: Increased GFAP and Iba1 level | |||||||
| Gschwind (2018) [38] | IHK model (Arctic Aβ male mice (12–15 weeks old)) | Temporal lobe (DG, CA1 and CA3), dorsal hippocampus, stradium lacunosum, strati pyramidale and radium of CA3 | Hallmarks of AD: Increased soluble Aβ in latent phase Inflammation: | Seizurogenic: Kainic acid/bicuculline | Decreased pyramidal cells in CA1 and CA3 | Increased seizures susceptibility, severity and frequency, and mortality | Unbiased |
| Increased CD68 expression in Hilus, CA1 and CA3 stratum radiatum | Decreased synaptic transmission strength in CA1 | Decreased hilar mossy cells in the DG | |||||
| Decreased IL-10 expression | Decreased cluster of activity (sIPSCs) with a higher number of events during baseline in AD mice | Decreased hippocampal expression in NPY | |||||
| No significant changes in IL-1 β and TNF-α | Increased NPY expression moderately in the hilus and CA3c during epileptogenesis | ||||||
| Increased Trem2 in latent phase of AD | Decreased neurogenesis in AD mice but no changes in the epileptogenesis-associated changes neurogenesis | ||||||
| Petrache (2019) [39] | APP knock-in model (APP NL-F/NL-F knock-in male mice (1-2 months old, 4–6 months old, and 10–18 months old) | LEC, cortex, cerebellum, CA1 and neocrotex | Hallmarks of AD: Increased Aβ in LEC in latent phase | Decreased canonical Wnt signaling activity progressively over time | Decreased pyramidal cell density in LEC and CA1 in the latent phase | No information available | Unbiased |
| Inflammation: Increased CD68 in latent phase | Impaired spont-aneous excitation and inhibition | ||||||
| Increased hyperexcitability state as AD progress in CA1 and neocortical region | |||||||
| No significant changes of GFAP in the LEC | |||||||
| Putra (2020) [40] | PTZ-induced model (Tau KO male and female mice (12 weeks old)) | Hippocampus (CA1, CA3, DG), ENT | Hallmarks of AD: Increased hyper-phosphorylated tau in the hippocampus during the first 24 hours | Seizurogenic: PTZ | Decreased neurodegeneration at 24 hours in hippocampus and ENT | Decreased convulsive seizures and cumulative seizure severity scores | Unbiased |
| Inflammation: Decreased IBA1 in CA3 and DG and GFAP in DG at 24 hours | Increased latency to convulsive seizure | ||||||
| Schultz (2018) [41] | AAV-hTau model (Tg4510 and Tau KO male and female mice (17 weeks old)) | Subiculum, DG | Hallmarks of AD: Increased neuronal activity | No information available | Increased neuronal activity Increased hippocampal neuronal loss severity | No information available | Unbiased |
| Increased⟶hTau and hippocampal tran-synaptic spread. | |||||||
| Hyperphosphorylated hTau in EC and DG | |||||||
| Inflammation: Increased IBA1 in hippocampal | |||||||
| Tzeng (2018) [42] | APP model (APP/PS1 mice (10–12 weeks old) | Hippocampal CA1, DG | Hallmarks of AD: Presence of amyloid deposition in 9-month-old AD mice | Seizurogenic: PTX | Increased NYP in DG of KO mice. | Increased seizure susceptibility, severity score and mortality in KO IL-18 | Unbiased |
| Increased excitatory synaptic proteins expression in KO IL-18 and basal excitatory synaptic transmission. | Altered genes related to seizure regulation | ||||||
| Inflammation: Increased IL1β levels in WT and KO of IL-18 | Increased hippocampal spine density in KO IL-18 |
AD, Alzheimer’s disease; MTLE, Mesial temporal lobe epilepsy; GFAP, Glial fibrillary acidic protein; Iba1, Ionized calcium-binding adaptor molecule 1; IL, Interleukin; TNF-α, tumor necrosis factor-alpha; CCL 12, Chemokine (C-C motif) ligand 12; PHF, paired helical filaments; Cdk5, Cyclin-dependent kinase 5; APP, amyloid precursor protein; BACE1, Beta-secretase 1; ADAM10, A Disintegrin and Metalloproteinase 10; PS1, Presenilin-1; GSK-3β, Glycogen synthase kinase-3 beta; SE, Seizure epilepticus; IHK, Intrahippocampal injection of kainic acid; CA, Cornu Ammonis; DG, Dentate gyrus; CD68, Cluster of differentiation 68; TREM2, Triggering receptor expressed on myeloid cells 2; sIPSPs, Spontaneous inhibitory postsynaptic potentials; NPY, Neuropeptide Y; LEC, Lateral entorhinal cortex; Wnt, Canonical Wingless/integrated; PTZ, Pentylenetetrazole; ENT, Entorhinal cortex; AAV, Adeno-associated viral vector; hTau, Human Tau; WT, Wildtype; KO, Knock-out; PTX, Picrotoxin; a Detailed items presented in Supplementary Table 1.
Role of neuroinflammation in AD-associated epilepsy
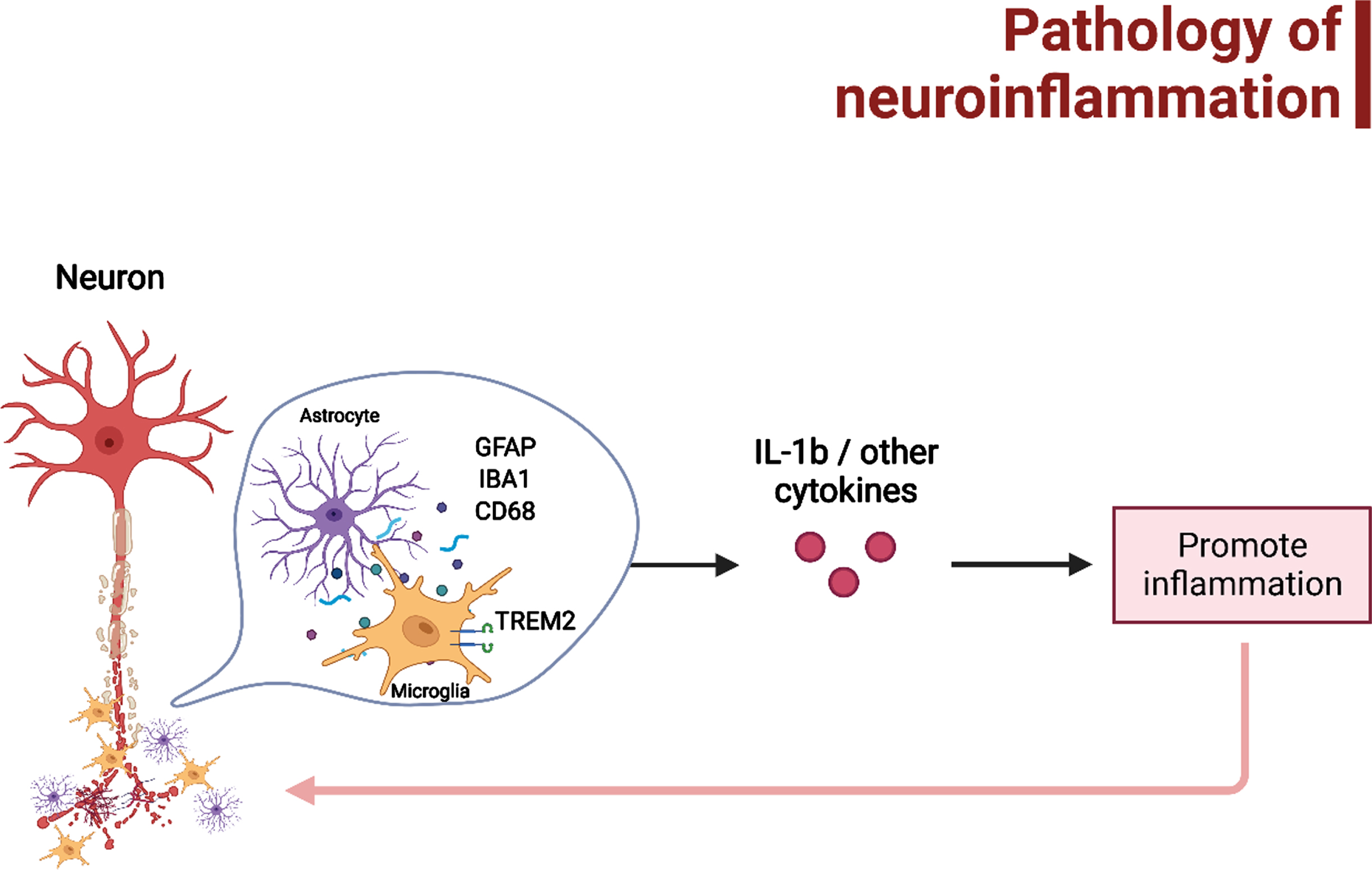
The direct and indirect correlation between AD and seizure progression in association with neuroinflammation were extracted from the findings of the included studies. Figure 2 depicted the pathology of neuroinflammation in AD and AD-associated epileptogenesis in relation to microgliosis and astrogliosis and their downstream mechanisms. A significant reduction in the level of inflammatory markers IL-1β in mice was detected after an episode of SE in transgenic mice without AD; however, changes in the cytokines and chemokines such as TNF-α, IL-6, C-C motif chemokine ligand 2 (CCL2), and C-C motif chemokine ligand 12 (CCL12) were not significant in comparison in mice with and without AD [37]. On the contrary, one other study has found an increase in IL-1β levels in AD transgenic mice as AD progressed among the other cytokines (IL-1α, MIP2, RANTES, and KC) [42]. Moreover, overexpression in IL-18 cytokine has demonstrated an increased risk of seizures in AD, so as under-expression, thus a balance of inflammatory cytokine is critical in suppressing the development of seizures in AD. Although no significant differences were detected in IL-1β and TNF-α in both wildtype and transgenic mice with AD, the study investigating AD pathology and neuroinflammation demonstrated a decreasing trend of expression in IL-10 in both wildtype and transgenic mice with AD [38]. Other neuroinflammation makers include glial fibrillary acidic protein (GFAP), cluster of differentiation 68 (CD68) immunoreactivity, and ionized calcium-binding adaptor molecule 1 (IBA1). The increase in the aforementioned neuroinflammatory markers indicates higher activation of glial cells, leading to heighten inflammation response. Two studies using both AD and epileptic mice models displayed a raise in GFAP levels [36, 37]. A study investigating signaling dysregulation in canonical wingless/integrated (Wnt) associated with AD pathology and neuroinflammation in knock-in transgenic AD mouse model showed an increase of CD 68 during the latent phase of AD [39], similarly with another study utilizing Arctic Aβ mice AD model has also demonstrated an increase CD 68 staining and expression of triggering receptor expressed on myeloid cells 2 (TREM2), but not inflammatory cytokines during a seizure [38]. Correspondingly, an increase of microgliosis was seen collectively in the mice after seizurogenic administration in an epileptic model, however, a significantly lesser number of microglial and astroglial were observed in tau KO mice [40]. An AD mice model induced with neuronal activity showed to have increased IBA1 intensity which was associated with the worsening of AD hallmark [41].
Fig. 2
The possible pathology of neuroinflammation that may lead to AD and/or AD-associated epileptogenesis, which have been mainly related to microgliosis and astrogliosis. Figure was created in BioRender.com.
DISCUSSION
The evidence from the rodent studies has suggested a possible relation of neuroinflammation in the pathogenesis of AD and AD-associated epilepsy. During neuroinflammation, glial activation produces functional and morphological changes within the cells that affect the neural-glial and glial-glial interactions [43]. Such alterations may lead to synaptic communication impairment, imbalance in neurotransmitter homeostasis, and potentially axonal degeneration and neuronal death [44]. The neurodegeneration process in AD is defined by synapse dysfunction and neuronal loss [8]. Recent findings have supported the notion on the abnormal progressive accumulation of Aβ oligomers is related to synaptic pathology and impaired neurogenesis in AD. Aβ oligomers might cause a cascade of events that triggers hyperexcitability and neuroinflammation that promotes neurotoxic effects, leading to a pernicious cycle of the neurodegenerative process [5].
In response to brain insult, astrocytes become activated [45]. Astrocytic activation happens as a result of hypertrophy and upregulation of intermediate filaments, such as glial fibrillary acidic protein (GFAP). Various cytokines are shown to involve either during the initiation or modulation of reactive astrogliosis, including IL-1β, TNF-α, and transforming growth factor beta-1β (TGF-1β), that are expressed on astrocytes and their interaction stimulates an astrocytic reactive response. Astrocyte-derived cytokines such as IL-1β and TNF-α are involved in the promotion of neurotoxicity meanwhile TGF-1β is believed to be neuroprotective. On the other hand, approximately a quarter of the cells within the brain are microglia, that actively screen and modulate the CNS environment to maintain homeostasis and physiological requirements, as well as respond to pathological affairs via brain immune cells for innate immune response coordination [46]. Microglial as pathological sensors of brain insult will be activated immediately and undergo critical morphological changes. In the initial phase, activation of the microglial is deemed as neuroprotective by migrating to the site of the insult and stimulating inflammatory response for healing. However, altered morphology accompanied by more reactive or activated microglia, in conjunction with increased activated microglia markers and proinflammatory cytokine levels (for example, IL-1β and TNF-α) disrupts brain function and neuronal dysfunction [47, 48].
On the other hand, a decrease in tau and neuroinflammation has been strongly associated with lessening seizure severity, improved cognitive functions, and mortality in many epilepsy models [38, 40, 49]. The range of cognitive and memory impairment in AD is similar to epilepsy, implicating a possible contribution of seizures in numerous cognitive and behavioral aspects of AD-related alterations [50]. Evidence from several studies has shown patients with AD suffer from acquiring and retaining memory effectively [51], which is consistent with the neuropathological and imaging findings that showed the vulnerability of the ENT and hippocampus in AD [52, 53], Correspondingly, seizures that affect the hippocampus, especially in temporal lobe epilepsy (TLE), have cumulative impacts on cognitive functions after recurrent seizures [50]. Circuit remodeling and alteration in the circuitry and molecular level of the hippocampus are possible mechanisms causing cognitive impairment in epilepsy and AD.
Applicability of targeting neuroinflammation as therapeutic strategy in AD and epilepsy
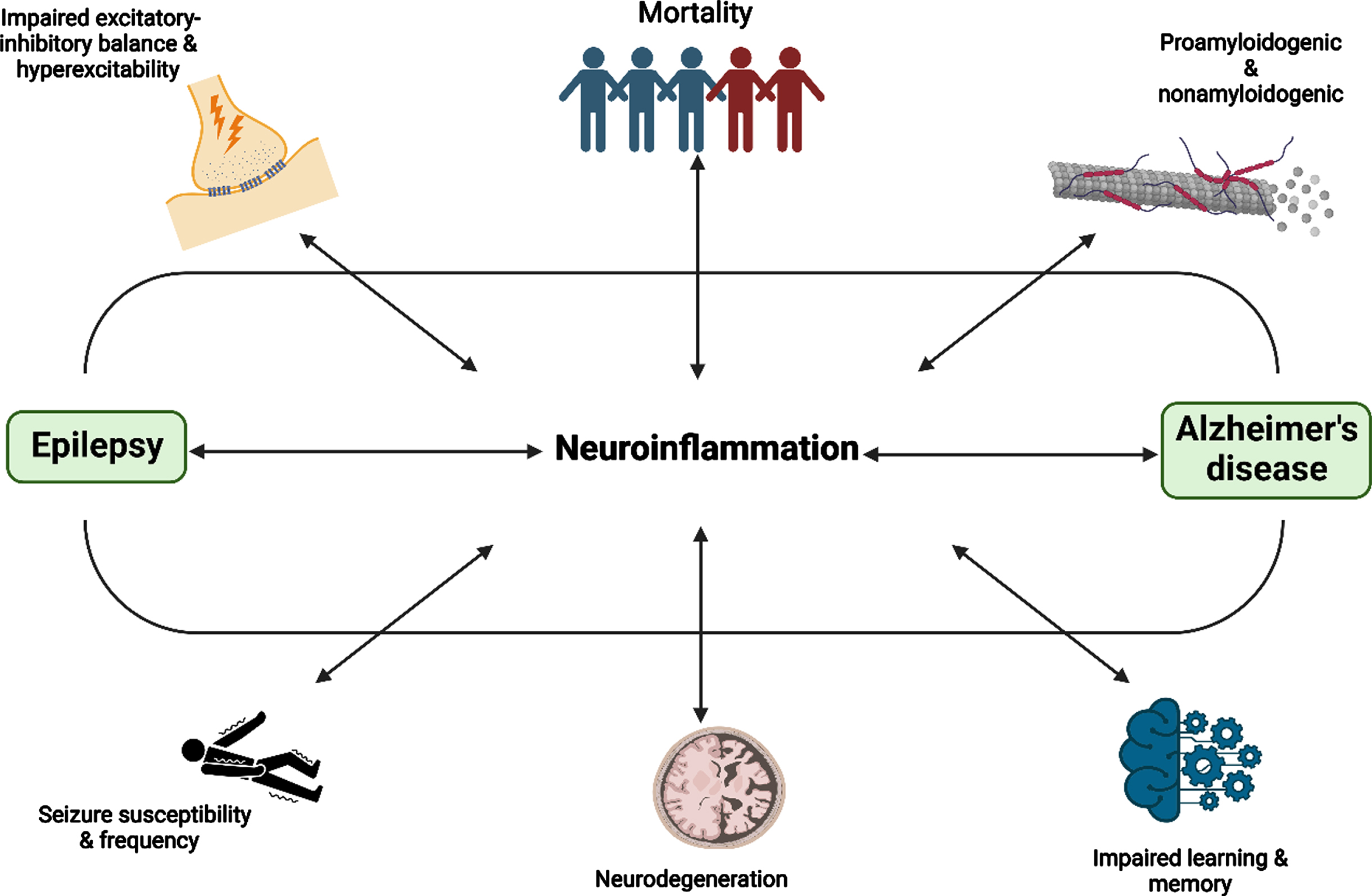
Recent literature has shown the presence of neuroinflammation in AD long before the deposition of amyloid plaques, which is also seen in some types of epilepsies, including TLE and SE [54, 55]. Microglial, astrocytes, and oligodendrocytes contribute to the regulation of neuroinflammation [56]. In AD, Aβ activates the glial and causes the release of several proinflammatory cytokines, leading to the manifestation of generalized neuroinflammation [2]. Consequently, this process would promote neurotoxic effects that may drive a vicious cycle of the neurodegenerative process due to heighten neuronal excitability. In both conditions, astrogliosis and microgliosis modify the glutamate-glutamine cycle [55]. The proinflammatory cytokines such as IL-6, IL-1β, and TNF-α that are released from microglial and astrocytes play a role in modulating the release of glutamate and altering its post-synaptic uptake, along with the signaling reduction of GABA [2, 55]. However, as the available literature that studied the stringent association of neuroinflammation on both AD and epilepsy is extremely limited, the effect of neuroinflammation represented in this systematic review may not reflect the true implication. In addition, the increase in neuronal hyperexcitability has also been postulated to increase the production of the Aβ via β- and γ-secretases stimulation, and promote the pathological expression of AD, causing a higher extent of neurodegeneration and cognitive deficit [54]. Furthermore, disruption in the BBB that happens in both AD and epilepsy was hypothesized as a consequence of the proinflammatory response [57]. The surge in toxic materials in the CNS due to the impaired BBB integrity that favors the perpetuation of a proinflammatory environment would further activate the microglia and proinflammatory cytokine release, thus, imbalance the excitatory-inhibitory system and pro-excitatory synaptic dysregulation [58]. As a result, this could potentially increase the occurrence of interictal epileptiform activity and also epileptic seizures [57]. The association and consequences of neuroinflammation in AD and epilepsy was shown in Fig. 3. As discussed, the aforementioned factors may depict a better correlation in the advancement of AD and epilepsy, but it is also important to acknowledge that the development of both conditions as one other comorbidity is an endless process and neuroinflammation is part of that process as an implication, and potentially a causal relationship.
Fig. 3
The association and consequences of neuroinflammation in AD and epilepsy. Figure was created in BioRender.com
More and more researchers are focusing on the effort in diminishing prolonged neuroinflammation in AD and epilepsy. In preclinical studies, therapeutic targets of neuroinflammation involving antibodies against inflammatory cytokine (TNF-α) and receptors (TREM2 and CD33) expressed in the microglia have shown favorable results in controlling neuroinflammation, and subsequently the progression of AD [18]. Meanwhile, inhibitors of TLR3 or TLR4 expressed by microglia [19] and anti-high motility group box-1 (HMGB-1) [59] in preclinical studies have also demonstrated a significant reduction in epileptic seizures by disrupting the inflammatory mechanisms. Inflammation-targeting small molecules in the management of AD and seizures were seen to produce positive neurologic outcomes due to their smaller size and low molecular weight that could penetrate the CNS domain [18, 19]. The therapeutic effects of some of the available nonsteroidal anti-inflammatory drugs (NSAID) have been postulated to target pathological hallmarks of AD and decrease the frequency of spontaneous seizures by interacting with inflammatory pathways [18, 19]. With this, the clinical benefits of anti-inflammatory treatment could be extrapolated as a potential therapeutic approach in patients with both AD and epilepsy, nonetheless, more studies are warranted as the aforementioned preclinical evidence focuses only on a condition, either AD or epilepsy, at a time.
Limitation and future direction
In this review, we have systematically summarized studies that evaluated the direct and indirect interaction of neuroinflammation between AD and seizure. The evidence presented in this systematic review can be used by researchers to enhance current understandings of the role of neuroinflammation in the progression of AD and epilepsy pathophysiology and to potentially identify and target inflammatory biomarkers as a new therapeutic approach for treatment. Moreover, the study result might be a good guide to employ and explore the other factors that may be involved in the trajectory of the conditions’ progression. However, the study models included in this systematic review solely utilized transgenic rodent models. Although transgenic models are useful in providing relevant information on the role neuroinflammation played in the pathophysiology of AD, such models do not accurately reflect the initiation and development of commonly seen sporadic AD in patients [60, 61]. Furthermore, the outcome measures were not standardized between the studies as various types of inflammatory markers and methods were used to assess the progression of AD and seizure. In addition, the studies that followed different types of study designs may also partially influence the conditions’ progressions and data presentation potentially causing a disparity in the context of the targeted inflammatory biomolecules.
Conclusion
This systematic review has summarized the evidence supporting the correlation between neuroinflammation in the progression of AD and epilepsy. The prospect of targeting neuroinflammation as a curative approach for both conditions may seem premature at this moment but noticeably promising. However, there is a need for more studies focusing on the common inflammatory biomarkers to develop standardized monitoring guidelines to prevent the manifestation of epilepsy in AD patients as well as AD initiation in epileptic patients.
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
The project was funded by the Ministry of Higher Education, Fundamental Research Grant Scheme (FRGS/1/2020/SKK0/MUSM/02/6).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230059.
REFERENCES
[1] | Buda O , Arsene D , Ceausu M , Dermengiu D , Curca GC ((2009) ) Georges Marinesco and the early research in neuropathology. Neurology 72: , 88. |
[2] | Giorgi FS , Saccaro LF , Busceti CL , Biagioni F , Fornai F ((2020) ) Epilepsy and Alzheimer’s disease: Potential mechanisms for an association. Brain Res Bull 160: , 107–120. |
[3] | Pandis D , Scarmeas N ((2012) ) Seizures in Alzheimer disease: Clinical and epidemiological data. Epilepsy Curr 12: , 184–187. |
[4] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[5] | Tiwari S , Atluri V , Kaushik A , Yndart A , Nair M ((2019) ) Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int J Nanomed 14: , 5541–5554. |
[6] | Tran L , Ha-Duong T ((2015) ) Exploring the Alzheimer amyloid-β peptide conformational ensemble: A review of molecular dynamics approaches. Peptides 69: , 86–91. |
[7] | Horwich A ((2002) ) Protein aggregation in disease: A role for folding intermediates forming specific multimeric interactions. J Clin Invest 110: , 1221–1232. |
[8] | Crews L , Masliah E ((2010) ) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19: , R12–R20. |
[9] | Chen JX , Yan SS ((2010) ) Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis 20: (Suppl 2), S569–578. |
[10] | Crews L , Masliah E ((2010) ) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19: , R12–20. |
[11] | Stafstrom CE , Carmant L ((2015) ) Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb Perspect Med 5: , a022426. |
[12] | Defination of Epilepsy (2014), https://www.ilae.org/guidelines/definition-and-classification/definition-of-epilepsy-2014https://www.ilae.org/guidelines/definition-and-classification/definition-of-epilepsy-2014, |
[13] | Lehmann L , Lo A , Knox KM , Barker-Haliski M ((2021) ) Alzheimer’s disease and epilepsy: A perspective on the opportunities for overlapping therapeutic innovation. Neurochem Res 46: , 1895–1912. |
[14] | Yang F , Chen L , Yu Y , Xu T , Chen L , Yang W , Wu Q , Han Y ((2022) ) Alzheimer’s disease and epilepsy: An increasingly recognized comorbidity. Front Aging Neurosci 14: , 940515. |
[15] | Giorgi FS , Biagioni F , Galgani A , Pavese N , Lazzeri G , Fornai F ((2020) ) Locus coeruleus modulates neuroinflammation in parkinsonism and dementia. Int J Mol Sci 21: , 8630. |
[16] | Rana A , Musto AE ((2018) ) The role of inflammation in the development of epilepsy. J Neuroinflammation 15: , 144. |
[17] | Vezzani A , French J , Bartfai T , Baram TZ ((2011) ) The role of inflammation in epilepsy. Nat Rev Neurol 7: , 31–40. |
[18] | Liu P , Wang Y , Sun Y , Peng G ((2022) ) Neuroinflammation as a potential therapeutic target in Alzheimer’s disease. Clin Interv Aging 17: , 665–674. |
[19] | Mukhtar I ((2020) ) Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 82: , 65–79. |
[20] | DiSabato DJ , Quan N , Godbout JP ((2016) ) Neuroinflammation: The devil is in the details. J Neurochem 139 Suppl 2: , 136–153. |
[21] | McGeer PL , McGeer EG ((1995) ) The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21: , 195–218. |
[22] | Rogers J , Webster S , Lue LF , Brachova L , Civin WH , Emmerling M , Shivers B , Walker D , McGeer P ((1996) ) Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging 17: , 681–686. |
[23] | Aisen PS , Davis KL ((1994) ) Inflammatory mechanisms in Alzheimer’s disease: Implications for therapy. Am J Psychiatry 151: , 1105–1113. |
[24] | Yu Y , Ye RD ((2015) ) Microglial Aβ receptors in Alzheimer’s disease. Cell Mol Neurobiol 35: , 71–83. |
[25] | Yan SD , Bierhaus A , Nawroth PP , Stern DM ((2009) ) RAGE and Alzheimer’s disease: A progression factor for amyloid-beta-induced cellular perturbation? J Alzheimers Dis 16: , 833–843. |
[26] | Heneka MT ((2017) ) Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol 27: , 220–222. |
[27] | Becher B , Spath S , Goverman J ((2017) ) Cytokine networks in neuroinflammation. Nat Rev Immunol 17: , 49–59. |
[28] | Giorgi FS , Saccaro LF , Galgani A , Busceti CL , Biagioni F , Frati A , Fornai F ((2019) ) The role of Locus Coeruleus in neuroinflammation occurring in Alzheimer’s disease. Brain Res Bull 153: , 47–58. |
[29] | Mandrekar-Colucci S , Landreth GE ((2010) ) Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets 9: , 156–167. |
[30] | Koh S ((2018) ) Role of neuroinflammation in evolution of childhood epilepsy. J Child Neurol 33: , 64–72. |
[31] | Probert L , Akassoglou K , Kassiotis G , Pasparakis M , Alexopoulou L , Kollias G ((1997) ) TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol 72: , 137–141. |
[32] | Campbell IL , Abraham CR , Masliah E , Kemper P , Inglis JD , Oldstone MB , Mucke L ((1993) ) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A 90: , 10061–10065. |
[33] | Vezzani A , Conti M , De Luigi A , Ravizza T , Moneta D , Marchesi F , De Simoni MG ((1999) ) Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J Neurosci 19: , 5054–5065. |
[34] | Hiragi T , Ikegaya Y , Koyama R ((2018) ) Microglia after Seizures and in Epilepsy. Cells 7: . |
[35] | Hooijmans CR , Rovers MM , de Vries RB , Leenaars M , Ritskes-Hoitinga M , Langendam MW ((2014) ) SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol 14: , 43. |
[36] | Aldabbagh Y , Islam A , Zhang W , Whiting P , Ali AB ((2022) ) Alzheimer’s disease enhanced tonic inhibition is correlated with upregulated astrocyte GABA transporter-3/4 in a knock-in APP mouse model. Front Pharmacol 13: , 822499. |
[37] | Canet G , Zub E , Zussy C , Hernandez C , Blaquiere M , Garcia V , Vitalis M , deBock F , Moreno-Montano M , Audinat E , Desrumaux C , Planel E , Givalois L , Marchi N ((2022) ) Seizure activity triggers tau hyperphosphorylation and amyloidogenic pathways. Epilepsia 63: , 919–935. |
[38] | Gschwind T , Lafourcade C , Gfeller T , Zaichuk M , Rambousek L , Knuesel I , Fritschy J-M ((2018) ) Contribution of early Alzheimer’s disease-related pathophysiology to the development of acquired epilepsy. Eur J Neurosci 47: , 1534–1562. |
[39] | Petrache AL , Rajulawalla A , Shi A , Wetzel A , Saito T , Saido TC , Harvey K , Ali AB ((2019) ) Aberrant excitatory-inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s disease. Cereb Cortex 29: , 1834–1850. |
[40] | Putra M , Puttachary S , Liu G , Lee G , Thippeswamy T ((2020) ) Fyn-tau ablation modifies PTZ-induced seizures and post-seizure hallmarks of early epileptogenesis. Front Cell Neurosci 14: , 592374. |
[41] | Schultz MK Jr., Gentzel R , Usenovic M , Gretzula C , Ware C , Parmentier-Batteur S , Schachter JB , Zariwala HA ((2018) ) Pharmacogenetic neuronal stimulation increases human tau pathology and trans-synaptic spread of tau to distal brain regions in mice. Neurobiol Dis 118: , 161–176. |
[42] | Tzeng T-C , Hasegawa Y , Iguchi R , Cheung A , Caffrey DR , Thatcher EJ , Mao W , Germain G , Tamburro ND , Okabe S , Heneka MT , Latz E , Futai K , Golenbock DT ((2018) ) Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proc Natl Acad Sci U S A 115: , 9002–9007. |
[43] | Chiu CC , Liao YE , Yang LY , Wang JY , Tweedie D , Karnati HK , Greig NH , Wang JY ((2016) ) Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods 272: , 38–49. |
[44] | Bal-Price A , Brown GC ((2001) ) Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 21: , 6480–6491. |
[45] | Buffo A , Rolando C , Ceruti S ((2010) ) Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79: , 77–89. |
[46] | Harry GJ ((2013) ) Microglia during development and aging. Pharmacol Ther 139: , 313–326. |
[47] | Streit WJ , Mrak RE , Griffin WS ((2004) ) Microglia and neuroinflammation: A pathological perspective. J Neuroinflammation 1: , 14. |
[48] | Greig NH , Tweedie D , Rachmany L , Li Y , Rubovitch V , Schreiber S , Chiang YH , Hoffer BJ , Miller J , Lahiri DK , Sambamurti K , Becker RE , Pick CG ((2014) ) Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement 10: , S62–S75. |
[49] | Kang JQ ((2021) ) Epileptic mechanisms shared by Alzheimer’s disease: Viewed via the unique lens of genetic epilepsy. Int J Mol Sci 22: , 7133. |
[50] | Chin J , Scharfman HE ((2013) ) Shared cognitive and behavioral impairments in epilepsy and Alzheimer’s disease and potential underlying mechanisms. Epilepsy Behav 26: , 343–351. |
[51] | Weintraub S , Wicklund AH , Salmon DP ((2012) ) The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006171. |
[52] | deToledo-Morrell L , Stoub TR , Wang C ((2007) ) Hippocampal atrophy and disconnection in incipient and mild Alzheimer’s disease. Prog Brain Res 163: , 741–753. |
[53] | Van Hoesen GW , Hyman BT , Damasio AR ((1991) ) Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1: , 1–8. |
[54] | Altuna M , Olmedo-Saura G , Carmona-Iragui M , Fortea J ((2022) ) Mechanisms involved in epileptogenesis in Alzheimer’s disease and their therapeutic implications. Int J Mol Sci 23: , 4307. |
[55] | Costa C , Romoli M , Liguori C , Farotti L , Eusebi P , Bedetti C , Siliquini S , Cesarini EN , Romigi A , Mercuri NB , Parnetti L , Calabresi P ((2019) ) Alzheimer’s disease and late-onset epilepsy of unknown origin: Two faces of beta amyloid pathology. Neurobiol Aging 73: , 61–67. |
[56] | Kwon HS , Koh S-H ((2020) ) Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener 9: , 42. |
[57] | Cai Z , Qiao PF , Wan CQ , Cai M , Zhou NK , Li Q ((2018) ) Role of blood-brain barrier in Alzheimer’s disease. J Alzheimers Dis 63: , 1223–1234. |
[58] | Milikovsky DZ , Ofer J , Senatorov VV Jr., Friedman AR , Prager O , Sheintuch L , Elazari N , Veksler R , Zelig D , Weissberg I , Bar-Klein G , Swissa E , Hanael E , Ben-Arie G , Schefenbauer O , Kamintsky L , Saar-Ashkenazy R , Shelef I , Shamir MH , Goldberg I , Glik A , Benninger F , Kaufer D , Friedman A ((2019) ) Paroxysmal slow cortical activity in Alzheimer’s disease and epilepsy is associated with blood-brain barrier dysfunction. Sci Transl Med 11: , eaaw8954. |
[59] | de Liyis BG , Tandy SG , Endira JF , Putri KA , Utami DKI ((2022) ) Anti-high mobility group box protein 1 monoclonal antibody downregulating P-glycoprotein as novel epilepsy therapeutics. Egypt J Neurol Psychiatr Neurosurg 58: , 121. |
[60] | Jankowsky JL , Zheng H ((2017) ) Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener 12: , 89. |
[61] | Kaushal A , Wani WY , Anand R , Gill KD ((2013) ) Spontaneous and induced nontransgenic animal models of AD: Modeling AD using combinatorial approach. Am J Alzheimers Dis Other Demen 28: , 318–326. |


