
An Alzheimer’s Disease Mechanism Based on Early Pathology, Anatomy, Vascular-Induced Flow, and Migration of Maximum Flow Stress Energy Location with Increasing Vascular Disease
Abstract
This paper suggests a chemical mechanism for the earliest stages of Alzheimer’s disease (AD). Cerebrospinal fluid (CSF) flow stresses provide the energy needed to induce molecular conformation changes leading to AD by initiating amyloid-β (Aβ) and tau aggregation. Shear and extensional flow stresses initiate aggregation in the laboratory and in natural biophysical processes. Energy-rich CSF flow regions are mainly found in lower brain regions. MRI studies reveal flow stress “hot spots” in basal cisterns and brain ventricles that have chaotic flow properties that can distort molecules such as Aβ and tau trapped in these regions into unusual conformations. Such fluid disturbance is surrounded by tissue deformation. There is strong mapping overlap between the locations of these hot spots and of early-stage AD pathology. Our mechanism creates pure and mixed protein dimers, followed by tissue surface adsorption, and long-term tissue agitation ultimately inducing chemical reactions forming more stable, toxic oligomer seeds that initiate AD. It is proposed that different flow stress energies and flow types in different basal brain regions produce different neurotoxic aggregates. Proliferating artery hardening is responsible for enhanced heart systolic pulses that drive energetic CSF pulses, whose critical maximum systolic pulse energy location migrates further from the heart with increasing vascular disease. Two glymphatic systems, carotid and basilar, are suggested to contain the earliest Aβ and tau AD disease pathologies. A key to the proposed AD mechanism is a comparison of early chronic traumatic encephalopathy and AD pathologies. Experiments that test the proposed mechanism are needed.
UNANSWERED AD QUESTIONS AND A PAPER ROADMAP
Although amyloid-β (Aβ) and tau protein have been identified as critically involved molecular participants in the etiology of Alzheimer’s disease (AD), clinical trials using anti-Aβ and anti-tau agents thus far have failed to produce a treatment that halts the progress of the disease [1, 2]. There is currently an AD research crisis and an urgent need to find a different approach that still incorporates Aβ and tau in any proposed disease mechanism. This paper suggests a combined energetic mechanical, anatomical, and chemical mechanism that we believe can provide that fresh approach. The mechanism relies on mechanical brain cerebrospinal fluid (CSF) flow stress to initiate conformation changes in Aβ and tau molecules that are ultimately responsible for initiating AD pathology.
Listed in Table 1 are questions regarding AD that are currently unanswered. We suggest that the discussion below justifying this new mechanism provides reasonable answers to many of these questions found at the end of this paper.
Table 1
Some currently unanswered questions
| •What initiates AD disease in the brain? |
| •Why is there such a long lag in early AD etiology? |
| •Why is there separation of time and space in the earliest Aβ and tau pathology? |
| •What is the mechanistic link between vascular disease and AD? |
| •Why is tau and not Aβ pathology correlated with AD symptoms? |
| •Why is AD such a highly complex disease? |
| •Why are prion-like seeds taken from the brain much more neurotoxic than those synthesized in the laboratory? |
| •What causes the similarities and differences between AD and CTE (chronic traumatic encephalopathy)? |
| •Why are the earliest pathologies of AD (Aβ only) and CTE (tau only) found in the same brain locations? |
| •Why do AD drug trials succeed so well in mice and fail so poorly in humans? |
Table 2
Current unanswered questions about AD and proposed answers
| Unanswered questions regarding AD | Suggested answers arising from the CSF flow stress energy hypothesis in this paper |
| What initiates AD disease in the brain? | Pulsed CSF flow in narrow dimension glymphatic paths, especially in the lower brain region where there are chaotic motion CSF “hot spots” in cistern subarachnoid spaces. |
| Why is there such a long lag in early AD etiology? | CSF flow is driven by vascular pulses. The hypothesis is that the fluid flow stress energy of the systolic pulse in any brain location is a critical variable in creating Aβ or tau prion-like seeds. Pulse energy is dependent upon glymphatic system parameters such as CSF flow path dimensions, flow rates, rate of energy transfer, and type of energy. Increasing age and poor health conditions help induce atherosclerosis of the large arteries causing the maximum systolic pulse energy location (MSPEL) to shift from the heart region very slowly first into the lower brain regions and then to the cortex. |
| What is the mechanistic link between vascular disease and AD? | Because of the influence of MSPEL on the etiology of the AD disease with increasing age-related atherosclerosis. |
| Why is there separation of time &space in the earliest Aβ and tau pathology? | There are two main glymphatic disease pathways in the brain, carotid for Aβ and basilar for tau. The shifting MSPEL apparently arrives at critical hydrodynamic sites at different times, early for Aβ and later for tau. Tau pathology apparently depends on maximum extensional flow stress located primarily in around the CA. |
| Why is tau and not Aβ pathology correlated with AD symptoms? | Probably because only small amounts of energy are needed to produce early Aβ aggregates, which are less toxic than tau aggregates. The latter need more energy to drastically rearrange the tau conformation to produce highly toxic tau seed aggregates. |
| Why are prion-like seeds taken from the brain much more neurotoxic than those synthesized in the laboratory? | Because the brain glymphatic system subjects Aβ and tau molecules to conformation-altering fluid flow stresses, most likely resulting from extensional CSF or ISF flow. |
| Why are the Aβ and tau pathologies so scattered in later AD stages? | Because of continually shifting MSPEL migration due to widespread CAA deposits and brain shrinkage significantly altering the local hydrodynamics as well as the local flow stress energy deposited. |
| What causes the similarities and differences among AD, CTE, and TBI? | Because each is initiated by a fluid flow pulse that becomes focused in narrow glymphatic pathways that produce different stress-induced toxic products. |
| Why are the initial pathologies of AD (Aβ only) and CTE (tau only) found in the same brain locations? | These are both formed by pulsed fluid stress. The brain is a nearly closed hydrodynamic system. Calculations show that in such a system, the energy of a pulse becomes focused in the deep sulci. Aβ42 is highly sensitive to fluid stresses. Tau is not and requires the higher energies of CTE and TBI to induce aggregation. The absence of Aβ pathology in CTE is explained by our chemical mechanism in which Aβ is involved in an intermediate and is ultimately released back into the CSF. |
| Why do AD drug trials succeed in mice and yet fail in humans? | The general anatomic makeup of the mouse and human may look qualitatively similar, but the hydrodynamics of the two are quite different. |
After a brief introductory review of the pertinent AD, Aβ, tau, and glymphatic system literature, we emphasize the importance of variable energy in causing conformation conversions that are needed to initiate Aβ and tau aggregation that ultimately produces neurotoxic products. Mechanical stresses from CSF flow through confined brain paths are emphasized, as is the idea that much of the basic research has been done on quiescent solutions despite recent indications that there is chaotic CSF flow in the critically important lower brain region. MRI studies have revealed in these brain cistern regions very high CSF energy “hot spots” [3]. Special emphasis is given extensional flow [4, 5] which has been shown to induce protein aggregation, especially in the presence of surfaces, even with proteins that do not spontaneously aggregate. We then demonstrate strong correlations among these hot spots and sites of early Aβ and tau pathology. The cerebral aqueduct region is suggested as one of the prime brain locations for the development of extensional fluid stress in tau protein [5].
Atherosclerosis in the major carotid and vertebrobasilar arteries causes strong pulsed systolic peak mechanical energy to be transferred to the carotid segment of the circle of Willis and vertebrobasilar system respectively. The displaced maximum systolic pulse energy location (MSPEL) provides new mechanical energy in these regions that activates Aβ and tau molecules dissolved in CSF, ultimately leading to formation of toxic seeds responsible for AD. A suggested chemical AD mechanism is finally presented in an illustrated format depicted with speculative conformation changes and high energy molecular states. Finally, possible answers are given to questions shown in Table 1 based on this mechanism and discussions in this paper.
However, please note that this suggested chemical mechanism applies only to the earliest events in the AD etiology. That is, the earliest chemical reactions that trigger a chain of complicated chemical reactions that are reflected in very early clinical AD symptoms.
INTRODUCTION
The chief toxin suspect in an explanation for AD for many decades, Aβ [6–9], has failed to explain the origin of AD. The roles of the other suspect, tau protein, are somewhat clearer [10–13]. This molecule is critical in maintaining the axon transport system and is therefore critical for axonal outgrowth and transport, as well as being involved in synaptic plasticity and membrane binding. Neuron dysfunction arises when tau is released from its binding site [14]. Tau becomes phosphorylated and migrates to the neuron soma and dendrite regions, where it is thought to hinder nerve transmission and memory processes. The presence of aggregated tau in neurons correlates with pre-AD dementia symptoms whereas Aβ aggregation does not. Yet, significant genetic data suggest that Aβ plays a vital role in AD [15, 16].
Not only is the ultimate scientific cause of AD unknown, but it is also a highly complex disease. AD is a common cause of cognitive impairment acquired in midlife and late life but its clinical impact is modified by other neurodegenerative and cerebrovascular conditions. “What is clearly emerging from studies is that AD is heterogeneous in each aspect, such as amyloid composition, tau distribution, relation between amyloid and tau, clinical symptoms, and genetic background, and thus it is probably impossible to explain AD with a single pathological process.” [17].
A radically different approach that still maintains Aβ and tau as major components and is based on the effects of brain fluid motion has been suggested [18] and expanded [5] based on the report of chaotic motions of the CSF within certain regions of the brain, especially in basal regions [3]. In this paper, this new approach is expanded to include anatomical correlations between Aβ/tau pathology and regions of energetic CSF motion observable by MRI. The correlations are striking and highly suggestive. But they are just that because of the limited experimental evidence available regarding the influence of brain CSF and interstitial fluid (ISF) flow stresses on the physical and chemical properties of Aβ and tau. Also introduced here is the concept that MSPEL migrates away from the heart with advancing atherosclerosis in the very early AD stages. It is suggested that early disease pathology appears in different locations because of the migrating MSPEL.
The misfolded protein misnomer
AD and many other related diseases are often classified as diseases caused by “misfolded proteins.” Both Aβ and tau are intrinsically disordered proteins (IDP) [19, 20]. These are proteins that do not maintain a well-defined, ordered structure in solution. Instead, they rapidly change their conformations, so that, in effect, they are continuously folding and refolding. Thus, what is meant by a “misfolded” protein in the case of Aβ and tau is probably a situation in which one ensemble, with predominantly alpha helical substructures, is transformed into another ensemble with predominantly cross beta structures. This conformation shift event involves a transitional ensemble of molecular structures that are precursors to Aβ or tau dimers or oligomers, some of which have been found to be highly neurotoxic [21–25] and are current drug targets.
The earliest steps in the mechanism for the initiation of such oligomer formation remains clouded. Except for certain well-defined genetic mutations [26], AD is a disease generally associated with aging. Yet, many healthy aged individuals die with high loads of Aβ-containing amyloid plaques without any AD symptoms. At the other end of the age spectrum, phosphorylated tau protein, the earliest tau pathology, is found in healthy young children [27, 28].
Whatever the chemical mechanism, the physical framework within which this mechanism operates is becoming increasingly understood with research on the glymphatic system.
The importance of the glymphatic brain fluid flow path
Aβ and tau molecules dissolved in CSF or ISF are contained in a highly complex brain environment. Depending on their location within the brain, these molecules are continuously jostled, squeezed, tumbled, distorted, stretched, adsorbed, and desorbed as they flow through their various crowded [29] glymphatic system pathways [30–37]. In all these locations, the CSF and ISF that contains dissolved Aβ and tau is shoved back and forth in its flow path in sync with the vascular heart pulse, the primary flow driver of these brain fluids. If in the ventricle region, the CSF origin, they are dragged through the very narrow cerebral aqueduct (CA). If in the basal brain region, they flow through and are temporarily trapped in narrow subarachnoid cisterns crowded with arteries and nerves. If in the cortex parenchyma, they continuously collide with molecules projecting into their very narrow flow paths or they undergo laminar shear in perivascular flow paths in the brain vascular system. As one of these molecules approaches a fluid-confining wall, it is subjected to shear, with resulting rotational and extensional flow components [38]. In some polymer solutions, a dissolved polymer molecule is accelerated toward the approaching wall [39]. As the flow path dimensions narrow, the extensional flow stretching component intensifies [18] (Fig. 3 in ref [18]). In narrow, confined lower brain subarachnoid spaces, the flowing CSF encounters a complex network of pulsing, CSF flow-blocking arteries and nerves or nerve bundles. Thus, within the basal brain region, dissolved Aβ and tau molecules are continuously subjected to significant fluid stress generated by CSF flow processes. However, ISF flow properties within the even narrower confines of the proposed cortex glymphatic path have not been measured and fully characterized [40], but these regions may nevertheless have important flow-induced chemical reactions [41].
Energy needed for Aβ and tau molecules to form toxic oligomers
To form neurotoxic oligomers, there must be a relatively drastic molecular conformation change. The activation energy for the transformation of Aβ from random coil to a cross beta conformation is estimated to be a rather high 23 kcal/mol [42]. In other words, this means that this conformational transition is thermally quite difficult, i.e., the fluid must be heated to a fairly high temperature, much higher than that in the human brain, to readily be accomplished. Either an in vivo catalyst must be present, or some other means is available in the brain to overcome this high activation energy.
It takes mechanical energy from vascular pulses to force CSF or ISF to flow in confined brain spaces. If Aβ or tau is dissolved in these fluids, some of this mechanical energy is transformed into chemical potential energy of these two molecules in the form of vibrational, rotational, and electronic energy. Electronic energy is stored in conformation changes, changes in the spatial relationships of atoms to each other within the molecule.
Previous papers have described the types of flow stress-induced conformation changes predicted for aggregation to proceed in the human brain [5, 18, 43]. Each of these changes is dependent on the amount of mechanical energy transferred to and converted into potential energy in each molecule. However, it is not just the amount of energy but the rate at which the energy is transferred. An example of this is in the case of extensional flow, in which the molecule is stretched much like a rubber band. As the stretch energy is increased, at some point, the rubber band will break. In this analogy, the energy at which it breaks corresponds to the activation energy needed to completely convert one type of ensemble into another to achieve a significant change in the average molecular conformation.
Thus, it has been proposed [5] that the directional nature of certain types of flow energy found within the brain have reduced the activation energy enough that conformational changes can take place because of these unusual fluid flow characteristics in different parts of the brain. Furthermore, it is proposed that these unusual mechanical-energy dependent conformation changes produce rare chemical compounds that are ultimately responsible for AD.
The need for a new chemical mechanism for AD
Much chemical experimentation has been focused on the chemical mechanism of the aggregation of these molecules in quiescent solutions [9]. Yet, many procedures used in purifying and preparing these quiescent solutions involve solution agitation. The limited number of experiments with carefully controlled flow conditions report an acceleration in the aggregation of Aβ [43, 44] or deposition of this aggregate on the confining walls in the case of capillary flow [45]. In one study [46], it was found that the kinetics of Aβ aggregation was qualitatively different under flow conditions, implying a different chemical mechanism from that with quiescent solutions. We have found no controlled shear studies on tau protein. Thus, careful experiments that study the kinetics of Aβ and tau aggregation under the various types of flow conditions found within the human brain glymphatic system need to be performed.
The role of this paper is to summarize the main experimental, pathological, and clinical evidence that is the foundation for a new suggested, quite general, quite tentative, chemical AD mechanism. This mechanistic hypothesis is meant to be a guide for future experiments that test the validity of the suggested mechanism. This mechanism is based on published literature from chemical and biological experiments as well as clinical observations.
FOUNDATION LITERATURE FOR THE MECHANISM
Shear and Aβ aggregation
A thermodynamic driving force for disease-induced protein “misfolding” is rarely suggested. We are led to believe that this is a random, spontaneous event. A sophisticated research program has yielded [46–50] a detailed quantitative mechanism for the aggregation of Aβ dissolved in quiescent aqueous solutions. It takes hours of lag time for the aggregation rate to finally accelerate and ultimately consume all the Aβ40 monomer in the solution in the formation of higher molecular weight aggregate.
Shear has been shown to accelerate this slow Aβ40 aggregation process [43, 44]. However, as has been demonstrated with other proteins [51, 52], the presence of an air-water interface or hydrophobic surface [53, 54] will also accelerate protein aggregation. It is still a matter of debate as to whether shear alone induces protein aggregation or accelerates secondary mechanisms that succeed the initial interface-induced nucleation step. In one other simple shear experiment in a stainless-steel capillary, with no air/water interface, the shear caused efficient aggregation of Aβ on the inner hydrophilic steel surface of the capillary at very low shear rates [45]. When flow was paused, Aβ-containing solute from the wall was released from the surface. Such a release has unwanted analytical clinical implications because needles with the same characteristics as this capillary are used in clinical studies monitoring Aβ concentrations in clinical spinal taps.
Shear has been shown by Raman spectroscopy [55] to alter protein conformation at the start of shear flow and return to its original conformation when flow is stopped. Shaking that presumably caused fragmentation of already formed Aβ fibrils also increased the rate of aggregation, but the shear rates in these experiments was not reported [46].
There are two major isoforms of Aβ, Aβ42 and Aβ40. Aβ42 is more prone to aggregate in solution than Aβ40 [56]. The only difference between these two is that Aβ42 has two extra hydrophobic amino acids at the C-terminus. The aggregated amyloid senile plaques in AD patients’ brains consist of mostly Aβ42, even though Aβ40 concentration is much higher more than that of Aβ42 [57]. It has been proposed [41] that Aβ40, a major component in cerebral amyloid angiopathy (CAA) deposits, is formed through laminar shear processes in the perivascular CSF flow around arteries penetrating the cortex parenchyma in the glymphatic system. In this system, ISF flow continues within the cortex parenchyma possibly exiting through perivascular spaces around venules into the lymphatic system [58]. It is within the parenchymal spaces that AD plaques containing Aβ are located.
Tau and tau oligomers
Tau is a much larger and more complex molecule than Aβ [13]. Tau has six isoforms created by alternative splicing. Its biochemistry is further complicated by having large numbers and types of post translational modifications, the most important being phosphorylation and acetylation [59, 60]. Its primary cellular function deals with the regulation of microtubules in CNS neuron axons. Hyperphosphorylation causes detachment of tau from microtubules and induction of tau toxicity [61].
The spontaneous aggregation of tau in quiescent solutions is much like that found with Aβ with a long lag time followed by exponential growth, a plateau, and the formation of fibrils. While the monomeric structure of tau is highly disordered, the structure of aggregated tau is formed by close packing of comparatively rigid cross β-stands. Monomer tau molecules have also been shown to aggregate in loose arrangements within the liquid phase in such a manner as to cause liquid-liquid phase separation [62]. Isoforms of tau have been isolated in the laboratory [63]. Tau samples extracted from patients’ brains have exhibited behavior that has caused them to be identified as multiple tau strains that contribute to the heterogeneity of pathogenesis in neurodegenerative diseases [64, 65].
The in vivo interaction of tau with Aβ resulting in tau aggregation has been demonstrated by several groups [24, 56, 66]. Tau conformation changes brought about in these cases could be caused by Aβ-tau attractive forces opening the “paper clip” tau structure to expose an inner binding group in the tau molecule that enables tau aggregation [67].
Tau pathology is complex. However, tau pathology tracks AD dementia very closely. Phosphorylation frees tau from its microtubule binding site and alters its conformation by opening the paperclip conformation [61, 67]. The resulting p-tau molecule is not toxic, is found in young people, and is designated as pre-tangle tau. The earliest p-tau pathology is found in the locus coeruleus (LC) near the cerebral aqueduct connecting the third and fourth ventricles. On the other hand, tau seed involvement in AD pathology begins with the appearance of these seeds in the entorhinal region, leading to neuron damage [67].
In the homologous seeding process, small amounts of tau seeds will accelerate tau aggregation [59]. Recent research has revealed many cases of heterologous seeding in which seeds from one compound initiate and accelerate the seeding of another. Many different cross seeding pairs of have been demonstrated, e.g., Aβ and tau, in a very active research area. Both the local replication of tau protein aggregates and their spreading throughout the brain regions are implicated in the progression of early AD.
The special role of extensional flow stress
Dobson et al. have demonstrated [4] that extensional flow and not laminar shear has the capability of initiating aggregation of proteins that do not aggregate spontaneously. Their experimental setup consisted of two syringes connected by a capillary with a much smaller inner diameter than that of the syringes. Protein solutions were passed back and forth between syringes. Extensional flow stress is created at the junction of the syringes and the capillary. The rate of energy transfer in the extensional flow process, which was dependent on the flow rate through their setup, was shown to be one of the most critical factors in causing the rapid initiation of protein aggregation. The forces in extensional flow caused the exposure to solvent of previously sequestered sequences ultimately responsible for the protein aggregation.
The question that arises from these experimental results is, where in the brain does a similar hydrodynamic arrangement exist? A recent analysis [5] concluded that the CA region, in which CSF passes back and forth between the third and fourth ventricles, comes closest to fitting this description. This is most interesting because the earliest tau pathology, the first appearance of phosphorylated tau, is found in this same region. On the other hand, the earliest AD pathology that involves prion-like spread through the brain is that involving Aβ in the frontal and temporal lobe regions. The earliest prion-like spread of tau seed pathology is later in the entorhinal region, quite distant from the Aβ spreading region. An unanswered question is, since there is evidence that Aβ and tau are both critical participants in AD, why is there such a large gap in time and space between the earliest prion-like pathologies of these two compounds?
MRI experiments and chaotic CSF motion in lower brain regions
Atsumi and colleagues have demonstrated with magnetic resonance imaging (MRI) measurements [3] that there are highly variable mechanical energy events within lower brain regions of the glymphatic CSF flow system. These measurements tag a CSF water molecule at a certain time and, after a defined time delay, measure the spatial coordinates of that same molecule. Thus, the data analysis in the MRI method used, Motion-sensitized Driven-equilibrium Steady-state Free Precession, reveals the speed and direction of travel of CSF water molecules in all parts of the brain as a function of time. What this analysis reveals even for healthy individuals is a chaotic flow of these water molecules, with surprisingly high energy spurts in the lower brainstem, certain brain cisterns, the third and fourth ventricles, and the connecting CA. We believe that chaotic events like this, when sufficient such energy transfer is properly oriented, can significantly alter a dissolved Aβ or tau molecule’s conformation.
As suggested in the Atsumi et al. [3] and related papers [68–70], these chaotic flows are dependent on the CSF flow properties and geometries of the specific lower brain region in which the molecule is located. Whether or not there are chemical reactions relating to these conformational perturbations will depend on their energy as well as opportunities for reactions of the excited tau or Aβ molecules with their surroundings such as other stretched molecules or nearby membrane surfaces [71]. Figure 3 below illustrates in shades of black and gray the mechanical energy generated by these chaotic motions in different brain regions. Those that are the blackest are designated as mechanical energy “hot spots.”
AD PATHOLOGY, ANATOMY, AND MECHANICAL ENERGY HOT SPOTS
Maps of hot spots, cisterns, and early pathology locations for three different diseases
Figure 1 shows a basal view map of the early disease phase locations for tau, Aβ, and α-synuclein pathology. These can be compared in Figs. 2 and 3 with the locations of the major subarachnoid brain cisterns and MRI-designated CSF hot spots respectively. In Fig. 1, major arteries serving the brain are shown in red. The two major arteries are the carotid pair feeding the upper part of the circle of Willis in Fig. 1 and the basilar artery that flows over the pons in Fig. 1. These two systems are weakly interconnected, forming the circle of Willis. The subarachnoid cisterns are formed by CSF flowing between the subarachnoid layer and the bottom layer in the artery-filled space depicted in Fig. 2. Parkinson’s disease stages in the table of Fig. 1 refer to early α-synuclein pathology. AD and chronic traumatic encephalopathy (CTE) stages in the table of Fig. 1 are early tau pathology, except where noted for Aβ pathology. Black and dark gray regions in Fig. 3 are designated CSF hot spots.
Fig. 1
Pathology locations by stage: AD (Alzheimer’s disease); Park (Parkinson’s disease); CTE (chronic traumatic encephalopathy). Underlying figure source: Alamy.com 2BEH8M2. Disease stages: from [27, 88, 91, 107].
![Pathology locations by stage: AD (Alzheimer’s disease); Park (Parkinson’s disease); CTE (chronic traumatic encephalopathy). Underlying figure source: Alamy.com 2BEH8M2. Disease stages: from [27, 88, 91, 107].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g001.jpg)
Fig. 2
A) A sketch of hot spot cisterns in the circle of Willis and midbrain regions. Pulsating arteries and regions surrounding them are in red. B) A sagittal view of the brain cistern locations.
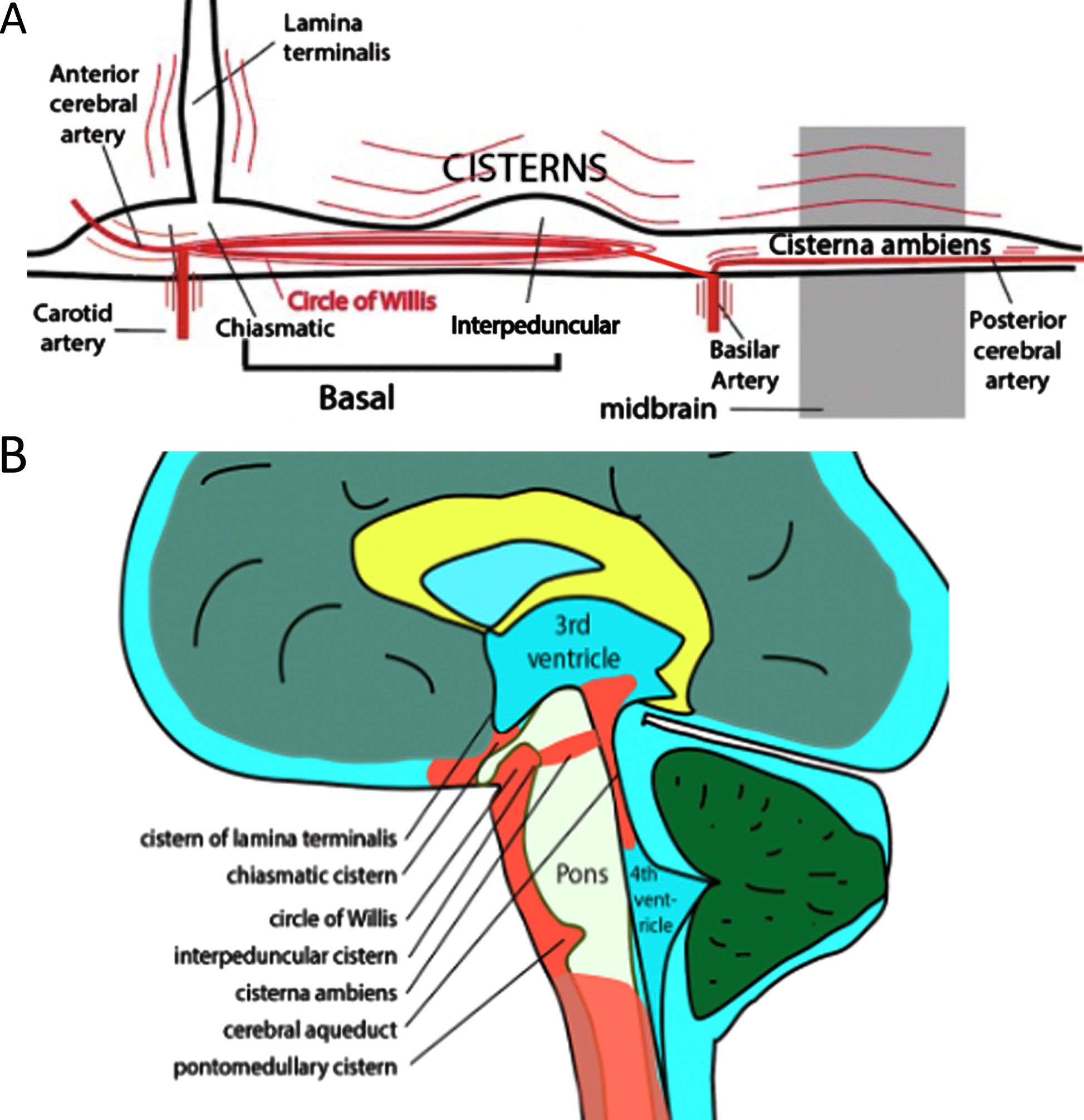
Fig. 3
Origins of Aβ and tau AD pathology correlates with black areas in this figure revealing regions in which CSF motion is found to be chaotic. Parkinson’s disease and amyotrophic lateral sclerosis (ALS) pathology also originate in the lower brainstem region where motor nerves and arteries jut into chaotic CSF. From [3] with modifications. Licensed and modified under a Creative Commons Attribution Non-Commercial-No Derivatives International.
![Origins of Aβ and tau AD pathology correlates with black areas in this figure revealing regions in which CSF motion is found to be chaotic. Parkinson’s disease and amyotrophic lateral sclerosis (ALS) pathology also originate in the lower brainstem region where motor nerves and arteries jut into chaotic CSF. From [3] with modifications. Licensed and modified under a Creative Commons Attribution Non-Commercial-No Derivatives International.](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g003.jpg)
Comparison of Figs. 1–3 demonstrates that nearly all the early pathological locations for the three diseases in the table of Fig. 1 are in the depicted cistern or brainstem hot spot regions. What is the connection between these hot spots and early AD pathology? Is one hot spot more important than the rest? If so, why?
The LC, CA, and CSF transit through the 3rd and 4th ventricles
CSF, generated in each of the ventricles, passes from the lateral to and through the 3rd and 4th ventricles. The CA, located between the latter two ventricles, is a very narrow tube whose ends act as quickly narrowing flow paths for the forward and reverse pulsing CSF. This means that with each heart pulse, there will also be two opposite direction CSF pulses, generating extensional flow stress at opposite ends of the CA. If the pulse has sufficient energy, there will be the possibility of generating a conformation change of any dissolved protein such as Aβ or tau caught in these regions that has the capability of forming a cross beta structured intermediate that can initiate the formation of a tau seed. There is another possibility, contained in the suggested mechanism below, of the stretched tau forming a high energy dimer with another stretched protein molecule other than Aβ in the CSF that is then adsorbed in that state on a nearby membrane [72] and, with time, forms a tau seed. Thus, a tau seed formed in this CA region would have the opportunity of interacting with the nearby LC.
LC is the first brain structure to exhibit pathology [73, 74]. Severe LC degeneration is a ubiquitous feature of AD, and the LC neurons are degenerated much earlier than when symptoms become clinically apparent. The major origin of noradrenergic modulation of the central nervous system by generating norepinephrine (NE), LC innervates extensive areas throughout the brain and is implicated in a variety of autonomic and cognitive functions [75, 76]. NE plays a role in neuroprotective functions that may reduce AD progression and underlies optimal memory performance. LC is a compact brainstem nucleus consisting of approximately 30,000–50,000 neurons in adult humans. While protein aggregates are thought to trigger neural degeneration in other brain regions, the LC appears to be spared from frank cell death until mid to late stages of disease [77].
The LC also plays a role in arousal and autonomic functioning such as wakefulness through connections with widespread areas of the brain and spinal cord and has been implicated in cognitive processes including attention, decision-making, and memory through connections with the frontal cortex and hippocampus [77]. The LC-NE system plays a major role in the sleep-wake cycle [78]. Sleep plays a major role in the disposal of brain waste including Aβ. Recent advances in in vivo imaging [76] in humans have enabled study of LC dysfunction in neurodegenerative processes, especially the development of tau aggregates that are apparently time-locked to prodromal symptoms of neurodegenerative diseases.
Early tau pathology is hypothesized to propagate in a prion-like manner along the LC-transentorhinal cortex white matter pathway, leading to atrophy of the entorhinal cortex and adjacent cortical regions in a progressive and stereotypical manner.
ATHEROSCLEROSIS AND THE CSF FLOW STRESS MAXIMUM
In the lower brain, there are major arteries such as the carotid and basilar that are comparatively close to the heart. These and other major arteries are not only in direct contact with the subarachnoid CSF but are primarily responsible for the forward and backward movement of the glymphatic CSF. The largest mechanical energy transfer from these arteries to surrounding tissues and CSF with its dissolved amyloid monomers should take place in these same major artery regions for healthy people.
If the suggestion that high systolic pulse energy is responsible for initiating amyloid aggregation, there is a critical possible prediction. With increasing age-related atherosclerosis, maximum systolic heart pulse energy is transferred from the early-affected aorta and carotid arteries to other downstream smaller arteries. Here this excess mechanical vascular energy is transferred to CSF surrounding these arteries both in their narrow subarachnoid flow paths and CSF in cisterns containing pulsating arteries [5, 79, 80]. Because of the longer residence time of dissolved Aβ and tau molecules in CSF cistern pools, they are subjected over long periods of time to cyclic, chaotic mechanical energy perturbation. MRI studies of basal cistern hot spots [3, 68, 70] demonstrate that such CSF becomes agitated in these cisterns to the point that they demonstrate locally increased recirculation. Such agitation, especially in the presence of hydrophobic surfaces, should be able to promote the aggregation of amyloid monomers [51]. There is a tight correlation between the locations of early neurodegenerative disease pathology and the intensity of this deposited mechanical energy, as seen in Figs. 1–3.
Putting all these facts together, it has been suggested that flow-induced stresses within the lower brain and cistern regions may be responsible for the initiation of Aβ and tau aggregation—and subsequent AD disease [5]. In this light, the decadal timing of this disease is of great interest. Why so slow and why so long in its etiology? The proposal [5] is that the key is the timing of the hardening of arteries. This process is a slow, decades-long evolution of vascular changes that can result in a fast-moving stroke or heart attack. But these vascular changes also bring changes in vascular pulse intensity that stimulate chaotic CSF movements in the lower brain, thereby connecting these vascular changes directly to the potential for inducing amyloid diseases. It is suggested that when the systolic CSF pulse energy finally reaches a certain critical energy level that stretches dissolved tau molecules and exposes a critical number of specific hydrophobic amyloid monomer molecule segments to CSF, the time from initiation of toxic oligomer precursor formation to early tau neuropathology can also be comparatively short.
As the vascular system changes with age that occur due to increasing atherosclerosis, arteries are in various stages of losses in their flexibility. The suggestion is that the maximum in the arterial and therefore CSF systolic pulse wave strength moves further and further away from the heart in the vascular tree system (Fig. 4). For example, maximum pulsatility initially moves from the aorta and carotid to the circle of Willis arteries and then beyond this circle with increasing atherosclerosis. Thus, the question is raised whether this migration of the MSPEL could also be responsible, at least in part, for the spread of early Aβ and tau pathology throughout the brain? This spread has been attributed to the toxic seed Aβ oligomer migration through neural circuits [81, 82]. Could at least some of this toxic spread be attributed to the resulting migrating location of the maximum systolic blood and CSF pulse pressure energy?
Fig. 4
Steady migration of Maximum Systolic Pulse Energy Location (MSPEL) for blood and CSF with age through the brain extending further from the heart with artery hardening. A similar diagram exists with “Basilar” substituted for “Carotid.” With permission [5].
![Steady migration of Maximum Systolic Pulse Energy Location (MSPEL) for blood and CSF with age through the brain extending further from the heart with artery hardening. A similar diagram exists with “Basilar” substituted for “Carotid.” With permission [5].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g004.jpg)
There are several effects taking place during this migration. One is the migration of the maximum energy deposition within the glymphatic CSF system that causes maximum stretch to an Aβ or a tau molecule in different brain locations with possibly different chemical products resulting from different energy inputs. Another effect is the mechanical stretch and strain of the tissue surrounding the glymphatic system. Pulse pressure transmitted to capillaries is reported to cause silent microbleeds caused by mechanical pulse pressure, breaking down the blood-brain barrier [83]. If the glymphatic perivascular CSF flow system extends to the capillary system, then any blood breakthrough of a capillary wall will mix with Aβ and tau dissolved in the surrounding CSF. This will expose these molecules to a sudden burst flow, possibly causing flow-induced stresses to these dissolved molecules. Such a process could be responsible for the observed Aβ plaque near the microbleeds [83].
In mice, toxic oligomers that formed in the same region as plaques are different from those found at a distance from plaques, implying differences in mechanisms of oligomer formation [84]. This has been attributed to differences in types of ISF flow within the parenchyma [85].
AMYLOID ETIOLOGY: MIGRATION OF THE CSF STRESS MAXIMUM? (MSPEL)
The above discussion leads to the question, could the various locations and stages of the pathologies shown in Fig. 1 be attributed to migration with increased atherosclerotic involvement of the maximum CSF systolic pulse energy throughout the brain as symbolized in Fig. 4? That is, does the MSPEL migrate with increasing vascular disease? Is there is a critical minimum amount of mechanical energy, especially for tau, arising from extensional flow stress that is needed from these pulses to produce flow-stressed molecular conformational change required to produce seeds that initiate neurodegenerative diseases? This is a complex question to answer since there are many of these diseases with quite different pathologies and etiologies in addition to unique individual medical histories within any given disease. How does MSPEL affect AD?
The earliest Aβ pathology in AD has an uncertain origin, but according to the ideas presented above, it would be reasonable to assume that early, comparatively low energy flow-stress events create low energy Aβ seeds in the lamina terminalis cistern region shown in Fig. 5. This reveals a massive confluence of arteries including the carotid which could be the origin of the earliest Aβ pathology. Such seeds would be rich in Aβ42, which is more sensitive to flow-induced aggregation in the laboratory than Aβ40. With aging and artery hardening, new Aβ seed formation could gradually spread throughout the interhemispheric cistern region and other lateral regions following its perivascular paths of the anterior cerebral and middle cerebral arteries [86–88]. Apparently both paths produce Aβ plaque [86].
Fig. 5
Lamina terminalis floor (center left) and basal cisterns (center to lower right). Used by permission from the Neurosurgical Atlas by Aaron Cohen-Gadol, MD.
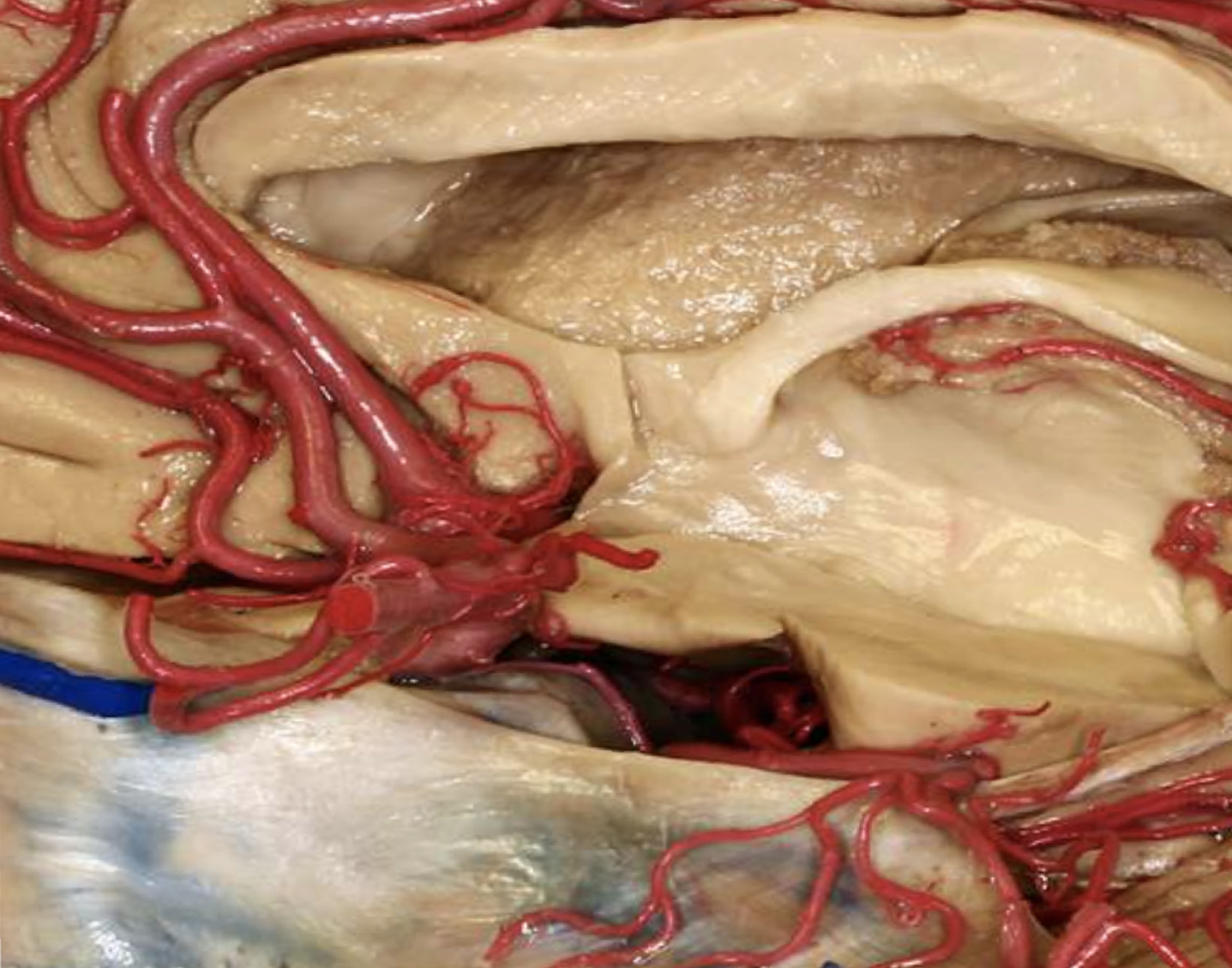
Meanwhile, Aβ seeds would be creating new seeds throughout the region in which they were first created. That is, initial seeding might spread through MSPEL migration and secondary seeding might spread through templated seeding around initial local seeding sites.
According to the above flow stress-induced mechanism, elaborated in detail below, a higher energy would be required for inducing tau seed because of tau’s more complex molecular structure. So, it is not surprising that a different tau pathology brain region might be required to attain the maximal pulsed extensional energy stress. Such a site is available in the CSF flow in the cerebral canal region. Flow stresses in this region could activate tau molecules sufficiently that when they approach the aqueduct wall (Fig. 6), the higher shear forces in this region help maintain or even enhance a stretched conformation. If the wall-shear stretched tau molecule attraction to the wall is strong enough, this stretched complex could be stable enough to adsorb and be maintained for a long period. Further adsorption of stretched tau molecules could ultimately build up oligomers and with further wall agitation create conditions favoring formation of a toxic, more thermodynamically stable oligomer state that ultimately triggers AD through LC neuron damage.
Fig. 6
Fluid dynamics calculation of the total strain within a cross sectional slice through the third ventricle and cerebral aqueduct (CA, lower right) during downward CSF flow. Figure courtesy of Vartan Kurtcuoglu based on data previously reported in reference [108].
![Fluid dynamics calculation of the total strain within a cross sectional slice through the third ventricle and cerebral aqueduct (CA, lower right) during downward CSF flow. Figure courtesy of Vartan Kurtcuoglu based on data previously reported in reference [108].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g006.jpg)
Tissue strain and amyloid disease
It is apparent from the above discussion that mechanical and fluid flow stresses are present in both brain tissue and CSF. Gangoda et al. [89] reported that increased vascular pulsatility can increase the amyloid-β protein precursor (AβPP) concentration as well as the enzyme β-secretase 1, causing an increase in Aβ production and secretion. Thus, an increase in systolic pulse energy can mechanically induce an increase in local Aβ concentration. This could increase the probability of flow stress-induced formation of Aβ-tau dimers within the CSF. At the same time, aging and hardening arteries cause a net decrease in the net CSF flow throughput, slowing down the rate at which Aβ and tau are removed from the brain if autonomic systems do not compensate, further increasing aggregation and disease probability.
Proteins experience larger shear stresses as they approach a confining wall or membrane surface causing an increased stretching of the molecule. Therefore, any protein within the brain that is adsorbed on that surface will initially probably be in a conformationally excited state. The molecule can remain in that state on that surface if intermolecular forces between the protein and surface are larger than the protein intramolecular forces attempting to relax the shear-induced stretch in the protein. In those hot spot regions where shear forces are highest, the tissue strain is also the highest as seen in Fig. 7, meaning that any adsorbed molecule in that region is also subjected to continuous oscillating tissue strain. Because the membrane surface has “floating” protein islands, if the adsorbed stretched molecule remains, it will probably encounter other similarly adsorbed proteins and can form island oligomers. If they retain the stretched conformation, they will be energetically and thermodynamically unstable. Continuous tissue agitation may cause ultimate molecular rearrangement and possible intramolecular chemical reaction leading to more thermodynamically stable molecular products. Such is the proposal in the suggested mechanism below.
Fig. 7
Whole brain tissue mechanical strain at the peak systole maximum in comparison with that at early systole. Yellow is maximum strain in the direction of the head and blue is strain at the same time in the direction of the feet. Strain intensity regions in this figure correlate spatially with chaotic flow region data in Fig. 3. Source [90] licensed under Creative Commons 4.0.
![Whole brain tissue mechanical strain at the peak systole maximum in comparison with that at early systole. Yellow is maximum strain in the direction of the head and blue is strain at the same time in the direction of the feet. Strain intensity regions in this figure correlate spatially with chaotic flow region data in Fig. 3. Source [90] licensed under Creative Commons 4.0.](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g007.jpg)
VASCULAR FLOWS AND TIMING OF Aβ AND TAU PATHOLOGY IN AD ETIOLOGY
Separation in time and space of the earliest Aβ and tau pathology?
One of the unsolved mysteries in AD is the reason for the timing and spatial separation of Aβ and tau pathologies. It is generally concluded that the earliest toxic tau seed pathology follows that of Aβ, but no conclusion has been reached as to why this is so. However, it is often reasoned that Aβ somehow is responsible for tau pathology. The MSPEL concept provides a possible reason. The blood supply for the brain is provided by two vascular systems, anchored by the carotid and the basil arteries. This provides two different general regions of the brain that will possibly be affected by two different MSPEL migrations induced by increasing atherosclerosis of two different arteries (carotid and basilar).
First, we have proposed above that Aβ pathology originates in or near the lamina terminalis (LT) and spreads from there through the carotid-based branch of the glymphatic system with its anterior and middle cerebral arteries. During this process, shear probably causes preferential Aβ42 seeds, because of its extreme sensitivity to flow stress, especially initially in any CSF flowing in and around any glymphatic flow-blocking objects in the LT floor (Fig. 5). Of all the “hot spots,” the LT region is probably the closest to the heart in the earliest Thal phase and could be the earliest brain region to be exposed to the MSPEL migration away from the heart with increasing atherosclerosis.
Later during MSPEL migration in the basilar-based glymphatic system branch, the maximum energy systolic CSF pulse energy location finally arrives in the CA region. The CA region is critical for hydrodynamic reasons because of its very large changes in CSF flow path dimensions, probably producing the strongest extensional flow stresses within either of the two glymphatic paths.
Why does Aβ pathology not also start in this region? When the MSPEL finally arrives in the CA region, these flow stresses are at last large enough to stretch the tau molecule to expose its critical repeat region to other similarly stretched protein solute molecules and to elevate the tau molecular energy enough to rearrange the tau structure to form a tau seed precursor. A suggested role of Aβ in this region will be shown by the mechanism below. Briefly it is this: the small stretched Aβ molecules are efficiently scavenged by the now-stretched large tau molecules forming dimers, thus preventing Aβ aggregation between stretched Aβ molecules.
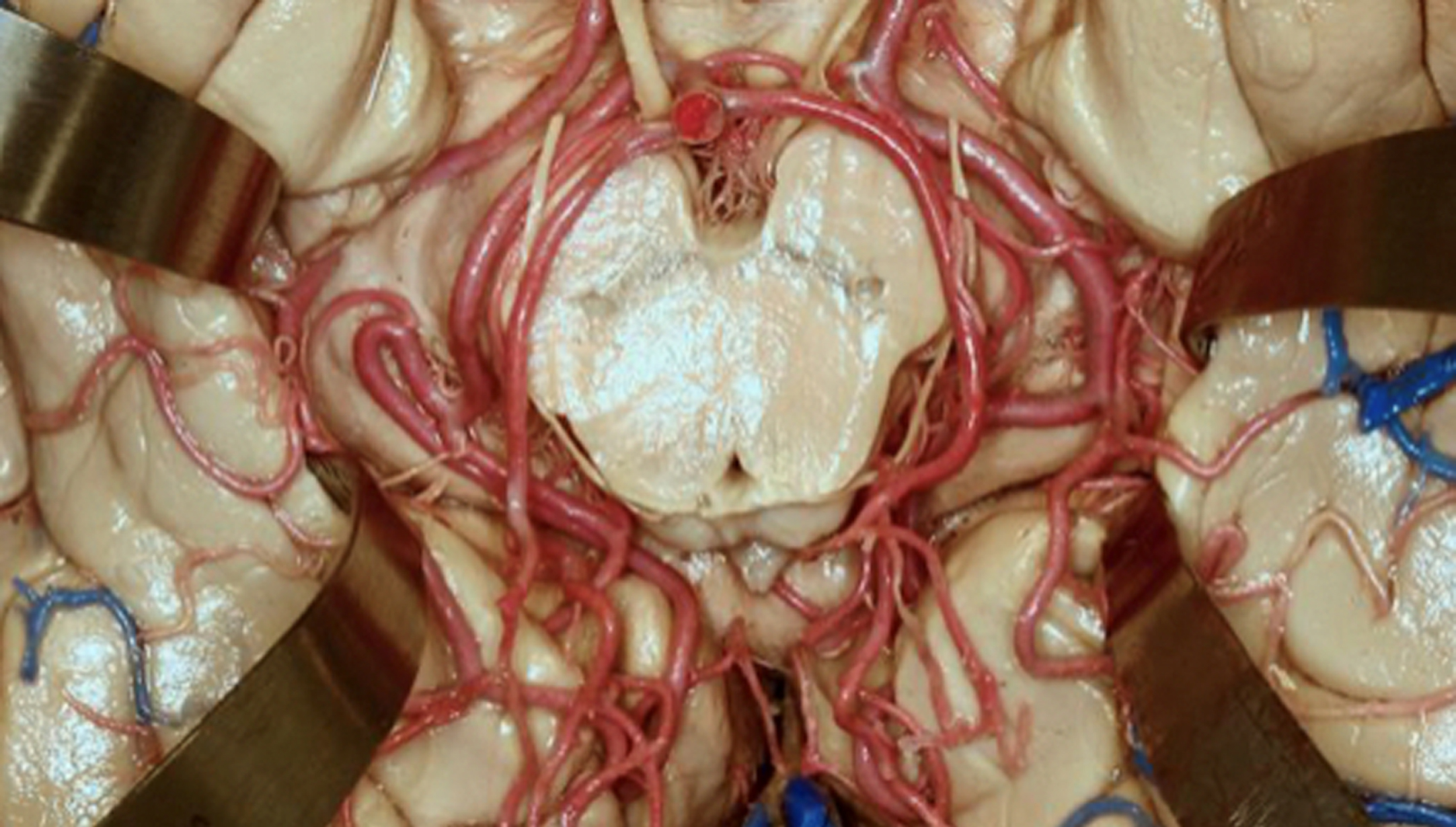
What regions are serviced by the basilar system? From an AD pathological perspective, regions of the brainstem and posterior cerebrum are the most important. What are the effects of changes brought about by the effects of atherosclerosis on MSPEL migration in these regions? An interesting vascular juncture is in the shown surrounding the midbrain region shown in Fig. 8. In this figure, the bottom center dot in the midbrain is the cross section of the CA. Any strong systolic CSF pulses will be felt by the tissue surrounding the CA imposed by the arteries surrounding it. This will be in addition to internal CA pulses arising from back-and-forth CSF flow entering the CA from the third and to a lesser extent fourth ventricles (during reverse CSF flow). Thus, a tau molecule stretched by extensional strain in the CA and adsorbed on the surface in or near the CA will be subjected to further oscillations on this surface. Such oscillation in addition to interaction with other adsorbed, stretched tau molecules or complex tau intermediates could well lead to formation of a toxic tau seed later.
Fig. 8
Vasculature surrounding the midbrain. Used by permission from the Neurosurgical Atlas by Aaron Cohen-Gadol, MD.
BRAIN INJURIES, PULSED CSF FLOW, AND AD
The earliest pathology resulting from CTE is limited to tau aggregation in the depths of cortex sulci (right two panels in Fig. 9). This is remarkably like the AD Aβ pathology like shown on the left side of Fig. 9. However, there are no signs of Aβ plaque in CTE other than that arising from separate AD disease [91, 92]. However, longer term CTE tau pathology is found at a distance from the cortex sulci. It presents in the same basal brain hot spot regions mentioned above for AD. Why is there this apparent spatial and time disconnect between the early and late CTE pathologies? And, in the earliest pathologies, why is there no Aβ in CTE when there is only Aβ in the earliest AD pathology in the same brain cortical sulci locations? The mechanism and discussion presented below attempts to explain these results.
Fig. 9
Earliest cortical pathological stages in AD (left) and CTE (right two images) phase 1 AD Aβ plaque pathology is noted by black arrows primarily in sulci. (There is no early cortical tau pathology in AD.) CTE tau pathology on the right is result of p-tau staining. (There is no CTE Aβ pathology other than that from coexisting CTE and AD). Sources: left [107] with permission; right two [91] with permission.
![Earliest cortical pathological stages in AD (left) and CTE (right two images) phase 1 AD Aβ plaque pathology is noted by black arrows primarily in sulci. (There is no early cortical tau pathology in AD.) CTE tau pathology on the right is result of p-tau staining. (There is no CTE Aβ pathology other than that from coexisting CTE and AD). Sources: left [107] with permission; right two [91] with permission.](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g009.jpg)
If one accepts the theory suggested in the previous sections that AD is caused by increasing flow stress generated by increased systolic heart pulse energy, then could this CTE etiology in some ways be caused by a reverse of much less energetic pulsed CSF flow in AD? That is, impacts on the head instantaneously create a sharp, local CSF pressure gradient at or near the impact site. CSF flowing in the glymphatic system is an incompressible fluid. The question is, where is/are the most critical pressure relief valve(s) that determine which direction CSF will flow to relieve this pressure? There is not a simple answer to this question. However, immediately following a severe blow to the head, an instantaneous CSF flow is probable in a reverse direction from the normal net CSF AD flow from cistern hot spots to the cortex.
Since CSF is thought to normally travel mainly in perivascular and subarachnoid cistern-containing routes from the ventricles to the cortex, is it not reasonable to assume that following a sudden severe blow to the head, a sudden CSF pressure wave will surge in the opposite direction from the impact site, possibly following a reverse perivascular path around minor and then major cortex arteries as well as subarachnoid paths around the brain surface? Why is early pathology found only in the depths of the sulci for such different pathologies in both very early AD and CTE?
The role of sulci in early AD and CTE pathology
Ghajari et al. [93] showed that brain tissue deformation induced by head impact loading is greatest in sulcal locations, where tau pathology is observed in cases of CTE. Human neuroimaging observations converged with computational predictions. Mechanical strain and strain rate are found to be greatest in sulci. Since the earliest observed tau pathology appears to be focused around vascular arteries or arterioles, it is suggested here that the high sulcal pressures suggested by the above findings cause a strong CSF pulse flow through the perivascular Virchow Robin space surrounding those penetrating arteries located within the sulci (Fig. 10).
Fig. 10
Virchow Robin space containing the CTE heavily pulsed CSF, rapidly flowing and narrowing perivascular CSF flow path that generates extensional flow that severely stretches dissolved Aβ and tau molecules and where the Aβ and tau can chemically combine into dimers that ultimately lead to tau seeds.
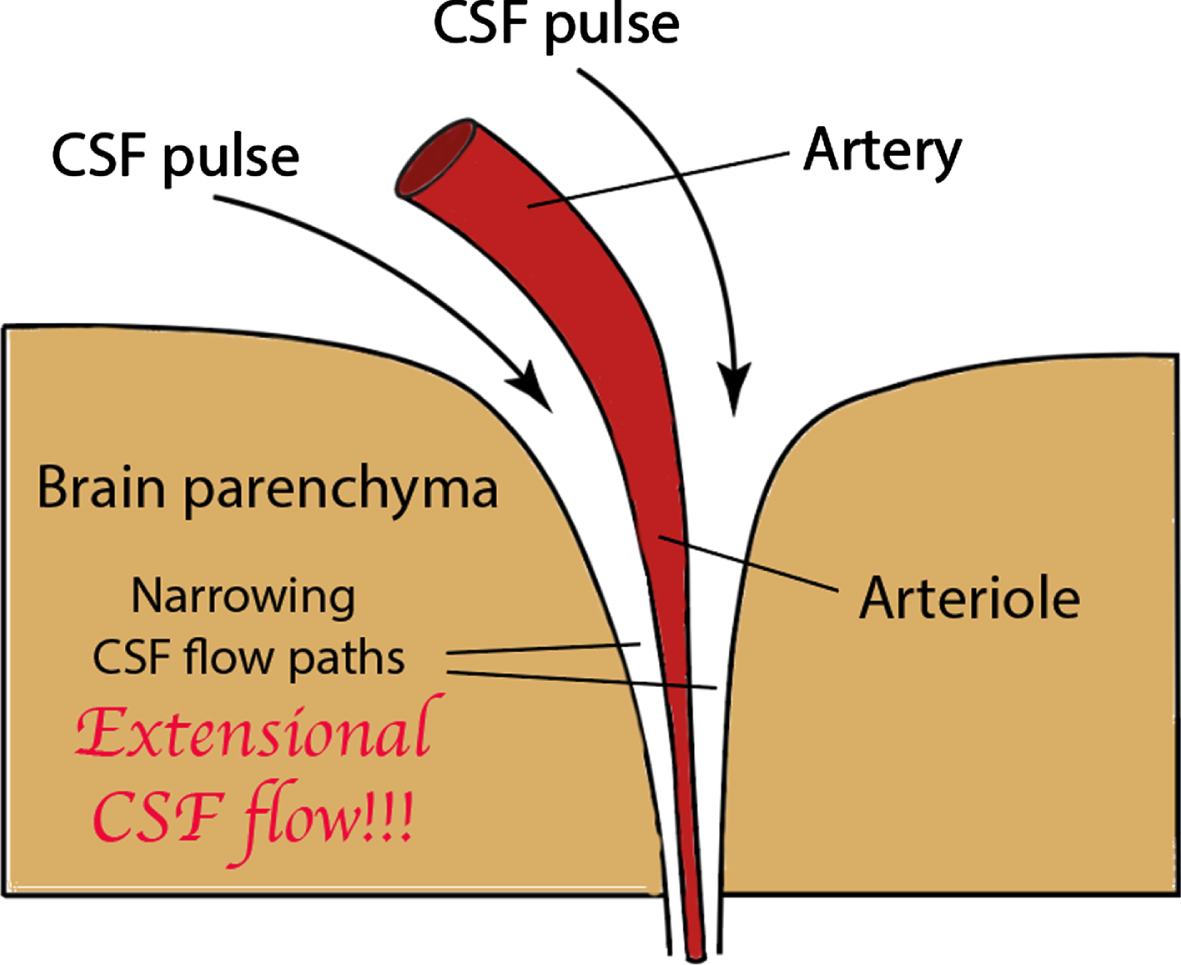
Note in Fig. 10 the narrowing dimensions in the CSF flow path as it is pushed through the perivascular space by the focused CSF pulse. This an ideal situation in which to generate extensional flow, especially with the indications of high strain rates, which are known to lead to formation of chemical intermediates that initiate protein aggregation [4]. Thus, we believe that damaging sulcal CSF flow stress is an integral process in the mechanism responsible for early CTE tau pathology.
However, the question now arises, could this process also be responsible for AD early Aβ pathology? The systolic pulse energy transmitted from the laminar terminalis and other arteries through the perivascular and subarachnoid spaces to cortical sulci is at least an order of magnitude lower than that arising from a TBI or CTE pulse. However, that same order of magnitude may also describe the increased sensitivity to extensional strain of Aβ42 when compared with tau. This would explain why in the early frontal and temporal AD, there is no tau pathology, only that of Aβ42.
PULSED FLOW ENERGY AND AD
The very strong pulses encountered in TBI and CTE should result in something like extreme AD, depending on where these pulses are experienced. This implies that, on average, both Aβ and tau conformations, as well as those of other CSF dissolved IDP—and perhaps non IDP—proteins, will be stretched to differing degrees. Thus, normally internally shielded hydrophobic regions on these protein molecules will be exposed to CSF and will also, at least for a short time, present greatly increased collision cross sections because of the imposed high strain levels. This will increase the probability of dimer formation during the collision between two stretched molecules.
However, because of the strength of the impact pulse and CSF incompressibility, the instantaneous flow pulse will be widespread throughout the brain, the extent depending on the earliest wall compliance of the confining CSF flow path. If sudden flow compliance is low in this path, the possibility for stretched protein dimer formation and adsorption may be widespread throughout the brain.
Much smaller CSF and tissue pulsations will still follow long after the TBI or CTE impact event(s) and keep applying comparatively low CSF pulsed flow and tissue stress energy levels on dissolved amyloid molecules and aggregated adsorbed proteins following traumatic brain events. Figure 11 illustrates the types of processes and energy changes that molecules are likely to undergo following a brain impact or strong systolic pulses in the CA region. Depending on the relaxation and reaction times of the respective excited amyloid monomer states, still-stressed Aβ and tau molecules can migrate toward the CSF flow path membrane walls and be adsorbed on them as monomers, mixed Aβ-protein dimers, or trimers, or Aβ dimers, if the Aβ concentration is elevated. If the adsorption is weak, as has been reported with some stressed Aβ aggregates [94], the excited molecular state can relax, and some monomers and/or shear-stressed products are released back into the CSF when stress ceases or diminishes. If the adsorption site is in a high tissue agitation hot spot zone, after long periods of time, there are possible interactions between adsorbed molecules. Furthermore, such tissue agitation may ultimately rearrange and reorganize the adsorbed molecules into more stable thermodynamic and possibly toxic amyloid disease states, as illustrated in Fig. 11.
Fig. 11
Suggested schematic of energetics of the sequence of events showing the flow stress-induced stretching of Aβ and tau molecules (Aβ * and Tau**), followed by the formation of an Aβ*-Tau** dimer, which is adsorbed on a membrane surface S, followed by the adsorption of another Aβ*-Tau** dimer, and finally, following long exposure to tissue strain in a cistern “hot zone,” with the formation of a thermodynamically more stable, surface-adsorbed toxic tau dimer. The released Aβ can be dissolved in CSF and eliminated. This same mechanism may be proposed for both CTE and AD, but with the extensional stress energy of both Aβ* and Tau** higher in CTE than in AD.
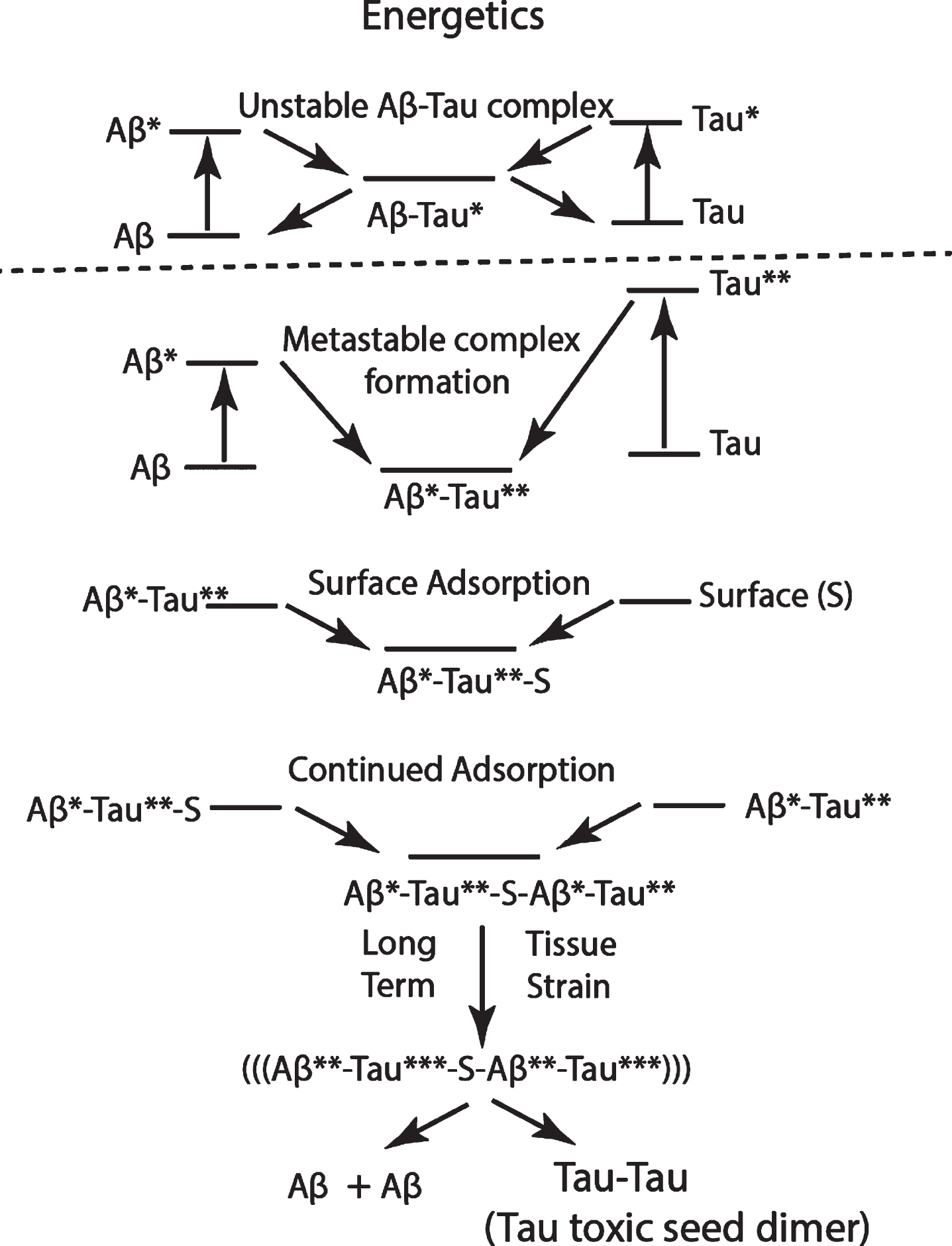
Why is there only early tau pathology and no early Aβ aggregate produced in CTE or TBI? We assume that the pulse energy is so high that Aβ, tau, and other CSF protein conformations are all very highly stretched within regions of extreme extensional strain, exposing intermolecular bonding regions normally shielded from CSF. Aβ is a much shorter molecule than tau. Therefore, if highly stretched tau molecules react with a single highly stretched Aβ molecule, only a short section of the tau molecule is intermolecularly bonded, meaning that more Aβ molecules could be bonded on a single flow-stressed tau-Aβ complex. If we consider a stressed tau molecule as a stressed Aβ scavenger, then the large collision cross section of a stressed tau molecule acts to lower the local Aβ concentration, resulting in a corresponding lowering of the Aβ*-Aβ* aggregation probability.
The Aβ and tau molecules represented below are very highly simplified cartoons of the average molecular structure of these IDPs, which are constantly changing their conformations in aqueous solutions. They are based on theoretical calculations with many simplifications to allow structural computations. We suggest that when they are exposed to pulsed brain CSF or ISF flow or tissue agitation conditions, these two molecules ultimately can assume a cross beta structure. Their IDP character is irreversibly changed, and they aggregate in complex ways, assuming much more rigid, often toxic structures.
PROPOSED MECHANISM FOR FLOW-STRESS INDUCED AD PATHOLOGY
Listed below are each of the proposed equations in a possible initial Aβ and tau aggregation mechanism accompanied by cartoon representations, however imperfect, of the molecules in that equation. The point in the cartoon is to give the reader a feeling for the types of mechanical fluid distortions of molecules and the proposed general types and timing of conformation changes responsible for the biological consequences of flow stress forces in AD. Locations and timing for each step in the Aβ segment of this mechanism for initial pathological stages are listed in front of each equation numeral. The carotid (GL-C) and basilar (GL-B) artery glymphatic systems are discussed above.
There can be no one single unqualified purely chemical mechanism for the following reasons: (a) the key Aβ and tau protein monomers are IDPs; (b) these monomers are attracted to and adsorbed by membrane surfaces [95]; (c) monomer protein aggregation is sensitive to the flow stress energy magnitude and types [7, 41, 96]; (d) it has been shown that the mechanism of Aβ aggregation is different under agitated conditions [46]; (e) in the human brain, Aβ and tau monomers are found in both ISF and CSF that flow through selected pathways in the brain; (f) flow energy and types of flow stress, both of which may well determine which flow-stress-induced chemical product is produced, vary in different parts of the brain’s glymphatic systems and may help determine the stage of AD disease; (g) Aβ and tau are not the only proteins that can form flow-stressed and energized intermediate products and combine with flow-stressed Aβ and/or tau molecules to form mixed protein aggregates; (h) there is currently no way of theoretically predicting what flow-stressed chemical intermediates might be.
Nevertheless, an energy-dependent mechanism is presented in the following sections that attempts to sum up the key parts of a general mechanism that can explain many critical experimental and clinical research results and answer many of the unanswered questions raised at this time regarding the origin and etiology of AD. What remains are experiments that test this mechanism!
The Aβ part of this mechanism takes place in the glymphatic-covid system, is initiated in the lamina terminalis hot spot at T1, the earliest time for either Aβ or tau flow-stress initiated aggregation, and spreads throughout the frontal and temporal lobes via Aβ flow-stress initiated seeds formed by the mechanistic steps [1] through [4] illustrated below.
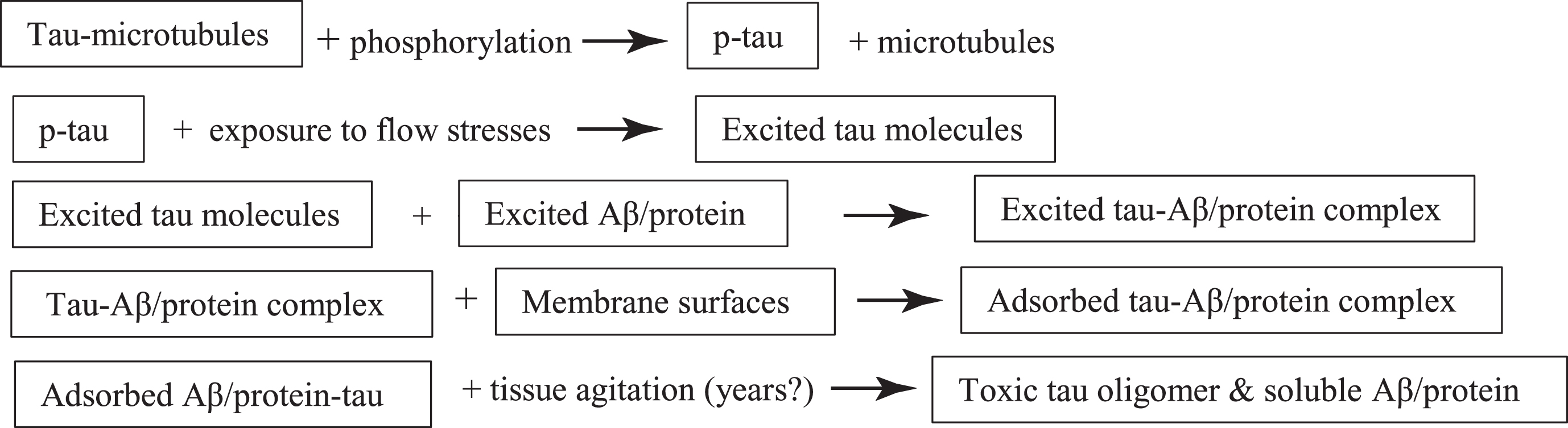
Tau protein’s role in any AD mechanism is much more complex than Aβ for the following reasons:
1) Tau is a more complex, IDP with some limited internal, relatively structured regions.
2) Tau is composed of around 400 amino acids, whereas Aβ is composed of only around 40. See Fig. 13 for the complexity of a calculated tau solution structure and its IDP character
3) Tau has 6 conformers with different physical and chemical properties because of different DNA splicing patterns.
4) Tau is subjected to post translational modifications (PTM) such as phosphorylation, e.g., p-tau, which alter the tau conformation.
5) In its different physiological roles, tau is in different conformations, which are critical to their functioning.
6) There are very few published papers on the effects of any types of flow stresses on tau molecules in any form.
7) For the above reasons, there is no one tau conformation structure to use in any mechanism. There are only many different possible structures, depending on their surroundings and which conformer is in which PTM state—and on their exposure to flow stresses.
Fig. 12
Energy levels for Aβ and tau proteins under normal quiescent solution conditions and under flow stress conditions with varying degrees of chemical excitation attained by transfer of mechanical energy of the stressed solvent to the Aβ or tau molecule. The asterisk (*) indicates comparatively low energy that is given off when the excited molecule relaxes. The double or triple asterisk (**/***) indicates the minimum energy needed to reach a critical level that allows for a significant conformation change, e.g., to a cross beta structure. The arrow indicates changes in electronic energy of the molecule and is not quantitative.
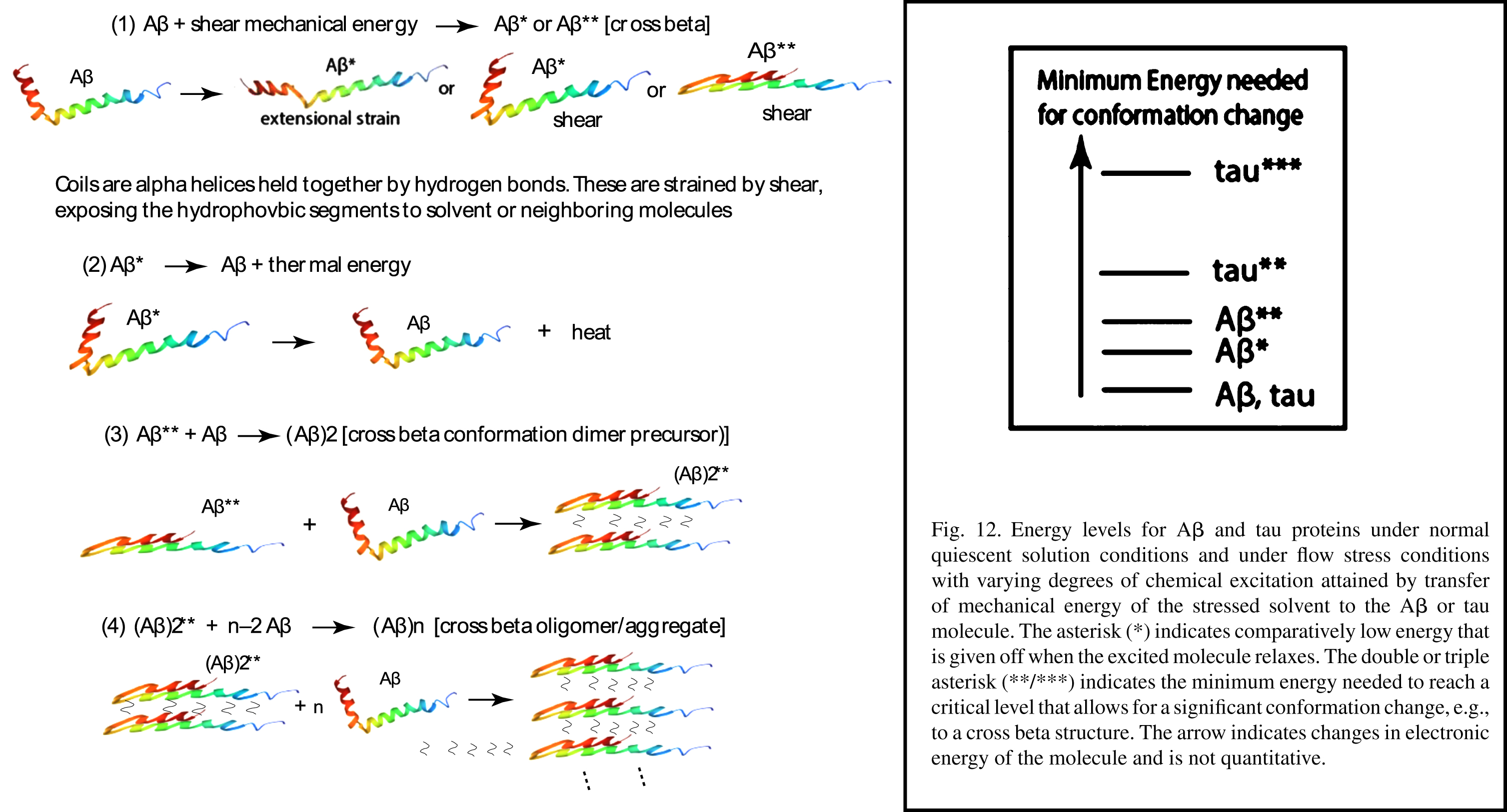
Fig. 13
Calculated lowest energy conformation state of tau in solution (color) and some representative other higher energy IDP conformation states (gray). The IDP tau is rapidly and constantly randomly cycling its conformation among these various states. With permission [109].
![Calculated lowest energy conformation state of tau in solution (color) and some representative other higher energy IDP conformation states (gray). The IDP tau is rapidly and constantly randomly cycling its conformation among these various states. With permission [109].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g013.jpg)
A prime focus in our paper is on whether in vivo flow stress changes the tau conformation to ultimately produce a different tau strain, especially one that ultimately produces a toxic tau seed. It is the toxic tau seed that initiates aggressive AD pathology. How is this seed formed? Are flow stresses involved? We suggest they are.
Figure 14 illustrates the effect of PTM partially opening the tau molecule to solvent in certain internal regions of the complex structure shown in Fig. 13. Figure 14 shows a much more simplified tau structure, now shown as a “paperclip” conformation. When thus exposed, the microtubule binding (MTB) repeating regions, shown in the center bottom of Fig. 14 as one red and three blue-gray blocks, can now take part in tau aggregation. Aggregation occurs primarily by joining together tau molecules in the MTB tau repeat region of each tau molecule as shown in the bottom left of Fig. 14. This opened paperclip molecule is attracted to other molecules with similarly exposed repeat regions and aggregates because of inter-digitating opposite strands (“steric zippers”) and comparatively weak intermolecular bonds [97].
![Paperclip tau model with some of its functions and reactions. With permission [67].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g014.jpg)
Phase separation alluded to in Fig. 14 concentrates tau molecules into a liquid-within-a-liquid spherical “bubble” region inside the neuron [98]. A question raised, given this increased tau concentration, is this bubble, when exposed to fluid flow stress inside the neuron, also a prime site for tau tangle aggregation? Another question raised is whether flow stressed-induced tau conformation changes produce a degradation-resistant toxic strain that would normally be degraded by the process alluded to in the lower right of Fig. 14.
In the upper right of Fig. 14, the microtubule binding tau molecule is shown as having two tails unassociated with the microtubules. Could internal cellular ISF flow induced by increased maximum systolic pulse tissue energy location agitation in CSF hot spots cause the displacement of tau molecules from their microtubule binding site through mechanical forces on these tails? If so, could subsequent MTB tau phosphorylation open its paper clip structure and, combined with energy transferred from ISF flow processes, lead to formation of tau tangles inside the neuron?
Illustrated below is a potential molecular mechanism for the formation of a critical tau seed starting with the paperclip tau structure in the same manner as used above with Aβ, recognizing that this structure is a purely hypothetical symbol of the great complexity of many possible tau molecule conformations. The mechanism builds on the hypotheses in our paper that the high flow stress brain locations of early tau pathology correspond with regions that are prime suspects for producing predominant extensional flow. Such flow is known to effectively change protein conformations in such a way that they more readily aggregate. Along the way in this protein chain aggregation process, it is suggested that toxic oligomers are formed, probably in conjunction with Aβ (and/or possibly other stretched CSF proteins), that ultimately wind up being adsorbed on membrane surfaces.
{We suggest that only tau extensional forces created by flow stresses in certain geometrical brain flow pathways such as in the CA region initiate aggregation. It is further suggested that, following the opening of the tau paperclip with phosphorylation, strong, pulsed back-and-forth flows create quasi-linear tau molecules with some residual structure. The degree of linearity depends directly on the amount of mechanical energy transferred to the tau molecule. These long molecules then present high collision cross sections for entanglement with other stretched dissolved protein molecules, especially highly stretched Aβ molecules, and markedly increase the probability of dimer formation from the reaction of tau with other linear IDP molecules.
After such multi-molecular complexes are adsorbed onto membrane surfaces, surface flow as well as cylic stretching and relaxation of the membrane itself are suggested to agitate these complexes over long periods of time. In such a process, it is proposed that much more thermodynamically stable toxic tau oligomers are ultimately formed. It is proposed in this mechanism that when this happens Aβ molecules are released back into the CSF liquid phase where they are either destroyed or transported out of the brain. If proteins other than Aβ are not released in this mechanism, they could be responsible for the reports of cross seeding aggregates with tau [99]. This complex process ultimately producing toxic prion-like seeds of tau and possibly other proteins is postulated to occur over a long period.
Thus, our proposed tau mechanism contains not only Aβ and tau reactants, but also: major types and variable amounts of flow energy, the PTM of the tau molecule, its current conformation, and the relative timing of these events with respect to times of initial Aβ and tau aggregation. Below is a block diagram of the overall scheme of the tau segment of the mechanism. This set of initial reactions is proposed to take place in and around the CA region of the basilar CSF lymphatic system.

Summing the above equations, we have:
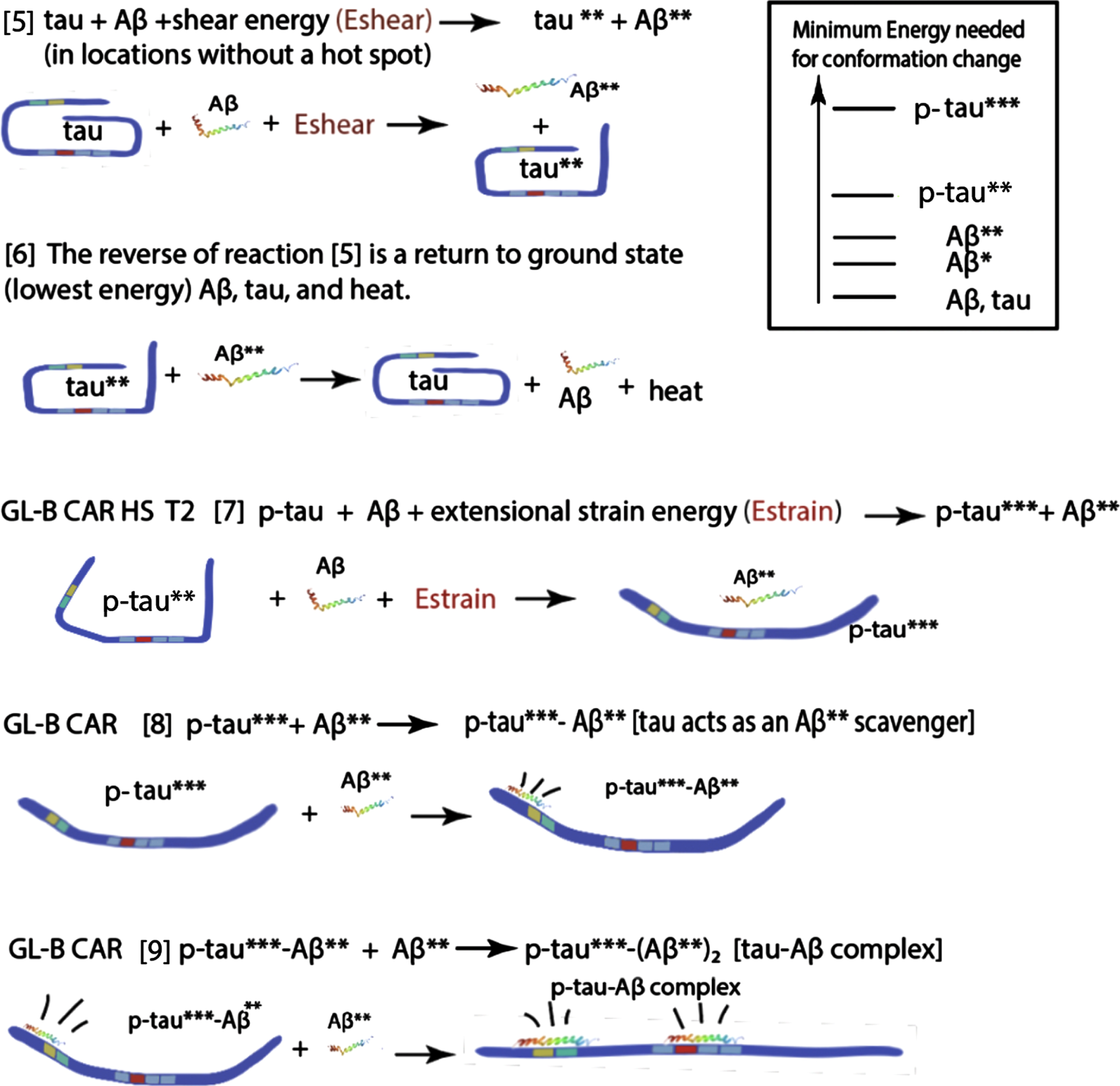
Locations and timing for each step in this tau-Aβ aggregation mechanism for the initial Braak and Thal stages are listed in front of each numeral: GL-B, glymphatic-basil system; CAR, cerebral aqueduct region; CAR-M, cerebral aqueduct region membrane; HS, CSF “hot spot”; T1, earliest time for Aβ aggregation; T2, earliest time for tau aggregate formation; LT, later than T2. In each equation, as an alternative, a CSF protein can substitute for Aβ.
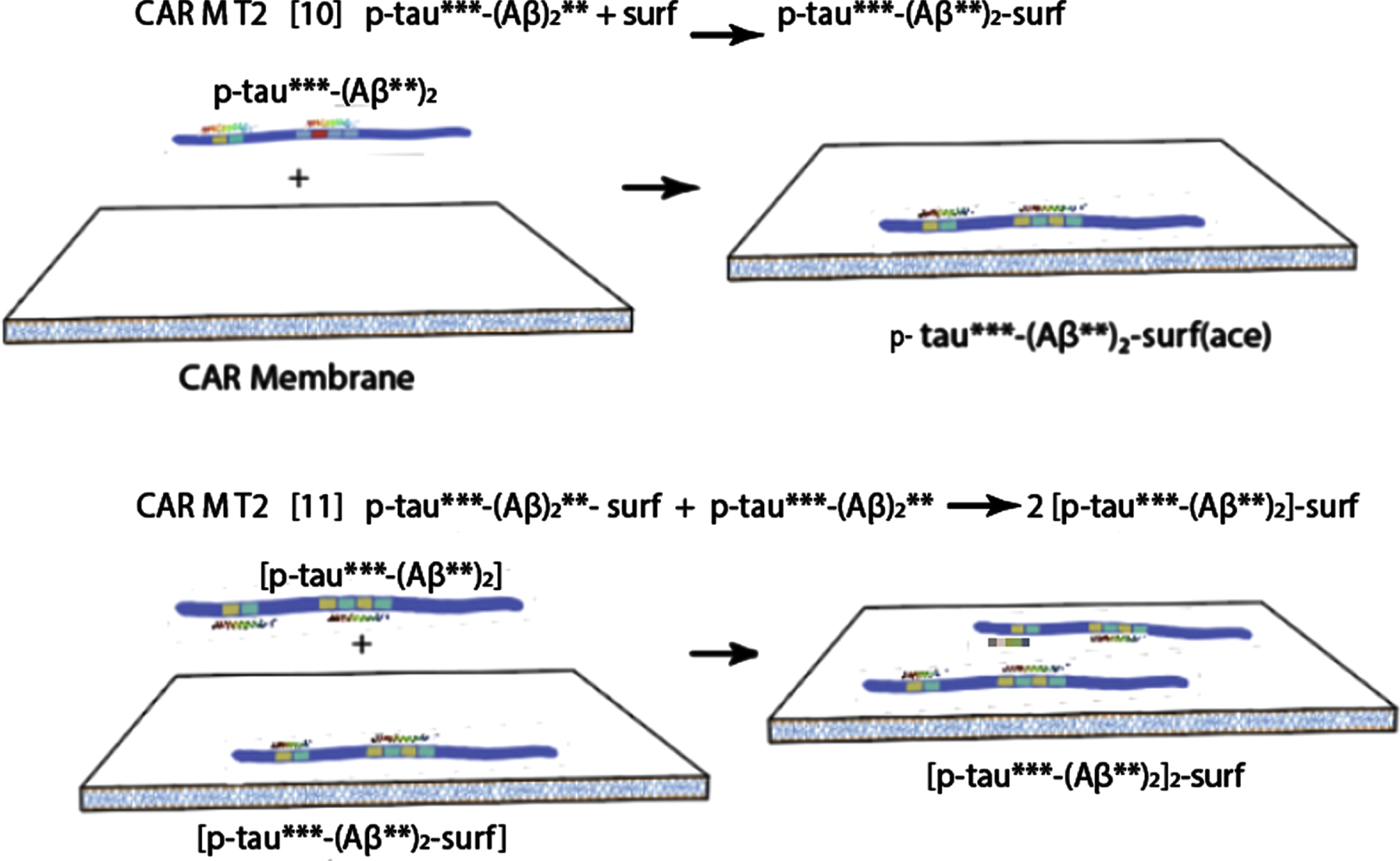
Attachment of tau-Aβ complex to the membrane, with long term tissue agitation following, ultimately produces a toxic tau oligomer precursor and soluble waste Aβ/protein molecules.
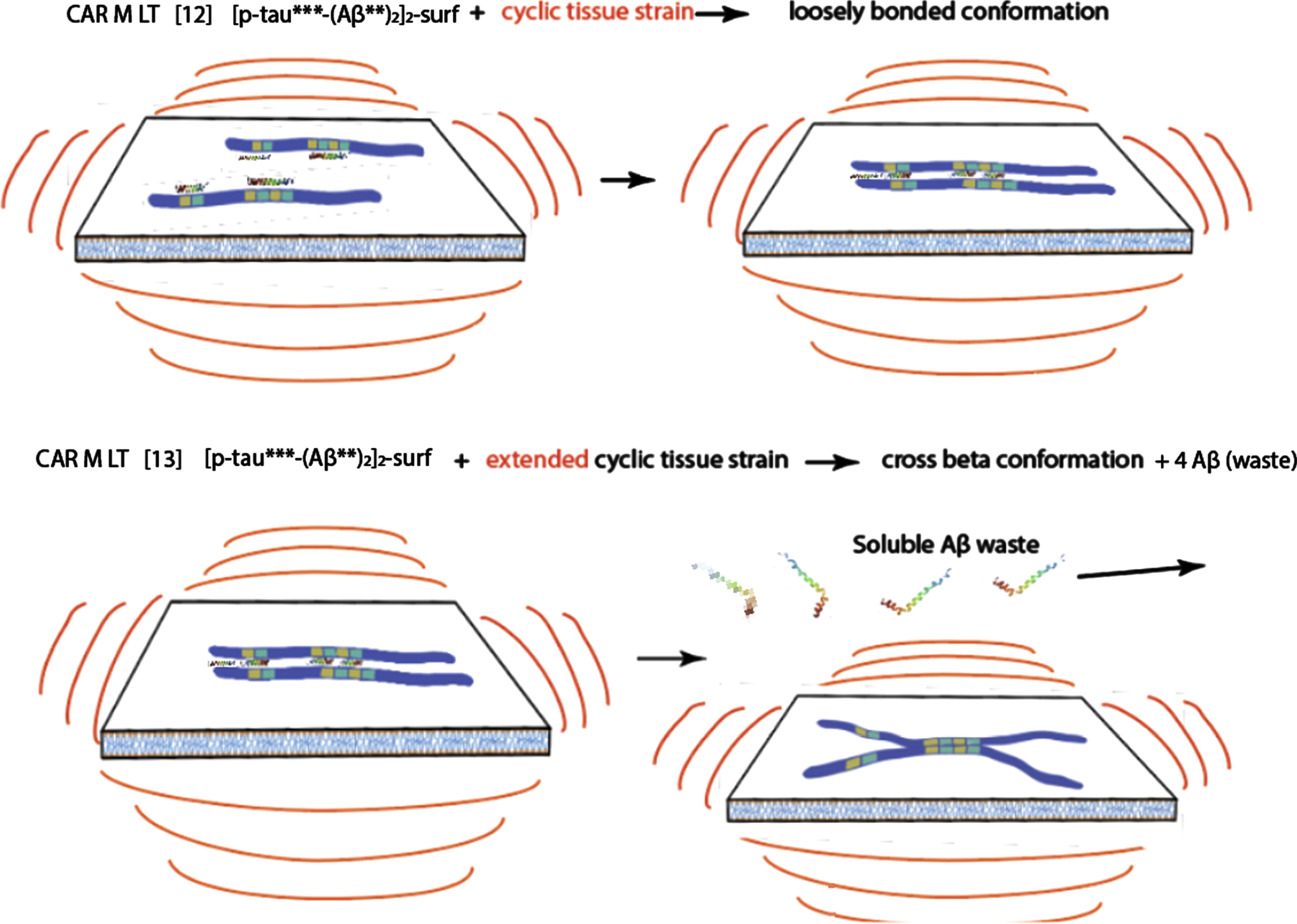
The last hypothetical membrane-tau complex depicted above (still in the preclinical phase, although the oscillations depicted above could take many years) is proposed to be a precursor to the ultimate toxic tau seed that initiates AD.
Several challenges to the mechanism
• Why are only associative areas involved in the cortex while sensorimotor regions are saved in the first phases of amyloid pathology?
This could be because of the comparatively long distance from the heart and thus the consequential reduced hydrodynamic pulse intensity in the sensorimotor region. The diameter of the arteries in that distant region may be smaller. In other words, if our mechanism is correct, the artery feed system in these regions during the early pathology phases have significantly different hydrodynamics from those found in the associative areas in those phases. Because of later migration of MSPEL to this region, pathology is initiated in these sensorimotor regions.
• Why does the tau pathology start accumulating close to a “hot spot", whereas amyloid does not?
Aβ and tau pathologies are proposed to arise responding to two different energy level and flow type events. Low level pulse energy is probably spread long range throughout the frontal lobe and focused in frontal sulci, resulting only in Aβ42 aggregates. On the other hand, tau pathology is dependent on achieving local high energy extensional flow around the CA, where distorted tau (and Aβ) molecules are quickly exposed to and adsorbed on local surfaces
• How does the mechanism integrate the proposed propagation mechanism with the main recognized way of neuropathological spreading, that is, through connected neurons, in a prion-like manner?
We are not denying the prion-like spread. What we are proposing is that MSPEL migration is occurring, possibly with consequences outlined in this paper that may appear to be very much like prion-like spread away from the heart.
• What explains cross seeding and coaggregation?
In coaggregation two or more monomer proteins are contained in the same aggregate. In cross-seeding, the monomer or aggregate of one protein act as a seed for the aggregation of another protein. In the case of coaggregation, two proteins can polymerize together to form a mixed aggregate [59, 100–102].
Given the above mechanism, there is ample opportunity for both coaggregation and cross seeding to take place. Mixed dimers proposed in the suggested mechanism can aggregate as a protein coaggregate on the membrane. The mechanism proposes the formation of the tau seed. However, if the protein in the dimer is, for example, α-synuclein rather than Aβ the ultimate product could be both tau and α-synuclein seeds resulting in cross seeding. On the other hand, any other protein, even a non IDP, can be stretched with sufficient chaotic energy, exposing hydrophobic bonding regions that can lead to aggregation with any other similarly distorted proteins such as tau, leading to coaggregation.
RECOMMENDATIONS
If the above discussion and mechanism have merit, the next question is, what can researchers and clinicians do to help prevent or cure AD? Our proposed answers:
1) Reduce the intensity of the systolic CSF pulse in the brain. If the earliest pathology of AD is dependent on the CSF systolic peak pulse energy, then reducing this peak energy would seem to be a primary research objective. Research has already demonstrated that reducing blood pressure reduces AD prevalence [79].
2) More laboratory research under flow conditions that mimic those in the brain! Most of the laboratory research on these diseases has been conducted under quiescent conditions. Is it reasonable to continue to emphasize this when there are clearly quite different conditions in the critical brain glymphatic system, with back-and-forth flow in nearly all parts of the brain and energetic chaotic flow in the CSF cistern hot spots and CA region described above, especially when it is known that flow stress induces protein aggregation, and particularly in the presence of destabilizing hydrophobic surfaces? Experiments need to be undertaken that leverage microfluidic geometries [103] and mimic the laminar shear and extensional flows, with an emphasis on effects of flow pulsatility and energy transfer rates, across the different CSF flow paths.
3) Change in drug research protocol. Will not all potential drugs also be exposed to these same energetic flow conditions in the brain as the Aβ and tau monomers? Are the current candidate drugs affected by shear, or extensional strain as are amyloid monomers? Many of these are proteins. What is needed is more research conducted on both pure drug solutions (like the protein-based aducanumab, a monoclonal antibody recently developed for AD) and mixtures of amyloid monomer and drug solutions subject to hydrodynamic stresses, with and without the destabilizing biological interfaces. Testing the flow properties of these drug solutions in the presence of amyloid monomers is particularly important as it has been shown that many stable protein-based antibodies that normally do not aggregate under quiescent conditions, aggregate when exposed to extensional strain conditions [4, 52, 104]. Again, it is important that these experiments be carried out that include effects of flow pulsatility.
4) Extensive MRI studies of the lower brain regions, especially more longitudinal clinical studies searching for early trends. For example, look for changes in flow patterns in regions of AD pathology, looking for early clues that detect buildup of possible amyloid coatings that may cause changes in flow path dimensions and hydrodynamics, and seek out any possible correlations with changes in disease symptoms or stages.
An intriguing example of such MRI studies is the cross section of the LC and CA shown in Fig. 15. In Fig. 15A, the white arrow is pointing to the LC at the edge of the CA near the 4th ventricle of a healthy volunteer. Figure 15B shows a similar cross section of a Parkinson’s disease patient. The CA hydrodynamics in the two panels obviously are quite different. As has been suggested previously [5], this region of the brain has the greatest opportunity for developing extensional flow strain that is capable of initiating tau aggregation which could nearly clog this CA segment and certainly change the CSF flow pattern that could indeed be responsible for tau pathology.
Fig. 15
Neuromelanin-sensitive MRI imaging of the locus coeruleus/subcoeruleus. Axial neuromelanin-sensitive T-weighted images of the locus in a healthy volunteer (A) and a patient with Parkinson’s disease (B). The locus area (arrows) is visible as an area of increased signal intensity. With permission from [105].
![Neuromelanin-sensitive MRI imaging of the locus coeruleus/subcoeruleus. Axial neuromelanin-sensitive T-weighted images of the locus in a healthy volunteer (A) and a patient with Parkinson’s disease (B). The locus area (arrows) is visible as an area of increased signal intensity. With permission from [105].](https://ip.ios.semcs.net:443/media/jad/2022/90-1/jad-90-1-jad220622/jad-90-jad220622-g015.jpg)
5) A more general need is for MRI studies exploring the anatomy of the CSF system, especially in basal cisterns, to verify if some anatomical features could represent risk or protective factors for extensional and shear flow and consequently for AD pathology.
6) The role of energy and energy dissipation in amyloid diseases has been raised above. The type of mechanical energy dissipation, e.g., predominant shear versus predominant extensional stresses, is also shown to be important in experiments and should also be a variable in any experiments. Variable energy pulsatile back-and-forth flow should also be experimental variables.
7) More experiments on amyloid monomer mixtures under pulsed flow conditions. The role of mixed amyloid aggregates has been recently emphasized in the literature and in the above mechanistic speculations. Given the apparent power of extensional flows in promoting protein aggregation, much more attention needs to be given to research with this variable energy source for experimentation on protein mixtures. For example, perhaps the increased ApoE4 protein concentration results in a competition with Aβ protein in the above proposed tau mechanism [106]. The resulting flow-stressed ApoE-tau intermediate could possibly make the conversion of the adsorbed tau/protein complex into the tau seed more efficient and shorten the time of conversion to toxic tau seed.
8) Explore the possibility that the above discussion and mechanism may also apply to Parkinson’s disease and the many other “misfolded protein” diseases.
9) See the following references for additional suggested experiments and clinical studies [41, 95].
What causes the extreme complexity of AD?
What has not been considered in the above discussion about the earliest AD pathology is the mechanism of the highly complex etiology of AD following early events that lead to the formation of initial Aβ and tau seeds. If the initiation of this disease, as proposed above, depends primarily on the hydrodynamics of CSF within different parts of the brain, it could be that the subsequent etiology of the disease also depends, at least in part, on the drastic physical changes in the brain architecture during later disease stages, where whole sections of the brain cortex shrink drastically—with hydrodynamic consequences.
It is very likely that both the cortical and sub-cortical CSF glymphatic path dimensions and geometry will also change, thus changing the hydrodynamic patterns throughout the brain, some readily observed with MRI, some smaller dimensioned ones not so easily detected. It is for this reason that we strongly urge researchers to undertake extensive MRI CSF studies of the type employed by Atsumi et al. [3] to try to provide experimental studies that will help test such a hypothesis. For example, MRI studies that delve more deeply into movements of Aβ and tau molecules trapped within CSF in potentially ever-changing confined spaces in or near the CA and especially in the basal cisterns. Could longitudinal studies then be correlated with the various phases identified as Braak and Thal stages? Could MRI studies of CSF hydrodynamic changes with time be correlated with changes in disease phases?
If the above proposed hypotheses have any validity, it becomes obvious that extensive MRI studies of many different parts of the brain seeking information of CSF flow dynamics need to be undertaken at all stages of the disease, especially in the lower brain region. MRI studies looking for any correlations between various disease stages and changes in CSF brain hydrodynamics in these brain locations would be an obvious test of our various mechanistic hypotheses. More hydrodynamic MRI studies are needed.
SUMMARY
A previously proposed hypothesis of flow-stressed induced protein aggregation is now expanded to include energetic CSF flows that create hot spots [5] in the lower brain regions that include brain cisterns and the cerebral aqueduct region. These regions are suggested to produce stretched Aβ and tau molecules that are capable of ultimately producing prion-like seeds that initiate AD.
The effects of energy variation of flow pulsatility on flow-induced amyloid aggregation is emphasized. The continuous migration of the atherosclerosis-induced, maximum heart systolic CSF pulse energy location in initiating amyloid disease pathology is suggested. The question is raised whether this migration is a driving force in AD etiology. Strong correlations between the anatomies of these hot spots and AD pathology would appear to be evidence that the earliest Aβ and tau pathology arise from CSF flow stresses.
Detailed experimental evidence for this suggested mechanism is admittedly rather thin and indirect. However, when taken as a whole, it is convincing enough to be taken seriously, especially because of the great need for new ideas in this research field. Thus, it is suggested that intensive experimental and clinical measurements are needed to test this flow stress mechanism using CSF flow conditions that mimic those found in the human brain. We believe that scientists, engineers, and medical personnel from a wide range of backgrounds ought to be engaged in this research area. Much can be learned from exploring the hydrodynamics, MRI, chemistry, anatomy, pathology, biophysics, and other relevant aspects of CSF flow processes, especially in the lower human brain regions. The upper cortex should not be abandoned in these studies, but because flow stress energies are much lower, there will probably be more challenges and a need for more sensitive methods in flow stress studies of that region. The role of variable fluid stress energy and different types of fluid stresses, especially pulsatile extensional flow stress, would appear to be critical in such research.
We believe that it is time for more research with a new focus: effects of pulsed flow stress as a key initiator of neurodegenerative diseases.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the editorial help of Jeffrey Iliff, Elinor Thomforde, and Susan Trumbore, and the reviewers of this paper for their helpful comments and suggestions.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0622r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220622.
REFERENCES
[1] | Oxford AE , Stewart ES , Rohn TT ((2020) ) Clinical trials in Alzheimer’s disease: A hurdle in the path of remedy, Int J Alzheimers Dis 2020: , 5380346. |
[2] | Reiss AB , Montufar N , DeLeon J , Pinkhasov A , Gomolin IH , Glass AD , Arain HA , Stecker MM ((2021) ) Alzheimer disease clinical trials targeting amyloid: Lessons learned from success in mice and failure in humans, Neurologist 26: , 52–61. |
[3] | Atsumi H , Horie T , Kajihara N , Sunaga A , Sakakibara Y , Matsumae M ((2020) ) Simple identification of cerebrospinal fluid turbulent motion using a dynamic improved motionsensitized driven-equilibrium steady-state free precession method applied to various types of cerebrospinal fluid motion disturbance, Neurol Med Chir (Tokyo) 60: , 30–36. |
[4] | Dobson J , Kumar A , Willis LF , Tuma R , Higazi DR , Turner R , Lowe DC , Ashcroft AE , Radford SE , Kapur N , David J , Brockwell DJ ((2017) ) Inducing protein aggregation by extensional flow, Proc Natl Acad Sci U S A 114: , 4673–4678. |
[5] | Trumbore CN ((2021) ) Shear-induced amyloid aggregation in the brain: V. Are Alzheimer’s and other amyloid diseases initiated in the lower brain and brainstem by cerebrosinal fluid flow stresses? J Alzheimers Dis 79: , 979–1002. |
[6] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med 8: , 595–608. |
[7] | Dobson CM , Knowles TPJ , Vendruscolo M ((2020) ) The amyloid phenomenon and its significance in biology and medicine, Cold Spring Harb Perspect Biol 12: , a033878. |
[8] | Karran E , De Strooper B ((2016) ) The amyloid cascade hypothesis: Are we poised for success or failure? J Neurochem 139: , 237–252. |
[9] | Cremades N , Dobson CM ((2018) ) The contribution of biophysical and structural studies of protein self assembly to the design of therapeutic strategies for amyloid diseases, Neurobiol Dis 109: , 178–190. |
[10] | Chiti F , Dobson CM ((2017) ) protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade, Annu Rev Biochem 86: , 27–68. |
[11] | Morris M , Maeda S , Vossel K , Mucke L ((2011) ) The many faces of tau, Neuron 12: , 410–426. |
[12] | Kumar H , Udgaonkar JB ((2019) ) Mechanistic approaches to understand the prion-like propagation of aggregates of the human tau protein, Biochim Biophys Acta Proteins Proteom 1867: , 922–932. |
[13] | Naseri NN , Wang H , Guo J , Sharma M , Luo W ((2019) ) The complexity of tau in Alzheimer’s disease, Neurosci Lett 705: , 183–194. |
[14] | Lim S , Haque MM , Kim D , Kim DJ , Kim YK ((2014) ) Cell-based models to investigate tau aggregation, Comput Struct Biotechnol J 12: , 7–13. |
[15] | Dourlen P , Kilinc D , Malmanche N , Chapuis J , Lambert JC ((2019) ) The new genetic landscape of Alzheimer’s disease: From amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol 138: , 221–236. |
[16] | Bloom GS ((2014) ) Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis, JAMA Neurol 71: , 505–508. |
[17] | Ferrari C , Sorbi S ((2021) ) The complexity of Alzheimer’s disease: An evolving puzzle, Physiol Rev 101: , 1047–1081. |
[18] | Trumbore CN ((2018) ) Shear-induced amyloid formation in the brain: III. The roles of shear energy and seeding in aroposed shear model. J Alzheimers Dis 65: , 47–70. |
[19] | Uversky VN , Gillespie JR , Fink AL ((2000) ) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41: , 415–427. |
[20] | Uversky VN ((2016) ) Dancing protein clouds: The strange biology and chaotic physics of intrin sically disordered proteins, J Biol Chem 291: , 6681–6688. |
[21] | Shafiei SS , Guerrero-Muñoz MJ , Castillo-Carranza DL ((2017) ) Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage, Front Aging Neurosci 9: , 83. |
[22] | Cline EN , Bicca MAB , Viola KL , Klein WL ((2018) ) The amyloid- hypothesis: Beginning of the third decade, J Alzheimers Dis 64: , S567–S610. |
[23] | Tanokashira D , Mamada N , Yamamoto F , Taniguchi K , Tamaoka A , Lakshmana MK , Araki W ((2017) ) The neurotoxicity of amyloid β-protein oligomers is reversible in a primary neuron model, Mol Brain 10: , 4. |
[24] | Rudenko LK , Wallrabe H , Periasamy A , Siller KH , Svindrych Z , Seward ME , Best MN , Bloom GS ((2019) ) Intraneuronal tau misfolding induced by extracellular amyloid-β oligomers, J Alzheimers Dis 71: , 1125–1138. |
[25] | Lasagna-Reeves CA , Castillo-Carranza DL , Guerrero-Muoz MJ , Jackson GR , Kayed R ((2010) ) Preparation and characterization of neurotoxic tau oligomers, Biochemistry 49: , 10039–10041. |
[26] | Latimer CS , Lucot KL , Keene CD , Cholerton B , Montine TJ ((2021) ) Genetic insights into Alzheimer’s disease, Annu Rev Pathol 16: , 351–376. |
[27] | Braak H , Del Tredici K ((2011) ) The pathological process underlying Alzheimer’s disease in individuals under thirty, Acta Neuropathol 121: , 171–181. |
[28] | Spires-Jones TL , Attems J , Thal DR ((2017) ) Interactions of pathological proteins in neurodegenerative diseases, Acta Neuropathol 134: , 187–205. |
[29] | Latshaw DC , Cheon M , Hall CK ((2014) ) Effects of macromolecular crowding on amyloid beta (16–22) aggregation using coarse-grained simulations, J Phys Chem B 118: , 13513–13526. |
[30] | Iliff JJ , Lee H , Yu M , Feng T , Logan J , Nedergaard M , Benveniste H ((2013) ) Brain-wide pathway for waste clearance captured by contrast-enhanced MRI, J Clin Invest 123: , 1299–309. |
[31] | Thomas JH ((2019) ) Fluid dynamics of cerebrospinal fluid flow in perivascular spaces, J R Soc Interface 16: , 20190572. |
[32] | Jessen NA , Munk ASF , Lundgaard I , Nedergaard M ((2015) ) The glymphatic system: A beginner’s guide, Neurochem Res 40: , 2583–2599. |
[33] | Plog BA , Nedergaard M ((2018) ) The glymphatic system in central nervous system health and disease: Past, present, and future, Annu Rev Pathol 13: , 379–394. |
[34] | Nedergaard M , Goldman SA ((2020) ) Glymphatic failure as a final common pathway to dementia, Science 370: , 50–56. |
[35] | Mestre H , Mori Y , Nedergaard M ((2020) ) The brain’s glymphatic system: Current controversies, Trends Neurosci 43: , 458–466. |
[36] | Raghunandan A , Ladron-de-Guevara A , Tithof J , Mestre H , Du T , Nedergaard M , Thomas JH , Kelley DH ((2021) ) Bulk flow of cerebrospinal fluid observed in periarterial spaces is not an artifact of injection, Elife 10: , e65958. |
[37] | Matsumoto S , Tsunematsu T ((2021) ) Association between sleep, Alzheimer’s, and Parkinson’s disease, Biology (Basel) 10: , 1127. |
[38] | Smith DE , Babcock HP , Chu S ((1999) ) Single-polymer dynamics in steady shear flow, Science 283: , 1724–1727. |
[39] | Usta O , Butler JE , Ladda AJC ((2006) ) Flow-induced migration of polymers in dilute solution, Phys Fluids 18: , 031703. |
[40] | Boster KAS , Tithof J , Cook DD , Thomas JH , Kelley DH ((2022) ) Sensitivity analysis on a network model of glymphatic flow, J R Soc Interface 19: , 20220257. |
[41] | Trumbore CN ((2016) ) Shear-induced amyloid formation in the brain. I. Potential vascular and parenchymal processes, J Alzheimers Dis 54: , 457–470. |
[42] | Kusumoto Y , Lomakin A , Teplow DB , Benedek GB ((1998) ) Temperature dependence of amyloid beta-protein fibrillization, Proc Natl Acad Sci U S A 95: , 12277–12282. |
[43] | Dunstan DE , Hamilton-Brown P , Asimakis P , Ducker W , Bertolini J ((2009) ) Shear flow promotes amyloid-β fibrilization, Protein Eng Des Sel 22: , 741–746. |
[44] | Sharma LG , Pandey LM ((2020) ) Shear-induced aggregation of amyloid β (1–40) in a parallel plate geometry, J Biomol Struct Dyn 39: , 6415–6423. |
[45] | Trumbore CN , Paik J , Fay D , Vachet RW ((2019) ) Preliminary capillary flow experiments with amyloid-beta, possible needle and capillary abeta adsorption, and a proposal for drug evaluation under shear conditions, J Alzheimers Dis 72: , 751–760. |
[46] | Cohen SI , Linse S , Luheshi LM , Hellstrand E , White A , Rajah L , Otzen DE , Vendruscolo M , Dobson CM , Knowles TP ((2013) ) Proliferation of amyloid- 42 aggregates occurs through a secondary nucleation mechanism, Proc Natl Acad Sci U S A 110: , 9758–9763. |
[47] | Meisl G , Yang X , Hellstrand E , Frohm B , Kirkegaard JB , Cohen SI , Dobson CM , Linse S , Knowles TP ((2014) ) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides, Proc Natl Acad Sci U S A 111: , 9384–9389. |
[48] | Michaels TCT , Šarić A , Habchi J , Chia S , Meisl G , Vendruscolo M , Dobson CM , Knowles TPJ ((2018) ) Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation, Annu Rev Phys Chem 69: , 273–298. |
[49] | Scheidt T , L—-apinska U , Kumita JR , Whiten DR , Klenerman D , Wilson MR , Cohen SIA , Linse S , Vendruscolo M , Dobson CM , Knowles TPJ , Arosio P ((2019) ) Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates, Sci Adv 5: , eaau3112. |
[50] | Michaels TCT , Šarić A , Curk S , Bernfur K , Arosio P , Meisl G , Dear AJ , Cohen SIA , Dobson CM , Vendruscolo M , Linse S , Knowles TPJ ((2020) ) Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide, Nat Chem 12: , 445–451. |
[51] | Banerjee S , Hashemi M , Lv Z , Maity S , Rochet JC , Lyubchenko YL ((2017) ) A novel pathway for amyloids self-assembly in aggregates at nanomolar concentration mediated by the interaction with surfaces, Sci Rep 7: , 45592. |
[52] | Grigolato F , Arosio P ((2021) ) The role of surfaces on amyloid formation, Biophys Chem 270: , 106533. |
[53] | McBride SA , Sanford SP , Lopez JM , Hirsa AH ((2016) ) Shear-induced amyloid fibrillization: The role of inertia, Soft Matter 12: , 3461–3467. |
[54] | Duerkop M , Berger E , Dürauer A , Jungbauer A ((2018) ) Impact of cavitation, high shear stress and air/liquid interfaces on protein aggregation, Biotechnol J 13: , 1800062. |
[55] | Ashton L , Dusting J , Imomoh E , Balabani S , Blanch EW ((2010) ) Susceptibility of different proteins to flow-induced conformational changes monitored with Raman spectroscopy, Biophys J 98: , 707–714. |
[56] | Qi R , Luo Y , Wei G , Nussinov R , Ma B ((2015) ) Aβ “Stretching-and-packing” cross-seeding mechanism can trigger tau protein aggregation, J Phys Chem Lett 6: , 3276–3282. |
[57] | Gu L , Guo Z ((2013) ) Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils, J Neurochem 126: , 305–311. |
[58] | Wardlaw JM , Benveniste H , Nedergaard M , Zlokovic BV , Mestre H , Lee H , Doubal FN , Brown R , Ramirez J , MacIntosh BJ , Tannenbaum A , Ballerini L , Rungta RL , Boido D , Sweeney M , Montagne A , Charpak S , Joutel A , Smith KJ , Black SE ; colleagues from the Fondation Leducq Transatlantic Network of Excellence on the Role of the Perivascular Space in Cerebral Small Vessel Disease ((2020) ) Perivascular spaces in the brain: Anatomy,hysiology and pathology. Nat Rev Neurol 16: , 137–153. |
[59] | Subedi S , Sasidharan S , Nag N , Saudagar P , Tripathi T ((2022) ) Amyloid cross-seeding: Mechanism, implication, and inhibition, Molecules 27: , 1776. |
[60] | Mueller RL , Combs B , Alhadidy MM , Brady ST , Morfini GA , Kanaan NM ((2021) ) Tau: A signaling hub protein, Front Mol Neurosci 19: , 647054. |
[61] | González A , Singh SK , Churruca M , Maccioni RB ((2022) ) Alzheimer’s disease and tau self-assembly: In the search of the missing link, Int J Mol Sci 23: , 4192. |
[62] | Rai SK , Savastano A , Singh P , Mukhopadhyay S , Zweckstetter M ((2021) ) Liquid-liquid phase separation of tau: From molecular biophysics to physiology and disease, Protein Sci 30: , 1294–1314. |
[63] | Mirbaha H , Chen D , Morazova OA , Ruff KM , Sharma AM , Liu X , Goodarzi M , Pappu RV , Colby DW , Mirzaei H , Joachimiak LA , Diamond MI ((2018) ) Inert and seed-competent tau monomers suggest structural origins of aggregation, Elife 7: , e36584. |
[64] | Sanders DW , Kaufman SK , DeVos SL , Sharma AM , Mirbaha H , Li A , Barker SJ , Foley AC , Thorpe JR , Serpell LC , Miller TM , Grinberg LT , Seeley WW , Diamond MI ((2014) ) Distinct tau prion strains propagate in cells and mice and define different tauopathies, Neuron 82: , 1271–1288. |
[65] | Gerson JE , Mudher A , Kayed R ((2016) ) Potential mechanisms and implications for the formation of tau oligomeric strains, Crit Rev Biochem Mol Biol 51: , 482–496. |
[66] | Busche MA , Hyman BT ((2020) ) Synergy between amyloid-β and tau in Alzheimer’s disease, Nat Neurosci 23: , 1183–1193. |
[67] | Ye H , Han Y , Li P , Su Z , Huang Y ((2022) ) The role of post-translational modifications on the structure and function of tau protein, J Mol Neurosci 72: , 1557–1571. |
[68] | Matsumae M , Hirayama A , Atsumi H , Yatsushiro S , Kuroda K ((2014) ) Velocity and pressure gradients of cerebrospinal fluid assessed with magnetic resonance imaging, J Neurosurg 120: , 218–227. |
[69] | Horie T , Kajihara N , Matsumae M , Obara M , Hayashi N , Hirayama A , Takizawa K , Takahara T , Yatsushiro S , Kuroda K ((2017) ) Magnetic resonance imaging technique for visualization of irregular cerebrospinal fluid motion in the ventricular system and subarachnoid space, World Neurosurg 97: , 523–531. |
[70] | Matsumae M , Kuroda K , Yatsushiro S , Hirayama A , Hayashi N , Takizawa K , Atsumi H , Sorimachi T ((2019) ) Changing the currently held concept of cerebrospinal fluid dynamics based on shared findings of cerebrospinal fluid motion in the cranial cavity using various types of magnetic resonance imaging techniques, Neurol Med Chir (Tokyo) 59: , 133–146. |
[71] | Shafrir Y , Durell S , Arispe N , Guy HR ((2010) ) Models of membrane-bound Alzheimer’s Abeta peptide assemblies, Proteins 78: , 3473–3487. |
[72] | Bok E , Leem E , Lee BR , Lee JM , Yoo CJ , Lee EM , Kim J ((2021) ) Role of the lipid membrane and membrane proteins in tau pathology, Front Cell Dev Biol 9: , 653815. |
[73] | Slater C , Wang Q ((2021) ) Alzheimer’s disease: An evolving understanding of noradrenergic involvement and the promising future of electroceutical therapies, Clin Transl Med 11: , e397. |
[74] | Beardmore R , Hou R , Darekar A , Holmes C , Boche D ((2021) ) The locus coeruleus in aging and Alzheimer’s disease: A postmortem and brain imaging review, J Alzheimers Dis 83: , 5–22. |
[75] | Liu KY , Marijatta F , Hämmerer D , Acosta-Cabronero J , Düzel E , Howard RJ ((2017) ) Magnetic resonance imaging of the human locus coeruleus: A systematic review, Neurosci Biobehav Rev 83: , 325–355. |
[76] | Ciampa CJ , Parent JH , Harrison TM , Fain RM , Betts MJ , Maass A , Winer JR , Baker SL , Janabi M , Furman DJ , D’Esposito M , Jagust WJ , Berry AS ((2022) ) Associations among locus coeruleus catecholamines, tau pathology, and memory in aging, Neuropsychopharmacology 47: , 1106–1113. |
[77] | Kelberman M , Keilholz S , Weinshenker D ((2020) ) What’s that (blue) spot on my MRI? Multimodal neuroimaging of the locus coeruleus in neurodegenerative disease, Front Neurosci 14: , 583421. |
[78] | Van Egroo M , Koshmanova E , Vandewalle G , Jacobs HIL ((2022) ) Importance of the locus coeruleus-norepinephrine system in sleep-wake regulation: Implications for aging and Alzheimer’s disease, Sleep Med Rev 62: , 101592. |
[79] | Tublin JM , Adelstein JM , Del Monte F , Combs CK , Wold LE ((2019) ) Getting to the heart of Alzheimer disease, Circ Res 124: , 142–149. |
[80] | Thorin-Trescases N , de Montgolfier O , Pincon A , Raignault A , Caland L , Labbe P , Thorin E ((2018) ) Impact of pulse pressure on cerebrovascular events leading to age-related cognitive decline, Am J Physiol Heart Circ Physiol 314: , H1214–H1224. |
[81] | Chen XQ , Mobley WC ((2019) ) Alzheimer disease pathogenesis: Insights from molecular and cellular biology studies of oligomeric Aβ and tau species, Front Neurosci 13: , 659. |
[82] | NguyenPH, RamamoorthyA, SahooBR, ZhengJ, FallerP, StraubJE, DominguezL, SheaJE, DokholyanNV, De SimoneA, MaB, NussinovR, NajafiS, NgoST, LoquetA, ChiricottoM, GangulyP, McCartyJ, LiMS, HallC, WangY, MillerY, MelchionnaS, HabensteinB, TimrS, ChenJ, HnathB, StrodelB, KayedR, LesnéS, WeiG, SterponeF, DoigAJ, DerreumauxP ((2021) ) Amyloid oligomers: A joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, Type II diabetes, and amyotrophic lateral sclerosis. Chem Rev 121: , 2545–2647. |
[83] | Stone J , Johnstone DM , Mitrofanis J , O’Rourke M ((2015) ) The mechanical cause of age related dementia (Alzheimer’s disease): The brain is destroyed by the pulse, J Alzheimers Dis 44: , 355–373. |
[84] | Pickett EK , Koffie RM , Wegmann S , Henstridge CM , Herrmann AG , Colom-Cadena M , Lleo A , Kay KR , Vaught M , Soberman R , Walsh DM , Hyman BT , Spires-Jones TL ((2016) ) Non-fibrillar oligomeric Amyloid-β within synapses, J Alzheimers Dis 53: , 787–800. |
[85] | Trumbore CN ((2018) ) Shear-induced amyloid formation in the brain: IV. Effects on synases surrounding senile plaque and in plaque-free regions. J Alzheimers Dis 66: , 57–73. |
[86] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol 82: , 239–259. |
[87] | Braak H , Del Tredici K ((2011) ) Alzheimer’s pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol 121: , 589–595. |
[88] | Braak H , Del Tredici K ((2015) ) Neuroanatomy and pathology of sporadic Alzheimer’s disease, Adv Anat Embryol Cell Biol 215: , 1–162. |
[89] | Gangoda SVS , Avadhanam B , Jufri NF , Sohn EH , Butlin M , Gupta V , Chung R , Avolio AP ((2018) ) Pulsatile stretch as a novel modulator of amyloid precursor protein processing and associated inflammatory markers in human cerebral endothelial cells, Sci Rep 8: , 1689. |
[90] | Sloots JJ , Biessel GJ , Zwanenburg JM ((2020) ) Cardiac and respiration-induced brain deformations in humans quantified with high-field MRI, Neuroimage 210: , 116581. |
[91] | McKee AC ((2020) ) The neuropathology of chronic traumatic encephalopathy: The status of the literature, Semin Neurol 40: , 359–369. |
[92] | Breen PW , Krishnan V ((2020) ) Recent preclinical insights into the treatment of chronic traumatic encephalopathy, Front Neurosci 14: , 616. |
[93] | Ghajari M , Hellyer PJ , Sharp DJ ((2017) ) Computational modelling of traumatic brain injury predicts the location of chronic traumatic encephalopathy pathology, Brain 140: , 333–343. |
[94] | Trumbore CN ((2019) ) Shear-induced amyloid formation of IDPs in the brain, Prog Mol Biol Transl Sci 166: , 225–309. |
[95] | Jones EM , Dubey M , Camp PJ , Vernon BC , Biernat J , Mandelkow E , Majewski J , Chi EY ((2012) ) Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption, Biochemistry 51: , 2539–2550. |
[96] | Meyer V , Dinkel PD , Rickman Hager E , Margittai M ((2104) ) Amplification of Tau fibrils from minute quantities of seeds, Biochemistry 53: , 5804–5809. |
[97] | Schmidt A , Annamalai K , Schmidt M , Grigorieff N , Fändrich M ((2016) ) Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril, Proc Natl Acad Sci U S A 113: , 6200–6205. |
[98] | Nesterov SV , Ilyinsky NS , Uversky VN ((2021) ) Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses, Biochim Biophys Acta Mol Cell Res 1868: , 119102. |
[99] | Tripathi T , Khan H ((2020) ) Direct interaction between the β-Amyloid core and tau facilitates cross-seeding: A novel target for therapeutic intervention, Biochemistry 59: , 341–342. |
[100] | Ivanova MI , Lin Y , Lee YH , Zheng J , Ramamoorthy A ((2021) ) Biophysical processes underlying cross-seeding in amyloid aggregation and implications in amyloid pathology, Biophys Chem 269: , 106507. |
[101] | Rahman MM , Lendel C ((2021) ) Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology, Mol Neurodegener 16: , 59. |
[102] | Ren B , Zhang Y , Zhang M , Liu Y , Zhang D , Gong X , Feng Z , Tang J , Chang Y , Zheng J ((2019) ) Fundamentals of cross-seeding of amyloid proteins: An introduction, J Mater Chem B 7: , 7267–7282. |
[103] | Choi YJ , Chae S , Kim JH , Barald KF , Park JY , Lee SH ((2013) ) Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow, Sci Rep 3: , 1921. |
[104] | Willis LF , Kumar A , Dobson J , Bond NJ , Lowe D , Turner R , Radford SE , Kapur N , Brockwell DJ ((2018) ) Using extensional flow to reveal diverse aggregation landscapes for three IgG1 molecules, Biotechnol Bioeng 115: , 1216–1225. |
[105] | García-Lorenzo D , Longo-Dos Santos C , Ewenczyk C , Leu-Semenescu S , Gallea C , Quattrocchi G , Pita Lobo P , Poupon C , Benali H , Arnulf I , Vidailhet M , Lehericy S ((2013) ) The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease, Brain 136: , 2120–2129. |
[106] | Ghosh S , Sil TB , Dolai S , Garai K ((2019) ) High-affinity multivalent interactions between apolipoprotein E and the oligomers of amyloid-β, FEBS J 286: , 4737–4753. |
[107] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD, Neurology 58: , 1791–1800. |
[108] | Siyahhan B , Knobloch V , de Zélicourt D , Asgari M , Schmid Daners M , Poulikakos D , Kurtcuoglu V ((2014) ) Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles, J R Soc Interface 11: , 20131189. |
[109] | Popov KI , Makepeace KAT , Petrotchenko EV , Dokholyan NV , Borchers CH ((2019) ) Insight into the structure of the “unstructured” tau protein, Structure 27: , 1710–1715.e4. |


