
miRNAs and Stem Cells as Promising Diagnostic and Therapeutic Targets for Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is a cumulative progressive neurodegenerative disease characterized mainly by impairment in cognitive functions accompanied by memory loss, disturbance in behavior and personality, and difficulties in learning. Although the main causes of AD pathogenesis are not fully understood yet, amyloid-β peptides and tau proteins are supposed to be responsible for AD onset and pathogenesis. Various demographic, genetic, and environmental risk factors are involved in AD onset and pathogenesis such as age, gender, several genes, lipids, malnutrition, and poor diet. Significant changes were observed in microRNA (miRNA) levels between normal and AD cases giving hope for a diagnostic procedure for AD through a simple blood test. As yet, only two classes of AD therapeutic drugs are approved by FDA. They are classified as acetylcholinesterase inhibitors and N-methyl-D-aspartate antagonists (NMDA). Unfortunately, they can only treat the symptoms but cannot cure AD or stop its progression. New therapeutic approaches were developed for AD treatment including acitretin due to its ability to cross blood-brain barrier in the brain of rats and mice and induce the expression of ADAM 10 gene, the α-secretase of human amyloid-β protein precursor, stimulating the non-amyloidogenic pathway for amyloid-β protein precursor processing resulting in amyloid-β reduction. Also stem cells may have a crucial role in AD treatment as they can improve cognitive functions and memory in AD rats through regeneration of damaged neurons. This review spotlights on promising diagnostic techniques such as miRNAs and therapeutic approaches such as acitretin and/or stem cells keeping in consideration AD pathogenesis, stages, symptoms, and risk factors.
BACKGROUND
According to the World Health Organization, more than 55 million people suffer from dementia worldwide and the cases are expected to increase every year by nearly 10 million new cases. Dementia can be defined as a syndrome associated with deterioration in cognition and expected consequences of biological aging. Dementia is ranked seventh in mortality rates worldwide and Alzheimer’s disease (AD) is considered as the most common form of dementia causing about 60% – 70% of cases.
AD is a cumulative progressive neurodegenerative disease that has an outstanding impact on cognitive functions such as memory loss, disturbance in behavior and personality, and difficulties in learning and memorizing accompanied by physical, economic, social, and psychological burdens not only to the patients but also to their families and societies [1]. Although the main causes of AD pathogenesis are not fully understood yet, the amyloid-β peptide (Aβ) and tau proteins are supposed to be responsible for AD onset and pathogenesis. Aβ results from the amyloidogenic cleavage of amyloid-β protein precursor (AβPP) by the β-secretase (BACE 1) combined with the γ-secretase and then accumulates forming Aβ plaques that lead to neurotoxicity, dementia, and AD development. Tau proteins are microtubule associated proteins that polymerizes tubulin into microtubules to maintain neuronal cytoskeleton including microtubule packing structure and stabilization [2–4]. When tau proteins are hyperphosphorylated, they clump together in different forms such as double-helix filaments, straight filaments, and neurofibrillary tangles (NFT) leading to neurodegenerative diseases and dementia.
The intensity of AD progression can be classified into different stages. Pre-symptomatic stage is the least severe form of AD with no apparent clinical symptoms [5, 6]. The mild stage of AD is characterized by several apparent AD symptoms including behavior and mood changes accompanied by memory and concentration loss [7]. Increased memory loss takes place in moderate AD stage as AD diffuses in cerebral cortex areas with difficulty in performing routine life activities [8]. Severe or late-stage AD is recognized through the accumulation of Aβ plaques and NFTs accompanied by progressive cognitive and functional impairment which may lead to death [5, 9].
According to many studies, various risk factors may lead to AD onset and pathogenesis. These factors are classified into demographic, genetic, as well as lifestyle, and environmental factors. Demographic risk factors include age, gender, and race. Genetic risk factors include AβPP, Presenilin 1 and 2 (PSEN1/2), Apolipoprotein E (APOE), and other genes. Lifestyle and environmental factors involve malnutrition, poor diet, smoking, and exposure to metals especially aluminum, copper, and zinc [10–14].
Currently, only five drugs are approved by FDA for AD treatment. These drugs act by two pathways. The first is acetylcholinesterase inhibition to prevent acetylcholine hydrolysis. This class of drugs includes donepezil, rivastigmine, and galantamine [15–17]. The second is e N-methyl-D-aspartate (NMDA) antagonism. This drug category is represented by memantine, which is a non-competitive NMDA channel blocker that decreases the activity of the neurotransmitter glutamate by preventing its binding to NMDA and thus reduces Ca2 + influx. Namzaric is a combination of Donepezil and Memantine that acts as acetylcholinesterase inhibitor and NMDA receptor antagonist to reduce the levels of both acetylcholine and glutamate [17, 18]. Unfortunately, these drugs can only treat the symptoms in mild, moderate, or severe AD cases but cannot cure AD or stop its progression.
AD was firstly diagnosed through some criteria that classified AD into probable AD, possible AD, and definite AD [19, 20]. For more accurate diagnosis, new criteria were established and the use of biomarker evidence through imaging, serum, and cerebrospinal fluid (CSF) was included to distinguish AD from other forms of dementia [21, 22]. Current diagnostic techniques include neuroimaging diagnosis through magnetic resonance imaging (MRI) for hippocampal atrophy measurement and positron emission tomography (PET) to detect the deposition of Aβ plaques in the brain [23–26]. Biomarkers screening in peripheral blood can be more applicable for monitoring disease progression. Many studies observed some variations in specific protein and microRNA (miRNA) levels between normal and AD cases in addition to differences in oxysterol levels according to AD stage giving hope for a diagnostic procedure with high potential in AD diagnosis through a simple blood test [27–29].
New therapeutic approaches were developed to treat AD and stop its progression. Firstly, acitretin was applied due to its ability to cross blood-brain barrier (BBB) in the brain of rats and mice and induce the expression of a disintegrin and metalloprotease 10 (ADAM 10) gene, the α-secretase of human AβPP, stimulating the non-amyloidogenic pathway for AβPP processing resulting in Aβ reduction [30, 31]. Also stem cell therapy was applied through different stem cell classes such as embryonic stem cells (ESCs), neural stem cells (NSCs), mesenchymal stem cells (MSCs), and induced pluripotent stem cells (iPSCs). ESCs can improve cognitive memory functions and learning in AD rats through differentiation into basal forebrain cholinergic neurons and γ-aminobutyric acid neurons [32]. The limited use of ESCs in therapy is due to their pluripotency which affects their differentiation and leads to the shift towards any direction resulting in neoplasia or teratoma [33, 34]. Growth factor secreting NSCs transplantation in rodent AD models can improve cognitive function and neurogenesis due to the therapeutic potential of NSCs paracrine effect [35]. This paracrine effect was exploited through NSC transplantation in rodent AD models resulting in neuro-inflammation reduction and neuronal differentiation through neuroprotective and immunomodulatory factors release [36].
MSCs can stimulate AD treatment through their various roles that include reduction of Aβ plaque load by Aβ degradation and internalization, immune regulation, and regeneration of damaged neurons [37]. MSCs transplantation also induced neurogenesis, secreted factors that provide support, and improved memory deficits and spatial learning [38–40]. iPSCs are able to differentiate into various cells, involving neurons, neutrospheres, and glial cells leading to reduction of plaque depositions in the 5XFAD transgenic AD mouse model and cognitive dysfunction improvement [41–43]. The use of iPSCs faces huge hurdles in clinical application including, long-term safety, effectiveness, and teratoma formation [44, 45]. In this review, many aspects of AD will be illustrated such as AD pathogenesis, stages, symptoms, risk factors as well as promising diagnostic procedures such as miRNA analysis. In addition, promising treatments such as acitretin and stem cell are also discussed increasing the probability of developing new approaches to treat AD and stop its progression resulting in the amelioration of behavior and health for people affected with AD.
ALZHEIMER’S DISEASE PATHOGENESIS AND MOLECULAR MECHANISMS
Although the main causes of AD pathogenesis are not fully understood yet, there are two factors that are supposed to play important roles in AD development. The first factor is Aβ, which is considered as a pathological by-product of the cleavage process of AβPP and the second is tau protein, which is a microtubule-associated protein that accumulates in cells in the form of NFT. AβPP is a transmembrane protein that has a crucial role in a wide range of biological activities including neuronal development, intracellular transport, and signaling besides neuronal homeostasis [17]. AβPP cleavage can be done either through the non-amyloidogenic or amyloidogenic pathway (Fig. 1). AβPP is cleaved in the non-amyloidogenic pathway by α-secretase producing extracellular soluble peptide alpha (sAβPPα) accompanied by C83 amino acid fragment that is cleaved by γ-secretase forming p3 fragment. Meanwhile, AβPP is cleaved in the amyloidogenic pathway by the action of β-secretase forming extracellular soluble peptide beta (sAβPPβ) accompanied by C99 amino acid fragment that is cleaved by γ-secretase leading to the formation of a 37– 49 amino acid residue known as Aβ [46]. sAβPPβ lacks region 1– 16 of carboxyl terminus that differentiates between sAβPPα and sAβPPβ. sAβPPβ functions as death receptor 6 ligand, resulting in axonal trimming and neuronal cell death [47]. Aβ has two isoforms which are Aβ1 - 40 and Aβ1 - 42. Aβ1 - 42 is considered as the essential constituent of the pathological amyloid plaques in AD [48]. Aβ monomers accumulation besides decreased clearance results in their aggregation into oligomers followed by regular amyloid fibrils forming Aβ plaques that cause neurotoxicity, dementia, as well as AD development. Aβ plaques are assumed to be involved in the stimulation of various activities including inflammation, oxidative stress, and tau hyperphosphorylation [17].
Fig. 1
AβPP splicing in non-amyloidogenic and amyloidogenic pathways.
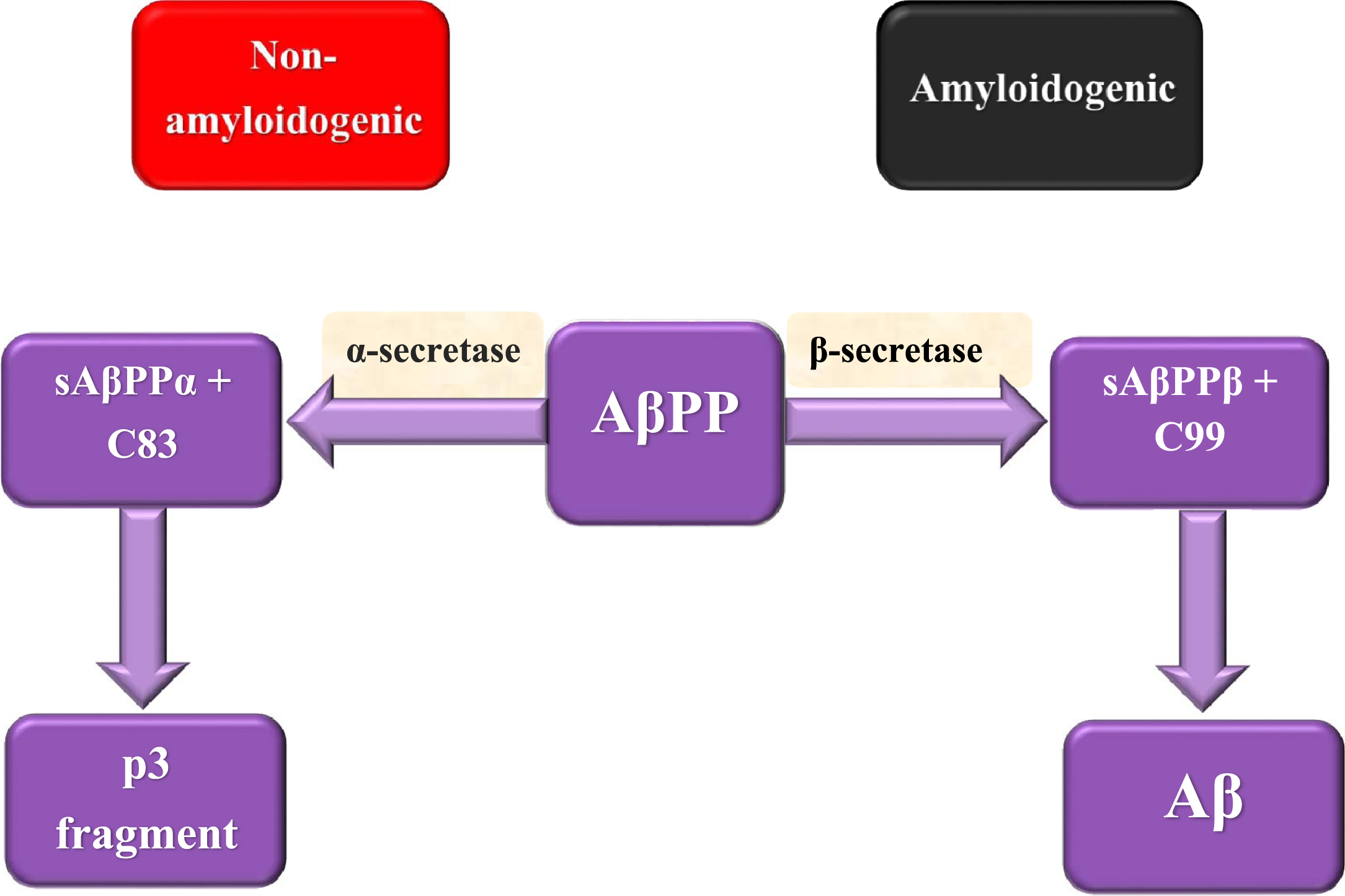
It was found that the extracellular domain of AβPP may have a critical role in triggering synapse formation. Trans-synaptic interactions between pre and postsynaptic AβPP take part in the adhesion of synapses [49]. Several reports recorded that AβPP and BACE1 knockout mice show up memory impairments which suggest that Aβ may have a crucial role in learning and memory [50]. Recently, it has been shown that a low Aβ concentration at the picomolar level reinforces hippocampal long-term potentiation accompanied by memory improvement, showing a new positive, modulatory function in neurotransmission and memory [51].
γ-secretase is a complex formed mainly from four proteins: presenilin, nicastrin, anterior pharynx-defective-1 (APH-1), and presenilin enhancer-2 (PEN-2) and located in the endoplasmic reticulum, Golgi complex and trans-Golgi network, endocytic and intermediate compartments [52]. There are two presenilin homologues in humans which are presenilin 1 and presenilin 2. More than 1000-point mutations in PSEN 1 and 2 are found to be involved in the onset of most of the AD familial forms [53].
Apolipoprotein E (ApoE) is a cholesterol transport protein that exists as a component of lipoprotein complexes along with other apolipoproteins and proteins in plasma and CSF. Three alleles (ɛ2, ɛ3, and ɛ4) were found in humans with different amino acids in two polymorphic sites [54]. The differences in amino acids modify protein charge and structural properties. Several studies reported that ApoE has a role in familial and sporadic late-onset AD [55]. Variation of ɛ4 allele is associated with a 2- to 3-fold increased risk of late-onset AD accompanied by cognitive impairment and having 2 altered copies rises the risk to a 5- to 10-fold increase. The same effect has been recorded for the cognitive impairment progression to dementia.
Individuals carrying APOE4 allele have elevated total and low-density lipoprotein cholesterol [56–58]. Elevated cholesterol levels are associated with Aβ overproduction leading to amyloid plaques formation as it is reported that one of Aβ physiological functions is to control cholesterol transport. Therefore, high cholesterol level dramatically reduces sAβPPα levels [54]. ADAM10 lacks the ability to cleave AβPP through the non-amyloidogenic pathway within a cholesterol-rich environment [59]. Alterations in cellular cholesterol levels in AD reduce the production of sAβPPα leading to neuronal degeneration [60]. Various pieces of evidence indicate that mutated cholesterol metabolism is strongly associated with AD development. Oxidized cholesterol which are named oxysterols mainly contribute to AD pathogenesis [61]. The most common oxysterols that are implicated in AD progression are 24-hydroxycholesterol and 27-hydroxycholesterol. Cholesterol is synthesized in situ in the brain which is considered the organ with the highest cholesterol load. To halt cholesterol accumulation within the brain, it is oxidized by the action of cytochrome p450 family enzymes producing oxysterols. Cholesterol is converted into 24-hydroxycholesterol (24-OH) by cholesterol 24-hydroxylase which is encoded by the CYP46A1 gene. 24-hydroxycholesterol has the ability to pass through the BBB unlike cholesterol and reaches the liver to be metabolized into bile acids [62–64]. Moreover, another oxysterol known as 27-hydroxycholesterol (27-OH) is generated through the action of 27-hydroxylase—encoded by CYP27A1 gene—and metabolized into 7α-hydroxy-3-oxo-4-cholestenoic acid (7-OH-4-C) through the action of CYP7B enzyme. 7-OH-4-C can pass through BBB reaching the liver to be eliminated [65–67].
In AD patients, it has been reported that plasma 24-OH levels vary according to the disease severity with increased levels in mild dementia patients and reduction in patients with severe dementia. The increase in plasma 24-OH in mild AD patients indicates elevated cholesterol transformation due to neuronal cell loss resulting in extraordinary cholesterol release [68, 69]. The decrease in plasma 24-OH in severe AD patients can be due to: 1) the reduction in neuronal degeneration rate in parallel with disease progression accompanied by a decrease in cholesterol levels that is essential for 24-OH production. Nevertheless, the hippocampal atrophy rate in AD is not correlated with the disease severity and remains stable indicating that the neuronal degeneration rate is also stable [70, 71]. 2) The reduction in the activity of factors that coordinate 24-OH production with AD progression. For instance, it could be due to the damage of nerve cells expressing cholesterol 24S-hydroxylase [72]. 3) Elevated levels of brain 24-OH may enhance a negative feedback loop leading to a decreased transformation of cholesterol to 24-OH to regulate cholesterol homeostasis and inhibit oxysterol production [73]. According to several studies, the elevated 27-OH accumulation in AD patients can be due to increased oxysterol flux across BBB resulting from hypercholesterolemia or BBB-damaged integrity [74]. In addition, 27-OH increased level can be a consequence of reduced activity of CYP7B enzyme that metabolizes 27-OH due to the downregulation in CYP7B expression in the brain of AD patients resulting from neuron loss [75]. The increase in oxysterols concentration may enhance cellular damage, neuron dysfunction, and degeneration, leading to the development of neuroinflammation and amyloidogenesis. These indications clarify that any disturbance in oxysterols levels may enhance AD progression and emphasize the relationship between hypercholesterolemia and AD pathogenesis (Fig. 2).
Fig. 2
Factors contribute to AD pathogenesis and development.
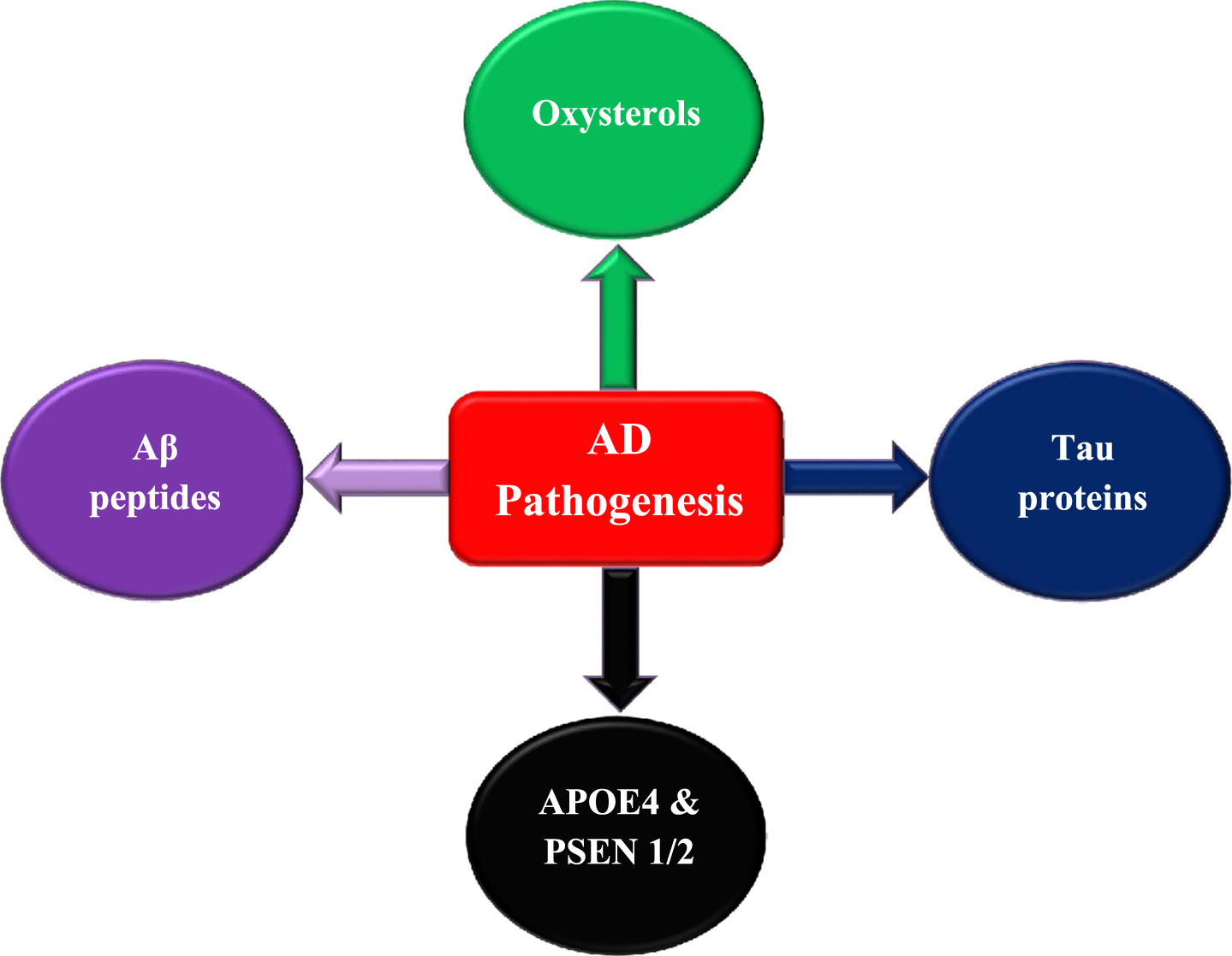
Several reports indicated that peroxisomes may contribute to specific fatty acids synthesis including docosahexaenoic acid that is necessary for the brain and plasmalogens. Reduction in docosahexaenoic acid was observed in the plasma of AD patients and may affect cognitive functions [76, 77]. Plasmalogens have important roles in neural cells and myelin synthesis. It is also reported that lower plasmalogen levels were indicated in patients suffering from severe dementia [78].
Tau proteins are microtubule-associated proteins that polymerize tubulin into microtubules and function mainly in the maintenance of the neuronal cytoskeleton in addition to microtubule packing structure and stabilization [2–4]. MAPT gene, the human gene for tau protein, is located on chromosome 17 and contains 15 exons [79]. Exons 2, 3, and 10 are alternatively spliced producing six isoforms. The longest isoform of tau contains 79 serine and threonine phosphate acceptor residues. Tau biological activity is regulated by the state of its phosphorylation as it has more than 30 phosphorylated sites [80]. It is supposed that AD onset may occur due to an imbalance in the protein kinase phosphorylation system forming abnormal and hyperphosphorylated tau protein [34]. Mitogen-activated protein kinases involve extracellular signal-related kinases, that are stimulated by growth factors, c-Jun N-terminal kinases, and p38 mitogen-activated protein kinases. The mentioned kinases can cause neuronal tau protein hyperphosphorylation leading to AD disease progression [81]. Hyperphosphorylated tau proteins accumulate together in different forms such as double-helix filaments, straight filaments, in addition to NFT causing instability of microtubules and abnormal axonal transport leading to neurodegenerative diseases and dementia. The degree of progression in tau pathology and brain atrophy illustrates the degree of dementia. Tau can be used as one of the clinical targets that improve AD early treatment [82].
ALZHEIMER’S DISEASE STAGES AND SYMPTOMS
AD clinical phases are classified into several stages. Firstly, AD undergoes pre-symptomatic stage which is known by mild memory loss accompanied by the absence of any functional impairment within daily activities or clinical symptoms of AD. The pre-symptomatic stage can persist for several years [5, 6]. Secondly, the mild stage of AD is recognized through the appearance of several AD symptoms including behavior and mood changes, impairment in performing daily routine activities accompanied by memory and concentration loss, difficulties in awareness and recognition in addition to depression development [7]. As AD progresses, patients develop moderate AD stage. In this stage, the patient suffers from increased memory loss due to disease spread to cerebral cortex areas with some troubles in routine skills such as reading, writing, and speaking as well as difficulties in recognizing his relatives, colleagues, and friends [8]. The final stage of AD is severe or late-stage AD which is characterized by progressive cognitive and functional impairment and difficulties in recognition of well-known members, places, and time as well as swallowing and urination due to more disease spread at cerebral cortex areas along with the accumulation of Aβ plaques and NFTs. Eventually, the accumulation of these symptoms may lead to the patient’s death [5, 9].
ALZHEIMER’S DISEASE RISK FACTORS
Several studies reported that AD onset may be caused by many risk factors including demographic risk factors, e.g., age, gender, and race in addition to genetic risk factors, e.g., AβPP, PSEN1/2, APOE, and other genes [10–14]. Also, lifestyle and environmental factors like malnutrition, poor diet, smoking, and exposure to metals especially aluminum, copper, and zinc accompanied by disturbance in lipid homeostasis may contribute to AD development.
Age
Age is considered as one of the most common risk factors in AD patients. Approximately 19% of individuals of age 75– 84 years and 35% of those who are older than 85 years may develop AD onset [10, 11]. In normal brains, there are some age-related changes in brain volume and weight as well as loss of synapses and dendrites accompanied by alterations within the density of senile plaques and NFT [83]. Moreover, Miller et al. observed senile plaques and NFT age-related density alterations and their severe increase in 199 individuals over 70 years old [84]. Additionally, Arrigada et al. observed senile plaques in most of the normal individuals over 55 years old suggesting that there is a pathological cascade from the aged normal non-demented brain to early AD that may progress to more severe AD stages [85, 86]. NFT appear promptly in the locus coeruleus (LC), which stimulates microglia to inhibit Aβ production by the supplement of noradrenaline to the cortex through terminal varicosities, due to aging, mild cognitive impairment (MCI), and AD. Cells injury within the LC may stimulate an age-related impairment of the BBB, thus including age-related vascular factors in AD [12–14].
Genes
As yet few genes were categorized as possible risk factors that contribute to AD development. This review will illustrate the role of genes that may be involved in AD development and progression including AβPP, PSEN1/2, APOE, and other genes.
Amyloid-β protein precursor (AβPP)
Mutations in AβPP gene are associated with the development of some forms of early-onset AD [87]. Cleavage of AβPP leads to the generation of various forms of Aβ that lead to AD onset including Aβ1 - 42 and Aβ1 - 40. Aβ1 - 42 accumulates forming senile plaques that deposit in the brain cortex and hippocampus beside the more soluble form Aβ1 - 40, which is associated with cerebral microvessels [88, 89]. Genome-wide association studies reported numerous genes that affect the metabolism of AβPP with eight of them take place within well-known AD susceptible loci. One of the genes is fermitin family homolog 2 gene (FERMT2) which may increase Aβ levels through the elevation of mature AβPP levels due to its downregulation and leads to alterations in Aβ levels in CSF [90].
Presenilin 1/2 (PSEN1/2)
It is assumed that mutations of PSEN1/2 genes might be related to the early onset familial AD most common form [89]. Full length PSEN consists of nine trans-membrane domains found on the endoplasmic reticulum. PSEN cleavage and accumulation into γ-secretase complex is pursued by transmission to the cell surface affecting AβPP processing [91]. Mutant PSEN1 may react with AβPP through the stimulation of γ-secretase cleavage for normal AβPP causing elevated Aβ deposition. Also, PSEN1 may contribute to AD development by a decrease in its role in neuroprotective mediated functions by ephrin-B [92]. PSEN1/2 may be implicated through the disturbance of cellular calcium homeostasis or interact with the transcriptional coactivator cAMP-response element binding (CREB-binding) protein which has an important role in gene expression regulation [93, 94].
Apolipoprotein E
Variation of allele ɛ4 in the APOE gene is considered as a main risk factor in AD as individuals with this variation have double to triple the frequency to develop AD in comparison with normal cases as well as its effect on cognitive functions, learning, and memory, having two copies may increase the risk of AD onset with a 5- to 10-fold [57, 58]. Expression of ɛ4 may also lead to elevated levels of Aβ, earlier AD onset, and faster progression of AD in aging brain [89, 95, 96].
Other genes
Other genes may be considered as possible risk factors for AD development including glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [97], evolutionarily conserved signaling intermediate in toll pathway (ECSIT) [98], genetic variation in the estrogen receptor (ESR) gene [99], a rare variant of the triggering receptor expressed on myeloid cells 2 (TREM2) gene [100], clusterin gene polymorphism [101], vitamin D receptor (VDR) gene polymorphism [102], and transferrin (Tf) gene [103].
Metals
Earlier reviews considered that aluminum was the only metal affecting AD onset and also more studies report that aluminum may play an important role in elevated mortality rates caused by AD among miners who are exposed to aluminum-rich dust [104–106]. Recently, reports suggested that a large number of metals along with aluminum such as copper, zinc, manganese, mercury, cadmium, and magnesium may be considered as risk factors involved in AD pathogenesis [107–114]. Many of these metals exhibited direct interactions with AβPP metabolism or ApoE [107]. Additionally, it is suggested that metal imbalance affecting cellular homeostasis could be a significant factor, therefore, AD plasma, serum, and brain is characterized by homeostasis deficiency among zinc, copper, and iron which are implicated in the AβPP and tau regulation in addition to ApoE interaction [115].
Malnutrition and obesity
Malnutrition might be considered as an AD risk factor as hypothesized by Abalan studies. This hypothesis was derived from the clinical observation for AD patients suffering from cachexia, urinary tract infections, terminal bronchopneumonia, and low triceps skinfold in addition to deficiency in the levels of serum albumin, iron, folate, tryptophan, and vitamin B12. These symptoms may result in a ‘protein-calorie malnutrition syndrome’ in AD which may lead to NFT formation as a result of chronic nutritional deficiency of calcium/magnesium. Additionally, AD patients developed vitamins A, E, D, and K deficiency in plasma indicating that regular supplements may improve their mental state and reduce Aβ deposition [116, 117].
Considering obesity as the main AD risk factor is controversial in many studies. On the other hand, different studies found a powerful link between obesity and AD development assuming that alterations in neuronal plasticity, which may lead to neuronal death by apoptosis or necrosis and nervous system damage, could be considered as consequences of metabolic changes associated with obesity. Additionally, obesity has been consistently correlated with dementia onset and higher body mass index increasing the risk of AD development [118, 119].
Lipids
Lipids are considered one of the main risk factors that contribute to AD progression. In the brain, lipids are secreted accompanied by ApoE which is formed by astrocytes. The ApoE-containing particles may increase the level of lipids within neurons. Lipid homeostasis within the brain is adjusted independently of peripheral transport with a firm regulation of lipid transport across BBB. ApoE4-mediated variations in lipid homeostasis may contribute to AD pathogenesis and neuropathology. Lipid composition in AD brains may be altered especially in sterol, phospholipid, and fatty acid profiles [120]. Additionally, the disturbance of oxysterols levels and homeostasis within the brain may accelerate AD progression.
CONVENTIONAL AND RECENT ALZHEIMER’S DISEASE DIAGNOSTIC TOOLS
Conventional AD diagnostic tools
The National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) formed a work group in 1984 to set up clinical diagnostic criteria for the classification of AD symptoms. These criteria classified AD into probable AD, that is diagnosed via dementia confirmed through neuropsychological tests, progressive memory loss, impaired daily life activity, and other symptoms without any systemic or brain disorders, possible AD, which is diagnosed by the presence of systemic or brain disorders and the absence of any neurologic or psychological disorders, and eventually definite AD, which is diagnosed through histopathological examinations applied to biopsy [19, 20].
In 2011, some modifications were applied to the 1984 NINCDS-ADRDA criteria by the National Institute on Aging-Alzheimer’s Association for more accuracy in the diagnosis of AD. These modifications established new criteria including probable and possible AD dementia to be used in clinical settings in addition to probable or possible AD dementia accompanied by pathophysiological evidence for research purposes besides clinical biomarkers. The usage of biomarker evidence through imaging, serum, and CSF was included to distinguish AD from other forms of dementia [21, 22].
Current diagnostic techniques include neuroimaging diagnosis represented by MRI for hippocampal atrophy measurement [23]. In addition to PET, which is considered as one of the non-invasive diagnostic techniques for AD patients. A specialized PET scan can be done after the injection of a radiolabeled tracer marker to detect Aβ plaques deposition within the brain. Currently, the use of Aβ PET imaging is still limited for patients because of its high cost [24–26].
Another technique for AD diagnosis is the detection of biomarkers through CSF. CSF is less costly but more invasive examination as it undergoes a lumbar puncture procedure under general anesthetic to assess Aβ1 - 42, hyperphosphorylated tau peptide (p-tau), and total tau protein content [121–123]. A promising less invasive approach is biomarkers screening in peripheral blood. Detecting biomarkers in peripheral blood can be more applicable for monitoring disease progression. It is reported by several studies that there are some variations in specific protein and miRNA levels between normal and AD cases giving hope for a diagnostic procedure with high potential in AD diagnosis through a simple blood test [27–29].
Recent diagnostic approaches
Currently, researchers try to develop new therapeutic approaches that can stop AD progression not only treat its symptoms and can also detect AD at its early stages. In this context, many previous reports hypothesize the role of some circulating miRNAs as effective diagnostic biomarkers and their role in AD detection.
miRNAs
miRNAs are small non-coding RNAs, ranging from 21 to 25 nucleotides, that have a critical role in the post-transcriptional regulation of gene expression either through binding to 3′ untranslated region (UTR) of the messenger RNA (mRNA) to inhibit translation or degrade target mRNA [124, 125]. The same mRNA can be regulated through different miRNAs and a single miRNA can bind to different mRNAs. miRNAs are synthesized from a long transcript with a double-stranded structure known as primary miRNA (Pri-miRNA). Pri-miRNAs are transcribed from miRNA-encoding genes by the action of RNA polymerase II due to its particular affinity [126]. Pri-miRNA is recognized in the nucleus by Pasha Protein which is associated with RNAase type III Drosha. The cleavage of pri-miRNAs is carried out through the enzymatic action of a micro precursor complex which is a combination of one subunit of Drosha (a class II RNase III enzyme), two subunits of DiGeorge syndrome chromosomal region eight proteins, and one subunit of SRp20 (a splicing factor) forming a hairpin-loop like structures nearly composed of 70 nucleotides known as precursor miRNA (pre-miRNA) [127, 128]. Therefore, pre-miRNAs form a complex with Ras-related nuclear and GTP (RAN-GTP) proteins and then transferred from the nucleus to reach the cytoplasm via the exportin-5 pathway [129, 130]. pre-miRNAs undergo secondary processing in the cytoplasm where a second cut for pre-miRNAs takes place by another RNAase III enzyme, Dicer, resulting in mature RNA duplexes of (21– 25 nucleotides) called miRNAs. miRNA duplexes are split by helicase enzyme forming two single miRNA strands called guide strand and passenger strand according to their 5’ to 3’ complementarity. After cleavage, guide strand is incorporated into RNA-induced silencing complex (RISC) while passenger strand is degraded. miRNAs form the RISC complex with Argonaute2 (Ago2) proteins which induces the post-transcriptional regulation of gene expression through two different mechanisms either by translation inhibition or mRNA degradation according to the complementarity between the miRNA and its target mRNA [128, 131, 132]. MiRNAs that bind totally to the complementary areas (3′ UTR) of their target mRNA stimulate its degradation through de-adenylation, cap removal, and exonucleolytic digestion. On the other hand, miRNAs that bind partially to the complementary areas of their target mRNA suppress translation at the initial or elongation phase [133].
Various studies illustrated miRNAs’ role by using AD mouse models, especially AβPP/PSEN transgenic mouse models in accordance with the discovery that AβPP and PSEN1 gene mutations play crucial roles in the onset of familial forms of AD [134, 135]. miRNA-29a, miRNA-29b, and miRNA-29c are known to have a crucial role in BACE 1 gene expression regulation and AD pathogenesis. MiRNA-29c role in AD was studied in peripheral blood. miRNA-29c expression level was downregulated in AD patients accompanied by a considerable increase in BACE 1 expression in comparison with normal cases [136, 137]. The researchers confirmed the in vitro results with in vivo studies when they obtained primary hippocampal neurons from senescence-accelerated mouse-resistant 1 (SAMR1) transfected with miRNA-29c or miRNA-29c inhibitors. It was observed that miRNA-29c significant upregulation lowered BACE 1 expression levels which were elevated in cells treated with miRNA-29c inhibitor. The verification of in vivo study to the results of in vitro study showed that miRNA-29c upregulation considerably decreased BACE-1 and Aβ proteins expression levels.
Jiang et al. conducted a study to examine miRNA-137 impact in AβPP/PSEN1 transgenic mice. This miRNA is involved in the regulation of both cognitive function and neuronal growth [138, 139]. The researchers studied the effect of miRNA-137 regulation on the transcription of calcium voltage-gated channel subunit alpha-1 C gene (CACNA1 C). CACNA1 C encodes the alpha 1 C subunit of the voltage-dependent calcium channel of type L CaV1.2 and regulates intracytoplasmic calcium (Ca2 +) in neurons. The studies showed that the reduction in miRNA-137 levels leads to the elevation in the levels of Aβ and CACNA1 C protein in AD mouse hippocampus and cerebral cortex resulting in an increase in the Ca2 + influx across CaV1.2 which causes neuronal dysfunctions [140, 141]. To verify these results, CACNA1 C expression was evaluated in human SH-SY5Y neuroblastoma cells treated with or without Aβ1–42 and then transfected with miRNA-137 mimics or inhibitors. The obtained results demonstrated that miRNA-137 downregulation is accompanied by an increase in Ca2 + levels and Aβ1–40 and Aβ1–42 decrease. Therefore, those outcomes suggest that an elevation in miRNA-137 could result in a reduction in Ca2 + levels within neurons, enhancing neuronal dysfunctions typical of AD.
Another study was done by Zhang et al. to evaluate the role of miRNA-200a-3p elevated expression in Aβ-induced neuronal apoptosis by suppressing the silent information regulator transcript-1 (SIRT1) in AβPP/PSEN1 mice [127]. SIRT1 is involved in the deacetylation of histone, non-histone proteins, and other transcription factors in addition to its role in gene transcription regulation, neuroprotection in neurodegenerative disorders, cellular senescence, energy balance, and oxidative stress [142]. In the hippocampus of AβPP/PSEN 1 mice treated with Aβ25–35, upregulation of miRNA-200a-3p, and a decreased SIRT1 level were recorded.
The study conducted by Jian et al. aimed to assess miRNA-34a role in Aβ production and clearance as it takes part in many AD pathways including Aβ deposition and cognitive dysfunction. The study compared AβPP/PSEN1 mice to miRNA-34a knockout mice. The level of miRNA-34a was elevated coordinated with the increase of Aβ level in AβPP/PSEN1 mice opposite to miRNA-34a knockout mice that showed fewer amyloid plaques with a considerable decrease in behavioral dysfunction. These observations demonstrate that miRNA-34a upregulation may increase the risk of AD pathogenesis [143]. These results agree with those of the studies that were performed by Xu et al. which recorded the miRNA-34a upregulation in AβPP/PSEN1 mice accompanied by elevated levels of Aβ, amyloid plaque deposition, and cognitive dysfunction. On the other hand, miRNA-34a knockout AβPP/PSEN1 mice exhibited improvement in cognitive functions and suppression of AβPP amyloidogenic processing [144].
Ghasemi-Kasman et al. performed a study to detect whether the miRNA-302/367 cluster can induce the transformation of astrocytes into neurons [145]. The miRNA-302/367 cluster consists of miRNA-367, miRNA-302a, miRNA-302b, miRNA-302c, and miRNA-302d. This cluster has an important role related to the regulation of cell proliferation, differentiation, and reprogramming [146]. An AD mouse model was used and induced by intracerebroventricular injection of streptozotocin. The injection of lentiviral particles loaded with miRNA-302/367 into the hippocampal dentate gyrus accompanied by valproate (VPA) intraperitoneal injection aimed to increase neuronal development, regulate neuronal genes expression, and decrease neural cell damage. Also, VPA can stimulate neprilysin, an Aβ degrading enzyme [147]. These outcomes suggest that the combination of miRNA-302/367 and VPA enhanced learning and memory deteriorated by the injection of streptozotocin.
The variation in expression of the miRNA-200 family in AD initial phases within the brain of Tg2576 mouse increasing AβPP protein level was clarified through a study done by Higaki et al. [135]. miRNA-200 family includes miRNA-200a, miRNA-200b, and miRNA-200c. Total RNA microarray analysis showed that miRNA-200a, miRNA-141, miRNA -429, miRNA-200b, and miRNA-200c were overexpressed only in 10 months Tg2576 mice. It is suggested that several miRNAs can act as diagnostic biomarkers for early Aβ accumulation. To detect whether miRNA-200b and miRNA-200c expressions are altered due to neuronal damage stimulated by Aβ1 - 42, an in vitro study was performed on primary murine neuronal cells detached from mice cortical tissues. This study illustrated that treatment of primary murine neuronal cells with Aβ stimulated miRNA-200b or – 200c overexpression. The cells were transfected with miRNA-200b and miRNA-200c confirming that their upregulation decreased secretion of Aβ in a conditioned medium [135].
Additionally, previous studies revealed that the level of serum miRNA-125b in AβPP/PSEN mice was downregulated in AD which makes miRNA-125b a possible biomarker for the disease [148]. It was also shown that the downregulation of miRNA-125b is associated with cognitive dysfunction. Moreover, the levels of miRNA-9 and miRNA-191-5p were also downregulated while miRNA-28-3p was upregulated. A study was performed to detect these miRNAs levels in AβPP/PSEN1 transgenic mice given epigallocatechin gallate (EGCG). EGCG is a constituent of green tea that can improve cognitive functions and reduce damage caused by oxidative stress making it a potential AD therapeutic agent [149,150]. miRNAs’ levels were inversed in mice treated with EGCG in comparison with untreated mice. miRNA-125b only seemed to be associated with cognitive function. To verify the correlation of miRNA-125b with AD, the level of this miRNA was detected in SH-SY5Y cells treated with EGCG demonstrating a considerable increase in miRNA-125b levels similar to those produced in an in vivo study.
Furthermore, a study was performed by Wang et al. aimed to illustrate the association between miRNA-146a, tau hyperphosphorylation, and rho-associated coiled-coil-containing protein kinase 1 (ROCK1) [151]. ROCK 1 protein binds to protein phosphatase and tensin homolog (PTEN) that contributes to tau dephosphorylation [152]. In the experiment, SH-SY5Y cells were treated with miRNA-146a whose upregulation led to a considerable increase in tau phosphorylation accompanied by inhibition in the ROCK1 protein translation. For the detection of PTEN role, SH-SY5Y cells were treated with siRNA ROCK1 resulting in reduced ROCK1 protein levels, lowered PTEN phosphorylation, in addition to elevated tau phosphorylation. This illustrates the impact of miRNA-146a expression that inhibits ROCK1 resulting in a decrease in the phosphorylation of PTEN and hyperphosphorylation of tau. In vivo study was performed through the treatment of 5xFAD mice with miRNA-146a inhibitor injections in the intra-hippocampal region. The observed results showed an elevation in the level of ROCK1 protein and tau hyperphosphorylation inhibition. Therefore, miRNA-146a has a critical role in AD pathogenesis and its inhibition could be effective in in vivo therapy.
Another study conducted by Alexandrov et al. revealed that NF-kB coupling and considerable upregulation of the inducible pro-inflammatory miRNA-146a and miRNA-155, containing multiple NF-kB DNA-binding and activation sites in their immediate promoters, caused deficits in complement factor H (CFH) expression and AD abundance. CFH is a soluble complement control glycoprotein that has an important role in the inhibition of the innate-immune response. Downregulation of CFH activates the complement system which is considered the main non-cellular component within the innate-immune system while transmitting neuro-inflammation. On contrast, the downregulation of miRNA-146a and miRNA-155 regulated mRNA targets including those encoding CFH. These results showed that miRNA-146a and miRNA-155 downregulation could be a potential therapeutic pathway for AD onset prevention and inflammation reduction [153]. We also hypothesized that miRNA-146a and miRNA-155 may have a role as diagnostic biomarkers for AD in rat model. In our work, we observed a significant upregulation in both miRNAs during the induction and withdrawal phases of aluminum chloride-induced rat AD model which had been corrected by MSCs transplantation and/or acitretin (unpublished data).
Sierksma et al. conducted a study using AβPP and tau transgenic mice to detect whether altered miRNAs are correlated with progressive memory impairment in AD [154]. miRNAseq showed that various miRNAs were commonly overexpressed in both APP and tau transgenic mice including miR-10a-5p, miR-142a-5p, miR-146a-5p, miR-155-5p, miR-211-5p, and miR-455-5p. Four of these, miR-142a-5p, miR-146a-5p, miR-155-5p, and miR-455-5p, showed overexpression in patients with AD. It was also reported that the upregulation of these miRNAs in tau transgenic mice can be observed in four months mice compared to ten months in AβPP transgenic mice which occurs when Aβ is accumulated. To confirm that the miRNAs upregulation is responsible for the onset of cognitive impairment, a single miRNA oligonucleotide or a mix of the six miRNAs were injected in the intracerebroventricular region in C57BL/6J mice. Overexpression of each miRNA took place in the hippocampus after these injections, but there were no serious cognitive impairments. The obtained results indicate that miRNAs could induce a protective effect halting AD progression.
Several studies reported that Aβ induces NF-κβ upregulation. NF-κβ transcription factor is a common risk factor associated with AD neurodegeneration [155]. Overexpression of NF-κβ results in the expression of many pro-inflammatory molecules including cytokines and chemokines which contribute to AD onset. NF-κβ activation resulted in the upregulation of AβPP/BACE1, thereby causing Aβ accumulation [156]. NF-κβ also has a crucial role in the upregulation of several miRNAs in the brain. These miRNAs involve miRNA-155, miRNA-146a, miRNA-9, and miRNA-125b which results in the downregulation of many protective proteins including synapsin 2, tetraspanin 12 (TSPAN12), and complement factor H. The decrease in synapsin 2 level causes deterioration in the synaptogenesis process, while TSPAN12 and complement factor H reduction induces Aβ accumulation and inflammatory response within the neuronal cells respectively [157].
Dong et al. conducted a study to examine serum miRNAs in AD patients, MCI, and vascular dementia patients, in addition to non-demented controls. Four miRNAs were identified including miRNA-93, miRNA-31, miRNA-143, and miRNA-146a. The expression of all miRNAs was reduced in the AD patients group only. Nevertheless, these miRNAs levels could not discriminate MCI and vascular dementia patients in comparison with controls as miRNA-146a and miRNA-93 were considerably upregulated in MCI individuals against control subjects while, miRNA-143 was downregulated, and miRNA-31 showed no change. Concerning vascular dementia patients, miRNA-31, miRNA-93, and miRNA-146a were considerably upregulated while miRNA-143 was downregulated. Consequently, variations in these miRNAs expression can distinguish between AD cases and control subjects, but cannot distinguish MCI and vascular dementia cases [158].
A study was performed by Denk et al. to detect the difference in miRNAs expression in CSF samples taken from AD patients and healthy individuals. Four ml CSF was collected via lumbar puncture and processed for miRNA isolation. 1,178 miRNAs were detected through Open Array qRT-PCR, based on miRbase version 14 (http://www.mirbase.org/). It was reported that seven miRNAs were upregulated in AD patients including (miRNA-146a, miRNA-100, miRNA-505, miRNA-4467, miRNA-766, miRNA-3622b-3p, and miRNA-296), while eight miRNAs were downregulated including (miRNA-449, miRNA-1274a, miRNA-4674, miRNA-335, miRNA-375, miRNA-708, miRNA-219, and miRNA-103). MiRNA-146a, miRNA-375, miRNA-103, and miRNA-100 showed considerable area under curve (AUC) values (0.97, 0.99, 0.87, and 0.72, respectively) [159].
Besides miRNAs’ role in Aβ production, they are involved in tau protein phosphorylation and dephosphorylation responsible for NFT formation. As known, miRNA-132 expression is downregulated in AD patients’ brains [160, 161]. Therefore, Salta et al. performed a study to detect the effect of miRNA-132 downregulation on AD pathogenesis. AβPP/PSEN mice were injected in the intracerebroventricular region with miRNA-132 and miRNA-132 inhibitor [162]. Researchers recorded that miRNA-132 downregulation led to the elevation of Aβ peptide and phosphorylated tau production in the AβPP/PSEN1 mice hippocampus while miRNA-132 upregulation is associated with Aβ reduction in addition to phosphorylated tau.
Smith et al. conducted another study to detect the contribution of downregulated miRNA-132/212 cluster in the onset of AD [163]. The use of miRNA-132/212 knockout mice in an in vivo study revealed elevated phosphorylation and tau accumulation in addition to autophagy which is a factor in tau accumulation [164]. Hernandez-Rapp et al. conducted a study to detect the effect of miRNA-132/212 absence on Aβ production, cleavage, and clearance. It was observed that miRNA-132/212 deletion elevated Aβ production and amyloid plaque formation in 3xTg-AD-miRNA-132/212 knockout mice [165]. The variation in the expression of miRNAs in serum, plasma, CSF, and brain suggests that miRNAs can be used effectively as potential biomarkers to differentiate patients with AD from healthy controls.
Fatty acids and oxysterols
As known, one of the most promising biomarkers to detect AD progression is the quantification of fatty acids and oxysterols levels within the circulation and brain. Fatty acids (FA) are analyzed by gas chromatography (GC) as fatty acid methyl esters (FAMEs). Total lipids are extracted and distributed into aliquots to be converted into methyl esters. FAMEs are analyzed in duplicate through the injection of a specific volume of each sample into the GC system to pass through the flame ionization detector and a polar fused silica capillary column. The FA composition appears as a relative percentage of the total peak area. Very-long-chain fatty acid were quantified in patients with dementia using GC/mass spectrometry (MS) set to selected-ion monitoring mode and detected in red blood cells and plasma of AD patients revealing their increase according to FA analyses [166]. Several studies reported the use of GC/MS to detect 24-OH and 27-OH levels in plasma [167]. Also, the use of oxysterols as potential biomarkers can be critical for the detection of AD progression especially 24-OH and 27-OH. At mild AD cases, 24-OH levels are elevated which dramatically decrease as the disease progresses reaching severe stage. Additionally, 27-OH level increases as a sign of AD development. The early detection of fatty acids and oxysterols levels could minimize BBB damage, neuron dysfunction and degeneration, neuroinflammation, and AD development [67, 168].
Zhang et al. performed a study where 27-OH treatment led downregulation in miRNA let-7g-5p expression indicating that miRNA let-7g-5p may have an important role in AD pathogenesis stimulated by 27-OHC [169]. Several studies showed that lipopolysaccharides may affect the upregulation of pro-inflammatory transcription factor complex NF-kB (p50/p65) and also related miRNAs, including miRNA-30b, miRNA-34a, miRNA-146a, and miRNA-155 [170]. These miRNAs are reported to be associated with AD pathogenesis. Therefore, oxysterols and lipopolysaccharides may contribute in altering the levels of miRNAs involved in inflammation and AD pathogenesis.
THERAPEUTIC APPROACHES
Conventional pharmaceutical treatment
Despite the urgent need for the discovery of drugs for AD treatment, only two classes of drugs are approved by FDA. These two classes include inhibitors to cholinesterase enzymes and antagonists to NMDA. Acetylcholinesterase inhibitors prevent acetylcholine (ACh) hydrolysis by blocking cholinesterase enzymes which leads to the elevation of ACh levels [171–173]. NMDA antagonists inhibit NMDAR glutamate receptor activity which decreases Ca2 + influx, prevents cell death and synaptic dysfunction, and restores normal activity [174]. Unfortunately, these two classes can only cure AD symptoms but cannot prevent disease progression.
Cholinesterase inhibitors
Cholinergic levels elevation represents an effective strategy that can improve cognition and neural cell function. Acetylcholinesterase inhibitors prevent ACh hydrolysis leading to ACh accumulation and cholinergic receptors activation [8]. The current FDA approved cholinesterase inhibitors are donepezil, rivastigmine, and galantamine.
Donepezil is an indanone benzyl piperidine derivative and the second generation of acetylcholinesterase inhibitors. Donepezil acts by binding to acetylcholinesterase which prevents acetylcholine breakdown and increases its level at the synapses. Donepezil is effective in the treatment of mild AD cases due to its ability to improve cognitive functions with some side effects in the gastrointestinal and nervous systems [175, 176].
Rivastigmine is a pseudo irreversible inhibitor for acetylcholinesterase and butyrylcholinesterase that acts through binding to the two acetylcholinesterase active sites which are anionic and estearic sites leading to the inhibition of ACh metabolism. Rivastigmine detaches slower than acetylcholinesterase, so it is named pseudo-irreversible inhibitor. The drug can be effective for mild and moderate AD cases as it ameliorates cognitive functions and routine activities. The drug is delivered orally but with some side effects including vomiting, nausea, and weight loss. There is a better way to deliver rivastigmine through transdermal patches for more dosage control and continual drug delivery through the skin, accompanied by enhanced tolerability and fewer side effects [177,178].
Galantamine (GAL) is a drug for mild and moderate AD cases. GAL is a selective tertiary isoquinoline alkaloid with a dual mechanism of action in which it acts as a competitive inhibitor of acetylcholinesterase and can also bind to the α-subunit of nicotinic acetylcholine receptors to stimulate them. GAL can enhance routine life activities and cognitive performance with good efficacy and tolerability, identical to other acetylcholinesterase inhibitors. GAL can be delivered through its attachment to ceria-containing hydroxyapatite particles for selective drug delivery to reach the affected regions in the brain or through the patch system to control released drug dosage [179–181].
N-methyl d-aspartate antagonists
NMDAR is thought to have a significant role in AD pathogenesis. Hyperactivity of NMDAR causes abnormal Ca2 + influx and overstimulation of glutamate, leading to neuronal cell death, synaptic dysfunction, and declined cognitive functions. Memantine is considered as the only approved drug as an NMDA antagonist for the treatment of moderate and severe AD [182–184].
Memantine is an uncompetitive antagonist of the NMDAR that inhibits over-activation of glutamate receptor implicated in moderate to severe AD cases neurotoxicity. The drug safety is high with good tolerability, it blocks the excitatory receptor with no interference with the normal synaptic transmission because of memantine’s low affinity [185,186].
Namzaric drug is a combination of donepezil and memantine, therefore, it acts as a cholinesterase inhibitor and NMDA receptor antagonist. It prevents acetylcholine hydrolysis and blocks glutamate receptor which results in an improvement in cognitive functions, learning, and routine activities [187].
New therapeutic approaches
Acitretin
Many reports showed that the bio-reactive metabolite of vitamin A, all-trans-retinoic acid (atRA), has an important role in AD [188,189]. Vitamin A (retinol) is taken in through food and transferred by chylomicrons reaching the liver. Retinol binds to the retinol-binding protein and transthyretin so it can move across the BBB reaching the neurons. Two-step oxidation occurs and retinoic acid is delivered in cells reaching the nucleus to bind to its receptors. These receptors are known as the dimeric receptors, and they are formed of retinoic X receptors (RXRs) only or RXRs combined with various receptors including retinoic acid receptor (RAR) or vitamin D receptor. It was reported that ADAM10 gene in human, mouse, and rat contains two RXR binding sites at the conserved region of the promoter [190]. More investigations were performed to detect the mechanism of ADAM10 gene regulation through vitamin A metabolites. Various human cell lines were treated with atRA from different tissues. Hepatic and kidney cells revealed no considerable response to the treatment while ADAM10 mRNA of neuroblastoma and monocyte precursors showed elevated ADAM10 transcription meaning that ADAM10 gene upregulation in the brain might be done with no general overexpression in peripheral tissue [191,192]. RAR/RXR heterodimers were identified as regulators of ADAM10 gene expression by using specific ligands for RXR dimerization partners. By co-expression studies, two RAR isotypes, which are RAR-α and RAR-β, were recognized to contribute to the induction of ADAM10 promoter. Additionally, these subtypes are upregulated in the hippocampus and may have a role in synaptic plasticity. It has been observed that a protein complex can bind directly to one of the two potential retinoid receptor binding sites which means that atRA acts directly on the ADAM10 promoter [190]. Vitamin A and retinoic acid cause numerous side effects, so they were substituted by synthetic retinoids utilized in clinical application in dermatology and oncology due to their ability to affect ADAM10 expression. The most commonly used synthetic retinoid was acitretin. Acitretin is a drug approved by FDA for psoriasis treatment since 1997 [193]. Using acitretin showed promising results in in vitro studies [192]. This synthetic retinoid was used as a candidate drug due to its ability to pass through BBB in the brain of rats and mice [30]. Acitretin can also induce the expression of ADAM 10 gene, the α-secretase of human AβPP, which activates the non-amyloidogenic pathway for AβPP processing and decreases Aβ levels [30, 31, 40]. Researchers applied acitretin to human and murine neuroblastoma cells resulting in the stimulation of ADAM10 promoter activity along with elevated levels of ADAM10 protein leading to a shift towards the non-amyloidogenic processing pathway. For more investigation in vivo, AβPP/PSEN 1 double-transgenic mice were treated with acitretin through intracerebral injections. After 48 hours of treatment, elevated AβPPs-α/β ratio and a 50% decrease in Aβ peptides were detected. Another study used acitretin for the treatment of f 5xFAD mice resulting in a significant reduction of Aβ and rebalance of AβPP processing through the stimulation of ADAM10 transcriptional activity. Acitretin treatment restored excitatory synapse counts and improved behavior [194]. Therefore, acitretin can be a precious therapeutic approach for AD treatment as it can restore cognitive functions and improve behavior by lowering Aβ levels in the brain.
Stem cell therapy
Before discussing stem cell therapy’s effectiveness on AD, we should represent different stem cell classes. Stem cells could be classified with regard to their potency or their origin. Recently, the most commonly used stem cells for AD research are ESCs, NSCs, iPSCs, and MSCs.
Stem cell classes differ in their ability to differentiate into various cell types. So, stem cells can be classified according to their potency as follows [195]. Totipotent stem cells can differentiate into embryonic and extraembryonic cells and form a complete organism. Pluripotent stem cells can differentiate only into the three germ cell layers (ectoderm, endoderm, and mesoderm) from which all cells and organs can be developed. Multipotent stem cells can differentiate into all cell types within one particular lineage. Oligopotent stem cells are more restricted than multipotent stem cells as they could only differentiate into closely related cells. Unipotent stem cells are the least potent stem cells as they can only differentiate into one cell type.
Stem cells could be also classified in respect to their origin. In this context, stem cell types and the strategies of their application as therapies in AD will be discussed.
Embryonic stem cells. ESCs are originated from embryonic blastocysts’ inner cell mass on day 5 or 6. ESCs are classified as pluripotent stem cells considering their massive ability in cell types of generation from the ectodermal, endodermal, and mesodermal germ layers in addition to their unlimited self-renewal capacity which makes them attractive candidates to be used in therapeutic strategies [196,197]. According to several studies, ESCs can improve cognitive memory functions and learning in AD rats through differentiation into basal forebrain cholinergic neurons and γ-aminobutyric acid neurons [32]. However, the use of ESCs in therapy is limited due to their pluripotency which is considered as one of the biggest advantages of ESCs but also one of the leading disadvantages as their differentiation can take place towards any direction leading to neoplasia or teratoma. ESCs can also develop abnormal immune response and rejection [33, 34, 198, 199]. Mouse ESCs (mESCs) were utilized to generate basal forebrain cholinergic neurons that were severely affected in AD patients. The transplanted neurons within AD rat models direct the differentiation of ESCs into neural precursor cells [200]. Human ESCs (hESCs) were used for the generation of cholinergic neurons in the vitreous and hippocampal tissues, which are linked to the neuronal network. Furthermore, mESCs and hESCs differentiated into mature basal forebrain cholinergic neurons and transplanted into AD mice resulting in improvement in learning and memory cognition [201,202].
Neural stem cells. Adult NSCs exist in the sub-granular zone of hippocampal dentate gyrus and in the subventricular zone of the lateral walls of the ventricles. They are self-renewing multipotent stem cells that are responsible for the production of neurons, oligodendrocytes, or astrocytes [203]. Growth factor secreting NSCs transplantation in rodent AD models can ameliorate cognitive function and neurogenesis due to the therapeutic potential of NSCs paracrine effect [35]. This paracrine effect was exploited through NSC transplantation in rodent AD models resulting in neuroinflammation reduction and neuronal differentiation by the secretion of neuroprotective and immunomodulatory factors [36]. According to several studies, the transplantation of human NSCs (hNSCs) from the embryonic telomere into the lateral ventricle of AD mice brains can result in NSCs migration and differentiation producing neurons and glial cells. This phenomenon reduces the levels of phosphorylated tau and Aβ1 - 42, decreases glial and astrocyte hyperplasia, stimulates endogenous synapse formation, rises synaptic and nerve fiber density, and ameliorates spatial memory in AD mouse models [204–206]. Many researchers transplanted NSCs within aged transgenic mice in order to express mutant presenilin, tau, and AβPP, and observed that transplanted NSCs results in spatial learning and memory function improvement in mice affected with dementia with no alteration in Aβ pathology. Brain-derived neurotrophic factor (BDNF) is a crucial NSCs neuroprotective factor. Through the increase of hippocampus synaptic density in addition to cholinergic neurons, BDNF could be utilized in the transplanted NSCs of AD rodents. In addition, the production of acetylcholine and retrieving cholinergic neurons integrity induce hNSCs to elevate BDNF levels and neurotrophins of nerve growth factor leading to cognitive and physical improvement in elder mice [35, 205, 207].
Mesenchymal stem cells. MSCs are considered as a type of adult multipotent stem cells but with a unique ability to transdifferentiate into cells not belonging to their tissue of origin, a property that has pushed some authors to classify them as pluripotent. MSCs are highly proliferative, easily handled, and easily accessed as they can be isolated from several tissues and organs including adipose tissues, bone marrow, umbilical cord, amniotic fluid, peripheral blood, muscle, and lung. MSCs can also be applied intravenously as they can cross BBB and travel to the site of injury, a property called homing, which is massively beneficial for patient treatments. Due to these advantages, MSCs are used widely in research and clinical application despite their relatively low neuronal differentiation rates [197, 208–210]. MSCs have various contributions to AD treatment including immune regulation, lowering of Aβ plaque levels through Aβ degradation, and regeneration of damaged neurons [37, 40]. MSCs transplantation also induced neurogenesis, provided support by secreted factors, and enhanced memory deficits and spatial learning [38, 39]. MSCs showed significant overexpression of neuroprotective and inhibition of pro-inflammatory cytokines that demonstrates their immune-modulating and anti-inflammatory effects. MSCs are associated with tissue repair through extracellular vesicle secretion, especially exosomes and microvesicles. Another approach for MSCs that they can be genetically modified and exploited to release extracellular vesicles accompanied by therapeutic agents that target Aβ deposits including siRNAs and enzymes [211, 212]. MSCs can be modulated to stimulate the expression of cytokines and vascular endothelial growth factor as they have regenerative effects in the AD model [213]. Several researchers reported that the application of umbilical cord MSCs in AD murine models showed improvement in memory deficits and learning. Green fluorescent protein– labeled bone marrow MSCs were injected regularly and showed a reduction in the Aβ plaques size within the hippocampus of AD animal models [214]. Placental-derived MSCs were transplanted in the lateral ventricle in AD mouse models showing memory improvement, and reduction in Aβ1–42, AβPP, and BACE1 expression levels [215]. MSCs were injected in AD animal models and induced neural precursor cell to produce hippocampal mature neurons through Wnt pathway activation [216]. Human MSCs were transplanted into aged rats and crossed BBB reaching the brain producing nerve cells and retrieving motor and cognitive activity [217]. Although there are some ethical issues concerning MSCs, particularly commercial cord blood banks, they are considered the major source of stem cells for AD research due to their easy access and handling if harvested after delivery [218, 219].
Induced pluripotent stem cells. iPSCs were first obtained from mouse fibroblasts in 2006 [220]. Differentiated somatic cells are reprogrammed into iPSCs by using specific reprogramming factors. This genetic modification enabled the production of pluripotent cells from patients for autologous transplantation [221]. It is reported that iPSCs can differentiate into various cells as neurons and neurospheres [41, 42]. Human iPSC– derived macrophage-like cells were genetically modified to differentiate into functional neurons generating an Aβ-degrading protease called neprilysin-2. This results in the reduction of Aβ levels in a 5xFAD transgenic mouse model [222]. iPSCs can differentiate into glial cells leading to plaque depositions reduction in addition to cognitive dysfunction improvement in the same previous mouse model [43]. Human iPSCs produced neuronal precursors that were transplanted into transgenic mice hippocampus suffering from severe Aβ deposition and spatial memory dysfunction, where they generated cholinergic neurons and resulted in improved memory dysfunctions [223]. Despite these successful studies, there are some unsolved issues regarding the usage of iPSCs that form massive hurdles in their clinical application. These include formation of teratoma, long-term safety and efficacy, immunogenicity, patient genetic disorders, and ideal reprogramming [44, 45].
CONCLUSION
AD is considered the most serious neurodegenerative problem that threaten the health and life of millions of people worldwide and this number increases every year. The diagnosis and treatment of AD represent a challenge as it is a multifactorial disease with different causes, stages, symptoms, and risk factors. The first criteria for AD diagnosis were set up by NINCDS-ADRDA in 1984 and updated in 2011 for more accuracy. Current diagnostic techniques include neuroimaging diagnosis represented by MRI for hippocampal atrophy measurement, PET to detect Aβ plaques deposition within the brain, and biomarker evidence through serum and CSF to distinguish AD from other forms of dementia. Detecting biomarkers in peripheral blood represent a promising diagnostic technique for monitoring disease progression as several studies reported that the variations in miRNA levels can distinguish between normal and AD cases giving hope for a diagnostic procedure with high potential in AD diagnosis through a simple blood test. Also, the quantification of fatty acids by GC/MS and detection of oxysterols levels could be essential for monitoring AD severity. The current pharmaceutical treatments, which include cholinesterase inhibitors and NMDA antagonists, treat only AD symptoms but do not cure the disease or stop its progression. Future therapies such as acitretin, stem cells, or a combination therapy between both of them can be promising because of the acitretin ability to pass through BBB and stimulate the non-amyloidogenic pathway for AβPP processing resulting in Aβ reduction along with stem cells contribution in regeneration of damaged neurons. In conclusion, AD treatment success relies on the development of very accurate prognostic and diagnostic procedures to estimate the candidates that can develop AD, detect AD at early stages, and monitor its progression at more advanced stages in addition to the development of an effective drug for the treatment of AD patients at different progression stages and those who may develop AD in the future.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | World Health Organization, (2021) . Key facts on dementia. https://www.who.int/news-room/fact-sheets/detail/dementia, Last updated September 20, 2021, Accessed on October 6, 2021. |
[2] | Goedert M , Spillantini MG ((2006) ) A century of Alzheimer’s disease. Science 314: , 777–781. |
[3] | Gao Y , Tan L , Yu JT , Tan L ((2018) ) Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr Alzheimer Res 15: , 283–300. |
[4] | Ashrafian H , Zadeh EH , Khan RH ((2021) ) Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int J Biol Macromol 167: , 382–394. |
[5] | De-Paula VJ , Radanovic M , Diniz BS , Forlenza OV ((2012) ) Alzheimer’s disease. Subcell Biochem 65: , 329–352. |
[6] | Dubois B , Hampel H , Feldman HH , Scheltens P , Aisen P , Andrieu S , Bakardjian H , Benali H , Bertram L , Blennow K , Broich K , Cavedo E , Crutch S , Dartigues JF , Duyckaerts C , Epelbaum S , Frisoni GB , Gauthier S , Genthon R , Gouw AA , Habert MO , Holtzman DM , Kivipelto M , Lista S , Molinuevo JL , O’Bryant SE , Rabinovici GD , Rowe C , Salloway S , Schneider LS , Sperling R , Teichmann M , Carrillo MC , Cummings J , Jack CR Jr ; Proceedings of the Meeting of the International Working Group (IWG) and the American Alzheimer’s Association on “The Preclinical State of AD”; July 23, 2015; Washington DC, USA ((2016) ) Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 12: , 292–323. |
[7] | Wattmo C , Minthon L , Wallin ÅK ((2016) ) Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimers Res Ther 8: , 7. |
[8] | Breijyeh Z , Karaman R ((2020) ) Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 25: , 5789. |
[9] | Apostolova LG ((2016) ) Alzheimer disease. Continuum 22: , 419–434. |
[10] | Knopman DS ((2001) ) An overview of common non-Alzheimer dementias. Clin Geriatr Med 17: , 281–301. |
[11] | Ferri CP , Prince M , Brayne C , Brodaty H , Fratiglioni L , Ganguli M , Hall K , Hasegawa K , Hendrie H , Huang Y , Jorm A , Mathers C , Menezes PR , Rimmer E , Scazufca M , Alzheimer’s Disease International ((2005) ) Global prevalence of dementia: A Delphi consensus study. Lancet 366: , 2112–2117. |
[12] | Grudzien A , Shaw P , Weintraub S , Bigio E , Mash DC , Mesulam MM ((2007) ) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28: , 327–335. |
[13] | Heneka MT , Nadrigny F , Regen T , Martinez-Hernandez A , Dumitrescu-Ozimek L , Terwel D , Jardanhazi-Kurutz D , Walter J , Kirchhoff F , Hanisch UK , Kummer MP ((2010) ) Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. U S A 107: , 6058–6063. |
[14] | Kalaria RN ((2010) ) Vascular basis for brain degeneration: Faltering controls and risk factors for dementia. Nutr Rev 68: (Suppl 2), 74–87. |
[15] | Rogers SL , Friedhoff LT ((1996) ) The efficacy and safety of donepezil in patients with Alzheimer’s disease: Results of a US multicentre, randomized, double-blind, placebo-controlled trial. The Donepezil Study Group. Dementia 7: , 293–303. |
[16] | Woodruff-Pak DS , Tobia MJ , Jiao X , Beck KD , Servatius RJ ((2007) ) Preclinical investigation of the functional effects of memantine and memantine combined with galantamine or donepezil. Neuropsychopharmacology 32: , 1284–1294. |
[17] | Chen GF , Xu TH , Yan Y , Zhou YR , Jiang Y , Melcher K , Xu HE ((2017) ) Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38: , 1205–1235. |
[18] | Riepe MW , Adler G , Ibach B , Weinkauf B , Gunay I , Tracik F ((2006) ) Adding memantine to rivastigmine therapy in patients with mild-to-moderate Alzheimer’s disease: Results of a 12-week, open-label pilot study. Prim Care Companion J Clin Psychiatry 8: , 258–263. |
[19] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[20] | Neugroschl J , Wang S ((2011) ) Alzheimer’s disease: Diagnosis and treatment across the of disease severity. Mt Sinai J Med 78: , 596–612 spectrum. |
[21] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[22] | Yaari R , Fleisher AS , Tariot PN ((2011) ) Updates to diagnostic guidelines for Alzheimer’s disease. Prim Care Companion CNS Disord 13: , PCC.11f01262. |
[23] | Burton EJ , Barber R , Mukaetova-Ladinska EB , Robson J , Perry RH , Jaros E , Kalaria RN , O’Brien JT ((2009) ) Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: A prospective study with pathological verification of diagnosis. Brain 132: (Pt 1), 195–203. |
[24] | Clark CM , Pontecorvo MJ , Beach TG , Bedell BJ , Coleman RE , Doraiswamy PM , Fleisher AS , Reiman EM , Sabbagh MN , Sadowsky CH , Schneider JA , Arora A , Carpenter AP , Flitter ML , Joshi AD , Krautkramer MJ , Lu M , Mintun MA , Skovronsky DM ; AV-45-A16 Study Group ((2012) ) Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: A prospective cohort study. Lancet Neurol 11: , 669–678. Erratum in: Lancet Neurol. 2012 Aug; 11(8):658. |
[25] | Saint-Aubert L , Barbeau EJ , Péran P , Nemmi F , Vervueren C , Mirabel H , Payoux P , Hitzel A , Bonneville F , Gramada R , Tafani M , Vincent C , Puel M , Dechaumont S , Chollet F , Pariente J ((2013) ) Cortical florbetapir-PET amyloid load in prodromal Alzheimer’s disease patients. EJNMMI Res 3: , 43. |
[26] | Weller J , Budson A ((2018) ) Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 7: , F1000 Faculty Rev-1161. |
[27] | Wang T , Xiao S , Liu Y , Lin Z , Su N , Li X , Li G , Zhang M , Fang Y ((2014) ) The efficacy of plasma biomarkers in early diagnosis of Alzheimer’s disease. Int J Geriatr Psychiatry 29: , 713–719. |
[28] | Hye A , Riddoch-Contreras J , Baird AL , Ashton NJ , Bazenet C , Leung R , Westman E , Simmons A , Dobson R , Sattlecker M , Lupton M , Lunnon K , Keohane A , Ward M , Pike I , Zucht HD , Pepin D , Zheng W , Tunnicliffe A , Richardson J , Gauthier S , Soininen H , Kłoszewska I , Mecocci P , Tsolaki M , Vellas B , Lovestone S ((2014) ) Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement 10: , 799–807.e2. |
[29] | Swarbrick S , Wragg N , Ghosh S , Stolzing A ((2019) ) Systematic review of miRNA as biomarkers in Alzheimer’s disease. Mol Neurobiol 56: , 6156–6167. |
[30] | Holthoewer D , Endres K , Schuck F , Hiemke C , Schmitt U , Fahrenholz F ((2012) ) Acitretin, an enhancer of alpha-secretase expression, crosses the blood-brain barrier and is not eliminated by P-glycoprotein. Neurodegener Dis 10: , 224–228. |
[31] | Endres K , Fahrenholz F , Lotz J , Hiemke C , Teipel S , Lieb K , Tüscher O , Fellgiebel A ((2014) ) Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83: , 1930–1935. |
[32] | Liu Y , Weick JP , Liu H , Krencik R , Zhang X , Ma L , Zhou GM , Ayala M , Zhang SC ((2013) ) Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat Biotechnol 31: , 440–447. |
[33] | Jin X , Lin T , Xu Y ((2016) ) Stem cell therapy and immunological rejection in animal models. Curr Mol Pharmacol 9: , 284–288. |
[34] | Liu XY , Yang LP , Zhao L ((2020) ) Stem cell therapy for Alzheimer’s disease. World J Stem Cells 12: , 787–802. |
[35] | Blurton-Jones M , Kitazawa M , Martinez-Coria H , Castello NA , Müller FJ , Loring JF , Yamasaki TR , Poon WW , Green KN , LaFerla FM ((2009) ) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106: , 13594–13599. |
[36] | Zhang Q , Wu HH , Wang Y , Gu GJ , Zhang W , Xia R ((2016) ) Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J Neurochem 136: , 815–825. |
[37] | Elia CA , Losurdo M , Malosio ML , Coco S ((2019) ) Extracellular vesicles from mesenchymal stem cells exert pleiotropic effects on amyloid-β, inflammation, and regeneration: A spark of hope for Alzheimer’s disease from tiny structures? Bioessays 41: , e1800199. |
[38] | Yang H , Xie Z , Wei L , Yang H , Yang S , Zhu Z , Wang P , Zhao C , Bi J ((2013) ) Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-beta deposition in an AβPP/PS1 transgenic mouse model. Stem Cell Res Ther 4: , 76. |
[39] | Matchynski-Franks JJ , Pappas C , Rossignol J , Reinke T , Fink K , Crane A , Twite A , Lowrance SA , Song C , Dunbar GL ((2016) ) Mesenchymal stem cells as treatment for behavioral deficits and neuropathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Transplant 25: , 687–703. |
[40] | Abu Nasra NO , Elzayat EM , Dawood KM , Hagag NM , Yehya MM , Hosney M ((2022) ) Regulatory effect of adipose-derived mesenchymal stem cells and/ or acitretin on Adam10 gene in Alzheimer’s disease rat model. Curr Stem Cell Res Ther 17: , 370–388. |
[41] | Cooper O , Hargus G , Deleidi M , Blak A , Osborn T , Marlow E , Lee K , Levy A , Perez-Torres E , Yow A , Isacson O ((2010) ) Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci 45: , 258–266. |
[42] | Nori S , Okada Y , Yasuda A , Tsuji O , Takahashi Y , Kobayashi Y , Fujiyoshi K , Koike M , Uchiyama Y , Ikeda E , Toyama Y , Yamanaka S , Nakamura M , Okano H ((2011) ) Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc Natl Acad Sci U S A 108: , 16825–16830. |
[43] | Cha MY , Kwon YW , Ahn HS , Jeong H , Lee YY , Moon M , Baik SH , Kim DK , Song H , Yi EC , Hwang D , Kim HS , Mook-Jung I ((2017) ) Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce Aβ deposition in a mouse model of Alzheimer’s disease. Stem Cells Transl Med 6: , 293–305. |
[44] | Hibaoui Y , Feki A ((2012) ) Human pluripotent stem cells: Applications and challenges in neurological diseases. Front Physiol 3: , 267. |
[45] | Araki R , Uda M , Hoki Y , Sunayama M , Nakamura M , Ando S , Sugiura M , Ideno H , Shimada A , Nifuji A , Abe M ((2013) ) Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 494: , 100–104. |
[46] | Soria Lopez JA , González HM , Léger GC ((2019) ) Alzheimer’s disease. Handb Clin Neurol 167: , 231–255. |
[47] | Nikolaev A , McLaughlin T , O’Leary DD , Tessier-Lavigne M ((2009) ) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457: , 981–989. |
[48] | Gu L , Guo Z ((2013) ) Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J Neurochem 126: , 305–311. |
[49] | Wang Z , Wang B , Yang L , Guo Q , Aithmitti N , Songyang Z , Zheng H ((2009) ) Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci 29: , 10788–10801. |
[50] | Kobayashi D , Zeller M , Cole T , Buttini M , McConlogue L , Sinha S , Freedman S , Morris RG , Chen KS ((2008) ) BACE1 gene deletion: Impact on behavioral function in a model of Alzheimer’s disease. Neurobiol Aging 29: , 861–873. |
[51] | Puzzo D , Privitera L , Leznik E , Fà M , Staniszewski A , Palmeri A , Arancio O ((2008) ) Picomolar amyloid-beta positively modulatessynaptic plasticity and memory in hippocampus. J Neurosci 28: , 14537–14545. |
[52] | Spasic D , Raemaekers T , Dillen K , Declerck I , Baert V , Serneels L , Füllekrug J , Annaert W ((2007) ) Rer1p competes with APH-1 for binding to nicastrin and regulates gamma-secretase complex assembly in the early secretory pathway. J Cell Biol 176: , 629–640. |
[53] | Obulesu M , Somashekhar R , Venu R ((2011) ) Genetics of Alzheimer’s disease: An insight into presenilins and apolipoprotein E instigated neurodegeneration. Int J Neurosci 121: , 229–236. |
[54] | Serý O , Povová J , Míšek I , Pešák L , Janout V ((2013) ) Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol 51: , 1–9. |
[55] | Daw EW , Payami H , Nemens EJ , Nochlin D , Bird TD , Schellenberg GD , Wijsman EM ((2000) ) The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet 66: , 196–204. |
[56] | Sing CF , Davignon J ((1985) ) Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am J Hum Genet 37: , 268–285. |
[57] | Saunders AM , Strittmatter WJ , Schmechel D , George-Hyslop PH , Pericak-Vance MA , Joo SH , Rosi BL , Gusella JF , Crapper-MacLachlan DR , Alberts MJ , Hulette C , Crain B , . Goldgaber D , Roses AD ((1993) ) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease . . Neurology 43: , 1467–1472. |
[58] | Liu G , Yao L , Liu J , Jiang Y , Ma G ; Genetic and Environmental Risk for Alzheimer’s disease (GERAD1) Consortium; Chen Z , Zhao B , Li K ((2014) ) Cardiovascular disease contributes to Alzheimer’s disease: Evidence from large-scale genome-wide association studies. Neurobiol Aging 35: , 786–792. |
[59] | Kojro E , Gimpl G , Lammich S , Marz W , Fahrenholz F ((2001) ) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A 98: , 5815–5820. |
[60] | Bodovitz S , Klein WL ((1996) ) Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem 271: , 4436–4440. |
[61] | Gamba P , Testa G , Gargiulo S , Staurenghi E , Poli G , Leonarduzzi G ((2015) ) Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front Aging Neurosci 7: , 119. |
[62] | Björkhem I , Cedazo-Minguez A , Leoni V , Meaney S ((2009) ) Oxysterols and neurodegenerative diseases. Mol Aspects Med 30: , 171–179. |
[63] | Hudry E , Van Dam D , Kulik W , De Deyn PP , Stet FS , Ahouansou O , Benraiss A , Delacourte A , Bougnères P , Aubourg P , Cartier N ((2010) ) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 18: , 44–53. |
[64] | Djelti F , Braudeau J , Hudry E , Dhenain M , Varin J , Bièche I , Marquer C , Chali F , Ayciriex S , Auzeil N , Alves S , Langui D , Potier MC , Laprevote O , Vidaud M , Duyckaerts C , Miles R , Aubourg P , Cartier N ((2015) ) CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138: (Pt 8), 2383–2398. |
[65] | Meaney S , Heverin M , Panzenboeck U , Ekström L , Axelsson M , Andersson U , Diczfalusy U , Pikuleva I , Wahren J , Sattler W , Björkhem I ((2007) ) Novel route for elimination of brain oxysterols across the blood-brain barrier: Conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J Lipid Res 48: , 944–951. |
[66] | Shafaati M , Marutle A , Pettersson H , Lövgren-Sandblom A , Olin M , Pikuleva I , Winblad B , Nordberg A , Björkhem I ((2011) ) Marked accumulation of 27-hydroxycholesterol in the brains of Alzheimer’s patients with the Swedish APP 670/671 mutation. J Lipid Res 52: , 1004–1010. |
[67] | Testa G , Staurenghi E , Zerbinati C , Gargiulo S , Iuliano L , Giaccone G , Fantò F , Poli G , Leonarduzzi G , Gamba P ((2016) ) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10: , 24–33. |
[68] | Zarrouk A , Vejux A , Mackrill J , O’Callaghan Y , Hammami M , O’Brien N , Lizard G ((2014) ) Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res Rev 18: , 148–162. |
[69] | Lütjohann D , Papassotiropoulos A , Björkhem I , Locatelli S , Bagli M , Oehring RD , Schlegel U , Jessen F , Rao ML , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41: , 195–198. |
[70] | Kaye JA , Swihart T , Howieson D , Dame A , Moore MM , Karnos T , Camicioli R , Ball M , Oken B , Sexton G ((1997) ) Volume loss of the hippocampus and temporal lobe in healthy elderly persons destined to develop dementia. Neurology 48: , 1297–1304. |
[71] | Jack CR Jr , Petersen RC , Xu Y , O’Brien PC , Smith GE , Ivnik RJ , Tangalos EG , Kokmen E ((1998) ) Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology 51: , 993–999. |
[72] | Papassotiropoulos A , Lütjohann D , Bagli M , Locatelli S , Jessen F , Rao ML , Maier W , Björkhem I , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol: A peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport 11: , 1959–1962. |
[73] | Saucier SE , Kandutsch AA , Gayen AK , Swahn DK , Spencer TA ((1989) ) Oxysterol regulators of 3-hydroxy-3-methylglutaryl-CoA reductase in liver. Effect of dietary cholesterol. J Biol Chem 264: , 6863–6869. |
[74] | Leoni V , Masterman T , Patel P , Meaney S , Diczfalusy U , Björkhem I ((2003) ) Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J Lipid Res 44: , 793–799. |
[75] | Yau JL , Rasmuson S , Andrew R , Graham M , Noble J , Olsson T , Fuchs E , Lathe R , Seckl JR ((2003) ) Dehydroepiandrosterone 7-hydroxylase CYP7B: predominant expression in primate hippocampus and reduced expressionin Alzheimer’s disease. Neuroscience 121: , 307–314. |
[76] | Hammouda S , Ghzaiel I , Khamlaoui W , Hammami S , Mhenni SY , Samet S , Hammami M , Zarrouk A ((2020) ) Genetic variants in FADS1 and ELOVL2 increase level of arachidonic acid and the risk of Alzheimer’s disease in the Tunisian population. Prostaglandins Leukot Essent Fatty Acids 160: , 102159. |
[77] | Astarita G , Jung KM , Berchtold NC , Nguyen VQ , Gillen DL , Head E , Cotman CW , Piomelli D ((2010) ) Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS One 5: , e12538. |
[78] | Lizard G , Rouaud O , Demarquoy J , Cherkaoui-Malki M , Iuliano L ((2012) ) Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J Alzheimers Dis 29: , 241–254. |
[79] | Neve RL , Harris P , Kosik KS , Kurnit DM , Donlon TA ((1986) ) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res 387: , 271–280. |
[80] | Iqbal K , Liu F , Gong CX , Grundke-Iqbal I ((2010) ) Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 7: , 656–664. |
[81] | Kins S , Kurosinski P , Nitsch RM , Götz J ((2003) ) Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol 163: , 833–843. |
[82] | Bakota L , Brandt R ((2016) ) Tau biology and tau-directed therapies for Alzheimer’s disease. Drugs 76: , 301–313. |
[83] | Imhof A , Kövari E , von Gunten A , Gold G , Rivara CB , Herrmann FR , Hof PR , Bouras C , Giannakopoulos P ((2007) ) Morphological substrates of cognitive decline in nonagenarians and centenarians: A new paradigm? J Neurol Sci 257: , 72–79. |
[84] | Miller FD , Hicks SP , D’Amato CJ , Landis JR ((1984) ) A descriptive study of neuritic plaques and neurofibrillary tangles in an autopsy population. Am J Epidemiol 120: , 331–341. |
[85] | Arriagada PV , Marzloff K , Hyman BT ((1992) ) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42: , 1681–1688. |
[86] | Armstrong RA ((2012) ) β-amyloid (Aβ) deposition in cognitively normal brain, dementia with Lewy bodies, and Alzheimer’s disease: A study using principal components analysis. Folia Neuropathol 50: , 130–139. |
[87] | Chartier-Harlin MC , Crawford F , Houlden H , Warren A , Hughes D , Fidani L , Goate A , Rossor M , Roques P , Hardy J , Mullan M ((1991) ) Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353: , 844–846. |
[88] | Delacourte A , Sergeant N , Champain D , Wattez A , Maurage CA , Lebert F , Pasquier F , David JP ((2002) ) Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 59: , 398–407. |
[89] | Armstrong RA ((2019) ) Risk factors for Alzheimer’s disease. Folia Neuropathol 57: , 87–105. |
[90] | Chapuis J , Flaig A , Grenier-Boley B , Eysert F , Pottiez V , Deloison G , Vandeputte A , Ayral AM , Mendes T , Desai S , Goate AM , Kauwe JSK , Leroux F , Herledan A , Demiautte F , Bauer C , Checler F , Petersen RC , Blennow K , Zetterberg H , Minthon L , Van Deerlin VM , Lee VM , Shaw LM , Trojanowski JQ , Albert M , Moghekar A , O’Brien R , Peskind ER , Malmanche N , Schellenberg GD , Dourlen P , Song OR , Cruchaga C , Amouyel P , Deprez B , Brodin P , Lambert JC; ADGC , Alzheimer’s Disease Neuroimaging Initiative ((2017) ) Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol 133: , 955–966. |
[91] | Honarnejad K , Herms J ((2012) ) Presenilins: Role in calcium homeostasis. Int J Biochem Cell Biol 44: , 1983–1986. |
[92] | Barthet G , Dunys J , Shao Z , Xuan Z , Ren Y , Xu J , Arbez N , Mauger G , Bruban J , Georgakopoulos A , Shioi J , Robakis NK ((2013) ) Presenilinmediates neuroprotective functions of ephrin-B and brain-derivedneurotrophic factor and regulates ligand-induced internalization andmetabolism of EphB2 and TrkB receptors. Neurobiol Aging 34: , 499–510. |
[93] | Francis YI , Stephanou A , Latchman DS ((2006) ) CREB-binding protein activation by presenilin 1 but not by its M146L mutant. Neuroreport 17: , 917–921. |
[94] | Zatti G , Burgo A , Giacomello M , Barbiero L , Ghidoni R , Sinigaglia G , Florean C , Bagnoli S , Binetti G , Sorbi S , Pizzo P , Fasolato C ((2006) ) Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 39: , 539–550. |
[95] | Oyama F , Shimada H , Oyama R , Ihara Y ((1995) ) Apolipoprotein E genotype, Alzheimer’s pathologies and related gene expression in the aged population. Brain Res Mol Brain Res 29: , 92–98. |
[96] | Beffert U , Poirier J ((1996) ) Apolipoprotein E, plaques, tangles and cholinergic dysfunction in Alzheimer’s disease. Ann N Y Acad Sci 777: , 166–174. |
[97] | Allen M , Cox C , Belbin O , Ma L , Bisceglio GD , Wilcox SL , Howell CC , Hunter TA , Culley O , Walker LP , Carrasquillo MM , Dickson DW , Petersen RC , Graff-Radford NR , Younkin SG , Ertekin-Taner N ((2012) ) Association and heterogeneity at the GAPDH locus in Alzheimer’s disease. Neurobiol Aging 33: , 203.e25–33. |
[98] | Soler-López M , Badiola N , Zanzoni A , Aloy P ((2012) ) Towards Alzheimer’s root cause: ECSIT as an integrating hub between oxidative stress, inflammation and mitochondrial dysfunction. Hypothetical role of the adapter protein ECSIT in familial and sporadic Alzheimer’s disease pathogenesis. Bioessays 34: , 532–541. |
[99] | Ryan J , Carrière I , Carcaillon L , Dartigues JF , Auriacombe S , Rouaud O , Berr C , Ritchie K , Scarabin PY , Ancelin ML ((2014) ) Estrogen receptor polymorphisms and incident dementia: The prospective 3C study. Alzheimers Dement 10: , 27–35. |
[100] | Ruiz A , Dols-Icardo O , Bullido MJ , Pastor P , Rodríguez-Rodríguez E , López de Munain A , de Pancorbo MM , Pérez-Tur J , Alvarez V , Antonell A , López-Arrieta J , Hernández I , Tárraga L , Boada M , Lleó A , Blesa R , Frank-García A , Sastre I , Razquin C , Ortega-Cubero S , Lorenzo E , Sánchez-Juan P , Combarros O , Moreno F , Gorostidi A , Elcoroaristizabal X , Baquero M , Coto E , Sánchez-Valle R , Clarimón J ; dementia genetic Spanish consortium (DEGESCO) ((2014) ) Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging 35: , 444.e1–4. |
[101] | Ponomareva N , Andreeva T , Protasova M , Shagam L , Malina D , Goltsov A , Fokin V , Mitrofanov A , Rogaev E ((2013) ) Age-dependent effect of Alzheimer’s risk variant of CLU on EEG alpha rhythm in non-demented adults. Front Aging Neurosci 5: , 86. |
[102] | Khorram Khorshid HR , Gozalpour E , Saliminejad K , Karimloo M , Ohadi M , Kamali K ((2013) ) Vitamin D Receptor (VDR) Polymorphisms and late-onset Alzheimer’s disease: An association study. Iran J Public Health 42: , 1253–1258. |
[103] | Wang Y , Xu S , Liu Z , Lai C , Xie Z , Zhao C , Wei Y , Bi JZ ((2013) ) Meta-analysis on the association between the TF gene rs1049296 and AD. Can J Neurol Sci 40: , 691–697. |
[104] | Armstrong RA ((2013) ) What causes Alzheimer’s disease? Folia Neuropathol 51: , 169–188. |
[105] | Peters S , Reid A , Fritschi L , de Klerk N , Musk AW ((2013) ) Long-term effects of aluminium dust inhalation. Occup Environ Med 70: , 864–868. |
[106] | Wang Z , Wei X , Yang J , Suo J , Chen J , Liu X , Zhao X ((2016) ) Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci Lett 610: , 200–206. |
[107] | Mutter J , Naumann J , Schneider R , Walach H ((2007) ) Quecksilber und Alzheimer-Erkrankung [Mercury and Alzheimer’s disease]. Fortschr Neurol Psychiatr 75: , 528–538. |
[108] | McCord MC , Aizenman E ((2014) ) The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front Aging Neurosci 6: , 77. |
[109] | Notarachille G , Arnesano F , Calò V , Meleleo D ((2014) ) Heavy metals toxicity: Effect of cadmium ions on amyloid beta protein 1-42. Possible implications for Alzheimer’s disease. Biometals 27: , 371–388. |
[110] | Pal A , Siotto M , Prasad R , Squitti R ((2015) ) Towards a unified vision of copper involvement in Alzheimer’s disease: A review connecting basic, experimental, and clinical research. J Alzheimers Dis 44: , 343–354. |
[111] | Min JY , Min KB ((2016) ) Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ Health 15: , 69. |
[112] | Veronese N , Zurlo A , Solmi M , Luchini C , Trevisan C , Bano G , Manzato E , Sergi G , Rylander R ((2016) ) Magnesium status in Alzheimer’s disease: A systematic review. Am J Alzheimers Dis Other Demen 31: , 208–213. |
[113] | Du K , Liu M , Pan Y , Zhong X , Wei M ((2017) ) Association of serum manganese levels with Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Nutrients 9: , 231. |
[114] | Hsu HW , Bondy SC , Kitazawa M ((2018) ) Environmental and dietary exposure to copper and its cellular mechanisms linking to Alzheimer’s disease. Toxicol Sci 163: , 338–345. |
[115] | Xu H , Finkelstein DI , Adlard PA ((2014) ) Interactions of metals and Apolipoprotein E in Alzheimer’s disease. Front Aging Neurosci 6: , 121. |
[116] | Abalan F ((1984) ) Alzheimer’s disease and malnutrition: A new etiological hypothesis. Med Hypotheses 15: , 385–393. |
[117] | Grimm MO , Mett J , Hartmann T ((2016) ) The impact of vitamin E and other fat-soluble vitamins on Alzheimer’s disease. Int J Mol Sci 17: , 1785. |
[118] | Tolppanen AM , Ngandu T , Kåreholt I , Laatikainen T , Rusanen M , Soininen H , Kivipelto M ((2014) ) Midlife and late-life body mass index and late-life dementia: Results from a prospective population-based cohort. J Alzheimers Dis 38: , 201–209. |
[119] | Mazon JN , de Mello AH , Ferreira GK , Rezin GT ((2017) ) The impact of obesity on neurodegenerative diseases. Life Sci 182: , 22–28. |
[120] | den Hoedt S , Janssen CIF , Astarita G , Piomelli D , Leijten FPJ , Crivelli SM , Verhoeven AJM , de Vries HE , Walter J , Martinez-Martinez P , Sijbrands EJG , Kiliaan AJ , Mulder MT ((2017) ) Pleiotropic effect of human ApoE4 on cerebral ceramide and saturated fatty acid levels. J Alzheimers Dis 60: , 769–781. |
[121] | Hampel H , Bürger K , Teipel SJ , Bokde AL , Zetterberg H , Blennow K ((2008) ) Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement 4: , 38–48. |
[122] | Peskind E , Nordberg A , Darreh-Shori T , Soininen H ((2009) ) Safety of lumbar puncture procedures in patients with Alzheimer’s disease. Curr Alzheimer Res 6: , 290–292. |
[123] | Mulder C , Verwey NA , van der Flier WM , Bouwman FH , Kok A , van Elk EJ , Scheltens P , Blankenstein MA ((2010) ) Amyloid-beta(1-42), totaltau, and phosphorylated tau as cerebrospinal fluid biomarkers forthe diagnosis of Alzheimer disease. Clin Chem 56: , 248–253. |
[124] | Bartel DP ((2004) ) MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: , 281–297. |
[125] | Ha M , Kim VN ((2014) ) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: , 509–524. |
[126] | Vishnoi A , Rani S ((2017) ) miRNA biogenesis and regulation of diseases: An overview. Methods Mol Biol 1509: , 1–10. |
[127] | Lee Y , Jeon K , Lee JT , Kim S , Kim VN ((2002) ) MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J 21: , 4663–4670. |
[128] | Connerty P , Ahadi A , Hutvagner G ((2015) ) RNA binding proteins in the miRNA pathway. Int J Mol Sci 17: , 31. |
[129] | Yi R , Qin Y , Macara IG , Cullen BR ((2003) ) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: , 3011–3016. |
[130] | Lund E , Güttinger S , Calado A , Dahlberg JE , Kutay U ((2004) ) Nuclear export of microRNA precursors. Science 303: , 95–98. |
[131] | Meijer HA , Smith EM , Bushell M ((2014) ) Regulation of miRNA strand selection: Follow the leader? Biochem Soc Trans 42: , 1135–1140. |
[132] | Zhu MY , Zhang W , Yang T ((2016) ) Diverse microRNAs with convergent functions regulate tumorigenesis. Oncol Lett 11: , 915–920. |
[133] | Valencia-Sanchez MA , Liu J , Hannon GJ , Parker R ((2006) ) Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 20: , 515–524. |
[134] | Zhang QS , Liu W , Lu GX ((2017) ) miR-200a-3p promotes β-amyloid-induced neuronal apoptosis through down-regulation of SIRT1 in Alzheimer’s disease. J Biosci 42: , 397–404. |
[135] | Higaki S , Muramatsu M , Matsuda A , Matsumoto K , Satoh JI , Michikawa M , Niida S ((2018) ) Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS One 13: , e0196929. |
[136] | Hébert SS , Horré K , Nicolaï L , Papadopoulou AS , Mandemakers W , Silahtaroglu AN , Kauppinen S , Delacourte A , De Strooper B ((2008) ) Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A 105: , 6415–6420. |
[137] | Yang G , Song Y , Zhou X , Deng Y , Liu T , Weng G , Yu D , Pan S ((2015) ) MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role and has a neuro-protective role in vitro and in vivo.. Mol Med Rep 12: , 3081–3088. |
[138] | Geekiyanage H , Chan C ((2011) ) MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J Neurosci 31: , 14820–14830. |
[139] | Geekiyanage H , Chan C ((2012) ) SPT, miR-137 and miR-181c: Therapeutic targets and noninvasive biomarkers. Alzheimers Dement 8: , 470–471. |
[140] | Davare MA , Hell JW ((2003) ) Increased phosphorylation of the neuronal L-type Ca (2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci U S A 100: , 16018–16023. |
[141] | Jakobsson J , Pålsson E , Sellgren C , Rydberg F , Ekman A , Zetterberg H , Blennow K , Landén M ((2016) ) CACNA1C polymorphism and altered phosphorylation of tau in bipolar disorder. Br J Psychiatry 208: , 195–196. |
[142] | Donmez G , Outeiro TF ((2013) ) SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol Med 5: , 344–352. |
[143] | Jian C , Lu M , Zhang Z , Liu L , Li X , Huang F , Xu N , Qin L , Zhang Q , Zou D ((2017) ) miR-34a knockout attenuates cognitive deficits in APP/PS1 mice through inhibition of the amyloidogenic processing of APP. Life Sci 182: , 104–111. |
[144] | Xu Y , Chen P , Wang X , Yao J , Zhuang S ((2018) ) miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci Lett 670: , 94–104. |
[145] | Ghasemi-Kasman M , Shojaei A , Gol M , Moghadamnia AA , Baharvand H , Javan M ((2018) ) miR-302/367-induced neurons reduce behavioral impairment in an experimental model of Alzheimer’s disease. Mol Cell Neurosci 86: , 50–57. |
[146] | Gao Z , Zhu X , Dou Y ((2015) ) The miR-302/367 cluster: A comprehensive update on its evolution and functions. Open Biol 5: , 150138. |
[147] | Sorial ME , El Sayed NSED ((2017) ) Protective effect of valproic acid in streptozotocin-induced sporadic Alzheimer’s disease mouse model: Possible involvement of the cholinergic system. Naunyn Schmiedebergs Arch Pharmacol 390: , 581–593. |
[148] | Tan L , Yu JT , Liu QY , Tan MS , Zhang W , Hu N , Wang YL , Sun L , Jiang T , Tan L ((2014) ) Circulating miR-125b as a biomarker of Alzheimer’s disease. J Neurol Sci 336: , 52–56. |
[149] | Unno K , Takabayashi F , Yoshida H , Choba D , Fukutomi R , Kikunaga N , Kishido T , Oku N , Hoshino M ((2007) ) Daily consumption of green tea catechin delays memory regression in aged mice. Biogerontology 8: , 89–95. |
[150] | Schaffer S , Asseburg H , Kuntz S , Muller WE , Eckert GP ((2012) ) Effects of polyphenols on brain ageing and Alzheimer’s disease: Focus on mitochondria. Mol Neurobiol 46: , 161–178. |
[151] | Wang G , Huang Y , Wang LL , Zhang YF , Xu J , Zhou Y , Lourenco GF , Zhang B , Wang Y , Ren RJ , Halliday GM , Chen SD ((2016) ) MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci Rep 6: , 26697. |
[152] | Vemula S , Shi J , Hanneman P , Wei L , Kapur R ((2010) ) ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood 115: , 1785–1796. |
[153] | Alexandrov P , Zhai Y , Li W , Lukiw W ((2019) ) Lipopolysaccharide-stimulated, NF-kB-, miRNA-146a- and miRNA-155-mediated molecular-genetic communication between the human gastrointestinal tract microbiome and the brain. Folia Neuropathol 57: , 211–219. |
[154] | Sierksma A , Lu A , Salta E , Vanden Eynden E , Callaerts-Vegh Z , D’Hooge R , Blum D , Buée L , Fiers M , De Strooper B ((2018) ) Deregulation of neuronal miRNAs induced by amyloid-β or TAU pathology. Mol Neurodegener 13: , 54. |
[155] | Jones SV , Kounatidis I ((2017) ) Nuclear factor-kappa B and Alzheimer disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol 8: , 1805. |
[156] | Bronzuoli MR , Iacomino A , Steardo L , Scuderi C ((2016) ) Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res 9: , 199–208. |
[157] | Kaur U , Banerjee P , Bir A , Sinha M , Biswas A , Chakrabarti S ((2015) ) Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: The NF-κB connection. Curr Top Med Chem 15: , 446–457. |
[158] | Dong H , Li J , Huang L , Chen X , Li D , Wang T , Hu C , Xu J , Zhang C , Zen K , Xiao S , Yan Q , Wang C , Zhang CY ((2015) ) Serum microRNA profiles serve as novel biomarkers for the diagnosis of Alzheimer’s disease. Dis Markers 2015: , 625659. |
[159] | Denk J , Boelmans K , Siegismund C , Lassner D , Arlt S , Jahn H ((2015) ) MicroRNA profiling of CSF reveals potential biomarkers to detect Alzheimer’s disease. PLoS One 10: , e0126423. |
[160] | Lau P , Bossers K , Janky R , Salta E , Frigerio CS , Barbash S , Rothman R , Sierksma AS , Thathiah A , Greenberg D , Papadopoulou AS , Achsel T , Ayoubi T , Soreq H , Verhaagen J , Swaab DF , Aerts S , De Strooper B ((2013) ) Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med 5: , 1613–1634. |
[161] | Wong HK , Veremeyko T , Patel N , Lemere CA , Walsh DM , Esau C , Vanderburg C , Krichevsky AM ((2013) ) De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet 22: , 3077–3092. |
[162] | Salta E , Sierksma A , Vanden Eynden E , De Strooper B ((2016) ) miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med 8: , 1005–1018. |
[163] | Smith PY , Hernandez-Rapp J , Jolivette F , Lecours C , Bisht K , Goupil C , Dorval V , Parsi S , Morin F , Planel E , Bennett DA , Fernandez-Gomez FJ , Sergeant N , Buée L , Tremblay MÈ , Calon F , Hébert SS ((2015) ) miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation. Hum Mol Genet 24: , 6721–6735. |
[164] | Ambegaokar SS , Jackson GR ((2012) ) The downward spiral of tau and autolysosomes: A new hypothesis in neurodegeneration. Autophagy 8: , 1144–1145. |
[165] | Hernandez-Rapp J , Rainone S , Goupil C , Dorval V , Smith PY , Saint-Pierre M , Vallée M , Planel E , Droit A , Calon F , Cicchetti F , Hébert SS ((2016) ) microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci Rep 6: , 30953. |
[166] | Zarrouk A , Riedinger JM , Ahmed SH , Hammami S , Chaabane W , Debbabi M , Ben Ammou S , Rouaud O , Frih M , Lizard G , Hammami M ((2015) ) Fatty acid profiles in demented patients: Identification of hexacosanoic acid (C26:0) as a blood lipid biomarker of dementia. J Alzheimers Dis 44: , 1349–1359. |
[167] | Zarrouk A , Hammouda S , Ghzaiel I , Hammami S , Khamlaoui W , Ahmed SH , Lizard G , Hammami M ((2020) ) Association between oxidative stress and altered cholesterol metabolism in Alzheimer’s disease patients. Curr Alzheimer Res 17: , 823–834. |
[168] | Zarrouk A , Debbabi M , Bezine M , Karym EM , Badreddine A , Rouaud O , Moreau T , Cherkaoui-Malki M , El Ayeb M , Nasser B , Hammami M , Lizard G ((2018) ) Lipid biomarkers in Alzheimer’s disease. Curr Alzheimer Res 15: , 303–312. |
[169] | Zhang X , Xi Y , Yu H , An Y , Wang Y , Tao L , Wang Y , Liu W , Wang T , Xiao R ((2019) ) 27-hydroxycholesterol promotes Aβ accumulation via altering Aβ metabolism in mild cognitive impairment patients and APP/PS1 mice. Brain Pathol 29: , 558–573. |
[170] | Zhao Y , Jaber VR , Pogue AI , Sharfman NM , Taylor C , Lukiw WJ ((2022) ) Lipopolysaccharides (LPSs) as potent neurotoxic glycolipids in Alzheimer’s disease (AD). Int J Mol Sci 23: , 12671. |
[171] | Eldufani J , Blaise G ((2019) ) The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimers Dement (N Y) 5: , 175–183. |
[172] | Sharma K ((2019) ) Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol Med Rep 20: , 1479–1487. |
[173] | Singh R , Sadiq NM ((2020) ) Cholinesterase Inhibitors. In StatPearls, StatPearls Publishing, Treasure Island, FL, USA, Available online: https://www.ncbi.nlm.nih.gov/books/NBK544336/, Updated July 18, 2022, Accessed on December 8, 2020. |
[174] | Wang R , Reddy PH ((2017) ) Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis 57: , 1041–1048. |
[175] | Cacabelos R ((2007) ) Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat 3: , 303–333. |
[176] | Kumar A , Gupta V , Sharma S ((2022) ) Donepezil. [Updated December 22, 2021]. In StatPearls [Internet]. StatPearls Publishing;, Treasure Island, FL, USA. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513257/ |
[177] | Birks J , Grimley Evans J , Iakovidou V , Tsolaki M , Holt FE ((2009) ) Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev 2: , CD001191. |
[178] | Khoury R , Rajamanickam J , Grossberg GT ((2018) ) An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther Adv Drug Saf 9: , 171–178. |
[179] | Scott LJ , Goa KL ((2000) ) Galantamine: A review of its use in Alzheimer’s disease. Drugs 60: , 1095–122. |
[180] | Wahba SM , Darwish AS , Kamal SM ((2016) ) Ceria-containing uncoated andcoated hydroxyapatite-based galantamine nanocomposites forformidable treatment of Alzheimer’s disease in ovariectomizedalbino-rat model. Eng C Mater Biol Appl 65: , 151–163. |
[181] | Kim JK , Park SU ((2017) ) Pharmacological aspects of galantamine for the treatment of Alzheimer’s disease. EXCLI J 16: , 35–39. |
[182] | Huang YJ , Lin CH , Lane HY , Tsai GE ((2012) ) NMDA neurotransmission dysfunction in behavioral and psychological symptoms of Alzheimer’s disease. Curr Neuropharmacol 10: , 272–285. |
[183] | Liu J , Chang L , Song Y , Li H , Wu Y ((2019) ) The role of NMDA receptors in Alzheimer’s disease. Front Neurosci 13: , 43. |
[184] | Companys-Alemany J , Turcu AL , Bellver-Sanchis A , Loza MI , Brea JM , Canudas AM , Leiva R , Vázquez S , Pallàs M , Griñán-Ferré C ((2020) ) A novel NMDA receptor antagonist protects against cognitive decline presented by senescent mice. Pharmaceutics 12: , 284. |
[185] | Folch J , Busquets O , Ettcheto M , Sánchez-López E , Castro-Torres RD , Verdaguer E , Garcia ML , Olloquequi J , Casadesús G , Beas-Zarate C , Pelegri C , Vilaplana J , Auladell C , Camins A ((2018) ) Memantine for the treatment of dementia: A review on its current and future applications. J Alzheimers Dis 62: , 1223–1240. |
[186] | Kuns B , Rosani A , Varghese D ((2022) ) Memantine. [Updated July 11, 2022]. In StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL, USA. Available from: https://www.ncbi.nlm.nih.gov/books/NBK500025/ (accessed on December 8, 2020). |
[187] | ((2015) ) Namzaric–a combination of 2 old drugs for Alzheimer’s disease. Med Lett Drugs Ther 57: , 105–106. |
[188] | Goodman AB , Pardee AB ((2003) ) Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Natl Acad Sci U S A 100: , 2901–2905. |
[189] | Goodman AB ((2006) ) Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J Cell Physiol 209: , 598–603. |
[190] | Prinzen C , Müller U , Endres K , Fahrenholz F , Postina R ((2005) ) Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J 19: , 1522–1524. |
[191] | Endres K , Postina R , Schroeder A , Mueller U , Fahrenholz F ((2005) ) Shedding of the amyloid precursor protein-like protein APLP2 by disintegrin-metalloproteinases. FEBS J 272: , 5808–5820. |
[192] | Tippmann F , Hundt J , Schneider A , Endres K , Fahrenholz F ((2009) ) Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J 23: , 1643–1654. |
[193] | Lee CS , Koo J ((2005) ) A review of acitretin, a systemic retinoid for the treatment of psoriasis. Expert Opin Pharmacother 6: , 1725–1734. |
[194] | Rosales Jubal E , Schwalm M , Dos Santos Guilherme M , Schuck F , Reinhardt S , Tose A , Barger Z , Roesler MK , Ruffini N , Wierczeiko A , Schmeisser MJ , Schmitt U , Endres K , Stroh A ((2021) ) Acitretin reverses early functional network degradation in a mouse model of familial Alzheimer’s disease. Sci Rep 11: , 6649. |
[195] | Ilic D , Polak JM ((2011) ) Stem cells in regenerative medicine: Introduction. Br Med Bull 98: , 117–126. |
[196] | Pera MF , Trounson AO ((2004) ) Human embryonic stem cells: Prospects for development. Development 131: , 5515–5525. |
[197] | Vasic V , Barth K , Schmidt MHH ((2019) ) Neurodegeneration and neuro-regeneration-Alzheimer’s disease and stem cell therapy. Int J Mol Sci 20: , 4272. |
[198] | Richards M , Fong CY , Chan WK , Wong PC , Bongso A ((2002) ) Human feeders support prolonged undifferentiated growth of human inner cell masses and embryonic stem cells. Nat Biotechnol 20: , 933–936. |
[199] | Fujikawa T , Oh SH , Pi L , Hatch HM , Shupe T , Petersen BE ((2005) ) Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am J Pathol 166: , 1781–1791. |
[200] | Moghadam FH , Alaie H , Karbalaie K , Tanhaei S , Nasr Esfahani MH , Baharvand H ((2009) ) Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation 78: , 59–68. |
[201] | Bissonnette CJ , Lyass L , Bhattacharyya BJ , Belmadani A , Miller RJ , Kessler JA ((2011) ) The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 29: , 802–811. |
[202] | Yue W , Li Y , Zhang T , Jiang M , Qian Y , Zhang M , Sheng N , Feng S , Tang K , Yu X , Shu Y , Yue C , Jing N ((2015) ) ESC-derived basal forebrain cholinergic neurons ameliorate the cognitive symptoms associated with Alzheimer’s disease in mouse models. Stem Cell Rep 5: , 776–790. |
[203] | Shimada IS , LeComte MD , Granger JC , Quinlan NJ , Spees JL ((2012) ) Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci 32: , 7926–7940. |
[204] | Ager RR , Davis JL , Agazaryan A , Benavente F , Poon WW , LaFerla FM , Blurton-Jones M ((2015) ) Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 25: , 813–826. |
[205] | Lee IS , Jung K , Kim IS , Lee H , Kim M , Yun S , Hwang K , Shin JE , Park KI ((2015) ) Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol Neurodegener 10: , 38. |
[206] | Li X , Zhu H , Sun X , Zuo F , Lei J , Wang Z , Bao X , Wang R ((2016) ) Human neural stem cell transplantation rescues cognitive defects in APP/PS1 model of Alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front Aging Neurosci 8: , 282. |
[207] | Xuan AG , Luo M , Ji WD , Long DH ((2009) ) Effects of engrafted neural stem cells in Alzheimer’s disease rats. Neurosci Lett 450: , 167–171. |
[208] | Lee J , Kuroda S , Shichinohe H , Ikeda J , Seki T , Hida K , Tada M , Sawada K , Iwasaki Y ((2003) ) Migration and differentiation of nuclear fluorescence-labeled bone marrow stromal cells after transplantation into cerebral infarct and spinal cord injury in mice. Neuropathology 23: , 169–180. |
[209] | Hass R , Kasper C , Böhm S , Jacobs R ((2011) ) Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal 9: , 12. |
[210] | Lykhmus O , Koval L , Voytenko L , Uspenska K , Komisarenko S , Deryabina O , Shuvalova N , Kordium V , Ustymenko A , Kyryk V , Skok M ((2019) ) Intravenously injected mesenchymal stem cells penetrate the brain and treat inflammation-induced brain damage and memory impairment in mice. Front Pharmacol 10: , 355. |
[211] | Alvarez-Erviti L , Seow Y , Yin H , Betts C , Lakhal S , Wood MJ ((2011) ) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 29: , 341–345. |
[212] | Katsuda T , Tsuchiya R , Kosaka N , Yoshioka Y , Takagaki K , Oki K , Takeshita F , Sakai Y , Kuroda M , Ochiya T ((2013) ) Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci Rep 3: , 1197. |
[213] | Garcia KO , Ornellas FL , Martin PK , Patti CL , Mello LE , Frussa-Filho R , Han SW , Longo BM ((2014) ) Therapeutic effects of the transplantation of VEGF overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of Alzheimer’s disease. Front Aging Neurosci 6: , 30. |
[214] | Naaldijk Y , Jäger C , Fabian C , Leovsky C , Blüher A , Rudolph L , Hinze A , Stolzing A ((2017) ) Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS1 Alzheimer mice. Neuropathol Appl Neurobiol 43: , 299–314. |
[215] | Yun HM , Kim HS , Park KR , Shin JM , Kang AR , il Lee K , Song S , Kim YB , Han SB , Chung HM , Hong JT ((2013) ) Placenta-derived mesenchymal stem cells improve memory dysfunction in an Aβ1 - 42-infused mouse model of Alzheimer’s disease. Cell Death Dis 4: , e958. |
[216] | Oh SH , Kim HN , Park HJ , Shin JY , Lee PH ((2015) ) Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the Wnt signaling pathway in an Alzheimer’s disease model. Cell Transplant 24: , 1097–1109. |
[217] | Park D , Yang G , Bae DK , Lee SH , Yang YH , Kyung J , Kim D , Choi EK , Choi KC , Kim SU , Kang SK , Ra JC , Kim YB ((2013) ) Human adipose tissue-derived mesenchymal stem cells improve cognitive function and physical activity in ageing mice. J Neurosci Res 91: , 660–670. |
[218] | Lee HJ , Lee JK , Lee H , Shin JW , Carter JE , Sakamoto T , Jin HK , Bae JS ((2010) ) The therapeutic potential of human umbilical cord blood-derived mesenchymal stem cells in Alzheimer’s disease. Neurosci Lett 481: , 30–35. |
[219] | Takahashi K , Yamanaka S ((2006) ) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: , 663–676. |
[220] | Yang H , Yang H , Xie Z , Wei L , Bi J ((2013) ) Systemic transplantation of human umbilical cord derived mesenchymal stem cells-educated T regulatory cells improved the impaired cognition in AβPPswe/PS1dE9 transgenic mice. PLoS One 8: , e69129. |
[221] | Ross CA , Akimov SS ((2014) ) Human-induced pluripotent stem cells: Potential for neurodegenerative diseases. Hum Mol Genet 23(R1): , R17–26. |
[222] | Takamatsu K , Ikeda T , Haruta M , Matsumura K , Ogi Y , Nakagata N , Uchino M , Ando Y , Nishimura Y , Senju S ((2014) ) Degradation of amyloid beta by human induced pluripotent stem cell-derived macrophages expressing Neprilysin-2. Stem Cell Res 13(3 Pt A): , 442–453. |
[223] | Fujiwara N , Shimizu J , Takai K , Arimitsu N , Saito A , Kono T , Umehara T , Ueda Y , Wakisaka S , Suzuki T , Suzuki N ((2013) ) Restoration of spatial memory dysfunction of human APP transgenic mice by transplantation of neuronal precursors derived from human iPS cells. Neurosci Lett 557 (Pt B): , 129–134. |


