
The Prevention of Inflammation and the Maintenance of Iron and Hepcidin Homeostasis in the Gut, Liver, and Brain Pathologies
Abstract
The human gut microbiome consists of a variety of microorganisms that inhabit the intestinal tract. This flora has recently been shown to play an important role in human disease. The crosstalk between the gut and brain axis has been investigated through hepcidin, derived from both hepatocytes and dendritic cells. Hepcidin could potentially play an anti-inflammatory role in the process of gut dysbiosis through a means of either a localized approach of nutritional immunity, or a systemic approach. Like hepcidin, mBDNF and IL-6 are part of the gut-brain axis: gut microbiota affects their levels of expression, and this relationship is thought to play a role in cognitive function and decline, which could ultimately lead to a number of neurodegenerative diseases such as Alzheimer’s disease. This review will focus on the interplay between gut dysbiosis and the crosstalk between the gut, liver, and brain and how this is mediated by hepcidin through different mechanisms including the vagus nerve and several different biomolecules. This overview will also focus on the gut microbiota-induced dysbiotic state on a systemic level, and how gut dysbiosis can contribute to beginnings and the progression of Alzheimer’s disease and neuroinflammation.
OVERVIEW
Our research question focused on the gut-brain axis (GBA) by exploring key organs and nerves that could potentially be involved in the crosstalk pathway. Our primary phase of literature review consisted of a PubMed search looking for all published literature around our research question. We focused on high fat-high sugar (HFHS) versus non HFHS diet animal models and narrowed down to a few key biomarkers that could be contributing to brain, gut, and liver pathologies including trimethylamine (TMA), trimethylamine N-oxide (TMAO), hepcidin, interleukin-6 (IL-6), and mature brain-derived neurotrophic factor (mBDNF). We then employed a review approach that examined over 80 papers with the key search terms of “Western Diet,” “high fat-high sugar diet,” “TMAO,” “TMA,” “brain,” “gut,” “mBDNF,” “IL-6,” and “gut dysbiosis.”
The next phase of our literature review involved compiling the papers that defined our key terms. We sought out studies using HFHS diet animal models and clinical trials. A state of “gut dysbiosis” was defined and we then focused on the gut microbiota distribution for each group, taking note of the differences between healthy and dysbiotic models. To understand more regarding the mechanism, we focused on papers that highlighted the role of each biomarker including TMA, TMAO, hepcidin, IL-6, and mBDNF. Once we established the pathways of each biomarker separately, we started focusing on reviewing papers that discussed how two or three biomarkers would interact with one another, like IL-6 and TMAO for instance.
After developing a stronger understanding of the relationship between these biomolecules, our last phase of the literature review was focused on formulating a hypothesis regarding two potential pathways in which the gut microbiota could be playing a role in neuroinflammation and neurologic disease. We created a Google document to track all possible crosstalk mechanisms and communications between these different biomarkers and ultimately laid down a basic framework that highlighted our two hypotheses. Finally, we compiled our findings from the literature review phase and constructed a comprehensive systematic review on the gut microbiota, different biomarkers, and the vagus nerve and how a HFHS diet shifts these factors through a lens of neurologic disease.
THE HUMAN GUT MICROBIOME
The human gut is one of the largest habitats for microorganisms in the body [1]. It supplies microbiota with the perfect niche, colonizing 70% of all total microbes in the body [1]. The gut microbiome is made up of archaea, bacteria, viruses, and eukaryota [2] and is dominated by two bacterial phylums that make up 80% of the microbiome: Bacteroidetes and Firmicutes [1]. Researchers are finding that 99% of the genes in the microbial communities harbored in the human gut are bacterial and equate to more than 1,000 bacterial species in each individual [2]. On a genetic level, 3.3 million non-redundant genes make up the human gut microbiome, having a gene set 150 times larger than the human gene complement [2].
Particularly over the past decade, researchers are finding that each individual’s microbiome varies in microbe composition [1]. While humans have a genome that is 99% similar to one another, each of our gut microbiomes are only 80–90% similar to one another [1]. One hypothesis insists on a “functional microbiome,” or “functional core” [3]. A functional core contains a complement of metabolic and molecular functions that are performed by the microbiome within the gut; however, it is not required for the specific organisms to be the same in each person [3]. While there is room for diversity and variety, a functional microbiome must have some degree of resilience to internal and external changes like age and medication changes [3]. The microorganisms must also resemble a symbiotic relationship between the microbes and the host and carry some of the key housekeeping pathways that cannot be done by humans themselves [3]. Without resilience, the microbiome would not be able to recover back to its healthy state and without a symbiotic relationship, the microbiome would not be practical for the host.
GUT DYSBIOSIS
Gut dysbiosis is defined as the disturbed state of the gut microbiome and its lack of diversity leading to an increased amount of pathogenic bacteria [4]. While this state could be a result of disease, diet change, non-modifiable genetic conditions, and medications, gut dysbiosis could also be a result of pro-inflammatory signaling bacteria that make up the gut microbiome [5]. Inflammation of the mucosal barrier in the intestinal lumen is dangerous, provided that 20% of cancers are preceded by chronic inflammation at the cancer site [4]. In healthy individuals, colonization resistance keeps pathogenic bacteria in low abundance, and prevents the pathogens from colonizing on the mucosa [4]. The diversity of the microflora assists in keeping the pathobiont in low abundance. In patients that are treated with antibiotics or anticancer treatments, the diversity of the microflora drastically decreases, and the mucosal barrier becomes inflamed [4]. The microflora of the sick individual is dominated by the expansion of the pathobiont, and these pathogens can escape into the systemic circulation by translocation through damaged epithelial tissue [2]. Once the pathogens escape, they cause a systemic infection that puts the host at a larger risk for cancers and disease.
Many diseases are thought to be associated with inflammation, including gastric cancers that are believed to be caused by infection with Helicobacter pylori that includes chronic gastritis, inflammatory bowel syndrome, and peptic ulcers [4]. Other non-gastric conditions that are believed to be associated with inflammation of the intestinal lumen wall include allergies, respiratory illness, and some neurologic conditions. The gut-brain communication axis modulates immune activity. An increase in chronic proinflammatory immune activity is recognized as an increasingly fundamental element in relation to neurodegenerative disorders [6]. A recent study assessing Parkinson’s disease identified the role of inflammation in the intestine is relevant in pathogenesis responsible for dysregulated immune activity [7]. Alterations in microbial composition of the gastrointestinal (GI) tract (dysbiosis) are believed to contribute to inflammatory and functional bowel disorders and psychiatric comorbidities [4]. Furthermore, a prominent study examined whether intestinal microbiota affects behavior and brain biochemistry in mice and determined the intestinal microbiota influences brain chemistry and behavior independently of the autonomic nervous system, GI-specific neurotransmitters, or inflammation [8]. Their findings suggest intestinal dysbiosis might contribute to psychiatric disorders in patients with bowel disorders.
DIET AND THE GUT MICROBIOME
Different diets can shape the composition of the microbes that ultimately change the function of the gut microbiome as a whole. A dietary shift between animal-based and plant-based, or HFHS diet (colloquially known as Western Diet) and vegan diet, respectively can implicate a significant change in microbial composition within just 24 hours of the change [9]. If the individual were to resort back to their original diet, the composition of gut bacteria would revert back in as little as 48 hours. While these dietary shifts may or may not be long-term, it has been proven that individuals with a primarily meat-based diet and others with a primarily plant-based diet have two completely different gut microbiome compositions [5]. The difference in diversity in regard to diet has much to do with the different macronutrients that are abundant in varying diets. Macronutrients include carbohydrates, proteins, and fats [1].
Carbohydrates are fermented by these colon microbes into organic acids and serve as an energy source for colon microbes. The major product of fermentation of carbohydrates are short-chain fatty acids which lower the pH of the colon and inhibit growth of pathogenic bacteria [10]. Diets high in fiber reduce hydrogen-sulfide producing microbes and increase the beneficial bacteria including Faecalibacterium prausnitzii, which has anti-inflammatory characteristics that play a role in preventing cancer and other GI conditions [4]. While proteins can be beneficial, a peaking interest has been focused on the harmful effects of proteins that come from red and processed meats [4]. Proteins from meat sources are shown to decrease levels of healthy Bifidobacterium adolescentis in the gut and increase Bacteroides and Clostridia [9]. Red and processed meats produce harmful hydrogen-sulfide byproducts when metabolized by these microbes, which can increase the individual’s risk of persistent inflammation in the gut [9]. HFHS diets are shown to increase the number of pro-inflammatory microbes in the gut, which stimulate the formation of taurine-conjugated bile acids. These acids promote the growth of pathogens and can cause illness [9]. A HFHS diet is particularly high in animal fat and studies have shown that individuals who eat this diet have decreased levels of bacterial diversity in the gut and less beneficial Bifidobacterium and Eubacterium [11].
HIGH FAT-HIGH SUGAR DIET
A HFHS diet is composed of high saturated fats and added sugars which are known to negatively impact cognitive function, particularly processes relying on the integrity of the hippocampus. Emerging evidence suggests that the gut microbiome influences cognitive function via the GBA, and that HFHS diet factors significantly alter the proportions of commensal bacteria in the GI tract [12]. Consumption of this diet negatively affects neurocognitive function, with a particular focus on recent evidence linking the gut microbiome with dietary- and metabolic-associated hippocampal impairment. Conlon and colleagues showed results displaying evidence linking gut bacteria to altered intestinal permeability and blood-brain barrier (BBB) integrity, thus making the brain more vulnerable to the influx of deleterious substances from the circulation. Consumption of a HFHS diet increased production of endotoxin by commensal bacteria, which promotes neuroinflammation and cognitive dysfunction. This prominent study showed that diet-induced alterations in gut microbiota impair peripheral insulin sensitivity, which is associated with hippocampal neuronal derangements and associated mnemonic deficits [12].
A HFHS diet was chosen to investigate the role of fat, carbohydrate, protein, vitamin, and mineral levels relating to diet and genotype models for AD [13]. Typically, a HFHS diet is rich in an amino acid, known as methionine. As methionine levels significantly increase, the body generates the amino acid homocysteine. Homocysteine is a thiol formed by the demethylation of methionine. Animal tissues are known to contain some free thiol but most of the compound occurs as homocysteine. When the level of homocysteine increases it damages the tissue structures of the arteries, initiating a release of cytokines, cyclins, and other inflammatory mediators. This accumulation leads to the loosening of arterial walls, the formation of local defects in the endothelium, which in turn lead to the deposition of cholesterol and calcium on the vascular wall. Vascular dysfunction represents a clinical preclinical step in the development of cardiovascular disease.
Finkelstein et al. [12] observed individuals whose plasma homocysteine values range from 15–50μM (with a normal limit approximating 12μM) are at an increased risk for vascular disorders. Their study consisted of folate supplementation given to patients with abnormal homocysteine metabolisms to eliminate a large percentage of neural tube defects through the enhancement of methionine synthase reaction. Additional factors found to be influencing homocysteine levels have been explored in several studies such as higher plasma homocysteine concentrations associated with an increased risk of extracranial carotid artery stenosis [11] and chronic venous disease in association with high serum homocysteine levels [13]. Elevated levels of plasma homocysteine and homocysteine thiolactone contribute to Alzheimer’s disease (AD) pathology by enhancing the interaction between fibrinogen and amyloid-β (Aβ) (i.e., Aβ-fibrin(ogen) interaction) that promotes the formation of tighter fibrin clots and delay clot fibrinolysis [14]. When Aβ interacts with fibrinogens, it is associated with increased abnormal clotting which damages neurons contributing to the cognitive decline expressed in AD patients. Recent articles assessed high homocysteine levels require therapeutic prevention. This intervention is necessary in order to prevent ischemic stroke [15]. The role of neuroprotective therapy in interrupting or slowing down the sequences responsible for damaging the biochemical and molecular processes initiate irreversible ischemic brain damage.
Excessive consumption of these diets high in saturated fats and sugars lead to obesity, metabolic syndrome, and impairment of cardiac functions; however, the underlying mechanisms are not fully understood [16]. TMAO is a primary gut microbiota-dependent metabolite of specific dietary nutrients and known as a key contributor to cardiovascular disease pathogenesis.
TMAO AND THE GUT MICROBIOME
TMAO is a compound that is produced in the liver that is derived from dietary precursors that are widely recognized as originating from HFHS, low fiber diets. TMA is produced exclusively by gut microbial enzymes from phosphatidylcholine and carnitine which is then oxidized in the liver by enzymes known as TMAO lyases. TMAO is responsible for a number of physiologic phenomena, including the upregulation of inflammatory pathways, promotion of foam cell formation, and enhancement of platelet reactivity which induces clotting. In a study focused on TMAO and its effects on the cardiovascular system, researchers found that higher levels of TMAO increased scavenger receptors in macrophages which ultimately results in the production of foam cells through the increase of macrophages binding low-density lipoprotein. Altogether, these macrophages would compile fatty deposits in blood vessels and disrupt several physiologic processes that resulted in a higher likelihood of developing inflammation and eventually cardiovascular disease. Studies have also shown that higher plasma levels of TMAO have been associated with higher concentrations of circulating inflammatory cytokines, indicating that this biomolecule potentially has a large effect on systemic inflammation levels in the body [1].
While TMAO can have implications on the cardiovascular system, it can also serve as a barometer of the health of the gut microbiome itself. Studies have found that TMA is produced by pathogenic bacteria found in a state of gut dysbiosis [4]. Connecting back to the HFHS diet, this specific profile of bacteria that have dominated the flora are responsible for breaking down foods rich in fat and carbohydrates into carnitine and betaine. An overproduction of TMAO indicates an overpopulation of pathogenic gut bacteria that can ultimately have poor outcomes for patients. Recent studies have looked into the effects of TMAO levels in AD patients and have found that mice with an increased level of TMAO displayed upregulation of scavenger receptor and CD68 expression (activated microglia marker for dementia), increased brain aging and cognitive impairment and elevated TMAO levels in their cerebrospinal fluid (CSF) [14]. This study also found that CSF TMAO was associated with CSF p-tau as well as p-tau/Aβ42 which could shed light on the potential relationship TMAO shares with tau pathology in AD patients.
IRON
Iron is an essential coenzyme required for several metabolic processes including oxygen transport, cellular respiration, and regulation of cell growth and differentiation [17]. Iron is also an essential component of gut microbial growth and can be indicative of inflammation. Iron serves as a critical metabolic source for pathogenic bacteria, as certain microbiota including Salmonella enterica serovar Typhimurium (S. Typhimurium) compete with other pathogens for iron in the setting of the inflamed gut [18]. A recent review paper highlighted studies that have shown that an excessive amount of iron in the GI tract will give rise to gut dysbiosis, which could have a number of physiological effects on the individual but will most directly lead to inflammation in the gut [19–21].
Iron is absorbed in the duodenum and partially in the colon. Excess iron in the GI tract can give rise to an overgrowth of pathogenic bacteria [22]. While most of the absorption happens in the small intestine, it has been hypothesized that iron is absorbed lastly in the colon as a defense mechanism to try to eliminate excess iron from the lumen as a means of nutritional immunity, ultimately starving pathogenic bacteria of their food source. Unabsorbed colonic iron is hypothesized to give rise to the growth of intestinal pathogens that make use of iron as their source to metabolize and reproduce [20, 21].
Excessive iron induces oxidative stress by generating reactive oxygen species (ROS). ROS damages the DNA and mtDNA, which affects DNA expression by epigenetic mechanisms and oxidizing proteins. ROS induces the release of iron from mitochondrial iron-sulfur cluster proteins of the respiratory chain and other storage proteins which stimulates interference with mitochondrial functions and disrupts iron homeostasis which accelerate the progression of neurodegenerative mechanisms. Neurodegenerative disorders include AD, Parkinson’s disease, multiple sclerosis, Friederichs ataxia, aceruloplasminemia, Huntington’s disease, and restless leg syndrome.
As iron is released into extracellular compartments it is taken up by cells such as astrocytes and neurons. It is still not fully understood how iron moves between neurons, microglia, and astrocytes. Jeong et al. observed astrocytes are ideally positioned to take up iron from the circulation and distribute it to other cells in the central nervous system (CNS) [18]. Astrocytes have the iron influx and efflux mechanisms needed for cell-to-cell transport of iron. DMT-1 is expressed by astrocytes and mediates iron influx into glial cells. Neurons and microglia can influx iron via transferrin [23]. Transferrin receptor mediates the uptake and intake of iron via ferroportin. IL-6 is now known to participate in neurogenesis (influencing both neurons and glial cells), and in the response of mature neurons and glial cells in normal conditions and following a wide arrangement [24]. Neurons, astrocytes, microglia, and endothelial cells are the essential sources of IL-6 in the CNS. IL-6 is the founder of neuropoetins, a prototypical four-helix bundle cytokine consisting of IL-6, IL-11, IL-27, and IL-31. Hepcidin’s antimicrobial properties are activated through inflammation [25]. The most common inflammatory cytokine is known as IL-6 (Fig. 1).
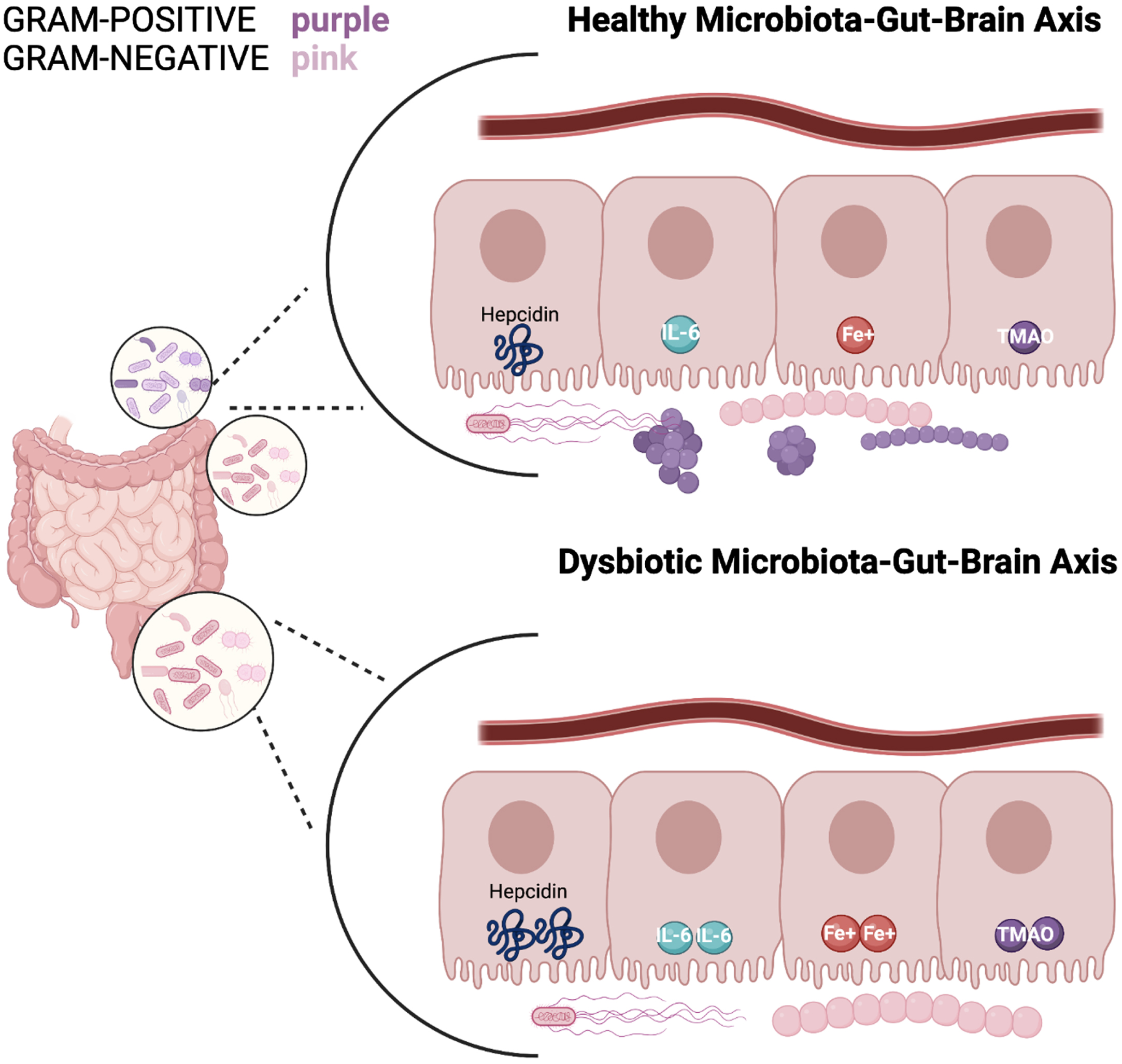
Fig. 1
A healthy microbiota consists of a balance of Gram-positive and Gram-negative bacteria; in which hepcidin, iron (Fe3+), IL-6, and TMAO levels express a normal state of balance. Whereas a dysbiosis state indicates an increase of gram-negative bacteria and elevated hepcidin levels resulting in excessive iron accumulation. This stimulates high ferritin levels which promotes less cognitive decline and therefore excess hepcidin production. The synthesis of hepcidin is rapidly increased by infection and inflammation in which elevated IL-6 levels initiate a pro-inflammatory cascade. With IL-6 upregulation occurring in the liver, the higher levels of TMA become oxidized to TMAO. Circulation of TMAO initiated in the small intestine initiates an inflammatory state.
HEPATOCYTE-DERIVED HEPCIDIN
Hepcidin, formally known as a liver expressed antimicrobial protein (LEAP-1), is secreted by hepatocytes from the liver, astrocytes, microglia, and macrophages and involves intestinal enterocytes [25]. Its primary role is to stop absorption of iron through enterocytes from the intestinal lumen, especially in inflammatory responses and antimicrobial properties. The influx of iron from digestion into enterocytes, and subsequent efflux from the cells into the blood stream is inhibited by hepcidin [25]. This mechanism occurs through direct hepcidin binding onto the iron import protein, known as divalent metal transporter-1 (DMT-1) [26]. DMT-1 is located in small intestine enterocytes. Comparably, cellular iron efflux prevention transpires when hepcidin binds to the iron export protein known as ferroportin. Hepcidin suppresses new absorption and restricts iron within the cell, ultimately halting all iron export [26] to capillaries.
Hepcidin is a peptide hormone produced primarily in hepatocytes that is a key regulator in iron homeostasis [22]. It is the primary regulator hormone for iron that was first reported in the early 2000 s and discovered in human urine and serum [22]. Hepcidin’s primary role functions in regulation of iron homeostasis via binding to the iron export channel, ferroportin, located on the basolateral surface of enterocytes and the plasma membrane of reticuloendothelial cells (macrophages). When serum iron levels are high, hepcidin binds to ferroportin to prevent the gut enterocytes from absorbing iron, and when iron levels are low, hepcidin release is inhibited. Hepcidin inhibition allows ferroportin channels to remain open, and iron is absorbed from the diet. Parmanand et al. have shown that the chelation of iron in the gut resulted in an overall reduction of potentially pathogenic bacteria within the gut, further implicating that bioavailability of iron may provide an advantageous environment for pathogens [27].
Hepcidin has been recently studied in its potential role of iron-homeostasis and inflammation crosstalk. In a review of patients with inflammatory bowel disease, it was shown that hepcidin-dependent changes in iron can impact inflammatory cytokine expression and on the other hand, inflammatory cytokines such as IL-6 can trigger hepcidin expression and alter iron homeostasis [27]. The upregulation of hepcidin induced by IL-6 during infections and inflammatory states impacts the amount of free iron circulating the body and is constantly in flux as it closely monitors the body’s need for iron in a state of health or disease (Fig. 2).
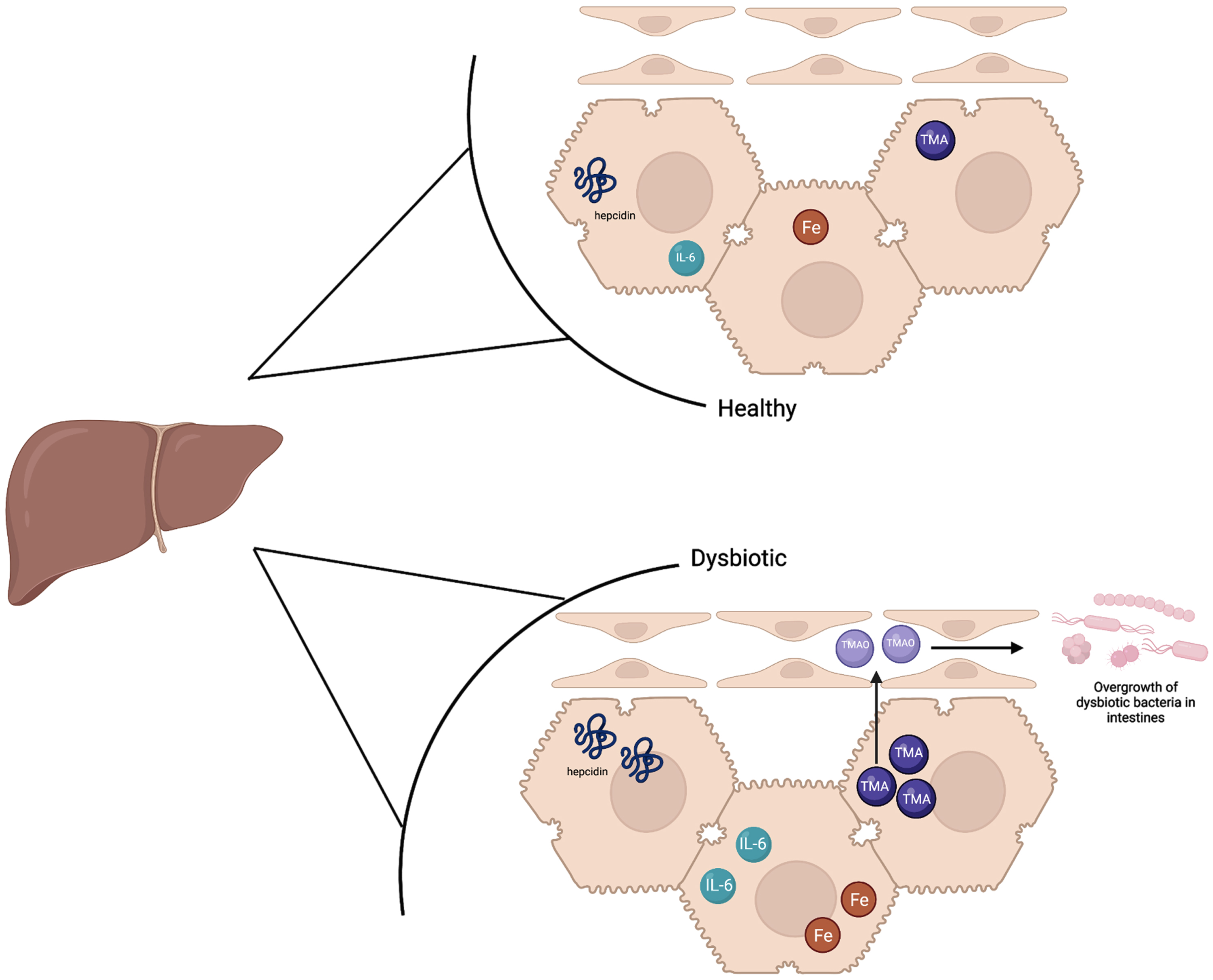
Fig. 2
The liver in a healthy state contains baseline hepcidin, IL-6, and TMA levels which supports a balance of healthy and dysbiotic bacteria. Iron levels are slightly higher in the healthy state than a dysbiotic state as hepcidin is upregulated in a dysbiotic state. In the dysbiotic state, IL-6 is also upregulated in the liver, and higher levels of TMA are sent to the liver to be oxidized to TMAO. TMAO is sent to the small intestines which ultimately causes inflammation and the overgrowth of dysbiotic bacteria.
Researchers have studied hepcidin in the setting of inflammation for years and the same pattern remains: hepcidin levels are increased in a state of inflammation [28]. In a state of gut dysbiosis, the brain has also been noted to endure some inflammation as well [8]. One potential explanation for this could be that the brain is acknowledging that there is inflammation elsewhere in the body, i.e., in the gut, and preventatively trying to protect itself. This could be happening through the expression of IL-6 in the brain which causes astrocytes and microglia to release hepcidin that ultimately sequesters iron in neurons. While this would systemically reduce bacterial growth in the intestines, locally in neurons the effect would “inadvertently” and ironically increase oxidative stress. It would be interesting to study the relationship of neuroinflammation and hepcidin production in more detail.
DENDRITIC CELL DERIVED HEPCIDIN
Hepcidin derived from the liver has been a key focus for researchers for years for its role in iron regulation and inflammation [22]. Recent studies have shown that dendritic cells (cDCs) are another source of hepcidin that could potentially have a separate but related role from the liver [29]. In their experiment, Bessman et al. added dextran sodium sulfate (DSS) to the drinking water of both wild type (Hamp + / +) mice and hepcidin-deficient (Hamp -/-) mice [29]. DSS is a toxin that causes inflammation and intestinal damage, and the goal of their study was to test the role of hepatocyte-derived hepcidin in the setting of intestinal inflammation. To their surprise, they found that while the Hamp + / + and Hamp -/- mice both exhibited similar weight loss secondary to gut inflammation, the Hamp-/- mice exhibited persistent weight loss, disruption of epithelial architecture, significantly shorter colon lengths, and overall lower systemic hepatocyte-derived hepcidin levels than Hamp + / + did, even after removal of DSS [30].
This new study exhibiting mucosal repair that is independent of hepatocyte hepcidin sheds light on the potential role of wound healing via cDC hepcidin. While hepcidin released from the liver could be sequestering iron systemically, the hepcidin from dendritic cells could potentially be working through a means of nutritional immunity by keeping a tight control on local iron and its availability to pathogenic bacteria to promote intestinal homeostasis by starving them of their food source. This ultimately allows tissue repair in the lumen at a much more localized level, analogous to a “first responder” to the area of inflammation directly [16]. Embracing their role, dendritic cells in the gut also function to sample luminal content to monitor for pathogens and enhance effector responses in response to infection and disease. One study showed that in inflamed tissue, these cDCs showed an increased expression in microbial recognition machinery, activation and production of critical immunological mediators, which further hints at a potential role of cDC-derived hepcidin in maintaining a state of homeostasis despite gut dysbiosis [23]. In conjunction with hepatocyte-derived hepcidin, these two very important biomolecules could be regulating inflammation in the body through two completely different, but equally necessary mechanisms.
HEPCIDIN-FERROPORTIN AXIS: RECEPTORS AT PLAY
The dietary pattern involved in the HFHS diet alters the composition of gut microbiota, influencing brain function by different mechanisms involving the GBA. Abreu et al. analyzed the role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells [26]. They found that different toll-like receptors (TLR) play a role in the immune system. TLR signaling induces intracellular iron sequestration in macrophages through two redundant mechanisms. TLR2 signaling downregulates ferroportin transcriptional expression, whereas TLR4 induces hepcidin secretion. Results also suggested iron increased lipopolysaccharide (LPS)-mediated hepcidin induction and TLR2 (pathway) downregulates ferroportin. LPS injected into mice causes the release of proinflammatory cytokines and triggers a well-characterized acute phase response, including induction of hepcidin by IL-6 [26]. These findings suggested that iron increases TLR4-mediated hepcidin expression, whereas TLR2 activation does not induce hepcidin expression. An additional study observed the endogenous expression of hepcidin by macrophages and neutrophils [25]. These myeloid cells produce hepcidin in response to the bacterial pathogen in the TLR4. Bacterial stimulation of macrophages triggered TLR4 dependent reduction in the iron exporter ferroportin.
In neutrophils, hepcidin induction was first observed as it migrated to the tissue site of the infection. A recent study suggested hepcidin expression in cultured hepatocytes or in the livers of mice infected with bacteria was independent of TLR4, suggesting the TLR4-hepcidin pathway is restricted to myeloid cell types [25]. Their findings identified endogenous myeloid cell hepcidin production as a previously unrecognized component of the host response to bacterial pathogens [25]. Myeloid cell types play an essential role in inflammation by producing hepcidin intrinsically, through the action of the TLR4 receptor, which is the key pattern recognition receptor for LPS. TLR4-specific hepcidin production by macrophages plays the largest role in localized foci of infection and inflammation or in reticuloendothelial organs, where these cell types are present in abundance.
BRAIN AND INFLAMMATORY RESPONSE: HEPCIDIN
Roy et al. (2005) suggested conditions in response to anemia of inflammation and found hepcidin antimicrobial peptides likely modulates iron transport from macrophages and enterocytes to red blood cell precursors as a consequence of its interaction with ferroportin, as it is the only transporter that facilitates iron emergence [28]. Their experiment suggested hepatocytes are not involved solely in iron storage but rather the command central for the maintenance of iron homeostasis because it receives multiple signals in relation to iron balance. These responses contribute to transcriptional control of hepcidin antimicrobial peptides.
A recent study analyzed the manipulation of hepcidin activity to recover neuronal damage due to brain inflammation in animal models and cultured cells [31]. The investigators found hepcidin produced by the brain cells in pathological conditions may limit iron transport through bovine brain microvascular endothelial cell system, and thereby also limit neuronal iron load. High iron load is detrimental for neuronal function. In addition, high iron load can also affect glial cell activity by turning them into cells that promote neuronal damage.
Vela et al. (2018) gave rise and expressed the importance of understanding hepcidin’s mechanistic insights of action in the brain in order to uncover its role in treating neuronal damage in neurodegenerative diseases [31]. Since iron is a critical element for maintaining iron homeostasis, it is used by the brain to synthesize myelin and neurotransmitter production. Excessive iron is toxic for brain cells and commonly observed in brain hemorrhages where inflammatory signaling is responsible for an increase in brain iron load which is associated with oxidative damage and cognitive decline [32]. During neuroinflammation, glial cells are activated, which perturbs iron homeostasis. Glial iron accumulation induces inflammatory states through the increasing release of pro-inflammatory cytokines, resulting in a self-propelling cycle of neuroinflammation and neurodegeneration. In addition, researchers proposed iron dysregulation as a pathogenic factor in neurodegenerative diseases [33]. The pathogenesis of glia and Aβ is the result of abnormal Aβ accumulation stimulated by the inflammatory cascade in AD [6]. Brain homeostasis is mediated by microglia that are residential immune cells regulating immune and tissue repair function, phagocytosis, and stimulation of microglial proliferation and activation. The BBB regulates the molecular transports of components to homeostatically regulate the neuronal extracellular environment. This regulation resides in and out of the CNS. The BBB is responsible for the production of preventing neurotoxic plasma components, pathogens, and blood cells from entering the brain. Damage to this area perpetuates an altered homeostasis within the CNS, indicating an increased risk of dementia and neurodegenerative diseases such as AD [6]. As iron crosses the vascular endothelial cells of the BBB, it binds to transferrin by the transferrin-TRF1 system that is expressed on the luminal side of endothelial cells (Fig. 3).
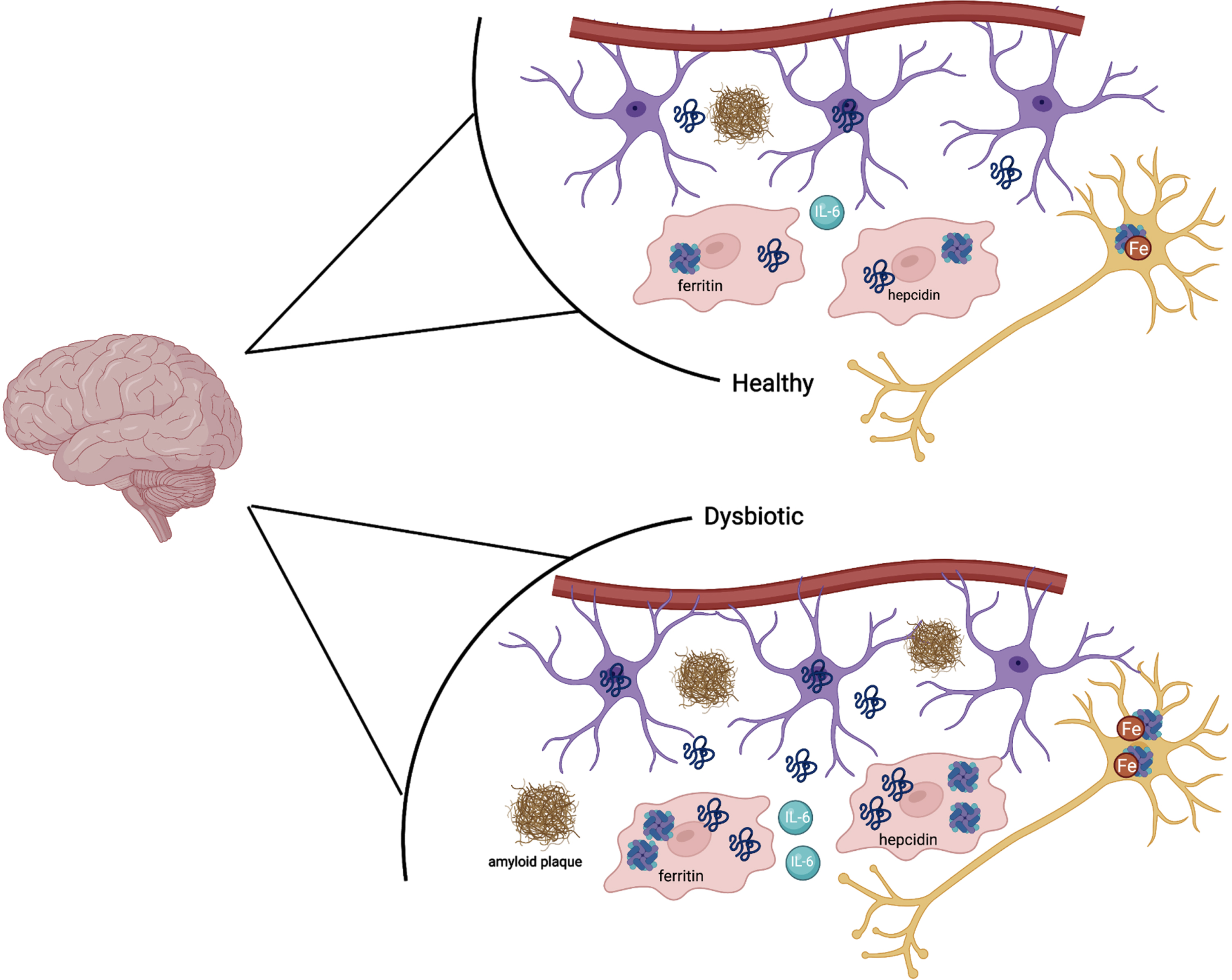
Fig. 3
In a state of a healthy microbiota, ferritin, hepcidin, IL-6, and iron are all at baseline levels. In a state of dysbiotic microbiota, the brain responds preventatively by upregulating ferritin and hepcidin levels, as well as IL-6. This is done via the astrocytes (purple) and microglia (red). In the neuron (dark yellow), iron is upregulated as well in the setting of neuroinflammation. Amyloid plaques can also be seen in the dysbiotic state as above.
BLOOD-BRAIN BARRIER CONNECTION WITH INTESTINAL DYSBIOSIS
HFHS consumption and gut microbiome alteration play a role in the activation of several neurobiological mechanisms that give rise to HFHS-mediated cognitive dysfunction, including BBB integrity and neuroinflammation. Consuming a HFHS diet promotes endotoxemia, which is linked to memory impairment. Previous studies suggest that HFHS diet impairs neuroprotective and anti-inflammatory effects in the gut through the weakening of the intestinal barrier via translocation of circulating gram-negative bacteria and impairment of permeability of the gut barrier [13]. The toxins that are permeating across the intestinal membrane trigger the immune response to release the proinflammatory cytokine of IL-6 peripherally, to thus influence the CNS by acting on the microglia. The microglia stimulate the neuroinflammation cascade. When microglia are activated, their morphology changes in association with increased secretion of pro-inflammatory cytokine IL-6, which can onset neuronal autophagy, loss of BBB integrity, brain damage, and activation of vagus nerve afferents [34].
INFLAMMATION: ONSET OF NEUROGENESIS, IL-6 MOST COMMON INFLAMMATORY CYTOKINE
IL-6 is an exceptional cytokine for its pleiotropic nature as it conforms to either pro or anti-inflammatory properties. IL-6 is a prototypical four-helix bundle cytokine that is the founder member of the neuropoietins [24]. IL-6 behaves in a neurotrophin-like manner and is known to participate in neurogenesis (influencing both neurons and glial cells), and in the response of mature neurons and glial cells in normal conditions and following a wide arrangement of injury models. Although IL-6 is often related to inflammatory and pathological situations, it is a factor that contributes decisively in the normal function of the brain. IL-6 is involved in the control of body weight, food intake, and energy expenditure [24]. Essential sources of IL-6 in the CNS include neurons, astrocytes, microglia, and endothelial cells. During inflammation, proinflammatory cytokine IL-6 is released from the intestinal mucosa and activates the vagus nerve afferents.
The production of IL-6 is increased by inflammation and associated with sepsis, endotoxemia, and treatment with pro-inflammatory cytokines. Hersherko et al. [35] recorded enterocytes have anti-inflammatory and cell protective effects of sepsis and endotoxemia. They suggested enterocyte IL6 production may be upregulated by cyclic-AMP (cAMP). Their studies reported treatments of cultured IEC-6 cells, a rat intestinal epithelial cell line, with cAMP augmented IL-61B induced IL-6 production. The role of cAMP and mechanisms by which cAMP regulates enterocyte IL-6 production are particularly significant, considering the role of cAMP in the regulation of multiple other functions of the intestinal mucosa. An upregulation of IL-6 related to neuroinflammation and hypoxia was seen in small intestine necrotizing enterocolitis pigs. The pigs expressed a common acute gut inflammatory disorder, known as necrotizing enterocolitis (NEC), an acute gut inflammatory disorder which is frequently observed in impaired neurodevelopmental stages. Sun et al. [36] found when NEC lesions are present in the small intestine, they are associated with changes in hippocampal gene expression which mediates neuroinflammation and disturbs neural circuit formation through enhanced neuronal differentiations. These findings give rise to future experimentations which would explore the neuroanatomical areas responsible for the changes in hippocampal gene expression. This future study could potentially uncover the importance of early brain protective interventions critical for infants affected by intestinal NEC lesions to ultimately reduce the individuals experienced neurological dysfunctions.
THE BI-DIRECTIONAL GUT-BRAIN AXIS
The gut, brain, and its microbes are complexly interconnected through a signaling network made up of neurons, hormones, immune cells, and microbial molecules known as the GBA. This bidirectional communication between the CNS, neuroendocrine system, autonomic nervous system, with the enteric nervous system and gut microbiota is mediated by signaling through the vagus nerve. The liver is a key component in our discussion as it produces hepcidin; however, it is not considered to be a part of the GBA directly. The bidirectional link regulates communication between the central and enteric nervous system, connecting the emotional and cognitive centers of our brain with peripheral intestinal functions. The GBA informs the brain through introception, our brain’s representation of sensations from our own bodies and the sensory consequence of this experienced activity (psychological science). The continuous internal changing of sensory conditions within the body informs the brain of the internal state and health of our body and is therefore of utmost importance in maintaining a homeostatic physiological balance in the body (Fig. 4).
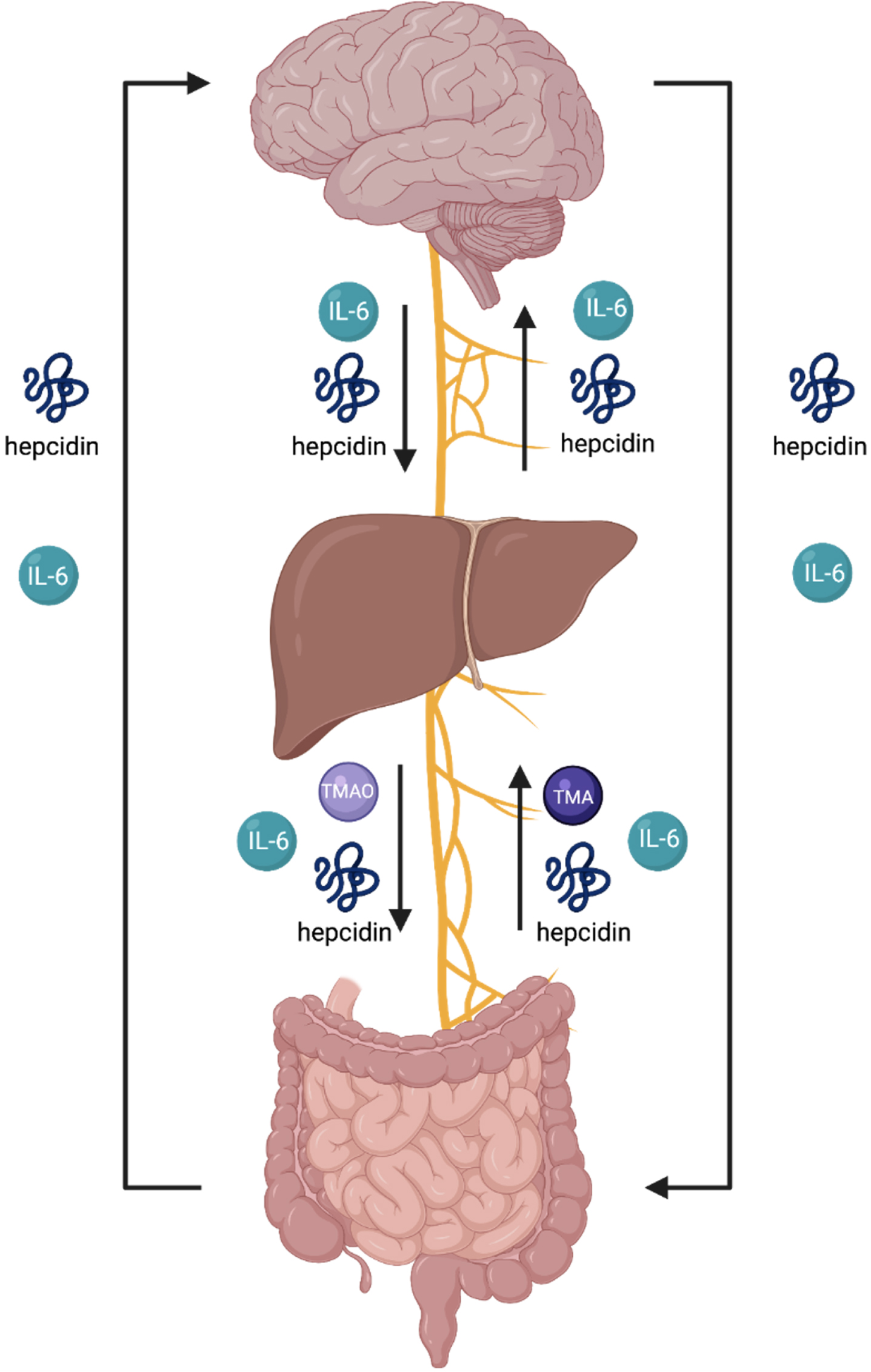
Fig. 4
The endocrine summary of the gut-brain axis (GBA) involves an endocrine and neural communication between the brain, liver and intestines via several biomolecules and the vagus nerve. The arrows in this figure indicate the flow of each biomolecule. An overproduction of TMAO signifies overdevelopment of pathogenic gut bacteria and induces activation of macrophages, onsetting the secretion of IL-6, thus, initiating collective activation of microcascade inflammatory responses to absorb bacteria in attempt to mediate gut dysbiosis. IL-6 is unregulated in inflammation and involved in the regulation of neural processes. As the liver produces hepcidin, the sustained overproduction of hepcidin and IL-6 is stimulated throughout blood circulation. The hepcidin ascending to the brain is derived from circulation and further expressed through the production of iron-load and inflammation. The brain’s reactivity in response to overproduction sequesters iron to neurons. The activation of hepcidin results in the shutdown of ferroportin in neural cells. The increase of iron in neural cells activates an oxidative state in the brain. The brain is highly susceptible to oxidative damage. Imbalance of gut dysbiosis, thus, plays a major role in the pathophysiology and pathogenic mechanism for neurodegenerative diseases as a primary contributor.
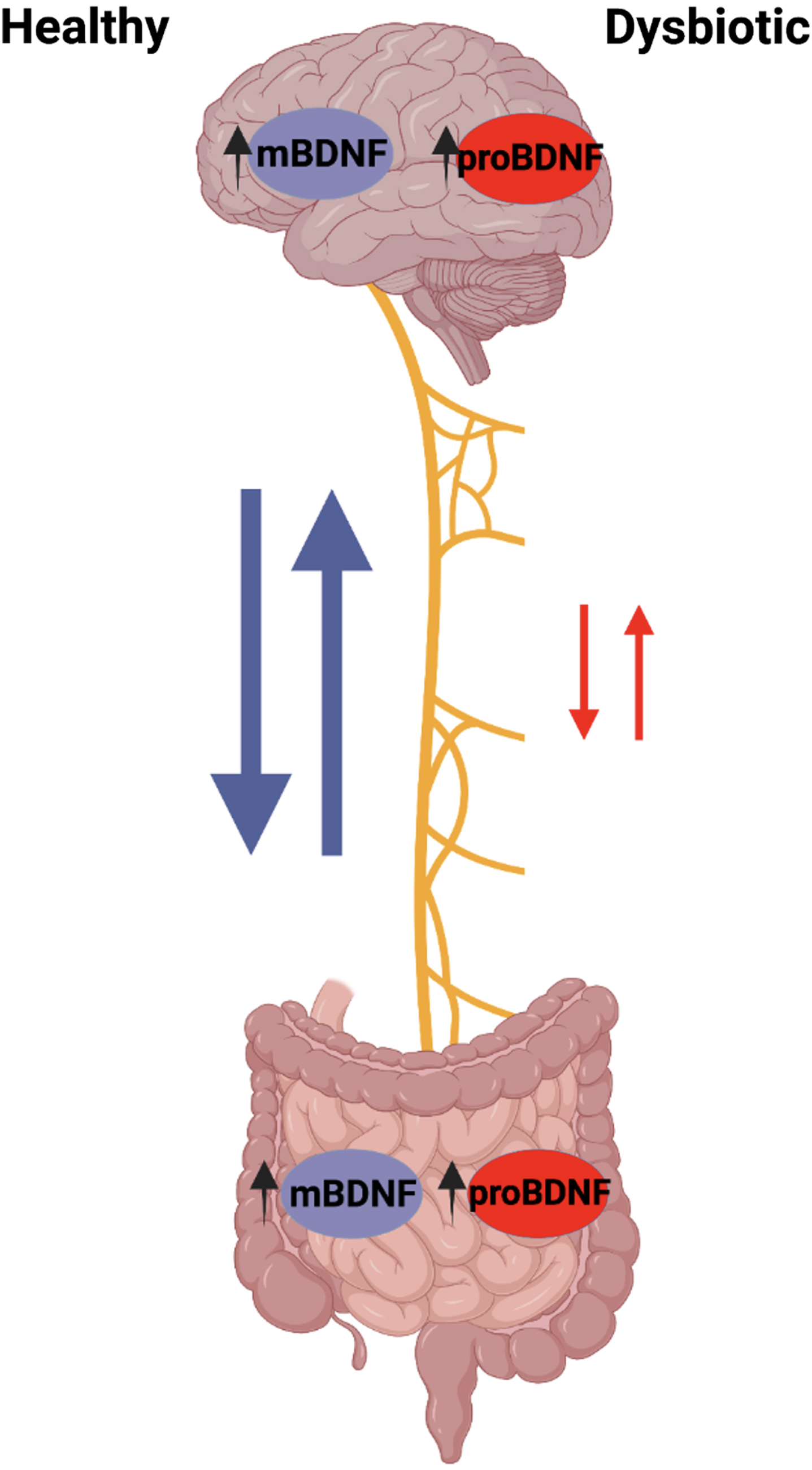
The GBA is becoming increasingly relevant in our study to understand the effects of GI microbiota on brain development and neurological diseases such as AD. In the brain, high levels of mBDNF can be found in the hippocampus, amygdala, cerebellum, and cerebral cortex with the highest levels found in hippocampal neurons [37, 38]. mBDNF is also expressed in other peripheral tissues, including that of the GI tract, some even higher levels than in the brain [39]. In our knowledge of the GBA, it may be interesting to see how levels of mBDNF in the gut may affect the mBDNF in the brain as well as the brain itself. The CNS communicates with microbiota in the brain-gut axis via the vagus nerve through cholinergic signaling. The vagus nerve afferent fibers can be directly or indirectly stimulated by microbiota through gut endocrine cells. These endocrine cells are activated by mBDNF or inhibited by pro-brain-derived neurotrophic factor (proBDNF), which in turn activate or inhibit vagal nerve communication to the brain. Furthermore, vagal nerve afferent fibers are able to stimulate efferent fibers through the inflammatory reflex. An increased presence of proBDNF, seen in the detriment of a HFHS diet, consequently affects the vagal nerve efferent cholinergic anti-inflammatory pathway, perpetuating further damage [40].
Across the brain, N-methyl-D-aspartate receptors (NMDAR) are important for regulating neuroplasticity and cognitive function. Due to this, mBDNF is believed to be associated with these NMDAR receptors [41]. In the absence of gut microbiota seen in germ-free mice, mBDNF levels in the CNS are reduced and thus inhibit the maintenance of NMDAR production. The reduced NMDAR input on GABA inhibitory interneurons causes the disinhibition of glutamatergic output, leading to many cognitive deficits [41]. This observation provides more evidence for gut microbiota influence on the brain through its ability to affect mBDNF expression.
Gut microbiota can modulate mBDNF expression and function in the brain; however, the exact mechanism is unclear. In one study, mBDNF levels are found to be lower in the cortex and hippocampus of germ-free mice compared to the controls, so GI microbiota does play some role in elevating brain mBDNF [42]. It is known that brain mBDNF is associated with learning and memory and is important for preserving cognitive abilities; however, little is known about how gut microbiota affects it through the GBA or even its relation to other peripheral mBDNF. Future studies are needed to explore gut mBDNF as it may uncover the unknown mechanism that bridges gut microbiota to mBDNF in the brain and may help in finding ways to prevent cognitive decline seen in many neurological diseases.
MBDNF: OVERVIEW
mBDNF is one of the most widely studied neurotrophic growth factors that is found in the brain and periphery. It is a key molecule that is involved with plastic changes in learning and memory. mBDNF is thought to help increase memory storage by helping stabilize long-term potentiation (LTP) through changes in spine morphology in neurons. mBDNF is encoded by the mBDNF gene and is synthesized as the precursor proBDNF, which then undergoes cleavage intra or extracellularly to produce the mature mBDNF (mBDNF) protein [43, 44]. The location for this cleavage is still unclear but is important because proBDNF and mBDNF have different functions. ProBDNF helps with long-term depression (LTD) induced by low-frequency stimulation, while mBDNF facilitates LTP induced by high frequency stimulation [45]. The expression of mBDNF is important for changes in synaptic plasticity and thus must be highly regulated.
mBDNF expression is highly regulated and is signaled through two membrane-bound receptors: p75 and TrkB [46]. ProBDNF binds with the p75 receptor to facilitate LTD and apoptosis. Mature mBDNF binds to tyrosine kinase receptors to promote cell survival and facilitate LTP. It has been found that when p75 is coexpressed with TrkB, and there is an increase in neurotrophin binding affinity, resulting in easier ligand discrimination [47]. ProBDNF is thought to help regulate mature mBDNF activity in non-pathological conditions. mBDNF has a complex genome that includes a range of promoters and at least eight 5’ exons and one 3’ exons. The promoters result in production and distribution of highly specific transcripts of mBDNF across the brain, which attributes to its diversity of functions in the brain including neuronal survival and differentiation [48]. Different levels of expressions for mBDNF and its precursor can be associated with differing degrees of affected cognitive abilities.
The mBDNF gene has a single nucleotide polymorphism that involves a valine to methionine amino acid substitution at position 66 (Val66Met). This impacts the packaging process of mature mBDNF to secretory vesicles and thus decreases its secretion and may relate to numerous psychiatric and neurological disorders such as schizophrenia [41]. Val66Met is correlated to lower serum mBDNF levels as well as volumetric decreases in specific brain regions such as the hippocampus, supporting the idea that mBDNF plays a role in memory performance and cognition. Numerous studies could not find an association between the Val66Met genotype and memory performance specifically, but the polymorphism may confer disadvantages in cognitive performance and episodic memory [49]. The mBDNF polymorphism has been a helpful comparison in our understanding of mBDNF’s effect on normal brain function and aging.
MBDNF AND THE BRAIN
Age is the major risk factor for the development of neurodegenerative diseases. Throughout the aging process, cognitive performance declines as synaptic plasticity is reduced in brain regions important for cognition. The most consistent deficit seen with aging is the decline in neurotrophic signaling, including mBDNF associated with LTP signaling. Therefore, it is inferred that age-related cognitive deficits can be correlated to the decrease in mBDNF expression in the affected brain regions seen in aging [50]. These decreased mBDNF levels are also accompanied by reductions in the expression and activation of tyrosine receptor kinase B (TrkB) receptors and the increase in levels of proBDNF and p75 associated with LTD and apoptosis [51]. TrkB receptors are activated by mBDNF and are an important part of a pathway that mediates cell survival and proliferation. The TrkB signaling pathway can be further divided into three main pathways: the MAPK/ERK pathway, the Pl3k/Akt pathway, and the PLC-γ pathway [52]. These three pathways work together to regulate important cognitive processes, explaining why decreased expression of mBDNF affects cognitive ability and function such as memory formation (Fig. 5).
Fig. 5
Normal mBDNF signaling pathway. mBDNF activates the TrkB signaling pathway which in turn activates the MAPK/ERK, PI3K/Akt, and PLC-γ pathways. These pathways work together to regulate important cognitive functions such as LTP formation and neurogenesis.
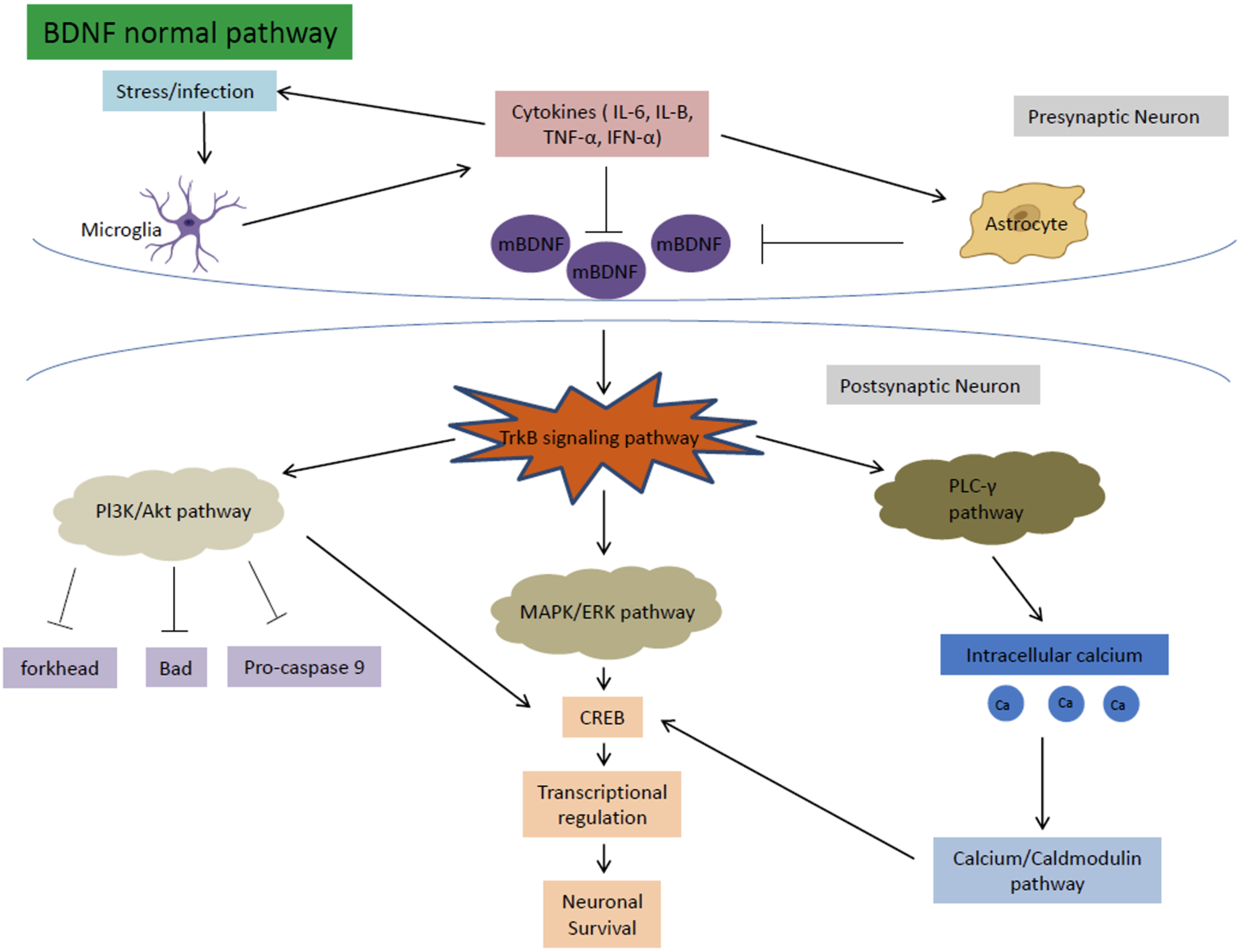
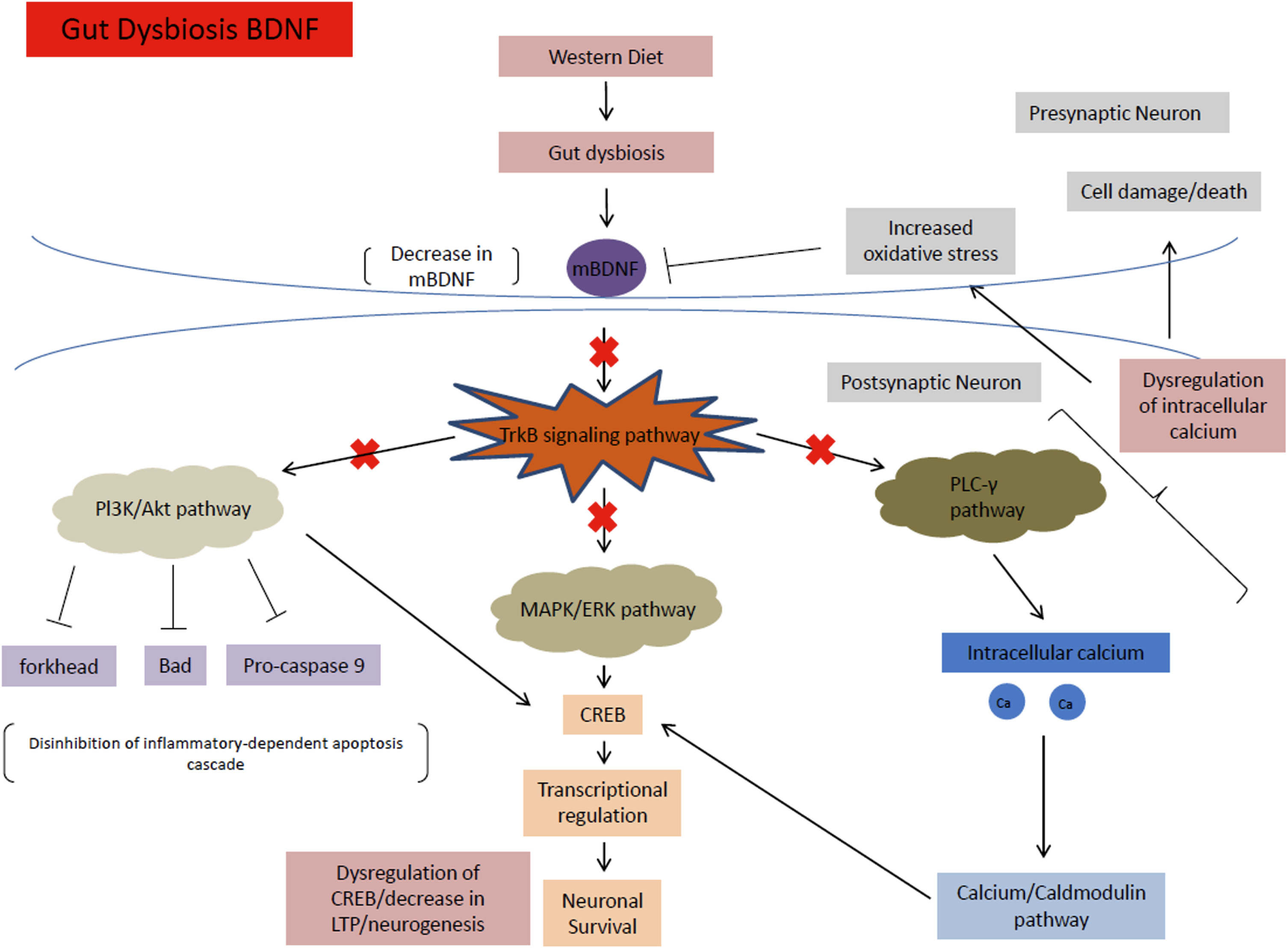
The MAPK/ERK pathway is responsible mainly for promoting neuronal growth, synaptic plasticity, cell survival, and differentiation [53]. The MAPK and ERK proteins regulate the cyclic AMP response element-binding protein (CREB), which is important for transcriptional regulation for a multitude of proteins associated with neuronal survival and LTP. In a HFHS diet, mBDNF is downregulated from gut dysbiosis. The decreased mBDNF downregulates the TrkB receptor and ultimately the MAPK/ERK pathway, leading to increased apoptosis and dysregulation of CREB.
The PI3k/Akt pathway is important for the inhibition of high glucose induced apoptosis. The pathway also reverses the effects of high glucose levels on the downregulation of synaptic plasticity-related proteins in hippocampal neurons [54]. Through the TrkB receptor, mBDNF upregulates the PI3K and Akt pathway to inhibit the forkhead, Bad, and Pro-caspase 9 apoptosis proteins [52]. PI3K/Akt also increases CREB activation to help with neuronal survival [54]. In a HFHS diet, decreased mBDNF causes the disinhibition of the PI3k/Akt pathway on apoptosis, leading to increased susceptibility for neuronal cell death.
The PLC-γ pathway is important for calcium signaling and synaptic plasticity [55]. The pathway is important for Ca2 + homeostasis. The levels of calcium ions maintained by the pathway activate kinases (calcium-calmodulin dependent kinases), that in turn activate transcription factors including CREB that help regulate neuroplasticity and neurogenesis [56]. In a HFHS diet, the downregulation of mBDNF disrupts the calcium ion homeostasis of the PLC-γ pathway causing a multitude of problems such as increased oxidative stress, apoptosis, and dysregulation of CREB and transcription factors that decrease LTP and neurogenesis (Fig. 6).
Fig. 6
mBDNF signaling pathway under HFHS diet conditions. This diet causes gut dysbiosis, which decreases levels of mBDNF. These decreased mBDNF levels downregulate the TrkB signaling pathway and ultimately the MAPK/ERK, PI3K/Akt, and PLC-γ pathways, causing a multitude of problems such as increased susceptibility to cell death, dysregulation of CREB and transcription factors, as well as a decrease in LTP and neurogenesis.
In addition to its role with synaptic plasticity, mBDNF is associated with other functions that may prove that decreased levels are detrimental to normal brain functioning. Mature mBDNF helps counteract noxious effects of neuronal cell death by exerting various trophic effects on hippocampal neurons [57]. In adult humans, high levels of mBDNF in the hippocampus have been seen to be related to survival and differentiation of dentate gyrus progenitor cells [58, 59]. In aged animals, low mBDNF levels have been associated with deficient neurogenesis [60]. More importantly, mBDNF has been seen to play a role in antioxidant defense during aging and increases with oxidative stress [50]. In one study with schizophrenia patients, it was found that both oxidative stress and decreased mBDNF levels may be implicated in the pathophysiology of the disease [58]. An inverse association between mBDNF and oxidative stress in schizophrenia could reflect a pathological mechanism affecting the mBDNF system to influence cognitive impairment [61]. In analyzing the brains of patients with psychiatric conditions and neurocognitive disorders, decreased mBDNF functionality has been found compared to healthy individuals, but it is still unclear whether or not this decrease is the direct cause of cognitive decline.
THE VAGUS NERVE AND ITS ROLE IN THE GUT-BRAIN AXIS
Up until this point, we have been reviewing the metabolic pathways of the brain, gut, and liver and the biomolecules that are produced in each as they relate to the pathophysiology of the HFHS diet and AD. Studying these pathways through the lens of an HFHS diet inducing an endocrine communication cascade involving TMA, TMAO, IL-6, hepcidin, and mBDNF and their effects on the brain, liver, small intestine, and large intestine collectively, reveals that there are still gaps in the literature with regard to understanding of how these organs and inflammatory signaling pathways communicate with each other in systemic metabolic disease. The literature hints that these same organs communicate via the vagus nerve, which has been shown to communicate directly with enteroendocrine cells [62].
The vagus nerve is responsible for modulating mBDNF expression and neurogenesis in the hippocampus while playing a pivotal role in gut microbiome to brain signaling which is significantly impacted by hippocampal neuroplasticity. mBDNF is known to promote neuronal survival, axonal guidance, and synaptic plasticity. A previous study determined the sensory processing of odorants is a dynamic process that requires plasticity at multiple levels [63]. For instance, in the olfactory bulb inhibitory interneurons undergo lifelong replacement through the process known as adult neurogenesis. Recent studies suggest vagal nerve activity influences neurogenesis and mBDNF mRNA expression in the adult hippocampus and therefore conducted a study verifying vagotomy decreased hippocampal neurogenesis [63, 64]. Although the mechanisms for this underlying process are not very clear, scientists discovered that a vagotomy decreases mBDNF, the neurotropic factor responsible for regulating adult hippocampal neurogenesis (Fig. 7).
Fig. 7
The neural summary of the gut-brain axis. Gut endocrine cells are activated by mature mBDNF or inhibited by proBDNF, which in turn activate or inhibit vagal nerve communication to the brain respectively. Additionally, afferent vagal nerve fibers stimulate efferent vagal nerve fibers via the inflammatory reflex.
The communication between the brain and microbiota is bidirectional through the stimulation of the neural pathways of the vagus nerve and through cytokines. Cytokine interleukins are key components of vagally-mediated immune-to-brain connection, thus novel mechanisms of future study as they serve a molecular link between the immune system and vagus nerve, and thus in the CNS. The vagus nerve stimulates interoceptive awareness through internal identification, recognition, and responses of microbiota metabolic signals through its visceral sensory afferents and motor efferents. The vagus nerve has anti-inflammatory properties in both its efferent and afferent fibers.
Yu and colleagues described neuropods, which are the specialized cells synapsing with nerves that sense mechanical, chemical and thermal stimuli of the gut lumen. Neuropod cells internally convert signals from stimuli into electrical pulses initiating propagation by synapsing onto afferent neurons of the vagus nerve. The brain and small intestine are linked by the vagal neurons carrying sensory information to the brain stem. The vagus nerve is responsible for sensing gut microbiota, transferring the gut information to the CNS for integration and generation of an adaptive or neuroinflammatory response [62]. The vagus nerve is thus responsible for neuron-to-neuron retrograde transmission in the enteric nervous system.
Where this becomes interesting would be the distal colon, which receives pelvic splanchnic nerve innervation instead of vagal nerve innervation. Not much is known about the neural connection between distal colon and the brain with respect to the GBA, but our preliminary results show that BDNF receptor presence as visualized with immunohistochemistry in APP mice on a HFHS diet in the distal colon was not statistically different between cohorts, but it was in the jejunum which does receive vagal innervation.
The vagus nerve is a critical component of the parasympathetic nervous system and operates through a cholinergic anti-inflammatory pathway [40]. The vagus nerve has anti-inflammatory properties in both its efferent and afferent fibers, and while these fibers are distributed throughout all layers of the digestive wall, they do not come in contact directly with gut microbiota [40]. Instead, the vagus nerve senses microbiota through diffusion of bacterial compounds or metabolites through its afferents, which are then integrated in the CNS to generate a response [40]. The communication between the microbiota and the brain is bidirectional and can occur through several different pathways, including neural through the vagus nerve and spinal cord, immune cytokines or metabolic signaling through different signaling molecules [40].
Macrophages and enterocytes are also involved in the sensation and detection of bacteria [65]. HFHS diets generate an over dominance of gram-negative bacteria of the microbiota and activate various responses. The negative bacteria circulating stimulates the vagus nerve to give afferent signals to the brain. The brain activated increased peristalsis in attempts to eliminate the bad bacteria. Enteroendocrine cells are also stimulating the vagus nerve assisting as support to fight the infection. Macrophages are cells engulfing pathogens which insist in the detection of phagocytosis and destruction of bacteria and necessary presentations of antigens to T-cells and initiate information by releasing molecules (cytokines, IL-6, mBDNF, TMA, TMAO) that activate other cells. Macrophages facilitate cytokine activation. In addition, the increased number of gram-negative bacteria produces TMA. TMA is considered toxic and thus adversely reacts with the liver causing an interference with liver production. As TMA is circulating through the blood stream it gets converted to TMAO. TMAO is pro inflammatory thus more TMAO circulating throughout the body will cause an increase in the secretion of IL-6 activating/driving inflammation in the body.
The liver’s recognition of bad bacteria because of increased TMA while simultaneously producing TMAO results in the overproduction of hepcidin. An increase of hepcidin sequesters more iron in the gut. TMAO is therefore dispatched to the gut to increase inflammation and sends hepcidin to the gut to evoke the enteroendocrine cells to retain the iron in the gut. However, hepcidin and IL-6 are not remaining in the gut as they are traveling throughout the enteroendocrine and cardiovascular system to the brain. An increase of IL-6 regulation in brain cells increases the inflammatory response and increases oxidative stress and neurodegeneration [66]. This gives rise to microglia activation as the brain assumes its inflamed and increases hepcidin. Hepcidin locks iron into all cells and hence neural cells. An increase of iron in neurons instigates the activation of the iron hypothesis to take effect. The overproduction of oxidative stress generates neuronal plaques and tangles which increase inflammation. APOE4 individuals are unable to handle the excessive amounts of iron production which onsets neurodegenerative diseases such as AD to develop.
In addition, the increase of IL-6 is causing a decrease in the amount of mBDNF produced. As mBDNF decreases and proBDNF increases, the resulting cascade terminates vagal nerve efferent fibers. Hence, the vagus nerve is unable to communicate with the gut in the efferent direction. Since mBDNF is also produced in the gut it thus cannot be activated in the afferent direction.
Bidirectionally the vagus nerve has been severed completely in both the afferent and efferent directions. This results in no communication happening between the brain, gut, and vagus nerve when normally the vagus nerve modulates all regulation. Thus, the only signaling remaining is the over-circulation sustained from iron overload as a result of endocrine dysfunction.
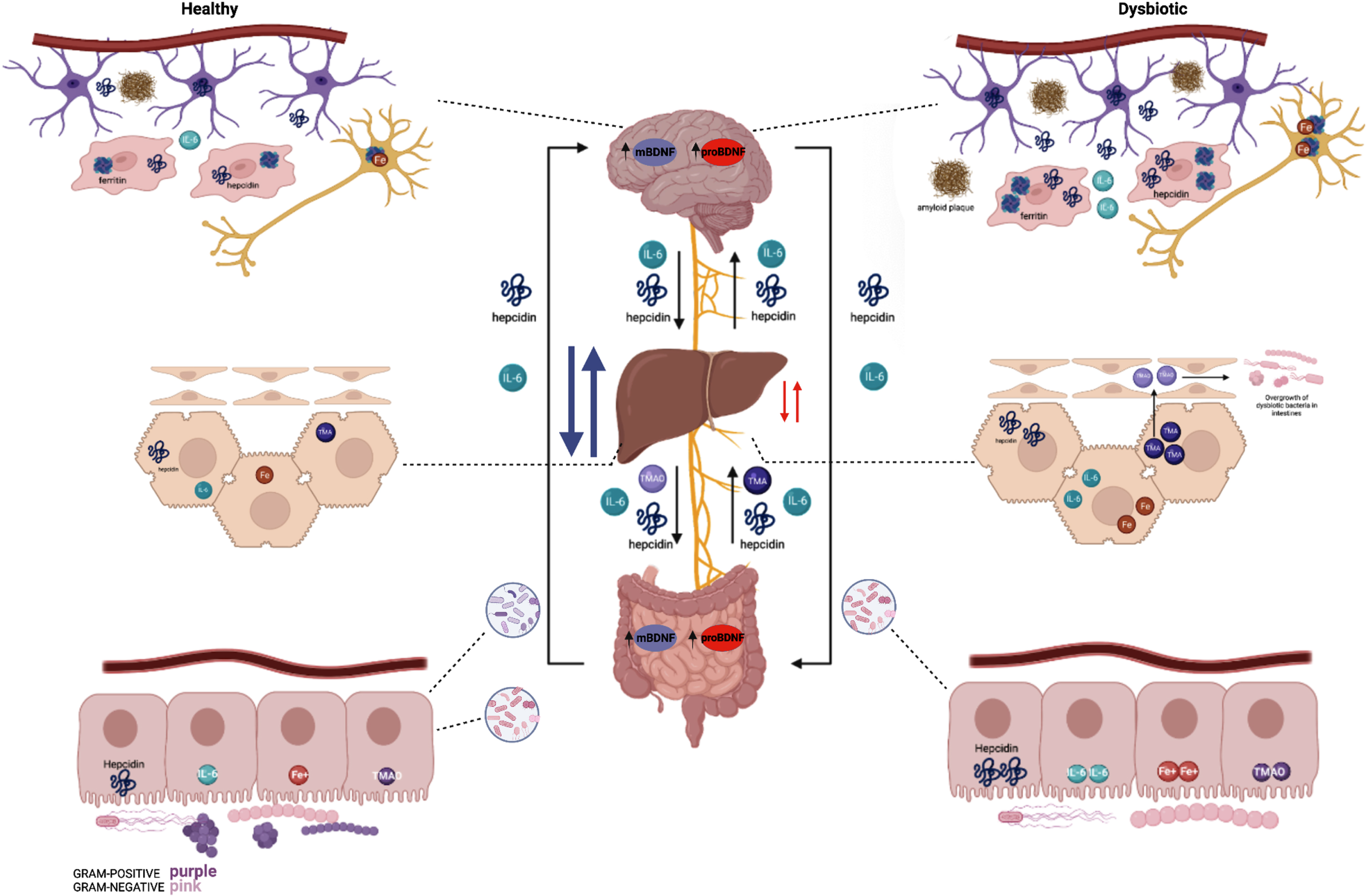
Enteroendocrine cells are located throughout the GI tract and have been shown to express voltage-gated sodium and calcium channels that interact with vagal afferents to modulate certain GI functions including motility, secretion, and food intake [40]. These cells are polymodal chemosensors that integrate both intrinsic and extrinsic signals within the gut and transmit this information to the nervous system [40]. Vagal chemoreceptors are also likely to be involved in the communication between the brain and gut microbiota through the sensation of short-chain fatty acids and gut hormones [40]. Vagal branches carry afferent satiety signals from the stomach and other areas of the GI tract to the brainstem and is an important modulator of appetite and food intake [67]. Vagal stimulation leads to a reduction in food intake, weight gain, and adipose tissue accumulation through increase CNS satiety signal [67]. We summarize the entire story of healthy versus dysbiotic states that regulated by the vagus nerve in Fig. 8.
Fig. 8
Summary of healthy (left) and dysbiotic (right) states of the gut-brain axis. The left region of the figure depicts the brain, liver, and gut at optimal health whereas the right region represents the brain, liver, and gut in dysbiotic states. The brain, in a state of a healthy microbiota, ferritin, hepcidin, IL-6, and iron are all at baseline levels. The liver in a healthy state contains baseline hepcidin, IL-6, and TMA levels which supports a balance of healthy and dysbiotic bacteria. Iron levels are slightly higher in the healthy state than a dysbiotic state as hepcidin is upregulated in a dysbiotic state. In addition, a healthy microbiota consists of a balance of Gram-positive and Gram-negative bacteria; in which hepcidin, iron (Fe3+), IL-6, and TMAO levels express a normal state of balance. The right region of the figure depicts the brain in a state of dysbiotic microbiota. The brain responds preventatively by upregulating ferritin and hepcidin levels, as well as IL-6. This is done via the astrocytes (purple) and microglia (red). In the neuron (dark yellow), iron is upregulated as well in the setting of neuroinflammation. Amyloid plaques can also be seen in the dysbiotic state as above. Below, the liver is illustrated in a state of dysbiosis. IL-6 is upregulated, and higher levels of TMA are sent to the liver to be oxidized to TMAO. TMAO is sent to the small intestines which ultimately causes inflammation and the overgrowth of dysbiotic bacteria. Most importantly, the dysbiotic microbiota indicates an increase of gram-negative bacteria and elevated hepcidin. Whereas a dysbiosis state indicates an increase of gram-negative bacteria and elevated hepcidin levels resulting in excessive iron accumulation. This stimulates high ferritin levels which promotes less cognitive decline and therefore excess hepcidin production. The synthesis of hepcidin is rapidly increased by infection and inflammation in which elevated IL-6 levels initiate a pro-inflammatory cascade. With IL-6 upregulation occurring in the liver, the higher levels of TMA become oxidized to TMAO. Circulation of TMAO initiated in the small intestine initiates an inflammatory state.
ADDITIONAL CONDITIONS THAT MAY AFFECT THE GUT-BRAIN AXIS
Several factors can alter the communication between the gut microbiome and the brain. Stress inhibits the vagus nerve by acting on G protein-coupled receptors in the brain and GI tract via corticotrophin-releasing factor and has deleterious effects on the gut microbiota as it counteracts its anti-inflammatory nature [40]. Stress-induced vagal nerve inhibition increases intestinal permeability and favors gut dysbiosis [40]. A low vagal tone has been studied and characterized in conditions including irritable bowel syndrome and irritable bowel disease, as well as a state of gut dysbiosis and low-grade inflammation in the GI tract [40]. Experimental studies with rat and human models show that increased sympathetic nervous system (SNS) activity results in increased lipolysis rate in adipose tissue, and that lipid accumulation could be due to reduced SNS activity in obese models rather than increased parasympathetic nervous activity [67].
Bariatric surgery and its effects on the GBA communication has been closely studied through the lens of gut microbiota and hormone shifts [67]. Also referred to as metabolic surgery, studies have shown sustainable weight loss, hypertension remission, remission of sleep apnea, and improvements in glycemic control and reduction of triglyceride levels following surgery [67]. Bariatric surgery has been hypothesized to have several effects on the nervous system, including potentially reducing SNS activity and significant downregulation of mBDNF in the hippocampus in patients with following surgery that displayed problematic eating behaviors with weight regain [67].
Metabolic surgery also has implications on the gut microbiota, which could ultimately impact the GBA as previously described. Over 75% of patients with severe obesity show low microbial gene richness which is associated with increased trunk fat-mass and metabolic comorbidities including type-II diabetes mellitus [67]. Patients diagnosed with obesity typically have a disrupted gut microbiome, reduced expression of tight junction proteins, increased gut permeability, increased leakage of lipopolysaccharides and bacterial metabolites (short-chain fatty acids, TMAO) into systemic circulation, mucosal inflammation, and bacterial translocation [67]. Following metabolic surgery, microbiota composition shifts occur including decreases in the abundance of Firmicutes and Actinobacteria, increases in Bacteroides, Proteobacteria, and phylum Verrucomicrobia, and overall increases in gut microbial diversity [67]. Increases in Bifidobacterium and Lactobacilli have been associated with increased vagal stimulation and reduced LPS-induced inflammation which ultimately enhances gut barrier function [67]. This shift in gut microbial composition both directly and indirectly influences the CNS as it has been shown that patients irritable bowel syndrome had decreased anxiety and stress, decreases in food cravings and depression, and increases in satiety and self-esteem following bariatric surgery with altered gut microbiome profiles [67]. In addition to a direct shift to the gut microbiome, an indirect food preference shift following surgery has been observed in patients that typically follow a pattern of low high-fat and high-sugar foods, which can impact the diversity of the gut microbiome and its impact on the brain as previously discussed in this review [67].
Experimentally, this axis opens an opportunity for further research regarding how the human gut microbiome is affecting these pathways individually, and altogether as an axis. Further research in microbiology can shed light on the processes that gut microbiota are undergoing in the setting of inflammation and oxidative stress, and how this can ultimately shift the production of mBDNF in the brain. Future studies in neuroscience can explore the implications of mBDNF and diet, and how gut microbiota composition and TMAO production affect other processes like learning and forming memories. Genomic scientists can further study the development of screening systems and how levels of TMAO, hepcidin, and other microbiota byproducts could shed light on disease detection. This multisystem, multi organismic interplay between the gut, brain, and liver is a complicated process that requires several disciplines in science to come together to formulate a better understanding of what is truly happening in the body as a result of TMAO, hepcidin, mBDNF, IL-6, and gut microbiota.
CONCLUSION
The gut microbiome was once considered a small component of the human body but as research emerges, its critical importance in human health is becoming more clear. A HFHS diet has been shown to provide precursor biomolecules that enable the overgrowth of pathogenic bacteria to produce TMAO, which can have a number of negative systemic effects on the body. While TMAO has been in the spotlight for years, it may not be the only contributing factor to the continued state of gut dysbiosis. We reviewed that IL-6 is often related to inflammatory and pathological situations and is therefore a factor that contributes decisively in the normal function of the brain. IL-6 can activate GI submucosal neurons through the GBA. Furthermore, mBDNF has become a more widely studied protein that plays a role in learning and memory. mBDNF is highly regulated by the p75 and TrkB receptors. However, gut microbiota has also been seen to modulate the mBDNF expression in the brain suggesting that the GBA may play a large role in regulating this important protein. The exact mechanism is still unclear, so more research is needed to study brain mBDNF as well as peripheral mBDNF in relation to the brain-gut axis. Excess iron levels in the gut provide an advantageous niche for pathogenic bacteria to dominate, which could be ameliorated by two different mechanisms of two different hepcidin molecules. Hepcidin derived from the liver could be combating inflammation in the body by sequestering iron in the brain and other areas of the body to prepare as a response to gut dysbiosis, versus dendritic-cell-derived hepcidin that may acting as the “first responder” in the intestinal tract, directly sequestering iron at the level of the mucosa to starve pathogens from their primary food source altogether. The microbiota-induced inflammation and oxidative stress is to be seen as a systemic condition rather than just a locally destructive event that occurs in the gut, and the role in hepcidin is critical in returning the body back to homeostasis to combat neuroinflammation. We still do not know the detailed mechanistic aspects of iron-induced regulation of hepcidin expression in brain cells; however, these two distinct variations of hepcidin molecules both contribute to a “healing” role that could prevent AD and other neurologic disease pathogenesis. Future side-by-side studies with both dendritic-cell-derived and hepatocyte-derived hepcidins must be conducted to solidify the roles of each through a lens of a HFHS diet and neurologic disease development and progression.
ACKNOWLEDGMENTS
The authors wish to thank Boston University Aram V. Chobanian & Edward Avedisian School of Medicine benefactors that contributed to the Department of Anatomy and Neurobiology Start-up Fund that supports undergraduate and graduate research, the John E. and Sarah M. McGinty Foundation, and the Campbell Foundation.
FUNDING
Department of Anatomy and Neurobiology Start-up Fund; John E. and Sarah M. McGinty Foundation; Campbell Foundation
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose with this work.
REFERENCES
[1] | Singh RK , Chang HW , Yan D , Lee KM , Ucmak D , Wong K , Abrouk M , Farahnik B , Nakamura M , Zhu TH , Bhutani T , Liao W ((2017) ) Influence of diet on the gut microbiome and implications for human health. J Transl Med 15: , 73. |
[2] | Wang WL , Xu SY , Ren ZG , Tao L , Jiang JW , Zheng SS ((2015) ) Application of metagenomics in the human gut microbiome. World J Gastroenterol 21: , 803–814. |
[3] | Gaines S , Williamson AJ , Hyman N , Kandel J ((2018) ) How the microbiome is shaping our understanding of cancer biology and its treatment. Semin Colon Rectal Surg 29: , 12–16. |
[4] | Akbar N , Khan NA , Muhammad JS , Siddiqui R ((2022) ) The role of gut microbiome in cancer genesis and cancer prevention. Health Sci Rev 2: , 100010. |
[5] | Shapira I , Sultan K , Lee A , Taioli E ((2013) ) Evolving concepts: How diet and the intestinal microbiome act as modulators of breast malignancy. ISRN Oncol 2013: , 693920. |
[6] | Guo T , Zhang D , Zeng Y , Huang TY , Xu H , Zhao Y ((2020) ) Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener 15: , 40. |
[7] | Paul B , Barnes S , Demark-Wahnefried W , Morrow C , Salvador C , Skibola C , Tollefsbol TO ((2015) ) Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin Epigenetics 7: , 112. |
[8] | Houser MC , Tansey MG ((2017) ) The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis 3: , 3. |
[9] | Beam A , Clinger E , Hao L ((2021) ) Effect of diet and dietary components on the composition of the gut microbiota. Nutrients 13: , 2795. |
[10] | Bercik P , Denou E , Collins J , Jackson W , Lu J , Jury J , Deng Y , Blennerhassett P , Macri J , McCoy KD , Verdu EF , Collins SM ((2011) ) The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice.. Gastroenterology 141: , 599–-609, 609.e1-3. |
[11] | Senghor B , Sokhna C , Ruimy R , Lagier JC ((2018) ) Gut microbiota diversity according to dietary habits and geographical provenance. Hum Microb J 7-8: , 1–9. |
[12] | Conlon MA , Bird AR ((2014) ) The impact of diet and lifestyle on gut microbiota and human health. Nutrients 7: , 17–44. |
[13] | Noble EE , Hsu TM , Kanoski SE ((2017) ) Gut to brain dysbiosis: Mechanisms linking western diet consumption, the microbiome, and cognitive impairment. Front Behav Neurosci 11: , 9. |
[14] | Chung YC , Kruyer A , Yao Y , Feierman E , Richards A , Strickland S , Norris EH ((2016) ) Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the β-amyloid fibrinogen interaction. J Thromb Haemost 14: , 1442–1452. |
[15] | Stampfer MJ , Malinow MR ((1995) ) Can lowering homocysteine levels reduce cardiovascular risk? N Engl J Med 332: , 328–329. |
[16] | Gao H , Xu H , Zhang J , Premaratne S , Su X , Zhang W , Wang S , Sun L , Yao J , Hao B , Yang T ((2021) ) Association of high serum homocysteine levels and severe chronic venous disease. Ann Vasc Surg 74: , 315–320. |
[17] | Makhmudovich AM , Sattarovna RG , Maksudovna VN , Azamatovich JS ((2021) ) Hyperhomocysteinemia and pathogenetic mechanisms of ischemic stroke. Am J Med Sci Pharm Res 3: , 66–76. |
[18] | Polage CR ((2013) ) Good and bad bacteria fight for iron in the gut.. Sci Transl Med 5: , 199ec140. |
[19] | Chen K , Zheng X , Feng M , Li D , Zhang H ((2017) ) Gut microbiota-dependent metabolite trimethylamine n-oxide contributes to cardiac dysfunction in western diet-induced obese mice. Front Physiol 8: , 139. |
[20] | Vogt NM , Romano KA , Darst BF , Engelman CD , Johnson SC , Carlsson CM , Asthana S , Blennow K , Zetterberg H , Bendlin BB , Rey FE ((2018) ) The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res Ther 10: , 124. |
[21] | Kiela PR , Ghishan FK ((2016) ) Physiology of intestinal absorption and secretion. Best Pract Res Clin Gastroenterol 30: , 145–159. |
[22] | Kortman GAM , Raffatellu M , Swinkels DW , Tjalsma H ((2014) ) Nutritional iron turned inside out: Intestinal stress from a gut microbial perspective. FEMS Microbiol Rev 38: , 1202–1234. |
[23] | Mills E , Dong XP , Wang F , Xu H ((2010) ) Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med Chem 2: , 51–64. |
[24] | Erta M , Quintana A , Hidalgo J ((2012) ) Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci 8: , 1254–1266. |
[25] | Schmidt PJ ((2015) ) Regulation of iron metabolism by hepcidin under conditions of inflammation. J Biol Chem 290: , 18975–18983. |
[26] | Abreu R , Quinn F , Giri PK ((2018) ) Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv 2: , 1089–1100. |
[27] | Parmanand BA , Kellingray L , Le Gall G , Basit AW , Fairweather-Tait S , Narbad A ((2019) ) A decrease in iron availability to human gut microbiome reduces the growth of potentially pathogenic gut bacteria; an colonic fermentation study. J Nutr Biochem 67: , 20–27. |
[28] | Roy CN , Andrews NC ((2005) ) Anemia of inflammation: The hepcidin link. Curr Opin Hematol 12: , 107–111. |
[29] | Bessman NJ , Mathieu JRR , Renassia C , Zhou L , Fung TC , Fernandez KC , Austin C , Moeller JB , Zumerle S , Louis S , Vaulont S , Ajami NJ , Sokol H , Putzel GG , Arvedson T , Sockolow RE , Lakhal-Littleton S , Cloonan SM , Arora M , Peyssonnaux C , Sonnenberg GF ((2020) ) Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 6487: , 186–189. |
[30] | Jeong SY , David S ((2003) ) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem 278: , 27144–27148. |
[31] | Vela D ((2018) ) The dual role of hepcidin in brain iron load and inflammation. Front Neurosci 12: , 740. |
[32] | Wu J , Hua Y , Keep RF , Nakamura T , Hoff JT , Xi G ((2003) ) Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 34: , 2964–2969. |
[33] | Ward RJ , Zucca FA , Duyn JH , Crichton RR , Zecca L ((2014) ) The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 13: , 1045–1060. |
[34] | Parker A , Fonseca S , Carding SR ((2020) ) Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 11: , 135–157. |
[35] | Hershko DD , Robb BW , Luo G , Hasselgren PO ((2002) ) Multiple transcription factors regulating the IL-6 gene are activated by cAMP in cultured Caco-2 cells. Am J Physiol Regul Integr Comp Physiol 283: , R1140–R1148. |
[36] | Sun J , Pan X , Christiansen LI , Yuan XL , Skovgaard K , Chatterton DEW , Kaalund SS , Gao F , Sangild PT , Pankratova S ((2018) ) Necrotizing enterocolitis is associated with acute brain responses in preterm pigs. J Neuroinflammation 15: , 180. |
[37] | Hofer M , Pagliusi SR , Hohn A , Leibrock J , Barde YA ((1990) ) Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 9: , 2459–2464. |
[38] | Timmusk T , Palm K , Metsis M , Reintam T , Paalme V , Saarma M , Persson H ((1993) ) Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10: , 475–489. |
[39] | Biddinger JE , Fox EA ((2014) ) Reduced intestinal brain-derived neurotrophic factor increases vagal sensory innervation of the intestine and enhances satiation. J Neurosci 34: , 10379–10393. |
[40] | Bonaz B , Bazin T , Pellissier S ((2018) ) The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci 12: , 49. |
[41] | Maqsood R , Stone TW ((2016) ) The gut-brain axis, BDNF, NMDA and CNS disorders. Neurochem Res 41: , 2819–2835. |
[42] | Sudo N , Chida Y , Aiba Y , Sonoda J , Oyama N , Yu XN , Kubo C , Koga Y ((2004) ) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558: , 263–275. |
[43] | Lee R , Kermani P , Teng KK , Hempstead BL ((2001) ) Regulation of cell survival by secreted proneurotrophins. Science 294: , 1945–1948. |
[44] | Mowla SJ , Farhadi HF , Pareek S , Atwal JK , Morris SJ , Seidah NG , Murphy RA ((2001) ) Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem 276: , 12660–12666. |
[45] | Nagappan G , Zaitsev E , Senatorov VV Jr , Yang J , Hempstead BL , Lu B ((2009) ) Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc Natl Acad Sci U S A 106: , 1267–1272. |
[46] | Chao MV ((2003) ) Neurotrophins and their receptors: A convergence point for many signaling pathways. Nat Rev Neurosci 4: , 299–309. |
[47] | Bibel M , Hoppe E , Barde YA ((1999) ) Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J 18: , 616–622. |
[48] | Adachi N , Numakawa T , Richards M , Nakajima S , Kunugi H ((2014) ) New insight in expression, transport, and secretion of brain-derived neurotrophic factor: Implications in brain-related diseases. World J Biol Chem 5: , 409–428. |
[49] | Tsai SJ , Gau YT , Liu ME , Hsieh CH , Liou YJ , Hong CJ ((2008) ) Association study of brain-derived neurotrophic factor and apolipoprotein E polymorphisms and cognitive function in aged males without dementia. Neurosci Lett 433: , 158–162. |
[50] | Mattson MP , Maudsley S , Martin B ((2004) ) BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 27: , 589–594. |
[51] | Miranda M , Morici JF , Zanoni MB , Bekinschtein P ((2019) ) Brain-derived neurotrophic factor: A key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci 13: , 363. |
[52] | Wurzelmann M , Romeika J , Sun D ((2017) ) Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen Res 12: , 7–12. |
[53] | Chen T , Wu Y , Wang Y , Zhu J , Chu H , Kong L , Yin L , Ma H ((2017) ) Brain-derived neurotrophic factor increases synaptic protein levels via the MAPK/Erk signaling pathway and Nrf2/Trx axis following the transplantation of neural stem cells in a rat model of traumatic brain injury. Neurochem Res 42: , 3073–3083. |
[54] | Zhong Y , Zhu Y , He T , Li W , Li Q , Miao Y ((2018) ) Brain-derived neurotrophic factor inhibits hyperglycemia-induced apoptosis and downregulation of synaptic plasticity-related proteins in hippocampal neurons via the PI3K/Akt pathway. Int J Mol Med 43: , 294–304. |
[55] | Numakawa T , Kumamaru E , Adachi N , Yagasaki Y , Izumi A , Kunugi H ((2009) ) Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc Natl Acad Sci U S A 106: , 647–652. |
[56] | Gulyaeva NV ((2017) ) Interplay between brain BDNF and glutamatergic systems: A brief state of the evidence and association with the pathogenesis of depression. Biochemistry (Moscow) 82: , 301–307. |
[57] | Almeida RD , Manadas BJ , Melo CV , Gomes JR , Mendes CS , Grãos MM , Carvalho RF , Carvalho AP , Duarte CB ((2005) ) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 12: , 1329–1343. |
[58] | Pencea V , Bingaman KD , Wiegand SJ ((2001) ) Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci 21: , 6706–6717. |
[59] | Shetty AK , Rao MS , Hattiangady B , Zaman V , Shetty GA ((2004) ) Hippocampal neurotrophin levels after injury: Relationship to the age of the hippocampus at the time of injury. J Neurosci Res 78: , 520–532. |
[60] | Apple DM , Solano-Fonseca R , Kokovay E ((2017) ) Neurogenesis in the aging brain. Biochem Pharmacol 141: , 77–85. |
[61] | Zhang XY , Chen DC , Tan YL , Tan SP , Wang ZR , Yang FD , Okusaga OO , Zunta-Soares GB , Soares JC ((2015) ) The interplay between BDNF and oxidative stress in chronic schizophrenia. Psychoneuroendocrinology 51: , 201–208. |
[62] | Yu CD , Xu QJ , Chang RB ((2020) ) Vagal sensory neurons and gut-brain signaling. Curr Opin Neurobiol 62: , 133–140. |
[63] | Bath KG , Akins MR , Lee FS ((2012) ) BDNF control of adult SVZ neurogenesis. Dev Psychobiol 54: , 578–589. |
[64] | O’Leary OF , Ogbonnaya ES , Felice D , Levone BR , Conroy CL , Fitzgerald P , Bravo JA , Forsythe P , Bienenstock J , Dinan TG , Cryan JF ((2018) ) The vagus nerve modulates BDNF expression and neurogenesis in the hippocampus. Eur Neuropsychopharmacol 28: , 307–316. |
[65] | Han X , Ding S , Jiang H , Liu G ((2021) ) Roles of macrophages in the development and treatment of gut inflammation. Front Cell Dev Biol 9: , 625423. |
[66] | Penkowa M , Molinero A , Carrasco J , Hidalgo J ((2001) ) Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience 102: , 805–818. |
[67] | Martinou E , Stefanova I , Iosif E , Angelidi AM ((2022) ) Neurohormonal changes in the gut-brain axis and underlying neuroendocrine mechanisms following bariatric surgery. Int J Mol Sci 23: , 3339. |


