
Understanding the Involvement of microRNAs in Mitochondrial Dysfunction and Their Role as Potential Biomarkers and Therapeutic Targets in Parkinson’s Disease
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting the elderly worldwide and causing significant movement impairments. The goal of PD treatment is to restore dopamine levels in the striatum and regulate movement symptoms. The lack of specific biomarkers for early diagnosis, as well as medication aimed at addressing the pathogenic mechanisms to decelerate the progression of dopaminergic neurodegeneration, are key roadblocks in the management of PD. Various pathogenic processes have been identified to be involved in the progression of PD, with mitochondrial dysfunction being a major contributor to the disease’s pathogenesis. The regulation of mitochondrial functions is influenced by a variety of factors, including epigenetics. microRNAs (miRNAs) are epigenetic modulators involved in the regulation of gene expression and regulate a variety of proteins that essential for proper mitochondrial functioning. They are found to be dysregulated in PD, as evidenced by biological samples from PD patients and in vitro and in vivo research. In this article, we attempt to provide an overview of several miRNAs linked to mitochondrial dysfunction and their potential as diagnostic biomarkers and therapeutic targets in PD.
INTRODUCTION
Parkinson’s disease (PD) is the world’s second most common neurological disorder, impacting millions of individuals worldwide [1]. It is an age-related progressive disorder in which the dopaminergic neurons in the Substantia Nigra pars Compacta region (SNpc) are degenerated [2]. Men are more commonly affected than women. As seen in postmortem PD brains, the disease is defined by the presence of α-synuclein aggregates and the presence of Lewy bodies in the surviving neurites [3]. It is characterized clinically by severe motor symptoms such as bradykinesia, rigidity, resting tremors, and an unstable gait, as well as non-motor symptoms such as cognitive impairment, sleep difficulties, olfactory impairment, constipation, and so on [2]. The current diagnosis of PD is primarily dependent on the presentation of motor symptoms. By the time PD is diagnosed, between 30–70% of dopaminergic neurons have degenerated [4]. All of the medications now available for the treatment of PD, including the gold standard levodopa, are aimed at symptomatic relief [5]. However, no medications that can delay or stop the progression of the disease are now available (i.e., disease modifying drug). As a result, recent research has been focused on finding molecular pathogenic mechanisms that may be implicated in the development and progression of PD. This could enable in the discovery of biomarkers that can aid in the early detection of the disease, as well as novel therapeutic targets that can be targeted to slow or stop the disease’s course. Neuroinflammation, α-synucleinopathy, protein aggregation and mishandling, oxidative stress, mitochondrial dysfunction, and other pathogenic processes have all been hypothesized as possible causes of neurodegeneration of dopaminergic neurons [6]. Of them, mitochondrial dysfunction has been recognized as a central mechanism in the development of PD [7]. Dopaminergic neurons depend on the mitochondria functioning for their huge energy requirements. The control of mitochondrial function is influenced by a variety of factors, including microRNAs (miRNAs). miRNAs are non-coding RNAs that are 19–24 nucleotides in length and play a critical role in regulation of gene expression and protein translation [8]. These short sequence RNAs are currently being investigated for their potential as early biomarkers as well as therapeutic targets in the treatment of a various diseases including PD. In this article, we will look at the involvement of several miRNAs involved in the regulation of mitochondrial function, as well as their potential as biomarkers and therapeutic targets in PD.
MITOCHONDRIA AND THEIR ROLE IN PD
Mitochondria are the double-membraned organelles found in the cell’s cytoplasm and have circular deoxyribonucleic acid (DNA) of their own [9]. They are known as the cell’s powerhouses due to their significant role in the production of adenosine triphosphate (ATP), the cell’s energy currency, via a highly integrated mechanism known as oxidative phosphorylation. In addition to ATP synthesis, mitochondria are involved in a variety of functions such as calcium homeostasis, apoptosis, redox signaling, carbohydrate and lipid metabolism, and so on [10, 11]. Mitochondria are extremely dynamic structures that alter their shape and number in response to the demands of the cells. Mitophagy and mitochondrial fusion and fission are important dynamic characteristics of mitochondria.
VULNERABILITY OF DOPAMINERGIC NEURONS TO NEURODEGENERATION
Many mechanisms were identified to be involved in the pathogenesis of PD, but mitochondrial dysfunction was thought to be the major player. A small subset of dopaminergic neurons in the SNpc region of brain were particularly vulnerable to mitochondrial dysfunction. These neurons in the mid brain are characterized by large and complex axonal arbor which requires massive amount of energy to maintain protein synthesis, axonal transportation, membrane potential, and propagation of action potential. The unmyelinated nature of these neurons further increases the energy demand. These neurons also have intrinsic pace-making activity which utilize the L-type calcium channels. The efflux of calcium ions requires much more ATPs than those required for the efflux of other ions like Na+. Hence, to meet all these energy demands, SNpc neurons depend on increased oxidative phosphorylation which on the other hand has negative consequences like increased reactive oxygen species (ROS) generation. Their energy requirement is very delicately balanced. Any condition that disturbs this balance make the SNpc neurons highly vulnerable to neurodegeneration [12–15]. From this, poor mitochondrial activity makes dopamine neurons more vulnerable to neurodegeneration owing to energy depletion and severe oxidative stress, which can lead to neuronal death. The fact that drugs like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and Rotenone cause Parkinsonism by inhibiting mitochondrial respiratory complex I demonstrates the role of mitochondrial dysfunction in PD. Furthermore, α-synuclein (SNCA), Parkinson juvenile disease protein (Parkin), PTEN-induced kinase 1 (PINK-1), DJ-1, leucine-rich repeat kinase 2 (LRRK2), and other PARK-designated PD-associated genes that are involved in the regulation of mitochondrial function have been found to be mutated in Familial PD [12]. In addition, the postmortem brains of PD patients showed the signs of oxidative damage in SN due to ROS, the primary source of which is mitochondria [11].
MITOCHONDRIAL DYSFUNCTION IN PD
Mitochondria dysfunction occurs due to various genetic and environmental factors. Mutations in various genes like PINK1, DJ-1, and LRRK2 which encode for the protein responsible for mitochondrial functioning are implicated in familial PD. Other factors like aging, exposure to pesticides and environmental toxins, mutations in mitochondrial genes, and epigenetic factors regulating various mitochondrial factors are some of the factors implicated in sporadic PD [16]. This impairment can be manifested by the accumulation of dysfunctional mitochondria, increased ROS production and oxidative stress, altered mitochondrial membrane potential, reduced ATP levels, and inhibition of the activity of respiratory complexes [17]. The following section briefly discusses various aspects of mitochondrial dysfunction in PD.
MITOCHONDRIAL RESPIRATORY COMPLEX I DEFICIENCY AND OXIDATIVE STRESS IN PD
Defects in the activity of the mitochondrial respiratory chain in PD have been explored in several studies [18]. Respiratory chain or electron transport chain is located in the inner mitochondrial membrane and consists of five complexes (complex I to complex V). Complex I to IV are involved in electron transfer in return to proton efflux creating a proton gradient across the membrane. Complex V is known as ATP synthase which couples the ATP production with the re-entry of protons through the complex [19]. Despite some individual heterogeneity, several findings consistently described a selective deficit of respiratory complex I, which is evident when the activity of various enzymes in the respiratory chain were assayed in the SN postmortem brain samples of PD patients. The activity of rotenone sensitive NADH CoQ1 reductase was assessed according to the method described by Hatefi and Rieske and was found to be significantly reduced in the samples [20]. Two neuropathological studies conducted on patients’ SN and striatum to examine the amount of respiratory chain complexes in these tissues have revealed a decrease in complex I subunits [21, 22]. The deficiency was also observed in frontal cortex, platelets, and skeletal muscles and can be attributed to oxidative damage or misassembly of complex I [21–23]. The deficit of additional respiratory chain complexes (namely complexes II, III, and IV) has been rarely reported [24, 25]. Complex III dysfunction has also been observed in platelets and lymphocytes of the PD patients [23]. MPTP and rotenone causes parkinsonism by inhibition of complex I and induced PD [22, 26]. Annonacin is another complex I inhibitor that is found to cause neurodegeneration in rats [27]. The mitochondrial complex I is composed of nearly 45 subunits including 14 core subunits. 7 of the core subunits were encoded by mitochondrial DNA and the other 7 by nuclear DNA. NDUFS2 gene encodes for a subunit of complex I which has the binding site for ubiquinone, the electron carrier as well as rotenone. It is also essential for the assembly of the other subunits to form complex I [28]. In a recent study, knockdown of NDUFS2 gene in the dopaminergic neurons resulted in the parkinsonism in the experimental mice [29, 30]. α-synuclein (a protein encoded by SNCA gene) which is the major component of Lewy bodies is found to be associated with the inner mitochondrial membrane and reduce the mitochondrial complex I activity. Accumulation of wild type α-synuclein in the mitochondria resulted in the reduction of complex I activity [31] The localization of A53T mutant form of α-synuclein with the mitochondrial membrane coincides with the age- related mitochondrial complex-1 inhibition in transgenic animals [32]. Damage to complex I or III results in the leakage of electrons from the electron transport chain into the mitochondrial matrix where they react with the molecular oxygen resulting in the generation of superoxides [33, 34]. These ROS cause lipid oxidation, protein oxidation, and mitochondrial DNA oxidation, all of which were found in the SN region of post-mortem brains of PD patients. In addition, ROS also promotes excitotoxicity, which further exacerbates the oxidative stress besides activating various neurotoxic mechanisms [23].
ALTERED MITOCHONDRIAL DYNAMICS IN PD
Mitochondria are a highly active organelle whose dynamics are critical to the proper functioning of neurons. Mitochondrial dynamics is a broad term used to include fission, fusion, trafficking, biogenesis, and degradation [35]. Mitochondrial fission is the division of a single mitochondrial cell into two daughter cells. It is necessary for mitochondrial clearance and transportation. The process is regulated by GTPase proteins, dynamin related protein 1 (Drp1), and mitochondrial fission (Fis1) protein [36]. LRRK2 is a GTPase and kinase domain containing protein present in cytosol. It phosphorylates Drp1 protein and promotes mitochondrial fission [37]. PTEN-induced putative kinase-1 (PINK1) is another protein kinase that promotes mitochondrial fission by indirect activation of Drp-1 protein [38]. Mitochondrial fusion is a dynamic process in which two mitochondria fuse together to form an elongated mitochondrion. Optic atrophy protein 1 (OPA1), and mitofusins 1 and 2 (Mfn1 and Mfn2) are the important proteins that regulate mitochondrial fusion [36, 39]. Mitofusins (Mfns) mediate the fusion of outer membranes of the neighboring mitochondria while OPA1 mediates the fusion of the inner mitochondrial membranes. PINK-1 and parkin proteins also play an important role in regulating fusion by promoting the ubiquitination subsequent degradation of Mfns [40]. These fission-fusion cycles are critical for maintaining homogeneous and healthy mitochondrial population in the cell [41]. They maintain mitochondrial integrity, mitochondrial turnover, and mitochondrial DNA (mtDNA) and are essential for the formation of synapses and dendritic spines [35].
Trafficking of the mitochondria through axons and dendrites to the sites of energy requirements is important to maintain the localization, maintenance, and function of the mitochondria [35]. Kinesins drive the anterograde (soma to synapse) transport whereas dyneins drive retrograde (synapse to soma) transport. Trafficking Kinesis Proteins 1 and 2 (TRAK 1 and 2) and Mitochondrial Rho GTPase 1 (MIRO-1) are the adaptor proteins involved in mitochondrial movement. PINK1 and parkin regulate the anterograde transport by the ubiquitination of (MIRO-1) [35, 42]. This movement depends on mitochondrial bioenergetics, ATP synthesis, and mitochondrial membrane potential [35, 43]. Mitophagy is the process in which mitochondria are engulfed in the double membrane autophagosomes that fuse with lysosomes, resulting in the destruction of defective mitochondria [44]. There are two types of mitophagy pathways: Parkin-dependent and Parkin-independent mitophagy. The former is a well-studied pathway in which recessive mutations have been linked to hereditary and idiopathic PD [45, 46]. In healthy neurons, PINK1 is incorporated into the mitochondrial outer membrane and is cleaved at its N-terminal by mitochondrial proteases such as presenilin. The cleaved PINK1 is re-translocated from the mitochondrial membrane into the cytoplasm and is destroyed via ubiquitin-mediated proteasome degradation [47]. During mitochondrial injury, the un-cleaved PINK1 becomes stabilized on the mitochondrial membrane and phosphorylate a variety of proteins, including ubiquitin and parkin [48]. Activation of parkin through phosphorylation of serine located in parkin’s N-terminal ubiquitin like domain and serine 65 of ubiquitin trigger mitophagy by ubiquitination of multiple outer mitochondrial membrane proteins [48–51]. LRRK2 which is associated with outer mitochondrial membrane (OMM) also promotes mitophagy [52]. DJ-I is a highly conserved protein performing various functions like modulation of transcription, chaperone-like action with antioxidant properties. Under oxidative stress, DJ-1 is converted to acidic variant and quenches ROS. This variant also localizes to OMM and promotes mitophagy [53]. Abnormal mitochondrial mitophagy and mitochondrial dynamics may arise due to the mutations in Parkin, PINK-1, LRRK2, DJ-1, and SNCA genes, accumulation of α-synuclein, and oxidative stress. This impairment results in accumulation of defective mitochondria, which ultimately leads to neurodegeneration [54].
MITOCHONDRIAL BIOGENESIS IN PD
Mitochondrial biogenesis is a highly integrated process which involves mtDNA replication, gene expression, protein synthesis, formation of mitochondrial membrane, and mitochondrial division. It occurs in the distal axons and the cell bodies of the dopaminergic neurons which require huge amounts of ATP [55]. Mitochondrial biogenesis occurs in response to oxidative stress, energy requirement, exercise, cell differentiation, etc. [56]. Even though, mitochondria have their own genetic material, they rely on approximately 1000–1500 proteins encoded by the nuclear genes for proper mitochondrial biogenesis. Of them, Peroxisome proliferator activated receptor gamma coactivator (PGC)-1α is considered as the master regulator of mitochondrial biogenesis [57]. It activates mitochondrial transcription factor A (TFAM) by inducing nuclear respiratory factors 1 and 2 (NRF 1 and 2). TFAM regulates the mtDNA transcription, packaging of mtDNA and copy number of mtDNA [58]. Mitochondrial biogenesis is impaired in PD due to the suppression of the above-mentioned regulators and leads to alteration in the mitochondrial dynamics and subsequent detrimental effects on the neurons [55]. This impairment in various aspects of mitochondrial homeostasis is a highly complex process and occurs in an interdependent manner. Impaired mitochondrial functions and homeostasis results in energy depletion, accumulation of defective mitochondria, severe oxidative stress, and excitotoxicity all together causing neuronal damage and ultimately neurodegeneration [11, 59].
miRNAs INVOLVED IN MITOCHONDRIAL DYSFUNCTION IN PD
Various factors were identified to be involved in the regulation of mitochondrial functions. Recently, the research has been focused on understanding the role of epigenetics in the regulation of mitochondrial functions. miRNAs are one of the epigenetic regulators which are involved in the maintenance of cellular homeostasis including mitochondrial functions [17, 60].
miRNAs AND THEIR ROLE IN REGULATION OF GENE EXPRESSION
miRNAs belong to non-coding single stranded RNA molecules that have been shown to control the expression of genes involved in cellular event, along with differentiation, proliferation, migration, and apoptosis [61–63]. They are about 18–22 nucleotides long and are termed as “master regulators of gene expression”. More than 5000 miRNAs have been found and described in the human genome to date [64]. miRNAs are encoded either in the non-coding regions or within the introns of protein coding genes. miRNAs encoded within the introns are known as mirtrons. miRNAs are transcribed from the DNA into primary miRNA (pri-miRNA) with the help of RNA polymerase II [65, 66] which is further processed by RNase III DROSHA and DiGeorge Syndrome critical region 8 (DGCR8) protein or pasha to form precursor-miRNA (pre-miRNA) [67]. The pre-miRNA is transported to the cytoplasm with the help of exportin-5 and Ran GTP. There it undergoes cleavage by Dicer (an endoribonuclease belonging to RNAse III family) and forms a double stranded short interfering RNA (siRNA) known as miRNA: miRNA* complex. This complex consisting of a guide strand and a passenger strand from which the guide miRNA is loaded into the RNA induced silencing complex (RISC) and passenger strand is removed and degraded. The RISC loading complex (RLC) along with the catalyst, Argonaute protein (AGO), is responsible for the final maturation of the miRNA. This form of biogenesis is known as the canonical miRNA biogenesis. Non-canonical biogenesis of miRNA is a process in which the DROSHA and DGCR8 pathway is bypassed. Mirtrons, miRNA derived from small nucleolar RNA (snRNA), endogenous short hairpin RNA and tRNA undergo this pathway [68]. The miRNAs bind to the 3’ Untranslated Region (3’-UTR) of the target mRNAs and they can also interact with 5’ UTR, coding sequences, gene promoters, and transcription factors [69]. Base complementarity between the seed region of the miRNA and 3’-UTR of the target mRNA is responsible for the regulation of gene expression. Based on their complementarity, miRNA binding to target mRNA in the RISC causes mRNA degradation, translational repression, or both [64]. Thus, miRNAs prevent protein translation through the degradation and sequestration of their target mRNAs [70]. Any dysregulation of those miRNAs may lead to various disorders ranging from cancer to neurodegenerative diseases like PD and Alzheimer’s disease [71, 72]. A plethora of in vitro and in vivo studies have found that several miRNAs were dysregulated in PD [72].
miRNAs ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION IN PD
Many miRNAs target the mRNAs of the proteins like LRRK2, DJ-1, PINK-1, Parkin, and voltage-dependent anion-selective channel-1 (VDAC1) which are associated with mitochondrial functioning as shown in Fig. 1. They were found to be dysregulated in various biological samples of the PD patients as well as in several in vitro and in vivo PD models as demonstrated in various studies [73]. The list of such miRNAs that are found to be dysregulated in PD are given below in the Table 1.
Fig. 1
miRNAs associated with mitochondrial dysfunction in PD. Various miRNAs were involved in the regulation of mitochondrial biogenesis, mitochondrial fission, fusion, mitophagy, electron transport chain by targeting key proteins like PGC-1α, LRRK2, PINK 1, Parkin, DJ-1, and SNCA. miRNAs in red are those that downregulate their respective target whereas those in green upregulate them. Green and red arrows indicate stimulation and inhibition of downstream pathway respectively. Created with BioRender.com.
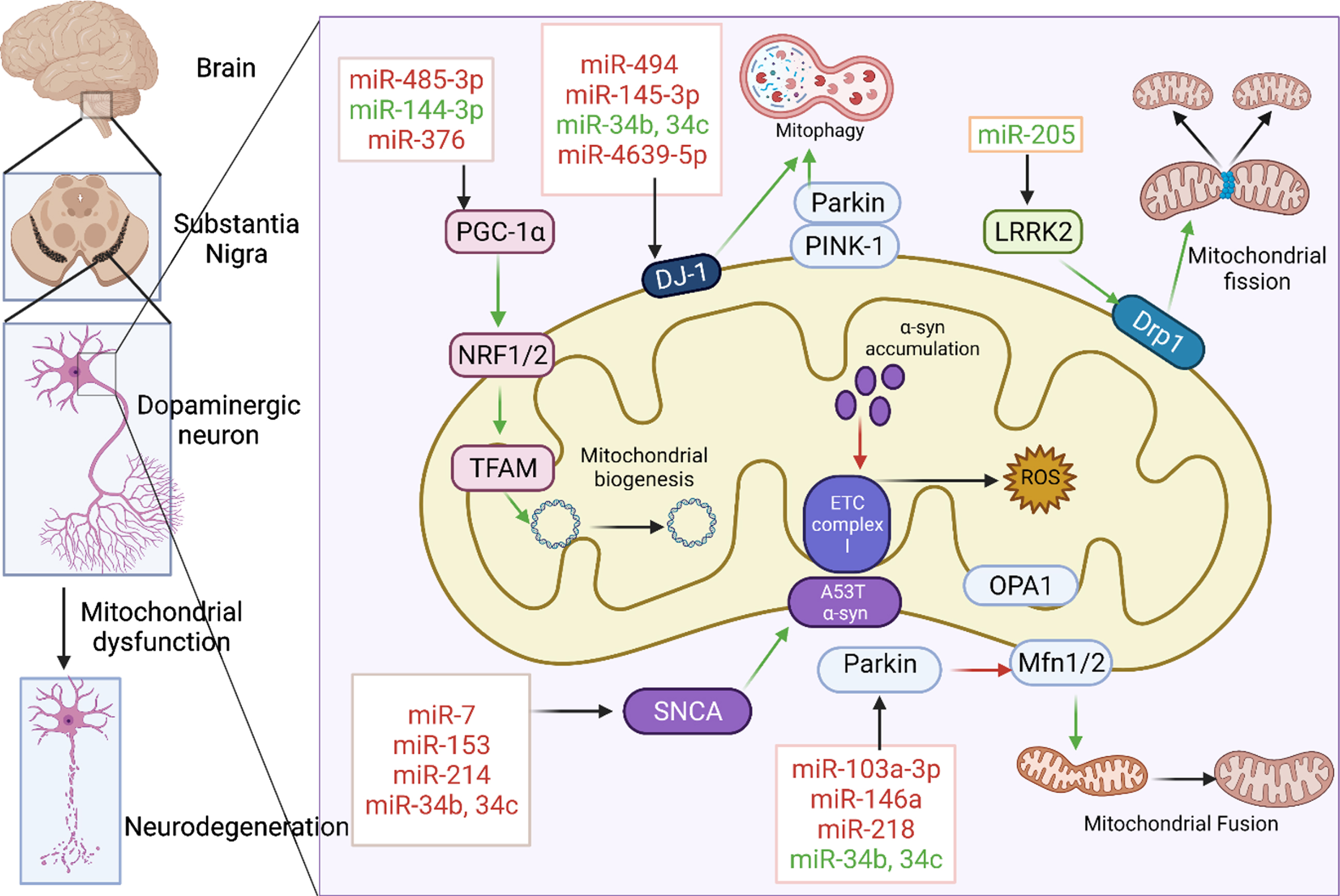
Table 1
miRNAs associated with mitochondrial dysfunction in PD
| Targeted mRNA | miRNA | Effect on mRNA | Region where miRNAs were identified | Reference |
| α-synuclein | miR-7 | ↓ | SNpc | [90] |
| miR-153 | ↓ | Plasma, saliva | [91] | |
| miR-214 | ↓ | Serum | [83] | |
| miR-34b, miR-34c | ↓ | AM, FC, SNpc, cerebellum | [92] | |
| Parkin | miR-103a-3p | ↓ | Brain, serum | [79] |
| miR-146a | ↓ | Serum, PBMC | [76] | |
| miR-218 | ↓ | FC | [78] | |
| miR-34b, mir-34c | ↑ | AM, FC, SNpc, cerebellum | [92] | |
| miR-181a | ↓ | Brain, CSF | [81] | |
| PINK1 | miR-27a, miR-27b | ↓ | PBMC | [89] |
| DJ-1 | miR-494 | ↓ | SNpc | [75] |
| miR-145-3p | ↓ | Saliva | [93] | |
| miR-34b, miR-34c | ↑ | AM, FC, SNpc, cerebellum | [74] | |
| miR-4639-5p | ↓ | Plasma | [82] | |
| LRRK2 | miR-205 | ↑ | Brain, CSF | [88] |
| VDAC1 | miR-7 | ↓ | SNpc | [84] |
| PGC1-α | MiR-485-3p | ↓ | SNpc | [94, 95] |
| MiR-144-3p | ↑ | FC, serum | [96] | |
| miR-376 | ↓ | PBMC | [97] | |
| Bim | miR-124 | ↑ | FC, SNpc | [98] |
“↓” indicates down regulation, “↑” indicates upregulation.
miR-34b and miR-34c indirectly target the parkin and DJ-1 proteins. miRNA profiling in the post-mortem brain samples PD patients (Braak stages 4 and 5) has revealed decrease levels of miR-34b and miR-34c in various regions like amygdala (AM), frontal cortex (FC), substantia nigra and cerebellum. Besides altered regulation of miR-34b/c was also identified in prodromal stage of PD. This miR-34b/c downregulation is associated with decreased DJ-1 and parkin expression in the differentiated SH-SY5Y cell line and also in the postmortem brain samples of PD patients [74]. miR-494 binds to the 3’-UTR of DJ-1 with complete complementarity and negatively regulates it translation. Transfection of Neuro 2a cell lines exposed to 1-Methyl-4-phenyl pyridinium (MPP+) and paraquat with miR-494 increased the susceptibility of the cells to oxidative stress due to down regulation of DJ-1. miR-494 was found to be highly expressed in SNpc region of the MPTP-induced PD mice model which resulted in decreased DJ-1 protein levels and exacerbation of neurodegeneration in the experimental mice [75]. Global miRNA profiling conducted by Jauhari et al. in a systemic rotenone induced PD rat model found that miR-146 is highly upregulated. Rotenone induced oxidative stress activated NF-kB which binds to the promoter region of miR-146 and upregulated its expression. miR-146 negatively regulates Parkin by binding to the 3’-UTR of PRKN. Over expression of miR-146 in SH-SY5Y cells resulted in increased mitochondrial fragmentation and the same was decreased when miR-146 was inhibited using synthetic inhibitors in rotenone exposed SH-SY5Y cells [76]. miR-218 binds to the 3’-UTR of PRKN, reduce the levels parkin and inhibit the mitophagy mediated by PINK1/PRKN. It was found to be downregulated in PD [77, 78]. miR-103a-3p was identified to be highly expressed in brain and plasma samples of PD patients. It binds to the 3’-UTR of PRKN and downregulate it. Parkin interacts with Ambra 1 and clears the dysfunctional mitochondria through mitophagy. Both Parkin and Ambra 1 were found to be downregulated and miR-103a-3p was upregulated in MPTP induced PD mice model and MPP+ exposed SH-SY5Y cells. The inhibition of miR-103a-3p exerted neuroprotective effect besides improving mitophagy in PD. The SH-SY5Y cell transfected with miR-103a-3p antagomir showed an increase in cell viability with no cytotoxicity as compared to the MPP+ treated group. Additionally, knock down of miR-103a-3p in mice by antagomir led to reduction in the loss of Tyrosine hydroxylase (TH) +ve cells in the SN due to improved mitophagy and alleviation of neurodegeneration [79]. miR-181a is abundantly expressed in brain and is found to be increased in postmortem brain samples and cerebrospinal fluid (CSF) of PD patients. The target mRNAs of miR-181a were found to be downregulated in aging and PD brains. miR-181a overexpression exacerbated α-synuclein induced neurotoxicity whereas its inhibition resulted in neuroprotection in mouse brains [80]. It downregulates parkin by binding to the 3’-UTR of PRKN and impacts mitophagy in a negative manner. SH-SY5Y cells co-transfected with GFP-LC3 (a fusion protein used to study autophagosome formation), mito-RFP (a lentiviral vector used to label mitochondria in the cells), and miR-181a showed a marked reduction in colocalization of mitochondria with autophagosomes/lysosomes in contrast to cell not transfected with miR-181a following FCCP (Carbonyl cyanide-p-trifluoro methoxy phenylhydrazone, mitochondrial uncoupler which induce mitophagy and stimulate PINK/Parkin pathway) treatment due to inhibition of parkin. In contrast, treatment with antagomir-181a enhanced mitophagy following FCCP treatment due to increased expression of parkin [81]. miR-4639-5p was found to be expressed in significantly higher levels in the plasma samples of early stage (Hoehn and Yahr stage 1-2.5) sporadic PD patients when compared to that of the healthy subjects. It negatively regulates DJ-1 by binding to its 3’-UTR and causes downregulation of DJ1 protein leading to severe increase in oxidative stress and mitochondrial fragmentation. SH-SY5Y cells treated with MPP+ or rotenone and transfected with miR-4639 mimics exhibited higher ROS and superoxide levels than those cells transfected with miR-4639 inhibitors [82]. miR-214 had been found to be involved in neuronal differentiation ad control neuronal outgrowth. miR-214 directly targets 3’-UTR of α-synuclein and decreases the expression of the protein. miR-214 levels were found to be significantly downregulated in MPTP induced PD mice model and MPP+ exposed SH-SY5Y cells. Transfection with miR-214 mimics and antagomirs in SH-SY5Y cells resulted in the reduction and elevation of α-synuclein aggregates in the cells. miR-214 was found to be the mediator of the neuroprotective effects of resveratrol by inhibiting α-synuclein. [83]. miR-7 has been shown to exhibit neuroprotective effect by inhibiting mitochondrial fragmentation, regulating the mitochondrial membrane potential and decreasing mitochondrial depolarization, regulating mitochondrial permeability transition pore (mPTP) and inhibiting cytochrome c release in MPP+ treated SH-SY5Y cells by directly targeting the 3’-UTR of VDAC1(an integral protein of mitochondrial OMM which forms channel for mPTP). Overexpression of miR-7 in the mice primary cortical neurons also led to downregulation of VDAC1 resulting in the modulation of mitochondrial morphology by regulation of mitochondrial membrane potential and mPTP. It also downregulated α-synuclein levels by directly targeting its 3’-UTR [84]. miR-7 and miR-153 are brain enriched miRNAs and they both target the 3’-UTR of α-synuclein [85]. miR-7 and miR-153 levels were found to be decreased in the substantia nigra and plasma samples of the PD patients respectively [86, 87]. LRRK2 levels were found to be upregulated in frontal cortex tissue of sporadic PD patients. miR-205 expression was found to be significantly downregulated in sporadic PD patients. miR-205 binds to the 3’-UTR of LRRK2 and down regulates its expression. Transfection of pre-miR-205 into HEK293T cells resulted in the suppression of LRRK2 protein. Transfection of neuronal cells with anti-miR-205 increased the expression of LRRK2 [88]. miR-27a and miR-27b directly target the 3’-UTR of PINK1 mRNA and downregulate its expression. Thereby they inhibit the PINK1 localization to damaged mitochondria, decrease in the phosphorylation of ubiquitin, Parkin translocation and LC3-II accumulation in mitochondria resulting in impaired mitophagy. Transfection of anti-miR-27a/b in HeLa cells and M17 cells increased the PINK1 protein level [89].
miR-124 is expressed abundantly in the dopaminergic neurons. Its neuroprotective effect has been demonstrated in various CNS diseases. miR-124 targets 3’-UTR of Bim (a proapoptotic protein associated with mitochondrial membrane which inhibits autophagy) and downregulates its expression. Thus, it mediates neuroprotection through the upregulation of autophagy and attenuation of apoptosis. mir-124 level was found to be significantly decreased in SNpc dopaminergic neurons of MPTP induced PD mice and MPP+ treated SH-SY5Y cells.
miR-124 mimics reduced the Bim levels in experimental animals and decreased neurodegeneration. Inhibition of miR-124 by transfection of ant-miR-124 into SH-SY5Y cells suppressed autophagy [98]. mir-485-3p and mir-485-5p were shown to exert tumor suppressor action by inhibiting the PGC-1α expression and subsequent mitochondrial respiration in breast cancer [94]. mir-485-3p is found to be elevated in the serum of PD patients and plays a role in neuroinflammation, suggesting that it could be used as a new diagnostic marker and therapeutic target in the disease [95]. miR-144-3p targets amyloid-β protein precursor (AβPP) and indirectly upregulates the genes involved in mitochondrial biogenesis, i.e., PGC-1α, NRF-1, and TFAM. Thus miR-144-3p plays an important role in mitochondrial functioning. In one study, miR-144-3p is found to be significantly down regulated in MPTP exposed SH-SY5Y cell lines. Transfection of MPTP treated SH-SY5Y cells with miR-144-3p mimics restored mitochondrial function [96]. miR-376 is a negative regulator of genes like PGC-1α, TFAM was found to be increased in MPP+ exposed SH-SY5Y cells. Peripheral blood mononuclear cells (PBMCs) of 33 PD subjects and 25 age-matched controls were assessed by measuring the mRNA levels of mitochondrial-related genes. A significant drop in both PGC-1α and TFAM levels occurred in PD patients compared with age matched controls. The results also represented that miR-376a was strongly upregulated in the PBMCs of the PD patients [97].
miRNAs REGULATING MITOCHONDRIAL FUNCTION AS POTENTIAL BIOMARKERS
Researchers have recently concentrated their efforts on developing miRNAs as biomarkers for the early detection of PD. The circulatory miRNAs which are secreted from cells and found in the various biological fluids such as Cerebrospinal fluid (CSF), plasma, serum, saliva, urine, and pleural fluid serve as biomarkers for the early detection of the disease [73].
CURRENT DIAGNOSTIC APPROACH AND IMPORTANCE OF EARLY DIAGNOSIS IN PD
Currently the diagnosis of PD is majorly based on the clinical symptoms like bradykinesia, dyskinesia, gait freezing, tremors of hand, foot, and jaw, decreased facial expression, and decreased arm swinging, along with frozen shoulder presented in those affected by the disease. Although motor symptoms are characteristic of PD, there are many non-motor functions associated with the disease which are seen right from the prodromal stage, about 20 years before the onset of the actual disease. These include constipation, REM sleep behavior disorder (RBD), olfaction impairment, depression, dysphagia, and neuropsychiatric disorders. The diagnosis of PD can be further assisted by other methods like neuroimaging, metabolite profiling, and analysis of some neurochemical biomarkers. The various neuroimaging techniques like magnetic resonance imaging, transcranial doppler ultrasonography, positron emission tomography (PET scan), and single photon emission computed tomography (SPECT) help in the differential diagnosis of the disease. PET and SPECT scans are used to assess the dopamine metabolism and deficiency [99]. There are certain neurochemicals and biochemicals present in the body that are altered due to the diseased state which can be used to confirm the disease. An exclusive characteristic of PD is the presence of Lewy bodies due to accumulation of α-synuclein. Several neurochemical biomarkers that can be useful in diagnosis of PD includes 8-Hydroxy-2’-Deoxyguanosine (a marker for oxidative stress), Orexin (a neuropeptide implicated in the non-motor manifestations of PD), peripheral proteosomes, dopamine, apolipoproteins, etc. Metabolic profiling also provides valuable information about the severity of PD. In the advanced stage of PD, lactate levels were found to be increased in striatum, and N-acetyl aspartate/citrate ratio levels were found to be decreased [100]. However, all these diagnoses are only possible after the establishment of the disease. Even though the non-motor symptoms appear at quite an early stage, they are often ignored and not seen seriously due to their non-specific nature. Many studies have shown that by the time the motor symptoms appear there is a 30-70% evident neuronal cell loss in the SNpc of the patient [4]. The reversal of the disease is difficult and only the rate of progression of the disease is merely mitigated. Hence, it becomes essential that early diagnostic techniques should be explored and established. Active research is ongoing to establish better biomarkers to solve the problem of early detection of PD. This will open the possibility of providing better medical care and even stopping the progression of the neurodegeneration. Many studies showed that miRNAs are implicated in occurrence and progression of PD. Besides gene regulation, miRNAs are also found to be involved in intercellular communication. To carry out the function they migrate outside the cell and circulate in various body fluids in the form of exosomes, protein complexes. These are termed as circulatory miRNAs and are being investigated extensively to be established as biomarkers for their non- or minimally invasive diagnostic potential [101].
miRNAs ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION IDENTIFIED IN BIOLOGICAL SAMPLES OF PD PATIENTS
Several miRNAs are associated with mitochondrial dysfunction and are found to be dysregulated in PD.
In a study conducted by Mo et al. to identify the miRNAs involved in α-synuclein aggregation, it was observed that miR-144-5p was upregulated in CSF of PD patients. There was a significant difference between the expression of mir-144-5p in PD patients when compared to that of the control group. The upregulation was consistent across all forms of severity of the PD patients [102]. A study done on the CSF miRNA and protein profiles showed that PD patients in early stages show a low level of miR-27a-3p as compared to control. This miRNA has potential to be a biomarker of PD in the early stages itself [103]. miR-145-3p regulates the expression of DJ-1 protein and maintains mitochondrial homeostasis. However, in PD patients an overexpression of miR-145-3p results in reduction of DJ-1 and hence, increase in oxidative stress. miR-145-3p has been detected in the saliva of PD patients and could serve as a potential auxiliary biomarker of PD [93].
In a study conducted by Shu et al., miR-146b-5p was found to be significantly decreased according to severity of PD in patient groups as compared to the healthy group [104]. miR-485-3p was shown to be significantly upregulated in serum of PD patients. A study compared the serum miR-485-3p levels in normal, Alzheimer’s disease, and PD patients and found that it was significantly higher in PD patients as compared to the other two and can be used for diagnostic screening of PD [95]. In a study conducted by Ding et al., miR-181a was found to be downregulated in the serum samples of sporadic PD patients along with alteration in the levels of four other miRNAs [105]. miR-214 was found to be upregulated in the serum samples of prodromal PD patients whereas it was found to be lower in advanced PD. Thus miR-214 levels negatively correlate with the severity of the disease and can be used as biomarker for diagnosing prodromal PD [106, 107]. miR-27a bind to the 3’-UTR of PINK1 protein and dysregulates mitophagy. It increases the phosphorylation of ubiquitin, Parkin translocation, and LC3-II accumulation in mitochondria. A study conducted by Chen et al. showed that miR-27a is upregulated and is detected in the plasma of PD patients [108]. miR-4639 levels were found to be significantly high in PD patients in a study conducted to explore a peripheral biomarker with a potential to be the therapeutic target. Increased miR-4639-5p levels in plasma comes from the exosomes derived from the CNS [82]. miR-153, miR-124 were found to be downregulated in different studies and could be used as potential biomarkers for the diagnosis of PD [87, 109]. miR-7 and miR-103-3p were identified to be significantly increased in the peripheral blood of the PD patients [110, 111]. Various miRNAs identified in the biological fluids of PD patients are given in Table 2.
Table 2
miRNAs identified in various biological fluids
| Biological fluid | MiRNAs | Expression | Reference |
| CSF | miR-144-5p | ↑ | [102] |
| miR-27a-3p (Early stages of PD) | ↓ | [103] | |
| Saliva | miR145-3p | ↑ | [112] |
| Serum | miR-146b-5p | ↓ | [104] |
| miR-485-3p | ↑ | [95] | |
| miR-181a | ↓ | [105] | |
| miR-214 (prodromal PD) | ↑ | [107] | |
| miR-214 (advanced PD) | ↓ | [106] | |
| Plasma | miR-27a | ↑ | [108] |
| miR-4639-5p | ↑ | [82] | |
| miR-153-3p | ↓ | [87] | |
| miR-124 | ↓ | [109] | |
| Peripheral blood | miR-7 | ↑ | [110] |
| miR-103-3p | ↑ | [111] |
miRNAs ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION AS POTENTIAL THERAPEUTIC OPTIONS IN PD
The treatment of PD available to date is aimed at restoring the dopamine levels in the striatum but long-term treatment is associated with severe side effects. Several new strategies which are aimed at controlling various pathogenic mechanisms are being explored. They include immunotherapy, regenerative therapy, gene therapy using CRISPR technology, viral vectors, use of natural products with neuroprotective action, and various targeted therapies [1, 113]. Recently small RNA drugs are also being explored as treatment options in various diseases. FDA approved the first siRNA drug, Patisiran, for a rare polyneuropathy which occurs due to hereditary transthyretin-mediated (hATTR) amyloidosis. Several miRNA drugs are being tested in clinical trials for various diseases [114]. miRNA based therapeutics can be categorized in to two groups: Agomirs or miRNA-mimics and antagomirs or antimiRs. The former over express the miRNA transcripts whereas the latter repress the function of corresponding miRNA [114]. The miRNA-based therapeutics are studied due to their different advantages. At first, one single miRNA has the ability to downregulate several targets by joining the 3’-UTR of a gene. Another advantage is its size of roughly 22 nucleotides aids in the development of miRNA medicines. Furthermore, they can be administered by a variety of drug delivery systems that have already been approved for human use. In peripheral tissues, miRNA-based medicines have proven clinical usefulness. The distribution of drugs into the brain, however, is a key barrier in the treatment of neurodegenerative illnesses like PD. Many viral vectors are available to deliver the mRNAs to the desired site of action but besides ensuring effective transfection, they may cause infection in the cells. Non-viral methods of delivery such as intranasal route, liposomes, nanoparticles, micelles, and others are being investigated [115]. Another limitation is degradation of miRNAs by endonucleases and circulating macrophages. Formulating the miRNA-based therapeutics in the form of micelles, liposomes, nanoparticles, etc., may overcome this limitation [116]. Various miRNAs associated with mitochondrial dysfunction in PD were studied for their therapeutic potential as agomirs and antagomirs in several preclinical models as given in Table 3.
Table 3
miRNAs evaluated for therapeutic potential in preclinical models
| Targeted miRNA | Agomir or AntagomiR | Biological model/tissue | Mode of neuroprotective action | Reference |
| miR-214 | Agomir | MPP+ treated SH-SY5Y cells, PD mice model | Downregulated α- synuclein | [83] |
| miR-144 | Agomir | MPTP exposed SH-SY5Y cells | Improved mitochondrial function by downregulating APP | [96] |
| miR-7 | Agomir | MPP+ treated SH-SY5Y cells | Reduced the mitochondrial fragmentation/ depolarization, mitigated oxidative stress and release of pre-apoptotic proteins. Also decreased pro-inflammatory cytokines by down regulating the NLRP3 inflammasome. | [84, 117] |
| miR-124 | Agomir | MPP+ treated SH-SY5Y cells | Regulated cell survival, autophagy, mitochondrial dysfunction in PD. | [118] |
| miR-146 | Antagomir | Rotenone exposed SH-SY5Y cells | Reduced mitochondrial fragmentation and oxidative stress | [76] |
| miR103a-3p | Antagomir | C57BL/6 mice model and SH-SY5Y cells | improved mitophagy invitro and in vivo through Parkin/ Ambra1 pathway. | [79] |
| miR-181a/b | Antagomir | SH-SY5Y human neuroblastoma cells | Stimulated mitochondrial biogenesis, regulate in vitro the expression of ATG5 and PARK2, which are involved in autophagy and mitophagy | [122] |
| miR-200a | Antagomir | MPTP treated SH-SY5Y cells | Increased SIRT1 which controls PGC-1α and exerted anti-oxidative effects and protected mitochondria | [124] |
| miR-376a | AntagomiR | MPTP treated SH-SY5Y cells | Improved PGC-1α and TFAM levels which are main players in mitochondrial biogenesis | [97] |
AGOMIRS OF miRNA ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION SHOWING NEUROPROTECTIVE EFFECT IN PRECLINICAL PD MODELS
miR-214 is a negative regulator of SNCA gene which is involved in the oxidative stress and mitochondrial impairment. Its levels were found to be downregulated in PD mouse model and MPP+ exposed SH-SY5Y cell line. Transfection of 100 nmol miR-214 mimic with lipofectamine 2000 downregulated the α-synuclein levels in in vitro model after 48 h of incubation and in vivo model after 21 days of injection [83]. miR-144-3p is a negative regulator of AβPP which decreases the mitochondrial functions by downregulation of PGC-1α, TFAM. Transfection of miR-144-3p mimics in MPTP exposed SH-SY5Y cells decreased AβPP expression and also alleviated MPTP induced mitochondrial dysfunction [96]. In MPP+ treated SH-SY5Y cells and primary cortical neurons, increased expression of miR-7 decreased the opening of mPTP channels and reduced the mitochondrial fragmentation/depolarization, mitigated oxidative stress and the release of pre-apoptotic proteins. In addition, it also decreased the level of pro-inflammatory cytokines by down regulating the NLRP3 inflammasome. These results show that miR-7 could be a promising target for the treatment of PD [117]. miR-124 which is abundantly expressed in brain and regulates cell survival, autophagy, mitochondrial dysfunction in PD. Through these functions, it exhibits a neuroprotective role and acts as a potential therapeutic candidate [118]. The role of miR-425 in neuroprotection has been explored in the C57BL/6 mice PD model. Its inhibition promotes mitochondrial accumulation in the cell body accompanied by increased ROS production. Therefore, miR-425 has protective role in PD and its mimics can be used as therapeutic option to treat PD [119].
ANTAGOMIRS OF miRNA ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION SHOWING NEUROPROTECTIVE EFFECT IN PRECLINICAL PD MODELS
miR-146a promotes mitochondrial fragmentation by inhibiting parkin. Its levels were found to be increased in PD. Transfection of rotenone-exposed SH-SY5Y cells with anti-miR-146a resulted in rescue of the cells from mitochondrial fragmentation and oxidative stress [76]. miR-103a-3p is a negative regulator of Parkin and regulates its expression by targeting a conserved binding site in the 3’-UTR of PRKN. miR-103a-3p inhibition by using antagomiR-103a-3p resulted in neuroprotective effects and improved mitophagy in vitro and in vivo. These findings suggested that miR-103a-3p inhibition has neuroprotective effects in PD by regulating mitophagy through the Parkin/Ambra1 pathway. Thus, modulating miR-103a-3p levels may be an applicable therapeutic strategy for PD [79]. Sirtuin 3 (SIRT3) is one of the sirtuins that are important for mitochondrial functioning. SIRT3 was identified as a target of miR-494-3p and this miRNA negatively regulated SIRT3 expression in SH-SY5Y cells. A study demonstrated that miR-494-3p expression and SIRT3 was increased and decreased respectively in SH-SY5Y cells following MPP+ treatment [120]. miR-494-3p inhibition promoted cell viability and superoxide dismutase activity and suppressed apoptotic rate and ROS generation in MPP+ induced SH-SY5Y cells. Additionally, it was found that upregulation of miR-494-3p suppressed SIRT3 expression and exacerbated motor impairment in MPTP induced PD mouse model. Thus miR-494-3p antagomir exerted a neuroprotective role in MPP+ induced PD by targeting SIRT3 and could be a therapeutic target in PD patients [121]. miR-181a and miR-181b regulate the key genes involved in mitochondrial biogenesis and autophagy. These miRNAs negatively target NRF1, Cytochrome oxygenase 11 (COX11), and Coenzyme Q binding protein 10B (COQ10B). Studies showed that downregulation of miR-181a/b stimulated mitochondrial biogenesis. In in vitro studies, miR-181a/b regulated the expression of Autophagy related gene 5 (ATG5) and PARK2 which encodes Parkin, which are the main player in autophagy and mitophagy respectively. An increase in mitophagy occurred in miR-181a/b antagomir treated PD mice model. It was also shown that miR-181a/b silencing protects cells from mitochondrial damage after FCCP treatment. These findings suggest that miR-181a/b could be a potential therapeutic method for PD [122]. Sirtuin 1 (SIRT1) is involved in various physiological processes. It activates PGC-1α to exert anti-oxidative effects and protect mitochondria and activates AMP-activated protein kinase (AMPK) mediated autophagy to remove abnormal proteins. miR-200a is a negative regulator of SIRT1. It binds to SIRT1 mRNA in its 3’-UTR. miR-200 was found to be overexpressed SH-SY5Y cells treated with MPP+ [123] resulting in the downregulation of SIRTI. Hence, therapeutic potential of AntagomiR-200a may be considered as a possible approach to prevent PD progression [124].
The above listed agomirs and antagomirs can be further explored for their clinical efficacy in treatment of PD and can be used as potential therapeutic options in PD. miRNAs are identified to play a major role in many pathogenic mechanisms involved in PD. Hence, they can be effectively targeted to manage PD. Various miRNAs are being evaluated for their effectiveness in PD in various preclinical and clinical models. Since PD is a chronic and progressive disorder, the miRNA-based drugs should have a durable concentration in the brain to act over a longer period of time. Chemical modification of the miRNAs may enhance their sustainable action. Site specific delivery is also an important factor. Various strategies like Stereotaxic injection, convention-enhanced delivery, magnetofection, etc., are being explored for the effective delivery of miRNAs to the target site [125].
CONCLUSION
PD is becoming more common, and there is a pressing need to address the limitations that PD patients face in order to improve their quality of life. Recent studies are focused on identifying the various pathogenic mechanisms involved in the occurrence and progression of PD. Mitochondrial dysfunction has been identified to play a central role in the progression of PD. Many factors were identified to be involved in the regulation of mitochondrial functions and miRNAs are one among them. Several miRNAs associated with mitochondrial functioning were found to be dysregulated in PD. Some of them were found to be present in various biological samples like cerebrospinal fluid, serum, plasma, and saliva. They can be utilized as potential biomarkers for the early diagnosis of PD. Some of the miRNA mimics and antagomirs were tested for their efficacy in the exerting neuroprotective action in various in vitro and in vivo PD models. Those agomirs and antagomirs could be explored further to be the potential therapeutic options in the treatment of PD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0449r1).
REFERENCES
[1] | Ntetsika T , Papathoma PE , Markaki I ((2021) ) Novel targeted therapies for Parkinson’s disease. Mol Med 27: , 17. |
[2] | Simon DK , Tanner CM , Brundin P ((2020) ) Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med 36: , 1–12. |
[3] | Cerri S , Mus L , Blandini F ((2019) ) Parkinson’s disease in women and men: What’s the difference? . J Parkinsons Dis 9: , 501–515. |
[4] | Rizek P , Kumar N , Mandar SJ ((2016) ) An update on the diagnosis and treatment of Parkinson disease. CMAJ 188: , 1157–1165. |
[5] | Paoletti FP , Tambasco N , Parnetti L ((2019) ) Levodopa treatment in Parkinson’s disease: Earlier or later? Ann Transl Med 7: , S189. |
[6] | Schapira AH , Jenner P ((2011) ) Etiology and pathogenesis of Parkinson’s disease. Mov Disord 26: , 1049–1055. |
[7] | Prasuhn J , Davis RL , Kumar KR ((2021) ) Targeting mitochondrial impairment in Parkinson’s disease: Challenges and opportunities. Front Cell Dev Biol 8: , 615461. |
[8] | Huemer F , Leisch M , Geisberger R , Zaborsky N , Greil R ((2021) ) miRNA-based therapeutics in the era of immune-checkpoint inhibitors. Pharmaceuticals (Basel) 14: , 89. |
[9] | Reddy PH , Tripathi R , Troung Q , Tirumala K , Reddy TP , Anekonda V , Shirendeb UP , Calkins MJ , Reddy AP , Mao P , Manczak M ((2012) ) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822: , 639–649. |
[10] | Xu J , Du W , Zhao Y , Lim K , Lu L , Zhang C , Li L ((2022) ) Mitochondria targeting drugs for neurodegenerative diseases— design, mechanism and application. Acta Pharm Sin B 12: , 2778–2789. |
[11] | Bose A , Beal MF ((2016) ) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139: , 216–231. |
[12] | Haddad D , Nakamura K ((2015) ) Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett 589: , 3702–3713. |
[13] | Pacelli C , Giguère N , Bourque MJ , Lévesque M , Slack RS , Trudeau LÉ ((2015) ) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25: , 2349–2360. |
[14] | Bolam JP , Pissadaki EK ((2012) ) Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov Disord 27: , 1478–1483. |
[15] | Mamelak M ((2018) ) Parkinson’s disease, the dopaminergic neuron and gammahydroxybutyrate. Neurol Ther 7: , 5–11. |
[16] | Moon HE , Paek SH ((2015) ) Mitochondrial dysfunction in Parkinson’s disease. Exp Neurobiol 24,: , 103–116. |
[17] | Chen Z , Rasheed M , Deng Y ((2021) ) The epigenetic mechanisms involved in mitochondrial dysfunction: Implication for Parkinson’s disease. , e. Brain Pathol 32: , 13012. |
[18] | Schapira AH ((2008) ) Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 7: , 97–109. |
[19] | Nolfi-Donegan D , Braganza A , Shiva S ((2020) ) Mitochondrial electron transport cha, Oxidativehosphorylation, oxidant production, and methods of measurement. Redox Biol 37: , 101674. |
[20] | Schapira AH , Cooper JM , Dexter D , Clark JB , Jenner P , Marsden CD ((1990) ) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54: , 823–827. |
[21] | Hattori N , Tanaka M , Ozawa T , Mizuno Y ((1991) ) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann Neurol 30: , 563–571. |
[22] | Mizuno Y , Ohta S , Tanaka M , Takamiya S , Suzuki K , Sato T , Oya H , Ozawa T , Kagawa Y ((1989) ) Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 163: , 1450–1455. |
[23] | Keane PC , Kurzawa M , Blain PG , Morris CM ((2011) ) Mitochondrial dysfunction in Parkinson’s disease. Parkinsons Dis 2011: , 716871. |
[24] | Shoffner JM , Watts RL , Juncos JL , Torroni A , Wallace DC ((1991) ) Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann Neurol 30: , 332–339. |
[25] | Winkler-Stuck K , Kirches E , Mawrin C , Dietzmann K , Lins H , Wallesch CW , Kunz WS , Wiedemann FR ((2005) ) Re-evaluation of the dysfunction of mitochondrial respiratory chain in skeletal muscle of patients with Parkinson’s disease. J Neural Transm (Vienna) 112: , 499–518. |
[26] | Jenner P ((2001) ) Parkinson’s disease, pesticides and mitochondrial dysfunction. Trends Neurosci 24: , 245–247. |
[27] | Champy P , Höglinger GU , Féger J , Gleye C , Hocquemiller R , Laurens A ((2004) ) Annonacin, a lipophilic inhibitor of mitochondrial complex I, induces nigral and striatal neurodegeneration in rats: Possible relevance for atypical parkinsonism in Guadeloupe. J Neurochem 88: , 63–69. |
[28] | Mimaki M , Wang X , McKenzie M , Thorburn DR , Ryan MT ((2012) ) Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta 1817: , 851–862. |
[29] | González-Rodríguez P , Zampese E , Stout KA , Guzman JN , Ilijic E , Yang B , Tkatch T , Stavarache MA , Wokosin DL , Gao L , Kaplitt MG , López-Barneo J , Schumacker PT , Surmeier DJ ((2021) ) Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599: , 650–656. |
[30] | Dunham-Snary KJ , Wu D , Potus F , Sykes EA , Mewburn JD , Charles RL , Eaton P , Sultanian RA , Archer SL ((2019) ) Ndufs2, a core subunit of mitochondrial complex I, Is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ Res 124: , 1727–1746. |
[31] | Devi L , Raghavendran V , Prabhu BM , Avadhani NG , Anandatheerthavarada HK ((2008) ) Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283: , 9089–9100. |
[32] | Chinta SJ , Mallajosyula JK , Rane A , Andersen JK ((2010) ) Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy. }. Neurosci Lett 486: , 235–239. |
[33] | Quinlan CL , Treberg JR , Perevoshchikova IV , Orr AL , Brand MD ((2012) ) Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic Biol Med 53: , 1807–1817. |
[34] | Kussmaul L , Hirst J ((2006) ) The mechanism of superoxide production by NADH: Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A 103: , 7607–7612. |
[35] | Van Laar VS , Berman SB ((2009) ) Mitochondrial dynamics in Parkinson’s disease. Exp Neurol 218: , 247–256. |
[36] | Frank S , Gaume B , Bergmann-Leitner ES , Leitner WW , Robert EG , Catez F , Smith CL , Youle RJ ((2001) ) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: , 515–525. |
[37] | Wang X , Yan MH , Fujioka H , Liu J , Wilson-Delfosse A , Chen SG , Perry G , Casadesus G , Zhu X ((2012) ) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 21: , 1931–1944. |
[38] | Pryde KR , Smith HL , Chau KY , Schapira AHV ((2016) ) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol 213: , 163–171. |
[39] | Pozo Devoto VM , Falzone TL ((2017) ) Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? . Dis Model Mech 10: , 1075–1087. |
[40] | Ziviani E , Tao RN , Whitworth AJ ((2010) ) Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc Natl Acad Sci U S A 107: , 5018–5023. |
[41] | Chen H , Chomyn A , Chan DC ((2005) ) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280: , 26185–26192. |
[42] | Wang X , Winter D , Ashrafi G , Schlehe J , Wong YL , Selkoe D , Rice S , Steen J , Lavoie MJ , Schwarz TL ((2011) ) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147: , 893–906. |
[43] | Valdinocci D , Simões RF , Kovarova J , Cunha-Oliveira T , Neuzil J , Pountney DL ((2019) ) Intracellular and intercellular mitochondrial dynamics in Parkinson’s disease. Front Neurosci 13: , 930. |
[44] | Ambros V ((1989) ) A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell 57: , 49–57. |
[45] | Sasaki S , Shirata A , Yamane K , Iwata M ((2004) ) Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63: , 678–682. |
[46] | Klein C , Westenberger A ((2012) ) Genetics of Parkinson’s diseas. Cold Spring Harb Perspect Med 2: , a008888. |
[47] | Fedorowicz MA , de Vries-Schneider RL , Rüb C , Becker D , Huang Y , Zhou C , Alessi Wolken DM , Voos W , Liu Y , Przedborski S ((2014) ) Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep 15: , 86–93. |
[48] | Narendra DP , Jin SM , Tanaka A , Suen DF , Gautier CA , Shen J , Cookson MR , Youle RJ ((2010) ) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: , e1000298. |
[49] | McWilliams TG , Barini E , Pohjolan-Pirhonen R , Brooks SP , Singh F , Burel S , Balk K , Kumar A , Montava-Garriga L , Prescott AR , Hassoun SM , Mouton-Liger F , Ball G , Hills R , Knebel A , Ulusoy A , di Monte DA , Tamjar J , Antico O , Fears K , Smith L , Brambilla R , Palin E , Valori M , Eerola-Rautio J , Tienari P , Corti O , Dunnett SB , Ganley IG , Suomalainen A , Muqit MMK ((2018) ) Phosphorylation of Parkin at serine 65 is essential for its activation. Open Biol 8: , 180108. |
[50] | Koyano F , Okatsu K , Kosako H , Tamura Y , Go E , Kimura M , Kimura Y , Tsuchiya H , Yoshihara H , Hirokawa T , Endo T , Fon EA , Trempe JF , Saeki Y , Tanaka K , Matsuda N ((2014) ) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: , 162–166. |
[51] | Narendra D , Tanaka A , Suen DF , Youle RJ ((2008) ) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: , 795–803. |
[52] | Plowey ED , Cherra Iii SJ , Liu Y-J , Chu CT ((2008) ) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105: , 1048–1056. |
[53] | Krebiehl G , Ruckerbauer S , Burbulla LF , Kieper N , Maurer B , Waak J , Wolburg H , Gizatullina Z , Gellerich FN , Woitalla D , Riess O , Kahle PJ , Proikas-Cezanne T , Krüger R ((2010) ) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One 5: , e9367. |
[54] | Liu J , Liu W , Li R , Yang H ((2019) ) Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 8: , 712. |
[55] | Van Laar VS , Arnold B , Howlett EH , Calderon MJ , St.Croix CM , Greenamyre JT , Sanders LH , Berman SB ((2018) ) Evidence for compartmentalized axonal mitochondrial biogenesis: Mitochondrial DNA replication increases in distal s as an early response to Parkinson’s disease-relevant stress. J Neurosci 38: , 7505–7515 |
[56] | Jornayvaz FR , Shulman GI ((2010) ) Regulation of mitochondrial biogenesis. Essays Biochem 47: , 69–84. |
[57] | Lehman JJ , Barger PM , Kovacs A , Saffitz JE , Medeiros DM , Kelly DP ((2000) ) Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106: , 847–856. |
[58] | Ngo HB , Lovely GA , Phillips R , Chan DC ((2014) ) Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 5: , 3077. |
[59] | Chen C , Turnbull DM , Reeve AK ((2019) ) Mitochondrial dysfunction in Parkinson’s disease— cause or consequence? . Biology (Basel) 8: , 38. |
[60] | Ambekar T , Pawar J , Rathod R , Patel M , Fernandes V , Kumar R , Singh SB , Khatri DK ((2021) ) Mitochondrial quality control: Epigenetic signatures and therapeutic strategies. Neurochem Int 148: , 105095. |
[61] | Fazi F , Nervi C ((2008) ) MicroRNA: Basic mechanisms and transcriptional regulatory networks for cell fate determination. Cardiovasc Res 79: , 553–561. |
[62] | Pandey A , Jauhari A , Singh T , Singh P , Singh N , Srivastava AK , Khan F , Pant AB , Parmar D , Yadav S ((2015) ) Transactivation of P53 by cypermethrin induced miR-200 and apoptosis in neuronal cells. Toxicol Res 4: , 1578–1586. |
[63] | Pandey A , Singh P , Jauhari A , Singh T , Khan F , Pant AB , Parmar D , Yadav S ((2015) ) Critical role of the miR-200 family in regulating differentiation and proliferation of neurons. J Neurochem 133: , 640–652. |
[64] | Griffiths-Jones S , Saini HK , van Dongen S , Enright AJ ((2008) ) miRBase: Tools for microRNA genomics. , D154-D. Nucleic Acids Res 36: , 158. |
[65] | Cai X , Hagedorn CH , Cullen BR ((2004) ) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10: , 1957–1966. |
[66] | Lee Y , Kim M , Han J , Yeom KH , Lee S , Baek SH , Kim VN ((2004) ) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: , 4051–4060. |
[67] | Gebert LFR , MacRae IJ ((2019) ) Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20: , 21–37. |
[68] | Abdelfattah AM , Park C , Choi MY ((2014) ) Update on non-canonical microRNAs. Biomol Concepts 5: , 275–287. |
[69] | Vishnoi A , Rani S ((2017) ) MiRNA biogenesis and regulation of diseases: An overview. Methods Mol Biol 1509: , 1–10. |
[70] | Dong H , Lei J , Ding L , Wen Y , Ju H , Zhang X ((2013) ) MicroRNA: Function, detection, and bioanalysis. Chem Rev 113: , 6207–6233. |
[71] | Palanichamy JK , Rao DS ((2014) ) miRNA dysregulation in cancer: Towards a mechanistic understanding. Front Genet 5: , 54. |
[72] | Briggs CE , Wang Y , Kong B , Woo TUW , Iyer LK , Sonntag KC ((2015) ) Midbrain dopamine neurons in Parkinson’s disease exhibit a dysregulated miRNA and target-gene network. Brain Res 1618: , 111–121. |
[73] | Nies YH , Mohamad Najib NH , Lim WL , Kamaruzzaman MA , Yahaya MF , Teoh SL ((2021) ) MicroRNA dysregulation in Parkinson’s disease: A narrative review. Front Neurosci 15: , 660379. |
[74] | Miñones-Moyano E , Porta S , Escaramís G , Rabionet R , Iraola S , Kagerbauer B , Espinosa-Parrilla Y , Ferrer I , Estivill X , Martí E ((2011) ) MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 20: , 3067–3078. |
[75] | Xiong R , Wang Z , Zhao Z , Li H , Chen W , Zhang B , Wang L , Wu L , Li W , Ding J , Chen S ((2014) ) MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol Aging 35: , 705–714. |
[76] | Jauhari A , Singh T , Mishra S , Shankar J , Yadav S ((2020) ) Coordinated action of miR-146a and parkin gene regulate rotenone-induced neurodegeneration. Toxicol Sci 176: , 433–445. |
[77] | Di Rita A , Maiorino T , Bruqi K , Volpicelli F , Bellenchi GC , Strappazzon F ((2020) ) miR-218 inhibits mitochondrial clearance by targeting PRKN E3 ubiquitin ligase. Int J Mol Sci 21: , 355. |
[78] | Xing RX , Li LG , Liu XW , Tian BX , Cheng Y ((2020) ) Down regulation ofmiR-218, miR-124, and miR-144 relates toParkinson’s disease via activating NF-κB signaling. Kaohsiung J Med Sci 36: , 786–792. |
[79] | Zhou J , Zhao Y , Li Z , Zhu M , Wang Z , Li Y , Xu T , Feng D , Zhang S , Tang F , Yao J ((2020) ) miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol Res 160: , 105197. |
[80] | Stein CS , McLendon JM , Witmer NH , Boudreau RL ((2022) ) Modulation of miR-181 influences dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Mol Ther Nucleic Acids 28: , 1–15. |
[81] | Cheng M , Liu L , Lao Y , Liao W , Liao M , Luo X , Wu J , Xie W , Zhang Y , Xu N ((2016) ) MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 7: , 42274–42287. |
[82] | Chen Y , Gao C , Sun Q , Pan H , Huang P , Ding J , Chen S ((2017) ) microRNA-4639 is a regulator of DJ-1 expression and a potential early diagnostic marker for Parkinson’s disease. Front Aging Neurosci 9: , 232. |
[83] | Wang Z , Zhang J , Duan Y , Zhang Q , Li G , Zheng D ((2015) ) microRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting α-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed Pharmacother 74: , 252–256. |
[84] | Chaudhuri AD , Choi DC , Kabaria S , Tran A , Junn XE ((2016) ) microRNA-7 regulates the function of mitochondrial permeability transition pore by targeting vdac1 expression. J Biol Chem 291: , 6483–6493. |
[85] | Doxakis E ((2010) ) Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J Biol Chem 285: , 12726–12734. |
[86] | McMillan KJ , Murray TK , Bengoa-Vergniory N , Cordero-Llana O , Cooper J , Buckley A , Wade-Martins R , Uney JB , O’Neill MJ , Wong LF , Caldwell MA ((2017) ) Loss of microRNA-7 regulation leads to α-Synuclein accumulation and dopaminergic neuronal loss. }. Mol Ther 25: , 2404–2414. |
[87] | Zhang J , Yang Y , Zhou C , Zhu R , Xiao X , Zhou B , Wan D ((2022) ) LncRNA miR-17-92a-1 cluster host gene (MIR17HG) promotes neuronal damage and microglial activation by targeting the microRNA-153-3p/alpha-synuclein axis in Parkinson’s disease. Bioengineered 13: , 4493–4516. |
[88] | Cho HJ , Liu G , Jin SM , Parisiadou L , Xie C , Yu J , Sun L , Ma B , Ding J , Vancraenenbroeck R ((2013) ) microRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 22: , 608–620. |
[89] | Kim J , Fiesel FC , Belmonte KC , Hudec R , Wang WX , Kim C , Nelson PT , Springer W , Kim J ((2016) ) miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol Neurodegener 11: , 55. |
[90] | Junn E , Lee KW , Jeong BS , Chan TW , Im JY , Mouradian MM ((2009) ) Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A 106: , 13052–13057. |
[91] | Kim H , Park G , Jeon BS , Park WY , Kim YE ((2013) ) A mir-153 binding site variation in SNCA in a patient with Parkinson’s disease. Mov Disord 28: , 1755–1756. |
[92] | Kabaria S , Choi DC , Chaudhuri AD , Mouradian MM , Junn E ((2015) ) Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett 589: , 319–325. |
[93] | Chen Y , Zheng J , Su L , Chen F , Zhu R , Chen X , Ye Q ((2020) ) Increased salivary microRNAs that regulate DJ-1 gene expression as potential markers for Parkinson’s disease. Front Aging Neurosci 12: , 210. |
[94] | Lou C , Xiao M , Cheng S , Lu X , Jia S , Ren Y , Li Z ((2016) ) miR-485-3p and mir-485-5p suppress breast cancer cell metastasis by inhibiting PGC-1α expression. , e. Cell Death Dis 7: , 2159. |
[95] | Lin X , Wang R , Li R , Tao T , Zhang D , Qi Y ((2021) ) Diagnostic performance of miR-485-3p in patients with Parkinson’s disease and its relationship with neuroinflammation. Neuromolecular Med 24: , 195–201. |
[96] | Li K , Zhang J , Ji C , Wang L ((2016) ) miR-144-3p and its target gene β-amyloid precursor protein regulate 1-methyl-4-phenyl-1,2-3,6-tetrahydropyridine-induced mitochondrial dysfunction. Mol Cells 39: , 543–549. |
[97] | Baghi M , Rostamian Delavar M , Yadegari E , Peymani M , Pozo D , Hossein Nasr-Esfahani M , Ghaedi K ((2020) ) Modified level of miR-376a is associated with Parkinson’s disease. J Cell Mol Med 24: , 2622–2634. |
[98] | Wang H , Ye Y , Zhu Z , Mo L , Lin C , Wang Q , Wang H , Gong X , He X , Lu G , Lu F , Zhang S ((2016) ) miR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to Bim. Brain Pathol 26: , 167–176. |
[99] | Li T , Le W ((2020) ) Biomarkers for Parkinson’s disease: How good are they? Neurosci Bull 36: , 183–194. |
[100] | Emamzadeh FN , Surguchov A ((2018) ) Parkinson’s disease: Biomarkers, treatment, and risk factors. Front Neurosci 12: , 612. |
[101] | Turchinovich A , Samatov TR , Tonevitsky AG , Burwinkel B ((2013) ) Circulating miRNAs: Cell-cell communication function? . Front Genet 4: , 119. |
[102] | Cristina M , Santos T , Barreto-Sanz MA , S BR , Bell R , Widnall C , Perez LT , Berteau C , Schulte C , Scheller D , Berg D , Maetzler W , Pedro AF , Nogueira A ((2018) ) miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget 9: , 17455–17465. |
[103] | Marques TM , Kuiperij HB , Bruinsma IB , van Rumund A , Aerts MB , Esselink RAJ , Bloem BR , Verbeek MM ((2017) ) microRNAs in cerebrospinal fluid as potential biomarkers for Parkinson’s disease and multiple system atrophy. Mol Neurobiol 54: , 7736–7745. |
[104] | Shu Y , Qian J , Wang C ((2020) ) Aberrant expression of microRNA-132-3p and microRNA-146a-5p in Parkinson’s disease patients. Open Life Sci 15: , 647–653. |
[105] | Ding H , Huang Z , Chen M , Wang C , Chen X , Chen J , Zhang J ((2016) ) Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Parkinsonism Relat Disord 22: , 68–73. |
[106] | Li L , Ren J , Pan C , Li Y , Xu J , Dong H , Chen Y , Liu W ((2021) ) Serum miR-214 serves as a biomarker for prodromal Parkinson’s disease. Front Aging Neurosci 13: , 700959. |
[107] | Yan JH , Hua P , Chen Y , Li LT , Yu CY , Yan L , Zhang H , He Y , Zheng H , Chen H , Zhang ZJ , Yao QH , Dong H , Liu WG ((2020) ) Identification of microRNAs for the early diagnosis of Parkinson’s disease and multiple system atrophy. J Integr Neurosci 19: , 429–436. |
[108] | Chen L , Yang J , Lü J , Cao S , Zhao Q , Yu Z ((2018) ) Identification of aberrant circulating miRNAs in Parkinson’s disease plasma samples. Brain Behav 8: , e00941. |
[109] | Li N , Pan X , Zhang J , Ma A , Yang S , Ma J , Xie A ((2017) ) Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol Sci 38: , 761–767. |
[110] | Li B , Jiang Y , Xu Y , Li Y , Li B ((2019) ) Identification of miRNA-7 as a regulator of brain-derived neurotrophic factor/A-synuclein axis in atrazine-induced Parkinson’s disease by peripheral blood and brain microRNA profiling. Chemosphere 233: , 542–548. |
[111] | Serafin A , Foco L , Zanigni S , Blankenburg H , Picard A , Zanon A , Giannini G , Pichler I , Facheris MF , Cortelli P , Pramstaller PP , Hicks AA , Domingues FS , Schwienbacher C ((2015) ) Overexpression of blood microRNAs 103a, 30b, and 29a in L-dopa-treated patients with PD. Neurology 84: , 645–653. |
[112] | Cressatti M , Juwara L , Galindez JM , Velly AM , Nkurunziza ES , Marier S , Canie O , Gornistky M , Schipper HM ((2020) ) Salivary microR-153 and microR-223 Levels as potential diagnostic biomarkers of idiopathic Parkinson’s disease. Mov Disord 35: , 468–477. |
[113] | Iarkov A , Barreto GE , Grizzell JA , Echeverria V ((2020) ) Strategies for the treatment of Parkinson’s disease: Beyond dopamine. Front Aging Neurosci 12: , 4. |
[114] | Hanna J , Hossain GS , Kocerha J ((2019) ) The potential for microRNA therapeutics and clinical research. Front Genet 10: , 478. |
[115] | Paul S , Bravo Vázquez LA , Pérez Uribe S , Roxana Reyes-Pérez P , Sharma A ((2020) ) Current status of microRNA-based therapeutic approaches in neurodegenerative disorders. Cells 9: , 1698. |
[116] | Rupaimoole R , Han HD , Lopez-Berestein G , Sood AK ((2011) ) MicroRNA therapeutics: Principles, expectations, and challenges. Chin J Cancer 30: , 368–370. |
[117] | Zhou Y , Lu M , Du RH , Qiao C , Jiang CY , Zhang KZ , Ding JH , Hu G ((2016) ) microRNA-7 targets Nod-like receptorprotein 3 inflammasome to modulate neuroinflammation in thepathogenesis of Parkinson’s disease.. Mol Neurodegener 11: , 28. |
[118] | Angelopoulou E , Paudel YN , Piperi C ((2019) ) miR-124 and Parkinson’s disease: A biomarker with therapeutic potential. Pharmacol Res 150: , 104515. |
[119] | Hu YB , Zhang YF , Wang H , Ren RJ , Cui HL , Huang WY , Cheng Q , Chen HZ , Wang G ((2019) ) miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson’s disease. Cell Death Dis 10: , 589. |
[120] | Meng H , Yan WY , Lei YH , Wan Z , Hou YY , Sun LK , Zhou JP ((2019) ) SIRT3 regulation of mitochondrial quality control in neurodegenerative diseases. Front Aging Neurosci 11: , 313. |
[121] | Geng L , Zhang T , Liu W , Chen Y ((2018) ) miR-494-3p modulates the progression of and Parkinson’s disease models by targeting SIRT3. Neurosci Lett 675: , 23–30. |
[122] | Indrieri A , Carrella S , Romano A , Spaziano A , Marrocco E , Fernandez-Vizarra E , Barbato S , Pizzo M , Ezhova Y , Golia FM ((2019) ) miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol Med 11: , e8734. |
[123] | Li X , Feng Y , Wang XX , Truong D , Wu Y-C ((2020) ) The critical role of SIRT1 in Parkinson’s disease: Mechanism and therapeutic considerations. Aging Dis 11: , 1608–1622. |
[124] | Salimian N , Peymani M , Ghaedi K , Esfahani MHN ((2018) ) Modulation in miR-200a/SIRT1axis is associated with apoptosis in MPP+-induced SH-SY5Y cells. Gene 674: , 25–30. |
[125] | Titze-de-Almeida SS , Soto-Sánchez C , Fernandez E , Koprich JB , Brotchie JM , Titze-de-Almeida R ((2020) ) The promise and challenges of developing miRNA-based therapeutics for Parkinson’s disease. Cells 9: , 841. |


