
Repetitive Transcranial Magnetic Stimulation Improves Brain-Derived Neurotrophic Factor and Cholinergic Signaling in the 3xTgAD Mouse Model of Alzheimer’s Disease
Abstract
Background:
Alzheimer’s disease (AD) is a debilitating disorder involving the loss of plasticity and cholinergic neurons in the cortex. Pharmaceutical treatments are limited in their efficacy, but brain stimulation is emerging as a treatment for diseases of cognition. More research is needed to determine the biochemical mechanisms and treatment efficacy of this technique.
Objective:
We aimed to determine if forebrain repetitive transcranial magnetic stimulation can improve cortical BDNF gene expression and cholinergic signaling in the 3xTgAD mouse model of AD.
Methods:
Both B6 wild type mice and 3xTgAD mice aged 12 months were given daily treatment sessions for 14 days or twice weekly for 6 weeks. Following treatment, brain tissue was extracted for immunological stains for plaque load, as well as biochemical analysis for BDNF gene expression and cholinergic signaling via acetylcholinesterase and choline acetyltransferase ELISA assays.
Results:
For the 3xTgAD mice, both 14 days and 6 weeks treatment regimens resulted in an increase in BDNF gene expression relative to sham treatment, with a larger increase in the 6-week group. Acetylcholinesterase activity also increased for both treatments in 3xTgAD mice. The B6 mice only had an increase in BDNF gene expression for the 6-week group.
Conclusion:
Brain stimulation is a possible non-invasive and nonpharmaceutical treatment option for AD as it improves both plasticity markers and cholinergic signaling in an AD mouse model.
INTRODUCTION
According to the World Health Organization, deaths by neurodegenerative disorders will soon surpass that of cancer and will become the second leading cause of death [1]. Among these brain disorders, Alzheimer’s disease (AD) is the most common cause of dementia and disability. In addition to amyloid plaques and neurofibrillary tangles, the number of synaptic densities, dendritic spines, and neuronal activity is drastically reduced in the AD brain. The underlying cause of this pathology is yet to be determined, and treatment options focusing on pharmaceutical interventions have limited effectiveness [2, 3]. For this reason, any therapeutic strategy with the ability to delay AD progression could make a lasting positive impact on the lives of affected individuals. Just the delay of disease onset by 5 years would substantially reduce the prevalence for those who would convert [4].
The cholinergic system is notably disrupted in AD, and thus cholinergic modulators serve as one primary pharmaceutical intervention [5] with some success in improving cognitive impairment in patients with AD [6]. Specifically, cholinergic loss in the forebrain and cortex has been well documented in AD and linked to disruptions in episodic memory and executive function early within the progression of the disease [7]. While cholinesterase inhibitors may provide symptom relief by decreasing the degradation of acetylcholine, they do not address the underlying cause in the loss of cholinergic function [2, 8]. Therefore, other treatment strategies that can maintain cholinergic neural health over simply increasing the circulation of acetylcholine may prove to be more fruitful.
One important component in maintaining cholinergic cell function and plasticity is brain-derived neurotrophic factor (BDNF) [9]. There has been documented evidence for a loss of regulation in frontal BDNF in patients with AD [10] and in mouse models of AD [11]. Indeed, BDNF has been implicated as an underlying mechanism of learning and memory, as well as brain health [12], especially within the realm of neurodegenerative disease [13]. Given that BDNF is implicated in maintaining the health of forebrain cholinergic cells and overall memory performance, and both of these have decreased signaling with AD [14], it is imperative to investigate treatment mechanisms that can mitigate these physiological alterations.
One promising new avenue of therapeutics for neurocognitive health is noninvasive brain stimulation, such as repetitive transcranial magnetic stimulation (rTMS). This technique modulates neural excitability and transmitter release through application of an alternating magnetic field to the head. Clinical trials have begun to show improved language [15, 16] and memory [17] in patients with AD and mild cognitive impairment (MCI). However, although there has been some insights into how rTMS influences brain function, this has yet to be fully investigated [18]. rTMS is thought to induce changes in plasticity primarily through BDNF [19, 20]. This technique is believed to influence cognition and BDNF signaling in aged mice [21] as well as cognitive function and cholinergic activity in a rat model of vascular dementia [20, 22]. Therefore, it is possible that rTMS improves cholinergic and neurotrophic signaling in AD. Thus, we aimed to determine such mechanisms in a mouse model of AD, where direct biochemical measurements can be made.
The aim of this study was to test the hypothesis that rTMS will alter BDNF expression and cholinergic signaling in the 3xTgAD mouse model of AD. The female 3xTgAD mouse readily exhibits both plaques and tangles as well as cognitive dysfunction by 12 months of age or sooner [23] and is therefore a relevant model in which to test this hypothesis. rTMS provides direct stimulation to cortical areas of the brain and could be expected to directly modulate cortical function [24]. We therefore focused our analyses on cortical samples. Additionally, although most treatment regimens are provided daily, some clinical trials show a delayed effect in cognition [25], while other show a benefit for either 4 weeks or 2 weeks of stimulation [26]. Treatment timing has yet to be fully explored and increasing the temporal allocation between treatment sessions may allow more time for the brain to respond to stimulation. Therefore, we completed two treatment protocols, one daily treatment for 2 weeks and one twice-weekly treatment over 6 weeks. Treating mice daily for 14 days fit with previous mouse rTMS protocols [24, 27, 28], whereas twice weekly for six weeks is designed to recapitulate clinical practice where patients cannot come to the clinic everyday but are encouraged to finish their regimen by six weeks [29]. This allowed us to test the secondary hypothesis that spacing treatments results in augmented biochemical effects compared to daily treatment.
MATERIALS AND METHODS
Stimulation
We used 12-month-old 3xTgAD mice and their B6129SF2/J wild type (B6) controls (Jackson Laboratories) for our experiments. All procedures were carried out in accordance with the approved guidelines of the institutional and animal care and use committee at the Department of Veterans Affairs, Palo Alto. Mice were housed in groups in a temperature and humidity-controlled environment that was maintained on a 12-h light/dark cycle, with ad libitum access to food and water. Analyses involving different brain regions were carried out on the same mice rather than having a different mouse per region.
12-month-old female 3xTgAD mice and their wild type B6 controls first underwent surgery to attach a coil support to the skull. This support is used to place the rTMS coil for stimulation in freely moving mice. The surgical procedure follows that of Madore et al. [24]. Briefly, mice are anesthetized using 3% isoflurane, and a 2–3 cm incision is made into the scalp. The peritoneum is scraped away, and the coil support is attached to the skull in a fixed location over Bregma as the frontal stimulation site via LockTite glue and dental cement. The skin was sutured together around the coil support, and the mice were allowed to recover for 5 days following surgery. Mice were then habituated to the coil over the course of three days prior to beginning the stimulation protocol. For stimulation, the coil is placed over the coil support and clipped into place. Stimulation ran at 10 Hz, and 20 V for 10 min. For the sham condition, the coil was attached to the coil support for 10 min but no stimulation was administered. Mice were stimulated every day for 2 weeks, or twice a week for 6 weeks. The total number of mice used in this study was 103: B6 rTMS 2 weeks = 15, B6 Sham 2 weeks = 13, B6 rTMS 6 weeks = 14, B6 Sham 6 weeks = 12, 3xTgAD rTMS 2 weeks = 12, 3xTgAD Sham 2 weeks = 12, 3xTgAD rTMS 6 weeks = 14, 3xTgAD Sham 6 weeks = 11. The 3xTgAD model produces fewer mice per litter and are less likely to live a full 12 months thus we had slightly less 3xTgAD mice than B6 for this study.
After finishing the stimulation protocol, we collected vaginal samples to determine estrus cycle stage of each mouse via vaginal cytology. One day after the end of the experimental protocol, mice were perfused with PBS and the brains extracted. The frontal and temporal cortices were separated, and flash frozen on dry ice. Alternatively, the brain was fixed in 4% paraformaldehyde for 48 h and transferred to 30% sucrose prior to slicing at 30μm on a cryostat.
A small number of 3xTgAD mice (n = 4) received daily injections of the TrkB blocker ANA12 (Sigma) at 0.5 mg/kg or vehicle control of saline 0.5% DMSO, 2 h prior to two weeks of rTMS or sham treatment. ANA12 was first dissolved to 5 mg/mL in DMSO at 60°C and then further diluted to 50μg/mL for injection. Other than the daily injections, these mice were treated the same as the 2-week stimulation group. Only the 3xTgAD mice were utilized for this condition because our results (below) showed that this was the only group that had consistent effects on of rTMS on both BDNF and acetylcholinesterase (AChE) measurements.
Additionally, we did all biochemistry analyses on a set of 4 naïve mice from each group to measure any baseline differences between the 3xTgAD and B6 mice that exist without the perturbations of surgery.
Immunohistochemistry
To ensure that the 3xTgAD mice did develop plaques and tangles, we performed a co-stain for Aβ and phospho-tau using Aβ peptides antibody (1:1200, Cell Signaling) and AT8 (1:1200, ThermoFisher), respectfully. Briefly, 30μM floating slices from the entire brain were rinsed in PBS prior to permeabilization in PBS containing 0.3% triton and blocking in 10% goat serum. Slices were incubated in the primary antibodies overnight at 4°C for 24 h, rinsed in PBS, and the secondary antibodies were applied for 1 hr at room temperature. DAPI was added at final rinse, then the slices were mounted and coverslipped (Fluoromount, Invitrogen) for imaging on a Keyence microscope. Quantification for plaques and tangles were carried out in ImageJ software, where the number of positive cells were tabulated in cortical regions and normalized to the total area of each region counted. Overall, the quantification resulted in skewed data (Kolmogorov-Smironov test), so we took the square root of the raw numbers to avoid violation of the normality assumption in subsequent statistical analyses. All regions were quantified in the same mice; thus we have multiple measurements per mouse.
BDNF measurements
For RNA extraction, temporal cortical pieces were first homogenized in Trizol (50 mg/mL) and spun at 12,000×g for 5 min at 4°C. To separate out the RNA, chloroform was added at 1/3 of the supernatant volume into a phase lock gel heavy separation column and spun at 12,000×g for 5 min at 4°C. The fraction containing RNA was removed and precipitated out with isopropanol. This RNA was rinsed with ethanol and resuspended in RNAase-free water containing 1% DNAse. RNA purity was quantified via nanodrop. 1μL of the RNA was subsequently converted to cDNA in 10μM oligo primers, 1x reverse transcriptase, and 10 mM dNTPs at 42°C for 1 h and 85°C for 5 min. For qPCR, pan-BDNF (Mm04239607_s1) and GADPH (Mm99999915_g1) primers from the TaqMan gene expression assay were utilized in accordance with the given protocol. The total reaction volume was 20μL (2μL cDNA, 10μL 2x Taqman Master Mix 1μL primer, 7μL nuclease free water) in a 96-well plate with coverfilm on a real-time qPCR cycler (Stratagene). qPCR was performed on these samples in the following phases: activation for 10 min at 95°C, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min. Standard curves and cycle threshold (Ct) were generated using standards obtained from total mouse brain RNA and the ddCt–2 method [30], using GADPH as the housekeeping gene, was utilized to determine relative gene expression differences between treatment and sham conditions.
Acetylcholine assays
The frontal cortex was homogenized in saline containing 0.1% tween at 20 mg/400μL (w/v) and spun at 5000×g for 30 min. The supernatant was used for both the Ellman’s acetylcholinesterase assay (ABCAM) and the choline acetyltransferase (ChAT) assay (AntibodiesOnline) in accordance with the instructions provided by the manufacturers. The Ellman’s assay was run in duplicate, where samples were mixed with acetylthiocholine and DNTP for 30 min before reading. Activity of AChE was calculated in comparison with a set of standards ranging from 1 mU/mL to 1000 mU/mL. Samples for ChAT were also run in duplicate using ELISA-based approach and compared to standards ranging from 0.78 ng/mL to 50 ng/mL. Measured ChAT and AChE levels were normalized to bicinchonic acid (BCA) as measured via a BCA assay kit according to the manufacturer’s instructions (Pierce BCA Protein Assay Kit, ThermoFisher).
RESULTS
3xTgAD model evaluation
Given that only female mice were utilized, we checked for an association between estrus cycle and our variables of interest. The estrus cycle stages were not associated with experimental condition (p = 0.499) nor any of the variables of interest (ChAT: p = 0.436, AChE: p = 0.915 BDNF qPCR: p = 0.747), so controlling for this variable was deemed unnecessary and thus was excluded from analysis.
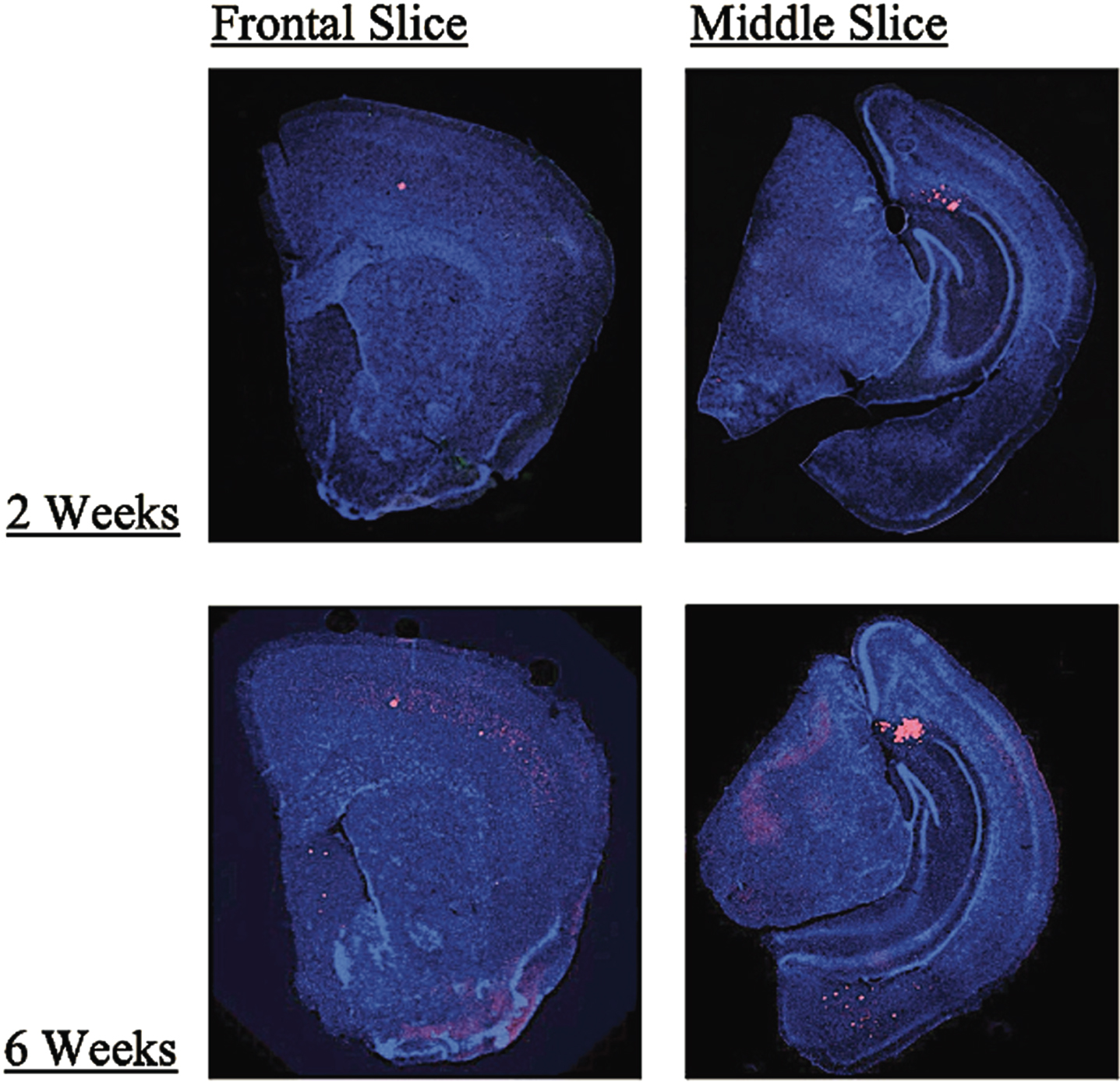
Additionally, we evaluated all our 3xTgAD mice for plaque and tangle load to ensure the model correctly exhibited the AD phenotype. Stain quantification for the dorsal cortex, ventral cortex, septum, hippocampus, amygdala, and subiculum for each mouse revealed that the 3xTgAD mice did develop both plaques and tangles in the expected regions. Quantification revealed there were no consistent differences between rTMS and sham groups for both treatment conditions. However, multivariate analysis showed an overall increase in plaque load for the 6-week group (M = 7.94, SD = 3.40) when compared to the 2-week stimulation group (M = 6.02, SD = 3.64), presumably because of the age difference (p < 0.0005, Pillai’s Trace = 0.50). Further investigation found that although there was a trend toward significance in the dorsal cortex (p = 0.068, etasq = 0.08) this result was driven by a significant effect in the septum (2 weeks: M = 0.60, SD = 0.57; 6 weeks: M = 4.04, SD = 2.79; p < 0.0001, etasq = 0.48). Figure 1 provides an example of the plaque accumulation in these mice in the 2-week and 6-week groups. No significant results were found for the tau quantification in any region.
Fig. 1
Examples front and midbrain coronal slices of 3xTgAD mice taken at 2x magnification in the 2-week and 6-week treatment groups showing greater Aβ plaque accumulation (red) in the 6-week group as compared to the 2-week group (blue = DAPI, red = Aβ). The largest change in plaque load found in the septum as shown in the frontal slices.
We measured BDNF gene expression and cholinergic function on a small number of naïve mice to measure a difference between the AD model and the B6 controls at baseline. BDNF gene expression for the naïve 3xTgAD mice was downregulated relative to the B6 mice (ddCT2 = 0.73, SD = 0.19). Additionally, 3xTgAD naive mice showed and overall lower level of ChAT (B6: M = 0.49, SD = 0.09; 3xTgAD: M = 0.37, SD = 0.05) and AChE (B6: M = 8.51, SD = 4.59; 3xTgAD: M = 7.75, SD = 4.15). These differences were not statistically significant due to the small sample size (n = 4 per group) of this preliminary data, but is consistent with the literature [9, 14, 31, 32]. Given the findings that our mice showed a trend for a loss of function that is consistent with previous findings and thus a suitable model, we assessed for differences between sham and rTMS in full experiment groups to determine if stimulation may help boost such deficits.
BDNF gene expression in sham versus rTMS conditions
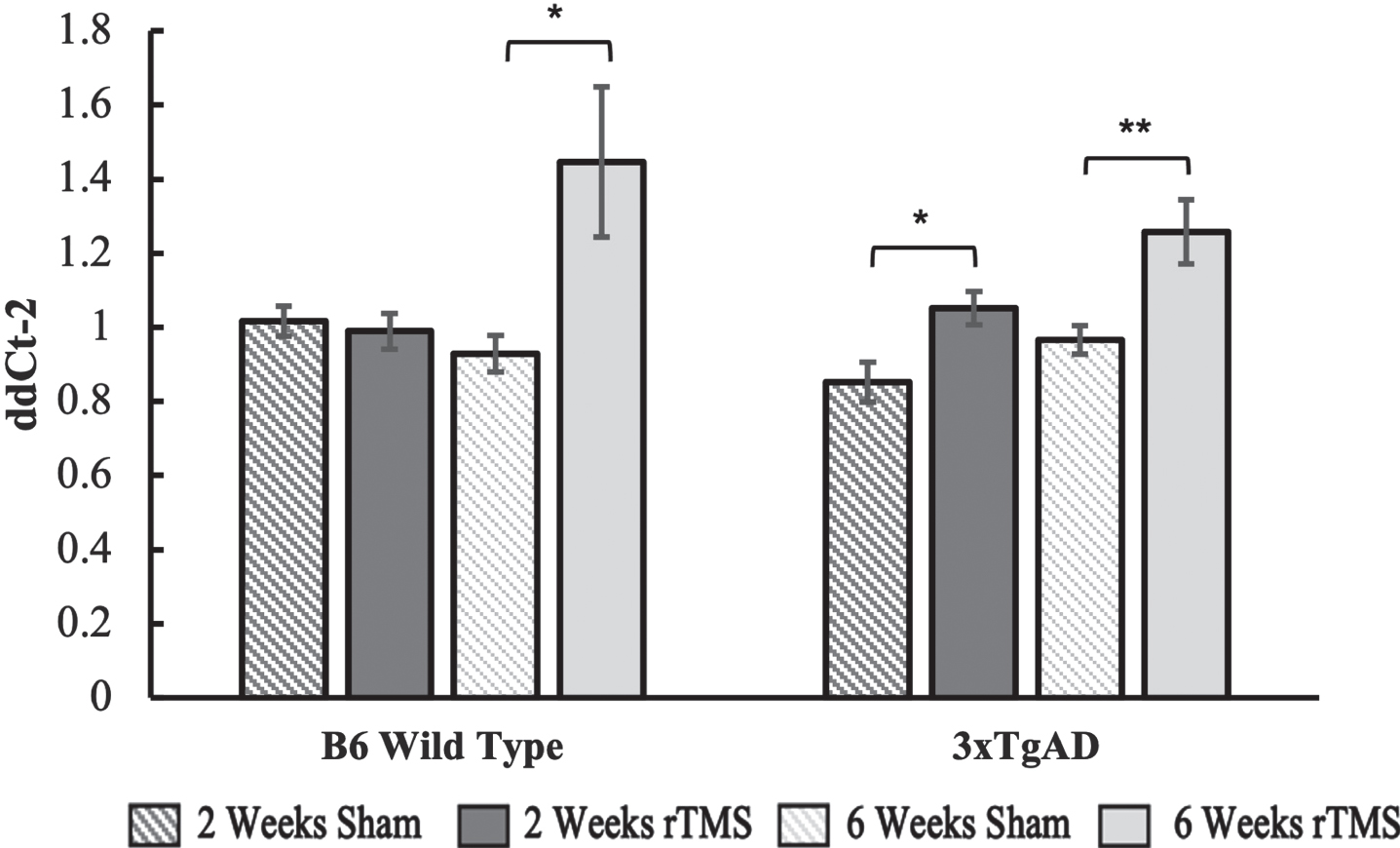
BDNF gene expression remained unchanged in the B6 group that received 2 weeks of daily rTMS treatment but was significantly upregulated in the 6-week rTMS group as compared to sham (p = 0.045). For the 3xTgAD group, there was an upregulation in both the 2-week (p = 0.048) and 6-week group (p = 0.007), with a trend towards a higher fold expression change in the 6-week group (Fig. 2).
Fig. 2
BDNF gene expression calculated using the ddCt2 method of the 2-week and 6-week groups for the B6 and 3xTgAD groups. Error bars represent the standard error; *p < 0.05, **p < 0.01. Sample size per group are as follows: B6 rTMS 2 weeks = 15, B6 Sham 2 weeks = 13, B6 rTMS 6 weeks = 14, B6 Sham 6 weeks = 12 3xTgAD rTMS 2 weeks = 12, 3xTgAD Sham 2 weeks = 12, 3xTgAD rTMS 6 weeks = 14, 3xTgAD Sham 6 weeks = 11.
Cholinergic function in sham versus rTMS conditions
There was a significantly lower level of AChE activity for the 3xTgAD 6-week sham group compared to the B6 sham group (p = 0.02), while the 2-week group was trending in the same direction (p = 0.18). This is consistent with the notion that there is a deficit in AChE in the 3xTgAD mice. No differences were found for the ChAT sham groups when comparing 3xTgAD and B6 mice for two (p = 0.83) and six weeks (p = 0.22).
Associations between BDNF gene expression and cholinergic function
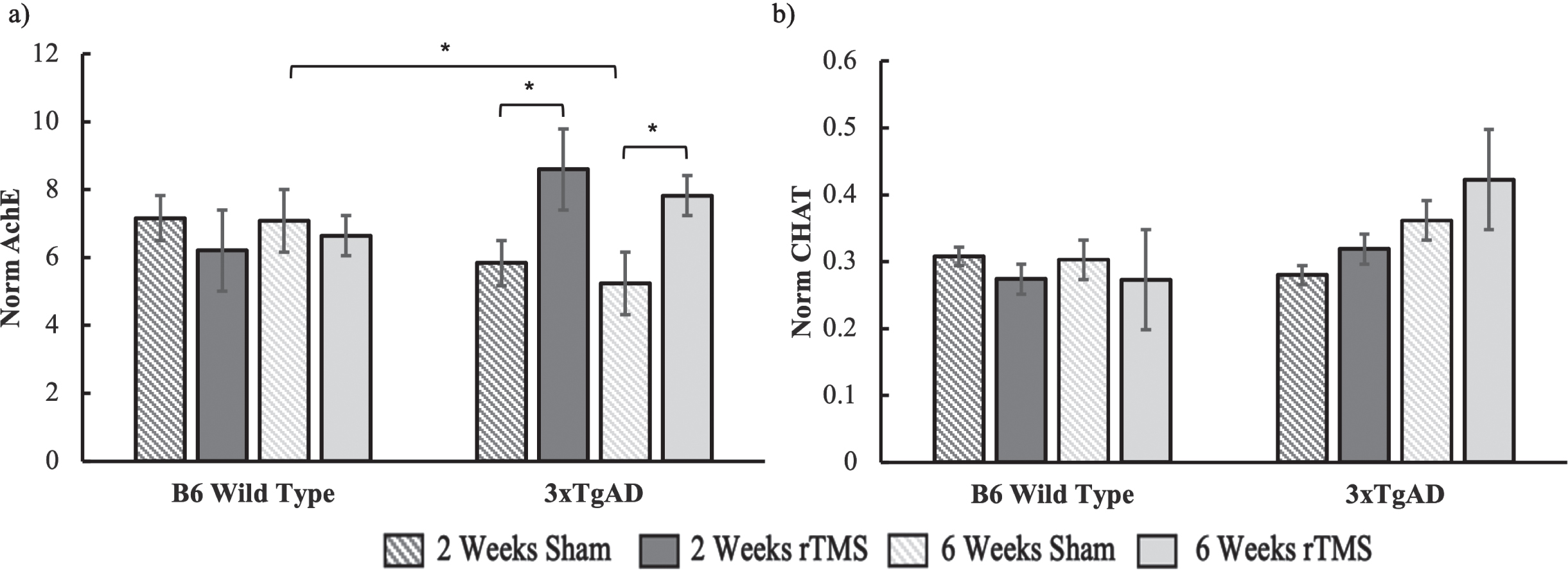
Under the notion that increased BDNF gene expression could be related to improved cholinergic functioning, we ran separate ANOVAs for the 2-week and 6-week groups on our acetylcholine measurements. For the AChE data (Fig. 3a), there was a significant interaction between condition and genotype for 2 weeks (F(1,46) = 5.44, p = 0.02), while the effect in the 6-week group was marginally significant (F(1,42) = 4.829, p = 0.06). These interactions reflected an increase in AChE activity in the 3xTgAD group who received rTMS as compared to sham, while there were no differences in the B6 groups (Fig. 3). There was no change in ChAT expression with brain stimulation in the 2-week group (all Fs < 1). The 6-week group also had no significant effects (all Fs < 1). Although the mean ChAT levels did show a trend for an increase in ChAT activity for the 3xTgAD group who received stimulation relative to sham, this was not significant (Fig. 3b).
Fig. 3
a) AChE and b) ChAT activity for both the B6 and 3xTgAD mice in the 2-week and 6-week groups. Error bars represent the standard error; *p < 0.05. Sample size per group are as follows: B6 rTMS 2 weeks = 15, B6 Sham 2 weeks = 13, B6 rTMS 6 weeks = 14, B6 Sham 6 weeks = 12 3xTgAD rTMS 2 weeks = 12, 3xTgAD Sham 2 weeks = 12, 3xTgAD rTMS 6 weeks = 14, 3xTgAD Sham 6 weeks = 11.
To further test if there is an association between BDNF and cholinergic signaling, we ran a simple correlation between the qPCR and AChE data. First, the values for the AChE rTMS groups were normalized to the mean of the B6 sham group to allow for a comparison with the calculated ddCt2 (Fig. 4a). The Pearson coefficient was significant in the positive direction (r = 0.32, p = 0.02). Including both genotype and stimulation time into a linear model did not alter the significance of the relationship between BDNF gene expression and AChE activity (p = 0.02), while both genotype (p = 0.83) and treatment time (p = 0.86) did not significantly contribute to the variance. Thus, regardless of genotype and treatment regimen, we found a positive relationship in the biochemical response to treatment for BDNF gene expression and AChE activity.
Fig. 4
a) Scatterplot showing the relationship between BDNF gene expression and AChE activity normalized to sham in response to rTMS treatment for both genotypes and treatment conditions. b) AChE activity normalized to total protein as measured via bicinchonic acid (BCA) for 3xTgAD mice (n = 4) treated with either the TrkB blocker ANA12 or Vehicle 2 hrs before rTMS or Sham daily for two weeks.
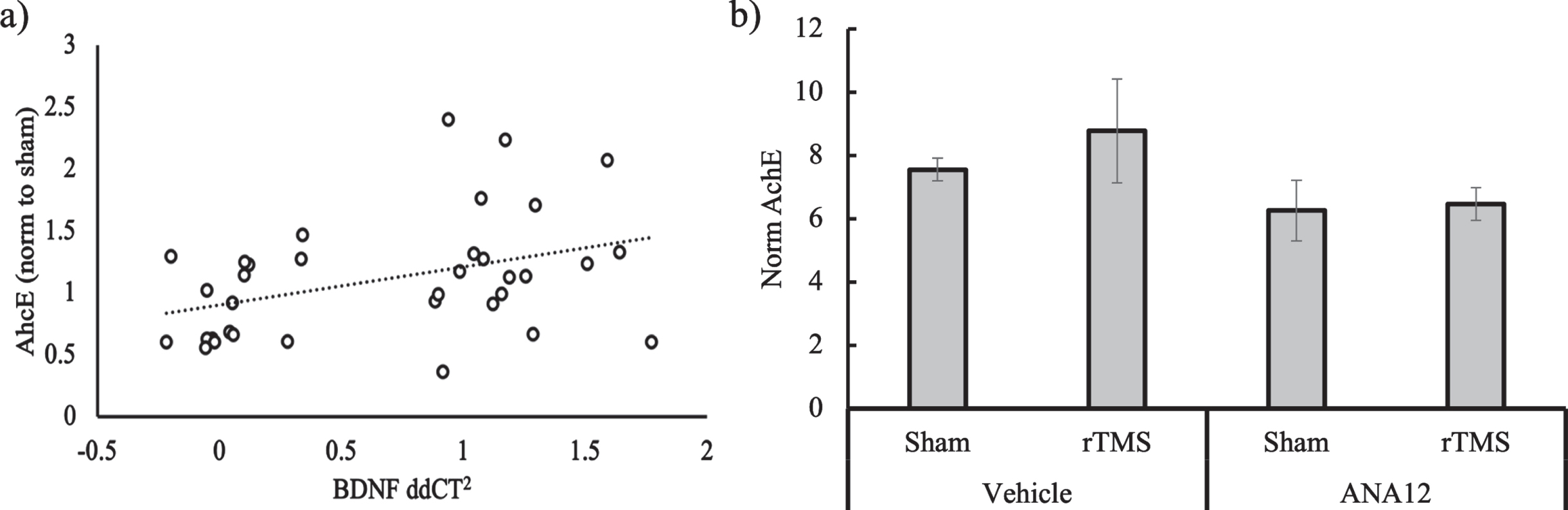
Finally, we measured AChE on the 3xTgAD mice who were injected with ANA12 or vehicle control before rTMS or sham treatment. The 3xTgAD mouse model was chosen because it showed a significant effect of rTMS on AChE as well as BDNF. With a small sample size (n = 4), no effects were significant, but overall, the ANA12 was associated with lower AChE levels as compared to the vehicle (Fig. 4b). There a was modest increase in AChE at 120% of sham in the Vehicle condition, but no difference between sham and rTMS in the ANA12 condition (103% of sham).
DISCUSSION
This study aimed to investigate if rTMS can be a useful treatment tool for AD through improving BDNF and cholinergic signaling. Analysis of cortical samples showed that rTMS does increase BDNF gene expression and AChE activity, regardless of the treatment schedule. For the B6 mice, rTMS did increase BDNF gene expression but only in the group that received two treatments a week for 6 weeks, and not in the group that received daily treatments for two weeks. Additionally, we found a correlation between BDNF gene expression and AChE in response to treatment, which was not modulated by genotype or treatment timing, while blocking the BDNF TrkB receptor trended towards a reduction in the AChE response to rTMS. Although future research is needed to solidify these findings, these data argue in favor for rTMS as a therapeutic option for AD, as it can increase BDNF gene expression and AChE activity, both which are related to cognitive decline in AD [33, 34].
BDNF has previously been shown to be a major factor in the effects of rTMS and is an important signaling component in cellular health and plasticity [35]. It has been shown to play a vital role in the health of cholinergic neurons, which degrade early in the onset of AD [36]. Although this study does not determine the specific pathway between rTMS, AChE, and BDNF, it does establish an important relationship that should be investigated further for mitigating the loss of neurons in AD. A noninvasive nonpharmaceutical alternative to AD treatment that can boost neurotransmitter signaling and, in turn, cognitive functioning would be critically important in improving the quality of life for patients.
Although we found an increase in cortical AChE activity, we did not find a concurrent significant increase in ChAT. This is consistent with the notion that rTMS increases acetylcholine levels, although future research should more directly measure changes in acetylcholine with brain stimulation. A larger sample size may be able to uncover a more consistent relationship between ChAT and rTMS. Indeed, previous studies have shown that AChE has higher activity levels than ChAT in rodents [37], so a larger sample size may be needed to uncover a more subtle effect. However, increasing acetylcholine levels by modulating AChE rather than ChAT is the main target of most AD drugs [38], as this enzyme is more abundant, while ChAT requires a regular supply of choline [39]. Therefore, our findings of increased AChE and a trend of increase ChAT remains an important effect of rTMS.
The association between AChE and BDNF in terms of brain stimulation is a novel finding but is well aligned with other potential AD treatments. For example, vitamin D and apelin-13 have been found to regulate AChE through the actions of BDNF in AD [40, 41]. Indeed, similarly to the present study, previous research has shown a correlation between cortical AChE and BDNF [40], and is in alignment with the notion that BDNF is important for cholinergic health [9]. Data from the current study also show that blocking the action of BDNF does has a negative effect on AChE activity. These results must be taken with caution given the small sample size and lack of statistical significance, but future studies will hopefully further elucidate this relationship. Therefore, methods that boost cellular health and plasticity through BDNF will likely lead to improved brain function in AD patients. One primary advantage for rTMS is it is less invasive and likely has fewer side effects than any pharmaceutical intervention.
The current study begins to demonstrate that a longer temporal allocation of treatment sessions is a viable option and has interesting implications for treatment regimens for human clinical trials. We recognize that the daily treatment group overall received two more treatment sessions than the 6-week group (14 versus 12), but our significant and positive results shed light on the importance of timing. Given the discrepancy in total treatment sessions, the current study is limited in its ability to directly and comprehensively compare the two treatment timings here, but the results from the 6-week group provide promising evidence that different timing options may be more efficacious. This opens the door for a dialogue related to treatment regimens in patients with AD or other diseases in terms of reducing cognitive decline and delaying the onset of late-stage AD. The next relevant step would be to test for behavioral or memory performance differences between a daily treatment, a twice weekly treatment, or even another periodic timeframe. Six weeks is relatively long in the mouse lifespan of approximately 2 years or less in the 3xTgAD model [42]. Given how important it is to delay the conversion from MCI to AD, future research should determine if rTMS could be used as a maintenance treatment once a week, or less often, to reduce decline and chances of conversion from MCI to AD.
Overall, this data provides encouraging evidence for the use of rTMS as a therapeutic tool for neurodegeneration. Increased cortical BDNF and acetylcholine signaling will likely be accompanied by improved neuroplasticity [43, 44] and even cognitive functioning [45]. The next steps include solidifying the biochemical chain of activation between BDNF and AChE and relating this to behavioral measurements. Additionally, the systematic investigation of the optimal treatment regimen, in terms of timing, location, and amplitude is very much needed [46, 47]. The combination of basic research, such as the current study, with clinical trials aimed at answering these issues will result in heightened treatment efficacy and thus the improvement in patient quality of life.
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Jennifer Roger from the University of Western Australia for guidance with the rTMS mouse model and Dr. Steven Ginsberg for guidance with the qPCR methodology. This project was funded by VA BLR&D 5IK2BX004105.
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States Government.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-5361r1).
REFERENCES
[1] | Gammon K ((2014) ) Neurodegenerative disease: Brain windfall. Nature 515: , 299–300. |
[2] | Epperly T , Dunay MA , Boice JL ((2017) ) Alzheimer disease: Pharmacologic and nonpharmacologic therapies for cognitive and functional symptoms. Am Fam Physician 95: , 771–778. |
[3] | Magierski R , Sobow T , Schwertner E , Religa D ((2020) ) Pharmacotherapy of behavioral and psychological symptoms of dementia: State of the art and future progress. Front Pharmacol 11: , 1168. |
[4] | Zissimopoulos J , Crimmins E , St.Clair P ((2015) ) The value of delaying alzheimer’s disease onset. Forum Heal Econ Policy 18: , 25–39. |
[5] | Birks J ((2006) ) Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 25: , CD005593. |
[6] | Bodick NC , Offen WW , Levey AI , Cutler NR , Gauthier SG , Satlin A , Shannon HE , Tollefson GD , Rasmussen K , Bymaster FP , Hurley DJ , Potter WZ , Paul SM ((1997) ) Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol 54: , 465–473. |
[7] | Sabri O , Meyer PM , Gräf S , Hesse S , Wilke S , Becker GA , Rullmann M , Patt M , Luthardt J , Wagenknecht G , Hoepping A , Smits R , Franke A , Sattler B , Tiepolt S , Fischer S , Deuther-Conrad W , Hegerl U , Barthel H , Schönknecht P , Brust P ((2018) ) Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain 141: , 1840–1854. |
[8] | Tamilselvan M , Tamilanban T , Chitra V ((2020) ) Unfolding remedial targets for Alzheimer’s disease. Res J Pharm Technol 13: , 3021–3027. |
[9] | Fahnestock M , Garzon D , Holsinger RMD , Michalski B ((2002) ) Neurotrophic factors and Alzheimer’s disease: Are we focusing on the wrong molecule? J Neural Transm Suppl, pp. 241–252. |
[10] | Aarons T , Bradburn S , Robinson A , Payton A , Pendleton N , Murgatroyd C ((2019) ) Dysregulation of BDNF in prefrontal cortex in Alzheimer’s disease. J Alzheimers Dis 69: , 1089–1097. |
[11] | Francis BM , Kim J , Barakat ME , Fraenkl S , Yücel YH , Peng S , Michalski B , Fahnestock M , McLaurin JA , Mount HTJ ((2012) ) Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging 33: , 555–563. |
[12] | Cunha C , Brambilla R , Thomas KL ((2010) ) A simple role for BDNF in learning and memory? Front Mol Neurosci 3: , 1. |
[13] | Blurton-Jones M , Kitazawa M , Martinez-Coria H , Castello NA , Müller FJ , Loring JF , Yamasaki TR , Poon WW , Green KN , LaFerla FM ((2009) ) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106: , 13594–13599. |
[14] | Iulita MF , Bistué Millón MB , Pentz R , Aguilar LF , Do Carmo S , Allard S , Michalski B , Wilson EN , Ducatenzeiler A , Bruno MA , Fahnestock M , Cuello AC ((2017) ) Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol Dis 108: , 307–323. |
[15] | Cotelli M , Manenti R , Cappa SF , Zanetti O , Miniussi C ((2008) ) Transcranial magnetic stimulation improves naming in Alzheimer disease patients at different stages of cognitive decline. Eur J Neurol 15: , 1286–1292. |
[16] | Cotelli M , Manenti R , Cappa SF , Geroldi C , Zanetti O , Rossini PM , Miniussi C ((2006) ) Effect of transcranial magnetic stimulation on action naming in patients with Alzheimer disease. Arch Neurol 63: , 1602–1604. |
[17] | Drumond Marra HL , Myczkowski ML , Maia Memória C , Arnaut D , Leite Ribeiro P , Sardinha Mansur CG , Lancelote Alberto R , Boura Bellini B , Alves Fernandes Da Silva A , Tortella G , Ciampi De Andrade D , Teixeira MJ , Forlenza OV , Marcolin MA ((2015) ) Transcranial magnetic stimulation to address mild cognitive impairment in the elderly: A randomized controlled study. Behav Neurol 2015: , 287843. |
[18] | Chervyakov AV , Chernyavsky AY , Sinitsyn DO , Piradov MA ((2015) ) Possible mechanisms underlying the therapeutic effects of transcranial magnetic stimulation. Front Hum Neurosci 9: , 303. |
[19] | Shang Y , Wang X , Shang X , Zhang H , Liu Z , Yin T , Zhang T ((2016) ) Repetitive transcranial magnetic stimulation effectively facilitates spatial cognition and synaptic plasticity associated with increasing the levels of BDNF and synaptic proteins in Wistar rats. Neurobiol Learn Mem 134: , 369–378. |
[20] | Zhang N , Xing M , Wang Y , Tao H , Cheng Y ((2015) ) Repetitive transcranial magnetic stimulation enhances spatial learning and synaptic plasticity via the VEGF and BDNF-NMDAR pathways in a rat model of vascular dementia. Neuroscience 311: , 284–291. |
[21] | Ma J , Zhang Z , Kang L , Geng D , Wang Y , Wang M , Cui H ((2014) ) Repetitive transcranial magnetic stimulation (rTMS) influences spatial cognition and modulates hippocampal structural synaptic plasticity in aging mice. Exp Gerontol 58: , 256–268. |
[22] | Zhang XQ , Li L , Huo JT , Cheng M , Li LH ((2018) ) Effects of repetitive transcranial magnetic stimulation on cognitive function and cholinergic activity in the rat hippocampus after vascular dementia. Neural Regen Res 13: , 1384–1389. |
[23] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 39: , 409–421. |
[24] | Madore MR , Poh E , Bollard SJ , Rivera J , Taylor J , Cheng J , Booth E , Nable M , Heath A , Yesavage J , Rodger J , McNerney MW ((2021) ) Moving back in the brain to drive the field forward: Targeting neurostimulation to different brain regions in animal models of depression and neurodegeneration. J Neurosci Methods 360: , 109261. |
[25] | Xiu MH , Guan HY , Zhao JM , Wang KQ , Pan YF , Su XR , Wang YH , Guo JM , Jiang L , Liu HY , Sun SG , Wu HR , Geng HS , Liu XW , Yu HJ , Wei BC , Li XP , Trinh T , Tan SP , Zhang XY ((2020) ) Cognitive enhancing effect of high-frequency neuronavigated rTMS in chronic schizophrenia patients with predominant negative symptoms: A double-blind controlled 32-week follow-up study. Schizophr Bull 46: , 1219–1230. |
[26] | Cotelli M , Calabria M , Manenti R , Rosini S , Zanetti O , Cappa SF , Miniussi C ((2011) ) Improved language performance in Alzheimer disease following brain stimulation. J Neurol Neurosurg Psychiatry 82: , 794–797. |
[27] | Rodger J , Mo C , Wilks T , Dunlop SA , Sherrard RM ((2012) ) Transcranial pulsed magnetic field stimulation facilitates reorganization of abnormal neural circuits and corrects behavioral deficits without disrupting normal connectivity. FASEB J 26: , 1593–606. |
[28] | Tang AD , Makowiecki K , Bartlett C , Rodger J ((2015) ) Low intensity repetitive transcranial magnetic stimulation does not induce cell survival or regeneration in a mouse optic nerve crush model. PLoS One 10: , e0126949. |
[29] | McClintock SM , Reti IM , Carpenter LL , McDonald WM , Dubin M , Taylor SF , Cook IA , O’Reardon J , Husain MM , Wall C , Krystal AD , Sampson SM , Morales O , Nelson BG , Latoussakis V , George MS , Lisanby SH ((2018) ) Consensus recommendations for the clinical application of repetitive transcranial magnetic stimulation (rTMS) in the treatment of depression. J Clin Psychiatry 79: , 35–48. |
[30] | Livak KJ , Schmittgen TD ((2001) ) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: , 402–408. |
[31] | Caccamo A , Maldonado MA , Bokov AF , Majumder S , Oddo S ((2010) ) CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 107: , 22687–22692. |
[32] | Hu Y , Chen X , Huang S , Zhu Q , Yu S , Shen Y , Sluiter A , Verhaagen J , Zhao J , Swaab D , Bao A ((2019) ) Early growth response-1 regulates acetylcholinesterase and its relation with the course of Alzheimer’s disease. Brain Pathol 29: , 502–512. |
[33] | Beeri MS , Sonnen J ((2016) ) Brain BDNF expression as a biomarker for cognitive reserve against Alzheimer disease progression. Neurology 86: , 702–703. |
[34] | Kuhl DE , Koeppe RA , Minoshima S , Snyder SE , Ficaro EP , Foster NL , Frey KA , Kilbourn MR ((1999) ) In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer’s disease. Neurology 52: , 691–699. |
[35] | Lu B , Nagappan G , Lu Y ((2015) ) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220: , 223–250. |
[36] | Hampel H , Mesulam MM , Cuello AC , Farlow MR , Giacobini E , Grossberg GT , Khachaturian AS , Vergallo A , Cavedo E , Snyder PJ , Khachaturian ZS ((2018) ) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141: , 1917–1933. |
[37] | Amenta F , Bronzetti E , Mancini M , Vega J , Zaccheo D ((1994) ) Choline acetyltransferase and acetylcholinesterase in the hippocampus of aged rats: Sensitivity to choline alphoscerate treatment. Mech Ageing Dev 74: , 47–58. |
[38] | Desmarais JE , Gauthier S ((2010) ) Alzheimer disease: Clinical use of cholinergic drugs in Alzheimer disease. Nat Rev Neurol 6: , 247–253. |
[39] | Stanciu GD , Luca A , Rusu RN , Bild V , Chiriac SIB , Solcan C , Bild W , Ababei DC ((2020) ) Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 10: , 40. |
[40] | Khairy EY , Attia MM ((2021) ) Protective effects of vitamin D on neurophysiologic alterations in brain aging: Role of brain-derived neurotrophic factor (BDNF). Nutr Neurosci 24: , 650–659. |
[41] | Luo H , Xiang Y , Qu X , Liu H , Liu C , Li G , Han L , Qin X ((2019) ) Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front Pharmacol 10: , 395. |
[42] | Kane AE , Shin S , Wong AA , Fertan E , Faustova NS , Howlett SE , Brown RE ((2018) ) Sex differences in healthspan predict lifespan in the 3xTg-AD Mouse model of Alzheimer’s disease. Front Aging Neurosci 10: , 172. |
[43] | Knipper M , da Penha Berzaghi M , Blöchl A , Breer H , Thoenen H , Lindholm D ((1994) ) Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci 6: , 668–671. |
[44] | Maurer SV , Williams CL ((2017) ) The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front Immunol 8: , 1489. |
[45] | Ko YH , Kwon SH , Lee SY , Jang CG ((2019) ) Isoorientin improves scopolamine-induced cognitive impairments by restoring the cholinergic system, antioxidant defense, and p-CREB/BDNF signaling in the hippocampus and frontal cortex. Arch Pharm Res 42: , 722–731. |
[46] | Heath A , Taylor JL , McNerney MW ((2018) ) rTMS for the treatment of Alzheimer’s disease: Where should we be stimulating? Expert Rev Neurother 18: , 903–905. |
[47] | Taylor JL , Hambro BC , Strossman ND , Bhatt P , Hernandez B , Ashford JW , Cheng JJ , Iv M , Adamson MM , Lazzeroni LC , McNerney MW ((2019) ) The effects of repetitive transcranial magnetic stimulation in older adults with mild cognitive impairment: A protocol for a randomized, controlled three-arm trial. BMC Neurol 19: , 326. |


