
High Soluble Amyloid-β42 Predicts Normal Cognition in Amyloid-Positive Individuals with Alzheimer’s Disease-Causing Mutations
Abstract
Background:
In amyloid-positive individuals at risk for Alzheimer’s disease (AD), high soluble 42-amino acid amyloid-β (Aβ42) levels are associated with normal cognition. It is unknown if this relationship applies longitudinally in a genetic cohort.
Objective:
To test the hypothesis that high Aβ42 preserves normal cognition in amyloid-positive individuals with Alzheimer’s disease (AD)-causing mutations (APP, PSEN1, or PSEN2) to a greater extent than lower levels of brain amyloid, cerebrospinal fluid (CSF) phosphorylated tau (p-tau), or total tau (t-tau).
Methods:
Cognitive progression was defined as any increase in Clinical Dementia Rating (CDR = 0, normal cognition; 0.5, very mild dementia; 1, mild dementia) over 3 years. Amyloid-positivity was defined as a standard uptake value ratio (SUVR) ≥1.42 by Pittsburgh compound-B positron emission tomography (PiB-PET). We used modified Poisson regression models to estimate relative risk (RR), adjusted for age at onset, sex, education, APOE4 status, and duration of follow-up. The results were confirmed with multiple sensitivity analyses, including Cox regression.
Results:
Of 232 mutation carriers, 108 were PiB-PET-positive at baseline, with 43 (39.8%) meeting criteria for progression after 3.3±2.0 years. Soluble Aβ42 levels were higher among CDR non-progressors than CDR progressors. Higher Aβ42 predicted a lower risk of progression (adjusted RR, 0.36; 95% confidence interval [CI], 0.19–0.67; p = 0.002) better than lower SUVR (RR, 0.81; 95% CI, 0.68–0.96; p = 0.018). CSF Aβ42 levels predicting lower risk of progression increased with higher SUVR levels.
Conclusion:
High CSF Aβ42 levels predict normal cognition in amyloid-positive individuals with AD-causing genetic mutations.
INTRODUCTION
Key support for the toxic amyloid hypothesis comes from the observation that mutations in any of three genes (APP, PSEN1, and PSEN2) lead to Alzheimer’s disease (AD) with complete penetrance [1]. The genetic evidence causally implicates the fibrillogenic 42-amino acid amyloid-β peptide (Aβ42). However, the disease pathogenesis may arise from either of two ends of the protein aggregation process: the increase in insoluble amyloid plaques or the depletion of the soluble Aβ42 peptide, which has important functions. While insoluble amyloid plaques can be present in normal individuals, low soluble levels of Aβ42 are an invariable feature of AD [2–4].
The hypothesis of Aβ toxicity has traditionally been supported by the notion that AD-causing mutation carriers must have high levels of soluble Aβ42 relative to non-mutation populations. In fact, mutation carriers have lower Aβ42 levels compared to non-mutation populations [3]. The reduction in soluble Aβ42 levels among mutation carriers begins as many as 25 years before the onset of cognitive symptoms [4]. Therefore, the toxicity in the process of accelerated protein aggregation among mutation carriers may conceivably be due to the depletion in soluble Aβ42 to a greater extent than the corresponding increase in amyloid [5]. This alternative hypothesis offers an explanation for the failures in translating amyloid reduction into cognitive improvement [6], even among mutation carriers [7], and for the paradoxes posed by the large proportion of amyloid-positive individuals without dementia and even of centenarians without history of cognitive abnormalities, half of whom have autopsy-confirmed AD pathology [8–10].
We recently observed that among amyloid positron emission tomography (PET)-positive individuals, higher levels of soluble Aβ42 were associated with normal cognition and brain volumes in all tertiles of brain amyloidosis, with an effect size greater than that of increases in brain amyloid burden [11]. We here tested the hypothesis that in amyloid PET-positive individuals with AD-causing APP, PSEN1, or PSEN2 mutations, higher CSF Aβ42 levels reduce the risk of cognitive progression to a greater extent than lower levels of brain amyloidosis and lower levels of CSF phosphorylated tau (p-tau) and total tau (t-tau).
METHODS
Overview
We conducted a retrospective longitudinal study among mutation carriers participating in the Dominantly Inherited Alzheimer Network (DIAN) cohort study. The DIAN study longitudinally evaluates families with dominantly inherited AD and non-carrier relatives of the probands. Details about the DIAN study design can be found in previous publications [1, 12]. We included all mutation carriers with at least two follow-up visits including baseline, without applying any other inclusion/exclusion criteria. All analyses were conducted in two cohorts: 1) amyloid PET-positive cohort, the most critical as these are individuals considered at greater risk for developing dementia; and 2) overall cohort, which included both amyloid PET-positive and amyloid PET-negative subjects.
Clinical assessment
Each assessment comprised a detailed medical history, neurological, and neuropsychological examination. Cognitive function was primarily quantified using the Clinical Dementia Rating scale (CDR = 0, indicates normal cognition; 0.5, very mild dementia; 1, mild dementia; 2, moderate dementia; and 3, severe dementia) [13]. Our primary endpoint was CDR progression, defined as any increase in CDR over the follow-up period. Secondary endpoints were progression to CDR ≥0.5 confirmed in two consecutive visits as per DIAN guidelines to minimize misclassification, progression to CDR ≥1, Mini-Mental State Examination (MMSE) score ≤24 at last visit (higher means better cognition) [14], and a CDR sum of boxes (CDR-SB) score ≥4.5 at last visit (lower means better cognition) [15]. CDR-SB = 4.5 corresponds to mild dementia [15].
CSF biomarkers
We evaluated CSF Aβ42, p-tau, and t-tau levels (INNO-BIA AlzBio3 from Fujirebio, Malvern, PA). In order to limit the variability due to measurement error, we considered only data from the same assay, as used in previous analyses [4]. While CSF Aβ40 was not available from this particular assay, precluding us from entering it into our analysis, it did not limit the testing of our hypothesis since Aβ40 exhibits lower fibrillogenicity and lesser depletion than Aβ42 [16], and is therefore less relevant to the process of protein aggregation than Aβ42.
Neuroimaging
Pittsburgh compound B PET (PiB-PET) was used to quantify the burden of insoluble brain amyloid plaques. To reduce partial volume effects, data were processed using a regional spread function (RSF), shown to enhance sensitivity [17]. We defined subjects as amyloid PiB-PET-positive if their standard uptake value ratio (SUVR) was ≥1.42 [18], validated by using +5% higher SUVR threshold (≥1.49) as a sensitivity analysis. At the last visit, we also used RSF-processed fluorodeoxyglucose (FDG)-PET to quantify the metabolism of the precuneus (average of both hemispheres) and brain magnetic resonance imaging (MRI) to quantify hippocampal volume (average of both hemispheres) adjusted for intracranial volume, using the DIAN protocol equation.
Aims and sample size calculation
Our primary aim was to test the hypothesis that higher CSF Aβ42 at baseline reduces the risk of CDR progression to a greater extent than lower SUVR, lower p-tau, and lower t-tau levels among PiB-PET-positive gene carriers. Secondarily, we tested whether compared with lower baseline SUVR, p-tau, and t-tau levels, higher baseline CSF Aβ42 predict lower risk of progression to CDR ≥0.5 in at least two consecutive visits, progression to CDR ≥1 at any visit, MMSE ≤24 at last visit, CDR-SB ≥4.5 at last visit, and higher FDG-PET metabolism and hippocampi volume at last visit. While these aims were focused on the PiB-PET-positive cohort, we conducted similar analyses on the full cohort, which included PiB-PET-negative participants.
Sample size calculation
In a previous cross-sectional study of PiB-PET-positive individuals, we found that higher CSF Aβ42 levels were associated with greater odds of normal cognition than AD (adjusted odds ratio [OR], 6.26; p < 0.001) or mild cognitive impairment (OR, 1.42; p = 0.006) [11]. Using these results, a sample size of 105 PiB-PET-positive mutation carriers was calculated to yield >80% power to detect a significant association (OR >1.85) between soluble Aβ42 levels (assuming continuous normal distribution) and the primary (CDR progression) and secondary endpoints (progression to CDR ≥0.5, progression to CDR ≥1, MMSE ≤24, CDR-SB ≥4.5) using multiple logistic regression with a two-sided Wald test after adjusting for other independent variables with a coefficient of determination (R2) of 15% and baseline probability of outcome of 50%. Such an estimated sample was also sufficient to explore weak effect-size associations (regression coefficient, 0.30–0.60) between CSF Aβ42 levels and other secondary outcomes (FDG-SUVR and hippocampal volume) with more than 80% power at a 5% alpha using multiple linear regression after adjusting for relevant covariates. Sample size estimation and simulations were carried out using PASS 2021 (Power Analysis and Sample Size Software [2021]. NCSS, LLC. Kaysville, UT, USA, ncss.com/software/pass).
Statistical analysis
Continuous standardized CSF and SUVR levels were considered primary predictors in the analyses for a direct comparison across effects of CSF and SUVR levels. For the primary outcome of CDR progression, we used a modified Poisson model regression [19] to determine the associations of baseline CSF Aβ42, CSF t-tau and p-tau, and SUVR levels with CDR progression after adjusting for age at onset (estimated mean age of symptom onset), sex, education, APOE4, and duration of follow-up. The age at onset was estimated for participants by the site investigators. We also validated our results by considering time to first CDR progression as an outcome using Cox proportional hazards regression analysis. A receiver operating characteristics curve analysis was used to determine the CSF Aβ42 cutoff along with its area under the curve, sensitivity, and specificity in predicting CDR progression. The binary secondary outcomes (progression to CDR ≥0.5, progression to CDR ≥1, MMSE ≤24, CDR-SB ≥4.5) were analyzed using relative risk (RR) regression [19], whereas the quantitative secondary outcomes (FDG-PET and normalized hippocampi volume) using multiple linear regression. Finally, in addition to adjusting for CDR-SB and MMSE at baseline, all the regression models were adjusted for the above-described covariates and validated by removing t-tau or p-tau to minimize the impact of multicollinearity. Huber Sandwich estimator was used to compute standard errors of the estimates in all the Cox, relative risk, and linear regression models. As a sensitivity analysis, we conducted the same analysis after removing subjects within 5% of the threshold (SUVR ≥1.49). The results of the Cox regression analysis were summarized with hazard ratio (HR) and 95% confidence interval (CI), those of relative risk regression with RR and 95% CI, and those of linear regression with regression coefficient (RC) and 95% CI. The effect sizes and p-values were qualitatively compared among CSF and SUVR levels. The fully-detailed statistical analysis, statistical analysis codes, and sample size estimation are provided in the Supplementary Material.
Ethics approval
The study protocol for DIAN was approved by the local ethical committees of all participating institutions. The DIAN study was conducted in accordance with the Declaration of Helsinki and written informed consent was obtained from each participant.
RESULTS
Of 534 subjects participating in the DIAN study (mean age, 38±11.1 years), 232 mutation carriers met eligibility criteria (mean age, 38.3±11 years). Among them, 191 had available PiB-PET data at baseline (mean SUVR, 1.9±1.0), of whom 108 were PiB-PET-positive (mean SUVR, 2.5±1.0) (Table 1). These subjects were followed for a mean of 3.3±2.0 years (range = 1, 9). Because of missing data on some CSF and SUVR levels, multivariable analyses were based on 93 PiB-PET-positive samples, 85 PiB-PET-positive samples (SUVR ≥1.49), and 162 overall samples which include PiB-PET-positive and negative samples (Supplementary Figure 1). No differences in the baseline characteristics except CDR at baseline were observed between cohorts with missing versus without missing data (Supplementary Table 1).
Table 1
Characteristics of study cohort
| PiB-PET-positive cohort (n = 108) | Overall cohort (n = 232) | |
| Age (y) | 41.0 (10.4) | 38.3 (11.0) |
| Sex (female) | 55 (50.9%) | 130 (56.0%) |
| Education (y) | 13.8 (3.0) | 14.3 (3.0) |
| APOE4 carriers | 40 (37.0%) | 72 (31.0%) |
| CSF Aβ42 (pg/ml) | 264.6 (107.9)a | 350.6 (186.8)a |
| SUVR (amyloid-PiB-PET) | 2.5 (1.0)b | 1.9 (1.0)b |
| t-tau (pg/ml) | 144.5 (89.4)c1 | 111.2 (81.6)c1 |
| p-tau (pg/ml) | 81.7 (38.1)c2 | 61.9 (37.6)c2 |
| CDR at baseline | ||
| 0 | 55 (50.9%) | 147 (63.4%) |
| 0.5 | 33 (30.6%) | 60 (25.9%) |
| 1 | 17 (15.7%) | 20 (8.6%) |
| 2 | 2 (1.9%) | 3 (1.3%) |
| 3 | 1 (0.9%) | 2 (0.9%) |
| CDR progression | ||
| No | 65 (60.2%) | 165 (71.1%) |
| Yes | 43 (39.8%) | 67 (28.9%) |
| CDR (two consecutive visits) | ||
| <0.5 | 53 (49.1%) | 150 (64.7%) |
| ≥0.5 | 55 (50.9%) | 82 (35.3%) |
| CDR (any visit) | ||
| <1 | 74 (68.5%) | 176 (75.9%) |
| ≥1 | 34 (31.5%) | 56 (24.1%) |
| CDR-SB at baseline | 1.8 (2.8)d | 1.2 (2.5)d |
| CDR-SB at last visit | 3.7 (5.0)d | 2.7 (4.7)d |
| ≥4.5 | 74 (68.5%) | 178 (76.7%) |
| <4.5 | 34 (31.5%) | 54 (23.3%) |
| MMSE at baseline | 26.4 (4.9)e1 | 27.1 (4.4)e1 |
| MMSE at last visit | 23.4 (7.7)e2 | 25.1 (7.2)e2 |
| ≤24 | 69 (63.9%) | 174 (75.0%) |
| >24 | 39 (36.1%) | 58 (25.0%) |
| FDG-PET at baseline (SUVR) | 1.8 (0.3)f1 | 1.8 (0.2)f1 |
| FDG-PET last visit (SUVR) | 1.7 (0.3)f2 | 1.8 (0.3)f2 |
| Average hippocampi baseline (mm3) | 4022.4 (708.7)g1 | 4174.6 (626.0)g1 |
| Average hippocampi last visit | 3795.2 (767.6)g2 | 4039.4 (741.3)g2 |
| Normalized hippocampi baseline (mm3) | 4022.3 (709.6)g1 | 4174.3 (627.0)g1 |
| Normalized hippocampi last visit (mm3) | 3795.2 (767.6)g2 | 4039.4 (741.3)g2 |
n, number of subjects; APOE4, Apolipoprotein ɛ4; CDR, clinical dementia rating; CDR-SB, CDR sum of boxes; CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; t-tau, total tau; p-tau, phosphorylated-tau; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; MMSE, Mini-Mental State Examination; FDG, fluorodeoxyglucose; pg, picogram; ml, milliliter; mm, millimeter. Data are expressed in mean±standard deviation (SD) or frequency (%). aCSF Aβ42 data were available for 191 subjects and 93 PiB-PET-positive. bSUVR data were available for 191 subjects and 108 PiB-PET-positive. c1CSF t-tau data were available for 194 subjects and 95 PiB-PET-positive. c2CSF p-tau data were available for 268 subjects and 122 PiB-PET-positive. dCDR-SB data were available for 232 subjects and 108 PiB-PET-positive. e1MMSE baseline data were available for 231 subjects and 107 PiB-PET-positive. e2MMSE last follow-up were available for 232 subjects and 108 PET-positive. f1FDG-PET data at baseline were available for 196 subjects and 102 PET-positive. f2FDG-PET at last visit were available for 172 subjects and 92 PET-positive. g1MRI baseline data were available for 215 subjects and 108 PET-positive. g2MRI last visit were available for 200 subjects and 98 PET-positive.
Primary endpoint
Amyloid PiB-PET-positive cohort
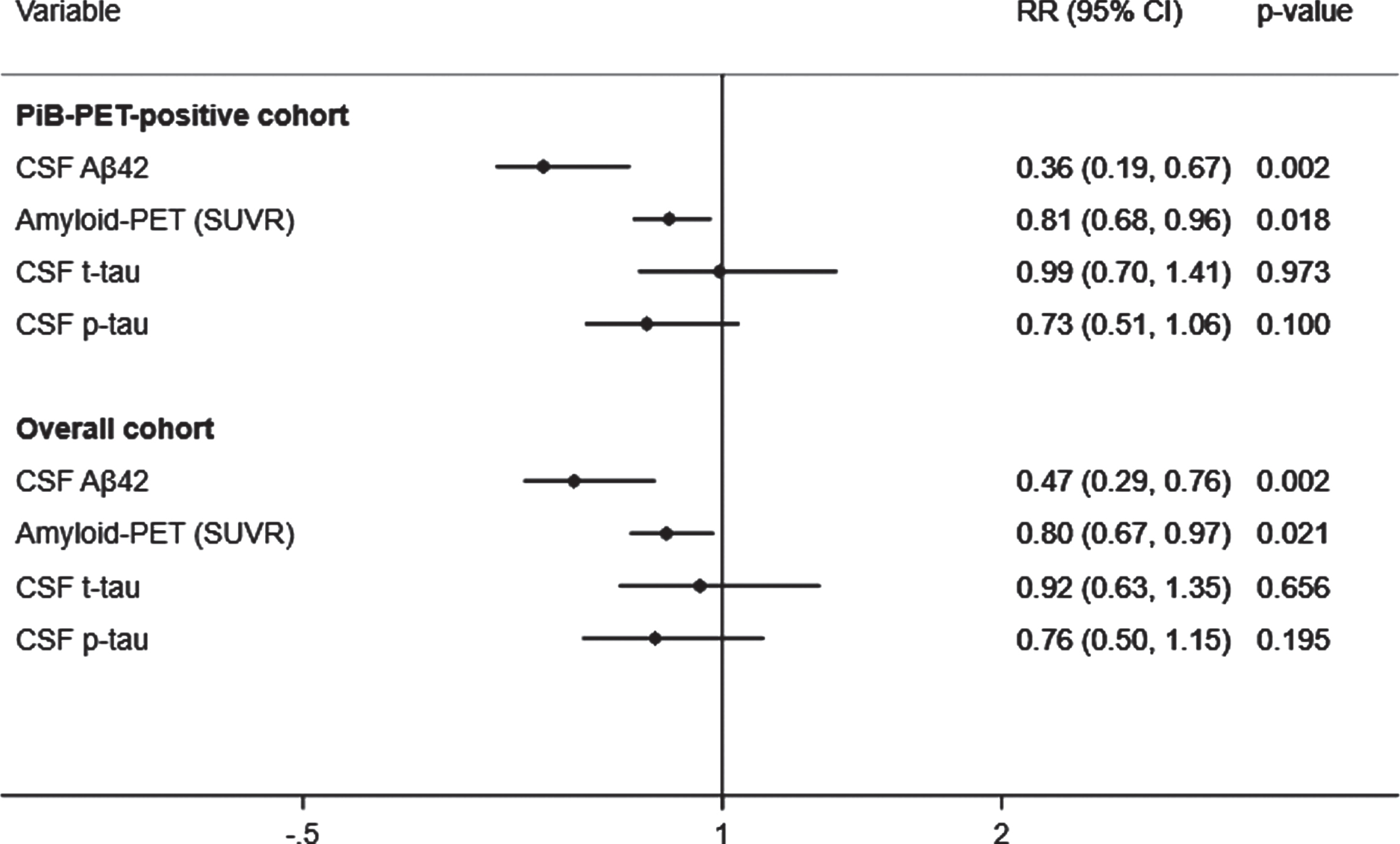
A total of 43 (39.8%) subjects met criteria for CDR progression. In adjusted analyses, the risk of progression was reduced to a greater extent by higher CSF Aβ42 (RR, 0.36; 95% CI, 0.19–0.67; p = 0.002) than lower SUVR (RR, 0.81; 95% CI, 0.68–0.96; p = 0.018) but not than lower t-tau or lower p-tau (Fig. 1, top). These results were unchanged after excluding 5% of the SUVR threshold. In addition, higher CSF Aβ42 levels were associated with a reduced hazard of time to first CDR progression (HR, 0.37; 95% CI, 0.18–0.77; p = 0.008) to a greater extent than lower SUVR (HR, 0.79; 95% CI, 0.62–1.02; p = 0.075) and lower p-tau (HR, 0.63; 95% CI, 0.40–0.98; p = 0.041). These results were confirmed in the stratified Cox model (Supplementary Table 2). CSF Aβ42 levels predicted progression to a greater extent than SUVR, CSF t-tau, or CSF p-tau levels in all analyses excluding p-tau or t-tau from the models to address multicollinearity, after additionally adjusting for baseline CDR and age, or after missing imputations (Supplementary Tables 3 and 4).
Fig. 1
Adjusted prediction of CDR progression with baseline CSF Aβ42, p-tau, t-tau, and amyloid (PiB)-PET SUVR levels. CSF, cerebrospinal fluid; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; CDR, Clinical Dementia Rating; Aβ42, 42-amino acid amyloid-beta peptide; p-tau, phospho-Tau; t-tau, total-Tau; HR, hazard ratio; CI, confidence interval. RR reflects effect size for the association with a one standard deviation higher in CSF Aβ42 levels and lower in SUVR, CSF t-tau and p-tau levels. Analyses were adjusted for age at onset, sex, education, APOE4, and duration of follow up. Overall cohort includes PiB-PET-positive and negative samples.
Overall cohort
A total of 67 (28.9%) subjects met criteria for CDR progression in the overall cohort. Higher CSF Aβ42 levels predicted a reduced risk of progression (RR, 0.47; 95% CI, 0.29–0.76; p = 0.002) to a greater extent than lower SUVR (RR, 0.80; 95% CI, 0.67–0.97; p = 0.021), but not than lower t-tau or p-tau (Fig. 1, bottom). The results were unchanged after additionally adjusting for baseline CDR and age in all analyses with or without p-tau or t-tau or after missing imputations (Supplementary Tables 3 and 4). Moreover, higher CSF Aβ42 levels were associated a reduced hazard of time to first CDR progression (HR, 0.51; 95% CI, 0.31–0.86; p = 0.011) than lower SUVR (HR, 0.79; 95% CI, 0.62–1.01; p = 0.062) and lower p-tau (HR, 0.62; 95% CI, 0.40–0.97; p = 0.037). These results were confirmed in the stratified Cox model (Supplementary Table 2).
CSF Aβ42 and CDR progression
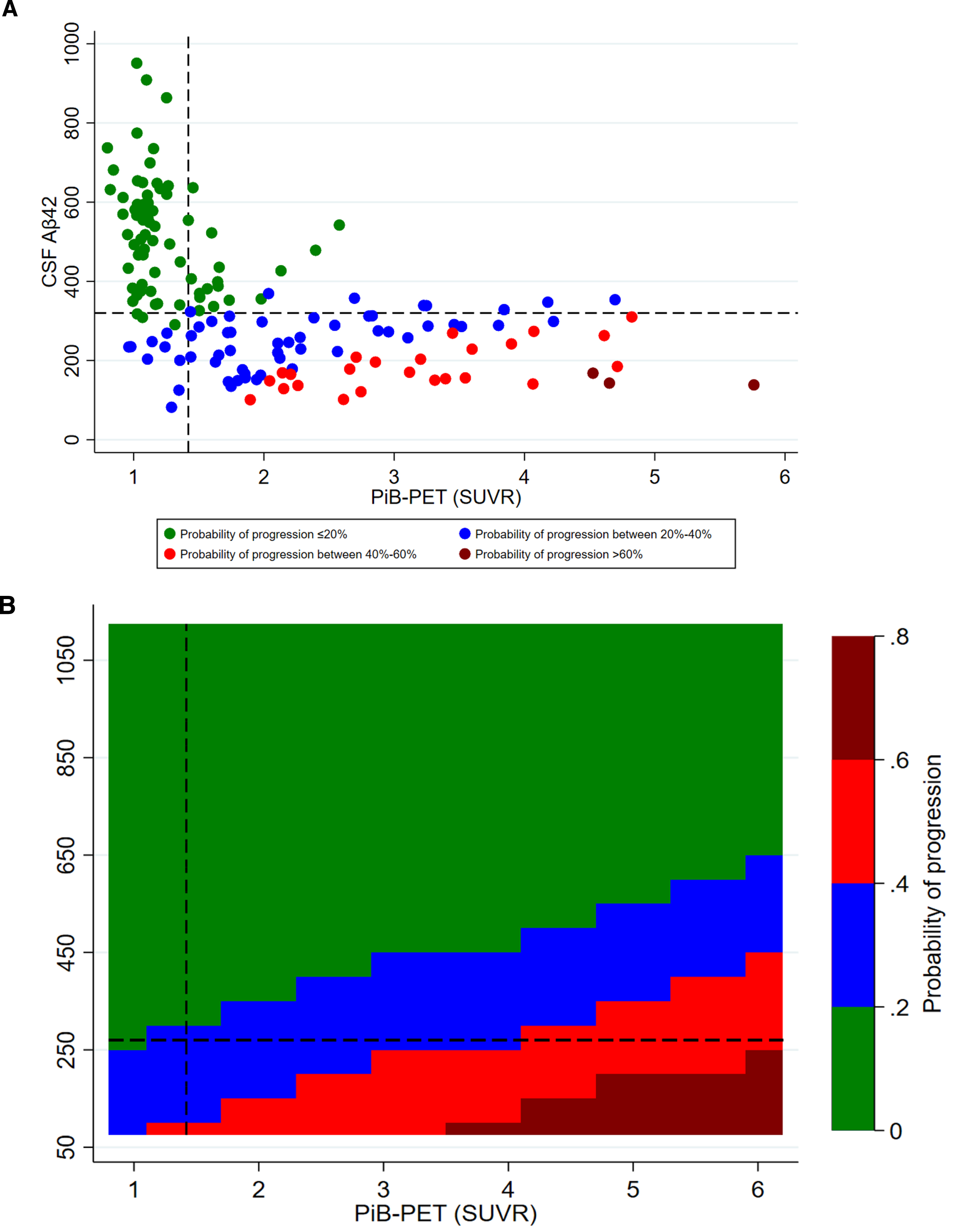
CSF Aβ42 levels were higher among CDR non-progressors than CDR progressors (Fig. 2). CSF Aβ42 <270 pg/ml predicted progression (area under the curve, 80.5%; sensitivity, 72.2%; specificity, 74.5%) regardless of increasing SUVR levels. The progression-free survival was longer with CSF Aβ42 ≥270 pg/ml compared to CSF Aβ42 <270 pg/ml over the follow-up period in both PiB-PET-positive (p = 0.002) and overall cohorts (p < 0.001) (Supplementary Figure 2). In adjusted analysis, higher CSF Aβ42 levels reduced the risk of progression even at very high PiB-PET SUVR levels. CSF Aβ42 levels predicting lower risk of progression increased with higher SUVR levels (Fig. 3).
Fig. 2
Comparison of CSF Aβ42 levels between non-progressors and progressors. PiB-PET-positive cohort: non-CDR progressors (297.73±13.66) versus CDR progressors (218.73±17.22); overall cohort: non-CDR progressors (380.83±14.5) versus CDR progressors (313.35±26.46). Error bar represents the standard error of mean. CSF, cerebrospinal fluid; PiB-PET, Pittsburgh compound B positron emission tomography; CDR, Clinical Dementia Rating; Aβ42, 42-amino acid amyloid-beta peptide. Overall cohort includes PiB-PET-positive and negative samples.
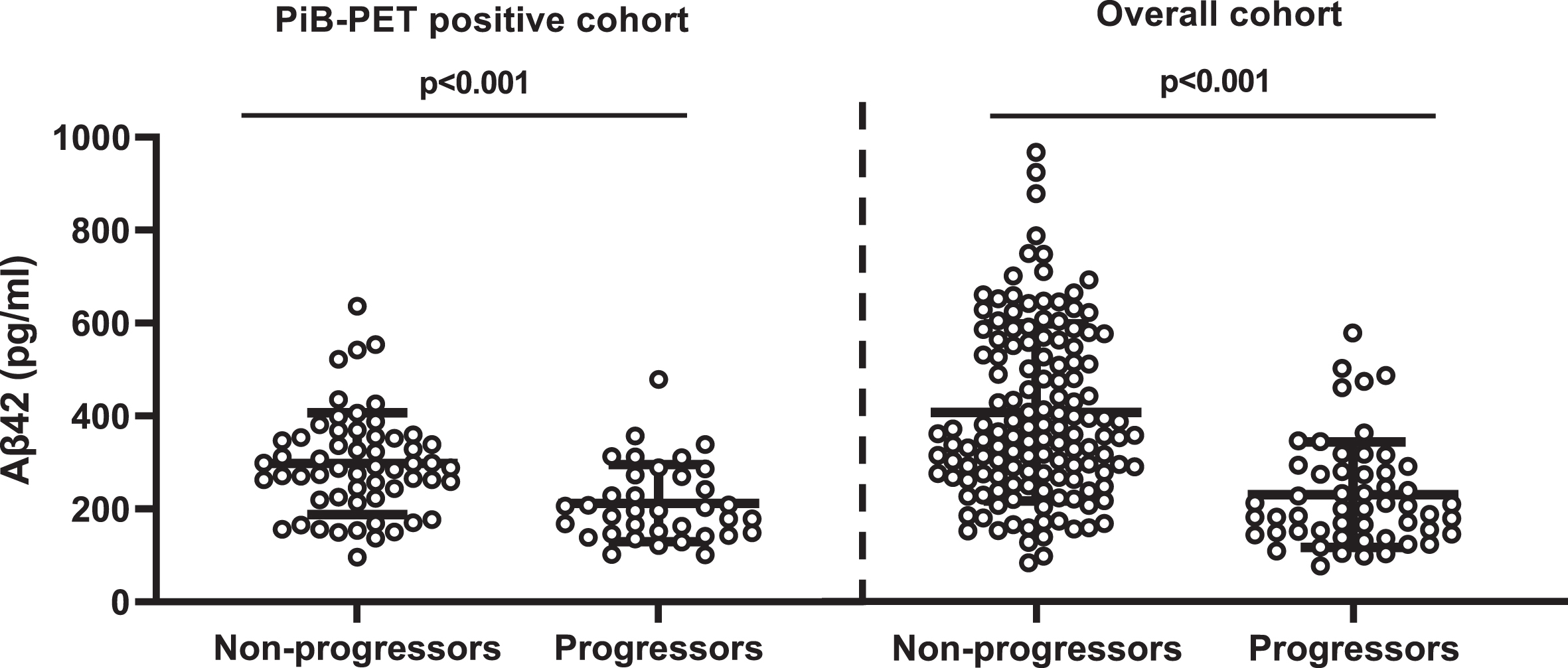
Fig. 3
Adjusted probability of CDR progression. A) scatter plot of CSF Aβ42 and PiB-PET SUVR levels; B) contour plot of CSF Aβ42 and PiB-PET SUVR levels. CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; PiB-PET, Pittsburgh compound B positron emission tomography; CDR, Clinical Dementia Rating; p-tau, phospho-Tau; t-tau, total-Tau. All models were adjusted for age at onset, sex, education, APOE4, p-tau, and t-tau levels. Overall cohort includes PiB-PET-positive and negative samples.
Secondary endpoints
Amyloid PiB-PET-positive cohort
Higher baseline CSF Aβ42 predicted a reduced risk of progression to CDR ≥0.5 (RR, 0.55; 95% CI, 0.36–0.83; p = 0.004) and to CDR ≥1 (RR, 0.27; 95% CI, 0.13–0.59; p = 0.001), and was associated with CDR-SB ≥4.5 (RR, 0.36; 95% CI, 0.36–0.18–0.70; p = 0.003) and MMSE ≤24 (RR, 0.30; 95% CI, 0.17–0.54; p < 0.001) better than lower SUVR, lower t-tau, or lower p-tau levels (Table 2). Higher CSF Aβ42 levels were also associated with larger hippocampi volume and higher FDG-PET uptake at baseline to a greater extent than lower SUVR, lower p-tau or lower t-tau levels (data not shown). These results were unchanged in age-adjusted models with other covariates and after removing t-tau and p-tau from the models (Supplementary Tables 5 and 6). Higher baseline CSF Aβ42 levels were associated with larger hippocampal volume (RC, 319.91; 95% CI, 73.97–565.86; p = 0.012) and higher FDG-PET uptake (RC, 0.14; 95% CI, 0.03–0.24; p = 0.011) to a greater extent than lower PiB-PET SUVR levels (Supplementary Table 7). The results were unchanged after excluding 5% of the SUVR threshold and in age-adjusted models with other covariates (Supplementary Table 7). The association between CSF Aβ42 levels and all secondary endpoints remained significant after excluding t-tau or p-tau (Supplementary Table 8).
Table 2
Adjusted associations of baseline CSF and SUVR with secondary cognitive outcomes
| PiB-PET-positive cohort | PiB-PET-positive cohort (SUVR ≥1.49) | Overall cohort | ||||||||||
| RR* | 95% CI | p | RR* | 95% CI | p | RR* | 95% CI | p | ||||
| Progression to CDR ≥0.5 | ||||||||||||
| CSF Aβ42 | 0.55 | 0.36 | 0.83 | 0.004 | 0.59 | 0.38 | 0.91 | 0.017 | 0.52 | 0.35 | 0.77 | 0.001 |
| SUVR (PiB-PET) | 0.75 | 0.63 | 0.90 | 0.002 | 0.78 | 0.65 | 0.93 | 0.006 | 0.70 | 0.59 | 0.84 | <0.001 |
| CSF t-tau | 0.97 | 0.78 | 1.19 | 0.764 | 0.96 | 0.78 | 1.19 | 0.693 | 0.98 | 0.78 | 1.23 | 0.876 |
| CSF p-tau | 0.67 | 0.52 | 0.86 | 0.002 | 0.69 | 0.53 | 0.89 | 0.005 | 0.60 | 0.46 | 0.79 | <0.001 |
| Progression to CDR ≥1 | ||||||||||||
| CSF Aβ42 | 0.27 | 0.13 | 0.59 | 0.001 | 0.29 | 0.13 | 0.62 | 0.002 | 0.26 | 0.13 | 0.51 | <0.001 |
| SUVR (PiB-PET) | 0.72 | 0.55 | 0.94 | 0.016 | 0.74 | 0.56 | 0.97 | 0.028 | 0.66 | 0.51 | 0.87 | 0.003 |
| CSF t-tau | 0.95 | 0.74 | 1.23 | 0.724 | 0.94 | 0.74 | 1.22 | 0.681 | 0.96 | 0.74 | 1.25 | 0.751 |
| CSF p-tau | 0.57 | 0.36 | 0.89 | 0.015 | 0.59 | 0.37 | 0.92 | 0.020 | 0.50 | 0.32 | 0.78 | 0.002 |
| CDR-SB ≥4.5 at last visit# | ||||||||||||
| CSF Aβ42 | 0.36 | 0.18 | 0.70 | 0.003 | 0.38 | 0.19 | 0.74 | 0.005 | 0.32 | 0.18 | 0.58 | <0.001 |
| SUVR (PiB-PET) | 0.78 | 0.63 | 0.96 | 0.018 | 0.79 | 0.64 | 0.98 | 0.030 | 0.73 | 0.59 | 0.90 | 0.004 |
| CSF t-tau | 1.09 | 0.85 | 1.39 | 0.492 | 1.09 | 0.85 | 1.37 | 0.520 | 1.12 | 0.88 | 1.43 | 0.366 |
| CSF p-tau | 0.47 | 0.29 | 0.75 | 0.002 | 0.48 | 0.30 | 0.76 | 0.002 | 0.40 | 0.25 | 0.64 | <0.001 |
| MMSE ≤24 at last visit# | ||||||||||||
| CSF Aβ42 | 0.30 | 0.17 | 0.54 | <0.001 | 0.32 | 0.18 | 0.59 | <0.001 | 0.26 | 0.15 | 0.45 | <0.001 |
| SUVR (PiB-PET) | 0.81 | 0.64 | 1.03 | 0.094 | 0.83 | 0.66 | 1.06 | 0.135 | 0.75 | 0.59 | 0.96 | 0.022 |
| CSF t-tau | 0.98 | 0.69 | 1.39 | 0.931 | 0.97 | 0.70 | 1.35 | 0.872 | 1.02 | 0.72 | 1.45 | 0.921 |
| CSF p-tau | 0.61 | 0.40 | 0.94 | 0.026 | 0.64 | 0.42 | 0.97 | 0.036 | 0.52 | 0.34 | 0.81 | 0.003 |
*Relative risk (RR) is with a one standard deviation higher in CSF Aβ42 levels and lower in CSF t-tau, CSF p-tau, and SUVR levels. CI, confidence interval; CDR, clinical dementia rating; CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; t-tau, total tau; p-tau, phospho-Tau; CDR-SB, CDR sum of boxes; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; MMSE, Mini-Mental State Examination; FDG, fluorodeoxyglucose. All CSF and SUVR values are standardized. Analysis adjusted for mean mutation age of symptom onset, sex, education, APOE4 status, and duration of follow up. #Analyses were also adjusted for baseline CDR-SB or MMSE; Overall cohort includes PiB-PET-positive and negative samples.
Overall cohort
Higher baseline CSF Aβ42 predicted a reduced risk of progression to CDR ≥0.5 (RR, 0.52; 95% CI, 0.35, 0.77; p = 0.001) and to CDR ≥1 (RR, 0.26; 95% CI, 0.13, 0.51; p < 0.001), and was associated with CDR-SB ≥4.5 (RR, 0.32; 95% CI, 0.18, 0.58; p < 0.001) and MMSE ≤24 (RR, 0.26; 95% CI, 0.15, 0.45; p < 0.001) better than lower SUVR, lower t-tau, or lower p-tau levels (Table 2). Similar results were observed in age-adjusted models with other covariates (Supplementary Table 5) and models with and without p-tau or t-tau (Supplementary Table 6). Larger hippocampal volume was inversely associated with SUVR levels (RC, –204.70; 95% CI, –341.07, –68.33; p = 0.004) and p-tau levels (RC, –219.06; 95% CI, –405.74, –32.37; p = 0.022) but not with CSF Aβ42 levels (Supplementary Table 7). Higher SUVR was associated with lower FDG-PET uptake (RC, –0.07; 95% CI, –0.13, –0.01; p = 0.022) but not with CSF Aβ42 (Supplementary Tables 7 and 8).
DISCUSSION
This longitudinal analysis of a genetic cohort showed that in amyloid PiB-PET-positive individuals with autosomal dominant AD-causing mutations higher levels of CSF Aβ42 were associated with reduced risk of progression to cognitive impairment, as well as larger hippocampal volume and higher brain metabolism in the precuneus. Conversely, cognitive deterioration was strongly predicted by lower levels of soluble Aβ42 but not by increases in SUVR, t-tau, or p-tau. These data are in agreement with those of a cross-sectional analysis of a sporadic AD cohort (in participants of the Alzheimer’s Disease Neuroimaging Inventory cohort study), in which higher CSF Aβ42 levels were associated with normal cognition regardless of (and despite increasing) SUVR levels among PiB-PET-positive individuals [11], making a reduction in CSF Aβ42 levels a better marker of progression than higher levels of CSF t-tau, p-tau, or amyloid PiB-PET SUVR.
A threshold of compensation can tentatively be drawn from these data, with further studies needed to validating it. We found that levels of CSF Aβ42 above 270 pg/ml predicted a reduced risk of CDR progression across any levels of PiB-PET SUVR. The better prediction of disease conversion by the reduction of CSF Aβ42 levels than by the increase in brain amyloidosis is in line with its observed decline as early as 25 years before the onset of cognitive impairment in genetic AD [4]. There is also support for a greater pathophysiologic importance of the loss of soluble Aβ42 over the accrual of insoluble PiB-PET amyloid from knockout animal models of the Aβ42 precursor, AβPP, which yields neurodegeneration without brain amyloidosis [20], and in AD patients treated with BACE-1 inhibitors, which reduced CSF Aβ42 levels and worsened cognitive symptoms regardless of changes in brain amyloid [21]. In vitro studies have shown that PSEN mutations reduce the levels of Aβ42 and Aβ40 [22], even if some mutations increase the Aβ42/40 ratio [23, 24]. Clinically, however, mutation carriers, including Down syndrome patients with APP duplications, have lower CSF levels of Aβ42 and Aβ42/40 ratio at the symptomatic stage compared to cognitively unimpaired individuals [3]. Supporting the importance of the soluble protein fraction, individuals with dementia associated with the rare APP E693del (Osaka) mutation exhibit low CSF Aβ42 despite no PiB-PET-amyloid positivity [25]. Importantly, the inverse correlation between the progressive loss of soluble Aβ42 along with the increase in insoluble Aβ is imperfect. In fact, we previously showed that soluble Aβ42 levels can still be high even in individuals with the highest amyloid-SUVR PiB-PET tertile, a relationship associated with normal cognition [11].
Amyloids represent the end product of an irreversible phase transition process of proteins, changing from a soluble to an insoluble state via a physicochemical process termed nucleation [26]. Nucleation is the rate-limiting step of the first stable amyloid cluster (nucleus), after which the phase transition proceeds spontaneously until all the available soluble substrate is consumed. While nucleation is not favorable under normal conditions, it becomes favorable at higher concentrations, such as those attained via gene duplication, or when the protein is rendered unstable due to structural mutations [27]. The net outcome is decreased availability of soluble Aβ42. This explains why their depletion is universal across all reported familial AD mutations, including APP duplications [3, 28]. Whereas an increase in amyloid plaque burden in the brain exhibits an inconsistent relationship with cognitive impairment, the depletion of soluble Aβ42 is universally associated with dementia in all familial and sporadic forms of AD, further suggesting it is pathophysiologically more relevant [1, 3, 4, 29]. This is supported by an extensive body of literature demonstrating the role of physiological (picomolar) concentrations of Aβ42 in memory and synaptic plasticity via alpha-7 nicotinic acetylcholine receptor (α7-nAChR) signaling (Supplementary Table 9). Very recently, synthetic Aβ42 monomers have been shown to improve impaired memory in conditional double knockout mice without plaque deposition and in the APP/PS1/Tau triple transgenic mice with Aβ42 deposition, effects that were mediated by the α7-nAChR [30]. These results not only indicate the importance of the loss of Aβ42 function as a pathogenic mechanism both in the presence and absence of plaques, but also suggest that Aβ42 replacement has therapeutic potential.
Major strengths of this analysis are its longitudinal design and the relatively large number of subjects with disease-causing mutations. The findings are supported by the use of several complementary analytic methods on cognitive assessments and neuroimaging data and confirmed by sensitivity analyses. Major limitations include the inability to adjust for the estimated year to symptom onset (highly correlated with CSF and SUVR levels and did not serve as a confounder), and the lack of data on CSF Aβ40 (and therefore of Aβ42/Aβ40 ratio; see below) and on oligomeric species. On the last point, subjects with normal cognition despite high PiB-PET plaque burden had high, not low CSF Aβ42 [11], opposite to the direction predicted by the hypothesis of oligomeric toxicity. Oligomers are very transient and the majority of them dissociate back to monomers rather than progress to aggregates [31]. Importantly, the reduction of soluble Aβ42 levels during the disease course reduces the substrate for the oligomers, further questioning any sustainable toxicity from them.
A note related to the issue of ratio versus absolute values is worth making. Early studies argued that absolute levels of Aβ42 decrease but the Aβ42/Aβ40 ratio increases due to the lower decrease of Aβ40 relative the more aggregation-prone Aβ42 [32, 33]. However, recent studies have found that the Aβ42/Aβ40 ratio decreases in AD [3, 34]. Pathophysiologically, amyloid aggregation is dependent on supersaturation [35–37], which is based on the absolute concentration of the peptide. Decreasing peptide concentration will decrease, not increase, the saturation and the related propensity to form any type of aggregates irrespective of its relative levels compared to other peptides [38]. Nearly 90% of AD-related mutations in the PSEN1 gene lead to reduced production of both Aβ42 and Aβ40 [22], a classical genetic loss-of-function mechanism. Further, there is no correlation between the Aβ42/Aβ40 ratio and the age of onset in mutation carriers [22].
In conclusion, higher soluble Aβ42 levels are associated with reduced risk of CDR progression, normal cognition, normal hippocampal volume, and normal precuneus metabolism to a greater extent than lower brain amyloid, lower p-tau, and lower t-tau levels in amyloid PiB-PET-positive individuals with autosomal dominant AD-causing genetic mutations. Brain toxicity in AD may be predominantly mediated by a reduction of the soluble protein pool, its functional fraction, rather than its accrual into amyloids.
DATA SHARING
All data are available upon request from the Dominantly Inherited Alzheimer Network (DIAN) through the study website, https://dian.wustl.edu/.
ACKNOWLEDGMENTS
We thank the Knight Alzheimer Disease Research Center (Knight ADRC) of the Dominantly Inherited Alzheimer Network (DIAN). The authors thank all the participants and their families and the support of all the research staff involved at each of the participating sites. We particularly thank the DIAN consortia statisticians for independently confirming the accuracy of the data source and replicating the results of the analysis conducted for this study.
Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA) and the Alzheimer’s Association (SG-20-690363-DIAN). Further funding came from the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec – Santé. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0808).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220808.
REFERENCES
[1] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC ; Dominantly Inherited Alzheimer Network ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease, N Engl J Med 367: , 795–804. Erratum in: N Engl J Med 367, 780. |
[2] | Fagan AM , Mintun MA , Mach RH , Lee SY , Dence CS , Shah AR , LaRossa GN , Spinner ML , Klunk WE , Mathis CA , DeKosky ST , Morris JC , Holtzman DM ((2006) ) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans, Ann Neurol 59: , 512–519. |
[3] | Fagan AM , Henson RL , Li Y , Boerwinkle AH , Xiong C , Bateman RJ , Goate A , Ances BM , Doran E , Christian BT , Lai F , Rosas HD , Schupf N , Krinsky-McHale S , Silverman W , Lee JH , Klunk WE , Handen BL , Allegri RF , Chhatwal JP , Day GS , Graff-Radford NR , Jucker M , Levin J , Martins RN , Masters CL , Mori H , Mummery CJ , Niimi Y , Ringman JM , Salloway S , Schofield PR , Shoji M , Lott IT ; Alzheimer’s Biomarker Consortium–Down Syndrome; Dominantly Inherited Alzheimer Network ((2021) ) Comparison of CSF biomarkers in Down syndrome and autosomal dominant Alzheimer’s disease: A cross-sectional study, Lancet Neurol 21: , 615–626. Erratum in: Lancet Neurol 2022; 21, e7. |
[4] | McDade E , Wang G , Gordon BA , Hassenstab J , Benzinger TLS , Buckles V , Fagan AM , Holtzman DM , Cairns NJ , Goate AM , Marcus DS , Morris JC , Paumier K , Xiong C , Allegri R , Berman SB , Klunk W , Noble J , Ringman J , Ghetti B , Farlow M , Sperling RA , Chhatwal J , Salloway S , Graff-Radford NR , Schofield PR , Masters C , Rossor MN , Fox NC , Levin J , Jucker M , Bateman RJ ; Dominantly Inherited Alzheimer Network ((2018) ) Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease, Neurology 91: , e1295–e1306. |
[5] | Espay AJ , Sturchio A , Schneider LS , Ezzat K ((2021) ) Soluble amyloid-β consumption in Alzheimer’s disease, J Alzheimers Dis 82: , 1403–1415. |
[6] | Panza F , Lozupone M , Seripa D , Imbimbo BP ((2019) ) Amyloid-β immunotherapy for Alzheimer disease: Is it now a long shot? Ann Neurol 85: , 303–315. |
[7] | Salloway S , Farlow M , McDade E , Clifford DB , Wang G , Llibre-Guerra JJ , Hitchcock JM , Mills SL , Santacruz AM , Aschenbrenner AJ , Hassenstab J , Benzinger TLS , Gordon BA , Fagan AM , Coalier KA , Cruchaga C , Goate AA , Perrin RJ , Xiong C , Li Y , Morris JC , Snider BJ , Mummery C , Surti GM , Hannequin D , Wallon D , Berman SB , Lah JJ , Jimenez-Velazquez IZ , Roberson ED , van Dyck CH , Honig LS , Sánchez-Valle R , Brooks WS , Gauthier S , Galasko DR , Masters CL , Brosch JR , Hsiung GR , Jayadev S , Formaglio M , Masellis M , Clarnette R , Pariente J , Dubois B , Pasquier F , Jack CR Jr , Koeppe R , Snyder PJ , Aisen PS , Thomas RG , Berry SM , Wendelberger BA , Andersen SW , Holdridge KC , Mintun MA , Yaari R , Sims JR , Baudler M , Delmar P , Doody RS , Fontoura P , Giacobino C , Kerchner GA , Bateman RJ ; Dominantly Inherited Alzheimer Network–Trials Unit ((2021) ) A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease, Nat Med 27: , 1187–1196. |
[8] | Chételat G , La Joie R , Villain N , Perrotin A , de La Sayette V , Eustache F , Vandenberghe R ((2013) ) Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease, Neuroimage Clin 2: , 356–365. |
[9] | Crystal H , Dickson D , Fuld P , Masur D , Scott R , Mehler M , Masdeu J , Kawas C , Aronson M , Wolfson L ((1988) ) Clinico-pathologic studies in dementia: Nondemented subjects with pathologically confirmed Alzheimer’s disease, Neurology 38: , 1682–1687. |
[10] | Polvikoski T , Sulkava R , Myllykangas L , Notkola IL , Niinistö L , Verkkoniemi A , Kainulainen K , Kontula K , Pérez-Tur J , Hardy J , Haltia M ((2001) ) Prevalence of Alzheimer’s disease in very elderly people: A prospective neuropathological study, Neurology 56: , 1690–1696. |
[11] | Sturchio A , Dwivedi AK , Young CB , Malm T , Marsili L , Sharma JS , Mahajan A , Hill EJ , Andaloussi SE , Poston KL , Manfredsson FP , Schneider LS , Ezzat K , Espay AJ ((2021) ) High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis, EClinicalMedicine 38: , 100988. |
[12] | Morris JC , Aisen PS , Bateman RJ , Benzinger TL , Cairns NJ , Fagan AM , Ghetti B , Goate AM , Holtzman DM , Klunk WE , McDade E , Marcus DS , Martins RN , Masters CL , Mayeux R , Oliver A , Quaid K , Ringman JM , Rossor MN , Salloway S , Schofield PR , Selsor NJ , Sperling RA , Weiner MW , Xiong C , Moulder KL , Buckles VD ((2012) ) Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network, Clin Investig (Lond) 2: , 975–984. |
[13] | Morris JC ((1993) ) The Clinical Dementia Rating (CDR): Current version and scoring rules, Neurology 43: , 2412–2414. |
[14] | Creavin ST , Wisniewski S , Noel-Storr AH , Trevelyan CM , Hampton T , Rayment D , Thom VM , Nash KJ , Elhamoui H , Milligan R , Patel AS , Tsivos DV , Wing T , Phillips E , Kellman SM , Shackleton HL , Singleton GF , Neale BE , Watton ME , Cullum S ((2016) ) Mini-Mental State Examination (MMSE) for the detection of dementia in clinically unevaluated people aged 65 and over in community and primary care populations, Cochrane Database Syst Rev 2016: , CD011145. |
[15] | O’Bryant SE , Waring SC , Cullum CM , Hall J , Lacritz L , Massman PJ , Lupo PJ , Reisch JS , Doody R ; Texas Alzheimer’s Research Consortium ((2008) ) Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: A Texas Alzheimer’s research consortium study, Arch Neurol 65: , 1091–1095. |
[16] | Maia LF , Kaeser SA , Reichwald J , Hruscha M , Martus P , Staufenbiel M , Jucker M ((2013) ) Changes in amyloid-β and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein, Sci Transl Med 5: , 194re2. |
[17] | Su Y , Blazey TM , Snyder AZ , Raichle ME , Marcus DS , Ances BM , Bateman RJ , Cairns NJ , Aldea P , Cash L , Christensen JJ , Friedrichsen K , Hornbeck RC , Farrar AM , Owen CJ , Mayeux R , Brickman AM , Klunk W , Price JC , Thompson PM , Ghetti B , Saykin AJ , Sperling RA , Johnson KA , Schofield PR , Buckles V , Morris JC , Benzinger TLS ; Dominantly Inherited Alzheimer Network ((2015) ) Partial volume correction in quantitative amyloid imaging, Neuroimage 107: , 55–64. |
[18] | Dincer A , Gordon BA , Hari-Raj A , Keefe SJ , Flores S , McKay NS , Paulick AM , Shady Lewis KE , Feldman RL , Hornbeck RC , Allegri R , Ances BM , Berman SB , Brickman AM , Brooks WS , Cash DM , Chhatwal JP , Farlow MR , la Fougère C , Fox NC , Fulham MJ , Jack CR Jr , Joseph-Mathurin N , Karch CM , Lee A , Levin J , Masters CL , McDade EM , Oh H , Perrin RJ , Raji C , Salloway SP , Schofield PR , Su Y , Villemagne VL , Wang Q , Weiner MW , Xiong C , Yakushev I , Morris JC , Bateman RJ , L S Benzinger T ; Dominantly Inherited Alzheimer Network DIAN ((2020) ) Comparing cortical signatures of atrophy between late-onset and autosomal dominant Alzheimer disease, Neuroimage Clin 28: , 102491. |
[19] | Dwivedi AK , Mallawaarachchi I , Lee S , Tarwater P ((2014) ) Methods for estimating relative risk in studies of common binary outcomes, J Appl Stat 41: , 484–500. |
[20] | Kent SA , Spires-Jones TL , Durrant CS ((2020) ) The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics, Acta Neuropathol 140: , 417–447. |
[21] | Egan MF , Kost J , Voss T , Mukai Y , Aisen PS , Cummings JL , Tariot PN , Vellas B , van Dyck CH , Boada M , Zhang Y , Li W , Furtek C , Mahoney E , Harper Mozley L , Mo Y , Sur C , Michelson D ((2019) ) Randomized trial of verubecestat for prodromal Alzheimer’s disease, N Engl J Med 380: , 1408–1420. |
[22] | Sun L , Zhou R , Yang G , Shi Y ((2017) ) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase, Proc Natl Acad Sci U S A 114: , E476–E485. |
[23] | Potter R , Patterson BW , Elbert DL , Ovod V , Kasten T , Sigurdson W , Mawuenyega K , Blazey T , Goate A , Chott R , Yarasheski KE , Holtzman DM , Morris JC , Benzinger TL , Bateman RJ ((2013) ) Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers, Sci Transl Med 5: , 189ra77. |
[24] | Kumar-Singh S , Theuns J , Van Broeck B , Pirici D , Vennekens K , Corsmit E , Cruts M , Dermaut B , Wang R , Van Broeckhoven C ((2006) ) Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40, Hum Mutat 27: , 686–695. |
[25] | Tomiyama T , Shimada H ((2020) ) APP Osaka mutation in familial Alzheimer’s disease-its discovery, phenotypes, and mechanism of recessive inheritance, Int J Mol Sci 21: , 1413. |
[26] | Malmberg M , Malm T , Gustafsson O , Sturchio A , Graff C , Espay AJ , Wright AP , El Andaloussi S , Lindén A , Ezzat K ((2020) ) Disentangling the amyloid pathways: A mechanistic approach to etiology, Front Neurosci 14: , 256. |
[27] | Chiti F , Calamai M , Taddei N , Stefani M , Ramponi G , Dobson CM ((2002) ) Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases, Proc Natl Acad Sci U S A 99: (Suppl 4), 16419–16426. |
[28] | Portelius E , Hölttä M , Soininen H , Bjerke M , Zetterberg H , Westerlund A , Herukka SK , Blennow K , Mattsson N ((2014) ) Altered cerebrospinal fluid levels of amyloid β and amyloid precursor-like protein 1 peptides in Down’s syndrome, Neuromolecular Med 16: , 510–516. |
[29] | Jack CR Jr , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers, Lancet Neurol 12: , 207–216. |
[30] | Duan Y , Lv J , Zhang Z , Chen Z , Wu H , Chen J , Chen Z , Yang J , Wang D , Liu Y , Chen F , Tian Y , Cao X ((2022) ) Exogenous Aβ1-42 monomers improve synaptic and cognitive function in Alzheimer’s disease model mice, Neuropharmacology 209: , 109002. |
[31] | Dear AJ , Michaels TCT , Meisl G , Klenerman D , Wu S , Perrett S , Linse S , Dobson CM , Knowles TPJ ((2020) ) Kinetic diversity of amyloid oligomers, Proc Natl Acad Sci U S A 117: , 12087–12094. |
[32] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med 8: , 595–608. |
[33] | Wolfe MS ((2007) ) When loss is gain: Reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8: , 136–140. |
[34] | Hansson O , Lehmann S , Otto M , Zetterberg H , Lewczuk P ((2019) ) Advantages and disadvantages of the use of the CSF amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s disease, Alzheimers Res Ther 11: , 34. |
[35] | Ciryam P , Kundra R , Morimoto RI , Dobson CM , Vendruscolo M ((2015) ) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases, Trends Pharmacol Sci 36: , 72–77. |
[36] | Freer R , Sormanni P , Ciryam P , Rammner B , Rizzoli SO , Dobson CM , Vendruscolo M ((2029) ) Supersaturated proteins are enriched at synapses and underlie cell and tissue vulnerability in Alzheimer’s disease, Heliyon 5: , e02589. |
[37] | Ciryam P , Tartaglia GG , Morimoto RI , Dobson CM , Vendruscolo M ((2013) ) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins, Cell Rep 5: , 781–790. |
[38] | Ezzat K , Sturchio A , Espay AJ ((2022) ) Proteins do not replicate, they precipitate: Phase transition and loss of function toxicity in amyloid pathologies, Biology (Basel) 11: , 535. |


